

Abstract
Synthetic antisense oligonucleotides (ASOs) have emerged as one of the most promising therapeutic approaches. So far, nine ASO drugs have received approval for clinical use, and four of them are based on splice-switching principles demonstrating the impact of ASO-mediated splice modulation. Notably, three among them (Exondys 51, Vyondys 53 and Viltepso) are based on phosphorodiamidate morpholino (PMO) chemistry whereas Spinraza is based on 2′-O-methoxyethyl phosphorothioate (2′-MOE PS) chemistry. Although systemic delivery of PMOs has displayed a good safety profile even at high doses, the 2′-O-methyl phosphorothioate modified (2′-OMe PS) ASO drug candidate (drisapersen) failed due to safety issues. The potency of 2′-modified RNA for splice-switching needs to be further improved by novel design strategies for broad applicability. Towards this goal, in this study, we evaluated the potential of incorporating DNA segments at appropriate sites in 2′-OMe PS and 2′-MOE PS ASOs to induce exon skipping. For this purpose, a four-nucleotide DNA segment was systematically incorporated into a 20-mer 2′-OMe PS and 2′-MOE PS ASO designed to skip exon 23 in mdx mouse myotubes in vitro. Our results demonstrated that 2′-modified RNA PS ASOs containing four or less PS DNA nucleotides at the 3′-end yielded improved exon 23 skipping efficacy in line with fully modified ASO controls. Based on these results, we firmly believe that the present study opens new avenues towards designing splice modulating ASOs with limited chemical modifications for enhanced safety and therapeutic efficacy.
We evaluated the potential of 2′-modified RNA antisense oligonucleotides (ASOs) incorporated with DNA segments to induce exon skipping. Results demonstrated that ASOs with 4 or less DNA nucleotides at the 3′-end induce more efficient exon skipping compared with the control.
Introduction
Over the last four decades, significant advances have been made towards the development of antisense therapeutics permitting the approval of a number of drugs. Primarily, there are two underlying mechanisms of antisense. In the first mechanism, synthetic antisense oligonucleotide (ASO) hybridizes to target RNA and activates RNase-H, which excises the target RNA and inhibits translation of the disease associated protein. These classes of ASOs are composed of two segments, a segment of phosphorothioate oligodeoxynucleotide (PS-DNA) and a segment of phosphorothioate or phosphodiester (PO) 2′-modified oligoribonucleotides (2′-PS/PO RNA) or other non-RNase-H activating modifications. This design of antisense is generally referred to as gapmer which has been studied broadly and to date, three drugs have been approved.1
In the second mechanism, ASO hybridizes to pre-mRNA and subsequently manipulate splicing events to correct genetic defects or manipulate gene function.1 Pre-mRNA splicing is an important process in regulating gene expression.2,3 By variably removing introns or exons from the primary gene transcript, splicing results in differential protein expression. However, splicing errors can arise from mutations which can lead to genetic conditions. Introduction of splice modulating ASOs to correct defective splicing was first described by Dominski and Kole in 1993 (ref. 4) and soon after shown to work in cells.5 In the last two decades, splice modulating ASO therapy has seen tremendous success for the treatment of various genetic conditions. The first splice modulating ASO drug Exondys 51 for the treatment of Duchenne muscular dystrophy (DMD) was approved by the US Food and Drug Administration (FDA) in September 2016, and later four additional drugs were also approved including Spinraza for the treatment of spinal muscular atrophy, and Vyondys 53, Viltepso and Amondys 45 again for DMD.6–10
For modulation of splicing, there are two types of modified ASOs that are being pursued. The first type is phosphorodiamidate morpholino (PMO). PMOs are non-ionic, very resistant to nucleolytic degradation, have very poor binding to serum proteins, and are rapidly eliminated following systemic administration. Four PMO ASO drugs for the treatment of DMD have been approved recently, which involve administration of a 60–80 milligrams per kg dose by intravenous infusion. Only a fraction of the administered dose reaches the muscles, the target organ, and the rest of PMO ASO is eliminated in urinary excretion.
