

Abstract
a. Purpose of Review
Mitochondrial dysfunction is a hallmark of aging. Mitochondrial genome (mtDNA) instability contributes to mitochondrial dysfunction, and mtDNA mutagenesis may contribute to aging. However, the origin of mtDNA mutations remains somewhat controversial. The goals of this review are to introduce and review recent literature on mtDNA mutagenesis and aging, address recent animal and epidemiological evidence for the effects of chemicals on mtDNA damage and mutagenesis, propose hypotheses regarding the contribution of environmental toxicant exposure to mtDNA mutagenesis in the context of aging, and suggest future directions and approaches for environmental health researchers.
b. Recent Findings
Stressors such as pollutants, pharmaceuticals, and ultraviolet radiation can damage the mitochondrial genome or disrupt mtDNA replication, repair, and organelle homeostatic processes, potentially influencing the rate of accumulation of mtDNA mutations. Accelerated mtDNA mutagenesis could contribute to aging, diseases of aging, and sensitize individuals with pathogenic mtDNA variants to stressors. We propose three potential mechanisms of toxicant-induced effects on mtDNA mutagenesis over lifespan: 1) increased de novo mtDNA mutations, 2) altered frequencies of mtDNA mutations, or 3) both.
c. Summary
There are remarkably few studies that have investigated the impact of environmental chemical exposures on mtDNA instability and mutagenesis, and even fewer in the context of aging. More studies are warranted because people are exposed to tens of thousands of chemicals, and are living longer. Finally, we suggest that toxicant-induced mtDNA damage and mutational signatures may be a sensitive biomarker for some exposures.
Keywords: mtDNA, mtDNA damage, mutagenesis, toxicology, aging, environmental health
Introduction
Pollution is the leading cause of premature death globally [1]. Regulation of pollutants that mandate clean air and water standards have vastly improved human health and resulted in humans living longer. However, increased longevity is often only in high income areas [2]. Only 15–30% of lifespan is estimated to be determined by genetics [3], therefore the environment, including pollution, in addition to other often congruent social determinants of health, likely plays a critical role in aging. Indeed, leukocyte telomere shortening is associated with psychosocial stress [4], and those living in proximity to high-traffic corridors with high PM2.5 exposure exhibit epigenetic markers of accelerated aging [5]. Although these effects are of significant public health concern, the contribution and mechanisms of exposure to environmental pollutants on human aging are not well understood [6]. Perhaps this is in part due to the complexity of interpreting changes in certain molecular biomarkers, such as the reported mixture of positive, negative, and no association with mitochondrial copy number variation and telomere length with levels of exposure to metals, organohalogens, and perfluorinated compounds [7].
A major hallmark of aging is mitochondrial dysfunction [8], and mitochondria are often dysfunctional in specific diseases of aging [9, 10]. For example, mitochondrial dysfunction in Parkinson’s Disease (PD) results from genetic (nuclear and mitochondrial) mutations [11], including age-related accumulation of mtDNA point mutations, mtDNA deletions, and mtDNA depletion in dopaminergic neurons of PD patients [12]. Exposure to the pesticides rotenone and paraquat, and a number of other chemicals [13] that cause mitochondrial dysfunction [14] and mtDNA damage, are major environmental factors contributing to idiopathic PD (eloquently reviewed by Gonzalez-Hunt and Sanders [15]). In animal and in vivo studies, mtDNA damage accumulated prior to dopaminergic neurodegeneration and mitochondrial respiration deficiencies after rotenone exposure [16] and paraquat exposure [17]. However, it remains unclear in humans whether mtDNA damage and mitochondrial dysfunction contribute to the etiology of aging and these diseases, or are a consequence of the disease (though see Sanders et al. [17]).
Recent reviews highlight the compelling possibility that mitochondrial genome instability, not just nuclear genome instability, may play a significant role in aging [18, 19], yet the origin of mtDNA mutations is still debated [20, 21] and the effect of exposures on mtDNA mutagenesis is understudied. This has not been addressed in the context of toxicology, in part likely due to the complexities of mitochondrial genetics and genomics. Advances in next-generation sequencing technologies capable of precisely detecting mtDNA mutations may not only resolve the debate on the origin of mtDNA mutations, but could permit the use of mtDNA mutations as a fingerprint for the cumulative effects of certain chemical exposures throughout life.
The particular vulnerability of mitochondria, and the mitochondrial genome specifically, to environmental exposures is of significant public health concern. Therefore, we review the role that chemical-induced mitochondrial genomic instability (particularly mtDNA damage and mutagenesis) may have in premature aging and diseases, and briefly recommend how future studies may use mtDNA stability as a biomarker for environmental exposures and aging. While this review focuses on the potential outcome of cumulative lifetime burden of exposure to environmental toxicants, it is critical to note that elderly individuals are also likely more sensitive to the effects of environmental exposures including mitochondrial toxicants [22], theoretically exacerbating the effects of exposure on health and longevity in a particularly vulnerable population in the near future (see other reviews in this special issue). One such study determined that short-term increases in mean ambient air temperature are associated with higher blood mtDNA lesions in older individuals [23], perhaps due to impaired mitochondrial turnover in aged individuals. Investigating whether or not elderly individuals may then accumulate higher levels mtDNA mutations remains an important area of future research, as will be discussed in this review.
Mitochondria, mtDNA replication and mutagenesis
Mitochondria are required for energy production and other important biological processes in almost all eukaryotes. Mitochondria contain multiple copies of their own genome [24–26] that encodes various proteins necessary for oxidative phosphorylation. The 16,569 bp human mitochondrial genome contains 13 protein coding genes (subunits of respiratory complexes I, III, IV, and V) in addition to 22 tRNAs, 2 rRNAs, and a non-coding region called the ‘D-loop’ [27]. Despite differences in genome size, gene orientation, genetic code, sequence, and even structure between species [28], mitochondrial genome function and biological processes are conserved across eukaryotes, providing robust evolutionary evidence for the importance of mtDNA function and maintenance.
The mitochondrial genome is replicated independently of the cell cycle by a sole DNA polymerase, Pol γ [27]. Multiple models of the mechanism of mtDNA replication exist within and between species [29–31]. However, most evidence supports a strand displacement mode of mtDNA replication in humans and other mammals [27, 32, 33], which has potential consequences for mtDNA mutational processes that we discuss below. Pol γ contains a catalytic subunit and 3’−5- exonuclease and 5’-dRP lyase activities which are required for proofreading and base excision repair. Initial theory suggested that the proximity of mtDNA to reactive oxygen species produced by the electron transport chain causes high levels of oxidative damage, explaining the ~10 to 20-fold higher mtDNA mutation rate compared to the nuclear genome [34]. However, current research suggests that endogenous Pol γ replication error is the major contributor to the high rate of mutation accumulation in mtDNA [20, 27]. Pol γ is a high-fidelity polymerase [35], which suggests that the absence of certain DNA repair pathways in mitochondria presumably contributes to the higher mtDNA mutation rate. For example, the rare event of nucleotide misincorporation by Pol γ cannot be repaired due to the absence of mismatch repair in mitochondria. The absence of other repair pathways, such as nucleotide excision repair, may increase the error rate of mtDNA replication at damaged sites. However, very few studies have questioned the effect of exogenous sources of mtDNA damage (e.g., genotoxicant exposures) on Pol γ processivity, fidelity, and exonuclease activity [36].
Mitochondrial genome stability is critical for human health. Mutations in POLG and two other crucial components of the mtDNA replication machinery (mitochondrial ssDNA binding protein and the mitochondrial helicase, Twinkle) result in mtDNA genomic instability and mitochondrial diseases in human patients. Error during mtDNA replication and exposure of susceptible single-stranded mtDNA ultimately leads to higher accumulation of mtDNA mutations or mtDNA depletion, resulting in disease [32, 37]. These mitochondrial diseases vary in pathology, but often present as metabolic dysfunction and myopathies or encephalopathies, as previously reviewed [38], and perhaps premature aging [39].
mtDNA mutagenesis and aging
Soon after the human mitochondrial genome was first sequenced (1988), mtDNA deletions and depletion disorders were proposed to contribute to aging and diseases of aging [40, 41]. Current research using advanced next-generation sequencing technologies with ultra-sensitive error-corrected sequencing demonstrates that mtDNA point mutations may also contribute to aging. For example, pre-frontal cortex brain tissue from healthy aged individuals (75+ years) had a five-fold increase in mtDNA mutation frequency compared to young individuals (<1 year) [42]. Early stage Alzheimer’s Disease patients harbored significantly higher mtDNA mutations in the hippocampus compared to healthy, aged adults and those with late-stage AD (where there is likely selection against cells and mitochondrial genomes containing harmful variants) [43]. Another study analyzed tumor-normal tissue pairs from 1,675 cancer patients and showed that the number of mtDNA base pair substitution mutations were significantly positively correlated with patient age [44]. Critically, these studies [42–44] confirmed an earlier result by Zheng et al.[45] that (in humans) the origin of mtDNA point mutations are Pol γ polymerase-mediated errors (based on high rates of C → T transition mutations), and not due to oxidative damage (which is considered to have a G → T transversion mutation signature), as previously thought [46, 47].
Analyses of mtDNA sequences from the same healthy, aged brain tissue from Kennedy et al. ([42]) discovered that C → T mutations exhibited significant heavy strand bias in a specific GG[C>T]G context [48]. In a ssDNA yeast model, C → T mutations dominated after exposure to hydrogen peroxide and paraquat, not G → T as observed in dsDNA [48]. This potentially uncovers a mechanism and signature of oxidative damage particularly relevant to mtDNA because the mtDNA heavy strand is often displaced during mtDNA replication, leaving it single-stranded and vulnerable to endogenous and exogenous damage. Therefore, Degtyareva et al. propose that cytosines may be sites for mutagenic lesions on persistent ss-mtDNA, though the biochemistry of the lesion remains unknown [49]. Relatedly, some have suggested that abasic sites and strand breaks prevail as the signature of oxidative damage over 8-oxo-dG lesions, resulting in targeted degradation of linearized mtDNA [21] by quality control processes such as mitophagy, components of the replication machinery including Pol γ, and mitochondrial genome maintenance exonuclease 1 [50, 51], or mechanisms of selective removal yet to be discovered [52]. This further emphasizes that the complex role of oxidative damage in mtDNA mutagenesis and aging, especially after exposure to toxicants that induce redox stress, merits further investigation [53].
Historically, mitochondria were thought to play a central role in aging because in most cells, mitochondria produce a majority of endogenous reactive oxygen species (ROS) that can damage DNA, protein, and affect other molecular and cellular processes (the ‘mitochondrial free radical theory of aging’) [46]. However, in addition to sequencing evidence against oxidative damage contributing to mtDNA mutagenesis, it is now known that mitochondrial ROS also play important endogenous roles in intracellular signaling that regulate cell senescence, metabolism, autophagy, and proteostasis. The mitochondrial theory of aging has been extensively reviewed (e.g. Pinto and Moraes [54]), but the contribution of the environment in the context of the mitochondrial free radical theory of aging has not been previously considered. Many chemicals and pollutants can cause molecular and cellular damage; therefore, it is reasonable to theorize that exposures such as those that disrupt the redox balance of cells or induce ROS production may cause oxidative damage.
