

Abstract
Motivation
With a large number of metagenomic datasets becoming available, eukaryotic metagenomics emerged as a new challenge. The proper classification of eukaryotic nuclear and organellar genomes is an essential step toward a better understanding of eukaryotic diversity.
Results
We developed Tiara, a deep-learning-based approach for the identification of eukaryotic sequences in the metagenomic datasets. Its two-step classification process enables the classification of nuclear and organellar eukaryotic fractions and subsequently divides organellar sequences into plastidial and mitochondrial. Using the test dataset, we have shown that Tiara performed similarly to EukRep for prokaryotes classification and outperformed it for eukaryotes classification with lower calculation time. In the tests on the real data, Tiara performed better than EukRep in analyzing the small dataset representing eukaryotic cell microbiome and large dataset from the pelagic zone of oceans. Tiara is also the only available tool correctly classifying organellar sequences, which was confirmed by the recovery of nearly complete plastid and mitochondrial genomes from the test data and real metagenomic data.
Availability and implementation
Tiara is implemented in python 3.8, available at https://github.com/ibe-uw/tiara and tested on Unix-based systems. It is released under an open-source MIT license and documentation is available at https://ibe-uw.github.io/tiara. Version 1.0.1 of Tiara has been used for all benchmarks.
Supplementary information
Supplementary data are available at Bioinformatics online.
1 Introduction
Microbial communities of unicellular eukaryotes (protists) and prokaryotes are an essential part of all ecosystems. Along with prokaryotes, protists are significant drivers in diverse nutrient cycling pathways (Worden et al., 2015). Autotrophic and mixotrophic protists fix carbon in aquatic environments, whereas heterotrophic protists catalyze nutrient cycling in aquatic and terrestrial ecosystems as selective consumers of bacteria and fungi (Caron et al., 2009).
Metagenomic studies changed our understanding of the prokaryotic communities and allowed us to uncover their taxonomic and functional diversity in various environments (Almeida et al., 2019; Sunagawa et al., 2015). However, even though microeukaryotes are key components of microbial communities, their study lags behind the study of prokaryotes (Keeling et al., 2017), and that is particularly true for the metagenomic studies. Until now, mainly metabarcoding (e.g. de Vargas et al., 2015), metatranscriptomics (e.g. Salazar et al., 2019) and single-cell genome sequencing (e.g. Strassert et al., 2018) were used to explore the diversity of microbial eukaryotes. In contrast to these methods, the utilization of the metagenomic approaches was hampered by the complexity and size of eukaryotic genomes, as well as a limited number of reference databases allowing further taxonomical or functional annotation. With a few exceptions, such as phytoplankton (Delmont et al., 2015; Duncan et al., 2020) or human microbiome (Olm et al., 2019) studies, eukaryotes have been rarely analyzed in metagenomic studies and neglected in some environments such as freshwaters or soil. Only recently, the metagenomic datasets from large sampling projects, such as the Tara Oceans expedition (Pesant et al., 2015) or Ocean Sampling Day (Kopf et al., 2015), were exploited to uncover the eukaryotic plankton biogeography (Leconte et al., 2020; Richter et al., 2020), taxonomy (Obiol et al., 2020) and functional diversity (Delmont et al., 2020). Metagenomic data often do not contain a sufficient amount of data to reconstruct nuclear genomes, but mitochondrial and plastid genomes, owing to their smaller size and a higher number of copies, maybe potentially reconstructed from those data. Most often the mitochondrial genomes (Andújar et al., 2015; Crampton-Platt et al., 2016) or only single genes, such as 16S rDNA (Piganeau et al., 2008; Piganeau and Moreau, 2007), are reconstructed from the metagenomic data. Organellar genomes have been shown to provide suitable data to address questions about microbial eukaryotes’ evolution and ecology (Cuvelier et al., 2010; Kim et al., 2011; Wideman et al., 2020). However, organellar data are mostly unexplored in the metagenomes, since they are often classified as bacterial sequences, and are thus removed from the eukaryotic genome assemblies (Delmont et al., 2020; Duncan et al., 2020). The main reason for this misclassification is the similarity of organellar genomes to bacterial genomes. Both mitochondria and plastids originated via endosymbiosis with bacteria, which results in overlapping gene content to bacterial genomes (Sibbald and Archibald, 2020).
Only a few approaches dedicated to the processing of the eukaryotic fraction from the metagenomic data exist. They might be split into those developed to analyze raw reads (Wood et al., 2019) or single genes (Schön et al., 2020), both of which strongly depends on the reference databases. Alternatively, the eukaryotic nuclear genomes might be reconstructed from the data using one of the two main existing pipelines. In the first approach, the assembled contigs are binned, visualized and manually refined using Anvio’O (Delmont and Eren, 2016; Eren et al., 2015), whereas the second approach, used in EukRep, assumes initial separation of contigs into two domains (Prokarya and Eukarya), and then binning within those two groups independently (West et al., 2018). Both approaches have been successfully used for obtaining partial nuclear eukaryotic genomes but failed to correctly classify the organellar fraction in the metagenomic data (Delmont et al., 2020; Duncan et al., 2020). Only one tool, MitoZ, was designed explicitly for the organellar data, but it is only applicable for the assembly, identification and analysis of the animals’ mitochondrial genomes (Meng et al., 2019).
The most widely used tools for biological sequence comparison are alignment-based methods such as Smith–Waterman algorithm (Smith and Waterman, 1981) and its further developments such as BLAST (Altschul et al., 1990) or BLAT (Kent, 2002). Several binning algorithms relying on the alignment-based approach, such as taxator-tk (Dröge et al., 2015), have been proposed for the taxonomic assignment of DNA sequences in metagenomes. Although the alignment-based methods are the most accurate for sequence comparisons, they fail if sequences are highly divergent or the reference database is limited (Ren et al., 2018). These methods are also computationally intensive, hence too time-consuming for large NGS genomic and metagenomic datasets (Yang et al., 2020). For those reasons, the use of alignment-based methods for metagenomic data is relatively confined. Alignment-free methods, based on k-mers or DNA substrings, provide promising alternatives to overcome the weaknesses of alignment-based methods (Ren et al., 2018). The usage of alignment-free methods is currently rapidly growing, and especially machine learning approaches have been used extensively for classification of various types of sequences from metagenomes (Krawczyk et al., 2018; Liang et al., 2020). The machine learning methods can leverage vast datasets to detect hidden structures and make accurate predictions. Their advantage is the ability to make predictions without strong assumptions about mechanisms underlying the biological data. The most promising approaches are based on deep learning, a family of machine learning methods exploiting artificial neural networks. They allow exploring large and multidimensional datasets (such as metagenomic data) by training complex networks with multiple layers. The learned networks perform better than traditional models and can discover high-level features (Angermueller et al., 2016).
