

Abstract
The retroviral protein Integrase (IN) catalyzes concerted integration of viral DNA into host chromatin to establish a permanent infection in the target cell. We learned a great deal about the mechanism of catalytic integration through structure/function studies over the previous four decades of IN research. As one of three essential retroviral enzymes, IN has also been targeted by antiretroviral drugs to treat HIV-infected individuals. Inhibitors blocking the catalytic integration reaction are now state-of-the-art drugs within the antiretroviral therapy toolkit. HIV-1 IN also performs intriguing non-catalytic functions that are relevant to the late stages of the viral replication cycle, yet this aspect remains poorly understood. There are also novel allosteric inhibitors targeting non-enzymatic functions of IN that induce a block in the late stages of the viral replication cycle. In this chapter, we will discuss the function, structure, and inhibition of retroviral IN proteins, highlighting remaining challenges and outstanding questions.
Keywords: hydrolase, enzyme, drug design, retrovirus, antiretroviral therapy
Introduction
The Retroviridae family describes a group of enveloped viruses that contain two copies of single-stranded, positive-sense RNA molecules. Retroviruses infect a wide range of animals, leading to blood and tissue pathologies. Genome and protein sequence analyses have divided the family into seven genera: Alpha-retrovirus, Beta-retrovirus, Gamma-retrovirus, Delta-retrovirus, Epsilon-retrovirus, Spumavirus, and Lentivirus (Figure 1). A defining feature of all retroviruses is their ability to integrate the complementary DNA copy (also known as viral DNA or vDNA) of their reverse transcribed RNA genome into host chromatin to establish a permanent infection in the target cell. Integration can occur in principle at any site in the genome, although there are preferences for certain regions of chromatin. This integration process is catalyzed by an essential retroviral enzyme, integrase (IN), whose primary function is to insert the vDNA into host chromosomal target DNA (tDNA) through endonucleolytic cut and paste reactions. IN has also become an important target for the latest antiretroviral therapeutics used in the clinic to treat HIV-infected individuals. In this chapter, we will focus on the structure, mechanism, and the development of inhibitors that target IN protein, highlighting some of the most relevant findings from HIV-1 and other retroviral INs.
Figure 1.

Phylogenetic tree of retroviral INs.
Enzymatic functions of retroviral IN
The discovery that a retroviral protein possesses DNA endonuclease activity was initially shown more than four decades ago with the first purification and enzymatic characterization of a 32 kDa protein, now termed IN [1]. Subsequently, numerous mechanistic aspects of the enzyme have been clarified through careful biochemical and structural studies. Retroviral IN possesses two distinct enzymatic activities and catalyzes two consecutive reactions. These reactions are termed 3´-processing and strand transfer [2]. During 3´-processing, di-or-trinucleotides are removed from the 3´ ends of vDNA, leaving exposed a reactive 3´-hydroxyl on each vDNA end [3-7]. These 3´-hydroxyls are activated to catalyze strand transfer into a tDNA. IN then joins the processed 3´-hydroxyl vDNA ends with a scissile phosphate on the opposing tDNA strands through SN2-type nucleophilic substitution [2]. Integration is irreversible if both ends of vDNA (U3 and U5 attachment sites of long terminal repeat sequences) are inserted into tDNA, termed “concerted integration”. However, if only one end is inserted, integration is incomplete and termed “half-site integration”, leaving one unintegrated end. Concerted integration is irreversible and establishes a permanent proviral infection within the target cell. The vDNA that is incorporated into the host cell genome is referred to as a provirus. In vitro, these reactions are performed in the context of a nucleoprotein assembly, which consists of multimers of IN bound to vDNA ends. The fact that a multimer of IN is required to catalyze integration into host DNA was immediately apparent upon consideration of the reaction, because two cuts in tDNA are necessary for concerted integration. Accordingly, early studies showed that multimers of IN assemble into a nucleoprotein complex for the integration reaction [8,9]. Once integrated in the host genome, the multimers of IN must be disassembled, and the two gaps and short single-stranded overhangs that remain following the integration reaction are repaired by cellular enzymes. These latter steps are among the less well studied processes in the retroviral replication cycle and have been reviewed elsewhere [10].
The functional stable association of integration activity (and by inference IN protein) with vDNA ends was first established by studies with preintegration complexes (PICs) [4,11-13], which are large nucleoprotein complexes that include the vDNA made by reverse transcription and a number of viral and cellular proteins. PICs are competent for efficient DNA integration in vitro when provided with a divalent metal ion and target DNA [11,12]. However, PICs are present in very low abundance in cell extracts and therefore not readily amenable to biochemical and structural studies. Biochemical studies took a major leap forward when it was shown that retroviral IN is both necessary and sufficient for the catalytic steps of DNA integration in the presence of linear DNA that mimics the ends of the vDNA, a divalent metal ion, and a target DNA (tDNA) [14-16]. Mutations in IN affected 3´ end processing and DNA strand transfer in parallel, suggesting a common active site for both reactions. However, these reaction systems lacked the full fidelity of biologically relevant concerted DNA integration because the reaction products were mostly the result of integration of a single DNA end into one strand of tDNA. These earlier reaction systems with purified IN also provided little evidence for the stable nucleoprotein intermediates that we now call intasomes.
An intasome is a collective term for the series of stable biochemically assembled nucleoprotein complexes on the integration reaction pathway (Figure 2). A pair of vDNA ends are first bridged by a multimer of IN to form the Stable Synaptic Complex (SSC) intasome. Two (or for some retroviruses three) nucleotides are cleaved from the 3´ ends of the vDNA within the SSC to form the Cleaved Synaptic Complex (CSC) (this is the 3´-processing step introduced above). The CSC then captures a target DNA to form the Target Capture Complex (TCC). Finally, nucleophilic attack by the 3´ hydroxyl ends of the vDNA on a pair of phosphodiester bonds in the tDNA result in covalent joining of the viral and target DNAs. IN remains stably associated with vDNA in the resulting Strand Transfer Complex (STC) (Figure 2). The sites of nucleophilic attack on the two tDNA strands are separated by a spacing that is characteristic for each retrovirus (typically four to six bases). The pathway for this type of reaction was first established with DNA transposition reactions where the corresponding stable complexes are called transpososomes [17-20]. In the case of bacteriophage Mu STC transpososome an active energy-dependent disassembly process is required to strip the transposase from the DNA product [21]; the mechanism of retroviral STC intasome disassembly, which is required for access of cellular enzymes to complete the integration process, remains to be determined.
Figure 2.

Schematic mechanism of intasome assembly. Under the right biochemical conditions, an oligomer of IN proteins assembles on the ends of vDNA to form the stable synaptic complex (SSC). IN then cleaves two nucleotides from the 3′ ends of vDNA, forming the cleaved synaptic complex (CSC) that exposes the conserved free 3′-OH groups of the catalytically competent CA dinucleotides. CSC intasomes can capture tDNA to form the target capture complex (TCC), which will rapidly catalyze strand transfer to form the post-catalytic strand transfer complex (STC) in which the tDNA and the integrated vDNA are still bound to the intasome. In this schematic, there are two oligonucleotides that mimic the two long-terminal repeats of vDNA ends, which facilitates in vitro assembly and biochemical/structural studies. However, in a cell, these would be replaced by a single vDNA genome containing long-terminal repeats on each end, respectively.
Structures of individual IN domains
The structures of retroviral IN domains provided initial insights into the function of the enzyme. Retroviral IN proteins range from ~280 to ~400 residues in length and have three structurally conserved domains characterized by low sequence identity (Figure 3). All retroviral INs include the N-terminal domain (NTD), the catalytic core domain (CCD) and the C-terminal domain (CTD), which are essential for performing the basic catalytic function of IN – to insert vDNA into tDNA. Some retroviral INs also have an N-terminal extension domain (NED), which is required for function in the specific retrovirus. There is a substantial variability in the length of the linkers connecting the three domains, and this appears to confer differences to the structure, organization, and function of different retroviral INs.
Figure 3.

Domain organization and conservation of retroviral IN proteins. (A) Schematic representation of different retroviral INs. The distinct domains (NED, NTD, CCD, and CTD) are indicated, along with amino acid positions. (B-D) sequence conservation among retroviral IN domains, including (B) the NTD, (C) the CCD, and (D) the CTD.
The NTD ranges in size from ~40-50 residues (Figure 4A-B). The first structures of retroviral IN NTDs were determined by NMR and described for HIV-1 and HIV-2 [22,23]. All retroviral IN NTDs share the highly conserved HHCC motif (HX3H … CX2C) that coordinates Zn2+ and is required for the proper folding of the domain into a three-helix bundle. In HIV-1 (PDB: 6put), the residues that coordinate Zn2+ are His12, His16, Cys40 and Cys43. Deletion or substitution of any residue within the HHCC motif or depletion of Zn+2 by chelating agents leads to unstructured NTD, which compromises IN activity [23,24]. Individually expressed NTDs are dimeric [22] and do not appear to bind DNA in vitro [25,26], but they do have interactions with DNA when part of higher-order nucleoprotein assemblies, which will be discussed later.
Figure 4.

Structures of individual IN domains. (A) The NTD from HIV-1 IN first solved by NMR, showing the basic organization and the Zn-binding HHCC motif. (B) Overlay of NTDs from different retroviral INs in the public domain. (C) Overlay of CCDs from different retroviral INs in the public domain. (D) Overlay of the CCD from HIV-1 IN with the catalytic domain from transposases, Metnase and Mos1. (E) Overlay of CTDs from different retroviral INs in the public domain. (F) The NED from PFV shown in its configuration interacting with vDNA. (G) Overlay of the NED from PFV and from MoMuLV.
The CCD comprises ~150 residues and is the most conserved IN domain (Figure 4C-D). It is responsible for the catalytic enzymatic activity, and all retroviral CCDs harbor the invariable catalytic triad motif D…DX35E [27], which is also present within a large superfamily of nucleases, recombinases and transposases [25,28]. In HIV-1, these residues are Asp64, Asp116, and Glu152. Collectively, they form a triad of negatively charged residues that coordinate a pair of divalent metal ions (Mg2+ or Mn2+) and participate in the enzymatic cleavage reaction catalyzed by IN. The mutation of any one of these residues will abolish enzymatic activity [27,29,30]. The first structure of the CCD was determined crystallographically for HIV-1 IN [31] and was facilitated by the mutation F185K, which improves the protein solubility [32,33]. This structure provided insights into the IN active site and the configuration of the residues, which led to a better understanding of the cleavage mechanism. There are many more CCD structures now available from different retroviral INs. A superposition of 11 CCD structures available in the PDB highlights the degree of conservation among them (Figure 4C), as well as among other members of the superfamily, such as the transposases Mos1 [34] and Metnase [35] (Figure 4D). This conservation reinforces the role of the CCD domain in the catalysis of nucleic acids more generally. The CCD can also catalyze the “disintegration” reaction – which is the reverse of the integration reaction – albeit much weaker than the forward reaction, and disintegration only occurs in the presence of Mn2+, but not with the biological cofactor Mg2+ [36,37]. Disintegration is likely particular to in vitro conditions and would be disfavored in a cell, because the enzyme, in the context of the Mg2+-bound intasome assembly, is tuned to drive the forward reaction (as will be discussed below). When expressed alone, the CCD forms stable dimers [31,38-41].
The CTD comprises ~50 residues and is the least conserved domain among INs. Retroviral CTDs are characterized by a Src homology 3 (SH3)-like β-barrel fold [42,43]. A superposition of the CTDs that are available in the PDB shows the overall similarity within the β-barrel, but also highlights the divergence in the loop regions, which play important roles in the structural organization of the higher-order nucleoprotein assemblies (Figure 4E). Retroviral CTDs have some correspondence to the Tudor family of chromatin binding domains, highlighting their involvement in DNA-binding, which is relevant to the enzymatic activity of IN. When expressed alone, the CTD can also adopt both monomeric and dimeric forms [42-44].
The NED that precedes the NTD is only found in members of Spumaviruses, Gamma-retroviruses, and Epsilon-retroviruses, and is characterized by ~50 residues. The structure of the NED has been initially described in the context of nucleoprotein intasome assemblies formed by PFV IN [45,46]. In these structures, the NED interacts with the phosphodiester backbone of the vDNA through a patch of positive residues (Figure 4F), which help to assemble and stabilize the nucleoprotein complex. The NED from MLV IN1-105 was described more recently [47], which shares 19% identity with the NED from PFV. Their secondary structures are somewhat conserved, whereas the tertiary structure shows important differences that are mostly associated with the length of the linkers connecting the alpha-helices to the beta-sheets within both structures (Figure 4G). The NED region of MLV is required for the proper folding of the NTD and also impacts integration activity [48]. Given the relevance of both, the combined NED+NTD domains have been referred to as the N-terminal region (NTR), to avoid confusion with retroviral INs that do not possess this domain [47].
IN multimerization
Retroviral INs are highly dynamic proteins that are capable of forming multiple oligomeric species in solution. The oligomerization properties depend on the fragment of the protein used, the strain, the protein concentration or buffer conditions, among other factors. For example, it has long been known that HIV-1 IN can form a range of oligomeric states, from monomer to dimer to tetramer, and the exact state will be dependent on the buffer conditions and IN concentration [32]. Other retroviral INs appear to either exist as monomers or dimers. Most of the individual domains described in the previous section (NTD, CCD, CTD) crystallized as dimers, or were dimeric under solution NMR conditions (Figure 5A-C). Lentiviral INs are currently the only genera where the IN can form multimers greater than dimer [49-51]. However, it appears that the tetrameric form, in solution, only exists when all three domains are present, and there are also many mutations that readily disrupt IN tetramerization [32,50,52,53]. This suggests that in biochemical assays the purified recombinant tetramer, at least in HIV-1, is somewhat unstable, and this instability may have interesting biological implications.
Figure 5.

Structures of oligomeric and multi-domain constructs of IN. (A-C) Individual domains of HIV-1 IN form homodimers. Each domain is colored accordingly to the domain representation on the top left of the panel. (D-F) Structures of the SIV, RSV and HIV-1 INs comprising the CCD-CTD domains and organized into dimers. (G) Structure of the NTD-CCD multi-domain construct from HIV-1 IN in a tetrameric configuration. The tetramer, or dimer of dimers, is maintained by CCD-CCD dimers that interact through the loop comprising residues ~186-195, as well as interactions involving NTD dimers. (H) Structure of the NTD-CCD multi-domain construct from MVV IN, also in a tetrameric configuration. This complex also had the IN-binding domain (IBD) of the protein LEDGF/p75 (discussed later) bound to each CCD dimer. This structure differs slightly by the manner in which the CCD-CCD dimers interact, as well as by the configuration of the NTDs. (I) Structure of the NTD-CCD multi-domain construct from HIV-2 IN, packed as a trimer of dimers in a distinct configuration from the other NTD-CCD constructs. This structure also had the IBD domain bound. (J) Structure of the NTD-CCD multi-domain construct from HIV-1 IN carrying a Phenylalanine at the N-terminus.
Structures of multi-domain constructs
The first multi-domain structure of IN that was determined experimentally encompassed the CCD-CTD domains. Structures from three distinct retroviral INs were determined around the same time – from Simian Immunodeficiency Virus (SIVmac) (Figure 5D) [54], Rous Sarcoma Virus (RSV) (Figure 5E) [55], and HIV-1 (Figure 5F) [56]. In all cases, the proteins crystallized as dimers and they all had their solubility optimized through point mutations. Both individual domains within these two-domain structures were similar to the isolated constructs resolved previously, but their arrangement with respect to one another was informative. In the CCD-CTD structures from SIVmac (IN50-293) and RSV (IN49-286), the CTDs bend back and interface with the core of the CCD dimer, connected by a flexible linker; in contrast, in the CCD-CTD structure from HIV-1 (IN52-288), the CTDs project from the core CCD dimer, connected by rigid extended alpha-helical linkers. These changes in the configurations of individual domains were the first indications of IN pliability, which likely plays an important role in IN function. In the following year, the structure of the HIV-1 NTD-CCD (IN1-212) was resolved (Figure 5G), which provided insight into the possible configuration within the nucleoprotein assembly [57]. Since then, three other NTD-CCD IN structures have been determined. The tetrameric (dimer of dimers) assembly of Maedi Visna Virus (MVV) IN1-219 (Figure 5H), which was also bound to the Integrase Binding Domain (IBD) of the host factor Lens Epithelium Derived Growth Factor (LEDGF, discussed later) revealed a similar overall organization, although there were differences in how the connectivity were interpreted between neighboring protomers in comparison to the HIV-1 structure [50]. The structure of the NTD-CCD (IN1-209) from HIV-2 was also determined in the presence of the IBD domain of LEDGF and crystalizes as a trimer of dimers with distinct NTD-NTD interfaces (Figure 5I) [58]. More recently, the crystal structure of the NTD-CCD (IN1-212) from HIV-1, which contains residue Phe at position 1 instead of Met (Met is typically used to facilitate bacterial overexpression of the protein) revealed a similar dimer of dimers arrangement, but again differences in the interfaces formed by the NTDs (Figure 5J) [59]. From the collective body of structures, the CCD-CCD interface is always conserved, but the interfaces among the other domains, which are connected by flexible linkers, frequently vary. These variations may arise from the sample itself, the conditions used for structure determination, or strategies for protein stabilization. Regardless, they suggest that the strong CCD-CCD dimerization interface scaffolds the IN multimer, but the final conformations and oligomeric configurations are dependent on the adjacent domains, among other factors. We should also expect INs from different genera to form distinct complexes that are context- and condition-dependent. Collectively, these data highlight IN plasticity and malleability, a hallmark of the protein. This plasticity is likely necessary for IN to assemble into nucleoprotein species and to perform its function. To date, there remain no structures of the full-length (FL) proteins in the absence of nucleic acid.
