

Abstract
This Article describes the development of a decarbonylative Pd-catalyzed aryl–fluoroalkyl bond-forming reaction that couples fluoroalkylcarboxylic acid-derived electrophiles [RFC(O)X] with aryl organometallics (Ar-M’). This reaction was optimized by interrogating the individual steps of the catalytic cycle (oxidative addition, carbonyl de-insertion, transmetalation, and reductive elimination) to identify a compatible pair of coupling partners and an appropriate Pd catalyst. These stoichiometric organometallic studies revealed several critical elements for reaction design. First, uncatalyzed background reactions between RFC(O)X and Ar–M’ can be avoided by using M’ = boronate ester. Second, carbonyl de-insertion and Ar–RF reductive elimination are the two slowest steps of the catalytic cycle when RF = CF3. Both steps are dramatically accelerated upon changing to RF = CHF2. Computational studies reveal that a favorable F2C–H----X interaction contributes to accelerating carbonyl de-insertion in this system. Finally, transmetalation is slow with X = difluoroacetate but fast with X = F. Ultimately, these studies enabled the development of an (SPhos)Pd-catalyzed decarbonylative difluoromethylation of aryl neopentylglycol boronate esters with difluoromethyl acetyl fluoride.
Graphical Abstract
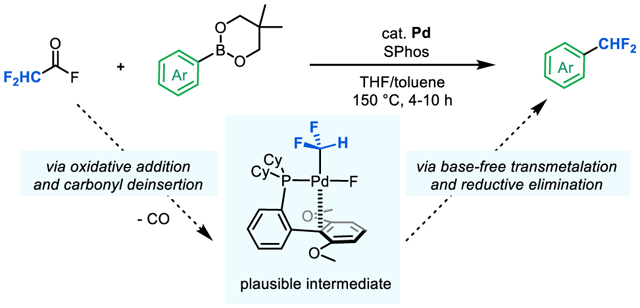
INTRODUCTION
Due to the prevalence of fluoroalkyl (RF) substituents in bio-active molecules, there is a high demand for reagents and synthetic methods for the formation of (heteroaryl)aryl–RF bonds.1,2 The prevailing approach involves transition metal-catalyzed cross-coupling of aryl halide electrophiles (ArX) with fluoroalkyl nucleophiles (RF–M).3,4 Despite extensive work in this area, the scope and broad utility of these transformations remain limited, largely due to challenges associated with the fluoroalkyl nucleophiles.4 The most common fluoroalkyl nucleophiles, R3SiRF, have limited availability for diverse RF substituents, undergo sluggish transmetalation in the absence of bases, and exhibit poor stability in the presence of the basic additives required for transmetalation.4,5 While some designer Ag and Zn-based fluoroalkyl nucleophiles have been developed to address these challenges, these reagents still have limitations with respect to synthetic accessibility and/or broad availability, particularly for diverse RF groups.4,6,7
A complementary cross-coupling approach to form (hetero)aryl–RF bonds would involve the reaction of fluoroalkyl carboxylic acid-derived electrophiles (RFC(O)X) with (hetero)aryl nucleophiles (Ar–M’, Scheme 1).8–13 This strategy eliminates the challenges associated with transmetalation from a weakly nucleophilic RF reagent.4,5 Furthermore, it leverages the abundance, low cost, and stability of fluoroalkyl carboxylic acid derivatives.13
Scheme 1.
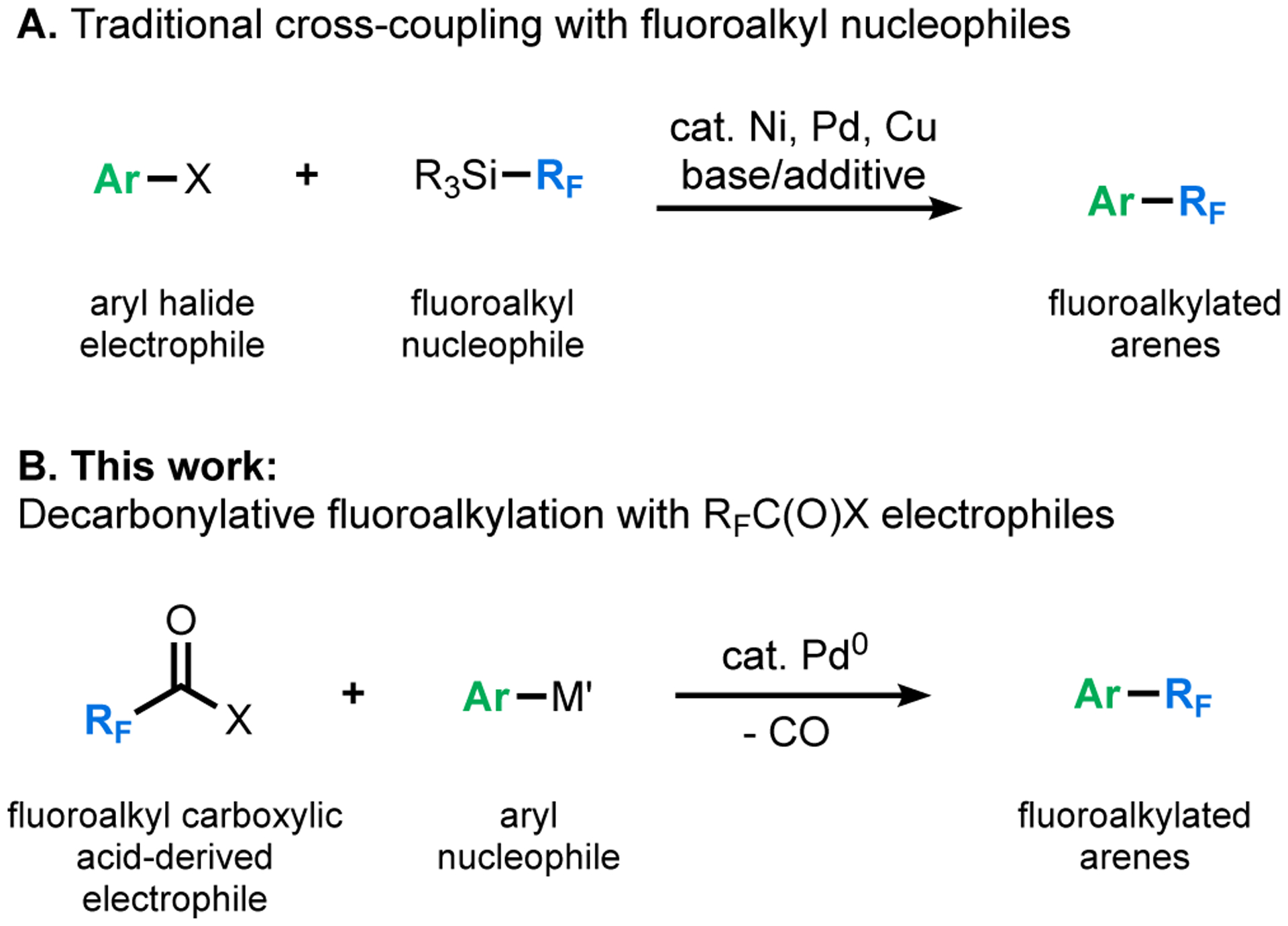
(A) Traditional fluoroalkylative cross-coupling using fluoroalkyl nucleophiles; (B) This work: decarbonylative fluoroalkylation with fluoroalkyl carboxylic acid-derived electrophiles.
A putative catalytic cycle for this transformation is shown in Scheme 2 and involves (i) oxidative addition of RFC(O)X to form [M]–acyl complex I, (ii) carbonyl de-insertion to generate [M]–RF intermediate II, (iii) transmetalation of the aryl nucleophile (Ar–M’) to form complex III, and (iv) aryl–RF bond-forming reductive elimination to release the product. An early study from our group established the feasibility of each of these individual steps using trifluoroacetic anhydride and diphenyl zinc as the coupling partners, and Pd[P(o-Tol)3]2/RuPhos as [M].13a However, catalytic turnover was not viable in this system due to a rapid uncatalyzed background reaction between the reagents to form trifluoromethyl ketones (Scheme 2, uncatalyzed acylation, v).
Scheme 2.
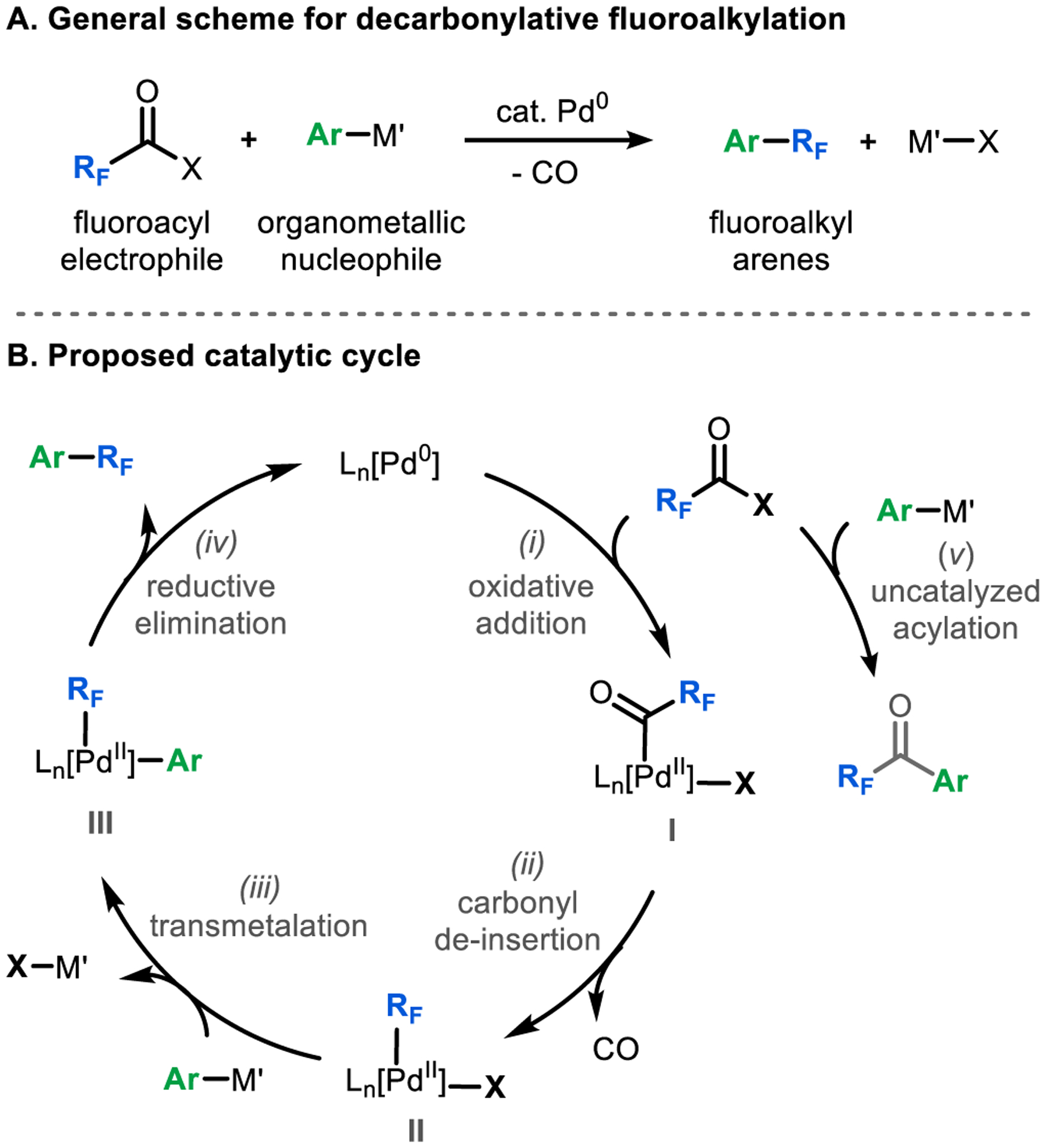
(A) General reaction scheme for decarbonylative fluoroalkylation and (B) proposed catalytic cycle with undesired acylation shown.
