

Abstract
Protein misfolding and aggregation is observed in many amyloidogenic diseases affecting either the central nervous system or a variety of peripheral tissues. Structural and dynamic characterization of all species along the pathways from monomers to fibrils is challenging by experimental and computational means because they involve intrinsically disordered proteins in most diseases. Yet understanding how amyloid species become toxic is the challenge in developing a treatment for these diseases. Here we review what computer, in vitro, in vivo, and pharmacological experiments tell us about the accumulation and deposition of the oligomers of the (Aβ, tau), α-synuclein, IAPP, and superoxide dismutase 1 proteins, which have been the mainstream concept underlying Alzheimer’s disease (AD), Parkinson’s disease (PD), type II diabetes (T2D), and amyotrophic lateral sclerosis (ALS) research, respectively, for many years.
Graphical Abstract

1. INTRODUCTION
The self-assembly of intrinsically disordered proteins (IDPs) into extracellular or intracellular transient oligomers and amyloid fibrils is shared by many human diseases, including Alzheimer’s (AD) and Parkinson’s (PD) diseases, type II diabetes (T2D), amyotrophic lateral sclerosis (ALS), and prion diseases.1 It is estimated that there about 50 and 10 million people worldwide living with AD and PD, respectively, and it was recently reported that AD can also be found in old chimpanzees.2 As citizens, we do not have an idea of its worldwide financial cost, but many of us know exactly the suffering for the patients and their families.
The history of senile dementia is very rich, dating back to the Greco-Roman period.3 However, AD was first described in 1906 by the German doctor Alois Alzheimer, PD was first described in 1817 by the English doctor James Parkinson, the French medical Professor Etienne Lancereaux is the pathfinder of T2D in 1877, and ALS, also named “maladie de Charcot” or “Lou Gehrig’s disease (American baseball player)”, was first described by Jean-Martin Charcot in Paris in 1874.
The key misfolded proteins are clearly identified for each disease: the Aβ protein (Aβ40 and Aβ42 with 40 and 42 amino acids) and the tau protein ranging from 352 to 421 amino acids in AD, the 140 amino acid α-synuclein (αS) protein in PD, the islet amyloid polypeptide (IAPP) or amylin of 37 amino acids in T2D, and the superoxide dismutase 1 of 32 kDa (SOD1), TAR DNA binding protein 43 (TPD-43), and 526 amino acid fused in sarcoma protein (FUS) in ALS. The first authors to correlate Aβ and αS proteins to AD and PD were Glenner and Wong,4 Goldberg and Lansbury,5 and Hardy and Selkoe,6 and the implications of the other proteins are reviewed in refs7–9. Their sequences do not have any homology and are very diverse in length, yet they all share the ability to form amyloid deposits or inclusions in the brain or the tissue (T2D) of patients, the exception being the wild-type SOD1 protein, but the monomeric apo-SOD1 in its disulfide-reduced state forms fibrillar aggregates under near quiescent conditions.10
In vitro, the Aβ, tau, αS, IAPP, TDP-43, and FUS proteins form readily cross-β structures with an aggregation kinetics profile typically displaying a sigmoidal curve where the proteins assemble into oligomers (lag-phase) prior to fibril elongation (growth phase) and a plateau where the fibrils and free monomers are in equilibrium (saturation phase), as shown in many reviews.11,12 Amyloid fibrils display an intermolecular hydrogen-bond (H-bond) network parallel to the fibril axis. The molecular mechanisms leading to amyloid fibrils are well described by primary nucleation, (fragmentation and surface-catalyzed) secondary nucleation, and elongation growth mechanisms as shown elsewhere.13 The aggregation kinetics and the lifetimes of the heterogeneous conformations of all oligomers along the amyloid fibril formation pathways are very sensitive to the amino acid length (e.g., Aβ42 vs Aβ40) and genetic risks, including several mutations in Aβ, αS, and SOD1 proteins and one unique mutation in IAPP, the level of hyperphorylation in tau and SOD1, and acetylation and glycosylation in tau. Experimental conditions modulate the self-assembly process, such as pH, T, peptide concentration, external applications resulting from agitation, electric field and shear forces, and the presence of membrane, metal ions, crowding, and heparin (for tau, in particular).14–16
Until 20 years ago, it was believed that the ability to form amyloid fibrils was restricted to a few proteins involved in diseases. However, there have been many more recent reports that nondisease proteins, short peptides, and even single amino acid homopolymers can form fibrils under appropriate conditions,17,18 and changing experimental conditions lead to nanotubes and ordered nanomaterials.19,20 Many studies have indicated the strategies and the selection pressures that protein sequences (either alone or helped by chaperones) have followed to avoid undesired aggregation, to adjust the kinetic and thermodynamic stability of their well folded three-dimensional (3D) structures, and to optimize the efficiency of their folding pathways.21
However, the emergence of amyloid folds is not a surprise for several reasons. They allow a variety of functional roles in both prokaryote and eukaryote organisms.22 Amyloids are able to replicate and catalyze their own formation, transmit information, and provide a scaffold for chemical reactions (e.g., ester hydrolysis) and enzyme-like activities.23–25 Even under early earth (prebiotic) conditions, peptides can form amyloid fibrils leading to the current amyloid world hypothesis in the origin of life26,27 and the possibility that all globular protein structures may have originated from amyloid fibrils.28
While SOD1 is a globular protein with a well-defined 3D structure, the Aβ, tau, and α-synuclein proteins belong to the class of intrinsically disordered proteins (IDPs). IDPs are also known to play a critical role in many cellular functions, such as signal transduction, cell growth, binding with DNA and RNA, and transcription, and are implicated in the development of cardiovascular problems and cancers.29 The IDPs involved in neurodegenerative diseases have a few aggregation-prone regions, and overall all IDPs have a low mean hydrophobicity and a high mean net charge.30
IDPs are structurally flexible and lack stable secondary structures in aqueous solution. When isolated, they behave as polymers in a good solvent and their radii of gyration are well described by the Flory scaling law.31 The insolubility and high self-assembly propensity of IDPs implicated in degenerative diseases have prevented high-resolution structural determination by solution nuclear magnetic resolution (NMR) and X-ray diffraction experiments. Local information at all aggregation steps can be, however, obtained by chemical shifts, residual coupling constants, and J-couplings from NMR, exchange hydrogen/deuterium (H/D) NMR, Raman spectroscopy, and secondary structure from fast Fourier infrared spectroscopy (FTIR) or circular dichroism (CD). Long-range tertiary contacts can be deduced from paramagnetic relaxation enhancement (PRE) NMR spectroscopy and single molecule Förster resonance energy transfer (sm-FRET), and short-range distance contacts can be extracted by cross-linked residues determined by mass spectrometry (MS). Low-resolution 3D information on monomers and oligomers can be obtained by ion-mobility mass-spectrometry data (IM/MS) providing cross-collision sections, dynamic light scattering (DLS), pulse field gradient NMR spectroscopy, and fluorescence correlation spectroscopy (FCS) providing hydrodynamics radius, small-angle X-ray scattering (SAXS) and small-angle neutron scattering (SANS), atomic force microscopy (AFM), and transmission electron microscopy (TEM) providing height features of the aggregates, as reported by some of the first and recent applications of these methods to IDPs.32–38 However, the information obtained from most experimental observables represents an average over the free energy landscape and gives time- and space-averaged properties. Experiments can also lead to different values of properties, for example the radius of gyration (Rg) as a result of the equilibrium between the monomeric and multimeric states of the IDPs under the conditions used. Fibril structures of long amyloid proteins are mainly proposed based on solid-state NMR (ssNMR) with the first high resolution structure of HET-s(218–289) prion39 and on cryo-electron miscroscopy (cryo-EM) experiments.40,41 Fibril structures of short amyloid peptides were also determined by X-ray diffraction analysis.42
Computer simulations at different time and length scales can in principle provide the dominant microstates of IDPs using multiple sampling techniques and various representations ranging from all-atom and coarse-grained (CG) to mesoscopic models.12,14,43–45 However, they are limited by the accuracy of the force field and the size of the energy landscape to be explored. Even on the fastest Anton computer, the simulation time using molecular dynamics (MD) simulations does not exceed 1 ms for a monomeric protein of 76 amino acids,46 i.e., several orders of magnitude less than the time scales of hours and days required for fibril formation in vitro at a μM concentration.11
The most currently accepted hypothesis is that accumulation of oligomers of the key proteins is the primary cause of AD, PD, T2D, and ALS diseases and initiates a series of events leading to neuronal or tissue death, a view pioneered by Golberg and Lansbury5 and reviewed more recently.47–50 There is also growing evidence from in vivo and in vitro studies of co-occurring pathologies across common neuogenerative diseases,51 indicating cross-talk between the amyloid proteins and interactions and cross-seeding between the Aβ, tau, and α-synuclein proteins which promote aggregation, generate different strains, and accelerate cognitive dysfunction.52–54
In summary, we provide an in-depth overview of our current knowledge on the biogenesis and domain organizations of Aβ, tau, α-synuclein, and IAPP related to their aggregation and binding properties, the molecular structures of amyloid monomers, oligomers and fibrils implicated in AD, T2D, and PD from experiments and simulations, as well as the early and final aggregation steps using coarse-grained simulations. We will not provide all the answers to the questions that we are facing, nor describe all the protein cellular partners interacting with these amyloid proteins, but we will discuss what experiments and simulations tell us about the role of liquid–liquid phase separation, the effect of crowding and shear flow, and the role played by the cell membranes and the Zn and Cu metal ions on protein aggregation. Next, we discuss what we know about ALS etiology and present a pharmacological perspective to cure these diseases considering small compounds, antibodies, or physical methods. This is followed by recent findings on crosstalk between amyloid proteins from in silico to in vivo experiments. We conclude with a series of unanswered questions that can potentially be handled by simulations and experiments, discuss the alternative hypotheses to amyloid oligomers causing human diseases, and list future directions.
