

Summary
Mitochondrial DNA (mtDNA) is present in multiple copies in human cells. We evaluated cross-sectional associations of whole-blood mtDNA copy number (CN) with several cardiometabolic disease traits in 408,361 participants of multiple ancestries in TOPMed and UK Biobank. Age showed a threshold association with mtDNA CN: each additional 10 years of age was associated with a 0.03 SD higher level of mtDNA CN (p = 0.0014) among younger participants (younger than 65 years) versus a 0.14 SD lower level of mtDNA CN (p = 1.82 × 10−13) among older participants (65 years and older). At lower mtDNA CN levels, we found age-independent associations with increased odds of obesity (p = 5.6 × 10−238), hypertension (p = 2.8 × 10−50), diabetes (p = 3.6 × 10−7), and hyperlipidemia (p = 6.3 × 10−56). The observed decline in mtDNA CN after 65 years of age may be a key to understanding age-related diseases.
Keywords: mitochondrial DNA copy number, cardiometabolic disease, whole-genome sequencing, whole-exom sequencing, aging, white blood cell counts, inflammation
Graphical abstract
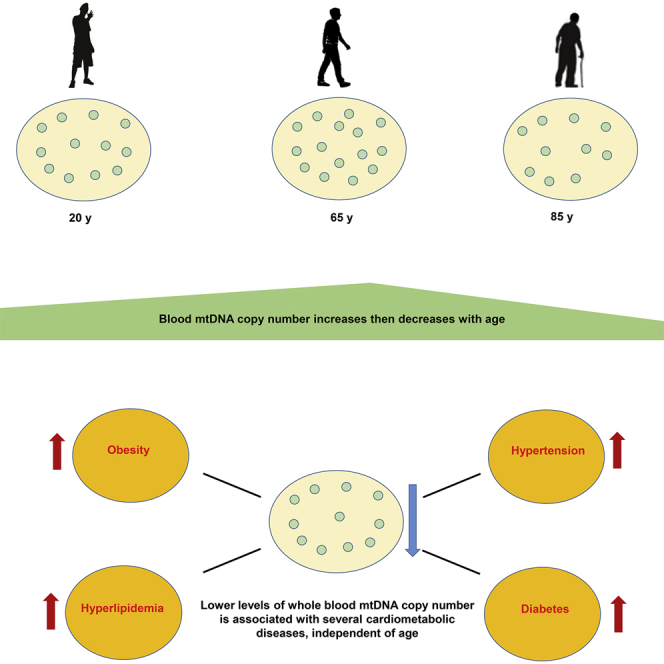
Highlights
mtDNA copy number in peripheral blood measured in large multi-ancestry cohorts
mtDNA copy number in blood cells declined with age after age 65
Lower mtDNA copy number was associated with cardiometabolic disease traits
Liu et al. examined the association of mitochondrial DNA (mtDNA) copy number (CN) with cardiometabolic traits in 408,361 individuals from TOPMed and UK Biobank, representing the most comprehensive cross-ancestry analyses for these traits. They identify a decline in mtDNA CN in participants older than 65 years and, at lower mtDNA CN levels, age-independent associations with obesity, diabetes, hypertension, and hyperlipidemia.
Introduction
Mitochondria convert dietary calories to molecular energy through oxidative phosphorylation (OXPHOS).1 In addition, mitochondria have essential roles in cellular differentiation, proliferation, reprogramming, and aging.2, 3, 4, 5, 6, 7 Mitochondria contain their own genome (mtDNA), which encodes 37 genes.1 Multiple copies of mtDNA are present per mitochondrion, and cells contain up to 7,000 mitochondria per cell.8 The mtDNA copy number (mtDNA CN) correlates with cellular energy generating capacity and metabolic status9 and, therefore, varies greatly across tissue and cell types.1,10,11
Several previous studies have identified a lower level of mtDNA CN in older individuals. This reduced mtDNA CN has been associated with a general decline in health12, 13, 14 and an increased risk of developing cardiovascular disease (CVD) outcomes.15 Cardiometabolic diseases (CMDs), including obesity, abnormal lipid level and glucose level in plasma, and high blood pressure, are known risk factors for the development of CVD.16,17 Thus, one mechanism by which a decrease in mtDNA CN could adversely affect not only CVD but also health in general is if the reduction in mtDNA CN was associated with an increase in CMDs. However, the associations of mtDNA CN with the CMD traits have not been consistently reported.18, 19, 20 We investigated associations of mtDNA CN with CMD traits in eight US cohorts from the Trans-Omics for Precision Medicine (TOPMed), representing the most comprehensive cross-ancestry analyses for these traits. These cohort studies included extensive cardiometabolic phenotyping and mtDNA CN estimated from whole-genome sequencing (WGS) data. For validation analyses, we analyzed individuals with whole-exome sequencing (WES) from the UK Biobank (Figure 1).
Figure 1.

Study design
Association analysis of mtDNA copy number (CN) with cardiometabolic disease traits was performed in cohorts from European Americans (n = 13,378), African Americans (n = 8,020), Chinese Americans (n = 601), and Hispanic and Latino Americans (n = 4,892) in TOPMed and from the UK Biobank (n = 381,470) of European ancestry participants. Meta-analysis was performed using the fixed-effects inverse-variance method to summarize the results among European Americans and among African Americans in TOPMed. WGS, whole-genome sequencing; WES, whole-exome sequencing. See also Tables S1–S4 and Figures S9–S11.
Results
Characteristics of study participants
The current study included 26,891 participants of eight cohorts from the TOPMed Consortium, including 13,378 European Americans, 8,020 African Americans, 601 Chinese Americans, and 4,892 Hispanic/Latino Americans, as well as 381,470 individuals of European ancestry from the UK Biobank.21, 22, 23, 24, 25, 26, 27, 28, 29, 30, 31, 32, 33 On average, 55% of the study participants were women, and the participants’ mean age was 57 years (range, 20–100 years; Table S1). We observed moderate to high heterogeneity in distributions of age, sex, and cardiometabolic phenotypes across cohorts and ancestries. For example, hypertension (HTN), obesity, diabetes, and hyperlipidemia were more prevalent in African Americans than in participants of other ancestry groups (Table S1).
A threshold effect between age and mtDNA CN
The standardized residuals of mtDNA CN were obtained by regressing mtDNA CN on “blood collection year” (see STAR Methods; Figure S1) to study the relationship of mtDNA CN (as the outcome) with age at blood collection. We observed a threshold effect of age on mtDNA CN (Figures 2A and S2–S4). On average, age was associated with a slightly increased level of mtDNA CN (0.032 SD/10 years [95% confidence interval (CI) = 0.013, 0.052], p = 0.0014) from age 20 to 65 years. However, after 65 years, every additional 10 years of age was associated with a 0.14 SD lower level of mtDNA CN (95% CI = −0.18, −0.10; p = 1.82 × 10−13). The relationship between mtDNA CN and age was similar in men and women, although women had higher mtDNA CNs than men had (β = 0.23; 95% CI = 0.20, 0.26; p = 7.4 × 10−60), as noted previously (Figure S5).14,34 The threshold effect between age and mtDNA CN remained similar after adjusting for white blood cell (WBC) compositions and platelet count (Figure S2).
Figure 2.

Association of mtDNA CN and cardiometabolic disease traits
(A) The relationship of mtDNA CN with age in TOPMed European American and African American participants (n = 21,398).
(B) Association and meta-analyses of mtDNA CN (n = 394,748) with obesity, hypertension (HTN), diabetes, and hyperlipidemia in TOPMed European Americans (n = 13,378) and in UK Biobank European ancestry participants (n = 381,470). ARIC, Atherosclerosis Risk in Communities study; CARDIA, Coronary Artery Risk Development in Young Adults Study; CHS, Cardiovascular Health Study; FHS, Framingham Heart Study; MESA, Multi-Ethnic Study of Atherosclerosis. A horizontal line represents the 95% confidence intervals of an odds ratio (represented by the box in the middle of the line) for a cohort or meta-analysis; N, sample size; X axis, odds ratio; a number with a square bracket represents an odds ratio with 95% confidence intervals for a meta-analysis.
(C) Comparison of odds ratio of cardiometabolic disease traits in participants with and without adjusting for white blood cell and platelet counts. Meta-analysis (n = 386,526) using inverse-variance weighting, combining the TOPMed European American participants and UK Biobank participants, with cell counts. The odds ratio (OR) corresponds to a 1-SD decrease in the mtDNA CN level.
(D) Age-specific meta-analysis (age younger than 65 years, n = 315,708; age 65 years and older, n = 79,782) combining European ancestry participants in TOPMed and UK Biobank. The effect size estimates are in units of cardiometabolic traits corresponding to a 1-SD decrease in mtDNA CN. See Tables S6–S8 and Figures S2–S8 and S12–S16.
