

ABSTRACT
T cell receptor (TCR) repertoire as a biomarker for predicting immunotherapy efficiency has been widely studied. However, its dynamics during radiotherapy combined with PD-1 blockade is little known. Using paired tumor and blood samples from the phase Ib clinical study (NCT03222440), we investigate the time-spatial TCR repertoire in esophageal squamous cell carcinoma (ESCC) patients treated with first-line definitive radiotherapy concurrently with anti-PD-1 antibody camrelizumab, and also evaluate the association between TCR repertoire and clinical outcomes. TCR sequencing was performed on tumor biopsies (n = 34, 15 pairs) and peripheral CD8+ T cells (n = 36, 18 pairs) collected at baseline and during treatment (after 40 Gy radiation and 2 rounds of camrelizumab). Whole exome sequencing was applied to estimate genomic mutations and tumor mutation burden. We show that the intratumoral TCR repertoire at baseline was correlated with tumor microenvironment and presented heterogeneity inter-individually. T-cell clones inflowed mutually between tumors and peripheral blood under combination treatment, resulting in an elevation of intratumoral TCR diversity. The peripheral CD8+ TCR diversity at baseline, increased tumor-peripheral Morisita-Horn overlap during treatment, and expansion of persistent intratumoral T-cell clones during treatment predicted improved survival. While it is unclear whether radiation contributed to the TCR changes versus PD-1 therapy alone, our results firstly reveal radiotherapy combined with PD-1 blockade greatly promoted time-spatial alteration of TCR repertoire between tumor and peripheral blood, which demonstrate the peripheral CD8+ TCR diversity at baseline and dynamic alteration of intratumoral TCRs acted as potential effective biomarkers of radiotherapy combined with immunotherapy in ESCC.
KEYWORDS: T cell receptor, radiotherapy, PD-1, esophageal carcinoma, T cell receptor repertoire
Introduction
Inhibitors of programmed cell death protein 1 (PD-1), such as nivolumab,1 pembrolizumab,2 and camrelizumab,3 have shown promising application in advanced/metastatic esophageal squamous cell carcinoma (ESCC). However, only 19%,1 16.7%2 and 20.2%3 of these ESCC patients had objective response from nivolumab, pembrolizumab and camrelizumab alone treatment. Identifying valid biomarkers for patient selection is urgently needed to promote the clinical outcomes in anti-PD-1 therapy.
Previous studies showed that the expression of programmed death-ligand 1 (PD-L1) was not eligible as a predictive biomarker for anti-PD-1/PD-L1 treatment in ESCC.1–4 Tumor mutational burden (TMB) was associated with the responseto camrelizumab in ESCC patients.5 Dendritic cells were related with better survival in ESCC patients after chemoradiotherapy and PD-1 blockade.6 Other checkpoint molecules, such as T-cell immunoglobulin and mucin-domain-containing-3 (TIM3) and T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain (TIGIT), were associated with poor survival in ESCC.7 Given that checkpoint blockade and TMB-induced neoantigens contributed to T cell antitumor immunoreactivity, T cell profiling is increasingly considered to have great predictive and prognostic value for immunotherapy.
Each T cell clones express a unique T cell receptor (TCR) that recognizes antigen in a unique cognate peptide, namely major histocompatibility complex (MHC). About 90–95% TCRs are composed of α- and β-subunits that are encoded by recombinant genes generated from somatic variable (V), diversity (D) and joining (J) segments (VJ rearrangement for α; V[D]J rearrangement for β), which results in the high diversities of complementarity determining region 3 (CDR3) and antigen-specific T cell repertoire. Recent studies have shown that deep sequencing of the TCR variable beta (Vβ) (TCRB) CDR3 region could monitor the dynamics of T-cell repertoire response to treatment.8,9 Moreover, TCR clonality and diversity showed association in response to a broad range of tumor-associated antigens.10,11 Nonetheless, the TCR profiling response to immunotherapy in ESCC is relatively sparse.
Concurrent chemoradiotherapy is the standard therapy for unresectable locally advanced ESCC patients. Chemo- and radiotherapy are supposed to promote antigen presentation by increasing the release of the danger signals and tumor antigens from dead and injured cells. A few studies have been reported that the relationship with T cell profiling and chemoradiotherapy in ESCC patients. For instance, one study revealed that clonal expansion of tumor-infiltrating lymphocytes (TILs) would be induced more within ESCCs after definitive chemoradiotherapy than those with surgery alone.12 Meanwhile, the increases of TCR diversities were significantly found in peripheral T cells but not in TILs after preoperative chemotherapy in ESCC patients.13
For unresectable locally advanced ESCC patients who were intolerant to or refuse chemotherapy, radiotherapy is the primary treatment. However, approximately 80% of patients treated with radiotherapy alone will die of recurrent or metastatic disease.14,15 Based on the hypothesis of synergetic effect of radiotherapy and PD-1 blockade on ESCC, we carried out a phase Ib study evaluating the safety and feasibility of combined definitive radiotherapy and anti-PD-1 antibody camrelizumab as first-line treatment in locally advanced ESCC patients who were treatment-naïve and ineligible or refused chemoradiotherapy from July 24, 2017 to January 25, 2018 (ClinicalTrials.gov NCT03222440).16 Nineteen patients were enrolled. Thirteen patients received full cycles (200 mg once every 2 weeks, total 32 weeks) of camrelizumab from the beginning of radiotherapy while 18 patients completed radiotherapy. We previously reported the clinical data of this phase Ib trial, which showed that radiotherapy combined with camrelizumab had antitumor efficacy with manageable toxicity for locally advanced ESCC.16
In order to investigate the TCR profiling in response to the combination of radiotherapy and immunotherapy in ESCC, we prospectively sequenced TCRB CDR3 in both intratumoral T cells and peripheral CD8+ T cells from patients at baseline and on-treatment (after 40 Gy radiation and 2 rounds of camrelizumab) in our study (ClinicalTrials.gov NCT03222440). Here, we illustrated the spatiotemporal dynamics of TCR diversities responding to radiotherapy combined with camrelizumab in ESCC, which provided biomarker candidates for predicting the outcome of the combination treatment.
Materials and methods
Study design and sample collection
The tumor biopsies and peripheral CD8+ T cells evaluated in this study were collected from patients enrolled in a phase Ib study evaluating the safety and feasibility of definitive radiotherapy combined with the anti-PD-1 antibody camrelizumab as first-line therapy for patients with locally advanced ESCC (ClinicalTrials.gov NCT03222440).16 Specifically, 19 patients with T3-4 N0M0 or T1-4N+M0 (stage II–IVa) who were naïve-treated, and were ineligible or declined concurrent chemoradiotherapy were enrolled. Camrelizumab was infused intravenously at a fixed dose of 200 mg once every 2 weeks from the beginning of radiotherapy for up to 32 weeks (i.e., for 16 cycles).5 Radiotherapy was delivered as RapidArc (volumetric arc) intensity-modulated RT with a simultaneous integrated boost (SIB-IMRT). The radiotherapy was given according to Chinese treatment guidelines for esophageal carcinoma17,18 and was prescribed to cover 95% of the pretreatment planning gross tumor volume (PGTV), given at 2.0 Gy per fraction, 5 fractions per week, to a total of 60 Gy over 6 weeks. Camrelizumab (also named as SHR-1210) is a selective, humanized, high-affinity IgG4-kappa PD-1 monoclonal antibody that blocks binding between PD-1 and PD-L1. Camrelizumab, alone or combined with chemotherapy has shown encouraging efficacy against recurrent or metastatic ESCC.5,19 The exploratory endpoints of this phase Ib study were changes in tumor immune microenvironment and peripheral immune cells. Baseline (before the first dose of radiation and camrelizumab) and on-treatment (after 40 Gy radiation and 2 rounds of camrelizumab) tumor biopsies and peripheral blood were prospectively collected (Supplementary Table 1). Deep biopsy samples of tumor tissues were collected under endoscopic ultrasonographic guidance20,21 and made into formalin-fixed paraffin-embedded (FFPE) tissue blocks. The study was approved by the institutional review board and ethics committee at Tianjin Medical University Cancer Institute & Hospital (E2017094) and conformed to the ethical principles outlined in the Declaration of Helsinki. All patients provided written informed consent to participate.
Purification of peripheral CD8+ T cells
6 mL EDTA-anticoagulant-treated peripheral blood samples were collected on the day before the first dose of radiation and camrelizumab and during treatment (after delivery of 40 Gy radiation and 2 rounds of camrelizumab). Within 2 hours after the fresh whole-blood collection, CD8+ T cells were isolated from the whole-blood samples by negative selection by using the RosetteSep™ Human CD8+ T Cell Enrichment Cocktail (StemCell, 15063) according to the manufacturer’s instruction. After being washed twice with cold phosphate-buffered saline (PBS), purified CD8+ T cells were resuspended in RNAlater (ThermoFisher, AM7020) and preserved at ‒80°C for further TCR analysis. The purification of CD8+ T cells was immediately confirmed by flow cytometry analysis after isolation. All purification of isolated CD8+ T cells was above 93%. We also isolated the peripheral blood monocyte cells (PBMCs) from 5 mL peripheral blood both at baseline and during treatment by Ficoll-Paque® density gradient centrifugation. The fresh PBMCs were tested by using flow cytometry.16 The baseline peripheral blood granulocytes on the upper layer of erythrocytes after Ficoll-Paque® density gradient centrifugation were preserved directly at ‒80°C, which were used as normal tissue control in TMB analysis.
Tissue processing and DNA extraction
Genomic DNA was isolated from FFPE tumor tissue samples with a commercially available Qiagen DNA FFPE kit (Maxwell® 16 FFPE Plus LEV DNA Purification, Qiagen, Hilden, Germany Kit. catalog: AS1135), and matched peripheral blood samples (CD8+ T cells and granulocytes) were extracted by using a DNeasy Blood & Tissue Kit (Qiagen, Hilden, Germany). DNA concentrations were measured with a Qubit fluorometer and the Qubit dsDNA HS (High Sensitivity) Assay Kit (Invitrogen, Carlsbad, CA, USA). The total DNA yield had to have been ≥1 µg. The purity of DNA was determined by the ratio of spectrophotometric absorbance of DNA at 260 nm to that of 280 nm, and the ratio at 260 nm to 230 nm; 260/280 had to have been ≥1.8 and 260/230 ≥ 2.
