

To the Editor:
During the clinical workup of an adolescent male with chronic active EBV disease, it was noted that natural killer (NK) cells of this individual did not express the Fc-receptor CD16A (Fig 1, A). The index patient, born to nonconsanguineous parents, was first seen when he was 11 years old, and an extended workup revealed persistently high levels of EBV-encoded small RNA (EBER) DNA in peripheral blood and multiple-organ infiltration with EBV-positive T lymphocytes including an IgA- and EBV-positive interstitial nephritis that eventually led to renal failure (see Case Report and Table E1 in this article’s Online Repository at www.jacionline.org).
FIG 1.
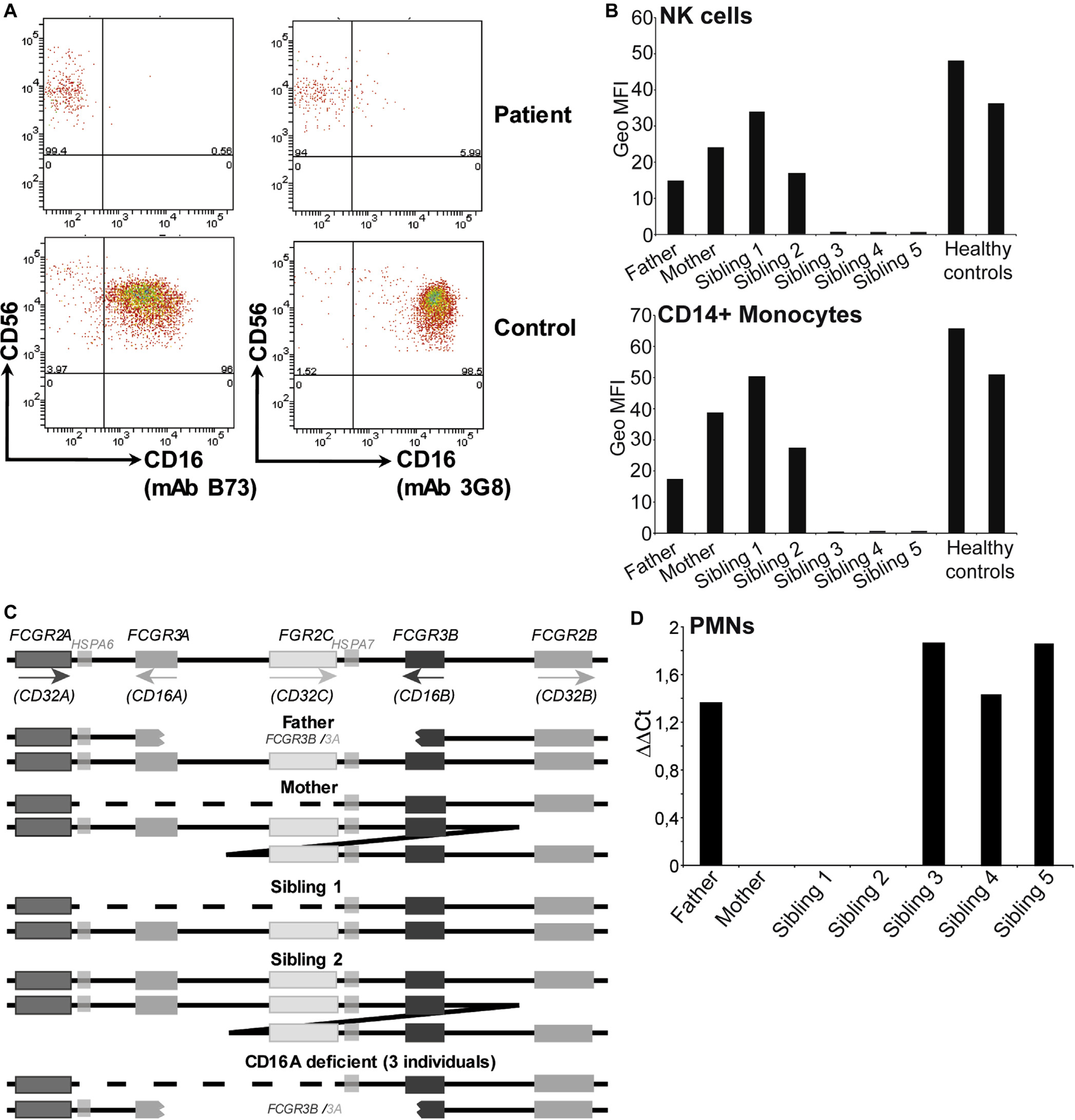
A, Representative cytometry plots showing the lack of CD16 expression by patient NK cells. B, CD16 expression on NK cells and monocytes of healthy, unrelated controls and family members. C, The structure of the low/medium- affinity FcγR locus on chromosome 1, compared with the same genomic region in the various members of the family studied. D, Low levels of CD16BA mRNA are found only in granulocytes of family members with this gene. MFI, Mean Fluorescence intensity; PMN, polymorphonuclear leukocyte.
