

Abstract
Mechanobiological cues influence chondrocyte biosynthesis and are often used in tissue engineering applications to improve the repair of articular cartilage in load-bearing joints. In this work, we explore the biophysical effects of an applied dynamic compression on chondrocytes encapsulated in viscoelastic hydrazone covalent adaptable networks (CANs). Here, hydrazone CANs exhibit viscoelastic loss tangents ranging from (9.03 ± 0.01) 10−4 to (1.67 ± 0.09) 10−3 based on the molar percentages of alkyl-hydrazone and benzyl-hydrazone crosslinks. Notably, viscoelastic alkyl-hydrazone crosslinks improve articular cartilage specific gene expression showing higher SOX9 expression in free swelling hydrogels and dynamic compression reduces hypertrophic chondrocyte markers (COL10A1, MMP13) in hydrazone CANs. Interestingly, dynamic compression also improves matrix biosynthesis in elastic benzyl-hydrazone controls but reduces biosynthesis in viscoelastic alkyl-hydrazone CANs. Additionally, intermediate levels of viscoelastic adaptability demonstrate the highest levels of matrix biosynthesis in hydrazone CANs, demonstrating on average 70 ± 4 μg of sulfated glycosaminoglycans per day and 31 ± 3 μg of collagen per day over one month in dynamic compression bioreactors. Collectively, the results herein demonstrate the role of matrix adaptability and viscoelasticity on chondrocytes in hydrazone CANs during dynamic compression, which may prove useful for tissue engineering applications in load-bearing joints.
Keywords: hydrazones, covalent adaptable networks, viscoelasticity, dynamic loading, cartilage tissue engineering
Table of contents
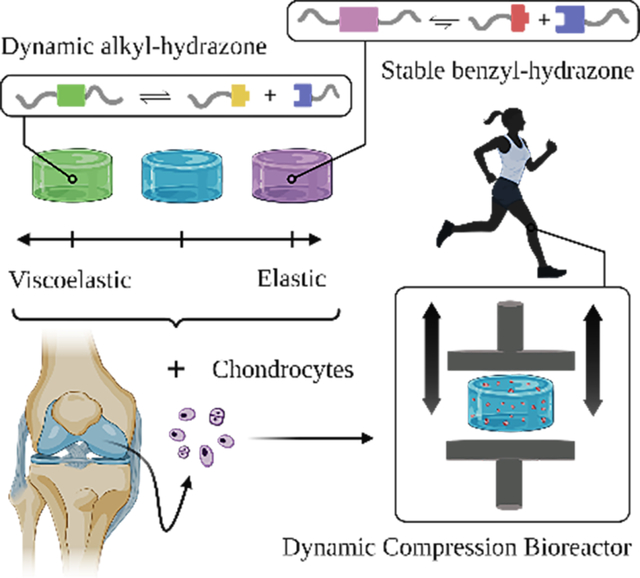
In recent years, growing emphasis has been placed on understanding how matrix mechanics and mechanobiology can be used to design better biomaterials. Here, we use specially designed bioreactors to elucidate how viscoelasticity of hydrazone covalent adaptable networks influences the behavior of chondrocytes during physiologically relevant dynamic compression and our results lend insights for cartilage tissue engineering in load-bearing joints.
Graphical Abstract
1. Introduction
Osteoarthritis is a painful condition where articular cartilage becomes severely degenerated.[1] Unfortunately, osteoarthritis affects more than 30 million people in the United States.[2] Additionally, arthritis-associated medical expenditures and loss of earnings in the United States are estimated to be >$300 billion annually.[3] Osteoarthritis is the most common form of arthritis,[2] and this economic burden is borne disproportionately by patients with osteoarthritis in load-bearing joints.[4]
Because of the need for reparative treatments prior to the onset of osteoarthritis, cartilage tissue engineering has emerged as one promising strategy to address these problems.[5] Cartilage tissue engineering strives to help regenerate damaged cartilage using cells, polymer matrices, and promotive cues.[6] In articulating joints, chondrocytes receive biomechanical cues in the form of dynamic compression,[7] which can increase chondrocyte biosynthesis,[8] when applied at physiologically relevant strains and frequencies.[9] In this way, dynamic compression bioreactors have been used to mimic the native cartilage environment and stimulate chondrocytes for cartilage tissue engineering in vitro. Efforts by tissue engineers to leverage this effect for cartilage tissue engineering have been comprehensively reviewed by Anderson and Johnstone.[10] Briefly, dynamic mechanical compression has been shown to influence chondrocyte anabolic and catabolic activities,[11] gene expression,[12] secretory properties,[13] nitric oxide production,[14] proliferation,[15] calcium signaling,[16] and the bulk mechanical properties of engineered tissues.[17] Further, the material properties of hydrogel scaffolds (e.g., incorporation of cell adhesive ligands,[18] crosslinking density,[19] network microarchitecture,[20] energy dissipation[21]) also influence chondrocyte responses to mechanical compression. These effects have been shown in the context of osteoarthritis, where dynamic compression can improve biosynthesis by chondrocytes isolated from patients with osteoarthritis.[22]
Hydrogel scaffolds used for cartilage tissue engineering in load-bearing joints must provide mechanical strength for withstanding biomechanical forces in articulating joints.[23] Many of the hydrogels used for cartilage tissue engineering are covalently crosslinked to provide resistance to compression in articulating joints.[24] Unfortunately, covalent bonds can limit cellular remodeling,[25] and confine extracellular matrix (ECM) deposition to the pericellular space.[26] While covalent hydrogels exhibit tailorable elastic mechanical properties,[27] native articular cartilage is viscoelastic.[28] Covalent adaptable networks (CANs),[29] are a growing area of interest within the biomaterials community, as these networks can adapt and dynamically reorganize in response to mechanical stresses and strains.[30] The reversibility of network crosslinks imparts viscoelastic material properties,[31] which can capture aspects of the mechanical properties of native tissues.[32] As one example, hydrazone CANs are well-suited for biological applications where the reversibility of the crosslinks occurs under physiological conditions (pH 7.4, 37°C).[33] McKinnon et al. developed viscoelastic hydrazone crosslinked poly(ethylene glycol) (PEG) hydrogels to study C2C12 myoblasts and ES-derived motor neurons.[34,35] Following this work, hydrazone crosslinked hyaluronic acid-based hydrogels have also been developed as viscoelastic cell culture platforms.[36,37] More recently, we used viscoelastic hydrazone CANs to encapsulate chondrocytes and study matrix deposition as a function of the network chemistry. We found that stress relaxation influenced ECM deposition,[38] and that creep compliance impacts chondrocyte morphology during mechanical deformation.[39]
Understanding how mechanobiological cues influence chondrocyte behavior is critical for improving cartilage tissue engineering strategies.[40] In this work, we sought to investigate mechanobiological interactions between viscoelastic matrix properties during dynamic mechanical compression using hydrazone CANs (Figure 1). To our knowledge this manuscript represents the first and only work to examine interactive effects between CAN viscoelasticity and physiologically relevant dynamic compressive loading for cartilage tissue engineering.
Figure 1.

Schematic illustration represents the experimental design for dynamic compression experiments. (a) The alkyl-hydrazone (green) crosslink equilibrium leads to more viscoelastic material properties (e.g., faster stress relaxation) than the more stable benzyl-hydrazone (purple) crosslinks. (b) The viscoelastic properties of hydrazone CANs are varied based on the molar percentage of alkyl-hydrazone versus benzyl-hydrazone crosslinks. (c) Articular chondrocytes were encapsulated in hydrazone hydrogels and allowed three days to recover from isolation and encapsulation stresses. (d) On Day 3, Chondrocyte-laden hydrazone hydrogels were transferred to dynamic compression bioreactors which were programed to apply a cyclic 15% strain at a rate of 1 Hz for one hour each day. Dynamically loaded hydrogels were compared to free swelling controls 3, 10, 17, 24 and 31 days after encapsulation.