Whole exome sequencing
Fragmented genomic DNA from tumor and peripheral blood granulocyte samples was used to construct a NEBNext Ultra II DNA Library (New England Biolabs, Ipswich, MA, USA), and exonic regions were captured in solution by using the SeqCap EZ Exome 64 M kit (Roche NimbleGen, Madison, WI, USA) according to the manufacturers’ instructions. Paired-end sequencing, resulting in 100 bases from each end of the fragments for the exome libraries, was done with a HiSeq 3000 Sequencing System (Illumina, San Diego, CA). The mean depth of coverage for the tumor samples was 203x. All samples had qualified QC.
Next generation sequencing analysis
Somatic mutations were detected in paired tissue DNA samples. From raw data, terminal adaptor sequences and low-quality reads were removed. BWA6 (version 0.7.12-r1039) was used to align the clean reads to the reference human genome (hg19). Picard (version 1.98) was used to mark PCR duplicates. Realignment and recalibration were done with GATK (version 3.4–46-gbc02625). MuTect2 (version 1.1.4) was used to “call” somatic insertion/deletions and single nucleotide variants. Identification of Copy Number Variants (CNV): CNV was expressed as the ratio of adjusted depth between tumor tissue DNA and germline DNA and was analyzed using FACETS-with log2 ratio thresholds of 0.322 and −0.415 for gain and loss, respectively.22
Mutations were considered as a candidate somatic mutation only when (i) the mutation had at least five high-quality reads (Phred score ≥30, mapping quality ≥30, and without paired-end reads bias) containing the particular base; (ii) the mutation was not present in >1% of the population in the 1,000 Genomes Project or the Single Nucleotide Polymorphism Database; and (iii) the mutation was not present in a local database of normal samples. For somatic mutations in tumors, a mutant allele must have been present in ≥3% of reads. Non-synonymous mutations annotated by variant effect predictor (VEP; www.ensembl.org) were used in tumor mutation burden (TMB) analysis.
PCR amplification of TCR β
We assessed TCR abundance in TCR-β chain CDR3 regions in both tumor and peripheral blood CD8+ T cells from 19 patients, before RT and at 4 weeks of RT (i.e., after delivery of 40 Gy and 2 rounds of camrelizumab). Multiplex PCR amplification of the CDR3 of the TRB was implemented, including PCR1 and PCR2 inclusively and semi-quantitatively, as described below. Unique molecular identifiers (UMIs) were used to enable the accurate bioinformatic identification of PCR duplicates. During the first round of PCR1, only 15 cycles were used to amplify CDR3 fragments using the specific primers against each V and J genes; in the second round, PCR was done with universal primers.
PCR1: 200ng of DNA (used as templates) were amplified after 25 µL of 2× QIAGEN Multiplex PCR Master Mix, 5 µL of 5 × Q solution, 1 μL of forward primers set pool, and 1 μL of reverse primers set pool were added to form a reaction system by using a Multiplex PCR Kit (QIAGEN, Germany. catalog: 206143). UMIs were added into the PCR mix. PCR was then done as follows: 1 cycle of 95°C for 15 minutes, 10 cycles of denaturation at 94°C for 30 seconds, and 15 cycles of both annealing at 60°C for 90s and extension for 30s at 72°C. After a final extension for 5 minutes at 72°C, the system was cooled down to 4°C. The target fragment of the multiplex-PCR products was purified on magnetic beads (Agencourt AMPure XP kit, Beckman Coulter, Beverly, MA, USA. catalog: A63882).
PCR2: The entire sample of the PCR1 product was used as a template for the second step of amplification after the addition of 2 μL communal primers, 25 μL phusion master mix prepared by using a Phusion® High-Fidelity PCR Kit (New England Biolabs, USA, catalog: E0553L), and nuclease-free water to reach a total volume of 50 μL. The reactions were then transferred to a thermal cycler that carried out the following program: one cycle of 98°C for 1 min; 25 cycles of denaturation at 98°C for 20 s, annealing at 65°C for 30s, and extension at 72°C for 30 s; and final extension at 72°C for 5 min. The samples were then held at 4°C.
Analysis of TRB sequencing data
Raw sequencing data were processed and analyzed with the following procedure. Briefly, (1) raw reads were filtered to remove undesired sequences that do not contain the primers for multi-PCR by using Cutadapt (https://github.com/marcelm/cutadapt); (2) the remaining high-quality paired reads were merged to obtain contigs by using Pear (Paired-End reAd merger, Scientific Computing Group, Heidelberg Institute for Theoretical Studies, Schloss-Wolfsbrunnenweg 35, D-69118 Heidelberg) and (3) aligned to reference TRB V/(D)/J gene sequences (http://www.imgt.org) using MiXCR (https://github.com/milaboratory/mixcr/) to determine the TRB V/(D)/J gene segment usage of each contig; (4) the CDR3 region was identified based on the conserved sequence of CDR3 region; (5) the CDR3 species were clustered to eliminate the sequencing errors according to base quality and sequence similarity.
Diversity, clonality, top frequency, and overlap index were used to characterize the immune repertoire. The diversity of the TCR repertoire is calculated with the Shannon–Wiener index (Shannon index), which is the function of both the relative number of clonotypes present and the relative abundance or distribution of each clonotype.23 The Shannon index is calculated as follows, where ni is the clonal size of the ith clonotype (that is, the number of copies of a specific clonotype), S is the number of different clonotypes, and N is the total number of TCR/BCR sequences analyzed:
Clonality is defined as 1 – (Shannon index)/ln(# of productive unique sequences).24 A maximally diverse population is associated with a clonality score of 0 and a perfectly monoclonal population with a clonality score of 1.
Simpson’s diversity index (Simpson’s index) measures the probability that two randomly selected individuals from a sample will be the same. The Simpson’s index is calculated as followed: where ni is the number of entities belonging to the ith type and N is the total number of entities in the dataset. Simpson’s index ranges between 0 and 1. The higher the value, the lower the diversity.
A separate metric was used to further characterize the diversity of the TCR repertoire – richness. Richness reflects how many different V-J rearrangements are present and is defined as the ratio (expressed as a percentage) between the number of observed rearrangements in a sample and the number of possible theoretical rearrangements between V families and J genes.25
The Morisita–Horn similarity index (MH overlap) for determining the similarity between samples.26 To further investigate the status of baseline tumor immune infiltration, we used the MH overlap to evaluate the composition and the abundance of T-cell rearrangements in tumor tissues versus peripheral blood CD8+ T cells:
where fi = n1i/N1 and gi = n2i/N2, n1i and n2i are the clone sizes of the ith clonotype (i.e., number of copies of each distinct CDR3 sequence for the TCR α- or β-chain) in samples 1 and 2, and N1 and N2 are the total number of TCRs in samples 1 and 2. The summations in the numerator and the denominator are over all c clonotypes in both samples. This index ranges between 0 and 1, with 0 representing minimum similarity and 1 maximum similarity.
TMB analysis
TMB was defined as the number of somatic non-synonymous single nucleotide variants and insertion/deletion mutations per megabase of the targeted region. Increasing values of TMB were expressed as the “fold” change of TMB along with the new regions joining.
Neoantigen prediction
We used predicted neoantigens to investigate potential associations of neoantigen burden and tumor response. Briefly, identified non-silent mutations from WES were used to generate a comprehensive list of peptides 8–11 amino acids in length with the mutated amino acid represented in each possible position. The binding affinity of every mutant peptide and its corresponding wild-type peptide to the patient’s germline HLA alleles were predicted using netMHCpan-3.0. Predicted neoantigens in correlation analysis were identified as those with a predicted binding strength of <50 nM and mutant peptide binding affinity less than 70% of wild-type binding affinity.
Multiplex immunofluorescence analysis
We performed tyramide signal amplification (TAS)-based multicolor immunofluorescence staining assay to identify the tumor immune microenvironment in baseline and on-treatment tumor tissues. Briefly, tumor tissue sections were deparaffinized. And different primary antibodies were sequentially applied, followed by incubation with horseradish peroxidase-conjugated secondary antibody and TSA. The slides were heated in a microwave after each TSA operation. PANO 7-plex IHC kit (Panovue, 0004100100) was used in multiplex immunofluorescence staining. Nuclei were stained with 4′-6′-diamidino-2-phenylindole (DAPI, SIGMA-ALDRICH, D9542) after all the human antigens had been labeled. Immune markers assessed included CD4, CD8, PD1, and PDL1. Tumor cells were identified with pan-cytokeratin (Pan-CK) antibodies. The stained slides were scanned with a Mantra System (PerkinElmer) and images were analyzed by using InForm image analysis software (PerkinElmer).
Statistical analyses
Analyses were performed using SPSS v.21.0 (STATA, College Station, TX, USA) or R 4.1.0 statistical software. Statistical significance between groups was compared using Fisher’s exact tests, non-parametric two-sided Mann–Whitney U tests for two independent samples or Wilcoxon Signed-Rank tests for paired samples, and correlations were evaluated assuming a non-Gaussian distribution (Spearman correlation) unless otherwise indicated. Statistical significance among multiple groups was compared using Kruskal–Wallis test. The Kaplan–Meier method was used to estimate OS and progressive-free survival (PFS). Differences in survival were compared with log-rank tests. The best cutoff of Kaplan–Meier survival analysis was calculated by the Youden index of the ROC curve. Reported P values were two-sided, and the significance level was set at 0.05. All heatmaps were created by using GenVisR (version 1.24.0) or ggplot2 (version 3.3.3) packages. The data cutoff date for all analyses was May 30, 2021.
Results
Genomic characteristics of tumors before radiotherapy combined with camrelizumab
Of the 19 patients enrolled, 12 were male and 7 were female, with a median age of 64 y (range, 46–74 y) (Figure 1a and Supplementary Table 2).16 At the data cutoff date of May 30, 2021, the median follow-up duration was 42.5 months (95% CI 38.5 to 46.5). Median OS was 16.7 months (95% CI 5.9‒27.9); OS rate was 31.6% at 24 months. Median PFS was 11.7 months (95% CI 0–30.3); PFS rate was 35.5% at 24 months.
Figure 1.
