

Abstract
Sterols are important lipid components of the plasma membrane (PM) in eukaryotic cells, but it is unknown how the PM retains sterols at a high concentration. Phospholipids are asymmetrically distributed in the PM, and phospholipid flippases play an important role in generating this phospholipid asymmetry. Here, we provide evidence that phospholipid flippases are essential for retaining ergosterol in the PM of yeast. A mutant in three flippases, Dnf1-Lem3, Dnf2-Lem3, and Dnf3-Crf1, and a membrane protein, Sfk1, showed a severe growth defect. We recently identified Sfk1 as a PM protein involved in phospholipid asymmetry. The PM of this mutant showed high permeability and low density. Staining with the sterol probe filipin and the expression of a sterol biosensor revealed that ergosterol was not retained in the PM. Instead, ergosterol accumulated in an esterified form in lipid droplets. We propose that ergosterol is retained in the PM by the asymmetrical distribution of phospholipids and the action of Sfk1. Once phospholipid asymmetry is severely disrupted, sterols might be exposed on the cytoplasmic leaflet of the PM and actively transported to the endoplasmic reticulum by sterol transfer proteins.
INTRODUCTION
Heterogeneity in the distribution of membrane phospholipids and sterols is essential for the diverse functions of cells. In the plasma membrane (PM) of eukaryotic cells, phosphatidylcholine (PC), sphingolipids, and gangliosides are predominantly distributed in the extracellular leaflet, whereas phosphatidylethanolamine (PE), phosphatidylserine (PS), and other charged lipids are mainly localized to the cytoplasmic leaflet (Zachowski, 1993; van Meer, 2011; Murate et al., 2015; Kobayashi and Menon, 2018). This asymmetric distribution of phospholipids is controlled by three types of lipid translocators: flippase, catalyzing inward phospholipid translocation (flip) (Hankins et al., 2015; Panatala et al., 2015; Andersen et al., 2016); floppase, catalyzing outward phospholipid translocation (flop) (Quazi and Molday, 2011; Hankins et al., 2015; Neumann et al., 2017); and scramblase, catalyzing bidirectional phospholipid translocation (Nagata et al., 2020).
Accumulating genetic and biochemical evidence indicates that flippases are integrally linked to phospholipid asymmetry of the organelle membrane from yeast to mammalian cells. Flippases, which are type 4 P-type ATPases (P4-ATPases), have the ability to translocate phospholipids from the extracellular leaflet of the PM or luminal leaflet of endomembranes to the cytoplasmic leaflet (Panatala et al., 2015). At the cellular level, flippases are associated with diverse physiological functions. Flippases in endomembranes function primarily in membrane trafficking processes (Chen et al., 1999; Gall et al., 2002; Hua et al., 2002; Pomorski et al., 2003; Saito et al., 2004; Furuta et al., 2007; Mioka et al., 2014; Lee et al., 2015; Tanaka et al., 2016), whereas those located in the PM are involved in multiple cellular processes: membrane trafficking (Hua et al., 2002; Pomorski et al., 2003; Furuta et al., 2007; Hachiro et al., 2013), apoptosis signaling (Segawa et al., 2014), mating signaling (Sartorel et al., 2015), the apical membrane barrier (Paulusma et al., 2006), cell polarity (Iwamoto et al., 2004; Saito et al., 2007; Das et al., 2012), and cell migration (Kato et al., 2013).
Flippases form heterodimeric complexes with noncatalytic subunits of the Cdc50 family. Budding yeast has five P4-ATPases: Drs2, Dnf1, Dnf2, Dnf3, and Neo1 (Tanaka et al., 2011) and three Cdc50 family member proteins: Cdc50, Lem3, and Crf1 (Saito et al., 2004; Furuta et al., 2007). Drs2 and Dnf3 interact with Cdc50 and Crf1, respectively, and are mainly localized to the endomembrane, such as the trans-Golgi network (TGN) and endosomes. On the other hand, both Dnf1 and Dnf2 form complexes with Lem3 and are mainly localized to the PM (Kato et al., 2002; Pomorski et al., 2003). Except for Neo1, interactions between the P4-ATPases and Cdc50 subunits are essential for endoplasmic reticulum (ER) exit and proper subcellular localization of the complexes but may also contribute to their lipid translocase activity and functions (Saito et al., 2004; Noji et al., 2006; Furuta et al., 2007; Bryde et al., 2010; Takahashi et al., 2011; Puts et al., 2012). Thus, phenotypes in P4-ATPase mutants are phenocopied by their subunit mutants (Saito et al., 2004; Furuta et al., 2007).
Dnf1/2-Lem3 complexes are endocytosed but recycled back to the PM through the endocytic recycling pathway (Saito et al., 2004; Furuta et al., 2007), maintaining the localization of these complexes to the PM. Genetic analyses suggested that the Dnf1/2-Lem3 complexes have PE and PS translocation activity (Kato et al., 2002; Pomorski et al., 2003; Parsons et al., 2006; Stevens et al., 2008). Considering the localization and activity of Dnf1/2-Lem3 complexes, they maintain phospholipid asymmetry predominantly at the PM. Compared to the other four P4-ATPases, little is known about the activity and function of the Dnf3-Crf1 complex. However, the deletion of DNF3 increases the sensitivity of the dnf1∆ dnf2∆ double mutant to the PE-binding peptide duramycin (Sartorel et al., 2015), and Dnf3 is implicated in the translocation of PS across the PM (Frøsig et al., 2020), suggesting possible functions of the Dnf3-Crf1 complex in PM phospholipid translocation.
In addition to Dnf1/2-Lem3, some regulators are involved in phospholipid asymmetry of the PM. Serine/threonine kinases Fpk1/2 up-regulate Dnf1/2 flippase activity via phosphorylation (Nakano et al., 2008). Pdr5p and Yor1p, two multidrug ABC transporters (Decottignies et al., 1998; Pomorski et al., 2003), and Opt2, a member of the oligopeptide transporter family (Yamauchi et al., 2015), are implicated in the flop of phospholipids. Recently, we isolated Sfk1 as a multicopy suppressor of the lem3∆ mutant; overexpression of Sfk1 suppressed PE and PS exposure in the PM (Mioka et al., 2018). Sfk1 is a conserved transmembrane protein belonging to the TMEM150/FRAG1/DRAM family (Chung et al., 2015). From genetic analyses, we proposed that Sfk1 might negatively regulate the transbilayer movement of phospholipids irrespective of direction in an unprecedented way. The lem3∆ sfk1∆ double mutant exhibits more severe defects in PE and PS asymmetry in the PM than the lem3∆ mutant, and the lem3∆ sfk1∆ mutant exhibits increased permeability of the PM (Mioka et al., 2018). However, these mutations do not affect cell growth. Given that PM phospholipid asymmetry is commonly observed in eukaryotes, it may be speculated that phospholipid asymmetry plays an important role (e.g., is essential for cell growth). Thus, there might be a gene that functions redundantly with LEM3 and SFK1 to control phospholipid asymmetry.
Another important feature of the PM is that this membrane is rich in sterols. Sterols such as mammalian cholesterol and the fungal ergosterol are essential membrane components with tightly controlled homeostasis (Lange and Steck, 2016). At the cellular level, the PM contains approximately 30–40 mol% cholesterol in PM lipids, whereas the ER contains approximately 5 mol% cholesterol (Radhakrishnan et al., 2008; Holthuis and Menon, 2014). Sterols are inserted into lipid membranes through the interaction between 3-hydroxyl groups and hydrocarbon rings of sterols and polar head groups and hydrocarbon chains of phospholipids, respectively (Ali et al., 2007). Each phospholipid has a different affinity for sterols, which determines the strength of their interaction with sterols (Lange and Steck, 2008; Almeida, 2009). Sphingolipids, PC, and PS interact strongly with sterol, whereas phospholipids with small polar head groups and unsaturated fatty acyl tails exhibit weaker interactions (Ramstedt and Slotte, 2006; Lange et al., 2013; Maekawa and Fairn, 2015). Numerous studies have suggested that these interactions contribute to the properties of the PM, including tight packing, high rigidity, and low permeability. However, it is unclear how the PM retains such a high concentration of sterols and whether the asymmetric distribution of PE and PS is involved in retaining sterols in the PM.
In this study, we searched for genes that functionally interact with LEM3 and SFK1 by synthetic lethal genetic screening and identified dnf3 and crf1 as interacting partners. The conditional crf1 lem3 sfk1 triple mutant cannot maintain ergosterol in the PM and instead accumulates esterified ergosterol in the lipid droplet (LD). Our results suggest that Dnf1/2-Lem3 and Dnf3-Crf1 flippases and Sfk1 function cooperatively to maintain the phospholipid asymmetry of the PM, which is essential for sterol retention in the PM and thus for the homeostatic control of sterol.
RESULTS
Dnf3-Crf1 flippase is involved in PM phospholipid asymmetry together with Dnf1/2-Lem3 flippases and Sfk1
To isolate genes involved in the regulation of phospholipid asymmetry of the PM in conjunction with Lem3 and Sfk1, we searched for mutations that display synthetic lethality with lem3∆ sfk1∆ mutations at 30°C. We isolated a new allele of the flippase noncatalytic subunit crf1 (Figure 1A). To confirm this synthetic lethality, we crossed the crf1∆ lem3∆ mutant to the lem3∆ sfk1∆ mutant, followed by tetrad analysis (Supplemental Figure 1, A and B). The crf1∆ lem3∆ sfk1∆ triple mutant did not germinate at 30°C but germinated at 25°C despite severe growth defects (Supplemental Figure 1B), which allowed us to obtain the crf1∆ lem3∆ sfk1∆ triple mutant for further analysis. We next tested the growth of the crf1∆ lem3∆ sfk1∆ triple mutant at 30 and 37°C. The triple mutant grew very slowly at 30°C and showed lethality at 37°C (Figure 1B). The deletion of DNF3, which encodes the catalytic subunit of Crf1 (Furuta et al., 2007), also grew poorly when combined with lem3∆ sfk1∆ (Supplemental Figure 1C).
FIGURE 1:

Synthetic growth defects of the crf1∆ lem3∆ sfk1∆ mutant. (A) Growth profiles on 5-fluoroorotic acid (5-FOA) plate medium in yeast plasmid shuffling assay. The crf1∆ lem3∆ sfk1∆ mutant harboring pRS316-SFK1 was transformed with YCplac111 (pLEU2-Con), YCplac111-LEM3 (pLEU2-LEM3), YCplac111-CRF1 (pLEU2-CRF1), or pRS315-SFK1 (pLEU2-SFK1). Transformants were streaked onto an SD-Leu + 5-FOA plate and grown at 30°C for 3 d. The cells that require pRS316-SFK1 for growth are sensitive to 5-FOA because pRS316 contains the URA3 gene (Boeke et al., 1984). The right panel illustrates this assay system. (B) Growth profiles by spot growth assay. As described in Materials and Methods, 10-fold serial dilutions of cell cultures were spotted onto YPDA and grown for 1.5 d at 30 or 37°C. (C) Localizations of Dnf3-3xGFP (triple tandem GFP) and Crf1-GFP in the endocytosis-defective vrp1∆ mutant. Cells were grown to mid–log phase in YPDA medium at 30°C. Arrows indicate the cells showing the PM localization of examined proteins. Bar, 5 µm. DIC, differential interference contrast.
Dnf3 is mainly localized to endosomal/Golgi membranes (Hua et al., 2002; Pomorski et al., 2003), but it was suggested that Dnf3 also functions at the PM (Frøsig et al., 2020). To examine whether Dnf3-Crf1 is transported to the PM, we used the endocytosis-deficient vrp1∆ mutant (Munn et al., 1995). Both Dnf3-3xGFP (triple tandem green fluorescent protein [GFP]) and Crf1-GFP were localized to intracellular structures but were barely detectable in the PM of the wild type. However, they were observed in the PM when endocytosis was inhibited (Figure 1C). This result indicates that the Dnf3-Crf1 flippase is transported between the PM and endomembranes, similar to Drs2-Cdc50 (Saito et al., 2004). These results raise the possibility that the synthetic growth defect of the crf1∆ lem3∆ sfk1∆ mutant is caused by defects in the PM, and this point was analyzed further.
We attempted to perform phenotypic analysis of the crf1∆ lem3∆ sfk1∆ triple mutant. However, the expression of some GFP-fused proteins resulted in lethality in the crf1∆ lem3∆ sfk1∆ background. Thus, we constructed temperature-sensitive (ts) mutants of SFK1 by random mutagenesis in the lem3∆ crf1∆ background as described in Materials and Methods. The crf1∆ lem3∆ sfk1-2 mutant exhibited acceptable growth at 30°C but a severe growth defect at 37°C (Figure 2A). From the growth profiles of the crf1∆ lem3∆ sfk1-2 mutant at 30 and 37°C (Supplemental Figure 2), we analyzed phenotypes of the triple mutant after culturing for 6 h after the shift to 37°C. DNA sequencing of the sfk1-2 mutant allele revealed that sfk1-2 contained one mutation that resulted in an amino acid substitution W16R (Figure 2B), which was located in the N-terminal transmembrane region.
