

Abstract
Background:
Total serum IgE (tIgE) is an important intermediate phenotype of allergic disease. Whole genome genetic association studies across ancestries may identify important determinants of IgE.
Objective:
By leveraging data from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program, the Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA) and the Atopic Dermatitis Research Network (ADRN), we aim to increase understanding of genetic variants affecting tIgE production across the ancestry and allergic disease spectrum (N=21,901).
Methods:
We performed genome-wide association within strata of study, disease, and ancestry groups, and combined results via a meta-regression approach that models heterogeneity attributable to ancestry. We also tested for association between HLA alleles called from whole genome sequence data and tIgE, assessing replication of associations in HLA alleles called from genotype array data. For details, please see the Methods section in this article’s Online Repository at www.jacionline.org.
Results:
We identified six loci at genome-wide significance (P<5×10−9), including four loci previously reported as genome-wide significant for tIgE, as well as new regions in chr11q13.5 and chr15q22.2, also identified in prior GWAS of atopic dermatitis and asthma. In the HLA allele association study, HLA-A*02:01 was associated with decreased tIgE (discovery P = 2×10−4, replication P = 5×10−4, discovery+replication P=4×10−7) and HLA-DQB1*03:02 was strongly associated with decreased tIgE in Hispanic/Latino ancestry populations (Hispanic/Latino discovery+replication P=8×10−8).
Conclusion:
We performed the largest GWAS and HLA association study of tIgE focused on ancestrally diverse populations and found several known tIgE and allergic disease loci that are relevant in non-European ancestry populations.
Keywords: total serum IgE, human leukocyte antigen, genome-wide association study, atopic dermatitis, asthma, multi-ethnic
Capsule Summary:
Known tIgE and allergic disease loci are relevant in non-European ancestry populations. HLA-A*02:01 and HLA-DQB1*03:02 are associated with decreased levels of tIgE.
Introduction
Total serum IgE (tIgE) is an important marker of atopy, estimated to affect between 27–36% of individuals living in developed countries1, 2. Total IgE is highly elevated in individuals suffering from atopic diseases such as asthma and is considered a risk factor for this disease. While several tIgE genome-wide association studies (GWAS) have been reported3–7, these have mostly been limited to European ancestry populations. The largest GWAS to date included 14,745 European ancestry samples5, and the only multi-ethnic GWAS to date included 4,292 samples4. Collectively, these GWAS have identified four genome-wide significant loci, with the human leukocyte antigen (HLA) region being the most consistently reported. In this study, we aimed to increase our understanding of genetic factors affecting tIgE across the ancestry and allergic disease spectrum.
Results and Discussion