Association analyses in European American participants
We generated cohort- and ancestry-specific mtDNA CN standardized residuals by regressing mtDNA CN on age, age-squared, sex, and “blood collection year” in primary analyses in TOPMed cohorts and UK Biobank (see STAR Methods). We then performed cohort- and ancestry-specific association analyses of the standardized mtDNA residuals (as the main predictor) with CMD traits (as the outcome), adjusting for age, age-squared (for blood pressure phenotypes), sex, body mass index (BMI, not for obesity), and smoking status (see STAR Methods).35, 36, 37 Meta-analysis was performed using the fixed-effects inverse-variance method to summarize results based on an a priori assumption that there is only one true treatment effect between studies (n = 13,378) (Table 1; Figures 2B, S6, and S7). Because low mtDNA CN was reported to be associated with an increased CMD risk,18, 19, 20,38 we reported β estimates as the change in a CMD outcome variable in response to 1 SD lower mtDNA CN in all analyses. We found that 1 SD decrease in mtDNA CN was significantly associated with 1.10-fold odds of obesity (95% CI = 1.05,1.15; p = 1.0 × 10−4), 1.08-fold odds of HTN (95% CI = 1.03, 1.12; p = 1.2 × 10−3), and 1.22-fold odds of diabetes (95% CI = 1.13, 1.30; p = 2.8 × 10−8), whereas it was not associated with hyperlipidemia (p = 0.13). For continuous traits, 1 SD decrease in mtDNA CN was significantly associated with a 0.030-unit (95% CI = 0.021, 0.039; p = 2.5 × 10−11) increase in log-transformed triglyceride (TRIG) value and a 0.20 kg/m2 (95% CI = 0.11, 0.29; p = 2.0 × 10−5) increase in BMI and was nominally associated with 0.43 mm Hg (95% CI = 0.077, 0.78; p = 0.019) increase in systolic blood pressure (SBP). In contrast, a 1 SD decrease in mtDNA CN was significantly associated with a 0.012-unit (95% CI = −0.017, −0.0071; p = 2.9 × 10−6) decrease in log-transformed high-density lipoprotein (HDL) value and a 0.0075 unit decrease in low-density lipoprotein (LDL) (95% CI= −0.013, −0.0022; p = 0.0058). mtDNA CN was not significantly associated with either diastolic blood pressure (DBP) or fasting plasma glucose (FBG; p > 0.05) in the initial meta-analysis (Table 1; Figures S6 and S7).
Table 1.
Cross-sectional association and meta-analysis of mtDNA CN with metabolic disease phenotypes
| Traits | Initial (TopMed, European ancestry) (N = 13,378) |
Validation (UK Biobank, European ancestry) (N = 381,470) |
Overall meta-analysis (N = 394,848) |
||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR | 95% CI | Β | SE | p value | OR | 95% CI | β | SE | p value | OR | 95% CI | β | SE | p value | |
| Binary outcomes | |||||||||||||||
| Obesity | 1.10 | 1.05–1.15 | – | – | 1.0 × 10−4 | 1.14 | 1.13–1.15 | – | – | 9.1 × 10−237 | 1.14 | 1.13–1.15 | – | – | 5.6 × 10−238 |
| HTN | 1.08 | 1.03–1.12 | – | – | 1.2 × 10−3 | 1.06 | 1.05–1.07 | – | – | 3.8 × 10−48 | 1.06 | 1.05–1.07 | – | – | 2.8 × 10−50 |
| Diabetes | 1.22 | 1.13–1.30 | – | – | 2.8 × 10−8 | 1.03 | 1.02–1.05 | – | – | 1.0 × 10−4 | 1.04 | 1.03–1.06 | – | – | 3.6 × 10−7 |
| Hyperlipidemia | 1.03 | 0.99–1.08 | – | – | 0.13 | 1.08 | 1.07–1.09 | – | – | 2.2 × 10−56 | 1.08 | 1.07–1.09 | – | – | 6.3 × 10−56 |
| Continuous outcomes | |||||||||||||||
| BMI | – | – | 0.2 | 0.046 | 2.0 × 10−5 | – | – | 0.3 | 0.0077 | 2.1 × 10−332 | – | – | 0.3 | 0.0076 | 1.0 × 10−335 |
| DBP | – | – | 0.042 | 0.1 | 0.69 | – | – | 0.24 | 0.019 | 5.9 × 10−39 | – | – | 0.24 | 0.018 | 4.6 × 10−38 |
| SBP | – | – | 0.43 | 0.18 | 0.019 | – | – | 0.64 | 0.033 | 7.5 × 10−86 | – | – | 0.64 | 0.032 | 9.5 × 10−87 |
| FBG | – | – | 0.054 | 0.093 | 0.56 | – | – | 0.23 | 0.019 | 1.1 × 10−32 | – | – | 0.22 | 0.019 | 6.5 × 10−32 |
| HDL | – | – | −0.012 | 0.0025 | 2.9 × 10−6 | – | – | 0.0034 | 0.0004 | 3.0 × 10−17 | – | – | 0.003 | 0.0004 | 2.2 × 10−14 |
| LDL | – | – | −0.0075 | 0.0027 | 0.0058 | – | – | 0.0066 | 0.00047 | 1.7 × 10−43 | – | – | 0.0061 | 0.0005 | 1.7 × 10−39 |
| TRIG | – | – | 0.03 | 0.0045 | 2.5 × 10−11 | – | – | 0.021 | 0.00082 | 4.5 × 10−149 | – | – | 0.022 | 0.0008 | 2.9 × 10−158 |
The β estimates are in units of metabolic traits corresponding to 1 SD lower mtDNA CN. Association analysis of mtDNA CN with metabolic traits was performed in cohorts of European Americans in TOPMed and also in European-ancestry participants in UK Biobank. Meta-analysis with the fixed or random-effects inverse-variance method was applied to summarize the results. See also Tables S2–S4, S6, and S8 and Figures S4–S13.
Meta-analysis with participants of European ancestry in UK Biobank
We tested seven associations with p < 0.01 from the initial meta-analysis in TOPMed for association in participants of European ancestry in the UK Biobank. Five of those seven associations were validated in the direction of association in the UK Biobank (Table 1; Figures 2B and S6–S8). Compared with those in the initial meta-analysis, analyses in the UK Biobank data yielded larger effect sizes for associations of mtDNA CN with five traits (Table 1). For example, a 1 SD decrease in mtDNA CN was associated with a 1.14-fold (95% CI = 1.13, 1.15) odds of obesity in the UK Biobank versus a 1.10-fold (95% CI = 1.05–1.15) odds of obesity in the initial meta-analysis in TOPMed. mtDNA CN was not significantly associated with hyperlipidemia in TOPMed, whereas it was significantly associated with 1.08-fold odds of hyperlipidemia (95% CI = 1.07, 1.09; p = 2.2 × 10−56) in the UK Biobank. Similarly, mtDNA CN displayed significant association with DBP (mm Hg) (β = 0.24; 95% CI = 0.20, 0.28; p = 5.9 × 10−39) and FBG (mg/dL) (β = 0.23; 95% CI = 0.19, 0.26; p = 1.1 × 10−32) in the UK Biobank, whereas it was not associated with those traits in TOPMed. A low level of mtDNA CN was associated with low levels of log-transformed LDL and HDL in TOPMed, but a 1 SD unit decrease in mtDNA CN was significantly associated with a 0.0034-unit (95% CI = 0.0026, 0.0042; p = 3.0 × 10−17) increase in log-transformed HDL and a 0.0066-unit increase in log-transformed LDL (95% CI = 0.0057, 0.0075; p = 1.7 × 10−43) in the UK Biobank. The association of mtDNA CN with TRIG, however, displayed consistent directionality in both TOPMed and the UK Biobank data (Table 1; Figures S4 and S5). Because of a much larger sample size in the UK Biobank, a meta-analysis combining all participants of European ancestry (total n = 394,848) in the TOPMed and UK Biobank yielded results similar to those of the UK-Biobank-only analysis for all 11 traits (Table 1; Figures S4 and S5).
Comparison of directionality of associations between European and other ancestries
The directionality of associations of mtDNA CN with CMD traits was consistent in African Americans (n = 8,020) (Table S2; Figure S9), Hispanic/Latino Americans (n = 4,892), and Asian Americans (n = 601) compared with that of the participants of European ancestry for most of the CMD traits (Table S3; Figures S10 and S11). In the meta-analysis of African American participants, 1 SD decrease in mtDNA CN was significantly associated with 1.14-fold odds of diabetes (95% CI = 1.07, 1.23; p = 2.0 × 10−4), a 0.75 mm Hg increase in SBP (95% CI = 0.27, 1.22; p = 2.0 × 10−3), and a 0.0077 unit increase in TRIG (95% CI = 0.0024, 0.013; p = 0.0039). In Asian-only participants (n = 601), 1 SD decrease in mtDNA CN was significantly associated with 1.43-fold odds of hyperlipidemia (95% CI = 1.19, 1.72; p = 0.00014). In Hispanic-only TOPMed participants (n = 4,892), 1 SD decrease in mtDNA CN was significantly associated with an increase of odds of diabetes (odds ratio [OR] = 1.14; 95% CI = 1.05, 1.24; p = 0.002), and significantly associated with a 0.016-unit decrease in LDL (95% CI = −0.027, −0.0050; p = 4.3 × 10−3) (Table S3). A pan-ancestry meta-analysis of all participants (n = 408,361), combining the TOPMed and the UK Biobank data, gave rise to similar results for all CMD traits to those using the UK-Biobank-only data because of a much larger sample size from the UK Biobank (Table S4).
