

SUMMARY
It is well established in the microbiome field that antibiotic (ATB) use and metabolic disease both impact the structure and function of the gut microbiome. But how host and microbial metabolism interacts with ATB susceptibility to affect the resulting dysbiosis remains poorly understood. In a streptozotocin-induced model of hyperglycemia (HG), we use a combined metagenomic, metatranscriptomic, and metabolomic approach to profile changes in microbiome taxonomic composition, transcriptional activity, and metabolite abundance both pre- and post-ATB challenge. We find that HG impacts both microbiome structure and metabolism, ultimately increasing susceptibility to amoxicillin. HG exacerbates drug-induced dysbiosis and increases both phosphotransferase system activity and energy catabolism compared to controls. Finally, HG and ATB co-treatment increases pathogen susceptibility and reduces survival in a Salmonella enterica infection model. Our data demonstrate that induced HG is sufficient to modify the cecal metabolite pool, worsen the severity of ATB dysbiosis, and decrease colonization resistance.
In brief
Wurster et al. find that streptozotocin-induced hyperglycemia significantly changes the metabolome and transcriptional behavior of the cecal microbiome. These changes potentially exacerbate amoxicillin-induced dysbiosis and decrease colonization resistance against Salmonella enterica. Together these data suggest that modifications to host metabolism may perturb microbiome metabolism and potentiate antibiotic susceptibility.
Graphical Abstract
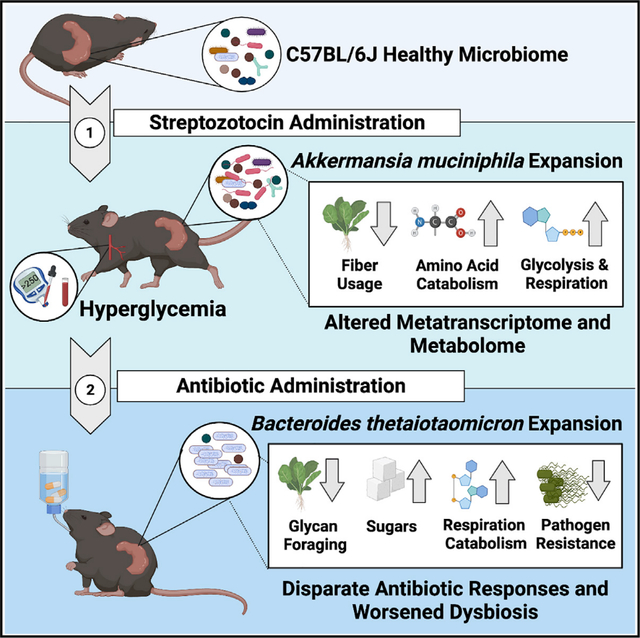
INTRODUCTION
Exposure to antibiotics (ATB) is one of the most significant known microbiome perturbations. Drug-induced dysbiosis occurs within hours of treatment, and is characterized by loss of total bacterial load, taxonomic diversity, and significant transcriptional changes (Cabral et al., 2019, 2020; Dethlefsen and Relman, 2011). This alters the intestinal metabolome, placing the host at higher risk for opportunistic infection (Bäumler and Sperandio, 2016; Buffie et al., 2012; Chang et al., 2008; Croswell et al., 2009; Kaiko and Stappenbeck, 2014; Rivera-Chávez et al., 2016; Theriot et al., 2016; Theriot and Young, 2015). Given the severity of ATBs on the microbiome and the near ubiquitous use of these drugs, it is critical to mechanistically understand ATB activity within the gut and the external factors that dictate susceptibility.
Microbial metabolism is a key determinant of ATB susceptibility (Stokes et al., 2019). Microbes performing ATP-generating processes like aerobic respiration have increased bactericidal drug sensitivity and experience a lethal respiratory burst during in vitro exposure (Adolfsen and Brynildsen, 2015; Belenky et al., 2015; Dwyer et al., 2014; Kohanski et al., 2007; Lam et al., 2020; Lobritz et al., 2015). Meanwhile, fermentation, diversion away from the tricarboxylic acid (TCA) cycle, or overall reduction in metabolism can confer drug tolerance in some species (Ahn et al., 2016; Conlon et al., 2016; Lobritz et al., 2015; Meylan et al., 2017; Thomas et al., 2013). We demonstrated that this trend holds true within the context of the microbiome, where ATB exposure dramatically reduces community metabolic capacity (Cabral et al., 2019). Surviving taxa like Bacteroides thetaiotaomicron (B. theta) can endure amoxicillin (AMX) exposure by transcriptional adaptation that prioritizes fiber fermentation over the utilization of simple sugars (Cabral et al., 2019). When considering mechanisms of in vivo susceptibility, it is important to consider the role of local nutrients on microbial metabolism. Host diet is likely one of the largest factors shaping the cecal nutrient pool; dietary changes can perturb microbiome diversity and activity, and thus may impact ATB susceptibility (Albenberg and Wu, 2014; Bisanz et al., 2019; Collins et al., 2018; David et al., 2014; Ley, 2014; Smits et al., 2017; Tanes et al., 2021). Congruently, we showed that added dietary glucose potentiates AMX toxicity within the cecum, reducing total bacterial load and B. theta’s drug tolerance (Cabral et al., 2019). This also occurs with the bactericidal drug ciprofloxacin, where consumption of a high fat/sugar diet increases mucus and simple sugar breakdown, increases gut glycolysis, and enhances microbiome drug susceptibility (Cabral et al., 2020). This suggests that the local nutrient pool can drive the severity of ATB activity in the microbiome by altering the metabolic rate of resident taxa.
Diet composition is not the sole determinant of nutrient availability within the gut. Normally, a small fraction of digested material reaches the dense communities of the lower gastrointestinal tract (GI). The composition of dietary molecules presented to the lower GI is impacted by multiple small intestinal (SI) digestive gradients and pancreaticobiliary secretions (Reese and Carmody, 2019; Shin et al., 2019). For example, the host controls colonic sugar concentrations via a combination of SI transporter expression, gastric emptying rate, and enteroendocrine function (Chen et al., 2016; Holst et al., 2016; Koepsell, 2020; Ussar et al., 2017). Disruptions of host metabolism, like digestive and metabolic disorders, are correlated with microbial dysbiosis, highlighting the connection between host and microbial systems in the GI (Brestoff and Artis, 2013; Westfall et al., 2015; Qin et al., 2012; Sabatino et al., 2017). For example, dysglycemic patients demonstrate bacterial infiltration of the intestinal epithelial mucosa, suggesting that dysglycemia triggers an inflammatory intestinal phenotype by prompting microbial breakdown of mucus glycoproteins (Chassaing et al., 2017).
Host hyperglycemia (HG) may cause potent modulation of the lower GI metabolic environment. Currently, the relationship between dysregulated host metabolism, the local metabolite environment of the GI, and the severity of ATB-induced dysbiosis remains relatively understudied. We hypothesize that changes in host metabolism associated with induced HG will alter the microbiota-accessible cecal metabolite pool and place the community in a metabolically permissive state that increases susceptibility to bactericidal ATBs. To test this, we used the single-dose streptozotocin (STZ) model rather than a diet- or genetically based model of glucose disruption (Deeds et al., 2011; Kobayashi et al., 2000; Wang and Liao, 2011). STZ is a glucosamine nitrosourea compound that induces HG via the selective and irreversible destruction of insulin-producing pancreatic β cells (Eleazu et al., 2013; Wu and Yan, 2015). STZ is quickly metabolized by the host, with a serum clearance time of about 15 minutes (Lee et al., 2010; Eleazu et al., 2013). Thus, STZ provides the benefit of rapidly-induced and irreversible HG without potentially microbiome-confounding factors like diet and host genetics (Deeds et al., 2011; Xiao et al., 2017; Yang et al., 2019). Existing research on glucose dysregulation and the microbiome is impacted by the use of dietary metabolic animal models like the high-fat diet-induced diabetes mouse (Fujisaka et al., 2016). In this study we used a multi-omic approach that profiled the taxonomic composition, transcriptional activity, and small molecule repertoire of the cecum to characterize the impact of STZ-induced HG on microbiome disruption during AMX treatment. We then profiled the effects of HG on AMX-induced pathogen susceptibility by challenging mice with Salmonella enterica. Our data show that HG is sufficient to modulate the cecal metabolite pool, and that these changes both potentiate ATB-induced dysbiosis and worsen the dysbiosis-related complication of opportunistic infection.
RESULTS
To examine the combined effect of HG and ATBs on microbiome structure and function, male C57BL/6J mice were given an intraperitoneal injection of either STZ or a sham (control). Mice were checked for HG 48 hours post injection and were then randomized. The next day, animals were given AMX or a sham (vehicle) for 24 hours ad libitum, which is sufficient to profile acute microbiome ATB responses without encountering significant extinction events (Cabral et al., 2020; 2019). After AMX delivery, mice were sacrificed and cecal contents were harvested for multi-omic profiling (Figure 1A).
Figure 1. STZ modifies glucose levels and impacts microbiome composition after AMX.

(A) Experimental design of this study. Figure was created with BioRender.com (BioRender, Toronto, Canada).
(B) Murine fasting blood glucose pre-STZ injection (Day 0), 2, and 5 days postinjection. Data represent mean ± SEM.
(C) Bray-Curtis Dissimilarity between 16S rRNA amplicons.
(D) α-diversity of WMGS experimental groups. Data represent mean ± SEM.
(E) Relative abundance of the five most-prominent bacterial phyla. Data represent mean ± SEM.
(F) Quantification of cecal glucose concentrations from experimental groups. Data represent mean ± SEM.
*p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. For (B): n = 44 control and 44 STZ-treated samples per time point; for (C): n = 8 to 11 per group; permutational ANOVA; for (D–F): N = 5 to 8 per group; Welch’s ANOVA with Dunnet T3 test for multiple hypothesis testing.
STZ caused significant and sustained HG (Figures 1B and S1A). Because STZ’s mechanism of action involves organ cytotoxicity, we quantified plasma cytokines to evaluate systemic inflammation (Eleazu et al., 2013). We found no statistical difference in cytokine levels prior to ATB administration (Figures S1B and S1C), but did observe an increase in IFN-γ 24 hours later (Figure S1D) that is likely correlated with the early stages of disease progression in this model (Hanafusa and Imagawa, 2008). We then assessed GI histopathology and quantified GI lipocalin-2 to profile for localized inflammation. STZ-treated mice had minimal to no edema or inflammation compared to controls (Figure S1E), and no difference in lipocalin-2 levels (Figure S1F). Together, these data show minimal differences in inflammatory phenotypes between STZ-treated and control mice during the time frame used in this study.
We profiled the effect of HG and AMX on microbiome structure using 16S rRNA sequencing. STZ treatment did not reduce diversity (Figure S1G) but did alter community composition when combined with AMX (Figures 1C, S1H, and S1I). Because 16S sequencing has limited phylogenetic resolution, we conducted the remaining analyses with whole metagenomic sequencing (WMGS) (Cabral et al., 2020; 2019; Clooney et al., 2016; Poretsky et al., 2014; Ranjan et al., 2016). WMGS showed that STZ alone did not impact α-diversity but bolstered the reduction in diversity and taxonomic shifts caused by AMX (Figures 1D and 1E).
Since STZ ablates insulin synthesis, and insulin helps regulate intestinal glucose absorption (Ussar et al., 2017), we asked if GI-localized glucose levels were altered by HG. However, we found that STZ-treated mice and controls had no significant difference in cecal glucose levels (Figure 1F). Therefore, glucose availability cannot explain the changes in community composition between experimental groups. Thus, we profiled the metabolome using quadrupole time-of-flight mass spectrometry (Q-TOF-MS) and liquid chromatography tandem mass spectrometry (LC-MS/MS) (Tables S1 and S2). By assessing cecal metabolite diversity using Principal Coordinates Analysis, we found that both STZ and AMX significantly impacted the cecal metabolome’s composition (Figure 2A). We hypothesized that STZ-induced HG establishes a transcriptional and metabolic environment that alters the microbiome’s response to ATB exposure. To confirm this, we assessed the impact of HG on microbiome function both pre- and post-AMX treatment.
Figure 2. STZ modifies the cecal metabolome and metatranscriptome.

(A) Bray-Curtis Dissimilarity of Q-TOF-MS extracts from experimental groups.
(B) Differentially abundant bacterial species following STZ treatment. Data represent log2 fold change ± SEM versus NG controls.
(C) Differentially abundant Q-TOF-MS metabolites following STZ treatment. Data represent log2 fold change ± SEM. See Table S1 for full results.
(D) Linear discriminant analysis of MetaCyc pathways following STZ treatment. See Table S6 for full results.
(E) Volcano plot of the cecal metatranscriptome following STZ treatment. Purple points represent differentially abundant transcripts. See Table S8 for full results.
(F) Taxon Stratified Community Metabolic Potential as calculated by MIMOSA. See Table S9 for full results.
*p < 0.05; **p < 0.01; ***p < 0.001. For (A and C): n = 6 per group, 2 replicates per sample; for (B): n = 5 to 8 per group; for (D–F): n = 4 per group.
HG significantly modifies the cecal metabolome and metatranscriptome
Unlike dietary models, STZ was associated with a single significant phylum-level change: the expansion of Verrucomicrobia (Figures 1E and S1I) (Xiao et al., 2017; Yang et al., 2019). We confirmed that this expansion was driven by Akkermansia muciniphila using differential abundance testing (Figure 2B) (Love et al., 2014). A. muciniphila forages carbon from epithelial mucins and has been proposed to breakdown gut lining integrity, which may contribute to cecal metabolome divergence via imbalances in the local carbon pool that impact microbial cross-feeding networks (Belzer et al., 2017; Cabral et al., 2020; Desai et al., 2016; Zhang et al., 2019). For example, we observed a reduction in the abundance of Blautia sp. YL58 after STZ (Figure 2B). Members of this genus are short-chain fatty acid (SCFA) producers that use mucin as a carbon substrate (Bui et al., 2019; Oliphant and Allen-Vercoe, 2019; Rey et al., 2010; Vacca et al., 2020). A. muciniphila may outcompete Blautia for mucins in the HG host, which would disrupt any syntropic reactions Blautia participates in. Because the pre-AMX community structure was similar between hosts, we felt that the disparity in ATB susceptibility was due to modified metabolic and transcriptional activity, rather than baseline taxonomic differences.
We paired differential abundance testing (Love et al., 2014) and pathway-level projection (Aggio et al., 2010) of our Q-TOF-MS data with random forest classification of LC-MS/MS features to identify distinctive metabolites between HG mice and controls (Figures S2A–S2C; Tables S1, S2, and S3). We then paired these findings with community- and species-level transcriptomics to better profile the microbiome’s functional capacity prior to ATB exposure. Despite consuming identical diets, HG and normoglycemic (NG) mice had varied levels of metabolites related to polysaccharide processing. We saw STZ-specific enrichment of the flavones apigenin, schaftoside, and daidzein; and significant reduction of major metabolites from apigenin breakdown such as 3–3-hydroxyphenyl propanoate (Figure 2C; Table S1). These metabolites can generate either hydroxyphenylacetic acids or phenolic intermediates that are converted to SCFAs by Firmicutes (Braune and Blaut, 2016), and their accumulation may indicate diminished SCFA generation. To that end, STZ-treatment reduced valerate levels (Figure 2C). HG also lowered phytate degradation and multiple polysaccharide-targeted carbohydrate-active enzymes (CAZymes; Figures 2C, 2D, and S2D; Tables S1, S4, and S6). Polysaccharide-fermenting taxa like B. theta (Martens et al., 2008; Sonnenburg et al., 2005) had reduced expression of targeted fiber import loci (Figure S2E: BT3086, BT3087, BT3090, and BT4581; Table S5). These data suggest that STZ may impair microbial fiber fermentation and alter the level of polysaccharide-derived carbon sources.
Amino acids (aa) are another significant bacterial carbon source (Wang et al., 2019a) that was impacted by STZ treatment. Multiple metabolites related to aromatic amino acid (AAA) generation, like 3-(3-hydroxyphenyl)propanoic acid and phenylethyl alcohol, were reduced by STZ (Figure 2C; Table S1). We saw enrichment of metabolites involved in aa catabolism like 6-methylnicotinamide, 2-ketoisocaproate and α-ketovaline, as well as pathway enrichment of AAA degradation and protein digestion, suggesting a shift toward aa catabolism rather than de novo synthesis (Figures S2A and S2B; Tables S1 and S3). Shikimate pathway intermediates like 3-dehydroquinate, 3-dehydroshikimate, and shikimate were enriched after STZ treatment, and likely caused by a block in a terminal component of the pathway, as transcription of both AAA and chorismate synthesis were reduced (Figures 2C and 2D; Tables S1 and S6). The shikimate pathway feeds directly into AAA generation via chorismate; thus, reduced transcription and accumulation of metabolic intermediates suggests a shift from anabolic to catabolic aa metabolism.
The shikimate pathway is also involved in B-vitamin generation and impacts the availability of energy carriers like coenzyme A (CoA) (Tzin and Galili, 2010). We observed enrichment of metabolites involved in pantothenate and CoA biosynthesis coupled with reduced pathway transcription (Figures 2D and S2B; Tables S3 and S6). STZ increased expression of thiazole biosynthesis, which is critical for generating vitamin B1 and thus key metabolic enzymes like pyruvate dehydrogenase, pyruvate decarboxylase, and a-ketoglutarate dehydrogenase (Andersen et al., 2015; Allaway et al., 2020; Yoshii et al., 2019) (Figure 2D; Table S6). We observed increased pyruvate, glycolysis, and gluconeogenesis-related metabolites, including glutamine and glycerol-3-phosphate (Figures S2A and S2B). This enrichment was coupled with elevated ATPase, phosphoenolpyruvate hydratase, and succinate dehydrogenase transcription (Figure 2E; Table S8), that, when considered in tandem with increased inosine and tRNA processing (Figure 2D; Tables S1 and S6), suggests that STZ bolsters respiration within the microbiome.