FIGURE 2:

The crf1∆ lem3∆ sfk1 triple mutants show severe defects in phospholipid asymmetry but not in membrane trafficking. (A) Isolation of the sfk1-2 ts mutant. Tenfold serial dilutions of cell cultures were spotted onto a YPDA plate, followed by incubation at 30 or 37°C for 1.5 d. (B) Amino acid substitution of the Sfk1-2 mutant protein. The W16R substitution occurs in the first transmembrane domain of Sfk1-2. (C) The crf1∆ lem3∆ sfk1-2 triple mutant was sensitive to PapB and duramycin. Tenfold serial dilutions were spotted onto a YPDA plate containing PapB or duramycin, followed by incubation at 30°C for 2 d. (D) PE was most exposed in the crf1∆ lem3∆ sfk1∆ mutant. Left panels: cells were cultured in YPDA at 30°C, and exposed PE was visualized by staining with Bio-Ro and Alexa Fluor 488–labeled streptavidin. Dashed lines indicate cell edges. Middle panel: the percentages of cells showing PE exposure were determined and are expressed as the mean ± SD of three independent experiments (n > 81 cells in total for each strain). An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05). Right panel: fluorescence intensity at the PM was quantitated as described in Materials and Methods. The ratio of the fluorescence at the PM (Fpm)/that of whole cell (Fwhole cell) was determined and is expressed in a boxplot (whiskers: maximum and minimum values; box: first quartile, median, and third quartile; circle: average). The numbers of cells analyzed were 23, 38, and 35 for the wild type, lem3∆ sfk1∆, and crf1∆ lem3∆ sfk1∆, respectively. An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (E) GFP-evt-2PH was mislocalized in the crf1∆ lem3∆ sfk1-2 mutant. Cells were grown in YPDA medium to mid–log phase at 30°C and then shifted to 37°C, followed by incubation for 6 h. Right panel: the percentage of cells with GFP-evt-2PH at the PM was determined and is expressed as the mean ± SD of three independent experiments (n >154 cells in total for each strain). An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (F) Normal localization of PM proteins in the crf1∆ lem3∆ sfk1-2 mutant. Cells were cultured as in E. Pma1 was detected by immunostaining as described in Materials and Methods. (G) Endocytosis was not significantly affected in the crf1∆ lem3∆ sfk1-2 mutant. Cells expressing the vacuole membrane marker Vph1-3xGFP were cultured as in E. Then, cells were incubated with FM4-64 on ice for 30 min, followed by incubation at 37°C for 30 min. Arrows indicate the colocalization of FM4-64 and Vph1-3xGFP. Bars, 5 µm.
Phospholipid asymmetry defects cause the exposure of PS and PE to the extracellular leaflet of the PM. The exposed PS and PE can be indirectly measured by examination of the growth sensitivities of the mutants to the PS-binding cyclodepsipeptide papuamide B (PapB) and PE-binding tetracyclic peptide duramycin. We previously reported that the lem3∆ sfk1∆ double mutant exhibited high sensitivities to both peptides (Mioka et al., 2018). Thus, we first tested the growth sensitivity of the crf1∆ lem3∆ sfk1-2 triple mutant to these peptides at 30°C (Figure 2C). The addition of either the crf1∆ or the dnf3∆ mutation to the lem3∆ mutant elevated the sensitivities to both peptides (Supplemental Figure 3A), consistent with a previous report on the dnf1∆ dnf2∆ dnf3∆ mutant (Sartorel et al., 2015). The crf1∆ lem3∆ sfk1-2 mutant did not grow at the concentrations at which the crf1∆ lem3∆ and lem3∆ sfk1-2 double mutants could grow (Figure 2C). This was confirmed by dose-response growth curve experiment (Supplemental Figure 3B). These results suggest that the crf1∆ lem3∆ sfk1-2 triple mutant exposed more PS and PE even at the permissive temperature than did the double mutants. To further confirm the defect in phospholipid asymmetry in the triple mutant, we next visualized the PE exposed to the extracellular surface using the PE-binding biotinylated Ro 09-0198 peptide (Bio-Ro). Fluorescence signals were not detected in either the wild type or the crf1∆ sfk1∆ double mutant but were detected in both the crf1∆ lem3∆ (45%) and lem3∆ sfk1∆ (58%) double mutants (Figure 2D, left and middle panels). In the crf1∆ lem3∆ sfk1∆ triple mutant, the proportion of cells with fluorescence signals increased to 85%. Furthermore, the average signal intensity in the triple mutant was 1.35-fold higher than that in the lem3∆ sfk1∆ mutant (Figure 2D, right panel).
Next, we examined PS distribution in the cytoplasmic leaflet of the PM in the crf1∆ lem3∆ sfk1-2 triple mutant. To visualize PS, we expressed PS biosensors, the C2 domain of lactadherin (Lact-C2) (Yeung et al., 2008) and the pleckstrin homology (PH) domain of evectin-2 (evt-2PH) (Uchida et al., 2011). GFP-Lact-C2 was mainly distributed in the PM of the examined cells, but intracellular localization was also observed in the triple mutant (Figure 2E, left panel; Supplemental Figure 3C). In contrast, GFP-evt-2PH was normally distributed only to the PM in the wild type and double mutants (more than 96% of cells with PM distribution), whereas the GFP-evt-2PH signal was lost or significantly reduced from the PM in the crf1∆ lem3∆ sfk1-2 triple mutant (59% of cells with PM distribution) (Figure 2E, middle and right panels; Supplemental Figure 3D). We speculate that Lact-C2 has a higher affinity for PS, resulting in the detection of a lower level of PS at the PM. Both GFP-Lact-C2 and GFP-evt-2PH were localized to intracellular structures in the triple mutant. However, they appeared to be localized to different structures, which may represent PS-containing membranes or nonspecific protein aggregations. Taken together, these results suggest that the asymmetric distribution of PE and PS was most disturbed in the crf1∆ lem3∆ sfk1 triple mutants.
As the Dnf3-Crf1 complex was mainly localized to endosomal/TGN compartments (Figure 1C) (Hua et al., 2002; Pomorski et al., 2003), the crf1∆ lem3∆ sfk1-2 mutant may exhibit a defect in membrane trafficking. We examined the localization of the endocytic recycling marker GFP-Snc1, which is mainly localized to polarized PM sites (Lewis et al., 2000), but its localization was not affected in the crf1∆ lem3∆ sfk1-2 triple mutant at 37°C (Figure 2F). Similarly, two PM proteins, Pdr5-GFP (ABC transporter) (de Thozee et al., 2007) and Pma1 (H+-ATPase) (Malinska et al., 2004), were normally transported to the PM in the crf1∆ lem3∆ sfk1-2 triple mutant (Figure 2F). We also examined endocytosis in the crf1∆ lem3∆ sfk1-2 triple mutant by uptake of the lipophilic dye FM4-64 (Vida and Emr, 1995). The FM4-64 signal was well colocalized to the vacuole membrane marker Vph1-3xGFP (Peters et al., 2001) in both the wild type and crf1∆ lem3∆ sfk1-2 triple mutant after 30 min of incubation, suggesting that the triple mutant did not have obvious defects in endocytosis (Figure 2G). These results suggest that the crf1∆ lem3∆ sfk1-2 triple mutant is not defective in membrane trafficking to or from the PM.
The PM shows high permeability and low density in the crf1∆ lem3∆ sfk1 triple mutants
Phospholipid asymmetry defects may have a profound effect on PM properties. Previously, we showed that the lem3∆ sfk1∆ double mutant exhibits an increase in membrane permeability by measuring rhodamine dye uptake (Mioka et al., 2018). This experiment was performed in the crf1∆ lem3∆ sfk1∆ triple mutant, and the results suggest that the permeability is further enhanced in the triple mutant compared with that in the lem3∆ sfk1∆ double mutant (Figure 3A). The large increase in membrane permeability prompted us to examine whether the lipid composition changes in the PM of the crf1∆ lem3∆ sfk1-2 triple mutant. We performed sucrose density gradient fractionation to isolate the PM. In the wild type, PM markers, both Pdr5-GFP (de Thozee et al., 2007) and Pma1 (Serrano et al., 1986; Bagnat et al., 2001), were recovered in high-density fractions, whereas Kex2, which is localized to endosomal/TGN compartments (Brickner and Fuller, 1997; Lewis et al., 2000), peaked at a lower density (Figure 3B). However, in the crf1∆ lem3∆ sfk1-2 triple mutant, Pdr5-GFP, Pma1, and Kex2 were recovered together in lower-density fractions (fractions 3–9) (Figure 3B). Pdr5-GFP and Pma1 were normally localized to the PM in the crf1∆ lem3∆ sfk1-2 triple mutant in microscopic analysis (Figure 2F), suggesting a decrease in PM density, which makes PM isolation from the triple mutant technically challenging. Thus, we measured the phospholipid composition in the total cellular lipids. No significant difference in lipid composition was found in the double and triple mutants (Supplemental Figure 4). These results suggest that a major change occurs in the PM of the crf1∆ lem3∆ sfk1 triple mutant.
FIGURE 3:
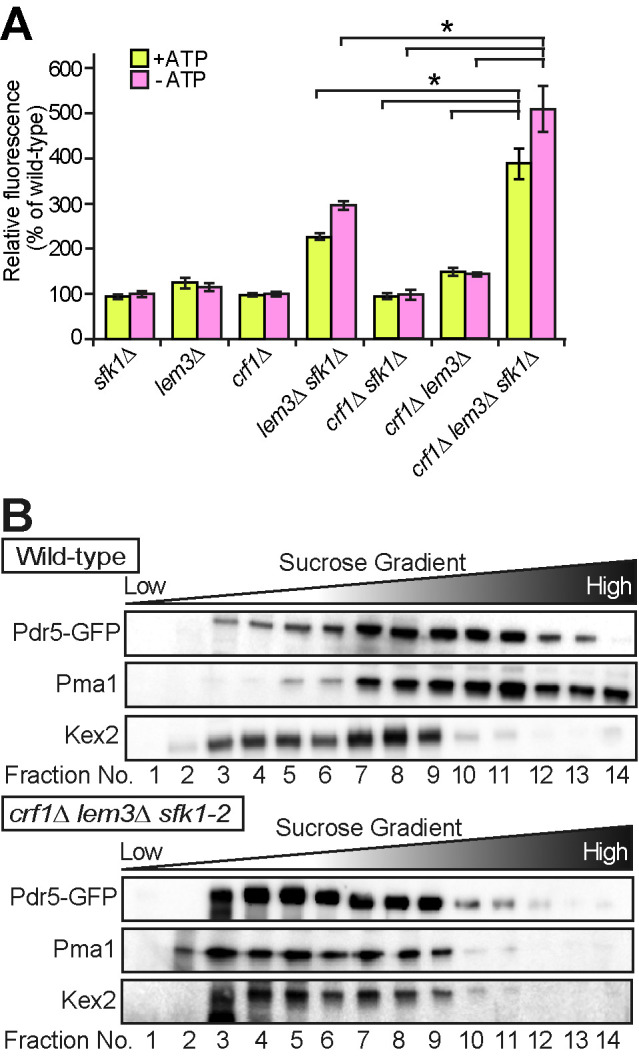
Increased permeability and decreased density of the PM in the crf1∆ lem3∆ sfk1 triple mutants. (A) Rhodamine uptake is increased in the crf1∆ lem3∆ sfk1∆ triple mutant. Cells were cultured in YPDA medium at 30°C, preincubated in SD medium in the absence (+ATP) or presence (–ATP) of 1 mM sodium azide for 30 min at 30°C, and incubated with rhodamine 6G for 60 min at 30°C. Rhodamine accumulation was measured as described in Materials and Methods. Values represent the mean ± SD from three independent experiments. Asterisks indicate a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (B) Sucrose density gradient centrifugation analysis of PM proteins, Pdr5-GFP and Pma1, and a TGN/endosome protein, Kex2, in the crf1∆ lem3∆ sfk1-2 triple mutant. Cells were cultured as in Figure 2E. Cell lysates were prepared from the wild type and the crf1∆ lem3∆ sfk1-2 triple mutant expressing Pdr5-GFP and fractionated in 22–60% sucrose step density gradients as described in Materials and Methods. Equivalent volumes from each fraction were subjected to SDS–PAGE, and proteins were detected by immunoblotting.
Isolation of KES1 as a multicopy suppressor of the crf1∆ lem3∆ sfk1-2 mutation
To explore the essential functions of phospholipid asymmetry in the PM, we screened for multicopy suppressors of the ts growth defect of the crf1∆ lem3∆ sfk1-2 triple mutant. Multicopy suppressor is a gene whose overexpression suppresses a mutant phenotype. We found that overexpression of KES1 suppressed the growth defect (Figure 4A). Kes1, also known as Osh4, is an oxysterol-binding protein (OSBP) homologue (Osh) that is implicated in sterol transport within cells (Antonny et al., 2018). Budding yeast contains seven Osh homologues, Osh1–7, that exchange specific lipids between organelles (Lev, 2010). We next tested whether overexpression of other Osh proteins, except for OSH1, which localizes to the nucleus–vacuole junction (Manik et al., 2017), could suppress the growth defect of the crf1∆ lem3∆ sfk1-2 triple mutant. Only KES1 overexpression suppressed the ts growth defect of the crf1∆ lem3∆ sfk1-2 triple mutant (Figure 4A). Increased rhodamine uptake was also suppressed by KES1 overexpression in the crf1∆ lem3∆ sfk1-2 triple mutant (Figure 4B)
FIGURE 4:

Overexpression of KES1 partially suppresses the phenotypes in the crf1∆ lem3∆ sfk1-2 triple mutant. (A) Suppression of the growth defect. Cell growth was examined in the crf1∆ lem3∆ sfk1-2 triple mutant carrying pRS316-SFK1, YEplac195, or YEplac195-OSH2-7. YEplac195 is a multicopy plasmid. After cells were cultured in SDA-Ura medium at 30°C overnight, 10-fold serial dilutions were spotted onto a YPDA plate, followed by incubation for 2 d at 30 or 37°C. (B) Suppression of high membrane permeability. Rhodamine uptake was examined in the wild type and crf1∆ lem3∆ sfk1-2 triple mutant harboring YEplac195 (pCon), YEplac195-KES1 (pKes1), or YEplac195-KES1L111D (pKes1m). Cells were cultured as in Figure 2E, except that SDA-Ura medium was used. The rhodamine uptake assay was performed as described in Materials and Methods. Rhodamine uptake is represented as a relative value of that (100%) in the wild type harboring YEplac195 incubated at 30°C. Values represent the mean ± SD from three independent experiments. Asterisks indicate a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (C) Overexpression of ergosterol binding–deficient KES1 mutants does not suppress ts growth of the crf1∆ lem3∆ sfk1-2 triple mutant. Cell growth was examined in the triple mutant transformed with pRS316-SFK1, YEplac195, YEplac195-KES1, or YEplac195-KES1 mutants (E117A, L111D, and Y97F). Cells were spotted and grown as in A.
