


Abstract
JASPAR (http://jaspar.genereg.net/) is an open-access database containing manually curated, non-redundant transcription factor (TF) binding profiles for TFs across six taxonomic groups. In this 9th release, we expanded the CORE collection with 341 new profiles (148 for plants, 101 for vertebrates, 85 for urochordates, and 7 for insects), which corresponds to a 19% expansion over the previous release. We added 298 new profiles to the Unvalidated collection when no orthogonal evidence was found in the literature. All the profiles were clustered to provide familial binding profiles for each taxonomic group. Moreover, we revised the structural classification of DNA binding domains to consider plant-specific TFs. This release introduces word clouds to represent the scientific knowledge associated with each TF. We updated the genome tracks of TFBSs predicted with JASPAR profiles in eight organisms; the human and mouse TFBS predictions can be visualized as native tracks in the UCSC Genome Browser. Finally, we provide a new tool to perform JASPAR TFBS enrichment analysis in user-provided genomic regions. All the data is accessible through the JASPAR website, its associated RESTful API, the R/Bioconductor data package, and a new Python package, pyJASPAR, that facilitates serverless access to the data.
INTRODUCTION
Transcription factors are proteins that interact with the DNA in a sequence-specific manner through recognition of their TF binding sites (TFBSs) located at cis-regulatory regions (promoters, enhancers) to regulate transcription (1). TF binding to these regions occurs through direct interactions between the DNA-binding domains (DBDs) of TFs and the DNA. DBDs are classified into structural classes and families, and TFs with related DBDs typically have similar DNA binding preferences (2). The binding of TFs to cis-regulatory regions promotes or inhibits the assembly of the transcription machinery, thereby controlling gene expression regulation (1,3–5).
Sequence-specific TF-DNA interactions at TFBSs can be experimentally determined either in vitro or in vivo. High-throughput in vitro methods include systematic evolution of ligands by exponential enrichment (SELEX) (6) and protein binding-microarrays (PBM) (7) where TFs are exposed to synthesized DNA sequences. High-throughput in vivo assays include chromatin immunoprecipitation-based methods such as ChIP-seq (8), ChIP-exo (9) and ChIP-nexus (10), and cleavage-based methods such as cleavage under targets and tagmentation (11) or cleavage under targets and release using nuclease (12). These high-throughput assays (reviewed in (1)) provide unprecedented means to characterize the binding properties of individual TFs. Nevertheless, a challenge lies in our understanding of how TFs interact cooperatively at regulatory elements, for instance by forming dimers (13). Recently, CAP-SELEX revealed that TF pairs can bind in a DNA-dependent manner and that the combined binding of TFs can alter their individual binding specificities (14).
Despite the establishment of a wide variety of experimental techniques that delineate TF-DNA binding interactions and TF binding specificities, experimentally identifying all TFBSs for all TFs in various systems and biological conditions is intractable. To address this challenge, researchers rely on computational modeling to predict and investigate TF-DNA interactions. Such methods are helpful for investigating results of experimental methods with low resolution. For instance, ChIP-seq peaks are typically an order of magnitude larger than the actual binding sites of a targeted TF, and therefore computational methods can be used to pinpoint the binding sites within the peaks (15,16).
Given the importance of understanding TF-DNA interactions in studying gene expression regulation, various computational methods have been devised to model and predict TFBSs. The methods utilize experimentally identified TFBSs to build models and computationally predict TFBSs in a given genomic sequence (5). These computational methods range from basic representations such as sequence consensus-based models and position frequency matrices (PFMs) to more complex representations such as Markov and deep learning-based models (reviewed in (13,17–18)). PFMs, which summarize occurrences of each nucleotide at each position in a set of observed TF-DNA interactions, are largely and most commonly used to capture TF binding specificities. Unlike the simple consensus-based models, PFMs can be transformed to probabilistic or energy-based models to obtain position weight matrices (PWMs) (or position-specific scoring matrices (PSSMs)) that can be used to scan any DNA sequence and predict TFBSs with sum weights above a defined threshold (reviewed in (17)). Hence, TF binding preferences can be represented as PFMs, which can be interpreted as TF binding profiles or motifs. In this manuscript, we will use the term PFM, motif, and TF binding profile interchangeably.
JASPAR is a popular and regularly maintained open-access and manually curated database storing TF binding preferences as PFMs. The JASPAR CORE collection provides non-redundant binding preferences for TFs (one versioned profile per TF per taxon, except when a TF has multiple DNA-binding preferences) across 6 taxa: urochordata, vertebrates, plants, insects, nematodes, and fungi. Inclusion of new profiles requires orthogonal evidence for the binding preferences of the TFs, which is rigorously evaluated by our expert curators. To complement the CORE collection, we previously introduced the Unvalidated collection to store high-quality TF-binding profiles that are lacking orthogonal supporting evidence in the literature (19). Beyond the high-quality TF binding profiles and metadata stored in JASPAR, the popularity of the database originates from its simplicity, the tools embedded in its web-interface, and the multitude of popular resources and tools directly integrating JASPAR profiles. Some of these tools include: (i) the MEME suite, allowing various motif enrichment and discovery analysis (20), (ii) TFBSshape allowing investigation of DNA shape features for TFBSs to provide insight on the mechanism of protein–DNA interaction (21,22), (iii) CiiiDER (23) for TFBS prediction and analysis such as enrichment assessment in DNA sequences, (iv) RSAT, allowing motif discovery, TFBS motif analyses (24) and (v) i-cisTarget, which allows the prediction of cis-regulatory modules and regulatory features (25,26).
In this paper, we present the 9th release of the JASPAR database, which provides a substantial update and expansion of TF binding profiles in the six taxonomic groups. The update includes not only binding profiles (as PFMs) but also revisited metadata. Additionally, we added word clouds to display enriched terms associated with TFs in the scientific literature. Furthermore, a rigorous structural classification of plant TF DBDs is provided to adequately consider the numerous plant-specific TFs. Finally, the update comes with a range of new or updated functionalities and resources such as a TFBS enrichment tool, the pyJASPAR package, new familial binding profiles, and native UCSC human and mouse genome tracks with TFBSs predicted from JASPAR TF binding profiles.