The second type of modified ASOs employed are 2′-PS/PO-RNA, in which 2′-substitutions include 2′-O-methyl (2′-OMe, Fig. 1 phosphorothioates form) and 2′-O-methoxyethyl (2′-MOE, Fig. 1 phosphorothioates form) that have been widely studied. These ASOs have desirable characteristics including high affinity to target RNA, increased nucleolytic stability, protein binding, and wide distribution to various organs following systemic administration. This increased stability does however lead to tissue accumulation and retention which needs to be addressed. Intrathecal administration of 2′-MOE PS-RNA ASO, as in Spinraza, has been successful for the treatment of spinal muscular atrophy, and administered at doses of 12 mg, every 4 months after initial 4 loading doses. On the other hand, systemic treatment with 2′-OMe PS-RNA ASO, drisapersen, (a DMD drug that advanced to Phase III clinical trials), has failed due to safety issues. In this trial, ASO was administered subcutaneously at doses of up to 6 mg per kg per week for 24 weeks.
Fig. 1. Structural representation of the nucleotide monomers used in this study. From left to right: DNA, 2′-OMe and 2′-MOE. All monomers are represented in their phosphorothioates (PS) form.

There have been continued effort towards improving the therapeutic index of splice modulating ASOs. Towards this goal, novel modifications of ASOs are being evaluated. Several of the modifications studied had significant improvement in affinity to the target RNA. These includes 2′-fluoro (2′-F), locked nucleic acid (LNA), twisted intercalating nucleic acid (TINA), anhydrohexitol nucleic acid (HNA), cyclohexenyl nucleic acid (CeNA), d-altritol nucleic acid (ANA), morpholino nucleic acid (MNA), serinol nucleic acid (SNA), 5-(phenyltriazol)-2′-deoxyuridine modified RNA or antisense containing segments of these modified nucleotides have been studied to modulate splicing.11–17 Overall goal of these studies has been to further increase nucleolytic stability and affinity of antisense to target RNA, thereby allowing improved potency of antisense. While increased affinity of these analogues have been shown to permit shorter lengths of the ASO,11 an improvement of the therapeutic index has not yet been achieved. Furthermore, with their very high affinity, for example with LNA, these antisense are known to form inter and intra-molecular structures, thereby limiting their therapeutic index.18
Of hundreds of chemical modifications introduced in DNA and RNA, very limited modifications have shown utility in the development of antisense.19,20 We wanted to exploit the lessons learned during the development of gapmer ASO to design ASO for splicing. In gapmer ASOs, the use of a segment of PS-DNA allows activation of RNase-H, while in addition providing additional characteristics such as protein binding. The next question we explored was whether we could use segments of PS-DNA appropriately in splice modulating ASO that would not activate RNase-H, but would provide other characteristics towards improving therapeutic index. In our earlier studies, we have shown that the affinity and RNase-H activation is impacted by incorporation of a segment of PS-DNA of varying length and at various positions in 2′-OMe PS RNA.21–29 Incorporation of a segment of 5-mer PS-DNA or less at the 3′-end of 2′-OMe RNA failed to activate RNase-H compared to if the segment was placed on the 5′-end or in the middle.
Based on these insights, we hypothesized that appropriate placement of a PS-DNA segment in splice modulating ASO would allow us to further enhance the potential of ASOs or even retain the potency of splice modulating ASOs. Most importantly, this strategy would permit to modulate their drug like properties such as protein binding, degradation, distribution, and elimination-very similar to what has been seen to date with gapmer antisense.
We believe that this is a novel and potential approach of constructing gapmer ASOs for splice switching applications that could limit the use of modified nucleotides without compromising the ASO efficacy. Herein, we evaluated the scope of this approach to induce exon 23 skipping in vitro in H-2Kb-tsA58 mdx mouse myotubes.30,31
Experimental
Design and synthesis of antisense oligonucleotides used in this study
ASO1-10 were synthesized by Microsynth (Balgach, Switzerland). ASO11 and 12, and two control ASOs were synthesized in-house on GE AKTA Oligopilot 10 oligonucleotide synthesizer (GE Healthcare Life Sciences, Parramatta, NSW, Australia) using standard phosphoramidite chemistry at 1 μmol scale unless specified. All in-house synthesis reagents were purchased from Merck Millipore (Bayswater, VIC, Australia). Synthesized oligonucleotides were deprotected and cleaved from the solid support by treatment with NH4OH (Merck Millipore) at 55 °C overnight, and the crude oligonucleotides were then purified by desalting through by illustra NAP-10 Columns (GE Healthcare Life Sciences).
