

SUMMARY
Obesity leads to chronic, systemic inflammation and can lead to insulin resistance (IR), β-cell dysfunction and ultimately type 2 diabetes (T2D). This chronic inflammatory state contributes to long-term complications of diabetes, including non-alcoholic fatty liver disease (NAFLD), retinopathy, cardiovascular disease and nephropathy, and may underlie the association of type 2 diabetes with other conditions such as Alzheimer’s disease, polycystic ovarian syndrome, gout, and rheumatoid arthritis. Here we review the current understanding of the mechanisms underlying inflammation in obesity, T2D and related disorders. We discuss how chronic tissue inflammation results in IR, impaired insulin secretion, glucose intolerance and T2D, and review the effect of inflammation on diabetic complications and on the relationship between T2D and other pathologies. In this context we discuss current therapeutic options for the treatment of metabolic disease, advances in the clinic and the potential of immune-modulatory approaches.
INTRODUCTION
Obesity along with genetic predisposition, as observed in some populations such as Pima Indians, Latinos, North Africans and Indians, is the most common cause of insulin resistance (IR). Additionally, IR is a key underlying etiology of type 2 diabetes (T2D). Before the onset of this disease, compensatory hyperinsulinemia is observed, which declines as a result of progressive β-cell dysfunction leading to deterioration of glucose homeostasis and eventually diabetes. In this way, the development of T2D is usually due to a combination of both, IR and β-cell dysfunction. Over time, T2D leads to a number of long-term complications, including microvascular disease (retinopathy, nephropathy, and neuropathy), macrovascular disease (stroke, myocardial infarction and peripheral arterial disease), heart failure and nonalcoholic fatty liver disease (NAFLD). Furthermore, it may contribute to the progression of Alzheimer’s disease, polycystic ovarian syndrome, gout and rheumatoid arthritis among other diseases promoted by inflammation.
Although there is a significant palette of anti-diabetic drugs available, most subjects do not achieve optimal levels of glucose control. Additionally, glucose control per se is not always sufficient to prevent the long-term complications of diabetes. Indeed, only three classes of glucose-lowering drugs have shown prevention of complications; metformin, glucagon-like 1 (GLP-1) analogs and sodium-dependent glucose transporter 2 (SGLT-2) inhibitors. While GLP-1 analogs mitigate cardiovascular diseases and SGLT-2 inhibitors prevent heart failure and nephropathy, none of these drugs has demonstrated efficiency against other complications such as retinopathy, nonalcoholic fatty liver disease (NAFLD) and diabetes-associated inflammatory diseases. Most importantly, except for metformin, progression of diabetes is not delayed by the available drugs. This highlights the importance of improved understanding of the underlying mechanisms of IR and β-cell dysfunction.
In this regard, chronic tissue inflammation has emerged as a key feature of obesity and T2D and is observed in insulin target tissues, such as adipose tissue, liver, muscle and pancreatic islets. These observations have led to the term “immunometabolism”, which incorporates the underlying interplay between immunologic processes and metabolic defects. The recruitment, accumulation and activation of pro-inflammatory macrophages in metabolic tissues is the ultimate driver of this chronic low-grade inflammation. While the macrophage is the major effector cell type, other types of immune cells participate in these inflammatory processes.
This review examines recent advances and underlying mechanisms of inflammation in obesity, T2D and related disorders. We describe how this chronic tissue inflammation includes multi-organ cross-talk causing IR, impaired insulin secretion, glucose intolerance and T2D. We also review the effect of inflammation on diabetic complications and how obesity and diabetes are connected to other inflammatory diseases. In particular, we span the spectrum from basic research to translational findings, including a review of current and potential immune-modulatory therapeutic options for metabolic disease aimed to treat disease progression and complications.
Inflammation in obesity and the pathogenesis of T2D
The potential contribution of chronic tissue inflammation to metabolic disease has been suggested for many years (Cook et al., 2000; Dandona et al., 2004; Donath and Shoelson, 2011). There are a number of key studies published over the past two decades which have provided foundational insights to the immunometabolism field. For example, Feingold et al. discovered that tumor necrosis factor (TNFα) leads to glucose intolerance in rodents (Feingold et al., 1989). The observation that adipose tissue in obesity expresses high levels of TNF-α and that its neutralization improves insulin sensitivity and glucose intolerance were key findings in establishing the association between immune cells and metabolic dysfunction (Hotamisligil et al., 1993; Yuan et al., 2001). Pro-inflammatory cytokines, like TNF-α, can activate a number of intracellular signaling molecules, such as JNK and IKKβ that are critical components of the inflammatory signaling system, leading to impaired insulin action (Arkan et al., 2005; Hirosumi et al., 2002). Activation of IKKβ leads to nuclear translocation of NF-κB which drives increased expression of inflammatory mediators, including chemokines and cytokines (Chen et al., 2012b; Shoelson et al., 2006; Shoelson et al., 2003).
An additional key component of inflammatory activation is a multimeric protein complex termed “inflammasome”, which is activated by cell nutrients, such as glucose and free fatty acids, inducing IL-1β production (Boni-Schnetzler et al., 2009; Maedler et al., 2002). Intracellularly, the inflammasome regulates the activation of caspase-1. Activated caspase-1 cleaves precursor cytokines, such as pro-IL-1β, leading to increased tissue levels of active IL-1β (Thornberry et al., 1992). Accordingly, binding of lipopolysaccharide (LPS) or saturated fatty acids (SFAs) to Toll-Like receptor-4 (TLR-4), and glucose via oxidative stress stimulate NLRP3, mediating activation of caspase-1 (Okla et al., 2018; Paik et al., 2021; Zhou et al., 2010). Although some studies have indicated that IL-1β can blunt insulin action, the most prominent effects of IL-1β are in the islets, where it modulates β-cell mass and function, see below.
Another key finding was the simultaneous discovery by Weisberg et al. and Xu et al. that macrophages accumulate in both mouse and human adipose tissue during the course of obesity (Weisberg et al., 2003; Xu et al., 2003). The pro-inflammatory polarization state of these cells leads to the release of a number of inflammatory cytokines and other factors that contribute to decreased insulin signaling. Unlike the classification of in vitro polarized pro-inflammatory (M1) and anti-inflammatory, alternatively activated (M2) macrophages (Martinez and Gordon, 2014; Orecchioni et al., 2019), these terms do not strictly apply to the in-vivo situation. In recognition of phenotypic, transcriptomic, and polarization differences between in-vitro and in-vivo macrophages, the terms “M1-like” and “M2-like” will be used as general descriptors throughout the rest of this review, when appropriate.
Adipose tissue inflammation
Following the initial reports of increased numbers of adipose tissue macrophages (ATMs) in both mouse and human obesity (Weisberg et al., 2003; Xu et al., 2003), additional studies showed that these cells contribute to the insulin resistant state (Bigornia et al., 2012; Lackey and Olefsky, 2016; Xu et al., 2003). Indeed, prevention of ATM accumulation or pro-inflammatory macrophage signaling, protects obese mice from glucose intolerance and IR (Desai et al., 2017; Dror et al., 2017; Lee and Olefsky, 2021; Takikawa et al., 2016). In addition to macrophages, there are other immune cell types which participate in adipose tissue inflammation during obesity. However, while certain T and B cell subsets play important regulatory roles, it is generally thought that macrophages are the major effector cells leading to decreased insulin signaling (Chawla et al., 2011; McLaughlin et al., 2017). In lean mice and humans, ATMs make up 10% to 15% of the stromovascular cells and largely display an M2-like polarization state (Weisberg et al., 2003; Xu et al., 2003) (Figure 1, lean AT). In obesity, the number of ATMs increases and can comprise up to 40% of all adipose tissue cells (Lumeng et al., 2007). Most of these obesity-induced ATMs are pro-inflammatory (M1-like) (Lumeng et al., 2007; Nguyen et al., 2007) (Figure 1, Obese AT) and similar findings have been shown in human ATMs (Fuchs et al., 2021; Wentworth et al., 2010). The phenotype of these macrophages is not fixed and can be modified by SFAs acting through a TLR-4 mechanism which stimulates the M1-like polarization state (Shi et al., 2006). Omega-3 fatty acids (fish oils) inhibit this inflammatory state by binding to the cell surface receptor GPR120 (Oh et al., 2010).
Figure 1. Chronic tissue inflammation in obesity.

In normal lean conditions, tissue macrophages in the liver, adipose tissue, pancreatic islets, intestine, and muscle generally display an anti-inflammatory M2-like (blue) polarization state. Obesity induces monocyte recruitment into the tissue and resident macrophage proliferation with a switch toward a more pro-inflammatory M1-like state (red), promoting systemic IR and glucose intolerance. Obesity also causes accumulation of lipid associated macrophages (green; Trem2+CD9+ LAMs). In the lean liver, Kupffer cells (KCs) represent ~10% of all cells. Obesity increases the recruitment of monocytes also into the liver, which differentiate into M1-like recruited hepatic macrophages (RHMs). In the progression to non-alcoholic steatohepatitis (NASH), KC genes involved in tissue repair, inflammation, and lipid metabolism, such as Trem2 and CD9, are upregulated.
An interesting study by Wernstedt Asterholm et al. (Wernstedt Asterholm et al., 2014) points out the marked functional differences between acute and chronic inflammation specifically in adipocytes. This paper used genetic manipulations to express specific anti-inflammatory proteins within adipocytes and demonstrated that this affected only adipocytes and not ATMs. They went on to show that inhibiting acute inflammation within adipocytes during high-fat diet (HFD) led to a marked decrease in adipogenesis and angiogenesis. This principle finding supports their main conclusion, which is that “acute” inflammation promotes adipogenesis under the challenge of HFD. Interestingly, this also led to an increase in ATMs and crown-like structures with a deterioration of glucose tolerance and insulin sensitivity. The changes in glucose homeostasis might be explained by the fact that these mice developed a “leaky” gut. The authors found that the anti-inflammatory effects in adipocytes were most marked in mesenteric WAT which, in turn, could affect the gastro-intestinal epithelium leading to increased permeability. They suggest that LPS and perhaps other factors that leak into the circulation could be responsible for worsening glucose tolerance (Wernstedt Asterholm et al., 2014).
ATMs are the most abundant immune cells in adipose tissue and tend to accumulate in crown-like structures, which are observed surrounding enlarged and dying adipocytes (Murano et al., 2008; Strissel et al., 2007). In obesity, ATMs span a wide polarization spectrum (Hill et al., 2018; Jaitin et al., 2019). Single cell-RNA sequencing studies have shown that ATMs classified as “M1” or “M2” by using specific surface markers, are actually quite heterogeneous and comprise several different discrete clusters of cells within each category in mouse and human adipose tissue (Cox et al., 2021; Hildreth et al., 2021; Hill et al., 2018; Jaitin et al., 2019; Kratz et al., 2014; Shaul et al., 2010; Xu et al., 2013). ATMs within obese adipose tissue are generally pro-inflammatory and CD11c is a typical marker used to identify these M1-like ATMs (Wentworth et al., 2010). However, Hill et al. showed that CD9 expression can be used to further define M1-like ATMs (Hill et al., 2018). These CD9+ ATMs exhibit unique locational and morphologic features, expressing high levels of pro-inflammatory cytokines and contain prominent intracellular lipid droplets. CD9+ ATMs are found within crown-like structures and are CD11c positive or negative. There is also a cluster of ATMs in obesity which display the monocyte marker Ly6C (Hill et al., 2018). These cells are derived from recently recruited monocytes and are undergoing macrophage differentiation (Yang et al., 2014; Yona et al., 2013). Over a period of several days, these cells lose Ly6C expression and become mature macrophages found within and outside of the crown-like structures (Hill et al., 2018; Oh et al., 2012). The great majority of these CD9+ or Ly6C+ cells are bone marrow-derived and form distinct clusters depending on whether they express the lipid receptor TREM2. Double positive TREM2+CD9+ ATMs actively participate in lipid metabolism and have been termed lipid-associated macrophages (LAMs) (Jaitin et al., 2019). Interestingly, accumulation of LAMs is functionally dependent on TREM2, since genetic deletion of TREM2 in obese mice promotes obesity, dyslipidemia, and glucose intolerance (Jaitin et al., 2019). These latter findings suggest that TREM2+ LAMs can actually mitigate obesity and the associated metabolic abnormalities. Thus, depending on the relative expression of CD11c, CD9, and TREM2, ATM show distinct phenotypes and further studies of the functional properties of these subsets will be of great interest.
There are several other immune cell types which participate in the overall state of adipose tissue inflammation in obesity. For example, CD8+ T cells are more abundant and can promote monocyte chemotaxis followed by differentiation into ATMs (Nishimura et al., 2009). Pro-inflammatory CD3+CD4+ T helper (Th1) cells are increased in obese adipose tissue and also contribute to the pro-inflammatory state via INF-y production (Winer et al., 2009). Of interest, several studies have examined the content and function of Tregs. These CD3+CD4+FOXP3+ Tregs are the most abundant (40–80%) adipose CD4+ T cell subset in normal adult mice (Bapat et al., 2015; Feuerer et al., 2009; Li et al., 2020). They can modulate the activity of other T cells and, importantly, inhibit monocyte immigration and drive them towards an anti-inflammatory polarization state (Romano et al., 2018). In obese fat, the number of Tregs decreases and this promotes increased ATM-mediated chronic inflammation (Feuerer et al., 2009). Interestingly, the transcriptomic profile of adipose Tregs is quite different from Tregs in other tissues. An important difference is the expression of peroxisome proliferator activated receptor γ (PPAR-γ), by adipose tissue Tregs (Cipolletta et al., 2012). Thiazolidinediones (TZDs) are insulin sensitizing PPAR-γ agonists and exert substantial anti-inflammatory effects via direct action on adipose Tregs. Interestingly, age appears to have independent effects on AT Tregs. Bapat et al. have found that Tregs in mouse visceral fat increase as a function of age, independent of obesity, and that Treg depletion in aged mice promotes insulin sensitivity (Bapat et al., 2015). Other immune cell types, such as mast cells, innate lymphoid cell type 1 (ILC1) cells, neutrophils, eosinophils, NK cells, invariant natural killer T (iNKT) cells, and B lymphocytes also participate in the overall inflammatory tone of adipose tissue largely by modifying ATM recruitment or phenotypes. Their role in the inflammatory process has been recently published and reviewed in detail elsewhere (Boulenouar et al., 2017; Kane and Lynch, 2019; Lee and Dixit, 2020; Lee et al., 2016; Lee and Olefsky, 2021; O’Sullivan et al., 2016; Wang et al., 2019; Wensveen et al., 2015; Zatterale et al., 2020).
Mechanism of adipose tissue macrophage accumulation
Increased monocyte chemotaxis into adipose tissue is the major mechanism leading to ATM accumulation (Kamei et al., 2006; Kanda et al., 2006; Nagareddy et al., 2014; Weisberg et al., 2006). As one example, injection of fluorescently labeled monocytes into obese mice, showed that enhanced monocyte uptake in obesity is induced via chemokines such as CCL2/MCP-1 and leukotriene B4 and galectin-3 (Amano et al., 2014; Li et al., 2016; Li et al., 2015). Another study shows that IL-6-mediated trans-signaling through the soluble form of the IL6 receptor (sIL-6R) interacting with glycoprotein 130 can also produce chemotactic effects for ATM accumulation (Kraakman et al., 2015). There are additional chemokines beyond these that can regulate macrophage recruitment and insulin resistance (Han and Levings, 2013; Xu et al., 2015). The majority of the literature show that genetic deletion or pharmacological inhibition of chemokine receptors reduces monocyte influx into obese adipose tissue (Kanda et al., 2006; Li et al., 2015; Lumeng et al., 2007; Spite et al., 2011; Weisberg et al., 2006). After arriving in the adipose tissue, monocytes can efflux into surrounding lymph nodes or back into the circulation (Auffray et al., 2007; Palframan et al., 2001). In obesity, there is increased adipose tissue expression of Semaphorin 3E and Netrin-1 which inhibit this emigration process, causing macrophage retention and further increasing ATM content (Nakano et al., 2013; Ramkhelawon et al., 2014; Shimizu et al., 2013; Wanschel et al., 2013). Resident ATMs, as well as blood monocyte-derived ATMs, also undergo increased proliferation in the context of obesity, further contributing to the increase in ATMs (Amano et al., 2014; Haase et al., 2014; Zamarron et al., 2017; Zheng et al., 2016). It will be of interest to assess the functional properties and transcriptomic profiles of proliferated vs. non-proliferated macrophages in comparison to ATMs derived from chemotaxis.
Initiating mechanisms for chronic tissue inflammation
Several initiating mechanisms have been proposed to generate chronic tissue inflammation and these mechanisms are not mutually exclusive and could work in a complementary manner. Regardless of the specific mechanism, the ultimate inflammation triggers would have to initiate either proliferation of ATMs or monocyte chemotaxis into the tissue. Increased influx of nutrients is an obvious feature of obesity, and can participate in the production and release of reactive oxygen species (ROS), resulting in oxidative stress (Houstis et al., 2006). Nutrient overload can also promote endoplasmatic reticulum (ER) stress and an unfolded protein response, activating intracellular inflammatory pathways (Hotamisligil and Erbay, 2008). Glucose and free fatty acids activate inflammasomes and toll-like receptors (TLRs), specifically TLR-4, with stimulation of the downstream inflammatory pathways (Boni-Schnetzler et al., 2009; Maedler et al., 2002; Shi et al., 2006; Zhou et al., 2010).
Hypoxia is another activator of inflammation and, particularly in obesity, it is a very early event in the initiation of adipose tissue inflammation (Halberg et al., 2009; Hosogai et al., 2007; Lee et al., 2014). It has been well described in both mouse and human adipose tissue that oxygen (O2) tension decreases in obesity (Halberg et al., 2009; Hosogai et al., 2007; Lawler et al., 2016; Pasarica et al., 2010; Pasarica et al., 2009; Seo et al., 2019; Smith et al., 2019). As adipocytes and adipose tissue expand, angiogenesis can lag behind with impaired capillary density leading to less interstitial perfusion of O2. Importantly, interstitial and intracellular O2 tension may not always go hand in hand, as the intracellular O2 levels represent the balance between O2 supply and demand. This is important since recent studies indicate that obesity leads to an increase in intracellular adipocyte O2 consumption, accounting for about 40% of the decrease in the interstitial O2 tension (Lee et al., 2014). The enhanced adipocyte O2 consumption is secondary to activated uncoupled oxidative phosphorylation, which, in turn, is due to increased free fatty acid stimulation of a mitochondrial protein termed Adenine nucleotide translocase (ANT2) (Lee et al., 2014; Shabalina et al., 2006). ANT2 is highly activated by the abundant SFAs in obese adipocytes, leading to proton leakage from the intermembrane space and to uncoupled mitochondrial respiration and greater O2 consumption. Therefore, stimulation of ANT2 serves to uncouple O2 utilization from ATP production in the mitochondria, lowering intracellular O2 levels. The intra-adipocyte hypoxic state triggers induction of hypoxia inducible factor 1 alpha (HIF-1α), primarily by inhibiting degradation of HIF-1α (Schodel and Ratcliffe, 2019; Semenza, 2019; Seo et al., 2019). HIF-1α within adipocytes can then initiate an inflammatory response by inducing transcription of chemokines (Imtiyaz and Simon, 2010). These chemokines initiate monocyte recruitment, and differentiation into pro-inflammatory M1-like ATMs. Supporting this mechanism, deletion of adipocyte ANT2 prevents increased oxygen consumption, blocks HIF-1α induction and protects from adipose tissue inflammation, glucose intolerance, and IR (Seo et al., 2019). Likewise, adipocyte-specific deletion of HIF-1α prevents the development of adipose tissue inflammation in obese mice mitigating IR and glucose intolerance (Jiang et al., 2011; Sun et al., 2013).
How does inflammation cause IR
The underlying mechanisms by which ATMs cause IR have been widely studied. The general idea is that ATMs produce factors that work in a paracrine or systemic manner and interrupt insulin signaling in target cells. Since these M1-like macrophages produce a variety of chemokines and cytokines, significant attention has been focused on these molecules as causes of decreased insulin sensitivity. Chemokines stimulate chemotaxis of circulating monocytes and other immune cells by providing a concentration gradient between the blood and the interstitial space. While some of these chemokines can leak into the circulation in obesity, this would not generate a necessary concentration gradient to stimulate monocyte chemotaxis. Furthermore, the circulating concentrations of chemokines are much lower than the biologically relevant levels in the interstitial space of obese adipose tissue, so circulating chemokines are a marker of tissue inflammation but are quite unlikely to cause IR.
Therefore, the effects of cytokines to mediate tissue inflammation have been intensively studied. TNF-α, the most well-examined of these cytokines, reduces insulin sensitivity (Hotamisligil et al., 1994). TNF-α promotes inhibitory phosphorylation of insulin receptor substrate (IRS) proteins, and enhances ceramide synthesis, adipocyte lipolysis and inhibits PPARγ expression (Guilherme et al., 2008; Stephens et al., 1997). Normal levels of PPARγ are necessary to maintain insulin sensitivity and the effects of ceramides to inhibit AKT phosphorylation and insulin action are well-known. However, the circulating levels of TNF-α in obese states are below the concentrations which impair insulin signaling (Amar et al., 2007; McGillicuddy et al., 2011; Stephens et al., 1997). Because of this, it is unlikely that TNF-α-“leakage” out of obese adipose tissue into the circulation is responsible for the decreased muscle and liver insulin sensitivity. However, locally-produced high levels of TNF-α in the liver and muscle tissues could cause IR (Lang et al., 1992). Another cytokine released by M1-like ATMs is IL-6. Unlike other cytokines, blood IL-6 circulates at biologically active levels. However, the effects of IL-6 on insulin signaling are unclear, since some studies have reported IL-6-mediated IR while others suggest that IL-6 have insulin-like actions with improved insulin sensitivity (Carey et al., 2006; Franckhauser et al., 2008). Another prominently released cytokine is IL-1β. It is well known that islet macrophage-derived IL-1β impairs insulin secretion contributing to the development of glucose intolerance and T2D (Eguchi et al., 2012; Maedler et al., 2002). Further, IL-1β was also shown to promote IR (Stienstra et al., 2010; Vandanmagsar et al., 2011; Wen et al., 2011; Zhou et al., 2010). In adipose tissue, IL-1β impairs adipocyte insulin signaling (Jager et al., 2007; Lagathu et al., 2006; Stienstra et al., 2010). In humans, IL-1β release from visceral ATMs is enhanced with glycemic deterioration and decreases after gastric bypass surgery (Dalmas et al., 2014). However, clinical evidence for a role of IL-1β in IR remains to be clarified. In a clinical study of IL-1Ra in patients with T2D no changes in insulin sensitivity could be found, although HbA1c decreased (Larsen et al., 2007). However, insulin sensitivity was only assessed in a small patient subgroup in this study. In contrast, in an elegant study in patients with T1D who also have IR caused by obesity, treatment with IL-1Ra improved insulin sensitivity and glycemic control (van Asseldonk et al., 2012). Finally, in a recent study of patients with rheumatoid arthritis and diabetes, IL-1Ra improved insulin sensitivity (Ruscitti et al., 2019). Therefore, it is likely that IL-1β plays also a role in IR.
Galectin-3, a member of the lectin family, is a factor which is exclusively macrophage-derived and achieves biologically effective concentrations in the circulation of obese mice and humans (Li et al., 2016). Studies have shown that galectin-3 can directly cause macrophage chemotaxis and impaired insulin signaling, while myeloid-specific deletion of galectin-3 inhibits HFD-induced glucose intolerance and IR (Li et al., 2016).