First, we formulated hydrazone PEG hydrogels with alkyl-hydrazone and benzyl-hydrazone crosslinks (Figure 1a) and quantified differences in their viscoelastic properties using small amplitude oscillatory shear (SAOS) rheometry (Figure 1b). Although chondrocytes do not require attachment sites for integrin binding in PEG hydrogels,[18] our group has previously incorporated benzaldehyde functionalized arginylglycylaspartic acid (RGD) in hydrazone CANs, which suggests that this platform could be easily modified to support the growth of anchorage dependent cells (e.g. MSCs).[34] In the present work, chondrocytes encapsulated in these networks (Figure 1c) were subjected to long-term culture in dynamic compression bioreactors designed to simulate mechanical stimuli experienced in articulating joints (Figure 1d). Gene expression was quantified by quantitative polymerase chain reaction (qPCR) to study how viscoelasticity and mechanical compression influence articular cartilage specific gene expression. Rates of biosynthesis were measured by quantifying the deposition of sulfated glycosaminoglycans (sGAGs) and collagen over time during dynamic compression. Histological analysis was employed to visualize the spatial distribution of these molecules. This study further validates the application of hydrazone CANs for cartilage tissue engineering and lends insight about how mechanobiological factors can affect chondrocytes and their matrix synthesis to improve cartilage tissue engineering.
2. Formulation of hydrazone CANs with consistent shear moduli
The alkyl-hydrazone and benzyl-hydrazone crosslinks have different forward (k1) and reverse (k−1) reaction rates, and this leads to differences in the chemical equilibria and the rates of crosslink reorganization in co-poly(alkyl-benzyl-hydrazone) CANs (Figure 2).[41] By varying the molar ratio of alkyl-hydrazone (Figure 2a) to benzyl-hydrazone (Figure 2b) crosslinks, the viscoelastic properties of the resulting hydrogels can be tuned in a user-defined manner.[38] To form hydrazone hydrogels, we synthesized 8-arm tripentaerythritol PEG macromers (Mn ~ 10 kDa) with reactive hydrazine, alkyl-aldehyde, and benzaldehyde end groups. Three CAN formulations were designed to achieve a range of viscoelastic properties, based on Flory-Stockmayer percolation theory (Equation 1).[42] Here the percolation threshold (pc) represents the fraction of crosslinks required to form a percolating network, which depends on the stoichiometric ratio (r = [El] / [Nu]) of electrophilic aldehydes [El] to nucleophilic [Nu] hydrazines and the functionality (f) of each type of reactive PEG macromer. The most viscoelastic condition (green) was designed with 78%:22% alkyl-hydrazone to benzyl-hydrazone crosslinks. Formulations were designed to account for network non-idealities and ensure that all three conditions contained percolating networks of stable benzyl-hydrazone crosslinks, as these have been previously shown to provide enhanced stability for long term chondrocyte culture experiments.[38] The most elastic condition was formulated with 0%:100% alkyl-hydrazone to benzyl-hydrazone) crosslinks (purple), and a mixed condition, composed of 39%:61% alkyl-hydrazone to benzyl-hydrazone crosslinks (blue), was included to further probe the experimental space.
Figure 2.

Gelation of alkyl-hydrazone and benzyl-hydrazone CANs. (a) Dynamic alkyl-hydrazone (green) crosslinks formed by reaction of alkyl-aldehyde (yellow) functionalized PEG with hydrazine (blue) functionalized PEG. (b) Stable benzyl-hydrazone (purple) crosslinks formed by the reaction of benzaldehyde (red) functionalized PEG with hydrazine (blue) functionalized PEG. (c) The final G∞ was statistically the same across all formulations. G∞ = [(G′)2 + (G′′)2](½) with plateau values (ΔG′/Δt ≈ ΔG′′/Δt ≈ 0) for each of the three hydrazone hydrogel conditions. (d) In situ gelation was monitored by a time sweep, showing shear storage (G’) and loss (G”) moduli over time. (e) Gelation points for hydrazone CANs were measured at ω = 1 rad s−1 and γ = 1%. Gelation point is defined here as the time required to measure a storage modulus greater than the loss modulus (G’ > G”), with an additional threshold of G’ ≥ 10 Pa to account for instrument error. Traces represent average measurements made in triplicate (n=3) with standard error where appropriate. Statistics represent the results of one-way ANOVA with Tukey’s multiple comparisons test (MCT) showing P ≥ 0.05 = ns, P < 0.05 = *, P < 0.01 = **, P < 0.001 = ***, P < 0.0001 = ****.
| (1) |
Hydrogels were formulated to minimize differences (Figure 2c) in the final shear moduli (G∞) and isolate the effects of viscoelastic crosslink rearrangement from the overall crosslinking density.[19,43] Gelation was monitored by time sweeps, measuring the shear storage (G’) and loss (G”) moduli as a function of time after mixing hydrazine and aldehyde functionalized PEGs. Data illustrate characteristic step-growth polymerizations of hydrazone CANs (Figure 2d). The gelation point is defined here as the time required to measure a storage modulus greater than the loss modulus (G’ > G”), with an additional threshold of G’ ≥ 10 Pa to account for instrument error. The gelation point was delayed for the elastic 0% CANs relative to the other two conditions, containing alkyl-hydrazone crosslinks (39% and 78%), due to the slower kinetics of formation of benzyl-hydrazone bonds at neutral pH (Figure 2e).[44] However, all three conditions consistently formed gels within two minutes of mixing precursor solutions, illustrating gelation timescales relevant for cell encapsulation.
3. The dynamic viscoelastic properties of hydrazone CANs
Viscoelasticity is defined as a materials tendency to elastically resist mechanical deformation while also dissipating energy.[45] Transient step-change rheometry was used to quantify differences in the viscoelastic behavior of the 78%, 39% and 0% alkyl-hydrazone CANs (Figure 3). First, stress relaxation was measured as a function of time (t) (Figure 3a). Normalized shear stress (σ/σmax) was fit to a two element stretched exponential model (Equation 2) to estimate time constants (τ) and stretching parameters (β) for alkyl (a) and benzyl (b) hydrazone crosslinks as a function of their molar composition (X).
Figure 3.

Shear rheometry was used to measure the viscoelastic responses of hydrazone CANs to step-changes. (a) Differences in viscoelastic stress relaxation between hydrazone CANs represented by variations in the normalized shear stress (σ/σmax) over time compared to model fits. (b) Average relaxation times (<τ>) represent the characteristic network reorganization timescales for each formulation. Stress relaxation data was fit with the Kohlrausch–Williams–Watts stretched exponential function (Equation 2) followed by integration to calculate the average relaxation times (Equation 3). (c) Hydrazone CANs relax different amounts of the initial stress over the course of 6 hours. (d) Differences in viscoelastic creep compliance (J) between hydrazone hydrogel formulations over time compared to model fits. (e) Linear average creep rates (<1/η>) as a function of hydrogel compositions. The creep compliance data were fit to (Equation 4), excluding initial creep ringing. (f) Hydrazone CANs were strained by different amounts over the course of 2 hour creep tests, and the final strain (γ) was plotted as a function of network composition. Traces represent average measurements made in triplicate (n=3) with standard error where appropriate. Statistics represent the results of one-way ANOVA with Tukey’s MCT showing P ≥ 0.05 = ns, P < 0.05 = *, P < 0.01 = **, P < 0.001 = ***, P < 0.0001 = ****.
| (2) |
Average relaxation times (<τ>) were then calculated (Equation 3) as characteristic relaxation timescales for each of the three hydrazone CAN formulations (Figure 3b).[46]
| (3) |
The average relaxation time for the most elastic 0% condition was statistically different from the two viscoelastic conditions (39%, 78%). The percentage of the maximum stress relaxed over a six hour period was also quantified to further illustrate differences in viscoelastic stress relaxation (Figure 3c). The 78%, 39% and 0% alkyl-hydrazone CANs relaxed 89 ± 0.36%, 69 ± 1.3%, and 28 ± 7.5% of the initial stress, respectively. Next, the creep compliance (J) was measured over time (Figure 3d), and the average creep compliance rate (1/<η>) for each network was calculated by subtracting the immediate elastic compliance response (J0) (Equation 4).
| (4) |
The results closely corresponded to differences observed during stress relaxation experiments. The viscoelastic conditions exhibited statistically significant creep compliance behavior relative to the elastic 0% condition (Figure 3e). The 78%, 39% and 0% CANs also demonstrated different levels of physical deformation in response to the constant shear stress, corresponding to 5.20 ± 0.33%, 2.46 ± 0.31%, and 0.44 ± 1.48% strain (γ), respectively (Figure 3f).