The most broadly used tool for the eukaryotic metagenomics is EukRep, which uses k-mer frequencies and linear SVMs for DNA sequences classification (West et al., 2018). It was shown to be useful for obtaining high-quality nuclear eukaryotic genomes from complex environmental samples (West et al., 2018), but lacks features which would enable proper organellar genomes classification. Here, we introduce Tiara, a deep-learning-based approach for identification of eukaryotic sequences in the metagenomic datasets. Its two-step classification process enables to classify nuclear and organellar eukaryotic fractions and subsequently divide organellar data into plastidial and mitochondrial classes. Tiara outperforms EukRep in terms of prediction accuracy and calculation time.
2 Materials and methods
Tiara is designed to classify assembled DNA sequences (contigs) into classes representing genomes of different origins (Fig. 1). In the first step, it classifies sequences into six classes: three representing prokaryotes (Archaea, Bacteria and Prokarya—sequences not distinguishable between Bacteria and Archaea), two representing eukaryotes (Eukarya—nuclear genomes and organelle—mitochondrial or plastidial genomes) and unknowns for sequences which could not be precisely classified. In the subsequent step, the organellar sequences are further classified into classes representing plastidial genomes, mitochondrial genomes and unknowns for unclassified.
Fig. 1.
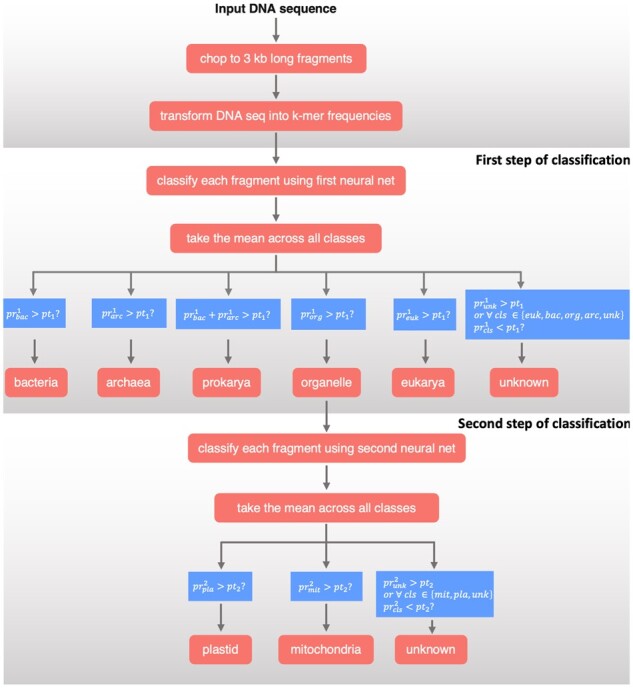
Scheme of the main steps of the dataflow implemented in Tiara
2.1 Training and test datasets
We took advantage of taxonomic information to emerge well balanced and not overlapping training and test datasets. We independently picked genomes for each category to obtain a diverse final dataset despite differences in genomes’ lengths and their representation in reference databases. The prepared dataset containing genomes from three domains of life was subsequently divided into non-overlapping training and test set. We downloaded data from the NCBI Genome database (Sayers et al., 2019) and the Joint Genome Institute (JGI; Grigoriev et al., 2012); the NCBI taxonomy was used to describe the data (Supplementary Tables S1 and S2). We prepared the training dataset based on 8220 genomic sequences (Supplementary Table S1) representing Eukarya (4381) [nuclear (73), plastid (2260) and mitochondrial genomes (2048)], Bacteria (1860) and Archaea (1979). Subsequently, for all genome sequences in the training dataset, 5 kb fragments were generated by splitting. Furthermore, 10% of fragments per bacterial genome were randomly picked to reduce the bacterial dataset size, but not diversity. The resulting training dataset contained a comparable number of genome fragments for prokaryotes and eukaryotes. Although we used many more prokaryotic genomes, their size was much smaller, so the overall ratio between prokaryotic and eukaryotic data (number of genomic fragments) in the training set was 3:2 (Supplementary Table S3). That allowed to achieve a better balance between the total number of fragments representing prokaryotes and eukaryotes classes. Fragments containing other letters than {A, T, G, C} were filtered out.
The test set of 550 genomes (Supplementary Table S2) contained 165 eukaryotic genomes (105 nuclear, 28 plastidial and 32 mitochondrial) and 385 prokaryotic genomes (306 Bacteria and 79 Archaea). Genomes selected as test set were not present in the EukRep training dataset or the Tiara training set and were selected with maximum overlap with the training genomes set at the genus level. The genomes with less than 20 contigs were chopped into 100 kb long chunks, and less contiguous assemblies remained unchanged to reflect the condition of metagenomic assemblies. Mitochondrial and plastidial genomes were fragmented into pieces in a range of 1–75 kb (Supplementary Methods).