ATMs release exosomes which work locally in a paracrine fashion and can also enter the circulation in sufficient concentrations to have distal effects on insulin sensitive tissues. Exosomes are a component of secreted extracellular vesicles (EVs) with a diameter of ~150nm and can be produced by almost all cell types (Pegtel and Gould, 2019). Exosomal cargo consists of a large number of proteins, lipids, micro RNAs (miRNAs), mRNAs, and noncoding RNA species. EV/exosome preparations obtained from obese human adipose tissue express increased levels of several miRNAs that negatively interact with the insulin signaling pathway (Kita et al., 2019). Consistent with this, exosomes prepared from obese adipose tissue cause IR and miR-141–3p has been implicated in this process (Dang et al., 2019; Kranendonk et al., 2014). However, since all of the cell types within adipose tissue release exosomes, studies using exosome preparations derived from whole adipose tissue do not reveal which cell type produces the biologically relevant exosomal particles. Recent studies, specifically focused on macrophages, have shown that ATM-derived exosomes isolated from obese mice directly cause decreased insulin signaling in adipocytes, myocytes, or primary hepatocytes in vitro (Ying et al., 2017) (Figure 2 adipose tissue). These “obese” exosomes contain high levels of miR-155, which represses PPAR-γ expression, thereby contributing to IR. Lean mice treated intravenously with these “obese” ATM exosomes develop glucose intolerance, hyperinsulinemia and IR comparable to obese mice, without changes in body weight. Interestingly, lean chow-fed mice produce ATM exosomes which cause the opposite effects. In other words, “lean” ATM exosomes directly enhance insulin signaling in vitro and, when given to obese mice, markedly improve insulin sensitivity and glucose tolerance. These effects were entirely attributed to the miRNA cargo of the exosomes, and miRNA-690 was identified as the predominant miRNA within M2-like macrophage exosomes causing these beneficial effects (Ying et al., 2021). Indeed, in vivo treatment with a miR-690 mimic recapitulates the insulin sensitizing effects of “lean” ATM exosomes, whereas treatment with an antagomir blocks miR-690-mediated effects. In this way, M2-like macrophage exosomes and miR-690 mimics have emerged as promising insulin sensitizers.
Figure 2. Exosomes as regulators of intercellular and interorgan crosstalk in metabolism.
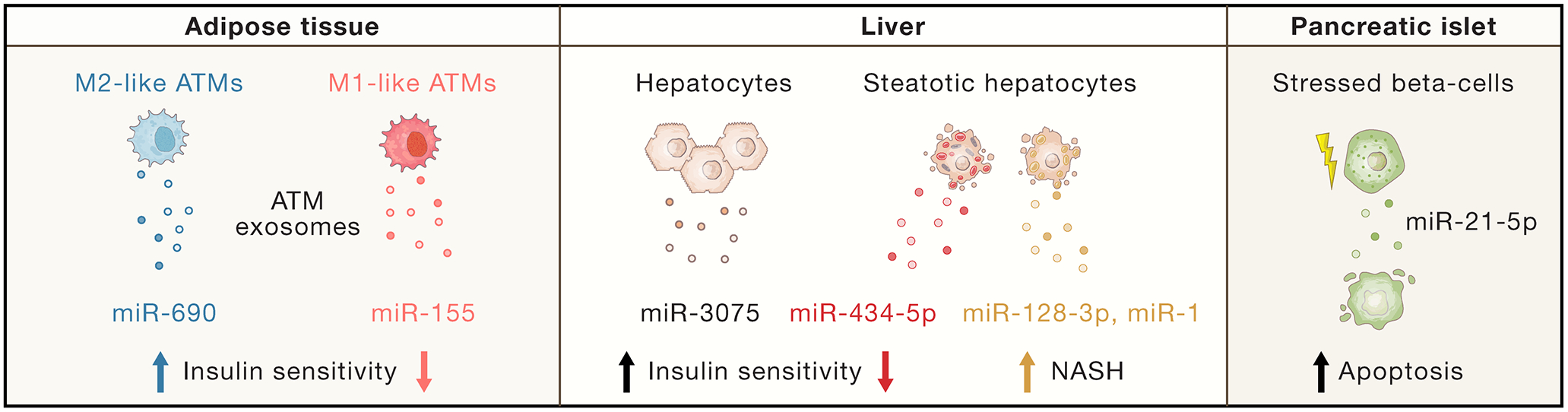
The release of exosomes from adipose tissue macrophages (ATMs), hepatocytes and islet β-cells have systemic metabolic effects. Exosomal miRNA (miR)-690 from M2-like ATMs improves insulin sensitivity, while miR-155 from M1-like ATMs can cause insulin resistance. In early stage HFD, hepatocytes secrete exosomes containing miR-3075, which produces beneficial metabolic effects. In contrast, the hepatocyte exosomes in chronically obese mice contain miR-434-5p, which promotes inflammation and insulin resistance. Exosomes from steatotic hepatocytes also contain pathogenic miRs, such as miR-128 and miR-1, that can induce inflammation or cause hepatic stellate cell activation, promoting NASH. β-cells stressed by pro-inflammatory stimuli release exosomes containing miR-21-5p, which can induce apoptosis in neighboring β-cells.
Hepatic inflammation
Obesity characteristically leads to hepatic steatosis and liver inflammation, commonly termed nonalcoholic fatty liver disease (NAFLD)(Polyzos et al., 2019). This is a very common condition and some estimates suggest one billion NAFLD patients worldwide (Younossi et al., 2019). Hepatocyte lipid accumulation is multi-factorial. In obesity adipocytes become insulin resistant, leading to impaired suppression of lipolysis with increased free fatty acid release. This enhanced influx of adipocyte-derived free fatty acids, as well as dietary fat entering the liver via gastrointestinal-derived chylomicrons are important pro-steatotic mechanisms (Donnelly et al., 2005; Meex and Watt, 2017). In addition, within the hepatocyte, de novo lipogenesis pathways are increased in obesity and this is coupled with decreased fatty acid oxidation (Song et al., 2018). All of these factors drive steatosis and NAFLD.
There are two major categories of liver macrophages: Kupfer cells (KCs) and recruited hepatic macrophages (RHMs) (Morinaga et al., 2015; Seidman et al., 2020). KCs are the resident liver macrophages and are derived from the embryonic yolk sac (Naito et al., 1989; Perdiguero et al., 2015) (Figure 1, lean liver). A second category of hepatic macrophages are bone marrow-derived and arise from circulating monocytes which enter the liver in response to chemokines, primarily CCL2/MCP-1, which can be secreted from steatotic hepatocytes and KCs (Morinaga et al., 2015; Obstfeld et al., 2010). These monocytes differentiate into M1-like pro-inflammatory macrophages, termed RHMs (Morinaga et al., 2015; Seidman et al., 2020). In obesity, KC numbers are relatively unchanged, but there is a large increase in RHMs (Morinaga et al., 2015; Obstfeld et al., 2010) (Figure 1, NAFLD). RHMs also produce chemokines, setting up a feed-forward loop, in which newly arrived RHMs produce chemokines, which attract additional monocytes into the liver (Morinaga et al., 2015). KCs perform scavenger and phagocytic functions, such as removing senescent red blood cells to ensure iron conservation, filtering gut-derived factors like LPS, and participate in tissue homeostasis and repair (Nguyen-Lefebvre and Horuzsko, 2015). These cells comprise up to 10 % of all liver cells and their transcriptomic profile does not fall into typical M1 or M2 categories (Bouwens et al., 1986; Morinaga et al., 2015). In obesity, RHMs are predominantly pro-inflammatory and are responsible for the major component of obesity-mediated hepatic inflammation (Morinaga et al., 2015). KCs and RHMs participate in intrahepatic signaling, since cytokines derived from hepatic macrophages can stimulate hepatocyte de novo lipogenesis and ceramide biosynthesis (Chavez and Summers, 2012; Holland et al., 2011; Sanders and Griffin, 2016). Macrophages are not the only immune cells participating in hepatic inflammation, since obesity also leads to an increase in liver neutrophils and CD8+ T-cells (Ghazarian et al., 2017; Talukdar et al., 2012). Neutrophil elastase can be released and taken up into hepatocytes (Talukdar et al., 2012), promoting intracellular degradation of insulin receptor substrate 2 (IRS-2) and thus potentiating hepatocyte IR.
Some estimates indicate that up to 37 % of obese NAFLD subjects progress to nonalcoholic steatohepatitis (NASH) (Abrams et al., 2004; Gholam et al., 2007; Machado et al., 2006; Ong et al., 2005), which potentially leads to cirrhosis with severely impaired liver function (Loomba et al., 2021). Further, NASH is increasingly recognized as a common complication of obesity (Akshintala et al., 2000). Given the commonality of obesity to NASH and T2D, NASH can also be thought of as a complication of T2D and studies have indicated that 20–40% of subjects with diabetes also have NASH (Younossi et al., 2019). Inflammation and ER stress are important drivers of NAFLD and the progression to NASH (Puri et al., 2008; Rutkowski et al., 2008). Fu et al. reported that hepatic ER stress is caused by significant changes in ER fatty acid and lipid composition, resulting in inhibition of calcium ATPase (SERCA) activity (Fu et al., 2011). In addition, the progression of NASH is partially dependent on pro-inflammatory hepatic macrophages producing TNF (Nakagawa et al., 2014; Todoric et al., 2020).
Recent studies using a variety of monocyte tracking techniques coupled with RNA-sequencing have revealed new insights into the phenotypes of KCs and RHMs in NASH (Seidman et al., 2020). As NASH develops, the original resident yolk sac-derived KCs undergo gradual apoptosis and are replaced by monocyte-derived RHMs which accumulate in NASH livers. These RHMs can enter the sinusoidal niche where they respond to local environmental clues and develop a KC-like transcriptomic profile. These “KC-like” cells in NASH livers are similar, but not identical, to original healthy KCs. Thus, apoptosis and defects in normal KC function represent an important feature of NASH. The exact pathophysiologic consequences of this form of KC dysfunction are currently unknown.
A key element of the NASH liver phenotype is collagen accumulation (Loomba et al., 2021; Loomba et al., 2018). Thus, it is important to understand the various determinants which convert normal stellate cells from the quiescent or inactive state into activated cells which proliferate and elaborate excess collagen that deposits in the extracellular matrix. Transforming growth factor beta (TGF-β) and connective tissue growth factor (CTGF) can be released by hepatocytes or hepatic macrophages and stimulate the stellate cell fibrogenic program (Gressner and Gressner, 2008; Hellerbrand et al., 1999). Indeed, a subpopulation of CD9+Trem2+ bone marrow-derived hepatic macrophages are found in close proximity to collagen fibrils and have been termed scar-associated macrophages (SAMs)(Seidman et al., 2020). These cells may play a role in promoting collagen production by stellate cells (Fallowfield et al., 2007). Hepatocytes under early HFD stress (4 weeks HFD) release exosomes that promotes insulin sensitizing effects via miR-3075 (Ji et al., 2021) (Figure 2 liver). In addition, several reports have shown that exosomes released by steatotic hepatocytes stimulate stellate cell fibrogenesis through their exosomal cargo, including miR-128-3p which suppress PPAR-γ signaling (Povero et al., 2015) and miR-1 via activation of the NF-κB pathway (Jiang et al., 2020). Apart from these intrahepatic signals, it is also likely that extrahepatic signals impinge on the liver to promote the NASH phenotype. For example, obesity is a frequent concomitant in subjects with NASH, and it is well known that obesity leads to a “leaky” gut (see section on intestinal inflammation), allowing LPS and other bacterial byproducts, or perhaps bacteria themselves to enter the bloodstream and target the liver. Normally, KCs filter out gut-derived products, but it is possible that the dysfunctional KCs observed in NASH livers are deficient in this function (Bennett et al., 2020). In this case, these gut-derived products may provide signals to other cells in the liver to promote NASH. Furthermore, adipose tissue is well-known to secrete adipocytokines such as leptin, adiponectin, and galectin-3, all of which influence steatosis and hepatic inflammation.
Muscle inflammation
The skeletal muscle (SM) is a key organ in the development of metabolic disease since it is responsible for the majority (70–80%) of insulin-stimulated glucose disposal (Ferrannini et al., 1988). Therefore, IR in SM is an important determinant of metabolic dysregulation in T2D (DeFronzo and Tripathy, 2009). Proposed mechanisms for SM IR include mitochondrial defects, ectopic fat accumulation, and intrinsic dysregulation of insulin action and these have been reviewed elsewhere (Di Meo et al., 2017; Samuel and Shulman, 2016). In this section, we will focus on the role of muscle inflammation as a possible mediator of IR. This subject is less well-studied compared to fat tissue and liver, but several reports suggest a role of chronic SM inflammation in IR (Boon et al., 2015; Varma et al., 2009). These reports demonstrated increased inflammatory markers, activated pro-inflammatory pathways as well as immune cell accumulation in SM in obesity and diabetes (Fink et al., 2014; Hong et al., 2009; Patsouris et al., 2014). Similar to visceral fat, muscle macrophages are increased in obesity in the intermyocellular/intermuscular adipose tissue (IMAT) between the muscle fibers as well as in perimuscular adipose tissue (PMAT) (Khan et al., 2015). These IMAT and PMAT macrophages exhibit a CD11c+ pro-inflammatory, M1-like phenotype (Fink et al., 2014; Khan et al., 2015; Patsouris et al., 2014). SM Th1, CD4+ and CD8+ T cells are also more abundant while Tregs are decreased, all of which characterize chronic SM inflammation and point to similar mechanisms as observed for visceral fat (Khan et al., 2015). These immune cells contribute to higher levels of pro-inflammatory cytokines, such as TNFα, IL-6, IL-1β and CCL2/MCP-1 in SM. SM can also be a secretory organ. Exercise and muscle contraction leads to increased secretion of “myokines” such as IL-6 with potential beneficial effects on lipid metabolism and inflammation (Eckardt et al., 2014; Pedersen and Febbraio, 2012). In the presence of the high SFA levels in obesity, myocyte TLR-2 and TLR-4 are stimulated (Wei et al., 2008). This causes them to secrete increased levels of chemokines, promoting monocyte migration and SM inflammation. In support of this, deletion of TLR-4 from cultured myocytes protects them from lipid-induced IR both in vitro and in vivo (Radin et al., 2008). IL-6 is probably the most well-studied myocyte cytokine and is a mediator of macrophage infiltration and polarization, as well as muscle repair (Munoz-Canoves et al., 2013). Several studies show that IL-6 has pro-inflammatory effects and can induce IR in obesity (Rehman et al., 2017; Rotter et al., 2003). However, acute IL-6 treatment increases both basal and insulin-stimulated SM glucose uptake and improves systemic glucose homeostasis (Carey et al., 2006; Pedersen and Febbraio, 2012). In vitro, TNFα, or conditioned media from Th1 cells or pro-inflammatory macrophages induce IR in myocyte cultures (de Alvaro et al., 2004). TNFα infusion in healthy humans also results in SM IR (Plomgaard et al., 2005). Interestingly, the anti-inflammatory cytokine IL-10 protects myocytes from the development of IR. Indeed, muscle-specific IL-10 expression attenuates muscle inflammation and IR in vivo, while genetic deletion of IL-10 promotes these metabolic abnormalities (Dagdeviren et al., 2016; Hong et al., 2009).
Intestinal inflammation
Obesity leads to several pro-inflammatory alterations in the gastrointestinal tract. The gut contains an extensive microbiome repertoire, which is altered in human and mouse obesity, a finding termed dysbiosis. Different analyses of microbiota composition have been extensively reported and the reader is referred to several excellent reviews on this subject (Choi et al., 2020; Fan and Pedersen, 2021; Khan et al., 2021).
The first mechanical barrier, preventing entry to the systemic circulation, is the intestinal epithelia, a single layer of cells, joined by tight junctions, as well as a mucosal layer, produced by goblet cells. In obesity, leakage of microbial factors or bacterial products, such as LPS occurs due to higher intestinal permeability (Amar et al., 2011; Cani et al., 2007; Cani et al., 2008; de La Serre et al., 2010). These microbial products and metabolites, such as short-chain FAs can activate G-protein-coupled receptors (GPCRs), or inflammasomes, triggering inflammation and impaired glucose metabolism (Kim et al., 2012; Kimura et al., 2013; Tolhurst et al., 2012). For instance, bile acids improve glucose and insulin sensitivity in obese and diabetic mice through activation of the nuclear farnesoid X receptor (FXR) and Takeda G-protein receptor (TGR5) (Swann et al., 2011; Thomas et al., 2009).
As the gut is the first organ exposed to food and microbial antigens, it contains a robust and complex immune system, located in the subepithelial lamina propria. Several changes in adaptive and intestinal innate immunity have been reported in obesity. IFN-γ and IL-1β-producing immune cells increase barrier permeability in obesity, while IL-22-secreting innate lymphoid cells (ILCs) and Tregs promote production of mucin and anti-microbial immunoglobulin A (IgA) (Al-Sadi and Ma, 2007; Cong et al., 2009; Luck et al., 2015; Wang et al., 2014). Furthermore, depletion of intestinal eosinophils by HFD points towards a protective function of these cells on the gut barrier (Johnson et al., 2015). In addition, HFD induces a pro-inflammatory shift in T cells, characterized by higher INF-γ+ Th1 and CD8+ T cells, along with reduced IL-10+ Tregs and IL17+ Th17 cells (Luck et al., 2015). Obese subjects also display an accumulation of intraepithelial CD8αβ+ T cells (Monteiro-Sepulveda et al., 2015). A lower level of IgA+ antibody-secreting cells and B cells in HFD-fed mice causes a reduction in colonic secretory IgA+ and might promote IR (Luck et al., 2019). Obesity is also associated with reduced IL-22- and IL-17-secreting intestinal group 3 innate lymphoid cells (ILC3s), which are important for barrier integrity (Luck et al., 2015). In contrast, ILC2s seem to have a detrimental effect, since mice deficient in ILC2 are resistant to diet-induced obesity (Sasaki et al., 2019). In humans, intestinal ILC1 and ILC2 numbers negatively correlate with body-mass index (Yudanin et al., 2019). Recent reviews (Febbraio and Karin, 2021; Herman and Birnbaum, 2021) suggest that fructose is a metabolic toxin causing impaired intestinal barrier integrity and endotoxemia. Fructose was thought to be exclusively metabolized in the liver, but it is now known that it can also be metabolized at its site of absorption in the small intestine (Goncalves et al., 2019; Jang et al., 2020; Taylor et al., 2021). Fructose enhances nutrient absorption (Taylor et al., 2021) and induces dyslipidemia and hepatic ER stress, contributing to steatosis and NASH via the gut-liver axis (see section on hepatic inflammation) (Jang et al., 2020; Todoric et al., 2020; Zhao et al., 2020).
HFD leads to a lack of CD103+CD11b+conventional dendritic cells (DCs) (Stagg, 2018) and a reduction in intestinal innate-like T cells, called mucosal-associated invariant T (MAIT) cells (Toubal et al., 2020). These MAIT cells regulate intestinal barrier function and the activated CD44+ phenotype of these cells in obesity promotes both intestinal and adipose tissue inflammation (Toubal et al., 2020; Varelias et al., 2018).
As in other tissues, intestinal macrophages are heterogeneous and are sub-grouped based on phenotypic markers, and functional differences. After circulating blood monocytes enter the gut lumen, they initially display high expression levels of Ly6C and CCR2, but gradually differentiate towards resident mature macrophages, losing Ly6C/CCR2 and gaining CD64/MHII expression (Bain et al., 2014; Bain and Mowat, 2014; Bain et al., 2013; Tamoutounour et al., 2013; Tamoutounour et al., 2012). These “monocyte-like” transition cells are characterized by upregulation of TNFα, IL-1β, or IL-6 and hyper-responsiveness to inflammatory stimuli (Bain et al., 2014; Bain et al., 2013). In obese mice, these gut CCR2+ pro-inflammatory monocytes/macrophages increase (Kawano et al., 2016; Rohm et al., 2017). In contrast, resident intestinal macrophages display anti-inflammatory features, such as IL-10 secretion, phagocytic activity, tissue repair, and anergy towards TLR stimulation (Bain et al., 2013). Mature CX3CR1+ macrophages are reduced in obesity (Hong et al., 2017; Luck et al., 2019). One of the initiating signals priming obesity-induced monocyte recruitment into the gut is mediated by the chemokine gradient generated by epithelial cells, which produce higher levels of CCL2/MCP-1. This was demonstrated in mouse models where macrophages lacked CCR2 and intestinal epithelial cells lacked CCL2/MCP-1, resulting in reduced colonic macrophage accumulation, intestinal permeability, inflammasome activation, and improved glucose metabolism (Kawano et al., 2016). In human obese subjects, CD14hi pro-inflammatory macrophages accumulate along the gastro-intestinal tract, due to higher blood monocyte recruitment (Rohm et al., 2021). Moreover, greater body-mass index or waist-to-height ratio, as well as sedentary lifestyle, positively correlate with intestinal inflammation (Rohm et al., 2021). This correlation was confirmed by colon-specific macrophage depletion improving insulin sensitivity and glucose intolerance in obese mice (Rohm et al., 2018). Bacteria might be a prerequisite for intestinal inflammation and glucose dysregulation since germ-free mice display lower colonic macrophage numbers (Bain et al., 2014) and are partially protected from adiposity and glucose intolerance (Backhed et al., 2004; Backhed et al., 2007).
Islet inflammation and impaired insulin production
Chronic low-grade inflammation in pancreatic islets is a hallmark of T2D, characterized by an increased number of innate immune cells, cytokines, and chemokines (Ehses et al., 2007; Maedler et al., 2002) (Figure 3). Following these initial reports, immunohistological and flow cytometric analysis confirmed and further characterized islet immune subsets in T2D (Butcher et al., 2014; Kamata et al., 2014; Martino et al., 2015; Nordmann et al., 2017; Richardson et al., 2009; Rodriguez-Calvo et al., 2014). Most cells express markers of M1-like macrophages but low numbers of other immune cells such as T and B cells may also be increased in islets in T2D (Boni-Schnetzler and Meier, 2019; Butcher et al., 2014; Ehses et al., 2007; Kamata et al., 2014; Martino et al., 2015; Nordmann et al., 2017; Richardson et al., 2009; Rodriguez-Calvo et al., 2014). Interestingly, increased frequency of immune cells (mostly CD8+ T cells) was also reported in the exocrine pancreas (Rodriguez-Calvo et al., 2014). Rodent models of obesity and T2D confirm these observations. Goto-Kakizaki rats, leptin receptor-deficient db/db mice and HFD-fed wildtype or KKAy mice show increased insulitis (Eguchi et al., 2012; Homo-Delarche et al., 2006). In the setting of HFD, the major mechanisms of increased intra-islet macrophage numbers seems to be proliferation of resident macrophages (Ying et al., 2019).
Figure 3. Islet inflammation.

In a normal state, pro-inflammatory cytokines released from macrophages and the anti-inflammatory IL-1 receptor antagonist (IL-1Ra) released from immune- and β-cells keep the IL-1 system balanced. In pre-diabetes, islet function is increased, the number of immune cells increases and the overall inflammatory balance is tilted towards pro-inflammation. A self-amplified prolonged pro-inflammatory milieu and amyloid polypeptide promotes deterioration of β-cell mass and function, eventually leading to type 2 diabetes.