Structures of nucleoprotein intasome assemblies
Trapping of intasomes required the development of higher fidelity in vitro systems that were capable of concerted integration of pairs of viral DNA ends [60-63]. Under these conditions, highly stable complexes of IN and vDNA ends (SSC intasomes) are formed, that mimic the tight association of IN with vDNA in the PICs [64-66]. Once formed in vitro, HIV-1 intasomes carry out 3´ end processing and DNA strand transfer with very high efficiency, and the two vDNA ends are transferred to target DNA sequentially [65]. The two active sites within the intasome process the chemical reactions independently. As the assembly events on each vDNA end appear to also be independent [67], and the assembly of HIV-1 intasomes is generally very inefficient in vitro, many intasomes are difficult to assemble in vitro. Lentiviral INs, especially from HIV-1, also have a high propensity to aggregate. These factors frustrated attempts at structural studies of intasomes for a long time. The key breakthrough in high-resolution structural studies of retroviral intasomes came from Cherepanov and colleagues finding that prototype foamy virus (PFV) IN is highly active in vitro and soluble [38]. Moreover, it efficiently assembles intasomes that are soluble and homogeneous.
Crystallization of PFV intasomes resulted in the first structures of any retroviral nucleoprotein assembly containing IN [45]. The structure revealed that IN binds to vDNA in a tetrameric configuration, with the two inner IN protomers fully resolved and wrapping around the ends of vDNA; the two outer IN protomers only had their CCDs resolved, indicating that they provide supporting function and leading to the hypothsis that the outer NTDs and CTDs may play auxiliary roles in the context of chromatin engagement. In this tetrameric PFV intasome structure, the DNA-protein interactions act as the glue that holds the intasome together, and all three canonical IN structural domains are involved in extensive protein–DNA and protein–protein interactions. Such a configuration could not have been predicted from the known structures of the protein domains alone. The structure of the pre-catalytic TCC and the post-catalytic STC intasomes were described shortly thereafter and defined the structural basis of DNA integration [46]. A key finding was that the cleft between IN dimers within the intasome accommodates chromosomal DNA in a severely bent conformation. Presumably, this bent DNA configuration helps to decrease the activation energy required for catalysis, driving the forward reaction and minimizing the possible reversal via the disintegration reaction. This conclusion could only be obtained from experimental structures of the nucleoprotein assemblies, and could not be determined from structures of individual domains or multi-domain constructs. Target DNA bending was subsequently also supported by bioinformatics analyses [44,68,69].
All IN proteins catalyze an identical set of reactions and share considerable sequence homology, especially in the CCD (Figure 3). This mechanistic and sequence similarity led to the general expectation that all retroviral intasomes would be fundamentally similar in structure. However, the PFV intasome structure raised a conundrum. The tetrameric PFV intasome structure requires a long linker between the catalytic and C-terminal terminal domain, but many retroviral INs have only a very short linker between these domains, making such an intasome architecture impossible. The surprising finding was when the intasome from Rous Sarcoma Virus (an alpharetrovirus) and from the Mouse Mammary Tumor Virus (a betaretrovirus) were found to be composed of eight IN protomers [70,71]. In these structures, the synaptic CTDs were contributed by the flanking protomers, rather than by the core protomers. Still, lentiviral intasomes remained refractory to structural biology efforts. Several key advances facilitated high-resolution structures of lentiviral intasomes. In the case of HIV-1, fusion of a non-specific DNA binding domain of a chromosomal protein derived from a Sulfolobus solfataricus to the N-terminus of HIV-1 IN resulted in a protein that, under appropriate conditions, assembles intasomes that are highly active and exhibit much less aggregation than intasomes assembled with wt IN [72]. In lieu of modifying the protein, an alternative approach was to identify a lentiviral IN that would be naturally better behaved under stringent biochemical conditions; the IN from MVV [50], a lentivirus that infects sheep, could assemble intasomes that were better behaved biochemically than its HIV-1 counterpart. Secondly, advances in cryo-EM [73] enabled high-resolution structures to be determined from populations that are not entirely homogeneous [74-76]. These collective advances led to structures of intasomes from both HIV-1 and MVV [77,78]. The structures revealed a dynamic organization containing a minimum of four INs and as much as sixteen INs, all arranged around the central vDNA ends. There are also now intasomes from the Human T-Cell Lymphotropic Virus (HTLV, a deltaretrovirus) and from the Simian Immunodeficiency Virus (SIV, another lentivirus). Collectively, the distinct structures reveal a remarkable pliability in the IN protein and the distinct manner of intasome assembly (Figure 6). All retroviral intasome structures to date have a common set of positionally conserved IN domains around the DNA substrate – referred to as the conserved intasome core (CIC) – but these domains can be contributed by different IN protomers, depending on the retrovirus [79]. The functional consequences of the different assemblies remain less well defined, but are of significant interest.
Figure 6.

Structures of intasomes from different retroviruses. Each IN protomer is colored with a different color. The oligomeric forms are tetramer (PFV and HTLV1), octamer (MMTV and RSV), hexadecamer (MVV) and both tetramer and dodecamer (HIV). The SIV structural form depicted here is the base unit of an intasome stack, which is a run-on oligomer of incompletely formed “proto-intasome” octamers containing 2 vDNAs per octameric unit. They are not biologically relevant as an oligomeric species, but should contain an identical active site and can therefore be successfully used to study INSTIs.
Molecular mechanism of retroviral integration
Biochemical and structural studies have collectively shed light on the molecular mechanism of retroviral integration. Early stereochemistry experiments using HIV-1 IN [2] indicated that both 3´-processing and strand transfer reactions occur by a common mechanism, using water as the nucleophile for 3´-end processing, and the 3´ hydroxyls at the ends of the vDNA as the nucleophile for DNA strand transfer. However, as already mentioned, intasomes formed with HIV-1 IN were not amenable to structural biology for a long time. Due to its favorable biochemical properties, the PFV intasome provided a better model system with which to elucidate the mechanics of these two reactions through careful structural and biochemical experiments. Crystallographically determined structures of the CSC intasome and the TCC intasome from PFV provided the first snapshots of intasome formation [45,46]. Later, the structures of uncleaved SSC intasomes and TCC intasomes, in their ground states committed to 3´-processing and strand transfer, respectively, elucidated the complete enzymatic process [80]. These latter structures were particularly important in clarifying our understanding of the atomic mechanism of integration. For 3´-processing, the structures of the SSC in its ground state revealed that a water molecule located in the active site and adjoining the metal ion is positioned for nucleophilic attack onto the vDNA scissile phosphate that coordinates the two metal ions (Figure 7A). For strand transfer, the structure of the TCC in its ground state revealed that the exposed 3′-hydroxyl of vDNA resides in a position poised to perform a nucleophilic attack on the nearby scissile phosphate of tDNA (Figure 7B). The putative pathway of nucleophilic attack by the water molecule and by the 3′-hydroxyl of vDNA are indicated by bold arrows in Figures 7A-B. Following the transesterification reaction, the viral—target DNA phosphodiester bond is relocated to its new configuration. The mechanism has important analogies to the classical two-metal ion mechanism, which was first proposed for divalent ion-dependent DNA hydrolytic cleavage based on structures of a 3´,5´ DNA exonuclease [81] and has since been used as a mechanistic model for many metal ion-dependent phosphoryl transfer enzymes [82]. These structures also provided an explanation as to why tDNA binding must occur following 3′-processing. The scissile dinucleotide end of the vDNA is sterically incompatible with the presence of tDNA, and may therefore serve as a natural strand transfer inhibitor. The requirement to complete 3′-processing prior to engaging substrate DNA may be an evolutionary adaptation to reduce the chance of suicidal auto-integrations.
Figure 7. Structural basis of integrase 3′ processing and strand transfer activities.

(A) Structure of the manganese-bound SSC (PDB 4E7I). The water labeled WNuc performs a nucleophilic attack on scissile phosphodiester bond of vDNA (bold red arrow), displacing two or three nucleotides from the end. The DNA and IN backbones are colored khaki and blue, respectively; red and orange sticks are oxygen and phosphorus, respectively. Purple and red spheres are manganese ions and water molecules, respectively, with the nucleophilic water labeled WNuc. Purple dashed lines indicate metal ion interactions. (B) Overlay of metal ion-bound TCC (PDB 4E7K); DNA and protein in green) and STC (PDB 4E7L; DNA and protein in pink) structures. The 3´OH performs a nucleophilic attack on the scissile phosphodiester bond of tDNA (bold red arrow) [80]. Both sets of metal ions are shown; the 3.8 Å spacing between ions in the TCC contracts to 3.2 Å in the STC. The curved black line indicates the displacement of the viral-target DNA phosphodiester bond after strand transfer relative to the scissile bond in tDNA. Other labeling is the same as in panel A.
In a cell, the same basic molecular mechanisms must occur for functional integration, but the process is more involved, and the timing of individual events is less clear. The cellular form of the complex – the PIC – remains a mysterious and poorly characterized entity. Minimally, the PIC consists of multimers of IN bound to the linear ends of vDNA. In reality, the PIC contains ~10k base pairs of vDNA, which are likely chromatinized very soon after nuclear entry [83-85], and for HIV-1, the viral capsid (CA) protein [86] and potentially host proteins (discussed below) including the barrier to autointegration factor (BAF) [87]. Notably, there have been substantial advances (and active ongoing research) in understanding the role of CA protein in nuclear integration targeting, and thus its relationship to the PIC [86,88,89]. The in situ data is supported by the recent breakthrough efforts to reconstitute the early events in the replication cycle using an in vitro system, which also highlighted the involvement of CA in endogenous reverse transcription and integration [90]. In PFV, Gag forms part of the PIC [91,92], and similarly, in MLV, the viral protein p12 (a cleavage product of the Gag precursor), is also a well-characterized component in the PIC [93,94] in addition to viral CA protein. Although we now know much about the basic biochemistry of integration, elucidating the mechanistic details, the PIC assembly pathway, and the spatiotemporal and structural organization of these events in cells would be of significant interest. In the meantime, the recent in vitro reconstituted system [90] provides an attractive alternative to cells that may be more tractable for studying certain processes.
Chromatin engagement by a retroviral intasome
The cellular target for integration is chromatinized DNA. It has long been appreciated that integration is favored in sites of chromatinized DNA and disfavored in naked DNA [95-97]. The structure of the PFV intasome complexed to a nucleosome but before integration (simulating the TCC) elucidated the basis for nucleosome capture by a retroviral intasome (Figure 8) [98]. This structure showed that intasomes form multivalent interactions with a nucleosome that involve both gyres of nucleosomal DNA and one H2A–H2B heterodimer. The DNA is lifted from the surface of the H2A–H2B heterodimer to allow integration at a single strongly preferred superhelical location at position +/−3.5 on the nucleosome. This lifting of the DNA was previously postulated through structures of the TCC and STC intasomes, which also showed similar DNA bending, and it is likely necessary to decrease the activation energy and promote the forward integration reaction. In a cell, nucleosome engagement events are probably more frequent than actual integration events. In fact, intasomes appear to be actively scanning host chromatin for a considerable time, and most attempts to integrate into host tDNA are unproductive [99], suggesting that chromatin engagement and integration are functionally uncoupled. In vitro, allowing the intasome to conduct strand transfer into the nucleosome resulted in a post-integration structure (simulating the STC) with important structural differences that are induced during strand transfer [100]. The post-integration structure revealed a twisting and sliding of the nucleosomal DNA arm by approximately two base pairs along the histone octamer, proximal to the integration site. This sliding is apparently necessary to lift the DNA from the histone H2A-H2B subunits to allow engagement with the intasome. These two structures highlight some of the dynamic rearrangements, in the context of chromatin, prior to and after catalytic integration. Although it’s clear that chromatin engagement is a general hallmark of retroviral intasomes, the preferred chromatin substrates for integration among the other intasomes needs to be clarified, and characterizing nucleosome preferences by other retroviral intasomes will surely reveal novel insights into the integration process.
Figure 8.

Structure of the PFV intasome bound to the nucleosome core particle. The intasome binds to superhelical location at position +/−3.5 on the nucleosome
The role of host factors in the enzymatic activity of IN
The integration reaction may be facilitated by cellular host factors, which affect the targeting and enzymatic properties of some retroviral INs. The primary host factor that both binds HIV-1 IN and impacts integration is the 60 kDa splicing variant of the transcriptional coactivator lens epithelium derived growth factor (LEDGF/p75), or LEDGF for short. Using a synthetic gene that stably and efficiently expresses HIV-1 IN in a line of 293T cells, it was shown that IN associates with LEDGF in nuclear extracts [49]. LEDGF, a nuclear protein that is involved in gene regulation and cellular stress response [101,102], binds tightly to HIV-1 IN, increases its solubility, and stimulates enzymatic activity in vitro, as measured by both conventional strand transfer of cleaved oligonucleotides and through concerted integration into circular plasmid DNA [49]. The structure of the IN-binding domain (IBD) of LEDGF in its free form [103] and bound to HIV-1 IN [104], provided insights into how the protein engages the integration complex. In cells, LEDGF also affects chromatin targeting and integration site specificity. It has been known that DNA integration is favored in genes marked by active transcription [105]. By analyzing global integration patterns, it was found that HIV-1 integration in cells depleted for LEDGF was less frequent in transcription units, less frequent in genes regulated by LEDGF, and more frequent in GC-rich DNA [106]. Since LEDGF is a nuclear factor that binds chromosomal DNA [107-109] through its PWWP domain and HIV-1 IN through its IBD domain, the factor may directly facilitate integration through a bimodal tethering mechanism to bring the PIC to regions characterized by active transcription. Replacing the PWWP domain with other chromatin-tethering domains alters genomic integration patters in cells [110,111]. In the absence of LEDGF, a second factor, HDGF2 (or HRP2), was shown to play an ancillary role in directing integration into actively transcribed genes [112]. Both LEDGF and HDGF2 directly bind lentiviral IN [49,113]. Even though depletion of both LEDGF and HDGF2 further reduced specificity for transcriptional targeting compared to LEDGF knock out cells, the preference for actively transcribed genes was not fully lost. It has been shown that HIV-1 capsid binding cellular protein cleavage and polyadenylation specificity factor subunit 6 (CPSF6) plays crucial role in integration site specificity, and deletion of this factor redistributes integration targeting toward lamina-associated domains [88,89]. Although CPSF6 mediates integration specificity, it binds to the capsid (CA) protein, and not to IN directly.
A variety of other host factors have been suggested to mediate functional integration through specific binding to HIV-1 IN [114]. The Snf5 protein of the mammalian Swi/Snf complex was initially reported to bind HIV-1 IN through a yeast-two hybrid analysis and became known as “Integrase Interactor 1” or Ini1 [115]. However, a bona fide role for this protein in integration has not been defined, and it was later suggested that Snf5/Ini1 may play a role in late stages of the retroviral replication cycle post-integration, when IN is part of Gag-Pol [116,117]. The human FACT complex, which consists of the two proteins Ssrp1 and Spt16 [118-120], apparently plays a similar role to LEDGF and HDGF2 in transcriptional activation [121], but does not contain an IN binding domain. The data on FACT and its involvement in retroviral integration is of interest, and multiple groups have reported results working with different retroviral INs. The FACT complex was suggested to bind ASLV IN (ASLV, like Rous Sarcoma Virus, belongs to the alpha-retroviral genus), but not HIV-1 IN, and knockdown of FACT components impaired ASLV integration into naked DNA in vitro and into cellular chromatin in vivo, but not HIV-1 integration [122]. In separate work, the lack of integration into naked DNA was reproduced, but FACT enhanced integration into chromatinized templates in vitro [123]. The conclusion from the latter work was that the FACT complex affects integration specificity by mediating local nucleosome dissociation and modulating the functional association between the HIV-1 intasome and the targeted nucleosome [123]. In separate work, the FACT complex was not implicated in viral integration, but instead was suggested to contribute to LTR-driven gene expression [124]. These collective data point to an interesting interplay between chromatin remodelers and retroviral INs, but also indicate that the role of FACT in viral integration remains an open question, and further experimental work needs to be conducted to clarify its role. Numerous other factors have been pulled down with HIV PICs (which contain numerous other proteins in addition to IN) [114,125]. An important consideration is whether the binding factor mediates function. Although in vitro biochemical assays, such as strand transfer and/or concerted integration, can shed some light on enzyme activity, whether stimulation of integration occurs, or is necessary, in vivo is unclear. IN is a non-processive enzyme and only needs to perform a single integration event during the retroviral replication cycle, which contrasts it from processive enzymes such as reverse transcriptase. Therefore, in vitro stimulation of activity only reveals part of the story, and it is important to examine whether a host factor affects integration in cells. With this criterion in mind, to date the only IN-binding proteins that play a clearly defined role in HIV-1 integration are LEDGF and HDGF2. Although other host factors, such as CPSF6, clearly affect integration targeting (reviewed in [86]), they do not directly bind IN, and are the mechanism is distinct.