We next examined whether the sterol-binding activity of Kes1 is required to suppress the growth defect of the crf1∆ lem3∆ sfk1-2 triple mutant. Overexpression of KES1E117A, KES1L111D, and KES1Y97F, which abolishes the binding of Kes1 to sterols (Im et al., 2005), did not suppress the growth defect of the triple mutant (Figure 4C). Correspondingly, overexpression of KES1L111D did not suppress rhodamine accumulation (Figure 4B, pKes1m). These results suggest that the crf1∆ lem3∆ sfk1-2 triple mutant may have a defect in intracellular transport, homeostasis, or distribution of ergosterol.
Ergosterol is reduced in the PM of the crf1∆ lem3∆ sfk1 triple mutants
We examined the distribution of ergosterol in the crf1∆ lem3∆ sfk1 triple mutants. Filipin is a polyene antibiotic that binds to cholesterol and ergosterol and is used as a probe for cellular sterol distribution (Beh and Rine, 2004; Kishimoto et al., 2016). Wild type and double mutants were evenly labeled with filipin at the PM (Figure 5A: Supplemental Figure 5). However, the crf1∆ lem3∆ sfk1∆ triple mutant drastically decreased filipin labeling to the PM and instead showed the enhancement of intracellular labeling (Figure 5A). Eighty-three percent of the crf1∆ lem3∆ sfk1∆ triple mutant cells clearly displayed the loss or reduction of filipin signal in the PM (n = 98 cells). Quantitative analysis of fluorescence images further confirmed the decrease in filipin intensity on the PM in the triple mutant (Figure 5A).
FIGURE 5:

Ergosterol is decreased in the PM of the crf1∆ lem3∆ sfk1 triple mutants. (A) Staining of ergosterol with filipin in the crf1∆ lem3∆ sfk1∆ triple mutant. Cells were grown in YPDA at 30°C, and filipin staining was performed as described in Materials and Methods. Right panel: fluorescence intensity at the PM was quantitated as described in Materials and Methods. The ratio of the fluorescence at the PM (Fpm)/that of whole cell (Fwhole cell) was determined and is expressed with a boxplot (whiskers: maximum and minimum values; box: first quartile, median, and third quartile; circle: average). The numbers of cells analyzed were 100, 100, 99, 97, and 98 for the wild type, lem3∆ sfk1∆, crf1∆ sfk1∆, crf1∆ lem3∆, and crf1∆ lem3∆ sfk1∆, respectively. An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (B) The distribution of GFP- and GFPenvy-D4H in wild-type cells. Cells harboring pRS316-GFP-D4H or -GFPenvy-D4H were grown in SDA-Ura medium at 30°C to mid–log phase. Right panel: the percentage of cells with GFP- or GFPenvy-D4H at the PM was determined and is expressed as the mean ± SD of three independent experiments (n > 155 cells in total for each strain). An asterisk indicates a significant difference, as determined by a two-tailed Student’s t test (p < 0.05). (C) GFPenvy-D4H binds to ergosterol in vitro. The liposome sedimentation assay was performed as described in Materials and Methods. Proteins were incubated with liposomes composed of DOPC or DOPC and 50% (mol) ergosterol (Ergo) or cholesterol (Chol) (with corresponding reduction in DOPC), followed by pelleting by centrifugation. Supernatant (Sup) and pellet (Ppt) fractions were separated by SDS–PAGE, followed by staining with Coomassie Brilliant Blue. Arrowheads indicate GFP-D4H or GFPenvy-D4H. A lower band appears to be an incomplete fragment. Right panel: proteins bound to liposomes were quantitated by an image analyzer. The percentage of the bound protein was determined as a relative value of that bound to cholesterol liposomes and is expressed as the mean ± SD of three independent experiments. Asterisks indicate a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (D) GFPenvy-D4H binds to DOPC liposomes containing more than 25% ergosterol. A liposome sedimentation assay was performed as in C with DOPC liposomes containing the indicated mol % ergosterol. An arrowhead indicates GFPenvy-D4H. A lower band appears to be an incomplete fragment. The amount of protein bound to liposomes is expressed as a relative value (percentage) of that bound to 60% ergosterol liposomes. Values represent the mean ± SD of three independent experiments. “S” and “P” indicate supernatant and pellet fractions, respectively. Asterisks and “n.s.” indicate significant and no significant differences as determined by the Tukey–Kramer test (*p < 0.05), respectively. (E) The PM localization of GFPenvy-D4H was dependent on ergosterol. ERG11 was expressed under the control of the glucose-repressible PGAL1 promoter. Cells were grown in SGA-Ura medium to mid–log phase at 30°C and then inoculated into fresh galactose (SGA-Ura) or glucose (SDA-Ura) medium, followed by incubation for 12 h at 30°C. Right panel: the percentage of cells with GFPenvy-D4H at the PM was determined and is expressed as the mean ± SD of three independent experiments (n > 108 cells in total for each condition). An asterisk indicates a significant difference, as determined by a two-tailed Student’s t test (p < 0.05). (F) GFPenvy-D4H was not localized to the PM in the crf1∆ lem3∆ sfk1-2 triple mutant. Cells were cultured as in Figure 2E, except that SDA-Ura medium was used. Right panel: the percentage of cells with GFPenvy-D4H at the PM was determined and is expressed as the mean ± SD of three independent experiments (n > 253 cells in total for each strain). An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05). Bars, 5 µm.
We also examined another sterol biosensor, D4H. A bacterial protein toxin, perfringolysin O, binds to cholesterol via its domain 4 (D4) (Shimada et al., 2002; Johnson et al., 2012). A D4 derivative, D4H (D4D434S), has been developed as a more sensitive probe; it binds to liposomes containing 20–30% cholesterol mole concentration (Johnson et al., 2012; Maekawa and Fairn, 2015). Although D4H has recently been used to detect the PM sterol in fission yeast (Marek et al., 2020), D4H has not been applied to budding yeast. We generated two fluorescent protein–conjugated D4Hs, GFP-D4H and GFPenvy-D4H, in which GFPenvy is a photostable dimeric GFP derivative (Slubowski et al., 2015; Bajar et al., 2016). When expressed in wild-type cells, GFP-D4H was localized to the PM in 35% of the cells (Figure 5B). In contrast, GFPenvy-D4H was localized to the PM in 94% of the cells. Interestingly, GFPenvy-D4H showed a characteristic localization pattern; it preferentially localized to daughter cells compared with mother cells. This localization pattern is described in the last part of Results in more detail.
We next examined the binding activity of recombinant GFP-D4H and GFPenvy-D4H to ergosterol in vitro by liposome sedimentation assay using 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) liposomes containing either 50% cholesterol or 50% ergosterol. Consistent with a previous report (Savinov and Heuck, 2017), GFP-D4H bound to ergosterol liposomes at an efficiency of 20% of that to cholesterol liposomes (Figure 5C). On the other hand, GFPenvy-D4H bound to ergosterol liposomes at an efficiency of 56% of that to cholesterol liposomes (Figure 5C), consistent with the results in living cells. The affinity of GFPenvy-D4H to ergosterol was examined with DOPC liposomes containing different concentrations of ergosterol from 10 to 60 mol%. Binding was detected when the ergosterol concentration was 25% or higher (Figure 5D). These results are comparable to the affinity of D4H to cholesterol (Maekawa and Fairn, 2015). The higher affinity of GFPenvy-D4H to ergosterol than GFP-D4H might be because GFPenvy forms a dimeric structure (Bajar et al., 2016).
We next confirmed that GFPenvy-D4H binds to ergosterol in living cells. The ERG11 gene encodes lanosterol demethylase, which is essential for ergosterol synthesis (Daum et al., 1998). The shutoff of ERG11 gene expression inhibited the distribution of GFPenvy-D4H to the PM, accompanied by a decrease in free ergosterol levels (Figure 5E; Supplemental Figure 6A). Treatment with the Erg11 inhibitor fluconazole confirmed this observation (Supplemental Figure 6B). We also examined the localization of GFPenvy-D4H in mutants of genes involved in the late steps of the ergosterol biosynthesis pathway (ERG2-6) (Munn et al., 1999; Heese-Peck et al., 2002). GFPenvy-D4H was not localized to the PM except for erg4∆, which catalyzes the last step (Supplemental Figure 6C). These results suggest that GFPenvy-D4H is localized to the PM by binding to ergosterol.
We examined the distribution of GFPenvy-D4H in the crf1∆ lem3∆ sfk1-2 triple mutant. The localization of GFPenvy-D4H to the PM decreased to some extent in the lem3∆ sfk1-2 and crf1∆ lem3∆ double mutants, but it drastically decreased to 16% in the crf1∆ lem3∆ sfk1-2 triple mutant (Figure 5F; Supplemental Figure 6D). Taking this together with the results of filipin staining, we concluded that ergosterol is significantly lost from the PM in the crf1∆ lem3∆ sfk1 triple mutants. Kes1 overexpression increased the PM localization of GFPenvy-D4H from 25 to 55% in the crf1∆ lem3∆ sfk1-2 triple mutant (Supplemental Figure 6E). These results suggest that the increased Kes1 enhances ergosterol transport to or inhibits loss of ergosterol from the PM in the triple mutant and that loss of ergosterol from the PM causes phenotypes of the triple mutant, including the growth defect.
We next examined whether exogenously added ergosterol would suppress the growth defect in the crf1∆ lem3∆ sfk1-2 triple mutant. We used strains carrying the gain-of-function mutation of a transcription factor UPC2, upc2-1 (G888D), which results in increased uptake of exogenous ergosterol under aerobic conditions (Lewis et al., 1988; Crowley et al., 1998). The exogenously added ergosterol, which was sufficient to suppress the growth defect of the ergosterol-deficient hem1∆ mutant (Georgiev et al., 2011), did not suppress the growth defect of the crf1∆ lem3∆ sfk1-2 triple mutant even when KES1 was overexpressed (Figure 6A). We presumed that the triple mutant cannot retain exogenously added ergosterol in the PM. Thus, we monitored the distribution of exogenously added TopFluor-cholesterol (TF-Chol), a fluorescent dye–conjugated cholesterol analogue (Barajas et al., 2014). TF-Chol was retained in the PM of the wild type and double mutants containing upc2-1 but was not in the crf1∆ lem3∆ sfk1-2 upc2-1 mutant; only 23% of the triple mutant showed TF-Chol in the PM (Figure 6B; Supplemental Figure 7). TF-Chol appeared to be internalized into the cell to be incorporated into cytoplasmic punctate structures in the crf1∆ lem3∆ sfk1-2 upc2-1 mutant (Figure 6B). These results suggest that ergosterol is not retained in the PM and that it is transported to intracellular punctate structures in the crf1∆ lem3∆ sfk1-2 mutant.
FIGURE 6:
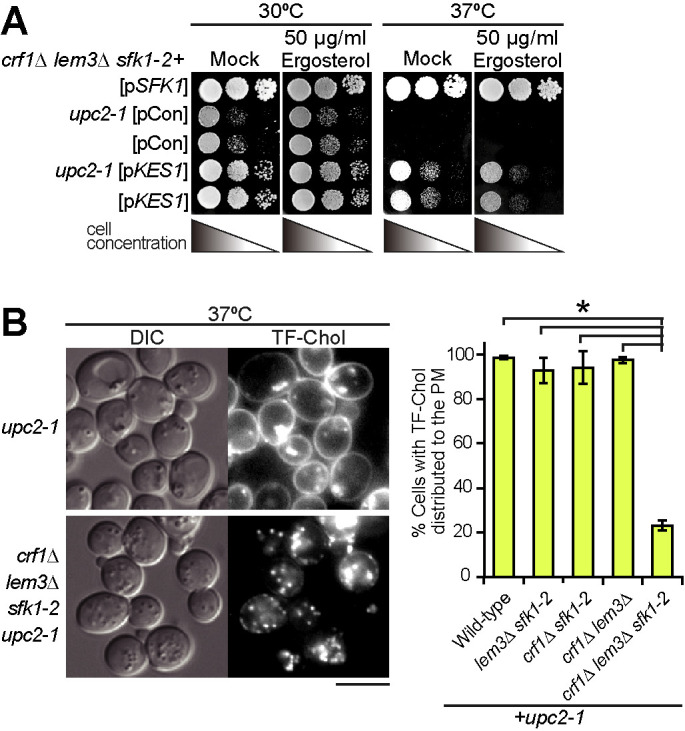
Exogenously added ergosterol appears to not be retained in the PM of the crf1∆ lem3∆ sfk1-2 upc2-1 mutant. (A) Exogenous ergosterol did not suppress ts growth in the crf1∆ lem3∆ sfk1-2 upc2-1 mutant. The crf1∆ lem3∆ sfk1-2 mutant with or without the upc2-1 mutation was transformed with pRS315-SFK1 (pSFK1), YEplac181 (pCon), or YEplac181-KES1 (pKES1). After cells were cultured in SD-Ura-Leu medium at 30°C overnight, 10-fold serial dilutions were spotted onto a YPDA plate containing 0.5% Tween-80 and 0.5% ethanol with or without 50 µg/ml ergosterol, followed by incubation for 1.5 d at 30 or 37°C. (B) TF-Chol is not retained in the PM of the crf1∆ lem3∆ sfk1-2 upc2-1 mutant. Left panel: cells were cultured and labeled with TF-Chol as described in Materials and Methods. Bar, 5 µm. Right panel: the percentage of cells with TF-Chol at the PM was determined and is expressed as the mean ± SD of three independent experiments (n > 290 cells in total for each strain). An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05).