These immune cells secrete cytokines and the first one described in islets from patients with T2D was IL-1β (Maedler et al., 2002). Islet IL-1β is derived from NLRP3 inflammasomes in intra-islet macrophages and in the setting of T2D mostly glucose, free fatty acids and islet amyloid polypeptide activate this sensor of metabolic stress (Boni-Schnetzler et al., 2009; Maedler et al., 2002; Masters et al., 2010; Wen et al., 2011). Further, insulin was shown to promote IL-1β maturation in a glucose-dependent manner (Dror et al., 2017). Blocking IL-1 signaling in human islet cultures using IL-1Ra prevents the induction of IL-6, IL-8, CXCL1, and TNF-α and the chemokine CCL2/MCP-1, suggesting that these pro-inflammatory mediators are downstream of IL-1 receptor activation (Boni-Schnetzler et al., 2009; Igoillo-Esteve et al., 2010). Thus, it makes sense that inhibition of IL-1 signaling also prevents immune cell infiltration into the islet (Ehses et al., 2009; Sauter et al., 2015). Immune cell-derived cytokines, such as IL-1β activate the highly expressed IL-1R1 on neighboring insulin-producing β-cells. Although, IL-1β acutely potentiates insulin secretion (Dror et al., 2017), prolonged activation of the IL-1 system in β-cells impairs insulin secretion and has a negative impact on β-cell mass, leading to β-cell failure and eventually hyperglycemia. Important for this concept, macrophage depletion, IL-1 and, to a certain extent, TNF-α blockade improve β-cell function and reduce glycemia in rodent models (Eguchi et al., 2012; Ehses et al., 2009; Nordmann et al., 2017; Sauter et al., 2015). This effect was also observed in a human study, in which the IL-1 pathway was blocked with IL-Ra (Larsen et al., 2007, 2009). This led to an increase in the ratio of circulating insulin/proinsulin in patients with T2D, indicative for improved β-cell function.
Islet resident macrophages are in a preactivated state even under normal conditions and IL-1β induces its own expression. Further, β-cell expression of the counterregulatory IL-1R antagonist (IL-1Ra) is reduced in T2D (Maedler et al., 2004), tilting the IL-1/IL-1Ra balance towards a pro-inflammatory state. Supporting this concept, β-cell-specific IL-1Ra ablation reduces β-cell proliferation and mass, and impairs insulin secretion (Boni-Schnetzler et al., 2018). Further, in response to inflammatory stimuli, β-cells secrete exosomes and miR-21-5p was identified to induce apoptosis in surrounding cells (Guay et al., 2015; Javeed et al., 2021; Lakhter et al., 2018) (Figure 2 pancreatic islet).
An important aspect of islet inflammation in the context of T2D is islet amyloidosis. The β-cell product islet amyloid polypeptide is a strong inducer of IL-1β in islets. It triggers the NLRP3 inflammasome to release mature IL-1β (Masters et al., 2010) and has the propensity to aggregate and form amyloid deposits. These are found in most patients with T2D (Clark et al., 1988) and are associated with β-cell apoptosis and a reduction in β-cell mass (Jurgens et al., 2011). Islet amyloid polypeptide secretion is increased in prediabetes because it is functionally coupled to the increased insulin demand. It not only activates immune cells but also recruits additional immune cells to the islet by triggering chemokine release from β-cells. Interestingly, in a human T2D study, only amyloid-positive islets showed increased immune cell infiltration (Kamata et al., 2014). The contribution of islet amyloidosis is generally underappreciated, as rodents do not show amyloid deposits due to differences in the polypeptide sequence. In support of this, transgenic expression of human islet amyloid polypeptide leads to increased chemokine and cytokine expression in a rodent model of long-term HFD feeding (Meier et al., 2014). Importantly, IL-1 blockade is able to mitigate islet amyloid pathogenesis (Westwell-Roper et al., 2015).
Taken together, in T2D islets show increased immune cell infiltration and cytokine release which in turn directly impairs β-cell mass and function. So far, IL-1β is identified as the major cytokine to mediate these pathologies since blocking IL-1 activation prevents deterioration of β-cell mass and function and improves glycemia. A detailed review of islet inflammation comparing human and rodent data was recently published (Boni-Schnetzler and Meier, 2019).
Beneficial physiological effects of inflammation on metabolism
Metabolism and the immune system are usually considered as two distinct entities. However, their functions are physiologically linked, supporting the concept of immunometabolism. Indeed, a key aspect of the immune system is to secure tissue integrity and repair, preserving homeostasis. In this function, immune cells consume a substantial amount of energy that needs to be mobilized and transported to these cells. Beyond the supply of energy, glucose also has a signaling function, activating a response to pathogens (Bantug et al., 2018; Murphy and O’Neill, 2018). If food is not available in sufficient quantities, as in starvation, glucose concentrations may be too low to support an immune response, thus saving energy at the risk of uncontrolled infection. Conversely, the immune system needs to control metabolism to secure its energy supply. Using infection as an example, an important component of the innate immune response to pathogens, is mediated through IL-1β, which is a strong stimulator of adrenocorticotropic hormone and cortisol release (Bataillard et al., 1992). Cortisol will induce IR with subsequent increased glucose concentrations that can fuel immune cells.
This competition or optimization of the distribution of cell nutrients regulated by the immune and endocrine-metabolic systems is apparent under several physiological situations such as during a meal or exercise. Indeed, following food ingestion, the gut flora and glucose stimulate the release of IL-1β by peritoneal macrophages (Dror et al., 2017; Traba et al., 2015). IL-1β then potentiates glucose-induced insulin secretion (Dror et al., 2017; Hajmrle et al., 2016; Maedler et al., 2002). Next, both insulin and IL-1β can regulate glucose disposal, since IL-1β facilitates glucose uptake into immune cells. Thereby, IL-1β alerts and fuels the immune system, possibly to prevent the dissemination of microorganisms contained in the food. Concomitantly, insulin signals the availability of glucose and regulates its distribution to the skeletal muscle, while surplus calories are stored as fat (Dror et al., 2017).
Another example of adaptive cross-talk between metabolism and immunity is the role of IL-6 during exercise. Beyond its function as a cytokine, IL-6 can be viewed as a myokine, since it is produced and released by contracting skeletal muscle during exercise (Pedersen and Febbraio, 2008). Interestingly, IL-6 released during physical activity also increases circulating GLP-1 levels (Adam and Westerterp-Plantenga, 2004; Ellingsgaard et al., 2011; Traub et al., 2017). Indeed, IL-6 stimulates GLP-1 secretion from intestinal L-cells and re-programs pancreatic α-cells to process proglucagon to GLP-1 via upregulation of the prohormone convertase 1/3 (Ellingsgaard et al., 2011; Traub et al., 2017). Thus, during exercise, IL-6-induced GLP-1 leads to metabolic adaptation by reducing gut motility and appetite. This also provides a trophic effect on the islets to prepare them for the insulin secretion needed for the expected following meal. Additionally, exercise-mediated mobilization of visceral fat requires IL-6 signaling (Wedell-Neergaard et al., 2018).
Several other cytokines contribute to the physiological regulation of metabolism. For example, IL-33 promotes insulin secretion via islet-resident group 2 innate lymphoid cells (Dalmas et al., 2017). Another example is IL-22, which contributes to the preservation of the gut mucosal barrier and, thus, mitigates endotoxemia-induced IR (Wang et al., 2014). Furthermore, IL-22 protects β-cells from oxidative and endoplasmic reticulum stress (Hasnain et al., 2014).
Finally, intra- and peri-islet macrophages participate in the adaptive increase of β-cell mass in response to high calorie intake by promoting β-cell proliferation (Ying et al., 2018). M2-like macrophages may promote β-cell replication by up-regulation of SMAD7 (Cao et al., 2014; Xiao et al., 2014).
Chronic effects of inflammation on the complications of diabetes
In contrast to the early beneficial regulatory effects of the immune system on metabolism described above, prolonged and exaggerated metabolic stress can lead to deleterious inflammatory reactions and precipitate autoinflammatory diseases. Although specific to each organ, a similar pattern is seen in many tissues affected by metabolic stress. Thus, increased nutrient exposure leads to cytokine and chemokine release, followed by immune cell recruitment and/or proliferation. In some cases, disproportional expansion of the tissue (e. g. adipocytes) or microvascular occlusions (retina, peripheral neurons), will lead to hypoxia and trigger an immune response. The inflammasome is a critical sensor of all these changes. Indeed, the NLRP3 inflammasome is activated by increased levels of glucose, fatty acids, cholesterol, and uric acid as well as by hypoxia (Duewell et al., 2010; Guo et al., 2015; Martinon et al., 2002; Martinon et al., 2006; Stienstra et al., 2010; Tschop and Thomas, 2006; Zhou et al., 2010). Depending on the specific tissue involved, this will lead to distinct pathologies, but the pattern of metabolic stress-induced inflammation remains similar. As detailed below, this can be observed in cardiovascular disease (CVD), NAFLD, retinopathy, nephropathy, neuropathy, and Alzheimer’s disease, among others.
Inflammation and cardiovascular disease
Almost 200 years ago, Carl von Rokitansky and Rudolf Virchow described atherosclerosis as an inflammatory process (Mayerl et al., 2006). Over the years, this view has been confirmed. Indeed, recent studies show that atherosclerosis is not only a complication of type 2 diabetes as part of the metabolic syndrome, but also shares a similar pattern of metabolic stress-induced inflammation. Myeloid cells play a critical role in CVD inflammation, and an initiating trigger can be the IL-1 system in sensing metabolic changes.
Early in the development of atherosclerosis, monocytes adhere to the endothelial surface and then migrate into the artery wall, where they can become foam cells that accumulate around endothelial lesions (Gerrity, 1981). This is initiated by the NLRP3 inflammasome, which is activated by crystalline cholesterol and SFAs, whereas, replacement of saturated by monounsaturated FAs reduces IL-1β priming and secretion (Bevilacqua et al., 1985; Duewell et al., 2010; Finucane et al., 2015; Libby et al., 1985; Wen et al., 2011). IL-β can then modulate chemotaxis and adhesion of monocytes (Dinarello, 2009). Further proof for the role of IL-1β in CVD has been generated by the CANTOS study (see below).
The role of IL-1α is less clear, although it has been implicated in the development of vascular smooth muscle cell senescence as a contributor to atherosclerosis (Gardner et al., 2015). IL-1α may also promote later stage development of ischaemic events (Cohen et al., 2010). Therefore, the beneficial effects of IL-1 receptor blockade after acute myocardial infarction may be due to antagonism of both IL-1β and IL-1α (Abbate et al., 2008).
The role of IL-6 in metabolism and in the context of CVD is complex (Febbraio, 2014). Since circulating IL-6 is elevated during obesity and correlates with the onset of diabetes (Spranger et al., 2003), it is often thought to have a negative effect on metabolism. This is supported by studies showing that IL-6 induces hepatic IR (Klover et al., 2003a; Klover et al., 2003b; Lagathu et al., 2003). This is in apparent contradiction to the physiological release of IL-6 by skeletal muscle cells in response to muscle contraction (Febbraio and Pedersen, 2002), since IR decreases with exercise (Wojtaszewski et al., 2000). Furthermore, IL-6 is associated with reduced visceral adipose tissue mass and blocking IL-6 leads to dyslipidemia (Wedell-Neergaard et al., 2018). A large-scale CV outcomes trial with an anti-IL-6 antibody is ongoing and should provide further answers on this subject (DOI: 10.1016/S0140-6736(21)00520-1).
In summary, the role of inflammation in CVD with a particular role for macrophages and IL-1β has been demonstrated. It is likely that other cytokines also contribute and ongoing clinical trials will provide further insights.
Inflammation and retinopathy, nephropathy, neuropathy and wound healing
The role of inflammation in the pathogenesis of diabetic retinopathy and macular edema is well established (Joussen et al., 2004; Mesquida et al., 2019). Chronic retinal inflammation is detectable from the early phases to the sight-threatening advanced stages of diabetic retinopathy (Rubsam et al., 2018). Key cytokines important to this process include IL-1β, IL-1 dependent IL-6, IL-8, and TNF-α, as well as monocyte adhesion to the endothelial wall followed by chemotaxis into the subendothelial space (Dong et al., 2013; Joussen et al., 2004; Simo-Servat et al., 2012). Important factors promoting these processes appear to be hypoxia due to microvasculature occlusions and hyperglycemia. The induced cytokines can damage the retinal vasculature, disrupting the blood-retinal barrier with subsequent macular edema and retinal neovascularization. A key mediator of these events is vascular endothelial growth factor (VEGF), which may be directly induced via hypoxia, IL-1β, IL-6, insulin, insulin-like growth factor-1 (IGF-1) and fibroblast growth factor (FGF) (Nagineni et al., 2012; Simo et al., 2014). The importance of cytokines in this process may explain why VEGF is overexpressed in diabetic macular edema despite the absence of overt hypoxia (Simo et al., 2014).
Innate immunity is also involved in the development and progression of diabetic nephropathy (Tang and Yiu, 2020). Mechanistically, TLR4 is overexpressed in kidneys of patients with T2D and correlates positively with HbA1c levels and negatively with renal function (Lin et al., 2012). Furthermore, animal studies support the participation of TLR2 and TLR4 in diabetic kidney disease (Jialal et al., 2014; Lin et al., 2012; Ma et al., 2014a; Ma et al., 2014b). In addition, NLRP3 inflammasome activation of IL-1β plays an important role by sensing metabolic stress in the diabetic kidney (Fang et al., 2013; Shahzad et al., 2016; Tang and Yiu, 2020; Vilaysane et al., 2010).
Although diabetic polyneuropathy has major clinical consequences such as diabetic foot ulcers and amputation, research on its pathogenesis is limited. Traditionally, diabetic neuropathy is classified as non-inflammatory in contrast to neuropathies such as Guillain–Barré syndrome or demyelinating neuropathy (Herder et al., 2019). However, several recent studies have uncovered strong correlations between inflammation and diabetic neuropathy (Bonhof et al., 2019; Herder et al., 2018; Herder et al., 2017; Herder et al., 2019). Nevertheless, mechanistic data are limited.
Wound healing is delayed in T2D and worldwide approximately 20% of patients with T2D develop diabetic wounds. Overactive pro-inflammatory cytokine pathways are a major contributor to this T2D co-morbidity (Geng et al., 2021).
Inflammation and Alzheimer’s disease
The correlation between T2D and the risk of Alzheimer’s disease (AD) is well-known and some have suggested that AD could be considered Type 3 Diabetes (Nguyen et al., 2020). Two major mechanisms have been widely reported in regards to the etiology of Alzheimer’s. One is the aggregation of Aβ monomers into amyloid fibrils or plaques following extracellular proteolytic cleavage of β-amyloid precursor protein (APP) (Braak and Braak, 1991; Kocahan and Dogan, 2017; Murphy and LeVine, 2010; Selkoe, 1994). Secondly, hyperphosphorylation of TAU leads to the formation of extracellular tangles as well as neuronal microtubular dysfunction (Alonso et al., 1996; Alonso et al., 1994; Alonso et al., 2018; Alonso et al., 1997). Chronic neuroinflammation is now recognized as an additional feature of Alzheimer’s and this is due to activation of brain resident macrophages, termed microglia (Akiyama et al., 2000; Kinney et al., 2018; Leng and Edison, 2021). These microglia release a variety of cytokines and other factors that might play a functional role in the pathogenesis of AD and have been referred to as disease associated microglia (Keren-Shaul et al., 2017). Although neuroinflammation with microglia activation may not be the initiating mechanism for AD, they appear to play a role in augmenting the Aβ and TAU etiologies (Dani et al., 2018; Hanisch, 2002; Hayes et al., 2002).
In the normal state, microglia are inactive and function to assess the local microenvironment for harmful molecules and communicate to neuronal cells (Arcuri et al., 2017). The increased presence of Aβ may serve as an activating signal for microglia and these cells then help clear excessive Aβ from the brain. During the chronically inflamed and activated state, the ability of the microglia to clear Aβ declines, which would promote the development of fibrils and plaques (Bolmont et al., 2008; Heneka et al., 2015a; Hickman et al., 2008). Several genetic studies show that a large proportion of the AD risk alleles are associated with genes that are primarily or exclusively expressed in microglia (Hansen et al., 2018). Most of these variants are located in noncoding areas and might function to promote the AD phenotype due to effects on enhancer and or promoter activity (Nott et al., 2019). Many of these genes reside in microglial pathways that involve phagocytosis, autophagy or lysosomal function (Nott et al., 2019). Reduced activity of these pathways can be a cause of increased cytokine production by microglia. There is also evidence that neuroinflammation can cause bone marrow-derived macrophage recruitment into the brain and participate in AD pathology (Theriault et al., 2015). These concepts are supported by the identification of genetic variants in the macrophage receptor TREM2 which cause enhanced risk for AD development (Guerreiro et al., 2013; Jonsson et al., 2013; Jonsson and Stefansson, 2013; Sims et al., 2017). TREM2 is highly expressed in microglia, but the exact mechanisms underlying the enhanced risk for AD remains unclear (Keren-Shaul et al., 2017). Since TREM2 is a receptor for a yet to be identified lipid species, it is possible that activation of TREM2 with a ligand or an activating antibody could represent a potential therapeutic approach.
Similar to macrophages throughout the body, activated microglia also release a number of cytokines which may participate in AD pathogenesis (Del Bo et al., 1995; Hanisch, 2002; Heneka et al., 2015b; Heneka et al., 2013; Smith et al., 2012). IL-1β might be the most important cytokine released in this context and is elevated in the brains of AD patients (Liu and Quan, 2018). IL-1β can cause increased secretion of Aβ precursor APP and promote enhanced processing to Aβ, further increasing fibril and plaque formation (Dash and Moore, 1995). Since Aβ also activates the NLRP3 inflammasome in microglia promoting the release of IL-1β, this could provide a feed-forward mechanism sustaining or increasing AD (Goldgaber et al., 1989; Yates et al., 2000). TNF-α and IL-6 are also released by activated microglia and lead to increased APP production, hyperphosphorylation of TAU and conversion of APP to Aβ by promoting β-secretase production (Chen et al., 2012a; Quintanilla et al., 2004). In addition to microglia, astrocytes can play a role in neuroinflammation, since activated microglial cells release cytokines that promote the development of reactive astrocytes that actively participate in neuronal cell death and damage (Liddelow et al., 2017).
The prevailing view is that Aβ monomers derived from APP proteolysis and hyperphosphorylated TAU protein are the prevailing mechanisms underlying the tangles and amyloid plaques which define AD brain pathology. However, the resident microglia, as well as brain astrocytes, appear to be important in promoting the processes which ultimately lead to AD and all of its clinical sequalae.
Additional obesity and diabetes-related inflammatory disorders
As discussed above, obesity and T2D have strong inflammatory components and chronic activation of the innate immune system may induce or precipitate other conditions driven by inflammation. Some examples are given below.
Polycystic ovary syndrome (PCOS) is a common disease defined by a combination of androgen excess and ovarian dysfunction, leading to hirsutism, anovulation and infertility (Escobar-Morreale, 2018). Interestingly, PCOS is strongly associated with IR and diabetes. A possible explanation for this is that inflammation might participate in PCOS. Indeed, women with PCOS show increased circulating markers of inflammation (Kelly et al., 2001). Furthermore, the ovary expresses IL-1β and IL-1 receptor type 1 with peri-ovulatory changes compatible with the notion that it has effects on ovulation (Kol et al., 1999). This is supported by several studies showing the impact of IL-1β on steroidogenesis (Popovic et al., 2019). Clinical intervention studies to determine whether IL-1β is causative in PCOS are ongoing (https://clinicaltrials.gov/ct2/show/NCT03578497).
Similar to the association of PCOS with T2D in women, obesity and T2D in men are frequently associated with hypogonadism (Rao et al., 2013). The association between low testosterone and T2D might also be partly explained by inflammation. Thus, TNF-α, IL-6 and IL-1β can indirectly inhibit the secretion of testosterone by reducing hypothalamic-pituitary gonadotropin production and can also directly inhibit testicular and androgen production (Jones and Kennedy, 1993). This idea has recently been supported by a clinical study showing that treatment with an IL-1 antagonist increased testosterone levels in obese men with testosterone deficiency (Ebrahimi et al., 2018).
Some inflammatory diseases with an aetiology unrelated to T2D may be exacerbated or precipitated by the concomitant occurrence of both conditions. A striking example is rheumatoid arthritis. Its primary pathology is autoimmune joint inflammation (Klareskog et al., 2009). Although the aetiology of rheumatoid arthritis is unrelated to T2D, the prevalence of both conditions together is increased (Ruscitti et al., 2017a; Ruscitti et al., 2017b). The molecular mechanism could involve cytokines, such as TNF-α and IL-1β, promoting both rheumatoid arthritis and T2D (Dinarello, 2011). Interestingly, monocytes from patients with rheumatoid arthritis and T2D display increased production of IL-1β via NLRP3-inflammasome activation compared to monocytes from patients with only one of the two conditions (Ruscitti et al., 2015). Accordingly, a recent clinical study of IL-1 antagonism in patients with rheumatoid arthritis and T2D showed a remarkable improvement in both glycemia, and joint disease activity [(Ruscitti, 2019), for details, see below]. Autoimmune skin diseases such as psoriasis are also associated with a profound increase in prevalence of T2D. A large longitudinal study found a prevalence of 11.4% of T2D in patients with psoriasis (Holm et al., 2019). Other examples of diseases which may be exacerbated by T2D, are gout, and Crohn’s disease (Donath, 2014).
An unexpected role of inflammation has been reported in postprandial hypoglycemia after bariatric surgery. This condition presents acutely after meal ingestion with disabling neuroglycopenic symptoms. This may result in a long-term increase in food intake and subsequent weight regain. The underlying mechanism is related to the rapid transit of food without gastric delay due to the anatomical changes, leading to an early peak in glycemia. This, in turn, causes an exaggerated induction of the postprandial rise in IL-1β-induced insulin secretion [see (Dror et al., 2017; Maedler et al., 2002) and above], leading to an overshoot of the insulin response, followed by hypoglycemia. This is supported by a proof-of-concept clinical study, which tackled this process at two steps (Hepprich et al., 2020). The initial hyperglycemic peak was reduced via SGLT-2-inhibition while the effect of IL-1β was blocked with an IL-1 receptor antagonist. Both drugs reduced postprandial insulin release and prevented episodes of hypoglycemia.
Therapeutic approaches, targeting inflammation
The first clinical evidence for the beneficial effect of anti-inflammatory treatment in diabetes was published in 1876 (Ebstein, 1876). This empirical study showed that sodium salicylate improved glycemia in patients with diabetes. More than a hundred years later, Shoelson and colleagues showed that this anti-diabetic effect is mediated via inhibition of the NF-κB pathway (Yuan et al., 2001). They went on to validate the effectiveness of salsalate to improve glycemia in proof-of-concept studies (Fleischman et al., 2008; Goldfine et al., 2008; Koska et al., 2009) (Faghihimani et al., 2013; Goldfine et al., 2013a) followed by larger placebo-controlled studies (Goldfine et al., 2013b; Goldfine et al., 2010). The only safety signal was a small increase in low-density lipoprotein cholesterol levels and a reversible rise in urinary albumin. These clinical studies add to the evidence for a role of chronic inflammation in diabetes.