Accounting for WBC compositions and platelets as covariates
WBC compositions and platelets were available in a subset of participants in TOPMed (n = 12,402) and in all participants of the UK Biobank (n = 381,470). We investigated the relationship of mtDNA CN with WBC compositions (e.g., neutrophil and lymphocyte) and platelet count (see STAR Methods).39, 40, 41, 42 mtDNA CN was inversely associated with the total WBC count and neutrophil count and positively associated with platelet count (Table S5). Moreover, we found that WBC compositions and platelets together explained about 10%–14% of the variation in mtDNA CN, and these blood cell components explained about 0.5%–6% of the variations in CMD traits across a few cohorts (Table S5). Therefore, WBC compositions and platelets are strong confounders for associations between mtDNA CN and several CMD traits. Thus, we compared results between models with and without WBC compositions and platelet count as additional covariates in the same participants. Directionality remained the same for all associations after adjusting for WBC and platelet count in the meta-analysis of participants of European ancestry in the TOPMed and UK Biobank data (Figure 2C; Table S6) and in the ancestry-specific participants and the UK-Biobank-only analyses (Figures S12–S15). Most non-lipid traits, e.g., HTN/SBP and obesity/BMI, displayed a great attenuation in their associations with mtDNA CN after adjusting for WBC compositions and platelet count. In contrast, the associations of mtDNA CN with hyperlipidemia, HDL, and LDL became moderately strengthened after adjusting for WBC and platelet counts (Figure 2C; Table S6).
Interaction analyses
Because of a large sample size, age showed significant interaction with mtDNA CN for multiple traits, including obesity/BMI, FBG, HDL, LDL, and TRIG, and sex showed a significant interaction with mtDNA CN for HTN and BMI in meta-analyses of all participants (Table S7). Because of the threshold effect of age on mtDNA CN, we further performed stratified analyses in younger (younger than 65 years) and older (65 years and older) participants. Hyperlipidemia/lipid traits and HTN displayed consistent effect sizes in associations of mtDNA CN between younger and older age groups. The effect sizes of obesity/BMI and DBP were larger in younger individuals, whereas the effect sizes for type 2 diabetes (T2D)/FBG and SBP were larger in older individuals, although the directionality remained the same between the two age groups (Figures 2D and S16; Table S8).
Discussion
We demonstrate associations of low levels of mtDNA CN in peripheral blood with an increased risk of CMDsin 408,361 individuals of multiple ancestries in TOPMed and UK Biobank, with adjustments made for traditional clinical covariates as well as for blood cell compositions. Cardiometabolic factors are known risks for the development of CVD. Therefore, our association findings further suggest that altered levels of mitochondrial energy production may be involved in the development of a cluster of conditions that increase the risk of CVD. More specifically, the CMD traits that were significantly associated with low levels mtDNA CN—increased odds for obesity, HTN, diabetes, and hyperglycemia—are all components of the metabolic syndrome.43 Because the metabolic syndrome is the clinical surrogate for insulin resistance, these data suggest that decreasing mtDNA CN may contribute to the insulin resistance accompanying aging.44
We identified a threshold effect of age on mtDNA CN, with a large decline in mtDNA CN observed from 65 years of age. Reduced mitochondrial function is considered one of the hallmarks of aging.45 Therefore, the observed age-related decline in mtDNA CN is potentially important in studying age-related diseases. Our stratified analysis in younger (younger than 65 years) and older (65 years and older) participants found that the effect sizes of associations varied by age groups for six traits in participants of European ancestry.
WBC compositions and platelets are blood biomarkers of systemic inflammation.8,46 It has been increasingly recognized that a chronic low-grade inflammatory state accompanies CMD risk47 and is associated with an increasing risk of obesity,48,49 diabetes,47,50,51 and HTN,52, 53, 54 whereas it is heterogeneously related to lipid levels.55 Complementary to those previous findings, this study found complex relationships between WBC compositions/platelets and CMD traits with respect to their heterogeneous directionalities and strengths in the associations of blood compositions with CMD traits. Similarly, WBC compositions (e.g., neutrophils and lymphocytes) and platelets displayed different directionalities in their associations with mtDNA CN, although we observed that a high WBC count was associated with a low level of mtDNA CN, which is consistent with previous findings.46,56, 57, 58, 59, 60 These results indicate that mtDNA CN, WBC compositions, and platelets may represent an interplay for CVD risk by contributing to those CMD risk factors that are components of the metabolic insulin-resistance syndrome. Further studies are needed to investigate the underlying molecular mechanisms and potential causal pathway among mtDNA CN, inflammation, and CMD risk.
Strengths of the study
This study included a large sample of men and women of multiple ancestries across a wide age range. In TOPMed, mtDNA CN was jointly estimated from WGS. A comprehensive examination showed that the mtDNA CN derived from WGS produced comparable or better results with known correlates (e.g., age and sex) compared with qPCR or other methods (e.g., mtDNA CN estimated from genotyping arrays and whole-exome sequencing).61 We also performed careful phenotype harmonization and examined several potential confounding variables of mtDNA CN in an association analysis with CMD traits. The UK Biobank, a large prospective cohort study, applied a range of approaches to its sample collection, processing, and assay data monitoring to minimize measurement error in traits and biomarker data.62 This may partially explain why we observed larger effect sizes in association of mtDNA CN with five of the CMD traits in the UK Biobank data than we did in the TOPMed data. Our combined analyses using TOPMed and UK Biobank provided a large dataset with the comprehensive data collection and quality control needed to enable testing of associations between mtDNA CN and CMD traits through the adult life.
Limitations of the study
Several limitations of the study should be noted. In this study, we used mtDNA CN estimated from whole blood, because peripheral blood is easily accessible, and changes in mtDNA in whole blood are likely to reflect metabolic health across multiple systems. However, this may not be the most-relevant tissue for cardiometabolic targets (e.g., cardiac muscle, skeletal muscle, or adipose tissue) and aging-related (e.g., brain) disease phenotypes. A previous study compared mtDNA CN in whole blood and plasma in the same participants with T2D and found a significant correlation between mtDNA CN of whole blood and plasma in those patients.63 Another study investigated mtDNA CN in skeletal muscle and cardiac muscle samples through autopsy and heart bypass surgery.8 However, none of those studies directly compared the mtDNA CN measured from both whole blood and skeletal muscles in the same human samples. A more recent study found that blood-derived mtDNA CN was associated with gene expression across multiple tissues and is predictive for incident neurodegenerative disease, which provides evidence supporting the hypothesis that changes in mtDNA in whole blood may reflect metabolic health across multiple systems.64
Second, although we accounted for confounders and known batch effects in mtDNA CN and harmonized metabolic traits, we still observed a moderate to high heterogeneity in the association coefficients in meta-analysis of most of the phenotypes in both ancestry-specific analyses and in TOPMed cohorts. Different distributions of age, sex, and phenotypes across study cohorts may partially explain the heterogeneity in those associations. Unobserved confounding factors, such as experimental conditions for blood draws, DNA extraction, and storage, may also have contributed to the heterogeneity. Finally, we were unable to determine causal relationships between mtDNA CN and CMD traits because of the cross-sectional nature of the study. A reverse causation from a CMD endpoint to mtDNA CN is also possible. A recent study found that mtDNA CN was associated with prevalent diabetes but not with incident diabetes, indicating that diabetes is likely to result in lower levels of mtDNA CN, rather than a lower level mtDNA CN resulting in diabetes.65 In further studies, it would be of interest to include analyses of mtDNA CN and CMD traits at two time points to provide further insight into associations of aging-related mtDNA CN change with CMD traits.