Native articular cartilage also demonstrates viscoelastic creep compliance and stress relaxation;[47] however, cartilage explants typically have viscoelastic timescales on the order of 100 −1800 s.[48] These timescales are much faster than the timescales reported here for our hydrazone CANs. While these specific hydrazone CAN formulations are not biomimetic,[49] incorporating time-dependent material properties, such as stress relaxation and creep, can still be valuable for controlling chondrocyte behavior for in vitro cartilage tissue engineering.[50] Cartilage tissue engineering is a complex process, which does not necessarily require matching the exact properties of the native tissue. For example, after Engler et al. discovered that matrix elasticity can direct stem cell lineage specification, early mechanobiological research focused on elastic moduli.[51] However, attempting to match the high elastic modulus of articular cartilage can inhibit the regenerative behavior of chondrocytes when encapsulated in non-degradable 3D hydrogels,[52] often impeding integration with the surrounding tissue.[53] Such discrepancies illustrate the need for materials platforms to better recapitulate the extracellular microenvironment, and further optimize viscoelastic mechanobiological cues specific for chondrocytes and cartilage tissue engineering. Hydrazone CANs offer one well-defined viscoelastic platform to study chondrocyte behavior for these purposes.
To further characterize the viscoelastic properties of hydrazone CANs, SAOS rheology was used to measure the storage (G’) and loss moduli (G”) as a function of frequency (ω) and amplitude (γ) (Figure 4). Here, the loss tangent (tan(δ) = G” / G’) is used to quantify the relative contributions of viscous and elastic behaviors, where higher loss tangents reflect greater viscoelasticity. Hydrazone CANs have higher loss tangents with decreasing angular frequency (Figure 4a), which suggests that hydrazone CANs behave more viscously over long timescales and more elastically over short timescales. For example, at high frequencies (ω = 10 rad s−1), the loss tangents were not significantly different and very small (< 2.0 × 10−3), indicating that these hydrazone CAN formulations may be approximated as ideal elastic solids during rapid deformation (Figure 4c). These results are useful for cartilage tissue engineering in load-bearing joints where hydrogel scaffolds must withstand rapid compressive forces from walking, running, and jumping.[54] To confirm this assumption, acellular 100% alkyl-hydrazone and 100% benzyl-hydrazone (i.e. 0% alkyl-hydrazone) CANs were subjected to a physiologically relevant dynamic compression cycle (20% strain, 1 Hz, 1 hour) and the uniaxial force response was measured over time (Figure S1). These tests showed relatively small declines in the final force response for both conditions, further suggesting that both alkyl-hydrazone and benzyl-hydrazone CANs respond elastically during rapid deformation. Importantly, this is not the case for shear rheology at low frequencies (ω = 0.001 rad s−1), where each of the three hydrazone CANs were significantly different from each other (Figure 4b). This property may also be beneficial for cartilage tissue engineering, as cellular processes such as matrix deposition occur over long timescales, which correspond to small frequencies.[55]
Figure 4.

SAOS rheometry to quantify frequency and strain dependent behavior of hydrazone CANs. (a) Frequency sweep spectra illustrate differences in the storage moduli (G’), loss moduli (G”), and loss tangents (tan(δ) = G” / G’) as a function of angular frequency (ω) at constant strain (γ = 1%). (b) Differences between the low frequency loss tangents of hydrazone CANs (ω = 0.001 rad s−1). (c) Measurements of the high frequency loss tangents of hydrazone CANs were not significantly different (ω = 10 rad s−1). (d) Amplitude sweeps illustrate the shear-thinning behavior of hydrazone CANs. Data represent the storage moduli (G’), loss moduli (G”), and loss tangent (tan(δ) = G” / G’) as a function of strain (γ) at constant angular frequency (ω = 1 rad s−1). (e) Each of the three hydrogel conditions showed similar strain-dependent crossovers (tan(δ) =1). (f) Quantification of the pre-strain modulus recovery illustrates that shear-thinning behavior is non-destructive. Percent recovery was calculated using the shear modulus after strain sweeps, expressed as a percentage of the shear modulus before strain sweeps as measured by time sweeps (ω = 1 rad s−1, γ = 1%). Traces represent average measurements made in triplicate (n=3) with standard error where appropriate. Statistics represent the results of one-way ANOVA with Tukey’s MCT showing P ≥ 0.05 = ns, P < 0.05 = *, P < 0.01 = **, P < 0.001 = ***, P < 0.0001 = ****.
All three of the hydrazone CAN formulations investigated in this work demonstrated strain-dependent behavior (Figure 4d). At low strains, the storage (G’) and loss moduli (G”) were constant. However, as the strain exceeded a critical threshold, the storage modulus declined and the loss modulus rose, resulting in strain-dependent crossovers (tan(δ) = 1). The crossover points were not significantly different between conditions, occurring between 200% and 300% strain (Figure 4e). Time sweeps were performed after the strain sweeps to ensure that the disassociation of dynamic hydrazone crosslinks was responsible for this shear-thinning behavior. All three conditions showed full recovery of the pre-strain moduli, illustrating that the shear-thinning behavior was non-destructive (Figure 4f) and not the result of breaking PEG chains. These results could be particularly relevant for enabling minimally invasive injectable delivery of chondrocytes for cartilage tissue engineering,[56] or may prove useful for 3D bioprinting,[57] and additive manufacturing.[58]
4. Hydrazone CANs maintain encapsulated chondrocyte populations during dynamic compression
After characterizing the viscoelastic properties of the hydrazone CANs, we next sought to verify if these formulations would maintain chondrocytes during physiologically relevant dynamic compression. Primary porcine chondrocytes were encapsulated at ~20 million cells per milliliter in hydrazone CANs as this density has been previously shown to resolve differences in matrix deposition by chondrocytes exposed to dynamic compression.[17] Chondrocyte-laden hydrazone CANs were exposed to physiologically relevant dynamic compression (1 hour/day, 1 Hz, 15% strain) in custom build bioreactors for 4 weeks.[9,59]
To track changes in chondrocyte populations caused by dynamic compression, double stranded DNA content was quantified for each condition relative to free swelling controls (Figure 5). This analysis revealed that dynamic compression increased chondrocyte populations over time in the elastic 0% controls, showing statistically significant differences after three weeks of dynamic compression (Day 24). Importantly, chondrocyte populations were generally stable in both the free-swelling controls (Figure S2a) and during dynamic compression culture (Figure S2b), implying that dynamic compression regime did not adversely affect chondrocyte survival in hydrazone CANs.
Figure 5.

Changes in double stranded DNA content due to dynamic compression. Data represents dynamically compressed samples normalized by free swelling controls to show relative changes caused by dynamic loading. Data from four CANs were averaged for each condition (n = 4) with standard error. Statistical significance represents the results of a two-way ANOVA with Tukey’s MCT where P ≥ 0.05 = ns, P < 0.05 = *, P < 0.01 = **, P < 0.001 = ***, P < 0.0001 = ****.
5. Viscoelasticity improves chondrocyte gene expression in free swelling hydrogels and dynamic loading downregulates hypertrophy markers in hydrazone CANs
Polymeric scaffolds with higher energy dissipation have been previously shown to improve expression of chondrogenic markers.[21] To test this phenomena in hydrazone CANs, we examined the relative expression of articular cartilage specific genes in order to establish how the viscoelastic properties of hydrazone CANs influence chondrocyte gene expression during dynamic compressive loading (Figure 6). We investigated three gene expression markers indicative of dedifferentiation; Collagen X (COL10A1), Collagen I (COL1A1), and Matrix Metalloproteinase 13 (MMP13). To complement these negative markers, we also studied the expression of two positive markers; SRY-box Transcription Factor 9 (SOX9) and Collagen II (COL2A1). The results of gene expression experiments were largely non-significantly different, however, notable exceptions illustrate potential benefits of hydrazone CANs for cartilage tissue engineering.