These data describe community-level changes to microbiome function. To identify species-specific contributors to metabolome variation, we taxon stratified our Q-TOF-MS data using MIMOSA (Figure 2F) (Noecker et al., 2016). A. muciniphila had the largest contribution to community metabolism, followed by B. theta (Figure 2F; Table S9). A. muciniphila significantly contributed to acetate variation between hosts, speaking to STZ-related difference in SCFAs. Metabolic signatures of increased metabolism, including glutamine, inosine, and glycerol-3-phosphate accumulation, could be explained by synthesis and degradation from A. muciniphila (glutamate and inosone) and synthesis from B. theta (inosine and glycerol-3-phosphate). Finally, variation in phosphatidylethanolamine, a major component of microbial cell walls, could be somewhat explained by A. muciniphila (Figure 2F), suggesting increased cell wall synthesis by this taxon. These data highlight that these two taxa are key in HG-specific changes to microbiome function. STZ has robust impacts on cecal microbiome function. While A. muciniphila and B. theta are involved in this phenotype, it is important to consider that there is redundancy in species function and in substrate utilization across biochemical pathways (Tian et al., 2020). Thus, it is likely that the cumulative effect of multiple metabolic disruptions incurred from STZ increases metabolic demand on the community and leads to increased AMX susceptibility.
HG modifies the composition of Bacteroidetes and Firmicutes after AMX exposure
Given the connection between microbial metabolism and ATB susceptibility (Belenky et al., 2015; Cabral et al., 2019; Lobritz et al., 2015; Stokes et al., 2019), we hypothesized that STZ-induced metabolic disruption bolstered AMX susceptibility. HG mice had a highly divergent microbial composition after ATB exposure compared to controls (Figures 3A and S3). Specifically, HG exacerbated the AMX-related reduction in α-diversity (Figure 1D). Only HG mice had a reduction in Verrucomicrobia, although this may be due to the pre-AMX expansion of A. muciniphila (Figures 1E and 3B). Interestingly, the loss of Actinobacteria, Firmicutes, and Proteobacteria, and the bloom in Bacteroidetes that was expected after AMX were greater in HG mice (Figures 3C–3F) (Cabral et al., 2019). Consistent with our previous work (Cabral et al., 2019), the Bacteroidetes bloom was driven by expansion of B. theta in both hosts (Figure 3G).
Figure 3. STZ and AMX treatment modify the composition of the cecal microbiome.

(A) Relative abundance of the 25 most-abundant species in our dataset. Data are represented as mean ± SEM for each species.
(B) Relative abundance of Bacteroidetes.
(C) Relative abundance of Actinobacteria.
(D) Relative abundance of Firmicutes.
(E) Relative abundance of Proteobacteria.
(F) Relative abundance of Verrucomicrobia.
(G) Relative abundance of B. theta.
(H) Differentially abundant bacterial species following AMX treatment in control and STZ mice, with interaction value. Data represent log2 fold change ± SEM versus vehicle-treated controls.
For all panels: n = 5 to 8 per group; for (B–G): *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; Welch’s ANOVA with Dunnet T3 test for multiple hypothesis testing.
Data represent mean ± SEM.
We calculated the interaction of HG and AMX to examine host-specific changes in species abundance (Love et al., 2014). In addition to B. theta, many members of the Bacteroides genus increased after AMX, with significantly elevated abundance in HG mice (Figures 3H and S3A). Meanwhile, the species with reduced abundance in HG mice after AMX treatment were primarily within the order Clostridiales (Figures 3H, S3B, S3C, S3D, S3E, S3F, S3G, S3H, and S3I). These taxa are key starch degraders and SCFA producers, and their reduction suggests an increased dysbiotic state in STZ and AMX co-treated mice (Bui et al., 2016; Iino et al., 2007; Kazemian et al., 2020; Newman et al., 2018). Overall, these data show that STZ-induced metabolic shifts can exacerbates the post-AMX bloom of Bacteroides and significantly worsen the loss of key SCFA-producing Firmicutes. This likely impacts the local metabolome and metatranscriptome, and thus AMX susceptibility, given the syntrophic nature of Bacteroides and Firmicutes metabolism (Fischbach and Sonnenburg, 2011).
HG exacerbates ATB dysbiosis and shifts microbial metabolism
As with taxonomic changes, the majority of detected functional AMX responses were highly host dependent. We used the same interaction calculation to profile host-specific changes in CAZyme and SEED subsystem transcript abundances (Figures 4A and 4B). Interestingly, HG animals lack the reduction in glycoside hydrolase (GH) 43 seen in controls, suggesting modified processing of hemicelluloses, pectins, xylans, and arabinose (Figure 4A; Table S4; Mewis et al., 2016). Given the reduced polysaccharide foraging in the STZ baseline, it is likely that the HG microbiota is unable to adapt its CAZyme expression in response to AMX. HG communities had overall fewer GH transcripts at the SEED subsystem level, and a greater loss of GH abundance relative to controls (Figures S4A and S4B; Tables S4 and S7). We saw STZ-specific accumulation of polyphenols and polysaccharides, providing further support for host-dependent modifications in polysaccharide metabolism (Figure 5A; Table S1). HG mice had accumulations of multiple phenylpropanoids, phenylacetic acids, polyphenols, alkaloids, flavonoids, and isoprenoids (Figure 5A; Table S1) and pathway-level enrichment of metabolites related to flavonoid/isoflavonoid synthesis after AMX treatment (Table S3). Because fiber metabolism can confer a protective phenotype to select gut microbes (Cabral et al., 2019), we anticipated that reduced fiber and polyphenol metabolism may directly contribute to the severity of AMX-induced dysbiosis in HG mice.
Figure 4. AMX differentially alters the cecal metatranscriptome.

(A) Differentially abundant CAZyme transcripts in control and STZ mice after AMX, with interaction values. See Table S4 for full results.
(B) Differentially abundant level 3 SEED Subsystem transcripts in control and STZ mice after AMX, with interaction values. See Table S7 for full results.
(C) Linear discriminant analysis of MetaCyc pathways following AMX treatment in STZ mice. See Table S6 for full results.
(D) Linear discriminant analysis of MetaCyc pathways following AMX treatment in control mice. See Table S6 for full results.
For all panels: n = 4 per group; for (A and B): Data represent log2 fold change ± SEM versus vehicle-treated controls. Blank panels are non-significant.
Figure 5. AMX differentially alters the cecal metabolome.

(A) Differentially abundant Q-TOF-MS metabolite features in control and STZ mice after AMX treatment with interaction value. Data represent log2 fold change ± SEM versus vehicle controls. See Table S1 for full results.
(B) KEGG pathway enrichment of differentially abundant Q-TOF-MS metabolites in STZ mice after AMX treatment versus the enrichment score in control andAMX-treated mice. Blank panels represent a lack of statistical significance. See Table S3 for full results.
(C) Taxon Stratified Community Metabolic Potential of control mice after AMX treatment as calculated by MIMOSA. See Table S9 for full results.
(D) Taxon Stratified Community Metabolic Potential of STZ-treated mice after AMX treatment as calculated by MIMOSA. See Table S9 for full results.
For (A and B): n = 6 per group, 2 replicates per sample; for (C and D): n = 4 per group; for (B): Significant = p < 0.05.
Mucus foraging by the microbiota after AMX was also perturbed. In our CAZyme dataset, HG mice had a loss of GHs that target the chitobiose core of mucins (GH115), and did not upregulate GH84, GH129, and GH89 which target N-acetylglucosamine, class-III mucins, and mucus glycoproteins, respectively (Figure 4A, Table S4). Simultaneously, STZ and AMX co-treatment downregulated expression of multiple pathways involved in mucin-derived carbon metabolism, including the Leloir pathway (foraging of mucus galactose residues), 4-deoxy-L-theo-hex-4-enopyranuronate degradation (breakdown of heparin and hyaluron into pyruvate), and D-galacturonate degradation (Figures 4C and 4D; Table S6; Tang et al., 2016). The sialic acid residue N-acetylneuraminic acid (NANA) was enriched in NG but not HG animals after AMX (Table S1). Because NANA is liberated by mucus breakdown (Crost et al., 2016), this suggested reduced muciniphilic activity by STZ and AMX co-treated communities. Ultimately, HG-related modifications in glycan foraging occur both before and after AMX, indicating that STZ-induced HG impacts the composition of the cecal carbon pool.
Further evidence of perturbed carbon foraging in the HG AMX-treated microbiome arose from examination of host and ATB interaction for SEED subsystem transcript abundances (Figure 4B; Table S7). We found an STZ-specific increase in phosphotransferase system (PTS) transcripts (Figure 4B, Table S7) as well as enrichment of PTS metabolites like mannitol 1-phosphate (Figure 5A; Table S1). PTS systems function to rapidly import target saccharides into bacterial cells, and increased PTS activity may be the result of higher environmental sugar concentrations (McCoy et al., 2015). Although AMX reduced cecal glucose concentrations in both hosts, HG mice had significantly higher glucose levels than controls (Figure 1F) and we observed STZ-specific enrichment of sugars like acetylated maltose (Figure 5A; Table S1).
Elevated sugars and PTS likely also increased catabolism. To that end, we observed significant HG-specific increases in glycolysis and gluconeogenesis (Figure 4B; Table S7) and pyruvate fermentation transcripts compared to controls (Figures S4A and S4B; Table S7). The metabolome of HG AMX-treated mice was specifically enriched in catabolism and catabolism-supporting pathways like 2-oxocarboxylic acid metabolism, glycolysis, starch/sucrose utilization, nicotinate/nicotinamide, and propanoate generation (Figure 5B; Table S3). The abundance of vitamin cofactors was also impacted; in our LC-MS/MS dataset, we saw HG-specific enrichment in riboflavin (Figure S4C: Cluster 699, Figure S4D; Table S2) (Steinert et al., 2020). The enrichment of nicotinate/nicotinamide, propanoate generation, and cofactor-related metabolites may impact the abundance of energy carriers (Belenky et al., 2007). Together these data suggest that carbon metabolism is disturbed in the HG AMX-treated microbiome, in part due to higher environmental concentrations of readily-metabolized sugars.
We stratified our metabolomics and transcriptional data with MIMOSA to identify taxonomic drivers of community behavior during AMX treatment (Figures 5C and 5D; Table S9). Regardless of host, B. theta was the major metabolome-contributing taxa, which is congruent with its dominance of the microbiome during AMX perturbation (Figures 3A, 3G, 5C, and 5D; Table S9). Of the metabolites correlated with differential community activity in HG mice, B. theta was responsible for enrichment of B-vitamins like riboflavin (via synthesis) and pantothenate (via synthesis and degradation). Additionally, sucrose and glycerol-3-phosphate levels could be partially explained by synthetic reactions from B. theta (Figure 5D; Table S9).
The fact that B. theta significantly shapes the metabolic function of the microbiome after AMX in both hosts prompted us to compare the post-ATB transcriptome and metabolome (Figure 6). We found that HG AMX-treated metabolomes were significantly enriched for metabolites involved in multiple aa generation pathways, nucleotide biosynthesis, and linoleic acids (Figure 6A; Table S3). Additionally, we saw significant enrichment of metabolites related to carbon processing (fructose/mannose metabolism, ABC transporters, PTS) and metabolic homeostasis (pyruvate metabolism, ubiquinone/terpenoid-quinone biosynthesis, and glutathione metabolism) (Figure 6A). At the MetaCyc pathway level, we observed HG-related increases in pyruvate fermentation and nucleotide biosynthesis (Figure 6B; Table S6). Unsurprisingly, B. theta was the major contributing taxon to this variation (Figure 6C; Table S9). Thus, we performed single-species transcriptomics on B. theta during AMX challenge in HG and NG mice. Interestingly, B. theta downregulated the expression of multiple polysaccharide utilization loci (BT4293-BT4299, BT4296–4298, BT3025, BT1761, and BT1762) and sugar import systems for fructose (BT1759–1763, and BT1759), ribose (BT2804), and fucose (BT3665) (Figure 6D; Table S5) only in HG mice (Figure 6D; Table S5; Lynch and Sonnenburg, 2012; Mardo et al., 2017; Mimee et al., 2015; Townsend et al., 2020). The combination of STZ and AMX treatment also coincided with significant upregulation of the NADH dehydrogenase complex (BT4058–4067) which is a primary redox balance locus (Fischbach and Sonnenburg, 2011). We also saw elevation of another NADH ubiquinone reductase operon (BT0616) (Goodman et al., 2009), and ATPase (BT1746) (Figure 6D; Table S5). Our community-level metabolomics indicated HG-specific loss of phosphoenolpyruvate (Table S1), which may be related to the differential regulation of respiration-related complexes in B. theta. Together these data show that HG, and the resultant changes in environmental metabolites, are sufficient to dramatically modify the transcriptional and metabolic behavior of B. theta during AMX treatment. This change ultimately impacts the AMX susceptibility of other taxa within the community and greatly perturbs the functional response of the larger community to ATB pressure.
Figure 6. STZ treatment modifies transcriptomic and metabolomic responses of the microbiome to AMX.

(A) KEGG pathway enrichment of differentially abundant Q-TOF-MS features in STZ AMX (+) mice compared to control AMX (+) mice. See Table S3 for full results.
(B) Linear discriminant analysis score of MetaCyc pathways in STZ AMX (+) mice compared to control AMX (+) mice. See Table S6 for full results.
(C) Taxon Stratified Community Metabolic Potential of STZ AMX (+) mice compared to control AMX (+) mice as calculated by MIMOSA. See Table S9 for full results.
(D) Differentially abundant B. theta transcripts after AMX treatment in STZ mice. Data represent log2 fold change ± SEM of STZ AMX (+) mice versus vehicle controls. See Table S5 for full results.
For (A): n = 6 per group with 2 technical replicates per sample; Significant = p < 0.05; for (B–D): n = 4 per group.
STZ and AMX co-treatment increases susceptibility to Salmonella enterica infection
We noticed that fatty acid metabolism was differentially impacted by STZ and AMX treatment. Specifically, co-treated communities were enriched for multiple N-acylethanolamines (Figure 5A; Table S1) and the precursor phosphatidylethanolamine (Table S1). Higher ethanolamine concentrations may suggest more fatty acid epoxidation and dysbiosis-associated inflammation within the GI (Ormsby et al., 2019; Thiennimitr et al., 2011). Ethanolamines are naturally generated by phosphatidylethanolamine breakdown during cell turnover, however, most of the microbiota is unable to ferment ethanolamines, and these compounds can increase the colonization and virulence of multiple enteric pathogens (Anderson et al., 2015, 2018; Nawrocki et al., 2018; Rowley et al., 2018; Garsin, 2010). Specifically, some Enterobacteriaceae are enriched for the genetic machinery required to use ethanolamines, and can funnel their breakdown products into both nitrogen metabolism and respiration (Anderson et al., 2015; Garsin, 2010; Srikumar and Fuchs, 2011; Thiennimitr et al., 2011). In Salmonella, exogenous ethanolamine signals a cascade of metabolic and virulence genes that promote intestinal colonization (Anderson and Kendall, 2016). ATB-induced dysbiosis is also associated with increased S. enterica colonization, likely through the induction of a respiratory-favorable environment and disruption of the endogenous microbiota (Yoon and Yoon, 2018; Zeng et al., 2017). The transcriptional changes we observed in B. theta are highly indicative of a respiratory-favorable environment in the HG AMX-treated GI (Figure 6D). Thus, we asked if the microbiome modifications in HG AMX-treated mice would increase infection susceptibility to S. enterica (Figure 7A).
Figure 7. STZ and AMX increase susceptibility to Salmonella enterica infection.

(A) Experimental design of pathogen challenge study. Figure was created with BioRender.com (BioRender, Toronto, Canada).
(B) S. enterica colony forming units (CFU) per gram of feces in control AMX(+/−), and STZ AMX(+/−) mice after infection with 1×103 cells. Data represent mean CFU ± SEM.
(C) Kaplan Meier survival curve of NG mice.
(D) Kaplan Meier survival curve of STZ mice.
(E) Bray-Curtis Dissimilarity between 16S rRNA amplicons from experimental groups.
(F) Taxonomic composition of the fecal microbiome based of genus-level 16S rRNA identity between 1 and 4 days post-infection with 1×103 cells. Data represented mean ± SEM.
(G) Contribution of Salmonella assigned reads in STZ AMX(+) and control AMX(+) mice 4 days post-infection. Data represent mean ± SEM.
*p < 0.05; **p < 0.01; ***p < 0.001; for (B): n = 8 to 10 per group, Mann-Whitney U test of STZ AMX(+) versus Control AMX(+); for (C and D): n = 4 to 5 per group; for (E–G): n = 8 to 10 per group; for (E): permutational ANOVA.
We found that co-treatment with STZ and AMX lowered the infective dose required to establish S. enterica colonization and significantly increased intestinal, hepatic, and splenic pathogen burden (Figures 7B and S5A). Host HG increased lethality by day 7 of infection (Figures 7C and 7D). In the control group, all vehicle-treated mice survived, and AMX-treated mice in the high dosage groups (1 × 104 and 1 × 105) experienced 75 and 50 percent survival respectively (Figure 7C). In the STZ group, vehicle-treated mice in the highest dosage groups (1 × 105 and 1 × 106) had 75 and 40 percent survival, while the AMX-treated mice experienced between 25 to 80 percent survival with lethality events starting as early as 24 hours (Figure 7D).
To check for any significant differences in GI physiology or immunocompetence, we assessed GI edema and inflammation between HG and control mice (Figure S5B). We saw no differences in GI physiology, but noticed slight differences in some serum cytokines (Figures S5C–S5I). For IL-1α, IL-6, and IL-12p70, variation was between vehicle-treated mice and may represent progression of the STZ model rather than responses to infection. For GM-CSF, IFN-β, IL-10, and IL-17A, plasma concentrations were only elevated in HG AMX-treated mice after infection and thus may be the result of differential susceptibility (Figure S5C–S5I).
Infection with S. enterica represents another form of microbiome dysbiosis in conjunction with ATBs. Thus, we profiled the fecal microbiome during the first 4 days of infection to assess if HG worsened infection-related dysbiosis (Figures 7E, 7F, 7G, S5J, S5K, and S5L). The initial infection significantly impacted microbiome β-diversity (Figure S5). However, diversity remained divergent in accordance with preinfection experimental treatment (Figure 7E). This indicates that regardless of infective dose, the microbiome changes induced by STZ or AMX remain the drivers of β-diversity. Interestingly, we noticed that only HG mice experienced significant reduction in α-diversity (Figure S5K). During the 4-day period following infection, Salmonella expansion was only detected in AMX-treated animals (Figure 7F). We then quantified the difference in Salmonella reads between control and HG mice after AMX, and found that HG mice had notably higher levels of Salmonella (Figure 7G). Together, these data suggest that the combination of STZ and AMX severely reduces the probability of survival after S. enterica challenge and increases pathogen burden and microbiome dysbiosis relative to NG controls. It is possible that the enrichment of favorable metabolites or change in respiratory potential in HG AMX-treated communities promotes the expansion and virulence of S. enterica, although more work is required to confirm this hypothesis.