RESULTS
Expansion and update of the JASPAR database
TF binding profiles
In the 9th release of JASPAR, we discarded unused collections introduced in early releases of the database (27–29) that either did not correspond to TF-specific binding profiles or were data-type specific; we maintained the CORE and Unvalidated collections. We computed and compiled TF binding profiles obtained from CAP-SELEX (14), NCAP-SELEX (30), SELEX-seq (31), PBMs (32), ChIP-seq (33–36) and DAP-seq experiments from ReMap 2022 (36) and GEO (37), and ChIP-exo (38) data (Supplementary Data 1 - Text for detailed list of datasets and method details). After manual curation of these profiles to confirm orthogonal supports in the literature, we augmented the CORE collection with 341 new binding profiles for TFs in four taxa (Table 1; Figure 1): 148 profiles in plants (a 24% expansion for this taxon), 101 profiles in vertebrates (a 13% expansion), 85 profiles in urochordates (only one motif was present since the second release of JASPAR in 2006 (27)), and seven profiles in insects (a 5% expansion). Out of these added profiles, 52 were upgraded from the Unvalidated to the CORE collection (27 and 25 for plants and vertebrates, respectively). Moreover, out of the newly introduced PFMs, 31 are associated with TF dimers. The literature that provides orthogonal evidence for the newly introduced TF binding profiles is provided in the metadata. Additionally, we updated 160 TF binding profiles across the six taxa with new PFMs (Table 1).
Table 1.
Growth overview of the CORE collection of JASPAR 2022 compared to the previous release
| Taxonomic Group | Non-redundant PFMs in JASPAR 2020 | New non-redundant PFMs in JASPAR 2022 | Removed profiles | Upgraded profiles (from Unvalidated to CORE) | Updated PFMs in JASPAR 2022 | Total PFMs (non-redundant) in JASPAR 2022 |
|---|---|---|---|---|---|---|
| Plants | 530 | 121 | 22 | 27 | 44 | 656 |
| Vertebrates | 746 | 76 | 6 | 25 | 102 | 841 |
| Urochordata | 1 | 85 | - | - | - | 86 |
| Insects | 143 | 7 | - | - | - | 150 |
| Nematodes | 43 | - | - | - | - | 43 |
| Fungi | 183 | - | 4 | - | 14 | 179 |
| CORE total | 1646 | 289 | 32 | 52 | 160 | 1955 |
Figure 1.
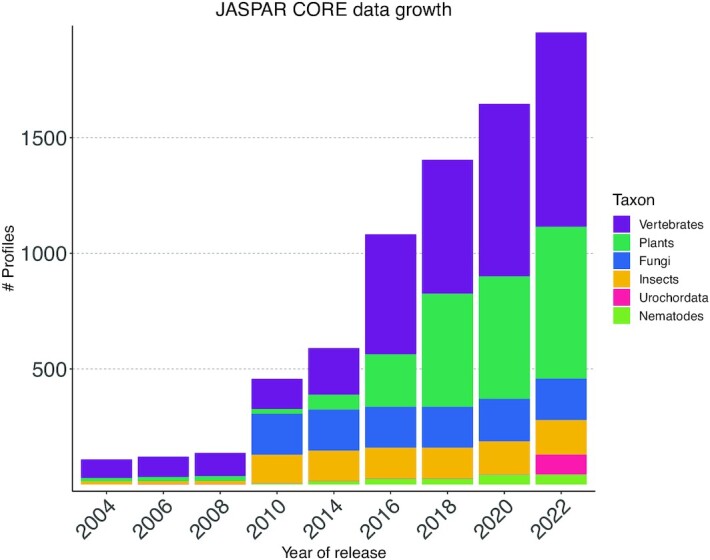
JASPAR CORE collection growth. The number of non-redundant profiles in each taxon (see legend) and overall through all JASPAR releases.
High-quality PFMs lacking orthogonal support were included in the Unvalidated collection (298 new profiles; Supplementary Data 1—Supplementary Figure S1, Supplementary Data 2—Supplementary Table S1). Specifically, 115 TF binding profiles are associated with zinc-finger TFs and 95 associated with TFs binding DNA as dimers. We provide the Unvalidated collection of TF binding profiles to the community to use with due caution since they are not yet supported with orthogonal evidence. We extend our invitation to the user community to be involved in the motif curation process by providing either new unvalidated profiles to consider or support to existing profiles in the collection.
We exhaustively revised the metadata to update information about the TF names, the structural class and family of the TF DBDs (following TFClass (39)), and links to external databases such as UniProt (40), ReMap (36), UniBind (15,16) and DNA Readout Viewer (41), whenever possible. Finally, we removed 32 profiles from the CORE collection (22 plant, 6 vertebrate and 4 fungi profiles) as they corresponded to synonyms of already present TF profiles, had low information content, or were derived from consensus strings (Table 1). In addition, we removed 85 profiles from the Unvalidated collection (44 vertebrate, 40 plant and 1 fungi profiles) because: (i) the corresponding profile or a new profile for the same TF was added to the CORE collection; (ii) the profile was of insufficient quality or (iii) the profile was misannotated (Supplementary Data 2—Supplementary Table S1; detailed list of all removed profiles at https://jaspar.genereg.net/changelog/).
The JASPAR 2022 CORE collection now stores 1955 non-redundant PFMs (841 for vertebrates, 656 for plants, 179 for fungi, 150 for insects, 43 for nematodes, and 86 for urochordates) (Table 1; Figure 1). Additionally, we maintained the associated collection of transcription factor flexible models (TFFMs; hidden Markov-based models capturing dinucleotide dependencies in TF–DNA interactions (42)) that were initialized using JASPAR CORE PFMs and trained on ChIP-seq data (Supplementary Data 1—Text). This process resulted in 303 new TFFMs (207 for vertebrates and 96 for plants).