In addition to the competing background reaction, our early work identified several other limitations associated with individual steps of the catalytic cycle.13a For instance, direct oxidative addition of trifluoroacetic anhydride at (RuPhos)Pd0 (step i) proved challenging. As such, a two-step sequence involving initial oxidative addition at Pd(P(o-Tol)3)2 followed by a separate ligand exchange between P(o-Tol)3 and RuPhos is required. Furthermore, both carbonyl de-insertion (step ii) and aryl–CF3 bond-forming reductive elimination (step iv) were slow and/or low yielding. Finally, transmetalation (step iii) was limited to strongly nucleophilic organometallic reagents like diphenyl zinc.13a
We hypothesized that these challenges could be addressed via a mechanistic-based redesign of the catalyst and coupling partners for this reaction. In this report, we initially identify fluoroalkyl anhydrides and aryl boronate esters as compatible RFC(O)X and Ar–M’ coupling partners. We then use this pair to interrogate each step of the cycle in Scheme 2 with (SPhos)Pd0 as the catalyst. These stoichiometric organometallic studies provide key insights into the impact of RF, X, and M’ on each step, ultimately informing the development of a Pd-catalyzed method for the difluoromethylation of aryl boronate esters.
RESULTS AND DISCUSSION
Identifying Compatible Coupling Partners.
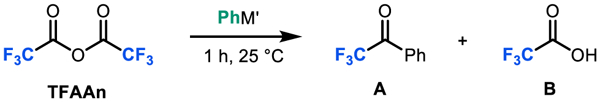
Our original attempts at Pd-catalyzed decarbonylative aryl fluoroalkylation were hampered by the uncatalyzed background addition of the diphenyl zinc nucleophile to the trifluoroacetic anhydride electrophile (TFAAn) to form phenyl trifluoromethyl ketone (A).13a This background reaction proceeds in 77% yield within 1 h at 25 °C (Table 1, entry 1), as determined by 19F NMR spectroscopic analysis. Our initial studies focused on identifying more compatible coupling partners for the proposed catalytic transformation. We hypothesized that aryl boron reagents, which are significantly less nucleophilic than their zinc counterparts,13a,14,22e would minimize ketone formation. Indeed, none of the ketone A was formed upon stirring a CDCl3 solution of trifluoroacetic anhydride (TFAAn) with phenyl boronic acid over 1 h at 25 °C. However, under these conditions a different undesired reaction, hydrolysis of the anhydride, proceeded to form trifluoroacetic acid (TFA, B) in quantitative yield (Table 1, entry 2). We next examined phenylboronic acid neopentylglycol ester (PhBneo) as the nucleophile, reasoning that it should minimize this hydrolysis process. Indeed, no detectable side product formation was observed upon stirring stirring a CDCl3 solution of TFAAn with PhBneo over 1 or 3 h at 25 °C (Table 1, entries 3 and 4). Compatibility was also observed when using phenylboronic acid pinacol ester (PhBpin) under otherwise identical conditions (entry 5). Furthermore, compatibility with PhBneo was maintained when moving to other fluoroalkyl anhydrides (e.g., difluoroacetic anhydride) was well as other fluoroalkyl carboxylic acid derivatives (e.g., difluoroacetyl fluoride; see Supporting Information for complete details).
Table 1.
Compatibility of TFAAn with different aryl nucleophiles.
| |||
|---|---|---|---|
| entry | PhM’ | yield A | yield B |
| 1 | Ph2Zn | 77% | 0% |
| 2 | PhB(OH)2 | <1% | >95% |
| 3 | PhBneo | <1% | <1% |
| 4a | PhBneo | <1% | <1% |
| 5 | PhBpin | <1% | <1% |
25 °C for 3 h.
Catalytic Cycle: Oxidative Addition and Carbonyl De-insertion.
With a pair of compatible reagents in hand, we next focused on challenges associated with the individual steps of the catalytic cycle. As described above, previous studies with RuPhos as the ligand accessed the TFAAn oxidative addition product in two discrete steps. First, Pd(P(o-Tol)3)2, was treated with TFAAn, and this was followed by a separate ligand exchange with RuPhos.13a We hypothesized that replacing the large isopropoxy-substituents of RuPhos with smaller methoxy groups (of SPhos) could accelerate oxidative addition and ligand substitution and facilitate the single-pot formation of SPhos-ligated trifluoroacetyl intermediate I-COCF3 (Figure 1). Indeed, the reaction of a THF solution of Pd[P(o-Tol)3]2/SPhos with TFAAn yielded I-COCF3 in 98% yield within 15 min at 25 °C (Figure 1B). Complex I-COCF3 was characterized in situ by 19F and 31P NMR spectroscopy, and the data are in excellent agreement with those for the reported RuPhos analogue.13a In particular, this complex can be clearly identified as a trifluoroacyl Pd intermediate (rather than a Pd–CF3 complex) based on the diagnostic chemical shift of the CF3 group (approximately −75 ppm Pd–C(O)CF3 versus −12 ppm for Pd–CF3).3a,13a,14
Figure 1.
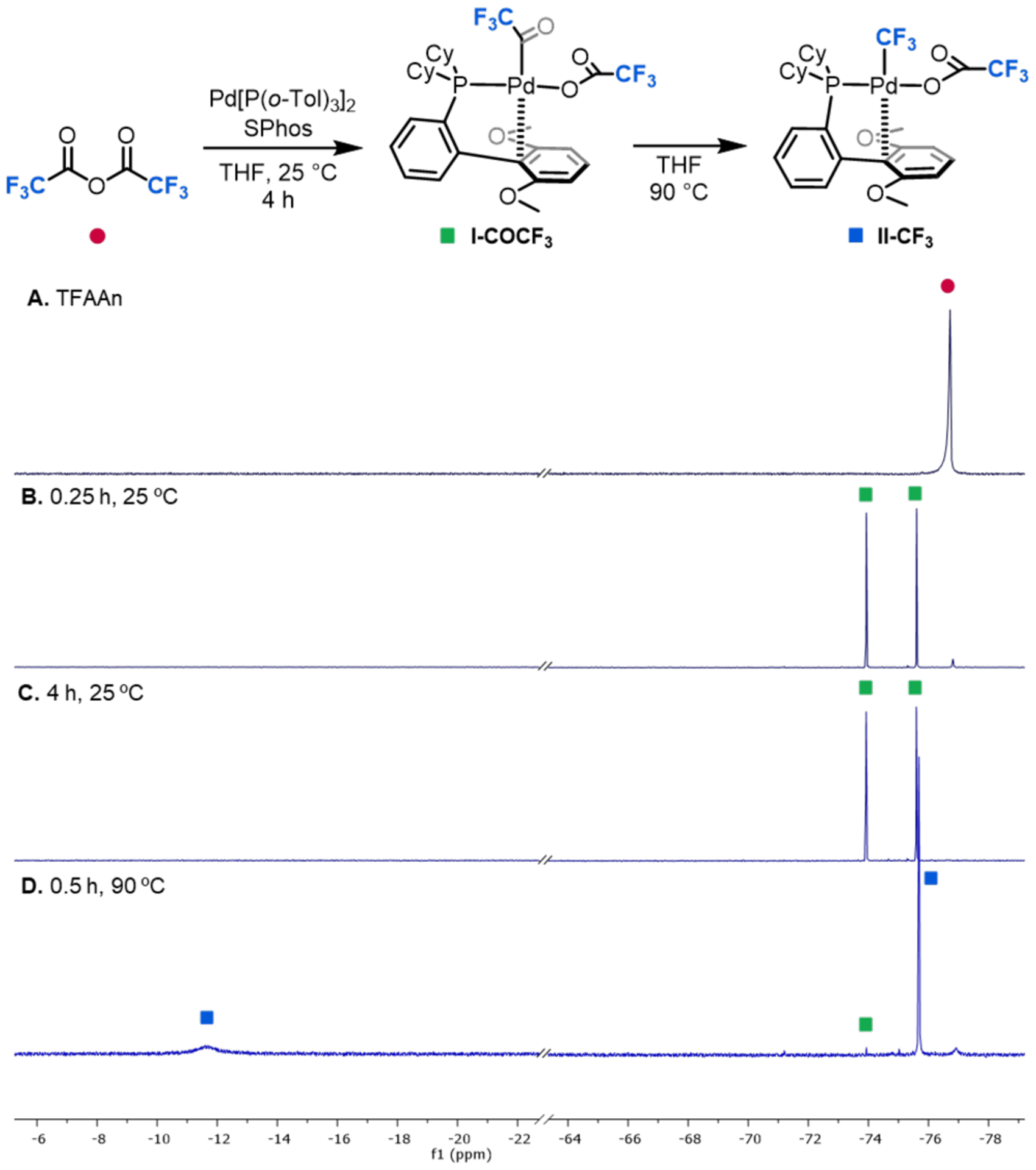
Oxidative addition and carbonyl de-insertion of TFAAn at SPhos/Pd0 in THF. 19F NMR spectra of (A) TFAAn; (B) Reaction of TFAAn with SPhos/Pd0 in THF after 0.25 h at room temperature; (C) Reaction of TFAAn with SPhos/Pd0 in THF after 4 h at room temperature; (D) Reaction heated to 90°C for 0.5 h. Spectra are referenced to 4-fluorotoluene (−119.85 ppm).
While Pd(P(o-Tol)3)2/SPhos proved highly reactive for oxidative addition of TFAAn at room temperature (Scheme 2, step i), carbonyl de-insertion (Scheme 2, step ii) at I-COCF3 remained slow in this system. After 4 h at 25 °C, no change in the 19F NMR spectrum was observed, and the decarbonylated intermediate, II-CF3, was not detected. CO de-insertion was only observed upon heating the reaction. After 30 min at 90 °C, I-COCF3 was nearly fully consumed with concomitant formation of II-CF3 in 91% yield (Figure 1C). Complex II-CF3 was characterized in situ (by analogy to the RuPhos analogue) based on its distinct broad Pd–CF3 19F NMR resonance at −11.6 ppm.3a,13a,15,16
We hypothesized that the rate of carbonyl de-insertion would be impacted by the nature of the migrating fluoroalkyl substituent.17a Thus we next explored the difluoromethyl analogue, in which a single fluorine atom is replaced by a hydrogen. This dramatically alters the size, nucleophilicity, dipole moment, and H-bond donor ability of the fluoroalkyl group,2a–d and all of these factors could potentially impact the carbonyl de-insertion step.17 Furthermore, it is well-documented that Ar–CHF2 bond-forming reductive elimination at PdII centers occurs under much milder conditions than analogous Ar–CF3 couplings.3,4,9a As such, this modification should accelerate this other challenging elementary step (iv) of the catalytic cycle in Scheme 2.
The reaction of a THF solution of Pd[P(o-Tol)3]2/SPhos with 1 equiv of difluoroacetic anhydride (DFAAn, Figure 2A) under otherwise identical conditions afforded >99% conversion of DFAAn within 15 min at room temperature. As shown in Figure 2B, the oxidative addition product I-COCHF2 was formed in 85% yield and characterized in situ via 19F NMR spectroscopy. This result demonstrates that oxidative addition remains fast in this system, despite the lower electrophilicity of DFAAn relative to that of TFAAn.
Figure 2.