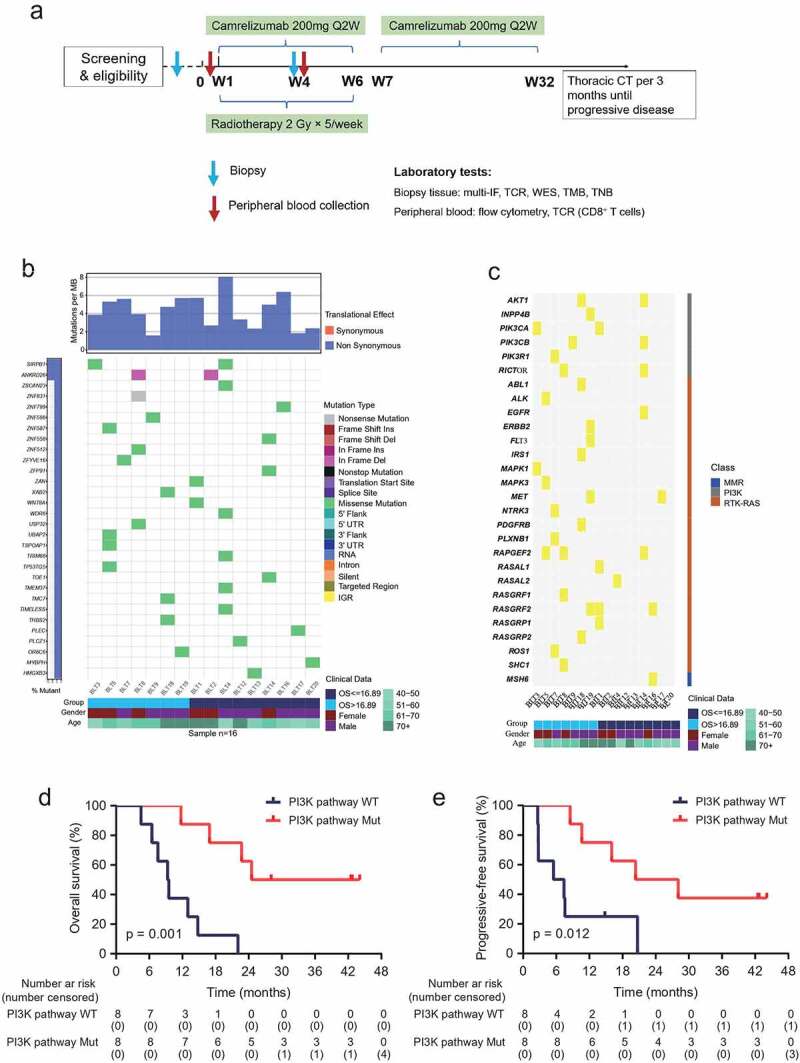
Whole-exome sequencing in baseline tumor tissues.
(a) Study workflow. (b) Mutation genes detected in baseline tumor samples. Upper graph, tumor mutation burden. Most mutations were non-synonymous. Middle graph, mutation genes at top 30 mutation occurrence in all samples were illustrated. A total of 16.89 months, median overall survival time. (c) Gene mutations enriched in representative signaling pathways in baseline tumor samples. (d and e) Kaplan–Meier estimates of overall survival (d) and progressive-free survival (e) among patients with or without PI3K pathway mutation. Cutoff value, Youden index of the ROC curve. N = 16. P < .05, significant difference. IF, immunofluorescence. TCR, T cell receptor. WES, whole-exome sequencing. TMB, tumor mutation burden. TNB, tumor neoantigen.
We performed whole-exome sequencing of tumor tissue samples from 16 patients at baseline. Analysis of baseline genomic data showed the TMB ranged from 1.59 to 8.06 Mutations/Mb, with a median of 4.35 Mutations/Mb (Figure 1b and Supplementary Figure S1); the predicted neoantigens ranged from 54 to 326, with a median of 152 (Supplementary Table 3). The CNV burden ranged from 76 to 3506, with a median of 1662 (Supplementary Figure S2), consistent with the previous report in ESCC.27 We did not find the association between TMB, the predicted neoantigens, CNV burden, and the objective response and OS/PFS. We also investigated potential enrichment of alterations in specific genes or pathways, including eight DNA damage response pathways, cell cycling pathways, the HIPPO pathway, the PI3K pathway, the RTK-RAS pathway, and the interferon-γ pathway (Figure 1c, Supplementary Table 4 and Figure S3). Then, we found that both OS and PFS were longer in patients with mutations enriched in the PI3K pathway than in patients without mutations (Figure 1d and e).
TCR repertoire associated with clinical outcome
We conducted TCRB sequencing in 18 baseline and 16 on-treatment tumor tissues including 15 pairs. The TCRB sequencing was also conducted in 18 pairs of baseline and on-treatment peripheral CD8+ T cells. The median total TCR reads and unique TCR were 4.33 × 106 and 470 in tumor tissues at baseline, and 3.20 × 106 and 3.25 × 104 during treatment, respectively (Supplementary Table 5 and 6). The median total and unique TCR reads were 4.73 × 106 and 3.19 × 104 in peripheral CD8+ T cells at baseline, and 5.89 × 106 and 2.70 × 104 peripheral CD8+ T cells during treatment, respectively (Supplementary Table 5 and 6). The median of total TCR reads in on-treatment tumor tissues was lower compared with that in baseline tumor tissues as well as that in peripheral CD8+ T cells both at baseline and during treatment (p = .017, p = .003, p < .005, respectively.).
To avoid bias from the sample size, we calculated TCR diversity indices, including Shannon index which was highly sensitive to low-frequency clones, clonality which was closely related to expanded clones, and Simpson’s index which emphasized high-frequency reads. Increased TCR diversity (higher Shannon index and lower Simpson’s index) in baseline tumors was found in patients with older age (Supplementary Fig. 4a and b). Other patient baseline characteristics, including gender, ECOG performance status score, smoking and drinking status, and disease stage, was not associated with TCR diversity in baseline tumors (Supplementary Table 2).
We analyzed the TCR diversity according to treatment response evaluated radiological and pathological responses, scored according to RECIST 1.1 by individual clinicians.16 The results showed objective responsive (partial and completed responsive) patients had lower baseline intratumoral clonality compared with non-responsive (stable and progressive disease) patients (Figure 2a). Responsive patients also had marginal higher TCR diversity (elevated Shannon index and lower Simpson’s index) in baseline tumors (Supplementary Fig. 4c and d). While analyzing on-treatment TCR diversity, we found responsive patients had significantly higher TCR diversity (increased Shannon index, lower clonality and Simpson’s index) in the on-treatment peripheral CD8+ T cells compared with non-responsive patients (Figure 2b).
Figure 2.
TCR diversity associated with treatment outcome of radiotherapy combined with camrelizumab.
(a) Responsive patients had lower clonality in baseline tumors. (b) Responsive patients had higher Shannon index, lower clonality and lower Simpson’s index in peripheral CD8+ T cells during treatment. (c and d) Landmark analysis of overall survival and progression-free survival among patients in TCR Shannon index (c) and clonality (d) of baseline peripheral CD8+ T cells. Responsive: partial + completed response. Non-responsive: stable + progressive disease. BLT, baseline tumor tissues. RTC, peripheral CD8+ T cells after 40 Gy radiation and 2 rounds of camrelizumab. BLC, baseline peripheral CD8+ T cells. Black solid line, landmark point. P < .05, significant difference.
We did not find that the TCR diversity in baseline tumors, on-treatment tumors, or on-treatment peripheral CD8+ T cells was associated with patient survival. However, landmark analysis revealed that patients with high baseline TCR diversity in peripheral CD8+ T cells had better OS and PFS (Figure 2c). On the contrary, high baseline clonality in peripheral CD8+ T cells was connected with worse OS and PFS (Figure 2d).
Baseline intratumoral TCR diversity associated with immune environment and heterogeneity
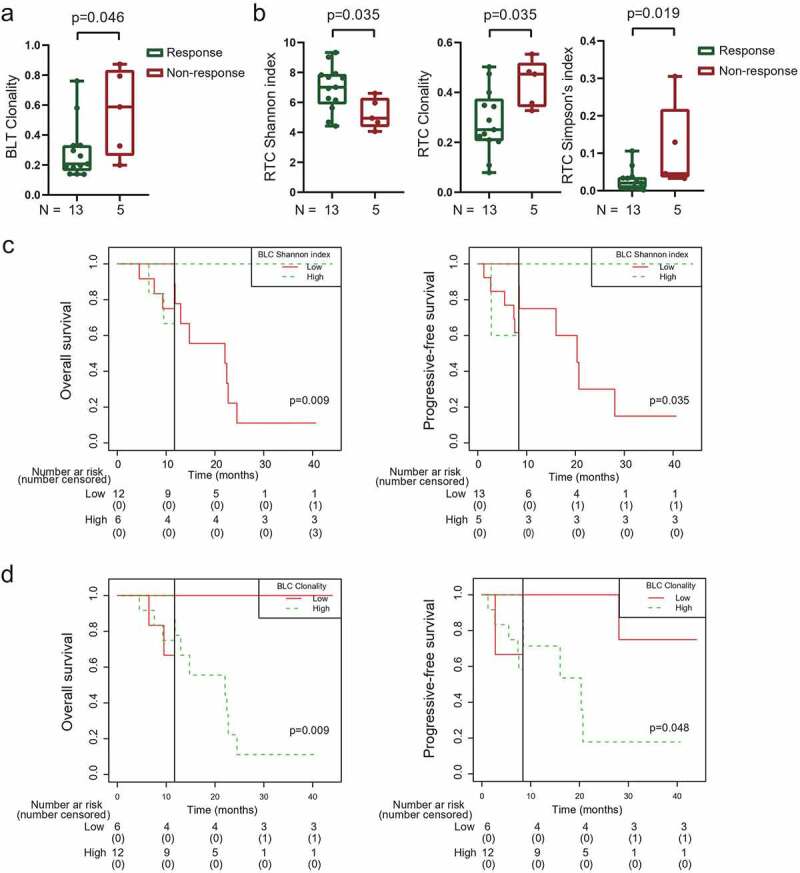
We previously reported that PD-1 expression on CD4+ and CD8+ TILs was associated with survival in these patients by using multicolor immunofluorescence staining.16 To explore the characteristics of intratumoral TCR diversity before treatment, we compared the TCR repertoire in 17 patients who had enough baseline tumor biopsies for both TCRB sequencing and multicolor immunofluorescence staining. We found that patients with high baseline PD-1+CD4+ TILs had increased TCR diversity and decreased clonality in baseline tumors (Figure 3a and Supplementary Figure S5). Similarly, patients with high baseline PD-1+CD8+ TILs also had higher TCR diversity and lower clonality in baseline tumors (Figure 3b).