STAR★Methods
Key resources table
| RESOURCES | SOURCE | IDENTIFIER |
|---|---|---|
| Deposited data | ||
| Atherosclerosis Risk in Communities study | TOPMed | phs001211.v3.p2.c1 |
| Coronary Artery Risk Development in Young Adults Study | TOPMed | phs001612 |
| The Cardiovascular Health Study | TOPMed | phs001368.v1.p1 |
| The Framingham Heart Study | TOPMed | phs000974 |
| The Genetic Epidemiology Network of Arteriopathy | TOPMed | phs001345.v1.p1 |
| Hispanic Community Health Study/Study of Latinos | TOPMed | phs001395 |
| The Jackson Heart Study | TOPMed | phs000964.v1.p1 |
| Multi-Ethnic Study of Atherosclerosis Study | TOPMed | phs001416.v1.p1 |
| The UK Biobank | UK Biobank | Application Number 17731 |
| Other | ||
| mtDNA copy number estimation by mitoAnalyzer software (https://lgsun.irp.nia.nih.gov/hsgu/software/mitoAnalyzer/) | Ding et al.34 | https://www.nia.nih.gov/ |
| Summary data | This paper | This paper |
| Analysis codes | This paper | https://github.com/chunyuliu1/mtDNA-copy-number-and-cardiometabolic-traits |
Resource availability
Lead contact
Further information and requests for data, resources, and reagents should be directed to and will be fulfilled by the lead contact, Chunyu Liu (liuc@bu.edu).
Materials availability
This study did not generate new unique reagents. mtDNA CN and phenotype data are available in the Database of Genotypes and Phenotypes (dbGaP) upon request (below).
Experimental model and subject details
This study only included human participants from prospective cohort studies. We included eight cohorts from the NHLBI’s TOPMed program.21 These eight cohorts included Atherosclerosis Risk in Communities study (ARIC) (n = 2,964), Coronary Artery Risk Development in Young Adults Study (CARDIA) (n = 3,452), The Cardiovascular Health Study (CHS) (n = 3,493), The Framingham Heart Study (FHS) (n = 4,124), The Genetic Epidemiology Network of Arteriopathy (GENOA) (n = 1,234), Hispanic Community Health Study/Study of Latinos (HCHS/SOL) (n = 3,868), The Jackson Heart Study (JHS) (n = 3,160), and Multi-Ethnic Study of Atherosclerosis Study (MESA) (n = 4,596). ARIC is a prospective epidemiologic study conducted in four communities which are Forsyth County, NC; Jackson, MS; the northwest suburbs of Minneapolis, MN; and Washington County, MD.33 Focusing on cardiovascular disease outcomes, event adjudication through 2017 consisted of expert committee review of death certificates, hospital records and telephone interviews. Buffy coat was purified using the Gentra Puregene Blood Kit (QIAGEN) using blood samples collected from several health exam visits. mtDNA-CN was available for 2,964 participants of European Americans and African Americans with WGS from TOPMed. CARDIA is a prospective cohort study which was initiated in 1984 to investigate life-style and other factors that influence cardiovascular disease and their risk factors during young adulthood. The study recruited and examined 5,116 African American and European ancestry women and men aged 18-30 years in four urban areas: Birmingham, Alabama; Chicago, Illinois; Minneapolis, Minnesota, and Oakland, California.22 The initial examination included carefully standardized measurements of major risk factors as well as assessments of psychosocial, dietary, and exercise-related characteristics that might influence them, or that might be independent risk factors. mtDNA CN was available for 3,452 participants with WGS sequencing in TOPMed. CHS is a population based, longitudinal, multicenter study of coronary heart disease and stroke in 5,888 elderly adults aged 65 years and older. The CHS originated in 1988 to recruit participants from four U.S. communities.23 The original cohort recruited 5,201 participants and 687 predominately African-American participants were recruited at three of the four field centers in 1992. The first exam began in June 1989. A second comprehensive exam began 3 years after the first exam. A total of n = 3,493 CHS participants (mean age 74 and 58% women) with WGS were included in this study. The FHS is a single-site, community-based, prospective study that was initiated in 1948 to investigate the risk factors for CVD.24 The second generation25 was recruited in 1971 and the third generation26 was recruited between 2002 and 2005. The first generation has been examined every two years. The second generation has been examined every 4-8 years. The third generation has had three examinations. A small number of spouse individuals of the second generation was examined at the same time when the third generation had their first examination. A total of 4,196 FHS participants were whole genome sequenced by TOPMed; of those, 376 were the first generation, 2218 were the second generation and 95 were spouses of the second generation participants; and 1507 were the third generation participants. This study included 4,124 FHS participants. GENOA study enrolled sibships in which at least 2 siblings had essential hypertension diagnosed prior to age 60 years. From 1995 to 2000, the first exam enrolled 1583 non-Hispanic white Americans from Rochester, Minnesota, and 1854 African Americans from Jackson, Mississippi.27 All siblings within the sibship were invited to participate, including both normotensive and hypertensive siblings. The second exam re-recruited 80% of participants from 2000 to 2005. The GENOA data consists of biological samples (DNA, serum, urine) as well as demographic, anthropometric, environmental, clinical, biochemical, physiological, and genetic data for understanding the genetic predictors of diseases of the heart, brain, kidney, and peripheral arteries. This study included 1,234 participants of African Americans. HCHS/SOL is a longitudinal cohort study established in 2008 following Hispanics/Latinos from four US cities: Bronx, NY; Chicago, IL; Miami, FL; San Diego, CA. This study was approved by the IRB in all field centers.28 This study included 3,868 participants with available whole-genome sequencing data from blood drawn in their first field center visit. Detailed information on HCHS/SOL was provided previously.28 Statistical methods used to analyze the present data account for the complex study design, including stratified sampling, clustering, and sampling probabilities. We also adjusted for 11 principal components, estimated from the TOPMed DCC. This study included 3,868 individuals with WGS and matched metabolic phenotypes. The JHS cohort is one of the largest prospective, epidemiologic investigation of CVD among African Americans residing in the three counties (Hinds, Madison, and Rankin) that make up the Jackson, Mississippi metropolitan area.29,30 Data and biologic materials have been collected from 5,306 participants, including a nested family cohort of 1,498 members of 264 families. The age at enrollment for the unrelated cohort was 35-84 years; the family cohort included related individuals > 21 years old. Participants provided extensive medical and social history and had an array of physical and biochemical measurements and diagnostic procedures during a baseline examination (2000-2004), two follow-up examinations (2005-2008 and 2009-2012), and ancillary studies. Samples for genomic DNA were collected during the first two examinations. Consent for genetic studies and broad sharing of genetic data was provided by 3,482 participants. After all quality control procedures, whole genome sequence data are available for 3,406 participants. Follow-up information on vital status, major illnesses or injuries, and hospitalizations to identify intervening clinical events is done annually by phone. Medical records of cardiovascular disease related hospitalizations and death certificates are abstracted and used for adjudication of cardiovascular events and related deaths. MESA (n = 4,596) is a study of the characteristics of subclinical cardiovascular disease and the risk factors that predict progression to clinically overt cardiovascular disease or progression of the subclinical disease. MESA researchers study a diverse, population-based sample of 6,814 men and women 45-84 years of age and free of prevalent clinical CVD when recruited from six field centers across the United States in 2000-2002.31 Event adjudication through 2015 consists of expert committee review of death certificates, hospital records and telephone interviews. DNA for mtDNA-CN analyses was isolated from exam 1 peripheral leukocytes using the Gentra Puregene Blood Kit. mtDNA-CN was available for 4,596 individuals (24.1% Black, 22.3% Hispanic, 13.1% Chinese, 40.5% White) derived from TOPMed WGS sequencing. Several of the TOPMed cohorts contained a small number of duplicated participants. After removing the duplicates, this study included 26,890 individuals with WGS from the TOPMed program (67.4% women; age range of 20-100 years; 45.4% European Americans, 32.6% African Americans, 19.6% Hispanic/Latino Americans and 2.4% Chinese Americans) (Table S1). Additionally, we included 381,470 participants of European ancestry from the UK Biobank with WES (54% women; 40-75 years) for validation (Table S1). The UK Biobank is a prospective cohort study, with the aim of improving the prevention, diagnosis and treatment of a wide range of serious and life-threatening illnesses – including cancer, heart diseases, stroke, diabetes, arthritis, osteoporosis, eye disorders, depression and forms of dementia. UK Biobank recruited 500,000 individuals from across the United Kingdom aged between 40-69 years in 2006-2010, who have undergone measures, provided blood, urine and saliva samples for future analysis.32 381,470 participants with whole exome sequencing and matched metabolic phenotypes were included in this study. This research has been conducted using the UK Biobank Resource under application number UKBB: 17731.
All study participants provided written informed consent for genetic studies. The protocols for WGS and WES were approved by the institutional review boards (IRB) of the participating institutions (supplemental information).