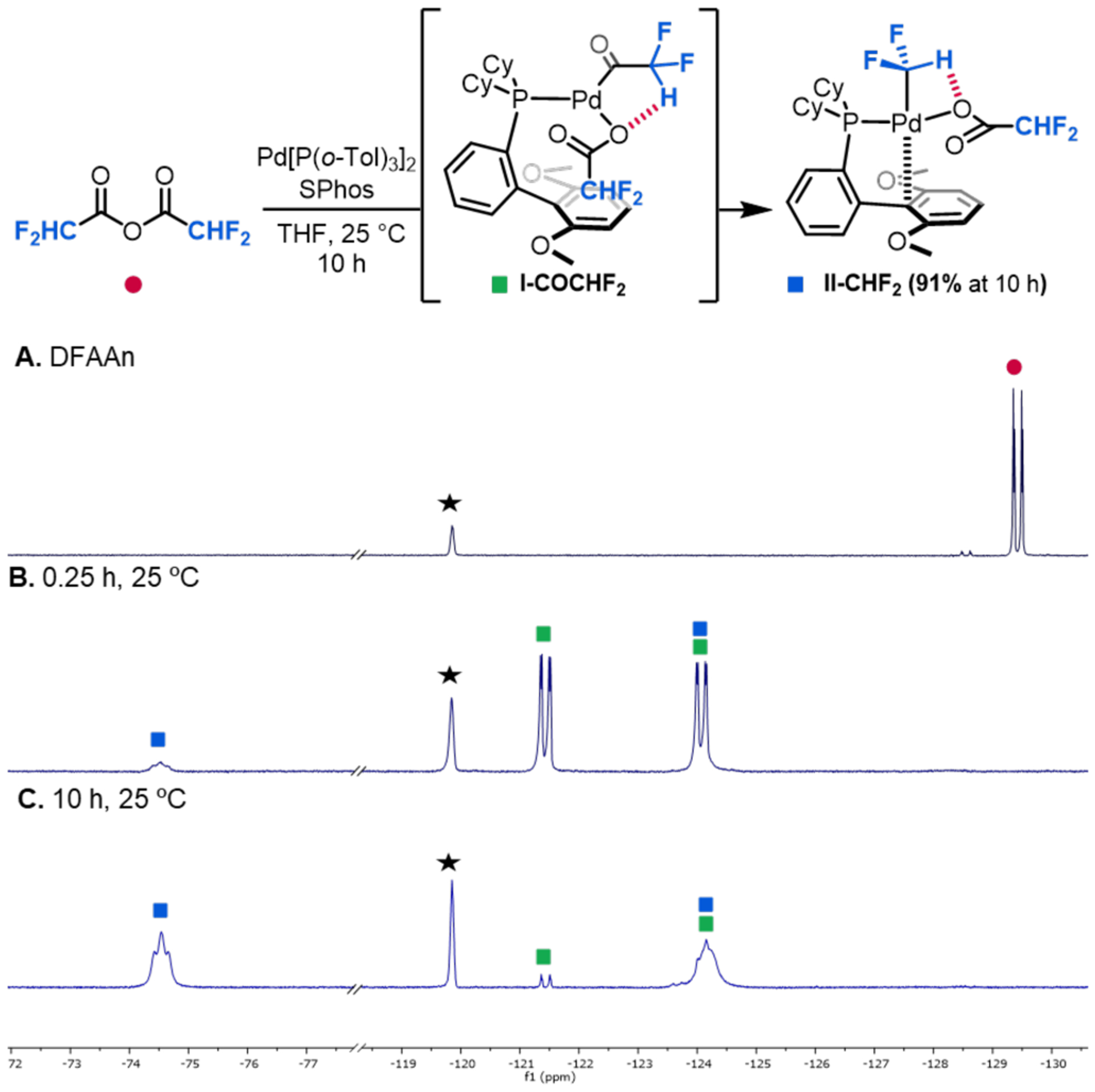
Oxidative addition and carbonyl de-insertion of DFAAn at SPhos/Pd0 in THF. 19F NMR spectrum of (A) DFAAn; (B) Reaction of DFAAn with SPhos/Pd0 in THF after 15 min at room temperature; (C) Reaction of DFAAn with SPhos/Pd0 in THF after 10 h at room temperature. Black star represents 4-fluorotoluene (−119.85 ppm, internal standard).
Interestingly, in marked contrast to the trifluoromethyl analogue, carbonyl de-insertion at I-COCHF2 also proceeded at room temperature. II-CHF2 was formed in 13% yield after 0.25 h, and the reaction was nearly complete within 10 h at 25 °C, affording II-CHF2 in 91% yield as determined by 19F NMR spectroscopy (Figure 2C). Complex II-CHF2 was isolated in 61% yield and was structurally characterized by X-ray crystal-lography. An ORTEP diagram of II-CHF2, along with representative bond distances and bond angles, are shown in Figure 3. A noteworthy feature of this structure is a short (2.38 Å) distance between H29 (from the CHF2 group) and O3 (of the difluoroacetate ligand). A significantly longer distance (3.60 Å) is observed between H29 and O4. The short (2.38 Å) distance as well as the C29-H29-O3 angle of 96.9° are consistent with the existence of an attractive interaction between H29 and O3.18
Figure 3.
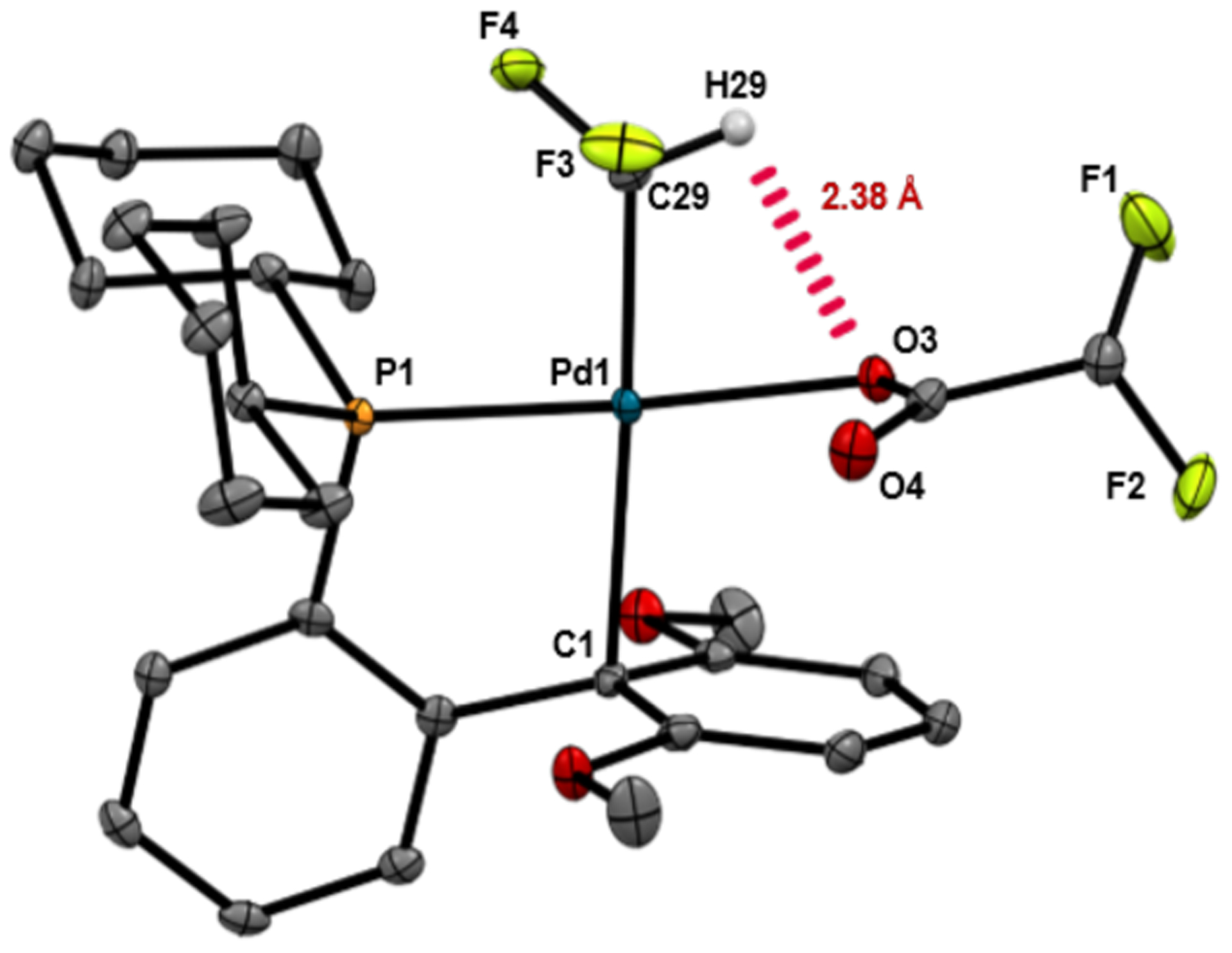
ORTEP diagram of II-CHF2. Select hydrogen atoms are omitted for clarity. Selected bond lengths (Å) and angle (deg): O3–Pd1 2.11, O4–Pd1 3.09, C29–Pd1 1.99, C1–Pd1 2.46; H29---O3 2.38, H29---O4 3.60; C29–Pd1–O3 81.7, C29–H29---O3 96.9.
Density functional theory (DFT) calculations19 (M06/LANL2DZ/6–311G**) were performed to interrogate the origin of the large rate enhancement for carbonyl de-insertion at I-COCHF2 relative to I-COCF3. This difference is counter to commonly accepted trends, where the rate is typically inversely proportional to the nucleophilicity of the migrating R group.17a Figure 4 shows an energy profile for 1,1-CO de-insertion at I-CORF proceeding through TS1-RF to initially form CO-bound complex (CO)Pd–RF. CO dissociation then generates the experimentally observed product II-RF. Consistent with the experimental observations, the calculations show a large (~16 kcal/mol) difference between the barrier for 1,1-de-insertion at I-COCHF2 versus I-COCF3. In addition, the overall thermodynamics associated with conversion of I-COCHF2 to II-CHF2 + CO (DG = −18.4 kcal/mol) is significantly more favorable than for the CF3 analogue (DG = −3.2 kcal/mol).
Figure 4.
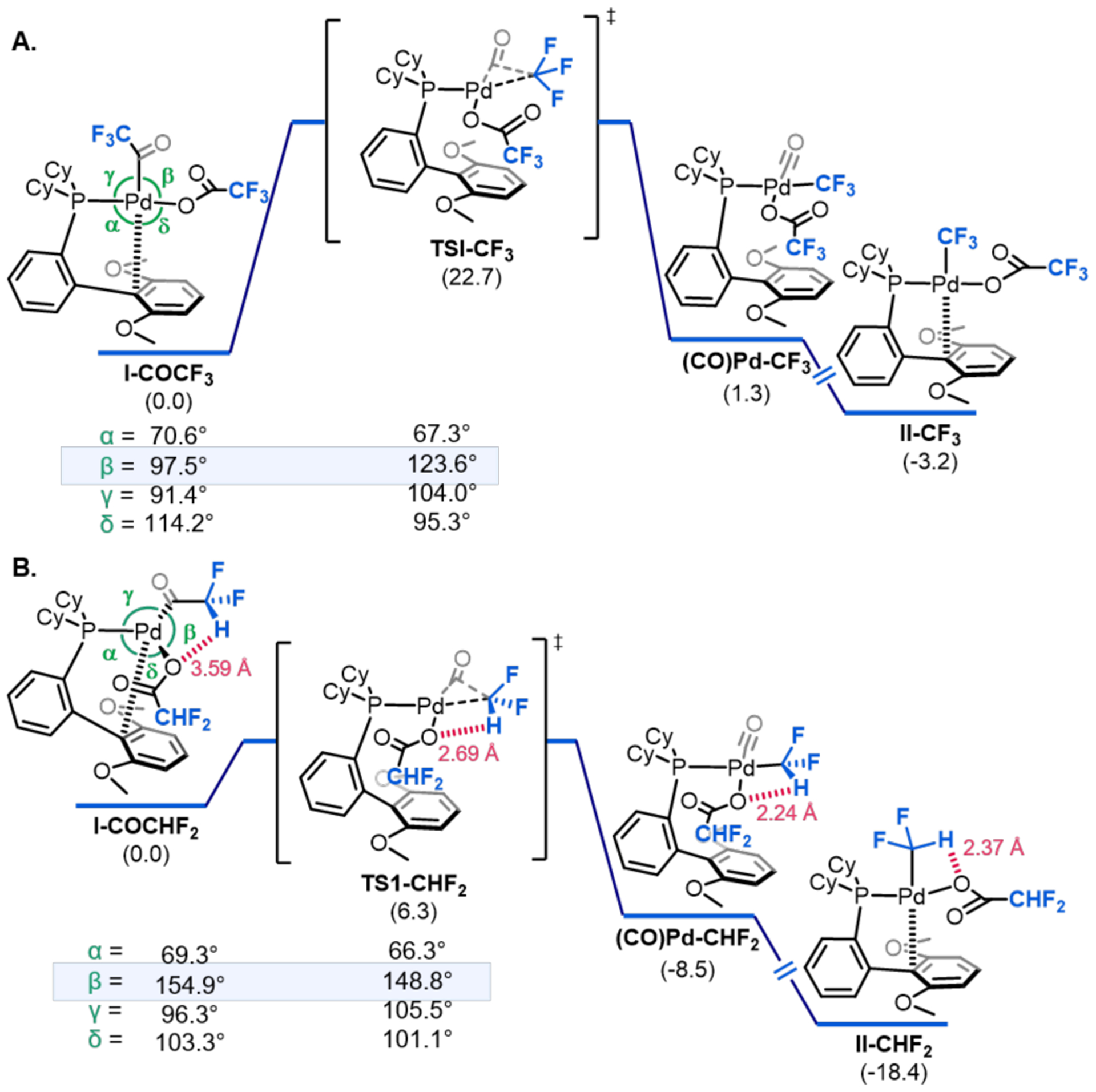
Energetics (the preferred binding mode highlighted as conformer A, see SI for details) for the carbonyl de-insertion process at (A) I-COCF3 and (B) I-COCHF2 with selected key angles α, β, γ, and δ for I-CORF and TS1-RF.