Figure 3.
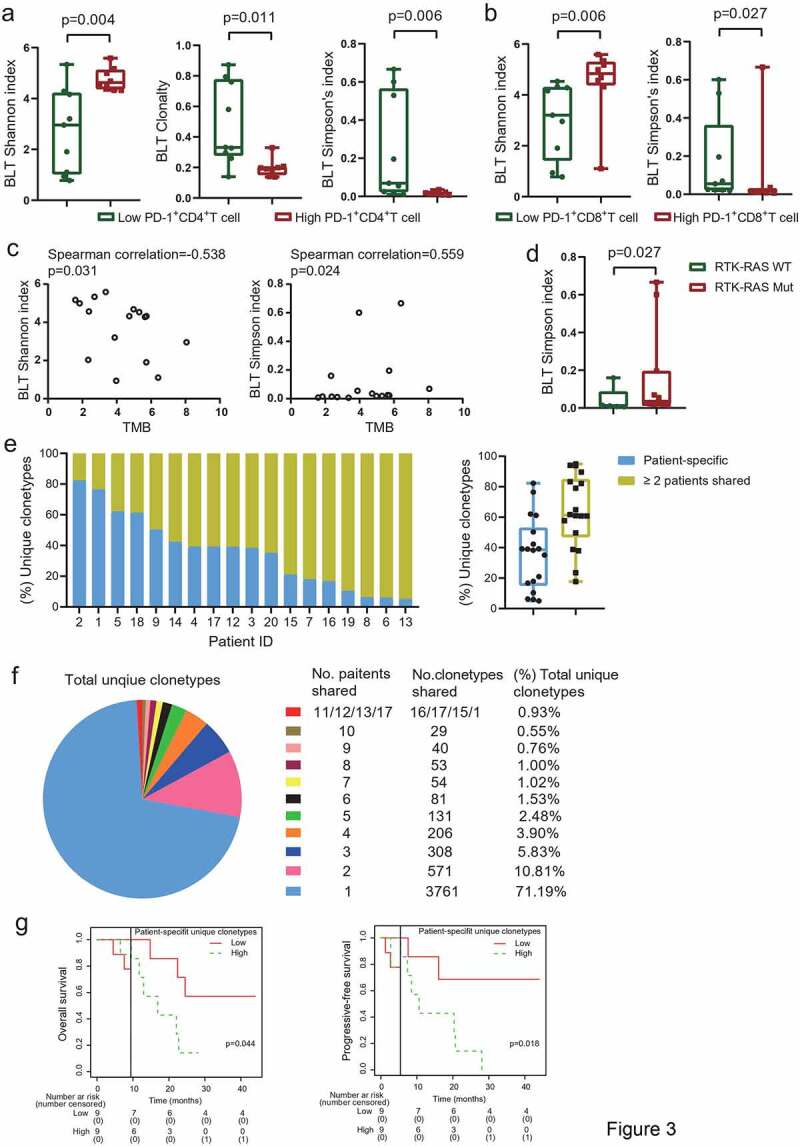
TCR repertoire in baseline tumor tissues associated with tumor microenvironment.
(a) Patients with high baseline PD-1+CD4+ TILs had higher Shannon index, lower clonality and lower Simpson’s index in baseline tumors. (b) Patients with high baseline PD-1+CD8+ TILs had higher Shannon index and lower Simpson’s index in baseline tumors. The median density (/mm2) of PD-1+CD4+ TILs and PD-1+CD8+ TILs was used as cutoffs in (a) and (b). (c) Association between TCR diversity and tumor mutation burden. (d) Patients with RTK-RAS pathway mutations had higher Simpson’s index in baseline tumors. (e) Patient-specific and shared unique T-cell clonotypes in individual patient. (f) Unique T-cell clonotypes shared among patients. (g) Landmark analysis of overall survival and progression-free survival among patients in patient-specific unique clonotypes in baseline tumors. TIL, tumor-infiltrated lymphocyte. TMB, tumor mutation burden. N = 17 (a and b), 16 (c and d), and 18 (e-g). P < .05, significant difference.
In analyzing the correlation between TCR diversity and tumor mutations in baseline tumors available from 16 patients, we found TCR diversity showed loosen correlation with TMB (Figure 3c), probably owing to limited samples. Furthermore, we found patients with mutation enriched in RTK-RAS pathway had higher TCR clonality (increased Simpson’s index) (Figure 3d and Figure 1c).
To investigate the heterogeneity of TCR repertoire among patients, we assessed the TCR clonotypes in the baseline tumors from 18 patients. We firstly classified the unique TCR clonotypes identified in the baseline tumors into two categories. The first category included the “patient-specific” clonotypes which were detected only in individual patients but not shared with the other patients. The second category included shared clonotypes which were identified in the baseline tumors from no less than 2 patients. We found a median of 181 (ranged from 8 to 795) of “patient-specific” clonotypes and 284 (ranged from 76 to 939) shared clonotypes in baseline tumors (Supplementary Fig. 6a). Both these two clonotype categories were positive associated with total unique TCR clonotypes (Supplementary Fig. 6b). The percentages of “patient-specific” and shared clonotypes covered wide range among each baseline tumors (5.06% to 82.27% and 17.73% to 94.94%, respectively). The median percentage of shared clonotypes was higher than “patient-specific” clonotypes (61.35% vs 38.65%, p = .039, Figure 3e). We then integrated all the unique clonotypes in all baseline tumors from the 18 patients (Supplementary Fig. 7). Of the total of 5283 unique clonotypes in these baseline tumor samples, the “patient-specific” clonotypes were 2.8 times more than those shared clonotypes (71.19% vs. 18.81%, Figure 3f). The numbers of TCR clonotypes decreased sharply with the increased numbers of patients shared (Figure 3f). Less than 1% of clonotypes were shared in more than 10 patients. Only one TCR clonotype was shared in 17 patients. No TCR clonotype was shared in 14–16 patients or all 18 patients (Figure 3f and Supplementary Fig. 7). Lastly, landmark analysis indicated patients with high percentages of “patient-specific” clonotypes in baseline tumors had shorter OS and PFS (Figure 3g). These results indicated the heterogeneity of TCR repertoire in baseline tumors among patients.
Combined radiotherapy with camrelizumab remodel TCR repertoire
To explore the variation of TCR repertoire during combination treatment, we initially compared the TCR repertoire in matched pre- and on-treatment tumors from 15 patients. The intratumoral TCR diversity increased remarkably during treatment compared with that at baseline (Figure 4a). On the contrary, the intratumoral clonality decreased significantly during treatment (Figure 4a). We also calculated the TCR richness index to monitor the actual number of unique TCR sequences.25 It was found that the intratumoral richness increased during treatment (P < .001, Supplementary Fig. 8a). Additionally, we found patients with MMR pathway mutation had higher intratumoral clonality during treatment than those without (Supplementary Fig. 8b). While, there was no association between TMB, TIL subsets and intratumoral TCR diversity during treatment. Opposite with the changes of TCR repertoire in tumors, the diversity of peripheral CD8+ TCRs decreased during treatment (Figure 4a and Supplementary Fig. 8a). The lymphocyte count deceased under the combination treatment compared with that at baseline in all 19 patients ([1.85 ± 0.56]×106/mL vs [0.51 ± 0.04]×106/mL, p < .001, paired t-test). Kaplan–Meier analysis did not show the association between lymphocyte count, objective response rate and survival.
Figure 4.

Radiotherapy plus camrelizumab remodel TCR diversity.
(a) Changes of Shannon index, clonality and Simpson’s index during combination treatment. (b) Intratumoral clonotypes identified at baseline expanded or contracted in on-treatment tumors. (c) Intratumoral expansion of persistent clonotypes during treatment was associated with patient survival. (d) Intratumoral clonotypes identified at baseline expanded or contracted in peripheral CD8+ T cells during treatment. Expanded, TCR frequency higher in on-treatment tumors (b and c) or in on-treatment peripheral blood (d) compared with their corresponding samples at baseline. Contracted, TCR frequency lower in on-treatment tumors (b) or in on-treatment peripheral blood (d) compared with their corresponding samples at baseline. RTT, intratumoral T cells after 40 Gy radiation and 2 rounds of camrelizumab. RTC, peripheral CD8+ T cells after 40 Gy radiation and 2 rounds of camrelizumab. N = 15 paired tumor tissues (a-c), 18 paired peripheral CD8+ T cells (a), and 17 (d). P < .05, significant difference.
We next evaluated the unique TCR clonotypes in tumors during treatment. The majority of clonotypes identified at baseline were lost during treatment across all paired tumors. Above 99.9% intratumoral clonotypes during treatment were newly detected that were not observed in tumors at baseline (Supplementary Table 7). Less than 0.1% of clonotypes existed persistently in tumors both before and during treatment (Supplementary Table 7). Because the on-treatment TCR richness increased exclusively in all tumors, it was feasible to categorize these persistent clonotypes into expanded or contracted, according to their frequencies in the on-treatment tumors were higher (expanded) or lower (contracted) than those in the baseline tumors. A median of 46% (ranged from 17% to 83%) persistent T-cell clones expanded in tumors during treatment (Figure 4b). And higher percentage of expansion of these persistent clones in on-treatment tumors was associated with both longer OS and PFS (Figure 4c). We also assessed a median of 19 (range from 3 to 65) T-cell clonotypes that were identified in baseline tumors persistently existed in the peripheral blood during treatment (Figure 4d and Supplementary Fig. 9). And a median of 50% (range from 18% to 91%) of these clones expanded in peripheral blood during treatment (Figure 4d).
Clonotype composition and variable-Joining (V-J) gene utilization profiles during combination treatment
To further characterize the TCR repertoire alteration during treatment, we evaluated the number of clonotypes with different frequencies. The total frequencies of the top 100 clones decreased in tumors, while increased in peripheral CD8+ T cells during treatment (Figure 5a). Patients with low frequencies of top 100 clones in baseline peripheral CD8+ T cells had longer OS (Figure 5b). We then divided the T-cell clones into high-frequency clones (clone fraction ≥0.1%), medium-frequency clones (0.01% to 0.1%), and low-frequency clones (≤0.01%) (Figure 5c). The proportions of high-frequency and medium-frequency clones decreased, whereas the proportions of low-frequency clones increased in all tumors during treatment compared with those in baseline tumors (Figure 5c). The high-frequency proportions of peripheral CD8+ T cells increased during treatment (Figure 5c). We found patients with higher low-frequency clones in the baseline tumors had better OS (Figure 5d). However, patients with higher high-frequency and higher medium-frequency clones in the baseline tumors had worse OS (Figure 5e and Supplementary Fig. 10).