Method details
mtDNA copy number estimation
mtDNA CN estimation in WGS
Whole blood derived DNA was used for WGS from TOPMed sequencing centers. In analyzing sequencing data, the coverage was defined as the number of reads that were mapped to a given nucleotide in the reconstructed sequence. The average coverage was ∼39x across samples in TOPMed. The program fastMitoCalc of the software package mitoAnalyzer34 was used to estimate mtDNA copy number across TOPMed participants. Because nuclear DNA (nDNA) is diploid, ordinarily inheriting the DNA from two parents, while mitochondrial DNA is haploid, coming only from the mother, the average mtDNA CN per cell was estimated as twice the ratio of the average coverage of mtDNA to the average coverage of the nuclear DNA (nDNA).34
mtDNA CN estimation in UK BioBank
Whole blood derived DNA was used for WES from the UK BioBank. In UK Biobank, we started with 49,997 Exome SPB CRAM files (version Jul 2018) downloaded from the UKB data repository, and used Samtools (ver1.9) to extract read summary statistics (‘idxstats’ command). A custom perl script was used to aggregate the summary statistics from each individual file into the following categories (see perl script and example stats file): 1) Total Reads (sum of columns 3 and 4, across all rows), 2) Mapped Reads (sum of column 3, across all rows), 3) Unmapped Reads (some of column 4 across all rows), 4) Autosomal Reads (sum of column 3, rows 1-22), 5) Chr X, 6) Chr Y, 7) Chr MT, 8) ‘Random’ Reads (sum of column 3, across rows 26-67), 9) ‘Unknown’ Reads (sum of column 3 across rows 68-194), 10) EBV Reads, 11) ‘Decoy1’ Reads (sum of column 3 across rows 196-582), 12) ‘Decoy2′ Reads (sum of column 3 across rows 583-2580). Linear regression models were used to adjust for total DNA and potential technical artifacts. Specifically, we used 10-fold cross validation for variable selection, using the ‘leaps’ R package (version 3.0), with an initial model with chrMT read count as the dependent variable, and ‘Total’, ‘Mapped’, ‘unknown’, ‘random’, ‘decoy1’ and ‘decoy2′ read counts as the independent variables. For each of the independent variables, we included a natural spline with df = 4 to allow for non-linear effects. The independent variables ‘Total’, ‘unknown’, ‘decoy1’ and ‘decoy2′ read counts were selected. We then increased the natural spline df to 15, and then used backward selection to reduction model complexity, requiring p < 0.005 to keep a term in the model. The final regression model residuals were generated with the following R (version 3.6.0) code: WES.mtDNA = residuals(lm(chrMT ∼ns(Total,df = 3) + ns(unknown,df = 4) + ns(decoy1,df = 7) + decoy2)). Mitochondrial SNP probe intensities were obtained from the “ukb_chrMT_l2r.txt” file downloaded from the UKBiobank, and samples were stratified by array type (UK BiLEVE, Axiom). To correct for potential artifacts and/or batch effects, we generated 250 principal components (PCs) using the ‘rpca’ command from the ‘rsvd’ package (version 1.0.3) from autosomal nuclear probes by randomly sampling 5% of probes from either even or odd chromosomes that were required to be present on both array types (n∼19,500 probes). Note that we generated the two independent sets of PCs so that we could ensure that probe selection for PCA did not bias results. Prior to PCA, all probe intensities were rank transformed to reduce the impact of any outliers. For each array type, all mitochondrial SNP probes (UKBelieve, n = 181; Axiom, n = 244) along with the 250 PCs were regressed on the ‘WES.mtDNA’ metric derived as described above. Beta estimates from these analyses were then used to generate fitted values in the full UKBiobank dataset using the ‘predict’ function (‘array.mtDNA’). Given the known impact of age, sex, and cell counts on mtDNA-CN, we first used visual inspection to identify outliers for cell counts: Log(WBC) ≤ 1.25 or ≥ 3; Log(RBC) ≤ 1.4 or ≥ 2; Platelet ≤ 10 or ≥ 500; Log(Lymphocyte) ≤ 0.10 or ≥ 2; Log(Mono) ≥ 0.9; Log(Neutrophil) ≤ 0.75 or ≥ 2.75; Log(Eos) ≥ 0.75; Log(Baso) ≥ 0.45. We then excluded non-Whites, related individuals (used.in.pca.calculation = 0), and cell count outliers and then adjusted for age, sex, and cell counts using a backward regression, starting with a natural spline (df = 4) for each covariate. The final model obtained was (“log_” indicates log-transformed variable):
Beta estimates from these analyses were then used to generate fitted values in the full UK Biobank dataset (n = 381,470) using the ‘predict’ function. For all analyses, mtDNA-CN was standardized by subtracting the mean and dividing by the standard deviation.
In ARIC, mtDNA CN has also been estimated from low-pass WGS and Affymetrix Genome-Wide Human SNP Array 6.0.61 We provided association results of mtDNA CN estimated from these two platforms to provide additional information on whether mtDNA CN estimated from different technologies gave rise to consistent results compared to that estimated from WGS (Table S9; supplemental information). These results were not included in any of the meta-analyses and comparisons in the main text.
Cardiometabolic disease phenotypes
Metabolic disease phenotypes were mapped to the health exams when blood was drawn for DNA extraction for mtDNA CN estimates. Our primary analysis focused on four CMD phenotypes – obesity, hypertension (HTN), diabetes, and hyperlipidemia. We analyzed binary traits in the primary analyses for reducing the multiple testing burden. Obesity was defined as body mass index (BMI) ≥ 30 (kg/m2). For the majority, but not all, of the TOPMed cohorts, a therapeutic indication was provided for a medication treatment, but this was not clear for the UK BioBank. T2D was defined as having a fasting blood glucose level of ≥ 126 mg/dL or currently receiving medications to lower blood glucose levels to treat diabetes. Hypertension (HTN) was defined as SBP ≥ 140 mmHg, or DBP ≥ 90 mmHg, or use of antihypertensive medication(s). Hyperlipidemia was defined as fasting total cholesterol (TC) ≥ 200 mg/dL or TRIG ≥ 150 mg/dL, or use of any lipid-lowering medication.
We also analyzed the association of mtDNA CN with continuous cardiometabolic traits that defined the binary traits: BMI, SBP, DBP, FBG, HDL cholesterol, LDL cholesterol, and TRIG levels. In the analysis of FBG, we excluded individuals with diabetes, defined as glucose value ≥ 126 mg/dL and/or taking glucose-lowering or diabetes medications.35 SBP and DBP values (mmHg) were derived from the averages of two measurements. We added 15 mmHg and 10 mmHg to SBP and DBP, respectively, for individuals taking any BP lowering medications.36 The TC measurements were divided by 0.8 for individuals using lipid treatment medications.37 LDL (mg/dL) was calculated as (TC - HDL - TRIG/5) in individuals with TRIG < 400 mg/dL using imputed TC values.37 In analyses of FBG and lipid levels, we excluded individuals whose fasting status was not established. TRIG, LDL and HDL values were log-transformed to approximate normality. Other continuous outcome variables were not transformed.
Quantification and statistical analysis
We used mtDNA CN as the primary independent variable in all association analyses with CMD traits. To identify confounders and covariates, we first examined whether mtDNA CN levels were associated with the ‘blood collection year’ (i.e., the year when blood was drawn, as a surrogate of batch effects for blood-derived DNA samples) in all participating cohorts. We discovered that ‘blood collection year’ explained a 0.9% to 16% variation in mtDNA CN (Figure S1). White blood cell (WBC) count, blood differential count and platelets were previously reported to be associated with mtDNA CN.14,39 To further understand possible confounding effects of these blood components on association analyses, we investigated whether mtDNA CN and CMD traits were associated with total WBC count, blood differential count, and platelet count that were measured or imputed using the Houseman method or a partial least-squares method40,41 (Table S5). We further examined the effect of age (Figure S2) and sex (Figure S3) on mtDNA CN after adjusting for ‘blood collection year’.
Based on observing significant associations of mtDNA CN in relation to ‘blood collection year’, age, and sex, we generated mtDNA CN residuals by regressing mtDNA CN on age, age squared, sex and blood collection year (as a factored variable) in each cohort for primary analyses. The residuals were standardized to a mean of zero and standard deviation (s.d.) of one, and used as the main predictor in all regression models. In the primary analysis, we used logistic regression (for unrelated individuals) and mixed effects logistic regression model (related individuals) to analyze binary outcomes (i.e., obesity, HTN) in relation to mtDNA CN residuals. Because age, sex, and BMI are important confounders or covariates for cardiometabolic traits, we further adjusted for sex and age as covariates in the analysis of obesity, and adjusted for sex, age, age-squared (only for HTN) and BMI as covariates in the analysis of T2D, hyperlipidemia, and HTN. Smoking was a traditional covariate in association analysis of mtDNA with disease phenotypes. Although we included smoking as an additional covariate, we found that the impact of smoking on associations of mtDNA CN with CMD traits was minimal (supplemental information; Figure S17). We excluded any value in mtDNA CN or a trait measurement if it was beyond 4 standard deviation of the mean of mtDNA CN residuals or a trait. We used linear effects models to analyze continuous outcome variables, adjusting for the same set of covariates as for the respective binary outcomes. For cohorts with family structure, we accounted for maternal lineage as random effects in linear or logistic mixed models. A maternal lineage was defined to include a founder woman with all of her children, and all grandchildren from daughters of the founder woman.42
We performed an initial meta-analysis in European American participants in TOPMed with fixed effects inverse variance method based on an a priori assumption that there is only one true treatment effect between studies.14,15 We performed validation analyses using data for European ancestry participants in the UK Biobank (Figure 1). We further compared meta-analysis results in participants of European ancestry to those from other ancestry origins in TOPMed cohorts. Finally, we performed fixed inverse-variance meta-analysis to combine results from the TOPMed and UK Biobank. We used p = 0.0125 (0.05/4) for significance to account for multiple testing for the primary results from the four binary traits, and used p = 0.05/9∼0.006 for significance in analysis of continuous outcomes.