Improved structural classification of plant TF DNA-binding domains
In JASPAR, TFs are classified based on TFClass (39), which provides a hierarchical structural classification (including superclass, class, and family) originally designed for human TFs and later extended to mammals. Since plant genomes contain many classes of TFs absent from TFClass, we expanded the TF structural classification using TFClass guidelines (39) and published structural evidence (Supplementary Data 2—Supplementary Table S2). In some rare cases (e.g. GARP and NF-Y TFs), we slightly diverged from TFClass so that the TF common name expected by users is provided in the structural class or family name. We arbitrarily decided to classify plant specific RAV TFs that contain two types of DBD (B3 and AP2) in the B3 Class. WRKY TFs that have a Zinc finger and a DBD derived from a GCM fold have been classified under the GCM domain factors class and WRKY family, and not in the Zinc-coordinating DNA-binding domains superclass. This homogenised classification introduced 27 novel entries in the TF DBD structural classification (Supplementary Data 2—Supplementary Table S2) and led to numerous corrections in the class and family fields compared to previous JASPAR releases.
Word clouds of terms associated with TFs in the scientific literature
Biological information about TFs, or genes in general, is scattered across many different resources, with PubMed possibly being the most extensive one. In an attempt to provide rich annotations for the TFs in JASPAR, we mined the corpus of article abstracts available in the PubMed database (43). We compiled sets of abstracts associated with each TF and weighted each word present by its relative importance when compared to all abstracts associated with other TFs in the same taxon (Supplementary Data 1—Text for method details). For each TF, the 200 highest weighted words were used to create a word cloud summarizing the annotations associated with that TF. As an example, Figure 2 illustrates the word cloud of terms associated with the PAX6 TF in the scientific literature. Among the most significant terms, we find ‘lens’, ‘iris’, and ‘foveal’ that are representative of the importance of PAX6 in the development of the eye, while the term ‘aniridia’ reflects the link between some PAX6 mutations and the genetic disorder aniridia (44,45).
Figure 2.

JASPAR TF word clouds. Webpage providing information about the binding profile associated with PAX6. The word cloud of terms obtained for PAX6 is highlighted in red, which supports the role of this TF in eye development and its implication in causing the genetic disorder aniridia.
TF binding profile clusters, familial binding profiles, and genomic tracks
We updated the hierarchical clustering of the JASPAR TF binding profiles for each taxon with the RSAT matrix-clustering tool (46). Users can explore the CORE and Unvalidated collections through radial trees, which highlight the TF DBD structural classes, and directly access the underlying profiles by clicking on the TF name (https://jaspar.genereg.net/matrix-clusters).
The hierarchical clustering of JASPAR PFMs was used to generate a collection of familial binding profiles (5,47), following previously published methodologies (16,48). Such familial motifs are useful in applications where motif redundancy (many TFs have similar binding preferences) is not desired. In brief, we defined clusters based on the DBD structural classes along the hierarchical clustering of PFMs. Next, we computed a familial binding profile for each cluster, summarizing the profiles within the clusters following (48) (Supplementary Data 1—Text for method details; Supplementary Data 1—Supplementary Figure S2). The familial binding profiles, also referred to as archetypes in (48), can be explored and downloaded at https://jaspar.genereg.net/matrix-clusters and https://jaspar.genereg.net/downloads/, respectively.
One of the primary uses of PFMs is to predict binding sites. To facilitate this, we created ready-made prediction tracks for genome visualization and interpretation. Specifically, we scanned the genomes of eight organisms (Arabidopsis thaliana, Caenorhabditis elegans, Ciona intestinalis, Danio rerio, Drosophila melanogaster, Homo sapiens, Mus musculus, and Saccharomyces cerevisiae) with the JASPAR CORE PFMs associated with the same taxon to predict TFBSs and update the JASPAR TFBS genomic tracks. Moreover, we created a collection of familial TFBSs by merging overlapping TFBSs that were predicted from PFMs associated with the same familial binding profile (Supplementary Data 1—Text for method details). The TFBS predictions associated with all PFMs are available at http://expdata.cmmt.ubc.ca/JASPAR/downloads/UCSC_tracks/2022/. The familial binding TFBSs are available at https://jaspar.genereg.net/downloads/. Finally, we provide JASPAR TFBS predictions as genomic tracks, which can be visualized in genome browsers. Notably, the UCSC Genome Browser (49) now presents predicted human (for the hg19 and hg38 genome assemblies) and mouse (for the mm10 and mm39 genome assemblies) JASPAR TFBS data as a native tracks for the human and mouse genomes with information such as TF names, TFBS prediction scores, and PFM logos (Supplementary Data 1 - Supplementary Figure S3).
A command-line tool to evaluate JASPAR TFBS enrichment in genomic regions
A common challenge in the field of transcriptional regulation is to predict the TF(s) most likely to control a set of cis-regulatory regions. This challenge is classically addressed by evaluating the enrichment for potential TFBSs associated with candidate TFs in the genomic regions of interest compared to background regions (16,26,50–53). We previously introduced an enrichment tool that evaluates the enrichment for sets of direct TF–DNA interactions from UniBind in user-provided DNA regions compared to background regions (16). Following the same strategy, we introduce a TFBS enrichment tool to predict TFs with an enrichment of JASPAR TFBSs using the Locus Overlap Analysis (LOLA) tool (54). The enrichment tool is available as a command-line tool (https://jaspar.genereg.net/enrichment/, https://bitbucket.org/CBGR/jaspar_enrichment/).