DISCUSSION
Recent estimates of HG global prevalence suggest that metabolic disruption occurs in approximately 10 percent of all people, with incidence increasing annually (Saeedi et al., 2019). Thus, understanding how host metabolism impacts ATB-induced dysbiosis is key to the development of microbiomeprotective therapeutic strategies. To address this knowledge gap, we used an integrated multi-omic strategy to examine how HG modifies the microbiome’s response to AMX. Specifically, we combined WMGS, metatranscriptomics, and untargeted metabolomics to examine differences in microbiome composition and function, both pre- and post-ATB treatment, and to characterize the severity of dysbiosis-related complications like enteric infection.
A key goal of this study was to profile the impact of altered host metabolism and the microbiota-accessible metabolite pool on microbiome function during ATB treatment. Since dietary modulation has inherent limitations involved in restructuring microbiome composition, we opted for a rapid-chemotherapeutic method to perturb the GI metabolite pool. We chose STZ, in part, due to its widespread use and quick onset of changes. However, it is critical to address limitations of the model, as it does not perfectly replicate the pathology of clinical HG. STZ has been used to replicate both type-1 and type-2 diabetic phenotypes in animals since the mid-1960s (Deeds et al., 2011; Eleazu et al., 2013). Models of STZ administration vary widely in their dosage concentration, injection frequency, and inclusion or exclusion of high-fat feed typically due to differences in research goals (Deeds et al., 2011; Furman, 2021). Because STZ does not perfectly mimic type-2 diabetes, the inclusion of a high-fat feeding period before injection was recently proposed as a method to induce hyperinsulinemia and insulin resistance in STZ-treated animals (Furman, 2021; Chao et al., 2018). Regardless, STZ consistently induces the characteristic symptoms of HG, insulin deficiency, polydipsia, and polyurea (Furman, 2021; Kolb, 1987). Although there is conservation of immunological responses to β cell ablation (Eleazu et al., 2013), one must consider that microbiome-related phenotypes derived from STZ-induced HG are likely specific to the submodel and may not readily translate across studies. For example, the only taxonomic changes we found after STZ treatment were the expansion of A. muciniphila and collapse of Blautia sp YL58. This contrasts with the many taxonomic shifts seen in existing work examining STZ-treated rats, but this may be due to inherent differences between mice and rats, use of multiple STZ doses, sample collection, sequencing depth, use of a diet in combination with STZ, or the time frame of weeks rather than days (Liu et al., 2019a; Ma et al., 2020; Patterson et al., 2015; Yin et al., 2020).
Interestingly, serum metabolomics in multiple low-dose STZ treated mice found enrichment of AAAs, bile acids, dipeptides, fatty acids, nucleotides, sphingolipids, and vitamins (Ugarte et al., 2012). These results are congruent with our pre-AMX metabolomics data and may represent true HG-related changes. Metabolomic studies focused on prediabetic patients have found shifts in aa catabolism as a potential biomarker of progression to type-1 diabetes, and spikes in both aromatic and branched-chain aa as predictive of type-2 diabetes (Wang et al., 2011; Neis et al., 2015), supporting our observed aa changes as a true HG phenotype. A potential explanation for this enrichment is that fiber use within the GI impacts the production of several aa-based metabolites by members of the Firmicutes phylum (Neis et al., 2015; Tanes et al., 2021), suggesting an intrinsic link between metabolic dysregulation and shifts in gut aa metabolism.
We found that STZ initiated a cascade of changes related to fiber and SCFA generation. Specifically, the loss of SCFA-producing Firmicutes may perturb syntropic reactions involved in fiber-fermentation (Bui et al., 2016; Oliphant and Allen-Vercoe, 2019; Rey et al., 2010; Vacca et al., 2020). Because the microbiome is responsible for this fermentation (Holscher, 2017), we anticipate that disturbances in polysaccharide processing are microbially-driven rather than host-derived. In the case of polyphenol substrates and metabolites, bacteria can coopt and liberate sugars from these compounds for use in their own metabolism (Fraser and Chapple, 2011; Braune and Blaut, 2016; Moore et al., 2002; Wang et al., 2019b; Lundgren and Thaiss, 2020; Vollmer et al., 2018) For example, some taxa can directly utilize flavones as a carbon source, fueling them into their respiratory cycle (Burlingame and Chapman, 1983). Thus, shifts in the abundance of dietary-derived polyphenols could modify microbial metabolism in the GI.
Reduced fiber use by the microbiota may be partially explained by a side-effect of STZ treatment. Rodents receiving a multiple low-dose regime (i.e., 50 mg/kg/day for 5 days) of STZ exhibit an initial reduction in food consumption (Motyl and McCabe, 2009). However animals exhibit hyperphagia one week post-injection (Motyl and McCabe, 2009; Zhang et al., 2008). It is possible that our experimental time point for AMX administration and sample collection (3 and 4 days-post injection, respectively) is associated with reduced food intake, which would lower the availability of fiber. Regardless, reduced fiber and polyphenol intake has been recognized as a form of dysbiosis, increasing susceptibility to bactericidal ATBs via modification of microbial metabolism (Cabral et al., 2020; 2019; Makki et al., 2018; Ng et al., 2019), and we propose that a similar disruption of the microbiome occurs here.
We observed that the HG microbiome was enriched for transcripts and metabolites involved in pyruvate metabolism and glycolysis. It is likely that the overall enrichment of aa catabolism directly contributes to increased community respiration because many of the altered amino acids are glucogenic (Berg et al., 2002). A key consideration of any ecological network is its taxonomic composition (Coyte and Rakoff-Nahoum, 2019). Polymicrobial interactions are a significant component of the microbiome’s ecology and changes to taxonomic structure or function will directly impact the overall activity (Coyte and Rakoff-Nahoum, 2019; Layeghifard et al., 2017; Boon et al., 2014). Because Firmicutes have been characterized to perform a bulk of aa, nitrogen, and sulfur metabolism reactions within the GI, it is possible that these taxa are driving the increased metabolic rate. This may prime the microbiome as a whole for increased AMX susceptibility, but more work is needed to confirm this hypothesis (Bernal et al., 2007; Böttcher et al., 2014; Gao et al., 2018; Meadows and Wargo, 2015). Overall, these data make a strong argument for the degree of control that changes in the baseline function of the microbiome has on compositional restructuring after ATB perturbation.
When comparing HG and NG communities during and after AMX treatment, the increased dysbiosis in HG mice was expected given both the increased basal metabolic rate and the elevation of simple sugars and PTS activity during AMX. Increased sugar availability and decreased polysaccharide utilization have been demonstrated to potentiate ATB toxicity within the GI (Cabral et al., 2019; 2020). The most striking finding to us was how divergent the transcriptional behavior of B. theta was between hosts. We previously identified that polysaccharide fermentation by B. theta functions as an amoxicillin tolerance response in NG animals (Cabral et al., 2019). However, this study complicates that understanding, as STZ-specific reductions in B. theta’s polysaccharide and mucus foraging suggests that these may not be universal amoxicillin tolerance responses. There may be non-mucosal or non-polysaccharide metabolite species that induce a protective phenotype to members of the Bacteroides genus. Alternatively, members of this genus possess b-lactamases, and differences in the expression of these resistance genes may be involved in the observed enrichment of Bacteroides in HG AMX-treated mice (Edwards, 1997). Regardless, reduction in fiber fermentation by Bacteroides disrupts the balance of nutrients available for syntrophic metabolism with Firmicutes and Actinobacteria (Fischbach and Sonnenburg, 2011). These changes may induce a proinflammatory state and contribute to the increased dysbiosis experienced by HG mice during ATB exposure. Given the total ecological complexity of the gut microbiome, a more robust understanding of cross-feeding networks will be integral to the full characterization of a given perturbation’s impact on the microbiome.
Lastly, we examined if the increased severity of AMX toxicity in HG mice would increase susceptibility to enteric infection. Overall, HG AMX-treated animals had both increased susceptibility to S. enterica and reduced overall survival after one-week of infection (Figure 7). Recent work by Thaiss et al. has shown that decreased barrier function caused by STZ increases S. enterica susceptibility (Thaiss et al., 2018). However, this study used a multiple-dose STZ model and did not infect mice until a few weeks after STZ treatment, thus these results may not translate to our study. For example, we found that, at low infection doses, STZ treatment had no impact on susceptibility in the absence of ATBs. Thus, it is possible that the HG ATB-treated microbiome is structurally, functionally, and metabolically perturbed in a way that promotes the pathogen colonization and expansion. For example, we found enrichment of multiple ethanolamines, which are a carbon source that cannot be used by the microbiota but can be utilized by Salmonella (Anderson et al., 2015; Srikumar and Fuchs, 2011; Thiennimitr et al., 2011). S. enterica has flexible metabolism compared to the bulk of the microbiota (Taylor and Winter, 2020), and can use inaccessible carbon sources like ethanolamines to promote colonization and niche adaptation in mammals (Anderson et al., 2015). Other metabolites that may have impacted S. enterica infection severity include acetyl-maltose, as Salmonella are equipped with tightly controlled maltose import systems and readily fuel this carbon source into their respiratory cycle (Erhardt and Dersch, 2015; Jain et al., 2020; Miller et al., 2013). Another metabolite of interest was pantetheine, which Salmonella can shunt into its CoA synthesis, potentially providing a fitness advantage through competitor exclusion (Ernst and Downs, 2015) (Table S1). An alternate explanation for the increased expansion of Salmonella is an overall increase in ATB-induced intestinal oxygenation. Salmonella are facultative anaerobes and can readily switch to aerobic respiration when needed (Rhen, 2019). Additionally, Salmonella can use inflammation-related metabolites like tetrathionate as terminal electron acceptors, and can coopt the oxygenated and inflamed gut for growth (Winter et al., 2010). Although more work is required to parse what components of the HG microbiome provide a competitive advantage to Salmonella after AMX treatment, our data provides strong preliminary evidence that STZ-induced HG can directly impact the acute consequences of ATB dysbiosis. Ultimately, our study shows that host-related physiology and metabolic state must be a key consideration of any current and future therapeutic strategy aimed at mitigating ATB-induced microbiome damage.
Limitations of the study
While our multi-omic approach robustly characterizes the cecal microbiome during dysglycemia and ATB perturbation, there are limitations in the study design and methodology that complicate the interpretation of the results. First, our study exclusively uses male mice. Female mice are partially resistant to STZ-induced HG and require significantly higher doses and (or) repetitive dosing regimens compared to males to induce a metabolic phenotype (Deeds et al., 2011; Goyal et al., 2016). An additional consideration is that STZ’s mechanism of action involves organ cytotoxicity (Deeds et al., 2011). Although STZ is rapidly eliminated from the host, it is nearly impossible to guarantee that off-target effects of pancreatic toxicity are not contributing to some microbiome phenotypes.
A key considerations of our metagenomic and metatranscriptomic-reliant analyses is the dependence on existing databases that possess annotation-based limitations and the need for imperfect alignment algorithms (HMP (Human Microbiome Project Consortium), 2012). While WMGS provides increased resolution over 16S rRNA sequencing, the taxonomic classification of sequencing reads is still subject to currently available reference genomes, which are biased toward some taxa over others (HMP (Human Microbiome Project Consortium), 2012; McLaren et al., 2019). Further, WMGS data is complicated by the fact that taxonomic levels are reported as relative abundances. Even metabolomic-focused pipelines like MIMOSA are limited by their reference databases. Specifically, full reaction annotations within the KEGG database are required for this pipeline (Noecker et al., 2016).
For untargeted metabolomics, ion annotation is still considered the primary bottleneck of analysis (Gertsman and Barshop, 2018; Schrimpe-Rutledge et al., 2016). The diversity in chemical modification, polarity, solubility, and ionization of chemical structures from complex biological samples often requires multiple analytical modes (i.e., positive versus negative ion mode) to be run in order to characterize all structures, and that can subsequently complicate ion identification (Gertsman and Barshop, 2018; Lei et al., 2011; Luan et al., 2019). While metabolomics offers a powerful examination of the small molecule repertoire of the cecum, it does not distinguish between bacterially-derived, fungal-derived and host-derived metabolites (Gertsman and Barshop, 2018). While pairing these data with metatranscriptomics and using networking models like MIMOSA helps improve inference of metabolite origin it does not eliminate the possibility of host-derived metabolites being mistaken for bacterially derived compounds and vice versa. Additionally, our metabolomics preparation is unable to separate intracellular- and extracellular-derived metabolites, potentially complicating biological interpretation. Ultimately, further work will be required to correlate STZ and AMX-induced metabolomic changes with individual taxa, and greater annotation of metabolic syntrophy in the gut will aid in the biological interpretation of subsequent metabolomic analyses.
STAR⋆METHODS
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Peter Belenky (peter_belenky@brown.edu).
Materials availability
This study did not generate new, unique reagents.