Figure 6.

Relative mRNA expression of articular cartilage specific genes. Gene expression was investigated three days (Day 3) after encapsulation under free swelling conditions (a) and after one week (Day 10) of dynamic compression (b). Dynamic loading data is normalized by free swelling controls at the same time point (Day 10). Data represent expression of genes encoding for collagen X (COL10A1), collagen I (COL1A1), matrix metalloproteinase 13 (MMP13), SRY-box transcription factor 9 (SOX9), and collagen II (COL2A1). Data from four hydrogels were averaged for each condition, excluding samples which did not provide reliable amounts of mRNA (n=3–4) showing mean and standard error. Data were normalized to ribosomal protein L30 (L30) using the ΔΔCq method.[65] Statistical significance represents the result of a two-way ANOVA where # = P < 0.05 between free swelling and dynamic compression at Day 10 and a one-way ANOVA with Tukey’s MCT where * = P < 0.05 for differences between hydrazone CAN formulations.
In order to establish a baseline for chondrocyte gene expression in hydrazone CANs with varied viscoelastic properties, we investigated expression levels during free swelling culture three days after encapsulation (Day 3) (Figure 6a). We found that chondrocytes in all conditions exhibited similar expression levels of the dedifferentiation markers COL10A1, COL1A1, and MMP13, suggesting that expression of these markers is largely independent of hydrazone CAN viscoelasticity. Interestingly, the expression of the positive marker SOX9 showed increased expression with increasing viscoelasticity. These results corroborate previous work which showed upregulation of SOX9 in polymers with higher energy dissipation.[21]
We next sought to investigate how these material properties would influence the transmission of biophysical cues to encapsulated chondrocytes in dynamic compression bioreactors (Figure 6b). Gene expression was analyzed after one week of dynamic compression (Day 10) normalized by free swelling controls (Day 10). This early time point was selected to minimize matrix deposition, as chondrocytes remodel their local microenvironments and can interfere with the transmission of biophysical cues at later time points.[60] We observed that dynamic loading significantly downregulated COL10A1 and MMP13 relative to free swelling controls and that this effect was largely independent of hydrogel viscoelasticity. These data are consistent with prior research suggesting that dynamic loading can help maintain chondrogenic phenotypes, as COL10A1 and MMP13 are markers of hypertrophy and are often associated with osteoarthritis.[61–64] COL2A1 expression was also significantly upregulated in the 78% condition compared to the 39% and 0% conditions, suggesting that viscoelasticity may also be beneficial for the chondrocyte phenotype during dynamic loading. Taken together, these findings indicate that hydrogel viscoelasticity may be beneficial for maintaining chondrocyte compared to elastic conditions, and that dynamic loading can help reduce hypertrophy in hydrazone CANs for cartilage tissue engineering applications.
6. Viscoelasticity of hydrazone CANs influences the effects of dynamic mechanical compression on chondrocyte biosynthesis
While scaffolds used for cartilage tissue engineering must provide mechanical support to resist physiological compression,[66] the embedded chondrocytes must also be able to deposit ECM in order to replace the scaffold with neocartilaginous tissue.[67] Here, we investigated ECM deposition by encapsulated chondrocytes to gauge whether hydrazone CANs would facilitate uniform cartilaginous matrix secretion during dynamic compression.
Collagens are fibrillar proteins which are primarily responsible for the shape and microarchitecture of articular cartilage.[68] sGAGs are a major component of proteoglycans, like aggrecan, which are responsible for retaining water and providing compressive strength.[69] Biochemical assays and histological staining were used to study the deposition of these two key cartilage matrix molecules and assess the development of neocartilaginous tissue over time (Figure 7). The effects of dynamic compression are often more pronounced at later time points, as it takes weeks for neotissue to develop within hydrogel networks.[8] We observed significant differences in both collagen and sGAG content between viscoelastic and elastic formulations (Figure S2) after four weeks of bioreactor culture.[38,50] We also observed increased matrix in the 39% alkyl-hydrazone condition with intermediate levels of crosslink adaptation during both free swelling and dynamic compression culture.
Figure 7.

Interactive effects of mechanobiological cues on chondrocyte ECM deposition. Here blue represents collagen (a-b) and red shows sGAGs (c-d) in hydrazone CANs. Linear regression was used to calculate matrix production rates over the experimental time course showing collagen (a) and sGAG (c) deposition rates (μg day−1) during free swelling and dynamic compression culture. To frame these results in the context of viscoelasticity, data are graphed by the viscoelasticity represented by tan(δ) at 15% strain and 1 rad s−1. This represents the dynamically applied strain at a frequency relevant for cellular mechanosensing,[71] which is useful for interpreting how viscoelasticity influences cell-matrix interactions in hydrazone CANs. Bright field microscopy was used to visualize 20 μm histological sections stained with (b) Masson’s Trichrome showing collagen (blue) and (d) Safranin O showing sGAGs (red) with cell nuclei (black/violet). Scale bars represent 15 μm. Data from four CANs were averaged for each condition (n = 4) with data representing mean values with standard error. Statistical significance represents the results of two-way ANOVA with Tukey’s MCT where P ≥ 0.05 = ns, P < 0.05 = *, P < 0.01 = **, P < 0.001 = ***, P < 0.0001 = **** for differences between hydrogel conditions and Sidak’s MCT where P ≥ 0.05 = ns, P < 0.05 = #, P < 0.01 = ##, P < 0.001 = ###, P < 0.0001 = #### for differences between dynamic compression and free swelling conditions.
To better understand biosynthesis by chondrocytes in hydrazone CANs, linear regression analysis (Figure S3) was performed to extract the rates of collagen (Figure 7a) and sGAG (Figure 7c) deposition over the experimental time course. Strikingly, we observed reversed effects from dynamic compression in viscoelastic and elastic hydrazone CANs (Table 1). Dynamic compression improved matrix biosynthesis in elastic 0% alkyl-hydrazone hydrogels, showing 127 ± 16% collagen deposition and 117 ± 22% sGAG deposition relative to free swelling controls. This effect was reversed for the viscoelastic 78% alkyl-hydrazone hydrogels, where dynamic mechanical compression reduced collagen deposition rates to 71 ± 12% and sGAG deposition rates to 75 ± 16% of free swelling levels. In 78% alkyl-hydrazone CANs, these differences resulted in a statistically significant reduction in collagen matrix production during dynamic compression. A similar phenomenon has been observed previously where degradation improved the deposition of collagen and sGAGs in (un)loaded constructs but not during dynamic compression.[59] However, we do not expect degradation to be the main factor in these networks as all three conditions (78%, 39%, and 0%) contain benzyl-hydrazone crosslinks well above the percolation threshold. To confirm this hypothesis, we measured compressive Young’s moduli for each condition over time (Figure S4). The moduli of chondrocyte-laden constructs subjected to dynamic compression were normalized by their respective free swelling controls. These data show that dynamic compression slightly increases the compressive modulus in all three conditions, although differences were not significant. These results imply that dynamic loading did not cause excessive degradation in the most viscoelastic 78% condition and cannot account for reduced matrix deposition rates. Further, this finding supports load-inhibition rather than transport phenomena as the primary mechanism behind matrix reduction in the 78% hydrogels during dynamic compression. Interestingly, we also observed decreased matrix synthesis rates during dynamic compression for the intermediate 39% condition. However, this effect was less pronounced, showing 93 ± 10% and 86 ± 14% of free swelling sGAG and collagen deposition rates during dynamic compression. These results match expectations, as both the effect of matrix mechanics,[38] and dynamic compression,[17] are typically more pronounced for collagens than sGAGs due the kinetics of matrix synthesis as well as diffusion and assembly considerations.[70]
Table 1.