We combined existing tIgE and genome-wide data from three large NIH-funded initiatives, including whole genome sequence (WGS) data from the NHLBI Trans-Omics for Precision Medicine (TOPMed) program8 and GWAS array data from the Consortium on Asthma among African-ancestry Populations in the Americas (CAAPA)9 and the Atopic Dermatitis Research Network (ADRN)10, and conducted a GWAS of tIgE in >20,000 subjects from 16 studies (23% African, 18% Hispanic/Latino, 59% European ancestry, Figure 1A). As expected, mean tIgE is higher in asthmatics and participants with atopic dermatitis (AD) compared to control groups and population-based studies, and different by ancestry group (Figure 1B, Table E1). Due to this heterogeneity of tIgE distribution, we defined GWAS strata by study, disease group, ancestry group and genotyping platform (WGS data vs. GWAS array data imputed using the TOPMed reference panel11). We performed GWAS separately for each stratum, and combined results via a meta-regression approach that models allelic effects along genetic axes of variation representative of ancestry as well as residual heterogeneity reflecting non-genetic differences between stratum (more detailed Methods in this article’s Online Repository at www.jacionline.org, Figure E3)8. This approach does not penalize effects that are ancestry-specific and quantifies heterogeneity attributable to aggregate genetic ancestry.
Figure 1: Clinical characteristics.
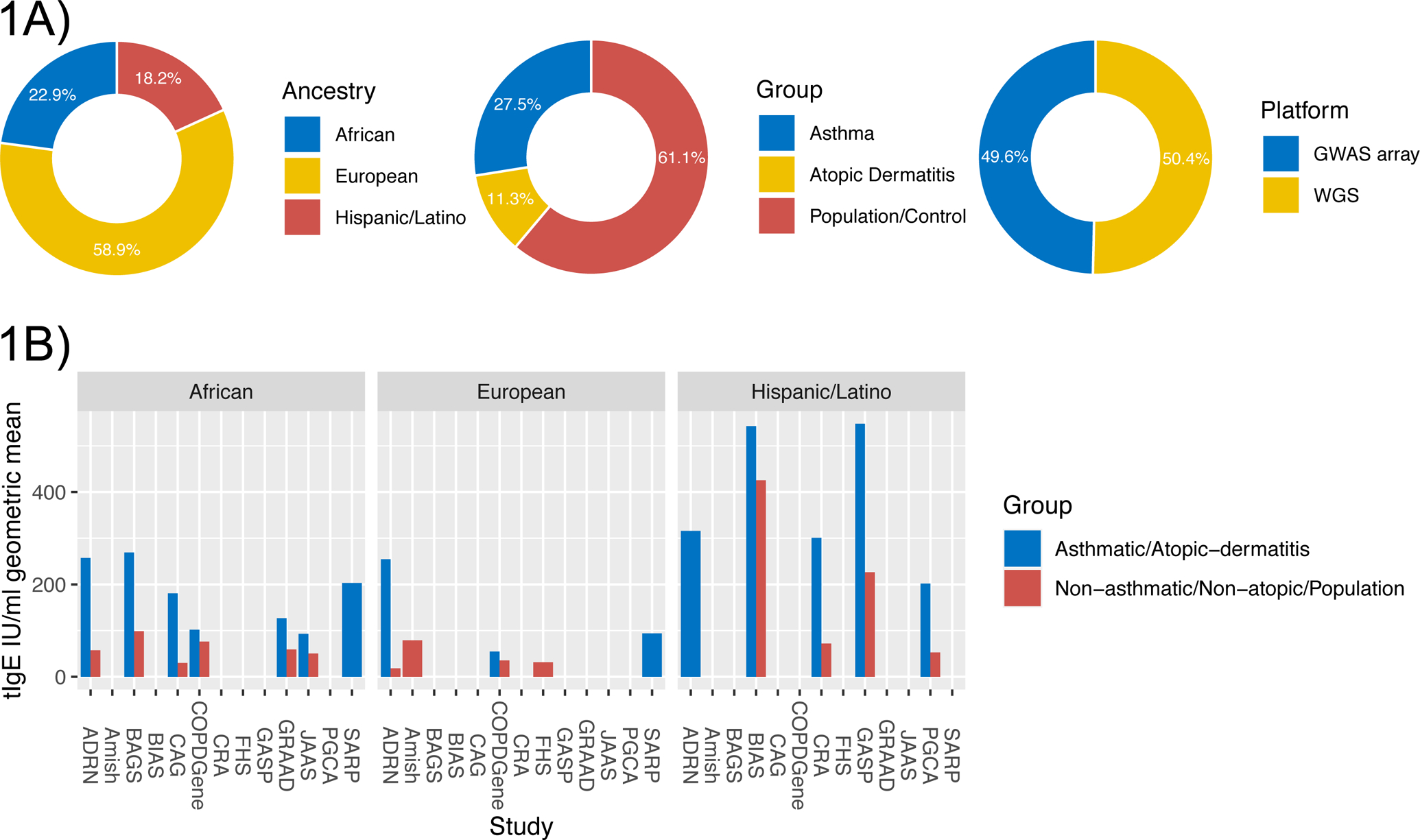
1A) Breakdown of ancestry, disease group and genotyping platform across studies. 1B) Barplot of the total serum IgE IU/ml geometric mean by study, stratified by allergic/non-allergic group, colored by ancestry.
Six loci reached genome-wide significance (P < 5×10−9 as recently recommended by Lin12, Figure 2, Table E2). All four loci previously reported as genome-wide significant by tIgE GWAS3, 5, 7 were identified, as well as two loci not previously reported in tIgE GWAS but reported by prior GWAS of AD and asthma: chr11q13.5 (EMSY+LRRC32), reported in early GWAS of AD13 and eosinophilic esophagitis14, and chr15q22.2 (RORA2+ANXA2+VPS13C), reported in an early GWAS of asthma5. Both these regions also reached genome-wide significance in a recent GWAS of asthma in the UK Biobank15. We contrast these findings with relatively weak associations observed for the epidermal differentiation complex (EDC) region and the chr17q12–21 locus, the strongest AD and asthma GWAS known genetic associations (pink dots in Figure 2). EMSY affects transcription of interferon genes16 and may shift the immune response to T helper Type 1, thereby reducing the T helper type 2 (TH2) immune response, while RORA2 regulates transcription of genes modifying TH2 cell responses17. Therefore, the chr11q13.5 (EMSY) and chr15q22.2 (RORA2) associations likely reflect changes in the TH2 immune response rather than other biological pathways conferring risk for developing allergic disease.
Figure 2: tIgE GWAS Manhattan plot.
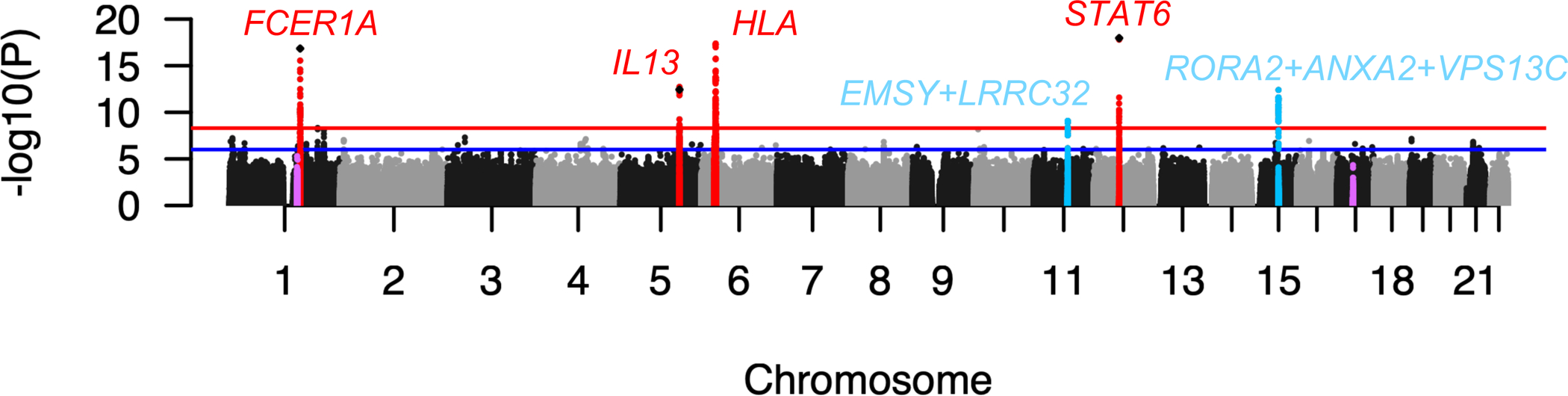
Red dots denote loci previously reported by the tIgE GWAS, light blue dots denote loci not previously reported by tIgE GWAS, and pink dots denote the chr1q21 EDC and chr17q12–21 regions. Diamonds denote lead SNPs from previous GWAS. The red line denotes genome-wide significance (P=5×10−9), the blue line denotes suggestive significance (P=10−6).