Eukaryotic nuclear genomes for both datasets were chosen manually and included genomes from large groups of eukaryotes representing all supergroups (sensu Burki et al., 2020): TSAR (Stramenopila, Alveolata, Rhizaria), Archaeplastida (Chloroplastida, Rhodophyta and Glaucophyta), Amorphea (Amoebozoa, Opisthokonta, Apusomonada), Haptista, Cryptista, Discoba and Metamonada. Overall, 178 nuclear genomes were selected (73 for training and 105 for test datasets). The test dataset (Supplementary Table S2) contained genomes belonging to groups such as Cryptista, Haptista, Metamonada and Glaucophyta, not included in the training dataset, to test Tiara’s ability to classify divergent genomic sequences correctly. Mitochondrial genomes marked as: ‘Fungi’, ‘Plants’, ‘Protist’, ‘Other Animals’, ‘Insects’ and ‘Other’ were downloaded from NCBI. In the case of animal mitochondrial genomes, we have chosen only two of the categories mentioned above, which represented this large group of small and homogeneous genomes in the best possible manner. For plastidial genomes representation in training datasets, we downloaded all available plastid genomes annotated as ‘Green algae’, ‘Protist’ and ‘Other’ in NCBI and one representative per each genus of ‘Land Plants’ to avoid overrepresentation of plants’ plastid genomes. Additional representatives of land plants genera were included in the test dataset. In the case of bacterial genomes, we selected one representative from each genus present in the NCBI database with the best assembly quality. Due to the insufficient number of complete archaeal genomes and a dominant number of low-quality genomes derived from metagenomic initiatives, we used the best quality genomes for each archaeal species present in the NCBI database. Selected representatives of large groups of Bacteria such as Candidate Phyla Radiation were placed solely in the test dataset.
2.2 Sequence representation
The tf-idf (short for term frequency–inverse document frequency) weighting scheme (Sammut and Webb, 2010) is commonly used in natural language processing (Yun-tao et al., 2005; Arroyo-Fernández et al., 2019) and information retrieval (Ramos, 2003). The tf-idf is a numerical statistic that summarizes two intuitions: that words common in a document are representative for this particular document and that words present in many documents are less informative than those unique for particular documents (Sebastiani, 2002). Here, we used the tf-idf to represent DNA sequences based on the analogy between our approach and text processing, where we treat DNA sequences as documents made of words (k-mers).
Each DNA sequence fragment was represented as a real-valued vector of length, where was the k-mer length. Let be a set of 5 kb non-overlapping sequence fragments coming from all DNA sequences used for training. Given , we defined as the oligonucleotide (k-mer) frequency vector for a sequence and as a vector describing the inverse document frequency of each k-mer:
where is equal to the number of sequence fragments that contain th k-mer (in the lexicographic order). Then the representation of the sequence was calculated as
where is the pointwise multiplication of vectors. The vectors are then normalized to sum to one. The effect of this representation is that k-mers that occur in many DNA fragments weigh less to the prediction, compared to k-mers present in only a few DNA fragments.
We have written our version of oligonucleotide frequency () calculation and an vector calculation method that works online (processing one sequence at a time).
2.3 Classification system
We used a two-stage classification method. In the first stage, the input sequences are classified into six classes: bacteria, archaea, prokaryote, eukaryote, organelle or unknown. The second stage differentiates between organelle subclasses: mitochondria, plastids and unknown (Fig. 1). This two-stage process relies on the two distinct two-layer feed-forward neural network architectures. Hyperparameter selection and training procedure are described in subsection 2.4.
During the classification process, we split the sequences into smaller fragments (5 kb). We then classify each fragment separately and take the mean probability for each class, resulting in five (in the first step of classification) or three (in the second step) values. We use notation to describe the mean output of the neural network at stage for a specific class. The classification is performed based on the probability thresholds, one for each classification stage. The threshold of probability, is set by the user in the range between 0.2 and 0.99. In the first stage, if the probability of a given class is higher than pt1, the sequence is assigned to this class (bacteria, archaea, eukarya or organelle). If it is lower, then the sequence is assigned as unknown, unless the sum of probabilities of bacteria and archaea is higher than, then the sequence is assigned to a more general class prokarya. In the second stage, if the probability of a given class of organellar sequences is higher than a, the sequence is assigned to this class (mitochondria or plastid), and if it is lower, the sequence is classified as unknown (Fig. 1).
2.4 Neural network architectures
2.4.1. Training
We implemented and trained our models using PyTorch (Paszke et al., 2019) and skorch (Tietz et al., 2017) packages using negative log-likelihood loss and an Adam optimizer (Kingma and Ba, 2015). We split the data into training (90%) and validation (10%) sets using a stratified splitting strategy: in each set, the proportion of sequences from each class was the same. The batch size was set to 128. A validation dataset was used to determine the best model.
2.4.2. Choosing the best models
To choose the best architectures, we performed a search over several hyperparameters: lengths of k-mers, number of nodes in the first and second neural network layer, dropout probability, learning rate and the number of epochs. We used several metrics to compare the models: accuracy, mean precision, mean recall and mean F1 score (Supplementary Methods). The means were taken across all classes. We evaluated the models on a validation set with a probability threshold of 0.5. For consistency, we picked the architecture with the highest average mean F1 score across all learning epochs. The results of the search are in Supplementary Tables S4 and S5. To choose the architectures for the first stage, 15 900 hyperparameter combinations were tested, whereas for the second stage 17 600 combinations were tested.
2.5 Implementation and availability
We developed our tool in Python 3.8, with use of the libraries skorch (Tietz et al., 2017), PyTorch (Paszke et al., 2019), biopython (Cock et al., 2009), numba (Lam et al., 2015), joblib (Varoquaux and Grisel, 2009) and tqdm. Tiara code is freely available under MIT license, and the code is stored on GitHub (https://github.com/ibe-uw/tiara). The models used in the program by default are the best models for each class (marked in bold in Supplementary Table S6), but the user can choose other optimal models for each k-mer length. By default, Tiara returns tabular output with a class assigned to each contig name but optionally allowing the user to output classified sequences to separate files in fasta format.
3 Results
We implemented in Tiara, a two-stage approach for classification of eukaryotic sequences from assembled metagenomic data. In the first stage, Tiara classifies sequences into six categories (Archaea, Bacteria, Prokarya, Eukaryota, organelles or unknown). In the second stage, putative organellar sequences are classified into plastids and mitochondria or unknown. Each stage of classification encapsulates trained neural network model. Next, we evaluated its performance and compared with EukRep using independent test dataset and showed usability using real metagenomic data.