2. Aβ BIOGENESIS AND DOMAIN ORGANIZATIONS OF TAU, α-SYNUCLEIN AND IAPP
2.1. Aβ Biogenesis: Role of Pathogenetic and Protective Mutations, Membrane Composition, and Detailed Interaction with γ-Secretase (gS)
2.1.1. Generation of Aβ Peptides by gS.
It is established that amyloid β protein (Aβ) contributes to the dysfunction and degeneration of neurons and the pathogenesis of AD.6,55–57 Aβ is derived from the proteolytic cleavage of the single-pass transmembrane amyloid precursor protein (APP) by secretases and may be processed along nonamyloidogenic and amyloidogenic pathways. The nonamyloidogenic pathway involves initial cleavage of APP by α-secretase (aS) leading to formation of sAPPα and the 83 amino acid fragment APP-C83. The amyloidogenic pathway involves initial cleavage of APP by β-secretase (bS) leading to formation of sAPPβ and the 99 amino acid fragment referred to as CTF99, APP-C99, or simply C99. Cleavage of C99 by γ-secretase is the last step in the production of Aβ (Figure 1).
Figure 1.
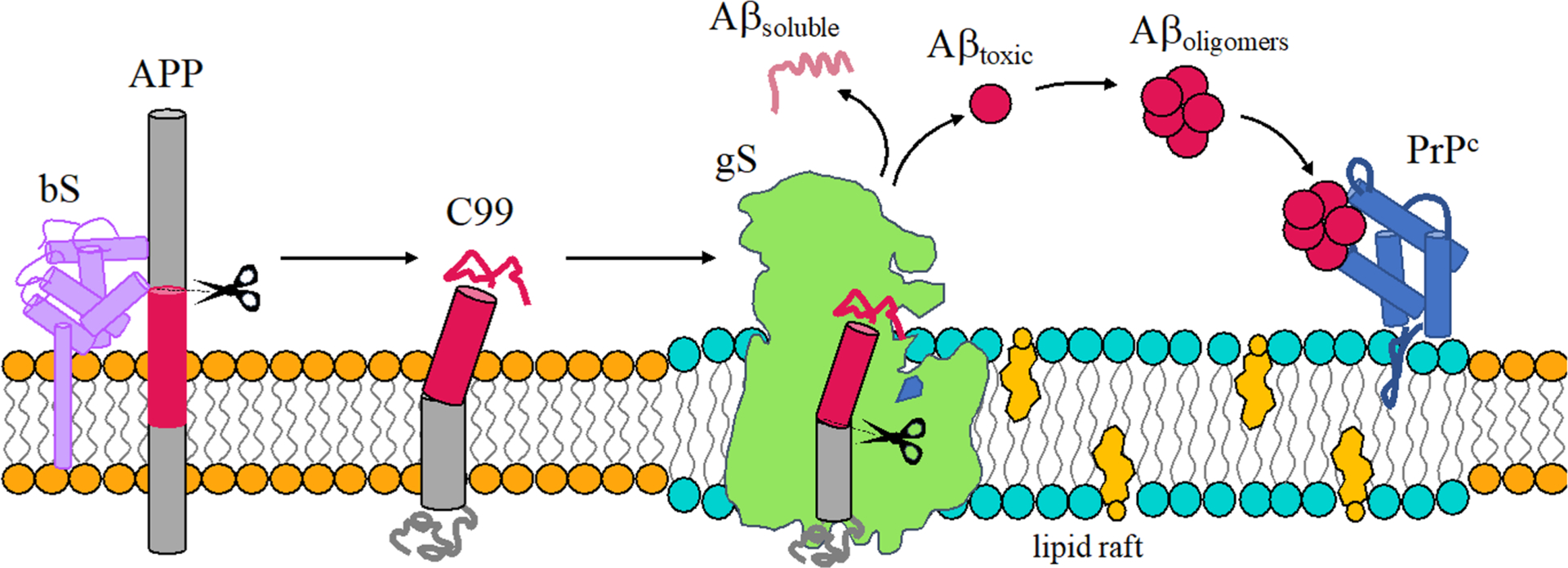
Schematic representation of the amyloidogenic processing of APP. Aβ is produced by sequential processing of APP by bS and gS to produce either a soluble and nontoxic mixture of Aβ or a more amyloidogenic Aβ mixture with a propensity to form oligomers that bind PrPc and potentially induce AD.
Cleavage of C99 by gS lacks fidelity.58–60 Cleavage of C99 by gS is initiated at the ε-sites (gS endopeptidase activity) to produce the amyloid intracellular domain (AICD) and Aβ49 or Aβ48 peptides. After the first cut, tri- and tetrapeptides are generated from sequential cleavage (gS carboxypeptidase-like activity) until Aβ proteins are released to the extracellular environment.61 Cleavage of C99 results in a variety of Aβ isoforms with Aβ40 being most prevalent and Aβ42 being a minor but more amyloidogenic form. Sequential cleavage by gS at points separated by roughly 0.5 nm results in a specific isoforms of Aβ. C99 is produced along two main cleavage lines Aβ49 > 46 > 43 > 40 and Aβ48 > 45 > 42 > 38 (Figure 2, panel 2D). The first is responsible for the release of the major isoform Aβ40 and the second leads to the minor isoforms Aβ42 and Aβ38.62
Figure 2.