P-values for heterogeneity attributable to genetic ancestry were used to assess evidence for effects that may differ by ancestry. For 3 of the 6 genome-wide significant loci (chr1q23.2, chr11q13.5, chr12q13.3) we observed no such evidence (P_anc>0.05 in Table E2), and only marginal evidence of heterogeneity attributable to ancestry for chr15q22.2 (minimum P_anc=0.03, Table E2). Effect sizes and direction of effect are generally consistent by ancestry for the lead variants in these loci (Figure E5). Heterogeneity attributable to ancestry at the chr5q31.1 locus is likely due to lack of association in the Hispanic/Latino ancestry group and a weaker effect in the African ancestry group (Figure E5). For the chr6p21.32 HLA locus, all three ancestries contributed to the association signal, but with varying effect sizes by ancestry group (P_anc<0.05 in Table E2) and gene locus (Figures E5, E7). However, we qualify that genetic ancestry is only captured in aggregate at the analysis stratum level, the ancestry spectrum in the Hispanic/Latino group is particularly diverse (Figure E3), and our study could not account for unmeasured environmental and socio-economic factors. Therefore, we caution that loci with association signals that are heterogeneous by ancestry should not be interpreted as biological differences between ancestry groups.
In addition to genetic heterogeneity between study strata, the wide array of geographical locations and clinical settings participants were recruited from also represents heterogeneity in environmental exposures. The meta-regression approach we used to combine association results also explicitly models residual heterogeneity8, thereby - at least in part - accounting for heterogeneity in environmental exposures, reducing the risk of over and under estimation of p-values. None of the lead variants in our study showed evidence of residual heterogeneity (P_het > 0.05 in Table E2, Figure E6), and only one of the credible set genome-wide significant variants showed evidence of residual heterogeneity (rs3024971 in the chr12q1.3 STAT6 locus, P_het=0.03, Table E2), which may be due to environmental exposure difference and/or disease group. However, several genome-wide significant variants not included in the credible set did show evidence of residual heterogeneity (P_het<=0.05 in Table E3), particularly variants in the HLA locus, perhaps reflecting varying exposure to environmental allergens.
We performed co-localization analysis using gene expression data available through GTEx (multiple tissues) and eQTLGen (blood) to test for causal variants in common between tIgE and gene expression18. This analysis showed expression of FCER1A, IL13 and STAT6 may be causally related to genetic control of tIgE production (posterior probability of one common causal variant [PPH4] > 0.99, Figure 3). Although the role of the protein products of these genes in the TH2 immune response is well known (i.e., FCER1A is an IgE receptor; IL13 is a TH2 cytokine, STAT6 mediates TH2 inflammation), it has previously been speculated the chr1q23.2 association may be related to DARC3, 19 and chr5q31.1 to RAD507; our results support the notion that FCER1A and IL13 are the more likely candidates.
Figure 3: Locus zoom plots of tIgE and gene expression regions with evidence for co-localization.
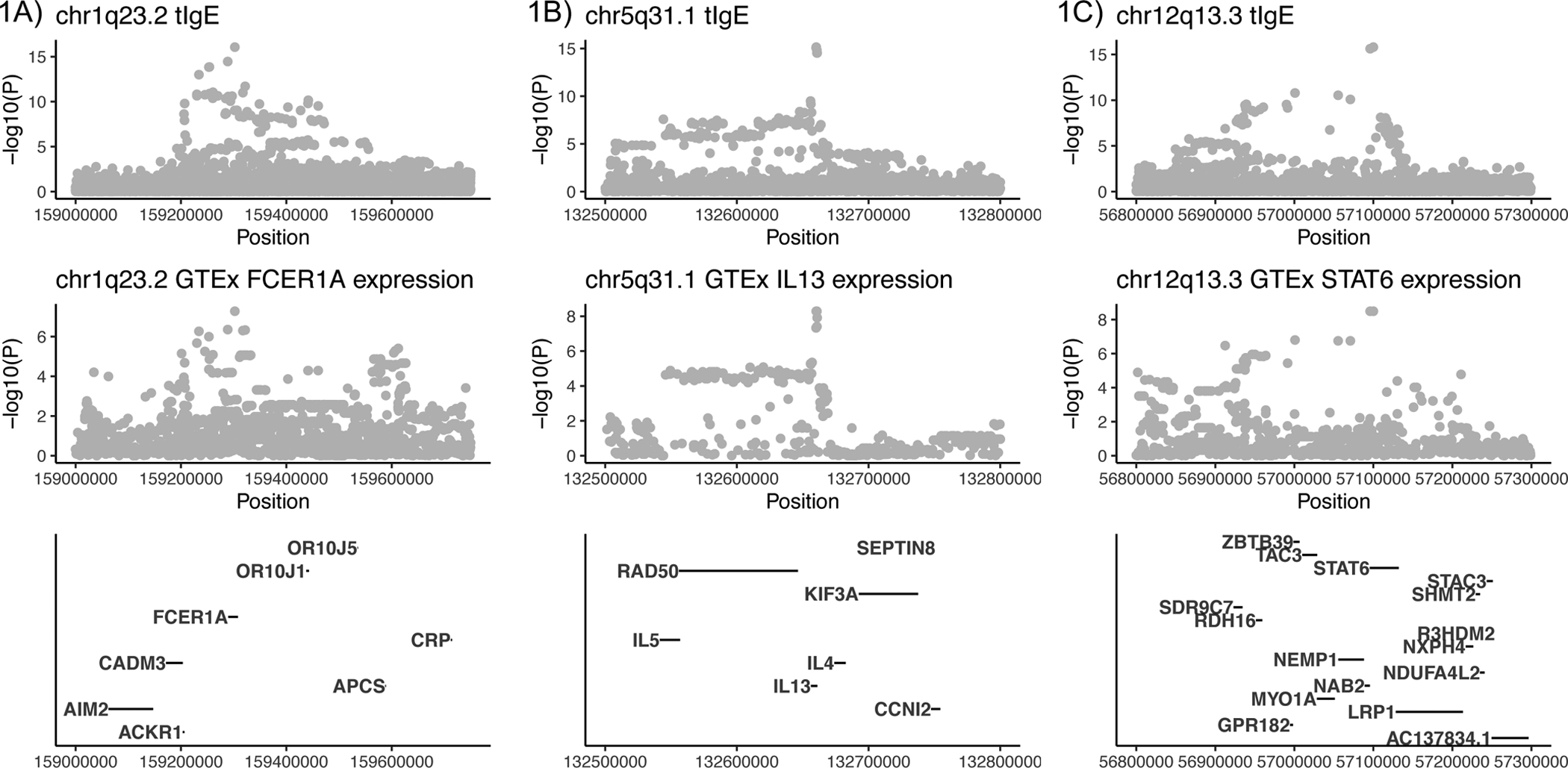
3A) FCER1A (ENSG00000179639.10) in Adipose Visceral Omentum (PPH4=0.9986), 3B) IL13 (ENSG00000169194) in Testis (PPH4=0.9961), 3C) STAT6 (ENSG00000166888) in Cultured Fibroblasts (PPH4=0.9984).