3.1 Performance comparison of different k-mer sizes
We searched hyperparameter space to obtain the best neural network models (nearly 35 000 models) using a validation dataset. The best hyperparameters for each k-mer length and classification stage (k = {4, 5, 6} for the first stage and k = {5, 6, 7} for the second stage) for an optimal number of epochs are shown in Supplementary Table S6. The best first stage neural network was trained with k-mer 6, and had two layers with 2048 and 1024 nodes, respectively. The best neural network in the second stage of classification used a k-mer 7 and had two layers with 128 and 64 nodes. For both stages, we used the dropout probability of 0.2. The first best model was trained for 41 epochs using a learning rate equal to 0.001, and the second for 47 epochs with a learning rate of 0.01. The comparison of the best models for each k-mer length (in bold) with a sub-optimal architecture (both layer sizes equal to 32, learning rate of 0.01 and 0.5 dropout probability—in italics) shows that larger neural networks are necessary to identify the biological signal present in the DNA sequences (Supplementary Table S6).
3.2 Probability threshold impact on the classification results
Probability thresholds pti is a parameter that can be set by the user, and its value might impact the classification accuracy to specific classes. We tested five pt1 and pt2 values ranging from 0.35 up to 0.95 to evaluate the accuracy of the Tiara classification (see Supplementary Methods) and choose the parameter for further analyses. The results (Supplementary Fig. S1; Supplementary Table S7) indicate that the higher the pt1, the more accurate is the classification to the eukaryote class because more sequences with low probabilities of classification to other classes end up in the class unknown. On the other hand, while increasing the probability threshold, classification to archaea and bacteria is achieved with lower accuracy. The pt2 value used in the second step of classification had no strong effect on the organellar classification. Based on those results, we set the default value of pti to 0.65. Moreover, Tiara allows to output probabilities for each DNA fragment, which could be parsed to answer specific questions.
3.3 Performance of trained models
3.3.1. Classification of eukaryotic and prokaryotic sequences
Depending on the k-mer length, Tiara achieved mean prediction accuracy between 98.65% and 98.93% for prokaryotic genomes and between 95.94% and 98.83% for nuclear genomes on the test dataset (Table 1).
Table 1.
Comparison of accuracy for Tiara and EukRep tools
| Software | k-mer | Average accuracy |
||
|---|---|---|---|---|
| Eukarya (n = 105) | Prokarya (n = 385) | organelles (n = 60) | ||
| Tiara | 4 | 0.9593 | 0.9871 | 0.9411mt/0.9708pt |
| Tiara | 5 | 0.9641 | 0.9865 | 0.9841mt/0.9981pt |
| Tiara | 6 | 0.9883 | 0.9893 | 0.9886mt/0.996pt |
| EukRep | 5 | 0.963 | 0.9849 | 0.4738mt/0.2979pt |
Note: In the case of EukRep, we calculated the ratio of organellar fragments classified as Eukaryote. All Tiara tests have been done with 0.65 probability cutoffs. EukRep was tested with default settings. The best model for a given class is shown in bold.
The best ratio between prediction accuracies for each class was noted for k-mer 6. Using this model, 96% of nuclear eukaryotic and 98% of prokaryotic genomes were classified with higher or equal prediction accuracy to 90%. Only three prokaryotic genomes have been classified with accuracy lower than 50%. However, most of the contigs derived from these genomes were assigned as ‘unknown’, and only two were classified as a eukaryote. All of these genomes were small and highly reduced, and they belonged to symbionts or parasites. This bias had also been previously observed for EukRep (West et al., 2018). We checked probability outputs for them and observed strong organellar signal for two endosymbionts, which might reflect the reductive evolution of their genomes. Importantly, Tiara achieved high accuracies (above 90%) for genomes from groups of taxa that were absent in the training dataset, like haptophytes and cryptophytes or prokaryotic CPR, which indicates that our models are not overfitting despite employing complex neural networks. Hence, Tiara will be able to classify contigs of novel evolutionary lineages correctly.
3.3.2. Classification of organellar sequences
Tiara achieved high prediction accuracy for a test set of organellar genomes (28 plastidial and 32 mitochondrial) with an average accuracy above 95% (Table 1). The best average accuracies were observed for k-mer 6 (pt: 99.60%; mt: 98.86%). Similar to the nuclear genomes’ classification, the accuracy of organellar genomes’ classification increased with k-mer length. (Supplementary Table S8).
3.3.3. DNA sequence length and the robustness of classification
Analysis of selected fragmented (1–75 kb) archaeal, bacterial and eukaryotic genomes showed a clear improvement of classification accuracy with increasing sequence length as it was previously reported for EukRep (West et al., 2018). Moreover, the increase of accuracy related to the length of the sequence seems more stable for Tiara than for EukRep (Supplementary Figs S2–S4 and, Supplementary Table S9). Since organellar genomes have not been classified and tested before, we checked a complete set of genomes and spectra of k-mers (Supplementary Methods). Analysis of fragmented organellar genomes (mitochondria: 1–5 kb, and plastids: 1–75 kb) for k-mers k = {4, 5, 6} showed that prediction accuracy increased with the fragment length (Supplementary Fig. S5). For organellar sequences longer than 3 kb, accuracy was higher than 90% and for sequences longer or equal to 5 kb—close to 100%. In the second stage, most of the sequences were assigned correctly to a given class (with accuracy higher than 90%) if the sequence was longer than 3 kb.
3.3.4. Impact of NUMTs and NUPTs on Tiara classification
We analyzed the classification of NUMTs and NUPTs (mitochondrial and plastidial fragments integrated into nuclear genomes) derived from nuclear genomes of Arabidopsis thaliana, Oryza sativa and Vitis vinifera and compared with the classification of randomly sampled sequences from corresponding genomes. Tiara classified NUPTs and NUMTs fractions as ‘organelle’ fraction more often than random genome fragments (minimum 1.6 times more often and maximum around 50 times). Finally, all differences in the probability of assignment to class ‘organelle’ between NUMTs or NUPTs sets and random fragments were statistically significant according to performed t-tests (see Supplementary Methods, Supplementary Tables S10 and S11).