NK-cell CD16 expression was not observed with either the B73 or the 3G8 mAbs that bind different epitopes on CD161. Moreover, CD16 protein was undetectable in western blot analysis of purified PBMCs (see Fig E1 in this article’s Online Repository at www.jacionline.org). Granulocytes of this individual did express the GPI-linked CD16B receptor, but at reduced levels compared with healthy controls (see Fig E2 in this article’s Online Repository at www.jacionline.org). Analysis of the immediate family identified 2 other siblings (a twin brother and 1 sister) who also lacked CD16A expression on peripheral blood NK cells and monocytes (Fig 1, B; a family tree is shown in Fig E3 in this article’s Online Repository at www.jacionline.org). Although these CD16A-deficient siblings did not present such severe clinical symptoms, detailed studies (Case Report and Table E1) have revealed frequently elevated levels of EBV DNAemia and their evolution is being monitored. Differences in the severity of disease between patients harboring similar mutations are not uncommon and as yet we have no definitive explanation as to why the clinical presentation of the index patient was more severe. However, because the marked divergence in clinical features between the twins occurred at age 14 to 15 years, it might be possible that at least some of these features were age-related. It may also be relevant that 1 of the 3 affected siblings was female.
Sequencing of patient CD16 cDNA clones identified both transmembrane and GPI-linked receptors (see Fig E4 in this article’s Online Repository at www.jacionline.org). However, analysis of the sequences revealed that the 5′ region of the cDNA clones encoding a transmembrane domain contained residues more typical of CD16B. The FcγR2/3 locus at chromosome 1q23.3 is known to be prone to recombination and unequal crossing over2 and detailed genetic analyses showed that the CD16A-deficient individuals had inherited a maternal chromosome where the FCGR3A gene had been deleted and a paternal chromosome that encoded a hybrid FCGR3B/A gene (Fig 1, C). Although not detected in cDNA from mononuclear cells, low levels of CD16B/A mRNA could be found in granulocyte mRNA (Fig 1, D), consistent with the 5′ regulatory elements being those of CD16B.
NK cells from the CD16A-deficient individuals did not mediate antibody-dependent cellular cytotoxicity (ADCC) (Fig 2, A). However, natural cytotoxicity against K562 cells or after stimulation of specific receptors including NKG2D and 2B4 was comparable to that mediated by CD16-expressing family members and unrelated healthy donors (Fig 2, B). These observations closely match the phenotype of NK cells from FcγR3A knockout mice,3 but were surprising because CD16A has been suggested to participate in processes of natural cytotoxicity, via association with CD2.4 However, NK-cell CD2 expression did not vary between CD16A-expressing and CD16A-deficient family members (see Fig E5 in this article’s Online Repository at www.jacionline.org) in multiple blood samples analyzed over an 18-month period. Mass cytometry analysis showed that the NK-cell populations found in CD16A deficiency were similar to those present in healthy donors (see Fig E6 in this article’s Online Repository at www.jacionline.org), suggesting that the absence of CD16A had only limited effects on general NK-cell differentiation. However, because all the family members were EBV- and human cytomegalovirus (HCMV)-seropositive, we assayed the levels of specific peripheral blood NK-cell subsets associated with these virus infections,5,6 to assess the ability of CD16A-deficient NK cells to participate in immune responses in vivo. EBV-reactive NK cells were present in all the individuals analyzed, but at markedly elevated levels in the 3 CD16A-deficient individuals studied (Fig 2, C), consistent with the higher levels of virus replication observed in these individuals compared with other family members. Interestingly, since the index patient received a hematopoietic stem-cell transplant in 2016, his EBV DNAemia is persistently negative and the frequency of the EBV-reactive NK cells has decreased to levels similar to those found in healthy EBV-seropositive individuals (around 1.2%, data not shown).