The role of the HLA locus in the TH2 immune response has been well established, but the relative importance of particular HLA genes is still a subject of debate. In our study, we observed the strongest association in the vicinity of the HLA-DRB+DQA+DQB gene region (an effect that appears relevant in all 3 ancestry groups) but note statistical signal peaks are also observed close to the HLA-A and HLA-B+C gene regions (Figure 4, Figure E7). We observed low levels of linkage disequilibrium (LD) between the HLA-A, HLA-B-C and HLA-DRB-DQA-DQB lead variants (Figure 4). Thus, these are likely independent association signals, and our finding suggests both major histocompatibility complex (MHC) class II and class I genes play a role in tIgE production.
Figure 4: Associations in the HLA locus.
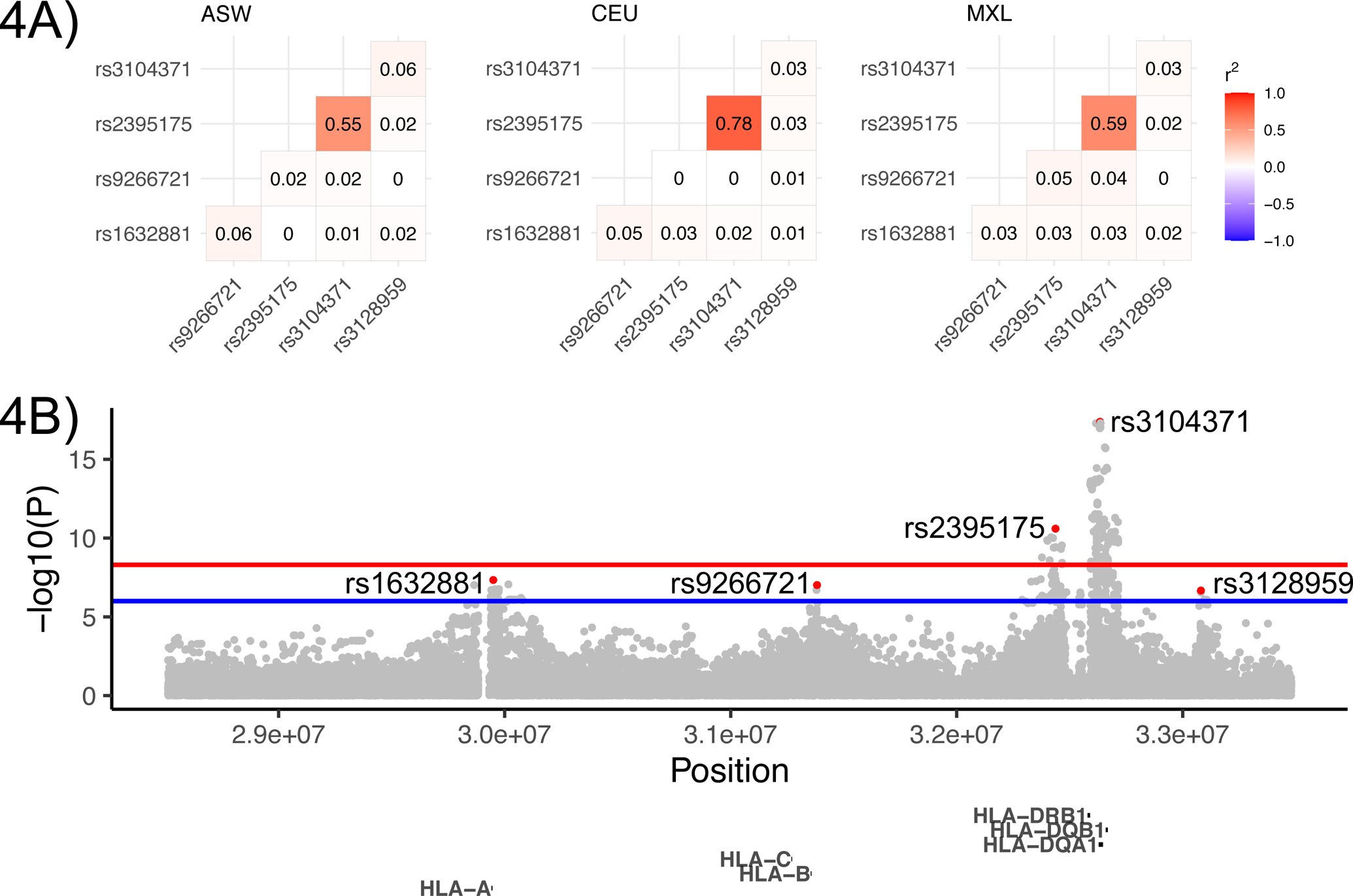
4A) Locus zoom plot. Lead variants in each peak are colored red. The red horizontal line denotes the 5×10−9 genome-wide significance threshold, the blue line denotes suggestive significance at 10−6. 4B) Linkage disequilibrium (r2) between lead variants in African (ASW), European (CEU) and Hispanic/Latino (MXL) 1000 Genomes populations.