Melting temperature (Tm) analysis of the antisense oligonucleotides
All ASOs were prepared as 2 μM concentration in buffer solution containing 10 mM NaCl, 0.01 mM EDTA and 10 mM sodium phosphate (pH 7.0). Before loaded onto a quartz cuvettes of 1 mm path-length, the ASOs were hybridized with the same molar concentration of a synthetic complementary RNA sequence 5′-r(AG GUA AGC CGA GGU UUG GCC)-3′, purchased from IDT (Coralville, Iowa, United States) in equal volume by denaturing at 95 °C followed by slow cooling to room temperature. Melting temperature measurement was then performed using Shimadzu UV-1800 UV spectrophotometer (Rydalmere, NSW, Australia) with a temperature range of 20–95 °C (ramp rate = 1.0 °C min−1). Tm values were calculated by the first derivative.
Cell culture and transfection
H-2K b -tsA58 (H2K) mdx myoblasts were cultured and differentiated as described previously.32,33 Briefly, when 60–80% confluent, myoblast cultures were treated with trypsin (Thermo Fisher Scientific, Scoresby, VIC, Australia) and seeded on 24 well plates pre-treated with 50 μg ml−1 poly-d-lysine (Merck Millipore), followed by 100 μg ml−1 Matrigel (Corning, supplied through In Vitro Technologies, Noble Park North, VIC, Australia) at a density of 2.5 × 104 cells per well. Cells were differentiated into myotubes in low glucose Dulbecco's Modified Eagle Medium (DMEM) (Thermo Fisher Scientific) containing 5% horse serum (Thermo Fisher Scientific) by incubating at 37 °C, 5% CO2 for 24 hours. Antisense oligonucleotides were complexed with lipofectin transfection reagent (Thermo Fisher Scientific) at the ratio of 2 : 1 (w/w) (lipofectin : ASO) and used in a final transfection volume of 500 μl per well in a 24-well plate as per the manufacturer's instructions, except that the solution was not removed after 3 hours.
RNA extraction and reverse transcription-polymerase chain reaction (RT-PCR)
RNA was extracted from transfected cells using ISOLATE II RNA Mini Kit (Bioline, Eveleigh, NSW, Australia) as per the manufacturer's instructions. The mouse dystrophin transcripts were amplified by nested-RT-PCR using SuperScript™ III Reverse Transcriptase and AmpliTaq Gold® 360 DNA Polymerase (Thermo Fisher Scientific) across exons 20–26 as described previously.32 The amount of RNA used for the primary amplification was 50 ng μL−1 using primer set Ex20Fo (5′-CAGAATTCTGCCAATTGCTGAG-3′) and Ex26Ro (5′-TTCTTCAGCTTGTGTCATCC-3′); PCR conditions are 55 °C for 30 min, 94 °C for 2 min before entering 31 cycles of 94 °C for 30 s, 55 °C for 30 s and 68 °C for 90 s. Then, 1 μL of this primary PCR products was subjected to the secondary PCR using primer set Ex20Fi (5′-CCCAGTCTACCACCCTATCAGAGC-3′) and Ex26Ri (5′-CCTGCCTTTAAGGCTTCCTT-3′); PCR conditions are 94 °C for 6 min before entering 33 cycles of 94 °C for 30 s, 55 °C for 1 min and 72 °C for 2 min. The secondary PCR products were separated on 2% agarose gels in Tris–acetate–EDTA buffer and the images were captured on a Fusion Fx gel documentation system (Vilber Lourmat, Marne-la-Vallee, France). Densitometry was performed by Image J software.34 The actual exon-skipping efficiency was determined by expressing the amount of exon skipped RT-PCR products as a percentage of total dystrophin transcript products.
Results and discussion
Establishing PS-DNA position is critical for splice modulation activity of the ASOs
To determine whether introduction of a PS-DNA segment can affect the splice modulation efficiency of a 2′-OMe PS ASO, we designed and synthesized ASOs 1–5 in which each ASO contains a four-nucleotide PS-DNA block positioned in different sections of the sequence (Table 1). The ASOs were designed based on the best performing exon 23 skipping ASO that was reported previously32,33 – this ASO was also used as control ASO in this study (Table 1, control 2′-OMe PS).