Data and code availability
Illumina sequencing read data have been deposited at the NCBI Short Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. LC-MS/MS and GNPS data have been deposited to MassIVE: https://massive.ucsd.edu and GNPS: https://gnps.ucsd.edu, respectively, and are publicly available as of the date of publication. DOIs are listed in the key sources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| Reagent or resouce | Source | Identifier |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Salmonella enterica Typhimurium SL1344 | Vanessa Sperandio, PhD (Univerisity of Texas, Southwestern) | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acetone, LCMS-grade | Fisher Scientific | K50738120840 |
| Acetonitrile, LCMS-grade | Fisher Scientific | PI86188 |
| Agar | Fisher Scientific | BP1423-2 |
| Agarose | Fisher Scientific | 14-223-040 |
| Agencourt AMPure XP Beads | Beckman Coulter | A63880 |
| Ammonium Fluoride | Sigma Aldrich | 338869 |
| Amoxicillin | Sigma Aldrich | A8523-5G |
| Deoxynucleotide (dNTP) Solution Mix | New England BioLabs | N0447S |
| Ethanol, 200 proof, molecular biology grade | Fisher Scientific | 07-678-003 |
| Formic acid | Fisher Scientific | RMB11202101 |
| Hematoxylin & Eosin Stain Solution | AbCam | Ab245880 |
| Heparin | Sigma Aldrich | H3393-25KU |
| Hydrochloric Acid | Fisher Scientific | A144-500 |
| Industrial Grade Nitrogen, Liquid | AirGas | NI240LT22 |
| Isopropanol, LCMS-grade | Fisher Scientific | A461-1 |
| Luria Bertani (LB) Broth | Fisher Scientific | BP1426-2 |
| Methacarn | Fisher Scientific | NC0547175 |
| Methanol, LCMS-grade | Fisher Scientific | A456-500 |
| Paraffin Wax | Fisher Scientific | 22-90-700 |
| Phosphate Buffered Saline, 10X | Fisher Scientific | BP399 |
| Sodium Citrate Dihydrate | Fisher Scientific | 5279-500 |
| Streptozotocin | Alfa Aesar | J61601-03 |
| Sucrose | Fisher Scientific | BP220-212 |
| TE Buffer, 1X Solution, pH 8.0, low EDTA | Fisher Scientific | AAJ75793AE |
| Tween20 | Fisher Scientific | BP337-100 |
| Water, LCMS-grade | Fisher Scientific | PI51140 |
| Water, Molecular Biology Grade | Fisher Scientific | BP2891-1 |
|
| ||
| Critical commercial assays | ||
|
| ||
| 10KD Spin Column | AbCam | Ab93349 |
| API-TOF Reference Mass Solution Kit | Agilent | AGG1969-85001 |
| DNA/RNA Shield Collection and Lysis Tube | Zymo Research | R1102 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R&D Systems | DY008 |
| Glucose Assay Kit - Reducing Agent Compatible | AbCam | Ab102517 |
| HiSeq X Ten Reagent Kit v2.5 | Illumina | FC-501-2501 |
| Legendplex Mouse Inflammation Panel 13-plex with Filter Plate | BioLegend | 740150 |
| MICROBExpress Bacterial mRNA Enrichment Kit | Thermo Fisher Scientific | AM1905 |
| MiSeq Reagent Kit v2 (500 cycles) | Illumina | MS-102-2003 |
| Mouse Lipocalin-2/NGAL DuoSet ELISA Kit | R&D Systems | DY1857-05 |
| NEBNEXT® rRNA Depletion Kit (Human/Mouse/Rat) (Includes Purification Beads) | New England BioLabs | E6350 |
| NEBNEXT® Ultra II Directional RNA Seq Library Prep Kit for Illumina (Includes Purification Beads) | New England BioLabs | E7765 |
| NEBNEXT® Ultra II FS DNA Library Prep Kit for Illumina (Includes Purification Beads) | New England BioLabs | E6177 |
| NucleoSpin® Gel and PCR Clean-up Kit | Machery-Nagel GmbH & Co | 740609 |
| Phusion High-Fidelity PCR Kit | Thermo Fisher Scientific | F553L |
| Qubit dsDNA Broad Range Assay Kit | Thermo Fisher Scientific | Q32850 |
| Qubit dsDNA High Sensitivity Assay Kit | Thermo Fisher Scientific | Q32851 |
| Qubit RNA High Sensitivity Assay Kit | Thermo Fisher Scientific | Q32852 |
| Sin-X UF 500 10k MWCO PES Spin Filter | Corning | 431478 |
| Spin-X Centrifuge Tube Flter, 0.22 μM | Costar | 8160 |
| ZymoBiomics Collection Tubes | Zymo Research | S6012-50 |
| ZymoBiomics DNA Miniprep Kit | Zymo Research | D4300 |
| ZymoBiomics DNA/RNA Miniprep Kit | Zymo Research | R2002 |
|
| ||
| Deposited data | ||
|
| ||
| 16S rRNA Reads | NCBI BioProject ID | PRJNA720755 |
| Metagenomic / Metatranscriptomic Reads | NCBI BioProject ID | PRJNA72012 |
| Q-TOF-MS Raw Data | This Study | Table S1 |
| GNPS Molecular Networking Data | GNPS.UCSD.edu | https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=e4efce0c33fb4ada96e373d53460f2d5 |
| LC-MS/MS Files | Massive.UCSD.edu | MSV000087093 |
| LC-MS/MS Data and Analysis Scripts | This Study | https://github.com/guziordo/Belenky-Brown-Diabetes-Antibiotics |
|
| ||
| Experimental models: Mouse Strains | ||
|
| ||
| C57BL/6 | Jackson Laboratories | Cat.# 000664 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Earth Microbiome Project: 806R | Caporaso et al., 2012 | https://earthmicrobiome.ucsd.edu/protocols-and-standards/primer-ordering-and-resuspension |
| Earth Microbiome Project: 515F with Barcode | Caporaso et al., 2012 | https://earthmicrobiome.ucsd.edu/protocols-and-standards/primer-ordering-and-resuspension |
|
| ||
| Software and algorithms | ||
|
| ||
| BBMap (version 37.96) | Bushnell, 2014 | https://sourceforge.net/projects/bbmap |
| Bowtie2 (version 2.2.0) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Bracken (version 2.0.0) | Lu et al., 2017 | http://ccb.jhu.edu/software/bracken/index.shtml?t=manual |
| BWA-Mem (version 0.7.15) | Li and Durbin, 2010 | http://bio-bwa.sourceforge.net/bwa.shtml |
| DIAMOND (version 0.9.11) | Buchfink et al., 2014 | https://github.com/bbuchfink/diamond |
| Global Natural Products Social Molecular Networking (GNPS) | Wang et al., 2016 | https://gnps.ucsd.edu |
| HUMAnN2 (version 0.11.1) | Franzosa et al., 2018 | https://bitbucket.org/biobakery/humann2/wiki/home |
| Kneaddata (version 0.6.1) | McIver et al., 2018 | https://bitbucket.org/biobakery/kneaddata/wiki/home |
| Kraken (version 2.0.7-beta) | Wood et al., 2019 | https://ccb.jhu.edu/software/kraken2 |
| LDA Effect Size (LEfSe, version) | Segata et al., 2011 | https://huttenhower.sph.harvard.edu/galaxy |
| MetaPhlan2 (version) | Segata et al., 2012 | https://bitbucket.org/biobakery/metaphlan2 |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| MATLAB Toolbox: Bioinformatics | MathWorks | https://www.mathworks.com/products/bioinfo.html |
| MZmine (version 2.52) | Pluskal et al., 2010 | http://mzmine.github.io/ |
| Paired-End Read Merger (PEAR; version 0.9.12) | Zhang et al., 2014 | https://cme.h-its.org/exelixis/web/software/pear |
| Prism (version 9.0.2) | GraphPad | https://www.graphpad.com/scientific-software/prism |
| R (version 4.0.3) | The R Project for Statistical Computing | https://www.r-project.org |
| R package: DADA2 (version 1.8.0) | Callahan et al., 2016 | https://bioconductor.org/packages/release/bioc/html/dada2.html |
| R package: DESeq2 (version 1.26.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R package: phyloseq (version 1.28.0) | McMurdie and Holmes, 2013 | https://bioconductor.org/packages/release/bioc/html/phyloseq.html |
| R package: randomForest (version 4.6-16) | Breiman, 2001 | https://cran.r-project.org/web/packages/randomForest/index.html |
| R package: vegan (version 2.5-7) | https://cran.r-project.org/web/packages/vegan/index.html | |
| R package: PAPi | Aggio et al., 2010 | https://www.bioconductor.org/packages//2.12/bioc/html/PAPi.html |
| R package: Mimosa (version 2.0) | Noecker et al., 2016 | https://borenstein-lab.github.io/MIMOSA2shiny |
| Rstudio (version) | Rstudio | https://www.rstudio.com/ |
| SAMSA2 (version 1.0) | Westreich et al., 2018 | https://github.com/transcript/samsa2 |
| Subread (featureCounts) (version 1.6.2) | Liao et al., 2014 | https://bioinf.wehi.edu/au/featureCounts |
| Trimmomatic (version 0.36) | Bolger et al., 2014 | https://www.usadellab.org/cms?page=trimmomatic |
|
| ||
| Other | ||
|
| ||
| 6550 iFunnel Q-TOF LC/MS | Agilent | G6550BA |
| Accuspin Micro17 Microcentrifuge | Fisher Scientific | 13-100-675 |
| Acquity UPLC columns, BEH C18, 1.7 μM (2.1×100mm) | Waters | 186002352 |
| Attune NxT Flow Cytometer | Invitrogen | N/A |
| ChemiDoc MP Imaging System | BioRad | 12003154 |
| Class II Type A2 Biological Safety Cabinet | Labguard | NU-540 |
| CleanPrep PCR Workstation | MyStaire | MY-DB24 |
| ContourNext® EZ Glucose Meter | Contour | N/A |
| ContourNext® EZ Glucose Meter Strips | Contour | N/A |
| Electrospray Ionization Source | Agilent | G1948B |
| GyroMax 737 Incubator | Amerex | N/A |
| Insulin Syringe, 0.5mL, 28G | BD | 329461 |
| Insulin Syringe, 1mL, 26G | BD | 329652 |
| Laboratory Rodent Diet 5001 | LabDiet | 0001319 |
| Lo-Bind 96-well plate, skirted | Eppendorf | 30129512 |
| Lo-Bind Microcentrifuge Tubes, nuclease-free | Eppendorf | 22431021 |
| Polypropylene Feeding Tubes, 20 ga × 30mm | Instech | FTP-20-38 |
| QE Basic, Exactive Hybrid Quadrupole-Orbitrap MS | Thermo Fisher Scientific | IQLAAEGAAPFALGMBDK |
| Qubit 3.0 Fluorometer | Thermo Fisher Scientific | Q33216 |
| SpectraMax M3 Multi-Mode Microplate Reader | Molecular Devices | 89429-536 |
| T100 Thermal Cycler | BioRad | 1861096 |
| Vanquish Autosampler | Thermo Fisher Scientific | 8308123 |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animal housing
Experimental procedures involving mice were conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Brown University. Five-week-old male C57BL/6J mice were purchased from the Jackson Laboratories (Bar Harbor, ME, USA) and given a two-week habituation period immediately following their arrival at Brown University. All animals were cohoused together in specific-pathogen-free (SPF), temperature controlled (21 ± 1.1°C), and 12-hour light/dark cycling conditions within Brown University’s animal care facility, while being fed a standard chow (Laboratory Rodent Diet 5001, LabDiet, St. Louis, MO, USA). After habituation, mice were randomized into new cages to reduce potential cage effects.
Bacterial strains
S. enterica Typhimurium SL1344 (GFP+, AmpR) was generously donated by Dr. Venessa Sperandio (University of Texas, Southwestern). Cells were grown at 37°C under shaking aerobic conditions in Luria-Bertani (LB) broth containing ampicillin (100 μg/mL). Colony forming units (CFU) were quantified on LB agar plates containing ampicillin (100 μg/mL). Because S. enterica Typhimurium SL1344 constitutively expresses green-fluorescent protein, CFU counts were confirmed by UV-imaging using the ChemiDoc Imaging System (Bio-Rad, Hercules, CA, USA).
METHOD DETAILS
Animal experiments
All animal work was conducted in accordance with protocols approved by the Institutional Animal Care and Use Committee (IACUC) of Brown University. To induce HG, 7-week-old male C57BL/6J mice were fasted for 4–6 hours, then given an intraperitoneal injection of either Na-Citrate buffered streptozotocin (STZ) (150 mg/kg, pH 4.5) or a Na-Citrate sham (pH 4.5). All mice were given overnight supplementation of 10% sucrose water to avoid post-procedural hypoglycemia. Sucrose water was then replaced with standard filter-sterilized water the following morning. Two days post-injection, fasting blood glucose was assessed in all mice using the CONTOUR®NEXT blood glucose monitoring system (Bayer AG, Whippany, NJ, USA). Mice with HG (fasting blood glucose ≥ 250 mg/dL) were selected for subsequent ATB treatment along with NG controls. 24-hours after glycemic assessment, all mice were randomized again to reduced potential cage effects and given either amoxicillin (25 mg/kg/day) or a pH-adjusted vehicle via filter-sterilized drinking water ad libitum for 24 hours (Cabral et al., 2019). Mice were subsequently sacrificed and dissected to collect blood, tissues, and cecal contents. Cecal contents were weighed then divided to be processed according to their downstream application (nucleic acid extraction, Q-TOF-MS, or LC-MS/MS). Exact processing methods are described in each application section below.
The 16S ribosomal RNA sequencing, whole metagenome sequencing, metatranscriptomic sequencing, metabolomics, and infection studies are the result of independent biological replicates conducted several months apart from one another. The 16S rRNA sequencing results were derived from two independent animal experiments performed in 2017 and 2018. Whole metagenomic sequencing results were derived from two independent animal experiments performed in 2018 and 2019. Metatranscriptomic results were paired from respective metagenomic samples. Metabolomics data were acquired from a separate animal experiment performed in 2020. Finally, infection data were acquired from two independent animal experiments performed in 2020 and 2021.
Multi-omic analysis: Pipelines/purpose/scope
Our multi-omic approach to microbiome analysis features the combinatory usage of the Kraken2 and Bracken annotation pipelines for whole metagenomic sequencing (Lu et al., 2017; Wood et al., 2019), and the HMP Unified Metabolic Analysis Network (HUMAnN2) (Franzosa et al., 2018) and Simple Annotation of Metatranscriptomes by Sequencing Analysis (SAMSA2) pipelines for metatranscriptomics (Westreich et al., 2018). Combined utilization of these pipelines facilitates examination of species-level taxonomic shifts (Kraken2/Bracken), community-level changes in transcript abundances (SAMSA2) and community-level gene expression that is normalized to the abundance of each taxon (HUMAnN2). We also used the pipeline developed by Deng et al. (Deng et al., 2018) to examine species-level transcriptional responses to STZ and amoxicillin challenge for high-abundance and transcriptionally active members of the microbiota.
Sequencing pipelines were used in conjunction with both quadrupole flow injection electrospray time-of-flight mass spectrometry (Q-TOF-MS) (Fuhrer et al., 2011) and liquid chromatography tandem mass spectrometry (LC-MS/MS) paired with spectral annotation and networking analysis via the Global Natural Products Social Metabolic Network (GNPS; http://gnps.ucsd.edu) (Wang et al., 2016). While recent advances in mass spectrometry methods have vastly increased the range and accuracy of metabolite detection, no single analytical method is currently capable of capturing the entirety of small molecules in a complex biological sample (Luan et al., 2019). Thus, we opted to increase our metabolite coverage through the combinatory use of a tandem (LC-MS/MS) and a high-resolution (Q-TOF-MS) method (Chen et al., 2019). The Q-TOF-MS data is presented at the metabolite level where unknown features are ignored. For pathway-level comparisons, available Kyoto Encyclopedia of Genes and Genomes compound identifiers were used to perform Pathway Activity Profiling (Aggio et al., 2010) of known features (Table S3). A deeper metabolome analysis including unknown molecules or related metabolites to known compounds is presented with the and data originating from our LC-MS/MS dataset using GNPS cluster identification.
Finally, integration of transcriptomic (HUMAnN2) and metabolomic (Q-TOF-MS) data was performed using the R implementation of Model-based Integration of Metabolite Observations and Species Abundances (Noecker et al., 2016). This software calculates the potential metabolic capacity of a microbiome by examining which enzymatic reactions are present in a community (i.e., the sum of all synthetic and degradation machinery present). This output is then compared against observed metabolite variations from KEGG-annotated metabolomics data.
Nucleic acid extraction and purification
For nucleic acid extraction, cecal contents were transferred to ZymoBIOMICS DNA/RNA Miniprep Kit (Zymo Research, Irvine, CA, USA) Collection Tubes containing DNA/RNA Shield. These tubes were then processed via vortex at maximum speed for 5 minutes to homogenize cecal contents, which were subsequently placed on ice until permanent storage at −80°C. Using the parallel extraction protocol as per the manufacturer’s instructions, the ZymoBIOMICS DNA/RNA Miniprep Kit was used to isolate total nucleic acids (DNA and RNA) from cecal slurry. Total DNA/RNA were eluted in nuclease-free water and quantified using the dsDNA-HS and RNA-HS kits on the Qubit 3.0 fluorometer (Thermo Fisher Scientific, Waltham, MA, USA).
16S rRNA amplicon generation and sequencing
The V4 hypervariable region of the 16S ribosomal RNA was amplified from extracted total DNA using the 806R and 515F barcoded primers published under the Earth Microbiome Project (Caporaso et al., 2012; Thompson et al., 2017). Amplicons were generated using Phusion high-fidelity polymerase and the following cycling protocol: 98°C for 30 s initial denaturation, then 25 cycles of 98°C for 10 s (denaturation), 57°C for 30 s (annealing), and 72°C for 30 s (extension). This was followed by a final extension of 72°C for 5 minutes. Amplicon libraries were submitted to the Rhode Island Genomics and Sequencing Center at the University of Rhode Island (Kingston, RI, USA) for pair-end sequencing (2×250 bp) on the Illumina MiSeq platform using the 500-cycle kit with standard protocols. We obtained an average of 11,511 ± 10,632 reads per sample for sequences related to Figures 1 and S1, and an average of 6,167 ± 3,498 reads per sample for sequences related to Figures 7 and S5.
16S rRNA read processing and analysis
Raw reads underwent quality filtering, trimming, de-noising and merging using the R (version 3.5.0) package implementation of DADA2 (version 1.8.0) (Cabral et al., 2020; 2019; Callahan et al., 2016). The resulting ribosomal sequence variants underwent taxonomic assignment by using the assignTaxonomy function in DADA2 with the RDP Classifier algorithm with RDP training set 18 (Wang et al., 2007). Both α (Shannon) and β (Bray-Curtis Dissimilarity) diversity were calculated using the R package phyloseq (version 1.24.2) (McMurdie and Holmes, 2013).
Metagenomic/transcriptomic library preparation
Libraries for metagenomics and metatranscriptomics were prepared as described in our recent work (Cabral et al., 2020). We prepared metagenomic libraries from DNA (100 ng) using the NEBNext® Ultra II FS DNA Library Prep Kit (New England BioLabs, Ipswich, MA, USA) and the > 100 ng input protocol as per the manufacturer’s instructions, which generated a pool of fragments whose average size was between 250 and 500 bp. Meanwhile, we prepared metatranscriptomic libraries from total RNA (≤1 ug) using a combination of the MICROBExpress kit (Invitrogen, Carlsbad, CA, USA), NEBNext® rRNA Depletion Kit for Human/Mouse/Rat (New England BioLabs, Ipswich, MA, USA), and the NEBNext® Ultra II Direction RNA Sequencing Prep Kit as per the manufacturers’ instructions. This generated a pool of fragments with an average size between 200 and 450 bp. Both metagenomic and metatranscriptomic libraries were pair-end sequenced (2×150 bp) on the Illumina HiSeq X Ten platform, yielding an average of 1,464,061 ± 728,330 reads per metagenomic sample and 35,884,874 ± 27,059,402 reads per metatranscriptomic sample.
Metagenomic/transcriptomic read processing
Raw metagenomic and metatranscriptomic reads underwent trimming and decontamination using KneadData (version 0.6.1) as previously described (Cabral et al., 2020; 2019; McIver et al., 2018). Illumina adaptor sequences were removed using Trimmomatic (version 0.36), then depleted of reads that mapped to C57BL/6J, murine mammary tumor virus (MMTV, accession NC_001503) and murine osteosarcoma virus (MOV, accession NC_001506.1) using Bowtie2 (version 2.2) (Bolger et al., 2014; Cabral et al., 2020; Langmead and Salzberg, 2012). Metatranscriptomic reads were additionally depleted of sequences that aligned to the SILVA 128 LSU and SSU Parc ribosomal RNA databases as previously described (Cabral et al., 2020; 2019; Pruesse et al., 2007).
Taxonomic classification of reads
We taxonomically classified trimmed and decontaminated metagenomic reads against a database of all completed bacterial, archaeal, and viral genomes contained within NCBI RefSeq using Kraken2 (version 2.0.7-beta, “Kraken2 Standard Database”) with a k-mer length of 35 (Wood et al., 2019). Bracken (version 2.0.0) was then used to calculate phylum- and species-level abundances from Kraken2 reports, and the R package phyloseq (version 1.28.0) was used to calculate α- and β-diversity metrics (Lu et al., 2017; McMurdie and Holmes, 2013).
We then performed differential abundance testing on species-level taxonomic assignments (Cabral et al., 2020; 2019). First, low-abundance taxa (< 1,000 reads in ≥ 20% of samples) were removed, then differential abundance testing of filtered counts was performed with the DESeq2 package (version 1.24.0) using default parameters (Love et al., 2014). All p values were corrected for multiple hypothesis testing using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). Features with an adjusted p value of less than 0.05 were considered statistically significant.
Metatranscriptomic analysis: SAMSA2
We used a modified version of the Simple Annotation of Metatranscriptomes by Sequences Analysis 2 (SAMSA2) pipeline to annotate trimmed and decontaminated metatranscriptomics reads as previously described (Cabral et al., 2020; 2019; Westreich et al., 2018). This modified pipeline involves implementation of the Paired-End Read Merger (PEAR) utility to generate merged reads and DIAMOND (version 0.9.12) aligner algorithm (Buchfink et al., 2014; Zhang et al., 2014) to generate alignments against RefSeq, SEED Subsystem, and CAZyme databases (Cantarel et al., 2009; Overbeek et al., 2013). The resulting alignments were subjected to differential abundance testing using DESeq2 (version 1.24.0) with standard parameters and Benjamini-Hochberg multiple hypothesis testing correction (Benjamini and Hochberg, 1995; Love et al., 2014). Features with an adjusted p value of less than 0.05 were considered statistically significant.
Metatranscriptomic analysis: HUMAnN2
We used the HMP Unified Metabolic Analysis Network 2 (HUMAnN2, version 0.11.1) pipeline to assess the impact of STZ-based HG and amoxicillin treatment on gene expression within the gut microbiome (Franzosa et al., 2018). We supplied the taxonomic profiles generated for each sample into the HUMAnN2 algorithm in order to assure consistent taxonomic assignment between paired samples (Segata et al., 2012; Cabral et al., 2020; 2019). Then, using HUMAnN2, we generated MetaCyc pathway abundances and used these to estimate community-level gene expression and normalized this to metagenomic abundance using the Witten-Bell method (Witten and Bell, 1991). Unstratified smoothed RPKM values were converted to relative abundances then analyzed using linear discriminant analysis as described (Cabral et al., 2020; 2019). This was performed with the LEfSe (version 1) toolkit hosted on the Huttenhower Galaxy server (Segata et al., 2011).