To further investigate the role of specific HLA alleles, we performed HLA allele calling from WGS data using HLA-LA20 and from GWAS array data using HIBAG21 followed by association analysis (see Online Repository Methods). We used the TOPMed WGS data sets for discovery and GWAS array data sets for replication. In this analysis, carriers of the HLA-A*02:01 allele had lower levels of tIgE (discovery P = 2×10−4, replication P = 5×10−4, discovery+replication P=4×10−7) (Table 1, Figures E13–E14). The HLA A2 serotype has previously been reported as associated with tIgE in subjects with ragweed sensitivity22. An association with HLA-DQA1*03:01 was also replicated, but we note a consistent lower allele frequency in GWAS array data sets compared to WGS data (Figure E12), which indicates potential bias in this allele call. Due to heterogeneity by ancestry at the HLA locus in our GWAS, we also performed an exploratory analysis combining discovery and replication data sets by ancestry group to identify any additional ancestry related associations. This revealed a strong association between HLA-DQB1*03:02 and tIgE in individuals with Hispanic/Latino ancestry (P = 7×10−8, Table 1), with some evidence of replication in individuals of European ancestry (P = 0.03, Table 1, Figure E12). HLA-DQB1*03:02 was recently reported by the Genetic susceptibility to Asthma and pollution in Peru (GASP) study (one of the constituent studies in our analysis) as associated with tIgE (P = 2×10−4)23, and this association was replicated in the other Hispanic/Latino ancestry populations in our study (Hispanic/Latino meta-analysis excluding GASP P=8×10−5).
Table 1: Significant HLA allele associations.
Associations in the discovery data set with P < 0.05/155 (the number of alleles tested), replicated associations with P < 0.05, and associations in the combined discovery+replication data set with P<0.05/155 in any of the ancestry groups, are included in this table. P-values < 0.05 are in bold font.
| All | African | European | Hispanic/Latino | |||||
|---|---|---|---|---|---|---|---|---|
| Allele | Beta [95% CI] | P | Beta [95% CI] | P | Beta [95% CI] | P | Beta [95% CI] | P |
| Discovery | ||||||||
| A*02:01 | −0.06 [−0.08,−0.03] | 2×10−4 | −0.06 [−0.15,0.03] | 2×10−1 | −0.05 [−0.09,−0.02] | 6×10 −4 | ||
| DQA1*03:01 | −0.07 [−0.10,−0.03] | 4×10 −5 | −0.02 [−0.10,0.06] | 6×10−1 | −0.06 [−0.10,−0.02] | 2×10 −3 | −0.16 [−0.25,−0.07] | 3×10 −4 |
| DQA1*05:01 | 0.06 [0.03,0.09] | 7×10 −5 | 0.04 [−0.02,0.11] | 2×10−1 | 0.06 [0.02,0.09] | 2×10 −3 | 0.12 [0.03,0.20] | 1×10 −2 |
| DQB1*03:01 | 0.06 [0.03,0.09] | 3×10 −5 | 0.05 [−0.01,0.11] | 8×10−2 | 0.06 [0.03,0.09] | 5×10 −4 | 0.07 [−0.02,0.16] | 1×10−1 |
| DQB1*03:03 | −0.08 [−0.13,−0.04] | 2×10 −4 | −0.18 [−0.34,−0.02] | 2×10 −2 | −0.07 [−0.12,−0.02] | 3×10 −3 | −0.19 [−0.43,0.06] | 1×10−1 |
| DRB1*04:04 | −0.13 [−0.19,−0.07] | 1×10 −5 | 0.00 [−0.30,0.31] | 1.00 | −0.15 [−0.21,−0.09] | 3×10 −6 | −0.01 [−0.23,0.21] | 9×10−1 |
| DRB1*11:01 | 0.08 [0.04,0.12] | 2×10 −4 | 0.10 [0.02,0.19] | 2×10 −2 | 0.07 [0.02,0.12] | 6×10 −3 | 0.10 [−0.06,0.26] | 2×10−1 |
| Replication | ||||||||
| A*02:01 | −0.05 [−0.08,−0.02] | 5×10 −4 | −0.08 [−0.15,−0.00] | 4×10 −2 | −0.06 [−0.09,−0.02] | 4×10 −3 | −0.03 [−0.07,0.02] | 3×10−1 |
| DQA1*03:01 | −0.08 [−0.12,−0.05] | 4×10 −6 | −0.11 [−0.24,0.02] | 1×10−1 | −0.06 [−0.11,−0.01] | 2×10 −2 | −0.10 [−0.16,−0.05] | 1×10 −4 |
| DQA1*05:01 | 0.03 [−0.01,0.06] | 2×10−1 | −0.03 [−0.11,0.05] | 5×10−1 | 0.02 [−0.02,0.07] | 3×10−1 | 0.07 [−0.00,0.14] | 7×10−2 |
| DQB1*03:01 | 0.02 [−0.01,0.04] | 2×10−1 | 0.06 [−0.01,0.13] | 1×10−1 | 0.03 [−0.01,0.07] | 2×10−1 | −0.01 [−0.05,0.04] | 7×10−1 |
| DQB1*03:03 | −0.04 [−0.08,0.01] | 2×10−1 | −0.10 [−0.30,0.10] | 3×10−1 | −0.03 [−0.10,0.03] | 3×10−1 | −0.03 [−0.10,0.05] | 5×10−1 |
| DRB1*04:04 | −0.01 [−0.10,0.08] | 8×10−1 | −0.10 [−0.76,0.55] | 8×10−1 | 0.04 [−0.07,0.15] | 5×10−1 | −0.13 [−0.29,0.04] | 1×10−1 |
| DRB1*11:01 | 0.03 [−0.01,0.07] | 2×10−1 | 0.03 [−0.06,0.12] | 5×10−1 | 0.05 [−0.01,0.11] | 8×10−2 | −0.00 [−0.07,0.07] | 9×10−1 |
| Ancestry | ||||||||
| A*02:01 | −0.07 [−0.13,−0.01] | 2×10 −2 | −0.05 [−0.08,−0.03] | 8×10 −6 | −0.03 [−0.07,0.02] | 3×10−1 | ||
| A*29:02 | 0.06 [−0.05,0.16] | 3×10−1 | 0.09 [0.04,0.14] | 2×10 −4 | 0.02 [−0.06,0.10] | 6×10−1 | ||
| B*40:01 | −0.11 [−0.26,0.03] | 1×10−1 | −0.09 [−0.14,−0.05] | 3×10 −5 | −0.03 [−0.23,0.18] | 8×10−1 | ||
| C*03:04 | −0.03 [−0.10,0.04] | 3×10−1 | −0.07 [−0.11,−0.04] | 1×10 −4 | −0.01 [−0.07,0.05] | 8×10−1 | ||
| DPB1*03:01 | −0.09 [−0.18,−0.01] | 3×10 −2 | −0.06 [−0.09,−0.03] | 2×10 −4 | −0.02 [−0.09,0.06] | 6×10−1 | ||
| DQA1*03:01 | −0.04 [−0.11,0.02] | 2×10−1 | −0.06 [−0.09,−0.03] | 1×10 −4 | −0.12 [−0.16,−0.07] | 3×10 −7 | ||
| DRB1*04:04 | −0.01 [−0.29,0.26] | 9×10−1 | −0.10 [−0.16,−0.05] | 2×10 −4 | −0.08 [−0.21,0.05] | 2×10−1 | ||
| DQB1*03:02 | −0.04 [−0.12,0.04] | 3×10−1 | −0.03 [−0.07,−0.00] | 3×10 −2 | −0.12 [−0.16,−0.08] | 8×10 −8 | ||
It is worth noting that, while tIgE is an important marker for atopic disease, it is a poor measure of symptoms with less clinical utility compared to measures of allergen-specific IgE. GWAS and especially HLA-association studies of allergen-specific IgE will likely yield better mechanistic insights into the genetic risk for atopic disease compared to tIgE. For example, a previous study reported a much stronger overlap in genetic association between asthma and allergen specific IgE compared to the overlap between asthma and tIgE24. While 5 of the 6 genome-wide significant loci in our study have been reported as genome-wide significant in allergic disease GWAS of asthma, atopic dermatitis, allergic rhinitis or eosinophilic esophagitis25, the FCER1A locus is a notable exception. This suggests that tIgE production is partly regulated by mechanisms other than the production of the more clinically relevant allergen-specific IgE. However, the limited availability of large publicly available genetic data sets with comparable between-study allergen-specific IgE measures curtail opportunities to conduct robust genetic association studies of allergen-specific IgE.