The initial discovery by Hotamisligil and Spiegelman on the effect of TNF-α in IR (Hotamisligil et al., 1993) was followed by a rapid attempt to translate the findings into the clinic. Unfortunately, these studies were not adequately designed, with low sample size and duration of treatment e. g. a single dose in 7–10 patients for a few days. (Bernstein et al., 2006; Dominguez et al., 2005; Ofei et al., 1996; Paquot et al., 2000). Taking into account the number of variable factors that can influence a metabolic response, including genetic background, body weight, food intake, and exercise, it is likely that the design of these studies was insufficient to detect changes, explaining the apparent lack of metabolic effects of TNF-α antagonism. For details on the limitations of these studies, see (Donath, 2014). However, indirect evidence supports the clinical potential of TNF-α antagonists in T2D. Thus, these drugs improve glycemia in obese subjects without diabetes as well as in patients with psoriasis, rheumatoid arthritis and Crohn’s disease (Gonzalez-Gay et al., 2006; Huvers et al., 2007; Kiortsis et al., 2005; Marra et al., 2007; Stanley et al., 2011; Timper et al., 2013; Yazdani-Biuki et al., 2006; Yazdani-Biuki et al., 2004). Furthermore, TNF-α antagonism reduces the incidence of T2D in patients with rheumatoid arthritis or psoriasis (Antohe et al., 2012; Solomon et al., 2011). However, it is unclear whether the beneficial effect on glycemia was direct or due to an improvement in the underlying disease with subsequent enhanced exercise capacity. On the other hand, a recent study directly compared TNF-α versus IL-1 blockade in patients with rheumatoid arthritis and T2D. TNF-α antagonism failed to decrease glycemia (Ruscitti, 2019). Therefore, a well-designed diabetes-focused study of TNF antagonism, ideally with clinically relevant outcomes, such as CVD, is warranted.
The most advanced clinical translation of inflammation and T2D has been achieved with IL-1 antagonism. An initial trial in patients with T2D showed that anakinra (a recombinant human IL-1Ra) improved β-cell secretory function and reduced glycemia (Larsen et al., 2007). The ability of IL-1 inhibitors to improve insulin secretion was confirmed in several follow up studies using anakinra (van Asseldonk et al., 2011; van Poppel et al., 2014). Similarly, treatment of T2D subjects with neutralizing anti-IL-1β antibodies demonstrated beneficial effects, although the magnitude varied depending on baseline HbA1c levels and sample size (Cavelti-Weder et al., 2012; Rissanen et al., 2012; Ruscitti, 2019; Sloan-Lancaster et al., 2013).
In a large CV outcome study (CANTOS), treatment with an anti-IL-1β antibody prevented CV events (Ridker et al., 2017a). A sub-analysis showed that blocking IL-1β also significantly decreased HbA1c during the first 6–9 months of treatment, with waning effect towards the end of the study (Everett et al., 2018b). This attenuation of the glucose lowering effect may be due to the design of the study, which allowed for lifestyle interventions and adaptations of standard anti-diabetic drugs. Supporting this assumption, in the non-diabetic patients, anti-IL-1β treatment decreased HbA1c for the duration of the study. Furthermore, IL-1 antagonism prevented new onset of diabetes for about 4 years. After 4 years, the number of patients followed decreased by 90% and the effect of anti-IL-1β could no longer be detected. The observed prevention of diabetes suggests an ongoing islet inflammatory process in patients with prediabetes. Most importantly, this outcome study showed that in a patient population with T2D, CV complications can be prevented (Everett et al., 2018b). An additional beneficial effect of IL-1β inhibition was prevention of heart failure, particularly in patients with higher body mass index and diabetes (Everett et al., 2018a), confirming previous clinical proof- of-concept studies (Abbate et al., 2015; Abbate et al., 2010; Van Tassell et al., 2017; Van Tassell et al., 2018). Although the CANTOS study showed the overall safety of prolonged IL-1β inhibition, in patients with severe infections necessitating inpatient treatment, IL-1β antagonism was associated with a higher incidence of fatal infections, which warrants caution in patients at risk.
Following these studies, a meta-analysis involving 2921 cases with 2TD treated with IL-1 blocking therapy, demonstrated a highly significant reduction in HbA1c (P<0.00001) (Kataria et al., 2019).
Recently, a new designer ligand for the gp130 receptor with CNTF-like properties that signals in a IL-6-receptor-dependent manner was shown to improve glycemia and prevent body weight gain in high-fat-diet-fed mice (Findeisen et al., 2019) showing that designer approaches targeting specific pathways but avoiding activation of others have great potential for future human studies.
A list of anti-inflammatory clinical trials targeting T2DM and its associated pathologies is given in Table 1.
Table 1.
Clinical studies of anti-inflammatory treatments in patients with type 2 diabetes and associated complications
CONCLUDING REMARKS
At this point, the only antidiabetic drugs demonstrating improvement in macrovascular disease are GLP-1 analogs (Marso et al., 2016) while SGLT-2 inhibitors are the only drug class demonstrating protective effects against heart failure and nephropathy (Zinman et al., 2015). This explains why these two classes of drugs are currently often used as first line antidiabetic treatment as opposed to insulin, sulfonylureas and DPP-4 inhibitors which only lower glucose. An anti-inflammatory drug may provide additional benefits beyond improving glycemia. Indeed, as detailed above, IL-1β antagonism mitigated β-cell dysfunction and improved glycemia, CV complications and heart failure (Abbate et al., 2015; Abbate et al., 2010; Cavelti-Weder et al., 2012; Donath, 2014; Everett et al., 2018a; Larsen et al., 2007; Ridker et al., 2017; Ridker et al., 2018b; Rissanen et al., 2012; Ruscitti, 2019; Sloan-Lancaster et al., 2013; Van Tassell et al., 2017; Van Tassell et al., 2018). Future studies are needed to determine whether anti-inflammatory treatment may counteract other complications of diabetes, including NASH, nephropathy, fatigue, polyneuropathy, retinopathy and macular oedema (Cavelti-Weder et al., 2011; Febbraio et al., 2018; Herder et al., 2013; Herder et al., 2018; Herder et al., 2017; Herder et al., 2009; Lehrskov et al., 2018; Mesquida et al., 2014; Schlesinger et al., 2018; Stahel et al., 2015; Sun and Karin, 2012; Tesch, 2017). Beyond IL-1β antagonism, other immunomodulatory drugs, either alone or in combination, may be more effective. However, the choice of an anti-inflammatory drug should be based on a pathophysiologic mechanistic understanding and not on a nonspecific effect, as with methotrexate, which did not decrease IL-1β and failed to prevent CV complications in patients with metabolic syndrome (Ridker et al., 2018a). An overview of validated and potential future pro-inflammatory targets in human T2DM and associated diseases is given in Figure 4.
Figure 4. Immunomodulatory targets for the treatment of diabetes and its complications.

Targeting the IL-1 pathway in patients with type 2 diabetes was shown to have beneficial effects on insulin sensitivity and secretion as well as atherosclerosis, heart failure and retinopathy. Future clinical studies building on preclinical work blocking TNF-α, the IL-1 system or other pro-inflammatory mediators may uncover novel routes to counteract type 2 diabetes and its complications.
The association of diabetes with other inflammatory diseases such as gout or rheumatoid arthritis allows treatment of two conditions with one therapeutic agent. This has been demonstrated by Ruscitti and Giacomelli in patients with rheumatoid arthritis and T2D (Ruscitti, 2019). Treatment of these patients with anakinra was highly effective, achieving a decrease in HbA1c by > 1% after 6 months, while simultaneously reducing rheumatoid disease activity. Therefore, anti-inflammatory treatment can improve both conditions and is already doable in countries where IL-1 antagonism is approved for rheumatoid arthritis or gout.
Several companies are developing inhibitors of the NLRP3-IL-1β pathway, including small molecules that can be taken orally, avoiding injection site reactions of IL-1Ra. The short half-life of these oral agents would also help in the management of severe infections. Hopefully, in the near future, these inhibitors will be tested in patients with T2D and associated complications. An encouraging phase 1b study using an oral NLRP3 inhibitor, dapansutrile, showed signs of improved heart failure and significantly decreased blood glucose levels (Wohlford et al., 2020).
ACKNOWLEGMENTS
Funding supporting the work in this review was from the Swiss National Science Foundation (SNF) Early Postdoc.Mobility grant (P2BSP3_200177 to T.R.) and by the U.S. National Institute of Diabetes and Digestive and Kidney Diseases (P30 DK063491 and DK101395 to J.M.O.).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
M.Y.D. is listed as the inventor on a patent filed in 2003 for the use of an IL-1 receptor antagonist for the treatment of or prophylaxis for type 2 diabetes.
REFERENCES
- Abbate A, Kontos MC, Abouzaki NA, Melchior RD, Thomas C, Van Tassell BW, Oddi C, Carbone S, Trankle CR, Roberts CS, et al. (2015). Comparative safety of interleukin-1 blockade with anakinra in patients with ST-segment elevation acute myocardial infarction (from the VCU-ART and VCU-ART2 pilot studies). The American journal of cardiology 115, 288–292. [DOI] [PubMed] [Google Scholar]
- Abbate A, Kontos MC, Grizzard JD, Biondi-Zoccai GG, Van Tassell BW, Robati R, Roach LM, Arena RA, Roberts CS, Varma A, et al. (2010). Interleukin-1 blockade with anakinra to prevent adverse cardiac remodeling after acute myocardial infarction (Virginia Commonwealth University Anakinra Remodeling Trial [VCU-ART] Pilot study). The American journal of cardiology 105, 1371–1377 e1371. [DOI] [PubMed] [Google Scholar]
- Abbate A, Salloum FN, Vecile E, Das A, Hoke NN, Straino S, Biondi-Zoccai GG, Houser JE, Qureshi IZ, Ownby ED, et al. (2008). Anakinra, a recombinant human interleukin-1 receptor antagonist, inhibits apoptosis in experimental acute myocardial infarction. Circulation 117, 2670–2683. [DOI] [PubMed] [Google Scholar]
- Abrams GA, Kunde SS, Lazenby AJ, and Clements RH (2004). Portal fibrosis and hepatic steatosis in morbidly obese subjects: A spectrum of nonalcoholic fatty liver disease. Hepatology 40, 475–483. [DOI] [PubMed] [Google Scholar]
- Adam TC, and Westerterp-Plantenga MS (2004). Activity-induced GLP-1 release in lean and obese subjects. Physiol Behav 83, 459–466. [DOI] [PubMed] [Google Scholar]
- Akiyama H, Barger S, Barnum S, Bradt B, Bauer J, Cole GM, Cooper NR, Eikelenboom P, Emmerling M, Fiebich BL, et al. (2000). Inflammation and Alzheimer’s disease. Neurobiol Aging 21, 383–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akshintala D, Chugh R, Amer F, and Cusi K (2000). Nonalcoholic Fatty Liver Disease: The Overlooked Complication of Type 2 Diabetes. In Endotext, Feingold KR, Anawalt B, Boyce A, Chrousos G, de Herder WW, Dhatariya K, Dungan K, Grossman A, Hershman JM, Hofland J, Kalra S, Kaltsas G, Koch C, Kopp P, Korbonits M, Kovacs CS, Kuohung W, Laferrere B, McGee EA, McLachlan R, Morley JE, New M, Purnell J, Sahay R, Singer F, Stratakis CA, Trence DL, and Wilson DP, eds. (South Dartmouth (MA)). [PubMed] [Google Scholar]
- Al-Sadi RM, and Ma TY (2007). IL-1beta causes an increase in intestinal epithelial tight junction permeability. J Immunol 178, 4641–4649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AC, Grundke-Iqbal I, and Iqbal K (1996). Alzheimer’s disease hyperphosphorylated tau sequesters normal tau into tangles of filaments and disassembles microtubules. Nat Med 2, 783–787. [DOI] [PubMed] [Google Scholar]
- Alonso AC, Zaidi T, Grundke-Iqbal I, and Iqbal K (1994). Role of abnormally phosphorylated tau in the breakdown of microtubules in Alzheimer disease. Proc Natl Acad Sci U S A 91, 5562–5566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AD, Cohen LS, Corbo C, Morozova V, ElIdrissi A, Phillips G, and Kleiman FE (2018). Hyperphosphorylation of Tau Associates With Changes in Its Function Beyond Microtubule Stability. Front Cell Neurosci 12, 338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alonso AD, Grundke-Iqbal I, Barra HS, and Iqbal K (1997). Abnormal phosphorylation of tau and the mechanism of Alzheimer neurofibrillary degeneration: sequestration of microtubule-associated proteins 1 and 2 and the disassembly of microtubules by the abnormal tau. Proc Natl Acad Sci U S A 94, 298–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amano SU, Cohen JL, Vangala P, Tencerova M, Nicoloro SM, Yawe JC, Shen Y, Czech MP, and Aouadi M (2014). Local proliferation of macrophages contributes to obesity-associated adipose tissue inflammation. Cell Metab 19, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermudez-Humaran LG, Smirnova N, Berge M, Sulpice T, Lahtinen S, et al. (2011). Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3, 559–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amar S, Zhou Q, Shaik-Dasthagirisaheb Y, and Leeman S (2007). Diet-induced obesity in mice causes changes in immune responses and bone loss manifested by bacterial challenge. Proc Natl Acad Sci U S A 104, 20466–20471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antohe JL, Bili A, Sartorius JA, Kirchner HL, Morris SJ, Dancea S, and Wasko MC (2012). Diabetes mellitus risk in rheumatoid arthritis: reduced incidence with anti-tumor necrosis factor alpha therapy. Arthritis care & research 64, 215–221. [DOI] [PubMed] [Google Scholar]
- Arcuri C, Mecca C, Bianchi R, Giambanco I, and Donato R (2017). The Pathophysiological Role of Microglia in Dynamic Surveillance, Phagocytosis and Structural Remodeling of the Developing CNS. Front Mol Neurosci 10, 191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arkan MC, Hevener AL, Greten FR, Maeda S, Li ZW, Long JM, Wynshaw-Boris A, Poli G, Olefsky J, and Karin M (2005). IKK-beta links inflammation to obesity-induced insulin resistance. Nat Med 11, 191–198. [DOI] [PubMed] [Google Scholar]
- Auffray C, Fogg D, Garfa M, Elain G, Join-Lambert O, Kayal S, Sarnacki S, Cumano A, Lauvau G, and Geissmann F (2007). Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 317, 666–670. [DOI] [PubMed] [Google Scholar]
- Backhed F, Ding H, Wang T, Hooper LV, Koh GY, Nagy A, Semenkovich CF, and Gordon JI (2004). The gut microbiota as an environmental factor that regulates fat storage. Proc Natl Acad Sci U S A 101, 15718–15723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Backhed F, Manchester JK, Semenkovich CF, and Gordon JI (2007). Mechanisms underlying the resistance to diet-induced obesity in germ-free mice. Proc Natl Acad Sci U S A 104, 979–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Bravo-Blas A, Scott CL, Perdiguero EG, Geissmann F, Henri S, Malissen B, Osborne LC, Artis D, and Mowat AM (2014). Constant replenishment from circulating monocytes maintains the macrophage pool in the intestine of adult mice. Nat Immunol 15, 929–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, and Mowat AM (2014). Macrophages in intestinal homeostasis and inflammation. Immunol Rev 260, 102–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, Guilliams M, Malissen B, Agace WW, and Mowat AM (2013). Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol 6, 498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bantug GR, Galluzzi L, Kroemer G, and Hess C (2018). The spectrum of T cell metabolism in health and disease. Nat Rev Immunol 18, 19–34. [DOI] [PubMed] [Google Scholar]
- Bapat SP, Myoung Suh J, Fang S, Liu S, Zhang Y, Cheng A, Zhou C, Liang Y, LeBlanc M, Liddle C, et al. (2015). Depletion of fat-resident Treg cells prevents age-associated insulin resistance. Nature 528, 137–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bataillard A, del Rey A, Klusman I, Arditi GM, and Besedovsky HO (1992). Interleukin-1 stimulates aldosterone secretion: involvement of renin, ACTH, and prostaglandins. The American journal of physiology 263, R840–844. [DOI] [PubMed] [Google Scholar]
- Bennett H, Troutman TD, Sakai M, and Glass CK (2020). Epigenetic Regulation of Kupffer Cell Function in Health and Disease. Front Immunol 11, 609618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein LE, Berry J, Kim S, Canavan B, and Grinspoon SK (2006). Effects of etanercept in patients with the metabolic syndrome. Arch Intern Med 166, 902–908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bevilacqua MP, Pober JS, Wheeler ME, Cotran RS, and Gimbrone MA Jr. (1985). Interleukin 1 acts on cultured human vascular endothelium to increase the adhesion of polymorphonuclear leukocytes, monocytes, and related leukocyte cell lines. The Journal of clinical investigation 76, 2003–2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigornia SJ, Farb MG, Mott MM, Hess DT, Carmine B, Fiscale A, Joseph L, Apovian CM, and Gokce N (2012). Relation of depot-specific adipose inflammation to insulin resistance in human obesity. Nutr Diabetes 2, e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolmont T, Haiss F, Eicke D, Radde R, Mathis CA, Klunk WE, Kohsaka S, Jucker M, and Calhoun ME (2008). Dynamics of the microglial/amyloid interaction indicate a role in plaque maintenance. J Neurosci 28, 4283–4292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonhof GJ, Herder C, Strom A, Papanas N, Roden M, and Ziegler D (2019). Emerging Biomarkers, Tools, and Treatments for Diabetic Polyneuropathy. Endocr Rev 40, 153–192. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler M, Boller S, Debray S, Bouzakri K, Meier DT, Prazak R, Kerr-Conte J, Pattou F, Ehses JA, Schuit FC, and Donath MY (2009). Free fatty acids induce a proinflammatory response in islets via the abundantly expressed interleukin-1 receptor I. Endocrinology 150, 5218–5229. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler M, Hauselmann SP, Dalmas E, Meier DT, Thienel C, Traub S, Schulze F, Steiger L, Dror E, Martin P, et al. (2018). beta Cell-Specific Deletion of the IL-1 Receptor Antagonist Impairs beta Cell Proliferation and Insulin Secretion. Cell Rep 22, 1774–1786. [DOI] [PubMed] [Google Scholar]
- Boni-Schnetzler M, and Meier DT (2019). Islet inflammation in type 2 diabetes. Semin Immunopathol 41, 501–513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon MR, Bakker LE, Haks MC, Quinten E, Schaart G, Van Beek L, Wang Y, Van Schinkel L, Van Harmelen V, Meinders AE, et al. (2015). Short-term high-fat diet increases macrophage markers in skeletal muscle accompanied by impaired insulin signalling in healthy male subjects. Clin Sci (Lond) 128, 143–151. [DOI] [PubMed] [Google Scholar]
- Boulenouar S, Michelet X, Duquette D, Alvarez D, Hogan AE, Dold C, O’Connor D, Stutte S, Tavakkoli A, Winters D, et al. (2017). Adipose Type One Innate Lymphoid Cells Regulate Macrophage Homeostasis through Targeted Cytotoxicity. Immunity 46, 273–286. [DOI] [PubMed] [Google Scholar]
- Bouwens L, Baekeland M, De Zanger R, and Wisse E (1986). Quantitation, tissue distribution and proliferation kinetics of Kupffer cells in normal rat liver. Hepatology 6, 718–722. [DOI] [PubMed] [Google Scholar]
- Braak H, and Braak E (1991). Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Butcher MJ, Hallinger D, Garcia E, Machida Y, Chakrabarti S, Nadler J, Galkina EV, and Imai Y (2014). Association of proinflammatory cytokines and islet resident leucocytes with islet dysfunction in type 2 diabetes. Diabetologia 57, 491–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, et al. (2007). Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56, 1761–1772. [DOI] [PubMed] [Google Scholar]
- Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, and Burcelin R (2008). Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57, 1470–1481. [DOI] [PubMed] [Google Scholar]
- Cao X, Han ZB, Zhao H, and Liu Q (2014). Transplantation of mesenchymal stem cells recruits trophic macrophages to induce pancreatic beta cell regeneration in diabetic mice. Int J Biochem Cell Biol 53, 372–379. [DOI] [PubMed] [Google Scholar]
- Carey AL, Steinberg GR, Macaulay SL, Thomas WG, Holmes AG, Ramm G, Prelovsek O, Hohnen-Behrens C, Watt MJ, James DE, et al. (2006). Interleukin-6 increases insulin-stimulated glucose disposal in humans and glucose uptake and fatty acid oxidation in vitro via AMP-activated protein kinase. Diabetes 55, 2688–2697. [DOI] [PubMed] [Google Scholar]
- Cavelti-Weder C, Babians-Brunner A, Keller C, Stahel MA, Kurz-Levin M, Zayed H, Solinger AM, Mandrup-Poulsen T, Dinarello CA, and Donath MY (2012). Effects of gevokizumab on glycemia and inflammatory markers in type 2 diabetes. Diabetes Care 35, 1654–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavelti-Weder C, Furrer R, Keller C, Babians-Brunner A, Solinger AM, Gast H, Fontana A, Donath MY, and Penner IK (2011). Inhibition of IL-1beta improves fatigue in type 2 diabetes. Diabetes care 34, e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chavez JA, and Summers SA (2012). A ceramide-centric view of insulin resistance. Cell Metab 15, 585–594. [DOI] [PubMed] [Google Scholar]
- Chawla A, Nguyen KD, and Goh YP (2011). Macrophage-mediated inflammation in metabolic disease. Nat Rev Immunol 11, 738–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen CH, Zhou W, Liu S, Deng Y, Cai F, Tone M, Tone Y, Tong Y, and Song W (2012a). Increased NF-kappaB signalling up-regulates BACE1 expression and its therapeutic potential in Alzheimer’s disease. Int J Neuropsychopharmacol 15, 77–90. [DOI] [PubMed] [Google Scholar]
- Chen Q, Wang H, Liu Y, Song Y, Lai L, Han Q, Cao X, and Wang Q (2012b). Inducible microRNA-223 down-regulation promotes TLR-triggered IL-6 and IL-1beta production in macrophages by targeting STAT3. PLoS One 7, e42971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi BS, Daoust L, Pilon G, Marette A, and Tremblay A (2020). Potential therapeutic applications of the gut microbiome in obesity: from brain function to body detoxification. Int J Obes (Lond) 44, 1818–1831. [DOI] [PubMed] [Google Scholar]
