

Abstract
Considering the limited information on the biology and molecular characteristics of disseminated tumor cells (DTCs) in head and neck squamous cell carcinoma (HNSCC), we examined the genomic alterations in DTCs from HNSCCs and their potential clinical relevance. To analyze both the lymphatic and hematogenous routes of tumor cell dissemination, we investigated samples from lymph nodes (LNs) and bone marrow (BM) of 49 patients using immunofluorescence double staining for epithelial cells expressing cytokeratin 18 (KRT18) and/or epithelial cell adhesion molecules (EpCAM, CD326). The identified marker‐positive cells were isolated by micromanipulation followed by single‐cell whole‐genome amplification and metaphase‐based comparative genomic hybridization (mCGH) to determine genome‐wide copy number alterations. The findings were correlated with clinical parameters and follow‐up data. We detected chromosomal aberrations in KRT18‐ and EpCAM‐positive cells from both compartments; BM‐derived cells showed a significantly higher percentage of aberrant genome (PAG) per cell than cells detected in LNs. No significant association was found between DTC data and clinical follow‐up. Genomic profiling of BM‐DTCs revealed genomic alterations typical for HNSCC, suggesting hematogenous dissemination of subclones around the time of surgery. In contrast, DTC data in LNs revealed that several marker‐positive cells were not of malignant origin, indicating the presence of epithelial glandular inclusions in parts of the processed neck LN samples. Therefore, DTC detection of LNs in the neck based only on epithelial markers is not advisable and requires detection of chromosomal instability (CIN), gene mutations, or additional markers, which have yet to be identified. Nevertheless, our investigation paves the way for larger studies to focus on HNSCC BM‐DTCs with high‐resolution methods to gain deeper insights into the biology of hematogenous metastasis in this cancer.
Keywords: bone marrow, disseminated tumor cells, genetic alterations, head and neck squamous cell carcinoma, lymph nodes, minimal residual disease
We isolated disseminated tumor cells from lymph nodes and bone marrow of patients with head and neck squamous cell carcinoma and comprehensively characterized them genetically at the single cell level. While the cells in the lymph nodes are harder to identify, those in the bone marrow showed up as highly aberrant subclones that could possibly become the starting point for distant metastases.

Abbreviations
- AAO‐HNS
American Academy of Otolaryngology ‐Head and Neck Surgery
- BM
bone marrow
- BM‐DTCs
bone marrow‐derived DTCs
- Chr.
chromosome
- CI
confidence interval
- CIN
chromosomal instability
- CIRC
cytokeratin‐positive interstitial reticulum cells
- CNA
copy number alteration
- CTC
circulating tumor cell
- cTNM
clinical classification of tumor (T), nodes (N), and metastases (M)
- DNA
deoxyribonucleic acid
- DPBS
Dulbecco's phosphate‐buffered saline
- DTC
disseminated tumor cell
- EC
esophageal cancer
- EGFR
epidermal growth factor receptor
- EMT
epithelial‐to‐mesenchymal transition
- EpCAM
epithelial cell adhesion molecule
- g
acceleration due to gravity g≈9.81m·s−2
- GO
Gene Ontology
- HNSCC
head and neck squamous cell carcinoma
- HR
hazard ratio
- IF
immunofluorescence
- ISCN
International System for Human Cytogenetic Nomenclature
- IU
international units
- KRT
cytokeratin
- LN
lymph node
- Mbp
mega base pairs
- mCGH
metaphase‐based comparative genomic hybridization
- MRD
minimal residual disease
- neg
negative
- NGS
next‐generation sequencing
- PAG
percentage of aberrant genome
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- pH
potential of hydrogen
- pos
positive
- pTNM
pathological classification of tumor (T), nodes (N), metastases (M)
- TCGA
The Cancer Genome Atlas
- UCSC
University of California Santa Cruz
- UICC
Union Internationale Contre le Cancer
- US
ultrasound
- WGA
whole‐genome amplification
1. Introduction
Annually, more than 550 000 new cases of malignant tumors are detected in the head and neck region which leads to ~ 300 000 deaths [1]. Head and neck squamous cell carcinoma (HNSCC) accounts for 95% of these cases [2, 3]. In the past 40 years, the generally poor prognosis for HNSCC has barely improved. With recent dramatic improvements in surgical techniques including microvascular reconstruction as the standard of care, precise radiotherapy, targeted anti‐EGFR therapy, and immunotherapy especially with pembrolizumab and nivolumab, long‐term cure seems achievable. For example, in early HNSCC stages without lymph node (LN) metastases, the 5‐year survival rate has increased to more than 80% [4]. However, ~ 20–30% of these patients develop locoregional relapse and even distant metastases at low frequency [5]. These rates are higher in locoregionally advanced stages, without distant metastases [6]. Such relapses seem to emerge from micro‐deposits and individual cancer cells that have disseminated before tumor resection and are termed minimal residual disease (MRD). These MRD cells escape routine diagnostics but can be detected using sensitive molecular detection assays in mesenchymal organs, lymph nodes (LNs), and bone marrow (BM). Immunodetection is commonly used to visualize disseminated tumor cells (DTCs) via epithelial antigens in mesenchymal indicator organs [9]. For HNSCC, the AE‐1/AE‐3 pan‐cytokeratin antibody has been the most widely used for DTC detection, as it recognizes a wide range of acidic and basic cytokeratins [6, 12]. EpCAM (epithelial cell adhesion molecule, CD326) has also been described as a reliable marker, especially for DTCs in LNs [7] or CTCs in blood [8], but has not been used in HNSCC.
Unlike other cancer entities (e.g., breast cancer or gastroesophageal cancer), DTCs are less well studied in HNSCC, but sufficient data have been published to conclude that with some margin, ~ 20–30% of patients harbor epithelial DTCs in the LNs and BM [9]. In addition, in contrast to other cancer entities, the prognostic relevance of DTCs in HNSCC is less well established and tends to be insignificant according to most available studies [9].
To date, the biology or molecular characteristics of DTCs in HNSCC remain unclear. Since the advent of single‐cell analysis, genomic profiling has been performed for DTCs in several cancer entities. For example, genomic DTC profiling in esophageal cancer (EC) revealed that some of the genomic alterations between DTCs from BM and LN diverge and that LN‐DTCs display significantly more aberrations than BM‐DTCs. The latter, in conjunction with findings in BM‐DTCs of breast and prostate cancer patients, was interpreted as a sign of early dissemination into the BM, as the chromosomal copy number alteration (CNA) burden of primary tumors steadily increases during their development and promotes further invasion and metastasis [10]. Interestingly, only highly aberrant DTCs in EC confer a poor prognosis [11].
To gain insight into the genomic makeup of DTCs in HNSCC patients, this study aimed to determine the genomic alterations and to test for differences between DTCs derived from BM and LN as well as their impact on overall and disease‐free survival.
2. Materials and methods
2.1. Study patients and sample collection
Patients with a primary diagnosis of HNSCC and without previous or simultaneous tumors in another region were included in the study. After routine staging (as reported earlier [12]) and consultation with a multidisciplinary tumor board, they were intended for primary surgical therapy with excision of all tumor‐affected tissues including a clinical safety margin of 10–15 mm, an elective or therapeutic neck dissection, and a defect reconstruction using primary wound closure, local, pedicled regional or free flaps, and of hard tissues by alloplastic reconstruction or free tissue transfer to our clinic for oral and maxillofacial surgery at the University Hospital Düsseldorf. Macroscopically, tumor‐free LNs and BM aspirates were harvested during the surgery. Written informed consent was obtained, and the Ethics Committee of the Medical Faculty of the Heinrich‐Heine‐University Düsseldorf (#3090) approved the study. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and research committee and with the 1964 Helsinki Declaration and its later amendments or comparable ethical standards. BM aspirates (18 mL) were retrieved after a small skin incision from each hip prior to the first incision for tumor surgery into a syringe containing 2 mL of heparin sodium (25 000 IU/5 mL; Ratiopharm®, Ulm, Germany; yielding 10 000 IU heparin) and were mixed thoroughly for 2 min. If necessary, the aspirates were stored overnight at 4 °C on a roller incubator. Neck LNs were investigated by ultrasound (US), depicted on a map containing the neck levels according to the American Academy of Otolaryngology‐Head and Neck Surgery (AAO‐HNS), and those most likely to be affected by metastasis but still clinically negative were then identified and harvested during neck dissection [13]. One half of a single LN was retained for assessment, and the second half was sent to the Institute of Pathology at the University Hospital Düsseldorf for routine evaluation. The retained parts of the LNs were again split into halves of which one part was placed into 1× DPBS solution (pH 7.4; Gibco, Invitrogen®, Karlsruhe, Germany), and the other part was snap‐frozen in liquid nitrogen.
