

Summary
Rift Valley Fever Virus (RVFV) is a zoonotic pathogen with pandemic potential. RVFV entry is mediated by the viral glycoprotein (Gn), but host entry factors remain poorly defined. Our genome-wide CRISPR screen identified low-density lipoprotein receptor-related protein 1 (mouse Lrp1/human LRP1), heat shock protein (Grp94), and receptor associated protein (RAP) as critical host factors for RVFV infection. RVFV Gn directly binds to specific clusters Lrp1 and is glycosylation independent. Exogenous addition of murine RAP domain 3 (mRAPD3) and anti-Lrp1 antibodies neutralize RVFV infection in taxonomically diverse cell lines. Mice treated with mRAPD3 and infected with pathogenic RVFV are protected from disease and death. A mutant mRAPD3 that binds Lrp1 weakly failed to protect from RVFV infection. Altogether, these data support Lrp1 as a host entry factor for RVFV infection and defines a new target to limit RVFV infections.
Keywords: Rift Valley Fever virus, Lrp1, viral entry, CRISPR screen
Graphical Abstract
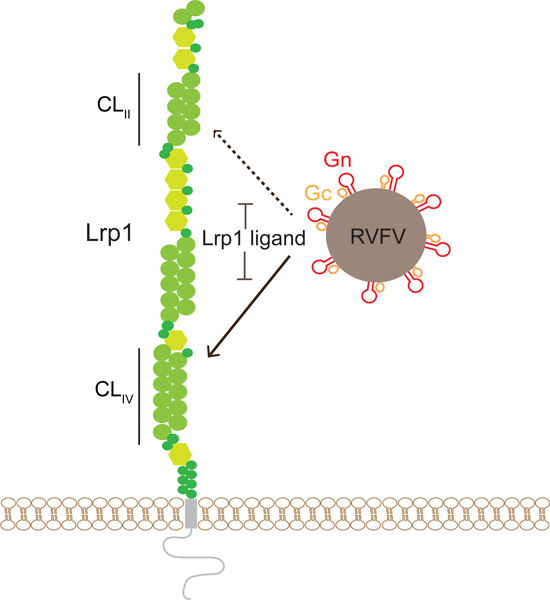
In Brief:
Lrp1 is identified as an essential host entry factor for Rift Valley Fever virus and potential target for therapy against this pathogen.
Introduction
Emerging viral diseases can be unpredictable and have a devastating impact on human health and the global economy, as evidenced by the recent outbreaks of Ebola virus (2014–15, 2018, and 2020), Zika virus (2015–16), and the ongoing SARS-CoV-2 pandemic. Rift Valley fever virus (RVFV) is a mosquito-borne phlebovirus that belongs to the Phenuiviridae family (formerly Bunyaviridae) of negative-sense RNA viruses. The geographic range of RVFV includes most of Africa, Madagascar, and the Saudi Arabian peninsula. Importantly, competent mosquito species are found in North America and Europe, and climate change is rapidly altering the natural habitat of RVFV-competent mosquito and reservoir species (Kraemer et al., 2019, Turell et al., 2008, Brustolin et al., 2017, Turell et al., 2013). RVFV causes severe disease in livestock, including sheep and cattle, dramatically impacting the socio-economic framework in endemic areas (Munyua et al., 2010, Rich and Wanyoike, 2010). RVFV is zoonotically transmitted from animals to people, and human infections can result in severe health consequences, including hepatitis, hemorrhagic fever, encephalitis, and retinal vasculitis (Madani et al., 2003, Hassan et al., 2011). The World Health Organization (WHO) prioritized research on RVFV owing to its public health risk and epidemic potential (Organization, 2018). Despite its significance to human health and the potential to negatively impact the economic landscape, there are no safe and efficacious prophylactic or therapeutic treatment options for human use. This gap is in part due to our lack of knowledge on host factors that contribute to cellular RVFV infection.
RVFV is an enveloped virus with a tripartite genome: L (large) segment, M (medium) segment, and S (small) segment. L encodes the viral RNA-dependent RNA polymerase (RdRp). M encodes the glycoprotein precursor (GPC) and the nonstructural protein NSm. S encodes for the nucleocapsid protein N and nonstructural protein NSs (Knipe and Howley, 2013). GPC is post-translationally cleaved into Gn and Gc. Gn forms the glycoprotein spikes, and Gc is a class II fusion protein that remains oriented away from the viral membrane. Gn and Gc together forms an icosahedral lattice on the virion surface (Freiberg et al., 2008). The viral Gn/Gc complexes mediate cell entry and fusion. However, the host proteins involved in the process of entry are not well understood.
RVFV infects the liver in animals including livestock, humans, and laboratory rodents. Cellular tropism of RVFV is very broad, and most cell types can become infected by RVFV, including neurons, epithelial cells, macrophages, granulocytes, pancreatic islet cells, adrenal glands, ovaries, testes, and placenta (Odendaal et al., 2020, Gommet et al., 2011, Gaudreault et al., 2015, Scharton et al., 2015, Hartman et al., 2014, McMillen et al., 2018). Early studies identified the lectin DC-SIGN on skin dendritic cells (DCs) as a factor for the internalization of RVFV through an incompletely defined mechanism (Léger et al., 2016, Lozach et al., 2011, Phoenix et al., 2016). Lectin molecules closely related to DC-SIGN, such as L-SIGN and DC-SIGNR, are found on RVFV-permissive cells, including hepatocytes. Although prior studies show that DC-SIGN is important for RVFV internalization, deglycosylation of the virus did not reduce infectivity, suggesting that attachment and entry may require distinct proteins (Hofmann et al., 2013). Another study identified heparan sulfate as a potential attachment factor for RVFV; however, removal of heparan sulfate reduced but did not eliminate RVFV infection (Riblett et al., 2016, de Boer et al., 2012). While these studies implicate several host factors in RVFV infection, definitive insights into host proteins that are essential for RVFV infection in different cell types and across taxonomically diverse species have yet to be described.
Given its broad cellular tropism, host factors that facilitate RVFV entry into cells from tissues relevant are yet to be understood for RVFV infection. To address this, we conducted an unbiased genome-wide CRISPR/Cas9 screen where infection by the virulent ZH501 strain of RVFV was used to identify host factors that are either pro-viral or anti-viral. We identified the surface receptor low-density lipoprotein (LDL) receptor-related protein 1 (Lrp1 in mice and LRP1 in humans) as an essential host factor. The screen also identified RAP and GRP94, both proteins that modulate Lrp1 surface presentation and function. Clonal knockout (KO) cells lacking Lrp1, RAP, or GRP94 show significantly reduced infection by both the pathogenic RVFV ZH501 strain and an attenuated vaccine strain, RVFV MP12GFP. Inhibition of the interaction between RVFV Gn glycoprotein and Lrp1 domain clusters using recombinantly purified mouse RAP domain 3 (mRAPD3) protein or anti-Lrp1 antibodies blocked RVFV entry to target cells from a range of host species. The significance of the Gn-Lrp1 interaction was evaluated in vivo in a mouse model, whereby intracranial infection (IC) of RVFV ZH501 with co-injection with mRAPD3 resulted in significant protection from disease and death. A mutant mRAPD3 has diminished binding to two of the four clusters within Lrp1 termed cluster II (CLII) and cluster IV (CLIV) and shows a correspondingly diminished ability to inhibit RVFV infection in cell culture and in mice. These results demonstrate that Lrp1 is an essential host factor for RVFV infection and that a direct interaction between Lrp1 and the viral Gn protein plays a key role in the infection process.
Results
CRISPR/Cas 9 screen identified multiple host factors essential for RVFV infection.
To identify host factors critical for RVFV infection, we used a previously described unbiased CRISPR/Cas9 library in a murine microglial BV2 cell line (Figure 1A) (Orvedahl et al., 2019). Using the pathogenic BSL-3 strain RVFV ZH501, we assessed the cytopathic effect (CPE) of RVFV infection in BV2 cells expressing Cas9 protein. Near 100% CPE was achieved within 48 hours post-infection (hpi) using multiplicities of infection (MOI) between 0.01–1.0 (Figure S1A-B). The BV2 library, consisting of 40 million cells transduced with lentiviruses expressing single guide RNAs (sgRNA) targeting 20,000 unique genes (approximately 4 guides/gene), was screened at an average of 500x redundancy. For the screen, initial infection of the library at MOI 0.1 and 0.01 resulted in significant CPE (Figure 1B). At around 8 days post-infection (dpi), we observed survivor clusters (colonies) in transduced but not control cells. Surviving cells were resistant to re-infection with RVFV (Figure 1C), suggesting that the resistant cells lacked pro-viral factor(s) required for RVFV infection.
Figure 1. A pooled genome-scale CRISPR screen identifies Lrp1 and Lrp1-associated proteins RAP and Grp94 as critical proteins for RVFV infection.
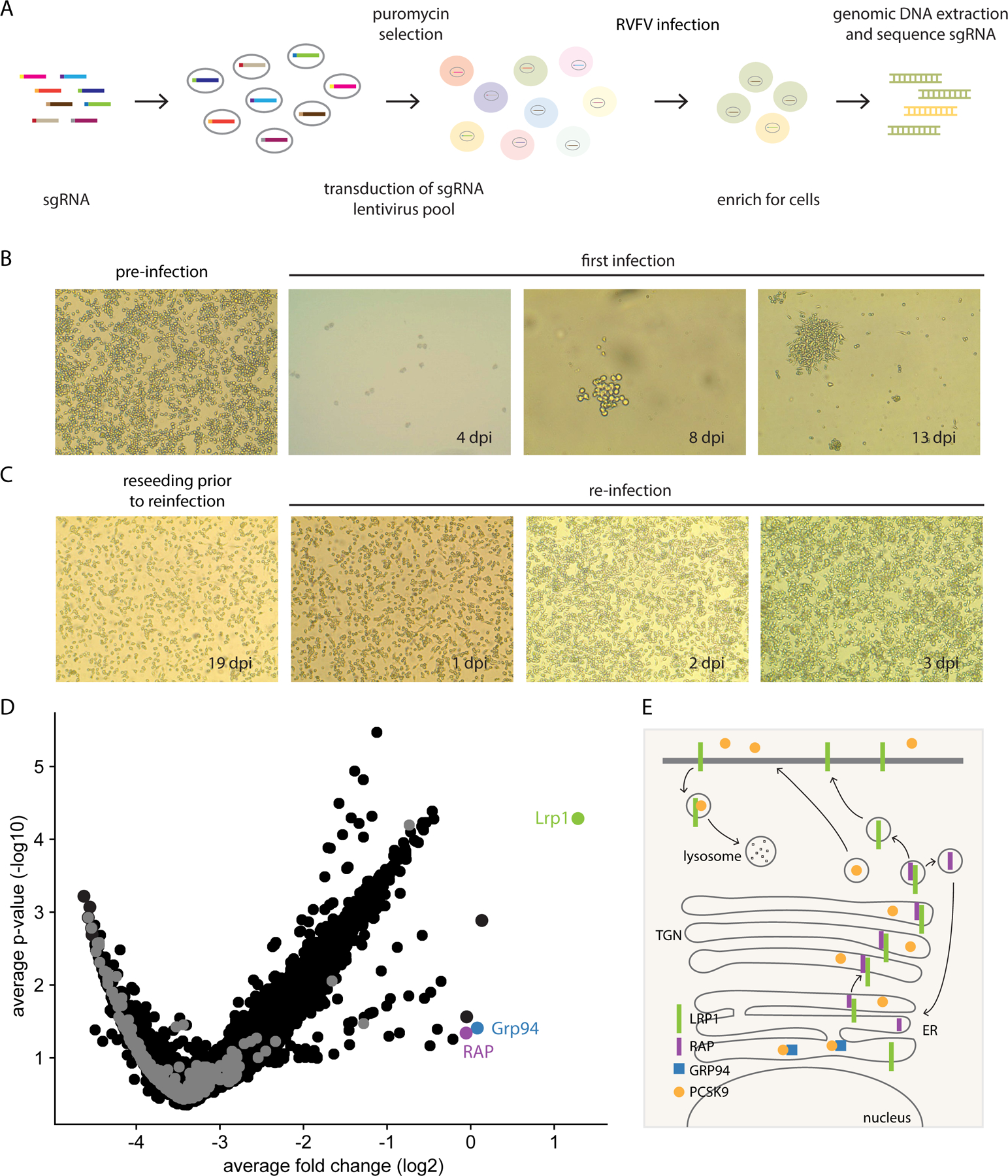
A. Schematic of the CRISPR/Cas9 screen in BV2 cells. B. Light microscope images (4X) of BV2 cells before infection and at 4 different time points post-infection. C. At 18 dpi, surviving cells were reseeded into new flasks for reinfection on 19 dpi and imaged at 4X by light microscopy. D. Volcano plot analysis of the BV2 screen results of surviving cells from the initial infection at an MOI 0.1. E. Summary of key interactions that modulate Lrp1 surface presentation, including RAP and Grp94.
Cells from the initial infections (MOI 0.1 or 0.01) and reinfections were subjected to next-generation sequencing (NGS) and analysis (Figure 1D). Among the candidate pro-viral genes, we identified the low-density lipoprotein (LDL) receptor-related protein 1 (Lrp1), lipoprotein receptor-related protein-associated protein 1 (Lrpap1; herein RAP) (Bu and Schwartz, 1998), and endoplasmin (Hsp90b1; herein Grp94) (Poirier et al., 2015) (Figure S1C). We also identified other known regulators of Lrp1 expression such as Pcsk9 (Figure S1C). Together, the connectivity of the genes from our screen suggests a key role for LRP1 and its regulators in RVFV infection.
Lrp1 is a member of the LDL receptor family (LDLR). LDLRs are highly conserved across species and plays roles in lipid metabolism, clearance of circulating lipoproteins including LDL, and in a variety of endocytic and inflammatory signaling processes relevant to lipid metabolism, atherosclerosis, and neurohomeostasis (Gonias and Campana, 2014, Heissig et al., 2020, Potere et al., 2019). Lrp1 is ubiquitously expressed, with higher levels of expression in the liver, placenta, and brain (Gonias and Campana, 2014, Herz and Strickland, 2001, Potere et al., 2019). Lrp1 is an essential gene as null mutations in the gene are embryonically lethal in mice (Herz et al., 1992). RAP is an important molecular chaperone of Lrp1 that universally inhibits ligand interaction to ensure passage of Lrp1 from the ER to the cell surface (Bu, 2001). Grp94 is an endoplasmic reticulum resident chaperone that controls Lrp1 expression by inhibiting degradation (Figure 1E) (Poirier et al., 2015).
RVFV infection is reduced in cells lacking Lrp1.
To determine the role of LRP1 in RVFV infection, we generated clonal BV2 cells with knockout alleles of Lrp1 (Lrp1KO C3 and R1-R6) by deleting 10 kb sequences containing exons 1 and 2 (Figure S2A-S2C and Figure 2A). Clone R3 displayed hypomorphic Lrp1 expression from an allele with an 8 kb deletion, leaving exon 1 intact and maintained the reading frame of the transcript (Figure S2D) whereas the other cell lines lacked detectable Lrp1 protein expression (Figure 2A). RVFV ZH501 infection of these Lrp1-deficient clones resulted in diminished viral RNA and Gn protein expression at 18 hpi (Figure 2B and Figure S2E). Based on these results, we selected Lrp1PKO R3 and Lrp1KO R4 clones for further characterization. Infection of these cells with the vaccine strain MP12 expressing GFP in place of NSs (MP12GFP) resulted in decreased infection as assayed by flow cytometry for GFP (Figure 2D-E). At earlier time points (3 and 6 hpi), MP12GFP infection was highly reduced in Lrp1KO R4 cells compared to BV2 WT as determined by examination of GFP expression by microscopy (Figure 2C and Figure S2F) or viral RNA levels by RT-qPCR analysis (Figure S2G). To control for non-specific effects of Lrp1 mutation on viral infection in general, we infected Lrp1KO R4 cells with influenza A (IAV) PR8 strain and measured viral RNA levels by RT-qPCR at 6 hpi (Figure S2H). There was no significant effect of mutation of Lrp1 on IAV infection indicating that Lrp1 is a critical host factor specific for RVFV infection.
Figure 2. LRP1 is essential for RVFV infection of BV2 cells.