Our study only investigated the association between individual HLA genes and tIgE. Due to long-range LD across the HLA region, multivariate haplotype association analysis of HLA alleles may provide more statistical power to detect HLA associations. However, such analysis require non-missing allele calls across all genes, which would reduce the available HLA discovery sample by ~10% and the HLA replication sample by ~50%. In addition, HLA haplotypes likely have even larger frequency differences between ancestry groups compared to individual HLA alleles, complicating cross-ancestry analysis. For these reasons, we opted to not perform multivariate HLA association analysis in our study.
In summary, we performed the largest GWAS and HLA association study for tIgE focused on ancestrally diverse populations. We conclude two of the four tIgE GWAS loci reported previously showed no evidence of heterogeneity by ancestry and that these loci are likely relevant in other ancestry groups. We also demonstrated chr11q13.5 and chr15q22.2, recently reported in a large European ancestry asthma GWAS15, are associated with tIgE production and may therefore play a role in allergic disease in non-European ancestry populations. Due to differing patterns of LD across population groups, multi-ethnic studies are particularly useful for identifying causal variants, and the lack of ancestry heterogeneity observed for credible variants in genome-wide significant loci (Table E2) suggest that these variants are plausible causal variants. Our results support the notion that both MHC class I and II genes can affect risk for allergic disease, and we identified replicated associations between tIgE and specific HLA alleles A*02:01 and DQB1*03:02.
Supplementary Material
Key messages:
This study is the largest genome-wide and HLA association study of tIgE performed to date in populations of diverse ancestry
We show that known tIgE and allergic disease loci, discovered primarily in European ancestry groups, are relevant to non-European ancestry groups
We also identified specific HLA alleles associated with tIgE with evidence of replication
Acknowledgments:
Molecular data for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung and Blood Institute (NHLBI). Genome Sequencing for “NHLBI TOPMed: Genetics of Cardiometabolic Health in the Amish” (phs000956.v4.p1) was performed at Broad Genomics (The Genomics Platform of the Broad Institute of Harvard and MIT, 3R01HL121007-01S1). Genome Sequencing for “NHLBI TOPMed: The Genetics and Epidemiology of Asthma in Barbados” (phs001143.v3.p1) was performed at Illumina (3R01HL104608-04S1). We gratefully acknowledge the contributions of Pissamai and Trevor Maul, Paul Levett, Anselm Hennis, P. Michele Lashley, Raana Naidu, Malcolm Howitt and Timothy Roach, and the numerous health care providers, and community clinics and co-investigators who assisted in the phenotyping and collection of DNA samples, and the families and patients for generously donating DNA samples to the Barbados Asthma Genetics Study (BAGS). Funding for BAGS was provided by National Institutes of Health (NIH) R01HL104608, R01HL087699, and HL104608 S1. Genome Sequencing for “NHLBI TOPMed: Genetic Epidemiology of COPD (COPDGene)” (phs000951.v4.p4) was performed at Broad Genomics (HHSN268201500014C). Genome Sequencing for “NHLBI TOPMed: The Genetic Epidemiology of Asthma in Costa Rica” (phs000988.v4.p1) was performed at NWGC (3R37HL066289-13S1). Measurement of IgE levels in the CRA study was supported by R37 HL066289 and P01HL132825. Genome Sequencing for “NHLBI TOPMed: Whole Genome Sequencing and Related Phenotypes in the Framingham Heart Study” (phs000974.v4.p3) was performed at Broad Genomics (3U54HG003067-12S2). Genome Sequencing for “NHLBI TOPMed: Severe Asthma Research Program (SARP)” (phs001446.v1.p1) was performed at NYGC Genomics (HHSN268201500016C). Core support including centralized genomic read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (3R01HL-117626-02S1; contract HHSN268201800002I). Core support including phenotype harmonization, data management, sample-identity QC, and general program coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393; U01HL-120393; contract HHSN268201800001I). The TOPMed component of the Amish Research Program was supported by NIH grants R01 HL121007, U01 HL072515, and R01 AG18728. Email Rhea Cosentino (rcosenti@som.umaryland.edu) for additional input. The COPDGene project was supported by Award Number U01 HL089897 and Award Number U01 HL089856 from the National Heart, Lung, and Blood Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Heart, Lung, and Blood Institute or the National Institutes of Health. The COPDGene project is also supported by the COPD Foundation through contributions made to an Industry Advisory Board comprised of AstraZeneca, Boehringer Ingelheim, GlaxoSmithKline, Novartis, Pfizer, Siemens and Sunovion. A full listing of COPDGene investigators can be found at: http://www.copdgene.org/directory. Measurement of IgE levels in COPDGene was supported by R01HL130512 and a grant from Novartis. The Framingham Heart Study (FHS) acknowledges the support of Contracts NO1-HC-25195, HHSN268201500001I and 75N92019D00031 from the National Heart, Lung and Blood Institute and grant supplement R01 HL092577-06S1 for this research. We also acknowledge the dedication of the FHS study participants without whom this research would not be possible. We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed, CAAPA and ADRN. We also thank all study participants and authors of the freely available software used in this study.