Viscoelastic properties and rates of chondrocyte matrix deposition for hydrazone CANs during free swelling and dynamic compression culture.
| Loss tangent [tan(δ)] | Free swelling control [μg day−1] | Dynamic compression [μg day−1] | |||
|---|---|---|---|---|---|
| γ = 15% ω = 1 rad s−1 | Collagen | sGAG | Collagen | sGAG | |
| 78% | (1.67 ± 0.09) 10−3 | 43 ± 3 | 52 ± 5 | 30 ± 3 | 47 ± 7 |
| 39% | (1.21 ± 0.14) 10−3 | 36 ± 4 | 75 ± 6 | 31 ± 3 | 70 ± 4 |
| 0% | (9.03 ± 0.05) 10−4 | 20 ± 2 | 38 ± 6 | 25 ± 3 | 45 ± 8 |
To better understand the mechanisms driving differences in neotissue quality we also calculated matrix deposition rates normalized by cell number (Table S1). These results corroborate the bulk matrix analysis for the viscoelastic hydrazone CAN formulations (78% and 39%), showing decreased rates of matrix deposition during dynamic compression relative to free swelling controls. However, for the elastic 0% formulation, accounting for cell number reduces differences in collagen deposition and reverses the effects of dynamic loading on sGAG deposition. These results suggest that cellularity is a primary mechanism driving increased matrix deposition during dynamic compression in the 0% elastic controls. Additionally, adjusting for cell number indicates that mechanically confined chondrocytes in more elastic constructs generate more matrix per cell despite resulting in lower quality neotissue overall. This phenomena has been previously observed by Lee et al. in viscoelastic calcium-alginate hydrogels under free swelling conditions.[50]
The spatial distribution of collagen (Masson’s Trichrome, Figure 7b) and sGAGs (Safranin O, Figure 7d) were also investigated qualitatively by histological sectioning and staining at the final time point (Day 31) but the differences between conditions were subtle, albeit consistent with the results from biochemical assays. Importantly, during both free swelling and dynamic compression culture the intermediate 39% condition with moderately viscoelastic properties demonstrated significantly higher sGAG deposition rates than either extreme. This condition also demonstrated robust collagen deposition surpassed only by the free swelling 78% condition. This effect has been previously observed in hydrazone CANs,[38] and these results further suggest that a balance between dynamic alkyl-hydrazone crosslinks and stable benzyl-hydrazone crosslinks is most applicable for cartilage tissue engineering.
7. Conclusion
Hydrazone covalent adaptable networks (CANs) were synthesized with varied viscoelastic properties (e.g., stress relaxation, creep compliance, frequency dependence, and shear-thinning) by tuning the molar ratio of alkyl-hydrazone to benzyl-hydrazone crosslinks in PEG hydrogels. Incorporation of adaptable alkyl-hydrazone crosslinks led to increased expression articular cartilage specific marker SOX9 under free swelling conditions and dynamic compression downregulated hypertrophy markers (COL10A1 and MMP13) independent of hydrogel viscoelasticity. These results suggest that both viscoelasticity and dynamic compressive loading may be beneficial for maintaining articular cartilage specific phenotypes in hydrazone CANs for cartilage tissue engineering.
We also found that viscoelasticity of hydrazone CANs altered how dynamic mechanical compression influenced rates of matrix deposition by encapsulated chondrocytes. Specifically, dynamic compression improved rates of sGAG and collagen biosynthesis in the elastic benzyl-hydrazone controls (0% alkyl-hydrazone), however, dynamic compression reduced rates of extracellular matrix deposition in viscoelastic 78% and 39% alkyl-hydrazone CANs. Interestingly, sGAG (70 ± 4 μg day−1) and collagen (31 ± 3 μg day−1) deposition rates were only slightly reduced in the 39% alkyl-hydrazone CANs. This resulted in biphasic behavior with respect to viscoelasticity which further illustrates that intermediate levels of adaptable viscoelasticity may be the most beneficial for cartilage tissue engineering in hydrazone CANs. These results provide insight for how mechanobiological cues influence chondrocytes in hydrazone CANs. Additionally, these experiments support the use of dynamic hydrazone CANs as advanced materials which could provide robust mechanical strength and adaptable viscoelastic properties that are well-suited for cartilage tissue engineering in load-bearing joints.
8. Experimental Section/Methods
Organic synthesis:
Chemicals and solvents used in this work were analytical grade and acquired from commercial sources unless otherwise described. PEG-hydrazine was synthesized by HATU (1-[Bis(dimethylamino)methylene]-1H-1,2,3-triazolo[4,5-b]pyridinium3-oxidhexafluorophosphate) coupling to form an amid bond using amine terminated PEG macromers (8 arm, 10 kDa) and tert-butyloxycarbonyl(boc)-protected hydrazinoacetic acid as previously described.[72] Frist, tri-boc-hydrazinoacetic acid (2.2 mol eq. / R-NH2) was reacted with HATU (2.0 mol eq. / R-NH2) and 4-methylmorpholine (5.0 mol eq. / R-NH2) in dimethylformamide (DMF) under argon for 10 minutes. In parallel, PEG amine was deprotonated with 4-methylmorpholine (5.0 mol eq. / R-NH2) in DMF. The two solutions were combined and the reaction was allowed to proceed overnight under argon at room temperature (23°C). Boc-protected PEG-hydrazine was then precipitated dropwise in cold (4°C) diethyl ether (Et2O) and dried in vacuo, before being dissolved in a 50:50 mixture of trifluoracetic acid (TFA) and dichloromethane (DCM). The deprotection reaction was allowed to proceed in a vented flask for 3 hours prior to purification. PEG-benzaldehyde was similarly synthesized using HATU reagent with PEG amine and 4-formylbenzoic acid as described above, excluding the deprotection reaction. Alkyl-PEG-aldehyde was synthesized using Dess-Martin Periodinane (DMP) to oxidize hydroxyl terminated PEG macromers.[73] Hydroxyl PEG (8 arm, 10 kDa) was dissolved with DMP (1.5 equiv. mol eq. / R-OH) in DMF containing catalytic dissolved water. The reaction was allowed to proceed for 3 hours at room temperature (23°C) prior to purification.
Chemical purification:
Crude reaction products were concentrated under reduced pressure, precipitated dropwise in cold diethyl ether (Et2O), centrifuged, and decanted. PEG products were washed in this way three times and dried in vacuo. Functionalized PEGs were then dissolved in deionized water and dialyzed in regenerated cellulose membranes (Spectra/Por MWCO 8,000 kDa) for 48 hours at room temperature (23°C). Polymer solutions were then lyophilized and dissolved in phosphate buffered saline (PBS). Each stock solution was carefully neutralized (pH = 7.0 ± 0.02) as small variations in pH can strongly influence hydrazone kinetics.[41]
1H-NMR spectroscopy:
Proton NMR spectroscopy was used to estimate PEG macromer functionalization (Bruker AV-III, CDCL3, 400 MHz). In each case, PEG chains were ≥ 80% functionalized with the intended products of the oxidation and coupling reactions. Functionalization was calculated by normalizing proton peak integrations to PEG protons in a single polymer arm (113.5 H). Under this normalization scheme, functional group integrations correspond to the protons within each functional unit. The average functionalization for each macromer was then calculated by dividing normalized functional peak integrations by the number of protons in each functional unit. Unmodified PEG-OH, δ = 3.73–3.54 (m, 113.5H). Functionalized PEG-CHO, δ = 9.85–9.64 (s, H), δ = 4.24–4.09 (s, 2H), δ = 3.73–3.54 (m, 113.5H). Unmodified PEG-NH2, δ = 3.73–3.54 (m, 113.5H). Functionalized PEG-Ar-CHO, δ = 10.12–10.02 (s, H), δ = 8.04–7.90 (m, 4H), δ = 3.73–3.54 (m, 113.5H). Functionalized PEG-NBoc-NBoc2, δ = 3.86–3.82 (s, 2H), δ = 3.73–3.54 (m, 113.5H), δ = 1.58–1.51 (d, 18H), δ = 1.51–1.44 (d, 9H). Functionalized PEG-NH-NH2, δ = 3.86–3.82 (s, 2H), δ = 3.73–3.54 (m, 113.5H).