3.4 Performance comparison between Tiara and EukRep
We compared Tiara with EukRep—a tool designed for the classification of eukaryotic and prokaryotic sequences from metagenomic data (West et al., 2018). EukRep was previously shown to outperform alignment-based methods. Thus, we compared Tiara only with EukRep, which is currently the state-of-the-art method. Finally, it enabled the fast identification of eukaryotic contigs and further forming eukaryotic MAGs (Metagenome Assembled Genomes). EukRep, similarly to Tiara, transforms DNA sequences into k-mer frequencies, but then uses linear-SVM (implemented in scikit-learn) for predictions, whereas Tiara uses sequential feed-forward neural networks. EukRep, as a binary classifier, separates data only into two classes (domains): eukaryotes and prokaryotes. Therefore, EukRep has not been trained on organellar DNA, so it was unclear how it classifies those sequences.
Tiara scored slightly better than EukRep, with the prediction accuracy of eukaryotic genomes 2.53% higher and prokaryotic genomes—0.44% higher (Supplementary Table S2). We also calculated the difference in prediction accuracy between Tiara and EukRep for each genomes pair (Fig. 2) to examine prediction accuracy in details. For eukaryotic genomes (105), Tiara got better accuracies in 63 cases and only for 25 genomes, the results were worse. Whereas prokaryotes were classified more evenly and for 275 genomes, both tools have the same results; in 89 cases, Tiara was better than EukRep, and in 21 cases, it was worse.
Fig. 2.
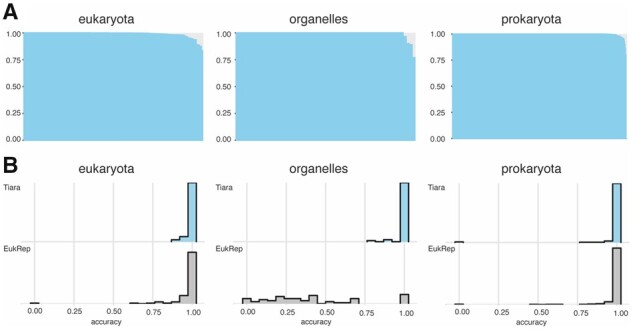
Efficiency of Tiara classification and comparison to EukRep using a set of test genomes. Test genomes were divided into three groups: eukaryotic nuclear genomes (eukaryota), plastid and mitochondrial genomes (organelles) and archaeal and bacterial genomes (prokaryota). (A) Accuracy of Tiara for each genome in three groups (B) Histogram of the density of accuracy for EukRep and Tiara for three groups of genomes. All tests have been performed using the model with k-mer 6 and 0.65 probability cutoff
To test the organellar genomes classification by EukRep, we used a set of fragmented plastid (10 kb) and mitochondrial (5 kb) genomes. EukRep assigned only 47% of mitochondrial and 30% of plastid contigs as eukaryotic sequences (Supplementary Table S2).
In addition, we checked the speed performance of both approaches (Supplementary Methods). Tiara supports parallel execution, whereas EukRep uses all cores available. However, for one core, Tiara was two times faster than EukRep. Finally, Tiara using 12 cores classified the test genome roughly five times faster than EukRep and reached a speed of 6.8 Mbp per second. (Supplementary Table S12).
3.5 Classification of sequences from the real data
3.5.1. Metagenome of Pseudoblepharisma tenue
To compare Tiara and EukRep performance, we used a well-described metagenomic dataset of microbiome of ciliate Pseudoblepharisma tenue (Supplementary Methods). Metagenomic and microscopic analysis of P.tenue proved the presence of two endosymbionts: purple bacteria Ca. Thiodictyon intracellulare (Chromatiales, Gammaproteobacteria) and eukaryotic alga Chlorella sp. (Chlorophyta; Munoz-Gomez et al., 2021). This low-complexity and highly controlled metagenome is a suitable and realistic model for testing the classification of DNA fragments of diverse origin (Bacteria, Eukaryota and organelles). The classification of contigs originated from assembled MAGs showed that Tiara was 10% more accurate than EukRep in classifying the nuclear genome of P. tenue and comparably accurate for endosymbionts’ genomes. Moreover, Tiara correctly predicted the origin of sequences in the mitochondrial fraction (Supplementary Table S13). Classification of total metagenomic data by Tiara revealed sequences previously identified in the mitochondrial fraction (three contigs) and additional five fragments of the plastid genome of Chlorella (around 110 kb in total) not mentioned in the original work (Supplementary Table S14).
3.5.2. Tara Oceans dataset
To test our approach on larger and more complex metagenomic data, we used datasets from the Tara Oceans—a large-scale initiative for studying marine plankton using meta-omics techniques (Pesant et al., 2015). We selected three samples from the same site (station) from the Mediterranean Sea (SRA: ERR1726574, ERR1726673, ERR868402), representing three different size fractions associated with protists (Supplementary Methods, Supplementary Table S15). Data were assembled (Supplementary Methods, Supplementary Table S8) and used for further analyses. We tested Tiara with three k-mer k = {4, 5, 6} and three minimum sequence lengths (1000, 3000, 5000 bp) for the first stage of classification (Supplementary Methods).
In the metagenome of the smallest size fraction (0.8–20 µm), prokaryotes seemed to prevail, as the majority of contigs were classified as Bacteria, Archaea or Prokarya (up to 97% for k = 5); however, datasets from larger size fractions (20–180 and 180–2000 µm) were dominated by eukaryotes (up to 96% for k = 6) (Supplementary Fig. S6 and Table S16). Contribution of contigs assigned to eukaryotes was the highest using model with the k-mer 6, which is in line with the results obtained from the test datasets. The organellar fraction’s overall contribution was low in assembled data and ranged between 0.26% and 4.2% across datasets and analysis variants. Nevertheless, organellar contigs were among the longest ones and exceeded 50 kb for sample ERR1726673 (Supplementary Table S10).