Single-species transcriptomics
We performed transcriptional analysis at the individual species level using a modified version of the pipeline developed by Deng et al. (Deng et al., 2018). First, species whose metagenomic abundance was subjected to an interaction between host glycemia and ATB usage were selected. We then calculated to total RNA read abundance for each of these species and performed transcriptional analysis only on those with 500,000 or greater reads per sample (Table S5). First, reads that mapped to candidate taxa were extracted from our metatranscriptomes using the BBSplit utility within BBMap (version 37.96) (Bushnell, 2014). Reads from B. theta, O. valericigenes, and O. spp. PEA192 were aligned to their corresponding reference genomes using BWA-MEM (version 0.7.15) (Cabral et al., 2020; Li and Durbin, 2010). Then, we used subread program (version 1.6.2) command featureCounts was used to generate a count table from alignments, and this count table was assessed for differential abundance using DESeq2 (Liao et al., 2014; Love et al., 2014). All p values were corrected for multiple hypothesis testing using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). Features with an adjusted p value of less than 0.05 were considered statistically significant.
Metabolite extraction and annotation: Q-TOF-MS
For untargeted Q-TOF-MS metabolomics, cecal samples were flash frozen upon collection and stored at −80°C until extraction. To extract metabolites, flash-frozen samples were removed from −80°C and placed on ice. A 10–20 mg sample was taken and submerged in 300 μl of fresh-made LC/MS-grade acetone:isopropanol (2:1) extraction solvent, then homogenized via vortex two times for 15 s each at 4°C. Supernatant extraction solvent was transferred to a new tube and was placed at −80°C temporarily. The 300 μl wash and homogenization was repeated, and this supernatant was then added to the original aliquot. Combined samples underwent centrifugation at 4°C for 10 minutes at 13,500 × G. After centrifugation, supernatant was moved to a fresh microcentrifuge tube, sealed with parafilm, and placed on dry ice before immediate delivery to General Metabolics Inc. (Boston, MA, USA) where samples were stored at −80°C.
Extracted metabolites were quantified as described in Fuhrer et al. (Fuhrer et al., 2011) using flow injection Time-of-Flight mass spectrometry on the Agilent G6550A iFunnel Quadrupole Time-of-Flight mass spectrometer (Agilent, Santa Clara, CA, USA) equipped with a dual AJS electrospray ionization source operated in negative ion mode. Samples were injected at a flowrate of 0.15 mL/minute in a mobile phase containing isopropanol and water (60%:40% ratio) buffered in 1mM Ammonium Fluoride, 15nM HP-0921, and 5μM homotaurine. Mass spectra data was recorded in 4 GHz high-resolution Ms mode at a rate of 1.4 spectra/second. We detected 714.3 ms/spectra and 9652 transients/spectra between 50 and 1000 m/z. Source operating parameters included a temperature of 225°C, drying gas rate of 11 L/min, nebulizer pressure of 20 psi, sheath gas temperature of 350°C and flow of 10 L/min. The source Vcap and Nozzle voltage were 3500V and 2000V. The ms TOF operating parameters include fragmentor, collision, RF peak-to-peak voltages of 350V, 0V, and 750V, respectively and the Skimmer was disabled.
Data processing and analysis was performed as described by Fuhrer et al. in MATLAB (The Mathworks, Natick, MA, USA) using functions from the following toolkits: Bioinformatics, Statistics, Database, and Parallel Computing (Fuhrer et al., 2011). Ions were additionally referenced against the Human Metabolome Database in addition to KEGG. Data analyses were run on an automated embedded platform by General Metabolics Inc. then delivered upon run completion. Finally, Principal Coordinate Analysis was performed on ion intensities by using Bray-Curtis dissimilarity paired with PERMANOVA analysis using the phyloseq (version 1.26.1) R package and subsequently visualized in Prism GraphPad (version 9.0.2) (McMurdie and Holmes, 2013).
Metabolite extraction and annotation: LC-MS/MS
For untargeted LC-MS/MS metabolomics, cecal samples were placed into 300 μl of LMCS-grade methanol then supplemented with 600 μl of 70% cold LC-MS-grade methanol. Samples were homogenized via vortex for 5 minutes, then placed at 4°C for an overnight incubation. Following incubation, samples were subjected to centrifugation at 1000 × G for 3 minutes. 500 μl of the supernatant was moved to a sterile microcentrifuge tube and stored at −80°C for long-term preservation.
Samples were thawed and diluted 1:1 (v/v) in 50% methanol prior to LC-MS/MS. Liquid chromatography was performed using a Vanquish Autosampler (Thermo Scientific, Waltham, MA, USA) and an Acquity UPLC column (Waters, Milford, MA, USA). Mass spectrometry was performed using a Q Exactive® Hybrid Quadrupole-Orbitrap Mass Spectrometer (Thermo Scientific, Waltham, MA, USA) in positive ion mode. All analysis used a 5 μL injection volume. Samples were eluted via water-acetonitrile gradient (98:2 to 2:98) containing 0.1% formic acid at a 0.4 mL min−1 flow rate. RAW files were converted via GNPS Vendor Conversion and mined with MZmine (ver. 2.52) prior to submission for feature based molecular networking (Pluskal et al., 2010; Nothias et al., 2020). Briefly, MS1 and MS2 feature extraction was performed for a centroid mass detector with a signal threshold of 5.0 × 105 and 5.0 × 104 respectively. Chromatogram builder was run with an m/z tolerance of 0.02 Da or 7 ppm and a minimum height of 1.0 × 105. Then, chromatograms were deconvoluted utilizing a baseline cut-off algorithm of 1.0 × 105 and a peak duration range of 0 to 1.00 minutes. Following this, isotopic peaks were then grouped with an m/z tolerance of 0.02 Da or 7 ppm and a retention time percentage of 0.1. The Join Aligner Module was then utilized with a 0.02 Da or 7 ppm m/z tolerance and a retention time tolerance of 0.1 minutes. Feature-based molecular networking on GNPS was performed with the following parameters: precursor and fragment ion mass tolerance 0.02 Da; minimum cosine of 0.7 and minimum matched peaks of 4, all others were defaults. Library searching was performed with the same parameters as described above.
Analysis of Q-TOF-MS metabolite data
Differentially abundant metabolites were identified using the DESeq2 package (version 1.22.2) with standard parameters (Love et al., 2014). All p values were corrected for multiple hypothesis testing using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995). Features with an adjusted p value of less than 0.05 were considered statistically significant. KEGG compound identifiers that were feature-matched by the Bioinformatics MATLAB toolkit were used to create a list of all KEGG IDs associated with differentially abundant metabolites. This list (and associated ion intensities) were used to perform KEGG pathway enrichment analysis using the PAPi R package (version 1.22.1) with standard parameters (Aggio et al., 2010). Pathways with an adjusted p value of less than 0.05 were considered statistically significant.
To link our transcriptional data and metabolomics data, we used the R package implementation of Mimosa (version 2.0.0) and the publicly availably KEGG reaction database (circa 2010) (Noecker et al., 2016). The configuration table settings were as follows: File1) taxon-stratified output from HUMAnN2 based of KEGG Orthology annotation. File2) per-sample ion counts of differentially abundant Q-TOF-MS metabolites. File1_type) “taxon stratified KO abundance (HUMAnN2 or PICRUSt/PICRUSt2.” Ref_choices) PICRUSt KO genomes and KEGG metabolic model. metType) KEGG compound ID. data_prefix) complete file path to the KEGG reaction database. Vsearch_path) complete file path to the vsearch executable (https://github.com/torognes/vsearch). The run_mimosa2 function was used with standard parameters to calculate the community metabolic potential within each sample, score this against the input metabolite table, and calculate the level of metabolic variation attributable to individual taxa using a linear rank regression as described (Noecker et al., 2016). All data tables produced by the run_mimosa2 function were then exported and data visualization was performed in Prism GraphPad (version 9.0.2). “Positive” metabolites have observed abundances that match the predictive model. “Negative” metabolites are those whose observed abundance diverges from the predictive model.
Analysis of LC-MS/MS metabolite data
First, principal coordinate analysis was performed on ion intensities by using Bray-Curtis dissimilarity paired with PERMANOVA analysis. These analyses were performed using the phyloseq (version 1.26.1) R package and subsequently visualized in Prism GraphPad (version 9.0.2) (McMurdie and Holmes, 2013). Random forest classification models on treatment mouse treatment group were then generated using the randomForest (version 4.6–16) R package (Breiman, 2001). Variable importance plots from the models were used to identify metabolites that best contributed to group classification. Each metabolite feature of interest was then checked for annotation in GNPS, if not directly annotated from MS/MS library searching, the node of interest was identified in the molecular network and assessed for spectral similarity to other annotated nodes. This provided a molecular family annotation of each unknown cluster. Models classifying HG mice treated with amoxicillin and not treated with amoxicillin resulted in out-of-bag prediction error of 2.7%. Classification of nonHG treated with amoxicillin and not treated with amoxicillin resulted in out-of-bag prediction error of 6.25%. Classification of HG mice and nonHG mice, both treated with ATBs, resulted in out-of-bag prediction error of 7.96%. Classification of HG mice and nonHG mice, neither treated with ATBs, resulted in out-of-bag prediction error of 16.67%.
Cecal glucose assessment
Cecal glucose levels were assessed using the Abcam Glucose Detection Kit (Abcam, Cambridge, United Kingdom). First, cecal material was weighed out and resuspended in glucose assay buffer at a concentration of 100 mg/mL, then homogenized via vortex until no visible clumps were present. Samples were spun at maximum speed for 1 minute to pellet any residual debris, and 500 μl of supernatant was transferred to a Corning Costar Spin-X 0.22 μM centrifuge tube filter (Corning Brand, Corning, New York, USA). The costar tubes containing supernatant were spun via centrifugation at 15,000 x G for 10 minutes, after which up to 500 μl of flow-through was transferred to an abcam 10kD spin column to deproteinize the samples. Samples were again spun at 15,000 x G for 10 minutes and flow-through was quantified using the Abcam Glucose Assay kit as per the manufacturer’s instructions.
Plasma cytokine profiling
Upon animal sacrifice, whole blood was collected via cardiac puncture and placed in a microcentrifuge tube containing up to 15μl of 1X heparin. Collection tubes were then spun via centrifugation at 13,000 x G for 10 minutes to isolate plasma. The plasma-containing supernatant was transferred to a new microcentrifuge tube and frozen at −80°C until ready to process. Once ready, samples were thawed on ice, split into a working aliquot and a re-frozen stock aliquot. The working aliquot was assessed for signatures of inflammation in mice using the LEGENDplex Mouse Inflammation Panel (13-plex) (BioLegend, San Diego, CA) flow cytometry kit as per the manufacturer’s instructions. Samples were processed on the Attune NxT Flow Cytometer (ThermoFisher, Waltham, MA) and subsequently analyzed using the LegendPlex cloud software cool (BioLegened, San Diego, CA). This panel allows for simultaneous profiling of IL-1α, IL-1β, IL-6, IL-10, IL-12p70, IL-17A, IL-23, MCP-1, IFN-β, IFN-γ, TNF-α, and GM-CSF. Cytokine concentrations were compared across samples using Welch’s ANOVA with Dunnet T3 test for multiple hypothesis testing. Only cytokines with a p value < 0.05 were included in the manuscript discussion.
Lipocalin-2 quantification
Cecal lipocalin-2 levels were assessed using the Mouse Lipocalin-2/NGAL DuoSet ELISA kit (R&D Systems, Minneapolis, MN). First, flash-frozen cecal contents were weighed and reconstituted into a freshly made working solution of 1X phosphate buffered saline (PBS) and 0.1% Tween 20 at a concentration of 100 mg/mL. This working solution was vigorously pipetted to aid in resuspension. Samples were mixed by vortex at max speed for at 5 minutes until fully homogenized, then spun via centrifugation at 12,000 rpm for 10 minutes. The supernatant was transferred to sterile microcentrifuge tubes and used as input for the DuoSet kit. Lipocalin-2 was quantified from these samples as per the manufacturer’s instructions. Concentrations were compared across samples using Welch’s ANOVA with Dunnet T3 test for multiple hypothesis testing.
GI histopathology assessment
During animal necropsy an approximate 1-inch section of the distal colon was collected and fixed in methacarn. Fixed tissues were incubated at room temperature for 24 hours, then washed twice with a 70% ethanol solution. Samples were placed in 70% ethanol and stored in a light-safe box at 4°C until ready to process. To process, tissues were transferred to histology cassettes, submerged in 70% ethanol, and submitted to the Molecular Pathology Core at Brown University. Core staff embedded the sample cassettes in paraffin, then sectioned the blocks at 4–5 μM thickness. Tissues sections were mounted on microscopy slides and stained with hematoxylin and eosin. Stained slides were dried for 24 hours before being shipped to the University of Texas MD Anderson Cancer Center for pathology scoring.
Enteric pathogen challenge
Salmonella enterica serovar Typhimurium SL1344 was grown overnight in 5 mL Luria-Bertani (LB) broth supplemented with fresh-made ampicillin (100 μg/mL) and grown at 37°C. This culture was diluted 1:1000 into fresh LB+ampicillin (100 μg/mL) the morning of infections and grown until cells were approximately at mid-log phase (OD600 = 0.3–0.4).
Rather than sacrificing animals after the 24-hours of amoxicillin treatment as outlined above (See Animal Procedures), animals were given an additional 48 hours of ad libitum amoxicillin within their drinking water followed by ATB-free filter-sterilized water for 24 hours. Subsequently, animals were moved to clean cages and placed under a 4-hour fast, at which point they were infected with an inoculum between 102 and 106 cells/dose via oral gavage (volume ≤ 200 μl). Animals were transferred to clean cages and weighed daily throughout the course of pathogen challenge. Fecal samples were collected daily then resuspended in 1 mL of 1X PBS and homogenized via vortex at maximum speed for at least 5 minutes. Fecal slurry was then serially diluted and plated onto ampicillin-supplemented (100 μg/mL) LB agar plates and grown at 37C for 24 hours. After growth, colonies were counted and the total colony forming units (CFU) were quantified per gram of feces to assess pathogen burden. To quantify non-intestinal S. enterica burden, fresh liver and spleen were collected during post-sacrifice necropsy, weighed, then placed into 1mL of 1X PBS, mixed via vortex for 5 minutes, serially diluted, and plated onto LB agar plates supplemented with ampicillin (100 mg/mL). CFUs were quantified 24 hours later.
During the course of infection, any animal that experienced a loss of ≥ 20 percent of total body weight was sacrificed as per our IACUC protocol. These qualified as “lethality events” and were logged accordingly.
QUANTIFICATION AND STATISTICAL ANALYSIS
Specific details of the statistical analyses for all experiments are outlined in the figure legends and Results section. Sample numbers represent biological replicates, and instances of technical replicates are specifically stated in corresponding figure legends. LEfSe (version 1.0) was used to analyze MetaCyc pathway abundance data generated by HUMAnN2 on the Galaxy web server using default settings (http://huttenhower.sph.harvard.edu/galaxy). Metatranscriptomic outputs generated by SAMSA2 and single-species sequencing, along with Q-TOF-MS abundances were subjected to differential abundance testing using the DESeq2 package (1.24.0) in R (version 3.5.2) under default parameters and included contrast:interaction comparisons (Love et al., 2014). All DESeq2 results were corrected using the Benjamini-Hochberg method (Benjamini and Hochberg, 1995) to account for multiple hypothesis testing and significance was considered when the adjusted p value was below 0.05. LC-MS/MS Random Forest testing was conducted using the R package implementation (Breiman, 2001). Permutational ANOVA calculations were made using the vegan R package (version 2.5.2). ANOVA, unpaired t tests, and Mann-Whitney U tests were performed in Prism Graphpad (version 9.0) without sample size estimation.
Supplementary Material
Highlights.
The cecal metatranscriptome and metabolome are altered in hyperglycemic mice
The hyperglycemic microbiota has more simple sugars and shows higher respiratory activity
Hyperglycemia-related changes are associated with worsened antibiotic dysbiosis
Amoxicillin promotes Salmonella susceptibility in hyperglycemic mice
ACKNOWLEDGMENTS
P.B. was supported by the U.S. Department of Defense through the Peer Reviewed Medical Research Program (W81XWH-18-1-0198), the National Institute of General Medical Sciences (P20GM121344), and the National Institute of Diabetes and Digestive and Kidney Diseases of the National Institutes of Health (R01DK125382). J.I.W. and S.P. were supported by the National Science Foundation Graduate Research Fellowship (1644760). This study made use of the Research Histology, Pathology, and Imaging core supported by the DHHS/NCI Cancer Center (P30CA16672) and computational resources from the Center for Computation and Visualization at Brown University. The funding agencies had no role in the study design, data collection, analysis, or interpretation. The opinions and interpretations presented here are not representative of the standing of the funding agencies.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110113.