Cellular host factors have also been identified that bind other retroviral INs and facilitate integration targeting. The bromodomain and extraterminal domain (BET) proteins were shown to preferentially bind gammaretroviral INs [126] and to direct integration to regions upstream of transcription start sites [127-130]. The primary forms of the BET proteins are Brd2, Brd3, and Brd4 (others exist in tissue-specific contexts). These proteins preferentially bind to acetylated histones, which are characteristic of upstream transcription start sites [131]. The association of BET proteins with gamma-retroviral IN provided mechanistic explanation to earlier observations that integration occurs preferentially within transcription start sites and upstream promoter regions [132]. Purified Brd4 protein binds tightly to Murine Leukemia Virus (MLV) IN and stimulates concerted integration, and small molecules targeting of BET proteins impaired MLV, but not HIV-1 integration in cells [127]. Like LEDGF, BET proteins appear to bring MLV intasomes to select chromatin locations through a bimodal tethering mechanism, binding IN and chromatin through two distinct locations on the proteins [133]. These collective data supported an earlier report showing that gamma-retroviral integration is preferred in transcription start sites and upstream promoter regions [132]. More recently, the cellular serine/threonine protein phosphatase 2A (PP2A) was identified as a binding partner of delta-retroviruses, which include the human T cell lymphotropic virus type 1 and 2 (HTLV-1 and HTLV-2), as well as bovine leukaemia virus (BLV) [134]. Structures of HTLV-1 intasomes in complex with PP2A were recently reported, and knockdown of the PP2A was shown to affect HTLV-1 infection [39,135]. However, the exact role of PP2A in delta-retroviral intasome function remains unclear, and other than binding IN, what this cellular host factor is doing in the context of integration remains to be determined. There are otherwise currently no known host factors for other retroviral genera, including beta-retroviruses, epsilon-retroviruses, and spumaviruses.
Non-catalytic functions of retroviral IN
Whereas the role of IN in the early stages of the viral replication cycle have been extensively studied, an unexpected role of IN within the late stages of the replication cycle has been brought to light. Recent discoveries showed that IN has non-enzymatic functions during virion maturation, following proteolytic processing of gag-pol. In HIV-1, IN binds the viral RNA genome in virions and exhibits distinct preference for select viral RNA structural elements, indicating specificity in this interaction [136]. Disruption of IN-mediated RNA binding leads to the formation of eccentric, non-infectious virions, and the viral genome is mislocalized outside of the conical capsid core [136-138]. These observations of mislocalized cores were reminiscent of early studies, which suggested that certain IN mutations – termed Class II mutations – led to the production of virions that were characterized by a broad range of defects at distinct stages of the replication cycle [139,140]. The Class II mutations are distinct from the Class I mutations, which selectively affect the integration step. By examining a wide body of mutations, it was clarified that disruption of IN-mediated RNA binding can occur through one of three possible mechanisms: (1) depletion of IN from virions, which would limit the amount of IN available to facilitate RNA packaging; (2) impairment of functional IN multimerization, which would disrupt proper IN/RNA binding; (3) direct disruption of IN/vRNA binding without affecting other biochemical or enzymatic properties, such as by obstructing the interaction interface [137]. Collectively, any inhibition of IN/vRNA interactions resulted in mislocalization of the viral ribonucleoprotein complexes outside the capsid lattice, which in turn led to premature degradation of the viral genome and IN in target cells. These data shed light on the functional phenotype of the Class II mutant subset. The data indicates that many of the defects that were traditionally ascribed to any of a number of early stages of the lifecycle, including defects in reverse transcription, nuclear import, or uncoating, may collectively result from an initial mislocalization of ribonucleoprotein complexes outside of the protective capsid shell during virion morphogenesis [141]. Treatment of virus producer cells with allosteric IN inhibitors (ALLINIs, to be described below) impairs IN-RNA binding in virions, also yielding mislocalization of ribonucleoprotein complexes [136,138,142]. Taken together, these exciting results suggest that HIV-1 IN plays a non-catalytic biological role in virion morphogenesis to ensure correct positioning of ribonucleoprotein complexes within protective capsid shell. Understanding how this process occurs at the molecular and biochemical level will be of interest. It is curious that with MLV, complete deletion of IN does not affect the viral replication cycle up to completion of reverse transcription [143]. A positive role of IN in morphogenesis may not therefore be a general feature of all retroviruses.
Catalytic Inhibition of Integration
The search for small molecule inhibitors targeting HIV-1 integration has been ongoing for nearly three decades. The early literature describing steps toward identifying IN inhibitors has been extensively reviewed [144,145]. Initial attempts identified small molecules that bind IN, but none of them potently inhibited integration in cells. This changed when the assay used to evaluate IN inhibitors was redesigned to evaluate strand transfer, rather than 3´-processing. In a series of papers, Hazuda and coworkers staged the complete integration reaction and defined the requirements for nucleoprotein assembly and the enzyme’s separate catalytic activities, namely 3´-processing and strand transfer [146,147]. This provided a means by which to screen compounds that inhibited IN strand transfer activity, specifically, which ultimately lead to the first potent IN inhibitors [148,149]. These compounds were characterized by a β-diketo acid (DKA) moiety as their pharmacophore, and subsequently became known as the IN strand transfer inhibitors, or INSTIs. There remained a major concern – that INSTIs would have exactly one shot at blocking the catalytic reaction, because integration needs to only occur once to establish infection in the target cell. This property contrasts IN with other processive enzymes, such as RT, and cast doubt on the promise of IN inhibition as a therapeutic strategy. However, eventually the new inhibitors turned out to be highly effective. It was later clarified that one reason for their efficacy is the very long dissociative half-life of the drug from the nucleoprotein assembly [150,151]. The early efforts lead to the first DKA INSTIs, and ultimately to clinically approved drugs. Below we will briefly describe the distinct compounds, their mechanisms of action, and pathways toward viral resistance. We note that this topic has been extensively reviewed over the last year, and we point the interested reader to this literature for further details [152-154].
Clinically approved INSTIs for use in antiretroviral therapy
There are now five INSTIs that are clinically approved by the US Food and Drug Administration (FDA) for use to treat HIV-infected individuals, and INSTIs are standard of care in ART regimens. These drugs include the first generation INSTIs Raltegravir (RAL, approved in 2007) and Elvitegravir (EVG, approved in 2011), as well as the second generation INSTIs Dolutegravir (DTG, approved in 2013), Bictegravir (BIC, approved in 2018) and Cabotegravir (CAB, approved in 2021). The chemical structures of these drugs are shown in Figure 9. With the exception of CAB, all the currently approved INSTIs are given as oral medications; CAB is the first injectable regimen for HIV-infected adults and is administered once monthly (an oral form of CAB was also approved to be used with Rilpivirine, a non-nucleoside reverse transcriptase inhibitor, for the first month prior to starting the injectable form of treatment) [155,156]. CAB was found to have an unusually long half-life, which led to its formulation as a long-acting injectable. The comparative safety and efficacy of these drugs has been recently discussed [157].
Figure 9.

Clinically approved drugs used to treat HIV-infected individuals. The first-generation drugs include Raltegravir and Elvitegravir, and the second generation drugs include Dolutegravir, Bictegravir, and Cabotegravir.
All clinically approved INSTIs have specific chemical features and a well-defined mechanism of inhibition. Early structures of the prototype foamy virus (PFV) intasome bound to both first and second generation INSTIs provided insights into the mode of INSTI binding [45,158,159]. The structures of PFV intasomes bound to INSTIs revealed commonalities between the different ligands. INSTIs are generally characterized by: (1) the presence of three electronegative atoms that chelate two Mg2+ ions in the active site of IN and that are typically embedded into or around a core scaffold of the ligand; (2) a halogenated benzene moiety that inserts into a pocket of the nucleoprotein assembly and makes a π-π stacking interaction with the penultimate base of viral DNA; (3) a linker that connects the core scaffold to the halogenated benzene [160]. The structural data also shed light on chemical strategies for INSTI improvement and modification, and there have been multiple improvements that conferred potency to the compounds, leading to the second-generation inhibitors [158]. First, the linker connecting the core scaffold to the halogenated benzene moiety was lengthened, which allowed the benzene moiety to extend deeper into a pocket and make a stronger interaction with the penultimate vDNA base. Second, the core scaffold was enlarged, and a third ring was appended to generate more surface contact with the IN active site, allowing the ligand to bind with increased stability in the active site. We now have structures of lentiviral intasomes from SIVrcm and from HIV-1 bound to second generation INSTI drugs [161,162] (Figure 10A). Since the active sites of lentiviral and PFV intasomes are <50% identical, these new structures highlighted small but significant differences between the two active sites, which confer some variability to the binding mode of certain drugs. The common feature of all INSTIs is that the vDNA forms part of the binding motif, which explains why INSTIs target intasomes specifically, and not free IN [148]. As a result, INSTIs are also fairly specific for their biological target, and they appear to have low off-target binding. Although there have been early reports that some INSTIs bind the Rag1/Rag2 recombinase enzymes [163], which form nucleoprotein assemblies with DNA that remotely resemble intasomes, the binding affinity of INSTIs toward Rag1/Rag2 assemblies is in the uM range [164,165], and whether such off-target effects are clinically significant remains unclear. In summary, INSTIs are potent and now established drugs with a well-defined binding mode and mechanism of inhibition.
Figure 10.

Binding modes of second generation and developmental naphthyridine-scaffold INSTIs to lentiviral intasomes. (A) Superposition of second generation INSTIs bound to SIVrcm and HIV-1 intasomes. (B) Superimposed binding modes of the second generation INSTI BIC and the developmental naphthyridine-scaffold compound 4d, bound to the HIV-1 intasome, are displayed. The compound 4d binds closer to the Mg2+ ions because the electronegative atoms are embedded into the core of the scaffold. The terminal adenine base of vDNA and all water molecules are omitted for clarity.
Developmental INSTIs containing the naphthyridine core scaffold
Nearly two decades ago, compounds containing a naphthyridine core were shown to contain IN inhibitory activity [166-168]. These compounds were developed into potent IN inhibitors that rival or even outcompete clinical drugs in terms of their ability to inhibit a broad range of viral resistant variants (VRVs) [169-172]. The best naphthyridine compounds were designed to include the features of the more potent second generation INSTIs, including: a halogenated benzyl moiety that base pairs with the penultimate cytosine of viral DNA; a long linker that connects the halogenated benzene to the core scaffold, while conferring flexibility to the INSTI against VRVs; modifications around the core scaffold that improve potency to the compounds. Overall, the binding mode of the naphthyridine compounds resembles that of all other INSTIs, with the exception that they bind closer to the IN active site by virtue of the fact that one of the three electronegative atoms is embedded directly into the core INSTI scaffold [162,169] (Figure 10B).
The structures of HIV-1 intasomes bound to naphthyridine-containing INSTIs provided novel insights into the small but significant variability of drug binding. Sequence divergence between HIV-1 IN complicates extrapolation of the PFV structural data to HIV-1, and many residues in HIV-1 that confer INSTI resistance when mutated are not conserved in PFV. This lack of conservation leads to important differences in how naphthyridine-containing INSTI binds to the active site of each IN. For example, the exact same ligand (either the developmental naphthyridine-based inhibitors 4f or 4c) binds distinctly to the different intasomes (Figure 11). The chemical substituent that is appended to the core scaffold of the INSTI and confers potency to the ligand extends out toward the solvent-exposed cleft when bound to the HIV-1 intasome, but bends back when bound to the PFV intasome; in the case of the sulfhydryl benzene moiety, the substituent makes an intramolecular π–π stacking interaction with the INSTI core and dislodges it from its preferred location. These recent data indicate that the HIV-1 intasome should now be used for subsequent efforts aimed at understanding drug binding and the acquisition of viral resistance, which will be discussed in the following section.
Figure 11.

INSTIs can bind differently to PFV and HIV intasomes. (A and B) Compound 4f bound to the (A) HIV (pink) and (B) PFV (gray) intasome. (C) Overlay of compound 4f binding modes. (D and E) Compound 4c, containing a 6-pentanol substituent, bound to the (D) HIV (green) and (E) PFV (gray, PDB 5FRN) intasome. (F) Overlay of compound 4c binding modes. Compound 4d, containing a 6-hexanol substituent, is also shown in its binding mode to the HIV (light blue) intasome. In (A), (B), (D), and (E), intasome active sites are shown as surface views, with labeled residues. R231 is poorly ordered in the map and is, therefore, displayed as an Ala stub. The terminal adenine is removed for clarity.
Viral resistance against clinically used INSTIs
All INSTIs are extremely potent inhibitors of WT IN, with affinities in the low nM range. The major difference between the first and second generation INSTIs primarily has to do with their ability to inhibit mutant forms of IN. Whereas both RAL and EVG can potently inhibit the WT enzyme, the virus quickly develops resistance against both compounds [173]. The first-generation INSTIs are susceptible to the emergence of viral resistance, and once resistant variants emerge, neither of these compounds is able to inhibit VRVs. In contrast, the second-generation INSTIs and the developmental naphthyridine-containing INSTIs can potently inhibit mutant forms of IN that arise during the course of treatment with the first-generation inhibitors [171,172,174]. Nonetheless, resistance to second generation INSTIs still occurs [175]. Resistance is particularly acute in HIV-1, because the virus rapidly selects for drug-resistant mutations, and viral sequences within an infected individual can differ by 10% or more [176]. Viral diversity and the rapid selection for drug-resistant mutations imposes challenges on the development of treatment strategies for people living with HIV. These challenges may also be exacerbated when INSTIs are administered sub-optimally, leading to further complications and genetic recombination in the infected cell [177,178].
The basic mechanism of resistance follows a well-defined pattern. First, an initiating mutation reduces the binding affinity of the drug but frequently confers a fitness penalty; secondary resistance substitutions arise after continued drug pressure and alleviate the negative effects of primary mutations and restore viral fitness, and they may also increase the level of INSTI resistance [179]. The quintessential pair of viral mutations that arises in response to continued INSTI treatment is the G140S/Q148H double mutant. Glu148 is initially mutated to His, which reduces the dissociative half-life of the compound and facilitates drug displacement, but this mutation comes with a fitness cost. Subsequently, Gly140 is mutated to Ser, and this mutation restores viral fitness while maintaining the decreased ligand binding affinity [180]. The mechanism of resistance was elucidated recently through structural studies using the SIVrcm intasome [161]. A change at residue 148 increases the local electropositivity of and redistributes the local charge around the Mg2+-ligand cluster, weakening the interaction between the drug heteroatoms and the metal ions. There are other mutations that can arise in these amino acids, and the collective set is typically defined as G140A/S + Q148H/K/R escape pathway. This general escape pathway is important to understand, because it underlies the binding mode of all INSTIs. There are also many complex mutations (“complex” is defined as having three or more residue changes compared to WT) that develop along this pathway, which may further impair binding and eventually lead to viral escape [171,172,174,181]. Several other escape pathways have been described. Resistance involving the residue Y143 is a frequent cause of RAL failure [182]. Resistance through the Y143 pathway can be readily explained, because RAL makes a putative π–π stacking interaction with Y143 in HIV-1 IN (the mechanism was defined with the prototype foamy virus IN, which contains Y212 at that position), and loss of this stacking interaction considerably reduces the binding affinity of the drug [45]. N155H is also frequently selected during INSTI treatment and appears to represent a separate pathway. Although an attempt was made to define the mechanism of resistance using the PFV intasome as a model system [159], given the considerable structural differences between the active sites of HIV-1 and PFV IN [161,162], the mechanistic basis for this pathway needs to be revisited with structures of lentiviral intasomes. Newer pathways that appear to be selective for second-generation INSTIs include the R263K and G118R variants [183-185]. There is currently no mechanistic explanation for these resistance pathways, and structures of mutant intasomes bound to second-generation INSTIs need to be determined. There are many other IN mutants that have been identified that exhibit decreased susceptibility to INSTIs (reviewed in [153]). Tissue culture experiments using serial viral passages have been helpful in identifying novel variants and may be good predictors of escape pathways that arise in patient populations [153,175,183]. There are multiple systematic reviews of mutations to INSTIs that arise in the clinic and/or in cell culture experiments, and the reader is pointed to those for further details [153,181].
One approach to combat resistance is to constrain the ligand to the regions characterized by the molecular substrate envelope of the enzyme. The substrate envelope is the molecular envelope of an enzyme that binds natural substrates. In the case of IN, the natural substrates are both viral and target DNA. If the ligand is maintained entirely within the natural substrate envelope of the enzyme, then a resistant variant that displaces the ligand must protrude into the natural substrate envelope. This may compromise viral fitness, and the resistant variant is less likely to persist in the viral population. Conversely, if the ligand protrudes out of the substrate envelope, then it is easier for resistant variants to displace the ligand and have no impact on the natural function of the enzyme, thus increasing the chance of maintaining normal viral fitness. The idea of maintaining a ligand within the substrate envelope to avoid the development of resistance was initially developed with and applied to the design of HIV-1 Protease inhibitors [186-188]. This idea was then adapted for use with INSTIs, initially using the atomic structure of the PFV intasome for defining the substrate envelope [169] and then using the atomic structure of the HIV-1 intasome [162]. Optimization of INSTIs should take into account this simple principle to increase the probability of identifying compounds that will remain potent against VRVs. The idea may be more broadly applicable to other studies of small molecule inhibition.