Funding:
Support for this work was provided by the National Institutes of Health, National Heart, Lung and Blood Institute, through the BioData Catalyst program (award 1OT3HL142479-01, 1OT3HL142478-01, 1OT3HL142481-01, 1OT3HL142480-01, 1OT3HL147154). Additional funding sources include NHLBI 1R01HL104608-01A14 and NIH/NIAID U19 AI117673.
Competing interests:
LAB is a consultant for Abbvie, Allakos, Astra-Zeneca, Benevolent AIBio, Incyte, Janssen, Leo Pharma, Lilly, Naos Bioderma, Novartis, Pfizer, Principia Biopharma, Rapt Therapeutics, Regeneron, Sanofi/Genzyme, Sanofi-Aventis, UCB and Vimalan and an investigator for Abbvie, Astra-Zeneca, Kiniksa, Leo Pharma, Pfizer, Regeneron and Sanofi and has stock in Medtronics, Moderna and Gilead. No conflict of interest RLG is a board member of MatriSys, Bioscience, has received a consulting fee from Sente, has pending grants through Novan and Regeneron, and has stock in Sente and MatriSys. CPH reports grants from NHLBI and Novartis related to this study, and grants from the Alpha-1 Foundation, Bayer, Boehringer-Ingelheim and Vertex, and personal fees from Takeda, outside of this study. DYML has been a consultant for Sanofi-Genzyme, Genentech, Janssen R & D, AbbVie, Amagma Therapeutics, Boehringer-Ingelheim, Leo Pharmaceuticals, Incyte Pharmaceuticals Emma Guttman-Yassky is an employee of Mount Sinai and has received research funds (grants paid to the institution) from Abbvie, Almirall, Amgen, AnaptysBio, Asana Biosciences, AstraZeneca,Boerhinger-Ingelhiem, Celgene, Dermavant, DS Biopharma, Eli Lilly, Galderma,Glenmark/Ichnos Sciences, Innovaderm, Janssen, Kiniksa, Kyowa Kirin, Leo Pharma, Novan, Novartis, Pfizer, Ralexar, Regeneron Pharmaceuticals, Inc., Sienna Biopharma, UCB and Union Therapeutics/Antibiotx; and is a consultant for Abbvie, Aditum Bio, Almirall, Alpine, Amgen, Arena, Asana Biosciences, AstraZeneca, Bluefin Biomedicine, Boerhinger-Ingelhiem, Boston Pharmaceuticals, Botanix, Bristol-Meyers Squibb, Cara Therapeutics, Celgene,Clinical Outcome Solutions, DBV, Dermavant, Dermira, Douglas Pharmaceutical, DS Biopharma, Eli Lilly, EMD Serono, Evelo Bioscience, Evidera, FIDE, Galderma, GSK, Haus Bioceuticals, Ichnos Sciences, Incyte, Kyowa Kirin, Larrk Bio, Leo Pharma, Medicxi, Medscape, Neuralstem, Noble Insights, Novan, Novartis, Okava Pharmaceuticals, Pandion Therapeutics, Pfizer, Principia Biopharma, RAPT Therapeutics, Realm, Regeneron Pharmaceuticals, Inc., Sanofi, SATO Pharmaceutical, Sienna Biopharma, Seanegy Dermatology, Seelos Therapeutics, Serpin Pharma, Siolta Therapeutics, Sonoma Biotherapeutics, Sun Pharma, Target PharmaSolutions and Union Therapeutics, Vanda Pharmaceuticals, Ventyx Biosciences, Vimalan. Consultant for Abbvie, Aditum Bio, Almirall, Amgen, Asana Biosciences, AstraZeneca, Boerhinger-Ingelhiem, Cara Therapeutics, Celgene, Concert, DBV, Dermira, DS Biopharma, Eli Lilly, EMD Serono, Galderma, Ichnos Sciences, Incyte Kyowa Kirin, Leo Pharma, Pandion Therapeutics, Pfizer, RAPT Therapeutics, Regeneron Pharmaceuticals, Inc., Sanofi, Sienna Biopharma, Target PharmaSolutions and Union Therapeutics. ASP has been a consultant for Abbvie, Arena, Bausch, Bristol Myers Squibb, Dermavant, Eli Lilly, Forte, Leo, Lifemax, Pfizer, Rapt, Regeneron, and Sanofi and an investigator (support to institution) for AbbVie, Eli Lilly, and Regeneron. LCS is an investigator for Regneneron and DBV Technologies, a consultant for Amagma, Alladapt, and Ukko and has grants from Genentech and Pfizer. KCB receives royalties from UpToDate. The rest of the authors declare that they have no relevant conflicts of interest.