2.2. Single‐cell preparation
Preparation of single‐cell suspensions from BM aspirates and LN samples was performed using a standardized protocol reported earlier [14, 15]. Ten milliliters of BM obtained from the operating room was suspended in 10 mL Hanks salt solution and centrifuged at 170 g for 10 min. The supernatant was removed, and the pellet was suspended in 20 mL of 1× DPBS buffer (pH 7.4). The cell suspension was then added to 20 mL Ficoll‐Paque (GE Healthcare, Chalfont St. Giles, UK) and centrifuged at 550 g for 30 min. The interphase containing peripheral blood mononuclear cells (PBMCs) was then removed, resuspended twice with 20 mL 1× DPBS buffer (pH 7.4), and centrifuged at 365 g for 10 min. The cell pellet was then resuspended in 2 mL of 1× DPBS buffer (pH 7.4) and counted, and the cell concentration was adjusted to 500 000 cells·mL−1 in 1× DPBS buffer (pH 7.4). Of these, suspensions, 0.5 mL each (corresponding to 250 000 cells), were placed on an adhesive slide (Menzel®, Braunschweig, Germany) containing two fields. After the cell suspension had settled for 30 min, the supernatant was removed, the adhesive slide was dried overnight at room temperature and then stored at −20 °C until the staining procedure.
The LN tissue obtained intraoperatively was freed from the fatty and connective tissue residues and necrotic areas and was cut into ~ 2‐mm3 pieces, placed in 1 mL 1× DPBS buffer (pH 7.4) in a Medicon (50 µL, BD Biosciences, San Jose, CA, USA) and, depending on the size of the fragments, the Medimachine (BD Biosciences) was run several times for 60 s. The resulting cell suspension was washed in 10 mL 1× PBS (pH 7.4), centrifuged for 10 min at 200 g , resuspended in 5 mL 1× PBS, filtered through a 70‐µm cell sieve (Greiner Bio‐One, Frickenhausen, Germany), processed, counted, and applied to the adhesive slides similar to the BM.
2.3. Double immunofluorescence (IF) staining
For double IF staining of 1 × 106 cells per LN and BM sample, a monoclonal mouse antibody against epithelial cell adhesion molecule (EpCAM; Clone: BerEp4, Dako®, Hamburg, Germany) together with a monoclonal rabbit anti‐cytokeratin 18 (CK18; Clone: E431‐1, Abcam®, Cambridge, UK) was applied. IF staining was performed using the protocol described by Driemel et al. [16]. DTCs were suspected to be among the KRT18‐ and/or EpCAM‐positive non‐granulated round cells with large nuclei located within the same focal plane of the adhesion slide as the peripheral blood lymphocytes to rule out any cross‐contamination.
Positively stained cells were isolated using a micromanipulator (Eppendorf®, Hamburg, Germany), and contamination with unstained cells was carefully avoided (Fig. 1). Specificity was ensured by double immunostaining a control cell line (LN1590) [14] and the SCC‐4‐cell line [17]), which was positive for both epithelial markers (Fig. 1). Single cells were whole‐genome amplified (WGA) using adapter‐linker/MseI‐PCR as previously described by Klein et al. [18], commercialized as the Ampli1 WGA Kit (Silicon Biosystems®, Bologna, Italy). To verify the quality of the primary PCR product, a control PCR was performed to detect specific MseI fragments. Two oligonucleotide pairs p53 exon 2/3 (375 bp) and KRT 19 (750 bp) were used for this purpose. Samples were regarded as suitable for mCGH examinations if at least one specific PCR product was successfully amplified.
Fig. 1.
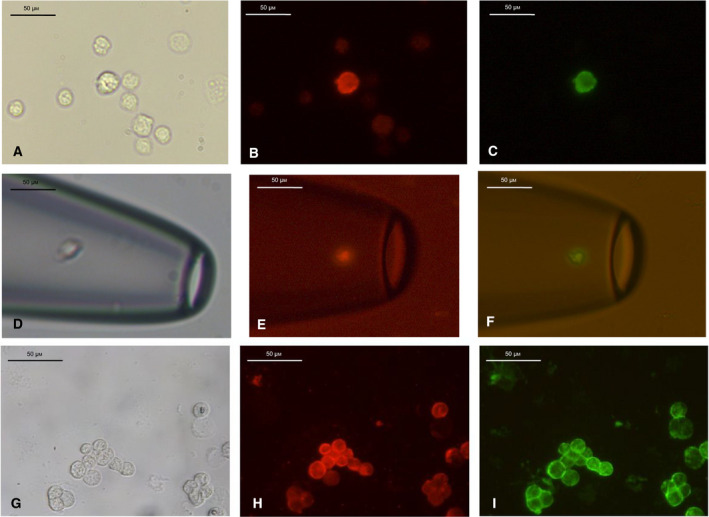
Example of a cytokeratin 18/epithelial cell adhesion molecule (KRT18/EpCAM) double‐positive cell, Pat. #30, LN 4, cell No. T3: (A) brightfield, (B) KRT18 (Cy3, red), and (C) EpCAM (Alexa 488, green). Disseminated tumor cell, detached from the adhesive slide, isolated, and captured in a microhematocrit capillary with the help of a micromanipulator (Eppendorf) at 40× magnification. (D) Bright‐field (from Pat. #49, LN 3, cell No. T3), (E) KRT18 (Cy3, red) and (F) EpCAM (Alexa 488, green). KRT18/EpCAM double immunofluorescence staining of the cell line LN1590 at 40× magnification served as a positive control. (G) Bright‐field, H. KRT18 (Cy3, red), and I. EpCAM (Alexa 488, green). Both epithelial antigens were detected in the control cell line LN1590. Scale bars correspond to 50 µm.
2.4. Metaphase‐based comparative genomic hybridization (mCGH)
mCGH of WGA single‐cell DNA was performed using a standardized protocol as previously reported [11, 19, 20]. The isis software (V 5.5.1; MetaSystems, Altlussheim, Germany) and a fluorescence microscope were used to generate and evaluate mCGH profiles. mCGH karyotypes were labeled according to the International System for Human Cytogenetic Nomenclature (ISCN) [21]. For each sample, the numbers and sites of alterations per cell were determined.
2.5. Statistical analysis
Hierarchical cluster analysis was performed with the r statistical Software [22] and the WECCA package for clustering of called aCGH data using default settings (distance measure = “agree”, linkage = “ward”, weight type = “all equal”) [23]. mCGH karyotypes were joined in a composite karyotype for primary HNSCC tumors and LN metastasis, for which more than one sample was analyzed. For each sample, the number of alterations was counted and the mean number of alterations was calculated for BM‐ and LN‐DTCs. The percentage of aberrant genomes per cell (PAG) was calculated to obtain a global measure of chromosomal instability (CIN) as described previously [11]. To this end, the number and size of alterations were considered, each alteration was translated into mega base pairs (Mbp), and the total length of the altered genome (based on hg38 positions) was divided by the total genome size from the UCSC table (3.088269832 Gb) (https://genome.ucsc.edu/cgi‐bin/hgTables?command=start). Alterations at the Chr. 9 and 1 pter‐1p33 were not included in the calculation because of known non‐tumor‐associated imbalances of the mCGH at these loci [24]. A Wilcoxon–Mann–Whitney U‐test was performed to compare the percentage of aberrant genomes (PAG) and number of aberrations per cell between the two sample groups. Finally, cells with more than 1% PAG were used as DTCs.