ASO names and their sequences used in this studya.
| ASO name | Sequence (5′-3′) | T m (°C) |
|---|---|---|
| ASO1 |
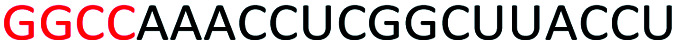
|
56.9 |
| ASO2 |
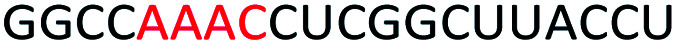
|
56.4 |
| ASO3 |

|
39.4 |
| ASO4 |
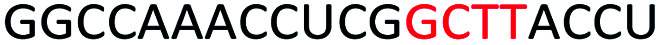
|
55.2 |
| ASO5 |

|
59.2 |
| ASO6 |
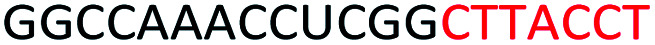
|
54.7 |
| ASO7 |
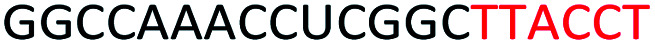
|
55.3 |
| ASO8 |
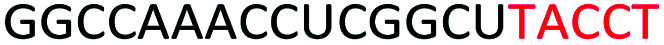
|
56.1 |
| ASO9 |

|
57.2 |
| ASO10 |

|
59.9 |
| ASO11 |

|
65.4 |
| ASO12 |

|
65.2 |
| Control 2′-OMe |

|
60.6 |
| Control 2′-MOE |

|
69.3 |
Red letters: DNA monomers, black letters: 2′-OMe monomers, blue letters: 2′-MOE monomers. All sequences were synthesized on a PS backbone. For Tm curves please see ESI Fig. S1.
First, we performed melting temperature (Tm) analysis of ASOs 1–5 against a 20-mer synthetic RNA sequence that is identical to the Dmd transcript target as described previously.32,33 The results showed that the control 2′-OMe PS ASO exhibited highest Tm (60.6 °C, Table 1) compared to all other tested ASOs. It is not surprising as the fully modified sequence was made up of all 2′-OMe monomers which has higher Tm than that of ASOs containing blocks of PS-DNA. Remarkably, ASO5 achieved 59.2 °C of Tm, highest among the five PS-DNA-containing ASOs at various positions, followed by ASO1 (56.9 °C), ASO2 (56.4 °C), ASO4 (55.2 °C) and ASO3 (39.4 °C). From the melting temperature results, we speculated that the PS-DNA segment when incorporated at 3′-end did not significantly affect the binding affinity of the ASO, given that only 1.4 °C decrease in Tm between ASO5 and control ASO. ASO1, 2 and 4 which has the DNA segments incorporated either directly at 5′-end, four nucleotides apart from 5′-end or four nucleotides apart from 3′-end, respectively, exhibits similar Tm (Table 1). However, the positions of the PS-DNA segments in these ASOs was critical to the affinity of the ASO/RNA duplex compared with control 2′-OMe PS ASO. This effect was clearly demonstrated in the case of ASO3 (lowest Tm) where PS-DNA segment was incorporated at the centre of the ASO.
Next, we tested the ability of the ASOs to induce splice modulation in an in vitro model system using H2Kb-tsA58 (H2K) mdx mouse myotubes. These cells contain a nonsense mutation in exon 23 of the dystrophin gene that leads to a disrupted reading frame and results in no dystrophin production.30,31 By recruiting splice modulating ASO to the mutated exon, the exon 23 is “skipped” so that the reading frame is restored.32,33 For this experiment, briefly, the ASOs were transfected using lipofectin transfection reagent at a ratio of 2 : 1 (2.5–100 nM) and incubated with cells for 24 h. Cells were then collected, extracted the RNA and RT-PCR was performed across exon 20–26 as previously described.33 The results showed that all five ASOs induces efficient exon 23 skipping, except for ASO3 where no skipping was recorded (688 bp band, Fig. 2A–F). Overall, ASO5 was the best performing ASO when compared with other ASOs in terms of exon 23 skipping (Fig. 2E). This is in line with the Tm results, except for the control ASO which exhibited highest Tm but did not perform as well as ASO5 (Fig. 2E and F).
Fig. 2. RT-PCR with sequence visualisation (A–F) and densitometry analysis (G) of RNA prepared from H2K mdx mouse myotubes transfected with ASO1 (A), 2 (B), 3 (C), 4 (D), 5 (E) and control 2′-OMe ASO (F). The triangles above the gel image indicate increasing ASO concentration (2.5, 5, 10, 25, 50 and 100 nM); FL (full-length) = 901 bp, represented in grey color; Δ23 (exon-23 skipped) = 688 bp, represented in blue color; Δ22-23 (exons-22 + 23 skipped) = 542 bp, represented in green color; UT = untreated, Ne = negative control.