FIG 2.
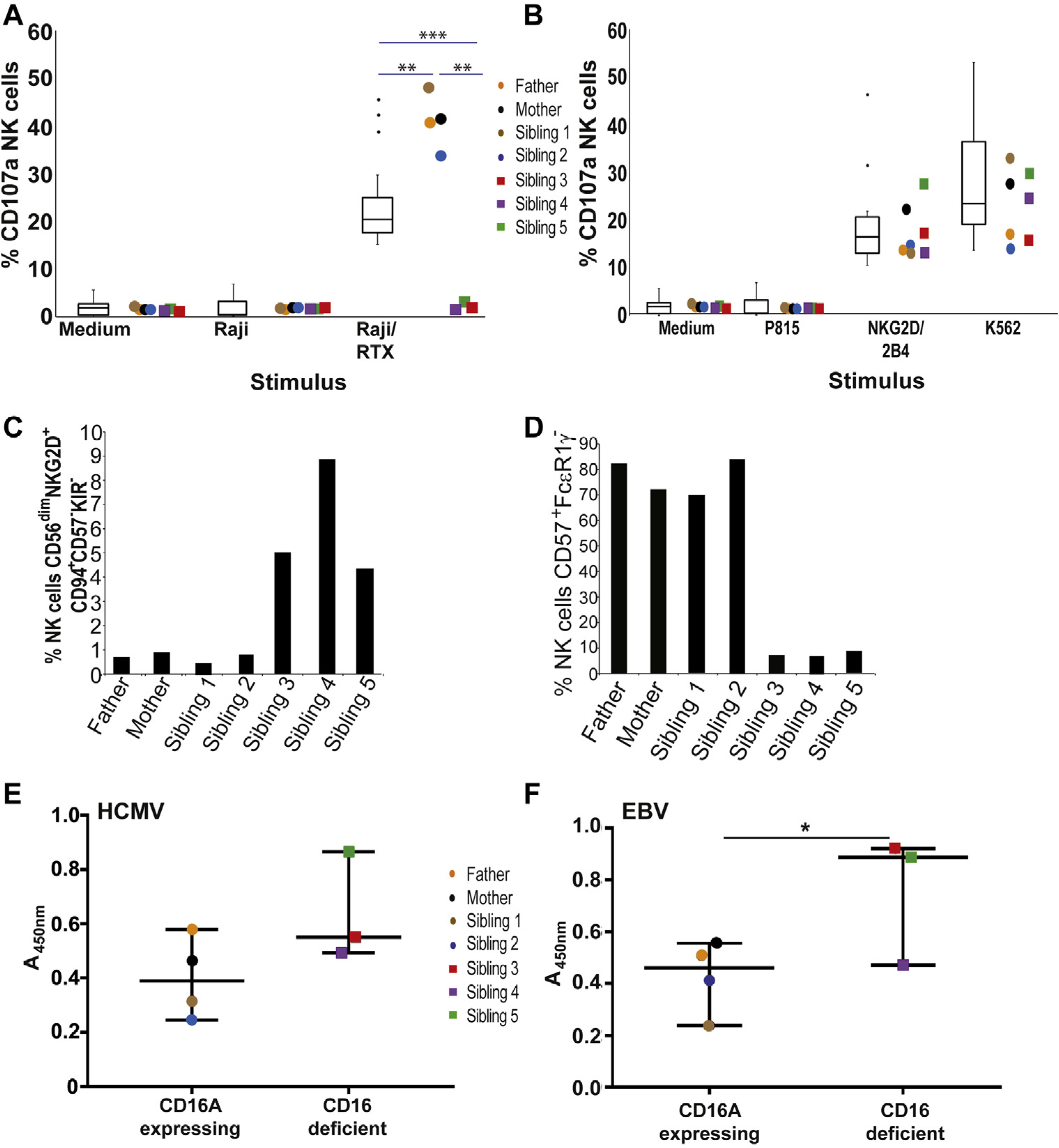
A, NK cells that do not express CD16A are unable to mediate ADCC. t test, median, and first and third quartiles are indicated. Error bars = median 95% CI. RTX, Rituximab. B, NK cells from CD16A-competent and -deficient family members mediate levels of natural cytotoxicity comparable to a large number of healthy controls. C, Increased frequency of EBV-reactive NK cells in PBMCs of CD16A-deficient family members. D, Reduced frequency of adaptive NK cells in PBMCs from CD16A-deficient family members. E, Both CD16A-expressing and CD16A-deficient family members make HCMV-specific antibodies able to activate CD16. F, CD16A-deficient family members make stronger EBV-specific, CD16-activating antibody responses than do CD16A-expressing relatives. Unpaired, nonparametric t test. Error bars = median 95% CI.