Ergosterol is esterified and accumulated in LDs in the crf1∆ lem3∆ sfk1-2 mutant
The loss of ergosterol in the PM raises a question: where does ergosterol distribute in the cell? We performed thin-layer chromatography (TLC) analysis of total sterols extracted from the cells. In double mutants, the free ergosterol level was approximately 80–85% of that in the wild type, but it decreased to 50% in the crf1∆ lem3∆ sfk1∆ triple mutant (Figure 7A). TLC analysis also showed a large increase in esterified ergosterol in the crf1∆ lem3∆ sfk1∆ triple mutant. We confirmed that this spot was observed in the wild type at the stationary phase, but not in the acyl-CoA:sterol acyltransferase–deficient are1∆ are2∆ mutant (Supplemental Figure 8) (Klug and Daum, 2014). Because esterified ergosterol is the main component of LDs, these results suggest that LDs are increased in the crf1∆ lem3∆ sfk1∆ triple mutant. To confirm this, we stained LDs with the lipophilic dye Nile red, which stains neutral lipids in LDs, triacylglycerol and esterified ergosterol (Greenspan et al., 1985). Neither the wild type nor the double mutants showed obvious staining of Nile red, whereas the crf1∆ lem3∆ sfk1∆ triple mutant exhibited a clear increase in the number of cells showing Nile red puncta (Figure 7B; Supplemental Figure 9A). We further examined the localization of GFP-tagged LD-related proteins, Tgl1 (steryl ester lipase) (Jandrositz et al., 2005) and Faa4 (long-chain fatty-acid-CoA ligase) (Kurat et al., 2006). The wild type and the double mutants contained a few puncta of these proteins, whereas the numbers of Tgl1-GFP and Faa4-GFP puncta increased 2.5- and 2.8-fold, respectively, in the crf1∆ lem3∆ sfk1-2 triple mutant compared with those in the wild type (Figure 7C; Supplemental Figure 9B). We confirmed that Tgl1-GFP and Faa4-GFP puncta were colocalized with Nile red–positive puncta; 85% of Tgl1-GFP (n = 377) and 88% of Faa4-GFP (n = 453) puncta were colocalized with Nile red in the crf1∆ lem3∆ sfk1-2 triple mutant (Figure 7C). These results suggest that a substantial amount of ergosterol was esterified and accumulated in LDs in the crf1∆ lem3∆ sfk1 triple mutants.
FIGURE 7:

LDs were increased in the crf1∆ lem3∆ sfk1 triple mutants. (A) TLC analysis of total sterols. Ergosterol contents were analyzed by TLC as described in Materials and Methods. Right panel: the percentage of free ergosterol relative to that of the wild type was determined and is expressed as the mean ± SD derived from the analysis of six independent samples. An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (B) Increase in Nile red–positive puncta in the crf1∆ lem3∆ sfk1∆ triple mutant. Cells were cultured in YPDA medium to mid–log phase at 30°C, followed by Nile red staining. Nile red staining was performed as described in Materials and Methods. Right panel: the percentage of cells with more than three Nile red puncta was determined and is expressed as the mean ± SD of three independent experiments (n > 315 cells in total for each strain). An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05). (C) Increase in LD marker (Tgl1-GFP and Faa4-GFP)-containing structures in the crf1∆ lem3∆ sfk1-2 triple mutant. Cells were cultured as in Figure 2E, followed by Nile red staining. Right panel: the Tgl1-GFP or Faa4-GFP puncta were counted in a single focal plane of each cell and are expressed with boxplots (whiskers: maximum and minimum values; box: first quartile, median, and third quartile; circle: average). The numbers of cells analyzed were 51 and 50 (wild type) and 53 and 51 (triple mutant) for Tgl1-GFP and Faa4-GFP, respectively. Asterisks indicate a significant difference, as determined by a two-tailed Student’s t test (p < 0.05). (D) Colocalization of TF-Chol puncta with Faa4-mCherry in the crf1∆ lem3∆ sfk1-2 upc2-1 mutant. Cells were cultured as in Figure 2E, except that YPDA medium containing TF-Chol was used. Bars, 5 µm.
TF-Chol was also detected in intracellular puncta in the crf1∆ lem3∆ sfk1-2 upc2-1 mutant (Figure 6B). We next examined whether TF-Chol colocalizes with Faa4-mCherry in the triple mutant. The crf1∆ lem3∆ sfk1-2 upc2-1 mutant contained approximately 9–14 TF-Chol puncta per cell, and 88% of these puncta (n = 603) were colocalized with Faa4-mCherry (Figure 7D).
Taken together, these results suggest that ergosterol is not retained in the PM and is transported to LDs in an esterified form, probably via the ER, in the crf1∆ lem3∆ sfk1 triple mutants.
The inhibition of sterol esterification partially suppresses growth defects and sterol retention in the PM of the crf1∆ lem3∆ sfk1-2 mutant
We examined whether inhibition of sterol esterification by mutations in ARE1/ARE2 suppresses the phenotypes of the crf1∆ lem3∆ sfk1-2 triple mutant. The growth defect of the triple mutant was partially suppressed by the are2∆ mutation but not by the are1∆ mutation (Figure 8A). Consistently, Are2 accounts for 65–75% of total cellular acyl-CoA:sterol acyltransferase activity (Yang et al., 1996; Yu et al., 1996). We then examined whether the are2∆ mutation restored sterol retention in the PM in the crf1∆ lem3∆ sfk1-2 triple mutant. The are2∆ mutation increased the number of cells showing the PM localization of GFPenvy-D4H from 17 to 60% in the crf1∆ lem3∆ sfk1-2 mutant (Figure 8B). These results are consistent with our notion that loss of ergosterol from the PM is responsible for the growth defect of the crf1∆ lem3∆ sfk1-2 triple mutant.
FIGURE 8:

The are2∆ mutation partially restores ergosterol in the PM of the crf1∆ lem3∆ sfk1-2 triple mutant. (A) Suppression of the growth defect. Tenfold serial dilutions were spotted onto a YPDA plate, followed by incubation for 1.5 d at 30 or 37°C. (B) Restoration of GFPenvy-D4H localization to the PM. Cells were cultured as in Figure 2E except that SDA-Ura medium was used. Bar, 5 µm. Right panel: the percentage of cells with GFPenvy-D4H at the PM was determined and is expressed as the mean ± SD of three independent experiments (n > 219 cells in total for each strain). Asterisks and “n.s.” indicate significant and not significant differences as determined by the Tukey–Kramer test (*p < 0.05), respectively.
Overexpression of Sfk1 alters the localization of GFPenvy-D4H
The molecular function of Sfk1 remains to be clarified, but our results described above may suggest that Sfk1 is functionally related to ergosterol in the PM. We examined whether a mutation or overexpression of SFK1 affects the localization of GFPenvy-D4H. Budding yeasts grow by budding, which is a polarized growth of the PM (Figure 9A, right panel). GFPenvy-D4H exhibited polarized localizations in many wild-type cells; it was localized to daughter cells (buds) or near the bud neck in medium- or large-budded cells (Figure 9A, yellow and pink arrows). These results suggest that the accessibility to ergosterol is different between bud and mother PMs because filipin, which was used in fixed cells, evenly stained ergosterol in daughter and mother cells. Interestingly, Sfk1 was mainly localized to mother cells but not to daughter cells, as described previously (Audhya and Emr, 2002), showing a localization pattern opposite to that of GFPenvy-D4H (Figure 9, B and C). The GFPenvy-D4H localizations in large-budded cells were categorized into three patterns: 1) localized throughout the PM (not polarized), 2) localized to the bud and mother cell PM near the bud neck (partially polarized), and 3) localized only to the bud (polarized) (Figure 9A, right panel). These differences may be because GFPenvy-D4H was expressed from a low-copy centromeric plasmid, whose copy number varies among individual cells (Gnügge and Rudolf, 2017). The fluorescence intensity profiles of Sfk1-3xmCherry and GFPenvy-D4H are shown for the “polarized” (Figure 9C) and “partially polarized” (Supplemental Figure 10A) patterns. The proportion of these localization patterns was not changed in the sfk1∆ mutant (Figure 9, A and D).
FIGURE 9:

Overexpression of Sfk1 excludes GFPenvy-D4H from the mother cell PM. (A) The polarized distribution of GFPenvy-D4H. Wild-type or sfk1∆ cells carrying pRS316-GFPenvy-D4H were grown in SD-Ura medium to mid–log phase at 30°C. “Polarized,” “partially polarized,” and “not polarized” localizations of GFPenvy-D4H are indicated with pink, yellow, and green arrows, respectively. Right panel illustrates the budding yeast cell cycle. Three patterns of the GFPenvy-D4H localization are shown. (B) Complementary localization of GFPenvy-D4H and Sfk1-3xmCherry to daughter (bud) and mother cells, respectively. Wild-type cells expressing these proteins were grown at 30°C. To show endogenously expressed Sfk1-3xmCherry clearly, the brightness was adjusted to make it brighter. (C) Fluorescence intensity profile of a cell showing the “polarized” pattern of GFPenvy-D4H. Fluorescence signals were quantified along the dotted line from the mother cell to the bud. The brightness of Sfk1-3xmCherry was adjusted as in B. (D) Quantification of three GFPenvy-D4H localization patterns. The cells in A were examined. The percentage of cells showing “polarized,” “partially polarized,” and “not polarized” localizations of GFPenvy-D4H was determined as described in Materials and Methods and is expressed as the mean ± SD of three independent experiments (n > 150 cells in total for each strain). “n.s.” indicates no significant difference between all combinations as determined by a two-tailed Student’s t test. (E) Heterogeneous (high and low) expression of Sfk1-mCherry by a multicopy plasmid. Wild-type cells carrying pRS316-GFPenvy-D4H and YEplac181-SFK1-mCherry were grown in SD-Leu-Ura medium to mid–log phase at 30°C. The brightness was not adjusted after background subtraction. Arrows indicate cells highly (yellow) and lowly (cyan) expressing Sfk1-mCherry. (F) High expression of Sfk1-mCherry significantly increased the “polarized” pattern of GFPenvy-D4H. Cells were examined and categorized as in D. Low or high expression of Sfk1-mCherry was determined as described in the legend of Supplemental Figure 10B. Bars: No, control plasmid; Endo, endogenous expression of Sfk1-3xmCherry; Low, multicopy plasmid of SFK1-mCherry but low expression of Sfk1-mCherry; High, multicopy plasmid of SFK1-mCherry and high expression of Sfk1-mCherry. The percentage of cells showing the indicated patterns is expressed as the mean ± SD of three independent experiments (n > 103 cells in total for each strain). An asterisk indicates a significant difference, as determined by the Tukey–Kramer test (p < 0.05), in the “polarized” and “not polarized” patterns. (G) GFPenvy-D4H was exclusively distributed to the bud in a cell highly expressing Sfk1-mCherry. The brightness is not adjusted after background subtraction. The right panel represents the fluorescence intensity profile quantified as in C. Bars, 3 µm.
We then examined the effect of SFK1 overexpression by using a multicopy plasmid carrying SFK1-mCherry. Expression from a multicopy plasmid generates heterogeneity in the level of gene expression among individual cells because of variation in plasmid copy number (Caunt et al., 1988). We took advantage of this expression characteristic to examine the correlation between the Sfk1 expression level and the D4H localization pattern. Cells were categorized into high- and low-expression groups based on the fluorescence intensity of Sfk1-mCherry. The relative expression level of SFK1-mCherry in highly expressing cells was more than threefold that in lowly expressing cells (Supplemental Figure 10B). In cells lowly expressing Sfk1-mCherry, the GFPenvy-D4H localization pattern was not changed (Figure 9, E, cyan arrows, and F). The mother cell–specific localization pattern of Sfk1-mCherry was not changed in highly expressing cells (Figure 9E, yellow arrows). Interestingly, in cells highly expressing Sfk1-mCherry, GFPenvy-D4H distribution was restricted exclusively to the daughter cells, and those cells that showed the “polarized” pattern were largely increased to 74% (Figure 9F). In these cells, the fluorescence intensity of GFPenvy-D4H was weak in the mother cell PM but increased sharply near the bud neck (Figure 9G). These results suggest that Sfk1 might maintain ergosterol in a state that is inaccessible to GFPenvy-D4H, although its function may be redundant with that of an unknown protein.
DISCUSSION
More than two decades have passed since the first report on the asymmetric distribution of phospholipids in the PM (Zachowski, 1993), but our understanding of its physiological significance is still limited. Our genetic screening reveals that the loss of Dnf1/2-Lem3 and Dnf3-Crf1 flippases and Sfk1 results in severe growth defects, probably due to loss of ergosterol from the PM. The growth defect could be due to increased permeability of the PM or abnormal regulation or/and function of PM proteins. Dnf3 was shown to be involved in some PM functions, including mating pheromone signaling (Sartorel et al., 2015) and pseudohyphal growth (Frøsig et al., 2020), but this is the first demonstration of Dnf3 involvement in essential cell function in a vegetative cell. Because Dnf3 is mainly localized to the TGN, it is also possible that Dnf3 indirectly regulates phospholipid asymmetry in the PM through transport of TGN-derived vesicles. Dnf3 might be also required for transport of an unknown regulator of phospholipid asymmetry to the PM.