Single round replication assays using clinical isolates of HIV-1 have enabled accurately defining the replication capacity of mutant viruses and the inhibitory potencies of INSTIs against resistant viruses [153,175]. These assays, in combination with structural biology and synthetic chemistry, have been essential for systematically comparing different INSTIs against broad panels of VRVs that arise either in the clinic or in tissue culture and establishing structure—activity relationships to guide INSTI design efforts. For example, it was possible to define how substituents could be optimally positioned around the naphthyridine core. The addition of an amino group at the 4- position yielded substantial improvements in potency against important VRVs [189]. Similarly, the addition of substituents at the 6- position typically resulted in improvements in potency, which could be attributed (at least in part) to the additional contacts between ligand and protein interface; 6- substituents could be titrated, for example the addition of a pentanol / hexanol was found to be more effective than the addition of an octanol or propanol in terms of minimizing the half maximal effective concentration (EC50) of ligand in cellular assays [169]. In contrast, substitutions at the 7- position yielded no benefits [170]. Substitutions at the 5- position have not been extensively explored, but some have yielded interesting findings [190]. There are ongoing efforts to combine the best chemical substitutions for each position to yield maximal benefits in terms of ligand potency against VRVs. Such systematic studies that include chemistry, structural biology, and virology have collectively yielded several compounds that are highly potent against many of the VRVs that arise in the clinic (e.g. Figure 11). Some of the best naphthyridine compounds, including 4c, 4d, and 4f, are at least as potent, if not more so, at inhibiting mutant forms of the virus and are frontrunners as next-generation drugs [162,169,172].
Future directions with INSTI development
The active site of the intasome bound by INSTIs can be conveniently described with a “roadmap” chemical representation depicting the important atomic interactions. INSTI interactions with IN residues and water molecules can be subdivided by their relative positions, with respect to the plane formed by the Mg2+-coordinating ligand scaffolds—respectively above, in-plane, and below the plane, as depicted in Figure 12 [162]. The INSTI core is engaged from above by the purine ring of the 3′-adenosine via a π–π stacking interaction. Below the core, there is a network of water molecules, which plays an important role in INSTI binding and the onset of resistance. Such depictions, in combination with structural biology data, can guide structure-based drug design. For example, there is a considerable chemical space on the side of the INSTI opposite the vDNA, which points toward the bulk solvent, on the left of the schematic. Both the second generation INSTIs and the developmental naphthyridine compounds have made use of some of this space, and their increased potency can be in part attributed to the additional interactions made in this general area [161,162]. Both of the recent structural reports describing HIV-1 and SIVrcm intasomes bound to the latest INSTIs also defined extensive water networks throughout the active site of the enzyme assembly (labeled “W” in Figure 12). Water molecules play important roles in ligand binding and contribute significantly to ligand affinity. For example, the displacement of water molecules can have drastic effects on ligand binding and potency, and the resulting effects have been described in both enthalpic and entropic terms [191-194]. The role of water, and the mechanisms through which water network reorganization contributes to ligand binding has gained considerable attention over the past decade within the drug design community. Despite the important advances in the recent structural biology data, experimental resolutions were still limited, and it is likely that more water molecules will be identified as experimental resolutions improve. An accurate understanding of water network topology and the interactions between neighboring water molecules or with the enzyme, coupled with an understanding of their dynamics and stability, is essential for gaining insight into mechanisms of viral resistance and for accurate drug design. The insights will lead to improved abilities to compute ligand binding free energies, to predict how changes in the active site affect ligand binding, and may help with designing improved compounds that make better use of the enzyme active site for potent inhibition. To complement efforts in identifying novel protein/ligand interfaces, it may also be possible to build into the empty cavity that is occupied by water molecules below the plane of the INSTI, as was recently proposed based on the cryo-EM structures of HIV-1 and SIVrcm intasomes bound to INSTIs [161,162].
Figure 12.
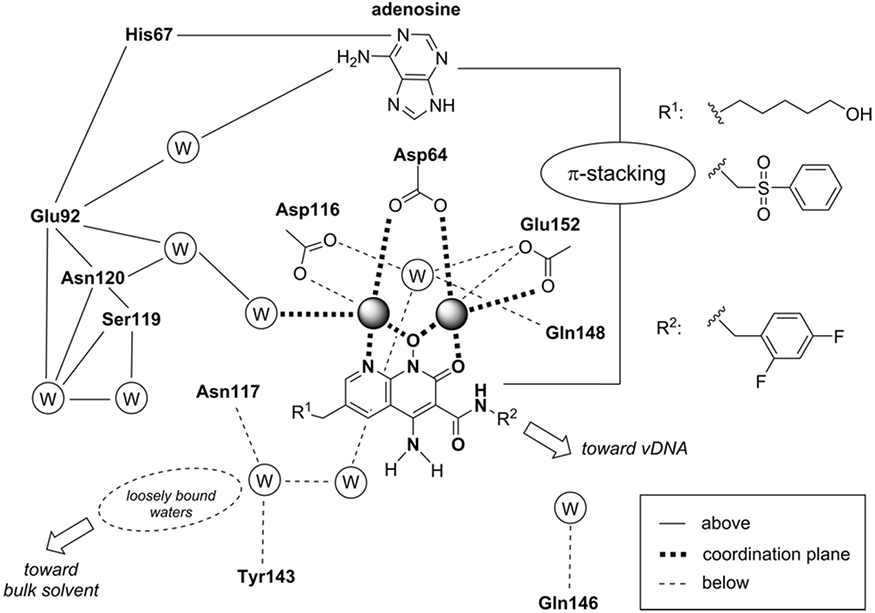
Schematic representation that recapitulates the receptor molecular environment and the water (W) networks with which the naphthyridine scaffold ligands interact when coordinating the Mg2+ ions. The scheme summarizes interactions by their locations with respect to the metal coordination plane of the naphthyridine scaffold (above, in-plane, or below). For clarity, the two water molecules coordinating the Mg2+ ions from above are not shown.
An important future consideration for all studies focusing on INSTI design is whether the compounds inhibit resistant variants of the virus. The most relevant variant appearing in patient samples is the G140S/Q148H double mutant, which arises in response to both first- and second-generation drugs and affects the potency of all INSTI regimens [153,175,181]. This double mutant exploits the INSTI Achiles’ heel – it affects the ability of all INSTIs to bind IN by altering the charge distribution around the metal ions and thus affecting the chelation the two Mg2+ ions [161]. Many of the complex resistant variants include the G140S/Q148H variants, but novel resistance pathways are also appearing [153]. It is important to begin investigating the drug resistant variants, because all currently available INSTIs, including 1st generation and 2nd generation drugs as well as developmental compounds, bind to the wild type enzyme with low nanomolar affinity and potently inhibit integration. There are extensive ongoing efforts to develop novel INSTI scaffolds that would improve upon the second-generation compounds, and some of these have shown impressive results in biochemical and cellular inhibition studies, with several compounds exhibiting picomolar affinities to the WT enzyme [195]. The latest efforts and successes have been described in a recent review [152]. However, once the virus evolves resistance, many currently available INSTIs fail. We argued that it is no longer productive to design compounds that accomplish the same goal in a slightly different manner. The more useful strategy will be to design compounds that target patient-derived VRVs, especially in response to the 2nd generation drugs. The rationale is that treatment targeting distinct variants that may arise in response to therapy restricts the viral population early on during therapy and reduces the number of variants that must be targeted in future therapeutic strategies [196]. Optimal ART will prevent future viral evolution and, in this way, it will be possible to stay ahead of the virus during treatment. Lessons from such studies with HIV can be extended to many human diseases.
Allosteric Integrase Inhibitors
A second class of drugs has been more recently developed that inhibits IN through a novel mechanism of action that is distinct from INSTIs. Using either high-throughput screening or structure-guided modeling that was intended to inhibit the interaction between IN and the cellular factor LEDGF, several groups reported on compounds that have inhibitory activity against IN [197,198]. Similar compounds have been reported in other work and given a variety of names, including Allosteric Integrase Inhibitors (ALLINIs), Non-Catalytic Integrase Inhibitors (NCINIs), LEDGF/p75-IN interaction site (LEDGINs) and IN-LAI (IN-LEDGF allosteric inhibitors), and multimeric IN inhibitors (MINIs). Below, we will give a brief account of the main properties of these compounds. There has been an extensive literature discussing the different ALLINI compounds, chemical optimizations, mechanism of action, and their utility, and the interested reader is pointed to those works for further details [199-202]. The class of allosteric inhibitors is characterized by the presence of heterocyclic cores, such as pyridine [203,204], thiophene [205], quinoline [197,198,206-209], isoquinoline [210], and thienopyridine [198,211]. All compounds belonging to this class bind to the interface formed by LEDGF and IN, effectively blocking this interaction. Although the blockage of this interaction can be clearly recapitulated in vitro [206,208,211], more recent data indicates that the compounds display the greatest potencies in the late phases of the viral replication cycle [138,203,207,209,212,213]. The basis of their mechanism of action is the rapid hyper-multimerization of IN [51,138,203,207,209,212-217]. Because IN has been implicated in RNA binding and packaging of the genomic RNA (see above), treatment with ALLINIs also leads to the improper localization of vRNA outside of the conical capsid core [51,136,138]. Early structural reports showed how ALLINI compounds bind at the CCD-CCD dimer interface, which would be normally occupied by the IBD of LEDGF/p75 [138,197]. These, as well as more recent structural reports [51,138,197,203,210,215,218-221], all used the CCD dimer to template different ALLINI compounds for structural studies. Several examples of ALLINI structures bound to the CCD dimer interface, and how their binding compares with that of the IBD domain of LEDGF, are shown in Fig. 13A-C. The sole structural report that used full-length IN revealed further inter-dimeric contacts spanning the CTD of IN, the ALLINI compound, and the CCD of IN, implying that regions beyond the CCD may also play a role in ALLINI potency [214] (Fig. 13D-E). These structural findings were supported by modeling experiments, which showed that ALLINIs facilitate the aberrant formation of inter-subunit interactions between an external CTD and the CCD-CCD dimer interface [217]. Notably, in the structural biology study, the NTD of IN was not observed in the structure, although it is known that the NTD may contribute to ALLINI potency and drug-mediated IN hyper-multimerization [51]. At least for the pyridine-based compounds, there actually appears to be selectivity for the tetrameric form of IN, whereas the quinoline-based compounds can hyper-multimerize both dimeric and tetrameric IN [51]. More recently, a comprehensive biophysical report showed that the quinoline-based compounds induce higher-order aggregates that have the characteristics of weak three-dimensional gels with a fractal-like character [216]. As with INSTIs, there are numerous ways in which resistance can occur, and several variants have been studied that arise to distinct drug classes [198,203,205,208,214,215,218]. Interestingly, ALLINIs appear to be selective for HIV-1 and do not potently inhibit SIV or HIV-2 [198,205,212], which is in stark contrast to the INSTIs that broadly bind to intasomes and inhibit viruses across different retroviral genera [45,135,161,162]. Taken together, the collective data is beginning to elucidate the intriguing structural, biophysical, and inhibitory properties of this developmental drug class. Although there are currently no FDA-approved ALLINI drugs, numerous ongoing efforts indicate that this is a promising class of selective compounds that targets a distinct genetic pathway of the HIV-1 replication cycle, and may therefore synergize with other treatment strategies, including INSTIs.
Figure 13.

Binding mode of ALLINIs to HIV IN. (A) The structure of the IBD of LEDGF/p75 (green) bound to a dimer of CCDs of HIV-1 IN (brown) (PDB 2B4J). Residues in contact distance are depicted. (B) The structure of compound KF116 (yellow surface) bound to the CCD dimer interface of HIV-1 IN (PDB 4O55). The chemical structure of KF116 is on the top left of the panel. (C) The structure of compound BI224436 (gray) bound to HIV-1 IN (PDB 4NYF). The chemical structure of BI224436 is on the top left of the panel. Green circles depict two critical regions of ALLINIs potency, the carboxylic acid, and tert-butoxy, that mimic the key residues Ile-365 and Asp-366 of LEDGF/p75, respectively. (D) The structure of the compound GSK1264 bound to the CCD:CTD dimer of HIV-1 IN (PDB 5HOT). Both the CCD and the CTD form part of the ALLINI interface. (E) Depiction of the CCD dimer interface where GSK1264 binds for comparison with panels A-C (here, the CTD has been omitted for clarity).
Concluding remarks
In this chapter, we have focused on the biochemical properties, structural biology, and inhibition of IN. Mechanistically, there is still much to learn about the interactions of retroviral INs with cellular proteins and with host chromatin, as well as the functional roles of these proteins. Excitingly, technologies for studying bona fide PICs, in and outside of their cellular context are also beginning to mature. The PIC remains a mysterious entity, and its constitution, structural organization, and interactions with viral/host components need to be clarified. There are active developments in the rapidly expanding fields of super-resolution florescence microscopy, focused ion beam milling, and cryo-electron tomography that are opening up opportunities for cellular structural biology, and at least some of these mysteries should be clarified in the next decade. There are also still numerous outstanding questions pertaining to the post-integration steps of the viral replication cycle, as well as an actively developing area of investigation detailing the mechanisms by which HIV-1 IN interacts with RNA to mediate correct packaging of the viral genome into virions. The RNA binding function of IN is a recently appreciated phenomenon and may open doors to entirely novel areas of investigation. In the context of inhibition, IN inhibitors are now a frontline treatment for HIV. The development of these drugs has been a decades long quest from the development of the first in vitro systems for retroviral DNA integration to large scale screening of compounds and the many uncertain steps between identifying lead compounds and finally bringing a drug to market. The early progress was made in the absence of high-resolution structural information because retroviral intasomes have been refractory to such studies. Advances in the biochemistry of retroviral IN systems, especially PFV IN and more recently SIVrcm and HIV-1 IN, together with prodigious advances in cryo-EM, have now opened the door to more systematic improvement of INSTIs and understanding of the mechanisms of drug resistance. Entirely different strategies of targeting HIV-1 integrase based on allosteric inhibitors, which compete with the RNA-binding function of IN, show great promise. During the next decade, we can anticipate exciting new advances that build on current structural knowledge of INSTI mechanism and progress with understanding some of the less well studied facets of IN and integration.
Acknowledgements
We thank Sriram Aiyer, Rick Bushman, and Mamuka Kvaratskhelia for critical reading of the manuscript. Molecular graphics and analyses were performed with the USCF Chimera package (supported by NIH P41 GM103311). This work was supported by NIH grants R01 AI136680, R01 AI146017, U54 AI150472, NSF Award MCB1933864 and MCB204809, the Margaret T. Morris Foundation, and the Intramural Program of the National Institute of Diabetes and Digestive Diseases of the National Institutes of Health.