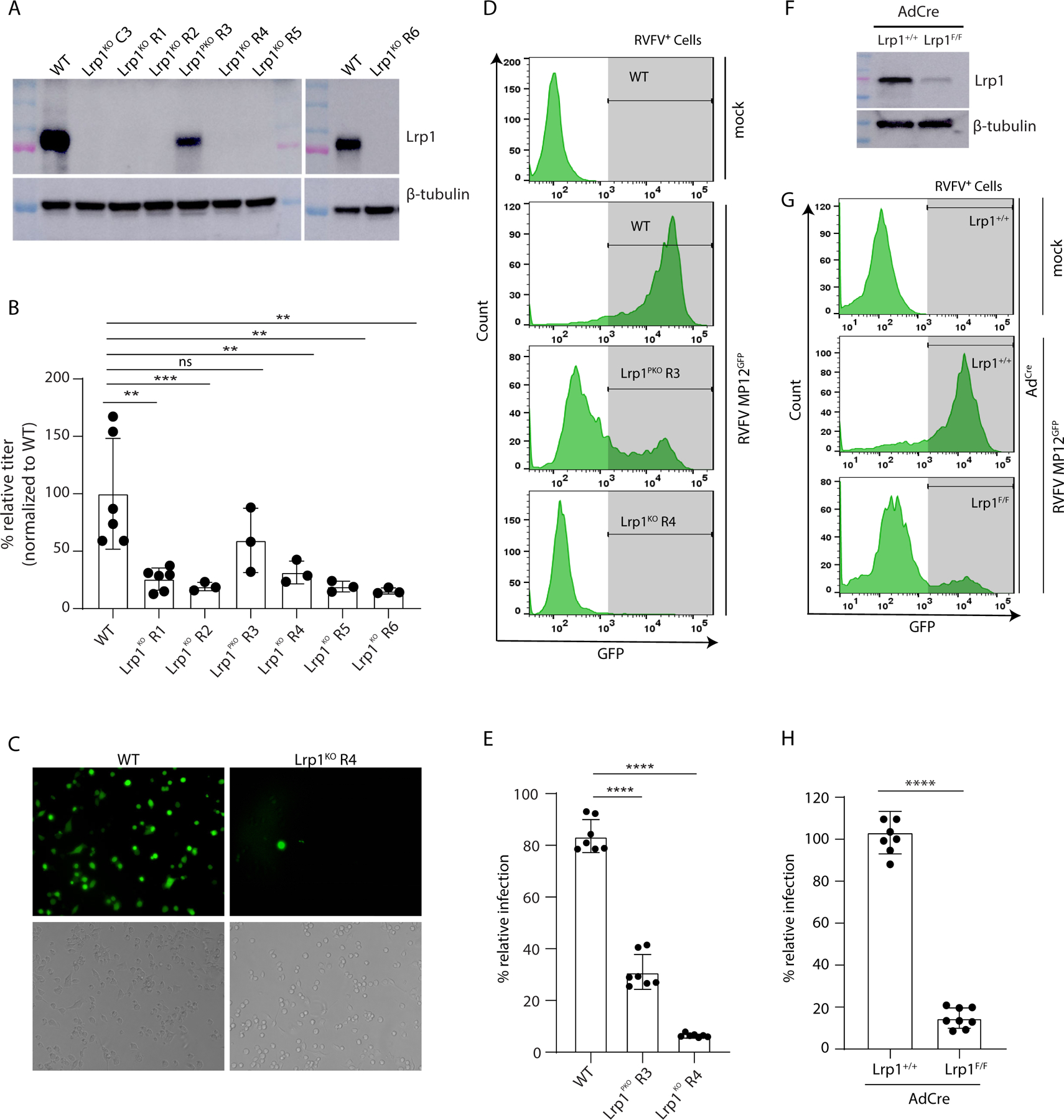
A. Western blot of BV2 Lrp1 knockout clones (Lrp1KO C3, Lrp1KO R1, Lrp1KO R2, Lrp1KO R4, Lrp1KO R5, and Lrp1KO R6), and partial knockout (Lrp1PKO R3) generated using either single gRNA or dual gRNA CRISPR/Cas9 approaches, as described in methods. B. BV2 wildtype (WT) and Lrp1KO clones were infected with RVFV ZH501 at a MOI of 0.1. After 18 hours, the cells were harvested for RNA extraction and subjected to q-RT-PCR analysis. Data shown are viral RNA (vRNA) titers normalized to wildtype BV2 cells. C. Microscopic images showing the WT and LRP1KO R4 cells infected with RVFV MP12GFP (MOI 5 for 6 hours) in fluorescence images (top panels) and DAPI-stained images (bottom panels). Images were taken at 20X magnification (For quantification, refer to Fig. S2H). D. Flow cytometry of WT, Lrp1PKO R3, and Lrp1KO R4 cells infected with RVFV MP12GFP. E. Corresponding analysis of flow cytometry histograms in D. F. Western blot of mouse embryonic fibroblasts (MEFs) from Lrp+/+ and Lrp1F/F mice infected with AdCre. G. Representative flow cytometry of MEFs Lrp1+/+ and LrpF/F cells infected for 5 days with AdCre and then infected with RVFV-MP12GFP at MOI of 1 for 15 hours. H. Corresponding analysis of flow cytometry histogram data in G.
Primary murine cells deficient for Lrp1 have reduced RVFV infection.
To test if Lrp1 is important in the infection of primary cells, we generated mouse embryonic fibroblasts (MEFs) from mice with floxed Lrp1 alleles. Infection of floxed MEFs (Lrp1F/F) with adenovirus expressing Cre recombinase (AdCre) resulted in a reduction in Lrp1 protein expression by western blot (Figure 2F). Infection of these Lrp1-deficient MEFs with RVFV MP12GFP resulted in >90% reduction in GFP levels, corresponding to RVFV infection (Figure 2G-H). IAV did not infect MEFs, and therefore as an alternative control, we tested infection of Lrp1-deficient MEFs with respiratory syncytial virus (RSV), which can readily infect MEFs. Resulting data showed no significant decrease in infection compared to non-floxed MEFs infected with AdCre (Figure S2I), suggesting that Cre-dependent deletion of Lrp1 contributed to reduced infection by RVFV MP12GFP, further supporting the significance of Lrp1 for RVFV infection.
Host proteins that regulate Lrp1 surface expression are also important for RVFV infection.
We next assessed the roles of RAP and Grp94 in RVFV infection. RAP is a critical chaperone for members of the LDL receptor family. We generated two BV2 RAPKO clones; RAPKO Clone A3 was hypomorphic and retained partial Lrp1 expression, while clone RAPKO A7 displayed near complete loss of Lrp1 expression (Figure 3A and Figure S3A). Infection of RAPKO A3 and RAP+ A7 clones with RVFV MP12GFP resulted in 30% and 80% reduction in infectivity, respectively (Figure 3B-C). These results highlight a correlation between reduced RAP and Lrp1 expression with a reduction in RVFV infection.
Figure 3. RAP and GRP94 knockout reduce RVFV infection indirectly by modulating Lrp1 levels.
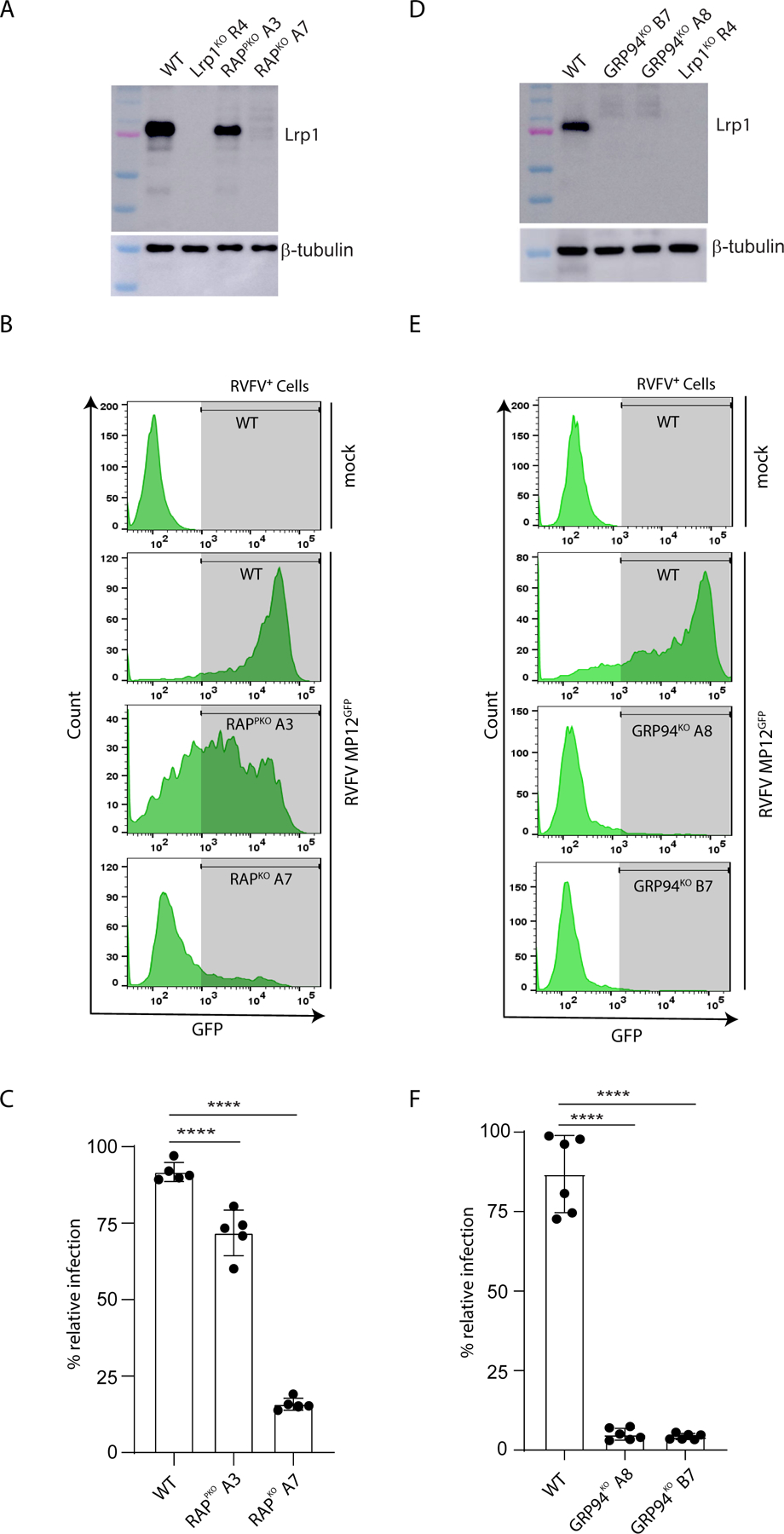
A. Western blot of BV2 knockout clones for RAP probed with an anti-Lrp1 antibody. B. BV2 knockout clones in A were infected with RVFV-MP12GFP at an MOI 1 for 15 hours. The cells were examined for virus infection (GFP) using flow cytometry. C. Quantitative analysis of flow data in B. Data are expressed as % infection relative to BV2 WT cells. D. Western blot of BV2 knockout clones for Grp94 probed with an anti-Lrp1 antibody. E. BV2 knockout clones in D were infected with RVFV-MP12GFP at an MOI 1 for 15 hours. The cells examined for virus infection (GFP) using flow cytometry. F. Quantification of the flow data in E. Data are expressed as % infection relative to BV2 WT cells. Experiments were done at least three times. ****, p<0.0001.
Loss of Grp94 expression and the concomitant enhancement of proprotein convertase subtilisin/kexin type 9 (Pcsk9) expression is known to increase the degradation of LDL receptors, including Lrp1 (Poirier et al., 2015, Canuel et al., 2013). We generated two clonal BV2 Grp94 KO lines; Grp94KO A8 and Grp94KO B7 (Figure S3B), which also lacked Lrp1 expression (Figure 3D). Infection of Grp94 KO lines with RVFV MP12GFP showed a ~95% reduction of infectivity (Figure 3E-F). Both RAPKO A7 and Grp94KO A8 were as permissive as the WT BV2 cells to IAV infection (Figure S3C-D). Together, these results suggests that multiple host factors regulating Lrp1 surface expression, including RAP and Grp94, are important for RVFV entry.
Lrp1 ligand binding clusters are essential for RVFV infection.
LRP1 is a large multidomain protein that consists of two chains, a 515 kDa extracellular alpha chain and an intracellular beta chain connected by an 85 kDa transmembrane domain. Within the alpha chain, there are four ligand-binding active regions with complement-like repeat clusters (CL; termed CLI, CLII, CLIII, and CLIV) that are separated by epidermal growth factor (EGF) repeats and β-propeller (YWTD) domains (Figure 4A). To determine the relative significance of LRP1 clusters for RVFV infection, we transcomplemented Lrp1KO R4 cells with each of the LRP1 mini-clusters. Infection of transduced Lrp1KO R4 cells expressing an individual LRP1 CL with RVFV MP12GFP revealed that LRP1 CLII and CLIV partially restored RVFV infection (Figure 4B). These results also identified LRP1 CLIV as making a greater contribution to infection, ~50% relative to WT. The expression of LRP1 CL in transduced Lrp1KO R4 was confirmed by flow cytometry and western blot, where all 4 clusters were detected via staining for HA antigen (Figure 4C and Figure S4A,B).
Figure 4. Lrp1 binds RVFV glycoprotein Gn.
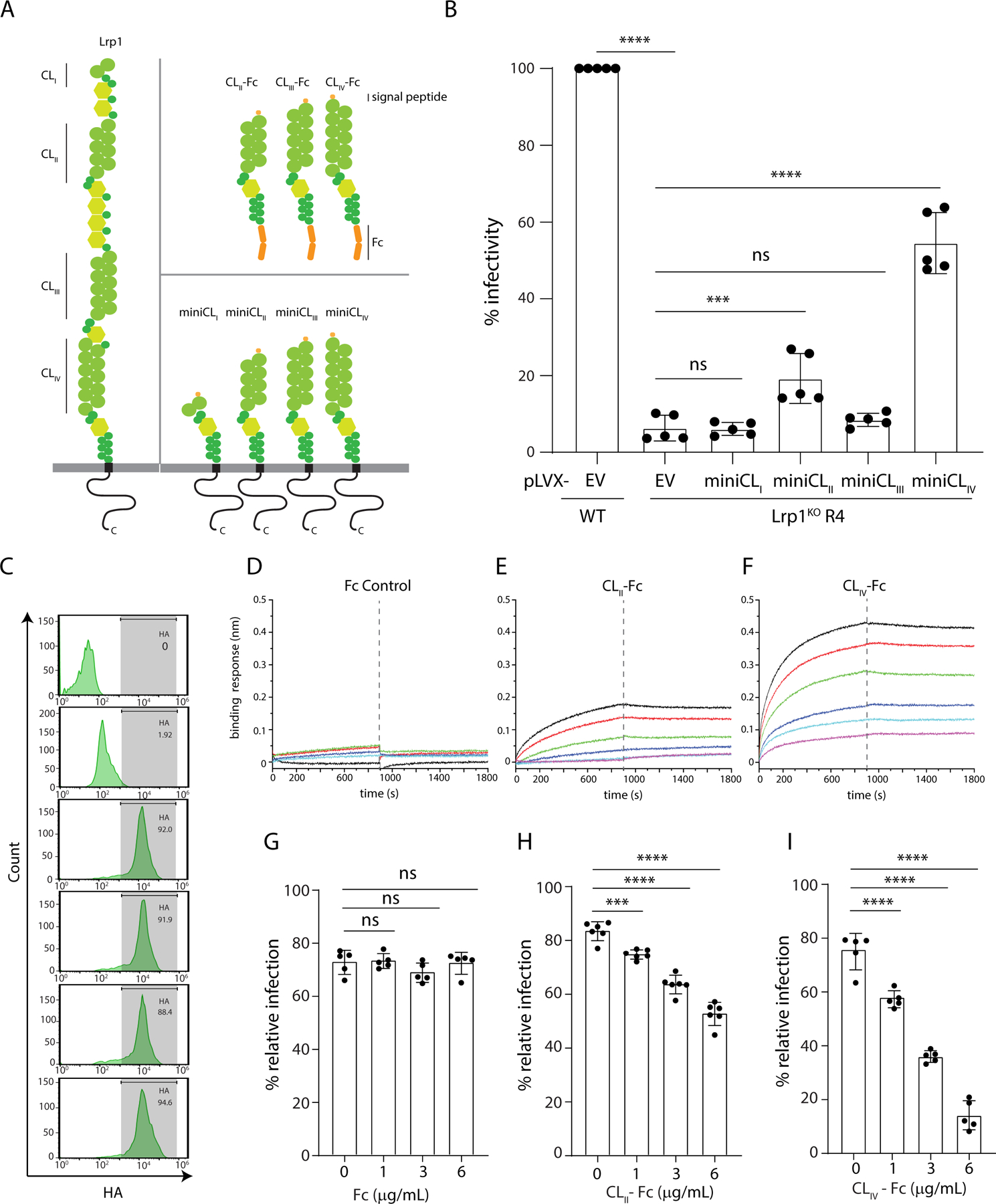
A. LRP1 is comprised of four clusters, CLI, CLII, CLIII, and CLIV, and the cytoplasmic and transmembrane domains (left). Mini-domains of CLI, CLII, CLIII, and CLIV were generated as -Fc fusions (top right). Lentiviruses carrying either pLVX-empty vector or pLVX-expressing HA-tagged minidomains mini-LRP1 CLI, CLII, CLIII, and CLIV were also generated (bottom right). B. BV2 WT and Lrp1KO cells were transduced with lentiviruses carrying either pLVX-empty vector (EV) or pLVX-expressing mini-LRP1 CLI, CLII, CLIII, and CLIV prior to infection with RVFV MP12GFP. The bar graph shows the quantification of % infectivity. C. Cell surface expression of mini-LRP1 Clusters in BV2 Lrp1KO cells using flow cytometry for HA. Biolayer interferometry sensograms of RVFV Gn binding to immobilized: D. Fc control, E. Fc-hLrp1 CLII, and F. Fc-hLrp1 CLIV. Neutraliztion of RVFV MP12GFP infection in the presence of exogenous G. Fc control, H. hLrp1 CLII, and I. hLrp1 CLIV.