- Cipolletta D, Feuerer M, Li A, Kamei N, Lee J, Shoelson SE, Benoist C, and Mathis D (2012). PPAR-gamma is a major driver of the accumulation and phenotype of adipose tissue Treg cells. Nature 486, 549–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark A, Wells CA, Buley ID, Cruickshank JK, Vanhegan RI, Matthews DR, Cooper GJ, Holman RR, and Turner RC (1988). Islet amyloid, increased A-cells, reduced B-cells and exocrine fibrosis: quantitative changes in the pancreas in type 2 diabetes. Diabetes Res 9, 151–159. [PubMed] [Google Scholar]
- Cohen I, Rider P, Carmi Y, Braiman A, Dotan S, White MR, Voronov E, Martin MU, Dinarello CA, and Apte RN (2010). Differential release of chromatin-bound IL-1alpha discriminates between necrotic and apoptotic cell death by the ability to induce sterile inflammation. Proceedings of the National Academy of Sciences of the United States of America 107, 2574–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Feng T, Fujihashi K, Schoeb TR, and Elson CO (2009). A dominant, coordinated T regulatory cell-IgA response to the intestinal microbiota. Proc Natl Acad Sci U S A 106, 19256–19261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook DG, Mendall MA, Whincup PH, Carey IM, Ballam L, Morris JE, Miller GJ, and Strachan DP (2000). C-reactive protein concentration in children: relationship to adiposity and other cardiovascular risk factors. Atherosclerosis 149, 139–150. [DOI] [PubMed] [Google Scholar]
- Cox N, Crozet L, Holtman IR, Loyher PL, Lazarov T, White JB, Mass E, Stanley ER, Elemento O, Glass CK, and Geissmann F (2021). Diet-regulated production of PDGFcc by macrophages controls energy storage. Science 373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dagdeviren S, Jung DY, Lee E, Friedline RH, Noh HL, Kim JH, Patel PR, Tsitsilianos N, Tsitsilianos AV, Tran DA, et al. (2016). Altered Interleukin-10 Signaling in Skeletal Muscle Regulates Obesity-Mediated Inflammation and Insulin Resistance. Mol Cell Biol 36, 2956–2966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalmas E, L.F. M, Dror E, Wueest S, Thienel C, Borsigova M, Marc S, Traunecker E, L.F. C, Dapito D, et al. (2017). Interleukin-33-Activated Islet-Resident Innate Lymphoid Cells Promote Insulin Secretion Through Myeloid Cell Retinoic Acid Production. Immunity 47, 1774–1786. [DOI] [PubMed] [Google Scholar]
- Dalmas E, Venteclef N, Caer C, Poitou C, Cremer I, Aron-Wisnewsky J, Lacroix-Desmazes S, Bayry J, Kaveri SV, Clement K, et al. (2014). T cell-derived IL-22 amplifies IL-1beta-driven inflammation in human adipose tissue: relevance to obesity and type 2 diabetes. Diabetes 63, 1966–1977. [DOI] [PubMed] [Google Scholar]
- Dandona P, Aljada A, and Bandyopadhyay A (2004). Inflammation: the link between insulin resistance, obesity and diabetes. Trends Immunol 25, 4–7. [DOI] [PubMed] [Google Scholar]
- Dang SY, Leng Y, Wang ZX, Xiao X, Zhang X, Wen T, Gong HZ, Hong A, and Ma Y (2019). Exosomal transfer of obesity adipose tissue for decreased miR-141–3p mediate insulin resistance of hepatocytes. Int J Biol Sci 15, 351–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dani M, Wood M, Mizoguchi R, Fan Z, Walker Z, Morgan R, Hinz R, Biju M, Kuruvilla T, Brooks DJ, and Edison P (2018). Microglial activation correlates in vivo with both tau and amyloid in Alzheimer’s disease. Brain 141, 2740–2754. [DOI] [PubMed] [Google Scholar]
- Dash PK, and Moore AN (1995). Enhanced processing of APP induced by IL-1 beta can be reduced by indomethacin and nordihydroguaiaretic acid. Biochem Biophys Res Commun 208, 542–548. [DOI] [PubMed] [Google Scholar]
- de Alvaro C, Teruel T, Hernandez R, and Lorenzo M (2004). Tumor necrosis factor alpha produces insulin resistance in skeletal muscle by activation of inhibitor kappaB kinase in a p38 MAPK-dependent manner. J Biol Chem 279, 17070–17078. [DOI] [PubMed] [Google Scholar]
- de La Serre CB, Ellis CL, Lee J, Hartman AL, Rutledge JC, and Raybould HE (2010). Propensity to high-fat diet-induced obesity in rats is associated with changes in the gut microbiota and gut inflammation. Am J Physiol Gastrointest Liver Physiol 299, G440–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeFronzo RA, and Tripathy D (2009). Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32 Suppl 2, S157–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Del Bo R, Angeretti N, Lucca E, De Simoni MG, and Forloni G (1995). Reciprocal control of inflammatory cytokines, IL-1 and IL-6, and beta-amyloid production in cultures. Neurosci Lett 188, 70–74. [DOI] [PubMed] [Google Scholar]
- Desai HR, Sivasubramaniyam T, Revelo XS, Schroer SA, Luk CT, Rikkala PR, Metherel AH, Dodington DW, Park YJ, Kim MJ, et al. (2017). Macrophage JAK2 deficiency protects against high-fat diet-induced inflammation. Sci Rep 7, 7653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Meo S, Iossa S, and Venditti P (2017). Skeletal muscle insulin resistance: role of mitochondria and other ROS sources. J Endocrinol 233, R15–R42. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2009). Immunological and inflammatory functions of the interleukin-1 family. Annu Rev Immunol 27, 519–550. [DOI] [PubMed] [Google Scholar]
- Dinarello CA (2011). Interleukin-1 in the pathogenesis and treatment of inflammatory diseases. Blood 117, 3720–3732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dominguez H, Storgaard H, Rask-Madsen C, Steffen Hermann T, Ihlemann N, Baunbjerg Nielsen D, Spohr C, Kober L, Vaag A, and Torp-Pedersen C (2005). Metabolic and vascular effects of tumor necrosis factor-alpha blockade with etanercept in obese patients with type 2 diabetes. J Vasc Res 42, 517–525. [DOI] [PubMed] [Google Scholar]
- Donath MY (2014). Targeting inflammation in the treatment of type 2 diabetes: time to start. Nature reviews. Drug discovery 13, 465–476. [DOI] [PubMed] [Google Scholar]
- Donath MY, and Shoelson SE (2011). Type 2 diabetes as an inflammatory disease. Nat Rev Immunol 11, 98–107. [DOI] [PubMed] [Google Scholar]
- Dong N, Xu B, Wang B, and Chu L (2013). Study of 27 aqueous humor cytokines in patients with type 2 diabetes with or without retinopathy. Mol Vis 19, 1734–1746. [PMC free article] [PubMed] [Google Scholar]
- Donnelly KL, Smith CI, Schwarzenberg SJ, Jessurun J, Boldt MD, and Parks EJ (2005). Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest 115, 1343–1351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror E, Dalmas E, Meier DT, Wueest S, Thevenet J, Thienel C, Timper K, Nordmann TM, Traub S, Schulze F, et al. (2017). Postprandial macrophage-derived IL-1[beta] stimulates insulin, and both synergistically promote glucose disposal and inflammation. Nature immunology 18, 283–292. [DOI] [PubMed] [Google Scholar]
- Duewell P, Kono H, Rayner KJ, Sirois CM, Vladimer G, Bauernfeind FG, Abela GS, Franchi L, Nunez G, Schnurr M, et al. (2010). NLRP3 inflammasomes are required for atherogenesis and activated by cholesterol crystals. Nature 464, 1357–1361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebrahimi F, Urwyler SA, Straumann S, Doerpfeld S, Bernasconi L, Neyer P, Schuetz P, Mueller B, Donath MY, and Christ-Crain M (2018). Interleukin-1 Antagonism in Men with Metabolic Syndrome and Low Testosterone - A Randomized Clinical Trial. The Journal of clinical endocrinology and metabolism. [DOI] [PubMed] [Google Scholar]
- Ebstein W (1876). Zur Therapie des Diabetes Mellitus, insbesondere über die Anwendung des Salicylsauren Natron bei demselben. In Berliner Klinische Wochenschrift, pp. 337–340. [Google Scholar]
- Eckardt K, Gorgens SW, Raschke S, and Eckel J (2014). Myokines in insulin resistance and type 2 diabetes. Diabetologia 57, 1087–1099. [DOI] [PubMed] [Google Scholar]
- Eguchi K, Manabe I, Oishi-Tanaka Y, Ohsugi M, Kono N, Ogata F, Yagi N, Ohto U, Kimoto M, Miyake K, et al. (2012). Saturated fatty acid and TLR signaling link beta cell dysfunction and islet inflammation. Cell Metab 15, 518–533. [DOI] [PubMed] [Google Scholar]
- Ehses JA, Lacraz G, Giroix MH, Schmidlin F, Coulaud J, Kassis N, Irminger JC, Kergoat M, Portha B, Homo-Delarche F, and Donath MY (2009). IL-1 antagonism reduces hyperglycemia and tissue inflammation in the type 2 diabetic GK rat. Proc Natl Acad Sci U S A 106, 13998–14003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehses JA, Perren A, Eppler E, Ribaux P, Pospisilik JA, Maor-Cahn R, Gueripel X, Ellingsgaard H, Schneider MK, Biollaz G, et al. (2007). Increased number of islet-associated macrophages in type 2 diabetes. Diabetes 56, 2356–2370. [DOI] [PubMed] [Google Scholar]
- Ellingsgaard H, Hauselmann I, Schuler B, Habib AM, Baggio LL, Meier DT, Eppler E, Bouzakri K, Wueest S, Muller YD, et al. (2011). Interleukin-6 enhances insulin secretion by increasing glucagon-like peptide-1 secretion from L cells and alpha cells. Nature medicine 17, 1481–1489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Escobar-Morreale HF (2018). Polycystic ovary syndrome: definition, aetiology, diagnosis and treatment. Nat Rev Endocrinol 14, 270–284. [DOI] [PubMed] [Google Scholar]
- Everett BM, Cornel J, Lainscak M, Anker SD, Abbate A, Thuren T, Libby P, Glynn RJ, and Ridker PM (2018a). Anti-Inflammatory Therapy with Canakinumab for the Prevention of Hospitalization for Heart Failure. Circulation. [DOI] [PubMed] [Google Scholar]
- Everett BM, Donath MY, Pradhan AD, Thuren T, Pais P, Nicolau JC, Glynn RJ, Libby P, and Ridker PM (2018b). Anti-Inflammatory Therapy With Canakinumab for the Prevention and Management of Diabetes. Journal of the American College of Cardiology 71, 2392–2401. [DOI] [PubMed] [Google Scholar]
- Faghihimani E, Aminorroaya A, Rezvanian H, Adibi P, Ismail-Beigi F, and Amini M (2013). Salsalate improves glycemic control in patients with newly diagnosed type 2 diabetes. Acta diabetologica 50, 537–543. [DOI] [PubMed] [Google Scholar]
- Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, and Iredale JP (2007). Scar-associated macrophages are a major source of hepatic matrix metalloproteinase-13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 178, 5288–5295. [DOI] [PubMed] [Google Scholar]
- Fan Y, and Pedersen O (2021). Gut microbiota in human metabolic health and disease. Nat Rev Microbiol 19, 55–71. [DOI] [PubMed] [Google Scholar]
- Fang L, Xie D, Wu X, Cao H, Su W, and Yang J (2013). Involvement of endoplasmic reticulum stress in albuminuria induced inflammasome activation in renal proximal tubular cells. PloS one 8, e72344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio MA (2014). Role of interleukins in obesity: implications for metabolic disease. Trends in endocrinology and metabolism: TEM 25, 312–319. [DOI] [PubMed] [Google Scholar]
- Febbraio MA, and Karin M (2021). “Sweet death”: Fructose as a metabolic toxin that targets the gut-liver axis. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Febbraio MA, and Pedersen BK (2002). Muscle-derived interleukin-6: mechanisms for activation and possible biological roles. FASEB J 16, 1335–1347. [DOI] [PubMed] [Google Scholar]
- Febbraio MA, Reibe S, Shalapour S, Ooi GJ, Watt MJ, and Karin M (2018). Preclinical Models for Studying NASH-Driven HCC: How Useful Are They? Cell metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feingold KR, Soued M, Staprans I, Gavin LA, Donahue ME, Huang BJ, Moser AH, Gulli R, and Grunfeld C (1989). Effect of tumor necrosis factor (TNF) on lipid metabolism in the diabetic rat. Evidence that inhibition of adipose tissue lipoprotein lipase activity is not required for TNF-induced hyperlipidemia. J Clin Invest 83, 1116–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E, Simonson DC, Katz LD, Reichard G Jr., Bevilacqua S, Barrett EJ, Olsson M, and DeFronzo RA (1988). The disposal of an oral glucose load in patients with non-insulin-dependent diabetes. Metabolism 37, 79–85. [DOI] [PubMed] [Google Scholar]
- Feuerer M, Herrero L, Cipolletta D, Naaz A, Wong J, Nayer A, Lee J, Goldfine AB, Benoist C, Shoelson S, and Mathis D (2009). Lean, but not obese, fat is enriched for a unique population of regulatory T cells that affect metabolic parameters. Nat Med 15, 930–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen M, Allen TL, Henstridge DC, Kammoun H, Brandon AE, Baggio LL, Watt KI, Pal M, Cron L, Estevez E, et al. (2019). Treatment of type 2 diabetes with the designer cytokine IC7Fc. Nature 574, 63–68. [DOI] [PubMed] [Google Scholar]
- Fink LN, Costford SR, Lee YS, Jensen TE, Bilan PJ, Oberbach A, Bluher M, Olefsky JM, Sams A, and Klip A (2014). Pro-inflammatory macrophages increase in skeletal muscle of high fat-fed mice and correlate with metabolic risk markers in humans. Obesity (Silver Spring) 22, 747–757. [DOI] [PubMed] [Google Scholar]
- Finucane OM, Lyons CL, Murphy AM, Reynolds CM, Klinger R, Healy NP, Cooke AA, Coll RC, McAllan L, Nilaweera KN, et al. (2015). Monounsaturated Fatty Acid-Enriched High-Fat Diets Impede Adipose NLRP3 Inflammasome-Mediated IL-1beta Secretion and Insulin Resistance Despite Obesity. Diabetes 64, 2116–2128. [DOI] [PubMed] [Google Scholar]
- Fleischman A, Shoelson SE, Bernier R, and Goldfine AB (2008). Salsalate improves glycemia and inflammatory parameters in obese young adults. Diabetes care 31, 289–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franckhauser S, Elias I, Rotter Sopasakis V, Ferre T, Nagaev I, Andersson CX, Agudo J, Ruberte J, Bosch F, and Smith U (2008). Overexpression of Il6 leads to hyperinsulinaemia, liver inflammation and reduced body weight in mice. Diabetologia 51, 1306–1316. [DOI] [PubMed] [Google Scholar]
- Fu S, Yang L, Li P, Hofmann O, Dicker L, Hide W, Lin X, Watkins SM, Ivanov AR, and Hotamisligil GS (2011). Aberrant lipid metabolism disrupts calcium homeostasis causing liver endoplasmic reticulum stress in obesity. Nature 473, 528–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuchs A, Samovski D, Smith GI, Cifarelli V, Farabi SS, Yoshino J, Pietka T, Chang SW, Ghosh S, Myckatyn TM, and Klein S (2021). Associations Among Adipose Tissue Immunology, Inflammation, Exosomes and Insulin Sensitivity in People With Obesity and Nonalcoholic Fatty Liver Disease. Gastroenterology 161, 968–981 e912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner SE, Humphry M, Bennett MR, and Clarke MC (2015). Senescent Vascular Smooth Muscle Cells Drive Inflammation Through an Interleukin-1alpha-Dependent Senescence-Associated Secretory Phenotype. Arteriosclerosis, thrombosis, and vascular biology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geng K, Ma X, Jiang Z, Huang W, Gao C, Pu Y, Luo L, Xu Y, Xu Y (2021). Innate Immunity in Diabetic Wound Healing: Focus on the Mastermind Hidden in Chronic Inflammatory. Front Pharmacol 12:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerrity RG (1981). The role of the monocyte in atherogenesis: I. Transition of blood-borne monocytes into foam cells in fatty lesions. The American journal of pathology 103, 181–190. [PMC free article] [PubMed] [Google Scholar]
- Ghazarian M, Revelo XS, Nohr MK, Luck H, Zeng K, Lei H, Tsai S, Schroer SA, Park YJ, Chng MHY, et al. (2017). Type I Interferon Responses Drive Intrahepatic T cells to Promote Metabolic Syndrome. Sci Immunol 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gholam PM, Flancbaum L, Machan JT, Charney DA, and Kotler DP (2007). Nonalcoholic fatty liver disease in severely obese subjects. Am J Gastroenterol 102, 399–408. [DOI] [PubMed] [Google Scholar]
- Goldfine AB, Conlin PR, Halperin F, Koska J, Permana P, Schwenke D, Shoelson SE, and Reaven PD (2013a). A randomised trial of salsalate for insulin resistance and cardiovascular risk factors in persons with abnormal glucose tolerance. Diabetologia 56, 714–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Fonseca V, Jablonski KA, Chen YD, Tipton L, Staten MA, Shoelson SE, and Targeting Inflammation Using Salsalate in Type 2 Diabetes Study, T. (2013b). Salicylate (salsalate) in patients with type 2 diabetes: a randomized trial. Annals of internal medicine 159, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Fonseca V, Jablonski KA, Pyle L, Staten MA, Shoelson SE, and Team T-TDS (2010). The effects of salsalate on glycemic control in patients with type 2 diabetes: a randomized trial. Annals of internal medicine 152, 346–357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldfine AB, Silver R, Aldhahi W, Cai D, Tatro E, Lee J, and Shoelson SE (2008). Use of salsalate to target inflammation in the treatment of insulin resistance and type 2 diabetes. Clin Transl Sci 1, 36–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldgaber D, Harris HW, Hla T, Maciag T, Donnelly RJ, Jacobsen JS, Vitek MP, and Gajdusek DC (1989). Interleukin 1 regulates synthesis of amyloid beta-protein precursor mRNA in human endothelial cells. Proc Natl Acad Sci U S A 86, 7606–7610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goncalves MD, Lu C, Tutnauer J, Hartman TE, Hwang SK, Murphy CJ, Pauli C, Morris R, Taylor S, Bosch K, et al. (2019). High-fructose corn syrup enhances intestinal tumor growth in mice. Science 363, 1345–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez-Gay MA, De Matias JM, Gonzalez-Juanatey C, Garcia-Porrua C, Sanchez-Andrade A, Martin J, and Llorca J (2006). Anti-tumor necrosis factor-alpha blockade improves insulin resistance in patients with rheumatoid arthritis. Clinical and experimental rheumatology 24, 83–86. [PubMed] [Google Scholar]
- Gressner OA, and Gressner AM (2008). Connective tissue growth factor: a fibrogenic master switch in fibrotic liver diseases. Liver Int 28, 1065–1079. [DOI] [PubMed] [Google Scholar]
- Guay C, Menoud V, Rome S, and Regazzi R (2015). Horizontal transfer of exosomal microRNAs transduce apoptotic signals between pancreatic beta-cells. Cell Commun Signal 13, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E, Cruchaga C, Sassi C, Kauwe JS, Younkin S, et al. (2013). TREM2 variants in Alzheimer’s disease. N Engl J Med 368, 117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilherme A, Virbasius JV, Puri V, and Czech MP (2008). Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol 9, 367–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo H, Callaway JB, and Ting JP (2015). Inflammasomes: mechanism of action, role in disease, and therapeutics. Nature medicine 21, 677–687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haase J, Weyer U, Immig K, Kloting N, Bluher M, Eilers J, Bechmann I, and Gericke M (2014). Local proliferation of macrophages in adipose tissue during obesity-induced inflammation. Diabetologia 57, 562–571. [DOI] [PubMed] [Google Scholar]
- Hajmrle C, Smith N, Spigelman AF, Dai X, Senior L, Bautista A, Ferdaoussi M, and MacDonald PE (2016). Interleukin-1 signaling contributes to acute islet compensation. JCI Insight 1, e86055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halberg N, Khan T, Trujillo ME, Wernstedt-Asterholm I, Attie AD, Sherwani S, Wang ZV, Landskroner-Eiger S, Dineen S, Magalang UJ, et al. (2009). Hypoxia-inducible factor 1alpha induces fibrosis and insulin resistance in white adipose tissue. Mol Cell Biol 29, 4467–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han JM, and Levings MK (2013). Immune regulation in obesity-associated adipose inflammation. J Immunol 191, 527–532. [DOI] [PubMed] [Google Scholar]
- Hanisch UK (2002). Microglia as a source and target of cytokines. Glia 40, 140–155. [DOI] [PubMed] [Google Scholar]
- Hansen DV, Hanson JE, and Sheng M (2018). Microglia in Alzheimer’s disease. J Cell Biol 217, 459–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasnain SZ, Borg DJ, Harcourt BE, Tong H, Sheng YH, Ng CP, Das I, Wang R, Chen AC, Loudovaris T, et al. (2014). Glycemic control in diabetes is restored by therapeutic manipulation of cytokines that regulate beta cell stress. Nature medicine 20, 1417–1426. [DOI] [PubMed] [Google Scholar]
- Hayes A, Thaker U, Iwatsubo T, Pickering-Brown SM, and Mann DM (2002). Pathological relationships between microglial cell activity and tau and amyloid beta protein in patients with Alzheimer’s disease. Neurosci Lett 331, 171–174. [DOI] [PubMed] [Google Scholar]
- Hellerbrand C, Stefanovic B, Giordano F, Burchardt ER, and Brenner DA (1999). The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol 30, 77–87. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, et al. (2015a). Neuroinflammation in Alzheimer’s disease. Lancet Neurol 14, 388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heneka MT, Golenbock DT, and Latz E (2015b). Innate immunity in Alzheimer’s disease. Nat Immunol 16, 229–236. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, et al. (2013). NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 493, 674–678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hepprich M, Wiedemann SJ, Schelker BL, Trinh B, Starkle A, Geigges M, Loliger J, Boni-Schnetzler M, Rudofsky G, and Donath MY (2020). Postprandial Hypoglycemia in Patients after Gastric Bypass Surgery Is Mediated by Glucose-Induced IL-1beta. Cell metabolism 31, 699–709 e695. [DOI] [PubMed] [Google Scholar]
- Herder C, Bongaerts BW, Rathmann W, Heier M, Kowall B, Koenig W, Thorand B, Roden M, Meisinger C, and Ziegler D (2013). Association of subclinical inflammation with polyneuropathy in the older population: KORA F4 study. Diabetes care 36, 3663–3670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herder C, Kannenberg JM, Carstensen-Kirberg M, Strom A, Bonhof GJ, Rathmann W, Huth C, Koenig W, Heier M, Krumsiek J, et al. (2018). A Systemic Inflammatory Signature Reflecting Cross Talk Between Innate and Adaptive Immunity Is Associated With Incident Polyneuropathy: KORA F4/FF4 Study. Diabetes 67, 2434–2442. [DOI] [PubMed] [Google Scholar]
- Herder C, Kannenberg JM, Huth C, Carstensen-Kirberg M, Rathmann W, Koenig W, Heier M, Puttgen S, Thorand B, Peters A, et al. (2017). Proinflammatory Cytokines Predict the Incidence and Progression of Distal Sensorimotor Polyneuropathy: KORA F4/FF4 Study. Diabetes care 40, 569–576. [DOI] [PubMed] [Google Scholar]
- Herder C, Lankisch M, Ziegler D, Rathmann W, Koenig W, Illig T, Doring A, Thorand B, Holle R, Giani G, et al. (2009). Subclinical inflammation and diabetic polyneuropathy: MONICA/KORA Survey F3 (Augsburg, Germany). Diabetes care 32, 680–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herder C, Roden M, and Ziegler D (2019). Novel Insights into Sensorimotor and Cardiovascular Autonomic Neuropathy from Recent-Onset Diabetes and Population-Based Cohorts. Trends in endocrinology and metabolism: TEM 30, 286–298. [DOI] [PubMed] [Google Scholar]