References:
- [1].Grandgenett DP, Vora AC, Schiff RD, A 32,000-Dalton nucleic acid-binding protein from avian retravirus cores possesses DNA endonuclease activity, Virology. 89 (1978) 119–132. 10.1016/0042-6822(78)90046-6. [DOI] [PubMed] [Google Scholar]
- [2].Engelman A, Mizuuchi K, Craigie R, HIV-1 DNA integration: Mechanism of viral DNA cleavage and DNA strand transfer, Cell. 67 (1991) 1211–1221. 10.1016/0092-8674(91)90297-c. [DOI] [PubMed] [Google Scholar]
- [3].Randolph CA, Champoux JJ, The Majority of Simian Immunodeficiency Virus/Mne Circle Junctions Result from Ligation of Unintegrated Viral DNA Ends That Are Aberrant for Integration, Virology. 194 (1993) 851–854. 10.1006/viro.1993.1329. [DOI] [PubMed] [Google Scholar]
- [4].Brown PO, Bowerman B, Varmus HE, Bishop JM, Retroviral integration: structure of the initial covalent product and its precursor, and a role for the viral IN protein, Proc National Acad Sci. 86 (1989) 2525–2529. 10.1073/pnas.86.8.2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Du Z, Ilyinskii PO, Lally K, Desrosiers RC, Engelman A, A mutation in integrase can compensate for mutations in the simian immunodeficiency virus att site., J Virol. 71 (1997) 8124–8132. 10.1128/jvi.71.11.8124-8132.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].ROTH MJ, SCHWARTZBERG PL, GOFF SP, Structure of the Termini of Dna Intermediates in the Integration of Retroviral Dna - Dependence on in Function and Terminal Dna-Sequence, Cell. 58 (1989) 47–54. [DOI] [PubMed] [Google Scholar]
- [7].Whitcomb JM, Hughes SH, The sequence of human immunodeficiency virus type 2 circle junction suggests that integration protein cleaves the ends of linear DNA asymmetrically., J Virol. 65 (1991) 3906–3910. 10.1128/jvi.65.7.3906-3910.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Jones KS, Coleman J, Merkel GW, Laue TM, Skalka AM, Retroviral integrase functions as a multimer and can turn over catalytically., J Biol Chem. 267 (1992) 16037–16040. 10.1016/s0021-9258(18)41960-6. [DOI] [PubMed] [Google Scholar]
- [9].Engelman A, Bushman FD, Craigie R, Identification of discrete functional domains of HIV-1 integrase and their organization within an active multimeric complex., Embo J. 12 (1993) 3269–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Skalka AM, Katz RA, Retroviral DNA integration and the DNA damage response, Cell Death and Differentiation. 12 (2005) 971–978. 10.1038/sj.cdd.4401573. [DOI] [PubMed] [Google Scholar]
- [11].Bowerman B, Brown PO, Bishop JM, Varmus HE, A nucleoprotein complex mediates the integration of retroviral DNA., Gene Dev. 3 (1989) 469–478. 10.1101/gad.3.4.469. [DOI] [PubMed] [Google Scholar]
- [12].Brown PO, Bowerman B, Varmus HE, Bishop JM, Correct integration of retroviral DNA in vitro, Cell. 49 (1987) 347–356. 10.1016/0092-8674(87)90287-x. [DOI] [PubMed] [Google Scholar]
- [13].Fujiwara T, Mizuuchi K, Retroviral DNA integration: Structure of an integration intermediate, Cell. 54 (1988) 497–504. 10.1016/0092-8674(88)90071-2. [DOI] [PubMed] [Google Scholar]
- [14].Bushman F, Fujiwara T, Craigie R, Retroviral DNA integration directed by HIV integration protein in vitro, Science. 249 (1990) 1555–1558. 10.1126/science.2171144. [DOI] [PubMed] [Google Scholar]
- [15].Craigie R, Fujiwara T, Bushman F, The IN protein of Moloney murine leukemia virus processes the viral DNA ends and accomplishes their integration in vitro, Cell. 62 (1990) 829–837. 10.1016/0092-8674(90)90126-y. [DOI] [PubMed] [Google Scholar]
- [16].Katz RA, Merkel G, Kulkosky J, Leis J, Skalka AM, The avian retroviral IN protein is both necessary and sufficient for integrative recombination in vitro, Cell. 63 (1990) 87–95. 10.1016/0092-8674(90)90290-u. [DOI] [PubMed] [Google Scholar]
- [17].Chaconas G, Harshey RM, Sarvetnick N, Bukhari AI, Mechanism of Bacteriophage Mu DNA Transposition, Cold Spring Harb Sym. 45 (1981) 311–319. 10.1101/sqb.1981.045.01.043. [DOI] [PubMed] [Google Scholar]
- [18].Chaconas G, Surette MG, Mechanism of Mu DNA transposition, Bioessays. 9 (1988) 205–208. 10.1002/bies.950090606. [DOI] [PubMed] [Google Scholar]
- [19].Mizuuchi K, Mechanism of transposition of bacteriophage mu: polarity of the strand transfer reaction at the initiation of transposition, Cell. 39 (1984) 395–404. 10.1016/0092-8674(84)90018-7. [DOI] [PubMed] [Google Scholar]
- [20].Mizuuchi K, Craigie R, Mechanism of Bacteriophage Mu Transposition, Annu Rev Genet. 20 (1986) 385–429. 10.1146/annurev.ge.20.120186.002125. [DOI] [PubMed] [Google Scholar]
- [21].Levchenko I, Luo L, Baker TA, Disassembly of the Mu transposase tetramer by the ClpX chaperone., Gene Dev. 9 (1995) 2399–2408. 10.1101/gad.9.19.2399. [DOI] [PubMed] [Google Scholar]
- [22].Cai M, Zheng R, Caffrey M, Craigie R, Clore GM, Gronenborn AM, Solution structure of the N-terminal zinc binding domain of HIV-1 integrase., Nat Struct Biol. 4 (1997) 567–577. [DOI] [PubMed] [Google Scholar]
- [23].Eijkelenboom APAM, van den Ent FMI, Vos A, Doreleijers JF, Hård K, Tullius TD, Plasterk RHA, Kaptein R, Boelens R, The solution structure of the amino-terminal HHCC domain of HIV-2 integrase: a three-helix bundle stabilized by zinc, Curr Biol. 7 (1997) 739–746. 10.1016/s0960-9822(06)00332-0. [DOI] [PubMed] [Google Scholar]
- [24].Zheng R, Jenkins TM, Craigie R, Zinc folds the N-terminal domain of HIV-1 integrase, promotes multimerization, and enhances catalytic activity, Proc National Acad Sci. 93 (1996) 13659–13664. 10.1073/pnas.93.24.13659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Khan E, Mack JPG, Katz RA, Kulkosky J, Skalka AM, Retroviral integrase domains: DNA binding and the recognition of LTR sequences, Nucleic Acids Res. 19 (1991) 851–860. 10.1093/nar/19.4.851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mumm SR, Grandgenett DP, Defining nucleic acid-binding properties of avian retrovirus integrase by deletion analysis., J Virol. 65 (1991) 1160–1167. 10.1128/jvi.65.3.1160-1167.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Kulkosky J, Jones KS, Katz RA, Mack JP, Skalka AM, Residues critical for retroviral integrative recombination in a region that is highly conserved among retroviral/retrotransposon integrases and bacterial insertion sequence transposases., Mol Cell Biol. 12 (1992) 2331–2338. 10.1128/mcb.12.5.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Haren L, Ton-Hoang B, Chandler M, INTEGRATING DNA: Transposases and Retroviral Integrases, Annu Rev Microbiol. 53 (1999) 245–281. 10.1146/annurev.micro.53.1.245. [DOI] [PubMed] [Google Scholar]
- [29].Engelman A, Craigie R, Identification of conserved amino acid residues critical for human immunodeficiency virus type 1 integrase function in vitro., Journal of Virology. 66 (1992) 6361–6369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].van Gent DC, Groeneger AA, Plasterk RH, Mutational analysis of the integrase protein of human immunodeficiency virus type 2., Proc National Acad Sci. 89 (1992) 9598–9602. 10.1073/pnas.89.20.9598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Dyda F, Hickman A, Jenkins T, Engelman A, Craigie R, Davies D, Crystal structure of the catalytic domain of HIV-1 integrase: similarity to other polynucleotidyl transferases, Science. 266 (1994) 1981–1986. 10.1126/science.7801124. [DOI] [PubMed] [Google Scholar]
- [32].Jenkins TM, Engelman A, Ghirlando R, Craigie R, A soluble active mutant of HIV-1 integrase: involvement of both the core and carboxyl-terminal domains in multimerization., Journal of Biological Chemistry. 271 (1996) 7712–7718. [DOI] [PubMed] [Google Scholar]
- [33].Jenkins TM, Hickman AB, Dyda F, Ghirlando R, Davies DR, Craigie R, Catalytic domain of human immunodeficiency virus type 1 integrase: identification of a soluble mutant by systematic replacement of hydrophobic residues., Proceedings of the National Academy of Sciences. 92 (1995) 6057–6061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Richardson JM, Colloms SD, Finnegan DJ, Walkinshaw MD, Molecular Architecture of the Mos1 Paired-End Complex: The Structural Basis of DNA Transposition in a Eukaryote, Cell. 138 (2009) 1096–1108. 10.1016/j.cell.2009.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Goodwin KD, He H, Imasaki T, Lee S-H, Georgiadis MM, Crystal Structure of the Human Hsmar1 -Derived Transposase Domain in the DNA Repair Enzyme Metnase , Biochemistry-Us. 49 (2010) 5705–5713. 10.1021/bi100171x. [DOI] [PubMed] [Google Scholar]
- [36].Bushman FD, Engelman A, Palmer I, Wingfield P, Craigie R, Domains of the integrase protein of human immunodeficiency virus type 1 responsible for polynucleotidyl transfer and zinc binding., Proc National Acad Sci. 90 (1993) 3428–3432. 10.1073/pnas.90.8.3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Chow S, Vincent K, Ellison V, Brown P, Reversal of integration and DNA splicing mediated by integrase of human immunodeficiency virus, Science. 255 (1992) 723–726. 10.1126/science.1738845. [DOI] [PubMed] [Google Scholar]
- [38].Valkov E, Gupta SS, Hare S, Helander A, Roversi P, McClure M, Cherepanov P, Functional and structural characterization of the integrase from the prototype foamy virus., Nucleic Acids Research. 37 (2009) 243–255. 10.1093/nar/gkn938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Barski MS, Minnell JJ, Hodakova Z, Pye VE, Nans A, Cherepanov P, Maertens GN, Cryo-EM structure of the deltaretroviral intasome in complex with the PP2A regulatory subunit B56γ, Nat Commun. 11 (2020) 5043. 10.1038/s41467-020-18874-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Galilee M, Alian A, Identification of Phe187 as a Crucial Dimerization Determinant Facilitates Crystallization of a Monomeric Retroviral Integrase Core Domain, Structure. 22 (2014) 1512–1519. 10.1016/j.str.2014.08.001. [DOI] [PubMed] [Google Scholar]
- [41].Shi K, Pandey KK, Bera S, Vora AC, Grandgenett DP, Aihara H, A Possible Role for the Asymmetric C-Terminal Domain Dimer of Rous Sarcoma Virus Integrase in Viral DNA Binding, Plos One. 8 (2013) e56892. 10.1371/journal.pone.0056892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Lodi PJ, Ernst JA, Kuszewski J, Hickman AB, Engelman A, Craigie R, Clore GM, Gronenborn AM, Solution structure of the DNA binding domain of HIV-1 integrase., Biochemistry. 34 (1995) 9826–9833. [DOI] [PubMed] [Google Scholar]
- [43].Eijkelenboom AP, Lutzke RA, Boelens R, Plasterk RH, Kaptein R, Hård K, The DNA-binding domain of HIV-1 integrase has an SH3-like fold., Nat Struct Biol. 2 (1995) 807–810. [DOI] [PubMed] [Google Scholar]
- [44].Aiyer S, Rossi P, Malani N, Schneider WM, Chandar A, Bushman FD, Montelione GT, Roth MJ, Structural and sequencing analysis of local target DNA recognition by MLV integrase, Nucleic Acids Res. 43 (2015) 5647–5663. 10.1093/nar/gkv410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Hare S, Gupta SS, Valkov E, Engelman A, Cherepanov P, Retroviral intasome assembly and inhibition of DNA strand transfer., Nature. 464 (2010) 232–236. 10.1038/nature08784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Maertens GN, Hare S, Cherepanov P, The mechanism of retroviral integration from X-ray structures of its key intermediates., Nature. 468 (2010) 326–329. 10.1038/nature09517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Guan R, Aiyer S, Cote ML, Xiao R, Jiang M, Acton TB, Roth MJ, Montelione GT, X-ray crystal structure of the N-terminal region of Moloney murine leukemia virus integrase and its implications for viral DNA recognition, Proteins Struct Funct Bioinform. 85 (2017) 647–656. 10.1002/prot.25245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Yang F, Seamon JA, Roth MJ, Mutational Analysis of the N-Terminus of Moloney Murine Leukemia Virus Integrase, Virology. 291 (2001) 32–45. 10.1006/viro.2001.1218. [DOI] [PubMed] [Google Scholar]
- [49].Cherepanov P, Maertens G, Proost P, Devreese B, Beeumen JV, Engelborghs Y, Clercq ED, Debyser Z, HIV-1 integrase forms stable tetramers and associates with LEDGF/p75 protein in human cells., Journal of Biological Chemistry. 278 (2003) 372–381. 10.1074/jbc.m209278200. [DOI] [PubMed] [Google Scholar]
- [50].Hare S, Nunzio FD, Labeja A, Wang J, Engelman A, Cherepanov P, Structural basis for functional tetramerization of lentiviral integrase., PLoS Pathog. 5 (2009) e1000515. 10.1371/journal.ppat.1000515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Koneru PC, Francis AC, Deng N, Rebensburg SV, Hoyte AC, Lindenberger J, Adu-Ampratwum D, Larue RC, Wempe MF, Engelman AN, Lyumkis D, Fuchs JR, Levy RM, Melikyan GB, Kvaratskhelia M, HIV-1 integrase tetramers are the antiviral target of pyridine-based allosteric integrase inhibitors., ELife. 8 (2019) 284. 10.7554/elife.46344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Lutzke RA, Plasterk RH, Structure-based mutational analysis of the C-terminal DNA-binding domain of human immunodeficiency virus type 1 integrase: critical residues for protein oligomerization and DNA binding., Journal of Virology. 72 (1998) 4841–4848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].McKee CJ, Kessl JJ, Shkriabai N, Dar MJ, Engelman A, Kvaratskhelia M, Dynamic modulation of HIV-1 integrase structure and function by cellular lens epithelium-derived growth factor (LEDGF) protein., 283 (2008) 31802–31812. 10.1074/jbc.m805843200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Chen Z, Yan Y, Munshi S, Li Y, Zugay-Murphy J, Xu B, Witmer M, Felock P, Wolfe A, Sardana V, Emini EA, Hazuda D, Kuo LC, X-ray structure of simian immunodeficiency virus integrase containing the core and C-terminal domain (residues 50-293) - an initial glance of the viral DNA binding platform11Edited by I. A. Wilson, J Mol Biol. 296 (2000) 521–533. 10.1006/jmbi.1999.3451. [DOI] [PubMed] [Google Scholar]
- [55].Yang ZN, Mueser TC, Bushman FD, Hyde CC, Crystal structure of an active two-domain derivative of Rous sarcoma virus integrase., Journal of Molecular Biology. 296 (2000) 535–548. 10.1006/jmbi.1999.3463. [DOI] [PubMed] [Google Scholar]
- [56].Chen JC, Krucinski J, Miercke LJ, Finer-Moore JS, Tang AH, Leavitt AD, Stroud RM, Crystal structure of the HIV-1 integrase catalytic core and C-terminal domains: a model for viral DNA binding., Proceedings of the National Academy of Sciences. 97 (2000) 8233–8238. 10.1073/pnas.150220297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang JY, Ling H, Yang W, Craigie R, Structure of a two-domain fragment of HIV-1 integrase: implications for domain organization in the intact protein., EMBO J. 20 (2001) 7333–7343. 10.1093/emboj/20.24.7333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hare S, Shun M-C, Gupta SS, Valkov E, Engelman A, Cherepanov P, A novel co-crystal structure affords the design of gain-of-function lentiviral integrase mutants in the presence of modified PSIP1/LEDGF/p75., PLoS Pathog. 5 (2009) e1000259. 10.1371/journal.ppat.1000259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Eilers G, Gupta K, Allen A, Zhou J, Hwang Y, Cory MB, Bushman FD, Duyne GV, Influence of the amino-terminal sequence on the structure and function of HIV integrase., Retrovirology. 17 (2020) 28–16. 10.1186/s12977-020-00537-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Hindmarsh P, Leis J, Reconstitution of Concerted DNA Integration with Purified Components, Adv Virus Res. 52 (1999) 397–410. 10.1016/s0065-3527(08)60308-5. [DOI] [PubMed] [Google Scholar]