The computed structures show the presence of an attractive interaction with electrostatic character between H29 (of the CHF2 group) and O3 (of the carboxylate ligand). This interaction appears to contribute significantly to both the kinetic and thermodynamic preference for carbonyl de-insertion at the CHF2 versus CF3 analogue20. In the ground state starting material, I-COCHF2, a weak H(d+)---O(d–) electrostatic contact (3.59 Å) contributes to a distortion of the coordination geometry at Pd away from square planar. For instance, the angle between the acyl and carboxylate ligands (β in Figure 4) is 97.5° in I-COCF3 (which cannot engage in this weak contact) versus 154.9° in I-COCHF2. Given that carbonyl de-insertion transition states involve a three-coordinate metal center,17b this distortion makes the geometry of I-COCHF2 much closer to that of the transition state, TS1-CHF2 than in the CF3 analogue. The H---O bond distance becomes significantly shorter moving from I-COCHF2 (3.59 Å) to TS1-CHF2 (2.69 Å) to II-CHF2 (2.37 Å). Notably, the latter closely matches that observed experimentally in the X-ray crystal structure of II-CHF2 (2.38 Å).
Further analysis of the electrostatic potential surfaces (EPSs) of I-COCHF2, TS1-CHF2, and II-CHF2 and of non-covalent interaction (NCI) maps of the II-RF adducts reveal the key role of various attractive interactions (Figure 5).20n–p Specifically, orbital donor-acceptor interactions, electrostatic interactions, and a series of weakly attractive non-covalent bonds (dipole-induced dipole) were all observed in the -CHF2 containing structures, most prominently in TS1-CHF2 (see Supporting Information for complete details). In contrast, the electrostatic potential surfaces of the CF3-analogues show more diffuse dispersive repulsive interactions between the highly electronegative trifluoromethyl groups. These repulsive interactions are most pronounced in TS1-CF3, providing further insights into the relatively high barrier to carbonyl de-insertion at I-COCF3.
Figure 5.
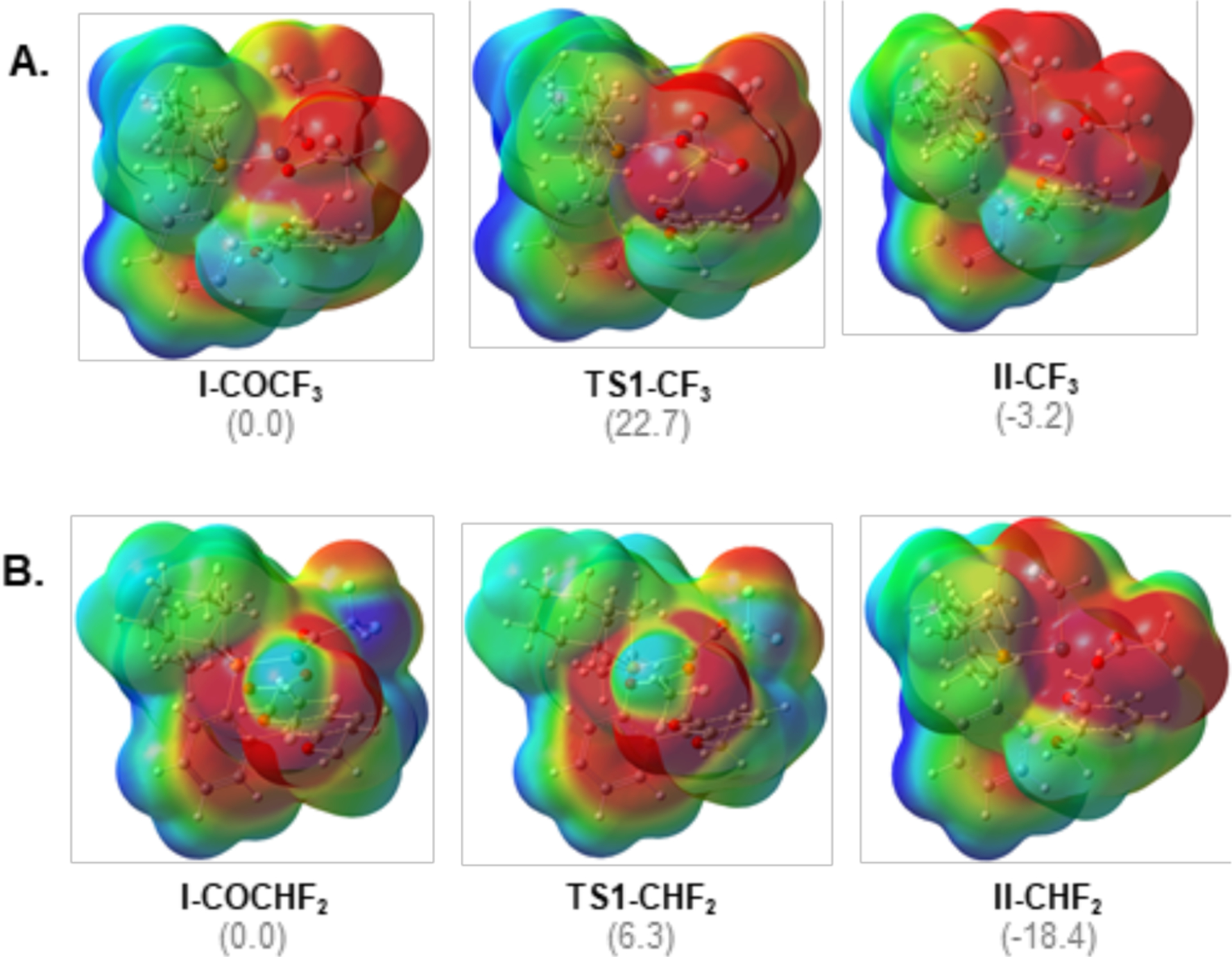
Electrostatic potential surfaces (for the optimal conformer A, see SI for details) generated for (A) I-COCF3, TS1-CF3, and II-CF3; (B) I-COCHF2, TS1-CHF2, and II-CHF2. Energies are in represented in kcal/mol.
Catalytic Cycle: Transmetalation and Reductive Elimination.
We next used complex II-CHF2 to interrogate the final two steps of the catalytic cycle: transmetalation and aryl–RF bond-forming reductive elimination (Figure 6). With boronic acid 1a as the nucleophile, 55% conversion of II-CHF2 was observed over 0.25 h at room temperature, with concomitant formation of the difluoromethylated organic product 1 in 40% yield. None of the PdII σ-aryl intermediate III-CHF2 was detected, indicating that Ar–CHF2 bond-forming reductive elimination is facile at room temperature.
Figure 6.
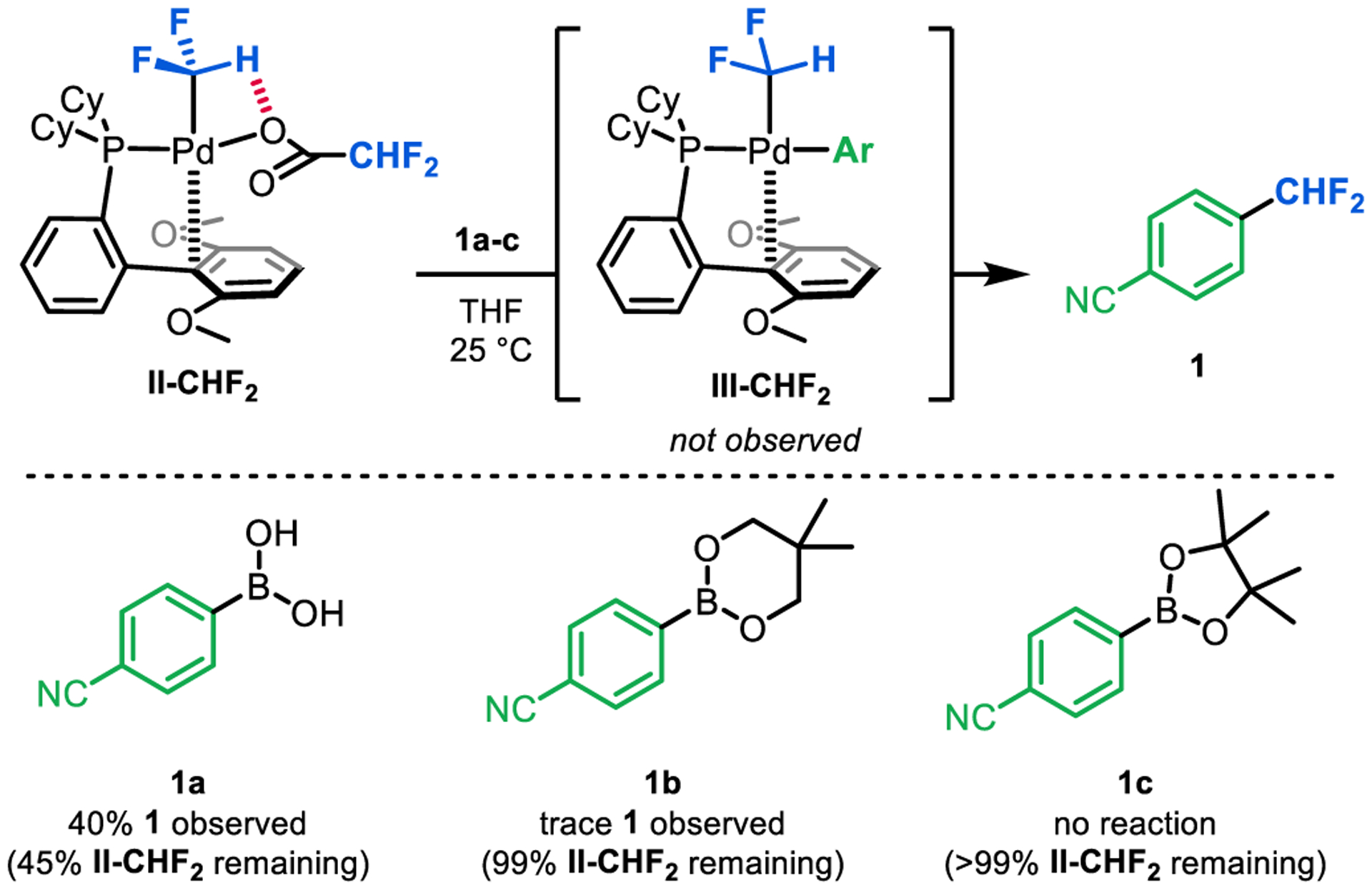
Transmetalation/reductive elimination sequence between II-CHF2 and aryl boron nucleophiles 1a-c.
In contrast, the boronate ester nucleophiles 1b and 1c showed low reactivity towards transmetalation with II-CHF2. In both cases, >99% of II-CHF2 remained after 0.25 h at 25 °C, and only traces of 1 were detected. These results indicate that transmetalation between the PdII–difluoroacetate intermediate II-CHF2 and aryl boronate esters is likely to be a key bottleneck in catalysis.