We compared associations between mtDNA CN and individual outcomes in the same participants with and without WBC count, differential count, and platelet count as additional covariates. We further investigated whether sex or age modified the association between mtDNA CN and outcome variables, adjusting for the same set of covariates described in the primary analyses. In these analyses, we generated mtDNA CN residuals by regressing the mtDNA CN on the blood collection year (as a factored variable) in each cohort for the primary analyses. We included an interaction term between mtDNA CN and sex/age in the association analyses. We also performed age-group stratified analyses between mtDNA CN and CMD traits in younger (< 65 years) and older (≥65 years) participants (supplemental information). The statistical software R (version 3.6.0) was used for all statistical analyses.
Acknowledgments
Detailed acknowledgment for each cohort is included in the supplemental information. In brief, the authors thank the staff and participants of the Atherosclerosis Risk in Communities study, the Cardiovascular Health Study, the Coronary Artery Risk Development in Young Adults Study, the Framingham Heart Study, the Jackson Heart Study, the Genetic Epidemiology Network of Arteriopathy Study, the Hispanic Community Health Study/Study of Latinos, the Multi-Ethnic Study of Atherosclerosis Study, and the UK Biobank for their important contributions. We gratefully acknowledge the studies and participants who provided biological samples and data for TOPMed. Whole-genome sequencing (WGS) for the Trans-Omics in Precision Medicine (TOPMed) program was supported by the National Heart, Lung, and Blood Institute (NHLBI). Centralized read mapping and genotype calling, along with variant quality metrics and filtering were provided by the TOPMed Informatics Research Center (R01HL-117626-02S1; contract HHSN268201800002I). Phenotype harmonization, data management, sample-identity QC, and general study coordination were provided by the TOPMed Data Coordinating Center (R01HL-120393-02S1; contract HHSN268201800001I). Additional phenotype harmonization was performed by the current study (R01AG059727). The views expressed in this manuscript are those of the authors and do not necessarily represent the views of the National Heart, Lung, and Blood Institute; the National Institutes of Health; or the US Department of Health and Human Services.
Author contributions
Data preparation, X.L., R.J.L., D.E.A., C.L., K.L.W., J.C.B., L.M.R., L.F.B., W.Z., J.A.S., A.P., J.Y., X.G., N.K., B.T., M.L.G., N.B.L., A.L.F., M.F., N.P., S.R.H., and T.S.; mtDNA CN estimation, T.W.B., J.D., G.A., and D.E.A.; statistical analyses, X.L., R.J.L., D.E.A., N.K., T.S., and C.L.; manuscript preparation and revision, X.L., R.J.L., J.C.B., D.E.A., C.L., A.L.F., S.S., S.R.H., D.L., J.I.R., and B.M.P.; funding support, C.L., C.L.S., S.S.R., K.D.T., P.A.P., L.A.C., E.B., R.S.V., J.G.W., M.F., J.I.R., A.C., and B.M.P.
Declaration of interests
The authors declare no competing interests, except for the disclosure from the following authors: Dr. Rocha Abecasis reports grants from National Heart Lung and Blood Institute (NIH) during the conduct of the study and personal fees and other from Regeneron Pharmaceuticals outside of the submitted work; Dr. Cupples reports personal fees from Dyslipidemia Foundation during the conduct of the study and personal fees from Veterans Administration outside of the submitted work; Dr. Psaty serves on the Steering Committee of the Yale Open Data Access Project funded by Johnson & Johnson outside of the submitted work.
Published: October 13, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.xgen.2021.100006.
Supplemental information
Data and code availability
This study did not generate any unique datasets. For the US cohorts, whole-genome sequencing data were generated by the Trans-Omics for Precision Medicine (TOPMed) program supported by National Heart, Lung, and Blood Institute. For TOPMed cohorts, mtDNA CN and phenotype data are available in the dbGaP upon request. The steps to request dbGAP access includes 1) obtain eRA Commons account, 2) obtain dbGaP access, 3) obtain access to Research Project through dbGaP, 4) grant access to individuals to your lab, and 5) log into GDC data portal. The detailed instructions can be found at the following link: (https://gdc.cancer.gov/access-data/obtaining-access-controlled-data). The access numbers are as follows: The Atherosclerosis Risk in Communities study, dbGaP: phs001211; The Coronary Artery Risk Development in Young Adults Study, dbGaP: phs001612; The Cardiovascular Health Study, dbGaP: phs001368; The Framingham Heart Study, dbGaP: phs000974; The Genetic Epidemiology Network of Arteriopathy, dbGaP: phs001345; The Hispanic Community Health Study/Study of Latinos, dbGaP: phs001395; The Jackson Heart Study, dbGaP: phs000964; The Multi-Ethnic Study of Atherosclerosis Study, dbGaP: phs001416; The UK Biobank data (whole-exome sequencing and phenotype data) were downloaded at https://www.ukbiobank.ac.uk/, UKBB: 17731. Codes for association analyses in TOPMed cohorts and estimation of mtDNA CN in the UK Biobank can be accessed at https://github.com/chunyuliu1/mtDNA-copy-number-and-cardiometabolic-traits.
References
- 1.Voet D.V.J., Pratt C.W. Second Edition. John Wiley and Sons; 2005. Fundamentals of Biochemistry; p. 547. [Google Scholar]
- 2.Osellame L.D., Blacker T.S., Duchen M.R. Cellular and molecular mechanisms of mitochondrial function. Best Pract. Res. Clin. Endocrinol. Metab. 2012;26:711–723. doi: 10.1016/j.beem.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Antico Arciuch V.G., Elguero M.E., Poderoso J.J., Carreras M.C. Mitochondrial regulation of cell cycle and proliferation. Antioxid. Redox Signal. 2012;16:1150–1180. doi: 10.1089/ars.2011.4085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Takahashi K., Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 5.Misko A.L., Sasaki Y., Tuck E., Milbrandt J., Baloh R.H. Mitofusin2 mutations disrupt axonal mitochondrial positioning and promote axon degeneration. J. Neurosci. 2012;32:4145–4155. doi: 10.1523/JNEUROSCI.6338-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Clayton D.A., Doda J.N., Friedberg E.C. The absence of a pyrimidine dimer repair mechanism in mammalian mitochondria. Proc. Natl. Acad. Sci. USA. 1974;71:2777–2781. doi: 10.1073/pnas.71.7.2777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Seo A.Y., Joseph A.M., Dutta D., Hwang J.C., Aris J.P., Leeuwenburgh C. New insights into the role of mitochondria in aging: mitochondrial dynamics and more. J. Cell Sci. 2010;123:2533–2542. doi: 10.1242/jcs.070490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller F.J., Rosenfeldt F.L., Zhang C., Linnane A.W., Nagley P. Precise determination of mitochondrial DNA copy number in human skeletal and cardiac muscle by a PCR-based assay: lack of change of copy number with age. Nucleic Acids Res. 2003;31:e61. doi: 10.1093/nar/gng060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.St John J.C. Mitochondrial DNA copy number and replication in reprogramming and differentiation. Semin. Cell Dev. Biol. 2016;52:93–101. doi: 10.1016/j.semcdb.2016.01.028. [DOI] [PubMed] [Google Scholar]
- 10.Alberts B., Johnson A., Lewis J., Raff M., Roberts K., Walter P. Garland Publishing; 1994. Molecular Biology of the Cell. [Google Scholar]
- 11.Moyes C.D., Battersby B.J., Leary S.C. Regulation of muscle mitochondrial design. J. Exp. Biol. 1998;201:299–307. [PubMed] [Google Scholar]