Shear rheology:
Hydrazone CANs were formed in situ between temperature-controlled Peltier plates (TA Instruments DH-R3). CANs were formulated to match conditions for cell culture, off-stoichiometry (r = [El] / [Nu] = 0.8) at 5.1, 5.3 and 5.5 w/v% to compensate for differences in macromer functionalization. Experimental conditions were informed by previous work indicating that a percolating network of benzyl-hydrazone bonds are required to maintain scaffold integrity and promote optimal neotissue development in hydrazone covalent adaptable networks.[38] Shear rheology was performed on acellular hydrogels, as chondrocytes were estimated to account ~1% total volume.[74] Mineral oil was applied to the hydrogel-air-instrument interface to prevent evaporative artifacts. Gelation was monitored by time sweep at 1 rad s−1, 1% strain, and 25°C. Gelation point was defined as the time for the instrument to register a storage modulus greater than the loss modulus with G’ ≥ 10 Pa to account for instrument limitations. For stress relaxation experiments, a 10% shear strain (γ) was applied and the shear stress (σ) was measured as a function of time. For creep experiments, a 100 Pa stress was constantly applied and the creep compliance (J) was measured as a function of time. Frequency sweeps were measured at 1% strain, and strain sweeps were performed at 1 rad s−1. Modulus recovery is defined here as the quotient of the shear modules (G) modulus before and after shear-thinning expressed as a percentage of the initial modulus. MATLAB was used to fit rheology data. Normalized shear stress was fit to a two-element Kohlrausch-Williams-Watts function (Equation 2) to account for heterogeneities such as addition and exchange reaction mechanisms.[75]
Chondrocyte isolation, encapsulation and culture:
Porcine chondrocytes were obtained by digesting cartilage from the femoral condyles and the patellar groove of Yorkshire swine stifle joints.[76] Chondrocytes were pooled prior to encapsulation within hydrogel replicates. Chondrocytes were given 3 days under free swelling conditions to recover from encapsulation stresses prior to dynamic compression culture. Chondrocyte-laden CANs were cultured in chondrocyte growth medium containing high-glucose DMEM (Gibco) supplemented with 10% fetal bovine serum (Gibco), 1% penicillin-streptomycin-fungizone (Gibco, Invitrogen), 50 mg mL−1 L-ascorbate-2-phosphate (Sigma-Aldrich), 40 mg mL−1 L-proline (Sigma-Aldrich), 100 mg mL−1 non-essential amino acids (Gibco), 100 mg mL−1 HEPES buffer (Sigma-Aldrich) and 50 mg mL−1 gentamicin (Invitrogen).
Dynamic compression bioreactors:
Primary porcine chondrocytes were encapsulated at ~20 million cells per milliliter in hydrazone CANs (D = 5 mm, t= 3 mm).[17] Chondrocyte-laden hydrazone CANs were loaded into custom-built dynamic compression bioreactors three days after encapsulation (Day 3).[14] Briefly, bioreactors were controlled by a programed sinusoidal waveform, precisely raising and lowering a platform equipped with permeable pins. Chondrocyte-laden hydrazone CANs (78%, 39%, and 0% alkyl-hydrazone crosslinks) were immersed in culture medium within 24-well plates on top of permeable well inserts. The compression regime was designed to be physiologically relevant, with daily cyclic compression for 1 hour at 1 Hz and 15% strain.[9] Compressive regiments were separated into two primary modes simulating activity and rest. The active mode was designed to simulate walking by applying a cyclic 15% unconfined compressive strain,[59] once a second (1 Hz), based on average walking speed and gait size.[77] Average daily step estimates translate to approximately two hours of walking per day.[78] To simulate reduced activity during patient recovery, this was reduced to 1 hour per day.[79] Chondrocyte-laden hydrazone CANs were exposed to this cycle for 4 weeks.[59]
Quantitative polymerase chain reaction (qPCR):
RNA was isolated by first mechanically homogenizing samples in RLT lysis buffer (Qiagen 79216) for 3 minutes with 5-mm steel beads shaking at 30 Hz (Qiagen TissueLyser). RNA was isolated from the homogenized samples using the RNeasy Mini Kit (Qiagen 74106). The quantity and purity of RNA were measured via spectrophotometry (ND-1000, NanoDrop). cDNA was synthesized from total RNA using the iScript Synthesis kit (Bio-Rad 1708841) and quantified via qRT-PCR using SYBR Green reagents (Bio-Rad 1708884) on an iCycler (Bio-Rad). Relative expression levels were quantified using the ΔΔCq method by normalizing to L30.[65] Primers are listed in Table 2. Relative expression reported in Figure 6a refer to free swelling controls three days after encapsulation (Day 3). Relative expression reported in Figure 6b show data for dynamically compressed samples (Day 10) normalized by free swelling controls at the same time point (Day 10).
Table 2.
Primers used for qRT-PCR.
| Gene | Forward primer (5′–3′) | Reverse primer (5′–3′) |
|---|---|---|
| L30 | AGATTTCCTCAAGGCTGGGC | GCTGGGGTACAAGCAGACTC |
| COL1A1 | GGGCAAGACAGTGATTGAATACA | GGATGGAGGGAGTTTACAGGAA |
| COL2A1 | CCTCAAGAAAGCCCTGCTCA | CCCCACTTACCGGTGTGTTT |
| MMP13 | TCATGCTTTTCCTCCCGGAC | GGGTCCTTGGAGTGGTCAAG |
| SOX9 | CACATCTCTCCCAACGCCAT | GTTGGTGGACCCTGGGATTG |
| COL10A1 | GCTGGTAGGACACCAACTCC | CAAAAGGGCTGTTTGTGGCA |
Biochemical assays:
Hydrogels were flash frozen in liquid nitrogen, lyophilized and then homogenized in digestion buffer by shaking for 10 minutes with 5-mm steel beads at 30 Hz (Qiagen TissueLyser). Homogenized samples were further enzymatically digested in a solution of 125 μg mL−1 papain (Worthington Biochemical) supplemented with 10 mM cysteine (Sigma Aldrich) overnight at 65°C. Sample solutions were centrifuged and the supernatant was used for dimethylmethylene blue (DMMB) assay and PicoGreen (Life Technologies) assay to quantify sGAG content,[80] and the double stranded DNA[81] respectively. A portion of each digest solution was hydrolyzed with an equal volume of 12 M hydrochloric acid for 15 hours at 120°C. Total collagen content was then estimated by hydroxyproline assay.[82]
Linear regression analysis:
Four hydrogel replicates (n=4) were analyzed from five time points (3, 10, 17, 24, and 31 days after encapsulation) to calculate matrix deposition rates for each experimental condition. Linear regression was used to extract linear matrix deposition rates. Linear regression was selected as most appropriate for interpreting chondrocyte behavior after comparing polynomials up to order 6 with extra sum-of-squares F tests where the simpler model is recommended unless the P values are less than 0.05.
Histological sectioning and staining:
Chondrocyte-hydrogel constructs were fixed at room temperature (23°C) for 30 minutes in 10% formalin. CANs were rinsed in DPBS (Gibco) for 30 minutes before incubation at 4°C in optimal cutting temperature (OCT) compound (Tissue-Tek) overnight. Samples were transferred to cryomolds with fresh OCT and flash frozen in liquid nitrogen. 20 μm sections (Leica Cryostat CM1850) were stained with Masson’s Trichrome for collagen and Safranin-O for sGAGs (Leica Autostainer-XL). Cover slides were applied with Permount (Fisher). And slides were imaged by bright field microscopy (Nikon TE-2000).