We hypothesized that this issue could be addressed by changing the X-type ligand on PdII from trifluoroacetate to a more reactive fluoride.21,22 Previous work23,24 has demonstrated that transition metal fluoride complexes exhibit high transmetalation activity towards various aryl boron nucleophiles. To generate a PdII–F intermediate, we treated a THF solution of II-CHF2 with anhydrous tetramethylammonium fluoride (Me4NF) for 0.5 h at 25 °C (Figure 7). This resulted in complete consumption of II-CHF2 and the appearance of a broad 19F NMR resonance at −349.5 ppm, which is diagnostic for a metal-fluo-ride.21 While this intermediate could not be isolated cleanly, the addition of boronate ester 1b resulted in consumption of the Pd–F signal within 15 min at 25 °C and formation of the reductive elimination product 1 in 27% yield (Figure 7). Again, the putative intermediate III-CHF2 was not detected. Overall, this sequence demonstrates that a fluoride ligand enables the targeted transmetalation/reductive elimination sequence with 1b, thus closing the formal catalytic cycle in this system.
Figure 7.
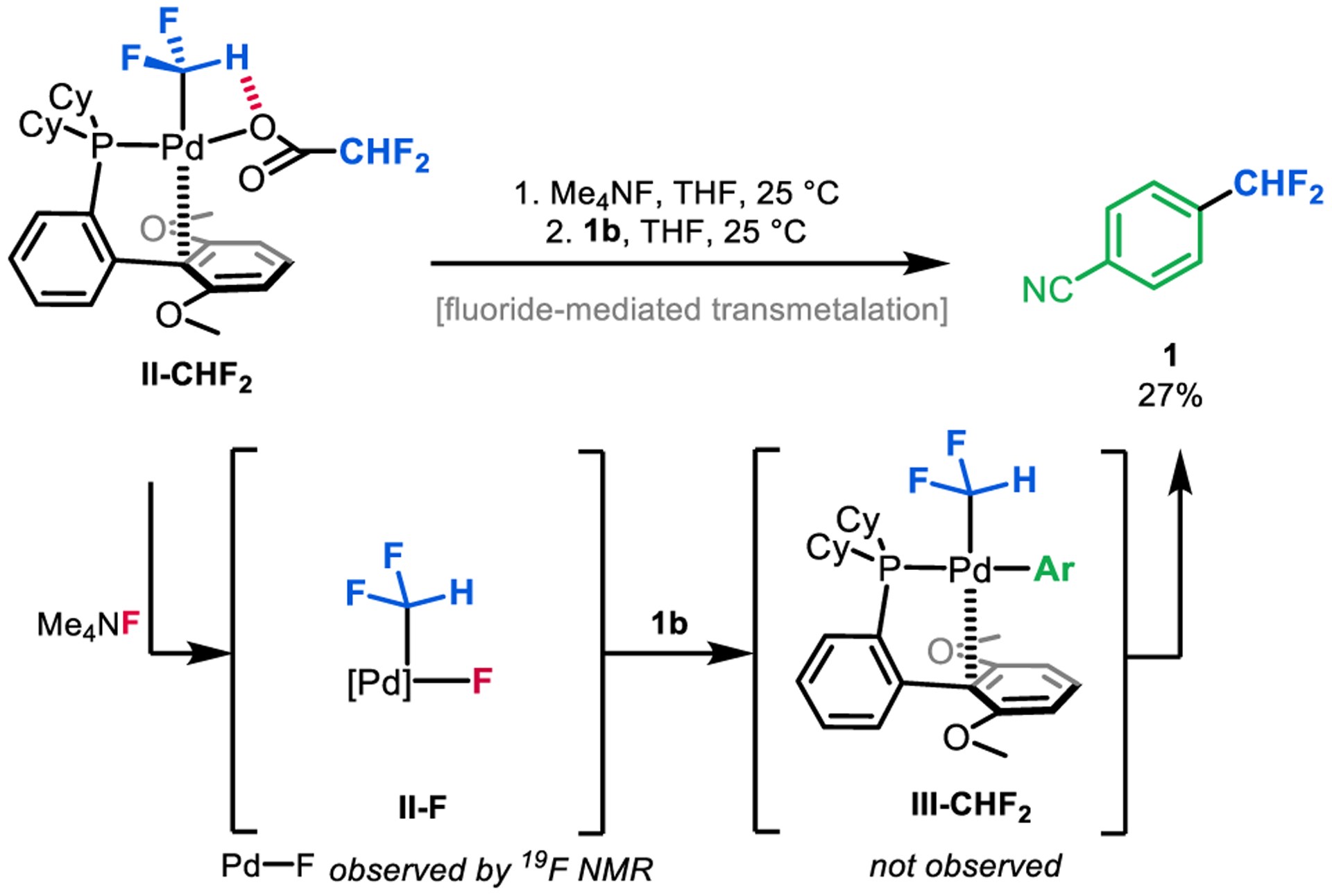
Generation of a Pd–F intermediate facilitates transmetalation with organoboron reagent 1b and subsequent reductive elimination (steps iii and iv in Scheme 2).
Development of Catalytic Reaction.
The organometallic studies described above demonstrate that each individual step of the catalytic cycle in Scheme 2 can proceed at room temperature. As such, our initial catalysis attempts focused on the room temperature SPhos/Pd[P(o-Tol)3]2-catalyzed decarbonylative coupling of DFAAn with boronate ester 1b in the presence of metal fluoride (MF) sources. As summarized in Figure 8, none of these reactions (with Me4NF, Bu4NF, or CsF) yielded the target difluoromethylated product 1. However, in the crude 19F NMR spectra of reactions that used CsF as the fluoride source, difluoroacetyl fluoride (DFAF) observed as a major byproduct.
Figure 8.
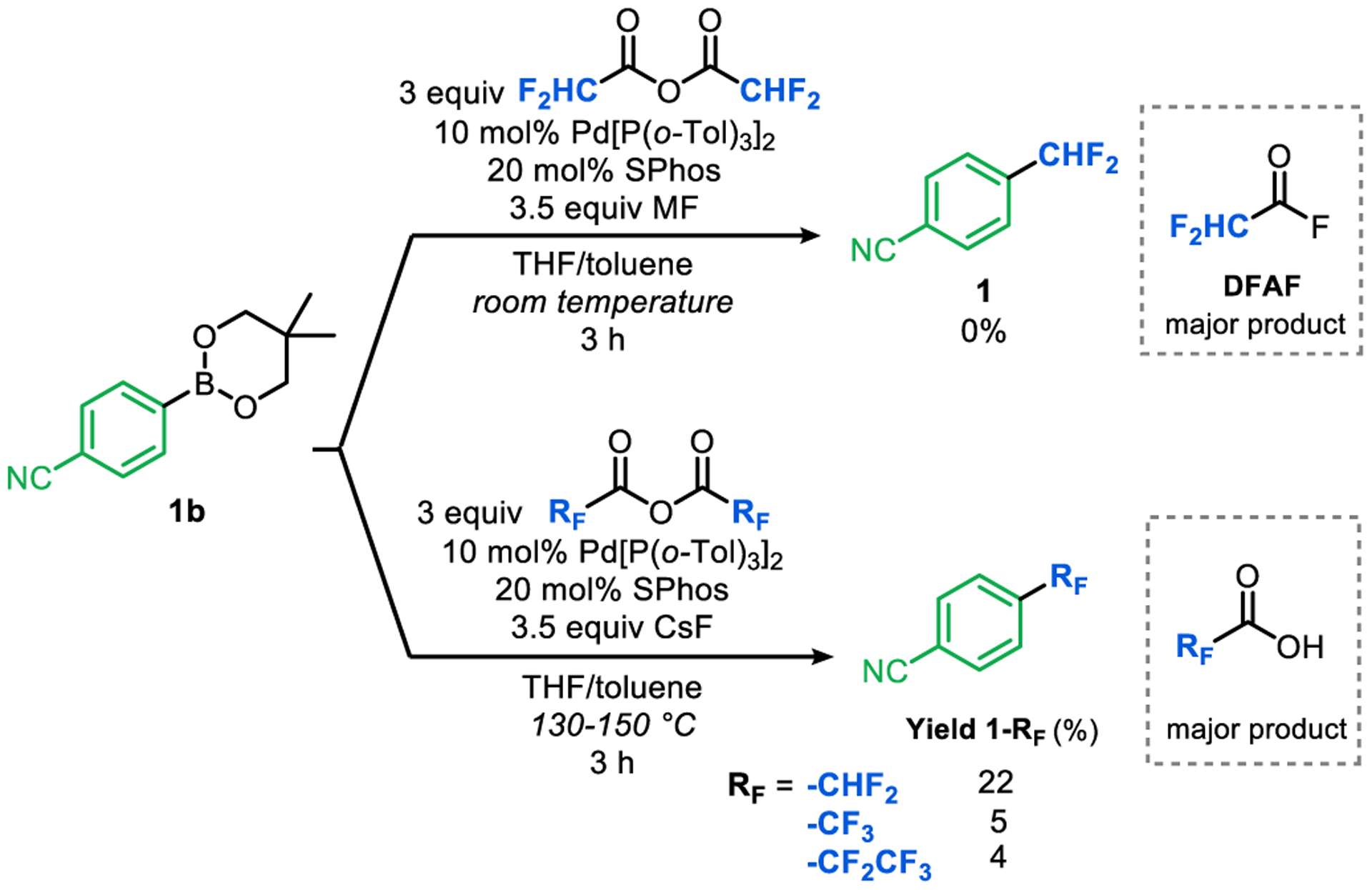
Initial attempts at catalysis using DFAAn and fluoride salts.
We noted that acid fluorides are significantly less electrophilic than their anhydride counterparts8j, which could result in slower oxidative addition. To address this potential issue, we next explored elevated temperatures. Gratifyingly, at 130 °C using excess CsF relative to DFAAn, the coupling product 1 was observed, albeit in modest (22%) yield (Figure 8). The stoichiometric studies suggest that at 130 °C carbonyl de-insertion should also be feasible for the −CF3 and −CF2CF3 analogues. As such, we explored the analogous catalytic reactions using TFAAn and pentafluoropropionic anhydride (PFPAn) at 130 °C. As shown in Figure 8, 4-trifluoromethylbenzonitrile and 4-pentafluoroethylbenzonitrile were formed in these transformations, in modest yields of 5% and 4%, respectively. In all three decarbonylative fluoroalkylation reactions a significant amount of the corresponding fluoroalkyl acid was observed in the crude mixture, consistent with competing decomposition of the anhydrides with traces of water in the fluoride salts.
Moving forward, we focused on optimizing catalytic decarbonylative difluoromethylation, since this was the highest yielding reaction among those in Figure 8. To eliminate the need for the hygroscopic fluoride additives, we independently synthesized anhydrous DFAF as a concentrated solution in THF (see SI for full details) and deployed it directly as the electrophile for cross-coupling.23,24 As shown in Table 2, entry 1, none of product 1 was detected at room temperature with DFAF as the electrophile, consistent with slow oxidative addition. However, upon heating this reaction to 130 °C, 1 was formed in 51% yield (entry 3). The reaction was further optimized with respect to reagent stoichiometry, temperature, solvent, and reaction time.25 Under the optimized conditions (10 mol % Pd[P(o-Tol)3]2, 20 mol % of SPhos, 5 equiv of DFAF, and 1 equiv of 1b in a mixture of THF:toluene at 150 °C for 5 h), 1 was formed in 92–93% yield as determined by 19F NMR spectroscopic analysis and was isolated in 77% yield (see SI for details).
Table 2.
Optimization of difluoromethylation of 1a with DFAF.
| ||||
|---|---|---|---|---|
| entry | X | T (°C) | solvent | yield (%)a |
| 1 | 1 | 25 | THF | <1 |
| 2 | 1 | 100 | THF | 29 |
| 3 | 1 | 130 | THF:toluene (1:1) | 51 |
| 4 | 3 | 130 | THF:toluene (1:1) | 58 |
| 5 | 5 | 130 | THF:toluene (1:1) | 67 |
| 6 | 5 | 150 | THF:toluene (1:1) | 86 |
| 7 | 5 | 150 | THF:dioxane (1:1) | 72 |
| 8b | 5 | 150 | THF:toluene (1:1) | 93 |
| 9b | 5 | 150 | THF:toluene (1:2) | 92 |
Yields determined by 19F NMR with 4-fluorotoluene internal standard.
Reaction was run for 5 h.