- Herman MA, and Birnbaum MJ (2021). Molecular aspects of fructose metabolism and metabolic disease. Cell Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hickman SE, Allison EK, and El Khoury J (2008). Microglial dysfunction and defective beta-amyloid clearance pathways in aging Alzheimer’s disease mice. J Neurosci 28, 8354–8360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildreth AD, Ma F, Wong YY, Sun R, Pellegrini M, and O’Sullivan TE (2021). Single-cell sequencing of human white adipose tissue identifies new cell states in health and obesity. Nat Immunol 22, 639–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill DA, Lim HW, Kim YH, Ho WY, Foong YH, Nelson VL, Nguyen HCB, Chegireddy K, Kim J, Habertheuer A, et al. (2018). Distinct macrophage populations direct inflammatory versus physiological changes in adipose tissue. Proc Natl Acad Sci U S A 115, E5096–E5105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirosumi J, Tuncman G, Chang L, Gorgun CZ, Uysal KT, Maeda K, Karin M, and Hotamisligil GS (2002). A central role for JNK in obesity and insulin resistance. Nature 420, 333–336. [DOI] [PubMed] [Google Scholar]
- Holland WL, Bikman BT, Wang LP, Yuguang G, Sargent KM, Bulchand S, Knotts TA, Shui G, Clegg DJ, Wenk MR, et al. (2011). Lipid-induced insulin resistance mediated by the proinflammatory receptor TLR4 requires saturated fatty acid-induced ceramide biosynthesis in mice. J Clin Invest 121, 1858–1870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holm JG, Thomsen SF (2019). Type 2 diabetes and psoriasis: links and risks. Psoriasis 9:1–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Homo-Delarche F, Calderari S, Irminger JC, Gangnerau MN, Coulaud J, Rickenbach K, Dolz M, Halban P, Portha B, and Serradas P (2006). Islet inflammation and fibrosis in a spontaneous model of type 2 diabetes, the GK rat. Diabetes 55, 1625–1633. [DOI] [PubMed] [Google Scholar]
- Hong CP, Park A, Yang BG, Yun CH, Kwak MJ, Lee GW, Kim JH, Jang MS, Lee EJ, Jeun EJ, et al. (2017). Gut-Specific Delivery of T-Helper 17 Cells Reduces Obesity and Insulin Resistance in Mice. Gastroenterology 152, 1998–2010. [DOI] [PubMed] [Google Scholar]
- Hong EG, Ko HJ, Cho YR, Kim HJ, Ma Z, Yu TY, Friedline RH, Kurt-Jones E, Finberg R, Fischer MA, et al. (2009). Interleukin-10 prevents diet-induced insulin resistance by attenuating macrophage and cytokine response in skeletal muscle. Diabetes 58, 2525–2535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosogai N, Fukuhara A, Oshima K, Miyata Y, Tanaka S, Segawa K, Furukawa S, Tochino Y, Komuro R, Matsuda M, and Shimomura I (2007). Adipose tissue hypoxia in obesity and its impact on adipocytokine dysregulation. Diabetes 56, 901–911. [DOI] [PubMed] [Google Scholar]
- Hotamisligil GS, and Erbay E (2008). Nutrient sensing and inflammation in metabolic diseases. Nat Rev Immunol 8, 923–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Murray DL, Choy LN, and Spiegelman BM (1994). Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A 91, 4854–4858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hotamisligil GS, Shargill NS, and Spiegelman BM (1993). Adipose expression of tumor necrosis factor-alpha: direct role in obesity-linked insulin resistance. Science 259, 87–91. [DOI] [PubMed] [Google Scholar]
- Houstis N, Rosen ED, and Lander ES (2006). Reactive oxygen species have a causal role in multiple forms of insulin resistance. Nature 440, 944–948. [DOI] [PubMed] [Google Scholar]
- Huvers FC, Popa C, Netea MG, van den Hoogen FH, and Tack CJ (2007). Improved insulin sensitivity by anti-TNFalpha antibody treatment in patients with rheumatic diseases. Ann Rheum Dis 66, 558–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igoillo-Esteve M, Marselli L, Cunha DA, Ladriere L, Ortis F, Grieco FA, Dotta F, Weir GC, Marchetti P, Eizirik DL, and Cnop M (2010). Palmitate induces a pro-inflammatory response in human pancreatic islets that mimics CCL2 expression by beta cells in type 2 diabetes. Diabetologia 53, 1395–1405. [DOI] [PubMed] [Google Scholar]
- Imtiyaz HZ, and Simon MC (2010). Hypoxia-inducible factors as essential regulators of inflammation. Curr Top Microbiol Immunol 345, 105–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jager J, Gremeaux T, Cormont M, Le Marchand-Brustel Y, and Tanti JF (2007). Interleukin-1beta-induced insulin resistance in adipocytes through down-regulation of insulin receptor substrate-1 expression. Endocrinology 148, 241–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaitin DA, Adlung L, Thaiss CA, Weiner A, Li B, Descamps H, Lundgren P, Bleriot C, Liu Z, Deczkowska A, et al. (2019). Lipid-Associated Macrophages Control Metabolic Homeostasis in a Trem2-Dependent Manner. Cell 178, 686–698 e614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Wada S, Yang S, Gosis B, Zeng X, Zhang Z, Shen Y, Lee G, Arany Z, and Rabinowitz JD (2020). The small intestine shields the liver from fructose-induced steatosis. Nat Metab 2, 586–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javeed N, Her TK, Brown MR, Vanderboom P, Rakshit K, Egan AM, Vella A, Lanza I, and Matveyenko AV (2021). Pro-inflammatory beta cell small extracellular vesicles induce beta cell failure through activation of the CXCL10/CXCR3 axis in diabetes. Cell Rep 36, 109613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ji Y, Luo Z, Gao H, Dos Reis FCG, Bandyopadhyay G, Jin Z, Manda KA, Isaac R, Yang M, Fu W, et al. (2021). Hepatocyte-derived exosomes from early onset obese mice promote insulin sensitivity through miR-3075. Nat Metab. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jialal I, Major AM, and Devaraj S (2014). Global Toll-like receptor 4 knockout results in decreased renal inflammation, fibrosis and podocytopathy. Journal of diabetes and its complications 28, 755–761. [DOI] [PubMed] [Google Scholar]
- Jiang C, Qu A, Matsubara T, Chanturiya T, Jou W, Gavrilova O, Shah YM, and Gonzalez FJ (2011). Disruption of hypoxia-inducible factor 1 in adipocytes improves insulin sensitivity and decreases adiposity in high-fat diet-fed mice. Diabetes 60, 2484–2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Chen Q, Wang W, Ling Y, Yan Y, and Xia P (2020). Hepatocyte-derived extracellular vesicles promote endothelial inflammation and atherogenesis via microRNA-1. J Hepatol 72, 156–166. [DOI] [PubMed] [Google Scholar]
- Johnson AM, Costanzo A, Gareau MG, Armando AM, Quehenberger O, Jameson JM, and Olefsky JM (2015). High fat diet causes depletion of intestinal eosinophils associated with intestinal permeability. PLoS One 10, e0122195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TH, and Kennedy RL (1993). Cytokines and hypothalamic-pituitary function. Cytokine 5, 531–538. [DOI] [PubMed] [Google Scholar]
- Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J, Bjornsson S, Huttenlocher J, Levey AI, Lah JJ, et al. (2013). Variant of TREM2 associated with the risk of Alzheimer’s disease. N Engl J Med 368, 107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonsson T, and Stefansson K (2013). TREM2 and neurodegenerative disease. N Engl J Med 369, 1568–1569. [DOI] [PubMed] [Google Scholar]
- Joussen AM, Poulaki V, Le ML, Koizumi K, Esser C, Janicki H, Schraermeyer U, Kociok N, Fauser S, Kirchhof B, et al. (2004). A central role for inflammation in the pathogenesis of diabetic retinopathy. FASEB J 18, 1450–1452. [DOI] [PubMed] [Google Scholar]
- Jurgens CA, Toukatly MN, Fligner CL, Udayasankar J, Subramanian SL, Zraika S, Aston-Mourney K, Carr DB, Westermark P, Westermark GT, et al. (2011). beta-cell loss and beta-cell apoptosis in human type 2 diabetes are related to islet amyloid deposition. Am J Pathol 178, 2632–2640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamata K, Mizukami H, Inaba W, Tsuboi K, Tateishi Y, Yoshida T, and Yagihashi S (2014). Islet amyloid with macrophage migration correlates with augmented beta-cell deficits in type 2 diabetic patients. Amyloid 21, 191–201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamei N, Tobe K, Suzuki R, Ohsugi M, Watanabe T, Kubota N, Ohtsuka-Kowatari N, Kumagai K, Sakamoto K, Kobayashi M, et al. (2006). Overexpression of monocyte chemoattractant protein-1 in adipose tissues causes macrophage recruitment and insulin resistance. J Biol Chem 281, 26602–26614. [DOI] [PubMed] [Google Scholar]
- Kanda H, Tateya S, Tamori Y, Kotani K, Hiasa K, Kitazawa R, Kitazawa S, Miyachi H, Maeda S, Egashira K, and Kasuga M (2006). MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J Clin Invest 116, 1494–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kane H, and Lynch L (2019). Innate Immune Control of Adipose Tissue Homeostasis. Trends Immunol 40, 857–872. [DOI] [PubMed] [Google Scholar]
- Kataria Y, Ellervik C, and Mandrup-Poulsen T (2019). Treatment of type 2 diabetes by targeting interleukin-1 – a meta-analysis of 2921 patients. Seminars in Immunopathology 41, 413–425. [DOI] [PubMed] [Google Scholar]
- Kawano Y, Nakae J, Watanabe N, Kikuchi T, Tateya S, Tamori Y, Kaneko M, Abe T, Onodera M, and Itoh H (2016). Colonic Pro-inflammatory Macrophages Cause Insulin Resistance in an Intestinal Ccl2/Ccr2-Dependent Manner. Cell Metab 24, 295–310. [DOI] [PubMed] [Google Scholar]
- Kelly CC, Lyall H, Petrie JR, Gould GW, Connell JM, and Sattar N (2001). Low grade chronic inflammation in women with polycystic ovarian syndrome. The Journal of clinical endocrinology and metabolism 86, 2453–2455. [DOI] [PubMed] [Google Scholar]
- Keren-Shaul H, Spinrad A, Weiner A, Matcovitch-Natan O, Dvir-Szternfeld R, Ulland TK, David E, Baruch K, Lara-Astaiso D, Toth B, et al. (2017). A Unique Microglia Type Associated with Restricting Development of Alzheimer’s Disease. Cell 169, 1276–1290 e1217. [DOI] [PubMed] [Google Scholar]
- Khan IM, Perrard XY, Brunner G, Lui H, Sparks LM, Smith SR, Wang X, Shi ZZ, Lewis DE, Wu H, and Ballantyne CM (2015). Intermuscular and perimuscular fat expansion in obesity correlates with skeletal muscle T cell and macrophage infiltration and insulin resistance. Int J Obes (Lond) 39, 1607–1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan S, Luck H, Winer S, and Winer DA (2021). Emerging concepts in intestinal immune control of obesity-related metabolic disease. Nat Commun 12, 2598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KA, Gu W, Lee IA, Joh EH, and Kim DH (2012). High fat diet-induced gut microbiota exacerbates inflammation and obesity in mice via the TLR4 signaling pathway. PLoS One 7, e47713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura I, Ozawa K, Inoue D, Imamura T, Kimura K, Maeda T, Terasawa K, Kashihara D, Hirano K, Tani T, et al. (2013). The gut microbiota suppresses insulin-mediated fat accumulation via the short-chain fatty acid receptor GPR43. Nat Commun 4, 1829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinney JW, Bemiller SM, Murtishaw AS, Leisgang AM, Salazar AM, and Lamb BT (2018). Inflammation as a central mechanism in Alzheimer’s disease. Alzheimers Dement (N Y) 4, 575–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiortsis DN, Mavridis AK, Vasakos S, Nikas SN, and Drosos AA (2005). Effects of infliximab treatment on insulin resistance in patients with rheumatoid arthritis and ankylosing spondylitis. Ann Rheum Dis 64, 765–766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kita S, Maeda N, and Shimomura I (2019). Interorgan communication by exosomes, adipose tissue, and adiponectin in metabolic syndrome. J Clin Invest 129, 4041–4049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klareskog L, Catrina AI, and Paget S (2009). Rheumatoid arthritis. Lancet 373, 659–672. [DOI] [PubMed] [Google Scholar]
- Klover P, Zimmers T, Koniaris L, Nowak I, Senn J, and Mooney R (2003a). Acute or chronic exposure to interleukin-6 induces hepatic insulin resistance in vivo. Diabetes 52, A290–A290. [DOI] [PubMed] [Google Scholar]
- Klover PJ, Zimmers TA, Koniaris LG, and Mooney RA (2003b). Chronic exposure to interleukin-6 causes hepatic insulin resistance in mice. Diabetes 52, 2784–2789. [DOI] [PubMed] [Google Scholar]
- Kluck V, Jansen T, Janssen M, Comarniceanu A, Efde M, Tengesdal IW, Schraa K, Cleophas MCP, Scribner CL, Skouras DB, et al. (2020). Dapansutrile, an oral selective NLRP3 inflammasome inhibitor, for treatment of gout flares: an open-label, dose-adaptive, proof-of-concept, phase 2a trial. Lancet Rheumatol 2, e270–e280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kocahan S, and Dogan Z (2017). Mechanisms of Alzheimer’s Disease Pathogenesis and Prevention: The Brain, Neural Pathology, N-methyl-D-aspartate Receptors, Tau Protein and Other Risk Factors. Clin Psychopharmacol Neurosci 15, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kol S, Ruutiainen-Altman K, Scherzer WJ, Ben-Shlomo I, Ando M, Rohan RM, and Adashi EY (1999). The rat intraovarian interleukin (IL)-1 system: cellular localization, cyclic variation and hormonal regulation of IL-1beta and of the type I and type II IL-1 receptors. Mol Cell Endocrinol 149, 115–128. [DOI] [PubMed] [Google Scholar]
- Koska J, Ortega E, Bunt JC, Gasser A, Impson J, Hanson RL, Forbes J, de Courten B, and Krakoff J (2009). The effect of salsalate on insulin action and glucose tolerance in obese non-diabetic patients: results of a randomised double-blind placebo-controlled study. Diabetologia 52, 385–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraakman MJ, Kammoun HL, Allen TL, Deswaerte V, Henstridge DC, Estevez E, Matthews VB, Neill B, White DA, Murphy AJ, et al. (2015). Blocking IL-6 trans-signaling prevents high-fat diet-induced adipose tissue macrophage recruitment but does not improve insulin resistance. Cell Metab 21, 403–416. [DOI] [PubMed] [Google Scholar]
- Kranendonk ME, Visseren FL, van Herwaarden JA, Nolte-’t Hoen EN, de Jager W, Wauben MH, and Kalkhoven E (2014). Effect of extracellular vesicles of human adipose tissue on insulin signaling in liver and muscle cells. Obesity (Silver Spring) 22, 2216–2223. [DOI] [PubMed] [Google Scholar]
- Kratz M, Coats BR, Hisert KB, Hagman D, Mutskov V, Peris E, Schoenfelt KQ, Kuzma JN, Larson I, Billing PS, et al. (2014). Metabolic dysfunction drives a mechanistically distinct proinflammatory phenotype in adipose tissue macrophages. Cell Metab 20, 614–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lackey DE, and Olefsky JM (2016). Regulation of metabolism by the innate immune system. Nat Rev Endocrinol 12, 15–28. [DOI] [PubMed] [Google Scholar]
- Lagathu C, Bastard JP, Auclair M, Maachi M, Capeau J, and Caron M (2003). Chronic interleukin-6 (IL-6) treatment increased IL-6 secretion and induced insulin resistance in adipocyte: prevention by rosiglitazone. Biochemical and biophysical research communications 311, 372–379. [DOI] [PubMed] [Google Scholar]
- Lagathu C, Yvan-Charvet L, Bastard JP, Maachi M, Quignard-Boulange A, Capeau J, and Caron M (2006). Long-term treatment with interleukin-1beta induces insulin resistance in murine and human adipocytes. Diabetologia 49, 2162–2173. [DOI] [PubMed] [Google Scholar]
- Lakhter AJ, Pratt RE, Moore RE, Doucette KK, Maier BF, DiMeglio LA, and Sims EK (2018). Beta cell extracellular vesicle miR-21-5p cargo is increased in response to inflammatory cytokines and serves as a biomarker of type 1 diabetes. Diabetologia 61, 1124–1134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang CH, Dobrescu C, and Bagby GJ (1992). Tumor necrosis factor impairs insulin action on peripheral glucose disposal and hepatic glucose output. Endocrinology 130, 43–52. [DOI] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Ehses JA, Donath MY, and Mandrup-Poulsen T (2009). Sustained effects of interleukin-1 receptor antagonist treatment in type 2 diabetes. Diabetes Care 32, 1663–1668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen CM, Faulenbach M, Vaag A, Volund A, Ehses JA, Seifert B, Mandrup-Poulsen T, and Donath MY (2007). Interleukin-1-receptor antagonist in type 2 diabetes mellitus. The New England journal of medicine 356, 1517–1526. 10.1056/NEJMoa065213 [DOI] [PubMed] [Google Scholar]
- Lawler HM, Underkofler CM, Kern PA, Erickson C, Bredbeck B, and Rasouli N (2016). Adipose Tissue Hypoxia, Inflammation, and Fibrosis in Obese Insulin-Sensitive and Obese Insulin-Resistant Subjects. J Clin Endocrinol Metab 101, 1422–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee AH, and Dixit VD (2020). Dietary Regulation of Immunity. Immunity 53, 510–523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee BC, Kim MS, Pae M, Yamamoto Y, Eberle D, Shimada T, Kamei N, Park HS, Sasorith S, Woo JR, et al. (2016). Adipose Natural Killer Cells Regulate Adipose Tissue Macrophages to Promote Insulin Resistance in Obesity. Cell Metab 23, 685–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, Chen A, Chung H, Murphy A, Watkins SM, et al. (2014). Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell 157, 1339–1352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, and Olefsky J (2021). Chronic tissue inflammation and metabolic disease. Genes Dev 35, 307–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lehrskov LL, Dorph E, Widmer AM, Hepprich M, Siegenthaler J, Timper K, and Donath MY (2018). The role of IL-1 in postprandial fatigue. Molecular metabolism 12, 107–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leng F, and Edison P (2021). Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17, 157–172. [DOI] [PubMed] [Google Scholar]
- Li C, Spallanzani RG, and Mathis D (2020). Visceral adipose tissue Tregs and the cells that nurture them. Immunol Rev 295, 114–125. [DOI] [PubMed] [Google Scholar]
- Li P, Liu S, Lu M, Bandyopadhyay G, Oh D, Imamura T, Johnson AMF, Sears D, Shen Z, Cui B, et al. (2016). Hematopoietic-Derived Galectin-3 Causes Cellular and Systemic Insulin Resistance. Cell 167, 973–984 e912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li P, Oh DY, Bandyopadhyay G, Lagakos WS, Talukdar S, Osborn O, Johnson A, Chung H, Maris M, Ofrecio JM, et al. (2015). LTB4 promotes insulin resistance in obese mice by acting on macrophages, hepatocytes and myocytes. Nat Med 21, 239–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Libby P, Wyler DJ, Janicka MW, and Dinarello CA (1985). Differential effects of human interleukin-1 on growth of human fibroblasts and vascular smooth muscle cells. Arteriosclerosis 5, 186–191. [DOI] [PubMed] [Google Scholar]
- Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, Bennett ML, Munch AE, Chung WS, Peterson TC, et al. (2017). Neurotoxic reactive astrocytes are induced by activated microglia. Nature 541, 481–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin M, Yiu WH, Wu HJ, Chan LY, Leung JC, Au WS, Chan KW, Lai KN, and Tang SC (2012). Toll-like receptor 4 promotes tubular inflammation in diabetic nephropathy. Journal of the American Society of Nephrology : JASN 23, 86–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, and Quan N (2018). Microglia and CNS Interleukin-1: Beyond Immunological Concepts. Front Neurol 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loomba R, Friedman SL, and Shulman GI (2021). Mechanisms and disease consequences of nonalcoholic fatty liver disease. Cell 184, 2537–2564. [DOI] [PubMed] [Google Scholar]
- Loomba R, Gindin Y, Jiang Z, Lawitz E, Caldwell S, Djedjos CS, Xu R, Chung C, Myers RP, Subramanian GM, et al. (2018). DNA methylation signatures reflect aging in patients with nonalcoholic steatohepatitis. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck H, Khan S, Kim JH, Copeland JK, Revelo XS, Tsai S, Chakraborty M, Cheng K, Tao Chan Y, Nohr MK, et al. (2019). Gut-associated IgA(+) immune cells regulate obesity-related insulin resistance. Nat Commun 10, 3650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luck H, Tsai S, Chung J, Clemente-Casares X, Ghazarian M, Revelo XS, Lei H, Luk CT, Shi SY, Surendra A, et al. (2015). Regulation of obesity-related insulin resistance with gut anti-inflammatory agents. Cell Metab 21, 527–542. [DOI] [PubMed] [Google Scholar]
- Lumeng CN, Bodzin JL, and Saltiel AR (2007). Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest 117, 175–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Chadban SJ, Zhao CY, Chen X, Kwan T, Panchapakesan U, Pollock CA, and Wu H (2014a). TLR4 activation promotes podocyte injury and interstitial fibrosis in diabetic nephropathy. PloS one 9, e97985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma J, Wu H, Zhao CY, Panchapakesan U, Pollock C, and Chadban SJ (2014b). Requirement for TLR2 in the development of albuminuria, inflammation and fibrosis in experimental diabetic nephropathy. Int J Clin Exp Pathol 7, 481–495. [PMC free article] [PubMed] [Google Scholar]
- Machado M, Marques-Vidal P, and Cortez-Pinto H (2006). Hepatic histology in obese patients undergoing bariatric surgery. J Hepatol 45, 600–606. [DOI] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ehses JA, Mathe Z, Bosco D, Berney T, Dayer JM, Reinecke M, Halban PA, and Donath MY (2004). Leptin modulates beta cell expression of IL-1 receptor antagonist and release of IL-1beta in human islets. Proc Natl Acad Sci U S A 101, 8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maedler K, Sergeev P, Ris F, Oberholzer J, Joller-Jemelka HI, Spinas GA, Kaiser N, Halban PA, and Donath MY (2002). Glucose-induced beta cell production of IL-1beta contributes to glucotoxicity in human pancreatic islets. J Clin Invest 110, 851–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marra M, Campanati A, Testa R, Sirolla C, Bonfigli AR, Franceschi C, Marchegiani F, and Offidani A (2007). Effect of etanercept on insulin sensitivity in nine patients with psoriasis. International journal of immunopathology and pharmacology 20, 731–736. [DOI] [PubMed] [Google Scholar]
- Marso SP, Daniels GH, Brown-Frandsen K, Kristensen P, Mann JF, Nauck MA, Nissen SE, Pocock S, Poulter NR, Ravn LS, et al. (2016). Liraglutide and Cardiovascular Outcomes in Type 2 Diabetes. The New England journal of medicine 375, 311–322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez FO, and Gordon S (2014). The M1 and M2 paradigm of macrophage activation: time for reassessment. F1000Prime Rep 6, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martino L, Masini M, Bugliani M, Marselli L, Suleiman M, Boggi U, Nogueira TC, Filipponi F, Occhipinti M, Campani D, et al. (2015). Mast cells infiltrate pancreatic islets in human type 1 diabetes. Diabetologia 58, 2554–2562. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, and Tschopp J (2002). The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 10, 417–426. [DOI] [PubMed] [Google Scholar]
- Martinon F, Petrilli V, Mayor A, Tardivel A, and Tschopp J (2006). Gout-associated uric acid crystals activate the NALP3 inflammasome. Nature 440, 237–241. [DOI] [PubMed] [Google Scholar]
- Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA, Becker C, Franchi L, Yoshihara E, Chen Z, et al. (2010). Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol 11, 897–904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayerl C, Lukasser M, Sedivy R, Niederegger H, Seiler R, and Wick G (2006). Atherosclerosis research from past to present--on the track of two pathologists with opposing views, Carl von Rokitansky and Rudolf Virchow. Virchows Arch 449, 96–103. [DOI] [PubMed] [Google Scholar]
- McGillicuddy FC, Harford KA, Reynolds CM, Oliver E, Claessens M, Mills KH, and Roche HM (2011). Lack of interleukin-1 receptor I (IL-1RI) protects mice from high-fat diet-induced adipose tissue inflammation coincident with improved glucose homeostasis. Diabetes 60, 1688–1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McLaughlin T, Ackerman SE, Shen L, and Engleman E (2017). Role of innate and adaptive immunity in obesity-associated metabolic disease. J Clin Invest 127, 5–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meex RCR, and Watt MJ (2017). Hepatokines: linking nonalcoholic fatty liver disease and insulin resistance. Nat Rev Endocrinol 13, 509–520. [DOI] [PubMed] [Google Scholar]
- Meier DT, Morcos M, Samarasekera T, Zraika S, Hull RL, and Kahn SE (2014). Islet amyloid formation is an important determinant for inducing islet inflammation in high-fat-fed human IAPP transgenic mice. Diabetologia 57, 1884–1888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesquida M, Drawnel F, and Fauser S (2019). The role of inflammation in diabetic eye disease. Semin Immunopathol 41, 427–445. [DOI] [PubMed] [Google Scholar]
- Mesquida M, Leszczynska A, Llorenc V, and Adan A (2014). Interleukin-6 blockade in ocular inflammatory diseases. Clin Exp Immunol 176, 301–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monteiro-Sepulveda M, Touch S, Mendes-Sa C, Andre S, Poitou C, Allatif O, Cotillard A, Fohrer-Ting H, Hubert EL, Remark R, et al. (2015). Jejunal T Cell Inflammation in Human Obesity Correlates with Decreased Enterocyte Insulin Signaling. Cell Metab 22, 113–124. [DOI] [PubMed] [Google Scholar]
- Morinaga H, Mayoral R, Heinrichsdorff J, Osborn O, Franck N, Hah N, Walenta E, Bandyopadhyay G, Pessentheiner AR, Chi TJ, et al. (2015). Characterization of distinct subpopulations of hepatic macrophages in HFD/obese mice. Diabetes 64, 1120–1130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Canoves P, Scheele C, Pedersen BK, and Serrano AL (2013). Interleukin-6 myokine signaling in skeletal muscle: a double-edged sword? FEBS J 280, 4131–4148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murano I, Barbatelli G, Parisani V, Latini C, Muzzonigro G, Castellucci M, and Cinti S (2008). Dead adipocytes, detected as crown-like structures, are prevalent in visceral fat depots of genetically obese mice. J Lipid Res 49, 1562–1568. [DOI] [PubMed] [Google Scholar]
- Murphy MP, and LeVine H 3rd (2010). Alzheimer’s disease and the amyloid-beta peptide. J Alzheimers Dis 19, 311–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murphy MP, and O’Neill LAJ (2018). Krebs Cycle Reimagined: The Emerging Roles of Succinate and Itaconate as Signal Transducers. Cell 174, 780–784. [DOI] [PubMed] [Google Scholar]
- Nagareddy PR, Kraakman M, Masters SL, Stirzaker RA, Gorman DJ, Grant RW, Dragoljevic D, Hong ES, Abdel-Latif A, Smyth SS, et al. (2014). Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab 19, 821–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagineni CN, Kommineni VK, William A, Detrick B, and Hooks JJ (2012). Regulation of VEGF expression in human retinal cells by cytokines: implications for the role of inflammation in age-related macular degeneration. J Cell Physiol 227, 116–126. [DOI] [PubMed] [Google Scholar]
- Naito M, Yamamura F, Nishikawa S, and Takahashi K (1989). Development, differentiation, and maturation of fetal mouse yolk sac macrophages in cultures. J Leukoc Biol 46, 1–10. [DOI] [PubMed] [Google Scholar]
- Nakagawa H, Umemura A, Taniguchi K, Font-Burgada J, Dhar D, Ogata H, Zhong Z, Valasek MA, Seki E, Hidalgo J, et al. (2014). ER stress cooperates with hypernutrition to trigger TNF-dependent spontaneous HCC development. Cancer Cell 26, 331–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakano Y, Chayama K, Ochi H, Toshishige M, Hayashida Y, Miki D, Hayes CN, Suzuki H, Tokuyama T, Oda N, et al. (2013). A nonsynonymous polymorphism in semaphorin 3A as a risk factor for human unexplained cardiac arrest with documented ventricular fibrillation. PLoS Genet 9, e1003364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen MT, Favelyukis S, Nguyen AK, Reichart D, Scott PA, Jenn A, Liu-Bryan R, Glass CK, Neels JG, and Olefsky JM (2007). A subpopulation of macrophages infiltrates hypertrophic adipose tissue and is activated by free fatty acids via Toll-like receptors 2 and 4 and JNK-dependent pathways. J Biol Chem 282, 35279–35292. [DOI] [PubMed] [Google Scholar]
- Nguyen TT, Ta QTH, Nguyen TKO, Nguyen TTD, and Giau VV (2020). Type 3 Diabetes and Its Role Implications in Alzheimer’s Disease. Int J Mol Sci 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen-Lefebvre AT, and Horuzsko A (2015). Kupffer Cell Metabolism and Function. J Enzymol Metab 1. [PMC free article] [PubMed] [Google Scholar]
- Nishimura S, Manabe I, Nagasaki M, Eto K, Yamashita H, Ohsugi M, Otsu M, Hara K, Ueki K, Sugiura S, et al. (2009). CD8+ effector T cells contribute to macrophage recruitment and adipose tissue inflammation in obesity. Nat Med 15, 914–920. [DOI] [PubMed] [Google Scholar]
- Nordmann TM, Dror E, Schulze F, Traub S, Berishvili E, Barbieux C, Boni-Schnetzler M, and Donath MY (2017). The Role of Inflammation in beta-cell Dedifferentiation. Sci Rep 7, 6285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Holtman IR, Coufal NG, Schlachetzki JCM, Yu M, Hu R, Han CZ, Pena M, Xiao J, Wu Y, et al. (2019). Brain cell type-specific enhancer-promoter interactome maps and disease-risk association. Science 366, 1134–1139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Sullivan TE, Rapp M, Fan X, Weizman OE, Bhardwaj P, Adams NM, Walzer T, Dannenberg AJ, and Sun JC (2016). Adipose-Resident Group 1 Innate Lymphoid Cells Promote Obesity-Associated Insulin Resistance. Immunity 45, 428–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Obstfeld AE, Sugaru E, Thearle M, Francisco AM, Gayet C, Ginsberg HN, Ables EV, and Ferrante AW Jr. (2010). C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 59, 916–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ofei F, Hurel S, Newkirk J, Sopwith M, and Taylor R (1996). Effects of an engineered human anti-TNF-alpha antibody (CDP571) on insulin sensitivity and glycemic control in patients with NIDDM. Diabetes 45, 881–885. [DOI] [PubMed] [Google Scholar]
- Oh DY, Morinaga H, Talukdar S, Bae EJ, and Olefsky JM (2012). Increased macrophage migration into adipose tissue in obese mice. Diabetes 61, 346–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh DY, Talukdar S, Bae EJ, Imamura T, Morinaga H, Fan W, Li P, Lu WJ, Watkins SM, and Olefsky JM (2010). GPR120 is an omega-3 fatty acid receptor mediating potent anti-inflammatory and insulin-sensitizing effects. Cell 142, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okla M, Zaher W, Alfayez M, and Chung S (2018). Inhibitory Effects of Toll-Like Receptor 4, NLRP3 Inflammasome, and Interleukin-1beta on White Adipocyte Browning. Inflammation 41, 626–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong JP, Elariny H, Collantes R, Younoszai A, Chandhoke V, Reines HD, Goodman Z, and Younossi ZM (2005). Predictors of nonalcoholic steatohepatitis and advanced fibrosis in morbidly obese patients. Obes Surg 15, 310–315. [DOI] [PubMed] [Google Scholar]
- Orecchioni M, Ghosheh Y, Pramod AB, and Ley K (2019). Macrophage Polarization: Different Gene Signatures in M1(LPS+) vs. Classically and M2(LPS−) vs. Alternatively Activated Macrophages. Front Immunol 10, 1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik S, Kim JK, Silwal P, Sasakawa C, and Jo EK (2021). An update on the regulatory mechanisms of NLRP3 inflammasome activation. Cell Mol Immunol 18, 1141–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palframan RT, Jung S, Cheng G, Weninger W, Luo Y, Dorf M, Littman DR, Rollins BJ, Zweerink H, Rot A, and von Andrian UH (2001). Inflammatory chemokine transport and presentation in HEV: a remote control mechanism for monocyte recruitment to lymph nodes in inflamed tissues. J Exp Med 194, 1361–1373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquot N, Castillo MJ, Lefebvre PJ, and Scheen AJ (2000). No increased insulin sensitivity after a single intravenous administration of a recombinant human tumor necrosis factor receptor: Fc fusion protein in obese insulin-resistant patients. J Clin Endocrinol Metab 85, 1316–1319. [DOI] [PubMed] [Google Scholar]
- Pasarica M, Rood J, Ravussin E, Schwarz JM, Smith SR, and Redman LM (2010). Reduced oxygenation in human obese adipose tissue is associated with impaired insulin suppression of lipolysis. J Clin Endocrinol Metab 95, 4052–4055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasarica M, Sereda OR, Redman LM, Albarado DC, Hymel DT, Roan LE, Rood JC, Burk DH, and Smith SR (2009). Reduced adipose tissue oxygenation in human obesity: evidence for rarefaction, macrophage chemotaxis, and inflammation without an angiogenic response. Diabetes 58, 718–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patsouris D, Cao JJ, Vial G, Bravard A, Lefai E, Durand A, Durand C, Chauvin MA, Laugerette F, Debard C, et al. (2014). Insulin resistance is associated with MCP1-mediated macrophage accumulation in skeletal muscle in mice and humans. PLoS One 9, e110653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedersen BK, and Febbraio MA (2008). Muscle as an endocrine organ: focus on muscle-derived interleukin-6. Physiol Rev 88, 1379–1406. [DOI] [PubMed] [Google Scholar]
- Pedersen BK, and Febbraio MA (2012). Muscles, exercise and obesity: skeletal muscle as a secretory organ. Nat Rev Endocrinol 8, 457–465. [DOI] [PubMed] [Google Scholar]
- Pegtel DM, and Gould SJ (2019). Exosomes. Annu Rev Biochem 88, 487–514. [DOI] [PubMed] [Google Scholar]
- Perdiguero EG, Klapproth K, Schulz C, Busch K, de Bruijn M, Rodewald HR, and Geissmann F (2015). The Origin of Tissue-Resident Macrophages: When an Erythro-myeloid Progenitor Is an Erythro-myeloid Progenitor. Immunity 43, 1023–1024. [DOI] [PubMed] [Google Scholar]
- Plomgaard P, Bouzakri K, Krogh-Madsen R, Mittendorfer B, Zierath JR, and Pedersen BK (2005). Tumor necrosis factor-alpha induces skeletal muscle insulin resistance in healthy human subjects via inhibition of Akt substrate 160 phosphorylation. Diabetes 54, 2939–2945. [DOI] [PubMed] [Google Scholar]
- Polyzos SA, Kountouras J, and Mantzoros CS (2019). Obesity and nonalcoholic fatty liver disease: From pathophysiology to therapeutics. Metabolism 92, 82–97. [DOI] [PubMed] [Google Scholar]
- Popovic M, Sartorius G, and Christ-Crain M (2019). Chronic low-grade inflammation in polycystic ovary syndrome: is there a (patho)-physiological role for interleukin-1? Semin Immunopathol 41, 447–459. [DOI] [PubMed] [Google Scholar]
- Povero D, Panera N, Eguchi A, Johnson CD, Papouchado BG, de Araujo Horcel L, Pinatel EM, Alisi A, Nobili V, and Feldstein AE (2015). Lipid-induced hepatocyte-derived extracellular vesicles regulate hepatic stellate cell via microRNAs targeting PPAR-gamma. Cell Mol Gastroenterol Hepatol 1, 646–663 e644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puri P, Mirshahi F, Cheung O, Natarajan R, Maher JW, Kellum JM, and Sanyal AJ (2008). Activation and dysregulation of the unfolded protein response in nonalcoholic fatty liver disease. Gastroenterology 134, 568–576. [DOI] [PubMed] [Google Scholar]
- Quintanilla RA, Orellana DI, Gonzalez-Billault C, and Maccioni RB (2004). Interleukin-6 induces Alzheimer-type phosphorylation of tau protein by deregulating the cdk5/p35 pathway. Exp Cell Res 295, 245–257. [DOI] [PubMed] [Google Scholar]
- Radin MS, Sinha S, Bhatt BA, Dedousis N, and O’Doherty RM (2008). Inhibition or deletion of the lipopolysaccharide receptor Toll-like receptor-4 confers partial protection against lipid-induced insulin resistance in rodent skeletal muscle. Diabetologia 51, 336–346. [DOI] [PubMed] [Google Scholar]
- Ramkhelawon B, Hennessy EJ, Menager M, Ray TD, Sheedy FJ, Hutchison S, Wanschel A, Oldebeken S, Geoffrion M, Spiro W, et al. (2014). Netrin-1 promotes adipose tissue macrophage retention and insulin resistance in obesity. Nat Med 20, 377–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramos-Zavala MG, Gonzalez-Ortiz M, Martinez-Abundis E, Robles-Cervantes JA, Gonzalez-Lopez R, and Santiago-Hernandez NJ (2011). Effect of diacerein on insulin secretion and metabolic control in drug-naive patients with type 2 diabetes: a randomized clinical trial. Diabetes care 34, 1591–1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao PM, Kelly DM, and Jones TH (2013). Testosterone and insulin resistance in the metabolic syndrome and T2DM in men. Nat Rev Endocrinol 9, 479–493. [DOI] [PubMed] [Google Scholar]
- Rehman K, Akash MSH, Liaqat A, Kamal S, Qadir MI, and Rasul A (2017). Role of Interleukin-6 in Development of Insulin Resistance and Type 2 Diabetes Mellitus. Crit Rev Eukaryot Gene Expr 27, 229–236. [DOI] [PubMed] [Google Scholar]
- Richardson SJ, Willcox A, Bone AJ, Foulis AK, and Morgan NG (2009). Islet-associated macrophages in type 2 diabetes. Diabetologia 52, 1686–1688. [DOI] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Pradhan A, MacFadyen JG, Solomon DH, Zaharris E, Mam V, Hasan A, Rosenberg Y, Iturriaga E, et al. (2018a). Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. The New England journal of medicine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ridker PM, Everett BM, Thuren T, MacFadyen JG, Chang WH, Ballantyne C, Fonseca F, Nicolau J, Koenig W, Anker SD, et al. (2017). Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. The New England journal of medicine 377, 1119–1131. [DOI] [PubMed] [Google Scholar]
- Ridker PM, MacFadyen JG, Everett BM, Libby P, Thuren T, Glynn RJ, and Group CT (2018b). Relationship of C-reactive protein reduction to cardiovascular event reduction following treatment with canakinumab: a secondary analysis from the CANTOS randomised controlled trial. Lancet 391, 319–328. [DOI] [PubMed] [Google Scholar]
- Rissanen A, Howard CP, Botha J, and Thuren T (2012). Effect of Anti-IL-1beta Antibody (Canakinumab) on Insulin Secretion Rates in Impaired Glucose Tolerance or Type 2 Diabetes: Results of a Randomized, Placebo-Controlled Trial. Diabetes, obesity & metabolism 14, 1088–1096. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Calvo T, Ekwall O, Amirian N, Zapardiel-Gonzalo J, and von Herrath MG (2014). Increased immune cell infiltration of the exocrine pancreas: a possible contribution to the pathogenesis of type 1 diabetes. Diabetes 63, 3880–3890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohm TV, Alasfoor S, Bosch AJ, and Cavelti-Weder C (2017). Increased Inflammatory Intestinal Macrophage Subpopulations after High-Fat Diet. Diabetes 2017 June; 66, page 51. [Google Scholar]
- Rohm TV, Alasfoor S, Bosch AJ, and Cavelti-Weder C (2018). Targeting Intestinal Macrophages as a Potential Therapeutic Option in Obesity. Diabetes Jul; 67(Supplement 1). [Google Scholar]
- Rohm TV, Fuchs R, Muller RL, Keller L, Baumann Z, Bosch AJT, Schneider R, Labes D, Langer I, Pilz JB, et al. (2021). Obesity in Humans Is Characterized by Gut Inflammation as Shown by Pro-Inflammatory Intestinal Macrophage Accumulation. Front Immunol 12, 668654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano M, Fanelli G, Tan N, Nova-Lamperti E, McGregor R, Lechler RI, Lombardi G, and Scotta C (2018). Expanded Regulatory T Cells Induce Alternatively Activated Monocytes With a Reduced Capacity to Expand T Helper-17 Cells. Front Immunol 9, 1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotter V, Nagaev I, and Smith U (2003). Interleukin-6 (IL-6) induces insulin resistance in 3T3-L1 adipocytes and is, like IL-8 and tumor necrosis factor-alpha, overexpressed in human fat cells from insulin-resistant subjects. J Biol Chem 278, 45777–45784. [DOI] [PubMed] [Google Scholar]
- Rubsam A, Parikh S, and Fort PE (2018). Role of Inflammation in Diabetic Retinopathy. Int J Mol Sci 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscitti P, Cipriani P, Di Benedetto P, Liakouli V, Berardicurti O, Carubbi F, Ciccia F, Alvaro S, Triolo G, and Giacomelli R (2015). Monocytes from patients with rheumatoid arthritis and type 2 diabetes mellitus display an increased production of interleukin (IL)-1beta via the nucleotide-binding domain and leucine-rich repeat containing family pyrin 3(NLRP3)-inflammasome activation: a possible implication for therapeutic decision in these patients. Clin Exp Immunol 182, 35–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscitti P, Ursini F, Cipriani P, Ciccia F, Liakouli V, Carubbi F, Guggino G, Berardicurti O, Grembiale R, Triolo G, et al. (2017a). Prevalence of type 2 diabetes and impaired fasting glucose in patients affected by rheumatoid arthritis: Results from a cross-sectional study. Medicine (Baltimore) 96, e7896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscitti P, Ursini F, Cipriani P, Greco M, Alvaro S, Vasiliki L, Di Benedetto P, Carubbi F, Berardicurti O, Gulletta E, et al. (2019). IL-1 inhibition improves insulin resistance and adipokines in rheumatoid arthritis patients with comorbid type 2 diabetes: An observational study. Medicine (Baltimore) 98, e14587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscitti P, Ursini F, Cipriani P, Liakouli V, Carubbi F, Berardicurti O, De Sarro G, and Giacomelli R (2017b). Poor clinical response in rheumatoid arthritis is the main risk factor for diabetes development in the short-term: A 1-year, single-centre, longitudinal study. PloS one 12, e0181203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruscitti P.a.A., Saverio and Airò Paolo and Battafarano Norma and Cantarini Luca and Cantatore Francesco Paolo and Carlino Giorgio and D’Abrosca Virginia and Frassi Micol and Frediani Bruno and Iacono Daniela and Maggio Roberta and Masedu Francesco and Mulé Rita and Pantano Ilenia and Prevete Immacolata and Sinigaglia Luigi and Valenti Marco and Viapiana Ombretta and Cipriani Paola and Giacomelli (2019). Anti-interleukin-1 treatment in patients with rheumatoid arthritis and type 2 diabetes (TRACK): a multicentre, randomised, open-label, prospective, controlled, parallel-group tria. PLoS Med In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutkowski DT, Wu J, Back SH, Callaghan MU, Ferris SP, Iqbal J, Clark R, Miao H, Hassler JR, Fornek J, et al. (2008). UPR pathways combine to prevent hepatic steatosis caused by ER stress-mediated suppression of transcriptional master regulators. Dev Cell 15, 829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samuel VT, and Shulman GI (2016). The pathogenesis of insulin resistance: integrating signaling pathways and substrate flux. J Clin Invest 126, 12–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanders FW, and Griffin JL (2016). De novo lipogenesis in the liver in health and disease: more than just a shunting yard for glucose. Biol Rev Camb Philos Soc 91, 452–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasaki T, Moro K, Kubota T, Kubota N, Kato T, Ohno H, Nakae S, Saito H, and Koyasu S (2019). Innate Lymphoid Cells in the Induction of Obesity. Cell Rep 28, 202–217 e207. [DOI] [PubMed] [Google Scholar]
- Sauter NS, Thienel C, Plutino Y, Kampe K, Dror E, Traub S, Timper K, Bedat B, Pattou F, Kerr-Conte J, et al. (2015). Angiotensin II induces interleukin-1beta-mediated islet inflammation and beta-cell dysfunction independently of vasoconstrictive effects. Diabetes 64, 1273–1283. [DOI] [PubMed] [Google Scholar]
- Schlesinger S, Herder C, Kannenberg JM, Huth C, Carstensen-Kirberg M, Rathmann W, Bonhof GJ, Koenig W, Heier M, Peters A, et al. (2018). General and Abdominal Obesity and Incident Distal Sensorimotor Polyneuropathy: Insights Into Inflammatory Biomarkers as Potential Mediators in the KORA F4/FF4 Cohort. Diabetes care. [DOI] [PubMed] [Google Scholar]
- Schodel J, and Ratcliffe PJ (2019). Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol 15, 641–659. [DOI] [PubMed] [Google Scholar]
- Seidman JS, Troutman TD, Sakai M, Gola A, Spann NJ, Bennett H, Bruni CM, Ouyang Z, Li RZ, Sun X, et al. (2020). Niche-Specific Reprogramming of Epigenetic Landscapes Drives Myeloid Cell Diversity in Nonalcoholic Steatohepatitis. Immunity 52, 1057–1074 e1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selkoe DJ (1994). Normal and abnormal biology of the beta-amyloid precursor protein. Annu Rev Neurosci 17, 489–517. [DOI] [PubMed] [Google Scholar]
- Semenza GL (2019). Pharmacologic Targeting of Hypoxia-Inducible Factors. Annu Rev Pharmacol Toxicol 59, 379–403. [DOI] [PubMed] [Google Scholar]
- Seo JB, Riopel M, Cabrales P, Huh JY, Bandyopadhyay GK, Andreyev AY, Murphy AN, Beeman SC, Smith GI, Klein S, et al. (2019). Knockdown of Ant2 Reduces Adipocyte Hypoxia And Improves Insulin Resistance in Obesity. Nat Metab 1, 86–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shabalina IG, Kramarova TV, Nedergaard J, and Cannon B (2006). Carboxyatractyloside effects on brown-fat mitochondria imply that the adenine nucleotide translocator isoforms ANT1 and ANT2 may be responsible for basal and fatty-acid-induced uncoupling respectively. Biochem J 399, 405–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shahzad K, Bock F, Al-Dabet MM, Gadi I, Kohli S, Nazir S, Ghosh S, Ranjan S, Wang H, Madhusudhan T, et al. (2016). Caspase-1, but Not Caspase-3, Promotes Diabetic Nephropathy. Journal of the American Society of Nephrology : JASN 27, 2270–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaul ME, Bennett G, Strissel KJ, Greenberg AS, and Obin MS (2010). Dynamic, M2-like remodeling phenotypes of CD11c+ adipose tissue macrophages during high-fat diet--induced obesity in mice. Diabetes 59, 1171–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, and Flier JS (2006). TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest 116, 3015–3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shimizu I, Yoshida Y, Moriya J, Nojima A, Uemura A, Kobayashi Y, and Minamino T (2013). Semaphorin3E-induced inflammation contributes to insulin resistance in dietary obesity. Cell Metab 18, 491–504. [DOI] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, and Goldfine AB (2006). Inflammation and insulin resistance. J Clin Invest 116, 1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoelson SE, Lee J, and Yuan M (2003). Inflammation and the IKK beta/I kappa B/NF-kappa B axis in obesity- and diet-induced insulin resistance. Int J Obes Relat Metab Disord 27 Suppl 3, S49–52. [DOI] [PubMed] [Google Scholar]