Figure 5.
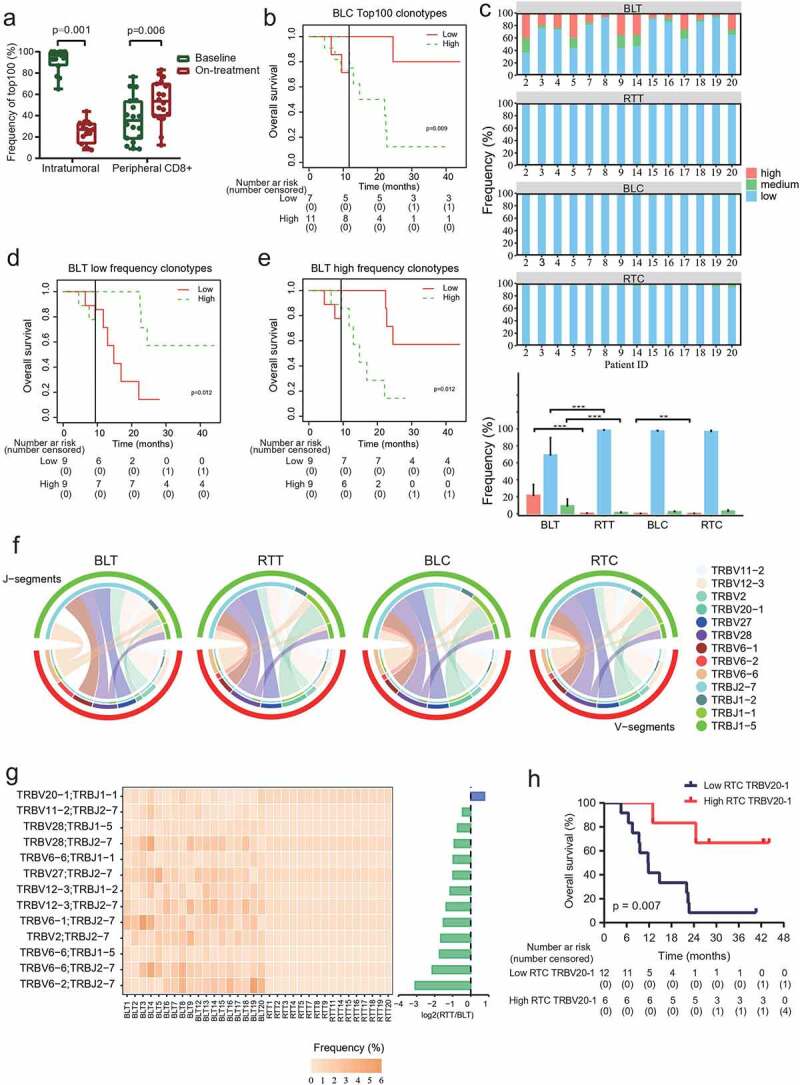
Clonotype redistribution during combination treatment.
(a) Total frequencies of top 100 clonotypes in tumors or peripheral CD8+ T cells before and during treatment. N = 15 paired tumor tissues, and 18 paired peripheral CD8+ T cells. (b) Landmark analysis of overall survival among patients in frequencies of top 100 clones in baseline peripheral CD8+ T cell. (c) Different frequencies of clonotypes during treatment. The upper four graphics showed the proportions of high-frequency, medium-frequency, and low-frequency clonotypes in tumors or peripheral CD8+ T cells from each patient before and during treatment. Low-frequency clones, ≤0.01%. Medium-frequency clones, 0.01% to 0.1%. High-frequency clones, ≥0.1%. N = 14. (d and e) Landmark analysis of overall survival among patients in low-frequency clonotypes (d) and high-frequency clonotypes (e) in baseline tumors. (f) Variable-Joining (V/J) gene utilization profiles of TCRβ (TRB) in tumors or peripheral CD8+ T cells before and during treatment. V-J pairs identified in all tumors and peripheral CD8+ T cell samples were included in the analysis. BLT, BLC and RTC, N = 18 each. RTT, N = 16. (g) Frequencies of V/J gene profiles of TRB in tumors from each patient before and during treatment. V-J pairs identified in all tumors were included in the analysis. (h) Kaplan–Meier estimates of overall survival among patients with TRBV20-1 in the peripheral CD8+ T cells during treatment. BLT and BLC, baseline tumor tissues and peripheral CD8+ T cells. RTT and RTC, tumor tissues and peripheral CD8+ T cells after 40 Gy radiation and 2 rounds of camrelizumab. P < .05, significant difference.
We then estimated the V-J gene utilization profiles of TRB pre- and during treatment. The segment TRBV12-3 and TRBJ2-7 were the most frequent for the baseline intratumoral T cells (Supplementary Fig. 11 and 12). The segment TRBV20-1 and TRBJ2-7 were the most frequent for the intratumoral T cells during treatment and peripheral CD8+ T cells both at baseline and during treatment (Supplementary Fig. 11 and 12). We also characterized the V-J pairing profile in all samples. The TRBV6-1/TRBJ2-7 pair was the most frequent for baseline intratumoral T cells, while its frequency decreased, and the TRBV20-1/TRBJ2-7 pair ranked to the most frequency during treatment (Figure 5f and Supplementary Fig. 13). Of the V-J pairing segments identified in baseline tumors, the TRBV20-1/TRBJ1-1 pair was the only one with increased frequency during treatment (Figure 5g). Patients with higher TRBV20-1 in the on-treatment peripheral CD8+ T cells had better OS (Figure 5h).
Mutual influx of T-cell clones between tumor and peripheral blood T cells
To investigate the potential migration of T-cell clones between local tumors and peripheral blood, we used Morisita-Horn (MH) overlap to evaluate the shared T-cell clones between tumor tissues and peripheral CD8+ T cells. Fourteen patients who had all four samples (tumors and peripheral CD8+ T cells both at baseline and during treatment) for TCRB sequencing were included in the analysis. The MH overlap elevated in 86% (12/14) patients during treatment (Figure 6a). And a higher increase in MH overlap (on-treatment MH overlap – baseline MH overlap) predicted improved PFS (Figure 6b). These results indicated the migration between local tumors and systematic blood.
Figure 6.
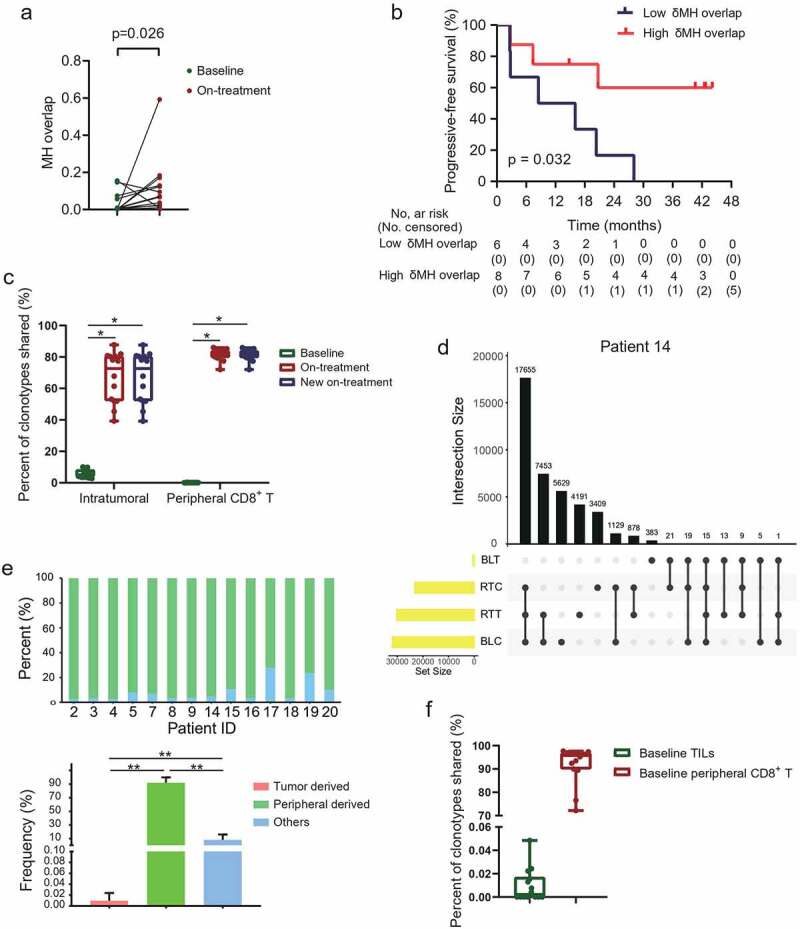
Mutual influx of T-cell clones between tumor and peripheral blood T cells.
(a) Morisita-Horn (MH) overlap index between tumors and peripheral CD8+ T cells. (b) Increased MH overlap index was associated with progressive-free survival. (c) Clonotypes shared between tumors and peripheral CD8+ T cells at baseline and during treatment. The new on-treatment TCR clonotypes firstly shared between tumors and peripheral CD8+ T cells during treatment. (d) Representative upset Venn diagram to assess unique TCR clonotypes in tumors and peripheral CD8+ T cells both at baseline and during treatment. Each row, unique clonotypes in individual tested samples. Each column, intersection set (shared clonotypes) or exception set (peculiar clonotypes) between samples. UpSetR (version 1.4.0) packages was used. (e) Different derivation of new shared clonotypes during treatment. (f) New shared clonotypes in baseline samples. N = 14. P < .05, significant difference.