REFERENCES
- Adolfsen KJ, and Brynildsen MP (2015). Futile cycling increases sensitivity toward oxidative stress in escherichia coli. Metab. Eng. 29, 26–35. 10.1016/j.ymben.2015.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aggio RBM, Ruggiero K, and Villas-Bôas SG (2010). Pathway activity profiling (PAPi): From the metabolite profile to the metabolic pathway activity. Bioinformatics 26, 2969–2976. 10.1093/bioinformatics/btq567. [DOI] [PubMed] [Google Scholar]
- Ahn S, Jung J, Jang I-A, Madsen EL, and Park W (2016). Role of glyoxylate shunt in oxidative stress response. J. Biol. Chem. 291, 11928–11938. 10.1074/jbc.M115.708149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albenberg LG, and Wu GD (2014). Diet and the intestinal microbiome: associations, functions, and implications for health and disease. Gastroenterology 146, 1564–1572. 10.1053/j.gastro.2014.01.058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allaway D, Haydock R, Lonsdale ZN, Deusch OD, O’Flynn C, and Hughes KR (2020). Rapid reconstitution of the fecal microbiome after extended diet-induced changes indicates a stable gut microbiome in healthy adult dogs. Appl. Environ. Microbiol. 86, e00562–20. 10.1128/AEM.00562-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen LW, Liu X, Peng TJ, Giberson TA, Khabbaz KR, and Donnino MW (2015). Pyruvate dehydrogenase activity and quantity decreases after coronary artery bypass grafting: A prospective observational sudy. Shock 43, 250–254. 10.1097/SHK.0000000000000306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CJ, and Kendall MM (2016). Location, location, location. Salmonella senses ethanolamine to gauge distinct host environments and coordinate gene expression. Microb. Cell 3, 89–91. 10.1371/journal.ppat.1005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CJ, Clark DE, Adli M, and Kemdall MM (2015). Ethanolamine signaling promotes Salmonella niche recognition and adaptation during infection. PLoS Pathog. 11, e1005278. 10.1371/journal.ppat.1005278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anderson CJ, Satkovich J, Köseoğlu VK, Agaisse H, and Kendall MM (2018). The ethanolamine permease EutH promotes vacuole adaptation of Salmonella enterica and Listeria monocytogenes during macrophage infection. Infect. Immun. 86, 1–12. 10.1128/IAI.00172-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bäumler AJ, and Sperandio V (2016). Interactions between the microbiota and pathogenic bacteria in the gut. Nature 535, 85–93. 10.1038/nature18849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belenky P, Bogan KL, and Brenner C (2007). NAD+ metabolism in health and disease. Trends Biochem Sci. 32, 12–9. 10.1016/j.tibs.2006.11.006. [DOI] [PubMed] [Google Scholar]
- Belenky P, Ye JD, Porter CBM, Cohen NR, Lobritz MA, Ferrante T, Jain S, Korry BJ, Schwarz EG, Walker GC, and Collins JJ (2015). Bactericidal antibiotics induce toxic metabolic perturbations that lead to cellular damage. Cell Rep. 13, 968–980. 10.1016/j.celrep.2015.09.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belzer C, Chia LW, Aalvink S, Chamlagain B, Piironen V, Knol J, and de Vos WM (2017). Microbial metabolic networks at the mucus layer lead to diet-independent butyrate and vitamin B12 production by intestinal symbionts. MBio 8, e00770–17. 10.1128/mBio.00770-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the false discovery rate: A practical and powerful approach to multiple testing. J. R. Stat. Soc. Ser. B Method. 57, 289–300. 10.2307/2346101. [DOI] [Google Scholar]
- Berg JM, Tymoczko JL, and Stryer L (2002). Carbon atoms of degraded amino acids emerge as major metabolic intermediates. In Biochemistry, 5th edition (W.H. Freeman; ), Section 23.5. [Google Scholar]
- Bernal V, Sevilla A, Cánovas M, and Iborra JL (2007). Production of L-carnitine by secondary metabolism of bacteria. Microb. Cell Fact. 6, 31. 10.1186/1475-2859-6-31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bisanz JE, Upadhyay V, Turnbaugh JA, Ly K, and Turnbaugh PJ (2019). Meta-analysis reveals reproducible gut microbiome alterations in response to a high-fat diet. Cell Host Microbe 26, 265–272.e4. 10.1016/j.chom.2019.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon E, Meehan CJ, Whidden C, Wong DH-J, Langille MGI, and Beiko RG (2014). Interactions in the microbiome: communities of organisms and communities of genes. FEMS Microbiol. Rev. 38, 90–118. 10.1111/1574-6976.12035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Böttcher C, Chapman A, Fellermeier F, Choudhary M, Scheel D, and Glawischnig E (2014). The biosynthetic pathway of indole-3-carbaldehyde and indole-3-carboxylic acid derivatives in arabidopsis. Plant Physiol. 165, 841–853. 10.1104/pp.114.235630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braune A, and Blaut M (2016). Bacterial species involved in the conversion of dietary flavonoids in the human gut. Gut Microbes 7, 216–234. 10.1080/19490976.2016.1158395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breiman L (2001). Random forests. Mach. Learn. 45, 5–32. 10.1023/A:1010933404324. [DOI] [Google Scholar]
- Brestoff JR, and Artis D (2013). Commensal bacteria at the interface of host metabolism and the immune system. Nat. Immunol. 14, 676–684. 10.1038/ni.2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buchfink B, Xie C, and Huson DH (2014). Fast and sensitive protein alignment using DIAMOND. Nat. Methods 12, 59–60. 10.1038/nmeth.3176. [DOI] [PubMed] [Google Scholar]
- Buffie CG, Jarchum I, Equinda M, Lipuma L, Gobourne A, Viale A, Ubeda C, Xavier J, and Pamer EG (2012). Profound alterations of intestinal microbiota following a single dose of clindamycin results in sustained susceptibility to Clostridium difficile-induced colitis. Infect. Immun. 80, 62–73. 10.1128/IAI.05496-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bui TPN, Shetty SA, Lagkouvardos I, Ritari J, Chamlagain B, Douillard FP, Paulin L, Piironen V, Clavel T, Plugge CM, and de Vos WM (2016). Comparative genomics and physiology of the butyrate-producing bacterium Intestinimonas butyriciproducens. Environ. Microbiol. Rep. 8, 1024–1037. 10.1111/1758-2229.12483. [DOI] [PubMed] [Google Scholar]
- Bui TPN, Schols HA, Jonathan M, Stams AJM, de Vos WM, and Plugge CM (2019). Mutual metabolic interactions in co-cultures of the intestinal Anaerostipes rhamnosivorans with an acetogen, methanogen, or pectin-degrader affecting butyrate production. Front. Microbiol. 10, 2449. 10.3389/fmicb.2019.02449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burlingame R, and Chapman PJ (1983). Catabolism of phenylpropionic acid and its 3-hydroxy derivative by Escherichia coli. J. Bacteriol. 155, 113–121. 10.1128/jb.155.1.113-121-1983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushnell B (2014). BBMap: A fast, accurate, splice-aware aligner. Presented at the 9th Annual Genomics of Energy Environment Meeting at Walnut Creek, CA March 17–20, 2014. [Google Scholar]
- Cabral DJ, Penumutchu S, Reinhart EM, Zhang C, Korry BJ, Wurster JI, Nilson R, Guang A, Sano WH, Rowan-Nash AD, et al. (2019). Microbial metabolism modulates antibiotic susceptibility within the murine gut microbiome. Cell Metab. 30, 800–823.e7. 10.1016/j.cmet.2019.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabral DJ, Wurster JI, Korry BJ, Penumutchu S, and Belenky P (2020). Consumption of a western-style diet modulates the response of the murine gut microbiome to Ciprofloxacin. mSystems 5, 51–19. 10.1128/mSystems.00317-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Callahan BJ, McMurdie PJ, Rosen MJ, Han AW, Johnson AJA, and Holmes SP (2016). DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 13, 581–583. 10.1038/nmeth.3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantarel BL, Coutinho PM, Rancurel C, Bernard T, Lombard V, and Henrissat B (2009). The carbohydrate-active enzymes database (CAZy): An expert resource for glycogenomics. Nucleic Acids Res. 37, D233–D238. 10.1093/nar/gkn663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso JG, Lauber CL, Walters WA, Berg-Lyons D, Huntley J, Fierer N, Owens SM, Betley J, Fraser L, Bauer M, et al. (2012). Ultrahigh-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 6, 1621–1624. 10.1038/ismej.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JY, Antonopoulos DA, Kalra A, Tonelli A, Khalife WT, Schmidt TM, and Young VB (2008). Decreased diversity of the fecal microbiome in recurrent Clostridium difficile-associated diarrhea. J. Infect. Dis. 197, 435–438. 10.1086/525047. [DOI] [PubMed] [Google Scholar]
- Chao P-C, Li Y, Chang C-H, Shieh JP, Cheng J-T, and Cheng K-C (2018). Investigation of insulin resistance in the popularly used four rat models of type-2 diabetes. Biomed. Pharmacother. 101, 155–161. 10.1016/j.biopha.2018.02.084. [DOI] [PubMed] [Google Scholar]
- Chassaing B, Raja SM, Lewis JD, Srinivasan S, and Gewirtz AT (2017). Colonic microbiota encroachment correlates with dysglycemia in humans. Cell. Mol. Gastroenterol. Hepatol. 4, 205–221. 10.1016/j.jcmgh.2017.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen L, Tuo B, and Dong H (2016). Regulation of intestinal glucose absorption by ion channels and transporters. Nutrients 8, 43. 10.3390/nu8010043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen MX, Wang S-Y, Kuo C-H, and Tsai I-L (2019). Metabolome analysis for investigating host-gut microbiota interactions. J. Formos. Med. Assoc. 118 (Suppl 1), S10–S22. 10.1016/j.jfma.2018.09.007. [DOI] [PubMed] [Google Scholar]
- Clooney AG, Fouhy F, Sleator RD, O’ Driscoll A, Stanton C, Cotter PD, and Claesson MJ (2016). Comparing apples and oranges?: Next generation sequencing and its impact on microbiome analysis. PLoS ONE 11, e0148028. 10.1371/journal.pone.0148028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins J, Robinson C, Danhof H, Knetsch CW, van Leeuwen HC, Lawley TD, Auchtung JM, and Britton RA (2018). Dietary trehalose enhances virulence of epidemic Clostridium difficile. Nature 553, 291–294. 10.1038/nature25178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conlon BP, Rowe SE, Gandt AB, Nuxoll AS, Donegan NP, Zalis EA, Clair G, Adkins JN, Cheung AL, and Lewis K (2016). Persister formation in Staphylococcus aureus is associated with ATP depletion. Nat. Microbiol. 1, 16051. 10.1038/nmicrobiol.2016.51. [DOI] [PubMed] [Google Scholar]
- Coyte KZ, and Rakoff-Nahoum S (2019). Understanding competition and cooperation within the mammalian gut microbiome. Curr. Biol. 29, R538–R544. 10.1016/j.cub.2019.04.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crost EH, Tailford LE, Monestier M, Swarbreck D, Henrissat B, Crossman LC, and Juge N (2016). The mucin-degradation strategy of Ruminococcus gnavus: The importance of intramolecular trans-sialidases. Gut Microbes 7, 302–312. 10.1080/19490976.2016.1186334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croswell A, Amir E, Teggatz P, Barman M, and Salzman NH (2009). Prolonged impact of antibiotics on intestinal microbial ecology and susceptibility to enteric Salmonella infection. Infect. Immun. 77, 2741–2753. 10.1128/IAI.00006-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- David LA, Maurice CF, Carmody RN, Gootenberg DB, Button JE, Wolfe BE, Ling AV, Devlin AS, Varma Y, Fischbach MA, et al. (2014). Diet rapidly and reproducibly alters the human gut microbiome. Nature 505, 559–563. 10.1038/nature12820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deeds MC, Anderson JM, Armstrong AS, Gastineau DA, Hiddinga HJ, Jahangir A, Eberhardt NL, and Kudva YC (2011). Single dose streptozotocin-induced diabetes: considerations for study design in islet transplantation models. Lab. Anim. 45, 131–140. 10.1258/la.2010.010090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng Z-L, Sztajer H, Jarek M, Bhuju S, and Wagner-Döbler I (2018). Worlds Apart - Transcriptome profiles of key oral microbes in the periodontal pocket compared to single laboratory culture reflect synergistic interactions. Front. Microbiol. 9, 124. 10.3389/fmicb.2018.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desai MS, Seekatz AM, Koropatkin NM, Kamada N, Hickey CA, Wolter M, Pudlo NA, Kitamoto S, Terrapon N, Muller A, et al. (2016). A dietary fiber-deprived gut microbiota degrades the colonic mucus barrier and enhances pathogen susceptibility. Cell 167, 1339–1353.e21. 10.1016/j.cell.2016.10.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dethlefsen L, and Relman DA (2011). Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc. Natl. Acad. Sci. USA 108 (Suppl 1), 4554–4561. 10.1073/pnas.1000087107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CTY, Lobritz MA, Braff D, Schwarz EG, et al. (2014). ATBs induce redox-related physiological alterations as part of their lethality. Proc. Natl. Acad. Sci. U.S.A. 111, E2100–9. 10.1073/pnas.1401876111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards R (1997). Resistance to beta-lactam antibiotics in Bacteroides spp. J. Med. Microbiol. 46, 979–986. 10.1099/00222615-46-12-979. [DOI] [PubMed] [Google Scholar]
- Eleazu CO, Eleazu KC, Chukwuma S, and Essien UN (2013). Review of the mechanism of cell death resulting from streptozotocin challenge in experimental animals, its practical use and potential risk to humans. J. Diabetes Metab. Disord. 12, 60. 10.1186/2251-6581-12-60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Erhardt M, and Dersch P (2015). Regulatory principles governing Salmonella and Yersinia virulence. Front. Microbiol. 6, 949. 10.3389/fmicb.2015.00949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ernst DC, and Downs DM (2015). The STM4195 gene product (PanS) transports coenzyme A precursors in Salmonella enterica. J. Bacteriol. 197, 1368–1377. 10.1128/JB.02506-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischbach MA, and Sonnenburg JL (2011). Eating for two: How metabolism establishes interspecies interactions in the gut. Cell Host Microbe 10, 336–347. 10.1016/j.chom.2011.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franzosa EA, McIver LJ, Rahnavard G, Thompson LR, Schirmer M, Weingart G, Lipson KS, Knight R, Caporaso JG, Segata N, and Huttenhower C (2018). Species-level functional profiling of metagenomes and metatranscriptomes. Nat. Methods 15, 962–968. 10.1038/s41592-018-0176-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraser CM, and Chapple C (2011). The phenylpropanoid pathway in Arabidopsis. Arabidopsis Book 9, e0152. 10.1199/tab.0152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuhrer T, Heer D, Begemann B, and Zamboni N (2011). High-throughput, accurate mass metabolome profiling of cellular extracts by flow injection-time-of-flight mass spectrometry. Anal. Chem. 83, 7074–7080. 10.1021/ac201267k. [DOI] [PubMed] [Google Scholar]
- Fujisaka S, Ussar S, Clish C, Devkota S, Dreyfuss JM, Sakaguchi M, Soto M, Konishi M, Softic S, Altindis E, et al. (2016). Antibiotic effects on gut microbiota and metabolism are host dependent. J. Clin. Invest. 126, 4430–4443. 10.1172/JCI86674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furman BL (2021). Streptozotocin-induced diabetic models in mice and rats. Curr Protoc 1, e78. 10.1002/cpz1.78. [DOI] [PubMed] [Google Scholar]
- Gao J, Xu K, Liu H, Liu G, Bai M, Peng C, Li T, and Yin Y (2018). Impact of the gut microbiota on intestinal immunity mediated by tryptophan metabolism. Front. Cell. Infect. Microbiol. 8, 13. 10.3389/fcimb.2018.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garsin DA (2010). Ethanolamine utilization in bacterial pathogens: roles and regulation. Nat. Rev. Microbiol. 8, 290–295. 10.1038/nrmi-cro2334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertsman I, and Barshop BA (2018). Promises and pitfalls of untargeted metabolomics. J. Inherit. Metab. Dis. 41, 355–366. 10.1007/s10545-017-0130-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodman AL, McNulty NP, Zhao Y, Leip D, Mitra RD, Lozupone CA, Knight R, and Gordon JI (2009). Identifying genetic determinants needed to establish a human gut symbiont in its habitat. Cell Host Microbe 6, 279–289. 10.1016/j.chom.2009.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal SN, Reddy NM, Patil KR, Nakhate KT, Ojha S, Patil CR, and Agrawal YO (2016). Challenges and issues with streptozotocin-induced diabetes - A clinically relevant animal model to understand the diabetes pathogenesis and evaluate therapeutics. Chem. Biol. Interact. 244, 49–63. 10.1016/j.cbi.2015.11.032. [DOI] [PubMed] [Google Scholar]
- Hanafusa T, and Imagawa A (2008). Insulitis in human type 1 diabetes. Ann. N Y Acad. Sci. 1150, 297–299. 10.1196/annals.1447.052. [DOI] [PubMed] [Google Scholar]
- Holscher HD (2017). Dietary fiber and prebiotics and the gastrointestinal microbiota. Gut Microbes 8, 172–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holst JJ, Gribble F, Horowitz M, and Rayner CK (2016). Roles of the gut in glucose homeostasis. Diabetes Care 39, 884–892. 10.2337/dc16-0351. [DOI] [PubMed] [Google Scholar]
- HMP (Human Microbiome Project Consortium) (2012). A framework for human microbiome research. Nature 486, 215–221. 10.1038/nature11209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iino T, Mori K, Tanaka K, Suzuki K-I, and Harayama S (2007). Oscillibacter valericigenes gen. nov., sp. nov., a valerate-producing anaerobic bacterium isolated from the alimentary canal of a Japanese corbicula clam. Int. J. Syst. Evol. Microbiol. 57, 1840–1845. 10.1099/ijs.0.64717-0. [DOI] [PubMed] [Google Scholar]