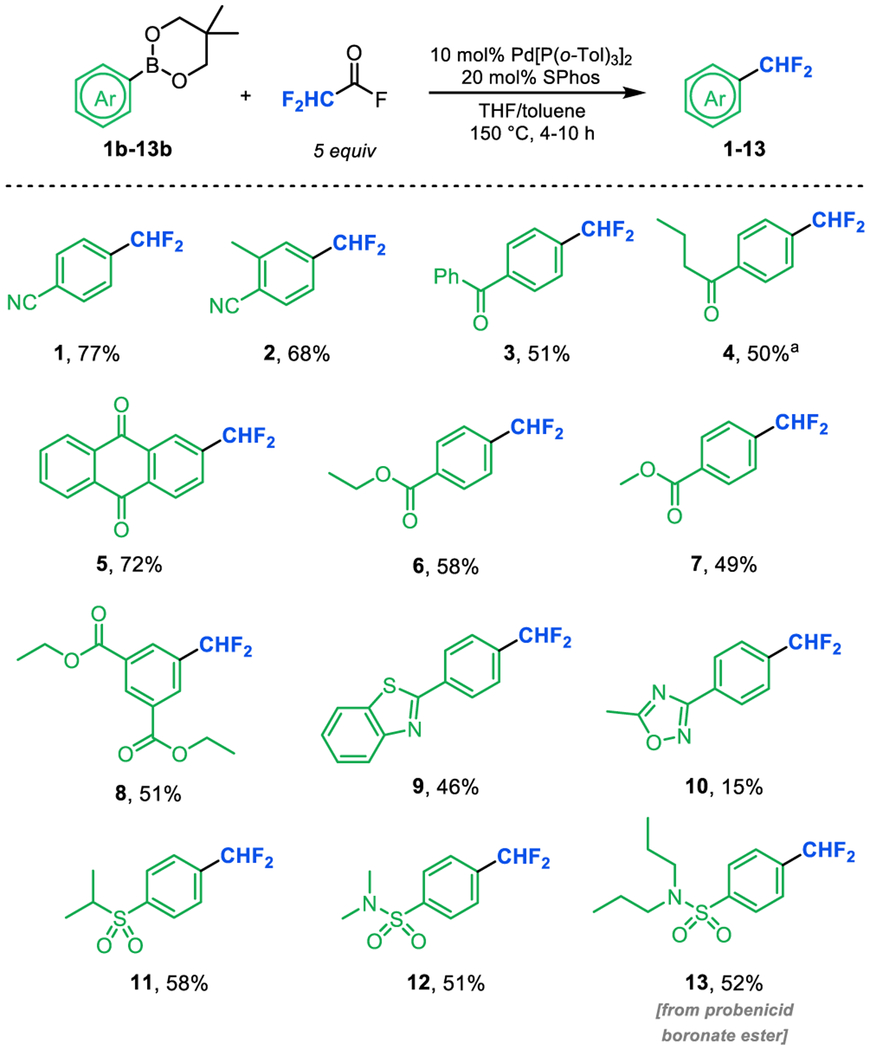
We next evaluated the scope of arene nucleophiles for this transformation. As summarized in Table 3, a variety of neopentylglycol boronate esters bearing electron withdrawing substituents (1–13) reacted to afford modest to excellent yields of difluoromethylarene products.26 Nitriles (1, 2), ketones (3–5), esters (6–8), sulfoxides (11), sulfonamides (12, 13) were well-tolerated under the reaction conditions. In addition, azole derivatives (9, 10) reacted in low to modest yields. Boronate esters bearing fluorinated substituents also underwent difluoromethylation in good to excellent yields but proved too volatile for isolation (see SI). Interestingly, boronate ester derivatives bearing electron donating substituents, such as methyl, phenyl, or benzyl ethers, showed low reactivity27 under these conditions (typically <5% yield). 19F NMR spectroscopic analysis of these low yielding reactions showed significant quantities of unreacted DFAF and no identifiable organic by-products. Ongoing efforts are focused on interrogating the mechanistic origin of this limitation and developing second generation catalysts to overcome it.
Table 3.
Scope of aryl boronate esters for the catalytic electrophilic decarbonylative difluoromethylation using difluoroacetyl fluoride.
|
15 mol% Pd[P(o-tol)3]2/30 mol% SPhos used for catalysis. See Supporting Information for details.
CONCLUSIONS
In summary, this Article presents a detailed investigation of decarbonylative cross-couplings between fluoroalkyl carboxylic acid-derived electrophiles and aryl boron nucleophiles. The combination of stoichiometric organometallic and computational studies unveiled several key findings that ultimately enabled the development of a catalytic difluoromethylation reaction. First, unusually low barriers are observed for the key carbonyl de-insertion step at (SPhos)PdII(C(O)CHF2)(X) complexes relative to their trifluoromethyl analogues. Several attractive non-covalent interactions involving the acidic CHF2 hydrogen appear to play a crucial role in lowering this barrier, a finding that could prove more broadly useful in the future development of decarbonylative couplings with these electrophiles.
The generation of a Pd–fluoride intermediate proved critical for promoting the challenging transmetalation step of the sequence. This finding led to the use of difluoroacetyl fluoride as the electrophile in catalysis to directly access a ‘transmetalation-active’ Pd-fluoride intermediate in situ and enable base-free transmetalation. While similar effects have been observed at nickel centers, this report is rare example of base-free cross-coupling of an acid fluoride derivative at Pd.24i Overall, we expect this study to engender interest in the unique properties of fluoroalkyl groups and the reactivity of metal–fluoroalkyl complexes in the context of catalytic reaction development.
Supplementary Material
ACKNOWLEDGMENT
We acknowledge Dr. Jeff Kampf for X-ray crystallographic analysis of compound II-CHF2. This work was supported by the NIH NIGMS (R35GM1361332). We also acknowledge computational resources and services provided by Compute Canada (SHARCNET: www.sharcnet.ca) and the Jaguar program supported by Dr. John F. Trant’s group facility at the University of Windsor.
ABBREVIATIONS
- THF
Tetrahydrofuran
- DCM
dichloromethane
- Et2O
diethyl ether
- DFAF
difluoroacetyl fluoride
- DFAAn
difluoroacetic anhydride
- TFAAn
trifluoroacetic anhydride
- DFA
difluoroacetic acid
- TFA
trifluoroacetic acid
- PFPAn
pentafluoropropionic anhydride
- PFPA
pentafluoropropionic acid
- TMAF
tetramethyl ammonium fluoride
- TBAF
tetrabutylammonium fluoride
- RT
room temperature
- SPhos
2-dicylcohexylphosphino-2’,6’-dimethoxybiphenyl
Footnotes
Supporting Information
The supporting information is available free of charge at the ACS publications website.
Detailed experimental procedures, analytical data, and computational data (PDF)
X-ray crystallographic data for II-CHF2 (CIF)
Deposition No.: 2099725
The authors declare no competing financial interest.
REFERENCES
- (1).On fluorine in molecules:; (a) Müller K; Faeh C; Diederich F Fluorine in pharmaceuticals: looking beyond intuition. Science 2007, 317, 1881–1886. [DOI] [PubMed] [Google Scholar]; (b) Purser S; Moore PR; Swallow S; Gouverneur V Fluorine in medicinal chemistry. Chem. Soc. Rev 2008, 37, 320–330. [DOI] [PubMed] [Google Scholar]; (c) Hagmann WK The many roles for fluorine in medicinal chemistry. J. Med. Chem 2008, 51, 4359–4369. [DOI] [PubMed] [Google Scholar]; (d) Wang J; Sanchez-Roselló M; Aceña JL; del Pozo C; Sorochinsky AE; Fustero S; Soloshonok VA; Liu H Fluorine in pharmaceutical industry: fluorine-containing drugs introduced to the market in the last decade (2001−2011). Chem. Rev 2014, 114, 2432–2506. [DOI] [PubMed] [Google Scholar]; (e) Liang T; Neumann CN; Ritter T Introduction of fluorine and fluorine-containing functional groups. Angew. Chem., Int. Ed 2013, 52, 8214–8264. [DOI] [PubMed] [Google Scholar]; (f) Caron S Where does fluorine come from? A review on the challenges associated with the synthesis of organofluorine compounds. Org. Process Res. Dev 2020, 24, 470–480. [Google Scholar]; (g) Shah P; Westwell AD; The role of fluorine in medicinal chemistry. J. Enzyme Inhib. Med. Chem 2007, 22, 527–540. [DOI] [PubMed] [Google Scholar]; (h) O’Hagan D Fluorine in health care: organofluorine containing blockbuster drugs. J. Fluor. Chem 2010, 131, 1071–1081. [Google Scholar]; (i) Jackel C; Salwiczek M Koksch B Fluorine in native protein environment - how the spatial demand and polarity of fluoroalkyl groups affect protein folding. Angew. Chem. Int. Ed 2006, 45, 4198–4203. [DOI] [PubMed] [Google Scholar]
- (2).On the properties and synthesis of difluoromethylarenes:; (a) Zafrani Y; Yeffet D; Sod-Moriah G; Berliner A; Amir D; Marciano D; Gershonov E; Saphier S Difluoromethyl ioisostere: examining the “lipophilic hydrogen bond donor” concept. J. Med. Chem 2017, 60, 797–804. [DOI] [PubMed] [Google Scholar]; (b) Zafrani Y; Saphier S; Gershonov E Utilizing the CF2H moiety as a H-bond-donated group in drug discovery. Future Med. Chem 2020, 12, 361–365. [DOI] [PubMed] [Google Scholar]; (c) Erickson JA; McLoughlin JI Hydrogen bond donor properties of the difluoromethyl group. J. Org. Chem 1995, 60, 1626–1631. [Google Scholar]; (d) Sessler CD; Rahm M; Becker S; Goldberg JM; Wang F; Lippard SJ CHF2, a hydrogen bond donor. J. Am. Chem. Soc 2017, 139, 9325–9332. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Rageot D; Beaufils F; Borsari C; Dall’Asen A; Neuberger M; Hebeisen P; Wymann MP Scalable, economical, and practical synthesis of 4-(difluoromethyl)pyridine-2-amine, a fey intermediate for lipid kinase inhibitors. Org. Process Res. Dev 2019, 23, 2416–2424. [Google Scholar]
- (3).Metal-catalyzed nucleophilic trifluoromethylation via cross-coupling:; (a) Cho EJ; Senecal TD; Kinzel T; Zhang Y; Watson DA; Buchwald SL The palladium-catalyzed trifluoromethylation of aryl chlorides. Science 2010, 328, 1679–1681. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Morimoto H; Tsubogo T; Litvinas ND; Hartwig JF A broadly applicable copper reagent for trifluoromethylations and perfluoroalkylations of aryl iodides and bromides. Angew. Chem., Int. Ed 2011, 50, 3793–3798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Metal-catalyzed nucleophilic fluoroalkylation:; (a) Fier PS; Hartwig JF Copper-mediated difluoromethylation of aryl and vinyl iodides. J. Am. Chem. Soc 2012, 134, 5524–5527. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Jiang X-L; Chen Z-H; Xu X-H; Qing F-L Copper-mediated difluoromethylation of electron-poor aryl iodides at room temperature. Org. Chem. Front 2014, 1, 774–776. [Google Scholar]; (c) Prakash GKS; Ganesh SK; Jones J-P; Kulkarni A; Masood K; Swabeck JK; Olah GA Copper-mediated difluoromethylation of (hetero)aryl iodides and β-styryl halides with tributyl(difluoromethyl)stannane. Angew. Chem., Int. Ed 2012, 51, 12090–12094. [DOI] [PubMed] [Google Scholar]; (d) Gu Y, Leng X & Shen Q Cooperative dual palladium/silver catalyst for direct difluoromethylation of aryl bromides and iodides. Nat Commun. 2014, 5, 1–7. [DOI] [PubMed] [Google Scholar]; (e) Chang D; Gu Y; Shen Q Pd-Catalyzed difluoromethylation of vinyl bromides, triflates, tosylates, and nonaflates. Chem. Eur. J 2015, 21, 6074–6078. [DOI] [PubMed] [Google Scholar]; (f) Nakamura Y; Fujiu M; Murase T; Itoh Y; Serizawa H; Aikawa K; Mikami K Cu-catalyzed trifluoromethylation of aryl iodides with trifluoromethylzinc reagent prepared in situ from trifluoromethyl iodide. Beilstein J. Org. Chem 2013, 9, 2404–2409. [DOI] [PMC free article] [PubMed] [Google Scholar]; (g) Aikawa K; Nakamura Y; Yokota Y; Toya W; Mikami K Stable but reactive perfluoroalkylzinc reagents: application in ligand-free copper-catalyzed perfluoroalkylation of aryl iodides. Chem. Eur. J 2015, 21, 96–100. [DOI] [PubMed] [Google Scholar]; (h) Aikawa K; Toya W; Nakamura Y; Mikami K Development of (trifluoromethyl)zinc reagent as trifluoromethyl anion and difluorocarbene sources. Org. Lett 2015, 17, 4996–4999. [DOI] [PubMed] [Google Scholar]; (i) Xu L; Vicic DA; Direct difluoromethylation of aryl halides via base metal catalysis at room temperature. J. Am. Chem. Soc 2016, 138, 2536–2539. [DOI] [PubMed] [Google Scholar]; (j) Serizawa H; Ishii K; Aikawa K; Mikami K Copper-Catalyzed difluoromethylation of aryl iodides with (difluoromethyl)zinc reagent. Org. Lett 2016, 18, 3686–3689. [DOI] [PubMed] [Google Scholar]