Another ongoing debate in the aging field is whether mtDNA mutations cause, or are merely a consequence of, aging. Evidence from studies in model organisms demonstrate the potential contribution of mtDNA mutations to aging. Homozygous Pol γ exonuclease activity-deficient “mutator” mice have increased mtDNA mutation frequencies and exhibit premature aging phenotypes compared to age-matched wild-type mice [55, 56]. Important to note, however, is that heterozygotes had a mutation frequency higher than aged wild-type mice, yet had no aging phenotypes [57]. In another study, one-year-old Pol γ-deficient mice also showed increased mtDNA transition mutations compared to wild-type in striatal tissue, and greater dopaminergic neurodegeneration when crossed with a Parkinson’s Disease mouse model [58]. An age-related hearing loss (AHL) mouse model crossed with a Pol γ-deficient mouse had a higher mtDNA point mutation frequency in inner ear tissue at both 5 and 17 months, and experienced accelerated hearing loss, premature death, and significant weight loss, hair loss, graying, and kyphosis as early as about 10 months of age compared to AHL mice with wild-type Pol γ [59]. There was no increase in mtDNA point mutation frequency from 5 to 17 months in the mutant mice, which could suggest that the rate of mtDNA mutation accumulation over lifespan may not contribute to aging directly, but that instead perhaps there is clonal expansion of mtDNA mutations early in life that result in an increased burden of mtDNA mutations later in life and contribute to aging pathologies [59]. However, Kim et al. did measure an increase in mtDNA deletion frequency from 5 months to 17 months of age, which could provide evidence that deletions contribute more to aging and aging pathologies than point mutations [59]. In fact, ultra-sensitive sequencing to detect low-frequency mtDNA deletion mutations (‘LostArc’) showed that levels of mtDNA deletions (not depletion) increased with age in human skeletal muscle biopsies, which correlated with impaired oxidative phosphorylation, and were exacerbated in POLG disease patients [32]. Lujan et al. hypothesize that impaired mtDNA degradation (mitophagy) over time may contribute to increased mtDNA deletions with age [32].
Other relevant mtDNA mutator models have been developed in Caenorhabditis elegans and Drosophila melanogaster [60–63]. These studies are consistent with the theory that increased frequencies of mtDNA mutations are associated with decreased longevity and other aging-related phenotypes compared to wild-type organisms (with the exception of Kauppila et al. [63]). An important caveat is that some of these studies have only sequenced specific regions of the mitochondrial genome (e.g., just the D-loop or a single gene) which could bias results, as there are likely different mutational and biological processes (such as selection) acting on different regions of the genome. Nevertheless, accelerated mtDNA mutation rates of error-prone Pol γ can cause aging and other aging-related pathologies, but raise doubt about whether normal rates of mtDNA mutagenesis contribute to aging [20]. Therefore, we believe that it is pertinent to investigate whether other factors, such as environmental exposures that cause mtDNA damage, sufficiently contribute to mtDNA mutagenesis and affect aging and aging-related diseases.
mtDNA heteroplasmy and aging
It is now clear that humans and many other eukaryotes retain low levels of mtDNA heteroplasmy, meaning mutant mtDNA is often present at variable frequencies within organelles, cells, and tissues [64]. This is of high relevance to human health and disease [65], as well as pathologies of aging. Heteroplasmic mutations are inherited maternally or arise and then expand in certain tissues during development and aging. It is thought that pathological phenotypes appear only when a mutant mitochondrial genome reaches a certain frequency, or “threshold”. Heteroplasmy often fluctuates, yet it still is incompletely understood what processes regulate mtDNA heteroplasmy through the germline and in the soma. In general, the redundant nature of mtDNA is predicted to 1) hinder the maternal transmission of mutant genomes through the germline [66–68], and 2) delay the accumulation of mtDNA mutations past a critical threshold through various processes [60, 68–75]. However, heteroplasmic mtDNA mutations are still associated with lifespan and give rise to diseases of aging – even those inherited maternally [76, 77].
The heteroplasmic nature of the mitochondrial genome contributes to the complexities of investigating mtDNA mutagenesis. Heteroplasmy is dependent on mtDNA copy number (mtCN), which is in part regulated by coupling of replication and degradation of mitochondrial genomes. However, mtCN varies by orders of magnitude between cell types, (e.g. myocardial muscle cells contain more than 6,000 copies per cell, while leukocytes have around 350 copies per cell) [26]. Though mtCN has been used widely as a biomarker for age, health, and exposure to environmental pollutants in peripheral tissues (e.g. [7, 78, 79]), there is high inter-individual variation in mtCN in matching tissues, further complicating interpretation of the biological consequences to changes (or not) in mtCN, and consequently heteroplasmy [26]. Many studies have had contradictory results when determining mtCN across age (no change, increase, or decrease), which is again highly dependent on tissue type. Perhaps this is due to the significant complexities involved with measuring and interpreting mtCN in laboratory and epidemiological studies. Issues such as methodological bias, specimen heterogeneity, cell type composition, and genetic and environmental factors (such as exercise and aging) must be considered for the precise quantification of mtCN and interpretation of literature [80]. Despite these factors, mitochondrial disease, aging, neurodegeneration, and cancer [80], as well as exposures (e.g., [81] and as reviewed previously [82, 83]), results in both increases and decreases to mtDNA copy number, complicating interpretation of changes. Therefore, the field would greatly benefit from an updated review exclusively focused on mtCN and chemical exposures.
Apart from mtCN change as a biomarker, turnover of mtDNA may be quite important in the context of interpreting findings of mtDNA mutations. For example, an adaptive response may result in increased turnover to facilitate removal of damaged or mutated mtDNA, theoretically resulting in no change in mtCN, and perhaps fewer mtDNA mutations and low levels of heteroplasmy. Conversely, perhaps increased rates of mitochondrial biogenesis and mtDNA replication result in overrepresentation of certain mtDNA molecules due to clonal expansion or selection [61]. Turnover rates may themselves be influenced by both environmental stressors and age, thus indirectly impacting age-related mutagenesis. For example, mild oxidative stress increases both mitochondrial biogenesis, degradation, and dynamics [83].
Another challenge in assessing mtDNA heteroplasmy is that mtDNA mutations are often present at frequencies that are orders of magnitude lower than the error rates of conventional sequencing technologies (1 in 1,000 base pairs) [84]. New technologies correct errors from mtDNA damage and PCR amplification during the preparation of sequencing libraries, and can accurately detect mtDNA mutations at frequencies as low as 1 in 107 base pairs [32, 85–87]. A recent mouse study conducted Duplex Sequencing of mtDNA from brain, muscle, single oocytes, and oocyte pools from aged dams (10-mo) and daughter pups (1-mo) to investigate the frequencies and spectrum of de novo germline and somatic mtDNA mutations across age and tissue type [88]. Remarkably, the majority of mtDNA mutations occurred below 1%, and in most cases, a variant was present in only one mtDNA molecule [88]. They observed ~2.6-fold increase in mtDNA mutation frequencies in the aged dams compared to the pups in all tissues except pooled oocytes, with an enrichment of G → A/C →T mutations, consistent with polymerase error, spontaneous deamination of cytosine on the heavy strand during replication, or oxidative damage to ssDNA [88]. In humans it is also likely that mtDNA mutations occur and persist at very low frequencies (perhaps within a single mitochondrial genome) [89], and these rare variants might have functional consequences on health and aging [64, 90].
Just as mtDNA mutations are present at low frequencies in cancers [91–94], exposure to environmental pollutants that cause mtDNA damage and mutations will likely result in very rare mutations, which will only be detected with ultra-sensitive sequencing technologies [95, 96]. Improved sequencing methods, including methods in single cell or single mitochondrion sequencing [97, 98], have revealed the pervasiveness of mtDNA heteroplasmy, yet the origin, nature, and significance of mtDNA mutations remain an exciting area of future research, particularly in the fields of toxicology and aging, as we discuss next.
mtDNA vulnerability to mutations and toxicants
The structure of the mitochondrial genome and the single-strand displacement model of mtDNA replication are thought to predispose mtDNA to damage or replication errors, though this is a very recent theory. The highly conserved double-stranded circular mitochondrial genome harbors significant strand asymmetry in regards to nucleotide composition. In humans and most mammals, the mitochondrial genome contains a heavy and a light strand (LS) in which the inner heavy strand (HS) contains twice as many guanine nucleotides. The origin of the asymmetrical composition of the mitochondrial genome is thought to be due to the displacement of the single-stranded parent HS, rendering it susceptible to endogenous damage and spontaneous deamination of cytosine (C → U; G → A), and adenine (A → hypoxanthine (hX); T → C) [99] or maybe oxidative damage to cytosine (C → T) [48]. It has recently been demonstrated that the guanine-rich HS may be highly permissive to G-quadruplex (G4) formation compared to the nuclear genome [86, 87]. G4 structures may play a functional role (such as increasing the binding efficiency of mitochondrial transcription factor A, TFAM) [102], but are also a likely source of genomic instability, as they are often detected in proximity to mtDNA deletion mutations [103]. Other structural motifs in mammalian mtDNA, such as triple-stranded DNA as a result of triplex repeats, are negatively associated with maximum lifespan [104]. Enrichment of secondary stem-loop structures that form on the LS, which may mimic origins of replication, is positively associated with longevity in mammals [105]. It is predicted that these structures result in lower accumulation of deletion mutations, which could “decelerate mitochondrial aging” due to fewer dysfunctional organelles, thus delaying cellular senescence [105]. These results are consistent with mitochondrial genome instability contributing to aging.
mtDNA has several other properties that may make it particularly susceptible to mutagenesis compared to the nuclear genome. First, mtDNA is located in the mitochondrial matrix in proximity to the electron transport chain, which is the primary site of ROS production in most cells [106]. However, the exact location of the mitochondrial genome and the type of ROS produced [107, 108] (which may also depend on the mechanism of toxicity) may factor in to mtDNA damage and mutagenesis. Furthermore, mitochondrial membrane potential is not uniform within an organelle, similar to localization of pH and voltage gradients, likely due to compartmentalization of ATP synthase and the ETC chain [109], potentially affecting which mtDNA molecules are exposed to ROS. This is important because the packaging and localization of mtDNA is heterogeneous across cell type and varies with cellular demands, be it mtDNA transcription, mtDNA replication, or being held in reserve, potentially resulting in altered vulnerability to ROS or other stressors. In general, oxidative stress plays a large, yet complex, role in theories of aging, and of mitochondrial dysfunction. It will be critical in future studies to continue to distinguish between the beneficial effects of low levels of oxidative stress (such as stimulation of mtDNA turnover), and higher levels, which cause mtDNA damage [110].