- Jain N, Mishra SK, Shankar U, Jaiswal A, Sharma TK, Kodgire P, and Kumar A (2020). G-quadruplex stabilization in the ions and maltose transporters gene inhibit Salmonella enterica growth and virulence. Genomics 112, 4863–4874. 10.1016/j.ygeno.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiko GE, and Stappenbeck TS (2014). Host-microbe interactions shaping the gastrointestinal environment. Trends Immunol. 35, 538–548. 10.1016/j.it.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kazemian N, Ramezankhani M, Sehgal A, Khalid FM, Kalkhoran AHZ, Narayan A, Wong GK-S, Kao D, and Pakpour S (2020). The trans-kingdom battle between donor and recipient gut microbiome influences fecal microbiota transplantation outcome. Sci Rep, 18349. 10.1038/s41598-020-75162-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi K, Forte TM, Taniguchi S, Ishida BY, Oka K, and Chan L (2000). The db/db mouse, a model for diabetic dyslipidemia: molecular characterization and effects of western diet feeding. Metabolism 49, 22–31. 10.1016/s0026-0495(00)90588-2. [DOI] [PubMed] [Google Scholar]
- Koepsell H (2020). Glucose transporters in the small intestine in health and disease. Pflügers Arch. Eur. J. Physiol. 472, 1207–1248. 10.1007/s00424-020-02439-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, and Collins JJ (2007). A common mechanism of cellular death induced by bactericidal antibiotics. Cell 130, 797–810. 10.1016/j.cell.2007.06.049. [DOI] [PubMed] [Google Scholar]
- Kolb H (1987). Mouse models of insulin dependent diabetes: Low-dose streptozocin-induced diabetes and nonobese diabetic (NOD) mice. Diabetes Metab. Rev. 3, 751–778. 10.1002/dmr.5610030308. [DOI] [PubMed] [Google Scholar]
- Lam PL, Wong RSM, Lam KH, Hung LK, Wong MM, Yung LH, Ho YW, Wong WY, Hau DKP, Gambari R, and Chui CH (2020). The role of reactive oxygen species in the biological activity of antimicrobial agents: An updated mini review. Chem. Biol. Interact. 320, 109023. 10.1016/j.cbi.2020.109023. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Layeghifard M, Hwang DM, and Guttman DS (2017). Disentangling interactions in the microbiome: A network perspective. Trends Microbiol. 25, 217–228. 10.1016/j.tim.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JH, Yang SH, Oh JM, and Lee MG (2010). Pharmacokinetics of drugs in rats with diabetes mellitus induced by alloxan or streptozocin: comparison with those in patients with type I diabetes mellitus. J. Pharm. Pharmacol. 62, 1–23. 10.1211/jpp.62.01.0001. [DOI] [PubMed] [Google Scholar]
- Lei Z, Huhman DV, and Sumner LW (2011). Mass spectrometry strategies in metabolomics. J. Biol. Chem. 286, 25435–25442. 10.1074/jbc.R111.238691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ley RE (2014). Harnessing microbiota to kill a pathogen: The sweet tooth of Clostridium difficile. Nat. Med. 20, 248–249. 10.1038/nm.3494. [DOI] [PubMed] [Google Scholar]
- Li H, and Durbin R (2010). Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics 26, 589–595. 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. 10.1093/bioinformatics/btt656. [DOI] [PubMed] [Google Scholar]
- Liu S, Qin P, and Wang J (2019a). High-fat diet alters the intestinal microbiota in streptozotocin-induced type 2 diabetic mice. Microorganisms 7, 1–12. 10.3390/microorganisms7060176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lobritz MA, Belenky P, Porter CBM, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, and Collins JJ (2015). Antibiotic efficacy is linked to bacterial cellular respiration. Proc. Natl. Acad. Sci. USA 112, 8173–8180. 10.1073/pnas.1509743112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu J, Breitwieser FP, Thielen P, and Salzberg SL (2017). Bracken: Estimating species abundance in metagenomics data. PeerJ Comput. Sci. 3, 1–17. 10.7717/peerj-cs.104. [DOI] [Google Scholar]
- Luan H, Wang X, and Cai Z (2019). Mass spectrometry-based metabolomics: Targeting the crosstalk between gut microbiota and brain in neurodegenerative disorders. Mass Spectrom. Rev. 38, 22–33. 10.1002/mas.21553. [DOI] [PubMed] [Google Scholar]
- Lundgren P, and Thaiss CA (2020). The microbiome-adipose tissue axis in systemic metabolism. Am. J. Physiol. Gastrointest. Liver Physiol. 318, G717–G724. 10.1152/ajpgi.00304.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch JB, and Sonnenburg JL (2012). Prioritization of a plant polysaccharide over a mucus carbohydrate is enforced by a Bacteroides hybrid two-component system. Mol. Microbiol. 85, 478–491. 10.1111/j.1365-2958.2012.08123.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Q, Li Y, Wang J, Li P, Duan Y, Dai H, An Y, Cheng L, Wang T, Wang C, et al. (2020). Investigation of gut microbiome changes in type 1 diabetic mellitus rats based on high-throughput sequencing. Biomed. Pharmacother. 124, 109873. 10.1016/j.biopha.2020.109873. [DOI] [PubMed] [Google Scholar]
- Makki K, Deehan EC, Walter J, and Bäckhed F (2018). The impact of dietary fiber on gut microbiota in host health and disease. Cell Host Microbe 23, 705–715. 10.1016/j.chom.2018.05.012. [DOI] [PubMed] [Google Scholar]
- Mardo K, Visnapuu T, Vija H, Aasamets A, Viigand K, and Alamäe T (2017). A highly active endo-levanase BT1760 of a dominant mammalian gut commensal Bacteroides thetaiotaomicron cleaves not only various bacterial levans, but also levan of timothy grass. PLoS ONE 12, e0169989. 10.1371/journal.pone.0169989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martens EC, Chiang HC, and Gordon JI (2008). Mucosal glycan foraging enhances fitness and transmission of a saccharolytic human gut bacterial symbiont. Cell Host Microbe 4, 447–457. 10.1016/j.chom.2008.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCoy JG, Levin EJ, and Zhou M (2015). Structural insight into the PTS sugar transporter EIIC. Biochim. Biophys. Acta 1850, 577–585. 10.1016/j.bbagen.2014.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McIver LJ, Abu-Ali G, Franzosa EA, Schwager R, Morgan XC, Waldron L, Segata N, and Huttenhower C (2018). bioBakery: A meta’omic analysis environment. Bioinformatics 34, 1235–1237. 10.1093/bioinformatics/btx754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaren MR, Willis AD, and Callahan BJ (2019). Consistent and correctable bias in metagenomic sequencing experiments. eLife 8, 46923. 10.7554/eLife.46923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McMurdie PJ, and Holmes S (2013). phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 8, e61217. 10.1371/journal.pone.0061217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meadows JA, and Wargo MJ (2015). Carnitine in bacterial physiology and metabolism. Microbiology (Reading) 161, 1161–1174. 10.1099/mic.0.000080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mewis K, Lenfant N, Lombard V, and Henrissat B (2016). Dividing the large glycoside hydrolase family 43 into subfamilies: A motivation for detailed enzyme characterization. Appl. Environ. Microbiol. 82, 1686–1692. 10.1128/AEM.03453-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meylan S, Porter CBM, Yang JH, Belenky P, Gutierrez A, Lobritz MA, Park J, Kim SH, Moskowitz SM, and Collins JJ (2017). Carbon sources tune antibiotic susceptibility in Pseudomonas aeruginosa via tricarboxylic acid cycle control. Cell Chem. Biol. 24, 195–206. 10.1016/j.chem-biol.2016.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller KA, Phillips RS, Mrázek J, and Hoover TR (2013). Salmonella utilizes D-glucosaminate via a mannose family phosphotransferase system permease and associated enzymes. J. Bacteriol. 195, 4057–4066. 10.1128/JB.00290-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mimee M, Tucker AC, Voigt CA, and Lu TK (2015). Programming a human commensal bacterium, Bacteroides thetaiotaomicron, to sense and respond to stimuli in the murine gut microbiota. Cell Syst. 1, 62–71. 10.1016/j.cels.2015.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moore BS, Hertweck C, Hopke JN, Izumikawa M, Kalaitzis JA, Nilsen G, O’Hare T, Piel J, Shipley PR, Xiang L, et al. (2002). Plant-like biosynthetic pathways in bacteria: From benzoic acid to chalcone. J. Nat. Prod. 65, 1956–1962. 10.1021/np020230m. [DOI] [PubMed] [Google Scholar]
- Motyl K, and McCabe LR (2009). Streptozotocin, type I diabetes severity and bone. Biol. Proced. Online 11, 296–315. 10.1007/s12575-009-9000-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nawrocki KL, Wetzel D, Jones JB, Woods EC, and McBride SM (2018). Ethanolamine is a valuable nutrient source that impacts Clostridium difficile pathogenesis. Environ. Microbiol. 20, 1419–1435. 10.1111/1462-2920.14048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neis EP, Dejong CH, and Rensen SS (2015). The role of microbial amino acid metabolism in host metabolism. Nutrients 7, 2930–2946. 10.3390/nu7042930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newman MA, Petri RM, Grüll D, Zebeli Q, and Metzler-Zebeli BU (2018). Transglycosylated starch modulates the gut microbiome and expression of genes related to lipid synthesis in liver and adipose tissue of pigs. Front. Microbiol. 9, 224. 10.3389/fmicb.2018.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ng KM, Aranda-Díaz A, Tropini C, Frankel MR, Van Treuren W, O’Loughlin CT, Merrill BD, Yu FB, Pruss KM, Oliveira RA, et al. (2019). Recovery of the gut microbiota after antibiotics depends on host diet, community context, and environmental reservoirs. Cell Host Microbe 26, 650–665.e4. 10.1016/j.chom.2019.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noecker C, Eng A, Srinivasan S, Theriot CM, Young VB, Jansson JK, Fredricks DN, and Borenstein E (2016). Metabolic model-based integration of microbiome taxonomic and metabolomic profiles elucidates mechanistic links between ecological and metabolic variation. mSystems 1, e00013–e00015. 10.1128/mSystems.00013-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nothias L-F, Petras D, Schmid R, Dührkop K, Rainer J, Sarvepalli A, Protsyuk I, Ernst M, Tsugawa H, Fleischauer M, et al. (2020). Feature-based molecular networking in the GNPS analysis environment. Nat. Methods 17, 905–908. 10.1038/s41592-020-0933-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliphant K, and Allen-Vercoe E (2019). Macronutrient metabolism by the human gut microbiome: Major fermentation by-products and their impact on host health. Microbiome 7, 91. 10.1186/s40168-019-0704-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ormsby MJ, Logan M, Johnson SA, McIntosh A, Fallata G, Papadopoulou R, Papachristou E, Hold GL, Hansen R, Ijaz UZ, et al. (2019). Inflammation associated ethanolamine facilitates infection by Crohn’s disease-linked adherent-invasive Escherichia coli. EBioMedicine 43, 325–332. 10.1016/j.ebiom.2019.03.071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overbeek R, Olson R, Pusch GD, Olsen GJ, Davis JJ, Disz T, Edwards RA, Gerdes S, Parrello B, Shukla M, et al. (2013). The SEED and the rapid annotation of microbial genomes using subsystems technology (RAST). Nucleic Acids Res. 42, D206–D214. 10.1093/nar/gkt1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson E, Marques TM, O’Sullivan O, Fitzgerald P, Fitzgerald GF, Cotter PD, Dinan TG, Cryan JF, Stanton C, and Ross RP (2015). Streptozotocin-induced type-1-diabetes disease onset in Sprague-Dawley rats is associated with an altered intestinal microbiota composition and decreased diversity. Microbiology (Reading) 161, 182–193. 10.1099/mic.0.082610-0. [DOI] [PubMed] [Google Scholar]
- Pluskal T, Castillo S, Villar-Briones A, and Oresic M (2010). MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinformatics 11, 395. 10.1186/1471-2105-11-395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poretsky R, Rodriguez-R LM, Luo C, Tsementzi D, and Konstantinidis KT (2014). Strengths and limitations of 16S rRNA gene amplicon sequencing in revealing temporal microbial community dynamics. PLoS ONE 9, e93827. 10.1371/journal.pone.0093827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pruesse E, Quast C, Knittel K, Fuchs BM, Ludwig W, Peplies J, and Glöckner FO (2007). SILVA: A comprehensive online resource for quality checked and aligned ribosomal RNA sequence data compatible with ARB. Nucleic Acids Res. 35, 7188–7196. 10.1093/nar/gkm864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, et al. (2012). A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490, 55–60. 10.1038/nature11450. [DOI] [PubMed] [Google Scholar]
- Ranjan R, Rani A, Metwally A, McGee HS, and Perkins DL (2016). Analysis of the microbiome: Advantages of whole genome shotgun versus 16S amplicon sequencing. Biochem. Biophys. Res. Commun. 469, 967–977. 10.1016/j.bbrc.2015.12.083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese AT, and Carmody RN (2019). Thinking outside the cereal box: Noncarbohydrate routes for dietary manipulation of the gut microbiota. Appl. Environ. Microbiol. 85, e02246–18. 10.1128/AEM.02246-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rey FE, Faith JJ, Bain J, Muehlbauer MJ, Stevens RD, Newgard CB, and Gordon JI (2010). Dissecting the in vivo metabolic potential of two human gut acetogens. J. Biol. Chem. 285, 22082–22090. 10.1074/jbc.M110.117713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhen M (2019). Salmonella and reactive oxygen species: A love-hate relationship. J. Innate Immun. 11, 216–226. 10.1159/000496370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rivera-Chávez F, Zhang LF, Faber F, Lopez CA, Byndloss MX, Olsan EE, Xu G, Velazquez EM, Lebrilla CB, Winter SE, and Bäumler AJ (2016). Depletion of butyrate-producing Clostridia from the gut microbiota drives an aerobic luminal expansion of Salmonella. Cell Host Microbe 19, 443–454. 10.1016/j.chom.2016.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowley CA, Anderson CJ, and Kendall MM (2018). Ethanolamine Influences human commensal Escherichia coli growth, gene expression, and competition with enterohemorrhagic E. coli O157:H7. MBio 9, 1–5. 10.1128/mBio.01429-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabatino A, Regolisti G, Cosola C, Gesualdo L, and Fiaccadori E (2017). Intestinal microbiota in type 2 diabetes and chronic kidney disease. Curr Diab. Rep. 17, 16. 10.1007/s11892-017-0841-z. [DOI] [PubMed] [Google Scholar]
- Saeedi P, Petersohn I, Salpea P, Malanda B, Karuranga S, Unwin N, Colagiuri S, Guariguata L, Motala AA, Ogurtsova K, et al. (2019). Global and regional diabetes prevalence estimates for 2019 and projections for 2030 and 2045: Results from the International Diabetes Federation Diabetes Atlas, 9th edition. Diabetes Research and Clinical Practice. 157, 107843. 10.1016/j.diabres.2019.107843. [DOI] [PubMed] [Google Scholar]
- Schrimpe-Rutledge AC, Codreanu SG, Sherrod SD, and McLean JA (2016). Untargeted metabolomics strategies-challenges and emerging directions. J. Am. Soc. Mass Spectrom. 27, 1897–1905. 10.1007/s13361-016-1469-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Izard J, Waldron L, Gevers D, Miropolsky L, Garrett WS, and Huttenhower C (2011). Metagenomic biomarker discovery and explanation. Genome Biol. 12, R60. 10.1186/gb-2011-12-6-r60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segata N, Waldron L, Ballarini A, Narasimhan V, Jousson O, and Huttenhower C (2012). Metagenomic microbial community profiling using unique clade-specific marker genes. Nat. Methods 9, 811–814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin A, Preidis GA, Shulman R, and Kashyap PC (2019). The gut microbiome in adult and pediatric functional gastrointestinal disorders. Clin. Gastroenterol. Hepatol. 17, 256–274. 10.1016/j.cgh.2018.08.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smits SA, Leach J, Sonnenburg ED, Gonzalez CG, Lichtman JS, Reid G, Knight R, Manjurano A, Changalucha J, Elias JE, et al. (2017). Seasonal cycling in the gut microbiome of the Hadza hunter-gatherers of Tanzania. Science 357, 802–806. 10.1126/science.aan4834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenburg JL, Xu J, Leip DD, Chen C-H, Westover BP, Weatherford J, Buhler JD, and Gordon JI (2005). Glycan foraging in vivo by an intestine-adapted bacterial symbiont. Science 307, 1955–1959. 10.1126/science.1108397. [DOI] [PubMed] [Google Scholar]
- Srikumar S, and Fuchs TM (2011). Ethanolamine utilization contributes to proliferation of Salmonella enterica serovar Typhimurium in food and in nematodes. Appl. Environ. Microbiol. 77, 281–290. 10.1128/AEM.01403-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinert RE, Lee Y-K, and Sybesma W (2020). Vitamins for the gut microbiome. Trends Mol. Med. 26, 137–140. 10.1016/j.molmed.2019.11.005. [DOI] [PubMed] [Google Scholar]
- Stokes JM, Lopatkin AJ, Lobritz MA, and Collins JJ (2019). Bacterial metabolism and antibiotic efficacy. Cell Metab. 30, 251–259. 10.1016/j.cmet.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanes C, Bittinger K, Gao Y, Friedman ES, Nessel L, Paladhi UR, Chau L, Panfen E, Fischbach MA, Braun J, et al. (2021). Role of dietary fiber in the recovery of the human gut microbiome and its metabolome. Cell Host Microbe 29, 394–407.e5. 10.1016/j.chom.2020.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang M, Etokidem E, and Lai K (2016). The Leloir pathway of galactose metabolism – A novel therapeutic target for hepatocellular carcinoma. Anticancer Res. 36, 6265–6272. 10.21873/anticanres.11221. [DOI] [PubMed] [Google Scholar]
- Taylor SJ, and Winter SE (2020). Salmonella finds a way: Metabolic versatility of Salmonella enterica serovar Typhimurium in diverse host environments. PLoS Pathog. 16, e1008540. 10.1371/journal.ppat.1008540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thaiss CA, Levy M, Grosheva I, Zheng D, Soffer E, Blacher E, Braverman S, Tengeler AC, Barak O, Elazar M, et al. (2018). Hyperglycemia drives intestinal barrier dysfunction and risk for enteric infection. Science 359, 1376–1383. 10.1126/science.aar3318. [DOI] [PubMed] [Google Scholar]