As a use case, we studied the differential enrichment of predicted TFBSs at DNase-seq peaks observed in A549 cells before and after 2 h treatment with 100 nM dexamethasone. DNase-seq is an assay capturing open chromatin regions (55). Dexamethasone is a known agonist of the glucocorticoid receptor (NR3C1), a nuclear receptor that binds the DNA upon ligand-based activation. Figure 3 provides a visual representation of the differential TFBS enrichment analysis results when considering DNase-seq peaks in treated versus untreated cells. As expected, NR3C1 (a member of the Steroid hormone receptors (NR3) family) was the top enriched TF (–log10(P) = 58.77). Among other TFs showing a high enrichment of TFBSs, we observed many members of the Three-zinc finger Kruppel-related family (e.g. KLF factors, SP3, and SP9) (Supplementary Data 2—Supplementary Table S3). In another example, we observed the enrichment of TFBSs for the TFs FOXA1 and GATA3 in regions surrounding CpGs that are hypomethylated in estrogen receptor positive (ER+) breast cancers (56) (Supplementary Data 1—Supplementary Figure S4, Supplementary Data 2—Supplementary Supplementary Table S4). These TFs are well established drivers of ER+ breast cancers binding to hypomethylated enhancers in ER+ breast cancers (56).
Figure 3.

TFBS differential enrichment analysis on DNase-seq data for A549 cells before and after 2 h of dexamethasone treatment. Enrichment significance for each JASPAR profile from the vertebrate CORE collection is shown in the y-axis as -log10(P) in this beeswarm plot. Each point depicts the Fisher exact test P-value (P) corresponding to a TF. The Points are colored based on the TF DBD structural family annotation, with a distinct color for each of the top 10 enriched families (see legend). Light yellow represents TF families outside of the top 10 enriched and with -log10(P) > 3 (Other) and brown represents TF families for which -log10(P) ≤ 3 (non significant, N.S.).
pyJASPAR—serverless pythonic interface to JASPAR data
All data is accessible through the JASPAR website (https://jaspar.genereg.net/), its associated RESTful API (https://jaspar.genereg.net/api/) (57), and the JASPAR2022 R/Bioconductor data package (source code at https://github.com/da-bar/JASPAR2022). The JASPAR database can also be accessed using Biopython (58) but it requires a local MySQL server to query the underlying database, which limits its access and use. To make access to JASPAR data easier, we introduce a new Python package, pyJASPAR (59), which allows users to query and access all JASPAR data without setting up the underlying MySQL database.
pyJASPAR is implemented in Python 3 using the Biopython motifs module and SQLite3 to provide a serverless Pythonic interface to the JASPAR database. The package allows users to query and access TF binding profiles across various releases of JASPAR. The releases currently available are: JASPAR2014, JASPAR2016, JASPAR2018, JASPAR2020, and JASPAR2022. The pyJASPAR package will be updated when future JASPAR releases become available. TF binding profiles can be retrieved using JASPAR matrix IDs, TF names, or other metadata information (Supplementary Data 1—Text for more details).
pyJASPAR is open source and the code is available at https://github.com/asntech/pyjaspar/ under the GPL-3.0 License. The module can easily be installed with Conda from the bioconda channel (https://anaconda.org/bioconda/pyjaspar) (60) or from the Python Package Index with the pip command. Detailed documentation with usage examples is available at https://pyjaspar.rtfd.io/.
CONCLUSIONS AND PERSPECTIVES
For the 9th release of the JASPAR database, we substantially expanded the JASPAR CORE collection by 19% (341 added motifs). The newly introduced TF binding profiles were obtained after manual curation of PFMs predicted de novo from >3500 ChIP-seq/-exo datasets (from ReMap 2022 (36) and GEO (61)) or retrieved from publically available repositories. While we continued our commitment to provide non-redundant, high-quality TF binding profiles for TFs across six taxa, this release comes with an important increase in the number of profiles for urochordata, with 86 PFMs available when JASPAR has contained a single one since 2006 (27). We now also provide TFBS predictions in Ciona intestinalis using the 86 JASPAR binding profiles. This increase exemplifies how the investigation of transcriptional regulation is expanding across more model organisms.
An important question is what fraction of TFs have a binding profile in JASPAR. For humans, the JASPAR vertebrates CORE collection contains a binding profile for 43% of the 1639 human TFs (1), 56% when including the Unvalidated collection. If we consider the 1717 reported TFs for A. thaliana (62), 21% of these TFs have a profile in the JASPAR plants CORE collection, 22% when including the Unvalidated collection.
From the previous version of the Unvalidated collection (19), we found literature support for 81 profiles. Unfortunately, our team of curators did not succeed in identifying orthogonal validation in the literature for several high-quality motifs found enriched at ChIP-seq/-exo peak summits. As a result, 298 of such profiles were added to the previously introduced Unvalidated collection (19). The lack of experimental support for these profiles indicates an opportunity for the research field to explore these understudied TFs (63). Notably, 61% of the profiles in the vertebrates Unvalidated collection is associated with C2H2 zinc finger factors. A potential contributing challenge to obtaining orthogonal evidence may be the fact that many zinc-fingers, which represent the largest class of TFs, have been reported to regulate a limited number or even a single gene (e.g. Zfp568 (64), ZNF558 (65), ZNF410 (66) and ZFP64 (67)).
This JASPAR update comes with a new tool to compute TFBS enrichment given user-provided input and background sequences, mimicking a similar tool available with the UniBind database (16). The tool relies on the genome-wide TFBSs predicted using PFMs from the JASPAR CORE collection. Even though JASPAR predicted TFBSs will contain a high number of false positives, the enrichment tool could be useful to suggest roles for TFs for which no direct TF-DNA interactions are available in UniBind (16).
Consistent with Weidemüller et al. (63), we noticed that limited scientific literature (i.e. at most a single manuscript in PubMed) exists for many TFs, which clearly impacts the utility of the JASPAR word clouds. This constraint varies between taxa. For example, while the average number of PubMed manuscripts per vertebrate TF was ∼500, urochordata TFs were associated with an average of only four manuscripts. Furthermore, a large number of TFs associated with individual PubMed manuscripts was observed. The average number of vertebrate TFs associated with PubMed IDs was ∼19 with some associated with hundreds of TFs. An example is PubMed ID 21873635 that describes methods development of the Gene Ontology database (822 TFs), PubMed ID 12477932 that describes the Mammalian Gene Collection (MGC) Program (805 TFs), and PubMed ID 15618518 that analyzes the expression of TFs in the mouse brain (722 TFs). These manuscripts include general information about TFs. Therefore, we see opportunities to further improve the literature annotation engine, by decreasing the influence of outlier manuscripts and incorporating emerging natural language processing methods.