Remarkably, at 2.5 nM, ASO5 induced 27% of exon 23 skipping (Fig. 2E and G), in comparison to no skipping of all other ASOs, except ASO4 which yielded the same amount of exon 23 skipping (Fig. 2D and G). At 5, 10 and 25 nM, ASO5 continued to perform exceptionally well, yielding 36, 42 and 50% of exon 23 skipped products, respectively, compared with all other ASOs. Surprisingly, at 50 nM, ASO1 showed higher exon 23 skipping efficiency than any other ASOs, recorded at 59% (Fig. 2A and G). It was also noted that ASO1 and 2 possess similar Tm data, however, ASO1 performed better than ASO2 at all concentrations, except at 25 nM (Fig. 2A and B). Although achieved distinctive Tm data, ASO2, 4 and 5 performed similarly at 50 nM, with 50, 48 and 49% of exon 23 skipping induced, respectively (Fig. 2B, D, E and G). Finally, at 100 nM, ASO4 and 5 both induced 54% of exon 23 skipping, while efficiency of ASO2 dropped to 36.5%, mainly due to the significant increase of the unwanted dual exon 22–23 skipped product (542 bp band, Fig. 2B). The Tm data and exon skipping results demonstrated that position of incorporation of PS DNA block in 2′-OMe ASO is critical towards the ASO splice modulation efficacy, as observed with the ASOs that have PS-DNA segment incorporated towards 3′-end of the ASO.
Length of PS-DNA segment on 3′-end affects splice modulation activity of the ASOs
Based on the results of the initial experiments, we believe that 3′-end is in fact a better position for the placement of PS-DNA segment for improving ASO efficacy. Our next step was to investigate whether the length of PS-DNA segment at 3′-end might affect the splice modulation activity of the ASO. We designed and synthesized a set of five additional ASOs (ASO6-10, Table 1) and tested its ability to induce exon 23 skipping in H2K mdx mouse myotubes. In line with previous experiment, we also performed Tm analysis for this ASO set. As predicted, the control ASO achieved highest Tm overall, followed by ASO10 (59.9 °C), 9 (57.2 °C), 8 (56.1 °C), 7 (55.3 °C) and 6 (54.7 °C) (Table 1). This is not surprising as ASO10 has only two PS-DNA nucleotides incorporated, compared with three, five, six and seven PS-DNA nucleotides in ASO9, 8, 7 and 6, respectively. ASO5, as measured above, achieved 59.2 °C of Tm, higher than any other ASOs except for the ASO10 and the control ASO. It is important to note that ASOs with more than five PS-DNA could activate RNase-H and excise targeted RNA thereby reduce splicing efficiency.
We then evaluated the exon skipping efficiency of ASO5-10 by transfecting the H2K mdx myotubes with the ASOs at 2.5–100 nM as described above. In this case, based on the ASO designs, we speculated that the exon skipping potency would be in the order of ASO10 > 9 > 5 > 8 > 7 > 6, given the increasing number of PS-DNA nucleotides in each sequence, however, the similar Tm data of ASO10 and 5 suggested that both ASOs could yield equivalent efficacy. In fact, ASO5 and 10 were found to be the best in inducing exon 23 skipping, in comparison to all other ASOs. The results showed that both ASOs perform equivalently at all concentrations, except at 10 and 50 nM, where ASO5 yielded 42 and 49% of exon 23 skipping, and ASO10 yielded 33 and 53%, respectively (Fig. 3D, F and H). This is quite remarkable since ASO5 was incorporated with four PS-DNA nucleotides at 3′-end, while ASO10 was only incorporated with two PS-DNA nucleotides.
Fig. 3. RT-PCR with sequence visualisation (A–G) and densitometry analysis (H) of RNA prepared from H2K mdx mouse myotubes transfected with ASO6 (A), 7 (B), 8 (C), 5 (D), 9 (E), 10 (F) and control 2′-OMe ASO (G). The triangles above the gel image indicate increasing ASO concentration (2.5, 5, 10, 25, 50 and 100 nM); FL (full-length) = 901 bp, represented in grey color; Δ23 (exon-23 skipped) = 688 bp, represented in blue color; Δ22–23 (exons-22 + 23 skipped) = 542 bp, represented in green color; UT = untreated, Ne = negative control.