Because the RVFV glycoprotein Gn mediates viral entry, we next determined whether Gn interacts with one or more of the LRP1 clusters through direct protein-protein interactions or if Gn-mediated entry requires additional host factors (Rusu et al., 2012). Co-immunoprecipitation (co-IP) assays with LRP1 CL proteins expressed as Fc fusions (termed CL-Fc; Figure 4A) demonstrated that recombinant RVFV Gn binds with LRP1 CLIV-Fc and CLII-Fc with high affinity, but not with CLIII-Fc or control Fc (Figure S4C). These results suggest that LRP1 CLII and CLIV contain binding regions for RVFV Gn protein, consistent with our transcomplementation data (Figure 4B). We further characterized the interaction between Gn and LRP1 domain clusters by biolayer interferometry (BLI), which revealed preferential binding of RVFV Gn to LRP1 CLIV-Fc, relative to Fc only, CLII-Fc (Figure 4D–4F), or CLIII-Fc (Figure S4D). The measured binding constant, KD of 96 ± 16 nM, was obtained with the steady state BLI data in the association phase (Figure S4F). Compared to LRP1 CLIV-Fc binding, RVFV Gn displayed weaker binding to LRP1 CLII-Fc with a KD of 485 ± 139 nM (Figure S4G). Next, we tested if exogenously added LRP1 CLII-Fc, CLIII-Fc, CLIV-Fc, and control Fc can inhibit RVFV MP12GFP infection. All three LRP1 clusters showed measurable neutralization (Figure 4G-I and Figure S4E), with LRP1 CLIV-Fc resulting in the most significant neutralization. Altogether, our data support a dominant role for LRP1 CLIV in MP12GFP infection through direct engagement of the viral Gn protein.
While the data for LRP1 cluster binding to RVFV Gn or RVFV MP12GFP neutralization were consistent with a role for CLII-Fc and CLIV-Fc, previous reports have shown that glycosylation alone may also provide viral attachment. Moreover, cluster-Fc binding analysis revealed slower off rates in the BLI studies (Figure 4E-F), potentially implicating avidity in the interaction between Lrp1 clusters and RVFV Gn proteins. To address these observations, we further characterized the CLII-Fc and CLIV-Fc fusion proteins by reducing and non-reducing SDS-PAGE (Figure S4H-I), which confirmed conformational changes due to the reducing agent (compare Figure S4H vs S4I), and upon deglycosylation (Figure S4H). In each protein, we observed a change in the SDS-PAGE mobility that is consistent with a loss of mass due to deglycosylation. Corresponding size exclusion chromatography further revealed that glycosylated and deglycosylated LRP1 CLII-Fc and CLIV-Fc were not misfolded or aggregated and eluted within the included volume of the column (Figure S4J-K).
Because cluster-Fc binding showed slower off rates in the BLI studies, we further characterized the CLII-Fc and CLIV-Fc fusion proteins by mass spectrometry (MS). Based on MS analysis under denaturing and native conditions, glycosylated CLII-Fc (Figure S5Aa-b and Figure S6A a-b) and CLIV-Fc (Figure S5Ba-b and Figure S6Ba-b) proteins have a mass higher than the expected molecular weight from the amino acid sequence and is consistent with additional mass contributions from glycosylation, while deglycosylated CLII-Fc (Figure S5Ac-h and Figure S6Ac-e) and CLIV-Fc (Figure S5Bc-h and Figure S6Bc-e) proteins resulted in a mass consistent with a CL-Fc dimer. Further analysis of the deglycosylated CLII-Fc and CLIV-Fc proteins under strong reducing conditions resulted in measurements of 68.1 and 76.7 kDa, respectively for CLII-Fc (Figure S5Ai-k ) and CLIV-Fc (Figure S5Bi-k), which are in agreement with the predicted masses of both the proteins in their monomeric forms. Since glycosylation can impact binding between LRP1 clusters and RVFV Gn, we further tested LRP1 CLII-Fc before (Figure S6C) and after deglycosylation (Figure S6D) in a pull down assay with RVFV Gn. A similar assay was carried out for LRP1 CLIV-Fc before (Figure S6E) and after deglycosylation (Figure S6F) with RVFV Gn. In each assay, we observed interaction between LRP1 clusters regardless of the glycosylation state, while the control Fc-only protein did not appear to interact with RVFV Gn (Figure S6G). Together these results show that while LRP1 proteins were glycosylated, the interaction between RVFV Gn and LRP1 clusters was glycosylation-independent.
RVFV entry is reduced in Lrp1-deficient cells.
To determine if RVFV attachment and internalization are compromised in Lrp1KO cells, we incubated BV2 WT and BV2 Lrp1KO R4 cells with RVFV-MP12GFP at 4 °C and 37 °C. RVFV virions bound to the cells (4 °C) or internalized (37 °C) were subjected to qRT-PCR analysis for quantification. Our results show a significant reduction in binding and internalization of the virus particles in BV2 Lrp1KO R4 cells, compared to BV2 WT cells (Figure 5A-B).
Figure 5. Lrp1 is critical for virus binding and internalization and anti-Lrp1 Abs inhibit RVFV infection.
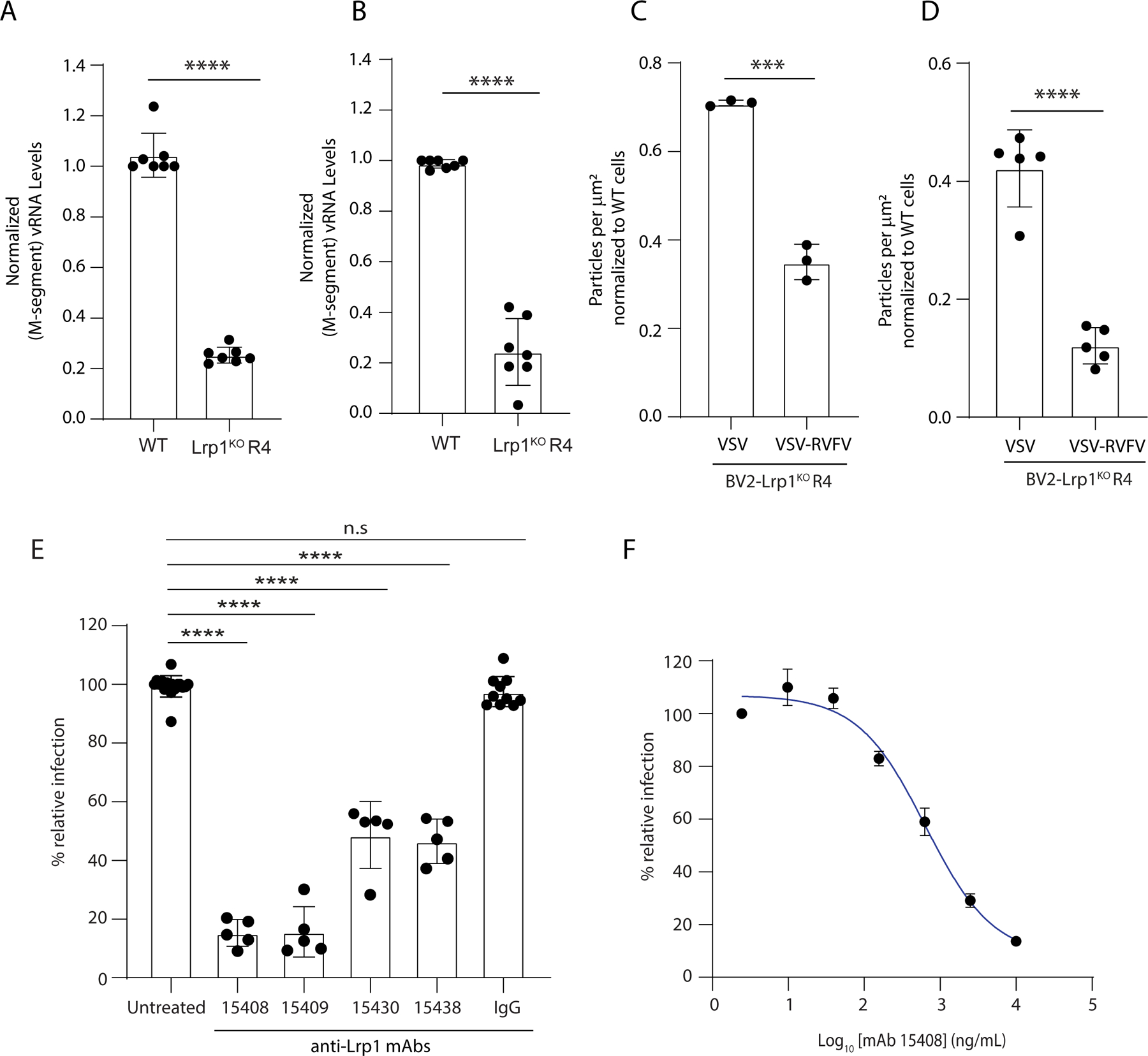
To evaluate binding vs internalization, BV2 WT and BV2 Lrp1KO R4 cells were incubated with RVFV MP12GFP at 4°C for binding assay A, and 37°C for internalization assay B. After washings, bound viral particles were quantified by RT-qPCR analysis. Quantification of Alexa Fluor labeled viral particles binding with BV2 WT and BV2 Lrp1KO R4 cells C. at 4°C and D. at 37°C were evaluated and normalized to respective levels of BV2 WT cells. E. BV2 WT cells were pre-incubated for 1 hour with 2.5 µg/mL of hLrp1 CLII-specific (15409), CLIV-specific antibodies (15438) and bi-specific (15408 and 15430) and then infected with RVFV MP12GFP. Cells were analyzed for virus infection after 16 hours. Bar graph represents % cells infected after the antibody treatment, compared to the infection of untreated cells. F. Dose-response curve showing the inhibition of RVFV MP12GFP infection of BV2 cells (y-axis) with EC50 936 ± 78 ng/mL after treatment with serial dilutions of IgG 15408 (x-axis).
Next we engineered recombinant vesicular somatitis virus (VSV) by replacing VSV glycoprotein with RVFV glycoproteins (VSV-RVFV) as glycoprotein spikes facilitate the virus attachment and internalization. We labeled VSV and VSV-RVFV with Alexa-flour 647 and Alexa-flour 588, respectively. Upon incubation of the labeled viruses with BV2 WT and Lrp1KO R4 cells at 4 °C and 37 °C, reduced binding occurred for VSV-RVFV to BV2 Lrp1KO R4 cells as compared to VSV, with the VSV glycoprotein on the viral surface (Figure 5C-D and Figure S7A). Taken together, our results demonstrate that Lrp1 is a critical host factor for RVFV attachment and entry and this interaction is dependent on the RVFV Gn protein.
RVFV Gn glycosylation and host glycosaminoglycans (GAGs) are dispensable for RVFV infection.
Viral glycoproteins are highly glycosylated, and previous studies revealed that the lectin DC-SIGN promotes RVFV internalization in dermal dendritic cells (Léger et al., 2016, Lozach et al., 2011). Related lectin molecules, such as L-SIGN and DC-SIGNR, are also found on RVFV-permissive cells, including hepatocytes. However, deglycosylating the virus did not reduce the infectivity, and interaction with lectin molecules was dependent on the glycosylation of Gn (Lozach et al., 2011). Here, we performed several binding assays with purified bacterial recombinant RVFV Gn protein, which lacks any glycosylation (Figure 4D-F, Figure S4C), and tested the ability to inhibit RVFV infection. Cells that were pre-treated with non-glycosylated Gn displayed dose-dependent inhibition of RVFV MP12GFP infection (Figure S7B), suggesting that viral Gn glycosylation is not critical for RVFV entry.
Host glycosaminoglycans (GAGs) such as heparan sulfate were also reported to play a role in RVFV infection, but removal of heparan sulfate did not eliminate RVFV infection (Riblett et al., 2016, de Boer et al., 2012). Consistent with this observation, our screen identified Ext2 encoding Exostosin-2 (Figure S1C), a key protein in the heparan sulfate biosynthesis pathway, as a host factor for RVFV entry. However, we found that deletion of Ext1 or Ext2 did not significantly impact virus infection (Figure S7C). Furthermore, pretreatment with surfen, a GAG inhibitor, did not result in a substantial change in virus infection (Figure S7D). Taken together, viral Gn glycosylation and host GAGs are not essential factors for RVFV infection.
CLII and CLIV-specific Abs reduce infection by RVFV.
To further evaluate the significance of Lrp1 as a potential receptor for RVFV, we used a phage-displayed library of synthetic human antigen-binding fragments (Fabs) to identify Fabs that specifically recognized Lrp1 CLII or CLIV. These efforts led to the identification of many unique Fabs with high affinity for Lrp1 CLII (Figure S7E) and for CLIV (Figure S7F). For each set of Fabs, we also evaluated their cross reactivity to CLII and CLIV. From these results, we identified four distinct Fab sequences for further evaluation in the full-length immunoglobulin (IgG) format, and their specificities for Lrp1 CLII-Fc and Lrp1 CLIV-Fc were evaluated (15408, 15409, 15430, and 15438) (Figure S7G-J). Fab15409 bound with an affinity and specificity to CLII (KD CLII=~2.3 nM and KD CLIV=ND) (Figure S7G), whereas Fab 15408 bound with high affinity to CLII and moderate affinity to CLIV (KD CLII=~1.0 nM and KD CLIV=11 nM) (Figure S7H). In comparison, Fabs 15430 and 15438, raised against LRP1 CLIV, bound with high affinity to CLIV (Fab 15430, KD CLII=~40 nM and KD CLIV=10 nM; Fab 15438, KD CLII=ND and KD CLIV=~1 nM) (Figure S7I-J). To further evaluate the impact of anti-Lrp1 antibodies on RVFV infection, we tested each of these Fabs in the context of a human IgG framework in cell-based neutralization assays of RVFV MP12GFP. The resulting data revealed >80% neutralization by the CLII-binding IgGs 15408 and 15409, and >50% neutralization by the CLIV-binding IgGs 15430 and 15438, compared with an isotype control (Figure 5E). As a follow-up, we evaluated bi-specific IgG 15408 in a dose-response neutralization assay, and the data revealed an EC50 of 936 ± 78 ng/mL (Figure 5F). IgG 15408 was selected on the basis that the Fab 15408 bound both Lrp1 CLII and CLIV (Figure S7H). Taken together these results support the specificity of Lrp1 for RVFV infection and suggest that antibodies targeting Lrp1 clusters, CLII in particular, have the potential to block access to RVFV entry and therefore present a potential therapeutic avenue to prevent RVFV infection.
mRAP binds Lrp1 and inhibits RVFV infection in cells derived from taxonomically diverse hosts.