Other properties that render mtDNA susceptible to environmental exposures and mutagenesis may be the lack of protective histones and the fact that several DNA repair pathways are absent from mitochondria [111, 112]. However, mtDNA is packaged into nucleoids, which may serve a protective function [113, 114]. In mammals, TFAM facilitates the compaction of mtDNA, thus regulating mtDNA replication and copy number, in addition to mtDNA transcription [115, 116]. Transcription of mtDNA may also be controlled by cytosine methylation [117, 118], or the more recently discovered adenosine methylation[119], although CpG methylation in mitochondria is still controversial [120]. Studies have demonstrated changes in mtDNA methylation after various exposures (tobacco smoke and air pollution for example [121, 122]) and mtDNA epigenetics may be a valuable biomarker for environmental exposure and health [123]. It is therefore possible that mtDNA epigenetic modifications that alter accessibility of mtDNA-damaging agents, and possibly mtDNA maintenance machinery, may result in heterogeneous accumulation of mutations across different cell types, different mitochondrial genomes, or potentially specific regions within the mitochondrial genome. On the other hand, it is accepted that mtDNA-nuclear DNA crosstalk is mediated via nuclear epigenetics [124]. Therefore, it is conceivable that chemical or age-induced changes to the nuclear epigenome that control mtDNA replication, repair, and transcription [125, 126] might also impact mtDNA mutagenesis.
Lastly, biological and physicochemical properties of the mitochondrion result in accumulation of cationic compounds, weak acids, and lipophilic compounds [33, 111, 127, 128]. All of these features in addition to the high copy number of mtDNA could render mtDNA a sensitive and reliable biomarker for toxicant-induced mtDNA damage, and thus exposure.
Evidence for effects of toxicants on mitochondrial genome integrity
Environmental toxicants likely contribute to mtDNA instability through a range of mechanisms such as direct mtDNA damage, depletion of nucleotide pools, redox stress, or interference with organelle dynamics (fusion/fission) and turnover (biogenesis and mitophagy), which are critical for health and healthy aging [129–132]. Here we will review the limited evidence of effects of chemical exposure on mtDNA mutagenesis in model and sentinel organisms, as well as human in vitro and population health studies (Table 1). Effects of toxicants on mtDNA copy number, biogenesis, degradation, and mitochondrial dynamics, all highly relevant to regulation of mtDNA mutational processes [71], have been reviewed elsewhere [83].
Table 1.
Summary of studies investigating the effects of toxicants on mtDNA mutagenesis.
| Toxicant | Lesion | Mutation | System | Method | Reference | |
|---|---|---|---|---|---|---|
| Laboratory studies | B[a]P 7,8-diol 9,10-epoxide | dG-N2-deoxyguanosine | G → T, mtDNA deletions, decrease mtDNA CN | Biochemical | Oligonucleotide substrates | Graziewicz et al. (2004)[133] |
| B[a]P; N-ethyl-N-nitrosourea (ENU) | dG-N2-deoxyguanosine; alkylation | None | M. musculus; bone marrow and liver | RMC; 12S rRNA and ND5 sequencing | Valente et al. (2016)[134] | |
| Methyl methanesulfonate (MMS) | Alkylation | C → G | S. cerevisiae | 16S rRNA sequencing | Stumpf and Copeland (2014) [135] | |
| Nucleoside reverse transcriptase inhibitors (NRTIs) | Nucleotide analog misincorporation | mtDNA depletion | - | - | Young (2017) [136] | |
| Cadmium Chloride | Unknown; hypothesize oxidative stress | Accumulation of common mtDNA deletion | Rat; renal cortex | PCR | Takaki et al. (2004) [137] | |
| Epidemiological studies | Lead | Unknown; hypothesize oxidative stress | Increased incidence of transition mutations (T:A → C:G) | Blood from male workers | Whole genome sequencing | Mani et al. (2020) [138] |
| Pesticides | Unknown; mixture | Inadequate statistical power | Blood and lung tissue from farmers | Whole genome sequencing | Wang and Zhao (2012) [139] | |
| Ultraviolet C | Pyrimidine dimers | Clonal expansion of common mtDNA deletion | Skin tissue from patients | Amplicon sequencing | Birket and Birch-Machin (2007) [140] | |
| Smoking, NRTIs, age | Not measured; bulky adducts, ROS, POLG inhibition | Age: T:A → C:G; smoking: ↑ heteroplasmy (clonal expansion) | Blood from smoking and non-smoking females, HIV+ patients | D-loop | Ziada et al. (2019) [141] | |
| Population genetics | Radiation | Break or oxidative damage | Increased haplotype diversity | Chernobyl bank vole | Whole genome sequencing | Kesäniemi et al. (2020) [142], Baker et al. (2017) [143] |
| Industrial pollution (heavy metals, PAHs) | Unknown; mixture | Reduced haplotype diversity | Azerbaijan marsh frog | CYTB, D-loop | Matson et al. (2006) [144] |
There is little to no direct evidence for a role of mtDNA damage in de novo point or deletion mutations, though mechanisms of mtDNA deletion formation have been speculated [145]. In addition to oxidative damage, stressors such as ultraviolet C radiation (UVC), or toxicants that form bulky adducts such as metabolites of benzo[a]-pyrene (B[a]P) and other polycyclic aromatic hydrocarbons, can cause direct damage to mtDNA in humans and other organisms [111, 112, 146–148]. Mitochondria lack nucleotide excision repair and mismatch repair mechanisms, rendering mtDNA susceptible to genotoxicant-induced mtDNA damage and mutations [112]. This is compelling and relevant to environmental health because a wide range of environmental pollutants can cause damage that requires nucleotide excision repair, while mismatch repair is required to correctly replace a misincorporated nucleotide resulting from polymerase error (potentially due to a damaged template during replication). If these lesions are not repaired and the organelle is not targeted for degradation, this could lead to nucleotide misincorporation during mtDNA replication, resulting in a mutation (where the absence of mismatch repair would presumably amplify this effect). For reference, an updated list of known mtDNA genotoxicants was recently curated by Copeland and Wallace [33].
Biochemical studies initially demonstrated that human Pol γ misincorporates purines (mostly dAMP, some dGMP) opposite N2-deoxyguanosine (dG) adducts derived from B[a]P 7,8-diol 9,10-epoxide exposure, with limited translesion synthesis [133]. This could result in a specific mutational signature (G → T), but more often is thought to result in mtDNA deletions and copy number depletion due to polymerase stalling and thus inhibition of mtDNA replication. In vitro exposure to UVC also causes mtDNA damage that blocks the processivity of Pol γ, but some evidence suggests bypass of pyrimidine dimers may result in nucleotide misincorporation [149]. In vivo, a recent study in Mus musculus showed that after 28 days of exposure to B[a]P or N-ethyl-N-nitrosourea (ENU), mice had significant mtDNA adduct formation in both bone marrow and liver compared to controls, but no increase in mtDNA point mutation frequency or deletion frequency in two mtDNA genes (12S rRNA and ND5) after either exposure [134]. It should be noted that a potential limitation in this study is the reliance on Random Mutation Capture, which can only assay mutations at TCGA sites. This offers an incomplete result when investigating effects of exposures because the trinucleotide context (5’ and 3’ base identity) cannot be investigated with this method. This highlights the need to conduct whole-genome studies. Nevertheless, mtDNA copy number did not decrease after ENU exposure as would be expected with polymerase stalling. This suggests perhaps mitochondrial-specific mechanisms to avoid mutation accumulation (which could even be tissue-dependent), such as targeted degradation of damaged or mutant mtDNA [51, 60, 132, 150]. These hypothesis remain an intriguing and important future area of research.
If mtDNA damage does not increase the frequency of point mutations alone, certain genetic backgrounds may predispose individuals to increased susceptibility to chemical-induced mtDNA mutagenesis [32, 110]. For example, in various Saccharomyces cerevisiae strains harboring genetic mutations in the POLG homolog MIP1, exposure to the alkylating agent methyl methanesulfonate (MMS) significantly increased the frequency of mtDNA mutations compared to wild-type [135]. Interestingly, the spectrum of MMS-induced mutation was similar in both wild-type and a MIP1 strain: C:G → G:C transversions in a PCR-amplified region of the mitochondrial 16S ribosomal subunit. Perhaps this mutation spectrum is due to the apparent presence of DNA Polymerase zeta in yeast mitochondria [152], which is an error-prone translesion synthesis polymerase that can result in G → C transversions at damaged and abasic sites [153]. Nevertheless, this demonstrates the potential for individual susceptibility to toxic exposures, particularly in individuals with deficiencies in POLG.
Off-target drug toxicity disrupts mtDNA integrity in humans, which is a significant concern in the geriatric population [22]. One of the most well-known examples is the effect of the nucleoside reverse transcriptase inhibitors (NRTIs), which are anti-viral drugs used to treat human immunodeficiency virus [136]. NRTIs are phosphorylated to active nucleotide analogs that lack a 3’ hydroxyl group, and when incorporated into the daughter strand, mtDNA replication is prematurely terminated resulting in significant mtDNA depletion. It is not inconceivable that other drugs and potentially environmental pollutants could similarly disrupt mitochondrial genome instability by interfering with nucleotide pool composition or perhaps creating nucleotide analogs or modifications that either stall the mtDNA replication machinery or result in error-prone replication, potentially resulting in copy number depletion, or even mtDNA mutations.
The origin of mitochondria from an ancient alpha (α)-proteobacteria ancestor explains the vulnerability of mitochondrial translation to many antibiotics. Individuals with a specific mutation in the 12S ribosomal RNA mitochondrial gene are at high risk to develop hearing loss after exposure to aminoglycosides compared to those without this mutation: however, not all individuals harboring this variant develop deafness, and not all those that develop deafness have had an exposure to aminoglycosides [151]. This suggests that there are other environmental factors involved, perhaps exposure to other drugs or chemicals with similar toxicities and highlights a compelling question for future investigation: why do patients with the same mutation (such as this 12S rRNA variant, or a nuclear POLG mutation) often have such variable clinical presentation of pathologies? These differences may be due to other genetic or environmental factors (social, diet, etc.), but perhaps variable environmental exposures play an important role in these aging-related diseases.
To our knowledge, there are only a few case studies that have investigated the effect of exposure to environmental pollutants on mtDNA mutations in human populations. Occupational workers in India with high lead blood levels had more mtDNA single nucleotide variants than those with low lead exposure. Most of these mtDNA mutations were transition mutations (T:A → C:G) [138]. Farmers from the Shandong province in China had more mtDNA mutations in lung tissue samples compared to peripheral blood, potentially suggesting mtDNA genomic instability from long-term inhalation to high levels of pesticides [139]. However, this study was only able to detect mutations at a minimum frequency of 25%, potentially missing the majority of low-frequency heteroplasmic mutations. Another more recent study investigated the effects of cigarette smoking on mtDNA mutagenesis in blood that was collected from a cohort of smoking and non-smoking females [141]. Of those who smoke, about 35% of individuals exhibited mtDNA heteroplasmy, compared to never smokers, where less than 20% of individuals exhibited mtDNA heteroplasmy (p < 0.01) [141]. Surprisingly, there was no association with smoking and frequency of de novo mutations, which suggests that this increase in heteroplasmy may be due to clonal expansion of pre-existing mutations. Perhaps either increased cellular turnover from stress, or perhaps the potential effects on mitochondrial fission and fusion dynamics as well as mitophagy by specific chemicals affects clonal expansion (heteroplasmy) [71]. Finally, although not an anthropogenic pollutant, there is epidemiological evidence that exposure to ultraviolet radiation contributes to increased age-related clonal expansion of a mtDNA point mutation and the common mtDNA deletion in skin cells [140], presumably due to effects on mtDNA replication or mtDNA homeostasis [157, 158] and mitochondrial dysfunction [159]. UVA and UVB wavelengths induce the same photolesions as UVC (though UVC cannot penetrate the ozone layer), in addition to a significant amount of oxidative mtDNA damage, likely contributing to mtDNA mutagenesis. This body of literature suggests that mtDNA damage can lead to mtDNA mutagenesis during aging, at least under certain conditions.