Disruption of phospholipid asymmetry is one main reason for the loss of ergosterol from the PM in the triple mutant. Phospholipids interact with sterol via their head groups and acyl chains, which contributes to ordering membrane lipids and securing lipid packing (Mesmin and Maxfield, 2009). In the crf1∆ lem3∆ sfk1 triple mutants, PS and PE are more exposed to the extracellular leaflet than in the double mutants and the level of PS in the cytoplasmic leaflet appears to be decreased. Sterols have a higher affinity to phospholipids containing saturated acyl chains than to those containing unsaturated acyl chains (Mesmin and Maxfield, 2009), and PS and PE species in the PM are more abundant in those containing saturated acyl chains than in other organelles in budding yeast (Schneiter et al., 1999). In addition, according to the umbrella model (Mesmin and Maxfield, 2009), phospholipid head groups in the membrane shield nonpolar cholesterol bodies from the aqueous phase. PS with a large head group has a higher affinity for cholesterol than other phospholipids (Maekawa and Fairn, 2015; Nyholm et al., 2019). Therefore, ergosterol, which is also enriched in the cytoplasmic leaflet (Solanko et al., 2018), loses favorable interacting partners in the triple mutant. This would result in a vast increase in “active ergosterol,” which may be actively removed from the PM by sterol transfer proteins (STPs) (Figure 10; see below).
FIGURE 10:

Model for the coordinated actions of flippases and Sfk1 in the control of PM sterol. PS and PE are retained in the cytoplasmic leaflet of the PM by flippases. PS has a high affinity for ergosterol because of its large head, which may shield sterol molecules from sterol transfer proteins (STPs). Sfk1 might decrease active ergosterol by unknown mechanisms. Thus, flippases and Sfk1 coordinately hold sterols within the membrane interior to limit the accessibility of sterols to the cytoplasm. In the crf1∆ lem3∆ sfk1 triple mutant, the simultaneous loss of these proteins results in reduced shielding by phospholipids, leading to a vast increase of active ergosterol, which is likely highly accessible to and easily extracted by STPs, resulting in the loss of ergosterol from the PM.
Although Sfk1 is implicated in the regulation of phospholipid asymmetry (Figure 2) (Mioka et al., 2018), its protein function remains unknown. Our results that overexpression of Sfk1 excludes GFPenvy-D4H from the mother cell PM suggest that Sfk1 may be functionally relevant to ergosterol. GFPenvy-D4H preferentially localized to the daughter cell PM. However, the ergosterol contents in the cytoplasmic leaflet did not seem to be significantly different between daughter and mother cell PMs because filipin uniformly stained both membranes and because ergosterol is predominantly localized to the cytoplasmic leaflet of the PM, including in mother cells (Solanko et al., 2018). Thus, the differential GFPenvy-D4H localization might reflect different physical states of ergosterol. When sterol levels exceed the interacting capacity of phospholipids in the membrane, a sterol molecule is predicted to be exposed to the surface of the membrane, which increases the chance of its interaction with sterol-sensing proteins. The model defines this behavior as the chemical activation of cholesterol (McConnell and Radhakrishnan, 2003; Maxfield and Menon, 2006; Nelson et al., 2008; Flanagan et al., 2009; Mesmin and Maxfield, 2009; Steck and Lange, 2010). It has been suggested that the D4-containing domain of perfringolysin O preferentially interacts with active cholesterol (Flanagan et al., 2009). Thus, we propose that the chemical activity of ergosterol is higher in the PM of daughter cells than in that of mother cells. The PM of daughter cells is mainly made from newly synthesized lipids by polarized vesicle transport (Pruyne et al., 2004). In this membrane, PE and PS would be exposed to the extracellular leaflet of PM when vesicles are fused with the PM, but they are rapidly flipped to the cytoplasmic leaflet by the action of Dnf1/2-Lem3 flippases, which is localized to the bud PM (Kato et al., 2002; Pomorski et al., 2003). Given the high affinity of PS with ergosterol, PS flipped to the cytoplasmic leaflet may effectively inhibit sterol activation. However, this ongoing process would not ensure that ergosterol molecules are fully kept in an inactive state. In addition, it is possible that ergosterol is also transported to the bud PM by STPs. Therefore, even with such a flippase action, lipids in the bud PM are somewhat agitated during membrane biogenesis, suggesting that sterol activation would not be fully controlled. On the other hand, Sfk1, but not Dnf1/2-Lem3 flippases, is localized to the mother cell PM. Our previous results suggest that Sfk1 represses spontaneous transbilayer movement of phospholipids (Mioka et al., 2018). An interesting possibility is that Sfk1 enhances interactions between ergosterol and phospholipids, which promotes lipid packing in the PM. In such a membrane, lipid movements, including transbilayer movement, would be less active, and sterols would be kept in an inactive state. Because exocytotic vesicle transport and membrane biogenesis do not actively occur in the mother cell PM, phospholipid asymmetry seems to be more established in this membrane. Sfk1 may maintain the established phospholipid asymmetry in a mechanism different from that of flippases and suppress the sterol activation.
The main localization sites were different between Dnf1/2-Lem3 and Sfk1, and the localization of Dnf3-Crf1 in the PM has not been clearly shown in the wild type. However, simultaneous loss of these proteins leads to severe disorganization of the PM, in which active ergosterol would be highly increased due to a reduced shielding effect by phospholipids and reduced lipid packing. The ergosterol in the triple mutant appears to be highly accessible and easily extracted by STPs, resulting in the loss of ergosterol from the PM (Figure 10). Some STPs, including those yet to be identified, seem to be involved in sterol transfer from the PM. These include oxysterol-binding protein homologues (Osh) and lipid transfer proteins anchored at a membrane contact site (LAMs) with StARkin domains (Antonny et al., 2018; Menon, 2018).
Our finding that ergosterol lost from the PM accumulated in LDs as esterified ergosterol is consistent with studies using exogenously added ergosterols (Li and Prinz, 2004; Georgiev et al., 2011). The PM has a much higher ergosterol concentration than the ER, but ergosterol transport by STPs between these membranes is kept in equilibrium because the active ergosterol concentrations seem to be similar in these membranes; the ER membrane contains fewer saturated phospholipids, and thus ergosterol in the ER is not shielded by surrounding phospholipids (Menon, 2018). In the crf1∆ lem3∆ sfk1 triple mutants, a vast increase in active ergosterol occurs in the PM, and STPs transport these ergosterols to the ER, in which ergosterol is esterified by Are1/Are2 to form LDs, until the active ergosterol concentration in the PM is balanced with that in the ER. The are2∆ mutation increases active ergosterol in the ER due to defective esterification, and this ergosterol would be transferred to the PM, resulting in the partial suppression of growth defects in the triple mutant.
How sterols are maintained at a high concentration in the PM has been a long-standing question in membrane biology. Phospholipid asymmetry, a conserved feature in the PM, has been implicated in this role (Panatala et al., 2015), but genetic analyses of flippases did not clearly demonstrate that they function in the retention of sterols in the PM. Our results have revealed that flippases actually play an essential role in retaining ergosterol in the PM, but the identification of an additional factor, Sfk1, which is totally different from flippases, was essential. It seems that yeast cells have acquired a robust system to retain an important molecule, ergosterol, in the PM. Our work indicates that unbiased genetic screening is a powerful approach to understanding cellular mechanisms that are regulated by a different set of proteins. Because Sfk1 is conserved as TMEM150A in mammalian cells (Chung et al., 2015), cholesterol might be retained in the PM via a similar mechanism.
MATERIALS AND METHODS
Request a protocol through Bio-protocol.
Media and chemicals
General chemicals were purchased from Wako Pure Chemicals Industry (Osaka, Japan) unless otherwise stated. Papuamide B was from the collection of R. Andersen (University of British Columbia, Canada). Duramycin was purchased from Sigma-Aldrich (St. Louis, MO). Yeast strains were grown in YPDA-rich medium (1% yeast extract [Difco Laboratories, Detroit, MI], 2% Bacto-peptone [Difco], 2% glucose, and 0.01% adenine). Strains carrying plasmids were grown in SD synthetic medium (0.67% yeast nitrogen base without amino acids [Difco] and 2% glucose) that contained the required nutritional supplements (Rose, 1990). The SDA medium was SD medium that contained 0.5% casamino acid (Difco). For the induction of the GAL1 promoter, 3% galactose and 0.2% sucrose were used as carbon sources (YPGA and SGA-Ura media).
Yeast strain manipulations and plasmid construction
The yeast strains and plasmids used in this study are listed in Supplemental Tables 1 and 2, respectively. Standard genetic manipulations of yeast strains were performed according to previously described methods (Guthrie and Fink, 1991). The PCR-based procedure was used to construct yeast strains carrying a complete gene deletion or a gene fusion with either GFP or mCherry (Longtine et al., 1998; Shaner et al., 2004). The amplified DNA fragments were introduced into the appropriate strains, and transformants were selected on appropriate plate media. Yeast transformations were performed using the lithium acetate method (Gietz and Woods, 2002; Gietz and Schiestl, 2007). All constructs that were produced by the PCR-based procedure were verified by colony PCR to confirm that the replacement or insertion occurred at the expected locus.
When the cell growth phenotype was examined by spot assay, cells were cultured in the appropriate medium overnight and adjusted to OD600 = 0.64, and then 10-fold serial dilutions were spotted onto the indicated plates. The dose-response growth curve experiment for sensitivity to duramycin was performed essentially as described previously (Takar et al., 2016). The sigmoidal curve fitting was analyzed by using an analysis tool in ImageJ.
Strains carrying 3xGFP or 3xmCherry at genomic loci were constructed as follows. pBluescript SK+ (pBSK)-3xGFP-Candida albicans URA3 (CaURA3) was constructed by subcloning 3xGFP from pBSK-SJL2-3xGFP (Lee et al., 2003; Sun et al., 2007) (a gift from D. G. Drubin), ADH1 terminator, and CaURA3 into pBSK. Then, a DNA fragment of VPH1 or DNF3, which encodes the C-terminal region, was inserted upstream of 3xGFP in pBSK-3xGFP-CaURA3. The resulting plasmids were linearized by cutting at a unique restriction enzyme site in the target gene, followed by transformation into yeast strains. Stable URA+ transformants were selected and screened for proper targeting by colony PCR. pBSK-SFK1-3xmCherry-CaURA3 was constructed by replacing DNF3 and 3xGFP with SFK1 and 3xmCherry, respectively. After stable SFK1-3xmCherry::CaURA3 transformants were obtained, CaURA3 was replaced with the KanMX6 cassette by marker fragment transformation. The functionality of DNF3-3xGFP/CRF1-GFP and SFK1-3xmCherry was confirmed by normal growth in the lem3∆ sfk1∆ and lem3∆ crf1∆ mutants containing these genes, respectively.
GFP-evt-2PH and upc2-1 were cloned into pRS306-based vectors and expressed at the URA3 locus as follows. pRS306-PTPI1-GFP-evt-2PH-TADH1 was constructed by replacing mCherry of pRS306-PTPI1-mCherry-evt-2PH-TADH1 (Miyasaka et al., 2020) with GFP. The upc2-1 (G888D) mutant fragment (–800 to +380 base pairs of the UPC2 gene) was generated by the standard two-step PCR mutagenesis technique and inserted into pRS306. These plasmids were linearized by cutting at a unique restriction enzyme site in URA3 and inserted into the URA3 locus.
To express OSH genes on a multicopy plasmid, DNA sequences encoding OSH genes were amplified by PCR and subcloned into either YEplac195, YEplac195-KanMX6, or YEplac181 plasmids. Sterol binding–deficient KES1 mutants (Im et al., 2005) were generated by the standard two-step PCR mutagenesis technique and subcloned into YEplac195. To express the SFK1-mCherry fusion gene on a multicopy plasmid, the SFK1-mCherry fragment was generated by overlap extension PCR and subcloned into YEplac181.
To express GFP-D4H and GFPenvy-D4H in Escherichia coli, the D4H (D4D434S) mutant fragment was generated by the standard two-step PCR mutagenesis technique using pColdI-mCherry-D4 as a template (Kishimoto et al., 2020). The GFP and D4H fragments were inserted into pColdI (Takara Bio, Shiga, Japan) to construct pColdI-GFP-D4H. To construct pColdI-GFPenvy-D4H, GFPenvy DNA (Slubowski et al., 2015) was newly synthesized with codon optimization for Saccharomyces cerevisiae (GeneArt Strings; Thermo Scientific, Carlsbad, CA) and amplified by PCR. pColdI-GFPenvy-D4H was constructed by replacing GFP in pColdI-GFP-D4H with this GFPenvy fragment. To express GFP-D4H and GFPenvy-D4H in yeast, the corresponding DNA fragments were inserted into pRS316-PTPI1-TADH1.
Schemes detailing the construction of plasmids are available on request.
Isolation of ts mutations of SFK1
The ts sfk1-2 strain was constructed by PCR-based random mutagenesis as follows. The approximately 1.2 kbp SFK1 DNA fragment, which corresponds to the region between the 40-base-pair upstream and 197-base-pair downstream sequences of the SFK1 gene, was PCR-amplified under a mutagenic condition (Toi et al., 2003) using the genomic DNA of the wild type (YKT38) as a template. On the other hand, the Kluyveromyces lactis LEU2 (KlLEU2) cassette DNA fragment was PCR-amplified under standard conditions using pUG73 (Euroscarf) as a template. In these PCRs, the primers contained additional sequences, so the SFK1 and KlLEU2 fragments had overlapping sequences at their 3′ and 5′ regions, respectively. Then, these fragments were used for overlap extension PCR with 5′ (SFK1) and 3′ (KlLEU2) primers to generate the SFK1-KILEU2 fragment. This fragment was introduced into YKT2386 (MATa crf1∆::HphMX4 lem3∆::TRP1 SFK1-GFP::KanMX6), and the transformants were selected at 30°C for LEU+ first and then for G418-sensitive phenotypes. Of these transformants, 265 clones were screened for those that showed growth defects at 37°C. Eight clones were isolated and back-crossed with YKT2332 (MATα crf1∆::HphMX4 lem3∆::TRP1) three times. The crf1∆::HphMX4 lem3∆::TRP1 sfk1-2::KlLEU2, which exhibited the tightest ts phenotype, was chosen for further analyses. Sequences of PCR primers used are available on request.