PFMs are still the most widely used models to represent TF binding preferences to DNA, despite their well-established caveats such as fixed-length and the failure to account for nucleotide interdependencies. A novel generation of computational models based on machine learning approaches such as deep learning are arising (68,69). Nevertheless, how to best share these models in a unified manner is still unclear despite some recent efforts (70) and will require discussion in the community. As the field moves towards a unified framework to share such models, we expect their inclusion in future JASPAR releases.
AUTHORS’ NOTE
The authors wish it to be known that, in their opinion, the first three authors should be regarded as joint first authors. The order of co-first authors provided here was decided through a mushroom picking competition around the Sognsvann lake, Oslo, Norway. Co-first authors can prioritise their names when adding this paper's reference to their résumés.
Supplementary Material
ACKNOWLEDGEMENTS
We thank the user community for useful input and the scientific community for performing experimental assays of TF-DNA interactions and for publicly releasing the data. We thank Shaun Mahony and Franklin Pugh for providing early access to ChIP-exo data from (38) and Emma Farley for pointers to urochordata data sets. We thank Vipin Kumar for contribution in the early curation sessions. We thank Walter Santana for his technical assistance to generate the motif radial trees, the UCSC Genome Browser project team for their assistance with the genome tracks, Harold Gutch and the NCMM IT team for their IT support, and Ingrid Kjelsvik for administrative support.
Contributor Information
Jaime A Castro-Mondragon, Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo, 0318 Oslo, Norway.
Rafael Riudavets-Puig, Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo, 0318 Oslo, Norway.
Ieva Rauluseviciute, Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo, 0318 Oslo, Norway.
Roza Berhanu Lemma, Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo, 0318 Oslo, Norway.
Laura Turchi, Laboratoire Physiologie Cellulaire et Végétale, Univ. Grenoble Alpes, CNRS, CEA, INRAE, IRIG-DBSCI-LPCV, 17 avenue des martyrs, F-38054, Grenoble, France.
Romain Blanc-Mathieu, Laboratoire Physiologie Cellulaire et Végétale, Univ. Grenoble Alpes, CNRS, CEA, INRAE, IRIG-DBSCI-LPCV, 17 avenue des martyrs, F-38054, Grenoble, France.
Jeremy Lucas, Laboratoire Physiologie Cellulaire et Végétale, Univ. Grenoble Alpes, CNRS, CEA, INRAE, IRIG-DBSCI-LPCV, 17 avenue des martyrs, F-38054, Grenoble, France.
Paul Boddie, Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo, 0318 Oslo, Norway.
Aziz Khan, Stanford Cancer Institute, Stanford University School of Medicine, Stanford, CA, 94305, USA.
Nicolás Manosalva Pérez, Department of Plant Biotechnology and Bioinformatics, Ghent University, Technologiepark 71, 9052 Ghent, Belgium; VIB Center for Plant Systems Biology, Technologiepark 71, 9052 Ghent, Belgium.
Oriol Fornes, Centre for Molecular Medicine and Therapeutics, Department of Medical Genetics, BC Children's Hospital Research Institute, University of British Columbia, 950 W 28th Ave, Vancouver, BC V5Z 4H4, Canada.
Tiffany Y Leung, Centre for Molecular Medicine and Therapeutics, Department of Medical Genetics, BC Children's Hospital Research Institute, University of British Columbia, 950 W 28th Ave, Vancouver, BC V5Z 4H4, Canada.
Alejandro Aguirre, Centre for Molecular Medicine and Therapeutics, Department of Medical Genetics, BC Children's Hospital Research Institute, University of British Columbia, 950 W 28th Ave, Vancouver, BC V5Z 4H4, Canada.
Fayrouz Hammal, Aix Marseille Univ, INSERM, TAGC, Marseille, France.
Daniel Schmelter, UCSC Genome Browser, University of California Santa Cruz, Santa Cruz, CA, 95060, USA.
Damir Baranasic, MRC London Institute of Medical Sciences, Du Cane Road, London, W12 0NN, UK; Institute of Clinical Sciences, Faculty of Medicine, Imperial College London, Hammersmith Hospital Campus, Du Cane Road, London W12 0NN, UK.
Benoit Ballester, Aix Marseille Univ, INSERM, TAGC, Marseille, France.
Albin Sandelin, The Bioinformatics Centre, Department of Biology & Biotech Research and Innovation Centre, University of Copenhagen, Ole Maaloes Vej 5, DK2200 Copenhagen N, Denmark.
Boris Lenhard, MRC London Institute of Medical Sciences, Du Cane Road, London, W12 0NN, UK; Institute of Clinical Sciences, Faculty of Medicine, Imperial College London, Hammersmith Hospital Campus, Du Cane Road, London W12 0NN, UK.
Klaas Vandepoele, Department of Plant Biotechnology and Bioinformatics, Ghent University, Technologiepark 71, 9052 Ghent, Belgium; VIB Center for Plant Systems Biology, Technologiepark 71, 9052 Ghent, Belgium; Bioinformatics Institute Ghent, Ghent University, Technologiepark 71, 9052 Ghent, Belgium.
Wyeth W Wasserman, Centre for Molecular Medicine and Therapeutics, Department of Medical Genetics, BC Children's Hospital Research Institute, University of British Columbia, 950 W 28th Ave, Vancouver, BC V5Z 4H4, Canada.
François Parcy, Laboratoire Physiologie Cellulaire et Végétale, Univ. Grenoble Alpes, CNRS, CEA, INRAE, IRIG-DBSCI-LPCV, 17 avenue des martyrs, F-38054, Grenoble, France.