To vividly track the compartmentalization influx of T-cell clones, we then focused on the T-cell clonotypes newly shared between tumors and peripheral CD8+ T cells during treatment but not shared between these two compartments at baseline. The new shared clonotypes occupied 72.63% and 81.88% of the total clonotypes in the intratumoral and peripheral CD8+ T cells during treatment, respectively (Figure 6c). Based on the baseline location of these new shared clonotypes, we grouped them into tumor-derived which were also detected in the baseline tumors, peripheral-derived which were also detected in baseline peripheral CD8+ T cells, and others which were detected in neither of the two biological compartments at baseline (Figure 6d). Of the total new shared clonotypes, more than 90% were peripheral-derived, tiny minority tumor-derived, and the rest less known the derivation in the present study (Figure 6e). Seven (7/14, 50%) patients had the new shared clonotypes shared with their baseline tumors (Figure 6f), constituting 0.015% of the new shared clonotypes in these patients. The median proportion of the new shared clonotypes shared with the baseline tumors and baseline peripheral CD8+ T cells were 0.002% and 95.78%, respectively (Figure 6f). These results demonstrated the mutual influx of T-cell clones between tumors and peripheral blood under combination treatment.
Discussion
In the present study, we illustrated the spatiotemporal variation of T-cell repertoire induced by radiotherapy combined with camrelizumab in ESCC patients for the first time. We found that the diversity of intratumoral TCR and the mutual influx between tumor and peripheral blood were greatly promoted by the combination treatment. Our results also revealed the baseline intratumoral TCR repertoire was closely associated with tumor immune microenvironment and had heterogeneity among patients. High diversity of baseline peripheral CD8+ T cells increased, on-treatment MH overlap between tumors and peripheral CD8+ T cells, and expansion of persistent intratumoral T-cell clones were the biomarker candidates for predicting the treatment outcome.
We found a positive correlation between TMB and TCR clonality, and a negative correlation between TMB and TCR diversity in baseline tumors. Higher TMB would elevate more production of neoantigens, which leading to enhanced expansion of neoantigen-specific T cells. However, we did not find the association between TMB and treatment response though what has been observed in advanced ESCCs with camrelizumab5 and multiple cancers with immunotherapy.28,29 The TMB was significantly higher in radioresistant cells than radiosensitive cells in breast cancer patients.30 Radiotherapy was associated with increases in deletion burden, which contributed to worse survival in patients with glioma.31 Combing with the close association between MMR pathway mutation and higher intertumoral clonality during treatment in our result, we inferred that tumor mutations during radiotherapy also played an important role in the treatment outcome. In addition, our results demonstrated that patients with mutations enriched in PI3K pathway may benefit from the treatment of radiotherapy combined with PD-1 blockade, which may attribute to the activation of PI3K/Akt/mTOR pathway32 and PI3K/Akt/STAT3 pathway in response to radiotherapy and immune therapy,33 promoting PD-L1 expression. These results supported the rational combination strategies of immunotherapy and radiotherapy, and suggested that the TMB during treatment and pathways enriched mutations would be potential biomarkers to predict the response to the combination treatment.
In analyzing the baseline intratumoral TCRs, we found that patients with high PD-1+CD4+ or PD-1+CD8+ TILs had lower T-cell clone expansion, and lower TCR clonality was associated with objective response. Besides strictly regulated in T cells, PD-1 is also affected by multiple cytokines in the tumor microenvironment; more inhibitory immune environment would induce higher level of PD-1 expression.34The ESCC patients with high PD-1 expressed TILs probably had more seriously inhibitory tumor immune microenvironment. It was reported that high PD-1 expressed CD8+ TILs exhibited exhausted traits and tumor-specific recognition compared with intermediated or no PD-1 expressed CD8+ TILs in non-small cell lung cancer.35 Applying anti-PD-1 antibody could block the PD-1/PD-L1 inhibitory signaling and improve the anti-tumor response. We found the heterogeneity of TCR repertoire existed in individuals. Furthermore, the patients with more “patient” specific clonotypes had worse survival. The different TCR repertoire in multiple regions in the same ESCC tumor contributed to intratumor heterogeneity of T-cell immune response.36 These “patient” specific clonotypes in baseline tumors probably included T cells that lacked the capacity to recognize the tumor cells.37 A large proportion of these cells might be bystander TILs that recognized unrelated tumor antigens, such as virus, resided in the tumor tissues during the process of tumorigenesis.38–40 Additionally, a part of these tumor-resident T cells may have specificities for MHC-peptide complex that previously existed in tumors, but subsequently lost due to deficiency of presenting MHC allele41 or tumor antigen editing.42 As a result, although high level of these cells presented in tumors, they could not be activated or revigorated effectively by radiotherapy combined with PD-1 blockade, thus uncapable as effector T cells under the combination treatment. We here could not determine whether the PD-1 expressed TILs represented tumor-specific T cells, or the specificities of “patient” specific T cells infiltrated in tumors. And we also need consider that different TCR clonotypes may recognize the same tumor antigen which would be impossible to discern in the present analysis. However, our findings indicated that heterogenous antitumor capability of T-cells in baseline ESCC tumors, which deserved further study in future.
Previous studies showed neoadjuvant nivolumab resulted in augmented TCR clonal diversity among TILs in resectable glioblastoma,43 while nivolumab induced temporal clonal expansion of TILs in advanced melanoma.44 In our study, we found a sharp elevation of TCR diversity occurred in the tumor T cells during the treatment, which was probably due to the release promotion of tumor antigens through radiotherapy. It was reported that T-cell clonal expansion could be induced in the ESCC patients who relapsed after definitive chemoradiotherapy, despite the clonal expansion was not associated with prognosis.12 However, we did not find the superiority of intratumoral clone expansion during treatment, partially because of the toxicity of radiation to proliferative T cells, as we collected the on-treatment tumor samples during radiotherapy. Intriguingly, more than 99.9% of T-cell clonotypes were newly detected in the on-treatment tumors, probably resulting from efficient attraction of novel functional T cells infiltrating into tumors.45,46 It may explain one of the mechanisms that radiotherapy combined with anti-PD-1 antibody enhance the anti-tumor response. Meanwhile, the result of expansion in persistent TILs during treatment enlightened another mechanism involved in the combination treatment, reinvigorating anti-tumor T cells that were dysfunctional before treatment.33,47,48 Additionally, the increased TCR clonality of peripheral blood during treatment indicated that combined radiotherapy with anti-PD-1 antibody also effectively activated the systematic anti-tumor immune response. Lastly, adjuvant nivolumab significantly prolonged PFS among patients with residual pathological disease after neoadjuvant chemoradiotherapy followed by surgery for locally advanced esophageal or gastroesophageal junction cancer.49 Sequential radiotherapy and PD-1 blockade could prevent the proliferative T cells from being killed by radiation, which could be a feasible option for the patients included in our study and sketch a specific TCR repertoire in tumor tissues as well as in peripheral blood.
Our results revealed the mutual influx of T-cell clones between tumors and peripheral blood under combined radiotherapy with anti-PD-1 therapy. It was reported that peripheral blood contributed to the construction of TILs in ESCC patients after preoperative chemotherapy.13 The unique clonotypes in post-treatment TILs shared with pre-treatment TILs and peripheral T cells were 55.6% and 48.8%, respectively.13 However, 95.78% of new shared clones derived from peripheral CD8+ T cells in our study. Radiation increased immunogenic cell death50 and presentation of tumor antigens.51 As an immune modulator,52 radiation induced secretion of chemokines, such as CXCL16, CXCL9 and CXCL10, which attracted T helper 1 cells and CD8+ T cells infiltrating into tumors.45,46,53 TCR repertoire in tumors was broadened by radiation.54 Our results indicated peripheral blood could act as a T-cell clone pool supporting the diversity of TCR repertoire in tumors, and this process was greatly promoted by combined radiotherapy and anti-PD-1 therapy. Consequently, we found TCR diversity of baseline peripheral CD8+ T cells was closely associated with both response and survival. And the higher peripheral-derived clonotypes migrated into the on-treatment tumors, the better survival was in the patients. Similar results were found in non–small cell lung cancer patients after PD-1/PD-L1 blocked.55,56 The TCR diversity may reflect the probability of neoantigen recognition.44,57 The peripheral cells with higher diversity provided more opportunities for tumor neoantigen recognition, subsequently resulting in enhanced antitumor immune response under the anti-PD-1 therapy.58 We supposed the tumor-specific CD8+ T cells circling peripherally in ESCC patients were capable to migrate into the local tumors and elicit anti-tumor responses to the combination treatment of radiotherapy combined with anti-PD-1 antibody, indeed. Our results also revealed that the intratumoral T-cell clones migrated into peripheral blood during treatment. However, the clonotypes migrated from the tumors into peripheral blood were much lower than those migrated from the peripheral blood into the tumors during treatment, which may be the cause of more serious local inflammation response in tumor tissues induced by radiotherapy.
Several limitations existed in our study. Due to the natural characteristics of the phase Ib clinical study, the sample size was limited. TCR metrics from normal esophageal tissues as control was not available. Additionally, TCR sequencing used to identify the TCR repertoire could not exactly represent the function of T cells. Although single-cell TCR sequencing had the inherent advantage that it could integrate the information of gene expression and TCR repertoire in one single cell, the ESCC tissues collected under endoscopic ultrasonography weighed less than 20 mg. Lack of adequate tissue samples limited the step of single-cell digestion for next single-cell TCR sequencing.
In conclusion, this is the first study to explore the time-spatial dynamics of TCR repertoire induced by radiotherapy combined with anti-PD-1 antibody in ESCC. Our results further revealed that the TCR diversity of peripheral CD8+ T cells could be an ideal predictive biomarker for this combination treatment, which deserved further evaluation in our ongoing phase III study (NCT04426955) and exploration the underlying mechanisms of anti-tumor immune response.
Supplementary Material
Acknowledgments
Author contributions: C.Y., W.Z., S.P. and P.W. contributed conception and design. C.Y. and W.Z. contributed development of methodology. C.Y., W.Z., X.M., Z.G., X.L., D.H., T.Z., X.C., F.C., G.Z., X.G., T.W. and Y.J. collected data. WZ and CY analyzed and interpreted the data. All authors participated in writing, reviewing, and approving the manuscript for submission.
Additional Information: We are grateful to all patients and their families and all members of the collaborative group involved in this trial. We thank Jiangsu HengRui Medicine Co., China, for kindly providing the anti-PD-1 antibody camrelizumab.
Funding Statement
This work was supported by Chinese National Key Research and Development Project (No. 2018YFC1315601) and National Natural Science Foundation of China (grants 81872462, 81972772 and 82073348).