Understanding the effect of exposures on mtDNA damage, copy number, and mutagenesis is a complex endeavor for environmental molecular epidemiologists. For example, air pollution, specifically PM2.5, is known to induce oxidative stress, mtDNA damage, and in some cases increase or decrease activity of mitochondrial homeostatic dynamics (fusion and fission) and mitophagy [154–156]. One could hypothesize that this could result in an increase or decrease in mtDNA mutations or heteroplasmy (see Figure 1 and Figure 2), and could depend on the dose and even be cell or tissue-specific [82, 83].
Figure 1. Schematic of effects of age and environmental toxicant exposures on mtDNA mutagenesis.
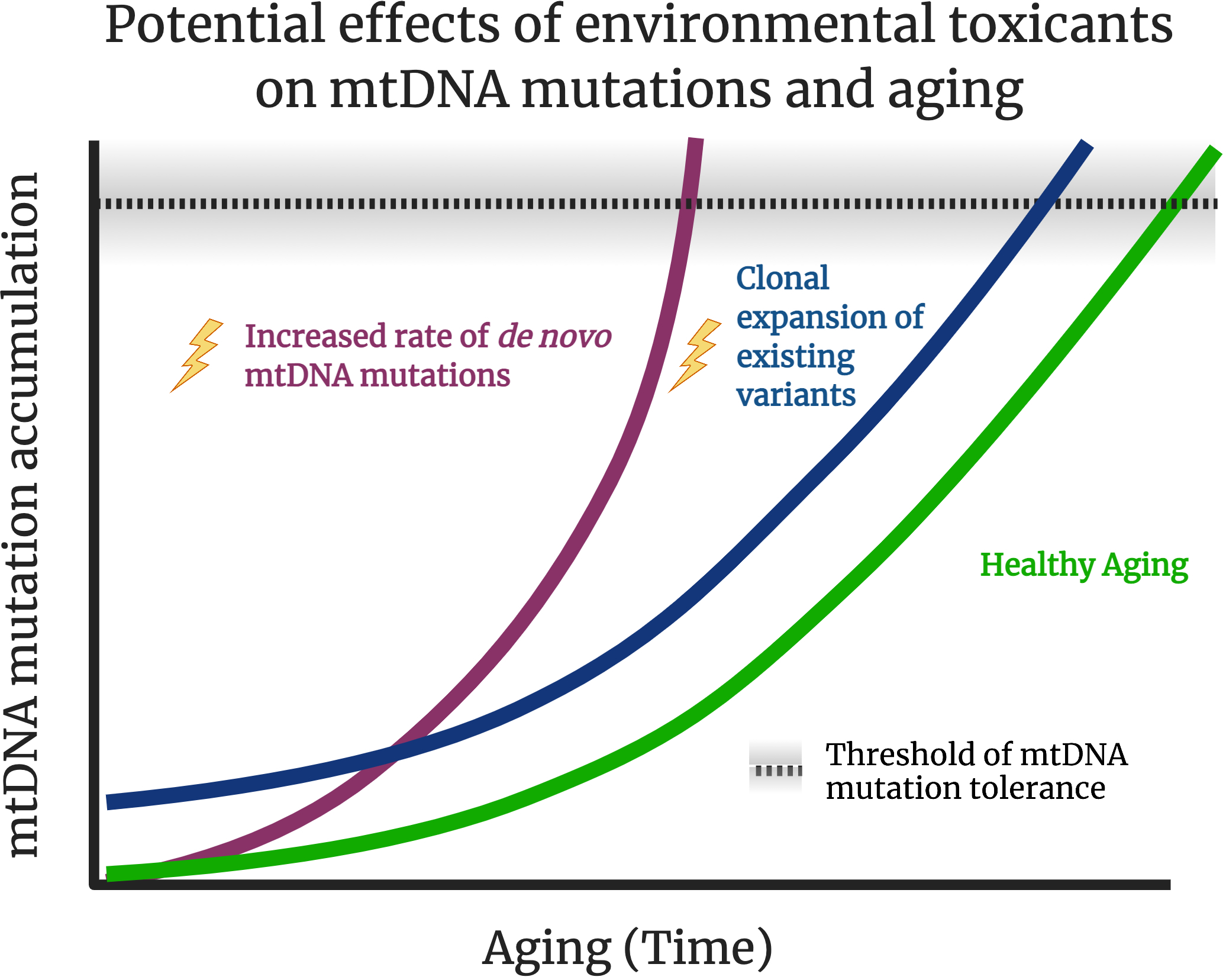
mtDNA mutations are thought to accumulation over time and contribute to healthy aging (green line). Exposure to exogenous stressors, such as environmental pollutants, may result in higher rates of mtDNA mutation accumulation (purple line). It is also hypothesized that many chemicals interfere with mtDNA replication, turnover, and organelle dynamics, potentially resulting in clonal expansion of existing mtDNA mutations, without increasing the rate of de novo mutation accumulation (blue line). Theoretically, if pathogenic mutations arise earlier or at great frequencies (or both; not shown), a threshold of tolerance (dashed line) of mtDNA mutations may be reached earlier in life, potentially resulting in premature aging phenotypes and accelerated onset of aging-related diseases such as neurodegeneration or cancer. It is likely that there will be high interindividual variation of the “threshold”, as shown in the shaded areas around the dashed line as some individuals are likely to be more sensitive and vulnerable, while others may be more tolerant to exposures and/or mtDNA mutation accumulation. Created with BioRender.com.
Figure 2. Schematic of effects of environmental toxicant exposures on mtDNA heteroplasmy in young versus old individuals or populations.
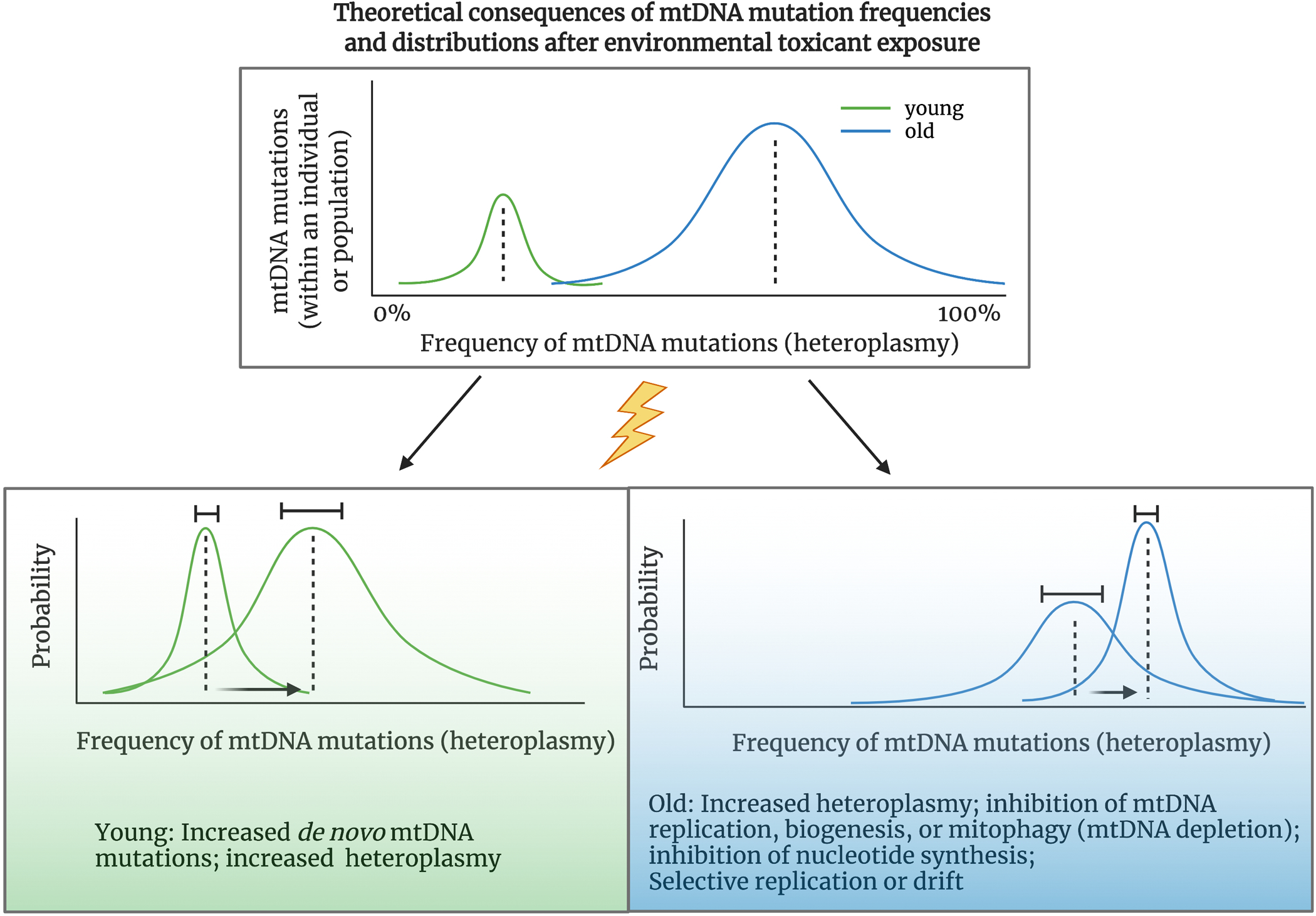
New evidence suggests mtDNA mutations undergo clonal expansion during aging. Theoretically, this results in more mutations at a higher frequency in aged versus young individuals. An old population also likely has a greater distribution in the frequency of mtDNA mutations compared to young populations. Exposure to stressors such as environmental pollutants may result in an increase in the rate of mutation accumulation or clonal expansion of existing variants, resulting in a shift and greater distribution of mtDNA heteroplasmy early in life, resulting in a premature “aged” mutational signature (green box) in individuals or a population with high exposures compared to low exposures. It is also likely that mtDNA mutation rates and heteroplasmy will vary when looking in within aged populations with variable environmental exposures. Those that have either endured a lifetime burden of exposures or recent exposure to chemicals may have greater accumulation of mtDNA mutations (i.e. more mutations within an individual, and more individuals with mtDNA mutations at higher frequencies), than healthy aged individuals and populations (blue box). Created with BioRender.com
Investigation of mtDNA mutagenesis in natural populations of sentinel organisms may also provide insight into the effects of exogenous stressors on mtDNA mutational processes [160]. A recent study of Chernobyl bank voles concluded that voles exposed to high levels of cesium and strontium radiation for 50 generations have increased accumulation of mtDNA mutations: the mitochondrial genome had more polymorphic sites and greater genetic diversity within the contaminated population compared to vole populations living in uncontaminated areas [143]. Perhaps mtDNA damage from radiation resulted in mtDNA mutations [143]. However, mutation as an evolutionary process does not act in isolation: there are other evolutionary forces that drive genetic diversity of a population, such as natural selection acting on a mutation, gene flow from individuals migrating between populations, or genetic drift. For example, contrary to the results from the Chernobyl voles, marsh frogs from heavy metal and PAH-contaminated sites in Azerbaijan exhibited lower genetic diversity compared to frogs from uncontaminated sites [144]. This is a result of either genetic drift acting on a declining population size due to heavy pollution (a genetic “sink”), or selection to tolerate pollution. These case studies provide some evidence of effects of pollution on mtDNA sequence variation and population health, but not conclusive proof of toxicant-induced mtDNA mutagenesis.