To obtain an approximation of potentially involved genes or pathways, Gene Ontology (GO) term enrichment analysis seemed too speculative because of the large number of genes located in the altered chromosomal regions (Fig. S1). Instead, oncogenes and tumor suppressor genes with a known role in HNSCC were identified. For this purpose, the 328 oncogenes and 82 tumor suppressor genes from the respective gene family of the Molecular Signatures Database v7.4 (https://www.gsea‐msigdb.org/gsea/msigdb/) were used [25]. Of the 328 oncogenes, 54 were selected as relevant for HNSCC based on a PubMed search under the search term "HNSCC AND oncogene" from the years 2001 to 2021 or presence among the 574 most frequently mutated genes in HNSCC in the TCGA database (https://portal.gdc.cancer.gov/exploration?filters; TableS1; the selected tumor suppressor genes are listed in Table S2). Chromosome band locations were extracted from the Ensembl BioMart database and were combined with information regarding the genes on chromosome bands from the UCSC genome table browser, using the reference genome GRCh38. Chromosome regions were matched to genes and gene sets using a custom R script in R version 4.0.5, and plots were created using ggplot2 version 3.3.3 and ggpubr version 0.4.0 [22]. Kaplan–Meier analysis was used to evaluate the influence of the primary tumor size (T‐category), LN status (N‐category), and occurrence of DTCs on disease‐related survival in months for a maximum of 5 years. The endpoint was patient death. Log‐rank tests were used to compare survival data, and Cox regression analysis was used to assess the predictive value of existing DTCs in LNs or BM. We also used Cox regression models to estimate the hazard ratios (HRs) and 95% CIs of the association between DTC detection and death adjusted for T and N stages. Statistical analysis was performed using spss statistics version 21 (SPSS Inc., Chicago, IL, USA). Results with a P value < 0.05 were considered significant.
3. Results
3.1. Detection and isolation of DTCs in HNSCC patients
Overall, 49 patients with HNSCC were included in this study. The locations of the respective primary tumors were as follows: lip mucosa (C00.4) three patients, tongue (C02.0, 1) 12 patients, upper and lower alveolus and gingiva (C03.0, 1) 11 patients, floor of the mouth (C04) 10 patients, hard palate (C05.0, 1) two patients, buccal mucosa (C06.0, 2) seven patients, parotid gland (C07.9), oropharynx (C10.2), nasopharynx (C11.1), and hypopharynx (C13.0) one patient each. The clinical follow‐up interval ranged from 0 to 169 months (average, 60 months; median, 38 months). Table 1 summarizes the remaining relevant clinical data. Even if the patients had LN metastases (cN+, pN+), only LNs that showed no evidence of metastasis in the preoperative clinical examinations (CT, ultrasound) or manual palpation, and dissection in the operating room and were actually tumor‐free on histopathology were selected.
Table 1.
Staging and grading of HNSCC patients included in this study; the number of patients did not add up to 100% as BM and LN samples were not available from all patients. However, we related the number of positive patients with the number of patients from whom BM and/or LN samples were available.
| Patients | N = 49 | Patients with marker‐positive cells BM | Patients with marker‐positive cells LN |
|---|---|---|---|
| Sex | |||
| Female (Ø73y/a) | 19 | 4/15 (26.7%) | 7/16 (43.8%) |
| Male (Ø64y/a) | 30 | 4/30 (13.33%) | 11/24 (45.83%) |
| pT‐Status | |||
| pT1 | 7 | 0/7 (0%) | 3/7 (42.86%) |
| pT2 | 25 | 4/21 (19.5%) | 11/21 (52.38%) |
| pT3 | 12 | 4/12 (33.3%) | 4/4 (100%) |
| pT4 | 5 | 0/5 (0%) | 0/4 (0%) |
| pN‐Status | |||
| pN0 | 25 | 4/25 (16%) | 12/20 (60%) |
| pN1‐2 | 24 | 4/20 (20%) | 6/20 (30%) |
| M‐Status | |||
| M0 | 44 | 7/42 (16.67%) | 15/35 (42.86%) |
| M1 | 5 | 1/3 (33.33%) | 3/5 (60%) |
| G‐Status | |||
| G1 | 1 | 0/1 (0%) | 0/1 (0%) |
| G2 | 38 | 6/37 (16.22%) | 16/38 (42.11%) |
| G3 | 10 | 2/7 (28.57%) | 2/10 (20%) |
| R‐Status | |||
| R0 | 31 | 4/28 (14.29%) | 12/15 (80%) |
| R1 | 16 | 3/15 (20%) | 6/13 (46.15%) |
| R2 | 2 | 1/2 (50%) | 0/2 (0%) |
Altogether, 47 LN preparations and 48 BM samples were available from 40 and 45 patients, respectively. In total, 22 of 47 (46.8%) LNs in 18 of 40 (45%) patients displayed marker‐positive cells. We examined 48 BM aspirates from 45 patients and detected marker‐positive cells in 17.8% (n = 8) of the cases. In terms of marker expression, 18 of 47 (38.3%) and 8 of 48 BM samples (16.7%) displayed KRT18pos/EpCAMneg‐DTCs. KRT18pos/EpCAMpos cells were observed in 6.8% (3/47) of the LN samples and 2.1% (1/48) of the BM samples. Only one of the LN samples showed a KRT18neg/EpCAMpos cell (2.1%, 1/47). In total, 179 KRT18‐ and/or EpCAM‐positive cells were detected by IF. Further, 113 KRT18pos/EpCAMneg, 3 KRT18pos/EpCAMpos, and 3 KRT18neg/EpCAMpos cells were found in the LN suspensions, 51 KRT18pos/EpCAMneg, 5 KRT18pos/EpCAMpos, and 5 KRT18neg/EpCAMpos cells were found in the BM samples (see Fig. S2).
3.2. mCGH analyses of single BM‐DTCs and LN‐DTCs
The number of manually micromanipulated cells was limited to three marker‐positive cells per sample. In total, 79 (LN: 52 + BM: 27) cells were isolated via micromanipulation. Of these, 20 cells (LN: 14 + BM: 6) were lost (25.3%; Fig. S2). Therefore, 59 (LN: 38+ BM: 21) cells were successfully isolated as single cells from adhesive slides and transferred to a microcentrifuge tube, accounting for a “pick‐rate” of 59/79 cells (74.7%). After primary amplification, 8/59 (13.6%) of the isolated single cells were positive for one and 32/59 (54.2%) were positive for two specific MseI fragments in the control PCR. Therefore, mCGH could be performed on 40 marker‐positive cells (25 LN‐derived and 15 BM‐derived; Fig. S2). Of these, 38 could be evaluated. In two of the LN‐derived cells, there were no detectable genomic aberrations; therefore, the amplifications and/or deletions in the genomes of 36 cells could be assessed. Amplifications in over 50% of single cells were found on chromosomes 1, 8, 11, 15–17, 19, and 20, those in over 25% of single cells were found on chromosomes 2, 3, 5, 12, 14, and 22, and those in over 10% of cells were found on chromosomes 9 and 10. Deletions were predominantly found on chromosomes 4q, 6q, 9p, 13, and 18 (Table 2). Cumulative mCGH plots of cells from the LN and BM compartments were created and compared (Fig. 2). The mean percentage of aberrations per cell in BM‐DTCs was 15.8%, which was significantly higher than that in LN‐DTCs (5.4%; P = 0.0002, Wilcoxon–Mann–Whitney U‐test). Accordingly, the PAG of BM‐derived cells was significantly higher than that of LN‐derived cells (P = 0.00003, Wilcoxon–Mann–Whitney U‐test, Fig. 2). In a previous immunohistochemical study, we found a large number of KRT5/14pos and/or CD44v6pos cells in LNs from pN0 HNSCC patients that did not show the morphological criteria of tumor cells but those of glandular cells, whereas some had a tubular arrangement or that of reticulum cells; we thus classified cells with no or only very low CIN (PAG ≤ 1%) as non‐neoplastic cells. This affected 9 of 23 marker‐positive LN‐derived cells. All 15 BM‐derived cells, except for one (#25, cell T4: 0.69% PAG) had a PAG of more than 1% [12]. Overall, we were able to detect LN‐DTCs in nine patients (7: pN0, 1: pN2a, 1: pN2b) and BM‐DTCs in five patients (4: cM0, 1: pM1). Thus, there was no correlation with a higher N‐ or M‐status of the patients.
Table 2.