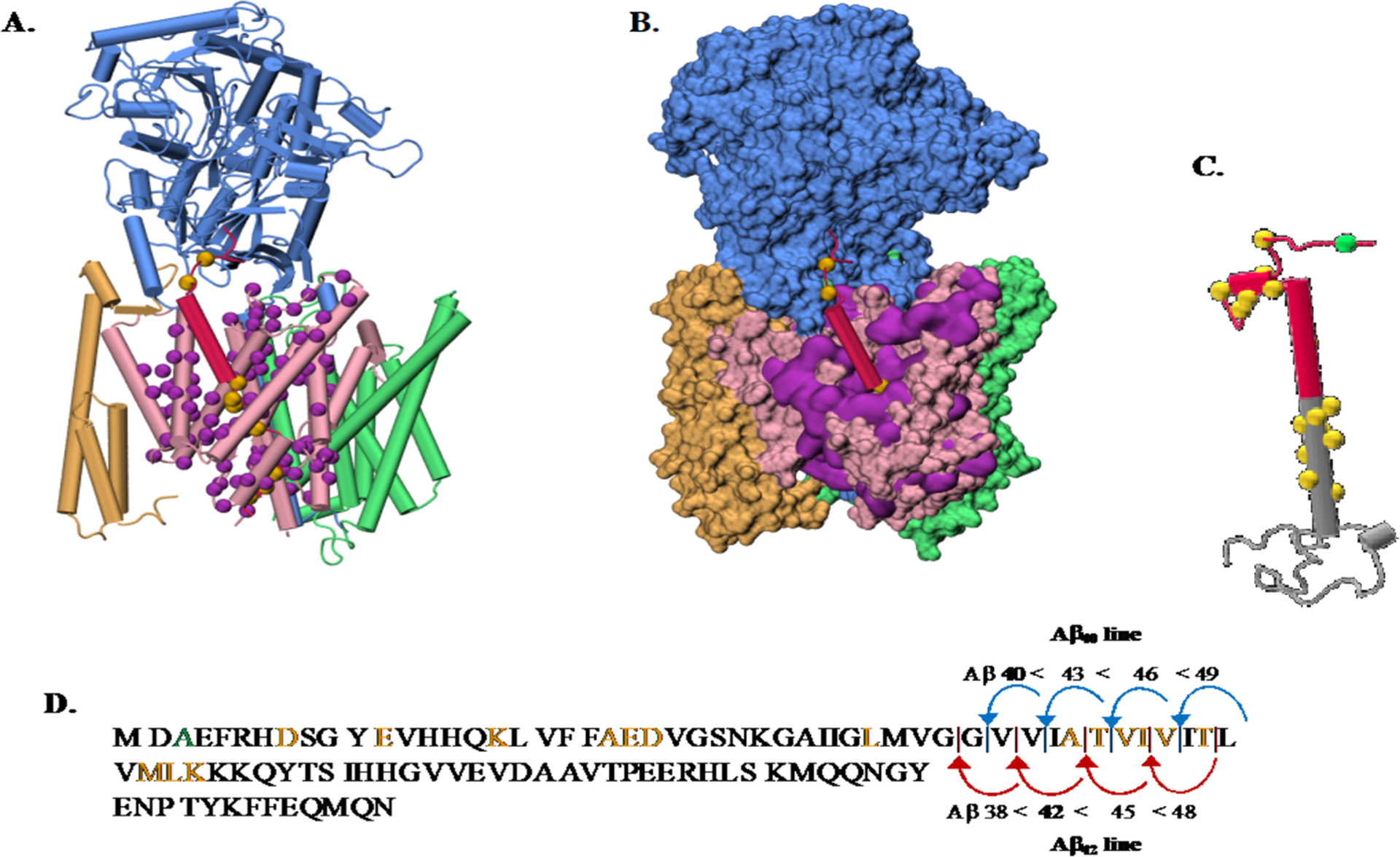
(A) Overview of the gS and APP structures and pathogenic AD mutations. (A) Depiction of the gS complex (PDB 6IYC).95 NCT subunit (blue), APH-1A (green), PEN-2 (yellow), and the catalytic PSN subunit (pink) bound to a C99 fragment (red). C-alpha atoms from the PSN disease-causing mutations139 that affect C99 processing are shown (purple spheres). C99 disease causing mutations available in the 6IYC structure are shown (yellow spheres). (B) gS structure with the same color code as part A in a surface representation. (C) Depiction of a C99 structure model (gray) with the Aβ sequence highlighted (red). C-alpha atoms from disease causing mutations are shown in yellow spheres with the protective Icelandic mutant (green). (D) C99 sequence with mutations highlighted in the same color code representation and showing the two main Aβ production lines. The structures in parts A, B, and C from the PDB were drawn using VMD.140
The observed infidelity in the cleavage of C99 may result from variation in the points of initiation and termination of cleavage, which in turn may be influenced by a variety of factors. Interestingly, gS is also known to process a wide variety of substrates,63–66 some of which are cleaved with high fidelity, suggesting that substrate sequence plays a primary role. A minimal model of the transmembrane domain of C99, Glu22-Lys55, is required for cleavage by gS, and importantly, negative charges on the extracellular side and positive charges on the intracellular side are required.67 The polybasic regions on the C-terminal side of TM helices are globally found in gS substrates68 and are conserved in both C99 and the Notch family. MD simulations have shown that following the first cleavage step the substrate is pulled deeper into the binding cavity of PS1. Negative charges at the N-terminus are observed to remain in place during the processing process.69 These observations support the conclusion that the N-terminal 21 amino acids of C99 are not required for gS cleavage.70
Evidence suggests that the mature gS complex is active at the plasma membrane and in endosomes71–73 and that cleavage of C99 by γ-secretase most commonly occurs when the enzyme and the substrate are colocalized in cholesterol-rich lipid raft domains. Therefore, while an important contributing factor to AD is mutation of APP or gS, alterations in the lipid membrane environment in which APP and gS are embedded also play a critical role. The actual products of APP cleavage depend on the specific membrane environment.74
While Aβ has long been implicated as a pathogenic agent in AD, the molecular mechanism by which Aβ induces neuronal dysfunction largely remains a mystery.75–79 One focus has been to characterize the structural ensembles of Aβ40 and Aβ42 monomers using experimental NMR and computational studies80,81 with an eye on identifying aggregation prone structures termed N* states.82 Given the role of the monomer ensemble in identifying aggregation prone sequences (N* conjecture), one promising putative mechanism for Aβ cytotoxicity involves the binding of Aβ oligomers with cellular prion protein PrPC83 leading to activation of a kinase and the abnormal phosphorylation of tau (see section 2.2).
2.1.2. Impact of Disease-Causing Mutations on APP and gS.
APP pathogenic mutations were the first to be recognized to cause early onset AD which led to the amyloid hypothesis.84–87 APP mutations are observed to be clustered near the aS, bS, and gS proteolytic cleavage sites and can be categorized according to location in the APP structure, including mutations located in the (1) APP extracellular region and (2) APP transmembrane fragment N-terminal to the Aβ42 cleavage site that typically have little effect on C99 cleavage. In addition, (3) the transmembrane C-terminal fragment below the Aβ42 cleavage site is a hotspot of familial AD (FAD) mutation and has been proposed to be important as the main gS recognition site. This latter region has also been proposed to be the region that determines the Aβ42:Aβ40 ratio by distorting the relative efficiencies of the ε-cleavage sites at C99 residues 48 or 49. Mutations that remove the positive charges from the invariant lysine or arginine residue located at the C99 TM junction greatly compromise the cleavage efficiency of gS.
2.1.3. Role of Mutations to APP and gS.
The majority of pathogenic APP mutations decrease the overall cleavage of C99 by gS, meaning that they are loss-of-function (LOF) mutations.68 Interestingly, the protective Icelandic mutation A673T was discovered in an individual with Down Syndrome (DS) who did not develop early onset AD related degeneration. Individuals with DS carry an extra copy of chromosome 21, and therefore, they also carry an extra copy of the gene for APP. This Aβ-lowering APP mutation is located in the APP extracellular region, close to the bS cleavage site and has been identified in the Islandic population.
PS1 pathogenic mutations modify substrate recognition, enzyme structure, or catalytic activity. These mutations result in partial or total loss-of-function of the enzyme. Before 2015, there was no structure available for gS. At this time, eight human gS structures have been determined by cryo-EM with a resolution range between 2.6 and 4.4 Å. gS is a transmembrane protein with four components.88–90 (1) Nicastrin (NCT) has been proposed to be in charge of substrate sorting, acting as a gatekeeper by sterically obstructing the access of proteins with large extracellular domains.91 (2) Anterior pharynx defective-1 (APH-1A) is required for the gS complex stability. Computational studies showed that it contains a water cavity able to transport water and store cations.92 (3) Presenilin (PS1) contains two aspartate residues that form the active site. (4) Presenilin enhancer 2 (PEN-2) is needed for the autocatalytic maturation.93,94 The recognition of APP by gS is illustrated in Figure 2A.95 180 mutations occurring in the PS1 subunit have been linked to familial AD.
Pathogenic PSEN-FAD mutations may affect gS endopeptidase activity (and the initial ε-cleavage site) and consistently impair the carboxypeptidase-like efficiency. This results in reduced processivity of Aβ49 or Aβ48 and the release of longer and more toxic Aβ43 or Aβ42 isoforms. These mutations result in the partial or total loss-of-function of the gS enzyme. PS1 mutants are dispersed among the whole gS structure, but those located at the gS complex surface may point to a substrate interaction/recognition site (Figure 2A and B).