- [61].Li M, Craigie R, Processing of viral DNA ends channels the HIV-1 integration reaction to concerted integration., Journal of Biological Chemistry. 280 (2005) 29334–29339. 10.1074/jbc.m505367200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Sinha S, Grandgenett DP, Recombinant human immunodeficiency virus type 1 integrase exhibits a capacity for full-site integration in vitro that is comparable to that of purified preintegration complexes from virus-infected cells., Journal of Virology. 79 (2005) 8208–8216. 10.1128/jvi.79.13.8208-8216.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Sinha S, Pursley MH, Grandgenett DP, Efficient Concerted Integration by Recombinant Human Immunodeficiency Virus Type 1 Integrase without Cellular or Viral Cofactors, J Virol. 76 (2002) 3105–3113. 10.1128/jvi.76.7.3105-3113.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Kotova S, Li M, Dimitriadis EK, Craigie R, Nucleoprotein intermediates in HIV-1 DNA integration visualized by atomic force microscopy., Journal of Molecular Biology. 399 (2010) 491–500. 10.1016/j.jmb.2010.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Li M, Mizuuchi M, Burke TR, Craigie R, Retroviral DNA integration: reaction pathway and critical intermediates., EMBO J. 25 (2006) 1295–1304. 10.1038/sj.emboj.7601005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Pandey KK, Bera S, Zahm J, Vora A, Stillmock K, Hazuda D, Grandgenett DP, Inhibition of human immunodeficiency virus type 1 concerted integration by strand transfer inhibitors which recognize a transient structural intermediate., Journal of Virology. 81 (2007) 12189–12199. 10.1128/jvi.02863-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bera S, Pandey KK, Vora AC, Grandgenett DP, Molecular interactions between HIV-1 integrase and the two viral DNA ends within the synaptic complex that mediates concerted integration., Journal of Molecular Biology. 389 (2009) 183–198. 10.1016/j.jmb.2009.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Serrao E, Krishnan L, Shun M-C, Li X, Cherepanov P, Engelman A, Maertens GN, Integrase residues that determine nucleotide preferences at sites of HIV-1 integration: implications for the mechanism of target DNA binding., Nucleic Acids Research. 42 (2014) 5164–5176. 10.1093/nar/gku136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Serrao E, Ballandras-Colas A, Cherepanov P, Maertens GN, Engelman AN, Key determinants of target DNA recognition by retroviral intasomes., Retrovirology. 12 (2015) 39. 10.1186/s12977-015-0167-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Yin Z, Shi K, Banerjee S, Pandey KK, Bera S, Grandgenett DP, Aihara H, Crystal structure of the Rous sarcoma virus intasome., Nature. 530 (2016) 362–366. 10.1038/nature16950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Ballandras-Colas A, Brown M, Cook NJ, Dewdney TG, Demeler B, Cherepanov P, Lyumkis D, Engelman AN, Cryo-EM reveals a novel octameric integrase structure for betaretroviral intasome function., Nature. 530 (2016) 358–361. 10.1038/nature16955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Li M, Jurado KA, Lin S, Engelman A, Craigie R, Engineered hyperactive integrase for concerted HIV-1 DNA integration., PloS One. 9 (2014) e105078. 10.1371/journal.pone.0105078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lyumkis D, Challenges and opportunities in cryo-EM single-particle analysis., The Journal of Biological Chemistry. 294 (2019) 5181–5197. 10.1074/jbc.rev118.005602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Punjani A, Fleet DJ, 3D Variability Analysis: Directly resolving continuous flexibility and discrete heterogeneity from single particle cryo-EM images, BioRxiv. 11 (2020) 2020.04.08.032466. 10.1101/2020.04.08.032466. [DOI] [PubMed] [Google Scholar]
- [75].Nakane T, Kimanius D, Lindahl E, Scheres SH, Characterisation of molecular motions in cryo-EM single-particle data by multi-body refinement in RELION., ELife. 7 (2018) 1485. 10.7554/elife.36861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Lyumkis D, Brilot AF, Theobald DL, Grigorieff N, Likelihood-based classification of cryo-EM images using FREALIGN., Journal of Structural Biology. 183 (2013) 377–388. 10.1016/j.jsb.2013.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Passos DO, Li M, Yang R, Rebensburg SV, Ghirlando R, Jeon Y, Shkriabai N, Kvaratskhelia M, Craigie R, Lyumkis D, Cryo-EM structures and atomic model of the HIV-1 strand transfer complex intasome., Science. 355 (2017) 89–92. 10.1126/science.aah5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Ballandras-Colas A, Maskell DP, Serrao E, Locke J, Swuec P, Jonsson SR, Kotecha A, Cook NJ, Pye VE, Taylor IA, Andresdottir V, Engelman AN, Costa A, Cherepanov P, A supramolecular assembly mediates lentiviral DNA integration, Science. 355 (2017) 93–95. 10.1126/science.aah7002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Engelman AN, Cherepanov P, Retroviral intasomes arising., Curr Opin Struct Biol. 47 (2017) 23–29. 10.1016/j.sbi.2017.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Hare S, Maertens GN, Cherepanov P, 3’-processing and strand transfer catalysed by retroviral integrase in crystallo., EMBO J. 31 (2012) 3020–3028. 10.1038/emboj.2012.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Beese LS, Steitz TA, Structural basis for the 3′-5′ exonuclease activity of Escherichia coli DNA polymerase I: a two metal ion mechanism., Embo J. 10 (1991) 25–33. 10.1002/j.1460-2075.1991.tb07917.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Steitz TA, Steitz JA, A general two-metal-ion mechanism for catalytic RNA, Proc National Acad Sci. 90 (1993) 6498–6502. 10.1073/pnas.90.14.6498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Wang GZ, Wang Y, Goff SP, Histones Are Rapidly Loaded onto Unintegrated Retroviral DNAs Soon after Nuclear Entry., Cell Host & Microbe. 20 (2016) 798–809. 10.1016/j.chom.2016.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Geis FK, Goff SP, Unintegrated HIV-1 DNAs are loaded with core and linker histones and transcriptionally silenced, Proc National Acad Sci. 116 (2019) 23735–23742. 10.1073/pnas.1912638116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Machida S, Depierre D, Chen H-C, Thenin-Houssier S, Petitjean G, Doyen CM, Takaku M, Cuvier O, Benkirane M, Exploring histone loading on HIV DNA reveals a dynamic nucleosome positioning between unintegrated and integrated viral genome, Proceedings of the National Academy of Sciences. 117 (2020) 6822–6830. 10.1073/pnas.1913754117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Bedwell GJ, Engelman AN, Factors that mold the nuclear landscape of HIV-1 integration, Nucleic Acids Res. 49 (2020) gkaa1207-. 10.1093/nar/gkaa1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Lee MS, Craigie R, A previously unidentified host protein protects retroviral DNA from autointegration., Proceedings of the National Academy of Sciences. 95 (1998) 1528–1533. 10.1073/pnas.95.4.1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Francis AC, Marin M, Singh PK, Achuthan V, Prellberg MJ, Palermino-Rowland K, Lan S, Tedbury PR, Sarafianos SG, Engelman AN, Melikyan GB, HIV-1 replication complexes accumulate in nuclear speckles and integrate into speckle-associated genomic domains, Nat Commun. 11 (2020) 3505. 10.1038/s41467-020-17256-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Achuthan V, Perreira JM, Sowd GA, Puray-Chavez M, McDougall WM, Paulucci-Holthauzen A, Wu X, Fadel HJ, Poeschla EM, Multani AS, Hughes SH, Sarafianos SG, Brass AL, Engelman AN, Capsid-CPSF6 Interaction Licenses Nuclear HIV-1 Trafficking to Sites of Viral DNA Integration., Cell Host & Microbe. 24 (2018) 392–404.e8. 10.1016/j.chom.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Christensen DE, Ganser-Pornillos BK, Johnson JS, Pornillos O, Sundquist WI, Reconstitution and visualization of HIV-1 capsid-dependent replication and integration in vitro, Science. 370 (2020) eabc8420. 10.1126/science.abc8420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Müllers E, Stirnnagel K, Kaulfuss S, Lindemann D, Prototype Foamy Virus Gag Nuclear Localization: a Novel Pathway among Retroviruses, J Virol. 85 (2011) 9276–9285. 10.1128/jvi.00663-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Tobaly-Tapiero J, Bittoun P, Lehmann-Che J, Delelis O, Giron M, Thé HD, Saïb A, Chromatin Tethering of Incoming Foamy Virus by the Structural Gag Protein, Traffic. 9 (2008) 1717–1727. 10.1111/j.1600-0854.2008.00792.x. [DOI] [PubMed] [Google Scholar]
- [93].Prizan-Ravid A, Elis E, Laham-Karam N, Selig S, Ehrlich M, Bacharach E, The Gag Cleavage Product, p12, is a Functional Constituent of the Murine Leukemia Virus Pre-Integration Complex, Plos Pathog. 6 (2010) e1001183. 10.1371/journal.ppat.1001183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Elis E, Ehrlich M, Prizan-Ravid A, Laham-Karam N, Bacharach E, p12 Tethers the Murine Leukemia Virus Pre-integration Complex to Mitotic Chromosomes, Plos Pathog. 8 (2012) e1003103. 10.1371/journal.ppat.1003103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Pruss D, Bushman FD, Wolffe AP, Human immunodeficiency virus integrase directs integration to sites of severe DNA distortion within the nucleosome core., Proceedings of the National Academy of Sciences. 91 (1994) 5913–5917. 10.1073/pnas.91.13.5913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Pryciak PM, Sil A, Varmus HE, Retroviral integration into minichromosomes in vitro., EMBO J. 11 (1992) 291–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Müller HP, Varmus HE, DNA bending creates favored sites for retroviral integration: an explanation for preferred insertion sites in nucleosomes., Embo J. 13 (1994) 4704–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Maskell DP, Renault L, Serrao E, Lesbats P, Matadeen R, Hare S, Lindemann D, Engelman AN, Costa A, Cherepanov P, Structural basis for retroviral integration into nucleosomes, Nature. 523 (2015) 366–369. 10.1038/nature14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Jones ND, L. MA Jr, Hanne J, Peake MB, Lee J-B, Fishel R, Yoder KE, Retroviral intasomes search for a target DNA by 1D diffusion which rarely results in integration, Nature Communications. 7 (2016) 11409. 10.1038/ncomms11409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wilson MD, Renault L, Maskell DP, Ghoneim M, Pye VE, Nans A, Rueda DS, Cherepanov P, Costa A, Retroviral integration into nucleosomes through DNA looping and sliding along the histone octamer., Nature Communications. 10 (2019) 4189–10. 10.1038/s41467-019-12007-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ge H, Si Y, Roeder RG, Isolation of cDNAs encoding novel transcription coactivators p52 and p75 reveals an alternate regulatory mechanism of transcriptional activation, EMBO J. 17 (1998) 6723–6729. 10.1093/emboj/17.22.6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Shinohara T, Singh DP, Fatma N, LEDGF, a survival factor, activates stress-related genes, Prog Retin Eye Res. 21 (2002) 341–358. 10.1016/s1350-9462(02)00007-1. [DOI] [PubMed] [Google Scholar]
- [103].Cherepanov P, Sun Z-YJ, Rahman S, Maertens G, Wagner G, Engelman A, Solution structure of the HIV-1 integrase-binding domain in LEDGF/p75., Nature Structural & Molecular Biology. 12 (2005) 526–532. 10.1038/nsmb937. [DOI] [PubMed] [Google Scholar]
- [104].Cherepanov P, Ambrosio ALB, Rahman S, Ellenberger T, Engelman A, Structural basis for the recognition between HIV-1 integrase and transcriptional coactivator p75., Proceedings of the National Academy of Sciences. 102 (2005) 17308–17313. 10.1073/pnas.0506924102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Schröder ARW, Shinn P, Chen H, Berry C, Ecker JR, Bushman F, HIV-1 Integration in the Human Genome Favors Active Genes and Local Hotspots, Cell. 110 (2002) 521–529. 10.1016/s0092-8674(02)00864-4. [DOI] [PubMed] [Google Scholar]
- [106].Ciuffi A, Llano M, Poeschla E, Hoffmann C, Leipzig J, Shinn P, Ecker JR, Bushman F, A role for LEDGF/p75 in targeting HIV DNA integration., Nature Medicine. 11 (2005) 1287–1289. 10.1038/nm1329. [DOI] [PubMed] [Google Scholar]
- [107].Wang H, Farnung L, Dienemann C, Cramer P, Structure of H3K36-methylated nucleosome–PWWP complex reveals multivalent cross-gyre binding, Nature Structural & Molecular Biology. 82 (2019) 1–6. 10.1038/s41594-019-0345-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Llano M, Delgado S, Vanegas M, Poeschla EM, Lens epithelium-derived growth factor/p75 prevents proteasomal degradation of HIV-1 integrase., Journal of Biological Chemistry. 279 (2004) 55570–55577. 10.1074/jbc.m408508200. [DOI] [PubMed] [Google Scholar]
- [109].Turlure F, Maertens G, Rahman S, Cherepanov P, Engelman A, A tripartite DNA-binding element, comprised of the nuclear localization signal and two AT-hook motifs, mediates the association of LEDGF/p75 with chromatin in vivo., Nucleic Acids Research. 34 (2006) 1653–1665. 10.1093/nar/gkl052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Ferris AL, Wu X, Hughes CM, Stewart C, Smith SJ, Milne TA, Wang GG, Shun M-C, Allis CD, Engelman A, Hughes SH, Lens epithelium-derived growth factor fusion proteins redirect HIV-1 DNA integration., Proceedings of the National Academy of Sciences. 107 (2010) 3135–3140. 10.1073/pnas.0914142107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Gijsbers R, Ronen K, Vets S, Malani N, Rijck JD, McNeely M, Bushman FD, Debyser Z, LEDGF Hybrids Efficiently Retarget Lentiviral Integration Into Heterochromatin, Mol Ther. 18 (2010) 552–560. 10.1038/mt.2010.36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Wang H, Jurado KA, Wu X, Shun M-C, Li X, Ferris AL, Smith SJ, Patel PA, Fuchs JR, Cherepanov P, Kvaratskhelia M, Hughes SH, Engelman A, HRP2 determines the efficiency and specificity of HIV-1 integration in LEDGF/p75 knockout cells but does not contribute to the antiviral activity of a potent LEDGF/p75-binding site integrase inhibitor., Nucleic Acids Research. 40 (2012) 11518–11530. 10.1093/nar/gks913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Cherepanov P, Devroe E, Silver PA, Engelman A, Identification of an evolutionarily conserved domain in human lens epithelium-derived growth factor/transcriptional co-activator p75 (LEDGF/p75) that binds HIV-1 integrase., Journal of Biological Chemistry. 279 (2004) 48883–48892. 10.1074/jbc.m406307200. [DOI] [PubMed] [Google Scholar]
- [114].Engelman AN, Maertens GN, Retrovirus-Cell Interactions, (2018) 163–198. 10.1016/b978-0-12-811185-7.00004-2. [DOI] [Google Scholar]
- [115].Kalpana GV, Marmon S, Wang W, Crabtree GR, GOFF SP, Binding and stimulation of HIV-1 integrase by a human homolog of yeast transcription factor SNF5., Science. 266 (1994) 2002–2006. [DOI] [PubMed] [Google Scholar]
- [116].Cano J, Kalpana GV, Inhibition of Early Stages of HIV-1 Assembly by INI1/hSNF5 Transdominant Negative Mutant S6, Journal of Virology. 85 (2011) 2254–2265. 10.1128/jvi.00006-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Porte AL, Cano J, Wu X, Mitra D, Kalpana GV, An Essential Role of INI1/hSNF5 Chromatin Remodeling Protein in HIV-1 Posttranscriptional Events and Gag/Gag-Pol Stability, Journal of Virology. 90 (2016) 9889–9904. 10.1128/jvi.00323-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Orphanides G, LeRoy G, Chang CH, Luse DS, Reinberg D, FACT, a factor that facilitates transcript elongation through nucleosomes., Cell. 92 (1998) 105–116. 10.1016/s0092-8674(00)80903-4. [DOI] [PubMed] [Google Scholar]
- [119].LeRoy G, Orphanides G, Lane WS, Reinberg D, Requirement of RSF and FACT for transcription of chromatin templates in vitro., Science. 282 (1998) 1900–1904. 10.1126/science.282.5395.1900. [DOI] [PubMed] [Google Scholar]
- [120].Orphanides G, Wu W-H, Lane WS, Hampsey M, Reinberg D, The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins, Nature. 400 (1999) 284–288. 10.1038/22350. [DOI] [PubMed] [Google Scholar]