- (5).Furuya T; Kamlet AS Catalysis for fluorination and trifluoromethylation. Nature 2011, 473, 470–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).On the reagent TMSCF3:; (a) Liu X; Wang M Liu Q. Trifluoromethyltrimethylsilane: nucleophilic trifluoromethylation and beyond. Chem. Rev 2015, 115, 683–730. [DOI] [PubMed] [Google Scholar]; (b) Krishnamoorthy S; Kothandaraman J; Saldana J; Surya Prakash GK Direct difluoromethylation of carbonyl compounds by using TMSCF3: the right conditions. Eur. J. Chem 2016, 4965–4969. [Google Scholar]; (c) Xie Q; Li L; Zhu Z; Zhang R; Ni C; Hu J From C1 to C2: TMSCF3 as a precursor to pentafluoroethylation. Angew. Chem. Int. Ed 2018, 57, 13211–13215. [DOI] [PubMed] [Google Scholar]; (d) Johnston CP; West TH; Dooley RE; Reid M; Jones AB; King EJ; Leach AG Lloyd-Jones GC Anion-initiated trifluoromethylation by TMSCF3: deconvolution of the siliconate-carbonanion dichotomy by stopped-flow NMR/IR. J. Am. Chem. Soc 2018, 140, 11112–11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Mudarra AL; Martinez de Salinas S; Perez-Temprano MH Beyond the traditional roles of Ag in catalysis: the transmetalating ability of organosilver(i) species in Pd-catalysed reactions. Org. Biomol. Chem 2019, 17, 1655–1667. [DOI] [PubMed] [Google Scholar]
- (8).For reviews on carboxylic acids and their derivatives as electrophiles in cross-coupling:; (a) Baudoin O New approaches for decarboxylative biaryl coupling. Angew. Chem. Int. Ed 2007, 46, 1373–1375. [DOI] [PubMed] [Google Scholar]; (b) Gooßen K, Rodriguez N, Gooßen LJ, Carboxylic acids as substrates in homogenous catalysis. Angew. Chem. Int. Ed 2008, 47, 3100–3120. [DOI] [PubMed] [Google Scholar]; (c) Yu DG.; B. J. Li.; Z. J. Shi. Exploration of new C−O electrophiles in cross-coupling reactions. Acc. Chem. Res 2010, 43, 1486–1495; [DOI] [PubMed] [Google Scholar]; (d) Rodriguez N, Gooßen LJ, Decarboxylative coupling reactions: a modern strategy for C–C-bond formation. Chem. Soc. Rev 2011, 10, 5030–5048; [DOI] [PubMed] [Google Scholar]; (e) Cornella J, Larrosa I, Decarboxylative carbon-carbon bond-forming transformations of (hetero)aromatic carboxylic acids. Synthesis 2012, 44, 653–6. [Google Scholar]; (f) Hoover JM Mechanistic aspects of copper-catalyzed decarboxylative coupling reactions of (hetero)aryl carboxylic acids. Comment. Inorg. Chem 2017, 37, 169–200. [Google Scholar]; (g) Takise R; Muto K; Yamaguchi J Cross-coupling of aromatic esters and amides. Chem. Soc. Rev 2017, 46, 5864–5888. [DOI] [PubMed] [Google Scholar]; (h) Guo L; Rueping M Decarbonylative cross-couplings: Nickel catalyzed functional group interconversion strategies for the construction of complex organic molecules. Acc. Chem. Res 2018, 51, 1185–1195. [DOI] [PubMed] [Google Scholar]; (i) Meng G; Szostak M N-Acyl-glutarimides: Privileged scaffolds in amide N–C bond cross-coupling. Eur. J. Org. Chem 2018, 2352–2365. [Google Scholar]; (j) Zhao Q; Szostak M Redox-neutral decarbonylative cross-couplings coming of age. ChemSusChem 2019, 12, 2983–2987. [DOI] [PubMed] [Google Scholar]; (k) Blanchard N; Bizet V Acid fluorides in transition-metal catalysis: a good balance between stability and reactivity. Angew. Chem. Int. Ed 2019, 58, 6814–6187. [DOI] [PubMed] [Google Scholar]; (l) Ogiwara Y; Sakai N Acyl fluorides in late transition-metal catalysis. Angew. Chem. Int. Ed 2020, 59, 574–594. [DOI] [PubMed] [Google Scholar]; (m) Wang Z; Wang X; Nishihara Y Nickel or palladium-catalyzed decarbonylative tranformations of carboxylic acid derivatives. Chem Asian J. 2020, 15, 1234–1247. [DOI] [PubMed] [Google Scholar]; (n) Zheng Y-L; Newman SG Cross-coupling reactions with esters, aldehydes, and alcohols. Chem. Commun 2021, 57, 2591–2604. [DOI] [PubMed] [Google Scholar]; (o) Fahandej-Sadi A; Lundgren RJ Copper-mediated synthesis of monofluoro aryl acetates via decarboxylative cross-coupling. Synlett 2017, 28, 2886–2890. [Google Scholar]; (p) Moon PJ; Yin S; Lundgren RJ Ambient decarboxylative arylation of malonate half-esters via oxidative catalysis. J. Am. Chem. Soc 2016, 138, 13826–13829. [DOI] [PubMed] [Google Scholar]
- (9).For relevant difluoromethylation examples:; (a) Pan F; Boursalian GB; Ritter T Palladium-catalyzed decarbonylative difluoromethylation of acid chlorides at room temperature. Angew. Chem. Int. Ed 2018, 57, 16871–16876. [DOI] [PubMed] [Google Scholar]; (b) Feng Z, Min Q, Fu X, An L, Zhang X Chlorodifluoromethane-triggered formation of difluoromethylated arenes catalysed by palladium. Nat. Chem 2017, 9, 918–923. [DOI] [PubMed] [Google Scholar]; (c) Hori K, Motohashi H, Saito D, Mikami K Precatalyst effects of Pd-catalyzed cross-coupling difluoromethylation of aryl boronic acids. ACS Catal. 2019, 9, 417–421. [Google Scholar]
- (10).Fluorinated carboxylic acids in decarboxylative coupling:; (a) Beatty JW; Douglas JJ; Cole KP; Stephenson CRJ A Scalable and operationally simple radical trifluoromethylation. Nat. Commun 2015, 6, 7919–7925. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kyohide M; Etsuko T; Midori A; Kiyosi K A convenient trifluoromethylation of aromatic halides with sodium trifluoroacetate. Chem. Lett 1981, 10, 1719–1720. [Google Scholar]; (c) Chen M; Buchwald SL Rapid and efficient trifluoromethylation of aromatic and heteroaromatic compounds using potassium trifluoroacetate enabled by a flow system. Angew. Chem. Int. Ed 2013, 52, 11628–11631. [DOI] [PMC free article] [PubMed] [Google Scholar]; (d) Ambler BR; Zhu L; Altman RA Copper-catalyzed synthesis of trifluoroethylarenes from benzylic bromodifluoroacetates. J. Org. Chem 2015, 80, 8449–8457. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Ambler BR; Santosh P; Altman RA Ligand-controlled regioselective copper-catalyzed trifluoromethylation to generate (trifluoromethyl)allenes. Org. Lett 2015, 17, 2506–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Carr GE; Chambers RD; Holmes TF; Parker DG Sodium perfluoroalkane carboxylates as sources of perfluoroalkyl groups. J. Chem. Soc., Perkin Trans 1988, 1, 921–926. [Google Scholar]; (g) Johansen MB; Lindhardt AT Copper-catalyzed and additive free decarboxylative trifluoromethylation of aromatic and heteroaromatic iodides. Org. Biomol. Chem 2020, 18, 1417–1425. [DOI] [PubMed] [Google Scholar]; (h) Zapf A; Beller M Fine chemical synthesis with homogeneous palladium catalysts: examples, status, and trends. Topics in Catalysis. 2002, 19, 101–109. [Google Scholar]
- (11).(a) Sun AC; McClain EJ; Beatty JW; Stephenson CRJ Visible light-mediated decarboxylative alkylation of pharmaceutically relevant heterocycles. Org. Lett 2018, 20, 3487–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Tung TT; Christensen SB; Nielsen J Diflluoroacetic acid as a new reagent for direct C–H difluoromethylation of heteroaromatic compounds. Chem. Eur. J 2017, 23, 18125–18128. [DOI] [PubMed] [Google Scholar]
- (12).For other examples of fluoroalkyl carboxylic acid derivatives being used as a fluoroalkyl sources, see:; (a) Yang M-H; Orsi DL; Altman RA Ligand-controlled regiodivergent palladium-catalyzed decarboxylative allylation reaction to access α, α-difluoroketones. Angew. Chem. Int. Ed 2015, 54, 2361–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Yang M-H; Hunt JR; Sharifi N; Altman RA Palladium catalysis enables benzylation of α, α-difluoroketone enolates. Angew. Chem. Int. Ed 2016, 55, 9080–9083. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Ambler BR; Yang M-H; Altman RA Metal-catalyzed decarboxylative fluoroalkylation reactions. Synlett 2016, 27, 2747–2755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Fluorinated carboxylic acids in decarbonylative coupling:; (a) Maleckis A; Sanford MS Catalytic cycle for palladium-catalyzed decarbonylative trifluoromethylation using trilfuoroacetic esters as the CF3 source. Organometallics 2014, 33, 2653–2660; [Google Scholar]; (b) Brigham CE; Malapit CA; Lalloo N; Sanford MS Nickel-catalyzed decarbonylative synthesis of fluoroalkyl thioethers. ACS Catal. 2020, 10, 8315–8320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bakhmutov VI; Bozoglian F; Gomez K; Gonzalez G; Grushin VV; Macgregor SA; Martin E; Miloserdov FM; Novikov MA; Panitier JA; Romashov LV CF3–Ph reductive elimination from [(XantPhos}Pd(CF3)(Ph)]. Organometallics 2012, 31, 1315–1328. [Google Scholar]
- (15).Lennox AJJ; Lloyd-Jones GC Selection of boron reagents for Suzuki–Miyaura coupling. Chem. Soc. Rev 2014, 43, 412–443. [DOI] [PubMed] [Google Scholar]
- (16). We conducted an analogous stoichiometric oxidative addition and carbonyl de-insertion experiment with pentafluoropropionic anhydride (PFPAn, RF = −CF2CF3). As summarized in Figures S9 and S10, the reaction with PPAn proceeded nearly identically to that with TFAAn, showing fast oxidative addition but very slow carbonyl de-insertion at room temperature [<1% yield of the carbonyl de-insertion product after 4 h at room temperature].