RAP binding to LRP1 was demonstrated biochemically in multiple previous studies (Migliorini et al., 2003). When recombinantly-expressed RAP is exogenously introduced in cell culture, RAP is known to bind to the LRP1 clusters and inhibit interactions with all known ligands (Bu and Schwartz, 1998). RAP contains three domains (D1-D3) (Figure 6A), and RAP D3 binds both LRP1 CLII and CLIV (Lazic et al., 2003, De Nardis et al., 2017, Prasad et al., 2015). Consistent with these studies, our BLI data revealed that both human LRP1 CLII and CLIV bind to mouse RAPD3 (mRAPD3) (Figure 6B and 6C), consistent with previous studies (De Nardis et al., 2017, Prasad et al., 2015). To establish if mRAPD3 and Gn have overlapping binding sites on LRP1, we tested Gn binding with LRP1 CLIV in the absence or presence of mRAPD3. At higher concentrations, we found that mRAPD3 competed with RVFV Gn for binding to LRP1 CLIV in vitro (Figure 6D). These results suggest that mRAPD3 can function as an inhibitor of Gn binding and as a probe to assess interactions with LRP1. We next performed inhibitory neutralization assays using mRAPD3. We found that mRAPD3 potently inhibited RVFV MP12GFP infection with an EC50 of 0.59 ± 0.2 μg/mL (Figure 6E-F). Consistent with previous studies (Migliorini et al., 2003, Rauch et al., 2020), mutant mRAPD3 (Figure 6A) showed weak interaction with LRP1 CLII (Figure 6G) and CLIV (Figure 6H). Upon incubation of mutant mRAPD3 with BV2 cells, RVFV MP12GFP infection was moderately affected (Figure 6I).
Figure 6. mRAPD3 competes with RVFV glycoprotein Gn for binding to Lrp1 and inhibits RVFV infection.
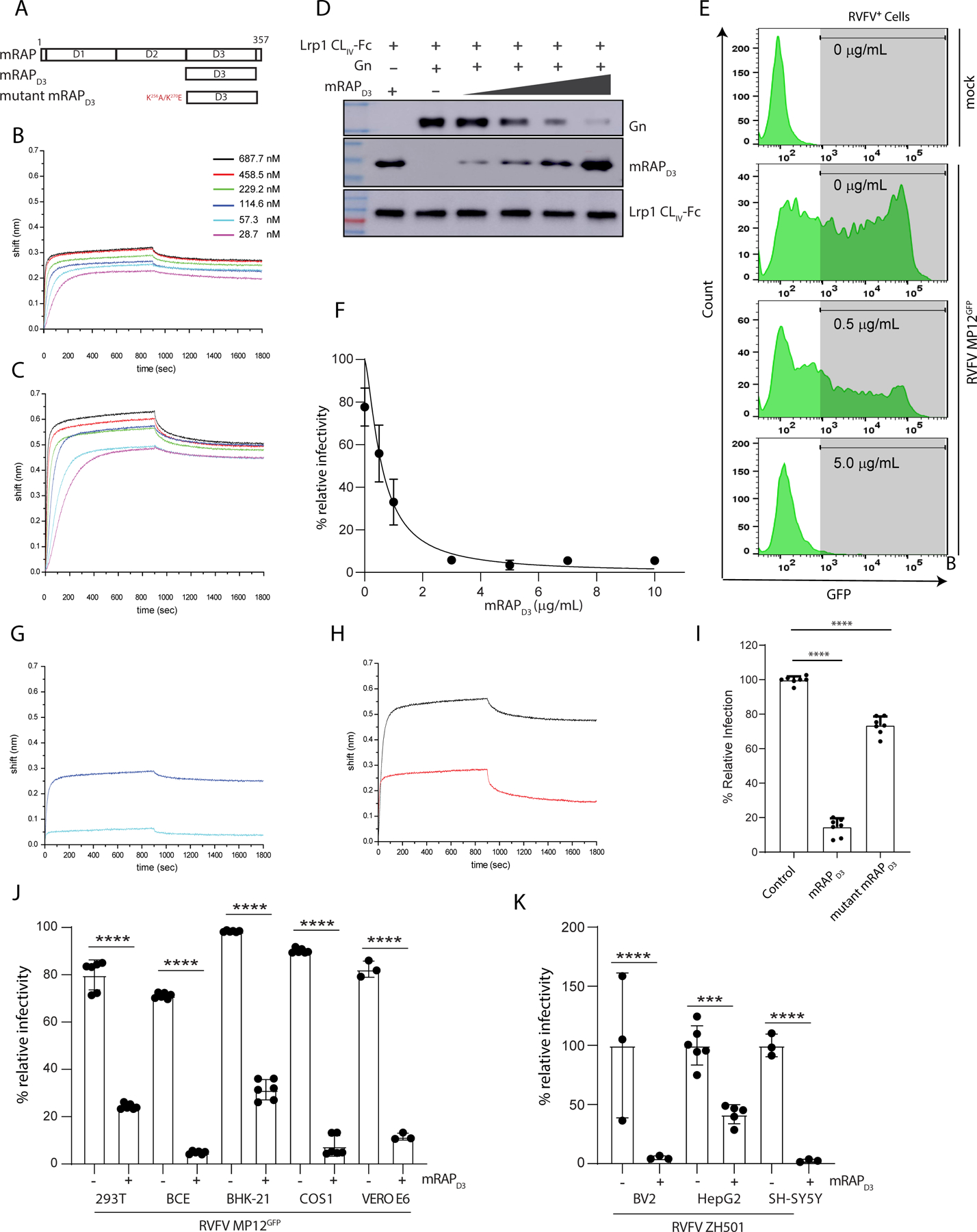
A. Domain organization of mouse RAP (mRAP) protein. BLI sensograms of mRAPD3 binding to immobilized B. LRP1 CLII immobilized C. LRP1 CLIV. D. mRAPD3 competition assay to assess relative binding of Gn to LRP1 CLIV in the presence of 1, 3, 6, or 10 μg/mL concentrations of mRAPD3. E. Flow cytometry data for BV2 cells infected with RVFV MP12GFP in the presence of increasing concentrations of mRAPD3. F. Analysis of relative infectivity as a function of mRAPD3 concentration. EC50 is 0.59 ± 0.2 μg/ml. BLI sensograms showing the binding of mRAPD3 and mutant mRAPD3 with G. LRP1 CLII and H. LRP1 CLIV. I. RVFV MP12GFP infection of BV2 cells in presence of mRAPD3 and mutant mRAPD3. J. Cell lines from different species were infected with RVFV-MP12GFP at an MOI 1 in the absence (−) or presence (+) of 5 μg/mL of mRAPD3 (10x EC50). Infection was assessed 15 hpi by flow cytometry. K. Mouse (BV2) and human (HepG2 and SH-SY5Y) cell lines were infected with RVFV ZH501 at an MOI 1 in the absence (−) or presence (+) of mRAPD3. Infection was assessed at 18 hpi by RT-qPCR on cell supernatants and intracellular flow cytometry for viral Gn protein.
To determine the relevance of LRP1 as an essential factor for RVFV infection in cells derived from other organisms (mice, hamsters, cows, monkeys, and humans), we treated cells with 5 μg/mL of mRAPD3 (10x EC50). In all cell lines tested, we observed a substantial inhibition of infection by RVFV MP12GFP (Figure 6J) and by the pathogenic RVFV ZH501 (Figure 6K). Importantly, we observed a dose-dependent reduction in RVFV infection across all cell lines, further supporting our observations (Figure S8A-I).
Since mRAPD3 can prevent infection in multiple cell types, we assessed whether mRAP inhibition of RVFV infection occurred at the level of virus binding or post-binding event. Pre-incubation of BV2 cells with mRAPD3 resulted in significant protection from RVFV MP12GFP infection, while post-infection treatment with mRAPD3 resulted in infection levels similar to control cells lacking mRAPD3 treatment (Figure S8J). These results support a model for mRAPD3 blocking RVFV Gn interaction with Lrp1 receptor as a pre-infection event. To assess the integrity of mutant mRAPD3 (Figure S8K), we used size exclusion chromatography to evaluate the proteins, which show similar elution profiles for mutant mRAPD3 and mRAPD3, suggesting that the physical properties, including hydrodynamic behavior of both proteins are similar (Figure S8L).
mRAP, an Lrp1 ligand, protects mice from lethal infection with RVFV ZH501.
To support Lrp1 as a critical factor for RVFV infection, we evaluated the effect of mRAPD3 treatment in vivo using a mouse model. Mice (C57BL/6) are extremely susceptible to RVFV infection, with an LD50 of <1 pfu or TCID50 after footpad injection (Dodd et al., 2012, Cartwright et al., 2020). Because mRAPD3 is highly effective at preventing RVFV ZH501 infection of neurons in cell culture (Figure 6K), we sought to determine whether mRAPD3 treatment can prevent RVFV infection of the brain using intracranial (IC) injection as an initial proof-of-concept experiment. Similar to footpad injection, the LD50 of RVFV ZH501 by IC injection is less than 1 pfu with an average survival time (AST) of 3.5 days (Figure S9A). We evaluated the effectiveness of administering 215 μg of mRAPD3 IC simultaneously with 10 pfu (Figure 7A) or 1 pfu (Figure S9B) of RVFV ZH501. Most mice lost weight within a day after IC injection but recovered and gained weight thereafter. Groups of infected, untreated mice succumbed to disease in both the 10 pfu group (13/13 died; AST 2.5 days) (Figure 7A) and 1 pfu group (11/14 died; AST 4.5 days) (Figure S9B), respectively. In contrast, co-administration of mRAPD3 along with IC infection with RVFV ZH501 resulted in a significant increase in survival in both dose groups and an increase in AST of those that succumbed (Figure 7A, Figure S9B). For the 10 pfu group, 12/17 mRAP-treated mice survived with an AST of 5.2 days for the five mice that died (Figure 7A). For the 1 pfu group, 11/17 mRAP-treated mice survived, with an AST 5.8 days for the six mice that succumbed to disease (Figure S9B). As controls, groups of mice were given equivalent amounts of either an irrelevant control protein (Ebola virus VP30 protein) or the mutant mRAPD3 that showed weaker interaction with Lrp1 and reduced neutralization of MP12 (Figure 6G-I). Mice in both control groups did not survive co-infection with 10 pfu of RVFV ZH501 and succumbed within an average of 3.5 days (Figure 7A). These results suggest that mRAP with Lrp1-binding capability is able to prevent lethal infection with RVFV.
Figure 7. mRAP binding to Lrp1 protects mice from intracranial infection of RVFV ZH501.
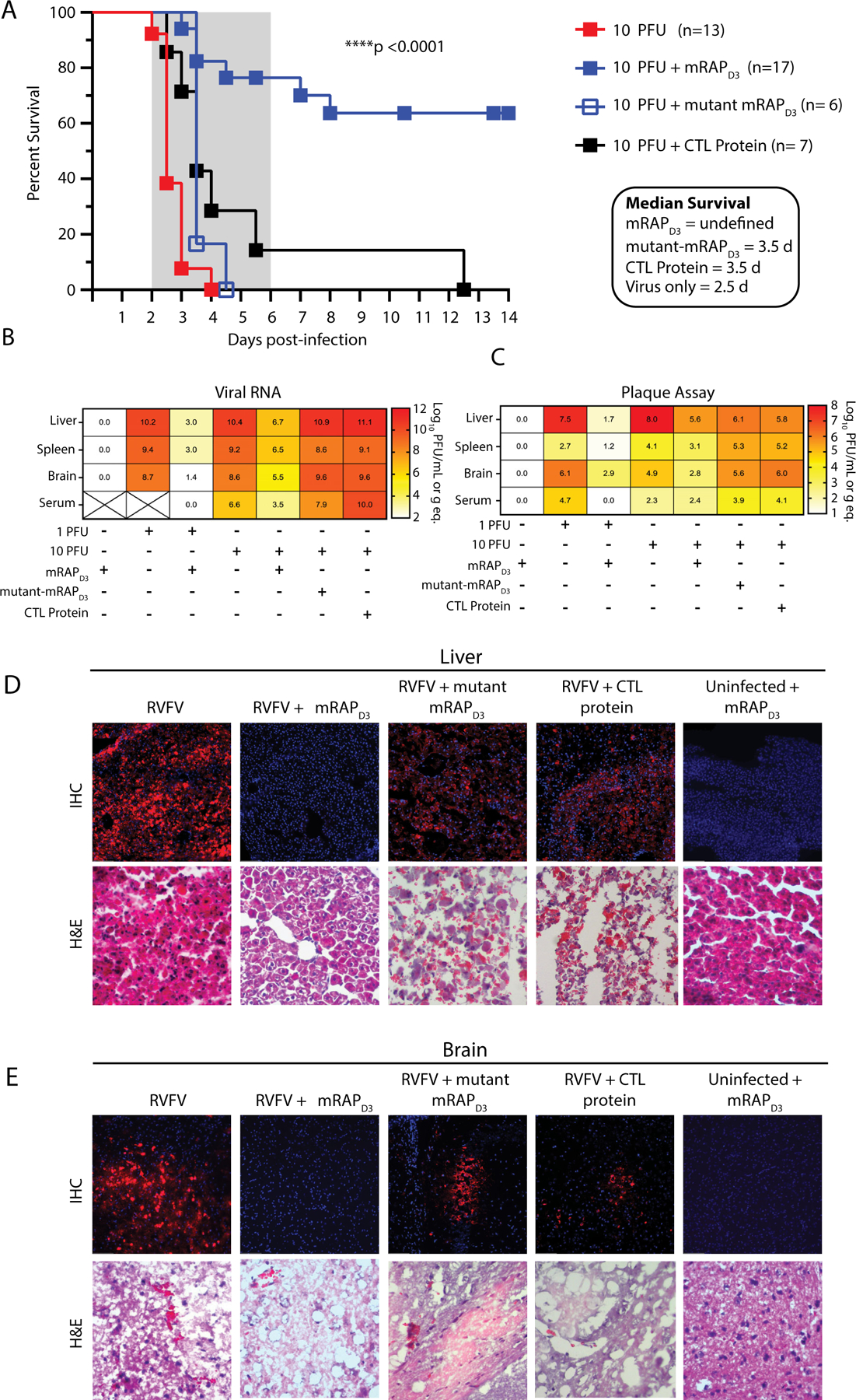
A. Survival of mice infected intracranially with 10 pfu of RVFV ZH501 in absence or presence of 215 μg of recombinant mRAPD3 protein, 210 μg of mutant mRAPD3, 250 μg of control protein (Ebola VP30). In a second experiment, 3 mice/group were euthanized at 3 dpi, and liver, spleen, brain and serum were harvested at necropsy and assessed for B. viral RNA levels by qRT-PCR or C. infectious virus by plaque assay. Heatmaps show average log-transformed titer for each tissue (indicated by the number in each cell of the heatmap) and are also represented visually by the color shading in the legend. X through the cell indicates samples that were not available for analysis. Pathology in D. liver and E. brain tissue was assessed by immunofluorescence for viral antigen using an anti-NP antibody (top panels) or H&E staining (lower panels) in presence or absence of the indicated proteins. Images were taken at 20X magnification. The liver and brain tissues shown in D and E are from respective animals; IF and H&E are from the same tissues. Additional representative animals and controls are shown in Figure S7.
In a follow-up experiment, groups of three mice from each treatment group underwent planned euthanasia at 3 dpi for direct comparison of tissue viral loads and pathology across groups. The liver, spleen, brain, and serum from mRAPD3-treated mice co-infected with 10 pfu of RVFV contained reduced, but not eliminated, levels of both viral RNA and infectious virus (Figure 7B-C) compared to untreated, mutant mRAP-treated, or control-protein treated RVFV-infected control mice. Infection levels of the tissues from 3 dpi were confirmed using immunofluorescence with an anti-NP antibody and histopathology. At 3 dpi, mice infected with 10 pfu contained widespread RVFV-antigen positive cells in both the liver and brain in untreated control, mutant mRAP, and control protein treated animals (Figure 7D-E). Hematoxylin and eosin (H&E) staining revealed classic indications of RVFV-mediated hepatic destruction and hemorrhage. Similar results were seen for control mice infected with 1 pfu of RVFV ZH501 (Figure S9C-D). In comparison, tissue sections from the mRAPD3 treated mice contained undetectable levels of viral antigen staining and no histological damage caused by viral infection (Figure 7D-E). The mRAPD3 treated mice that survived RVFV infection showed anti-RVFV serum titers consistent with infection and survival (Figure S9E). Collectively, these in vivo proof-of-concept experiments provide evidence of a significant reduction in viral infection in multiple tissues when mRAPD3 is co-administered with RVFV at the time of infection. Important controls including a mutant mRAP that shows reduced Lrp1 binding were not able to rescue mice from lethal infection. These results provide further support for a role for Lrp1 as a major cellular factor required for RVFV infection in a rodent model.