In contrast, the development of “adaptive” NK cells (CD56dim, CD57+, FcεR1γ−) in the CD16A-deficient individuals was markedly reduced compared with family members who possessed even a single copy of FCGR3A (Fig 2, D; representative flow cytometry plots are shown in Fig E7 in this article’s Online Repository at www.jacionline.org). Interestingly, the increased levels of CD2 expression reported on adaptive NK cells, compared with CD56dim NK cells, were also observed on the adaptive NK cells of the individuals studied here, including those scarce adaptive NK cells found in CD16A-deficient family members (see Fig E8 in this article’s Online Repository at www.jacionline.org).
Because it has been shown that the expansion, at least in vitro, of FcεR1γ-deficient NK cells requires coculture with virus-infected cells and human serum containing virus-specific antibodies,7 we tested for the presence of CD16-binding and CMV-specific antibodies, using a cell-based CD16 reporter assay.8 CD16 activation by CMV- and EBV-specific antibodies was not less in CD16A-deficient family members compared with those expressing CD16A (Fig 2, E and F), and at least for EBV was actually significantly stronger. These observations are consistent with the suggestion that the inability of NK cells from CD16A-deficient individuals to sense immune complexes might impair the normal expansion of these cells.
To our knowledge, this is the first description of CD16A-deficient humans. Our analyses show that this receptor is essential for the ADCC function of human NK cells. Increases in the frequency and number of NK cells in the blood of patients with infectious mononucleosis correlate inversely with disease severity and viral loads,9 supporting an important role for NK cells in control of EBV. That chronic EBV replication was a feature of the clinical presentation of CD16A deficiency strongly suggests that ADCC makes a critical contribution to the NK-cell response to EBV infection.
Our data also clearly imply that, in vivo, the interaction between CD16A and Ig-Fc markedly influences the development of the adaptive NK cells that are specialized to mediate ADCC.7 The frequency of adaptive NK cells in the CD16A-deficient individuals was more than 10-fold less than that in CD16A-expressing family members. Not all HCMV-seropositive subjects in the general population present with an adaptive NK-cell expansion, perhaps because of differences between HCMV isolates, or perhaps because of genetic variation not yet formally associated with this phenotype. Nevertheless, it seems plausible that the failure of adaptive NK cells to expand in 3 CD16A-deficient individuals of a single family is due directly to the absence of CD16A rather than other factors. The more than 10-fold expansion of the adaptive NK-cell population observed in the index patient after successful hematopoietic stem-cell transplantation, in the absence of detectable virus reactivation, also argues that the reduced numbers of adaptive NK cells in CD16A deficiency reflects a defect intrinsic to the hematopoetic system. Given that adaptive cells can be found, albeit in reduced numbers, in CD16A-deficient individuals, we favor the hypothesis that “sensing” of immune complexes via CD16A is of major importance for the expansion of this population. In this small sample there is no clear correlation between antibody titers and adaptive NK-cell expansion, but analysis of a larger population of HCMV- and EBV-seropositive individuals will be necessary to properly explore this issue. A detailed biochemical and functional characterization of how antiviral IgG influences adaptive NK-cell development in vivo could open new avenues of IgG-based intervention strategies against multiple infectious and malignant diseases.
Supplementary Material
Acknowledgments
We thank patients and family members for kindly donating blood samples, Dr Elvira Ramil from the DNA sequencing facility, Instituto de Investigación Sanitaria Puerta de Hierro, for expert support, constructive advice, and help, and Professor J. L. Strominger, Harvard University, for the gift of hybridomas.