- 12.Mengel-From J., Thinggaard M., Dalgård C., Kyvik K.O., Christensen K., Christiansen L. Mitochondrial DNA copy number in peripheral blood cells declines with age and is associated with general health among elderly. Hum. Genet. 2014;133:1149–1159. doi: 10.1007/s00439-014-1458-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang R., Wang Y., Ye K., Picard M., Gu Z. Independent impacts of aging on mitochondrial DNA quantity and quality in humans. BMC Genomics. 2017;18:890. doi: 10.1186/s12864-017-4287-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ashar F.N., Moes A., Moore A.Z., Grove M.L., Chaves P.H.M., Coresh J., Newman A.B., Matteini A.M., Bandeen-Roche K., Boerwinkle E., et al. Association of mitochondrial DNA levels with frailty and all-cause mortality. J. Mol. Med. (Berl.) 2015;93:177–186. doi: 10.1007/s00109-014-1233-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Ashar F.N., Zhang Y., Longchamps R.J., Lane J., Moes A., Grove M.L., Mychaleckyj J.C., Taylor K.D., Coresh J., Rotter J.I., et al. Association of mitochondrial DNA copy number with cardiovascular disease. JAMA Cardiol. 2017;2:1247–1255. doi: 10.1001/jamacardio.2017.3683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leon B.M., Maddox T.M. Diabetes and cardiovascular disease: Epidemiology, biological mechanisms, treatment recommendations and future research. World J. Diabetes. 2015;6:1246–1258. doi: 10.4239/wjd.v6.i13.1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tune J.D., Goodwill A.G., Sassoon D.J., Mather K.J. Cardiovascular consequences of metabolic syndrome. Transl. Res. 2017;183:57–70. doi: 10.1016/j.trsl.2017.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Xu F.X., Zhou X., Shen F., Pang R., Liu S.M. Decreased peripheral blood mitochondrial DNA content is related to HbA1c, fasting plasma glucose level and age of onset in type 2 diabetes mellitus. Diabet. Med. 2012;29:e47–e54. doi: 10.1111/j.1464-5491.2011.03565.x. [DOI] [PubMed] [Google Scholar]
- 19.Lee J.Y., Lee D.C., Im J.A., Lee J.W. Mitochondrial DNA copy number in peripheral blood is independently associated with visceral fat accumulation in healthy young adults. Int. J. Endocrinol. 2014;2014:586017. doi: 10.1155/2014/586017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guyatt A.L., Burrows K., Guthrie P.A.I., Ring S., McArdle W., Day I.N.M., Ascione R., Lawlor D.A., Gaunt T.R., Rodriguez S. Cardiometabolic phenotypes and mitochondrial DNA copy number in two cohorts of UK women. Mitochondrion. 2018;39:9–19. doi: 10.1016/j.mito.2017.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Taliun D., Harris D.N., Kessler M.D., Carlson J., Szpiech Z.A., Torres R., Taliun S.A.G., Corvelo A., Gogarten S.M., Kang H.M., et al. Sequencing of 53,831 diverse genomes from the NHLBI TOPMed Program. bioRxiv. 2019:563866. doi: 10.1038/s41586-021-03205-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Friedman G.D., Tekawa I., Grimm R.H., Manolio T., Shannon S.G., Sidney S. The leucocyte count: correlates and relationship to coronary risk factors: the CARDIA study. Int. J. Epidemiol. 1990;19:889–893. doi: 10.1093/ije/19.4.889. [DOI] [PubMed] [Google Scholar]
- 23.Fried L.P., Borhani N.O., Enright P., Furberg C.D., Gardin J.M., Kronmal R.A., Kuller L.H., Manolio T.A., Mittelmark M.B., Newman A., et al. The Cardiovascular Health Study: Design and rationale. Ann. Epidemiol. 1991;1:263–276. doi: 10.1016/1047-2797(91)90005-w. [DOI] [PubMed] [Google Scholar]
- 24.Dawber T.R., Meadors G.F., Moore F.E., Jr. Epidemiological approaches to heart disease: the Framingham Study. Am. J. Public Health Nations Health. 1951;41:279–281. doi: 10.2105/ajph.41.3.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Feinleib M., Kannel W.B., Garrison R.J., McNamara P.M., Castelli W.P. The Framingham Offspring Study: Design and preliminary data. Prev. Med. 1975;4:518–525. doi: 10.1016/0091-7435(75)90037-7. [DOI] [PubMed] [Google Scholar]
- 26.Splansky G.L., Corey D., Yang Q., Atwood L.D., Cupples L.A., Benjamin E.J., D’Agostino R.B., Sr., Fox C.S., Larson M.G., Murabito J.M., et al. The third generation cohort of the National Heart, Lung, and Blood Institute’s Framingham Heart Study: Design, recruitment, and initial examination. Am. J. Epidemiol. 2007;165:1328–1335. doi: 10.1093/aje/kwm021. [DOI] [PubMed] [Google Scholar]
- 27.Daniels P.R., Kardia S.L., Hanis C.L., Brown C.A., Hutchinson R., Boerwinkle E., Turner S.T., Genetic Epidemiology Network of Arteriopathy study Familial aggregation of hypertension treatment and control in the Genetic Epidemiology Network of Arteriopathy (GENOA) study. Am. J. Med. 2004;116:676–681. doi: 10.1016/j.amjmed.2003.12.032. [DOI] [PubMed] [Google Scholar]
- 28.Lavange L.M., Kalsbeek W.D., Sorlie P.D., Avilés-Santa L.M., Kaplan R.C., Barnhart J., Liu K., Giachello A., Lee D.J., Ryan J., et al. Sample design and cohort selection in the Hispanic Community Health Study/Study of Latinos. Ann. Epidemiol. 2010;20:642–649. doi: 10.1016/j.annepidem.2010.05.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sempos C.T., Bild D.E., Manolio T.A. Overview of the Jackson Heart Study: a study of cardiovascular diseases in African American men and women. Am. J. Med. Sci. 1999;317:142–146. doi: 10.1097/00000441-199903000-00002. [DOI] [PubMed] [Google Scholar]
- 30.Wilson J.G., Rotimi C.N., Ekunwe L., Royal C.D., Crump M.E., Wyatt S.B., Steffes M.W., Adeyemo A., Zhou J., Taylor H.A., Jr., et al. Study design for genetic analysis in the Jackson Heart Study. Ethn. Dis. 2005;15(suppl 6):S6–S37. [PubMed] [Google Scholar]
- 31.Bild D.E., Bluemke D.A., Burke G.L., Detrano R., Diez Roux A.V., Folsom A.R., Greenland P., Jacob D.R., Jr., Kronmal R., Liu K., et al. Multi-ethnic study of atherosclerosis: Objectives and design. Am. J. Epidemiol. 2002;156:871–881. doi: 10.1093/aje/kwf113. [DOI] [PubMed] [Google Scholar]
- 32.Sudlow C., Gallacher J., Allen N., Beral V., Burton P., Danesh J., Downey P., Elliott P., Green J., Landray M., et al. UK Biobank: An open access resource for identifying the causes of a wide range of complex diseases of middle and old age. PLoS Med. 2015;12:e1001779. doi: 10.1371/journal.pmed.1001779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.ARIC Investigators The Atherosclerosis Risk in Communities (ARIC) Study: Design and objectives. Am. J. Epidemiol. 1989;129:687–702. [PubMed] [Google Scholar]
- 34.Ding J., Sidore C., Butler T.J., Wing M.K., Qian Y., Meirelles O., Busonero F., Tsoi L.C., Maschio A., Angius A., et al. Assessing mitochondrial DNA variation and copy number in lymphocytes of ∼2,000 Sardinians using tailored sequencing analysis tools. PLoS Genet. 2015;11:e1005306. doi: 10.1371/journal.pgen.1005306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Scott R.A., Lagou V., Welch R.P., Wheeler E., Montasser M.E., Luan J., Mägi R., Strawbridge R.J., Rehnberg E., Gustafsson S., et al. DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium Large-scale association analyses identify new loci influencing glycemic traits and provide insight into the underlying biological pathways. Nat. Genet. 2012;44:991–1005. doi: 10.1038/ng.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Levy D., Ehret G.B., Rice K., Verwoert G.C., Launer L.J., Dehghan A., Glazer N.L., Morrison A.C., Johnson A.D., Aspelund T., et al. Genome-wide association study of blood pressure and hypertension. Nat. Genet. 2009;41:677–687. doi: 10.1038/ng.384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Willer C.J., Schmidt E.M., Sengupta S., Peloso G.M., Gustafsson S., Kanoni S., Ganna A., Chen J., Buchkovich M.L., Mora S., et al. Global Lipids Genetics Consortium Discovery and refinement of loci associated with lipid levels. Nat. Genet. 2013;45:1274–1283. doi: 10.1038/ng.2797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lee H.K., Song J.H., Shin C.S., Park D.J., Park K.S., Lee K.U., Koh C.S. Decreased mitochondrial DNA content in peripheral blood precedes the development of non-insulin-dependent diabetes mellitus. Diabetes Res. Clin. Pract. 1998;42:161–167. doi: 10.1016/s0168-8227(98)00110-7. [DOI] [PubMed] [Google Scholar]