- [121].LeRoy G, Oksuz O, Descostes N, Aoi Y, Ganai RA, Kara HO, Yu J-R, Lee C-H, Stafford J, Shilatifard A, Reinberg D, LEDGF and HDGF2 relieve the nucleosome-induced barrier to transcription in differentiated cells, Science Advances. 5 (2019) eaay3068. 10.1126/sciadv.aay3068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Winans S, Larue RC, Abraham CM, Shkriabai N, Skopp A, Winkler D, Kvaratskhelia M, Beemon KL, The FACT Complex Promotes Avian Leukosis Virus DNA Integration., Journal of Virology. 91 (2017) e00082–17. 10.1128/jvi.00082-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Matysiak J, Lesbats P, Mauro E, Lapaillerie D, Dupuy J-W, Lopez AP, Benleulmi MS, Calmels C, Andreola M-L, Ruff M, Llano M, Delelis O, Lavigne M, Parissi V, Modulation of chromatin structure by the FACT histone chaperone complex regulates HIV-1 integration., Retrovirology. 14 (2017) 39. 10.1186/s12977-017-0363-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Lopez AP, Kugelman JR, Garcia-Rivera J, Urias E, Salinas SA, Fernandez-Zapico ME, Llano M, The Structure-Specific Recognition Protein 1 Associates with Lens Epithelium-Derived Growth Factor Proteins and Modulates HIV-1 Replication., Journal of Molecular Biology. 428 (2016) 2814–2831. 10.1016/j.jmb.2016.05.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Raghavendra NK, Shkriabai N, Graham RL, Hess S, Kvaratskhelia M, Wu L, Identification of host proteins associated with HIV-1 preintegration complexes isolated from infected CD4 + cells, Retrovirology. 7 (2010) 66. 10.1186/1742-4690-7-66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Studamire B, Goff SP, Host proteins interacting with the Moloney murine leukemia virus integrase: Multiple transcriptional regulators and chromatin binding factors, Retrovirology. 5 (2008) 48. 10.1186/1742-4690-5-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Sharma A, Larue RC, Plumb MR, Malani N, Male F, Slaughter A, Kessl JJ, Shkriabai N, Coward E, Aiyer SS, Green PL, Wu L, Roth MJ, Bushman FD, Kvaratskhelia M, BET proteins promote efficient murine leukemia virus integration at transcription start sites., Proceedings of the National Academy of Sciences of the United States of America. 110 (2013) 12036–12041. 10.1073/pnas.1307157110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Gupta SS, Maetzig T, Maertens GN, Sharif A, Rothe M, Weidner-Glunde M, Galla M, Schambach A, Cherepanov P, Schulz TF, Bromo- and Extraterminal Domain Chromatin Regulators Serve as Cofactors for Murine Leukemia Virus Integration, Journal of Virology. 87 (2013) 12721–12736. 10.1128/jvi.01942-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Rijck JD, de Kogel C, Demeulemeester J, Vets S, Ashkar SE, Malani N, Bushman FD, Landuyt B, Husson SJ, Busschots K, Gijsbers R, Debyser Z, The BET family of proteins targets moloney murine leukemia virus integration near transcription start sites., Cell Reports. 5 (2013) 886–894. 10.1016/j.celrep.2013.09.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Aiyer S, Swapna GVT, Malani N, Aramini JM, Schneider WM, Plumb MR, Ghanem M, Larue RC, Sharma A, Studamire B, Kvaratskhelia M, Bushman FD, Montelione GT, Roth MJ, Altering murine leukemia virus integration through disruption of the integrase and BET protein family interaction, Nucleic Acids Res. 42 (2014) 5917–5928. 10.1093/nar/gku175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Filippakopoulos P, Picaud S, Mangos M, Keates T, Lambert J-P, Barsyte-Lovejoy D, Felletar I, Volkmer R, Müller S, Pawson T, Gingras A-C, Arrowsmith CH, Knapp S, Histone Recognition and Large-Scale Structural Analysis of the Human Bromodomain Family, Cell. 149 (2012) 214–231. 10.1016/j.cell.2012.02.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Wu X, Li Y, Crise B, Burgess SM, Transcription start regions in the human genome are favored targets for MLV integration., Science. 300 (2003) 1749–1751. 10.1126/science.1083413. [DOI] [PubMed] [Google Scholar]
- [133].Larue RC, Plumb MR, Crowe BL, Shkriabai N, Sharma A, DiFiore J, Malani N, Aiyer SS, Roth MJ, Bushman FD, Foster MP, Kvaratskhelia M, Bimodal high-affinity association of Brd4 with murine leukemia virus integrase and mononucleosomes, Nucleic Acids Res. 42 (2014) 4868–4881. 10.1093/nar/gku135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Maertens GN, B’-protein phosphatase 2A is a functional binding partner of delta-retroviral integrase., Nucleic Acids Research. 44 (2016) 364–376. 10.1093/nar/gkv1347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Bhatt V, Shi K, Salamango DJ, Moeller NH, Pandey KK, Bera S, Bohl TE, Kurniawan F, Orellana K, Zhang W, Grandgenett DP, Harris RS, Sundborger-Lunna AC, Aihara H, Structural basis of host protein hijacking in human T-cell leukemia virus integration, Nature Communications. 11 (2020) 1–9. 10.1038/s41467-020-16963-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Kessl JJ, Kutluay SB, Townsend D, Rebensburg S, Slaughter A, Larue RC, Shkriabai N, Bakouche N, Fuchs JR, Bieniasz PD, Kvaratskhelia M, HIV-1 Integrase Binds the Viral RNA Genome and Is Essential during Virion Morphogenesis., Cell. 166 (2016) 1257–1268.e12. 10.1016/j.cell.2016.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Elliott JL, Eschbach JE, Koneru PC, Li W, Puray-Chavez M, Townsend D, Lawson DQ, Engelman AN, Kvaratskhelia M, Kutluay SB, Integrase-RNA interactions underscore the critical role of integrase in HIV-1 virion morphogenesis., ELife. 9 (2020). 10.7554/elife.54311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Jurado KA, Wang H, Slaughter A, Feng L, Kessl JJ, Koh Y, Wang W, Ballandras-Colas A, Patel PA, Fuchs JR, Kvaratskhelia M, Engelman A, Allosteric integrase inhibitor potency is determined through the inhibition of HIV-1 particle maturation., Proceedings of the National Academy of Sciences of the United States of America. 110 (2013) 8690–8695. 10.1073/pnas.1300703110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Engelman A, Englund G, Orenstein JM, Martin MA, Craigie R, Multiple effects of mutations in human immunodeficiency virus type 1 integrase on viral replication., J Virol. 69 (1995) 2729–2736. 10.1128/jvi.69.5.2729-2736.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Engelman A, In Vivo Analysis of Retroviral Integrase Structure and Function, Adv Virus Res. 52 (1999) 411–426. 10.1016/s0065-3527(08)60309-7. [DOI] [PubMed] [Google Scholar]
- [141].Elliott JL, Kutluay SB, Going beyond Integration: The Emerging Role of HIV-1 Integrase in Virion Morphogenesis, Viruses. 12 (2020) 1005. 10.3390/v12091005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Fontana J, Jurado KA, Cheng N, Ly NL, Fuchs JR, Gorelick RJ, Engelman AN, Steven AC, Distribution and Redistribution of HIV-1 Nucleocapsid Protein in Immature, Mature, and Integrase-Inhibited Virions: a Role for Integrase in Maturation, J Virol. 89 (2015) 9765–9780. 10.1128/jvi.01522-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Schwartzberg P, Colicelli J, Goff SP, Construction and analysis of deletion mutations in the pol gene of moloney murine leukemia virus: A new viral function required for productive infection, Cell. 37 (1984) 1043–1052. 10.1016/0092-8674(84)90439-2. [DOI] [PubMed] [Google Scholar]
- [144].Hazuda D, Felock PJ, Hastings JC, Pramanik B, Wolfe AL, Discovery and analysis of inhibitors of the human immunodeficiency integrase., Drug Des Discov. 15 (1997) 17–24. [PubMed] [Google Scholar]
- [145].Pommier Y, Johnson AA, Marchand C, Integrase inhibitors to treat HIV/Aids, Nature Reviews. Drug Discovery. 4 (2005) 236–248. 10.1038/nrd1660. [DOI] [PubMed] [Google Scholar]
- [146].Hazuda DJ, Felock PJ, Hastings JC, Pramanik B, Wolfe AL, Differential divalent cation requirements uncouple the assembly and catalytic reactions of human immunodeficiency virus type 1 integrase., J Virol. 71 (1997) 7005–7011. 10.1128/jvi.71.9.7005-7011.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Hazuda D, Felock P, Hastings J, Pramanik B, Wolfe A, Goodarzi G, Vora A, Brackmann K, Grandgenett D, Equivalent inhibition of half-site and full-site retroviral strand transfer reactions by structurally diverse compounds., J Virol. 71 (1997) 807–811. 10.1128/jvi.71.1.807-811.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Espeseth AS, Felock P, Wolfe A, Witmer M, Grobler J, Anthony N, Egbertson M, Melamed JY, Young S, Hamill T, Cole JL, Hazuda DJ, HIV-1 integrase inhibitors that compete with the target DNA substrate define a unique strand transfer conformation for integrase., Proceedings of the National Academy of Sciences. 97 (2000) 11244–11249. 10.1073/pnas.200139397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Hazuda DJ, Felock P, Witmer M, Wolfe A, Stillmock K, Grobler JA, Espeseth A, Gabryelski L, Schleif W, Blau C, Miller MD, Inhibitors of Strand Transfer That Prevent Integration and Inhibit HIV-1 Replication in Cells, Science. 287 (2000) 646–650. 10.1126/science.287.5453.646. [DOI] [PubMed] [Google Scholar]
- [150].Hightower KE, Wang R, DeAnda F, Johns BA, Weaver K, Shen Y, Tomberlin GH, Carter HL, Broderick T, Sigethy S, Seki T, Kobayashi M, Underwood MR, Dolutegravir (S/GSK1349572) Exhibits Significantly Slower Dissociation than Raltegravir and Elvitegravir from Wild-Type and Integrase Inhibitor-Resistant HIV-1 Integrase-DNA Complexes, Antimicrobial Agents and Chemotherapy. 55 (2011) 4552–4559. 10.1128/aac.00157-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].White KL, Osman N, Cuadra-Foy E, Brenner BG, Shivakumar D, Campigotto F, Tsiang M, Morganelli PA, Novikov N, Lazerwith SE, Jin H, Niedziela-Majka A, Long Dissociation of Bictegravir from HIV-1 Integrase-DNA Complexes, Antimicrob Agents Ch. (2021). 10.1128/aac.02406-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Jóźwik IK, Passos DO, Lyumkis D, Structural Biology of HIV Integrase Strand Transfer Inhibitors, Trends Pharmacol Sci. 41 (2020) 611–626. 10.1016/j.tips.2020.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Smith SJ, Zhao XZ, Passos DO, Lyumkis D, Burke TR, Hughes SH, Integrase Strand Transfer Inhibitors Are Effective Anti-HIV Drugs, Viruses. 13 (2021) 205. 10.3390/v13020205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Engelman AN, Cherepanov P, Close-up: HIV/SIV intasome structures shed new light on integrase inhibitor binding and viral escape mechanisms, Febs J. 288 (2021) 427–433. 10.1111/febs.15438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Orkin C, Arasteh K, Hernández-Mora MG, Pokrovsky V, Overton ET, Girard P-M, Oka S, Walmsley S, Bettacchi C, Brinson C, Philibert P, Lombaard J, St. Clair M, Crauwels H, Ford SL, Patel P, Chounta V, D’Amico R, Vanveggel S, Dorey D, Cutrell A, Griffith S, Margolis DA, Williams PE, Parys W, Smith KY, Spreen WR, Long-Acting Cabotegravir and Rilpivirine after Oral Induction for HIV-1 Infection, New Engl J Med. 382 (2020) 1124–1135. 10.1056/nejmoa1909512. [DOI] [PubMed] [Google Scholar]
- [156].Swindells S, Andrade-Villanueva J-F, Richmond GJ, Rizzardini G, Baumgarten A, Masiá M, Latiff G, Pokrovsky V, Bredeek F, Smith G, Cahn P, Kim Y-S, Ford SL, Talarico CL, Patel P, Chounta V, Crauwels H, Parys W, Vanveggel S, Mrus J, Huang J, Harrington CM, Hudson KJ, Margolis DA, Smith KY, Williams PE, Spreen WR, Long-Acting Cabotegravir and Rilpivirine for Maintenance of HIV-1 Suppression, New Engl J Med. 382 (2020) 1112–1123. 10.1056/nejmoa1904398. [DOI] [PubMed] [Google Scholar]
- [157].Scarsi KK, Havens JP, Podany AT, Avedissian SN, Fletcher CV, HIV-1 Integrase Inhibitors: A Comparative Review of Efficacy and Safety, Drugs. 8 (2020) 1–28. 10.1007/s40265-020-01379-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Hare S, Smith SJ, Métifiot M, Jaxa-Chamiec A, Pommier Y, Hughes SH, Cherepanov P, Structural and functional analyses of the second-generation integrase strand transfer inhibitor dolutegravir (S/GSK1349572)., Molecular Pharmacology. 80 (2011) 565–572. 10.1124/mol.111.073189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Hare S, Vos AM, Clayton RF, Thuring JW, Cummings MD, Cherepanov P, Molecular mechanisms of retroviral integrase inhibition and the evolution of viral resistance., Proceedings of the National Academy of Sciences of the United States of America. 107 (2010) 20057–20062. 10.1073/pnas.1010246107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Cherepanov P, Maertens GN, Hare S, Structural insights into the retroviral DNA integration apparatus, Curr Opin Struc Biol. 21 (2011) 249–256. 10.1016/j.sbi.2010.12.005. [DOI] [PubMed] [Google Scholar]
- [161].Cook NJ, Li W, Berta D, Badaoui M, Ballandras-Colas A, Nans A, Kotecha A, Rosta E, Engelman AN, Cherepanov P, Structural basis of second-generation HIV integrase inhibitor action and viral resistance, Science. 367 (2020) 806–810. 10.1126/science.aay4919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Passos DO, Li M, Jóźwik IK, Zhao XZ, Santos-Martins D, Yang R, Smith SJ, Jeon Y, Forli S, Hughes SH, Burke TR, Craigie R, Lyumkis D, Structural basis for strand-transfer inhibitor binding to HIV intasomes., Science. 367 (2020) 810–814. 10.1126/science.aay8015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Melek M, Jones JM, O’Dea MH, Pais G, Burke TR, Pommier Y, Neamati N, Gellert M, Effect of HIV integrase inhibitors on the RAG1/2 recombinase, Proc National Acad Sci. 99 (2002) 134–137. 10.1073/pnas.012610699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Nishana M, Nilavar NM, Kumari R, Pandey M, Raghavan SC, HIV integrase inhibitor, Elvitegravir, impairs RAG functions and inhibits V(D)J recombination., Cell Death & Disease. 8 (2017) e2852. 10.1038/cddis.2017.237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Nilavar NM, Paranjape AM, Raghavan SC, Biochemical activity of RAGs is impeded by Dolutegravir, an HIV integrase inhibitor, Cell Death Discov. 6 (2020) 50. 10.1038/s41420-020-0281-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Zhuang L, Wai JS, Embrey MW, Fisher TE, Egbertson MS, Payne LS, Guare JP, Vacca JP, Hazuda DJ, Felock PJ, Wolfe AL, Stillmock KA, Witmer MV, Moyer G, Schleif WA, Gabryelski LJ, Leonard YM, Lynch JJ, Michelson SR, Young SD, Design and Synthesis of 8-Hydroxy-[1,6]Naphthyridines as Novel Inhibitors of HIV-1 Integrase in Vitro and in Infected Cells, J Med Chem. 46 (2003) 453–456. 10.1021/jm025553u. [DOI] [PubMed] [Google Scholar]
- [167].Hazuda DJ, Anthony NJ, Gomez RP, Jolly SM, Wai JS, Zhuang L, Fisher TE, Embrey M, Guare JP, Egbertson MS, Vacca JP, Huff JR, Felock PJ, Witmer MV, Stillmock KA, Danovich R, Grobler J, Miller MD, Espeseth AS, Jin L, Chen I-W, Lin JH, Kassahun K, Ellis JD, Wong BK, Xu W, Pearson PG, Schleif WA, Cortese R, Emini E, Summa V, Holloway MK, Young SD, A naphthyridine carboxamide provides evidence for discordant resistance between mechanistically identical inhibitors of HIV-1 integrase, P Natl Acad Sci Usa. 101 (2004) 11233–11238. 10.1073/pnas.0402357101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Embrey MW, Wai JS, Funk TW, Homnick CF, Perlow DS, Young SD, Vacca JP, Hazuda DJ, Felock PJ, Stillmock KA, Witmer MV, Moyer G, Schleif WA, Gabryelski LJ, Jin L, Chen I-W, Ellis JD, Wong BK, Lin JH, Leonard YM, Tsou NN, Zhuang L, A series of 5-(5,6)-dihydrouracil substituted 8-hydroxy-[1,6]naphthyridine-7-carboxylic acid 4-fluorobenzylamide inhibitors of HIV-1 integrase and viral replication in cells, Bioorg Med Chem Lett. 15 (2005) 4550–4554. 10.1016/j.bmcl.2005.06.105. [DOI] [PubMed] [Google Scholar]
- [169].Zhao XZ, Smith SJ, Maskell DP, Métifiot M, Pye VE, Fesen K, Marchand C, Pommier Y, Cherepanov P, Hughes SH, Burke TR, HIV-1 Integrase Strand Transfer Inhibitors with Reduced Susceptibility to Drug Resistant Mutant Integrases., ACS Chemical Biology. 11 (2016) 1074–1081. 10.1021/acschembio.5b00948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Zhao XZ, Smith SJ, Maskell DP, Métifiot M, Pye VE, Fesen K, Marchand C, Pommier Y, Cherepanov P, Hughes SH, Burke JTR, Structure-Guided Optimization of HIV Integrase Strand Transfer Inhibitors, Journal of Medicinal Chemistry. 60 (2017) 7315–7332. 10.1021/acs.jmedchem.7b00596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Smith SJ, Zhao XZ, B. TR Jr, Hughes SH, HIV-1 integrase inhibitors that are broadly effective against drug-resistant mutants, Antimicrobial Agents and Chemotherapy. (2018) AAC.01035-18. 10.1128/aac.01035-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Smith SJ, Zhao XZ, Passos DO, Lyumkis D, Burke TRJ, Hughes SH, HIV-1 Integrase Inhibitors That Are Active against Drug-Resistant Integrase Mutants., Acs Infect Dis. 64 (2020) 430. 10.1128/aac.00611-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Blanco J-L, Varghese V, Rhee S-Y, Gatell JM, Shafer RW, HIV-1 Integrase Inhibitor Resistance and Its Clinical Implications, J Infect Dis. 203 (2011) 1204–1214. 10.1093/infdis/jir025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Smith SJ, Zhao XZ, Burke TR, Hughes SH, Efficacies of Cabotegravir and Bictegravir against drug-resistant HIV-1 integrase mutants, Retrovirology. 15 (2018) 37. 10.1186/s12977-018-0420-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Anstett K, Brenner B, Mesplede T, Wainberg MA, HIV drug resistance against strand transfer integrase inhibitors., Retrovirology. 14 (2017) 36. 10.1186/s12977-017-0360-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [176].Korber B, Gaschen B, Yusim K, Thakallapally R, Kesmir C, Detours V, Evolutionary and immunological implications of contemporary HIV-1 variation, Brit Med Bull. 58 (2001) 19–42. 10.1093/bmb/58.1.19. [DOI] [PubMed] [Google Scholar]