Anthony Mathelier, Centre for Molecular Medicine Norway (NCMM), Nordic EMBL Partnership, University of Oslo, 0318 Oslo, Norway; Department of Medical Genetics, Institute of Clinical Medicine, University of Oslo and Oslo University Hospital, Oslo, Norway.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
Norwegian Research Council [187615]; Helse Sør-Øst; University of Oslo through the Centre for Molecular Medicine Norway (NCMM) (to Mathelier group); Norwegian Research Council [288404 to R.R.P., J.A.C.M., Mathelier group]; Norwegian Cancer Society [197884 to R.B.L., Mathelier group]; GRAL program [ANR-10-LABX-49-01] with the frame of the CBH-EUR-GS [ANR-17-EURE-0003 to Parcy group]; PhD fellowship from CNRS Prime80 (to L.T.); NHGRI [5U41HG002371-20 to D.S.]; BOF grant from Ghent University [BOF24Y2019001901 to N.M.P.]; PhD Fellowship from the Provence-Alpes-Côte d’Azur Regional Council (Région SUD); Institut National de la Santé et de la Recherche Médicale (INSERM) (to F.H.); Novo Nordisk Foundation [NNF20OC0059951, NNF19OC0058262]; Danish Cancer Foundation [R204-A12359]; Danish Independent Research Fund [6110-00207B, 7014-00120B]; Carlsberg Foundation; ERC the European Union's Horizon 2020 research and innovation programme (MSCA ITN pHioniC) (to Sandelin group and collaborators); Canadian Institutes of Health Research [PJT-162120]; Natural Sciences and Engineering Research Council of Canada (NSERC) Discovery Grant [RGPIN-2017-06824]; BC Children's Hospital Foundation and Research Institute (to Wasserman group). The open access publication charge for this paper has been waived by Oxford University Press – NAR Editorial Board members are entitled to one free paper per year in recognition of their work on behalf of the journal.
Conflict of interest statement. None declared.
REFERENCES
- 1. Lambert S.A., Jolma A., Campitelli L.F., Das P.K., Yin Y., Albu M., Chen X., Taipale J., Hughes T.R., Weirauch M.T.. The human transcription factors. Cell. 2018; 172:650–665. [DOI] [PubMed] [Google Scholar]
- 2. Weirauch M.T., Yang A., Albu M., Cote A.G., Montenegro-Montero A., Drewe P., Najafabadi H.S., Lambert S.A., Mann I., Cook K.et al.. Determination and inference of eukaryotic transcription factor sequence specificity. Cell. 2014; 158:1431–1443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Reiter F., Wienerroither S., Stark A.. Combinatorial function of transcription factors and cofactors. Curr. Opin. Genet. Dev. 2017; 43:73–81. [DOI] [PubMed] [Google Scholar]
- 4. Venters B.J., Pugh B.F.. How eukaryotic genes are transcribed. Crit. Rev. Biochem. Mol. Biol. 2009; 44:117–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Wasserman W.W., Sandelin A.. Applied bioinformatics for the identification of regulatory elements. Nat. Rev. Genet. 2004; 5:276–287. [DOI] [PubMed] [Google Scholar]
- 6. Jolma A., Kivioja T., Toivonen J., Cheng L., Wei G., Enge M., Taipale M., Vaquerizas J.M., Yan J., Sillanpää M.J.et al.. Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 2010; 20:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Berger M.F., Philippakis A.A., Qureshi A.M., He F.S., Estep P.W. 3rd, Bulyk M.L.. Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 2006; 24:1429–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Johnson D.S., Mortazavi A., Myers R.M., Wold B.. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007; 316:1497–1502. [DOI] [PubMed] [Google Scholar]
- 9. Franklin Pugh B. Ultra-high resolution mapping of protein-genome interactions using ChIP-exo. BMC Proc. 2012; 6:O27. [Google Scholar]
- 10. He Q., Johnston J., Zeitlinger J.. ChIP-nexus enables improved detection of in vivo transcription factor binding footprints. Nat. Biotechnol. 2015; 33:395–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kaya-Okur H.S., Wu S.J., Codomo C.A., Pledger E.S., Bryson T.D., Henikoff J.G., Ahmad K., Henikoff S.. CUT&Tag for efficient epigenomic profiling of small samples and single cells. Nat. Commun. 2019; 10:1930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Skene P.J., Henikoff S.. An efficient targeted nuclease strategy for high-resolution mapping of DNA binding sites. Elife. 2017; 6:e21856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Slattery M., Zhou T., Yang L., Dantas Machado A.C., Gordân R., Rohs R.. Absence of a simple code: how transcription factors read the genome. Trends Biochem. Sci. 2014; 39:381–399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Jolma A., Yin Y., Nitta K.R., Dave K., Popov A., Taipale M., Enge M., Kivioja T., Morgunova E., Taipale J.. DNA-dependent formation of transcription factor pairs alters their binding specificity. Nature. 2015; 527:384–388. [DOI] [PubMed] [Google Scholar]
- 15. Gheorghe M., Sandve G.K., Khan A., Chèneby J., Ballester B., Mathelier A.. A map of direct TF-DNA interactions in the human genome. Nucleic Acids Res. 2019; 47:e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Puig R.R., Boddie P., Khan A., Castro-Mondragon J.A., Mathelier A.. UniBind: maps of high-confidence direct TF-DNA interactions across nine species. BMC Genomics. 2021; 22:482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Stormo G.D. Modeling the specificity of protein-DNA interactions. Quant Biol. 2013; 1:115–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Koo P.K., Ploenzke M.. Deep learning for inferring transcription factor binding sites. Curr Opin Syst Biol. 2020; 19:16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranašić D.et al.. JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic Acids Res. 2020; 48:D87–D92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Bailey T.L., Johnson J., Grant C.E., Noble W.S.. The MEME Suite. Nucleic Acids Res. 2015; 43:W39–W49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang L., Zhou T., Dror I., Mathelier A., Wasserman W.W., Gordân R., Rohs R.. TFBSshape: a motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2014; 42:D148–D155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Chiu T.