Controlled laboratory studies may provide insight into the mechanisms and effects of toxicants on mtDNA mutagenesis. For decades, evolutionary biologists have used mutation accumulation line (MA) experiments to investigate mtDNA mutational processes [161]. MA experiments are often conducted in model organisms with rapid generation times that reproduce clonally (such as species of the keystone freshwater crustacean Daphnia) or via selfing (such as C. elegans) [162–165]. However, there are still challenges to directly estimate mtDNA mutation rates such as ability to detect very low frequency mutations, and the presence of selection acting to favor or purge mtDNA mutations on different levels (organelle, cell, tissue, germline bottleneck) [166]. Until recently, no MA experiment had investigated the effect of chemical exposure on nuclear genome mutational processes [167–169], and to our knowledge no MA experiment has investigated the effect of exposures on mtDNA mutagenesis. Though MA experiments are laborious and time intensive, with the advancement of sequencing technologies and dropping costs of sequencing, we propose this to be a relevant and useful approach to not only investigate the effect of exposures on mtDNA mutagenesis, but potentially provide resolution as to what cellular processes (and possible interventions in treating human diseases) contribute to the origin and transmission of mtDNA mutations.
mtDNA as a biomarker for exposure and health: summary and recommendations for future studies
However large or small the contribution of mtDNA mutations in aging and diseases of aging, because environmental toxicants can interfere with mitochondrial genome integrity and because a relatively high level of mtDNA mutations can be tolerated, mtDNA mutagenesis may be a good biomarker for footprints of exposure and potential health consequences. From our review of the evidence provided above, we consider possible theoretical outcomes of future investigations, as illustrated in Figure 1. First, exposures may result in increased mtDNA mutation rates. Exposed individuals may have higher frequencies of mtDNA mutations than those with lower exposures, and on a population level, there may be a larger distribution (more variation) in the frequencies of mtDNA mutations (Figure 2). It is conceivable that this accelerated rate of mtDNA mutagenesis increases the risk of a pathogenic variant appearing earlier in life, potentially only in a subset of cells. Second, exposures may affect the replication machinery and organelle dynamics and turnover, which may result in significant clonal expansion of existing variants (Figure 1). Exposed individuals may have more mutations at higher frequencies compared to low exposure (Figure 2). On a population level, there may be more individuals with greater mtDNA heteroplasmy, which may be indicative of clonal expansion of existing mutations (Figure 2). As mutant mitochondrial genomes escape degradation and proliferate, it is possible that pathogenic mutations pass a critical threshold earlier, resulting in accelerated aging and earlier onset of aging diseases (Figure 1). A third possibility is that both mechanisms are occurring, as different chemicals exert different mechanisms of toxicity, and the human exposome is vast.
It is critical to better understand the effects of the environment on aging, particularly on diseases of aging such as cancer and neurodegenerative diseases, as the proportion of people over the age of 65 will almost double globally by 2050, from 9% to 16% [170]. The search for both drug and lifestyle interventions for treatment of mitochondrial diseases is very active, and such approaches may be beneficial [171, 172]. However, from a public health perspective, pollution prevention is a more equitable approach as the WHO predicts 80% of older people will be living in low- and middle-income countries [173]. Robust molecular biomarkers are therefore imperative to understand exposure and potential health outcomes for regulation. Research to better understand the associations of mtDNA mutations (including specific signatures of mtDNA mutagenesis) and heteroplasmy in epidemiological studies, as well as uncovering the mechanisms that regulate, and consequences of exposures on, mtDNA mutational processes in laboratory research remain exciting areas of future environmental health research.
Acknowledgements
The authors would like to thank Bill Copeland and Scott Kennedy for thoughtful discussions and for providing critical feedback on this manuscript.
Funding Information:
TCL and JNM were supported by the National Institutes of Health (F31 ES030588 and P42ES010356, respectively)
Footnotes
Conflict of Interest
Tess Leuthner and Joel Meyer declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
References
- 1.Landrigan PJ, Fuller R, Acosta NJR, et al. (2018) The Lancet Commission on pollution and health. Lancet 391:462–512. 10.1016/S0140-6736(17)32345-0 [DOI] [PubMed] [Google Scholar]
- 2.Chetty R, Stepner M, Abraham S, et al. (2016) The association between income and life expectancy in the United States, 2001–2014. JAMA - J Am Med Assoc 315:1750–1766. 10.1001/jama.2016.4226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Graham Ruby J, Wright KM, Rand KA, et al. (2018) Estimates of the heritability of human longevity are substantially inflated due to assortative mating. Genetics 210:1109–1124. 10.1534/genetics.118.301613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Meier HCS, Hussein M, Needham B, et al. (2019) Cellular Response to Chronic Psychosocial Stress: Ten-year Longitudinal Changes in Telomere Length in the Multi-Ethnic Study of Atherosclerosis. Psychoneuroendocrinology 107:70–81. 10.1016/j.psyneuen.2019.04.018.Cellular [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ward-Caviness CK, Russell AG, Weaver AM, et al. (2020) Accelerated epigenetic age as a biomarker of cardiovascular sensitivity to traffic-related air pollution. Aging (Albany NY) 12:24141–24155. 10.18632/aging.202341 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sorrentino JA, Sanoff HK, Sharpless NE (2014) Defining the toxicology of aging. Trends Mol Med 20:375–384. 10.1016/j.molmed.2014.04.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Vriens A, Nawrot TS, Janssen BG, et al. (2019) Exposure to Environmental Pollutants and Their Association with Biomarkers of Aging: A Multipollutant Approach. Environ Sci Technol 53:5966–5976. 10.1021/acs.est.8b07141 [DOI] [PubMed] [Google Scholar]
- 8.van der Rijt S, Molenaars M, McIntyre RL, et al. (2020) Integrating the Hallmarks of Aging Throughout the Tree of Life: A Focus on Mitochondrial Dysfunction. Front Cell Dev Biol 8:1–12. 10.3389/fcell.2020.594416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sun N, Youle RJ, Finkel T (2016) The Mitochondrial Basis of Aging. Mol Cell 61:654–666. 10.4028/0-87849-999-7.758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nunnari J, Suomalainen A (2012) Mitochondria: In Sickness and in Health. Cell 148:1145–1159. 10.1016/j.cell.2012.02.035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Helley MP, Pinnell J, Sportelli C, Tieu K (2017) Mitochondria: A common target for genetic mutations and environmental toxicants in Parkinson’s disease. Front Genet 8:1–24. 10.3389/fgene.2017.00177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bose A, Beal MF (2016) Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139:216–231. 10.1111/jnc.13731 [DOI] [PubMed] [Google Scholar]
- 13.Breckenridge CB, Berry C, Chang ET, et al. (2016) Association between Parkinson’s disease and cigarette smoking, rural living, well-water consumption, farming and pesticide use: Systematic review and meta-analysis. PLoS One 11:1–42. 10.1371/journal.pone.0151841 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Goldman SM (2014) Environmental toxins and Parkinson’s disease. Annu Rev Pharmacol Toxicol 54:141–164. 10.1146/annurev-pharmtox-011613-135937 [DOI] [PubMed] [Google Scholar]
- 15.Gonzalez-Hunt CP, Sanders LH (2021) DNA damage and repair in Parkinson’s disease: Recent advances and new opportunities. J Neurosci Res 99:180–189. 10.1002/jnr.24592 [DOI] [PubMed] [Google Scholar]
- 16.Sanders LH, Howlett EH, McCoy J, Timothy Greenamyre J (2014) Mitochondrial DNA damage as a peripheral biomarker for mitochondrial toxin exposure in rats. Toxicol Sci 142:395–402. 10.1093/toxsci/kfu185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sanders LH, Paul KC, Howlett EH, et al. (2017) Base excision repair variants and pesticide exposure increase Parkinson’s disease risk. Toxicol Sci 158:188–198. 10.1093/toxsci/kfx086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Schumacher B, Pothof J, Vijg J, Hoeijmakers JHJ (2021) The central role of DNA damage in the ageing process. Nature 592:695–703. 10.1038/s41586-021-03307-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yousefzadeh M, Henpita C, Vyas R, et al. (2021) Dna damage—how and why we age? Elife 10:1–17. 10.7554/eLife.62852 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Szczepanowska K, Trifunovic A (2017) Origins of mtDNA mutations in ageing. Essays Biochem 61:325–337. 10.1042/EBC20160090 [DOI] [PubMed] [Google Scholar]
- 21.Zsurka G, Peeva V, Kotlyar A, Kunz W (2018) Is There Still Any Role for Oxidative Stress in Mitochondrial DNA-Dependent Aging? Genes (Basel) 9:175. 10.3390/genes9040175 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Will Y, Shields JE, Wallace KB (2019) Drug-induced mitochondrial toxicity in the geriatric population: Challenges and future directions. Biology (Basel) 8:1–14. 10.3390/biology8020032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Peng C, Sanchez-guerra M, Wilson A, et al. (2017) Short-term effects of air temperature and mitochondrial DNA lesions within an older population. Environ Int 23–29. 10.1016/j.envint.2017.03.017.Short-term [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Clay Montier LL, Janice D, Yidong B (2009) Number matters: control of mammalian mitochondrial DNA copy number. J Genet Genomics 36:125–131. 10.1080/00497878.2016.1120114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Moraes CT (2001) What regulates mitochondrial DNA copy number in animal cells? Trends Genet 17:199–205. 10.1016/S0168-9525(01)02238-7 [DOI] [PubMed] [Google Scholar]
- 26.Wachsmuth M, Hübner A, Li M, et al. (2016) Age-Related and Heteroplasmy-Related Variation in Human mtDNA Copy Number. PLOS Genet 12:e1005939. 10.1371/journal.pgen.1005939 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gustafsson CM, Falkenberg M, Larsson NG (2016) Maintenance and Expression of Mammalian Mitochondrial DNA. Annu Rev Biochem 85:133–160. 10.1146/annurev-biochem-060815-014402 [DOI] [PubMed] [Google Scholar]
- 28.Burger G, Gray MW, Lang BF (2003) Mitochondrial genomes: Anything goes. Trends Genet 19:709–716. 10.1016/j.tig.2003.10.012 [DOI] [PubMed] [Google Scholar]