In terms of concentration, at 2.5 nM, all ASOs yielded exon 23 skipped products, except for the control ASO (no skipping, Fig. 3G) and ASO6 (17% of dual exon skipping, Fig. 3A and H). Notably, unlike our speculation above, ASO7 induced 10% of exon 23 skipping at 2.5 nM, better than 4% of ASO8 (Fig. 3B, C and H). At 5 nM, ASO9 yielded 12% of exon 23 skipped product, lower than any other ASOs except ASO6 (3%). This is also not in line with our prediction from the Tm data as ASO9 should perform better than ASO7 (27%) and 8 (30%). However, at 10 nM, ASO9 induced 34% of exon 23 skipping, higher than any other ASOs except for the control ASO and ASO5 (Fig. 3E and H). In addition, ASO6, 7 and 8 performed similarly at this concentration (25, 25 and 27%, respectively). At 25 nM, ASO6, 7 and 8 continued to induce a similar amount of exon 23 skipped products (34, 36 and 34%, respectively), while ASO9 efficacy dropped to 30% (Fig. 3A–H). Remarkably, ASO5 and 10 achieved the highest efficacy with 50 and 49% of exon 23 skipping induced, 1.3 times higher than the control ASO (31%) (Fig. 3D, F, G and H). Surprisingly, at 50 nM, ASO6 yielded higher exon 23 skipped product than ASO7, 8 and 9 (38% compared with 30, 28 and 28%, respectively, Fig. 3A–C, E and H). The results also showed that ASO10 performed slightly better than ASO5 at this concentration (53% compared with 49%, respectively, Fig. 3D, F and H), while the control ASO induced less skipping than both ASOs (41%). Finally, at 100 nM, ASO6, 7 and 9 yielded similar amount of exon 23 skipping (38, 39 and 38%, respectively), while ASO8 yielded slightly less skipping (36%) (Fig. 3A–C, E and H). Notably, ASO5 and 10 induced the same amount of exon 23 skipped products at this concentration (54%, Fig. 3D, F and H), compared with 57% of the control ASO. We also observed the dual exon 22,23 skipped products with all ASOs in an overall dose-dependent manner, ranging from 5–43% (Fig. 3A–H).
Effect of PS-DNA segment position in a 2′-MOE ASO sequence
Encouraged by the promising data achieved in the first two sets of experiments, we also wanted to explore whether our approach could have similar effects on an alternative 2′-modified RNA chemistry. In this case, we designed and synthesized three additional ASOs with PS-DNA segment incorporated directly at either 5′-end (ASO11) or 3′-end (ASO12) of the ASO, while the fully modified 2′-MOE ASO served as the control (2′-MOE control, Table 1). We also performed Tm analysis for the ASOs and as predicted, the data showed that the control 2′-MOE achieved highest Tm (69.3 °C, Table 1), followed by ASO11 (65.4 °C) and ASO12 (65.2 °C). Unlike the case of ASO1 and 5 where the Tm was significant different (56.9 °C compared with 59.2 °C), ASO11 and ASO12 achieved fairly equal Tm data, suggested that although sharing similar design concept in which the PS-DNA segment was introduced at either 3′-end (ASO1 and 11) or 5′-end (ASO5 and 12), alternative 2′-modified RNA chemistry could affect the ASO target binding affinity.
Next, we tested the ASO potency in inducing exon 23 skipping in H2K mdx cells as described above. Upon transfection and collection, the RT-PCR results showed that overall, despite having equal Tm results, ASO12 induced higher amount of total exon skipping products (including the dual exon 22, 23 skipping) than ASO11 (Fig. 4A, B and D). However, in terms of exon 23 skipping alone, ASO11 performed slightly better than ASO12 except at 100 nM (Fig. 4A and B). This is quite surprising as from the previous experiment where ASO5 was superior in inducing exon 23 skipping compared with ASO1 at all concentrations, we expected that ASO12 would outperform ASO11, but not in this case. Interestingly, both ASOs did not induce any skipping at 2.5 and 5 nM, while the control 2′-MOE ASO induced 35% of exon 23 skipping at 5 nM (Fig. 4C and D).
Fig. 4. RT-PCR with sequence visualisation (A–C) and densitometry analysis (D) of RNA prepared from H2K mdx mouse myotubes transfected with ASO11 (A), 12 (B) and control 2′-MOE (C). The triangles above the gel image indicate increasing ASO concentration (2.5, 5, 10, 25, 50 and 100 nM); FL (full-length) = 901 bp, represented in grey color; Δ23 (exon-23 skipped) = 688 bp, represented in blue color; Δ22–23 (exons-22 + 23 skipped) = 542 bp, represented in green color; UT = untreated, Ne = negative control.