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
References
- 1.Kato K, Cho BC, Takahashi M, Okada M, Lin C-Y, Chin K, Kadowaki S, Ahn M-J, Hamamoto Y, Doki Y, et al. Nivolumab versus chemotherapy in patients with advanced oesophageal squamous cell carcinoma refractory or intolerant to previous chemotherapy (ATTRACTION-3): a multicentre, randomised, open-label, phase 3 trial. Lancet Oncol. 2019;20(11):1506–16. doi: 10.1016/S1470-2045(19)30626-6. [DOI] [PubMed] [Google Scholar]
- 2.Kojima T, Shah MA, Muro K, Francois E, Adenis A, Hsu C-H, Doi T, Moriwaki T, Kim S-B, Lee S-H, et al. Randomized phase III KEYNOTE-181 study of pembrolizumab versus chemotherapy in advanced esophageal cancer. J Clin Oncol. 2020;38(35):4138–4148. doi: 10.1200/JCO.20.01888. [DOI] [PubMed] [Google Scholar]
- 3.Huang J, Xu J, Chen Y, Zhuang W, Zhang Y, Chen Z, Chen J, Zhang H, Niu Z, Fan Q, et al. Camrelizumab versus investigator’s choice of chemotherapy as second-line therapy for advanced or metastatic oesophageal squamous cell carcinoma (ESCORT): a multicentre, randomised, open-label, phase 3 study. Lancet Oncol. 2020;21(6):832–842. doi: 10.1016/S1470-2045(20)30110-8. [DOI] [PubMed] [Google Scholar]
- 4.Lee J, Kim B, Jung HA, La Choi Y, Sun J-M.. Nivolumab for esophageal squamous cell carcinoma and the predictive role of PD-L1 or CD8 expression in its therapeutic effect. Cancer Immunol Immunother. 2021;70(5):1695–1703. doi: 10.1007/s00262-020-02766-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Huang J, Xu B, Mo H, Zhang W, Chen X, Wu D, Qu D, Wang X, Lan B, Yang B, et al. Safety, activity, and biomarkers of SHR-1210, an anti-PD-1 antibody, for patients with advanced esophageal carcinoma. Clin Cancer Res. 2018;24(6):1296–1304. doi: 10.1158/1078-0432.CCR-17-2439. [DOI] [PubMed] [Google Scholar]
- 6.Zhang W, Yan C, Zhang T, Chen X, Dong J, Zhao J, Han D, Wang J, Zhao G, Cao F, et al. Addition of camrelizumab to docetaxel, cisplatin, and radiation therapy in patients with locally advanced esophageal squamous cell carcinoma: a phase 1b study. Oncoimmunology. 2021;10:1971418. doi: 10.1080/2162402X.2021.1971418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang P, Chen Y, Long Q, Li Q, Tian J, Liu T, Wu Y, Ding Z. Increased coexpression of PD-L1 and TIM3/TIGIT is associated with poor overall survival of patients with esophageal squamous cell carcinoma. J Immunother Cancer. 2021;9 e002836 doi: 10.1136/jitc-2021-002836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zhang J, Ji Z, Smith KN.. Analysis of TCR beta CDR3 sequencing data for tracking anti-tumor immunity. Methods Enzymol. 2019;629:443–464. [DOI] [PubMed] [Google Scholar]
- 9.Li S, Sun J, Allesoe R, Datta K, Bao Y, Oliveira G, Forman J, Jin R, Olsen LR, Keskin DB, et al. RNase H-dependent PCR-enabled T-cell receptor sequencing for highly specific and efficient targeted sequencing of T-cell receptor mRNA for single-cell and repertoire analysis. Nat Protoc. 2019;14:2571–2594. doi: 10.1038/s41596-019-0195-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang H, Liu L, Zhang J, Chen J, Ye J, Shukla S, Qiao J, Zhan X, Chen H, Wu CJ, et al. Investigation of antigen-specific T-cell receptor clusters in human cancers. Clin Cancer Res. 2020;26(6):1359–1371. doi: 10.1158/1078-0432.CCR-19-3249. [DOI] [PubMed] [Google Scholar]
- 11.Spear TT, Evavold BD, Baker BM, Nishimura MI. Understanding TCR affinity, antigen specificity, and cross-reactivity to improve TCR gene-modified T cells for cancer immunotherapy. Cancer Immunol Immunother. 2019;68:1881–1889. doi: 10.1007/s00262-019-02401-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Mori T, Kumagai K, Nasu K, Yoshizawa T, Kuwano K, Hamada Y, Kanazawa H, Suzuki R. Clonal expansion of tumor-infiltrating T cells and analysis of the tumor microenvironment within esophageal squamous cell carcinoma relapsed after definitive chemoradiation therapy. Int J Mol Sci . 2021;22:1098 doi: 10.3390/ijms22031098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang C, Palashati H, Tan Q, Ku W, Miao Y, Xiong H, Lu Z. Immediate and substantial evolution of T-cell repertoire in peripheral blood and tumor microenvironment of patients with esophageal squamous cell carcinoma treated with preoperative chemotherapy. Carcinogenesis. 2018;39:1389–1398. doi: 10.1093/carcin/bgy116. [DOI] [PubMed] [Google Scholar]
- 14.Wu KL, Chen GY, Xu ZY, Fu XL, Qian H, Jiang GL. Three-dimensional conformal radiation therapy for squamous cell carcinoma of the esophagus: a prospective phase I/II study. Radiother Oncol. 2009;93:454–457. doi: 10.1016/j.radonc.2009.10.014. [DOI] [PubMed] [Google Scholar]
- 15.Cooper JS, Guo MD, Herskovic A, Macdonald JS, Martenson JA Jr., Al-Sarraf M, Byhardt R, Russell AH, Beitler JJ, Spencer S, et al. Chemoradiotherapy of locally advanced esophageal cancer: long-term follow-up of a prospective randomized trial (RTOG 85-01). Radiation Therapy Oncology Group. JAMA. 1999;281(17):1623–1627. doi: 10.1001/jama.281.17.1623. [DOI] [PubMed] [Google Scholar]
- 16.Zhang W, Yan C, Gao X, Li X, Cao F, Zhao G, Zhao J, Er P, Zhang T, Chen X, et al. Safety and feasibility of radiotherapy plus camrelizumab for locally advanced esophageal squamous cell carcinoma. The Oncologist. 2021;26(7):e1110–e24. doi: 10.1002/onco.13797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen Y, Ye J, Zhu Z, Zhao W, Zhou J, Wu C, Tang H, Fan M, Li L, Lin Q, et al. Comparing paclitaxel plus fluorouracil versus cisplatin plus fluorouracil in chemoradiotherapy for locally advanced esophageal squamous cell cancer: a randomized, multicenter, phase III clinical trial. J Clin Oncol. 2019;37(20):1695–1703. doi: 10.1200/JCO.18.02122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gao X-S, Esophageal Carcinoma Cooperative Group of Radiation Oncology Society of Chinese Medical A . Treatment guideline of radiotherapy for Chinese esophageal carcinoma (draft). Chin J Cancer. 2010;29:855–859. doi: 10.5732/cjc.010.10250. [DOI] [PubMed] [Google Scholar]
- 19.Luo H, Lu J, Bai Y, Mao T, Wang J, Fan Q, Zhang Y, Zhao K, Chen Z, Gao S, et al. Effect of camrelizumab vs placebo added to chemotherapy on survival and progression-free survival in patients with advanced or metastatic esophageal squamous cell carcinoma: the ESCORT-1st randomized clinical trial. JAMA. 2021;326(10):916–925. doi: 10.1001/jama.2021.12836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Noordman BJ, Spaander MCW, Valkema R, Wijnhoven BPL, van Berge Henegouwen MI, Shapiro J, Biermann K, van der Gaast A, van Hillegersberg R, Hulshof MCCM, et al. Detection of residual disease after neoadjuvant chemoradiotherapy for oesophageal cancer (preSANO): a prospective multicentre, diagnostic cohort study. Lancet Oncol. 2018;19(7):965–974. doi: 10.1016/S1470-2045(18)30201-8. [DOI] [PubMed] [Google Scholar]
- 21.Shapiro J, Ten Kate FJ, van Hagen P, Biermann K, Wijnhoven BP, van Lanschot JJ. Residual esophageal cancer after neoadjuvant chemoradiotherapy frequently involves the mucosa and submucosa. Ann Surg. 2013;258:678–688. discussion 88-9. doi: 10.1097/SLA.0b013e3182a6191d. [DOI] [PubMed] [Google Scholar]
- 22.Shen R, Seshan VE. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Keylock C. Simpson diversity and the Shannon–Wiener index as special cases of a generalized entropy. Oikos. 2005;109:203–207. doi: 10.1111/j.0030-1299.2005.13735.x. [DOI] [Google Scholar]
- 24.Cooper ZA, Frederick DT, Juneja VR, Sullivan RJ, Lawrence DP, Piris A, Sharpe AH, Fisher DE, Flaherty KT, Wargo JA, et al. BRAF inhibition is associated with increased clonality in tumor-infiltrating lymphocytes. Oncoimmunology. 2013;2(10):e26615. doi: 10.4161/onci.26615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Postow MA, Manuel M, Wong P, Yuan J, Dong Z, Liu C, Perez S, Tanneau I, Noel M, Courtier A, et al. Peripheral T cell receptor diversity is associated with clinical outcomes following ipilimumab treatment in metastatic melanoma. J Immunother Cancer. 2015;3(1):23. doi: 10.1186/s40425-015-0070-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Venturi V, Kedzierska K, Tanaka MM, Turner SJ, Doherty PC, Davenport MP. Method for assessing the similarity between subsets of the T cell receptor repertoire. J Immunol Methods. 2008;329:67–80. doi: 10.1016/j.jim.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 27.Nagashima T, Yamaguchi K, Urakami K, Shimoda Y, Ohnami S, Ohshima K, Tanabe T, Naruoka A, Kamada F, Serizawa M, et al. Japanese version of the cancer genome atlas, JCGA, established using fresh frozen tumors obtained from 5143 cancer patients. Cancer Science. 2020;111(2):687–699. doi: 10.1111/cas.14290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Samstein RM, Lee C-H, Shoushtari AN, Hellmann MD, Shen R, Janjigian YY, Barron DA, Zehir A, Jordan EJ, Omuro A, et al. Tumor mutational load predicts survival after immunotherapy across multiple cancer types. Nat Genet. 2019;51(2):202–206. doi: 10.1038/s41588-018-0312-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Janjigian YY, Sanchez-Vega F, Jonsson P, Chatila WK, Hechtman JF, Ku GY, Riches JC, Tuvy Y, Kundra R, Bouvier N, et al. Genetic predictors of response to systemic therapy in esophagogastric cancer. Cancer Discov. 2018;8(1):49–58. doi: 10.1158/2159-8290.CD-17-0787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jang BS, Han W, Kim IA. Tumor mutation burden, immune checkpoint crosstalk and radiosensitivity in single-cell RNA sequencing data of breast cancer. Radiother Oncol. 2020;142:202–209. doi: 10.1016/j.radonc.2019.11.003. [DOI] [PubMed] [Google Scholar]