Statistics:
Traces represent averages of measurements made in triplicate (n=3). Bar graphs show mean values ± standard error. Standard significance thresholds were used to define statistical differences (e.g., P < 0.05 = *, P < 0.01 = **, P < 0.001 = ***, P < 0.0001 = ****). Comparisons of three or more independent groups were analyzed by ordinary 1-way or 2-way ANOVA with multiple comparison tests as noted. Statistical analysis was performed with GraphPad Prism 8 software.
Supplementary Material
Acknowledgements
Some graphics were created using BioRender.com. The authors would like to acknowledge Alex Anderson and Sarah Schoonraad for lending advice concerning dynamic compression equipment. This work was funded by NIH grants (R01 DK120921 & R01 DE016523). B.M.R. was partially supported by a DoE GAANN Fellowship (P200A150211). C.J.W was supported by NIH Predoctoral Fellowship (F31HL142223).
Footnotes
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Benjamin M. Richardson, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA. The BioFrontiers Institute, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA.
Cierra J. Walker, The BioFrontiers Institute, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA. Materials Science and Engineering Program, University of Colorado Boulder, 4001 Discovery Drive, Boulder, CO 80303, USA
Mollie M. Maples, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA. The BioFrontiers Institute, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA.
Mark A. Randolph, Department of Orthopedic Surgery, Massachusetts General Hospital, Harvard Medical School, 55 Fruit St, WAC 435, Boston, MA 02114, USA Division of Plastic Surgery, Massachusetts General Hospital, Harvard Medical School, 15 Parkman St, WACC 453, Boston, MA 02114, USA.
Stephanie J. Bryant, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA. The BioFrontiers Institute, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA. Materials Science and Engineering Program, University of Colorado Boulder, 4001 Discovery Drive, Boulder, CO 80303, USA
Kristi S. Anseth, Department of Chemical and Biological Engineering, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA. The BioFrontiers Institute, University of Colorado Boulder, 3415 Colorado Ave, Boulder, CO 80303, USA.
References
- [1].Altman R, Asch E, Bloch D, Bole G, Borenstein D, Brandt K, Christy W, Cooke TD, Greenwald R, Hochberg M, et al. , Arthritis Rheum. 1986, DOI 10.1002/art.1780290816. [DOI] [PubMed] [Google Scholar]
- [2].Cisternas MG, Murphy L, Sacks JJ, Solomon DH, Pasta DJ, Helmick CG, Alternative Methods for Defining Osteoarthritis and the Impact on Estimating Prevalence in a US Population-Based Survey, NIH Public Access, 2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Murphy LB, Cisternas MG, Pasta DJ, Helmick CG, Yelin EH, Arthritis Care Res. 2018, DOI 10.1002/acr.23425. [DOI] [PubMed] [Google Scholar]
- [4].Institute for Health Metrics and Evaluation, Lancet 2017. [DOI] [PubMed]
- [5].Kon E, Verdonk P, Condello V, Delcogliano M, Dhollander A, Filardo G, Pignotti E, Marcacci M, Di Matteo B, Perdisa F, et al. , Bone Joint Res. 2009, 37, 1565. [Google Scholar]
- [6].Hunziker EB, Osteoarthr. Cartil. 2001, 10, 432. [Google Scholar]
- [7].Chen C, Tambe DT, Deng L, Yang L, Am. J. Physiol. - Cell Physiol 2013, DOI 10.1152/ajpcell.00242.2013. [DOI] [PubMed] [Google Scholar]
- [8].Buschmann MD, Gluzband YA, Grodzinsky AJ, Hunziker EB, J. Cell Sci 1995. [DOI] [PubMed] [Google Scholar]
- [9].Lee DA, Bader DL, J. Orthop. Res 1997, 15, 181. [DOI] [PubMed] [Google Scholar]
- [10].Anderson DE, Johnstone B, Front. Bioeng. Biotechnol 2017, 5, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Nicodemus GD, Bryant SJ, Osteoarthr. Cart 2010, 18, 126. [DOI] [PubMed] [Google Scholar]
- [12].Mauck RL, Byers BA, Yuan X, Tuan RS, Biomech. Model. Mechanobiol. 2007, DOI 10.1007/s10237-006-0042-1. [DOI] [PubMed] [Google Scholar]
- [13].Schneider MC, Barnes CA, Bryant SJ, Biotechnol. Bioeng. 2017, DOI 10.1002/bit.26320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Villanueva I, Hauschulz DS, Mejic D, Bryant SJ, Osteoarthr. Cartil. 2008, 16, 909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Lee DA, Noguchi T, Frean SP, Lees P, Bader DL, in Biorheology, 2000. [PubMed] [Google Scholar]
- [16].O’Conor CJ, Leddy HA, Benefield HC, Liedtke WB, Guilak F, Proc. Natl. Acad. Sci. U. S. A. 2014, DOI 10.1073/pnas.1319569111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Mauck RL, Seyhan SL, Ateshian GA, Hung CT, Ann. Biomed. Eng. 2002, DOI 10.1114/1.1512676. [DOI] [PubMed] [Google Scholar]
- [18].Villanueva I, Weigel CA, Bryant SJ, Acta Biomater. 2009, 5, 2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Bryant SJ, Chowdhury TT, Lee DA, Bader DL, Anseth KS, Ann. Biomed. Eng. 2004, 32, 407. [DOI] [PubMed] [Google Scholar]
- [20].Roberts JJ, Bryant SJ, Biomaterials 2013, 34, 9969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Abdel-Sayed P, Darwiche SE, Kettenberger U, Pioletti DP, Biomaterials 2014, 35, 1890. [DOI] [PubMed] [Google Scholar]
- [22].Jeon JE, Schrobback K, Hutmacher DW, Klein TJ, Osteoarthr. Cartil. 2012, DOI 10.1016/j.joca.2012.04.019. [DOI] [PubMed] [Google Scholar]
- [23].Martínez-Moreno D, Jiménez G, Gálvez-Martín P, Rus G, Marchal JA, Biochim. Biophys. Acta - Mol. Basis Dis 2019, DOI 10.1016/j.bbadis.2019.03.011. [DOI] [PubMed] [Google Scholar]
- [24].Bas O, Catelas I, De-Juan-Pardo EM, Hutmacher DW, Adv. Drug Deliv. Rev. 2018, DOI 10.1016/j.addr.2018.07.015. [DOI] [PubMed] [Google Scholar]
- [25].Nicodemus GD, Skaalure SC, Bryant SJ, Acta Biomater. 2011, 7, 492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Bryant SJ, Anseth KS, J. Biomed. Mater. Res. 2002, 59, 63. [DOI] [PubMed] [Google Scholar]
- [27].Roberts JJ, Earnshaw A, Ferguson VL, Bryant SJ, J. Biomed. Mater. Res. Part B Appl. Biomater 2011, 99B, 158. [DOI] [PubMed] [Google Scholar]
- [28].Spirt AA, Mak AF, Wassell RP, J. Orthop. Res. 1989, 7, 43. [DOI] [PubMed] [Google Scholar]
- [29].Mcbride MK, Worrell BT, Brown T, Cox LM, Sowan N, Wang C, Podgorski M, Martinez AM, Bowman CN, Annu. Rev. Chem. Biomol. Eng 2019, 10, 175. [DOI] [PubMed] [Google Scholar]
- [30].Zou W, Dong J, Luo Y, Zhao Q, Xie T, Adv. Mater. 2017, 29, 1606100. [DOI] [PubMed] [Google Scholar]
- [31].Rosales AM, Anseth KS, Nat. Publ. Gr. 2016, 1, 1. [Google Scholar]
- [32].Tang S, Richardson BM, Anseth KS, 2020, DOI 10.1016/j.pmatsci.2020.100738. [DOI] [Google Scholar]