Abbreviations:
- tIgE
total serum IgE
- GWAS
genome-wide association study
- HLA
human leukocyte antigen
- WGS
whole genome sequence
- TOPMed
Trans-Omics for Precision Medicine
- CAAPA
Consortium on Asthma among African-ancestry Populations in the Americas
- ADRN
Atopic Dermatitis Research Network
- AD
atopic dermatitis
- FHS
Framingham Heart Study
- EDC
epidermal differentiation complex
- TH2
T helper type 2
- GTEx
Genotype-Tissue Expression
- MHC
major histocompatibility complex
- GASP
Genetic susceptibility to Asthma and pollution in Peru
- LD
linkage disequilibrium
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Tariq SM, Matthews SM, Hakim EA, Stevens M, Arshad SH, Hide DW. The prevalence of and risk factors for atopy in early childhood: a whole population birth cohort study. J Allergy Clin Immunol 1998; 101:587–93. [DOI] [PubMed] [Google Scholar]
- 2.Wuthrich B, Schindler C, Leuenberger P, Ackermann-Liebrich U. Prevalence of atopy and pollinosis in the adult population of Switzerland (SAPALDIA study). Swiss Study on Air Pollution and Lung Diseases in Adults. Int Arch Allergy Immunol 1995; 106:149–56. [DOI] [PubMed] [Google Scholar]
- 3.Granada M, Wilk JB, Tuzova M, Strachan DP, Weidinger S, Albrecht E, et al. A genome-wide association study of plasma total IgE concentrations in the Framingham Heart Study. J Allergy Clin Immunol 2012; 129:840–5 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Levin AM, Mathias RA, Huang L, Roth LA, Daley D, Myers RA, et al. A meta-analysis of genome-wide association studies for serum total IgE in diverse study populations. J Allergy Clin Immunol 2013; 131:1176–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med 2010; 363:1211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pino-Yanes M, Gignoux CR, Galanter JM, Levin AM, Campbell CD, Eng C, et al. Genome-wide association study and admixture mapping reveal new loci associated with total IgE levels in Latinos. J Allergy Clin Immunol 2015; 135:1502–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Weidinger S, Gieger C, Rodriguez E, Baurecht H, Mempel M, Klopp N, et al. Genome-wide scan on total serum IgE levels identifies FCER1A as novel susceptibility locus. PLoS Genet 2008; 4:e1000166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Magi R, Horikoshi M, Sofer T, Mahajan A, Kitajima H, Franceschini N, et al. Trans-ethnic meta-regression of genome-wide association studies accounting for ancestry increases power for discovery and improves fine-mapping resolution. Hum Mol Genet 2017; 26:3639–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Daya M, Rafaels N, Brunetti TM, Chavan S, Levin AM, Shetty A, et al. Association study in African-admixed populations across the Americas recapitulates asthma risk loci in non-African populations. Nat Commun 2019; 10:880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao L, Bin L, Rafaels NM, Huang L, Potee J, Ruczinski I, et al. Targeted deep sequencing identifies rare loss-of-function variants in IFNGR1 for risk of atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol 2015; 136:1591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kowalski MH, Qian H, Hou Z, Rosen JD, Tapia AL, Shan Y, et al. Use of >100,000 NHLBI Trans-Omics for Precision Medicine (TOPMed) Consortium whole genome sequences improves imputation quality and detection of rare variant associations in admixed African and Hispanic/Latino populations. PLoS Genet 2019; 15:e1008500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lin DY. A simple and accurate method to determine genomewide significance for association tests in sequencing studies. Genet Epidemiol 2019; 43:365–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Esparza-Gordillo J, Weidinger S, Folster-Holst R, Bauerfeind A, Ruschendorf F, Patone G, et al. A common variant on chromosome 11q13 is associated with atopic dermatitis. Nat Genet 2009; 41:596–601. [DOI] [PubMed] [Google Scholar]
- 14.Sleiman PM, Wang ML, Cianferoni A, Aceves S, Gonsalves N, Nadeau K, et al. GWAS identifies four novel eosinophilic esophagitis loci. Nat Commun 2014; 5:5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pividori M, Schoettler N, Nicolae DL, Ober C, Im HK. Shared and distinct genetic risk factors for childhood-onset and adult-onset asthma: genome-wide and transcriptome-wide studies. Lancet Respir Med 2019; 7:509–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ezell SA, Tsichlis PN. Akt1, EMSY, BRCA2 and type I IFN signaling: a novel arm of the IFN response. Transcription 2012; 3:305–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee JE, Choi G, Cho M, Kim D, Lee MO, Chung Y. A critical regulation of Th2 cell responses by RORalpha in allergic asthma. Sci China Life Sci 2020. [DOI] [PubMed] [Google Scholar]
- 18.Giambartolomei C, Vukcevic D, Schadt EE, Franke L, Hingorani AD, Wallace C, et al. Bayesian test for colocalisation between pairs of genetic association studies using summary statistics. PLoS Genet 2014; 10:e1004383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Vergara C, Tsai YJ, Grant AV, Rafaels N, Gao L, Hand T, et al. Gene encoding Duffy antigen/receptor for chemokines is associated with asthma and IgE in three populations. Am J Respir Crit Care Med 2008; 178:1017–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Dilthey AT, Mentzer AJ, Carapito R, Cutland C, Cereb N, Madhi SA, et al. HLA*LA-HLA typing from linearly projected graph alignments. Bioinformatics 2019; 35:4394–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zheng X, Shen J, Cox C, Wakefield JC, Ehm MG, Nelson MR, et al. HIBAG--HLA genotype imputation with attribute bagging. Pharmacogenomics J 2014; 14:192–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marsh DG, Chase GA, Freidhoff LR, Meyers DA, Bias WB. Association of HLA antigens and total serum immunoglobulin E level with allergic response and failure to respond to ragweed allergen Ra3. Proc Natl Acad Sci U S A 1979; 76:2903–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Akenroye AT, Brunetti T, Romero K, Daya M, Kanchan K, Shankar G, et al. Genome-wide Association study of Asthma, Total IgE, and Lung function in a Cohort of Peruvian Children. J Allergy Clin Immunol 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bonnelykke K, Matheson MC, Pers TH, Granell R, Strachan DP, Alves AC, et al. Meta-analysis of genome-wide association studies identifies ten loci influencing allergic sensitization. Nat Genet 2013; 45:902–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.MacArthur J, Bowler E, Cerezo M, Gil L, Hall P, Hastings E, et al. The new NHGRI-EBI Catalog of published genome-wide association studies (GWAS Catalog). Nucleic Acids Res 2017; 45:D896–D901. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.