- 31.Kocakavuk E, Anderson KJ, Varn FS, Johnson KC, Amin SB, Sulman EP, Lolkema MP, Barthel FP, Verhaak RGW. Radiotherapy is associated with a deletion signature that contributes to poor outcomes in patients with cancer. Nat Genet. 2021;53(7):1088–1096. doi: 10.1038/s41588-021-00874-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lastwika KJ, Wilson W 3rd, Li QK, Norris J, Xu H, Ghazarian SR, Kitagawa H, Kawabata S, Taube JM, Yao S, et al. Control of PD-L1 expression by oncogenic activation of the AKT-mTOR pathway in non-small cell lung cancer. Cancer Res. 2016;76:227–238. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 33.Gong X, Li X, Jiang T, Xie H, Zhu Z, Zhou F, Zhou C. Combined radiotherapy and Anti-PD-L1 antibody synergistically enhances antitumor effect in non-small cell lung cancer. J Thorac Oncol. 2017;12:1085–1097. doi: 10.1016/j.jtho.2017.04.014. [DOI] [PubMed] [Google Scholar]
- 34.Jiang X, Wang J, Deng X, Xiong F, Ge J, Xiang B, Wu X, Ma J, Zhou M, Li X, et al. Role of the tumor microenvironment in PD-L1/PD-1-mediated tumor immune escape. Mol Cancer. 2019;18(1):10. doi: 10.1186/s12943-018-0928-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Thommen DS, Koelzer VH, Herzig P, Roller A, Trefny M, Dimeloe S, Kiialainen A, Hanhart J, Schill C, Hess C, et al. A transcriptionally and functionally distinct PD-1+ CD8+ T cell pool with predictive potential in non-small-cell lung cancer treated with PD-1 blockade. Nat Med. 2018;24(7):994–1004. doi: 10.1038/s41591-018-0057-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen Z, Zhang C, Pan Y, Xu R, Xu C, Chen Z, Lu Z, Ke Y. T cell receptor beta-chain repertoire analysis reveals intratumour heterogeneity of tumour-infiltrating lymphocytes in oesophageal squamous cell carcinoma. J Pathol. 2016;239:450–458. doi: 10.1002/path.4742. [DOI] [PubMed] [Google Scholar]
- 37.Scheper W, Kelderman S, Fanchi LF, Linnemann C, Bendle G, de Rooij MAJ, Hirt C, Mezzadra R, Slagter M, Dijkstra K, et al. Low and variable tumor reactivity of the intratumoral TCR repertoire in human cancers. Nat Med. 2019;25:89–94. doi: 10.1038/s41591-018-0266-5. [DOI] [PubMed] [Google Scholar]
- 38.Snell LM, MacLeod BL, Law JC, Osokine I, Elsaesser HJ, Hezaveh K, Dickson RJ, Gavin MA, Guidos CJ, McGaha TL, et al. CD8+ T cell priming in established chronic viral infection preferentially directs differentiation of memory-like cells for sustained immunity. Immunity. 2018;49(4):678–94 e5. doi: 10.1016/j.immuni.2018.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Simoni Y, Becht E, Fehlings M, Loh CY, Koo S-L, Teng KWW, Yeong JPS, Nahar R, Zhang T, Kared H, et al. Bystander CD8+ T cells are abundant and phenotypically distinct in human tumour infiltrates. Nature. 2018;557(7706):575–579. doi: 10.1038/s41586-018-0130-2. [DOI] [PubMed] [Google Scholar]
- 40.McKinney EF, Lee JC, Jayne DR, Lyons PA, Smith KG. T-cell exhaustion, co-stimulation and clinical outcome in autoimmunity and infection. Nature. 2015;523:612–616. doi: 10.1038/nature14468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.McGranahan N, Rosenthal R, Hiley CT, Rowan AJ, Watkins TBK, Wilson GA, Birkbak NJ, Veeriah S, Van Loo P, Herrero J, et al. Allele-Specific HLA loss and immune escape in lung cancer evolution. Cell. 2017;171(6):1259–71 e11. doi: 10.1016/j.cell.2017.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Verdegaal EM, de Miranda NF, Visser M, Harryvan T, van Buuren MM, Andersen RS, Hadrup SR, van der Minne CE, Schotte R, Spits H, et al. Neoantigen landscape dynamics during human melanoma–T cell interactions. Nature. 2016;536(7614):91–95. doi: 10.1038/nature18945. [DOI] [PubMed] [Google Scholar]
- 43.Schalper KA, Rodriguez-Ruiz ME, Diez-Valle R, Lopez-Janeiro A, Porciuncula A, Idoate MA, Inoges S, de Andrea C, Lopez-Diaz de Cerio A, Tejada S, et al. Neoadjuvant nivolumab modifies the tumor immune microenvironment in resectable glioblastoma. Nat Med. 2019;25:470–476. doi: 10.1038/s41591-018-0339-5. [DOI] [PubMed] [Google Scholar]
- 44.Riaz N, Havel JJ, Makarov V, Desrichard A, Urba WJ, Sims JS, Hodi FS, Martín-Algarra S, Mandal R, Sharfman WH, et al. Tumor and microenvironment evolution during immunotherapy with nivolumab. Cell. 2017;171(4):934–49 e16. doi: 10.1016/j.cell.2017.09.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Spranger S, Dai D, Horton B, Gajewski TF. Tumor-residing Batf3 dendritic cells are required for effector T cell trafficking and adoptive T cell therapy. Cancer Cell. 2017;31:711–23 e4. doi: 10.1016/j.ccell.2017.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Matsumura S, Wang B, Kawashima N, Braunstein S, Badura M, Cameron TO, Babb JS, Schneider RJ, Formenti SC, Dustin ML, et al. Radiation-induced CXCL16 release by breast cancer cells attracts effector T cells. J Immunol. 2008;181(5):3099–3107. doi: 10.4049/jimmunol.181.5.3099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Thommen DS, Schumacher TN. T cell dysfunction in cancer. Cancer Cell. 2018;33:547–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kordbacheh T, Honeychurch J, Blackhall F, Faivre-Finn C, Illidge T. Radiotherapy and anti-PD-1/PD-L1 combinations in lung cancer: building better translational research platforms. Ann Oncol. 2018;29:301–310. doi: 10.1093/annonc/mdx790. [DOI] [PubMed] [Google Scholar]
- 49.Kelly RJ, Ajani JA, Kuzdzal J, Zander T, Van Cutsem E, Piessen G, Mendez G, Feliciano J, Motoyama S, Lievre A, et al. Adjuvant nivolumab in resected esophageal or gastroesophageal junction cancer. N Engl J Med. 2021;384:1191–1203. doi: 10.1056/NEJMoa2032125. [DOI] [PubMed] [Google Scholar]
- 50.Golden EB, Frances D, Pellicciotta I, Demaria S, Helen Barcellos-Hoff M, Formenti SC. Radiation fosters dose-dependent and chemotherapy-induced immunogenic cell death. Oncoimmunology. 2014;3:e28518. doi: 10.4161/onci.28518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Sharabi AB, Nirschl CJ, Kochel CM, Nirschl TR, Francica BJ, Velarde E, Deweese TL, Drake CG . Stereotactic radiation therapy augments antigen-specific PD-1-mediated antitumor immune responses via cross-presentation of tumor antigen. Cancer Immunol Res. 2015;3:345–355. doi: 10.1158/2326-6066.CIR-14-0196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Burnette B, Weichselbaum RR. Radiation as an immune modulator. Semin Radiat Oncol. 2013;23:273–280. doi: 10.1016/j.semradonc.2013.05.009. [DOI] [PubMed] [Google Scholar]
- 53.Dillon MT, Bergerhoff KF, Pedersen M, Whittock H, Crespo-Rodriguez E, Patin EC, Pearson A, Smith HG, Paget JTE, Patel RR, et al. ATR inhibition potentiates the radiation-induced inflammatory tumor microenvironment. Clin Cancer Res. 2019;25(11):3392–3403. doi: 10.1158/1078-0432.CCR-18-1821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rudqvist NP, Pilones KA, Lhuillier C, Wennerberg E, Sidhom JW, Emerson RO, Robins HS, Schneck J, Formenti SC, Demaria S, et al. Radiotherapy and CTLA-4 blockade shape the TCR repertoire of tumor-infiltrating T cells. Cancer Immunol Res. 2018;6:139–150. doi: 10.1158/2326-6066.CIR-17-0134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Han J, Yu R, Duan J, Li J, Zhao W, Feng G, Bai H, Wang Y, Zhang X, Wan R, et al. Weighting tumor-specific TCR repertoires as a classifier to stratify the immunotherapy delivery in non-small cell lung cancers. Sci Adv. 2021;7. doi: 10.1126/sciadv.abd6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Han J, Duan J, Bai H, Wang Y, Wan R, Wang X, Chen S, Tian Y, Wang D, Fei K, et al. TCR repertoire diversity of peripheral PD-1+CD8+T cells predicts clinical outcomes after immunotherapy in patients with non–small cell lung cancer. Cancer Immunol Res. 2020;8:146–154. doi: 10.1158/2326-6066.CIR-19-0398. [DOI] [PubMed] [Google Scholar]
- 57.Hebeisen M, Allard M, Gannon PO, Schmidt J, Speiser DE, Rufer N. Identifying individual T cell receptors of optimal avidity for tumor antigens. Front Immunol. 2015;6:582. doi: 10.3389/fimmu.2015.00582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang AC, Postow MA, Orlowski RJ, Mick R, Bengsch B, Manne S, Xu W, Harmon S, Giles JR, Wenz B, et al. T-cell invigoration to tumour burden ratio associated with anti-PD-1 response. Nature. 2017;545(7652):60–65. doi: 10.1038/nature22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.