We annotated 21 contigs assigned as organellar and longer than 10 kb (Supplementary Methods). Among those 21 contigs, 13 were annotated as mitochondrial and seven as plastidial, and for one, blastN reported no significant hits (Supplementary Table S17). For the smallest size fraction (0.5-20 µm), all five analyzed contigs were derived from two plastid genomes, belonging to the dictyochophycean Florenciella parvula and the green alga Pycnococcus provasoli. Three fragments of the Pycnococcus plastid genome together accounted for the 58.2% of its estimated size and carried 38 genes. The largest taxonomic diversity of contigs was detected in the size fraction 20–180 µm; organellar genomes of nine protists (diatoms, ciliates) and animals (crustaceans, insects and molluscs) were identified. For the largest size fraction (180–2000 µm), we identified three partial mitochondrial genomes that belonged to animals (crustaceans and hydrozoans).
4 Discussion
We developed Tiara, a machine learning-based tool, which can efficiently and accurately separate eukaryotic sequences from the prokaryotic ones to overcome difficulties with eukaryotic data classification in the metagenomic data. Tiara does not rely on large reference databases and can be efficiently utilized in pipelines for identifying eukaryotic scaffolds and binning into MAGs. Tiara is also the first tool designed to consider organellar sequences as a separate class, allowing their further analyses.
Trained models encapsulated within Tiara scored high accuracy for validation and test dataset, suggesting that models are not overfitting. Moreover, longer k-mers coupled with large networks resulted in the best performance in both classification stages, confirming that complex neural networks can better identify informative signal within DNA sequences. By manipulation of the probability thresholds (pt1 and pt2), it is possible to customize Tiara for different tasks and maximize detection of eukaryotes (by increasing the pt1) or prokaryotes (by decreasing the pt1). The length of sequences is another critical factor affecting the accuracy of classification. For all tested types of genomes, accuracy increased exponentially with the length of fragments in the range between 1 and 10 kb. Thus, we proposed to use a minimum sequence length of 3 kb as default. Lowering this value can increase the number of false positives but might allow detection of rare organisms. On the other hand, increasing this value can speed up the process and minimize the risk of misclassification while losing the information about less-abundant taxa.
Using a test dataset, we have shown that Tiara performs similarly to EukRep (representing the current state-of-the-art) in terms of prokaryotes classification, and outperformed it in terms of classification of eukaryotes with considerably lower calculation time. Tiara was trained on a much larger dataset than EukRep and employed neural networks, which allowed longer k-mer (k = 6) usage and performed better with more complex data. EukRep uses linear-SVMs, which are less effective when dealing with multidimensional data. Crucially, Tiara correctly classifies sequences from organellar genomes (up to nearly 100% of plastid sequences and 99% of mitochondrial sequences). In contrast, EukRep recovered only small portion of organellar fragments (approximately 33%), classifying them as eukaryotic ones (Table 1).
The tests also confirmed the performance of Tiara on the real metagenomic data from Pseudoblepharisma tenue microbiome. Tiara classification accuracy was 10% higher than EukRep for the eukaryotic genome (Supplementary Table S13), and it also successfully identified the organellar fraction present in the data (Supplementary Table S14), which allowed to find a nearly complete plastid genome of Chlorella. Analysis of the metagenomic data from the Mediterranean Sea allowed the classification and reconstruction of the organellar genomes. Tiara classified most of the contigs as nuclear genomes for the fraction larger than 20 μm, and the smallest fraction was dominated by prokaryotes, as already reported in previous studies (Tully et al., 2018). Analysis of a range of k-mers and a sequence length cut-off confirmed that the lowest false-positive rate is achieved for k-mer 6 with a minimum sequence length of 3 kb. Even though organellar fragments constituted less than 10% of contigs, they were among the longest ones. Thanks to Tiara, we reconstructed three partial plastid genomes and twelve almost complete mitochondrial genomes from the Mediterranean Sea dataset. Only one plastid genome was classified to the species level (99% of identity); however, it most likely represents a different strain than those deposited as reference data in databases. For all mitochondrial genomes, the classification was restricted by the NCBI database’s lack of close reference. Six of the identified genomes had only a moderate similarity to crustacean genomes of Undinula vulgaris (∼72%) and Paracyclopina nana (∼76%). This result suggests that we recovered mitochondrial genomes of crustacean species currently not represented in the NCBI database.
Still, some challenges remain. The classification of shorter sequences might be wrong because those sequences are less informative. Thus, we recommend analyzing sequences that are longer than 3 kb to reduce the false-positive rate. The classification of eukaryotic sequences can also be disturbed by the existence of NUMTs and NUPTs—fragments of mitochondrial or plastid genomes localized in nuclear genomes (Kim and Lee, 2018). Those fragments might be misassigned as organellar sequences, which we confirmed by tests on genomes of model species of plants (Supplementary Tables S10 and S11). Another problem constitutes introns and other extremely divergent non-coding regions, which might significantly disturb k-mer frequencies locally, resulting in the wrong prediction if a given sequence is too short to retain distinctive signal. Finally, the relatively high similarity of rDNA operons between groups can result in misclassification of those regions. Thus, we suggest using additional tools like Phyloflash, which is designed to reconstruct and explore the phylogenetic composition of rDNA sequences, to analyze those regions (Gruber-Vodicka et al., 2020).
In the current analysis, the class ‘unknown’ from the first stage of classification has been added to the class ‘eukaryotic’ to maximize eukaryotic sequences’ recovery. This decision was based on the assumption that ambiguous predictions will less likely fall into the class prokaryote since the set of prokaryotic genomes used for training was extensive, diverse and evenly sampled. Consequently, there is a high chance that those fragments belong to eukaryotic genomes. The prokaryotic and viral sequences that might end up in the class ‘unknown’ can also be easily removed during the preprocessing step like binning and bin refinement. However, our assumption might slightly increase the number of false positives in the first stage of classification.
Currently, sequences shorter than the given threshold remain unclassified and should be treated separately using gene-centric approaches. Those sequences are less informative and might significantly increase the number of false positives. However, it is still worth analyzing them to detect mitochondrial and plastid genomes of rare protists.