Locations of amplifications and deletions in bone marrow‐ or lymph node‐derived disseminated tumor cells (BM‐ and LN‐DTCs) determined in a relevant number of cases. Locations are linked to the genes and products that may be involved. The GISTIC module identifies regions of the genome that are significantly amplified or deleted across a set of samples (https://www.genepattern.org/modules/docs/GISTIC_2.0).
| BM‐DTC (%) | LN‐DTC (%) | HNSCC GISTIC Q‐values [33] | Gene involved/affected product | |
|---|---|---|---|---|
| Location of amplification | ||||
| 11q13 | 60 | 4.3 | 0 |
CCND1 (Cyclin D1) FGF 3 and FGF 4 FADD |
| 8q24 | 53.3 | 2.3 | 5.3246e‐52 |
MYC (C‐myc) POU5F1B (Oct‐4) |
| 3q26 | 46.7 | 8.7 | 5.5054e‐119 | PIK3CA (PI3K‐AKT signaling cascade) |
| 17q22 | 33.3 | 8.7 | NA | RAD51C |
| 9q34 | 33.3 | 8.7 | 9.73e‐18 | NOTCH 1 |
| 5p15 | 9.1 | 8 | 2.092e‐53 |
PDCD6 (programmed cell death protein 6) CEP72 (Centrosomal Protein 72) |
| Location of deletion | ||||
| 4q35.2 | 40 | 4.3 | 3.5081e‐66 | FAT 1 |
| 18q12qter | 27.2 | 16 | 1.8465e‐65 |
ADNP2 (Activity‐dependent neuroprotector homeobox) PARD6G (Par‐6 Family Cell Polarity Regulator Gamma) |
| 9p21 | 26.7 | 4.3 | 7.4273e‐160 | CDKN2A (cyclin‐dependent kinase inhibitor 2A, p16/INK4A) |
| 13q12q14, 13q21q22 13q31q32 | 26.7 | 4.3 | 7.6449e‐16 |
BRCA2, RB1 (Dysplasia into CIS (carcinoma in situ)) |
Fig. 2.
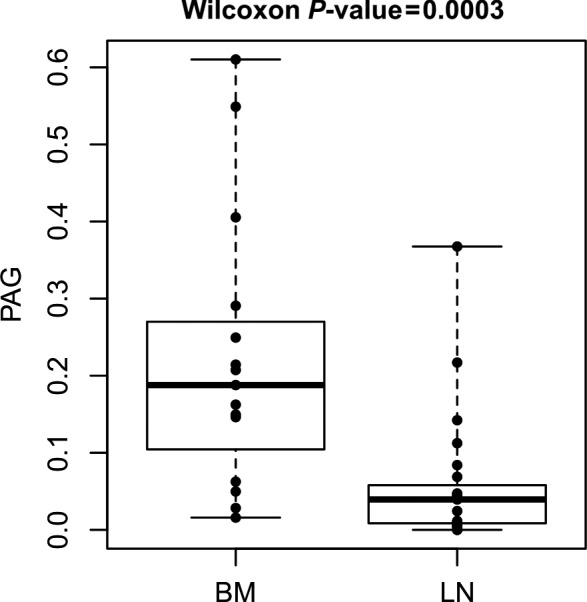
Percentage of aberrant genome per cell (PAG) showing significantly higher values in marker‐positive cells derived from bone marrow (BM) than in those derived from lymph nodes (LN; Wilcoxon–Mann–Whitney U‐test: P = 0.0003). Box plot with median and interquartile box.
Finally, 14 DTCs per compartment were used for further analyses.
To distinguish whether BM‐derived cells showed more aberrations than LN‐derived cells because of their origin (LN vs. BM) or because of their expression status (KRT18pos/EpCAMneg, KRT18pos/EpCAMpos, and KRT18neg/EpCAMpos), the number of aberrations and PAG was correlated with the expression status. However, there were no significant differences in the number of aberrations and PAG between the three groups. Considering the small number of KRT18neg/EpCAMpos cells (only one cell), these cells could not be evaluated. Further, we evaluated whether this difference would also occur in a patient‐specific manner. Marker‐positive cells and DTCs of BM (23 marker‐positive cells/14 DTCs) and LN (15 marker‐positive cells/9 DTCs) of the same five patients (# 22, 25, 48, 49, and 50) could be isolated and evaluated by mCGH, also showing significantly more genomic aberrations in BM‐DTCs than in LN‐DTCs (P = 0.0105, Wilcoxon–Mann–Whitney U‐test; Fig. S3A,B). Hierarchical analyses were carried out using the r software (R Core Team, Vienna, Austria), which determined the clonal relationship of BM‐ and LN‐DTCs. The similarities of genomic changes were shown by their close proximity in the dendrogram (Fig. 3) and indicated that DTCs were grouped roughly according to their origins, that is, LN and BM. DTCs from BM and LN of the same patient also showed a clonal relationship (pointing to an intratumoral homogeneity) and tended to have a stronger relationship as compared to DTCs from different patients (intertumoral heterogeneity). Of the five patients for whom DTCs were available from both compartments, the cells of patient # 22 (UICC II) partially clustered together, those of patients # 25 (UICC II) and 49 (UICC III) were found in different clusters and those of patients # 48 (UICC IV) and 50 (UICC III) in the immediate vicinity (see Fig. 3).
Fig. 3.
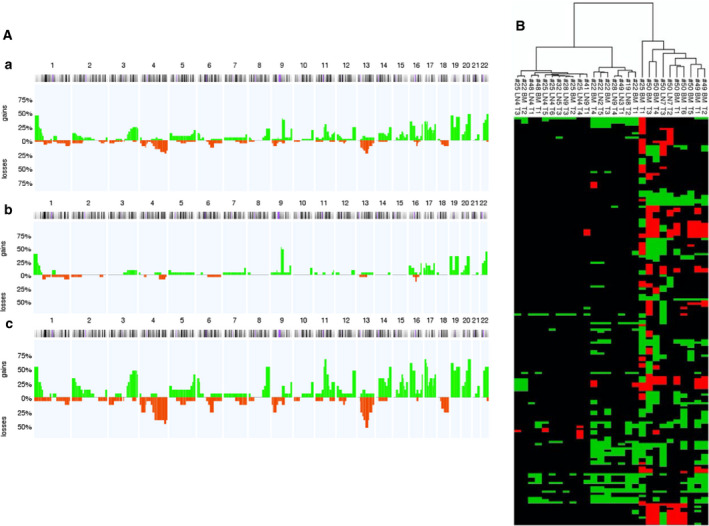
(A) Metaphase‐based comparative genomic hybridization (mCGH) analysis of marker‐positive cells showing genomic gains and losses allocated to chromosomes. Cumulative mCGH plot of (a) all analyzed marker‐positive cells from bone marrow (BM) and lymph node (LN) samples, (b) 23 LN‐derived marker‐positive cells, and (c) 15 BM‐derived marker‐positive cells. Horizontal axis = chromosome number, vertical axis = percentage of genomic aberrations, green = amplifications and red = deletions. (B) Dendrogram of similarity analyses of all disseminated tumor cells (DTCs; percentage of aberrant genome (PAG) > 1%) from bone marrow (BM) and lymph nodes (LNs) using r software. In the dendrogram, the chromosomes are in the ascending order on the y‐axis from top to bottom (no visual numbering). The respective DTC is shown on the x‐axis. The dendrogram is on the top of the x‐axis. Green boxes indicate amplifications and red box indicates deletions. The first sample number (#) corresponds to the patient number; LN = LNs with the corresponding numbering; BM = BM with the corresponding numbering T = tumor cell with the corresponding numbering.
A comparison of affected genes within the chromosomal segments showed that BM‐DTCs have more amplifications in HNSCC‐relevant oncogenes Table 3, P = 0.012; paired Wilcoxon rank‐sum test) and deletions in tumor suppressor genes (Table 4, P = 2.e‐06; paired Wilcoxon rank‐sum test) compared to LN‐DTCs (Fig. 4). For example, the most frequently affected oncogenes BAX and SH3GL1 were amplified in 7/14 (50%) BM‐DTCs, but only in 2/14 (14%) LN‐DTCs. The tumor suppressor genes RB1 and TET2 were lost in 6/14 (42.8%) BM‐DTCs, whereas only one LN‐DTC showed RB1 loss, and no LN‐DTC showed TET2 loss.
Table 3.