-P., Xin B., Markarian N., Wang Y., Rohs R.. TFBSshape: an expanded motif database for DNA shape features of transcription factor binding sites. Nucleic Acids Res. 2020; 48:D246–D255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Gearing L.J., Cumming H.E., Chapman R., Finkel A.M., Woodhouse I.B., Luu K., Gould J.A., Forster S.C., Hertzog P.J.. CiiiDER: a tool for predicting and analysing transcription factor binding sites. PLoS One. 2019; 14:e0215495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Nguyen N.T.T., Contreras-Moreira B., Castro-Mondragon J.A., Santana-Garcia W., Ossio R., Robles-Espinoza C.D., Bahin M., Collombet S., Vincens P., Thieffry D.et al.. RSAT 2018: regulatory sequence analysis tools 20th anniversary. Nucleic Acids Res. 2018; 46:W209–W214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Herrmann C., Van de Sande B., Potier D., Aerts S.. i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 2012; 40:e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Imrichová H., Hulselmans G., Atak Z.K., Potier D., Aerts S.. i-cisTarget 2015 update: generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Res. 2015; 43:W57–W64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Vlieghe D., Sandelin A., De Bleser P.J., Vleminckx K., Wasserman W.W., van Roy F., Lenhard B.. A new generation of JASPAR, the open-access repository for transcription factor binding site profiles. Nucleic Acids Res. 2006; 34:D95–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bryne J.C., Valen E., Tang M.-H.E., Marstrand T., Winther O., da Piedade I., Krogh A., Lenhard B., Sandelin A.. JASPAR, the open access database of transcription factor-binding profiles: new content and tools in the 2008 update. Nucleic Acids Res. 2008; 36:D102–D106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Portales-Casamar E., Thongjuea S., Kwon A.T., Arenillas D., Zhao X., Valen E., Yusuf D., Lenhard B., Wasserman W.W., Sandelin A.. JASPAR 2010: the greatly expanded open-access database of transcription factor binding profiles. Nucleic Acids Res. 2010; 38:D105–D110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zhu F., Farnung L., Kaasinen E., Sahu B., Yin Y., Wei B., Dodonova S.O., Nitta K.R., Morgunova E., Taipale M.et al.. The interaction landscape between transcription factors and the nucleosome. Nature. 2018; 562:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Brozovic M., Dantec C., Dardaillon J., Dauga D., Faure E., Gineste M., Louis A., Naville M., Nitta K.R., Piette J.et al.. ANISEED 2017: extending the integrated ascidian database to the exploration and evolutionary comparison of genome-scale datasets. Nucleic Acids Res. 2018; 46:D718–D725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lambert S.A., Yang A.W.H., Sasse A., Cowley G., Albu M., Caddick M.X., Morris Q.D., Weirauch M.T., Hughes T.R.. Similarity regression predicts evolution of transcription factor sequence specificity. Nat. Genet. 2019; 51:981–989. [DOI] [PubMed] [Google Scholar]
- 33. Ricardi M.M., González R.M., Zhong S., Domínguez P.G., Duffy T., Turjanski P.G., Salgado Salter J.D., Alleva K., Carrari F., Giovannoni J.J.et al.. Genome-wide data (ChIP-seq) enabled identification of cell wall-related and aquaporin genes as targets of tomato ASR1, a drought stress-responsive transcription factor. BMC Plant Biol. 2014; 14:29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Du M., Zhao J., Tzeng D.T.W., Liu Y., Deng L., Yang T., Zhai Q., Wu F., Huang Z., Zhou M.et al.. MYC2 orchestrates a hierarchical transcriptional cascade that regulates jasmonate-mediated plant immunity in tomato. Plant Cell. 2017; 29:1883–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Liu Y., Shi Y., Zhu N., Zhong S., Bouzayen M., Li Z.. SlGRAS4 mediates a novel regulatory pathway promoting chilling tolerance in tomato. Plant Biotechnol. J. 2020; 18:1620–1633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Hammal F., de Langen P., Bergon A., Lopez F., Ballester B.. ReMap 2022: a database of Human, Mouse, Drosophila and Arabidopsis regulatory regions from an integrative analysis of DNA-binding sequencing experiments. Nucleic Acids Res. 2021; 10.1093/nar/gkab996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Barrett T., Wilhite S.E., Ledoux P., Evangelista C., Kim I.F., Tomashevsky M., Marshall K.A., Phillippy K.H., Sherman P.M., Holko M.et al.. NCBI GEO: archive for functional genomics data sets–update. Nucleic Acids Res. 2013; 41:D991–D995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rossi M.J., Kuntala P.K., Lai W.K.M., Yamada N., Badjatia N., Mittal C., Kuzu G., Bocklund K., Farrell N.P., Blanda T.R.et al.. A high-resolution protein architecture of the budding yeast genome. Nature. 2021; 592:309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Wingender E., Schoeps T., Haubrock M., Krull M., Dönitz J.. TFClass: expanding the classification of human transcription factors to their mammalian orthologs. Nucleic Acids Res. 2018; 46:D343–D347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. UniProt Consortium UniProt: the universal protein knowledgebase in 2021. Nucleic Acids Res. 2021; 49:D480–D489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Adam K., Gyorgypal Z., Hegedus Z.. DNA Readout Viewer (DRV): visualization of specificity determining patterns of protein-binding DNA segments. Bioinformatics. 2020; 36:2286–2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mathelier A., Wasserman W.W.. The next generation of transcription factor binding site prediction. PLoS Comput. Biol. 2013; 9:e1003214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Sayers E.W., Beck J., Bolton E.E., Bourexis D., Brister J.R., Canese K., Comeau D.C., Funk K., Kim S., Klimke W.et al.. Database resources of the National Center for Biotechnology Information. Nucleic Acids Res. 2021; 49:D10–D17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Jordan T., Hanson I., Zaletayev D., Hodgson S., Prosser J., Seawright A., Hastie N., van Heyningen V.. The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1992; 1:328–332. [DOI] [PubMed] [Google Scholar]
- 45. Gehring W.J., Ikeo K.. Pax 6: mastering eye morphogenesis and eye evolution. Trends Genet. 1999; 15:371–377. [DOI] [PubMed] [Google Scholar]