Discussion
Given the broad tropism of mosquito-transmitted zoonotic viruses such as RVFV, host factors that mediate entry are critical in order to fully understand viral emergence, zoonosis, and spread. Previous studies have implicated several cellular factors in RVFV binding and entry. The glycosaminoglycan (GAG) heparan sulfate was identified in a genetic screen as essential for RVFV infection (de Boer et al., 2012, Riblett et al., 2016). Although the studies showed that heparan sulfate proteoglycan (HSPG) inhibition resulted in inhibition of RVFV infection in some cell types, the exact role of HSPG in RVFV infection was unclear. Interestingly, our screen also identified an HSPG-related gene, Ext2, a gene involved in the synthesis of GAGs. We found, however, that deletion of Ext1 or Ext2 did not have a significant impact on RVFV infection in mouse BV2 cells and that treatment with the GAG inhibitor surfen also did not inhibit RVFV infection. These results support a minimal role for HSPGs in BV2 cells despite their importance as a receptor for macromolecular endocytic cargo; instead, they may play a role in augmentation of RVFV infection. Like HSPGs, C-type lectins such as DC-SIGN were identified as mediating RVFV infection of dermal dendritic cells and some other cell types (Léger et al., 2016, Phoenix et al., 2016, Lozach et al., 2011). RVFV has broad tropism and infects a wide range of tissues. Since DC-SIGN was not expressed in many cell types, including BV2 cells in which these assays were conducted, our results do not directly address the significance of the previous findings for DC-SIGN. It is important to note that DC-SIGN interaction with RVFV glycoprotein was glycosylation-dependent, which suggests that DC-SIGN is unlikely to be a proteinaceous receptor for RVFV (Lozach et al., 2011). In contrast, our results here, including biochemical studies using recombinant bacterially expressed non-glycosylated RVFV Gn and deglycosylated LRP1, show that the interaction between Gn and Lrp1 does not depend on the glycosylation state of LRP1 or RVFV Gn.
Given the limitations of previous findings and to address the need to better understand tropism and entry of RVFV, we conducted a pooled genome-scale screen using the CRISPR/Cas9 system. Our studies identified an LDL receptor family protein, Lrp1, as an essential host factor capable of mediating RVFV infection across cell lines from multiple species. A combination of Lrp1 KO cells and cells lacking key chaperones for Lrp1 processing and surface presentation, including RAP and Grp94, provide support for Lrp1 as a proteinaceous entry factor. In cells lacking RAP or Grp94, we observed reduced Lrp1 expression and concomitantly demonstrated reduced binding by two strains of RVFV. Cells lacking Lrp1 expression also showed reduced binding by a chimeric VSV expressing the RVFV glycoproteins, demonstrating that interaction between Lrp1 and RVFV is at the level of glycoprotein binding and entry. Lrp1 is also important for RVFV infection of primary cells, as primary MEFs from Lrp1F/F mice transduced with AdCre showed reduced infectivity by RVFV. Our biochemical analysis revealed a direct interaction between RVFV Gn with some but not all complement-like repeat clusters in the Lrp1 ectodomain. Notably, Lrp1 CLIV has emerged as an important site of interaction. Exogenous addition of Lrp1 CLIV-Fc resulted in potent neutralization of RVFV infection in vitro.
The D3 domain from mouse RAP is a known Lrp1-interacting protein, and when added exogenously, it serves as an inhibitor of all known Lrp1 ligands. Our biochemical studies showed that mouse RAPD3, like RVFV Gn, bound to Lrp1 domain CLIV with higher affinity than CLII. We also showed that RAPD3 competed with Gn for binding to Lrp1. Blocking the LRP1 receptor with RAPD3 inhibited RVFV infection in cells derived from a variety of species including rodents, ruminants, and primates. Notably, RAPD3 was also effective in human SH-SY5Y neuronal cells, where infection with ZH501 was rendered undetectable, further supporting the broad importance of Lrp1 in RVFV infection with implications for understanding neuropathogenesis. As an important control, we show that mRAPD3 containing two point mutations that reduce binding to CLIV and CLII can no longer effectively block RVFV infection, thus supporting the model that Gn binding to CLIV as an important interaction. Finally, we showed that human antibodies that target Lrp1 are also potent inhibitors of RVFV infection.
Similar to RAP, Grp94 impacts Lrp1 cell surface levels and Lrp1 recycling via an indirect mechanism. Pcsk9 is expressed as a pro-protease and is eventually secreted where it binds LRP1 to enhance LRP1 endocytosis. In the endoplasmic reticulum, Grp94 binds to Pcsk9 and prevents its release from the cell. As discussed above, our screen identified Lrp1, Grp94, and RAP as proviral factors. The factors were identified as hits in our screen based on log2-fold enrichment relative to an untreated pooled cell population. Importantly, the relative levels of sgRNA relative to an untreated pooled cell population indicates sgRNA targeting Pcsk9 is inversely correlated with RVFV infectivity, suggesting loss of Pcsk9 gene product results in higher levels of Lrp1 and consequent higher levels of infection. Taken together, these observations provide further evidence that a pathway regulating Lrp1 biosynthesis and surface presentation is essential for RVFV infection.
In this study, we established that RVFV glycoprotein Gn interacts directly with host factor, Lrp1, and this interaction is largely driven through direct binding to Lrp1 CLIV, with a weaker interaction with Lrp1 CLII. This observation is consistent with previous observations in unrelated studies as Lrp1 CLII and CLIV are largely responsible for binding over 100 ligands reported thus far for Lrp1 (Neels et al., 1999). Our data showed that bacterially-expressed Gn, lacking glycosylation, binds directly to Lrp1 and also competes with virus and inhibits infection of cells. Similarly, deglycosylatied LRP1 also interacted with RVFV Gn lacking glycosylation. Thus, our results point to a model where RVFV Gn interaction with Lrp1 functions as a proteinaceous entry factor for RVFV infection. The exact mechanism by which Lrp1 functions in RVFV entry is under further investigation. Since Lrp1 expression is ubiquitous and the RVFV receptor is conserved across taxonomically diverse species, our results support Lrp1 as a potential host factor that can promote infection in multiple cell types and would explain the broad tropism of RVFV across species. The discovery of Lrp1 as a major cellular factor for RVFV provides a framework to better understand the molecular basis for RVFV attachment and internalization.
In addition to biochemical and in vitro evidence presented above, we provide compelling data on the in vivo relevance of Lrp1 as a critical factor for RVFV infection through proof-of-concept mouse experiments. Simultaneous intracranial administration of mRAPD3 and RVFV ZH501 significantly enhanced survival from this otherwise highly lethal infection. In contrast, a mutant mRAPD3, which binds weakly to Lrp1, failed to protect mice, supporting the specificity for the role of Lrp1 in viral entry. We hypothesize that mRAPD3 was able to block infection of cells in the brain, similar to our results from the exogenously treated cell lines, and thereby reduce dissemination from the brain to the liver and spleen resulting in enhanced survival of the mice. These experiments pave the way for further exploration of the role of Lrp1 in dissemination and tropism in vivo.
In summary, we identified Lrp1 as a novel proteinaceous host factor important for RVFV entry with potential to support infection. Conservation of Lrp1 across cell types and species, including mosquitos, a vector host for RVFV, highlight the significance of our finding in the context of broad tropism observed for RVFV. While the exact mechanism by which Lrp1 mediates RVFV entry require further study, our findings provide a foundation for answering many open questions related to RVFV, including mechanisms associated with zoonotic transmission, tropism, spread, and pathogenesis. Knowledge gained from these studies positions us to explore Lrp1, a conserved cell-surface protein, as a target for prophylactic and therapeutic development of RVFV infections.
Limitations of the study
In this study, we identify Lrp1 as a host factor for RVFV infection using genetic deletion of Lrp1.We also show that key proteins that facilitate processing of Lrp1, such as RAP (Lrpap1) and Grp94 (HSP90b1) are important for RVFV infection. In support, we block Lrp1 interaction with viral glycoprotein with an Lrp1 ligand, RAP, and show that RVFV infection is neutralized in cells derived from taxonomically distinct hosts. Our biochemical studies support a direct interaction between Lrp1 and RVFV glycoprotein Gn that is independent of glycosylation status of Gn or Lrp1 Clusters. However, Lrp1 interaction with RVFV Gn is likely to be complex and may include receptor binding and signaling. Lrp1 and RVFV Gn proteins are glycosylated and complete characterization of glycosylation is important to determine the molecular mechanisms that drive viral entry. Future work, including ongoing structural analysis of the interaction, is needed to fully define the entry mechanisms for RVFV infection.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Gaya Amarasinghe (gamarasinghe@wustl.edu).
Materials availability
New reagents generated in this study are available through the lead contact, Gaya Amarasinghe (gamarasinghe@wustl.edu).
Data and code availability
CRISPR screen gRNA enrichment data were analyzed to generate the volcano plots using the following software: https://github.com/mhegde/volcano_plots.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Biosafety and Regulatory Requirements.
All work with RVFV ZH501 was performed in the Regional Biocontainment Laboratory (RBL) at the University of Pittsburgh under BSL-3 conditions. Personnel wore powered-air purifying respirators (PAPRs) for respiratory protection (3M VersaFlo TR-300). The Pitt RBL is a registered entity with the Federal Select Agent program (FSAP) and is approved for work with RVFV. Inactivation protocols were approved by Pitt’s institutional biosafety oversight committees.
Animal study oversight.
All mouse work performed either at Washington University School of Medicine (WUSM) or at the University of Pittsburgh (Pitt) adhered to the highest level of humane animal care standards. Each institution is fully accredited by the Association for Assessment and Accreditation of Laboratory Animal Care (AAALAC). All animal work was performed under the standards of the Guide for the Care and Use of Laboratory Animals published by the National Institutes of Health (NIH) and according to the Animal Welfare Act guidelines. All animal studies adhered to the principles stated in the Public Health Services Policy on Humane Care and Use of Laboratory Animals. The WUSM and University of Pittsburgh Institutional Animal Care and Use Committee (IACUC) approved and oversaw the animal work conducted at WUSM and the University of Pittsburgh, respectively.
Animal procedures.
Mice were anesthetized with isoflurane followed by intracranial (IC) injection of diluted virus inoculum in a total volume of 10 ul using a Hamilton syringe and a 27g ½ inch needle. For assessment of mRAP treatment, virus was diluted and mixed with 215 ug of mRAPD3, mutant mRAPD3, or control protein Ebola VP30, followed by injection in a volume of 10 ul. Mice were monitored for disease and were euthanized when IACUC-approved euthanasia criteria were met. Surviving mice were necropsied at the end of each experiment. A subset of mice were euthanized at 3 dpi for comparison of tissue viral load across treated and untreated groups. At necropsy, blood was drawn for serum, and then liver, spleen and brain tissue were harvested. Half of each tissue was saved for pathology and half was homogenized for virological analysis. Infectious virus was assessed by viral plaque assay described. RNA was extracted and RT-qPCR was used to quantify the amount of vRNA in each tissue, as described previously (McMillen et al., 2018).
METHODS DETAILS
Cells.
All cell lines were cultured in Dulbecco’s Modified Eagle Medium (DMEM) (Gibco, Cat. 11965084) supplemented with 10% fetal bovine serum (FBS) (Sigma Millipore, Cat. F2442) in a humidified incubator at 37 °C and 5% CO2. For the murine microglial BV2 cell lines, the media was supplemented with 10 mM HEPES (Corning, Cat 25–060-CI) and 1 mM sodium pyruvate (Corning, Cat. 25–000-CI). HEK293T (CRL-3216), VeroE6 (CRL-1586), HepG2 (HB-8065), SH-SY5Y (CRL-2266), and COS1 (CRL-1650) cells were obtained from American Type Culture Collection (ATCC). BCE C/D-1b (ATCC 2048), BHK-21 (ATCC CCL-10), and BV2 cells (Blasi et al., 1987, Stansley et al., 2012) were provided by M. Diamond (WUSM) and S. Whelan (WUSM), respectively.
Preparation of primary mouse embryonic fibroblasts:
Lrp1-flox mice were purchased from the Jackson Laboratory (B6;129S7-Lrp1tm2Her/J, Stock# 012604). E14.5 embryos were obtained by timed mating of Lrp1F/+ mice and were genotyped by polymerase chain reaction using genomic DNA from tissue digested with 0.5 mg/ml proteinase K in DirectPCR Lysis Reagent (Viagen, 101-T) for 30 min at 55 °C. Genotyped embryos were minced into small pieces and digested with 0.25% Trypsin/0.02% EDTA (Millipore Sigma T4049) for 25 min, followed by culture in DMEM supplemented with 10% FBS and cryostock after two days of culture.
Viruses.
RVFV ZH501 (provided by S. Nichol, CDC) was generated from reverse genetics plasmids containing the WT ZH501 sequence, which was confirmed by sequencing. RVFV ZH501 is a select agent and is handled at BSL-3 in the Pitt RBL. Virus was amplified in VeroE6 cells and p2 stock was used for this study (titer 1×107 pfu/mL). A standard viral plaque assay (VPA) was used to measure infectious titers; VPAs used an agarose overlay (1x minimum essential medium, 2% FBS, 1% penicillin/streptomycin, HEPES buffer, and 0.8% SeaKem agarose) and were incubated for 3 days at 37 °C, followed by visualization using crystal violet. RVFV MP12GFP (provided by M. Diamond, WUSM) was amplified in VeroE6 cells. The virus was collected 5 dpi and then filtered through 0.45 μm, aliquoted, and frozen at −80 °C. The titer of the virus stock was calculated (~ 6.5 × 107 IU/mL) and all experiments in this study were performed using the same stock of the virus. Adenoviruses Ad-mCherry (Cat #1767) and Ad-mCherry Cre recombinase (Cat #1773) were purchased from Vector Biolabs. Adenoviruses were used for infection of mouse embryonic fibroblasts (MEFs). Lentiviruses were used to transduce the sgRNA to generate BV2 library cells (Orvedahl et al., 2019). Influenza A virus, strain PR8 (IAV PR8) was provided by J. Boon (WUSM). Respiratory syncytial virus, RSV GFP5 (Cat# R125) was purchased from Viratree.
Antibodies.
The following antibodies were used in the study: rabbit anti-LRP1 (Cell Signaling, cat. 64099), rabbit anti-His antibody (Cell Signaling, Cat. 2365), anti-β tubulin (Sigma Aldrich, Cat. T8328–200UL), anti-RVFV clone 4–39-CC (BEI Resources; NR-43195), custom rabbit anti-RVFV NP from GenScript (Cartwright et al., 2020).
CRISPR Cas9 Screen.
BV2 Cas9 library cells were generated as described previously (Orvedahl et al., 2019). Briefly, Cas9 activity was evaluated in BV2-Cas9 cells by transducing pXPR 011 plasmid (Addgene 59702) expressing eGFP and sgRNA targeting eGFP. Further, the BV2-Cas9 cells were transduced with the Brie library (Addgene #73633) targeting 19,674 mouse genes with 78,637 gRNAs (~4 gRNAs for each gene) (Doench et al., 2016). 160 × 106 cells were transduced with the library at 0.25 infectivity rate to achieve a coverage of 500x and two days post-transduction, puromycin (Sigma Aldrich, Cat. P833) was added and the cells were selected in puromycin for 5 days. Library cells were expanded and coverage of 500 per sgRNA (40×106 transduced cells). Two vials of library cells each containing 25 × 106 cells were seeded in 150 cm2 flasks and infected with RVFV ZH501 at MOIs of 0.1 and 0.01 in the University of Pittsburgh RBL BSL-3 facility. Infections were carried out using DMEM with 2% FBS. Cells were observed daily for cytopathic effect (CPE). Dead floating cells were removed and replaced with fresh DMEM/2%FBS. By 4 dpi, the majority of the dead cells were removed. Surviving cells were cultured in DMEM with 10% FBS for an additional 14 days, during which time colonies developed. At 18 dpi, all remaining cells were then trypsinized; half of the cells were treated with TRIzol for genomic DNA extraction and the other half were re-infected with RVFV ZH501 at MOIs of 0.1 and 0.01. Three days after re-infection, remaining live cells were treated with TRIzol for DNA extraction.