List of oncogenes potentially affected due to their position on an amplified chromosome segment per sample (cell). LK stands for LN, and KM stands for BM. The nomenclature of the individual cells is for example: # 19 LK8 T2: patient 19, lymph node 8, tumor cell 2. Each gene represents a point in Fig. 4A.
| sample ID | HNSCC oncogenes on amplified region |
|---|---|
| #19 LK8 T2 | SH3GL1, BAX |
| #22 LK2 T5 | CCND3, ERBB2 |
| #25 LK4 T3 | |
| #25 LK4 T4 | |
| #25 LK4 T5 | |
| #25 LK4 T6 | |
| #28 LK9 T3 | |
| #28 LK9 T4 | |
| #32 LK5 T3 | |
| #41 LK9 T1 | |
| #48 LK4 T1 | SH3GL1, BAX |
| #49 LK3 T1 | |
| #50 LK7 T2 | CREB3L2, MET |
| #50 LK7 T3 | NFIB, JAK2, GNAQ, NOTCH1, ERBB2 |
| #22 KM T1 | NRAS, ERBB2, SH3GL1, BAX |
| #22 KM T2 | |
| #22 KM T3 | SH3GL1, BAX |
| #22 KM T4 | CCND3, FGF3, CCND1, FGF4, SH3GL1, BAX |
| #25 KM T1 | HRAS, FGF3, CCND1, FGF4, SH3GL1 |
| #25 KM T2 | |
| #48 KM T1 | SH3GL1, BAX |
| #49 KM T1 | FGF3, CCND1, FGF4, SH3GL1, BAX |
| #49 KM T2 | SH3GL1, BAX |
| #50 KM T1 | FOS, RAD51B |
| #50 KM T3 | MDM2, FOS, RAD51B |
| #50 KM T4 | KDM5A, CCND2, KRAS, FOS, RAD51B |
| #50 KM T5 | KDM5A, CCND2, KRAS, BAX |
| #50 KM T6 |
Table 4.
List of tumor suppressor genes potentially affected due to their position on a lost chromosome segment per sample (cell). LK stands for LN, and KM stands for BM. The nomenclature of the individual cells is for example: # 19 LK8 T2: patient 19, lymph node 8, tumor cell 2. Each gene represents a point in Fig. 4B.
| Sample ID | Tumor suppressor genes on regions with chromosomal losses |
|---|---|
| #19 LK8 T2 | |
| #22 LK2 T5 | |
| #25 LK4 T3 | CYLD |
| #25 LK4 T4 | CYLD, CDH1 |
| #25 LK4 T5 | CYLD |
| #25 LK4 T6 | |
| #28 LK9 T3 | |
| #28 LK9 T4 | |
| #32 LK5 T3 | |
| #41 LK9 T1 | FBXW7 |
| #48 LK4 T1 | |
| #49 LK3 T1 | |
| #50 LK7 T2 | CDC73, SDHC, FH, PHOX2B, FBXW7, KDM5C, KDM6A |
| #50 LK7 T3 | FH, MSH2, MSH6, TNFAIP3, RB1, BRCA2 |
| #22 KM T1 | |
| #22 KM T2 | |
| #22 KM T3 | |
| #22 KM T4 | RB1 |
| #25 KM T1 | SDHB, CDKN2C, MUTYH, CDC73, SDHC, FH, MSH2, MSH6, PMS1, APC, PIK3R1, GATA3, KLF6, ERCC5, EP300 |
| #25 KM T2 | |
| #48 KM T1 | |
| #49 KM T1 | PHOX2B, FBXW7, TET2, CDKN2A, RB1, BRCA2, SMAD4 |
| #49 KM T2 | FBXW7, TET2, CDKN2A, FANCG, RB1, BRCA2, SMAD4 |
| #50 KM T1 | FBXW7, TET2, RB1, BRCA2, AMER1, KDM5C, KDM6A |
| #50 KM T3 | MLH1, VHL, XPC, FANCD2, SETD2, PHOX2B, FBXW7, TET2, TNFAIP3, CDKN2A, FANCG, FANCF, WT1, DDB2, EXT2, SDHD, ATM, RB1, BRCA2, ERCC5, SMAD4, AMER1, KDM5C, KDM6A |
| #50 KM T4 | FH, VHL, XPC, FANCD2, FBXW7, TET2, WRN, RB1, BRCA2, CYLD, AMER1, KDM5C, KDM6A |
| #50 KM T5 | FBXW7, TET2, APC, CDKN2A |
| #50 KM T6 | SMAD4, KDM5C, KDM6A |
Fig. 4.
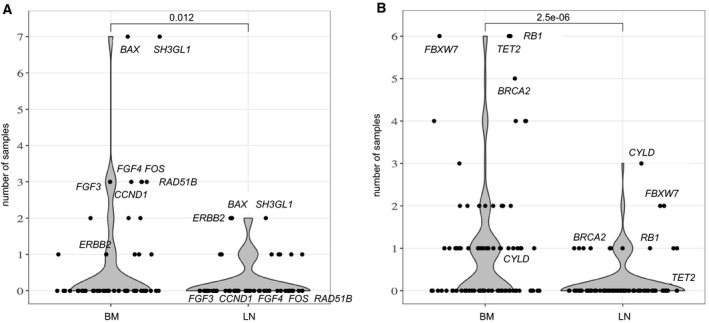
Number of disseminated tumor cells with copy number alterations in chromosomal regions where known oncogenes and tumor suppressor genes for head and neck squamous cell carcinoma (HNSCC) are located. (A) Number of bone marrow‐derived (BM) and lymph node‐derived (LN) cells (y‐axis) with amplifications in HNSCC oncogenes (A) or losses in tumor suppressors (B) (Tables 3 and 4). Each point corresponds to a gene locus and the most frequently amplified, and deleted gene loci are labeled. P‐values are calculated using a paired Wilcoxon rank‐sum test.
3.3. Prognostic significance of DTC detection
Forty‐nine patients were included in the analysis, and disease‐related survival was evaluated in months for a period of at least 10 years. Significant differences were detected using log‐rank tests. Disease‐related survival dropped significantly (P = 0.0054, log‐rank test) with tumor size. The median survival rates decreased with tumor stage. Patients with LN metastasis (N1‐3) showed shorter survival rates than those in patients without LN metastasis (P = 0.012, log‐rank test, Fig. S4). For survival analysis in correlation with marker‐positive cells, patients with at least one KRT18pos and/or EpCAMpos cell in the BM or LNs were compared with patients without the detection of these cells. The median survival of patients with marker‐positive cells was longer (80.4 months ± 16.8) than that of patients without marker‐positive cells (48.4 months ± 9), but this difference was not statistically significant (P = 0.220, log‐rank test). Patients harboring marker‐positive cells in the BM had a shorter disease‐related survival (52.9 months ± 19.9) compared to that in patients without marker‐positive cells in the BM (64.4 months ± 10.3, P = 0.64, log‐rank test) whereas patients with positive cells in LNs seemed to live longer (90.9 months ± 18.5) than patients without marker‐positive cells found in the LNs (45.9 months ± 8.5, P = 0.13, log‐rank test) (Fig. S4). In the multivariable analysis using multiple Cox regressions, the pT and pN stages were also included in addition to the detection of marker‐positive cells. Detection of marker‐positive cells did not influence patient survival with respect to tumor size (P = 0.344, Cox regression), LN status (P = 0.449, Cox regression), or both (P = 0.474, Cox regression). Furthermore, for the maximum number of aberrations in the marker‐positive cells from the BM (P = 0.281, Cox regression), the LNs (P = 0.386, Cox regression), or their combination (P = 0.154, Cox regression), a significant association with 5‐year survival could not be established. Even if only the DTCs are considered (PAG > 1%), there is no significant correlation with the 5‐year survival for the detection of BM‐DTCs (P = 0.508; Cox regression), LN‐DTCs (P = 0.099; Cox regression) or the detection of DTCs from both compartments (P = 0.118, Cox regression).
4. Discussion
In this study, we evaluated the genome‐wide CNAs of DTCs derived from BM and LN samples of HNSCC patients via micromanipulation using a low‐resolution method. We found that half of the DTCs showed alterations typical of primary HNSCC. As observed in previous genomic DTC profiling studies, several DTCs harbored no or very few CNAs. However, this was a typical finding in BM samples from patients with non‐metastatic cancer, whereas DTCs isolated from LN samples showed a genomic profile expected from the tumor entity and stage, with a significantly higher number of alterations compared to BM‐DTCs [19]. As we used the same experimental workflow for our study, including the anti‐KRT18/EpCAM‐immunodetection assay as previously published [11, 16], the observation of very few alterations in our LN‐DTCs was rather surprising. However, to isolate marker‐positive DTCs from LNs, they were mechanically disintegrated to generate single‐cell suspensions. The cell suspensions were then sedimented on adhesive slides for subsequent immunodetection and isolation. We analyzed samples from 49 patients with primary HNSCC; While most DTCs in our study were KRT18pos/EpCAMneg (n = 164), only eight DTCs displayed the KRT18pos/EpCAMpos or KRT18neg/EpCAMpos (n = 8) phenotype. Notably, 94.9% of LN‐DTCs were negative for EpCAM. This is in contrast to previous studies on esophageal cancer [7], in which most LN‐DTCs were EpCAM‐positive. This was interpreted as a sign of an active and proliferating phenotype in these DTCs, as corroborated by in vitro data, and its association with poor survival [11]. There are several potential explanations for the observed low EpCAM expression in HNSCC‐DTCs. Similar to esophageal cancer DTCs, environmental cues from the BM may drive cancer cells in a non‐proliferative quiescent/dormant state, which agrees with an EpCAM‐poor/negative phenotype [9]. Furthermore, the low detection frequency of circulating tumor cells (CTCs) in HNSCC with EpCAM‐based methods also indicates low or absent EpCAM expression in DTCs. Interestingly, the CTC detection frequency in HNSCC can be increased by more than 300% when an EpCAM‐independent method is applied in a side‐by‐side comparison with the EpCAM‐based CellSearch system [26]. Considering that EpCAM positivity in primary HNSCCs is correlated with good prognosis and epithelial differentiation [27], it is conceivable that isolated HNSCC‐DTCs tend to be EpCAM‐low or EpCAM‐negative, for example, triggered by epithelial‐to‐mesenchymal transition (EMT) [28].