While mutations to APP and gS mutations are known to modify C99 processing by gS, the details of how various mutations impact the cleavage process are still unknown. The disperse distribution of the mutation sites suggests that there should be a variety of mechanisms. (1) Disturbed PS1/APP interactions96 act by changing the preference of the initial position of the ε-cleavage site (gS endopeptidase activity). It has been proposed that APP mutations modify the tilting of the TMD helix, thereby altering the presentation of substrate to the proteolytic enzyme changing the initial ε-cleavage site. Alternatively, the mechanism of substrate docking and displacement toward the catalytic site may be impacted, as there is evidence that the substrate binding site is distinct from the catalytic active site. Once the substrate is docked, it is subsequently displaced to the catalytic site of the enzyme.97 (2) Loss of gS carboxypeptidase activity interferes with the catalytic efficiency, releasing premature intermediate and longer APP products.98 (3) Inhibitory effects at the initial ε-cleavage sites may lead to changes in the distribution of Aβ isoforms critically impacting aggregation kinetics and toxic effects.99 (4) Finally, catalytic cycle impairment may lead to alterations in the gS product lines.98
2.1.4. Role of Membrane in Aβ Genesis and Aggregation.
Early experimental63 and computational100 studies demonstrated that C99 consists of an extracellular region, including asparagine glycosylation sites, an extracellular juxtamembrane (JM) helical domain Q15–V21 (Q686–V695), an TM domain K28–K53 (K699–K724), and an intracellular C-terminal domain in model membranes (Figure 2C). Using ssNMR experiments, Tycko and co-workers suggested that, for the construct containing 27 residues K28–K55 (K699–K726) in multilamellar vesicles, the TM domain of APP adopts a mixture of helical and nonhelical structures,101 which varies as temperature is altered. Smith and workers reported NMR and FTIR data of the wild type (WT) construct N1–K55 (N672–K726) and the Flemish mutant A21G (A692G).102 They observed that at least part of the JM domain assumes a β-strand structure. Recent simulation results provide some support for this intriguing observation.103
Variations in sequence of C99 impact dimerization of the C99 TM domain, altering not only lateral mobility of the protein but also its TM helical structure (tilt and kink), C99 dimer structure and stability, and the structure of the TM helices within the dimer.100,104 There is evidence that changes in the stability of the C99 dimer influence its cleavage by gS and the resulting overall level of Aβ protein and its isoform distribution.101,104,105 Multhaup et al. first recognized that modifications in sequence that reduced homodimer affinity impacted cleavage of C99 by gS.106 Subsequent studies of homodimer formation in WT and mutant C99 congeners supported the view that C99 homodimerization is critical to C99 processing by gS and Aβ formation.107,108 However, it has been argued that C99 homodimerization is weak and may be largely irrelevant in vivo, suggesting that C99 monomer is the sole substrate for gS in the production of Aβ.109 There is little doubt that C99 homodimer is an essential species in the overall ensemble of C99 structures.107–111 More recently, Sanders et al. reported an ensemble of coexisting C99 monomer, dimer, and large-scale oligomers in lipid bicelles.112
2.1.5. Role of Membrane and Cholesterol on APP.
The role of membrane and cholesterol on APP has been examined in computational studies of C99 monomer and dimer structure in membrane and micelle environments as a function of protein sequence and composition of the lipid environment.103,110,113,114 Many of the observations derived from simulations have been validated using NMR experiments, including the existence of a flexible “hinge” region in the C99 monomer structure,113,114 the first C99 homodimer structure in a bilayer environment,113 and the ability to “environmentally select” C99 topologically distinct homodimer structures in membrane environments of varying lipid composition.114
Enhanced levels of cholesterol resulting from diet, genetic predisposition, or aging are positively correlated with early onset of AD.115–117 A variety of theories have been proposed to explain these observations. Lower levels of cholesterol promote membrane fluidity and nonamyloidogenic cleavage of APP by aS.118,119 In addition, decreased levels of cholesterol diminish both bS and gS activity and deplete cholesterol rich lipid raft microdomains deemed important for colocalization of gS and its substrate C99.62,120,121 Finally, site-specific binding of cholesterol to C99 protein has been observed. It has been proposed that elevated levels of cholesterol may increase the population of C99–cholesterol dimers,109,122 thus enhancing the partitioning of C99 to lipid raft domains and the proteolytic cleavage of C99 by gS to produce Aβ.
Membranes comprising these distinct cellular domains are composed of a mixture of lipids, including glycerolipids, sphingolipids, and cholesterol. The complex lipid mixtures are characterized not by a uniform mixture but by a heterogeneous mosaic of liquid-disordered regions and liquid-ordered microdomains of varying lipid composition, including regions rich in saturated lipids, sphingomyelin, and cholesterol referred to as detergent-resistant membranes or lipid rafts. Many studies120,123–127 have proposed a role for raft domains in the biogenesis of Aβ. However, their influence on the mechanism of creation of Aβ is not understood.
Current evidence suggests that the mature gS complex is active at the plasma membrane and in endosomes. However, the actual product of APP cleavage depends on the specific membrane environment. In addition, there is substantial evidence that cleavage of C99 by gS most commonly occurs when the enzyme and substrate are colocalized in cholesterol-rich lipid raft domains.
The enzyme bS possesses a single transmembrane anchoring domain. It has been observed in the plasma membrane and endosomes colocalized with APP in regions where the membrane is enriched in lipid raft domains. Like bS, APP possesses a single transmembrane domain and is found in a variety of locations in the cell including in lipid raft domains, colocalized with the enzymes bS and gS. It was recently proposed that a complex of βS and gS formed in cholesterol rich membrane domains might lead to substrate shuttling and enhanced efficiency in the biogenesis of Aβ.128 Taken together, these observations demonstrate the important role played by membrane spatial heterogeneity in Aβ genesis.
2.1.6. Role of Membrane on gS.
gS is accountable for the final step in the regulated intramembrane proteolysis of APP to generate Aβ. As such, substantial attention has focused on understanding how the lipid environment modulates gS activity, including variations in the membrane lipid composition and the presence of cholesterol and sphingolipids.66,129 Clinical and experimental studies suggest that lipid composition is altered in AD brain tissue130 and that the production of Aβ peptides varies with the membrane composition.131 The mechanism explaining these observations and the exact cause or consequence have not been determined. Holmes et al. reported that the composition of lipid bilayers has profound and complicated effects on the production of Aβ peptides by gS.132 Both the fatty acyl chains and headgroups of the bilayer can regulate proteolysis of the intramembrane protease. gS has very low activity when embedded in fatty acyl chain lengths below 14 carbons then activity rises in a bell-shaped form and decreases again at 22 carbons. It has also been proposed that the Aβ42:Aβ40 ratio decreases as the FA chain length increases. Thicker hydrophobic membranes, together with a reduced fluidity induced by modification of the head groups, retain longer Aβ species and allow subsequent gS cleavage to shorter isoforms. In the same study, Holmes et al. found that the double bond isomer in a phospholipid fatty acyl chain also influences the gS activity modifying the Aβ42:Aβ40 ratios. Similarly, the presence of cholesterol in a membrane leads to a considerable increase in gS activity that decreased at higher concentrations producing a bell-shaped form in the gS activity.132
Computational modeling approaches have made an effort to characterize changes in the gS conformational states while varying the bilayer lipid composition.69,133 Aguayo et al. found that bilayer lipid composition has a large impact on the gS structural ensemble and proposed that lateral pressure across the bilayer and the protein-bilayer hydrophobic mismatch regulate the proteolytic enzyme.133 Higher transmembrane lateral pressures restrain the gS dynamics and favor active state conformations of the PS1 catalytic subunit, which may explain experimentally observed differences in gS activity when varying, for example, cis/trans unsaturations of lipids.133 Thinner bilayers reduce the distances between the PS1 catalytic ASP residues in the active site.