- 29.Brown TA, Cecconi C, Tkachuk AN, et al. (2005) Replication of mitochondrial DNA occurs by strand displacement with alternative light-strand origins, not via a strand-coupled mechanism. Genes Dev 1–11. 10.1101/gad.1352105.other [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jõers P, Jacobs HT (2013) Analysis of Replication Intermediates Indicates That Drosophila melanogaster Mitochondrial DNA Replicates by a Strand-Coupled Theta Mechanism. PLoS One 8:. 10.1371/journal.pone.0053249 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lewis SC, Joers P, Willcox S, et al. (2015) A rolling circle replication mechanism produces multimeric lariats of mitochondrial DNA in Caenorhabditis elegans. PLoS Genet 11:e1004985. 10.1371/journal.pgen.1004985 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lujan SA, Longley MJ, Humble MH, et al. (2020) Ultrasensitive deletion detection links mitochondrial DNA replication, disease, and aging. Genome Biology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Copeland WC, Wallace KB (2018) Mitochondrial Genomics and Targeted Toxicities, Third Edit. Elsevier [Google Scholar]
- 34.Brown WM, George M, Wilson AC (1979) Rapid evolution of animal mitochondrial DNA. Proc Natl Acad Sci U S A 76:1967–71. 10.1073/pnas.76.4.1967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Longley MJ, Prasad R, Srivastava DK, et al. (1998) Identification of 5′-deoxyribose phosphate lyase activity in human DNA polymerase γ and its role in mitochondrial base excision repair in vitro. Proc Natl Acad Sci U S A 95:12244–12248. 10.1073/pnas.95.21.12244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Gustafson MA, Sullivan ED, Copeland WC (2020) Consequences of compromised mitochondrial genome integrity. DNA Repair (Amst) 93:102916. 10.1016/j.dnarep.2020.102916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.DeBalsi KL, Hoff KE, Copeland WC (2017) Role of the mitochondrial DNA replication machinery in mitochondrial DNA mutagenesis, aging and age-related diseases. Ageing Res Rev 33:89–104. 10.1016/j.arr.2016.04.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Taylor RW, Turnbull DM (2005) Mitochondrial DNA mutations in human disease. Nat Rev Genet 6:389–402. 10.1038/nrg1606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Nguyen K V, Sharief FS, Chan SSL, et al. (2006) Molecular diagnosis of Alpers syndrome. J Hepatol 45:108–116. 10.1016/j.jhep.2005.12.026 [DOI] [PubMed] [Google Scholar]
- 40.Larsson NG, Clayton DA (1995) Molecular genetic aspects of human mitochondrial disorders. Annu Rev Genet 29:151–178. 10.1146/annurev.ge.29.120195.001055 [DOI] [PubMed] [Google Scholar]
- 41.Bender A, Krishnan KJ, Morris CM, et al. (2006) High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat Genet 38:515–517. 10.1038/ng1769 [DOI] [PubMed] [Google Scholar]
- 42.Kennedy SR, Salk JJ, Schmitt MW, Loeb LA (2013) Ultra-sensitive sequencing reveals an age-related increase in somatic mitochondrial mutations that are inconsistent with oxidative damage. PLoS Genet 9:e1003794. 10.1371/journal.pgen.1003794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoekstra JG, Hipp MJ, Montine TJ, Kennedy SR (2016) Mitochondrial DNA mutations increase in early stage Alzheimer disease and are inconsistent with oxidative damage. Ann Neurol 80:301–306. 10.1002/ana.24709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ju YS eo., Alexandrov LB, Gerstung M, et al. (2014) Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 3:1–29. 10.7554/eLife.02935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zheng W, Khrapko K, Coller HA, et al. (2006) Origins of human mitochondrial point mutations as DNA polymerase γ-mediated errors. Mutat Res - Fundam Mol Mech Mutagen 599:11–20. 10.1016/j.mrfmmm.2005.12.012 [DOI] [PubMed] [Google Scholar]
- 46.Harman D (1956) Aging: A Theory on Free Radical Radiation Chemistry. J Gerontol 11:298–300 [DOI] [PubMed] [Google Scholar]
- 47.Miquel J (1998) An update on the oxygen stress-mitochondrial mutation theory of aging: Genetic and evolutionary implications. Exp Gerontol 33:113–126. 10.1016/S0531-5565(97)00060-0 [DOI] [PubMed] [Google Scholar]
- 48.Degtyareva NP, Saini N, Sterling JF, et al. (2019) Mutational signatures of redox stress in yeast single-strand DNA and of aging in human mitochondrial DNA share a common feature. PLoS Biol 17:1–28. 10.1371/journal.pbio.3000263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Degtyareva NP, Saini N, Sterling JF, et al. (2019) Mutational signatures of redox stress in yeast single-strand DNA and of aging in human mitochondrial DNA share a common feature. PLoS Biol 17:1–28. 10.1371/journal.pbio.3000263 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nissanka N, Bacman SR, Plastini MJ, Moraes CT (2018) The mitochondrial DNA polymerase gamma degrades linear DNA fragments precluding the formation of deletions. Nat Commun 9:. 10.1038/s41467-018-04895-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Peeva V, Blei D, Trombly G, et al. (2018) Linear mitochondrial DNA is rapidly degraded by components of the replication machinery. Nat Commun 9:1–11. 10.1038/s41467-018-04131-w [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Moretton A, Morel F, Macao B, et al. (2017) Selective mitochondrial DNA degradation following double-strand breaks. PLoS One 12:1–17. 10.1371/journal.pone.0176795 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Nissanka N, Moraes CT (2018) Mitochondrial DNA damage and reactive oxygen species in neurodegenerative disease. FEBS Lett 592:728–742. 10.1002/1873-3468.12956.Mitochondrial [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pinto M, Moraes CT (2015) Mechanisms linking mtDNA damage and aging. Free Radic Biol Med 250–258. 10.1016/j.freeradbiomed.2015.05.005.Mechanisms [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Trifunovic A, Wredenberg A, Falkenberg M, et al. (2004) Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429:417–423. 10.1038/nature02517 [DOI] [PubMed] [Google Scholar]
- 56.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, & Prolla TA (2005) Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science (80- ) 309:481. [DOI] [PubMed] [Google Scholar]
- 57.Vermulst M, Bielas JH, Kujoth GC, et al. (2007) Mitochondrial point mutations do not limit the natural lifespan of mice. Nat Genet 39:540–543. 10.1038/ng1988 [DOI] [PubMed] [Google Scholar]
- 58.Pickrell AM, Huang C-H, Kennedy SR, et al. (2015) Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 87:371–81. 10.1016/j.neuron.2015.06.034 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kim M, Haroon S, Chen G, et al. (2019) Increased burden of mitochondrial DNA deletions and point mutations in early-onset age-related hearing loss in mitochondrial mutator mice. Exp Gerontol 125:1–28. 10.1016/j.exger.2019.110675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Haroon S, Li A, Weinert JL, et al. (2018) Multiple Molecular Mechanisms Rescue mtDNA Disease in C. elegans. Cell Rep 22:3115–3125. 10.1016/j.celrep.2018.02.099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Samstag CL, Hoekstra JG, Huang CH, et al. (2018) Deleterious mitochondrial DNA point mutations are overrepresented in Drosophila expressing a proofreading-defective DNA polymerase γ. PLoS Genet 14:1–27. 10.1371/journal.pgen.1007805 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Andreazza S, Samstag CL, Sanchez-Martinez A, et al. (2019) Mitochondrially-targeted APOBEC1 is a potent mtDNA mutator affecting mitochondrial function and organismal fitness in Drosophila. Nat Commun 10:. 10.1038/s41467-019-10857-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kauppila TES, Bratic A, Jensen MB, et al. (2018) Mutations of mitochondrial DNA are not major contributors to aging of fruit flies. Proc Natl Acad Sci U S A 115:E9620–E9629. 10.1073/pnas.1721683115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Stewart JB, Chinnery PF (2021) Extreme heterogeneity of human mitochondrial DNA from organelles to populations. Nat Rev Genet 22:106–118. 10.1038/s41576-020-00284-x [DOI] [PubMed] [Google Scholar]
- 65.Stewart JB, Chinnery PF (2015) The dynamics of mitochondrial DNA heteroplasmy: implications for human health and disease. Nat Rev Genet 16:530–542. 10.1038/nrg3966 [DOI] [PubMed] [Google Scholar]
- 66.Stewart JB, Larsson NG (2014) Keeping mtDNA in Shape between Generations. PLoS Genet 10:. 10.1371/journal.pgen.1004670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Hill JH, Chen Z, Xu H (2014) Selective propagation of functional mitochondrial DNA during oogenesis restricts the transmission of a deleterious mitochondrial variant. Nat Genet 46:389–392. 10.1038/ng.2920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Lieber T, Jeedigunta SP, Palozzi JM, et al. (2019) Mitochondrial fragmentation drives selective removal of deleterious mtDNA in the germline. Nature. 10.1038/s41586-019-1213-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aryaman J, Bowles C, Jones NS, Johnston IG (2018) Mitochondrial network fragmentation modulates mutant mtDNA accumulation independently of absolute fission-fusion rates
- 70.Aryaman J, Johnston IG, Jones NS (2019) Mitochondrial heterogeneity. Front Genet 10:1–16. 10.3389/fgene.2018.00718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Aryaman J, Bowles C, Jones NS, Johnston IG (2019) Mitochondrial network state scales mtDNA genetic dynamics. Genetics 212:1429–1443. 10.1101/409128 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gitschlag BL, Kirby CS, Samuels DC, et al. (2016) Homeostatic Responses Regulate Selfish Mitochondrial Genome Dynamics in C.??elegans. Cell Metab 24:91–103. 10.1016/j.cmet.2016.06.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Knorre DA (2020) Intracellular quality control of mitochondrial DNA: evidence and limitations. Philos Trans R Soc B Biol Sci 375:20190176. 10.1098/rstb.2019.0176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lin Y-F, Schulz AM, Pellegrino MW, et al. (2016) Maintenance and propagation of a deleterious mitochondrial genome by the mitochondrial unfolded protein response. Nature 533:416–9. 10.1038/nature17989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Valenci I, Yonai L, Bar-Yaacov D, et al. (2015) Parkin modulates heteroplasmy of truncated mtDNA in Caenorhabditis elegans. Mitochondrion 20:64–70. 10.1016/j.mito.2014.11.001 [DOI] [PubMed] [Google Scholar]
- 76.Ross JM, Coppotelli G, Hoffer BJ, Olson L (2014) Maternally transmitted mitochondrial DNA mutations can reduce lifespan. Sci Rep 4:1–3. 10.1038/srep06569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Ross JM, Stewart JB, Hagström E, et al. (2013) Germline mtDNA mutations aggravate ageing and can impair brain development. Nature 501:412–415. 10.1038/nature12474.Germline [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Boovarahan SR, Kurian GA (2018) Mitochondrial dysfunction: A key player in the pathogenesis of cardiovascular diseases linked to air pollution. Rev Environ Health 33:111–122. 10.1515/reveh-2017-0025 [DOI] [PubMed] [Google Scholar]