However, in the present study on HNSCC, no correlation was observed with clinical follow‐up data beyond a non‐significant trend toward reduced survival in BM‐DTC‐positive patients, which is in contrast with data from other studies on HNSCC [9, 29].
At first glance, the low aberration of epithelial marker‐positive LN cells appears puzzling. A simple explanation could be that these cells are ectopic KRT‐positive salivary gland derivatives, which embryologically develop together with neck LNs and can be inclusions of glandular tissue in LNs [12]. In tissue slides, irregularly stained non‐malignant epithelial marker‐positive cells can be identified to some extent by their morphology [12] and excluded from further analysis. In our study, these cells posed a problem for our approach when working with LN suspensions without the morphologic context of histological tissue sections. Most likely, KRT‐positive cells with very few small alterations (maximum > 0–1% PAG; n = 7) or no alteration at all (n = 2) were normal epithelial cells derived from such inclusions. The very few mCGH alterations are most likely noise, which despite all control experiments [24], is not untypical for this method. Furthermore, mesenchymal cells such as fibroblastic reticulum cells (CK‐positive interstitial reticulum cells, CIRCs) can also express KRT18 in reactive LNs [30] and occur in large numbers in tumor‐draining LNs that are subcapsular in the paracortical regions [31, 32]. However, 11 of the 14 LN‐DTCs displayed typical HNSCC copy number alterations similar to BM‐DTCs [33]. In addition, we saw in the five patients with available material from both compartments that the LN‐DTCs sometimes cluster very closely with the BM‐DTCs of the same patient. Occurring in 53.3% of BM‐DTCs, the most frequently observed alterations were gains on chromosome 8q24 containing the MYC gene coding for the transcription factor c‐myc, which is in accordance with the current literature [34]. C‐myc regulates ~ 15% of human genes and induces gene expression. Aberrant c‐myc expression may result in uncontrolled gene expression, even in protooncogenes [35]. C‐Myc belongs to the mitogenic signaling pathway downstream of EGFR and may contribute to the limited clinical effectiveness of EGFR inhibitors despite frequent EGFR‐overexpression in HNSCC [36]. Aberrations in 8q24 have also been associated with poor prognosis in patients with other malignancies, such as breast cancer [37]. In the present study, gains on chromosome 11q13 were detected in 28.9% of LN‐DTCS and BM‐DTCs. This region includes the CCND1 gene that encodes cyclin D1, which can be found in 30–60% of HNSCC cases [38, 39]. CCND1 gains and CDKN2A loss constitute two of the most common genomic alterations in HNSCC and facilitate cell cycle progression and cell survival [40]. Amplifications on chromosome 3 were observed in 46.7% of BM‐DTCs and only in 8.7% of LN‐DTCs. Speicher et al. [41] observed amplifications primarily on 3q26qter in primary HNSCC tumors. Genes located at 3q26 are involved in the PI3K‐AKT‐signaling pathway and play a role in regulating cell growth, proliferation, and motility. Kozaki et al. [42] identified amplifications in subunit alpha of the PIK3CA‐gene encoding phosphatidylinositol‐4,5‐bisphosphate‐3‐kinase in HNSCC patients. Gains on 3q26 in primary HNSCC tissues are associated with the final stages of invasive carcinoma [43]. Further amplifications were found on chromosome 17q22. This region harbors RAD51C, which plays a significant role in DNA double‐strand repair [44]. Scheckenbach et al. [45] recently showed that amplifications in RAD51C represent a genetic risk profile for HNSCC. Further frequent deletions on BM‐DTCs targeted 9p21 that harbors the locus of CDKN2A (p16/INK4A), encoding p16, which inhibits cyclin‐dependent kinases such as CDK4 and CDK6 (cyclin‐dependent kinase 4 and 6). They phosphorylate retinoblastoma protein (pRb), leading to uncontrolled switching of the cell cycle from the G1‐phase to the S phase and uncontrolled DNA replication [46]. Cyclin D1 activates CDK4 and CDK6. These findings demonstrate the complexity and interaction of amplifications in the chromosome region 11q3 (activating cyclin D1) and deletion of chromosome region 9p21 (inactivation of p16), which can lead to uncontrolled proliferation and tumor progression [47]. In turn, losses at 9p21 are mainly found in the stage of tumor development, which marks the transition from normal mucosa to benign squamous hyperplasia or an alternate precursor lesion [48].
The relevance of comparing the number of DTCs with altered oncogenes and tumor suppressors is limited, as we only observed large chromosomal rearrangements and missed smaller, local amplifications or deletions. Furthermore, our data warrant careful interpretation because of the small number of cases in this study and the low resolution of mCGH used here. Future studies will need to apply modern NGS‐based technologies for genomic profiling; further, additional markers are needed to better identify LN‐DTCs in HNSCC. Clearly, our study demonstrates that this will be a challenging task because immunodetection in LNs commonly relies on the epithelial phenotype, which can be misleading in neck LNs and needs to be carefully considered before applying expensive modern genomic technologies.
5. Conclusions
Taken together, we gained insight into the genomic characteristics of DTCs isolated from BM and LN samples. On the one hand, our study revealed that reliable detection of DTCs in cell suspension is impossible with the established marker‐based detection systems because of irregular glandular tissue enclosed in neck LNs. On the other hand, BM‐DTCs displayed aberrations in the expected range typical for HNSCC, demonstrating early hematogenous dissemination of aberrant subclones to distant sites, which might have the proclivity to form metastases. Thus, future studies with larger patient cohorts should focus on characterizing these BM‐DTCs to identify vulnerabilities enabling better prevention of metastasis, which is an increasing clinical problem in patients with HNSCC after successful multimodal local treatment.
Conflict of interest
The authors declare no conflict of interest.
Author contributions
CS provided the BM and LN samples and clinical follow‐up data; BR, SS, RN, and BB conducted the experiments and collected the data; and LS, JL, MB, and KH analyzed the data. LS, CS, and NHS wrote the manuscript; NRK, WTK, and KH critically revised the paper. NHS and CS initiated the research, supervised experiments, and had the idea of publishing this paper.
Supporting information
Fig. S1. Overview of the number of genes present in the respective chromosomal segment with amplifications (A) or losses (B) of each of the 48 disseminated tumor cells. Each point corresponds to an amplified (A) or deleted (B) chromosomal region containing the indicated genes on the y‐axis. The nomenclature of the individual cells is for example: # 22 KM T1: patient 22, cell from the bone marrow (KM), tumor cell 1. Gray box plots with median, interquartile range (IQR)), and whiskers with a maximum IQR of 1.5.
Fig. S2. Classification and enumeration of the cells according to the marker constellation (cytokeratin 18 (KRT18)pos/epithelial cell adhesion molecule (EpCAM)neg, KRT18pos/EpCAMpos and KRT18neg/EpCAMpos) and stage in the protocol (visual screening of the staining, successful isolation by micromanipulation and successful amplification). Blue bars: lymph node‐derived cells (LN), orange bars: bone marrow‐derived cells (BM).