Variations in lipid headgroups can impact the mobility of the enzyme. In particular, a reduced flexibility is observed in transmembrane helix 2 related to substrate entry. Inactivation of gS resulting from the presence of charged lipids has been observed. This may be due to high PS1 structural restriction caused by bilayer rigidity that disturbs substrate recruitment and entry into the gS active site, as proposed by Holmes et al. Alternatively, it has been proposed that charged lipids interacting with the catalytic aspartates may lead to inactivation of the enzyme.134,135
Since C99 cleavage by gS most commonly occurs when the enzyme and its substrates are colocalized in cholesterol-rich lipid raft domains, Aguayo et al. studied the structural properties of gS in the presence of cholesterol rich bilayers and a mixture of lipids mimicking a lipid raft.133 They found that higher cholesterol concentrations lead to increased lateral pressures favoring active enzyme conformations. Interestingly, gS in the presence of lipid mixtures does not show high lateral pressures but does favor dynamic structural transitions between active and inactive states of the GS complex. These dynamics were also observed in more compact NCT conformations. In that case, lipid headgroups interact with and retain the NCT extracellular domain,133 which is folded over the gS active site. This allows for the steric sorting of gS substrates, similar to what has been observed in other studies. Of note, the omega-3 and omega-6 polyunsaturated fatty acids are two components of brain cell membranes, with omega-3 slowing the progression of AD and omega-6 increasing the AD risk,137,138 but their impact on Aβ biogenesis and the conformational ensemble remains poorly understood.
2.2. Domain Organization, Isoforms of Tau, and Minimal Sequence for Tau Aggregation and Toxicity
The tau protein forms paired helical filaments (PHFs) in neurofibrillary tangles central to the development of AD and other tauopathies. Tau is a microtubule-associated protein that plays an important role in axonal stabilization, neuronal development, and neuronal polarity. Full-length tau (htau40) includes a projection domain, a microtubule-binding domain (MBD) of four imperfect sequence repeats (R1 to R4), and a C-terminal domain (Figure 3A). The projection domain protrudes from the microtubule surface and it contains an N-terminal region (with two N-terminal inserts 2N) and a proline-rich region. The MBD has high affinity for microtubules. All 4 repeats end with a PGGG sequence, and there are two hexapeptide regions (PHF6*: 275VQIINK280 and PHF6: 306VQIVYK311) located in repeats 2 (R2) and 3 (R3). The MBD and the proline-rich regions are both positively charged. The N-terminal part and a short region at the C-terminus are acidic. Alternative splicing leads to the generation of six major isoforms of tau in the human brain: htau40-(2N4R), htau32(1N4R), htau24(0N4R), htau39(2N3R), htau37(1N3R), and htau23(0N3R), ranging from 352 to 441 amino acid residues in length (Figure 3B). The MBD itself reproduces much of the aggregation behavior of tau in cells and animal models. Therefore, the peptides only encompassing the repeat region including K18 (4R) and K19 (3R) were often used to study tau aggregation to provide important insights into the amyloidogenesis of tau. The K18 and K19 are prone to aggregation since they do not contain the flanking regions that inhibit amyloidogenesis and they correspond to an amyloid core of PHFs.141
Figure 3.
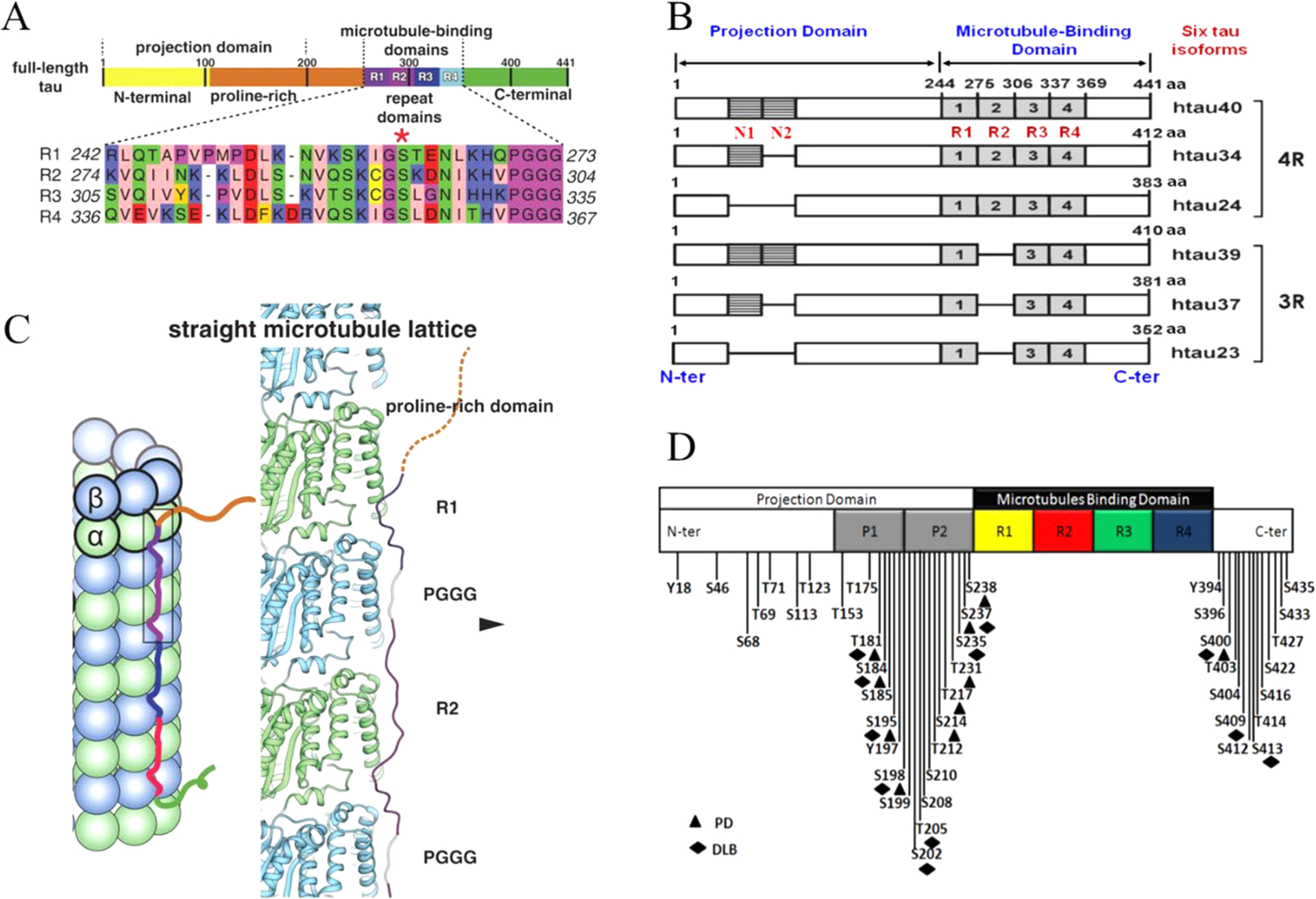
Domain organization, isoforms of tau, and binding to microtubules. (A) Schematic of tau domain architecture and assigned functions. The MT-binding domain of four repeats is defined as residues 242 to 367. The inset shows the sequence alignment of the four repeat sequences, R1 to R4, that make up the repeat domain. Ser262 is marked by the asterisk.142 (B) Schematic representation of the six human tau isoforms and two tau constructs. Tau isoforms differ by the absence or presence of one or two 29-amino acid inserts in the amino-terminal part, in combination with either three (R1, R3, and R4) or four (R1–R4) repeat regions (black boxes) in the carboxy-terminal part. Isoform sizes range from 352 amino acids (aa) to 441 aa. (C) Model of full-length tau binding to microtubules and tubulin oligomers.142 (D) Schematic representation of the largest isoform of tau with specific phosphorylation sites. Serine, threonine, and tyrosine residues that can be phosphorylated in AD, PD, and dementia with Lewy bodies (DLB) are indicated.