This work was supported by grants SAF2014-58752-R and SAF2017-83265-R to H.T.R. and grant SAF2016-80363-C2-2-R to C.V. (Spanish Science agency/European Regional Development Fund, European Union, AEI/FEDER, EU). The group of M.V.-G. has also received funding from the Spanish Ministry of Science and Innovation (grant nos. RTC-2017-6379-1 and RTI2018-093569-B-I00 [MCIU/AEI/FEDER, EU]) and the regional government of Madrid (grant no. S2017/BMD-3733-2). The group of M.V.-G. also belongs to the research network TENTACLES (RED2018-102411-T) funded by the Spanish Ministry of Science. A.P.P. and A.B.M. acknowledge PhD studentships funded by the National Secretary of Higher Education, Science, Technology and Innovation (SENESCYT, Ecuador) and Spanish Ministry of Economy and Competitivity (MINECO), respectively (SVP-2014-068263). M.M. was supported by grant GCB15152947MELE from Fundación de la Asociacion Española contra el Cáncer. Work in the laboratory of H.H. is supported by Infect-ERA grant TANKACY, BMBF 031L0090, and German Research Foundation (DFG) grant HE2526/9-1. E.L.G. has grants from the Spanish Association Against Cancer (AECC) and Carlos III Health Institute (grant no. PI16/01605). C.A.B. acknowledges support from the National Institutes of Health (grant no. DP2AI11219301). The agencies who provided financial support for the conduct of the research and preparation of the article played no role in the study design; in the collection, analysis, and interpretation of data; in the writing of the report; or in the decision to submit the article for publication. The rest of the authors declare that they have no relevant conflicts of interest.
REFERENCES
- 1.Perussia B, Trinchieri G, Jackson A, Warner NL, Faust J, Rumpold H, et al. The Fc receptor for IgG on human natural killer cells: phenotypic, functional, and comparative studies with monoclonal antibodies. J Immunol 1984;133:180–9. [PubMed] [Google Scholar]
- 2.Hargreaves CE, Rose-Zerilli MJ, Machado LR, Iriyama C, Hollox EJ, Cragg MS, et al. Fcgamma receptors: genetic variation, function, and disease. Immunol Rev 2015;268:6–24. [DOI] [PubMed] [Google Scholar]
- 3.Hazenbos WL, Gessner JE, Hofhuis FM, Kuipers H, Meyer D, Heijnen IA, et al. Impaired IgG-dependent anaphylaxis and Arthus reaction in Fc gamma RIII (CD16) deficient mice. Immunity 1996;5:181–8. [DOI] [PubMed] [Google Scholar]
- 4.Grier JT, Forbes LR, Monaco-Shawver L, Oshinsky J, Atkinson TP, Moody C, et al. Human immunodeficiency-causing mutation defines CD16 in spontaneous NK cell cytotoxicity. J Clin Invest 2012;122:3769–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Azzi T, Lunemann A, Murer A, Ueda S, Beziat V, Malmberg KJ, et al. Role for early-differentiated natural killer cells in infectious mononucleosis. Blood 2014; 124:2533–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guma M, Angulo A, Vilches C, Gomez-Lozano N, Malats N, Lopez-Botet M. Imprint of human cytomegalovirus infection on the NK cell receptor repertoire. Blood 2004;104:3664–71. [DOI] [PubMed] [Google Scholar]
- 7.Lee J, Zhang T, Hwang I, Kim A, Nitschke L, Kim M, et al. Epigenetic modification and antibody-dependent expansion of memory-like NK cells in human cytomegalovirus-infected individuals. Immunity 2015;42:431–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corrales-Aguilar E, Trilling M, Reinhard H, Merce-Maldonado E, Widera M, Schaal H, et al. A novel assay for detecting virus-specific antibodies triggering activation of Fcgamma receptors. J Immunol Methods 2013;387:21–35. [DOI] [PubMed] [Google Scholar]
- 9.Williams H, McAulay K, Macsween KF, Gallacher NJ, Higgins CD, Harrison N, et al. The immune response to primary EBV infection: a role for natural killer cells. Br J Haematol 2005;129:266–74. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.