Fig. S3. A. Cumulative mCGH plots of marker‐positive cells of five patients for whom both, lymph node (LN)‐ and bone marrow (BM)‐samples were available. a. 12 cytokeratin 18/epithelial cell adhesion molecule (KRT18pos/EpCAMneg) cells from LN samples and b. 15 KRT18pos/EpCAMneg and KRT18pos/EpCAMpos cells from the BM samples of the five patients # 22, 25, 48, 49, and 50. Horizontal axis = chromosome number; vertical axis = percentage of genomic aberrations; green = amplification and red = deletion. B. Number of aberrations (y‐axis) in marker‐positive cells of the same five patients (# 22, 25, 48, 49, and 50) with both LN and BM samples (x‐axis) in a dot plot diagram. BM‐derived cells displayed significantly (Mann–Whitney U‐test: p = 0.0105) more genomic aberrations than LN‐derived cells. A black dot stands for a LN‐derived cell, a gray one for a BM‐derived cell. The spread is indicated by the lower and upper crossbars. The mean value by the middle crossbar.
Fig. S4. Disease‐related survival depending on a: tumor size and b: lymph node (LN) metastasis (pN‐status), detected disseminated tumor cells (DTCs) in c: bone marrow (BM), d: DTCs in LNs, and e: DTCs in BM or LNs of head and neck squamous cell carcinoma patients.
Table S1. List of the identified head and neck squamous cell carcinoma (HNSCC)‐relevant 54 oncogenes that were used to search for potentially relevant genes on altered chromosome sections of the DTCs.
Table S2. List of the identified head and neck squamous cell carcinoma (HNSCC)‐relevant 82 tumor suppressor genes that were used to search for potentially relevant genes on altered chromosome sections of the DTCs.
Acknowledgements
The authors wish to thank Mrs. Marianne Hölbling, Mrs. Dorothee Köhler, and Dr. Pia Valeska Kersten‐Thiele for their excellent technical assistance. We thank Dr. Leila Telan M.D. for critically reviewing the manuscript. No specific funding was provided for this work. Open Access funding enabled and organized by Projekt DEAL.
Data accessibility
Data will be made available by the corresponding author upon reasonable request.
References
- 1. Jemal A, Bray F, Center MM, Ferlay J, Ward E & Forman D (2011) Global cancer statistics. CA Cancer J Clin 61, 69–90. [DOI] [PubMed] [Google Scholar]
- 2. Chin D, Boyle GM, Porceddu S, Theile DR, Parsons PG & Coman WB (2006) Head and neck cancer: past, present and future. Expert Rev Anticancer Ther 6, 1111–1118. [DOI] [PubMed] [Google Scholar]
- 3. Gath HJ & Brakenhoff RH (1999) Minimal residual disease in head and neck cancer. Cancer Metastasis Rev 18, 109–126. [DOI] [PubMed] [Google Scholar]
- 4. Cramer JD, Burtness B, Le QT & Ferris RL (2019) The changing therapeutic landscape of head and neck cancer. Nat Rev Clin Oncol 16, 669–683. [DOI] [PubMed] [Google Scholar]
- 5. Kulasinghe A, Perry C, Jovanovic L, Nelson C & Punyadeera C (2015) Circulating tumour cells in metastatic head and neck cancers. Int J Cancer 136, 2515–2523. [DOI] [PubMed] [Google Scholar]
- 6. Szturz P, Nevens D & Vermorken JB (2020) Oligometastatic disease management: finding the sweet spot. Front Oncol 10, 617793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Izbicki JR, Hosch SB, Pichlmeier U, Rehders A, Busch C, Niendorf A, Passlick B, Broelsch CE & Pantel K (1997) Prognostic value of immunohistochemically identifiable tumor cells in lymph nodes of patients with completely resected esophageal cancer. N Engl J Med 337, 1188–1194. [DOI] [PubMed] [Google Scholar]
- 8. Cristofanilli M, Budd GT, Ellis MJ, Stopeck A, Matera J, Miller MC, Reuben JM, Doyle GV, Allard WJ, Terstappen LW et al. (2004) Circulating tumor cells, disease progression, and survival in metastatic breast cancer. N Engl J Med 351, 781–791. [DOI] [PubMed] [Google Scholar]
- 9. Sproll C, Fluegen G & Stoecklein NH (2018) Minimal residual disease in head and neck cancer and esophageal cancer. Adv Exp Med Biol 1100, 55–82. [DOI] [PubMed] [Google Scholar]
- 10. Polzer B & Klein CA (2013) Metastasis awakening: the challenges of targeting minimal residual cancer. Nat Med 19, 274–275. [DOI] [PubMed] [Google Scholar]
- 11. Schumacher S, Bartenhagen C, Hoffmann M, Will D, Fischer JC, Baldus SE, Vay C, Fluegen G, Dizdar L, Vallbohmer D et al. (2017) Disseminated tumour cells with highly aberrant genomes are linked to poor prognosis in operable oesophageal adenocarcinoma. Br J Cancer 117, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Sproll C, Freund AK, Hassel A, Holbling M, Aust V, Storb SH, Handschel J, Teichmann C, Depprich R, Behrens B et al. (2017) Immunohistochemical detection of lymph node‐DTCs in patients with node‐negative HNSCC. Int J Cancer 140, 2112–2124. [DOI] [PubMed] [Google Scholar]
- 13. Robbins KT, Clayman G, Levine PA, Medina J, Sessions R, Shaha A, Som P & Wolf GT (2002) Neck dissection classification update: revisions proposed by the American Head and Neck Society and the American Academy of Otolaryngology‐Head and Neck Surgery. Arch Otolaryngol Head Neck Surg 128, 751–758. [DOI] [PubMed] [Google Scholar]
- 14. Hosch S, Kraus J, Scheunemann P, Izbicki JR, Schneider C, Schumacher U, Witter K, Speicher MR & Pantel K (2000) Malignant potential and cytogenetic characteristics of occult disseminated tumor cells in esophageal cancer. Cancer Res 60, 6836–6840. [PubMed] [Google Scholar]
- 15. Klein CA, Blankenstein TJ, Schmidt‐Kittler O, Petronio M, Polzer B, Stoecklein NH & Riethmuller G (2002) Genetic heterogeneity of single disseminated tumour cells in minimal residual cancer. Lancet 360, 683–689. [DOI] [PubMed] [Google Scholar]
- 16. Driemel C, Kremling H, Schumacher S, Will D, Wolters J, Lindenlauf N, Mack B, Baldus SA, Hoya V, Pietsch JM et al. (2014) Context‐dependent adaption of EpCAM expression in early systemic esophageal cancer. Oncogene 33, 4904–4915. [DOI] [PubMed] [Google Scholar]
- 17. Rheinwald JG & Beckett MA (1981) Tumorigenic keratinocyte lines requiring anchorage and fibroblast support cultured from human squamous cell carcinomas. Cancer Res 41, 1657–1663. [PubMed] [Google Scholar]
- 18. Klein CA, Schmidt‐Kittler O, Schardt JA, Pantel K, Speicher MR & Riethmuller G (1999) Comparative genomic hybridization, loss of heterozygosity, and DNA sequence analysis of single cells. Proc Natl Acad Sci USA 96, 4494–4499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Schmidt‐Kittler O, Ragg T, Daskalakis A, Granzow M, Ahr A, Blankenstein TJ, Kaufmann M, Diebold J, Arnholdt H, Muller P et al. (2003) From latent disseminated cells to overt metastasis: genetic analysis of systemic breast cancer progression. Proc Natl Acad Sci USA 100, 7737–7742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Stoecklein NH, Erbersdobler A, Schmidt‐Kittler O, Diebold J, Schardt JA, Izbicki JR & Klein CA (2002) SCOMP is superior to degenerated oligonucleotide primed‐polymerase chain reaction for global amplification of minute amounts of DNA from microdissected archival tissue samples. Am J Pathol 161, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shaffer LG, Slovak ML & Campbell L (2009) ISCN 2009: An International System for Human Cytogenetic Nomenclature. Karger, Basel, Switzerland. [Google Scholar]
- 22. R Core Team (2021) R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna, Austria. [Google Scholar]
- 23. Van Wieringen WN, Van De Wiel MA & Ylstra B (2008) Weighted clustering of called array CGH data. Biostatistics 9, 484–500. [DOI] [PubMed] [Google Scholar]
- 24. du Manoir S, Schrock E, Bentz M, Speicher MR, Joos S, Ried T, Lichter P & Cremer T (1995) Quantitative analysis of comparative genomic hybridization. Cytometry 19, 27–41. [DOI] [PubMed] [Google Scholar]
- 25. Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES et al. (2005) Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Garrel R, Mazel M, Perriard F, Vinches M, Cayrefourcq L, Guigay J, Digue L, Aubry K, Alfonsi M, Delord JP et al. (2019) Circulating tumor cells as a prognostic factor in recurrent or metastatic head and neck squamous cell carcinoma: the CIRCUTEC prospective study. Clin Chem 65, 1267–1275. [DOI] [PubMed] [Google Scholar]