- Simo R, Sundstrom JM, and Antonetti DA (2014). Ocular Anti-VEGF therapy for diabetic retinopathy: the role of VEGF in the pathogenesis of diabetic retinopathy. Diabetes care 37, 893–899. [DOI] [PubMed] [Google Scholar]
- Simo-Servat O, Hernandez C, and Simo R (2012). Usefulness of the vitreous fluid analysis in the translational research of diabetic retinopathy. Mediators Inflamm 2012, 872978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sims R, van der Lee SJ, Naj AC, Bellenguez C, Badarinarayan N, Jakobsdottir J, Kunkle BW, Boland A, Raybould R, Bis JC, et al. (2017). Rare coding variants in PLCG2, ABI3, and TREM2 implicate microglial-mediated innate immunity in Alzheimer’s disease. Nat Genet 49, 1373–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sloan-Lancaster J, Abu-Raddad E, Polzer J, Miller JW, Scherer JC, De Gaetano A, Berg JK, and Landschulz WH (2013). Double-blind, randomized study evaluating the glycemic and anti-inflammatory effects of subcutaneous LY2189102, a neutralizing IL-1beta antibody, in patients with type 2 diabetes. Diabetes care 36, 2239–2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith GI, Mittendorfer B, and Klein S (2019). Metabolically healthy obesity: facts and fantasies. J Clin Invest 129, 3978–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith JA, Das A, Ray SK, and Banik NL (2012). Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res Bull 87, 10–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- So A, De Smedt T, Revaz S, and Tschopp J (2007). A pilot study of IL-1 inhibition by anakinra in acute gout. Arthritis Res Ther 9, R28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solomon DH, Massarotti E, Garg R, Liu J, Canning C, and Schneeweiss S (2011). Association between disease-modifying antirheumatic drugs and diabetes risk in patients with rheumatoid arthritis and psoriasis. JAMA : the journal of the American Medical Association 305, 2525–2531. [DOI] [PubMed] [Google Scholar]
- Song Z, Xiaoli AM, and Yang F (2018). Regulation and Metabolic Significance of De Novo Lipogenesis in Adipose Tissues. Nutrients 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spite M, Hellmann J, Tang Y, Mathis SP, Kosuri M, Bhatnagar A, Jala VR, and Haribabu B (2011). Deficiency of the leukotriene B4 receptor, BLT-1, protects against systemic insulin resistance in diet-induced obesity. J Immunol 187, 1942–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spranger J, Kroke A, Mohlig M, Hoffmann K, Bergmann MM, Ristow M, Boeing H, and Pfeiffer AF (2003). Inflammatory cytokines and the risk to develop type 2 diabetes: results of the prospective population-based European Prospective Investigation into Cancer and Nutrition (EPIC)-Potsdam Study. Diabetes 52, 812–817. [DOI] [PubMed] [Google Scholar]
- Stagg AJ (2018). Intestinal Dendritic Cells in Health and Gut Inflammation. Front Immunol 9, 2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahel M, Becker M, Graf N, and Michels S (2015). SYSTEMIC INTERLEUKIN 1beta INHIBITION IN PROLIFERATIVE DIABETIC RETINOPATHY: A Prospective Open-Label Study Using Canakinumab. Retina. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanley TL, Zanni MV, Johnsen S, Rasheed S, Makimura H, Lee H, Khor VK, Ahima RS, and Grinspoon SK (2011). TNF-alpha antagonism with etanercept decreases glucose and increases the proportion of high molecular weight adiponectin in obese subjects with features of the metabolic syndrome. J Clin Endocrinol Metab 96, E146–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stephens JM, Lee J, and Pilch PF (1997). Tumor necrosis factor-alpha-induced insulin resistance in 3T3-L1 adipocytes is accompanied by a loss of insulin receptor substrate-1 and GLUT4 expression without a loss of insulin receptor-mediated signal transduction. J Biol Chem 272, 971–976. [DOI] [PubMed] [Google Scholar]
- Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, Rensen PC, Voshol PJ, Fantuzzi G, Hijmans A, et al. (2010). The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab 12, 593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strissel KJ, Stancheva Z, Miyoshi H, Perfield JW 2nd, DeFuria J, Jick Z, Greenberg AS, and Obin MS (2007). Adipocyte death, adipose tissue remodeling, and obesity complications. Diabetes 56, 2910–2918. [DOI] [PubMed] [Google Scholar]
- Sun B, and Karin M (2012). Obesity, inflammation, and liver cancer. J Hepatol 56, 704–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun K, Halberg N, Khan M, Magalang UJ, and Scherer PE (2013). Selective inhibition of hypoxia-inducible factor 1alpha ameliorates adipose tissue dysfunction. Mol Cell Biol 33, 904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swann JR, Want EJ, Geier FM, Spagou K, Wilson ID, Sidaway JE, Nicholson JK, and Holmes E (2011). Systemic gut microbial modulation of bile acid metabolism in host tissue compartments. Proc Natl Acad Sci U S A 108 Suppl 1, 4523–4530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takikawa A, Mahmood A, Nawaz A, Kado T, Okabe K, Yamamoto S, Aminuddin A, Senda S, Tsuneyama K, Ikutani M, et al. (2016). HIF-1alpha in Myeloid Cells Promotes Adipose Tissue Remodeling Toward Insulin Resistance. Diabetes 65, 3649–3659. [DOI] [PubMed] [Google Scholar]
- Talukdar S, Oh DY, Bandyopadhyay G, Li D, Xu J, McNelis J, Lu M, Li P, Yan Q, Zhu Y, et al. (2012). Neutrophils mediate insulin resistance in mice fed a high-fat diet through secreted elastase. Nat Med 18, 1407–1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamoutounour S, Guilliams M, Montanana Sanchis F, Liu H, Terhorst D, Malosse C, Pollet E, Ardouin L, Luche H, Sanchez C, et al. (2013). Origins and functional specialization of macrophages and of conventional and monocyte-derived dendritic cells in mouse skin. Immunity 39, 925–938. [DOI] [PubMed] [Google Scholar]
- Tamoutounour S, Henri S, Lelouard H, de Bovis B, de Haar C, van der Woude CJ, Woltman AM, Reyal Y, Bonnet D, Sichien D, et al. (2012). CD64 distinguishes macrophages from dendritic cells in the gut and reveals the Th1-inducing role of mesenteric lymph node macrophages during colitis. Eur J Immunol 42, 3150–3166. [DOI] [PubMed] [Google Scholar]
- Tang SCW, and Yiu WH (2020). Innate immunity in diabetic kidney disease. Nat Rev Nephrol 16, 206–222. [DOI] [PubMed] [Google Scholar]
- Taylor SR, Ramsamooj S, Liang RJ, Katti A, Pozovskiy R, Vasan N, Hwang SK, Nahiyaan N, Francoeur NJ, Schatoff EM, et al. (2021). Dietary fructose improves intestinal cell survival and nutrient absorption. Nature 597, 263–267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesch GH (2017). Diabetic nephropathy - is this an immune disorder? Clin Sci (Lond) 131, 2183–2199. [DOI] [PubMed] [Google Scholar]
- Theriault P, ElAli A, and Rivest S (2015). The dynamics of monocytes and microglia in Alzheimer’s disease. Alzheimers Res Ther 7, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas C, Gioiello A, Noriega L, Strehle A, Oury J, Rizzo G, Macchiarulo A, Yamamoto H, Mataki C, Pruzanski M, et al. (2009). TGR5-mediated bile acid sensing controls glucose homeostasis. Cell Metab 10, 167–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, Miller DK, Molineaux SM, Weidner JR, Aunins J, and et al. (1992). A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature 356, 768–774. [DOI] [PubMed] [Google Scholar]
- Timper K, Hruz P, Beglinger C, and Donath MY (2013). Infliximab in the treatment of Crohn disease and type 1 diabetes. Diabetes care 36, e90–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todoric J, Di Caro G, Reibe S, Henstridge DC, Green CR, Vrbanac A, Ceteci F, Conche C, McNulty R, Shalapour S, et al. (2020). Fructose stimulated de novo lipogenesis is promoted by inflammation. Nat Metab 2, 1034–1045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J, Reimann F, and Gribble FM (2012). Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61, 364–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toubal A, Kiaf B, Beaudoin L, Cagninacci L, Rhimi M, Fruchet B, da Silva J, Corbett AJ, Simoni Y, Lantz O, et al. (2020). Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat Commun 11, 3755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traba J, Kwarteng-Siaw M, Okoli TC, Li J, Huffstutler RD, Bray A, Waclawiw MA, Han K, Pelletier M, Sauve AA, et al. (2015). Fasting and refeeding differentially regulate NLRP3 inflammasome activation in human subjects. The Journal of clinical investigation 125, 4592–4600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traub S, Meier DT, Schulze F, Dror E, Nordmann TM, Goetz N, Koch N, Dalmas E, Stawiski M, Makshana V, et al. (2017). Pancreatic alpha Cell-Derived Glucagon-Related Peptides Are Required for beta Cell Adaptation and Glucose Homeostasis. Cell Rep 18, 3192–3203. [DOI] [PubMed] [Google Scholar]
- Tschop M, and Thomas G (2006). Fat fuels insulin resistance through Toll-like receptors. Nature medicine 12, 1359–1361. [DOI] [PubMed] [Google Scholar]
- van Asseldonk EJ, Stienstra R, Koenen TB, Joosten LA, Netea MG, and Tack CJ (2011). Treatment with Anakinra improves disposition index but not insulin sensitivity in nondiabetic subjects with the metabolic syndrome: a randomized, double-blind, placebo-controlled study. The Journal of clinical endocrinology and metabolism 96, 2119–2126. [DOI] [PubMed] [Google Scholar]
- van Asseldonk EJP, Van Popper PCM, Ballak DB, Stienstra R, Netea MG, and Tack CJ (2012). One week of treatment with the IL-1 receptor antagonist anakinra improves insulin sensitivity in patients with type 1 diabetes mellitus: results from a clinical trial. 48th Annual Meeting of the European Association for the Study of Diabetes Abstract 560. [Google Scholar]
- van Poppel PC, van Asseldonk EJ, Holst JJ, Vilsboll T, Netea MG, and Tack CJ (2014). The interleukin-1 receptor antagonist anakinra improves first-phase insulin secretion and insulinogenic index in subjects with impaired glucose tolerance. Diabetes, obesity & metabolism 16, 1269–1273. [DOI] [PubMed] [Google Scholar]
- Van Tassell BW, Canada J, Carbone S, Trankle C, Buckley L, Oddi Erdle C, Abouzaki NA, Dixon D, Kadariya D, Christopher S, et al. (2017). Interleukin-1 Blockade in Recently Decompensated Systolic Heart Failure: Results From REDHART (Recently Decompensated Heart Failure Anakinra Response Trial). Circ Heart Fail 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Tassell BW, Lipinski MJ, Appleton D, Roberts CS, Kontos MC, Abouzaki N, Melchior R, Mueller G, Garnett J, Canada J, et al. (2018). Rationale and design of the Virginia Commonwealth University-Anakinra Remodeling Trial-3 (VCU-ART3): A randomized, placebo-controlled, double-blinded, multicenter study. Clinical cardiology 41, 1004–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, Ravussin E, Stephens JM, and Dixit VD (2011). The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nature medicine 17, 179–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelias A, Bunting MD, Ormerod KL, Koyama M, Olver SD, Straube J, Kuns RD, Robb RJ, Henden AS, Cooper L, et al. (2018). Recipient mucosal-associated invariant T cells control GVHD within the colon. J Clin Invest 128, 1919–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varma V, Yao-Borengasser A, Rasouli N, Nolen GT, Phanavanh B, Starks T, Gurley C, Simpson P, McGehee RE Jr., Kern PA, and Peterson CA (2009). Muscle inflammatory response and insulin resistance: synergistic interaction between macrophages and fatty acids leads to impaired insulin action. Am J Physiol Endocrinol Metab 296, E1300–1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vilaysane A, Chun J, Seamone ME, Wang W, Chin R, Hirota S, Li Y, Clark SA, Tschopp J, Trpkov K, et al. (2010). The NLRP3 inflammasome promotes renal inflammation and contributes to CKD. Journal of the American Society of Nephrology : JASN 21, 1732–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitale A, Cantarini L, Rigante D, Bardelli M, and Galeazzi M (2015). Anakinra treatment in patients with gout and type 2 diabetes. Clinical rheumatology 34, 981–984. [DOI] [PubMed] [Google Scholar]
- Wang H, Shen L, Sun X, Liu F, Feng W, Jiang C, Chu X, Ye X, Jiang C, Wang Y, et al. (2019). Adipose group 1 innate lymphoid cells promote adipose tissue fibrosis and diabetes in obesity. Nat Commun 10, 3254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X, Ota N, Manzanillo P, Kates L, Zavala-Solorio J, Eidenschenk C, Zhang J, Lesch J, Lee WP, Ross J, et al. (2014). Interleukin-22 alleviates metabolic disorders and restores mucosal immunity in diabetes. Nature 514, 237–241. [DOI] [PubMed] [Google Scholar]
- Wanschel A, Seibert T, Hewing B, Ramkhelawon B, Ray TD, van Gils JM, Rayner KJ, Feig JE, O’Brien ER, Fisher EA, and Moore KJ (2013). Neuroimmune guidance cue Semaphorin 3E is expressed in atherosclerotic plaques and regulates macrophage retention. Arterioscler Thromb Vasc Biol 33, 886–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wedell-Neergaard AS, Lang Lehrskov L, Christensen RH, Legaard GE, Dorph E, Larsen MK, Launbo N, Fagerlind SR, Seide SK, Nymand S, et al. (2018). Exercise-Induced Changes in Visceral Adipose Tissue Mass Are Regulated by IL-6 Signaling: A Randomized Controlled Trial. Cell metabolism. [DOI] [PubMed] [Google Scholar]
- Wei Y, Chen K, Whaley-Connell AT, Stump CS, Ibdah JA, and Sowers JR (2008). Skeletal muscle insulin resistance: role of inflammatory cytokines and reactive oxygen species. Am J Physiol Regul Integr Comp Physiol 294, R673–680. [DOI] [PubMed] [Google Scholar]
- Weisberg SP, Hunter D, Huber R, Lemieux J, Slaymaker S, Vaddi K, Charo I, Leibel RL, and Ferrante AW Jr. (2006). CCR2 modulates inflammatory and metabolic effects of high-fat feeding. J Clin Invest 116, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisberg SP, McCann D, Desai M, Rosenbaum M, Leibel RL, and Ferrante AW Jr. (2003). Obesity is associated with macrophage accumulation in adipose tissue. J Clin Invest 112, 1796–1808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen H, Gris D, Lei Y, Jha S, Zhang L, Huang MT, Brickey WJ, and Ting JP (2011). Fatty acid-induced NLRP3-ASC inflammasome activation interferes with insulin signaling. Nat Immunol 12, 408–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wensveen FM, Jelencic V, Valentic S, Sestan M, Wensveen TT, Theurich S, Glasner A, Mendrila D, Stimac D, Wunderlich FT, et al. (2015). NK cells link obesity-induced adipose stress to inflammation and insulin resistance. Nat Immunol 16, 376–385. [DOI] [PubMed] [Google Scholar]
- Wentworth JM, Naselli G, Brown WA, Doyle L, Phipson B, Smyth GK, Wabitsch M, O’Brien PE, and Harrison LC (2010). Pro-inflammatory CD11c+CD206+ adipose tissue macrophages are associated with insulin resistance in human obesity. Diabetes 59, 1648–1656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wernstedt Asterholm I, Tao C, Morley TS, Wang QA, Delgado-Lopez F, Wang ZV, and Scherer PE (2014). Adipocyte inflammation is essential for healthy adipose tissue expansion and remodeling. Cell Metab 20, 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westwell-Roper CY, Chehroudi CA, Denroche HC, Courtade JA, Ehses JA, and Verchere CB (2015). IL-1 mediates amyloid-associated islet dysfunction and inflammation in human islet amyloid polypeptide transgenic mice. Diabetologia 58, 575–585. [DOI] [PubMed] [Google Scholar]
- Winer S, Chan Y, Paltser G, Truong D, Tsui H, Bahrami J, Dorfman R, Wang Y, Zielenski J, Mastronardi F, et al. (2009). Normalization of obesity-associated insulin resistance through immunotherapy. Nat Med 15, 921–929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wohlford GF, Van Tassell BW, Billingsley HE, Kadariya D, Canada JM, Carbone S, Mihalick VL, Bonaventura A, Vecchie A, Chiabrando JG, et al. (2020). Phase 1B, Randomized, Double-Blinded, Dose Escalation, Single-Center, Repeat Dose Safety and Pharmacodynamics Study of the Oral NLRP3 Inhibitor Dapansutrile in Subjects With NYHA II-III Systolic Heart Failure. J Cardiovasc Pharmacol 77, 49–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wojtaszewski JF, Hansen BF, Gade, Kiens B, Markuns JF, Goodyear LJ, and Richter EA (2000). Insulin signaling and insulin sensitivity after exercise in human skeletal muscle. Diabetes 49, 325–331. [DOI] [PubMed] [Google Scholar]
- Xiao X, Gaffar I, Guo P, Wiersch J, Fischbach S, Peirish L, Song Z, El-Gohary Y, Prasadan K, Shiota C, and Gittes GK (2014). M2 macrophages promote beta-cell proliferation by up-regulation of SMAD7. Proceedings of the National Academy of Sciences of the United States of America 111, E1211–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, and Chen H (2003). Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest 112, 1821–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu L, Kitade H, Ni Y, and Ota T (2015). Roles of Chemokines and Chemokine Receptors in Obesity-Associated Insulin Resistance and Nonalcoholic Fatty Liver Disease. Biomolecules 5, 1563–1579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X, Grijalva A, Skowronski A, van Eijk M, Serlie MJ, and Ferrante AW Jr. (2013). Obesity activates a program of lysosomal-dependent lipid metabolism in adipose tissue macrophages independently of classic activation. Cell Metab 18, 816–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Zhang L, Yu C, Yang XF, and Wang H (2014). Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark Res 2, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yates SL, Burgess LH, Kocsis-Angle J, Antal JM, Dority MD, Embury PB, Piotrkowski AM, and Brunden KR (2000). Amyloid beta and amylin fibrils induce increases in proinflammatory cytokine and chemokine production by THP-1 cells and murine microglia. J Neurochem 74, 1017–1025. [DOI] [PubMed] [Google Scholar]
- Yazdani-Biuki B, Mueller T, Brezinschek HP, Hermann J, Graninger W, and Wascher TC (2006). Relapse of diabetes after interruption of chronic administration of anti-tumor necrosis factor-alpha antibody infliximab: a case observation. Diabetes Care 29, 1712–1713. [DOI] [PubMed] [Google Scholar]
- Yazdani-Biuki B, Stelzl H, Brezinschek HP, Hermann J, Mueller T, Krippl P, Graninger W, and Wascher TC (2004). Improvement of insulin sensitivity in insulin resistant subjects during prolonged treatment with the anti-TNF-alpha antibody infliximab. Eur J Clin Invest 34, 641–642. [DOI] [PubMed] [Google Scholar]
- Ying W, Gao H, Dos Reis FCG, Bandyopadhyay G, Ofrecio JM, Luo Z, Ji Y, Jin Z, Ly C, and Olefsky JM (2021). MiR-690, an exosomal-derived miRNA from M2-polarized macrophages, improves insulin sensitivity in obese mice. Cell Metab 33, 781–790 e785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, Seo JB, Yang BH, Wollam J, Riopel M, et al. (2018). Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity. Cell metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Lee YS, Dong Y, Seidman JS, Yang M, Isaac R, Seo JB, Yang BH, Wollam J, Riopel M, et al. (2019). Expansion of Islet-Resident Macrophages Leads to Inflammation Affecting beta Cell Proliferation and Function in Obesity. Cell Metab 29, 457–474 e455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying W, Riopel M, Bandyopadhyay G, Dong Y, Birmingham A, Seo JB, Ofrecio JM, Wollam J, Hernandez-Carretero A, Fu W, et al. (2017). Adipose Tissue Macrophage-Derived Exosomal miRNAs Can Modulate In Vivo and In Vitro Insulin Sensitivity. Cell 171, 372–384 e312. [DOI] [PubMed] [Google Scholar]
- Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, Strauss-Ayali D, Viukov S, Guilliams M, Misharin A, et al. (2013). Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38, 79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Younossi Z, Tacke F, Arrese M, Chander Sharma B, Mostafa I, Bugianesi E, Wai-Sun Wong V, Yilmaz Y, George J, Fan J, and Vos MB (2019). Global Perspectives on Nonalcoholic Fatty Liver Disease and Nonalcoholic Steatohepatitis. Hepatology 69, 2672–2682. [DOI] [PubMed] [Google Scholar]
- Yuan M, Konstantopoulos N, Lee J, Hansen L, Li ZW, Karin M, and Shoelson SE (2001). Reversal of obesity- and diet-induced insulin resistance with salicylates or targeted disruption of Ikkbeta. Science 293, 1673–1677. [DOI] [PubMed] [Google Scholar]
- Yudanin NA, Schmitz F, Flamar AL, Thome JJC, Tait Wojno E, Moeller JB, Schirmer M, Latorre IJ, Xavier RJ, Farber DL, et al. (2019). Spatial and Temporal Mapping of Human Innate Lymphoid Cells Reveals Elements of Tissue Specificity. Immunity 50, 505–519 e504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamarron BF, Mergian TA, Cho KW, Martinez-Santibanez G, Luan D, Singer K, DelProposto JL, Geletka LM, Muir LA, and Lumeng CN (2017). Macrophage Proliferation Sustains Adipose Tissue Inflammation in Formerly Obese Mice. Diabetes 66, 392–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zatterale F, Longo M, Naderi J, Raciti GA, Desiderio A, Miele C, and Beguinot F (2020). Chronic Adipose Tissue Inflammation Linking Obesity to Insulin Resistance and Type 2 Diabetes. Front Physiol 10, 1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao S, Jang C, Liu J, Uehara K, Gilbert M, Izzo L, Zeng X, Trefely S, Fernandez S, Carrer A, et al. (2020). Dietary fructose feeds hepatic lipogenesis via microbiota-derived acetate. Nature 579, 586–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng C, Yang Q, Cao J, Xie N, Liu K, Shou P, Qian F, Wang Y, and Shi Y (2016). Local proliferation initiates macrophage accumulation in adipose tissue during obesity. Cell Death Dis 7, e2167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou R, Tardivel A, Thorens B, Choi I, and Tschopp J (2010). Thioredoxin-interacting protein links oxidative stress to inflammasome activation. Nat Immunol 11, 136–140. [DOI] [PubMed] [Google Scholar]
- Zinman B, Wanner C, Lachin JM, Fitchett D, Bluhmki E, Hantel S, Mattheus M, Devins T, Johansen OE, Woerle HJ, et al. (2015). Empagliflozin, Cardiovascular Outcomes, and Mortality in Type 2 Diabetes. The New England journal of medicine 373, 2117–2128. [DOI] [PubMed] [Google Scholar]