Notably, at 10 and 25 nM, ASO11 and 12 both performed better than the control ASO, yielded 45 and 35% (ASO11), 33 and 27% (ASO12) of exon 23 skipped products, compared with 30 and 25% of the control ASO (Fig. 4A–D). At 50 nM, all three ASOs induced similar amount of exon 23 skipping, recorded at 58, 54 and 56%, respectively for ASO11, 12 and control ASO. Remarkably, at 100 nM, ASO12 achieved highest exon 23 skipping with 78%, in comparison to 65% of the ASO11 and 56% of the control ASO (Fig. 4A–D). This could be due to an increase in the undesired dual exon 22–23 skipped products induced by ASO11 and control ASO at 100 nM. These products were also detected with all ASOs at various concentrations, ranging from 23 to 41% (Fig. 4). This experiment again validates our theory that improved efficiency can be achieved with PS-DNA segment incorporated at 3′-end of the ASO sequence, but different 2′-modified RNA chemistry can result in variable exon skipping.
Conclusions
In summary, we report a novel approach to design splice modulating ASO by incorporating a PS-DNA segment into a 2′-modified RNA sequence. Our results demonstrated that 2′-modified PS ASO with a four PS-DNA segment or less at the 3′-end substantially improved exon 23 skipping potential. Based on our results, we firmly believe that drug-like properties of 2′-modified ASOs for splice modulation could be modulated with use of an appropriately placed segment of PS-DNA as has been achieved in the use of gapmer antisense for RNase H mediated mechanism. Our study significantly contributes in the future design of splice modulating ASO towards developing ASO therapy with better a safety profile and improved therapeutic efficacy.
Author contributions
R. N. V. and S. A. conceived the research; B. T. L. performed the experiments. All authors analysed the results, co-wrote and reviewed the manuscript.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
RNV acknowledges funding from the McCusker Charitable Foundation and Department of Health, Western Australia Merit Award Scheme. BTL and RNV acknowledges funding from Perron Institute for Neurological and Translational Science and Murdoch University. We thank Prof. Steve Wilton and Prof. Sue Fletcher and their research group for providing H2K mdx cells. We thank Dr Tamer Kosbar for the synthesis of ASO11, 12, and two control ASOs.
Electronic supplementary information (ESI) available: Tm curves, EC50 data and MS and HPLC analysis data of the ASOs. See DOI: 10.1039/d1ra00878a
References
- Le B. T. Raguraman P. Kosbar T. R. Fletcher S. Wilton S. D. Veedu R. N. Mol. Ther.–Nucleic Acids. 2019;14:142–157. doi: 10.1016/j.omtn.2018.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y. Rio D. C. Annu. Rev. Biochem. 2015;84:291–323. doi: 10.1146/annurev-biochem-060614-034316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daoutsali E. and Aartsma-Rus A., in Advances in Nucleic Acid Therapeutics, The Royal Society of Chemistry, 2019, pp. 103–125, 10.1039/9781788015714-00103 [DOI] [Google Scholar]
- Dominski Z. Kole R. Proc. Natl. Acad. Sci. U. S. A. 1993;90:8673–8677. doi: 10.1073/pnas.90.18.8673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sierakowska H. Sambade M. J. Agrawal S. Kole R. Proc. Natl. Acad. Sci. U. S. A. 1996;93:12840–12844. doi: 10.1073/pnas.93.23.12840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Syed Y. Y. Drugs. 2016;76:1699–1704. doi: 10.1007/s40265-016-0657-1. [DOI] [PubMed] [Google Scholar]
- Hoy S. M. Drugs. 2017;77:473–479. doi: 10.1007/s40265-017-0711-7. [DOI] [PubMed] [Google Scholar]
- Heo Y. A. Drugs. 2020;80:329–333. doi: 10.1007/s40265-020-01267-2. [DOI] [PubMed] [Google Scholar]
- Dhillon S. Drugs. 2020;80:1027–1031. doi: 10.1007/s40265-020-01339-3. [DOI] [PubMed] [Google Scholar]