A recent cryo-EM study revealed different tau constructs on microtubules, and complemental atomic models of tau–tubulin interactions were generated by Rosetta modeling (Figure 3C).142 The conserved tubulin-binding repeats within tau adopt similar extended structures along the crest of the protofilament, spanning both intra- and interdimer interfaces, centered on α-tubulin and connecting three tubulin monomers. The cryo-EM structures suggest that all four tau repeats are likely to associate with the MT surface in tandem, through adjacent tubulin subunits along a PF. This modular structure explains how alternatively spliced variants can have essentially identical interactions with tubulin but different affinities according to the number of repeats present.142
Besides the six tau isomers, other tau fragments were also studied for their roles in tau aggregation. There is considerable interest in discovering the minimal sequence and active conformational nucleus that defines tau aggregation events. Truncation of tau may play a causative role in tauopathies. The long-running research of truncated tau has led to the generation of the first active tau vaccine that has entered clinical trials.143 Studies have shown that proteolytic fragments of tau can drive neurodegeneration in a fragment-dependent manner. Proteolytic fragments of tau have been found in the cerebrospinal fluid and plasma of patients with different tauopathies, providing an opportunity to develop these fragments as novel disease progression biomarkers.144 For example, tau (297–391) forms filaments that structurally mimic the core of paired helical filaments in AD brain.145 A fragment from the proteolytically stable core of the PHF, tau 297–391 known as “dGAE”, spontaneously forms cross-β-containing PHFs and straight filaments under physiological conditions. The comparison of the structures of the filaments formed by dGAE in vitro with those deposited in the brains of individuals diagnosed with AD found that they share a similar macromolecular structure.145 Cleavage of tau by legumain (LGMN) has been proposed to be crucial for aggregation of tau into fibrils.136 Using an in vitro enzymatic assay and nontargeted mass spectrometry, four putative LGMN cleavage sites were identified at tau residues N167-, N255-, N296-, and N368. Cleavage at N368 generates variously sized N368-tau fragments that are aggregation prone in the Thioflavin T assay in vitro. Both N368-cleaved tau and uncleaved tau were significantly increased in AD because of the accumulation of pathological tau inclusions. However, most of N368-cleaved tau remains largely soluble and is present only in low proportion in tau insoluble aggregates compared to uncleaved tau. This suggests that LGMN-cleaved tau has a limited role in the progressive accumulation of tau inclusions in AD.146
It is well-known that two hexapeptide regions, PHF6* and PHF6, located in R2 and R3 are top amyloidogenic motifs. The AcPHF6 may promote Aβ fibrillogenesis.147 However, in the longer tau sequence, tau local structure shields the PHF6 motif.148 When Aβ acts as tau aggregation seeds, Aβ fibril can promote the exposure of these hexapeptide regions.149 However, the PHF6 peptide lacks the ability to seed aggregation of tau244–372 in cells.150 But as the hexapeptide is gradually extended to 31 residues, the peptides aggregate more slowly and gain potent activity to induce aggregation of tau244–372 in cells.150 Further characterizations narrow down the β-forming region to a 25-residue sequence, indicating that the nucleus for self-propagating aggregation of tau244–372 in cells is packaged in a remarkably small peptide. Disease-associated mutations, isomerization of a critical proline, or alternative splicing are all sufficient to destabilize this local structure and trigger spontaneous aggregation.148 Numerous MD simulations have been conducted to study the PHP6 conformation and aggregation. In a recent MD and Markov state model (MSM) study,151 PHF6 can spontaneously aggregate to form multimers enriched with β-sheet structure, and the β-sheets in multimers prefer to exist in a parallel way. The residues Ile308, Val309, and Tyr310 play an essential role in the dimerization. MSM analysis shows that the formation of dimer mainly occurs in three steps. The separated monomers collide with each other at random orientations, and then a dimer with short β-sheet structure at the N-terminal forms; finally, β-sheets elongate to form an extended parallel β-sheet.151
Many studies have targeted these hexapeptide regions to prevent tau aggregation and toxicity. Using the model peptide, Ac-PHF6-NH2, the substitution of its amino acids with proline is shown to reduce self-assembly. Two of these modified inhibitors also disassemble preformed Ac-PHF6-NH2 fibrils, and one inhibits induced cytotoxicity of the fibrils.152 Inhibitors based on the peptide SVQIVY, shifted by −1 residue compared with PHF6 (VQIVYK), can also block proteopathic seeding by patient-derived fibrils.153 It was suggested that inhibitors based on the structure of the PHF6 segment only partially inhibit full-length tau aggregation and are ineffective at inhibiting seeding by full-length fibrils and that the PHF6* segment is the more powerful driver of tau aggregation.154 The PHF6* based-inhibitors not only inhibit tau aggregation but also inhibit the ability of exogenous full-length tau fibrils to seed intracellular tau in HEK293 biosensor cells into amyloid.154
Tau constructs can self-aggregate to PHFs directly; however, hyperphosphorylated tau appears to aggregate more readily and may sequester normal tau at lower concentrations.155,156 The regulation of tau primarily involves post-translational modifications (PTMs) including phosphorylation, truncation, and acetylation. The most common tau PTM is phosphorylation. In the AD brain, tau is excessively phosphorylated, at least ~3-fold over normal brain, leading to the disruption of the MTs and the promotion of filament formation.157 Other PTMs can also regulate tau aggregation; for example, tau truncation may take place after tau hyperphosphorylation with subsequent glycation.158 Methylation has been shown to suppress tau aggregation propensity whereas glycation and acetylation promote pathological tau aggregation.159,160 The distribution of phosphorylation sites along the sequence of full-length tau is uneven. Htau40 has 80 serine/threonine and 5 tyrosine residues that can be phosphorylated. Most of them are in either the N- or C-terminal regions. Mass spectrometry identified around 36 sites in purified PHF-tau from human Alzheimer brain.161 Figure 3D illustrates the known phosphorylation sites in tau.
Cryo-EM structures of tau fibers in four distinct diseases, AD,140 Pick’s disease, chronic traumatic encephalopathy, and Corticobasal degeneration (CBD) have different conformations.41,162–164 These structures are discussed in section 3. The tau protein and its fragments have an intrinsic ability to assemble into amyloid structures of large dimensions in the absence of aggregation-enhancing species, such as heparin or alternative polyanions. Luo et al. have shown that heating K18 to a high temperature leads to tau aggregation, but it disassociates to monomeric state reversibly when cooling down.165 Their replica exchange molecular dynamics (REMD) simulations predicted that tau proteins could form amyloid fibrils at a high temperature of 343 K, and the tau amyloid fibrils may cold dissociate at 275 K. This intriguing feature was then confirmed by fluorescence experiments, and it was observed that K18 fibrils cold dissociate when cooled. They also found that heparin locks the tau fibril and prevents its reversion. Adamcik et al. demonstrated that in the absence of heparin, the tau306–327 R3 fragment is able to self-assemble into large, flat, multistranded ribbons consisting of up to 45 laterally assembled protofilaments.166
Besides heparin, other polyanions have also been employed to study the structural consequences; aggregation through interaction with a physiologically relevant aggregation inducer is important. The formation of AD filaments is routinely modeled in vitro by mixing tau with heparin. Heparin promotes tau aggregation and recently has been shown to be involved in the cellular uptake of tau aggregates.167 Polyphosphate initiates tau aggregation through intra- and intermolecular scaffolding, most notably breaking long-range interactions between the termini.168 Different from heparin, polyglutamic acid does not immediately convert tau into oligomers.169 Tau is predominantly monomeric in the presence of polyglutamic acid at low temperature and only aggregates into oligomers and fibrils at higher temperature and longer incubation time. Utilizing this feature, through a combined NMR spectroscopy and molecular ensemble calculation method, Akoury et al. examined the conformational ensembles of K18 in the presence and absence of the polyglutamic acid and found that binding of polyglutamic acid to tau remodels the conformational ensemble of tau.170
Using a heparin-immobilized chip, surface plasmon resonance (SPR) revealed that tau K18 and K19 bind heparin with a Kd of 0.2 and 70 μM, respectively. In SPR competition experiments, N-desulfation and 2-O-desulfation had no effect on heparin binding to K18, whereas 6-O-desulfation severely reduced binding, suggesting a critical role for 6-O-sulfation in the tau-heparin interaction. The tau-heparin interaction became stronger with longer-chain heparin oligosaccharides. As expected for an electrostatics-driven interaction, a moderate amount of salt (0.3 M NaCl) abolished binding.171 However, it was found that heparin-induced tau filaments are structurally heterogeneous and differ from AD filaments,172 as discussed in section 3.