Genomic DNA Extraction, Next-Generation Sequencing, and Analysis.
Genomic DNA was extracted from TRIzol treated samples as previously (Chomczynski, 1993). Briefly, 20 μL of chloroform was added to each TRIzol treated sample (~1 mL), incubated at 25 °C for 2–3 mins, and then centrifuged at 12,000 x g for 15 mins at 4 °C. The upper aqueous phase containing RNA was discarded. 300 μL of ethanol was added to each sample and mixed by inverting several times. Samples were incubated for 2–3 mins and then centrifuged at 2000 x g for 5 mins at 4 °C to pellet the DNA. The pellet was resuspended in 1 mL of 100 mM sodium citrate (pH 8.5) in 10% ethanol, incubated for 30 mins, and centrifuged at 2,000 × g for 5 mins at 4 °C. The supernatant was discarded and the process was repeated twice. The pellet was washed with 75% ethanol and gDNA pellets were air-dried and solubilized in 500 μL of 8 mM NaOH. After centrifugation at 12,000 × g at 4 °C for 10 mins, the supernatant was transferred to a new tube, and the pH was adjusted to 7.5 with HEPES. The DNA purity and concentration were determined using the NanoDrop 2000c spectrophotometer (Thermo Scientific).
Illumina sequencing was performed at the Broad Institute at the Massachusetts Institute of Technology, similar to previous studies (Orvedahl et al., 2019). Briefly, gDNA was PCR amplified in a 96-well plate, each well containing up to 10 μg of the DNA, using primers amplifying barcodes associated with each sgRNA in the integrated vector. PCR products were purified and sequenced on Illumina HiSeq 2000. Barcodes were deconvoluted and mapped to the reference file. An array of read counts were generated and normalized to 107 total reads per sample as scores files. The data was then log2-transformed to generate log2-norm files. The abundance of perturbations was calculated as log2 fold change (LFC) by subtracting the average of log2 normalized values of each infection condition with the uninfected log2-normalized values. Volcano plots were generated to display the primary screening data where the x-axis represents average log2 fold change of all perturbations of a gene and the y-axis represents average p-values on the log10 scale (github.com/mhegde/volcano_plots).
Generation of BV2 Knockout cell lines.
All knockout cell lines were generated at Genome Engineering and iPSC center (GEiC) at Washington University. Briefly, BV2 cells were nucleofected with Cas9 complexed with gene-specific gRNAs. For Lrp1 KO cells, a deletion strategy was employed to remove the first two exons with flanking sequences (Extended Figure 2). A total of four gRNAs were used, and the target sites are g1: 5’-GAGTAAACAGGGACACCCGCGGG; g2: 5’-CGGCTCGGGACCCCACTGAGGGG; g3: TCTGATTACACCACTTATTGGGG; and g4: GGTTATCAAGGGTAACATGTAGG. g1 and g4 RNPs were co-transfected first, and after the cells recovered, g2 and g3 RNPs were co-transfected into the same pool to increase the chance of introducing the deletion of the first two coding exons in every allele. The cells were subjected to single-cell sorting, and DNA was extracted from each clone for amplification in deletion-specific PCRs. PCR products were sequenced to reveal deletion junctions. Lack of PCR amplification of fragments within the deleted region confirmed that all alleles contain a deletion, and the deleted sequence did not invert or randomly integrate to elsewhere in the genome. For RAP and Grp94 knockout cells, a single gRNA was used to target each gene. The gRNA target sites 5’-CTCCCCGGACTCGCGCTTGGCGG and 5’-AAGACCACTCAAATCGAACACGG were used to target RAP and Grp94 genes, respectively. Single-cell clones were analyzed via next-generation sequencing for out-of-frame indels.
Neutralization assays with mRAPD3 and soluble LRP1 CLII, CLIII, and CLIV domains.
Cell lines (e.g. BV2, HEK-293T, BHK-21, BCE, HepG2, SH-SY5Y, VeroE6) from different species were seeded in 24-well plates and cultured overnight. The next day, media was removed and cells were incubated with recombinant mRAPD3 protein at concentrations as defined in the figure in DMEM media supplemented with 2% FBS. After 45–60 mins of mRAPD3 treatment, the cells were infected with either RVFV MP12GFP or RVFV ZH501. 15–24 hpi, cells were assessed for virus infection through GFP expression by flow cytometry, intracellular Gn expression by flow cytometry (Albe et al., 2019), or viral RNA synthesis by qRT-PCR analysis (McMillen et al., 2018). In LRP1 neutralization assays, Fc and Fc-fused LRP1 CLII, CLIII, and CLIV domains were pre-incubated with the RVFV MP12GFP virus in serum-free media at increasing concentrations as described in the figure. After 1 hr of incubation at 37 °C, the preparations were used to infect the BV2 cells. Virus infection was examined 15 hpi by flow cytometry.
Plasmids.
RVFV glycoprotein Gn ectodomain (amino acid 1 – 316; accession number DQ380200) (Wu et al., 2017) derived from ZH501 and mRAPD3 (amino acid 243–360; NM_013587.2) were cloned into a pET28 vector (Novagen). Mouse Lrp1 CLI (residues 26–114), CLII (residues 804–1184), CLIII (residues 2482–2943), and CLIV (residues 3294–3784) domains were cloned into a modified pLVX-EF1 α-vector (Takara) containing the Lrp1 transmembrane (TM) and cytoplasmic tail (CT) domains (residues 3785–4545).
Protein expression and purification.
Gn316 expression plasmids were transformed in BL21(DE3) E. coli cells (Novagen). Colonies were cultured in Luria Broth media at 37 °C to an OD600 of 0.6 and induced with 0.5 mM isopropyl-β-D-thiogalactoside (IPTG) for 12 hr at 18 °C. Cells were harvested and resuspended in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 5 mM 2-mercaptoethanol. Cells were lysed using an EmulsiFlex-C5 homogenizer (Avestin). The pellet was resuspended in 30 mL cold 2 M urea, 20 mM Tris-HCl (pH8.0), 500 mM NaCl, 2% Triton™ X-100 prior to centrifugation at 47,000 x g at 4 °C for 10 mins. Inclusion bodies were isolated after repeated rounds of resuspension in urea and centrifugation. The final pellet was resuspended in 20 mM Tris-HCl (pH 8.0), 500 mM NaCl, 5 mM imidazole, 8 M urea, and 1 mM 2-mercaptoethanol. Gn316 was refolded on a NiFF (GE Healthcare) column using a reverse linear urea gradient and eluted with imidazole. Gn316 was further purified using a size exclusion column (SD200 10/300L, GE Healthcare). mRAPD3 was expressed in BL21(DE3) E. coli cells (Novagen), cultured in Luria Broth media at 37 °C, induced with 0.5 mM IPTG, and grown for 12 hr at 18 °C. Cells were harvested and resuspended in lysis buffer containing 20 mM Tris-HCl (pH 8.0), 150 mM NaCl, 20 mM imidazole, 5 mM 2-mercaptoethanol, and were lysed using an EmulsiFlex-C5 homogenizer (Avestin). Lysates were clarified by centrifugation at 47,000 x g at 4 °C for 40 min. Proteins were purified using a series of chromatographic columns and a size exclusion column as a final step. Protein purity was determined by Coomassie staining of SDS-PAGE. Mutant mRAPD3 was purified similarly as Gn. Endochrome-K™ kit (Charles River) was used, following the manufacturer’s instructions to determine endotoxin levels for purified mRAPD3 proteins and the control protein.
Biolayer Interferometry.
BLI assays were conducted at 30 °C at 1,000 rpm (Octet Red, ForteBio). Anti-Human IgG Fc Capture biosensors were hydrated in kinetics buffer (Phosphate Buffer Saline (PBS) containing 0.02% Tween-20, 1 mg/mL BSA) for 15 min. Recombinant human LRP1 CLIV-Fc Chimera (R&D SYSTEMS, #5395-L4–050), recombinant human LRP-1 CLII-Fc chimera (R&D SYSTEMS, #2368-L2–050), or recombinant human IgG1 Fc (R&D SYSTEMS, #110-HG-100) were loaded at 200 nM in buffer for 600s prior to baseline equilibration for 300 s. Association of RVFV Gn or mRAPD3 at various concentrations (0.5, 1, 4, 8, and 12 µg/mL) was carried out for 900 s prior to dissociation for 900 s. Data were baseline subtracted to the buffer only controls. Experiments were done in triplicate.
Competition and pull-down assays.
Competition assay was performed using rProtein A Sepharose® Fast Flow resin (GE Healthcare, #17–1279-03). Human LRP-1 CLIV-Fc Chimera (R&D SYSTEMS, #5395-L4–050) was immobilized on resin prior to incubation with RVFV Gn, mRAPD3, or fixed concentration of RVFV Gn in the presence of increasing concentrations of mRAPD3 (1 −10 µg/mL). After a 1 h incubation at 25 °C, beads were washed six times with PBS-T buffer prior to elution of bound proteins in 2X-laemmli sample buffer. Samples were run on SDS–PAGE and analyzed by western blotting using an anti-His-tag antibody (Cell Signaling, Cat 2365) or anti-human Fc antibody (Abcam, Cat ab98624). Similarly, pulldown assay was performed by incubating RVFV Gn with human IgG1 Fc and recombinant human LRP-1 CLII, CLIII and CLIV Fc chimera using rProtein A beads. After washings, the elutions were analyzed by western blotting with anti-His and anti-human Fc antibodies (see above).
Deglycosylation of Lrp1 CLII-Fc and CLIV-Fc.
For deglycosylation reactions, 50 µg of CLII-Fc and CLIV-Fc were incubated with New England Biolabs Deglycosylation Mix II (Cat. #P6044S) under non-denaturing conditions in phosphate buffer initially at 25 ºC for 30 minutes followed by 37 ºC for 16 hours. Deglycosylation Mix II contains a mixture of glycosidases optimized for both N- and O-linked glycans including O-Glycosidase, PNGase F, α2–3,6,8,9 Neuraminidase A, β1–4 Galactosidase S and β-N-Acetylhexosaminidasef. Deglycosylation was confirmed by gel mobility assays and analytical SEC.
Analytical Size Exclusion Chromatography- Multi-angle Light Scattering (SEC-MALS).
Analytical SEC was performed using an Agilent 1260 Infinity II HPLC System equipped with a Superdex 200 Increase 5/150 GL column coupled to a multi-angle light scattering system (DAWN HELEOS-II, Optilab T-rEX, and Eclipse DualTec - Wyatt Technologies). For all experiments, 10 µL of protein at a concentration of 1 mg/mL in PBS was injected onto the column. Chromatograms were processed using Astra 7.3 (Wyatt Technologies).
Mass Spectrometry.
For reduced samples, 20 pmols of protein in PBS was incubated with ~700 mM tris(2-carboxyethyl)phosphine hydrochloride (TCEP-HCl) for 5 minutes. The solution was then diluted ~7-fold with 94.9:5:0.1 water/acetonitrile (ACN)/formic acid (FA) (%v/v/v) for a total of a 50 µL injection. For nonreduced samples, 20 pmols of protein was diluted ~25-fold with 94.9:5:0.1 water/ACN/FA (%v/v/v) for a total of a 50 µL injection. For denaturing MS, the sample was injected into a custom-built liquid chromatography (LC) apparatus for LC-MS analysis. The sample was desalted on a reversed phase C4 column (ACQUITY UPLC Protein BEH C4, 300Å, 1.7 µm, 2.1 mm X 50 mm) (Waters, Milford, MA), 60 °C in a column oven, for 4.5 minutes with water/FA (0.1% FA) at 200 µL/min and eluted by a 7-minute linear gradient 16 – 80 % ACN/0.1% FA (flow rate 200 µL/min). The samples were directly infused into a Bruker Maxis HM Q-TOF MS (Billerica, MA) for mass analysis tuned for mid-range or high-range m/z measurements, with an adjusted 7 mbar funnel pressure for high m/z and desolvation with a collection range of 500–3000 m/z. For extended mass range analysis, the C4-eluted proteins were directly infused into a Thermo Exactive Extended Mass Range Orbitrap Mass Spectrometer (Waltham, MA) with an acquisition range of 1500–12000 m/z; instrumental settings were as described by VanAernum et al. unless otherwise noted: in-source dissociation off, HCD off, injection flatapole DC 15 V, interflatapole lens 12 V, bent flatapole DC 10 V, C-trap pressure setting 1. For SEC Native experiments, samples were diluted in 200 mM ammonium acetate and separated on a size-exclusion column (Waters BEH SEC, 2.1×50mm, 200Å) and directly infused onto the Thermo Exactive Extended Mass Range Orbitrap Mass Spectrometer (Waltham, MA) with an acquisition range of 1500–12000 m/z, in-source dissociation 50 V, HCD 50 V, source DC offset 18 V, injection flatapole DC 12 V, interflatapole lens 10 V, bent flatapole 5 V, transfer multipole DC 4 V, C-trap pressure setting 4. Data were analyzed using Intact Mass™ deconvolution software (PMI Protein Metrics, Cupertino, CA, version 3.11).
Flow Cytometry.
RVFV MP12GFP and RSVGFP infected cells were analyzed by flow cytometry (BD LSR Fortessa™ X-20 and BD LSR Fortessa™) and the data were analyzed using BD FACS Diva software, as described previously (Ganaie et al., 2017). All flow experiments were done at the Flow Cytometry Facility, Department of Pathology and Immunology, WUSM. For flow experiments with RVFV ZH501, infected cells were harvested at the indicated time points, stained with LIVE/DEAD™ Fixable Blue Dead Cell Stain Kit for UV excitation (Invitrogen L34961), permeabilized with BD fix/perm, then stained with RVFV anti-Gn monoclonal antibody (BEI NR-43195) followed by a FITC-conjugated anti-mouse secondary antibody. Samples were acquired using BD LSRII flow cytometer and analyzed with FlowJo at the University of Pittsburgh Flow core facility. Uninfected cells were run in parallel for subtraction of background.
Reverse transcription- quantitative PCR for influenza virus.
cDNA was synthesized using SuperScript™ III (Invitrogen) by following the manufacturer’s instructions. A multiplex RT-qPCR system was used to detect IAV PR8 M segment using the following primers: Forward 5’- AAGACCAATCCTGTCACCTCTGA-3’ and reverse 5’- CAAAGCGTCTACGCTGCAGTCC-3’.
Virus binding and internalization assays.
BV2-WT and BV2-Lrp1 KO (R4) cells (5 × 105) were seeded in 12-well plates and incubated with GAG antagonist surfen (10 µM) for 30 mins. Next, the cells were moved to 4 °C for 30 mins and then incubated with RVFV-MP12GFP virus (MOI 0.5) for 1 hr at 4 °C. The cells were washed 5 times with PBS supplemented with 3% bovine serum albumin and 0.02 % tween-20. For virus binding assay, the cells were collected and lysed in RLT buffer (Qiagen) for RNA extraction using RNeasy Mini Kit (Qiagen). For internalization assay, the cells were incubated at 37 °C for 1 more hour. The cells were again washed and collected for RNA extraction using RNeasy kit (Qiagen). The RT-qPCR was performed using Power SYBR Green Master Mix (Thermo Scientific) with mouse hprt as a control. RVFV MP12 M-segment was amplified using forward primer: GTCAGCTCATCACCTCAACAA and reverse primers: CACCTGTCATCTGCCTACAAA and host gene hprt was amplified using forward primer: CTGGTGAAAAGGACCTCTCGAAG and reverse primer: CCAGTTTCACTAATGACACAAACG.
Virus Particle Binding Assay.