- [177].Varadarajan J, McWilliams MJ, Hughes SH, Treatment with suboptimal doses of raltegravir leads to aberrant HIV-1 integrations., Proceedings of the National Academy of Sciences of the United States of America. 110 (2013) 14747–14752. 10.1073/pnas.1305066110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [178].Varadarajan J, McWilliams MJ, Mott BT, Thomas CJ, Smith SJ, Hughes SH, Drug resistant integrase mutants cause aberrant HIV integrations., Retrovirology. 13 (2016) 71–10. 10.1186/s12977-016-0305-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Quashie PK, Mesplede T, Wainberg MA, Evolution of HIV integrase resistance mutations., Current Opinion in Infectious Diseases. 26 (2013) 43–49. 10.1097/qco.0b013e32835ba81c. [DOI] [PubMed] [Google Scholar]
- [180].Delelis O, Malet I, Na L, Tchertanov L, Calvez V, Marcelin A-G, Subra F, Deprez E, Mouscadet J-F, The G140S mutation in HIV integrases from raltegravir-resistant patients rescues catalytic defect due to the resistance Q148H mutation, Nucleic Acids Res. 37 (2009) 1193–1201. 10.1093/nar/gkn1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Rhee S-Y, Grant PM, Tzou PL, Barrow G, Harrigan PR, Ioannidis JPA, Shafer RW, A systematic review of the genetic mechanisms of dolutegravir resistance., Journal of Antimicrobial Chemotherapy. 32 (2019) 1551. 10.1093/jac/dkz256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Malet I, Delelis O, Valantin M-A, Montes B, Soulie C, Wirden M, Tchertanov L, Peytavin G, Reynes J, Mouscadet J-F, Katlama C, Calvez V, Marcelin A-G, Mutations Associated with Failure of Raltegravir Treatment Affect Integrase Sensitivity to the Inhibitor In Vitro▿, Antimicrob Agents Ch. 52 (2008) 1351–1358. 10.1128/aac.01228-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Quashie PK, Mesplede T, Han Y-S, Oliveira M, Singhroy DN, Fujiwara T, Underwood MR, Wainberg MA, Characterization of the R263K Mutation in HIV-1 Integrase That Confers Low-Level Resistance to the Second-Generation Integrase Strand Transfer Inhibitor Dolutegravir, Journal of Virology. 86 (2012) 2696–2705. 10.1128/jvi.06591-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Quashie PK, Mesplede T, Han Y-S, Veres T, Osman N, Hassounah S, Sloan RD, Xu H-T, Wainberg MA, Biochemical analysis of the role of G118R-linked dolutegravir drug resistance substitutions in HIV-1 integrase., Antimicrobial Agents and Chemotherapy. 57 (2013) 6223–6235. 10.1128/aac.01835-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Lübke N, Jensen B, Hüttig F, Feldt T, Walker A, Thielen A, Däumer M, Obermeier M, Kaiser R, Knops E, Heger E, Sierra S, Oette M, Lengauer T, Timm J, Häussinger D, Failure of Dolutegravir First-Line ART with Selection of Virus Carrying R263K and G118R., The New England Journal of Medicine. 381 (2019) 887–889. 10.1056/nejmc1806554. [DOI] [PubMed] [Google Scholar]
- [186].King NM, Prabu-Jeyabalan M, Nalivaika EA, Schiffer CA, Combating susceptibility to drug resistance: lessons from HIV-1 protease., Chemistry & Biology. 11 (2004) 1333–1338. 10.1016/j.chembiol.2004.08.010. [DOI] [PubMed] [Google Scholar]
- [187].Yilmaz NK, Swanstrom R, Schiffer CA, Improving Viral Protease Inhibitors to Counter Drug Resistance., Trends in Microbiology. 24 (2016) 547–557. 10.1016/j.tim.2016.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Shen Y, Altman MD, Ali A, Nalam MNL, Cao H, Rana TM, Schiffer CA, Tidor B, Testing the substrate-envelope hypothesis with designed pairs of compounds., 8 (2013) 2433–2441. 10.1021/cb400468c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Zhao XZ, Smith SJ, Métifiot M, Marchand C, Boyer PL, Pommier Y, Hughes SH, B. TR Jr, 4-Amino-1-hydroxy-2-oxo-1,8-naphthyridine-Containing Compounds Having High Potency against Raltegravir-Resistant Integrase Mutants of HIV-1, Journal of Medicinal Chemistry. 57 (2014) 5190–5202. 10.1021/jm5001908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Smith SJ, Zhao XZ, Passos DO, Pye VE, Cherepanov P, Lyumkis D, Burke TR, Hughes SH, HIV-1 Integrase Inhibitors with Modifications That Affect Their Potencies against Drug Resistant Integrase Mutants, Acs Infect Dis. (2021). 10.1021/acsinfecdis.0c00819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [191].Biela A, Sielaff F, Terwesten F, Heine A, Steinmetzer T, Klebe G, Ligand Binding Stepwise Disrupts Water Network in Thrombin: Enthalpic and Entropic Changes Reveal Classical Hydrophobic Effect, Journal of Medicinal Chemistry. 55 (2012) 6094–6110. 10.1021/jm300337q. [DOI] [PubMed] [Google Scholar]
- [192].Biela A, Nasief NN, Betz M, Heine A, Hangauer D, Klebe G, Dissecting the hydrophobic effect on the molecular level: the role of water, enthalpy, and entropy in ligand binding to thermolysin., Angewandte Chemie (International Ed. in English). 52 (2013) 1822–1828. 10.1002/anie.201208561. [DOI] [PubMed] [Google Scholar]
- [193].Cramer J, Krimmer SG, Heine A, Klebe G, Paying the Price of Desolvation in Solvent-Exposed Protein Pockets: Impact of Distal Solubilizing Groups on Affinity and Binding Thermodynamics in a Series of Thermolysin Inhibitors, Journal of Medicinal Chemistry. 60 (2017) 5791–5799. 10.1021/acs.jmedchem.7b00490. [DOI] [PubMed] [Google Scholar]
- [194].Krimmer SG, Betz M, Heine A, Klebe G, Methyl, ethyl, propyl, butyl: futile but not for water, as the correlation of structure and thermodynamic signature shows in a congeneric series of thermolysin inhibitors., ChemMedChem. 9 (2014) 833–846. 10.1002/cmdc.201400013. [DOI] [PubMed] [Google Scholar]
- [195].Ivashchenko AA, Ivanenkov YA, Koryakova AG, Karapetian RN, Mitkin OD, Aladinskiy VA, Kravchenko DV, Savchuk NP, Ivashchenko AV, Synthesis, biological evaluation and in silico modeling of novel integrase strand transfer inhibitors (INSTIs), European Journal of Medicinal Chemistry. (2020) 112064. 10.1016/j.ejmech.2020.112064. [DOI] [PubMed] [Google Scholar]
- [196].Simonetti FR, Kearney MF, Review: Influence of ART on HIV genetics., Current Opinion in HIV and AIDS. 10 (2015) 49–54. 10.1097/coh.0000000000000120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Fader LD, Malenfant E, Parisien M, Carson R, Bilodeau F, Landry S, Pesant M, Brochu C, Morin S, Chabot C, Halmos T, Bousquet Y, Bailey MD, Kawai SH, Coulombe R, LaPlante S, Jakalian A, Bhardwaj PK, Wernic D, Schroeder P, Amad M, Edwards P, Garneau M, Duan J, Cordingley M, Bethell R, Mason SW, Bös M, Bonneau P, Poupart M-A, Faucher A-M, Simoneau B, Fenwick C, Yoakim C, Tsantrizos Y, Discovery of BI 224436, a Noncatalytic Site Integrase Inhibitor (NCINI) of HIV-1, Acs Med Chem Lett. 5 (2014) 422–427. 10.1021/ml500002n. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Christ F, Voet A, Marchand A, Nicolet S, Desimmie BA, Marchand D, Bardiot D, der Veken NJV, Remoortel BV, Strelkov SV, Maeyer MD, Chaltin P, Debyser Z, Rational design of small-molecule inhibitors of the LEDGF/p75-integrase interaction and HIV replication, Nature Chemical Biology. 6 (2010) 442–448. 10.1038/nchembio.370. [DOI] [PubMed] [Google Scholar]
- [199].Demeulemeester J, Chaltin P, Marchand A, Maeyer MD, Debyser Z, Christ F, LEDGINs, non-catalytic site inhibitors of HIV-1 integrase: a patent review (2006 – 2014), Expert Opin Ther Pat. 24 (2014) 609–632. 10.1517/13543776.2014.898753. [DOI] [PubMed] [Google Scholar]
- [200].Engelman AN, Multifaceted HIV integrase functionalities and therapeutic strategies for their inhibition, Journal of Biological Chemistry. (2019) jbc.REV119.006901. 10.1074/jbc.rev119.006901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Feng L, Larue RC, Slaughter A, Kessl JJ, Kvaratskhelia M, HIV-1 Integrase Multimerization as a Therapeutic Target, in: Torbett BE, Richman Douglas D, Goodsell DS (Eds.), Springer International Publishing, 2015: pp. 93–119. 10.1007/82_2015_439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Engelman A, Kessl JJ, Kvaratskhelia M, Allosteric inhibition of HIV-1 integrase activity, Curr Opin Chem Biol. 17 (2013) 339–345. 10.1016/j.cbpa.2013.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Sharma A, Slaughter A, Jena N, Feng L, Kessl JJ, Fadel HJ, Malani N, Male F, Wu L, Poeschla E, Bushman FD, Fuchs JR, Kvaratskhelia M, A New Class of Multimerization Selective Inhibitors of HIV-1 Integrase., PLoS Pathog. 10 (2014) e1004171. 10.1371/journal.ppat.1004171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Fader LD, Bailey M, Beaulieu E, Bilodeau F, Bonneau P, Bousquet Y, Carson RJ, Chabot C, Coulombe R, Duan J, Fenwick C, Garneau M, Halmos T, Jakalian A, James C, Kawai SH, Landry S, LaPlante SR, Mason SW, Morin S, Rioux N, Simoneau B, Surprenant S, Thavonekham B, Thibeault C, Trinh T, Tsantrizos Y, Tsoung J, Yoakim C, Wernic D, Aligning Potency and Pharmacokinetic Properties for Pyridine-Based NCINIs, Acs Med Chem Lett. 7 (2016) 797–801. 10.1021/acsmedchemlett.6b00194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Amadori C, van der Velden YU, Bonnard D, Orlov I, van Bel N, Rouzic EL, Miralles L, Brias J, Chevreuil F, Spehner D, Chasset S, Ledoussal B, Mayr L, Moreau F, García F, Gatell J, Zamborlini A, Emiliani S, Ruff M, Klaholz BP, Moog C, Berkhout B, Plana M, Benarous R, The HIV-1 integrase-LEDGF allosteric inhibitor MUT-A: resistance profile, impairment of virus maturation and infectivity but without influence on RNA packaging or virus immunoreactivity, Retrovirology. 14 (2017) 50. 10.1186/s12977-017-0373-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Kessl JJ, Jena N, Koh Y, Taskent-Sezgin H, Slaughter A, Feng L, de Silva S, Wu L, Grice SFJL, Engelman A, Fuchs JR, Kvaratskhelia M, Multimode, cooperative mechanism of action of allosteric HIV-1 integrase inhibitors., 287 (2012) 16801–16811. 10.1074/jbc.m112.354373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Rouzic EL, Bonnard D, Chasset S, Bruneau J-M, Chevreuil F, Strat FL, Nguyen J, Beauvoir R, Amadori C, Brias J, Vomscheid S, Eiler S, Lévy N, Delelis O, Deprez E, Saïb A, Zamborlini A, Emiliani S, Ruff M, Ledoussal B, Moreau F, Benarous R, Dual inhibition of HIV-1 replication by integrase-LEDGF allosteric inhibitors is predominant at the post-integration stage, Retrovirology. 10 (2013) 144. 10.1186/1742-4690-10-144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Tsiang M, Jones GS, Niedziela-Majka A, Kan E, Lansdon EB, Huang W, Hung M, Samuel D, Novikov N, Xu Y, Mitchell M, Guo H, Babaoglu K, Liu X, Geleziunas R, Sakowicz R, New Class of HIV-1 Integrase (IN) Inhibitors with a Dual Mode of Action, J Biol Chem. 287 (2012) 21189–21203. 10.1074/jbc.m112.347534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Gupta K, Brady T, Dyer BM, Malani N, Hwang Y, Male F, Nolte RT, Wang L, Velthuisen E, Jeffrey J, Duyne GDV, Bushman FD, Allosteric inhibition of human immunodeficiency virus integrase: late block during viral replication and abnormal multimerization involving specific protein domains., The Journal of Biological Chemistry. 289 (2014) 20477–20488. 10.1074/jbc.m114.551119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Wilson TA, Koneru PC, Rebensburg SV, Lindenberger JJ, Kobe MJ, Cockroft NT, Adu-Ampratwum D, Larue RC, Kvaratskhelia M, Fuchs JR, An Isoquinoline Scaffold as a Novel Class of Allosteric HIV-1 Integrase Inhibitors, Acs Med Chem Lett. 10 (2019) 215–220. 10.1021/acsmedchemlett.8b00633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Christ F, Shaw S, Demeulemeester J, Desimmie BA, Marchand A, Butler S, Smets W, Chaltin P, Westby M, Debyser Z, Pickford C, Small-molecule inhibitors of the LEDGF/p75 binding site of integrase block HIV replication and modulate integrase multimerization., Antimicrobial Agents and Chemotherapy. 56 (2012) 4365–4374. 10.1128/aac.00717-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Desimmie BA, Schrijvers R, Demeulemeester J, Borrenberghs D, Weydert C, Thys W, Vets S, Remoortel BV, Hofkens J, Rijck JD, Hendrix J, Bannert N, Gijsbers R, Christ F, Debyser Z, LEDGINs inhibit late stage HIV-1 replication by modulating integrase multimerization in the virions, Retrovirology. 10 (2013) 57. 10.1186/1742-4690-10-57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Balakrishnan M, Yant SR, Tsai L, O’Sullivan C, Bam RA, Tsai A, Niedziela-Majka A, Stray KM, Sakowicz R, Cihlar T, Non-Catalytic Site HIV-1 Integrase Inhibitors Disrupt Core Maturation and Induce a Reverse Transcription Block in Target Cells, Plos One. 8 (2013) e74163. 10.1371/journal.pone.0074163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [214].Gupta K, Turkki V, Sherrill-Mix S, Hwang Y, Eilers G, Taylor L, McDanal C, Wang P, Temelkoff D, Nolte RT, Velthuisen E, Jeffrey J, Duyne GDV, Bushman FD, Structural Basis for Inhibitor-Induced Aggregation of HIV Integrase., PLoS Biology. 14 (2016) e1002584. 10.1371/journal.pbio.1002584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [215].Feng L, Sharma A, Slaughter A, Jena N, Koh Y, Shkriabai N, Larue RC, Patel PA, Mitsuya H, Kessl JJ, Engelman A, Fuchs JR, Kvaratskhelia M, The A128T Resistance Mutation Reveals Aberrant Protein Multimerization as the Primary Mechanism of Action of Allosteric HIV-1 Integrase Inhibitors*, J Biol Chem. 288 (2013) 15813–15820. 10.1074/jbc.m112.443390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Gupta K, Allen A, Giraldo C, Eilers G, Sharp R, Hwang Y, Murali H, Cruz K, Janmey P, Bushman F, Duyne GDV, Allosteric HIV Integrase Inhibitors Promote Formation of Inactive Branched Polymers via Homomeric Carboxy-Terminal Domain Interactions, Structure. 29 (2021) 213–225.e5. 10.1016/j.str.2020.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [217].Deng N, Hoyte A, Mansour YE, Mohamed MS, Fuchs JR, Engelman AN, Kvaratskhelia M, Levy R, Allosteric HIV-1 integrase inhibitors promote aberrant protein multimerization by directly mediating inter-subunit interactions: Structural and thermodynamic modeling studies., Protein Science : A Publication of the Protein Society. 25 (2016) 1911–1917. 10.1002/pro.2997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [218].Slaughter A, Jurado KA, Deng N, Feng L, Kessl JJ, Shkriabai N, Larue RC, Fadel HJ, Patel PA, Jena N, Fuchs JR, Poeschla E, Levy RM, Engelman A, Kvaratskhelia M, The mechanism of H171T resistance reveals the importance of Nδ-protonated His171 for the binding of allosteric inhibitor BI-D to HIV-1 integrase., Retrovirology. 11 (2014) 100. 10.1186/s12977-014-0100-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Hoyte AC, Jamin AV, Koneru PC, Kobe MJ, Larue RC, Fuchs JR, Engelman AN, Kvaratskhelia M, Resistance to pyridine-based inhibitor KF116 reveals an unexpected role of integrase in HIV-1 Gag-Pol polyprotein proteolytic processing, J Biol Chem. 292 (2017) 19814–19825. 10.1074/jbc.m117.816645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [220].Patel PA, Kvaratskhelia N, Mansour Y, Antwi J, Feng L, Koneru P, Kobe MJ, Jena N, Shi G, Mohamed MS, Li C, Kessl JJ, Fuchs JR, Indole-based allosteric inhibitors of HIV-1 integrase, Bioorg Med Chem Lett. 26 (2016) 4748–4752. 10.1016/j.bmcl.2016.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Nakamura T, Nakamura T, Amano M, Miyakawa T, Yamagata Y, Matsuoka M, Nakata H, A Conformational Escape Reaction of HIV-1 against an Allosteric Integrase Inhibitor, J Virol. 94 (2020). 10.1128/jvi.00486-20. [DOI] [PMC free article] [PubMed] [Google Scholar]