- FDA NEWS RELEASE, FDA Approves Targeted Treatment for Rare Duchenne Muscular Dystrophy Mutation, https://www.fda.gov/news-events/press-announcements/fda-approves-targeted-treatment-rare-duchenne-muscular-dystrophy-mutation-0, accessed on 25 Mar 2021
- Chen S. Le B. T. Chakravarthy M. Kosbar T. R. Veedu R. N. Sci. Rep. 2019;9:6078. doi: 10.1038/s41598-019-42523-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le B. T. Adams A. M. Fletcher S. Wilton S. D. Veedu R. N. Mol. Ther.–Nucleic Acids. 2017;9:155–161. doi: 10.1016/j.omtn.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le B. T. Filichev V. V. Veedu R. N. RSC Adv. 2016;6:95169–95172. doi: 10.1039/C6RA22346J. [DOI] [Google Scholar]
- Le B. T. Chen S. Abramov M. Herdewijn P. Veedu R. N. Chem. Commun. 2016;52:13467–13470. doi: 10.1039/C6CC07447B. [DOI] [PubMed] [Google Scholar]
- Chen S. Le B. T. Rahimizadeh K. Shaikh K. Mohal N. Veedu R. N. Molecules. 2016;21:1582. doi: 10.3390/molecules21111582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le B. T. Murayama K. Shabanpoor F. Asanuma H. Veedu R. N. RSC Adv. 2017;7:34049–34052. doi: 10.1039/C7RA06091B. [DOI] [Google Scholar]
- Le B. T. Hornum M. Sharma P. K. Nielsen P. Veedu R. N. RSC Adv. 2017;7:54542–54545. doi: 10.1039/C7RA10964D. [DOI] [Google Scholar]
- Shimo T. Tachibana K. Kawawaki Y. Watahiki Y. Ishigaki T. Nakatsuji Y. Hara T. Kawakami J. Obika S. Chem. Commun. 2019;55:6850–6853. doi: 10.1039/C8CC09648A. [DOI] [PubMed] [Google Scholar]
- Bao T. L. Veedu R. N. Fletcher S. Wilton S. D. Expert Opin. Orphan Drugs. 2016;4:139–152. doi: 10.1517/21678707.2016.1122517. [DOI] [Google Scholar]
- Gait M. J. Arzumanov A. A. McClorey G. Godfrey C. Betts C. Hammond S. Wood M. J. A. Nucleic Acid Ther. 2019;29:1–12. doi: 10.1089/nat.2018.0747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metelev V. and Agrawal S., US Pat., 5652355A, 1992
- Metelev V. and Agrawal S., US Pat., 6143881, 1995
- Metelev V. and Agrawal S., US Pat., 6346614B1, 1995
- Agrawal S. and Gait M. J., in Advances in Nucleic Acid Therapeutics, The Royal Society of Chemistry, 2019, pp. 1–21, 10.1039/9781788015714-00001 [DOI] [Google Scholar]
- Agrawal S. Trends Mol. Med. 2020;26:1061–1064. doi: 10.1016/j.molmed.2020.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S. Mayrand S. H. Zamecnik P. C. Pederson T. Proc. Natl. Acad. Sci. U. S. A. 1990;87:1401–1405. doi: 10.1073/pnas.87.4.1401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agrawal S. Jiang Z. Zhao Q. Shaw D. Cai Q. Roskey A. Channavajjala L. Saxinger C. Zhang R. Proc. Natl. Acad. Sci. U. S. A. 1997;94:2620–2625. doi: 10.1073/pnas.94.6.2620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen L. X. Kandimalla E. R. Agrawal S. Bioorg. Med. Chem. 1998;6:1695–1705. doi: 10.1016/S0968-0896(98)00131-X. [DOI] [PubMed] [Google Scholar]
- Bhagat L. Putta M. R. Wang D. Yu D. Lan T. Jiang W. Sun Z. Wang H. Tang J. X. La Monica N. Kandimalla E. R. Agrawal S. J. Med. Chem. 2011;54:3027–3036. doi: 10.1021/jm200113t. [DOI] [PubMed] [Google Scholar]
- Bulfield G. Siller W. G. Wight P. A. Moore K. J. Proc. Natl. Acad. Sci. U. S. A. 1984;81:1189–1192. doi: 10.1073/pnas.81.4.1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rando T. A. Blau H. M. J. Cell Biol. 1994;125:1275–1287. doi: 10.1083/jcb.125.6.1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilton S. D. Lloyd F. Carville K. Fletcher S. Honeyman K. Agrawal S. Kole R. Neuromuscul. Disord. 1999;9:330–338. doi: 10.1016/S0960-8966(99)00010-3. [DOI] [PubMed] [Google Scholar]
- Mann C. J. Honeyman K. Cheng A. J. Ly T. Lloyd F. Fletcher S. Morgan J. E. Partridge T. A. Wilton S. D. Proc. Natl. Acad. Sci. U. S. A. 2001;98:42–47. doi: 10.1073/pnas.98.1.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider C. A. Rasband W. S. Eliceiri K. W. Nat. Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.