Despite its advantages, organellar genomes so far have not been widely used compared to metabarcoding or single-cell approaches. We hope that Tiara will enable researchers to make more use of metagenomic data. Organellar data can be employed for phylogenomic reconstruction and uncover new eukaryotic lineages, as already have been shown for mitochondrial genomes of marine heterotrophic protists (Wideman et al., 2020). Organellar sequences, similarly to barcodes, are also applicable for diversity assessment and biogeographic studies. Even partial organellar genomes might be successfully used to study organellar genomes’ structure and content (Cuvelier et al., 2010; Hovde et al., 2014). Ultimately, all these approaches enable a deeper understanding of diversity and evolution of eukaryotes.
The reconstructed underrepresented genomes can be used to supplement existing databases that would further reduce the false positives and allow for more precise classification. Tiara could also be broadly used for metagenomic data preprocessing to remove eukaryotic contamination, including more difficult to distinguish from prokaryotic data organellar sequences.
Our analyses have shown that despite the low proportion of organellar DNA fragments in publicly available metagenomic datasets, Tiara allows us to identify and correctly classify plastidial and mitochondrial sequences and use them for further analyses like phylogenomics, comparative genomics and population genomics. Among analyzed datasets, we were able to identify organellar sequences of previously unreported plastid and mitochondrial genomes. Our results also suggest that even a limited amount of data, insufficient for nuclear genome assembly, could be used to reconstruct almost complete organellar genomes.
Supplementary Material
Acknowledgements
The authors thank A.Z. Worden and C.-M. Yung for access to the genomic data of Minidiscus variabilis. Work on the M. variabilis genome was conducted by the U.S. Department of Energy Joint Genome Institute, a DOE Office of Science User Facility, is supported by the Office of Science of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. They also thank Stanisław Dunin-Horkawicz and Kacper Maciszewski for critical reading of the manuscript and many colleagues for carrying out beta tests of the software.
Funding
This work was supported by the European Molecular Biology Organization [EMBO Installation Grant 4150 to A.K.].
Conflict of Interest: none declared.
Data availability
The data underlying this article are available in the online supplementary materials and from https://github.com/ibe-uw/tiara.
Contributor Information
Michał Karlicki, Institute of Evolutionary Biology, Faculty of Biology & Biological and Chemical Research Centre, University of Warsaw, Warszawa 02-089, Poland.
Stanisław Antonowicz, Institute of Evolutionary Biology, Faculty of Biology & Biological and Chemical Research Centre, University of Warsaw, Warszawa 02-089, Poland.
Anna Karnkowska, Institute of Evolutionary Biology, Faculty of Biology & Biological and Chemical Research Centre, University of Warsaw, Warszawa 02-089, Poland.
References
- Almeida A. et al. (2019) A new genomic blueprint of the human gut microbiota. Nature, 568, 499–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S.F. et al. (1990) Basic local alignment search tool. J. Mol. Biol., 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Andújar C. et al. (2015) Phylogenetic community ecology of soil biodiversity using mitochondrial metagenomics. Mol. Ecol., 24, 3603–3617. [DOI] [PubMed] [Google Scholar]
- Angermueller C. et al. (2016) Deep learning for computational biology. Mol. Syst. Biol, 12, 878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo-Fernández I. et al. (2019) Unsupervised sentence representations as word information series: revisiting TF–IDF. Comput. Speech Lang., 56, 107–129. [Google Scholar]
- Burki F. et al. (2020) The new tree of eukaryotes. Trends Ecol. Evol., 35, 43–55. [DOI] [PubMed] [Google Scholar]
- Caron D.A. et al. (2009) Protists are microbes too: a perspective. ISME J., 3, 4–12. [DOI] [PubMed] [Google Scholar]
- Cock,P.J.A. et al. (2009) Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics, 25, 1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crampton-Platt A. et al. (2016) Mitochondrial metagenomics: letting the genes out of the bottle. Gigascience, 5, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuvelier M.L. et al. (2010) Targeted metagenomics and ecology of globally important uncultured eukaryotic phytoplankton. Proc. Natl. Acad. Sci. USA, 107, 14679–14684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Vargas C. et al. ; Tara Oceans Coordinators. (2015) Ocean plankton. Eukaryotic plankton diversity in the sunlit ocean. Science, 348, 1261605. [DOI] [PubMed] [Google Scholar]
- Delmont T.O. et al. (2021) Functional repertoire convergence of distantly related eukaryotic plankton lineages revealed by genome-resolved metagenomics. BiorXiv, doi:10.1101/2020.10.15.341214, 23 January 2021 , preprint: not peer reviewed.
- Delmont T.O. et al. (2015) Genome reconstructions indicate the partitioning of ecological functions inside a phytoplankton bloom in the Amundsen Sea, Antarctica. Front. Microbiol., 6, 1090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmont T.O., Eren A.M. (2016) Identifying contamination with advanced visualization and analysis practices: metagenomic approaches for eukaryotic genome assemblies. PeerJ, 4, e1839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dröge J. et al. (2015) Taxator-tk: precise taxonomic assignment of metagenomes by fast approximation of evolutionary neighborhoods. Bioinformatics, 31, 817–824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan A. et al. (2020) Metagenome-assembled genomes of phytoplankton communities across the Arctic Circle. BiorXiv, doi:10.1101/2020.06.16.154583, 17 June 2020, preprint: not peer reviewed.