- 39.Tin A., Grams M.E., Ashar F.N., Lane J.A., Rosenberg A.Z., Grove M.L., Boerwinkle E., Selvin E., Coresh J., Pankratz N., Arking D.E. Association between mitochondrial DNA copy number in peripheral blood and incident CKD in the Atherosclerosis Risk in Communities Study. J. Am. Soc. Nephrol. 2016;27:2467–2473. doi: 10.1681/ASN.2015060661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Houseman E.A., Accomando W.P., Koestler D.C., Christensen B.C., Marsit C.J., Nelson H.H., Wiencke J.K., Kelsey K.T. DNA methylation arrays as surrogate measures of cell mixture distribution. BMC Bioinformatics. 2012;13:86. doi: 10.1186/1471-2105-13-86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Abdi H. Partial least squares regression and projection on latent structure regression (PLS Regression) Wiley Interdiscip. Rev. Comput. Stat. 2010;2:97–106. [Google Scholar]
- 42.Liu C., Dupuis J., Larson M.G., Levy D. Association testing of the mitochondrial genome using pedigree data. Genet. Epidemiol. 2013;37:239–247. doi: 10.1002/gepi.21706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Huang P.L. A comprehensive definition for metabolic syndrome. Dis. Model. Mech. 2009;2:231–237. doi: 10.1242/dmm.001180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barzilai N., Ferrucci L. Insulin resistance and aging: A cause or a protective response? J. Gerontol. A Biol. Sci. Med. Sci. 2012;67:1329–1331. doi: 10.1093/gerona/gls145. [DOI] [PubMed] [Google Scholar]
- 45.López-Otín C., Blasco M.A., Partridge L., Serrano M., Kroemer G. The hallmarks of aging. Cell. 2013;153:1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Esposito K., Giugliano D. The metabolic syndrome and inflammation: association or causation? Nutr. Metab. Cardiovasc. Dis. 2004;14:228–232. doi: 10.1016/s0939-4753(04)80048-6. [DOI] [PubMed] [Google Scholar]
- 47.Vozarova B., Weyer C., Lindsay R.S., Pratley R.E., Bogardus C., Tataranni P.A. High white blood cell count is associated with a worsening of insulin sensitivity and predicts the development of type 2 diabetes. Diabetes. 2002;51:455–461. doi: 10.2337/diabetes.51.2.455. [DOI] [PubMed] [Google Scholar]
- 48.Dixon J.B., O’Brien P.E. Obesity and the white blood cell count: Changes with sustained weight loss. Obes. Surg. 2006;16:251–257. doi: 10.1381/096089206776116453. [DOI] [PubMed] [Google Scholar]
- 49.Kullo I.J., Hensrud D.D., Allison T.G. Comparison of numbers of circulating blood monocytes in men grouped by body mass index (<25, 25 to <30, > or =30) Am. J. Cardiol. 2002;89:1441–1443. doi: 10.1016/s0002-9149(02)02366-4. [DOI] [PubMed] [Google Scholar]
- 50.Ohshita K., Yamane K., Hanafusa M., Mori H., Mito K., Okubo M., Hara H., Kohno N. Elevated white blood cell count in subjects with impaired glucose tolerance. Diabetes Care. 2004;27:491–496. doi: 10.2337/diacare.27.2.491. [DOI] [PubMed] [Google Scholar]
- 51.Twig G., Afek A., Shamiss A., Derazne E., Tzur D., Gordon B., Tirosh A. White blood cells count and incidence of type 2 diabetes in young men. Diabetes Care. 2013;36:276–282. doi: 10.2337/dc11-2298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Orakzai R.H., Orakzai S.H., Nasir K., Santos R.D., Rana J.S., Pimentel I., Carvalho J.A.M., Meneghello R., Blumenthal R.S. Association of white blood cell count with systolic blood pressure within the normotensive range. J. Hum. Hypertens. 2006;20:341–347. doi: 10.1038/sj.jhh.1001992. [DOI] [PubMed] [Google Scholar]
- 53.Karthikeyan V.J., Lip G.Y.H. White blood cell count and hypertension. J. Hum. Hypertens. 2006;20:310–312. doi: 10.1038/sj.jhh.1001980. [DOI] [PubMed] [Google Scholar]
- 54.Gillum R.F., Mussolino M.E. White blood cell count and hypertension incidence. The NHANES I Epidemiologic Follow-up Study. J. Clin. Epidemiol. 1994;47:911–919. doi: 10.1016/0895-4356(94)90195-3. [DOI] [PubMed] [Google Scholar]
- 55.Lai Y.C., Woollard K.J., McClelland R.L., Allison M.A., Rye K.A., Ong K.L., Cochran B.J. The association of plasma lipids with white blood cell counts: Results from the Multi-Ethnic Study of Atherosclerosis. J. Clin. Lipidol. 2019;13:812–820. doi: 10.1016/j.jacl.2019.07.003. [DOI] [PubMed] [Google Scholar]
- 56.Wu I.C., Lin C.C., Liu C.S., Hsu C.C., Chen C.Y., Hsiung C.A. Interrelations between mitochondrial DNA copy number and inflammation in older adults. J. Gerontol. A Biol. Sci. Med. Sci. 2017;72:937–944. doi: 10.1093/gerona/glx033. [DOI] [PubMed] [Google Scholar]
- 57.Knez J., Marrachelli V.G., Cauwenberghs N., Winckelmans E., Zhang Z., Thijs L., Brguljan-Hitij J., Plusquin M., Delles C., Monleon D., et al. Peripheral blood mitochondrial DNA content in relation to circulating metabolites and inflammatory markers: A population study. PLoS ONE. 2017;12:e0181036. doi: 10.1371/journal.pone.0181036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Sharma P. Inflammation and the metabolic syndrome. Indian J. Clin. Biochem. 2011;26:317–318. doi: 10.1007/s12291-011-0175-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Saltiel A.R., Olefsky J.M. Inflammatory mechanisms linking obesity and metabolic disease. J. Clin. Invest. 2017;127:1–4. doi: 10.1172/JCI92035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Paoletti R., Bolego C., Poli A., Cignarella A. Metabolic syndrome, inflammation and atherosclerosis. Vasc. Health Risk Manag. 2006;2:145–152. doi: 10.2147/vhrm.2006.2.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Longchamps R.J., Castellani C.A., Yang S.Y., Newcomb C.E., Sumpter J.A., Lane J., Grove M.L., Guallar E., Pankratz N., Taylor K.D., et al. Evaluation of mitochondrial DNA copy number estimation techniques. PLoS ONE. 2020;15:e0228166. doi: 10.1371/journal.pone.0228166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.UK Biobank . 2019. Biomarker assay quality procedures: Approaches used to minimise systematic and random errors (and the wider epidemiological implications). Version 1.2.https://biobank.ctsu.ox.ac.uk/crystal/crystal/docs/biomarker_issues.pdf [Google Scholar]
- 63.Rosa H.S., Ajaz S., Gnudi L., Malik A.N. A case for measuring both cellular and cell-free mitochondrial DNA as a disease biomarker in human blood. FASEB J. 2020;34:12278–12288. doi: 10.1096/fj.202000959RR. [DOI] [PubMed] [Google Scholar]
- 64.Yang S.Y., Castellani C.A., Longchamps R.J., Pillalamarri V.K., O’Rourke B., Guallar E., Arking D.E. Blood-derived mitochondrial DNA copy number is associated with gene expression across multiple tissues and is predictive for incident neurodegenerative disease. Genome Res. 2021;31:349–358. doi: 10.1101/gr.269381.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.DeBarmore B., Longchamps R.J., Zhang Y., Kalyani R.R., Guallar E., Arking D.E., Selvin E., Young J.H. Mitochondrial DNA copy number and diabetes: the Atherosclerosis Risk in Communities (ARIC) study. BMJ Open Diabetes Res. Care. 2020;8:e001204. doi: 10.1136/bmjdrc-2020-001204. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This study did not generate any unique datasets. For the US cohorts, whole-genome sequencing data were generated by the Trans-Omics for Precision Medicine (TOPMed) program supported by National Heart, Lung, and Blood Institute. For TOPMed cohorts, mtDNA CN and phenotype data are available in the dbGaP upon request. The steps to request dbGAP access includes 1) obtain eRA Commons account, 2) obtain dbGaP access, 3) obtain access to Research Project through dbGaP, 4) grant access to individuals to your lab, and 5) log into GDC data portal. The detailed instructions can be found at the following link: (https://gdc.cancer.gov/access-data/obtaining-access-controlled-data). The access numbers are as follows: The Atherosclerosis Risk in Communities study, dbGaP: phs001211; The Coronary Artery Risk Development in Young Adults Study, dbGaP: phs001612; The Cardiovascular Health Study, dbGaP: phs001368; The Framingham Heart Study, dbGaP: phs000974; The Genetic Epidemiology Network of Arteriopathy, dbGaP: phs001345; The Hispanic Community Health Study/Study of Latinos, dbGaP: phs001395; The Jackson Heart Study, dbGaP: phs000964; The Multi-Ethnic Study of Atherosclerosis Study, dbGaP: phs001416; The UK Biobank data (whole-exome sequencing and phenotype data) were downloaded at https://www.ukbiobank.ac.uk/, UKBB: 17731. Codes for association analyses in TOPMed cohorts and estimation of mtDNA CN in the UK Biobank can be accessed at https://github.com/chunyuliu1/mtDNA-copy-number-and-cardiometabolic-traits.