- 27. Baumeister P, Hollmann A, Kitz J, Afthonidou A, Simon F, Shakhtour J, Mack B, Kranz G, Libl D, Leu M et al. (2018) High expression of EpCAM and Sox2 is a positive prognosticator of clinical outcome for head and neck carcinoma. Sci Rep 8, 14582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gires O, Pan M, Schinke H, Canis M & Baeuerle PA (2020) Expression and function of epithelial cell adhesion molecule EpCAM: where are we after 40 years? Cancer Metastasis Rev 39, 969–987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Grobe A, Blessmann M, Hanken H, Friedrich RE, Schon G, Wikner J, Effenberger KE, Kluwe L, Heiland M, Pantel K et al. (2014) Prognostic relevance of circulating tumor cells in blood and disseminated tumor cells in bone marrow of patients with squamous cell carcinoma of the oral cavity. Clin Cancer Res 20, 425–433. [DOI] [PubMed] [Google Scholar]
- 30. Moll R, Divo M & Langbein L (2008) The human keratins: biology and pathology. Histochem Cell Biol 129, 705–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Gould VE, Bloom KJ, Franke WW, Warren WH & Moll R (1995) Increased numbers of cytokeratin‐positive interstitial reticulum cells (CIRC) in reactive, inflammatory and neoplastic lymphadenopathies: hyperplasia or induced expression? Virchows Arch 425, 617–629. [DOI] [PubMed] [Google Scholar]
- 32. Jacob PM, Nair RA, Nair SP & Jayasudha AV (2016) Cytokeratin‐positive interstitial reticulum cells in the lymph node: a potential pitfall. Indian J Pathol Microbiol 59, 128–129. [DOI] [PubMed] [Google Scholar]
- 33. Yang J, Chen Y, Luo H & Cai H (2020) The landscape of somatic copy number alterations in head and neck squamous cell carcinoma. Front Oncol 10, 321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Patmore HS, Cawkwell L, Stafford ND & Greenman J (2005) Unraveling the chromosomal aberrations of head and neck squamous cell carcinoma: a review. Ann Surg Oncol 12, 831–842. [DOI] [PubMed] [Google Scholar]
- 35. Nilsson JA & Cleveland JL (2003) Myc pathways provoking cell suicide and cancer. Oncogene 22, 9007–9021. [DOI] [PubMed] [Google Scholar]
- 36. Leemans CR, Braakhuis BJ & Brakenhoff RH (2011) The molecular biology of head and neck cancer. Nat Rev Cancer 11, 9–22. [DOI] [PubMed] [Google Scholar]
- 37. Pereira B, Chin SF, Rueda OM, Vollan HK, Provenzano E, Bardwell HA, Pugh M, Jones L, Russell R, Sammut SJ et al. (2016) The somatic mutation profiles of 2,433 breast cancers refines their genomic and transcriptomic landscapes. Nat Commun 7, 11479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Berenson JR, Yang J & Mickel RA (1989) Frequent amplification of the bcl‐1 locus in head and neck squamous cell carcinomas. Oncogene 4, 1111–1116. [PubMed] [Google Scholar]
- 39. Wang X, Pavelic ZP, Li YQ, Wang L, Gleich L, Radack K, Gluckman JL & Stambrook PJ (1995) Amplification and overexpression of the cyclin D1 gene in head and neck squamous cell carcinoma. Clin Mol Pathol 48, M256–M259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Pickering CR, Zhang J, Yoo SY, Bengtsson L, Moorthy S, Neskey DM, Zhao M, Ortega Alves MV, Chang K, Drummond J et al. (2013) Integrative genomic characterization of oral squamous cell carcinoma identifies frequent somatic drivers. Cancer Discov 3, 770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Speicher MR, Howe C, Crotty P, du Manoir S, Costa J & Ward DC (1995) Comparative genomic hybridization detects novel deletions and amplifications in head and neck squamous cell carcinomas. Cancer Res 55, 1010–1013. [PubMed] [Google Scholar]
- 42. Kozaki K, Imoto I, Pimkhaokham A, Hasegawa S, Tsuda H, Omura K & Inazawa J (2006) PIK3CA mutation is an oncogenic aberration at advanced stages of oral squamous cell carcinoma. Cancer Sci 97, 1351–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Argiris A, Karamouzis MV, Raben D & Ferris RL (2008) Head and neck cancer. Lancet 371, 1695–1709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Chen JJ, Silver D, Cantor S, Livingston DM & Scully R (1999) BRCA1, BRCA2, and Rad51 operate in a common DNA damage response pathway. Cancer Res 59, 1752s–1756s. [PubMed] [Google Scholar]
- 45. Scheckenbach K, Baldus SE, Balz V, Freund M, Pakropa P, Sproll C, Schafer KL, Wagenmann M, Schipper J & Hanenberg H (2014) RAD51C–a new human cancer susceptibility gene for sporadic squamous cell carcinoma of the head and neck (HNSCC). Oral Oncol 50, 196–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Leemans CR, Snijders PJF & Brakenhoff RH (2018) The molecular landscape of head and neck cancer. Nat Rev Cancer 18, 269–282. [DOI] [PubMed] [Google Scholar]
- 47. Stransky N, Egloff AM, Tward AD, Kostic AD, Cibulskis K, Sivachenko A, Kryukov GV, Lawrence MS, Sougnez C, McKenna A et al. (2011) The mutational landscape of head and neck squamous cell carcinoma. Science 333, 1157–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Califano J, van der Riet P, Westra W, Nawroz H, Clayman G, Piantadosi S, Corio R, Lee D, Greenberg B, Koch W et al. (1996) Genetic progression model for head and neck cancer: implications for field cancerization. Cancer Res 56, 2488–2492. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Overview of the number of genes present in the respective chromosomal segment with amplifications (A) or losses (B) of each of the 48 disseminated tumor cells. Each point corresponds to an amplified (A) or deleted (B) chromosomal region containing the indicated genes on the y‐axis. The nomenclature of the individual cells is for example: # 22 KM T1: patient 22, cell from the bone marrow (KM), tumor cell 1. Gray box plots with median, interquartile range (IQR)), and whiskers with a maximum IQR of 1.5.
Fig. S2. Classification and enumeration of the cells according to the marker constellation (cytokeratin 18 (KRT18)pos/epithelial cell adhesion molecule (EpCAM)neg, KRT18pos/EpCAMpos and KRT18neg/EpCAMpos) and stage in the protocol (visual screening of the staining, successful isolation by micromanipulation and successful amplification). Blue bars: lymph node‐derived cells (LN), orange bars: bone marrow‐derived cells (BM).
Fig. S3. A. Cumulative mCGH plots of marker‐positive cells of five patients for whom both, lymph node (LN)‐ and bone marrow (BM)‐samples were available. a. 12 cytokeratin 18/epithelial cell adhesion molecule (KRT18pos/EpCAMneg) cells from LN samples and b. 15 KRT18pos/EpCAMneg and KRT18pos/EpCAMpos cells from the BM samples of the five patients # 22, 25, 48, 49, and 50. Horizontal axis = chromosome number; vertical axis = percentage of genomic aberrations; green = amplification and red = deletion. B. Number of aberrations (y‐axis) in marker‐positive cells of the same five patients (# 22, 25, 48, 49, and 50) with both LN and BM samples (x‐axis) in a dot plot diagram. BM‐derived cells displayed significantly (Mann–Whitney U‐test: p = 0.0105) more genomic aberrations than LN‐derived cells. A black dot stands for a LN‐derived cell, a gray one for a BM‐derived cell. The spread is indicated by the lower and upper crossbars. The mean value by the middle crossbar.
Fig. S4. Disease‐related survival depending on a: tumor size and b: lymph node (LN) metastasis (pN‐status), detected disseminated tumor cells (DTCs) in c: bone marrow (BM), d: DTCs in LNs, and e: DTCs in BM or LNs of head and neck squamous cell carcinoma patients.
Table S1. List of the identified head and neck squamous cell carcinoma (HNSCC)‐relevant 54 oncogenes that were used to search for potentially relevant genes on altered chromosome sections of the DTCs.
Table S2. List of the identified head and neck squamous cell carcinoma (HNSCC)‐relevant 82 tumor suppressor genes that were used to search for potentially relevant genes on altered chromosome sections of the DTCs.
Data Availability Statement
Data will be made available by the corresponding author upon reasonable request.