- Eren A.M. et al. (2015) Anvi’o: an advanced analysis and visualization platformfor ’omics data. PeerJ, 3, e1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grigoriev I.V. et al. (2012) The genome portal of the department of energy joint genome institute. Nucleic Acids Res., 40, D26–D32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber-Vodicka,H.R. et al. (2020) phyloFlash: Rapid Small-Subunit rRNA Profiling and Targeted Assembly from Metagenomes. mSystems, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hovde,B.T. et al. (2014) The mitochondrial and chloroplast genomes of the haptophyte Chrysochromulina tobin contain unique repeat structures and gene profiles. BMC Genomics, 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling P.J. et al. (2017) Marine protists are not just big bacteria. Curr. Biol., 27, R541–R549. [DOI] [PubMed] [Google Scholar]
- Kent W.J. (2002) BLAT—the BLAST-like alignment tool. Genome Res., 12, 656–664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E. et al. (2011) Newly identified and diverse plastid-bearing branch on the eukaryotic tree of life. Proc. Natl. Acad. Sci. USA, 108, 1496–1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim,H.T. and Lee,J.M. (2018) Organellar genome analysis reveals endosymbiotic gene transfers in tomato. PLoS One, 13, e0202279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma,D.P. and Ba,J.L. (2015) Adam: A method for stochastic optimization. In, 3rd International Conference on Learning Representations, ICLR 2015 -- Conference Track Proceedings. [Google Scholar]
- Kopf A. et al. (2015) The ocean sampling day consortium. Gigascience, 4, 27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk P.S. et al. (2018) PlasFlow: predicting plasmid sequences in metagenomic data using genome signatures. Nucleic Acids Res., 46, e35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lam,S.K. et al. (2015) Numba: a LLVM-based Python JIT compiler. Proc. Second Work. LLVM Compil. Infrastruct. HPC - LLVM ' 15, 1–6. [Google Scholar]
- Leconte J. et al. (2020) Genome resolved biogeography of mamiellales. Genes (Basel), 11, 66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Q. et al. (2020) DeepMicrobes: taxonomic classification for metagenomics with deep learning. NAR Genomics Bioinf., 2, lqaa009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng G. et al. (2019) MitoZ: a toolkit for animal mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res., 47, e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Gomez,S.A. et al. (2021) A microbial eukaryote with a unique combination of purple bacteria and green algae as endosymbionts. Sci. Adv., 7, eabg4102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obiol A. et al. (2020) A metagenomic assessment of microbial eukaryotic diversity in the global ocean. Mol. Ecol. Resour., 20, 718–731. [DOI] [PubMed] [Google Scholar]
- Olm M.R. et al. (2019) Genome-resolved metagenomics of eukaryotic populations during early colonization of premature infants and in hospital rooms. Microbiome, 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paszke,A. et al. (2019) PyTorch: An Imperative Style, High-Performance Deep Learning Library. Adv. Neural Inf. Process. Syst., 32. [Google Scholar]
- Pesant S. et al. ; Tara Oceans Consortium Coordinators. (2015) Open science resources for the discovery and analysis of Tara Oceans data. Sci. Data, 2, 150023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piganeau G. et al. (2008) Picoeukaryotic sequences in the Sargasso Sea metagenome. Genome Biol., 9, R5–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piganeau G., Moreau H. (2007) Screening the Sargasso Sea metagenome for data to investigate genome evolution in Ostreococcus (Prasinophyceae, Chlorophyta). Gene, 406, 184–190. [DOI] [PubMed] [Google Scholar]
- Ramos J.E. (2003) Using TF-IDF to determine word relevance in document queries. In: Proceedings of the first instructional conference on machine learning, pp. 29–48.
- Ren J. et al. (2018) Alignment-free sequence analysis and applications. Annu. Rev. Biomed. Data Sci, 1, 93–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter D. et al. (2020) Genomic Evidence for Global Ocean Plankton Biogeography Shaped by Large-Scale Current Systems, BiorXiv, doi:10.1101/867739, 24 December 2020, preprint: not peer reviewed. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salazar G. et al. ; Tara Oceans Coordinators. (2019) Gene expression changes and community turnover differentially shape the global ocean metatranscriptome. Cell, 179, 1068–1083.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sammut C., Webb G.I. (2010) TF–IDF. In: Sammut C., Webb G.I. (eds.) Encyclopedia of Machine Learning. Springer US, Boston, MA, pp. 986–987. [Google Scholar]
- Sayers E.W. et al. (2019) Database resources of the National Center for Biotechnology Information. Nucleic Acids Res., 47, D23–D28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schön M.E. et al. (2020) PhyloMagnet: fast and accurate screening of short-read meta-omics data using gene-centric phylogenetics. Bioinformatics, 36, 1718–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebastiani F. (2002) Machine learning in automated text categorization. ACM Comput. Surv., 34, 1–47. [Google Scholar]
- Sibbald S.J., Archibald J.M. (2020) Genomic insights into plastid evolution. Genome Biol. Evol., 12, 978–990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith T.F., Waterman M.S. (1981) Identification of common molecular subsequences. J. Mol. Biol., 147, 195–197. [DOI] [PubMed] [Google Scholar]
- Strassert J.F.H. et al. (2018) Single cell genomics of uncultured marine alveolates shows paraphyly of basal dinoflagellates. ISME J., 12, 304–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sunagawa S. et al. ; Tara Oceans coordinators. (2015) Ocean plankton. Structure and function of the global ocean microbiome. Science, 348, 1261359. [DOI] [PubMed] [Google Scholar]
- Tietz,M. et al. (2017) skorch: A scikit-learn compatible neural network library that wraps PyTorch. [Google Scholar]
- Tully,B.J. et al. (2018) The reconstruction of 2,631 draft metagenome-assembled genomes from the global oceans. Sci. Data, 5, 170203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varoquaux,G. and Grisel,O. (2009) Joblib: running python function as pipeline jobs. Packag. python. org/joblib. [Google Scholar]
- West P.T. et al. (2018) Genome-reconstruction for eukaryotes from complex natural microbial communities. Genome Res., 28, 569–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wideman J.G. et al. (2020) Unexpected mitochondrial genome diversity revealed by targeted single-cell genomics of heterotrophic flagellated protists. Nat. Microbiol., 5, 154–165. [DOI] [PubMed] [Google Scholar]
- Wood D.E. et al. (2019) Improved metagenomic analysis with Kraken 2. Genome Biol., 20, 257–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Worden A.Z. et al. (2015) Rethinking the marine carbon cycle: factoring in the multifarious lifestyles of microbes. Science, 347, 1257594–1257594. [DOI] [PubMed] [Google Scholar]
- Yang A. et al. (2020) Review on the application of machine learning algorithms in the sequence data mining of DNA. Front. Bioeng. Biotechnol., 8, 1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yun-Tao Z. et al. (2005) An improved TF-IDF approach for text classification. J. Zhejiang Univ. A, 6, 49–55. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data underlying this article are available in the online supplementary materials and from https://github.com/ibe-uw/tiara.