The heparin induced tau structures illustrate the structural versatility of amyloid filaments and raise questions about the relevance of in vitro assembly.173 Our understanding of this question is that polyanion induced tau structure and those found in patients’ brain so far present favorable states in the complex amyloid formation energy landscape. As the structure of Aβ fibril, the tau fibril may exist in different forms from different patients. This can be supported by the study of PHP6 based inhibitors. Donors with progressive supranuclear palsy exhibited more variation in inhibitor sensitivity, suggesting that fibrils from these donors were more polymorphic and potentially vary within individual donor brains.153 Exploring the interplay between fibrillization and amorphous aggregation channels on the energy landscapes of 3R and 4R tau provided a global view of polymorphic tau aggregates.174 A coarse-grained protein force field was used to study the energy landscapes of nucleation of the 3R and 4R fibrils derived from patients with Pick’s and Alzheimer’s diseases. The landscapes for nucleating both fibril types contain amorphous oligomers leading to branched structures as well as prefibrillar oligomers. These two classes of oligomers differ in their structural details: The prefibrillar oligomers have more parallel in-register β-strands, which ultimately lead to amyloid fibrils, while the amorphous oligomers are characterized by a near random β-strand stacking, leading to a distinct amorphous phase.174 The feature of the energy landscape connecting the tau oligomers and fibrils was reflected in the sm-FRET studies of oligomer diversity during aggregation of K18, although also in the presence of heparin.175 Kjaergaard et al. observed that the shortest growing filaments only represent a small population of transient oligomers with cross-β structure, while the two largest oligomer populations are structurally distinct from fibrils and are both kinetically and thermodynamically unstable. The first electrostatic driven population is in rapid exchange with monomers; the second kinetically more stable one is probably off-pathway to fibril formation.175
In vitro, 0N4R tau fibrils contain a monomorphic β-sheet core enclosed by dynamically heterogeneous fuzzy coat segments.176 A variety of experiments indicate that 0N4R tau fibrils exhibit heterogeneous dynamics. Outside the rigid R2-R3 core, the R1 and R4 repeats are semirigid even though they exhibit β-strand character and the proline-rich domains undergo large-amplitude anisotropic motions, whereas the two termini are nearly isotropically flexible. It has been suggested that the N- and C-termini differentially associate with PHFs177 and play distinct roles in the stability and consequently neurotoxicity of tau filaments.178 The N-terminal fragment (residues 1–15) did not affect tau polymerization179 whereas a fragment consisting of residues 1–196 could inhibit polymerization of full-length tau.180 The tau C-terminus is crucial in the formation of tau PHFs,181 and it also modulates the cross-seeding barrier between 4R and 3R tau.182 Xu et al. performed multiscale MD simulations to study the structure and dynamics of full-length tau filaments, especially the effects of the flanking regions on the stability of the filament core.183 They found that full-length tau filaments consist of one dense core and two sparse layers, consistent with the structural model derived from the experimental observations. The relative stability of the filaments not only depends on the core morphology but can also shift by interactions among different domains.
2.3. α-Synuclein and IAPP Domains and Aggregation Properties
αS is a 14 kDa neuronal protein that is predominantly localized at the presynaptic termini.184 In its physiological form, αS is monomeric and disordered,185 although some studies have generated a debate186 on whether it adopts a helical tetramer in vivo.187 The aggregation of αS is inherently connected with PD, as its aggregates are major components of intracellular inclusions known as Lewy bodies forming in dopaminergic neurons of PD patients.188 There are also links between the αS-encoding gene and familial forms of PD, with mutations, duplications, and triplications being found in patients affected by early onset forms of PD.189 Fibrillar aggregates of the relevant aggregation-prone region of αS, the nonamyloid-β component (NAC), are also found in AD patients190 and in the context of other neurodegenerative disorders, including dementia with Lewy bodies,191 multiple system atrophy,192 and other synucleinopathies.193
While the pathological relevance of αS is generally acknowledged, its function remains unclear.194 The abundance of αS at the synaptic termini has suggested that it may be involved in neuronal processes and studies have indicated possible roles in synaptic plasticity195 and learning.196 A number of pieces of evidence have been collected on a possible function of αS in the trafficking of synaptic vesicles (SVs) during neurotransmitter release.197,198 αS binds SVs in vitro and colocalizes with SVs in synaptosomes in a calcium responsive manner.199 Key evidence indicates that αS has a role of chaperone for the assembly of the SNARE complex;200 the machinery promoting the fusion of SVs with the plasma membrane. αS was indeed shown to rescue the formation of the SNARE complex in knockout mice lacking CSPα,201 whereas knockout mice lacking the three synucleins (α-, β-, and γ-) showed neuropathological phenotypes that are indicative of impaired SNARE activity.202 The interaction with SVs by αS has also been implicated in the promotion of SV-clustering,203,204 a key process in the maintenance of pools regulating SV homeostasis during neurotransmitter release.198 In addition to its interaction with SVs, αS has also been implicated in the regulation of the vesicle trafficking from the endoplasmic reticulum (ER) to the Golgi205 and the mitigation of oxidative stress in mitochondria.206
The domain organization of αS is defined on the basis of its sequence properties and in relation to the biological context (Figure 4). The major biological form of αS in vivo features the binding with cellular membranes.207 Membrane interactions by αS are promoted by a lipophilic domain (residues 1 to 90) and featuring 7 imperfect sequence repeats. These 11 residue repeats induce binding via a disorder-to-order transition into amphipathic class A2 α-helical segments that promote the membrane binding.208 This domain also has genetic links with inherited forms of early onset PD, as it hosts all the PD-related αS mutations (A30P, E46K, H50Q, G51D, and A53T).189
Figure 4.
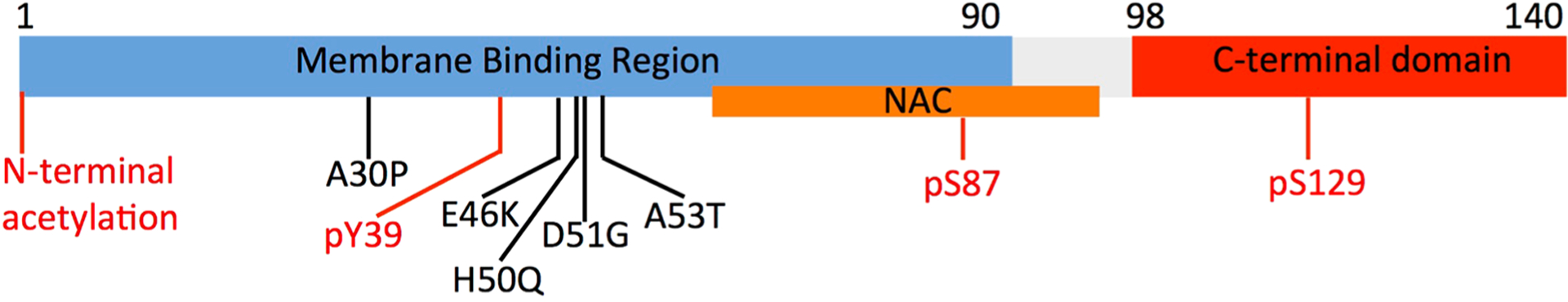
Key αS domains having a role in functional and pathological contexts. The membrane-binding domain (residues 1 to 90), the NAC region (residues 61 to 95), and the C-terminal domain (residues 98 to 140) are shown in blue, orange, and red, respectively. The diagram also shows the main pathological mutations (black) and key post-translational modifications (red) such as the N-terminal acetylation and phosphorylation of residues Ser87, Ser129, and Tyr39.
Another domain of αS, the NAC (residues 61–95), has relevance in the context of self-assembly and aggregation. NAC is essential for the kinetics of αS aggregation209 and is the main component of the core of αS fibrils. Fibrils of the NAC region have been isolated in the context of PD as well as in other neurodegenerative diseases.190 Finally, the negatively charged C-terminal domain of αS spanning residues 99 to 140 (net charge of −9) is implicated in calcium binding199 and in protein–protein interactions at the surface of synaptic vesicles.197,200
IAPP or amylin is a 37-residue hormone produced by pancreatic β-cells to regulate the response to high glucose levels in the blood in conjunction with insulin, which is cosecreted.210 The biogenesis of amylin requires the production of the proIAPP, a peptide composed of 67 residues whose cleavage generates IAPP.211 In addition to his role as a hormone, IAPP is well-known for its connection with T2D. Indeed, fibrillar aggregates of IAPP are the major constituents of deposits in pancreatic islets that are found in the majority of patients suffering from this condition. Despite the fact that the toxicity of IAPP amyloids has been shown in vitro,212,213 it remains to be established whether its aggregation is a causative factor or a downstream effect of TD2. Indeed, in contrast to Aβ protein where many familial mutations exist, there is only one unique disease-causing mutation in IAPP (S20G), and in the majority of the cases, no specific alterations or mutations of IAPP are found in diabetic individuals, suggesting that other factors, such as the failure of control mechanisms to prevent protein misfolding, may be involved. Islet amyloids are de facto found also in small populations of nondiabetic elderly individuals,214 likely owing to a reduced efficiency in the mechanisms of clearance of protein aggregation. In vitro IAPP has been shown to aggregate in a concentration dependent manner, and it rapidly self-assembles into amyloids when concentrated in the millimolar range, a condition that by contrast is well managed in the secretory granules of β-cells of healthy individuals.