Gradient purified VSV-RVFV and VSV particles were labeled with AlexaFluor 594 and 647, respectively, as previously described (Cureton et al, 2009). Both viruses were added to indicated cells and incubated at either 37 °C for 15 min or 4 C for 1 h. Three minutes before the end of the incubation, 1 ug/mL Alexa 488 labeled wheat germ agglutinin was added to the media. Cells were then washed two times with ice cold PBS and fixed with 2% PFA for 10 minutes at room temperature. Samples were imaged using a Nikon Ti2 inverted microscope outfitted with a spinning disc head (Yokogawa), Andor Zyla 4.2 Plus sCMOS monochrome camera, and piezo Z stage (Physik Instrument). Images were acquired using Nikon Elements Acquisition Software AR 5.02. Image analysis was performed using Arivis Vision4D. Briefly, cells were masked, and the volume was determined using membrane-based segmentation. Bound viral particles were counted for each image and particle binding per area was calculated by dividing particle counted by the determine cellular volume. At least 3 images were acquired and analyzed for each sample.
Antibody selections by phage display.
A synthetic phage-displayed Fab library (Persson et al., 2013) was used for binding selections with immobilized Fc-tagged LRPI-CII (R&D SYSTEMS, #2368-L2–050) or LRPI-CIV (R&D SYSTEMS, #5395-L4–050), as described (Kuruganti et al., 2016). Following 4 rounds of selections, individual clones were characterized for binding to target and control proteins by phage ELISA. Phagemid DNA from binding clones was amplified by the PCR and sequenced to decode the antibody variable region sequences.
IgG production.
DNA encoding the variable regions of phage-derived antibodies was amplified from phagemid DNA by the PCR and sub-cloned in to separate light and heavy chain expression vectors. Equal amounts of DNA from heavy and light chain expression vectors were mixed, diluted in Opti-MEM medium (Gibco), and complexed with FectoPro transfection reagent (Polyplus Transfection) for 10 minutes. Complexed DNA was transfected in to Expi-293F cells in Expi293 medium and and the cultures were incubated for 5 days at 37 °C in a humidified, 8% CO2 environment with shaking. Secreted IgG protein was purified from supernatants with Protein A sepharose (GE Healthcare), eluted in IgG elution buffer (Thermo), neutralized with 1 M Tris buffer pH 8.0 (Invitrogen), and exchanged in to PBS using centrifugal concentrators.
Enzyme-linked immunosorbent assays.
Binding of Fab-phage or IgGs to antigen was measured by ELISA. Wells of microplates (Nunc) were coated overnight at 4 °C with a 2 μg/mL antigen solution in PBS pH 7.4 and blocked with PBS, 0.2% BSA for 1 hour at room temperature. Blocking solution was removed, plates were washed 4 times with PBS, 0.05% Tween, and phage or IgG was added and incubated for 30 minutes. Plates were washed, incubated for 30 minutes with an appropriate secondary antibody, and developed with TMB substrate (KPL Laboratories).
Histology and immunofluorescence (IF).
For histology, tissues were fixed and inactivated in 4% PFA prior to removal from the BSL-3 lab. Tissues were incubated in 15% sucrose in PBS for 24 hrs at 4 °C, followed by 30% sucrose in PBS for another 24 hrs prior to flash freezing. For flash freezing, sucrose saturated tissues were submerged in liquid nitrogen-cooled 2-methylbutane for 30 seconds then stored at −80 °C until cryosectioning. For sectioning, frozen tissues were embedded in Fisher Healthcare™ Tissue-Plus™ O.C.T. compound, re-flash frozen, then cryosectioned on a Thermo Scientific Microm HM 550 at 6–8 um thickness. Frozen sections were placed on charged slides and stored at −80 °C until staining. Following an alcohol rehydration series, slides were stained following standard hematoxylin and eosin (H&E) staining procedures. Images were taken at 20x on an Olympus CX41 microscope with a Levenhuk microscope digital camera (M base series). For immunofluorescence staining, OCT media was washed off of the slides with 1xPBS + 0.5% BSA (PBB). Washes were followed by a 15-minute permeabilization step using 0.1% Triton X-100 detergent + 1x PBS at room temperature. Following permeabilization, slides were blocked using normal donkey serum for 45 minutes at room temperature. The samples were incubated with a custom anti-rabbit RVFV NP antibody (1:50) (Cartwright et al., 2020) for 1 hour at room temperature, followed by incubation with a donkey anti-rabbit Cy3 secondary antibody (Jackson ImmunoResearch). The slides were counterstained with Hoescht and mounted using Gelvatol. Fluorescent slides were imaged using a Leica DMI8 inverted fluorescent microscope, and denoised using the Leica Application Suite X software provided by the Center for Vaccine Research.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed by Prism Version 8.0 (GraphPad). Statistical significance was determined by using 1-way ANOVA analysis, followed by Dunnett’s test for comparison of three or more groups and unpaired (Student) t-test for comparison of two groups. Error bars show mean and standard deviation (Mean ± SD) unless otherwise specified. The number of animals (n), mean values and statistical comparison groups are described in the figures and figure legends.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit anti-Lrp1 antibody | Cell Signaling | Cat# 64099, RRID: AB_2799654 |
| Rabbit anti-His antibody | Cell Signaling | Cat# 2365 |
| Mouse anti-tubulin antibody | Sigma Aldrich | Cat# T8328–200UL RRID: AB_1844090 |
| Anti-RVFV clone 4–39-CC | This Paper | N/A |
| Anti-human IgG Fc (HRP) | Abcam | Cat# ab98624 RRID: AB_10673832 |
| Anti-HA antibody | SantaCruz | sc-805 RRID: AB_631618 |
| Anti-rabbit secondary antibody Alex Fluor-764 | Jackson Immuno Research | NC025445 |
| Rabbit anti-RVFV NP | Genescript | N/A |
| Donkey anti-rabbit Cy3 secondary antibody | Jackson Immuno Research | Cat# 711–165-152 RRID: AB_2307443 |
| Peroxidase-conjugated Donkey Anti-Mouse IgG | Jackson Immuno Research | Cat# 715–035-150 RRID: AB_2340770 |
| Bacterial and virus strains | ||
| E. coli BL21 (DE3) | Novagen | Cat# 69450 |
| E. coli Stable3 | Thermo Fisher Scientific | Cat# C737303 |
| RVFV ZH501 (reverse genetics generated) | US CDC | N/A |
| RVFV MP12 GFP | Zhang et al., 2018 | N/A |
| Adenovirus mCherry | Vector Biolabs | Cat# 1767 |
| Adenodvirus mCherry Cre | Vector Biolabs | Cat# 1773 |
| IAV PR8 strain | Williams et al., 2018 | N/A |
| RSV GFP5 | Viratree | Cat# R125 |
| VSV-RVFV-eGFP | S. Whelan WashU | N/A |
| Lentivirus | Addgene vectors | Cat# 8454 RRID: Addgene_8454 Cat# 12260 RRID: Addgene_12260 |
| VSV-eGFP | S. Whelan WashU | N/A |
| Biological samples | ||
| Chemicals, peptides, and recombinant proteins | ||
| RVFV Gn (aa 1–316) | this manuscript | N/A |
| RVFV Gn (aa 1–410) | Genscript | N/A |
| mRAPD3 | this manuscript | N/A |
| mutant mRAPD3 | this manuscript | N/A |
| human LRP1 CLII- Fc | R & D Systems | Cat# 2368-L2–050 |
| human LRP1 CLIII- Fc | R & D Systems | Cat# 4824-L3–50 |
| human LRP1 CLIV- Fc | R & D Systems | Cat# 5395-L4–050 |
| human IgG1 Fc | R & D Systems | Cat# 110-HG-100 |
| Control VP30 protein | Xu et al., 2017 | N/A |
| Puromycin | Sigma Aldrich | Cat# P8833 |
| Blasticidin | Gibco | Cat# R21001 |
| Lipofectamine 2000 | Invitrogen | Cat# 11668019 |
| Surfen (GAG Inhibitor) | Sigma Aldrich | Cat# 362330 100MG |
| Fetal Bovine Serum | Gibco | Cat# F2442 |
| Fetal Bovine Serum | Gibco | Cat# F2442 |
| HEPES | Sigma Millipore | Cat# 25–060-CI |
| Sodium pyruvate | Corning | Cat# 25–000-CI |
| Protein deglycosylation Mix II | New England Biolabs | Cat# P6044S |
| DMEM Media | Gibco | Cat# 11965084 |
| ammonium acetate, 99.999% purity | Millipore Sigma | 372331–100G; CAS 631–61-8 |
| Tris-2(carboxyethyl)phosphine hydrochloride | Millipore Sigma | C4706–10G; CAS 115–96-8 |
| LCMS grade formic acid | CovaChem | 11202–10×1; CAS 64–18-6 |
| Triton X-100 | Sigma | X100 |
| Permount | Fisher Chemical | Sp15–100 |
| TRIzol Reagent | Ambion | 15596018 |
| Seracare TMB 2-C ELISA HRP Substrate | Fisher Scientific | 50–674-21 |
| Seracare TMB STOP Solution | Fisher Scientific | 50–674-44 |
| Paraformaldehyde | MP Biochemicals | 150146 |
| Critical commercial assays | ||
| RNeasy Mini Kit | Qiagen | Cat# 74004 |
| Endochrome-K LAL Chromogenic kit | Charles River | Lot# M1153EK1 |
| Power SYBR™ Green PCR Master Mix | ThermoFisher Scientific | Cat# 4367659 |
| SuperScript III Platinum One-Step qRT-PCR Kit | Invitrogen | Cat# 11732088 |
| Deposited data | ||
| CRISPR Screen data files | This paper | |
| Experimental models: Cell lines | ||
| Human: HEK-293T | ATCC | CRL-3216 RRID: CVCL_0063 |
| Human: SH-SY5Y | ATCC | CRL-2266 RRID: CVCL_0019 |
| Human: HepG2 | ATCC | HB-8065 RRID: CVCL_0027 |
| Mouse: BV2 | Orvedahl et al., 2019 | N/A |
| African Green Monkey: VeroE6 | ATCC | CRL-1586 RRID: CVCL_0574 |
| African Green Monkey: COS1 | ATCC | CRL-1650 RRID: CVCL_0223 |
| Bovine: BCE C/D-1b | ATCC | CRL- 2048 RRID: CVCL_2865 |
| Baby Hamster: BHK-21 | ATCC | CCL-10 RRID: CVCL_1915 |
| BV2 Lrp1 KO (R1 - R6) | this manuscript | N/A |
| BV2 Lrp1 PKO R3 | this manuscript | N/A |
| BV2 Lrpap1 KO A7 | this manuscript | N/A |
| BV2 Lrpap1 PKO A3 | this manuscript | N/A |
| BV2 Grp KO (A8, B7) | this manuscript | N/A |
| BV2 Ext1, Ext2 | M. Diamond, Wash U | N/A |
| Experimental models: Organisms/strains | ||
| Mice | The Jackson Laboratory | C57BL/6 |
| Oligonucleotides | ||
| GAGTAAACAGGGACACCCGCNGG | Lrp1 5’ gRNA1 | N/A |
| CGGCTCGGGACCCCACTGAGNGG | Lrp1 5’ gRNA2 | N/A |
| GGTTATCAAGGGTAACATGTNGG | Lrp1 3’ gRNA1 | N/A |
| TCTGATTACACCACTTATTGNGG | Lrp1 3’ gRNA2 | N/A |
| CTCCCCGGACTCGCGCTTGGNGG | Lrpap1 (RAP) gRNA | N/A |
| AAGACCACTCAAATCGAACANGG | Grp94 gRNA | N/A |
| TGAAAATTCCTGAGACACATGG | RVFV-2912Fwd | N/A |
| ACTTCCTTGCATCATCTGATG | RVFV-2981Rev | N/A |
| 56FAM/CAATGTAAGGGGCCTGTGTGGACTTGTG/3BHQ_1 | RVFV-2950-Probe | N/A |
| 5’- AAGACCAATCCTGTCACCTCTGA-3’ | IAV-PR8-M-For | N/A |
| 5’- CAAAGCGTCTACGCTGCAGTCC-3’. | IAV-PR8-M-Rev | N/A |
| 5’-GTCAGCTCATCACCTCAACAA-3’ | RVFV-MP12- M seg For | N/A |
| 5’-CACCTGTCATCTGCCTACAAA-3’ | RVFV-MP12- M seg Rev | N/A |
| 5’-CTGGTGAAAAGGACCTCTCGAAG-3’ | Mouse hprt Fw | N/A |
| 5’-CCAGTTTCACTAATGACACAAACG-3’ | Mouse hprt Rev | N/A |
| pLVX-EF1a | Takara | Cat# 631253 |
| pLVX-mini LRP1 CLI | this manuscript | N/A |
| pLVX-mini LRP1 CLI | this manuscript | N/A |
| pLVX-mini LRP1 CLI | this manuscript | N/A |
| pLVX-mini LRP1 CLI | this manuscript | N/A |
| pET-mRAP D3 | this manuscript | N/A |
| pET-mutant mRAP D3 | this manuscript | N/A |
| pET-Gn316 | this manuscript | N/A |
| Prism 8.3.0 | Graphpad | N/A |
| BD FACS Diva software™ | BD Biosciences | N/A |
| Octet software | Forte Bio | N/A |
| FlowJo v10.6.2 | FlowJo LLC | N/A |
| Origin 7 | Origin Lab Corporation | N/A |
| oyager Chromatography Data system v2.0.2.9 | Wyatt Technology Corporation | N/A |
| Astra 7.3 | Wyatt Technology Corporation | N/A |
| Byos® v 3.11 | Protein Metrics Inc. | N/A |
| Freestyle™ v 1.4 | Thermo Fisher Scientific Inc. | N/A |
| DataAnalysis v 4.4 | Bruker Corp. | N/A |
| Leica Application Suite X | Leica | N/A |
| Other | ||
Highlights:
Unbiased genome-wide screen identifies Lrp1 as a host factor for RVFV infection.
RVFV surface glycoprotein Gn directly binds to Lrp1 and blocks RVFV infection.
Lrp1 ligands inhibit RVFV infection in cells derived from taxonomically distinct hosts.
Lrp1 ligands protects from lethal RVFV infection in vivo.
Acknowledgements
We thank members of our groups for discussions and support. We also thank Drs. J. Boon, M. Diamond, and S. Khader for providing critical reagents and for discussion. We thank Dr. A. Schwartz (Washington University School of Medicine) and J. Herz (UT Southwestern) for critical discussions and for their encouragement to embark on studies on Lrp1. We also thank R. Ridings and S. Barrick for coordinating studies between WUSM and Pitt, and E.O.L. Amarasinghe and E.E.L. Amarasinghe for assistance with Figure 1. Research was supported by NIH grants (R01AI161765 to G.K.A and A.L.H; R01NS101100 to A.L.H; P01AI120943 and R01AI123926 to G.K.A.; R01AI107056 to D.W.L.; U19AI142784 and U19AI10972505 to H.W.V.; R01AI130152 to T.E.; T32AI060525 to C.M.M.; T32AI106688 and 1K08AI144033 to A.O.; R37 AI059371 to S.P.J.W.; R24GM136766 and P41GM103422 to M.L.G.). A.K.M was supported by a Burroughs Wellcome CAMS award (1013362.02). Additional support was provided by the Pediatric Infectious Diseases Society, St. Jude Children’s Research Hospital Fellowship Program in Basic Research, and Society for Pediatric Research Physician Scientist Bridging to Success Award to A.O.; Alzheimer’s Association Research Grant (AARG-16–441560) to T.J.B; and The Leukemia and Lymphoma Society Scholar Award to T.E. We wish to thank the Microscopy Resources on the North Quad (MicRoN) core at Harvard Medical School for imaging and image analysis assistance. We would like to thank Bruker for technical and instrument support, and PMI who donated the software used for deconvolution (R42GM1213302 supported the software). We also thank the Genome Engineering and iPSC Center (GEiC) at the Washington University in St. Louis for gRNA validation services.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Competing interest statement:
HWV is a founder of Casma therapeutics and pierianDx and is employed by Vir Biotechnology. None of these companies funded the work reported here. Invention disclosures for method of use for Lrp1 interaction with RVFV Gn (G.K.A., A.L.H., D.W.L., H.W.V. and S.G.) and for the use of anti-Lrp1 antibodies by U Toronto (S.S.S., S.M., G.K.A.) have been filed.
Inclusion and Diversity
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. One or more of the authors of this paper self-identifies as an underrepresented ethnic minority in science. One or more of the authors of this paper self-identifies as a member of the LGBTQ+ community. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list.