- (17).For relative rates in carbonylation reactions, see:; (a) Shusterman AJ; Tamir I; Pross A The mechanism of organometallic migration reactions. A configuration mixing approach. J. Organomet. Chem 1988, 340, 203–222. [Google Scholar]; (b) Ortuno MA; Busra D; Cramer CJ Mechanism of Pd-catalyzed decarbonylation of biomass-derived hydrocinnamic acid to styrene following activation as an anhydride. Inorg. Chem 2016, 55, 4124–4131 [DOI] [PubMed] [Google Scholar]
- (18).(a) Johnston RC; Cheong HY C-H---O non-classical hydrogen bonding in the stereomechanics of organic transformations: theory ad recognition. Org. Biomol. Chem 2013, 11, 5057–5064. [DOI] [PubMed] [Google Scholar]; (b) Gilli P; Pretto L; Bertolasi V; Gilli G Predicting hydrogen-bond strengths from acid-base molecular properties. The pKa slide rule: toward the solution of a long-lasting problem. Acc. Chem. Res 2009, 42, 33–44. [DOI] [PubMed] [Google Scholar]
- (19).For DFT methods, see:; (a) Schrödinger Release 2019–2: Macro-Model, Schrödinger, LLC, New York, NY, 2019. [Google Scholar]; b) Schrödinger Release 2019–2: Maestro, Schrödinger, LLC, New York, NY, 2019. [Google Scholar]; (b) Frisch MJ; Trucks GW; Schlegel HB; Scuseria GE; Robb MA; Cheeseman JR; Scalmani G; Barone V; Mennucci B; Petersson GA; Nakatsuji H; Caricato M; Li X; Hratchian HP; Izmaylov AF; Bloino J; Zhang G; Sonnenberg JL; Hada M; Ehara M; Toyota K; Fukuda R; Hasegawa J; Ishida M; Nakajima T; Honda Y; Kitao O; Nakai H; Vreven T; Montgomery JA; Peralta JE Jr.; Ogliaro F; Bearpark M; Heyd JJ; Brothers E; Kudin KN; Staroverov VN; Kobayashi R; Normand J; Raghavachari K; Rendell A; Burant JC; Iyengar SS; Tomasi J; Cossi M; Rega N; Millam JM; Klene M; Knox JE; Cross JB; Bakken V; Adamo C; Jaramillo J; Gomperts R; Stratmann RE; Yazyev O; Austin AJ; Cammi AR; Pomelli C; Ochterski JW; Martin RL; Morokuma K; Zakrzewski VG; Voth GA; Salvador P; Dannenberg JJ; Dapprich S; Daniels AD; Farkas Ö; Foresman JB; Ortiz JV; Cioslowski J; Fox DJ Gaussian 09, Revision C.02; Gaussian, Inc.: Wallingford, CT, 2009. [Google Scholar]; (c) Roy LE; Hay PJ; Martin RL Revised Basis Sets for the LANL Effective Core Potentials. J. Chem. Theory Comput 2008, 4, 1029–1031. [DOI] [PubMed] [Google Scholar]; (d) Zhao Y, Truhlar DG. Density Functionals with Broad Applicability in Chemistry. Acc. Chem. Res 2008, 41, 157–167. [DOI] [PubMed] [Google Scholar]; (e) Huzinaga S, Gaussian Basis Sets for Molecular Calculations, Elsevier Science Pub. Co., Amsterdam, 1984. 15 P. J. Hay and W. R. Wadt, J. Chem. Phys, 1985, 82, 299–310. [Google Scholar]; (f) Hay PJ and Wadt WR, Ab initio effective core potentials for molecular calculations. Potentials for the transition metal atoms Sc to Hg. J. Chem. Phys 1985, 82, 270–283. [Google Scholar]; (g) Gonzalez C; Schlegel HB An improved algorithm for reaction path following. J. Chem. Phys, 1989, 90, 2154–2161; [Google Scholar]; (h) Gonzelez C; Schlegel HB Reaction Path Following in Mass-Weighted Internal Coordinates. J. Phys. Chem, 1990, 94, 5523–5527. [Google Scholar]; (i) Barone V; Cossi M Quantum Calculation of Molecular Energies and Energy Gradients in Solution by a Conductor Solvent Model. J. Phys. Chem. A, 1998, 102, 1995–2001; [Google Scholar]; (j) Cossi M; Rega N; Scalmani G; Barone VJ Energies, structures, and electronic properties of molecules in solution with the C-PCM solvation model. Comput. Chem, 2003, 24, 669–681. [DOI] [PubMed] [Google Scholar]; (k) Reed AE; Weinstock RB; Weinhold F Natural population analysis. J. Chem. Phys, 1985, 83, 735–746; [Google Scholar]; (l) Reed AE; Weinhold F Natural localized molecular orbitals. J. Chem. Phys, 1985, 83, 1736–1740; [Google Scholar]; (m) Reed AE; Curtiss LA; Weinhold F Intermolecular interactions from a natural bond orbital, donor-acceptor viewpoint. Chem. Rev, 1988, 88, 899–926. [Google Scholar]; (n) Glendening ED; Reed AE; Carpenter JE; Weinhold F NBO Version 3.1; [Google Scholar]; (o) Contreras-García J; Johnson ER; Keinan S; Chaudret R; Piquemal J-P; Beratan DN; Yang W NCIPLOT: A program for plotting noncovalent regions. J. Chem. Theory Comput 2011, 7, 625–632. [DOI] [PMC free article] [PubMed] [Google Scholar]; (p) Shakourian-Fard M; Kamath G; Jamshidi Z Trends in physisorption of ionic liquids on boron-nitride sheets. J. Phys. Chem. C 2014, 118, 26003–26016. [Google Scholar]
- (20). Trifluoroacetic anhydride (TFAAn) and pentafluoropropionic anhydride (PFPAn) both undergo rapid oxidative addition at room temperature but require elevated temperatures for carbonyl de-insertion. Both TFAAn and PFPAn lack the acidic hydrogen found in DFAAn and require high temperatures for carbonyl de-insertion. See the Supporting Information (p. S8–13) for more details.
- (21).(a) Fraser SL, Antipin M. Yu., Khroustalyov VN, Grushin VV, Molecular fluoro palladium complexes J. Am. Chem. Soc 1997, 119, 4769–4770. [Google Scholar]; (b) Beweries T; Brammer L; Jasim NA; McGrady JE; Perutz RN, Whitwood AC Energetics of halogen bonding of group 10 metal fluoride complexes. J. Am. Chem. Soc 2011, 133, 14338–14348. [DOI] [PubMed] [Google Scholar]
- (22).For recent example of studies on transmetalation-active organometallic complexes with organoboron and silicon, see:; (a) Carrow BP; Hartwig JF Distinguishing between pathways for transmetalation in Suzuki-Miyaura reactions. J. Am. Chem. Soc 2011, 133, 2116–2119. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Amatore C, Jutand A & Le Duc G The triple role of fluoride ions in palladium-catalyzed Suzuki–Miyaura reactions: unprecedented transmetalation from [ArPdFL2] complexes. Angew. Chem. Int. Ed 2012, 51, 1379–1382. [DOI] [PubMed] [Google Scholar]; (c) Thomas AA & Denmark SE Pre-transmetalation intermediates in the Suzuki–Miyaura reaction revealed: the missing link. Science 2016, 352, 329–332. [DOI] [PubMed] [Google Scholar]; (d) Malapit CA; Bour JR; Brigham CE; Sanford MS Base-free nickel-catalysed decarbonylative Suzuki-Miyaura coupling of acid fluorides. Nature 2018, 563, 100–104. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Malapit CA; Bour JR; Laursen SR; Sanford MS Mechanism and scope of nickel-catalyzed decarbonylative borylation of carboxylic acid fluorides. J. Am. Chem. Soc 2019, 141, 17322–17330. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Malapit CA; Borrell M; Milbauer MW; Brigham CE; Sanford MS Nickel-catalyzed decarbonylative amination of carboxylic acid esters. J. Am. Chem. Soc 2020, 142, 5918–5923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23). For reviews on utility of acid fluorides in decarbonylative cross-coupling, see refs 8k–m.
- (24).For examples of utility of acid fluorides in decarbonylative cross-coupling, see refs 20d, 20f, and:; (a) Keaveney ST; Schoenebeck F Palladium-catalyzed decarbonylative trifluoromethylation of acid fluorides. Angew. Chem. Int. Ed 2018, 57, 4073–4077. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Wang Z; Wang X; Nishihara Y Nickel-catalysed decarbonylative borylation of aroyl fluorides. Chem. Commun 2018, 54, 13969–13972. [DOI] [PubMed] [Google Scholar]; (c) Ogiwara Y; Sakurai Y; Hattori H; Sakai N Palladium-catalyzed reductive conversion of acyl fluorides via ligand-controlled decarbonylation. Org. Lett 2018, 20, 4204–4208. [DOI] [PubMed] [Google Scholar]; (d) Okuda Y; Xu J; Ishida T; Wang C.-a.; Nishihara Y Nickel-catalyzed decarbonylative alkylation of aroyl fluorides assisted by Lewis-acidic organoboranes. ACS Omega 2018, 3, 13129–13140. [DOI] [PMC free article] [PubMed] [Google Scholar]; (e) Sakurai S; Yoshida T; Tobisu M Iridium-catalyzed decarbonylative coupling of acyl fluorides with arenes and heteroarenes via C-H activation. Chem. Lett 2019, 48, 94–97. [Google Scholar]; (f) Ogiwara Y; Iino Y; Sakai N Catalytic C–H/C–F coupling of azoles and acyl fluorides. Chem. Eur. J 2019, 25, 6513–6516. [DOI] [PubMed] [Google Scholar]; (g) Wang X; Wang Z; Liu L; Asanuma Y; Nishihara Y Nickel-catalyzed decarbonylative stannylation of acyl fluorides under ligand-free conditions. Molecules 2019, 24, 1671–1682. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Wang X; Wang Z; Nishihara Y Nickel/copper-cocatalyzed decarbonylative silylation of acyl fluorides. Chem. Commun 2019, 55, 10507–10510. [DOI] [PubMed] [Google Scholar]; (i) Kayumov M; Zhao J-N; Mirzaakhmedov S; Wang D-Y; Zhang A Synthesis of arylstannanes via palladium-catalyzed decarbonylative coupling of aroyl fluorides. Adv. Synth. Catal 2020, 362, 776–781. [Google Scholar]; (j) Chen Q; Fu L; Nishihara Y Palladium/copper-cocatalyzed decarbonylative alkynylation of acyl fluorides with alkynylsilanes: synthesis of unsymmetrical diarylethynes. Chem. Commun 2020, 56, 7977–7980. [DOI] [PubMed] [Google Scholar]; (k) Wang Z; Wang X; Ura Y; Nishihara Y Nickel-catalyzed decarbonylative cyanation of acyl chlorides. Org. Lett 2019, 21, 6779–6784. [DOI] [PubMed] [Google Scholar]; (l) Sakurai Y; Oqiwara Y; Sakai N Palladium-catalyzed annulation of acyl fluorides with norbornene via decarbonylation and CO reinsertion. Chem. Eur. J 2020, 26, 12972–12977. [DOI] [PubMed] [Google Scholar]; (m) He B; Liu X; Li H; Zhang X; Ren Y; Su W Rh-Catalyzed general method for directed C–H functionalization via decarbonylation of in-situ-generated acid fluorides from carboxylic acids. Org. Lett 2021, 23, 4191–4196. [DOI] [PubMed] [Google Scholar]
- (25). Optimal yields are obtained by conducting this reaction in tall, 10-mL scintillation vials, and other vessels examined resulted in in significantly diminished yields. We hypothesize that having additional head space for the reaction is beneficial due to the gaseous nature of DFAF and THF under the reaction conditions. See p. S34 for complete details about the selection of reaction vessel.
- (26). The mass balance in the catalytic reaction was evaluated via 19F NMR spectroscopy using 2-(4-fluorophenyl)-5,5-dimethyl-1,3,2-dioxaborinane as the substrate. After 3 h, 17% of the difluoromethylated product [1-(difluoromethyl)-4-fluorobenzene], 14% of the protodeboronation product [fluorobenzene], and 43% of the aryl boron starting material were observed, accounting for 74% of the mass balance. Neither 4-fluorophenylboronic acid nor 4,4’-difluorobiphenyl were detected. See p. S32–33 for additional details.
- (27). Given the poor reactivity of electron-rich aryl boronate nucleophiles in the catalytic reaction, we evaluated the fluoride-mediated transmetalation of II-CHF2 with the p-OCH3 substituted substrate 19b. The difluoromethylated product 19 was formed in 26% yield (nearly identical to the 27% yield of 1 in Figure 7), suggesting that the transmetalation step is not the origin of the poor reactivity of electron-rich boronate esters in the catalytic reaction. See the Supporting Information (pages S18–19) for additional details.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.