- 79.Brunst KJ, Baccarelli AA, Wright RJ (2015) Integrating mitochondriomics in children’s environmental health. J Appl Toxicol 35:976–991. 10.1002/jat.3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Filograna R, Mennuni M, Alsina D, Larsson NG (2021) Mitochondrial DNA copy number in human disease: the more the better? FEBS Lett 595:976–1002. 10.1002/1873-3468.14021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Yuan H, Zhang Q, Guo J, et al. (2016) A PGC-1α-Mediated Transcriptional Network Maintains Mitochondrial Redox and Bioenergetic Homeostasis against Doxorubicin-Induced Toxicity in Human Cardiomyocytes: Implementation of TT21C. Toxicol Sci 150:400–417. 10.1093/toxsci/kfw006 [DOI] [PubMed] [Google Scholar]
- 82.Leuthner TC, Hartman JH, Ryde IT, Meyer JN (2021) PCR-Based Determination of Mitochondrial DNA Copy Number in Multiple Species. In: Methods in Molecular Biology. pp 91–111 [DOI] [PubMed] [Google Scholar]
- 83.Meyer JN, Leuthner TC, Luz AL (2017) Mitochondrial fusion, fission, and mitochondrial toxicity. Toxicology 391:42–53. 10.1016/j.tox.2017.07.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Fox EJ, Reid-Bayliss KS, Emond MJ, Loeb LA (2014) Accuracy of Next Generation Sequencing Platforms. Next Gener Seq Appl. 10.1007/978-1-4939-0715-1_7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schmitt MW, Kennedy SR, Salk JJ, et al. (2012) Detection of ultra-rare mutations by next-generation sequencing. Proc Natl Acad Sci U S A 109:14508–13. 10.1073/pnas.1208715109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kou R, Lam H, Duan H, et al. (2016) Benefits and Challenges with Applying Unique Molecular Identifiers in Next Generation Sequencing to Detect Low Frequency Mutations. PLoS One 11:e0146638. 10.1371/journal.pone.0146638 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Marquis J, Lefebvre G, Kourmpetis YAI, et al. (2017) MitoRS, a method for high throughput, sensitive, and accurate detection of mitochondrial DNA heteroplasmy. BMC Genomics 18:326. 10.1186/s12864-017-3695-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Arbeithuber B, Hester J, Cremona MA, et al. (2020) Age-related accumulation of de novo mitochondrial mutations in mammalian oocytes and somatic tissues. PLoS Biol 1–37. 10.1371/journal.pbio.3000745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Morris J, Na YJ, Zhu H, et al. (2017) Pervasive within-Mitochondrion Single-Nucleotide Variant Heteroplasmy as Revealed by Single-Mitochondrion Sequencing. Cell Rep 21:2706–2713. 10.1016/j.celrep.2017.11.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Shtolz N, Mishmar D (2019) The Mitochondrial Genome–on Selective Constraints and Signatures at the Organism, Cell, and Single Mitochondrion Levels. Front Ecol Evol 7:342. 10.3389/fevo.2019.00342 [DOI] [Google Scholar]
- 91.Hertweck KL, Dasgupta S (2017) The Landscape of mtDNA Modifications in Cancer: A Tale of Two Cities. Front Oncol 7:262. 10.3389/fonc.2017.00262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ju YS eo., Alexandrov LB, Gerstung M, et al. (2014) Origins and functional consequences of somatic mitochondrial DNA mutations in human cancer. Elife 3:1–28. 10.7554/eLife.02935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Gammage PA, Frezza C (2019) Mitochondrial DNA: The overlooked oncogenome? BMC Biol 17:1–10. 10.1186/s12915-019-0668-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Patel J, Baptiste BA, Kim E, et al. (2020) DNA damage and mitochondria in cancer and aging. Carcinogenesis 41:1625–1634. 10.1093/carcin/bgaa114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Salk JJ, Schmitt MW, Loeb LA (2018) Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet 19:269–285. 10.1038/nrg.2017.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Salk JJ, Kennedy SR (2020) Next-Generation Genotoxicology: Using Modern Sequencing Technologies to Assess Somatic Mutagenesis and Cancer Risk. Environ Mol Mutagen 61:135–151. 10.1002/em.22342 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yao Y-G, Kajigayab S, Young NS (2015) Mitochondrial DNA mutations in single human blood cells. Mutat Res 779:68–77. 10.1016/j.mrfmmm.2015.06.009.Mitochondrial [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Atilano SR, Udar N, Satalich TA, et al. (2021) Low frequency mitochondrial DNA heteroplasmy SNPs in blood, retina, and [RPE +choroid] of age-related macular degeneration subjects. PLoS One 16:1–24. 10.1371/journal.pone.0246114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Reyes A, Gissi C, Pesole G, Saccone C (1998) Asymmetrical directional mutation pressure in the mitochondrial genome of mammals. Mol Biol Evol 15:957–966. 10.1093/oxfordjournals.molbev.a026011 [DOI] [PubMed] [Google Scholar]
- 100.Bedrat A, Lacroix L, Mergny JL (2016) Re-evaluation of G-quadruplex propensity with G4Hunter. Nucleic Acids Res 44:1746–1759. 10.1093/nar/gkw006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Falabella M, Fernandez RJ, Johnson FB, Kaufman BA (2018) Potential Roles for G-Quadruplexes in Mitochondria. Curr Med Chem 26:2918–2932. 10.2174/0929867325666180228165527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lyonnais S, Tarrés-Soler A, Rubio-Cosials A, et al. (2017) The human mitochondrial transcription factor A is a versatile G-quadruplex binding protein. Sci Rep 7:1–15. 10.1038/srep43992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Bharti SK, Sommers JA, Zhou J, et al. (2014) DNA sequences proximal to human mitochondrial DNA deletion breakpoints prevalent in human disease form G-quadruplexes, a class of DNA structures inefficiently unwound by the mitochondrial replicative Twinkle helicase. J Biol Chem 289:29975–93. 10.1074/jbc.M114.567073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pabis K (2021) Triplex and other DNA motifs show motif-specific associations with mitochondrial DNA deletions and species lifespan. Mech Ageing Dev 194:111429. 10.1016/j.mad.2021.111429 [DOI] [PubMed] [Google Scholar]
- 105.Khaidakov M (2016) Species-specific lifespans: Can it be a lottery based on the mode of mitochondrial DNA replication? Mech Ageing Dev 155:1–6. 10.1016/j.mad.2016.02.012 [DOI] [PubMed] [Google Scholar]
- 106.Lee SR, Han J (2017) Mitochondrial Nucleoid: Shield and Switch of the Mitochondrial Genome. Oxid Med Cell Longev 2017:8060949. 10.1155/2017/8060949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hernansanz-Agustín P, Enríquez JA (2021) Generation of reactive oxygen species by mitochondria. Antioxidants 10:1–18. 10.3390/antiox10030415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Brand MD (2016) Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 100:14–31. 10.1016/j.freeradbiomed.2016.04.001 [DOI] [PubMed] [Google Scholar]
- 109.Burke Peter J. (2017) Mitochondria, Bioenergetics & Apoptosis in Cancer Peter. Trends Cancer 3:857–870. 10.1016/j.trecan.2017.10.006.Mitochondria [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Sies H (2021) Oxidative eustress: On constant alert for redox homeostasis. Redox Biol 41:101867. 10.1016/j.redox.2021.101867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Meyer JN, Leung MCK, Rooney JP, et al. (2013) Mitochondria as a target of environmental toxicants. Toxicol Sci 134:1–17. 10.1093/toxsci/kft102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Zhao L, Sumberaz P (2020) Mitochondrial DNA Damage: Prevalence, Biological Consequence, and Emerging Pathways. Chem Res Toxicol 33:2491–2502. 10.1021/acs.chemrestox.0c00083 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kukat C, Wurm CA, Spåhr H, et al. (2011) Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA. Proc Natl Acad Sci U S A 108:13534–13539. 10.1073/pnas.1109263108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kukat C, Davies KM, Wurm CA, et al. (2015) Cross-strand binding of TFAM to a single mtDNA molecule forms the mitochondrial nucleoid. Proc Natl Acad Sci U S A 112:11288–11293. 10.1073/pnas.1512131112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Farge GÉR, Falkenberg M (2019) Organization of DNA in mammalian mitochondria. Int J Mol Sci 20:1–14. 10.3390/ijms20112770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Mishmar D, Levin R, Naeem MM, Sondheimer N (2019) Higher Order Organization of the mtDNA: Beyond Mitochondrial Transcription Factor A. Front Genet 10:1–9. 10.3389/fgene.2019.01285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sharma Nidhi; Pasala Monica; Prakash A (2016) Mitochondrial DNA: Epigenetics and Environment. 25:289–313. 10.1002/em.22319.Mitochondrial [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Patil V, Cuenin C, Chung F, et al. (2019) Human mitochondrial DNA is extensively methylated in a non-CpG context. Nucleic Acids Res 47:10072–10085. 10.1093/nar/gkz762 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hao Z, Wu T, Cui X, et al. (2020) N6 -Deoxyadenosine Methylation in Mammalian Mitochondrial DNA. Mol Cell 78:382–395. 10.1016/j.molcel.2020.02.018.N [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Matsuda S, Yasukawa T, Sakaguchi Y, et al. (2018) Accurate estimation of 5-methylcytosine in mammalian mitochondrial DNA. Sci Rep 8:1–13. 10.1038/s41598-018-24251-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Janssen BG, Gyselaers W, Byun HM, et al. (2017) Placental mitochondrial DNA and CYP1A1 gene methylation as molecular signatures for tobacco smoke exposure in pregnant women and the relevance for birth weight. J Transl Med 15:1–10. 10.1186/s12967-016-1113-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Breton CV, Song AY, Xiao J, et al. (2019) Effects of air pollution on mitochondrial function, mitochondrial DNA methylation, and mitochondrial peptide expression. Mitochondrion 22–29. 10.1016/j.mito.2019.04.001.Effects [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Zhou Z, Goodrich JM, Strakovsky RS (2020) Mitochondrial epigenetics and environmental health: Making a case for endocrine disrupting chemicals. Toxicol Sci 178:16–25. 10.1093/toxsci/kfaa129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Weinhouse C (2017) Mitochondrial-epigenetic crosstalk in environmental toxicology. Toxicology 391:5–17. 10.1016/j.tox.2017.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Saki M, Prakash A (2017) DNA damage related crosstalk between the nucleus and mitochondria. Free Radic Biol Med 107:216–227. 10.1016/j.freeradbiomed.2016.11.050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kelly RDW, Mahmud A, McKenzie M, et al. (2012) Mitochondrial DNA copy number is regulated in a tissue specific manner by DNA methylation of the nuclear-encoded DNA polymerase gamma A. Nucleic Acids Res 40:10124–10138. 10.1093/nar/gks770 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Meyer JN, Hartman JH, Mello DF (2018) Mitochondrial Toxicity. Toxicol Sci 162:15–23. 10.1093/toxsci/kfy008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Roubicek DA, de Souza-Pinto NC (2017) Mitochondria and mitochondrial DNA as relevant targets for environmental contaminants. Toxicology 391:100–108. 10.1016/j.tox.2017.06.012 [DOI] [PubMed] [Google Scholar]