Isolation of mutants synthetically lethal with the lem3∆ sfk1∆ mutations
Mutants synthetically lethal with lem3∆ sfk1∆ were isolated according to the procedures described previously (Kishimoto et al., 2005). From 1 × 104 mutagenized cells screened, three single recessive mutations were identified by genetic analyses, and the corresponding wild-type genes were cloned. These genes encode CRF1, DNF3, and ANY1/CFS1 (van Leeuwen et al., 2016; Yamamoto et al., 2017). Null mutations of these genes were confirmed to be synthetically sick or lethal with lem3∆ sfk1∆.
Isolation of multicopy suppressors of the crf1Δ lem3Δ sfk1-2 mutant
The crf1∆ lem3∆ sfk1-2 mutant (YKT2340) was transformed with a yeast genomic DNA library constructed in the multicopy plasmid YEp24 (Botstein et al., 1979). Transformants were selected on SDA-Ura plates. The plates were incubated at 25°C for 2 d and then shifted to 37°C, followed by incubation for 3 d. Approximately 1 × 106 transformants were screened, and 186 clones were isolated. To exclude clones that carried LEM3 or SFK1, the sensitivity of the clones to duramycin and cycloheximide was examined (Mioka et al., 2018). Plasmids were recovered from yeast and reintroduced into the original mutant to confirm the suppression of growth defects. As a result, 10 different genomic regions were found to be responsible for suppression by DNA sequencing. The clones that contained a gene relevant to phospholipid asymmetry or lipid metabolism were further analyzed, and KES1, CHO1, and CFS1 were identified as suppressors.
Microscopic observations
For observation of proteins fused to a fluorescent protein in living cells, cells were grown under the indicated conditions to mid–log phase (OD600 of 0.8–1.2), collected, mounted on a microslide glass, and immediately observed. Cells were observed under a Nikon ECRIPS E800 microscope (Nikon Instech, Tokyo, Japan) as described previously (Saito et al., 2004).
Staining of PE exposed to the extracellular leaflet of the PM was performed using the Bio-Ro as described previously (Mioka et al., 2018). Immunofluorescence staining of Pma1 was performed as described previously (Martinez-Munoz and Kane, 2008). For staining with filipin, cells were grown in YPDA to mid–log phase and fixed with 3.8% formaldehyde for 10 min at room temperature. The fixed cells were washed twice with phosphate-buffered saline (PBS) and resuspended in PBS containing 2.5 mg/ml filipin complex (Sigma-Aldrich). After incubation at room temperature for 15 min in the dark, cells were washed with PBS once and observed with a UV filter set. For TF-Chol labeling, the cells harboring the upc2-1 mutation were precultured overnight in YPDA and diluted into YPDA containing 0.5% Tween-80, 0.5% ethanol, and 10 µg/ml TF-Chol (Avanti Polar Lipids, Alabaster, AL). The cells were incubated at 30°C for 3 h and then shifted to 37°C, followed by 6 h of incubation. Cells were collected, washed twice with fresh SD medium, resuspended in SD medium, and observed with a GFP filter set. Nile red staining of LDs was performed as described previously with minor modifications (Verstrepen et al., 2004). Five OD600 units of cell culture were collected and resuspended in 100 µl PBS containing 50 µg/ml Nile red (Sigma-Aldrich). After brief mixing, the cell suspension was incubated for 15 min at room temperature in the dark. Cells were collected, washed five times with PBS, resuspended in PBS, and observed with a G-2A filter set.
Endocytosis was examined by internalization of FM4-64 as described previously with minor modifications (Kishimoto et al., 2005). Cells were incubated in YPDA at 30°C for 3 h and then shifted to 37°C, followed by 6 h of incubation. Four OD600 units of the cells were labeled with 32 µM FM4-64 (Invitrogen, Madison, WI) in YPDA on ice for 30 min and then washed once with ice-cold YPDA. Internalization of FM4-64 was initiated by the addition of prewarmed YPDA, and the cells were incubated at 37°C for 30 min, followed by microscopic observation.
Image analysis
When the PM localization of lipid biosensors (GFP-evt-2PH and TF-Chol) was examined, cells that exhibited the mean fluorescence intensity in the PM larger than 120% of the mean fluorescence intensity of cytoplasmic space were classified as showing the PM localization. In the case of GFPenvy-D4H, which exhibited the polarized distribution, the PM of the polarized region was selected and examined as described above. The PM fluorescence intensity (filipin, Bio-Ro, and Sfk1-mCherry) was analyzed using programmed macros in ImageJ as follows. 1) The background was subtracted, 2) a cell was selected and its mean fluorescence intensity was quantified (Fwhole cell), 3) the cell periphery (0.2 µm width, two pixels) was selected as the PM and its mean fluorescence intensity was quantified (Fpm), and 4) the signal ratio of the mean Fpm/Fwhole cell was calculated.
To analyze the intensity profile of GFPenvy-D4H, the cell periphery was traced with a two-pixel-wide freehand line tool along the PM of the bud and mother cell. Then, the fluorescence intensity was measured and plotted. The GFPenvy-D4H localization pattern was categorized into three patterns as follows. The PM of budded cells was divided into three regions, the bud PM (bud), the mother cell PM proximal to the bud neck (proximal to bud), and the mother cell PM distal to the bud neck (distal to bud), and the mean fluorescence intensity of each region was calculated as Fbud, Fproximal to bud, and Fdistal to bud, respectively. Then, the cells were categorized as “polarized” (Fproximal to bud is smaller than 20% of Fbud), “partially polarized” (Fproximal to bud is larger than, but Fdistal to bud is smaller than, 20% of Fbud), or “not polarized” (Fdistal to bud is larger than 20% of Fbud).
Rhodamine uptake assay
The rhodamine uptake assay was performed essentially as described previously (Mioka et al., 2018).
Sucrose density gradient fractionation
Sucrose density gradient fractionation was performed as described previously (Furuta et al., 2007; Georgiev et al., 2011) with minor modifications. Cells were grown at 30°C to mid–log phase in 200 ml YPDA medium and collected. Cells were converted to spheroplasts with zymolyase (Nacalai Tesque, Kyoto, Japan) and broken using a multibead shocker (Yasui-Kikai, Osaka, Japan) in break buffer (0.8 M sorbitol, 20 mM HEPES at pH 7.5, 1 mM EDTA, and protease inhibitor cocktail [Nacalai Tesque]). The step gradient of sucrose was prepared with the following concentrations: 0.5 ml 60%, 2.5 ml 44%, 2 ml 40%, 1.5 ml 37%, 2 ml 34%, 2 ml 32%, 0.5 ml 29%, and 0.5 ml 22% (wt/wt) sucrose in the break buffer. The pellet was resuspended in 0.5 ml of the break buffer and loaded on top of the gradient and then centrifuged at 200,000 × g in the P40ST rotor (Hitachi, Tokyo, Japan) for 16 h at 4°C. Fractions (0.9 ml) were manually collected from the top of the samples. Pdr5-GFP, Pma1, and Kex2 were detected in each fraction by Western blotting with anti-GFP (Nacalai Tesque), anti-Pma1 (a gift from R. Serrano), and anti-Kex2 (a gift from S. Nothwehr) antibodies, respectively.
Lipid analysis
Cells were grown at 30°C to mid–log phase in 250 ml YPDA medium and collected. Total lipids were extracted by the Bligh and Dyer method (Bligh and Dyer, 1959). Phospholipid amounts were determined by phosphorus assay (Rouser et al., 1970). For the phospholipid analysis, samples containing 200 nmol phosphates were subjected to TLC plates (Merck, Darmstadt, Germany), and phospholipids were detected as described previously (Mioka et al., 2018). To detect free and esterified ergosterol, lipid extracts containing 20 nmol phosphates were subjected to high-performance TLC (Merck) separation with hexane/diethyl ether/formic acid (40:10:2, vol:vol:vol). Ergosterols were stained with a mixture of ferric chloride/sulfuric acid/acetic acid by heating (Lowry, 1968), and the spots were scanned by an image analyzer. The ergosterol content was determined by TLC-densitometric analysis using ImageJ.
Liposome sedimentation assay
Recombinant GFP-D4H and GFPenvy-D4H proteins were prepared from E. coli as described previously (Kishimoto et al., 2020). The protein concentrations were determined by bicinchoninic acid assay. Multilamellar liposomes were prepared by combining DOPC (NOF Corporation, Tokyo, Japan) with cholesterol or ergosterol from chloroform stocks. The lipid mixture was evaporated under a stream of nitrogen gas. Then, liposome buffer (0.1 M sucrose, 20 mM HEPES at pH 7.5, 100 mM KCl, and 1 mM EDTA) was added to the dry lipids, and the suspension was vortexed to produce liposomes. D4H binding to liposomes was analyzed as described previously (Ishitsuka et al., 2011) with minor modifications. Recombinant GFP- or GFPenvy-D4H protein (200 nmol) was incubated with liposomes (final total lipid concentration is 100 μM) in HEPES-buffered saline (pH 7.5) for 30 min at room temperature. Then, the mixtures were centrifuged at 21,600 × g for 10 min at 25°C. The pellets were washed with HEPES-buffered saline twice. The pellets were subjected to SDS–PAGE followed by Coomassie Brilliant Blue staining. For the quantification of the protein, the stained gel was scanned and analyzed by ImageJ.
Statistical analysis
Statistical comparisons of means from two samples were performed using a two-tailed Student’s t test. To compare the means of multiple groups, statistical analyses were performed using one-way analysis of variance followed by Tukey–Kramer multiple comparisons. A p value <0.05 was regarded as significant.
Supplementary Material
Acknowledgments
We thank Shan Gao for her contribution to the initial stage of this work. We also thank David G. Drubin (University of California, Berkeley) and Toshihide Kobayashi (University of Strasbourg) for plasmids, Ramon Serrano (Polytechnic University of Valencia) for the anti-Pma1 antibody, and Steven F. Nothwehr (University of Missouri) for the anti-Kex2 antibody. We thank Masato Umeda (Kyoto University) for providing Bio-Ro, Akiko Yamaji-Hasegawa (RIKEN) and Francoise Hullin-Matsuda (INSERM) for technical advice on the sterol quantification, and Tomohiko Taguchi (Tohoku University) for helpful comments on evt-2PH. This work was supported by Japanese Society for the Promotion of Science (JSPS) KAKENHI Grants JP18K06104 (T. K.), JP 21K06076 (T. K.), JP18K14645 (T. M.), and JP19K06536 (K. T.) and by the Institute for Fermentation, Osaka (T. K.). This work was partly supported by the Photo-excitonix Project at Hokkaido University.
Abbreviations used:
- Bio-Ro
biotinylated Ro09-0198 peptide
- CaURA3
Candida albicans URA3
- D4
domain 4
- DIC
differential interference contrast
- DOPC
1,2-dioleoyl- sn-glycero-3-phosphocholine
- ER
endoplasmic reticulum
- evt-2PH
pleckstrin homology domain of evectin-2
- 5-FOA
5-fluoroorotic acid
- GFP
green fluorescent protein
- KlLEU2
Kluyveromyces lactis LEU2
- Lact-C2
C2 domain of lactadherin
- LD
lipid droplet
- Osh
oxysterol-binding protein homologue
- P4-ATPases
type 4 P-type ATPases
- PapB
papuamide B
- pBSK
pBluescript SK+
- PBS
phosphate-buffered saline
- PC
phosphatidylcholine
- PE
phosphatidylethanolamine
- PH
pleckstrin homology
- PM
plasma membrane
- PS
phosphatidylserine
- STPs
sterol transfer proteins
- TF-Chol
TopFluor-cholesterol
- TGN
trans-Golgi network
- TLC
thin-layer chromatography
- ts
temperature sensitive
- 3x
triple tandem.
Footnotes
This article was published online ahead of print in MBoC in Press (http://www.molbiolcell.org/cgi/doi/10.1091/mbc.E20-11-0699) on May 26, 2021.