References:
- ALBE JR, BOYLES DA, WALTERS AW, KUJAWA MR, MCMILLEN CM, REED DS & HARTMAN AL 2019. Neutrophil and macrophage influx into the central nervous system are inflammatory components of lethal Rift Valley Fever encephalitis in rats. PLoS Pathog, In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- BLASI E, RADZIOCH D, DURUM SK & VARESIO L 1987. A murine macrophage cell line, immortalized by v-raf and v-myc oncogenes, exhibits normal macrophage functions. Eur J Immunol, 17, 1491–8. [DOI] [PubMed] [Google Scholar]
- BRUSTOLIN M, TALAVERA S, NUNEZ A, SANTAMARIA C, RIVAS R, PUJOL N, VALLE M, VERDUN M, BRUN A, PAGES N & BUSQUETS N 2017. Rift Valley fever virus and European mosquitoes: vector competence of Culex pipiens and Stegomyia albopicta (= Aedes albopictus). Med Vet Entomol, 31, 365–372. [DOI] [PubMed] [Google Scholar]
- BU G 2001. The roles of receptor-associated protein (RAP) as a molecular chaperone for members of the LDL receptor family. Int Rev Cytol, 209, 79–116. [DOI] [PubMed] [Google Scholar]
- BU G & SCHWARTZ AL 1998. RAP, a novel type of ER chaperone. Trends Cell Biol, 8, 272–6. [DOI] [PubMed] [Google Scholar]
- CANUEL M, SUN X, ASSELIN MC, PARAMITHIOTIS E, PRAT A & SEIDAH NG 2013. Proprotein convertase subtilisin/kexin type 9 (PCSK9) can mediate degradation of the low density lipoprotein receptor-related protein 1 (LRP-1). PLoS One, 8, e64145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CARTWRIGHT HN, BARBEAU DJ & MCELROY AK 2020. Rift Valley Fever Virus Is Lethal in Different Inbred Mouse Strains Independent of Sex. Front Microbiol, 11, 1962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CHOMCZYNSKI P 1993. A reagent for the single-step simultaneous isolation of RNA, DNA and proteins from cell and tissue samples. Biotechniques, 15, 532–4, 536–7. [PubMed] [Google Scholar]
- DE BOER SM, KORTEKAAS J, DE HAAN CA, ROTTIER PJ, MOORMANN RJ & BOSCH BJ 2012. Heparan sulfate facilitates Rift Valley fever virus entry into the cell. J Virol, 86, 13767–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DE NARDIS C, LOSSL P, VAN DEN BIGGELAAR M, MADOORI PK, LELOUP N, MERTENS K, HECK AJ & GROS P 2017. Recombinant Expression of the Full-length Ectodomain of LDL Receptor-related Protein 1 (LRP1) Unravels pH-dependent Conformational Changes and the Stoichiometry of Binding with Receptor-associated Protein (RAP). J Biol Chem, 292, 912–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DODD KA, BIRD BH, METCALFE MG, NICHOL ST & ALBARINO CG 2012. Single-dose immunization with virus replicon particles confers rapid robust protection against Rift Valley fever virus challenge. J Virol, 86, 4204–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DOENCH JG, FUSI N, SULLENDER M, HEGDE M, VAIMBERG EW, DONOVAN KF, SMITH I, TOTHOVA Z, WILEN C, ORCHARD R, VIRGIN HW, LISTGARTEN J & ROOT DE 2016. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat Biotechnol, 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FREIBERG AN, SHERMAN MB, MORAIS MC, HOLBROOK MR & WATOWICH SJ 2008. Three-Dimensional Organization of Rift Valley Fever Virus Revealed by Cryoelectron Tomography. Journal of Virology, 82, 10341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GANAIE SS, ZOU W, XU P, DENG X, KLEIBOEKER S & QIU J 2017. Phosphorylated STAT5 directly facilitates parvovirus B19 DNA replication in human erythroid progenitors through interaction with the MCM complex. PLoS Pathog, 13, e1006370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GAUDREAULT NN, INDRAN SV, BRYANT PK, RICHT JA & WILSON WC 2015. Comparison of Rift Valley fever virus replication in North American livestock and wildlife cell lines. Front Microbiol, 6, 664. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GOMMET C, BILLECOCQ A, JOUVION G, HASAN M, ZAVERUCHA DO VALLE T, GUILLEMOT L, BLANCHET C, VAN ROOIJEN N, MONTAGUTELLI X, BOULOY M & PANTHIER JJ 2011. Tissue tropism and target cells of NSs-deleted rift valley fever virus in live immunodeficient mice. PLoS Negl Trop Dis, 5, e1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- GONIAS SL & CAMPANA WM 2014. LDL receptor-related protein-1: a regulator of inflammation in atherosclerosis, cancer, and injury to the nervous system. Am J Pathol, 184, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HARTMAN AL, POWELL DS, BETHEL LM, CAROLINE AL, SCHMID RJ, OURY T & REED DS 2014. Aerosolized rift valley fever virus causes fatal encephalitis in african green monkeys and common marmosets. J Virol, 88, 2235–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HASSAN OA, AHLM C, SANG R & EVANDER M 2011. The 2007 Rift Valley fever outbreak in Sudan. PLoS Negl Trop Dis, 5, e1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HEISSIG B, SALAMA Y, TAKAHASHI S, OSADA T & HATTORI K 2020. The multifaceted role of plasminogen in inflammation. Cell Signal, 75, 109761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HERZ J, CLOUTHIER DE & HAMMER RE 1992. LDL receptor-related protein internalizes and degrades uPA-PAI-1 complexes and is essential for embryo implantation. Cell, 71, 411–21. [DOI] [PubMed] [Google Scholar]
- HERZ J & STRICKLAND DK 2001. LRP: a multifunctional scavenger and signaling receptor. J Clin Invest, 108, 779–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- HOFMANN H, LI X, ZHANG X, LIU W, KÜHL A, KAUP F, SOLDAN SS, GONZÁLEZ-SCARANO F, WEBER F, HE Y & PÖHLMANN S 2013. Severe Fever with Thrombocytopenia Virus Glycoproteins Are Targeted by Neutralizing Antibodies and Can Use DC-SIGN as a Receptor for pH-Dependent Entry into Human and Animal Cell Lines. Journal of Virology, 87, 4384–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KNIPE DM & HOWLEY P 2013. Fields Virology, Philadelphia, Wolters Kluwer. [Google Scholar]
- KRAEMER MUG, REINER RC, BRADY OJ, MESSINA JP, GILBERT M, PIGOTT DM, YI D, JOHNSON K, EARL L, MARCZAK LB, SHIRUDE S, DAVIS WEAVER N, BISANZIO D, PERKINS TA, LAI S, LU X, JONES P, COELHO GE, CARVALHO RG, VAN BORTEL W, MARSBOOM C, HENDRICKX G, SCHAFFNER F, MOORE CG, NAX HH, BENGTSSON L, WETTER E, TATEM AJ, BROWNSTEIN JS, SMITH DL, LAMBRECHTS L, CAUCHEMEZ S, LINARD C, FARIA NR, PYBUS OG, SCOTT TW, LIU Q, YU H, WINT GRW, HAY SI & GOLDING N 2019. Past and future spread of the arbovirus vectors Aedes aegypti and Aedes albopictus. Nature Microbiology, 4, 854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- KURUGANTI S, MIERSCH S, DESHPANDE A, SPEIR JA, HARRIS BD, SCHRIEWER JM, BULLER RM, SIDHU SS & WALTER MR 2016. Cytokine Activation by Antibody Fragments Targeted to Cytokine-Receptor Signaling Complexes. J Biol Chem, 291, 447–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LAZIC A, DOLMER K, STRICKLAND DK & GETTINS PG 2003. Structural organization of the receptor associated protein. Biochemistry, 42, 14913–20. [DOI] [PubMed] [Google Scholar]
- LÉGER P, TETARD M, YOUNESS B, CORDES N, ROUXEL RN, FLAMAND M & LOZACH PY 2016. Differential Use of the C-Type Lectins L-SIGN and DC-SIGN for Phlebovirus Endocytosis. Traffic, 17, 639–656. [DOI] [PubMed] [Google Scholar]
- LOZACH PY, KUHBACHER A, MEIER R, MANCINI R, BITTO D, BOULOY M & HELENIUS A 2011. DC-SIGN as a receptor for phleboviruses. Cell Host Microbe, 10, 75–88. [DOI] [PubMed] [Google Scholar]
- MADANI TA, AL-MAZROU YY, AL-JEFFRI MH, MISHKHAS AA, AL-RABEAH AM, TURKISTANI AM, AL-SAYED MO, ABODAHISH AA, KHAN AS, KSIAZEK TG & SHOBOKSHI O 2003. Rift Valley fever epidemic in Saudi Arabia: epidemiological, clinical, and laboratory characteristics. Clin Infect Dis, 37, 1084–92. [DOI] [PubMed] [Google Scholar]
- MCMILLEN CM, ARORA N, BOYLES DA, ALBE JR, KUJAWA MR, BONADIO JF, COYNE CB & HARTMAN AL 2018. Rift Valley fever virus induces fetal demise in Sprague-Dawley rats through direct placental infection. Science Advances, 4, eaau9812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MIGLIORINI MM, BEHRE EH, BREW S, INGHAM KC & STRICKLAND DK 2003. Allosteric modulation of ligand binding to low density lipoprotein receptor-related protein by the receptor-associated protein requires critical lysine residues within its carboxyl-terminal domain. J Biol Chem, 278, 17986–92. [DOI] [PubMed] [Google Scholar]
- MUNYUA P, MURITHI RM, WAINWRIGHT S, GITHINJI J, HIGHTOWER A, MUTONGA D, MACHARIA J, ITHONDEKA PM, MUSAA J, BREIMAN RF, BLOLAND P & NJENGA MK 2010. Rift Valley fever outbreak in livestock in Kenya, 2006–2007. Am J Trop Med Hyg, 83, 58–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NEELS JG, VAN DEN BERG BM, LOOKENE A, OLIVECRONA G, PANNEKOEK H & VAN ZONNEVELD AJ 1999. The second and fourth cluster of class A cysteine-rich repeats of the low density lipoprotein receptor-related protein share ligand-binding properties. J Biol Chem, 274, 31305–11. [DOI] [PubMed] [Google Scholar]
- ODENDAAL L, DAVIS AS, FOSGATE GT & CLIFT SJ 2020. Lesions and Cellular Tropism of Natural Rift Valley Fever Virus Infection in Young Lambs. Veterinary Pathology, 57, 66–81. [DOI] [PubMed] [Google Scholar]
- ORGANIZATION, W. H. 2018. 2018 Annual review of diseases prioritized under the Research and Development Blueprint. Workshop on Prioritization of Pathogens. [Google Scholar]
- ORVEDAHL A, MCALLASTER MR, SANSONE A, DUNLAP BF, DESAI C, WANG YT, BALCE DR, LUKE CJ, LEE S, ORCHARD RC, ARTYOMOV MN, HANDLEY SA, DOENCH JG, SILVERMAN GA & VIRGIN HW 2019. Autophagy genes in myeloid cells counteract IFNgamma-induced TNF-mediated cell death and fatal TNF-induced shock. Proc Natl Acad Sci U S A, 116, 16497–16506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PERSSON H, YE W, WERNIMONT A, ADAMS JJ, KOIDE A, KOIDE S, LAM R & SIDHU SS 2013. CDR-H3 diversity is not required for antigen recognition by synthetic antibodies. J Mol Biol, 425, 803–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PHOENIX I, NISHIYAMA S, LOKUGAMAGE N, HILL TE, HUANTE MB, SLACK OAL, CARPIO VH, FREIBERG AN & IKEGAMI T 2016. N-glycans on the rift valley fever virus envelope glycoproteins Gn and Gc redundantly support viral infection via DC-SIGN. Viruses, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- POIRIER S, MAMARBACHI M, CHEN WT, LEE AS & MAYER G 2015. GRP94 Regulates Circulating Cholesterol Levels through Blockade of PCSK9-Induced LDLR Degradation. Cell Rep, 13, 2064–71. [DOI] [PubMed] [Google Scholar]
- POTERE N, DEL BUONO MG, MAURO AG, ABBATE A & TOLDO S 2019. Low Density Lipoprotein Receptor-Related Protein-1 in Cardiac Inflammation and Infarct Healing. Front Cardiovasc Med, 6, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PRASAD JM, MIGLIORINI M, GALISTEO R & STRICKLAND DK 2015. Generation of a Potent Low Density Lipoprotein Receptor-related Protein 1 (LRP1) Antagonist by Engineering a Stable Form of the Receptor-associated Protein (RAP) D3 Domain. J Biol Chem, 290, 17262–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RAUCH JN, LUNA G, GUZMAN E, AUDOUARD M, CHALLIS C, SIBIH YE, LESHUK C, HERNANDEZ I, WEGMANN S, HYMAN BT, GRADINARU V, KAMPMANN M & KOSIK KS 2020. LRP1 is a master regulator of tau uptake and spread. Nature, 580, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RIBLETT AM, BLOMEN VA, JAE LT, ALTAMURA LA, DOMS RW, BRUMMELKAMP TR & WOJCECHOWSKYJ JA 2016. A Haploid Genetic Screen Identifies Heparan Sulfate Proteoglycans Supporting Rift Valley Fever Virus Infection. J Virol, 90, 1414–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RICH KM & WANYOIKE F 2010. An assessment of the regional and national socio-economic impacts of the 2007 Rift Valley fever outbreak in Kenya. Am J Trop Med Hyg, 83, 52–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- RUSU M, BONNEAU R, HOLBROOK MR, WATOWICH SJ, BIRMANNS S, WRIGGERS W & FREIBERG AN 2012. An assembly model of rift valley Fever virus. Front Microbiol, 3, 254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- SCHARTON D, VAN WETTERE AJ, BAILEY KW, VEST Z, WESTOVER JB, SIDDHARTHAN V & GOWEN BB 2015. Rift Valley fever virus infection in golden Syrian hamsters. PLoS One, 10, e0116722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- STANSLEY B, POST J & HENSLEY K 2012. A comparative review of cell culture systems for the study of microglial biology in Alzheimer’s disease. J Neuroinflammation, 9, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TURELL MJ, BRITCH SC, ALDRIDGE RL, KLINE DL, BOOHENE C & LINTHICUM KJ 2013. Potential for mosquitoes (Diptera: Culicidae) from Florida to transmit Rift valley fever virus. Journal of Medical Entomology, 50, 1111–1117. [DOI] [PubMed] [Google Scholar]
- TURELL MJ, DOHM DJ, MORES CN, TERRACINA L, WALLETTE DL JR., HRIBAR LJ, PECOR JE & BLOW JA 2008. Potential for North American mosquitoes to transmit Rift Valley fever virus. J Am Mosq Control Assoc, 24, 502–7. [DOI] [PubMed] [Google Scholar]
- WILLIAMS GD, TOWNSEND D, WYLIE KM, KIM PJ, AMARASINGHE GK, KUTLUAY SB & BOON ACM 2018. Nucleotide resolution mapping of influenza A virus nucleoprotein-RNA interactions reveals RNA features required for replication. Nat Commun, 9, 465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- WU Y, ZHU Y, GAO F, JIAO Y, OLADEJO BO, CHAI Y, BI Y, LU S, DONG M, ZHANG C, HUANG G, WONG G, LI N, ZHANG Y, LI Y, FENG WH, SHI Y, LIANG M, ZHANG R, QI J & GAO GF 2017. Structures of phlebovirus glycoprotein Gn and identification of a neutralizing antibody epitope. Proc Natl Acad Sci U S A, 114, E7564–E7573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- XU W, LUTHRA P, WU C, BATRA J, LEUNG DW, BASLER CF & AMARASINGHE GK 2017. Ebola virus VP30 and nucleoprotein interactions modulate viral RNA synthesis. Nat Commun, 8, 15576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- ZHANG R, KIM AS, FOX JM, NAIR S, BASORE K, KLIMSTRA WB, RIMKUNAS R, FONG RH, LIN H, PODDAR S, CROWE JE JR., DORANZ BJ, FREMONT DH & DIAMOND MS 2018. Mxra8 is a receptor for multiple arthritogenic alphaviruses. Nature, 557, 570–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
CRISPR screen gRNA enrichment data were analyzed to generate the volcano plots using the following software: https://github.com/mhegde/volcano_plots.