- [33].Kölmel DK, Kool ET, Kö DK, Kool ET, Kölmel DK, Kool ET, Chem. Rev. 2017, 117, 10358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].McKinnon DD, Domaille DW, Cha JN, Anseth KS, Adv. Mater. 2014, 26, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].McKinnon DD, Domaille DW, Brown TE, Kyburz KA, Kiyotake E, Cha JN, Anseth KS, Soft Matter 2014, 10, 9230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Lou J, Stowers R, Nam S, Xia Y, Chaudhuri O, Biomaterials 2018, 154, 213. [DOI] [PubMed] [Google Scholar]
- [37].Lou J, Liu F, Lindsay CD, Chaudhuri O, Heilshorn SC, Xia Y, Adv. Mater. 2018, 30, 1705215. [DOI] [PubMed] [Google Scholar]
- [38].Richardson BM, Wilcox DG, Randolph MA, Anseth KS, Acta Biomater. 2019, 83, 71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Richardson B, Walker C, Macdougall L, Hoye J, Randolph M, Bryant S, Anseth KS, Biomater. Sci. 2020, DOI 10.1039/d0bm00860e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sanchez-Adams J, Leddy HA, McNulty AL, O’Conor CJ, Guilak F, Curr. Rheumatol. Rep. 2014, DOI 10.1007/s11926-014-0451-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].McKinnon DD, Domaille DW, Cha JN, Anseth KS, Chem. Mater. 2014, 26, 2382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Sahini M, Sahimi M, Applications Of Percolation Theory, 1994. [Google Scholar]
- [43].Anseth KS, Bowman CN, Brannon-Peppas L, Biomaterials 1996, 17, 1647. [DOI] [PubMed] [Google Scholar]
- [44].Kool ET, Crisalli P, Chan KM, Org. Lett. 2014, 16, 1454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ferry JD, Viscoelastic Properties of Polymers, 1980. [Google Scholar]
- [46].Brown TE, Carberry BJ, Worrell BT, Dudaryeva OY, McBride MK, Bowman CN, Anseth KS, Biomaterials 2018, 178, 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Mow VC, Kuei SC, Lai WM, Armstrong CG, J. Biomech. Eng. 1980, 102, 73. [DOI] [PubMed] [Google Scholar]
- [48].June RK, Neu CP, Barone JR, Fyhrie DP, Mater. Sci. Eng. C 2011, DOI 10.1016/j.msec.2010.11.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Shin H, Jo S, Mikos AG, Biomaterials 2003, DOI 10.1016/S0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- [50].Lee HP, Gu L, Mooney DJ, Levenston ME, Chaudhuri O, Nat. Mater. 2017, 16, 1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Engler AJ, Sen S, Sweeney HL, Discher DE, Cell 2006, 126, 677. [DOI] [PubMed] [Google Scholar]
- [52].Guimarães CF, Gasperini L, Marques AP, Reis RL, Nat. Rev. Mater. 2020, 5, 351. [Google Scholar]
- [53].Yang YHK, Ogando CR, Barabino GA, J. Funct. Biomater. 2020, DOI 10.3390/jfb11010005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mann RA, Hagy J, Am. J. Sports Med. 1980, DOI 10.1177/036354658000800510. [DOI] [PubMed] [Google Scholar]
- [55].Cheema U, Nazhat SN, Alp B, Foroughi F, Anandagoda N, Mudera V, Brown RA, Biotechnol. Bioprocess Eng. 2007, DOI 10.1007/BF02931797. [DOI] [Google Scholar]
- [56].Liu M, Zeng X, Ma C, Yi H, Ali Z, Mou X, Li S, Deng Y, He N, Bone Res. 2017, 514, DOI 10.1038/boneres.2017.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Wang LL, Highley CB, Yeh Y-C, Galarraga JH, Uman S, Burdick JA, J. Biomed. Mater. Res. Part A 2018, 106, 865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Malda J, Visser J, Melchels FP, Jüngst T, Hennink WE, Dhert WJA, Groll J, Hutmacher DW, Adv. Mater. 2013, DOI 10.1002/adma.201302042. [DOI] [PubMed] [Google Scholar]
- [59].Roberts JJ, Nicodemus GD, Greenwald EC, Bryant SJ, Clin. Orthop. Relat. Res. 2011, 469, 2725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Knight MM, Lee DA, Bader DL, Biochim. Biophys. Acta - Mol. Cell Res 1998, 1405, 67. [DOI] [PubMed] [Google Scholar]
- [61].Aigner T, Reichenberger E, Bertling W, Kirsch T, Stöß H, von der Mark K, Virchows Arch. B Cell Pathol. Incl. Mol. Pathol. 1993, DOI 10.1007/BF02899263. [DOI] [PubMed] [Google Scholar]
- [62].D’Angelo M, Yan Z, Nooreyazdan M, Pacifici M, Sarment DS, Billings PC, Leboy PS, J. Cell. Biochem. 2000, DOI . [DOI] [PubMed] [Google Scholar]
- [63].Aisenbrey EA, Bryant SJ, J. Mater. Chem. B 2016, DOI 10.1039/c6tb00006a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Aisenbrey EA, Bryant SJ, Biomaterials 2019, 190–191, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Livak KJ, Schmittgen TD, Methods 2001, 25, 402. [DOI] [PubMed] [Google Scholar]
- [66].Akalp U, Bryant SJ, Vernerey FJ, Soft Matter 2016, DOI 10.1039/c6sm00583g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Huey DDJ, Hu JJC, Athanasiou KAK, Science (80-. ). 2012, 6933, 917. [Google Scholar]
- [68].Sophia-Fox AJ, Bedi A, Rodeo SA, Sport. Heath 2009, 1, 461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Hardingham TE, Fosang AJ, FASEB J. 1992, DOI 10.1096/fasebj.6.3.1740236. [DOI] [PubMed] [Google Scholar]
- [70].Dhote V, Vernerey FJ, Biomech. Model. Mechanobiol. 2014, DOI 10.1007/s10237-013-0493-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Charrier EE, Pogoda K, Wells RG, Janmey PA, Nat. Commun. 2018, 9, DOI 10.1038/s41467-018-02906-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Carpino LA, J. Am. Chem. Soc. 1993, 115, 4397. [Google Scholar]
- [73].Meyer SD, Schreiber SL, J. Org. Chem. 1994, 59, 7549. [Google Scholar]
- [74].Sasazaki Y, Seedhom BB, Shore R, Rheumatology 2008, DOI 10.1093/rheumatology/ken341. [DOI] [PubMed] [Google Scholar]
- [75].Berry GC, Plazek DJ, Rheol. Acta 1997, 36, 320. [Google Scholar]
- [76].Yoo JJ, Bichara DA, Zhao X, Randolph MA, Gill TJ, J. Biomed. Mater. Res. A 2011, 99, 102. [DOI] [PubMed] [Google Scholar]
- [77].Mohler BJ, Thompson WB, Creem-Regehr SH, Pick HL, Warren WH, Exp. Brain Res. 2007, 181, 221. [DOI] [PubMed] [Google Scholar]
- [78].Tudor-Locke C, Bassett DR, Sport. Med. 2004, 34, 1. [DOI] [PubMed] [Google Scholar]
- [79].Hamel MB, Toth M, Legedza A, Rosen MP, Arch. Intern. Med. 2008, DOI 10.1001/archinte.168.13.1430. [DOI] [PubMed] [Google Scholar]
- [80].Farndale RW, Sayers CA, Barrett AJ, Connect. Tissue Res. 1982, 9, 247. [DOI] [PubMed] [Google Scholar]
- [81].Singer VL, Jones LJ, Yue ST, Haugland RP, Anal. Biochem. 1997, DOI 10.1006/abio.1997.2177. [DOI] [PubMed] [Google Scholar]
- [82].Woessner JFF, Arch. Biochem. Biophys. 1961, 93, 440. [DOI] [PubMed] [Google Scholar]
- [83].Park S, Hung CT, Ateshian GA, Osteoarthr. Cartil. 2004, DOI 10.1016/j.joca.2003.08.005. [DOI] [PubMed] [Google Scholar]
- [84].Sridhar BV, Brock JL, Silver JS, Leight JL, Randolph MA, Anseth KS, Adv. Healthc. Mater. 2015, 4, 702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Stegemann H, Stalder K, Clin. Chim. Acta 1967, 18, 267. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.