- Theriot CM, Bowman AA, and Young VB (2016). Antibiotic-induced alterations of the gut microbiota alter secondary bile acid production and allow for Clostridium difficile spore germination and outgrowth in the large intestine. mSphere. 1, e00045–15. 10.1128/mSphere.00045-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Theriot CM, and Young VB (2015). Interactions Between the Gastrointestinal Microbiome and Clostridium difficile. Annu. Rev. Microbiol. 69, 445–461. 10.1146/annurev-micro-091014-104115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiennimitr P, Winter SE, Winter MG, Xavier MN, Tolstikov V, Huseby DL, Sterzenbach T, Tsolis RM, Roth JR, and Bäumler AJ (2011). Intestinal inflammation allows Salmonella to use ethanolamine to compete with the microbiota. Proc. Natl. Acad. Sci. USA 108, 17480–17485. 10.1073/pnas.1107857108/-/DCSupplemental/pnas.201107857SI.pdf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas VC, Kinkead LC, Janssen A, Schaeffer CR, Woods KM, Lindgren JK, Peaster JM, Chaudhari SS, Sadykov M, Jones J, et al. (2013). A dysfunctional tricarboxylic acid cycle enhances fitness of Staphylococcus epidermidis during β-lactam stress. MBio 4, e00437–e13. 10.1128/mBio.00437-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson LR, Sanders JG, McDonald D, Amir A, Ladau J, Locey KJ, Prill RJ, Tripathi A, Gibbons SM, Ackermann G, et al. ; Earth Microbiome Project Consortium (2017). A communal catalogue reveals earth’s multiscale microbial diversity. Nature 551, 457–463. 10.1038/nature24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian L, Wang X-W, Wu A-K, Fan Y, Friedman J, Dahlin A, Waldor MK, Weinstock GM, Weiss ST, and Liu Y-Y (2020). Deciphering functional redundancy in the human microbiome. Nat. Commun. 11, 6217. 10.1038/s41467-020-19940-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Townsend GE 2nd, Han W, Schwalm ND 3rd, Hong X, BencivengaBarry NA, Goodman AL, and Groisman EA (2020). A master regulator of Bacteroides thetaiotaomicron gut colonization controls carbohydrate utilization and an alternative protein synthesis factor. MBio 11, e03221–19. 10.1128/mBio.03221-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzin V, and Galili G (2010). The biosynthetic pathways for shikimate and aromatic amino acids in Arabidopsis thaliana. Arabidopsis Book 8, e0132. 10.1199/tab.0132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ugarte M, Brown M, Hollywood KA, Cooper GJ, Bishop PN, and Dunn WB (2012). Metabolomic analysis of rat serum in streptozotocin-induced diabetes and after treatment with oral triethylenetetramine (TETA). Genome Med. 4, 35. 10.1186/gm334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ussar S, Haering M-F, Fujisaka S, Lutter D, Lee KY, Li N, Gerber GK, Bry L, and Kahn CR (2017). Regulation of glucose uptake and enteroendocrine function by the intestinal epithelial insulin receptor. Diabetes 66, 886–896. 10.2337/db15-1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vacca M, Celano G, Calabrese FM, Portincasa P, Gobbetti M, and De Angelis M (2020). The Controversial role of human gut Lachnospiraceae. Microorganisms 8, 573. 10.3390/microorganisms8040573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer M, Esders S, Farquharson FM, Neugart S, Duncan SH, Schreiner M, Louis P, Maul R, and Rohn S (2018). Mutual interaction of phenolic compounds and microbiota: Metabolism of complex phenolic apigenin-C- and kaempferol-O-derivatives by human fecal samples. J. Agric. Food Chem. 66, 485–497. 10.1021/acs.jafc.7b04842. [DOI] [PubMed] [Google Scholar]
- Wang C-Y, and Liao JK (2011). A mouse model of diet-induced obesity and insulin resistance. In Mouse Models of Innate Immunity, Methods in Molecular Biology, Allen IC, ed. (Totowa, NJ: Humana Press; ), pp. 421–433. 10.1007/978-1-61779-430-8_27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Garrity GM, Tiedje JM, and Cole JR (2007). Naive Bayesian classifier for rapid assignment of rRNA sequences into the new bacterial taxonomy. Appl. Environ. Microbiol. 73, 5261–5267. 10.1128/AEM.00062-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, et al. (2011). Metabolite profiles and the risk of developing diabetes. Nat. Med. 17, 448–453. 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Carver JJ, Phelan VV, Sanchez LM, Garg N, Peng Y, Nguyen DD, Watrous J, Kapono CA, Luzzatto-Knaan T, et al. (2016). Sharing and community curation of mass spectrometry data with Global Natural Products Social Molecular Networking. Nat. Biotechnol. 34, 828–837. 10.1038/nbt.3597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Xia K, Yang X, and Tang C (2019a). Growth strategy of microbes on mixed carbon sources. Nat. Commun. 10, 1279. 10.1038/s41467-019-09261-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang M, Firrman J, Liu L, and Yam K (2019b). A review on flavonoid apigenin: Dietary intake, ADME, antimicrobial effects, and interactions with human gut microbiota. BioMed Res. Int. 2019, 7010467. 10.1155/2019/7010467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westfall S, Lomis N, Singh SP, Dai SY, and Prakash S (2015). The gut microflora and its metabolites regulate the molecular crosstalk between diabetes and neurodegeneration. J. Diabetes Metab. 06, 1000577. 10.4172/2155-6156.1000577. [DOI] [Google Scholar]
- Westreich ST, Treiber ML, Mills DA, Korf I, and Lemay DG (2018). SAMSA2: A standalone metatranscriptome analysis pipeline. BMC Bioinformatics 19, 175. 10.1186/s12859-018-2189-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter SE, Thiennimitr P, Winter MG, Butler BP, Huseby DL, Crawford RW, Russell JM, Bevins CL, Adams LG, Tsolis RM, et al. (2010). Gut inflammation provides a respiratory electron acceptor for Salmonella. Nature 467, 426–429. 10.1038/nature09415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Witten IH, and Bell TC (1991). The zero-frequency problem: Estimating the probabilities of novel events in adaptive text compression. IEEE Trans. Inf. Theory 37, 1085–1095. [Google Scholar]
- Wood DE, Lu J, and Langmead B (2019). Improved metagenomic analysis with Kraken 2. Genome Biol. 20, 257. 10.1186/s13059-019-1891-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, and Yan L-J (2015). Streptozotocin-induced type 1 diabetes in rodents as a model for studying mitochondrial mechanisms of diabetic β cell glucotoxicity. Diabetes Metab. Syndr. Obes. 8, 181–188. 10.2147/DMSO.S82272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao L, Sonne SB, Feng Q, Chen N, Xia Z, Li X, Fang Z, Zhang D, Fjære E, Midtbø LK, et al. (2017). High-fat feeding rather than obesity drives taxonomical and functional changes in the gut microbiota in mice. Microbiome 5, 43. 10.1186/s40168-017-0258-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang M, Liu Y, Xie H, Wen Z, Zhang Y, Wu C, Huang L, Wu J, Xie C, Wang T, et al. (2019). Gut microbiota composition and structure of the Ob/Ob and Db/Db mice. Int. J. Endocrinol. 2019, 1394097. 10.1155/2019/1394097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin R, Xue Y, Hu J, Hu X, and Shen Q (2020). The effects of diet and streptozotocin on metabolism and gut microbiota in a type 2 diabetes mellitus mouse model. Food Agric. Immunol. 31, 723–739. 10.1080/09540105.2020.1761302. [DOI] [Google Scholar]
- Yoon MY, and Yoon SS (2018). Disruption of the gut ecosystem by antibiotics. Yonsei Med. J. 59, 4–12. 10.3349/ymj.2018.59.1.4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshii K, Hosomi K, Sawane K, and Kunisawa J (2019). Metabolism of dietary and microbial vitamin B family in the regulation of host immunity. Front. Nutr. 6, 48. 10.3389/fnut.2019.00048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng MY, Inohara N, and Nuñ ez G (2017). Mechanisms of inflammationdriven bacterial dysbiosis in the gut. Mucosal Immunol. 10, 18–26. 10.1038/mi.2016.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Lv X-Y, Li J, Xu ZG, and Chen L (2008). The characterization of high-fat diet and multiple low-dose streptozotocin induced type 2 diabetes rat model. J. Diabetes Res. 2008, 704045. 10.1155/2008/704045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J, Kobert K, Flouri T, and Stamatakis A (2014). PEAR: a fast and accurate Illumina Paired-End reAd mergeR. Bioinformatics 30, 614–620. 10.1093/bioinformatics/btt593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Li Q, Cheng L, Buch H, and Zhang F (2019). Akkermansia muciniphila is a promising probiotic. Microb. Biotechnol. 12, 1109–1125. 10.1111/1751-7915.13410. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Illumina sequencing read data have been deposited at the NCBI Short Read Archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. LC-MS/MS and GNPS data have been deposited to MassIVE: https://massive.ucsd.edu and GNPS: https://gnps.ucsd.edu, respectively, and are publicly available as of the date of publication. DOIs are listed in the key sources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| Reagent or resouce | Source | Identifier |
|---|---|---|
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Salmonella enterica Typhimurium SL1344 | Vanessa Sperandio, PhD (Univerisity of Texas, Southwestern) | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Acetone, LCMS-grade | Fisher Scientific | K50738120840 |
| Acetonitrile, LCMS-grade | Fisher Scientific | PI86188 |
| Agar | Fisher Scientific | BP1423-2 |
| Agarose | Fisher Scientific | 14-223-040 |
| Agencourt AMPure XP Beads | Beckman Coulter | A63880 |
| Ammonium Fluoride | Sigma Aldrich | 338869 |
| Amoxicillin | Sigma Aldrich | A8523-5G |
| Deoxynucleotide (dNTP) Solution Mix | New England BioLabs | N0447S |
| Ethanol, 200 proof, molecular biology grade | Fisher Scientific | 07-678-003 |
| Formic acid | Fisher Scientific | RMB11202101 |
| Hematoxylin & Eosin Stain Solution | AbCam | Ab245880 |
| Heparin | Sigma Aldrich | H3393-25KU |
| Hydrochloric Acid | Fisher Scientific | A144-500 |
| Industrial Grade Nitrogen, Liquid | AirGas | NI240LT22 |
| Isopropanol, LCMS-grade | Fisher Scientific | A461-1 |
| Luria Bertani (LB) Broth | Fisher Scientific | BP1426-2 |
| Methacarn | Fisher Scientific | NC0547175 |
| Methanol, LCMS-grade | Fisher Scientific | A456-500 |
| Paraffin Wax | Fisher Scientific | 22-90-700 |
| Phosphate Buffered Saline, 10X | Fisher Scientific | BP399 |
| Sodium Citrate Dihydrate | Fisher Scientific | 5279-500 |
| Streptozotocin | Alfa Aesar | J61601-03 |
| Sucrose | Fisher Scientific | BP220-212 |
| TE Buffer, 1X Solution, pH 8.0, low EDTA | Fisher Scientific | AAJ75793AE |
| Tween20 | Fisher Scientific | BP337-100 |
| Water, LCMS-grade | Fisher Scientific | PI51140 |
| Water, Molecular Biology Grade | Fisher Scientific | BP2891-1 |
|
| ||
| Critical commercial assays | ||
|
| ||
| 10KD Spin Column | AbCam | Ab93349 |
| API-TOF Reference Mass Solution Kit | Agilent | AGG1969-85001 |
| DNA/RNA Shield Collection and Lysis Tube | Zymo Research | R1102 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R&D Systems | DY008 |
| Glucose Assay Kit - Reducing Agent Compatible | AbCam | Ab102517 |
| HiSeq X Ten Reagent Kit v2.5 | Illumina | FC-501-2501 |
| Legendplex Mouse Inflammation Panel 13-plex with Filter Plate | BioLegend | 740150 |
| MICROBExpress Bacterial mRNA Enrichment Kit | Thermo Fisher Scientific | AM1905 |
| MiSeq Reagent Kit v2 (500 cycles) | Illumina | MS-102-2003 |
| Mouse Lipocalin-2/NGAL DuoSet ELISA Kit | R&D Systems | DY1857-05 |
| NEBNEXT® rRNA Depletion Kit (Human/Mouse/Rat) (Includes Purification Beads) | New England BioLabs | E6350 |
| NEBNEXT® Ultra II Directional RNA Seq Library Prep Kit for Illumina (Includes Purification Beads) | New England BioLabs | E7765 |
| NEBNEXT® Ultra II FS DNA Library Prep Kit for Illumina (Includes Purification Beads) | New England BioLabs | E6177 |
| NucleoSpin® Gel and PCR Clean-up Kit | Machery-Nagel GmbH & Co | 740609 |
| Phusion High-Fidelity PCR Kit | Thermo Fisher Scientific | F553L |
| Qubit dsDNA Broad Range Assay Kit | Thermo Fisher Scientific | Q32850 |
| Qubit dsDNA High Sensitivity Assay Kit | Thermo Fisher Scientific | Q32851 |
| Qubit RNA High Sensitivity Assay Kit | Thermo Fisher Scientific | Q32852 |
| Sin-X UF 500 10k MWCO PES Spin Filter | Corning | 431478 |
| Spin-X Centrifuge Tube Flter, 0.22 μM | Costar | 8160 |
| ZymoBiomics Collection Tubes | Zymo Research | S6012-50 |
| ZymoBiomics DNA Miniprep Kit | Zymo Research | D4300 |
| ZymoBiomics DNA/RNA Miniprep Kit | Zymo Research | R2002 |
|
| ||
| Deposited data | ||
|
| ||
| 16S rRNA Reads | NCBI BioProject ID | PRJNA720755 |
| Metagenomic / Metatranscriptomic Reads | NCBI BioProject ID | PRJNA72012 |
| Q-TOF-MS Raw Data | This Study | Table S1 |
| GNPS Molecular Networking Data | GNPS.UCSD.edu | https://gnps.ucsd.edu/ProteoSAFe/status.jsp?task=e4efce0c33fb4ada96e373d53460f2d5 |
| LC-MS/MS Files | Massive.UCSD.edu | MSV000087093 |
| LC-MS/MS Data and Analysis Scripts | This Study | https://github.com/guziordo/Belenky-Brown-Diabetes-Antibiotics |
|
| ||
| Experimental models: Mouse Strains | ||
|
| ||
| C57BL/6 | Jackson Laboratories | Cat.# 000664 |
|
| ||
| Oligonucleotides | ||
|
| ||
| Earth Microbiome Project: 806R | Caporaso et al., 2012 | https://earthmicrobiome.ucsd.edu/protocols-and-standards/primer-ordering-and-resuspension |
| Earth Microbiome Project: 515F with Barcode | Caporaso et al., 2012 | https://earthmicrobiome.ucsd.edu/protocols-and-standards/primer-ordering-and-resuspension |
|
| ||
| Software and algorithms | ||
|
| ||
| BBMap (version 37.96) | Bushnell, 2014 | https://sourceforge.net/projects/bbmap |
| Bowtie2 (version 2.2.0) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| Bracken (version 2.0.0) | Lu et al., 2017 | http://ccb.jhu.edu/software/bracken/index.shtml?t=manual |
| BWA-Mem (version 0.7.15) | Li and Durbin, 2010 | http://bio-bwa.sourceforge.net/bwa.shtml |
| DIAMOND (version 0.9.11) | Buchfink et al., 2014 | https://github.com/bbuchfink/diamond |
| Global Natural Products Social Molecular Networking (GNPS) | Wang et al., 2016 | https://gnps.ucsd.edu |
| HUMAnN2 (version 0.11.1) | Franzosa et al., 2018 | https://bitbucket.org/biobakery/humann2/wiki/home |
| Kneaddata (version 0.6.1) | McIver et al., 2018 | https://bitbucket.org/biobakery/kneaddata/wiki/home |
| Kraken (version 2.0.7-beta) | Wood et al., 2019 | https://ccb.jhu.edu/software/kraken2 |
| LDA Effect Size (LEfSe, version) | Segata et al., 2011 | https://huttenhower.sph.harvard.edu/galaxy |
| MetaPhlan2 (version) | Segata et al., 2012 | https://bitbucket.org/biobakery/metaphlan2 |
| MATLAB | MathWorks | https://www.mathworks.com/products/matlab.html |
| MATLAB Toolbox: Bioinformatics | MathWorks | https://www.mathworks.com/products/bioinfo.html |
| MZmine (version 2.52) | Pluskal et al., 2010 | http://mzmine.github.io/ |
| Paired-End Read Merger (PEAR; version 0.9.12) | Zhang et al., 2014 | https://cme.h-its.org/exelixis/web/software/pear |
| Prism (version 9.0.2) | GraphPad | https://www.graphpad.com/scientific-software/prism |
| R (version 4.0.3) | The R Project for Statistical Computing | https://www.r-project.org |
| R package: DADA2 (version 1.8.0) | Callahan et al., 2016 | https://bioconductor.org/packages/release/bioc/html/dada2.html |
| R package: DESeq2 (version 1.26.1) | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| R package: phyloseq (version 1.28.0) | McMurdie and Holmes, 2013 | https://bioconductor.org/packages/release/bioc/html/phyloseq.html |
| R package: randomForest (version 4.6-16) | Breiman, 2001 | https://cran.r-project.org/web/packages/randomForest/index.html |
| R package: vegan (version 2.5-7) | https://cran.r-project.org/web/packages/vegan/index.html | |
| R package: PAPi | Aggio et al., 2010 | https://www.bioconductor.org/packages//2.12/bioc/html/PAPi.html |
| R package: Mimosa (version 2.0) | Noecker et al., 2016 | https://borenstein-lab.github.io/MIMOSA2shiny |
| Rstudio (version) | Rstudio | https://www.rstudio.com/ |
| SAMSA2 (version 1.0) | Westreich et al., 2018 | https://github.com/transcript/samsa2 |
| Subread (featureCounts) (version 1.6.2) | Liao et al., 2014 | https://bioinf.wehi.edu/au/featureCounts |
| Trimmomatic (version 0.36) | Bolger et al., 2014 | https://www.usadellab.org/cms?page=trimmomatic |
|
| ||
| Other | ||
|
| ||
| 6550 iFunnel Q-TOF LC/MS | Agilent | G6550BA |
| Accuspin Micro17 Microcentrifuge | Fisher Scientific | 13-100-675 |
| Acquity UPLC columns, BEH C18, 1.7 μM (2.1×100mm) | Waters | 186002352 |
| Attune NxT Flow Cytometer | Invitrogen | N/A |
| ChemiDoc MP Imaging System | BioRad | 12003154 |
| Class II Type A2 Biological Safety Cabinet | Labguard | NU-540 |
| CleanPrep PCR Workstation | MyStaire | MY-DB24 |
| ContourNext® EZ Glucose Meter | Contour | N/A |
| ContourNext® EZ Glucose Meter Strips | Contour | N/A |
| Electrospray Ionization Source | Agilent | G1948B |
| GyroMax 737 Incubator | Amerex | N/A |
| Insulin Syringe, 0.5mL, 28G | BD | 329461 |
| Insulin Syringe, 1mL, 26G | BD | 329652 |
| Laboratory Rodent Diet 5001 | LabDiet | 0001319 |
| Lo-Bind 96-well plate, skirted | Eppendorf | 30129512 |
| Lo-Bind Microcentrifuge Tubes, nuclease-free | Eppendorf | 22431021 |
| Polypropylene Feeding Tubes, 20 ga × 30mm | Instech | FTP-20-38 |
| QE Basic, Exactive Hybrid Quadrupole-Orbitrap MS | Thermo Fisher Scientific | IQLAAEGAAPFALGMBDK |
| Qubit 3.0 Fluorometer | Thermo Fisher Scientific | Q33216 |
| SpectraMax M3 Multi-Mode Microplate Reader | Molecular Devices | 89429-536 |
| T100 Thermal Cycler | BioRad | 1861096 |
| Vanquish Autosampler | Thermo Fisher Scientific | 8308123 |