- 129.Liu YJ, McIntyre RL, Janssens GE, Houtkooper RH (2020) Mitochondrial fission and fusion: A dynamic role in aging and potential target for age-related disease. Mech Ageing Dev 186:111212. 10.1016/j.mad.2020.111212 [DOI] [PubMed] [Google Scholar]
- 130.Pickles S, Vigié P, Youle RJ (2018) Mitophagy and Quality Control Mechanisms in Mitochondrial Maintenance. Curr Biol 28:R170–R185. 10.1016/J.CUB.2018.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kleele T, Rey T, Winter J, et al. (2020) Distinct molecular signatures of fission predict mitochondrial degradation or proliferation. bioRxiv 593:. 10.1101/2020.11.11.372557 [DOI] [PubMed] [Google Scholar]
- 132.Babbar M, Basu S, Yang B, et al. (2020) Mitophagy and DNA damage signaling in human aging. Mech Ageing Dev 186:111207. 10.1016/j.mad.2020.111207 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Graziewicz MA, Sayer JM, Jerina DM, Copeland WC (2004) Nucleotide incorporation by human DNA polymerase γ opposite benzo[a]pyrene and benzo[c]phenanthrene diol epoxide adducts of deoxyguanosine and deoxyadenosine. Nucleic Acids Res 32:397–405. 10.1093/nar/gkh213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Valente WJ, Ericson NG, Long AS, et al. (2016) Mitochondrial DNA exhibits resistance to induced point and deletion mutations. Nucleic Acids Res gkw 716. 10.1093/nar/gkw716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Stumpf JD, Copeland WC (2014) MMS Exposure Promotes Increased MtDNA Mutagenesis in the Presence of Replication-Defective Disease-Associated DNA Polymerase γ Variants. PLoS Genet 10:. 10.1371/journal.pgen.1004748 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Young MJ (2017) Off-Target Effects of Drugs that Disrupt Human Mitochondrial DNA Maintenance. Front Mol Biosci 4:. 10.3389/fmolb.2017.00074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Takaki A, Jimi S, Segawa M, et al. (2004) Long-term cadmium exposure accelerates age-related mitochondrial changes in renal epithelial cells. Toxicology 203:145–154. 10.1016/j.tox.2004.06.005 [DOI] [PubMed] [Google Scholar]
- 138.Mani MS, Chakrabarty S, Mallya SP, et al. (2019) Whole mitochondria genome mutational spectrum in occupationally exposed lead subjects. Mitochondrion 48:60–66. 10.1016/j.mito.2019.04.009 [DOI] [PubMed] [Google Scholar]
- 139.Wang CY, Zhao ZB (2012) Somatic mtDNA mutations in lung tissues of pesticide-exposed fruit growers. Toxicology 291:51–55. 10.1016/j.tox.2011.10.018 [DOI] [PubMed] [Google Scholar]
- 140.Birket MJ, Birch-Machin MA (2007) Ultraviolet radiation exposure accelerates the accumulation of the aging-dependent T414G mitochondrial DNA mutation in human skin. Aging Cell 6:557–564. 10.1111/j.1474-9726.2007.00310.x [DOI] [PubMed] [Google Scholar]
- 141.Ziada AS, Lu MY, Ignas-Menzies J, et al. (2019) Mitochondrial DNA somatic mutation burden and heteroplasmy are associated with chronological age, smoking, and HIV infection. Aging Cell 18:1–15. 10.1111/acel.13018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kesäniemi J, Lavrinienko A, Tukalenko E, et al. (2020) Exposure to environmental radionuclides alters mitochondrial DNA maintenance in a wild rodent. Evol Ecol 34:163–174. 10.1007/s10682-019-10028-x [DOI] [Google Scholar]
- 143.Baker RJ, Dickins B, Wickliffe JK, et al. (2017) Elevated mitochondrial genome variation after 50 generations of radiation exposure in a wild rodent. Evol Appl 10:784–791. 10.1111/eva.12475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Matson CW, Lambert MM, Mcdonald TJ, et al. (2006) Evolutionary Toxicology : Population-Level Effects of Chronic Contaminant Exposure on the Marsh Frogs ( Rana ridibunda ) of Azerbaijan. Environ Health Perspect 114:547–552. 10.1289/ehp.8404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Fontana GA, Gahlon HL (2020) Mechanisms of replication and repair in mitochondrial DNA deletion formation. Nucleic Acids Res 48:11244–11258. 10.1093/nar/gkaa804 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Leung MCK, Rooney JP, Ryde IT, et al. (2013) Effects of early life exposure to ultraviolet C radiation on mitochondrial DNA content, transcription, ATP production, and oxygen consumption in developing Caenorhabditis elegans. BMC Pharmacol Toxicol 14:9. 10.1186/2050-6511-14-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Jung D, Cho Y, Collins LB, et al. (2009) Effects of benzo[a]pyrene on mitochondrial and nuclear DNA damage in Atlantic killifish (Fundulus heteroclitus) from a creosote-contaminated and reference site. Aquat Toxicol 95:44–51. 10.1016/j.aquatox.2009.08.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.González-Hunt CP, Leung MCK, Bodhicharla RK, et al. (2014) Exposure to Mitochondrial Genotoxins and Dopaminergic Neurodegeneration in Caenorhabditis elegans. PLoS One 9:e114459. 10.1371/journal.pone.0114459 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Kasiviswanathan R, Collins TRL, Copeland WC, et al. (2012) The interface of transcription and DNA replication in the mitochondria. Biochim Biophys Acta - Gene Regul Mech 1819:970–978. 10.1016/j.bbagrm.2011.12.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Bess AS, Crocker TL, Ryde IT, Meyer JN (2012) Mitochondrial dynamics and autophagy aid in removal of persistent mitochondrial DNA damage in Caenorhabditis elegans. Nucleic Acids Res 40:7916–7931. 10.1093/nar/gks532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Cohen BH (2010) Pharmacologic effects on mitochondrial function. Dev Disabil Res Rev 16:189–199. 10.1002/ddrr.106 [DOI] [PubMed] [Google Scholar]
- 152.Krasich R, Copeland WC (2017) DNA polymerases in the mitochondria: A critical review of the evidence. Front Biosci (Landmark Ed 22:692–709. 10.2741/4510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Alexander MP, Begins KJ, Crall WC, Holmes MP (2013) High Levels of Transcription Stimulate Transversions at GC Base Pairs. 54:44–53. 10.1002/em.21740.High [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wang Y, Zhang M, Li Z, et al. (2019) Fine particulate matter induces mitochondrial dysfunction and oxidative stress in human SH-SY5Y cells. Chemosphere 218:577–588. 10.1016/j.chemosphere.2018.11.149 [DOI] [PubMed] [Google Scholar]
- 155.Li R, Kou X, Geng H, et al. (2015) Effect of ambient PM2.5 on lung mitochondrial damage and fusion/fission gene expression in rats. Chem Res Toxicol 28:408–418. 10.1021/tx5003723 [DOI] [PubMed] [Google Scholar]
- 156.Li R, Kou X, Geng H, et al. (2015) Mitochondrial damage: An important mechanism of ambient PM2.5 exposure-induced acute heart injury in rats. J Hazard Mater 287:392–401. 10.1016/j.jhazmat.2015.02.006 [DOI] [PubMed] [Google Scholar]
- 157.Gebhard D, Mahler B, Matt K, et al. (2014) Mitochondrial DNA copy number - but not a mitochondrial tandem CC to TT transition - is increased in sun-exposed skin. Exp Dermatol 23:209–211. 10.1111/exd.12327 [DOI] [PubMed] [Google Scholar]
- 158.Reimann V, Krämer U, Sugiri D, et al. (2008) Sunbed use induces the photoaging-associated mitochondrial common deletion. J Invest Dermatol 128:1294–1297. 10.1038/sj.jid.5701151 [DOI] [PubMed] [Google Scholar]
- 159.Stout R, Birch-Machin M (2019) Mitochondria’s role in skin ageing. Biology (Basel) 8:. 10.3390/biology8020029 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Jayasundara N (2017) Ecological significance of mitochondrial toxicants. Toxicology 391:64–74. 10.1016/J.TOX.2017.07.015 [DOI] [PubMed] [Google Scholar]
- 161.Katju V, Bergthorsson U (2019) Old trade, new tricks: Insights into the spontaneous mutation process from the partnering of classical mutation accumulation experiments with high-throughput genomic approaches. Genome Biol Evol 11:136–165. 10.1093/gbe/evy252 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Xu S, Schaack S, Seyfert A, et al. (2012) High Mutation Rates in the Mitochondrial Genomes of Daphnia pulex. Mol Biol Evol 29:763–769. 10.1093/molbev/msr243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xu S, Van Tran K, Neupane S, et al. (2017) Single-sperm sequencing reveals the accelerated mitochondrial mutation rate in male Daphnia pulex (Crustacea, Cladocera). Proceedings Biol Sci 284:. 10.1098/rspb.2017.1548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Konrad A, Thompson O, Waterston RH, et al. (2017) Mitochondrial mutation rate, spectrum and heteroplasmy in Caenorhabditis elegans spontaneous mutation accumulation lines of differing population size. Mol Biol Evol 34:1319–1334. 10.1093/molbev/msx051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Wernick RI, Estes S, Howe DK, Denver DR (2016) Paths of Heritable Mitochondrial DNA Mutation and Heteroplasmy in Reference and gas-1 Strains of Caenorhabditis elegans. Front Genet 7:51. 10.3389/fgene.2016.00051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Schaack S, Ho EKH, Macrae F (2020) Disentangling the intertwined roles of mutation, selection and drift in the mitochondrial genome. Philos Trans R Soc B Biol Sci 375:20190173. 10.1098/rstb.2019.0173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Chain FJJ, Flynn JM, Bull JK, Cristescu ME (2019) Accelerated rates of large-scale mutations in the presence of copper and nickel. Genome Res 29:64–73. 10.1101/gr.234724.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Meier B, Cooke SL, Al E (2014) C. elegans whole genome sequencing reveals mutational signatures related to carcinogens and DNA repair deficiency. Genome Res 1:1624–1636. 10.1101/gr.175547.114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Volkova N V, Meier B, González-Huici V, et al. (2020) Mutational signatures are jointly shaped by DNA damage and repair. Nat Commun 11:. 10.1038/s41467-020-15912-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.(2011) Global health and ageing
- 171.Russell OM, Gorman GS, Lightowlers RN, Turnbull DM (2020) Mitochondrial Diseases: Hope for the Future. Cell 181:168–188. 10.1016/j.cell.2020.02.051 [DOI] [PubMed] [Google Scholar]
- 172.Parikh S, Goldstein A, Koenig MK, et al. (2015) Diagnosis and management of mitochondrial disease: A consensus statement from the Mitochondrial Medicine Society. Genet Med 17:689–701. 10.1038/gim.2014.177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Naviaux RK (2020) Perspective: Cell danger response Biology—The new science that connects environmental health with mitochondria and the rising tide of chronic illness. Mitochondrion 51:40–45. 10.1016/j.mito.2019.12.005 [DOI] [PubMed] [Google Scholar]