REFERENCES
- Ali MR, Cheng KH, Huang J (2007). Assess the nature of cholesterol-lipid interactions through the chemical potential of cholesterol in phosphatidylcholine bilayers. Proc Natl Acad Sci USA 104, 5372–5377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almeida PF (2009). Thermodynamics of lipid interactions in complex bilayers. Biochim Biophys Acta 1788, 72–85. [DOI] [PubMed] [Google Scholar]
- Andersen JP, Vestergaard AL, Mikkelsen SA, Mogensen LS, Chalat M, Molday RS (2016). P4-ATPases as phospholipid flippases—structure, function, and enigmas. Front Physiol 7, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antonny B, Bigay J, Mesmin B (2018). The oxysterol-binding protein cycle: burning off PI(4)P to transport cholesterol. Annu Rev Biochem 87, 809–837. [DOI] [PubMed] [Google Scholar]
- Audhya A, Emr SD (2002). Stt4 PI 4-kinase localizes to the plasma membrane and functions in the Pkc1-mediated MAP kinase cascade. Dev Cell 2, 593–605. [DOI] [PubMed] [Google Scholar]
- Bagnat M, Chang A, Simons K (2001). Plasma membrane proton ATPase Pma1p requires raft association for surface delivery in yeast. Mol Biol Cell 12, 4129–4138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bajar BT, Wang ES, Lam AJ, Kim BB, Jacobs CL, Howe ES, Davidson MW, Lin MZ, Chu J (2016). Improving brightness and photostability of green and red fluorescent proteins for live cell imaging and FRET reporting. Sci Rep 6, 20889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barajas D, Xu K, de Castro Martin IF, Sasvari Z, Brandizzi F, Risco C, Nagy PD (2014). Co-opted oxysterol-binding ORP and VAP proteins channel sterols to RNA virus replication sites via membrane contact sites. PLoS Pathog 10, e1004388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beh CT, Rine J (2004). A role for yeast oxysterol-binding protein homologs in endocytosis and in the maintenance of intracellular sterol-lipid distribution. J Cell Sci 117, 2983–2996. [DOI] [PubMed] [Google Scholar]
- Bligh EG, Dyer WJ (1959). A rapid method of total lipid extraction and purification. Can J Biochem Physiol 37, 911–917. [DOI] [PubMed] [Google Scholar]
- Boeke JD, LaCroute F, Fink GR (1984). A positive selection for mutants lacking orotidine-5’-phosphate decarboxylase activity in yeast: 5-fluoro-orotic acid resistance. Mol Gen Genet 197, 345–346. [DOI] [PubMed] [Google Scholar]
- Botstein D, Falco SC, Stewart SE, Brennan M, Scherer S, Stinchcomb DT, Struhl K, Davis RW (1979). Sterile host yeasts (SHY): a eukaryotic system of biological containment for recombinant DNA experiments. Gene 8, 17–24. [DOI] [PubMed] [Google Scholar]
- Brickner JH, Fuller RS (1997). SOI1 encodes a novel, conserved protein that promotes TGN-endosomal cycling of Kex2p and other membrane proteins by modulating the function of two TGN localization signals. J Cell Biol 139, 23–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryde S, Hennrich H, Verhulst PM, Devaux PF, Lenoir G, Holthuis JC (2010). CDC50 proteins are critical components of the human class-1 P4-ATPase transport machinery. J Biol Chem 285, 40562–40572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caunt P, Impoolsup A, Greenfield PF (1988). Stability of recombinant plasmids in yeast. J Biotechnol 8, 173–192. [DOI] [PubMed] [Google Scholar]
- Chen CY, Ingram MF, Rosal PH, Graham TR (1999). Role for Drs2p, a P-type ATPase and potential aminophospholipid translocase, in yeast late Golgi function. J Cell Biol 147, 1223–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung J, Nakatsu F, Baskin JM, De Camilli P (2015). Plasticity of PI4KIIIalpha interactions at the plasma membrane. EMBO Rep 16, 312–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley JH, Leak FW Jr, Shianna KV, Tove S, Parks LW (1998). A mutation in a purported regulatory gene affects control of sterol uptake in Saccharomyces cerevisiae. J Bacteriol 180, 4177–4183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Das A, Slaughter BD, Unruh JR, Bradford WD, Alexander R, Rubinstein B, Li R (2012). Flippase-mediated phospholipid asymmetry promotes fast Cdc42 recycling in dynamic maintenance of cell polarity. Nat Cell Biol 14, 304–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum G, Lees ND, Bard M, Dickson R (1998). Biochemistry, cell biology and molecular biology of lipids of Saccharomyces cerevisiae. Yeast 14, 1471–1510. [DOI] [PubMed] [Google Scholar]
- Decottignies A, Grant AM, Nichols JW, de Wet H, McIntosh DB, Goffeau A (1998). ATPase and multidrug transport activities of the overexpressed yeast ABC protein Yor1p. J Biol Chem 273, 12612–12622. [DOI] [PubMed] [Google Scholar]
- de Thozee CP, Cronin S, Goj A, Golin J, Ghislain M (2007). Subcellular trafficking of the yeast plasma membrane ABC transporter, Pdr5, is impaired by a mutation in the N-terminal nucleotide-binding fold. Mol Microbiol 63, 811–825. [DOI] [PubMed] [Google Scholar]
- Flanagan JJ, Tweten RK, Johnson AE, Heuck AP (2009). Cholesterol exposure at the membrane surface is necessary and sufficient to trigger perfringolysin O binding. Biochemistry 48, 3977–3987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frøsig MM, Costa SR, Liesche J, Østerberg JT, Hanisch S, Nintemann S, Sørensen H, Palmgren M, Pomorski TG, López-Marqués RL (2020). Pseudohyphal growth in Saccharomyces cerevisiae involves protein kinase-regulated lipid flippases. J Cell Sci 133, jcs.235994. [DOI] [PubMed] [Google Scholar]
- Furuta N, Fujimura-Kamada K, Saito K, Yamamoto T, Tanaka K (2007). Endocytic recycling in yeast is regulated by putative phospholipid translocases and the Ypt31p/32p-Rcy1p pathway. Mol Biol Cell 18, 295–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gall WE, Geething NC, Hua Z, Ingram MF, Liu K, Chen SI, Graham TR (2002). Drs2p-dependent formation of exocytic clathrin-coated vesicles in vivo. Curr Biol 12, 1623–1627. [DOI] [PubMed] [Google Scholar]
- Georgiev AG, Sullivan DP, Kersting MC, Dittman JS, Beh CT, Menon AK (2011). Osh proteins regulate membrane sterol organization but are not required for sterol movement between the ER and PM. Traffic 12, 1341–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH (2007). Quick and easy yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2, 35–37. [DOI] [PubMed] [Google Scholar]
- Gietz RD, Woods RA (2002). Transformation of yeast by lithium acetate/single-stranded carrier DNA/polyethylene glycol method. Methods Enzymol 350, 87–96. [DOI] [PubMed] [Google Scholar]
- Gnügge R, Rudolf F (2017). Saccharomyces cerevisiae shuttle vectors. Yeast 34, 205–221. [DOI] [PubMed] [Google Scholar]
- Greenspan P, Mayer EP, Fowler SD (1985). Nile red: a selective fluorescent stain for intracellular lipid droplets. J Cell Biol 100, 965–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guthrie C, Fink GR (1991). Guide to Yeast Genetics and Molecular Biology, San Diego: Academic Press. [Google Scholar]
- Hachiro T, Yamamoto T, Nakano K, Tanaka K (2013). Phospholipid flippases Lem3p-Dnf1p and Lem3p-Dnf2p are involved in the sorting of the tryptophan permease Tat2p in yeast. J Biol Chem 288, 3594–3608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hankins HM, Baldridge RD, Xu P, Graham TR (2015). Role of flippases, scramblases and transfer proteins in phosphatidylserine subcellular distribution. Traffic 16, 35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heese-Peck A, Pichler H, Zanolari B, Watanabe R, Daum G, Riezman H (2002). Multiple functions of sterols in yeast endocytosis. Mol Biol Cell 13, 2664–2680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holthuis JC, Menon AK (2014). Lipid landscapes and pipelines in membrane homeostasis. Nature 510, 48–57. [DOI] [PubMed] [Google Scholar]
- Hua Z, Fatheddin P, Graham TR (2002). An essential subfamily of Drs2p-related P-type ATPases is required for protein trafficking between Golgi complex and endosomal/vacuolar system. Mol Biol Cell 13, 3162–3177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im YJ, Raychaudhuri S, Prinz WA, Hurley JH (2005). Structural mechanism for sterol sensing and transport by OSBP-related proteins. Nature 437, 154–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishitsuka R, Saito T, Osada H, Ohno-Iwashita Y, Kobayashi T (2011). Fluorescence image screening for chemical compounds modifying cholesterol metabolism and distribution. J Lipid Res 52, 2084–2094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwamoto K, Kobayashi S, Fukuda R, Umeda M, Kobayashi T, Ohta A (2004). Local exposure of phosphatidylethanolamine on the yeast plasma membrane is implicated in cell polarity. Genes Cells 9, 891–903. [DOI] [PubMed] [Google Scholar]
- Jandrositz A, Petschnigg J, Zimmermann R, Natter K, Scholze H, Hermetter A, Kohlwein SD, Leber R (2005). The lipid droplet enzyme Tgl1p hydrolyzes both steryl esters and triglycerides in the yeast, Saccharomyces cerevisiae. Biochim Biophys Acta 1735, 50–58. [DOI] [PubMed] [Google Scholar]
- Johnson BB, Moe PC, Wang D, Rossi K, Trigatti BL, Heuck AP (2012). Modifications in perfringolysin O domain 4 alter the cholesterol concentration threshold required for binding. Biochemistry 51, 3373–3382. [DOI] [PubMed] [Google Scholar]
- Kato U, Emoto K, Fredriksson C, Nakamura H, Ohta A, Kobayashi T, Murakami-Murofushi K, Kobayashi T, Umeda M (2002). A novel membrane protein, Ros3p, is required for phospholipid translocation across the plasma membrane in Saccharomyces cerevisiae. J Biol Chem 277, 37855–37862. [DOI] [PubMed] [Google Scholar]
- Kato U, Inadome H, Yamamoto M, Emoto K, Kobayashi T, Umeda M (2013). Role for phospholipid flippase complex of ATP8A1 and CDC50A proteins in cell migration. J Biol Chem 288, 4922–4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kishimoto T, Ishitsuka R, Kobayashi T (2016). Detectors for evaluating the cellular landscape of sphingomyelin- and cholesterol-rich membrane domains. Biochim Biophys Acta 1861, 812–829. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Tomishige N, Murate M, Ishitsuka R, Schaller H, Mely Y, Ueda K, Kobayashi T (2020). Cholesterol asymmetry at the tip of filopodia during cell adhesion. FASEB J 34, 6185–6197. [DOI] [PubMed] [Google Scholar]
- Kishimoto T, Yamamoto T, Tanaka K (2005). Defects in structural integrity of ergosterol and the Cdc50p-Drs2p putative phospholipid translocase cause accumulation of endocytic membranes, onto which actin patches are assembled in yeast. Mol Biol Cell 16, 5592–5609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klug L, Daum G (2014). Yeast lipid metabolism at a glance. FEMS Yeast Res 14, 369–388. [DOI] [PubMed] [Google Scholar]
- Kobayashi T, Menon AK (2018). Transbilayer lipid asymmetry. Curr Biol 28, R386-R391. [DOI] [PubMed] [Google Scholar]
- Kurat CF, Natter K, Petschnigg J, Wolinski H, Scheuringer K, Scholz H, Zimmermann R, Leber R, Zechner R, Kohlwein SD (2006). Obese yeast: triglyceride lipolysis is functionally conserved from mammals to yeast. J Biol Chem 281, 491–500. [DOI] [PubMed] [Google Scholar]
- Lange Y, Steck TL (2008). Cholesterol homeostasis and the escape tendency (activity) of plasma membrane cholesterol. Prog Lipid Res 47, 319–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lange Y, Steck TL (2016). Active membrane cholesterol as a physiological effector. Chem Phys Lipids 199, 74–93. [DOI] [PubMed] [Google Scholar]
- Lange Y, Tabei SM, Ye J, Steck TL (2013). Stability and stoichiometry of bilayer phospholipid-cholesterol complexes: relationship to cellular sterol distribution and homeostasis. Biochemistry 52, 6950–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S, Uchida Y, Wang J, Matsudaira T, Nakagawa T, Kishimoto T, Mukai K, Inaba T, Kobayashi T, Molday RS, et al. (2015). Transport through recycling endosomes requires EHD1 recruitment by a phosphatidylserine translocase. EMBO J 34, 669–688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee WL, Oberle JR, Cooper JA (2003). The role of the lissencephaly protein Pac1 during nuclear migration in budding yeast. J Cell Biol 160, 355–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lev S (2010). Non-vesicular lipid transport by lipid-transfer proteins and beyond. Nat Rev Mol Cell Biol 11, 739–750. [DOI] [PubMed] [Google Scholar]
- Lewis MJ, Nichols BJ, Prescianotto-Baschong C, Riezman H, Pelham HR (2000). Specific retrieval of the exocytic SNARE Snc1p from early yeast endosomes. Mol Biol Cell 11, 23–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis TL, Keesler GA, Fenner GP, Parks LW (1988). Pleiotropic mutations in Saccharomyces cerevisiae affecting sterol uptake and metabolism. Yeast 4, 93–106. [DOI] [PubMed] [Google Scholar]
- Li Y, Prinz WA (2004). ATP-binding cassette (ABC) transporters mediate nonvesicular, raft-modulated sterol movement from the plasma membrane to the endoplasmic reticulum. J Biol Chem 279, 45226–45234. [DOI] [PubMed] [Google Scholar]
- Longtine MS, McKenzie A 3rd, Demarini DJ, Shah NG, Wach A, Brachat A, Philippsen P, Pringle JR (1998). Additional modules for versatile and economical PCR-based gene deletion and modification in Saccharomyces cerevisiae. Yeast 14, 953–961. [DOI] [PubMed] [Google Scholar]
- Lowry RR (1968). Ferric chloride spray detector for cholesterol and cholesteryl esters on thin-layer chromatograms. J Lipid Res 9, 397. [PubMed] [Google Scholar]
- Maekawa M, Fairn GD (2015). Complementary probes reveal that phosphatidylserine is required for the proper transbilayer distribution of cholesterol. J Cell Sci 128, 1422–1433. [DOI] [PubMed] [Google Scholar]
- Malinska K, Malinsky J, Opekarova M, Tanner W (2004). Distribution of Can1p into stable domains reflects lateral protein segregation within the plasma membrane of living S. cerevisiae cells. J Cell Sci 117, 6031–6041. [DOI] [PubMed] [Google Scholar]
- Manik MK, Yang H, Tong J, Im YJ (2017). Structure of yeast OSBP-related protein Osh1 reveals key determinants for lipid transport and protein targeting at the nucleus-vacuole junction. Structure 25, 617–629.e613. [DOI] [PubMed] [Google Scholar]