- 46. Castro-Mondragon J.A., Jaeger S., Thieffry D., Thomas-Chollier M., van Helden J.. RSAT matrix-clustering: dynamic exploration and redundancy reduction of transcription factor binding motif collections. Nucleic Acids Res. 2017; 45:e119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Mahony S., Auron P.E., Benos P.V.. DNA familial binding profiles made easy: comparison of various motif alignment and clustering strategies. PLoS Comput. Biol. 2007; 3:e61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Vierstra J., Lazar J., Sandstrom R., Halow J., Lee K., Bates D., Diegel M., Dunn D., Neri F., Haugen E.et al.. Global reference mapping of human transcription factor footprints. Nature. 2020; 583:729–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Navarro Gonzalez J., Zweig A.S., Speir M.L., Schmelter D., Rosenbloom K.R., Raney B.J., Powell C.C., Nassar L.R., Maulding N.D., Lee C.M.et al.. The UCSC Genome Browser database: 2021 update. Nucleic Acids Res. 2021; 49:D1046–D1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kwon A.T., Arenillas D.J., Worsley Hunt R., Wasserman W.W.. oPOSSUM-3: advanced analysis of regulatory motif over-representation across genes or ChIP-Seq datasets. G3. 2012; 2:987–1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Puente-Santamaria L., Wasserman W.W., Del Peso L.. TFEA.ChIP: a tool kit for transcription factor binding site enrichment analysis capitalizing on ChIP-seq datasets. Bioinformatics. 2019; 35:5339–5340. [DOI] [PubMed] [Google Scholar]
- 52. Roopra A. MAGIC: A tool for predicting transcription factors and cofactors driving gene sets using ENCODE data. PLoS Comput. Biol. 2020; 16:e1007800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Arenillas D.J., Forrest A.R.R., Kawaji H., Lassmann T., Wasserman W.W., Mathelier AConsortium FANTOM Consortium . CAGEd-oPOSSUM: motif enrichment analysis from CAGE-derived TSSs. Bioinformatics. 2016; 32:2858–2860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Sheffield N.C., Bock C.. LOLA: enrichment analysis for genomic region sets and regulatory elements in R and Bioconductor. Bioinformatics. 2015; 32:587–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Song L., Crawford G.E.. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold Spring Harb. Protoc. 2010; 2010:db.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Fleischer T., Tekpli X., Mathelier A., Wang S., Nebdal D., Dhakal H.P., Sahlberg K.K., Schlichting E., Børresen-Dale A.-L.Oslo Breast Cancer Research Consortium (OSBREAC) et al.. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat. Commun. 2017; 8:1379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Khan A., Mathelier A.. JASPAR RESTful API: accessing JASPAR data from any programming language. Bioinformatics. 2017; 34:1612–1614. [DOI] [PubMed] [Google Scholar]
- 58. Cock P.J.A., Antao T., Chang J.T., Chapman B.A., Cox C.J., Dalke A., Friedberg I., Hamelryck T., Kauff F., Wilczynski B.et al.. Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 2009; 25:1422–1423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Khan A. pyJASPAR: a Pythonic interface to JASPAR transcription factor motifs. 2021; 10.5281/zenodo.5062370. [DOI] [Google Scholar]
- 60. Grüning B., Dale R., Sjödin A., Chapman B.A., Rowe J., Tomkins-Tinch C.H., Valieris R., Köster J.. Bioconda: sustainable and comprehensive software distribution for the life sciences. Nat. Methods. 2018; 15:475–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Edgar R. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002; 30:207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Jin J., Tian F., Yang D.-C., Meng Y.-Q., Kong L., Luo J., Gao G.. PlantTFDB 4.0: toward a central hub for transcription factors and regulatory interactions in plants. Nucleic. Acids. Res. 2017; 45:D1040–D1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Weidemüller P., Kholmatov M., Petsalaki E., Zaugg J.B.. Transcription factors: Bridge between cell signaling and gene regulation. Proteomics. 2021; e2000034. [DOI] [PubMed] [Google Scholar]
- 64. Yang P., Wang Y., Hoang D., Tinkham M., Patel A., Sun M.-A., Wolf G., Baker M., Chien H.-C., Lai K.-Y.N.et al.. A placental growth factor is silenced in mouse embryos by the zinc finger protein ZFP568. Science. 2017; 356:757–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Johansson P.A., Brattås P.L., Douse C.H., Hsieh P., Pontis J., Grassi D., Garza R., Jönsson M.E., Atacho D.A.M., Pircs K.et al.. A human-specific structural variation at the ZNF558 locus controls a gene regulatory network during forebrain development. 2020; bioRxiv doi:18 August 2020, preprint: not peer reviewedhttps://www.biorxiv.org/content/10.1101/2020.08.18.255562. [DOI] [PubMed]
- 66. Lan X., Ren R., Feng R., Ly L.C., Lan Y., Zhang Z., Aboreden N., Qin K., Horton J.R., Grevet J.D.et al.. ZNF410 uniquely activates the NuRD component CHD4 to silence fetal hemoglobin expression. Mol. Cell. 2021; 81:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Lu B., Klingbeil O., Tarumoto Y., Somerville T.D.D., Huang Y.-H., Wei Y., Wai D.C., Low J.K.K., Milazzo J.P., Wu X.S.et al.. A transcription factor addiction in leukemia imposed by the MLL promoter sequence. Cancer Cell. 2018; 34:970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Avsec Ž., Weilert M., Shrikumar A., Krueger S., Alexandari A., Dalal K., Fropf R., McAnany C., Gagneur J., Kundaje A.et al.. Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat. Genet. 2021; 53:354–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Minnoye L., Taskiran I.I., Mauduit D., Fazio M., Van Aerschot L., Hulselmans G., Christiaens V., Makhzami S., Seltenhammer M., Karras P.et al.. Cross-species analysis of enhancer logic using deep learning. Genome Res. 2020; 30:1815–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Avsec Ž., Kreuzhuber R., Israeli J., Xu N., Cheng J., Shrikumar A., Banerjee A., Kim D.S., Beier T., Urban L.et al.. The Kipoi repository accelerates community exchange and reuse of predictive models for genomics. Nat. Biotechnol. 2019; 37:592–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.