- Marek M, Vincenzetti V, Martin SG (2020). Sterol biosensor reveals LAM-family Ltc1-dependent sterol flow to endosomes upon Arp2/3 inhibition. J Cell Biol 219, e202001147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Munoz GA, Kane P (2008). Vacuolar and plasma membrane proton pumps collaborate to achieve cytosolic pH homeostasis in yeast. J Biol Chem 283, 20309–20319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maxfield FR, Menon AK (2006). Intracellular sterol transport and distribution. Curr Opin Cell Biol 18, 379–385. [DOI] [PubMed] [Google Scholar]
- McConnell HM, Radhakrishnan A (2003). Condensed complexes of cholesterol and phospholipids. Biochim Biophys Acta 1610, 159–173. [DOI] [PubMed] [Google Scholar]
- Menon AK (2018). Sterol gradients in cells. Curr Opin Cell Biol 53, 37–43. [DOI] [PubMed] [Google Scholar]
- Mesmin B, Maxfield FR (2009). Intracellular sterol dynamics. Biochim Biophys Acta 1791, 636–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mioka T, Fujimura-Kamada K, Mizugaki N, Kishimoto T, Sano T, Nunome H, Williams DE, Andersen RJ, Tanaka K (2018). Phospholipid flippases and Sfk1p, a novel regulator of phospholipid asymmetry, contribute to low permeability of the plasma membrane. Mol Biol Cell 29, 1203–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mioka T, Fujimura-Kamada K, Tanaka K (2014). Asymmetric distribution of phosphatidylserine is generated in the absence of phospholipid flippases in Saccharomyces cerevisiae. Microbiologyopen 3, 803–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miyasaka M, Mioka T, Kishimoto T, Itoh E, Tanaka K (2020). A complex genetic interaction implicates that phospholipid asymmetry and phosphate homeostasis regulate Golgi functions. PLoS One 15, e0236520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn AL, Heese-Peck A, Stevenson BJ, Pichler H, Riezman H (1999). Specific sterols required for the internalization step of endocytosis in yeast. Mol Biol Cell 10, 3943–3957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munn AL, Stevenson BJ, Geli MI, Riezman H (1995). end5, end6, and end7: mutations that cause actin delocalization and block the internalization step of endocytosis in Saccharomyces cerevisiae. Mol Biol Cell 6, 1721–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murate M, Abe M, Kasahara K, Iwabuchi K, Umeda M, Kobayashi T (2015). Transbilayer distribution of lipids at nano scale. J Cell Sci 128, 1627–1638. [DOI] [PubMed] [Google Scholar]
- Nagata S, Sakuragi T, Segawa K (2020). Flippase and scramblase for phosphatidylserine exposure. Curr Opin Immunol 62, 31–38. [DOI] [PubMed] [Google Scholar]
- Nakano K, Yamamoto T, Kishimoto T, Noji T, Tanaka K (2008). Protein kinases Fpk1p and Fpk2p are novel regulators of phospholipid asymmetry. Mol Biol Cell 19, 1783–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson LD, Johnson AE, London E (2008). How interaction of perfringolysin O with membranes is controlled by sterol structure, lipid structure, and physiological low pH: insights into the origin of perfringolysin O-lipid raft interaction. J Biol Chem 283, 4632–4642. [DOI] [PubMed] [Google Scholar]
- Neumann J, Rose-Sperling D, Hellmich UA (2017). Diverse relations between ABC transporters and lipids: an overview. Biochim Biophys Acta 1859, 605–618. [DOI] [PubMed] [Google Scholar]
- Noji T, Yamamoto T, Saito K, Fujimura-Kamada K, Kondo S, Tanaka K (2006). Mutational analysis of the Lem3p-Dnf1p putative phospholipid-translocating P-type ATPase reveals novel regulatory roles for Lem3p and a carboxyl-terminal region of Dnf1p independent of the phospholipid-translocating activity of Dnf1p in yeast. Biochem Biophys Res Commun 344, 323–331. [DOI] [PubMed] [Google Scholar]
- Nyholm TKM, Jaikishan S, Engberg O, Hautala V, Slotte JP (2019). The affinity of sterols for different phospholipid classes and its impact on lateral segregation. Biophys J 116, 296–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panatala R, Hennrich H, Holthuis JC (2015). Inner workings and biological impact of phospholipid flippases. J Cell Sci 128, 2021–2032. [DOI] [PubMed] [Google Scholar]
- Parsons AB, Lopez A, Givoni IE, Williams DE, Gray CA, Porter J, Chua G, Sopko R, Brost RL, Ho CH, et al. (2006). Exploring the mode-of-action of bioactive compounds by chemical-genetic profiling in yeast. Cell 126, 611–625. [DOI] [PubMed] [Google Scholar]
- Paulusma CC, Groen A, Kunne C, Ho-Mok KS, Spijkerboer AL, Rudi de Waart D, Hoek FJ, Vreeling H, Hoeben KA, van Marle J, et al. (2006). Atp8b1 deficiency in mice reduces resistance of the canalicular membrane to hydrophobic bile salts and impairs bile salt transport. Hepatology 44, 195–204. [DOI] [PubMed] [Google Scholar]
- Peters C, Bayer MJ, Buhler S, Andersen JS, Mann M, Mayer A (2001). Trans-complex formation by proteolipid channels in the terminal phase of membrane fusion. Nature 409, 581–588. [DOI] [PubMed] [Google Scholar]
- Pomorski T, Lombardi R, Riezman H, Devaux PF, van Meer G, Holthuis JC (2003). Drs2p-related P-type ATPases Dnf1p and Dnf2p are required for phospholipid translocation across the yeast plasma membrane and serve a role in endocytosis. Mol Biol Cell 14, 1240–1254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruyne D, Legesse-Miller A, Gao L, Dong Y, Bretscher A (2004). Mechanisms of polarized growth and organelle segregation in yeast. Annu Rev Cell Dev Biol 20, 559–591. [DOI] [PubMed] [Google Scholar]
- Puts CF, Panatala R, Hennrich H, Tsareva A, Williamson P, Holthuis JC (2012). Mapping functional interactions in a heterodimeric phospholipid pump. J Biol Chem 287, 30529–30540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quazi F, Molday RS (2011). Lipid transport by mammalian ABC proteins. Essays Biochem 50, 265–290. [DOI] [PubMed] [Google Scholar]
- Radhakrishnan A, Goldstein JL, McDonald JG, Brown MS (2008). Switch-like control of SREBP-2 transport triggered by small changes in ER cholesterol: a delicate balance. Cell Metab 8, 512–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramstedt B, Slotte JP (2006). Sphingolipids and the formation of sterol-enriched ordered membrane domains. Biochim Biophys Acta 1758, 1945–1956. [DOI] [PubMed] [Google Scholar]
- Rose M (1990). Methods in Yeast Genetics: A Laboratory Course Manual, ed. Winston F., Hieter P., Cold Spring Harbor, NY: Cold Spring Harbor Laboratory Press. [Google Scholar]
- Rouser G, Fkeischer S, Yamamoto A (1970). Two dimensional thin layer chromatographic separation of polar lipids and determination of phospholipids by phosphorus analysis of spots. Lipids 5, 494–496. [DOI] [PubMed] [Google Scholar]
- Saito K, Fujimura-Kamada K, Furuta N, Kato U, Umeda M, Tanaka K (2004). Cdc50p, a protein required for polarized growth, associates with the Drs2p P-type ATPase implicated in phospholipid translocation in Saccharomyces cerevisiae. Mol Biol Cell 15, 3418–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito K, Fujimura-Kamada K, Hanamatsu H, Kato U, Umeda M, Kozminski KG, Tanaka K (2007). Transbilayer phospholipid flipping regulates Cdc42p signaling during polarized cell growth via Rga GTPase-activating proteins. Dev Cell 13, 743–751. [DOI] [PubMed] [Google Scholar]
- Sartorel E, Barrey E, Lau RK, Thorner J (2015). Plasma membrane aminoglycerolipid flippase function is required for signaling competence in the yeast mating pheromone response pathway. Mol Biol Cell 26, 134–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savinov SN, Heuck AP (2017). Interaction of cholesterol with perfringolysin O: what have we learned from functional analysis? Toxins (Basel) 9, 381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneiter R, Brugger B, Sandhoff R, Zellnig G, Leber A, Lampl M, Athenstaedt K, Hrastnik C, Eder S, Daum G, et al. (1999). Electrospray ionization tandem mass spectrometry (ESI-MS/MS) analysis of the lipid molecular species composition of yeast subcellular membranes reveals acyl chain-based sorting/remodeling of distinct molecular species en route to the plasma membrane. J Cell Biol 146, 741–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K, Kurata S, Yanagihashi Y, Brummelkamp TR, Matsuda F, Nagata S (2014). Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science 344, 1164–1168. [DOI] [PubMed] [Google Scholar]
- Serrano R, Kielland-Brandt MC, Fink GR (1986). Yeast plasma membrane ATPase is essential for growth and has homology with (Na++ K+), K+- and Ca2+-ATPases. Nature 319, 689–693. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Campbell RE, Steinbach PA, Giepmans BN, Palmer AE, Tsien RY (2004). Improved monomeric red, orange and yellow fluorescent proteins derived from Discosoma sp. red fluorescent protein. Nat Biotechnol 22, 1567–1572. [DOI] [PubMed] [Google Scholar]
- Shimada Y, Maruya M, Iwashita S, Ohno-Iwashita Y (2002). The C-terminal domain of perfringolysin O is an essential cholesterol-binding unit targeting to cholesterol-rich microdomains. Eur J Biochem 269, 6195–6203. [DOI] [PubMed] [Google Scholar]
- Slubowski CJ, Funk AD, Roesner JM, Paulissen SM, Huang LS (2015). Plasmids for C-terminal tagging in Saccharomyces cerevisiae that contain improved GFP proteins, Envy and Ivy. Yeast 32, 379–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanko LM, Sullivan DP, Sere YY, Szomek M, Lunding A, Solanko KA, Pizovic A, Stanchev LD, Pomorski TG, Menon AK, Wustner D (2018). Ergosterol is mainly located in the cytoplasmic leaflet of the yeast plasma membrane. Traffic 19, 198–214. [DOI] [PubMed] [Google Scholar]
- Steck TL, Lange Y (2010). Cell cholesterol homeostasis: mediation by active cholesterol. Trends Cell Biol 20, 680–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens HC, Malone L, Nichols JW (2008). The putative aminophospholipid translocases, DNF1 and DNF2, are not required for 7-nitrobenz-2-oxa-1,3-diazol-4-yl-phosphatidylserine flip across the plasma membrane of Saccharomyces cerevisiae. J Biol Chem 283, 35060–35069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun Y, Carroll S, Kaksonen M, Toshima JY, Drubin DG (2007). PtdIns(4,5)P2 turnover is required for multiple stages during clathrin- and actin-dependent endocytic internalization. J Cell Biol 177, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi Y, Fujimura-Kamada K, Kondo S, Tanaka K (2011). Isolation and characterization of novel mutations in CDC50, the non-catalytic subunit of the Drs2p phospholipid flippase. J Biochem 149, 423–432. [DOI] [PubMed] [Google Scholar]
- Takar M, Wu Y, Graham TR (2016). The essential Neo1 protein from budding yeast plays a role in establishing aminophospholipid asymmetry of the plasma membrane. J Biol Chem 291, 15727–15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, Fujimura-Kamada K, Yamamoto T (2011). Functions of phospholipid flippases. J Biochem 149, 131–143. [DOI] [PubMed] [Google Scholar]
- Tanaka Y, Ono N, Shima T, Tanaka G, Katoh Y, Nakayama K, Takatsu H, Shin HW (2016). The phospholipid flippase ATP9A is required for the recycling pathway from the endosomes to the plasma membrane. Mol Biol Cell 27, 3883–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toi H, Fujimura-Kamada K, Irie K, Takai Y, Todo S, Tanaka K (2003). She4p/Dim1p interacts with the motor domain of unconventional myosins in the budding yeast, Saccharomyces cerevisiae. Mol Biol Cell 14, 2237–2249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida Y, Hasegawa J, Chinnapen D, Inoue T, Okazaki S, Kato R, Wakatsuki S, Misaki R, Koike M, Uchiyama Y, et al. (2011). Intracellular phosphatidylserine is essential for retrograde membrane traffic through endosomes. Proc Natl Acad Sci USA 108, 15846–15851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Leeuwen J, Pons C, Mellor JC, Yamaguchi TN, Friesen H, Koschwanez J, Usaj MM, Pechlaner M, Takar M, Usaj M, et al. (2016). Exploring genetic suppression interactions on a global scale. Science 354, aag0839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Meer G (2011). Dynamic transbilayer lipid asymmetry. Cold Spring Harb Perspect Biol 3, a004671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verstrepen KJ, Van Laere SD, Vercammen J, Derdelinckx G, Dufour JP, Pretorius IS, Winderickx J, Thevelein JM, Delvaux FR (2004). The Saccharomyces cerevisiae alcohol acetyl transferase Atf1p is localized in lipid particles. Yeast 21, 367–377. [DOI] [PubMed] [Google Scholar]
- Vida TA, Emr SD (1995). A new vital stain for visualizing vacuolar membrane dynamics and endocytosis in yeast. J Cell Biol 128, 779–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto T, Fujimura-Kamada K, Shioji E, Suzuki R, Tanaka K (2017). Cfs1p, a novel membrane protein in the PQ-Loop family, isinvolved in phospholipid flippase functions in yeast. G3 (Bethesda) 7, 179–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamauchi S, Obara K, Uchibori K, Kamimura A, Azumi K, Kihara A (2015). Opt2 mediates the exposure of phospholipids during cellular adaptation to altered lipid asymmetry. J Cell Sci 128, 61–69. [DOI] [PubMed] [Google Scholar]
- Yang H, Bard M, Bruner DA, Gleeson A, Deckelbaum RJ, Aljinovic G, Pohl TM, Rothstein R, Sturley SL (1996). Sterol esterification in yeast: a two-gene process. Science 272, 1353–1356. [DOI] [PubMed] [Google Scholar]
- Yeung T, Gilbert GE, Shi J, Silvius J, Kapus A, Grinstein S (2008). Membrane phosphatidylserine regulates surface charge and protein localization. Science 319, 210–213. [DOI] [PubMed] [Google Scholar]
- Yu C, Kennedy NJ, Chang CC, Rothblatt JA (1996). Molecular cloning and characterization of two isoforms of Saccharomyces cerevisiae acyl-CoA:sterol acyltransferase. J Biol Chem 271, 24157–24163. [DOI] [PubMed] [Google Scholar]
- Zachowski A (1993). Phospholipids in animal eukaryotic membranes: transverse asymmetry and movement. Biochem J 294 (Pt 1), 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.