Among the factors that inhibit the aggregation of IAPP in β-cells are the acidic pH (~5.0), shown to be largely unfavorable to the misfolding of IAPP in vitro,215 and the presence of high levels of Zn(II) in β-cells. The binding to Zn(II) indeed promotes conformations of the otherwise intrinsically disordered IAPP that are aggregation resistant, presumably by shielding the two amyloidogenic sequences of IAPP. Indeed, two regions have been found to be crucial for the fibrillization of IAPP. These are included in the N- and C-strands forming the fibrillar core of IAPP amyloids and spanning respectively residues 8 to 17 and 28 to 37. The two strands form a single stack in the cross-β arrangement, generating two facing β-sheets stabilized by a steric-zipper dry interface.216
3. STRUCTURES OF Aβ, TAU, α-SYNUCLEIN, IAPP OLIGOMERS, AND FIBRILS FROM EXPERIMENTS
3.1. Experimental Techniques for Fibrils
Amyloids are proteineous deposits associated with peculiar physical and chemical features: (i) they exhibit a very low degree of solubility, (ii) they form nanoobjects of various ultrastructural shapes and multiple sizes ranging from a few nanometers to microns, and (ii) they can represent objects of heterogeneous morphologies at the macroscopic scale (e.g., aggregates, fibrils, or oligomers) and heterogeneous conformation or polymorphic nature at the atomic level.217 Additionally, amyloid deposits tend to be difficult to isolate, biochemically handle, and purify due to their insolubility. Their biochemical features constitute major hurdles for biophysical approaches and structural biology toward determining atomic resolution structural models. The development of innovative methods in structural biology during the past two decades has been very fruitful, and more than 100 structures of protein and peptide fibrils (~40 from full length constructs) have been deposited at the PDB218 using different techniques such as X-ray diffraction, NMR, and EM.210 3D model determination of amyloid architecture relies on a multistep process to elucidate structural features at various levels:
3.1.1. Length of the Amyloid Core.
The amino acid sequence of the amyloid core usually covers only a fraction of the full-length protein; for example, the amyloid core of α-synuclein does not comprise its C-terminal domain.219 A simple biochemical maneuver to delineate the length of the amyloid core is the enzymatic digestion of protein segments that are not involved in the core region. H/D exchange performed by mass spectrometry220 or solution NMR221 is an experimental approach that is commonly used to delineate the amyloid core at a residue-specific resolution.
3.1.2. Secondary Structure.
Amyloid deposits are rich in β-sheet secondary structure; this information can be rapidly extracted with CD and FTIR. A key step toward determining the amyloid structure is the identification of the number and delineations of the β-sheets as well as the localization of β-sheet breakers or turns. This can be achieved by high-resolution techniques, i.e. ssNMR and cryo-EM. Using solid-state, the secondary structure can be predicted from chemical shifts using computational routines such as TALOS222 or by the secondary chemical shifts.223 Because the chemical exchange of amide hydrogens with the buffer is remarkably slowed down if the involved residue is comprised in the amyloid core, H/D exchange techniques offer a powerful approach to distinguish amino-acids involved in the hydrophobic amyloid core and thus identify β-sheet positioning. Recent advances in cryo-EM methodology, including software developments and new electron detectors, have facilitated structure determination of amyloid fibrils, backbone conformation, and secondary structure elements.224 They can be recognized after single particle EM analysis, recently demonstrated for αS225 and Aβ.226
3.1.3. Three-Dimensional Fold and Intermolecular Packing.
Although most amyloids share the generic cross-β architecture, the number of β-sheet elements and their placement relative to each other can be very diverse. Several generic architectures have been proposed, such as the β-helix, β-sandwich, superpleated β-sheet structure, or β-roll. The supramolecular arrangement of amyloid fibrils is often regular in a protofilament and has been observed as β-sheets that run antiparallel or parallel in-register along the fibril axis. The β-solenoid fold was the first experimentally determined amyloid fold at high resolution, from the amyloid prion HET-s by ssNMR.39 This architecture is characterized by specific amino acid sequence patterns composed of hydrophobic residues pointing inside the core that sequentially alternate with polar residues pointing outside. Although originally associated with functional amyloids, this fold has been recently seen in tau fibrils by cryo-EM,40 suggesting this fold to be generic in the context of pathological and functional amyloids.
The cross-β nature of the sample is essentially characterized by X-ray diffraction,227 and additional information relative to high-order symmetry can be derived from scanning transmission EM228 and tilted-beam transmission EM.229 These two approaches provide the so-called mass-per-length measurement, a crucial structural parameter to determine how many protein monomers are stacked per fibril layer. 3D structure determination of amyloids using ssNMR relies on the collection of internuclear distance restraints, typically in the range of 2–8 Å.230 Distinction between intramolecular proximities (i.e., two nuclei in two β-sheets within the same molecule) and intermolecular proximities (i.e., two nuclei in two β-sheets in adjacent molecules) requires the use of strategic isotope labeling231 and is still a major bottleneck for ssNMR-based structure determination. In analogy to distance measurements by ssNMR, double electron–electron resonance spectroscopy (DEER) and continuous-wave electron paramagnetic resonance (EPR) are powerful approaches to provide residue–residue structural restraints in a distance range not accessible to ssNMR > 10 Å.232
In the case of short amyloid-forming peptides that crystallize, conformational studies at atomic resolution can be performed by X-ray diffraction analysis42 and microelectron diffraction.233 Numerous fibril-forming peptides have been crystallized to uncover numerous intermolecular stacking arrangements. Notably, these studies have highlighted the role of steric zipper motifs that consist of complementary side chains interdigitation in a dry interface, resulting in stable packing of high density.
Cryo-EM has opened an avenue to obtain both intramolecular and intermolecular arrangement of amyloid fibrils at atomic resolution.224 A major advantage of cryo-EM is the ability to obtain high-resolution maps from patient-derived samples, reducing artifacts associated with in vitro aggregation of recombinant proteins. Amyloid fibrils are usually observed as bundles of individual filaments termed protofilaments. Ultrastructural analysis of such bundles has revealed the presence of particular high order architectures, such as twisted or straight morphologies. AFM and EM are used to decipher the morphology of amyloid fibrils, but their resolution is limited. Cryo-EM has recently proven to be a unique technique to characterize the quaternary arrangement of protofilaments, as exemplified by the determination of the staggering of nonplanar β-strands in the case of Aβ42226 and tau.40
3.2. Diversity of Fibril Structures in Tau, α-Synuclein, Aβ, and IAPP
3.2.1. Tau Filaments.
Tau filaments were first observed in paired helical PHF234 and then a mixing of PHF and straight filaments (SF) by EM.235 Structure determination of tau filaments has been limited for a long time by the important length of the amyloid core and the particular nature of the filament that is composed of a rigid core and a fuzzy coat lacking structural order. Moreover, in vitro aggregation of recombinant tau requires the use of cofactors such as heparin, and these cofactors might modulate the details and the obtained polymorphism236 of the cryo-EM structures of tau in AD and Pick’s disease.
In AD, both structures of PHF and SF (Figures 5A and B) are composed of eight β-sheets in a C-shaped fold, although lateral protofilament contacts revealed a different intermolecular organization between the protofilaments, suggesting an important role of ultrastructural polymorphism in the context on in vivo tau aggregation. Filament cores are made of two identical protofilaments comprising residues 306–378 of tau protein, which adopt a combined cross-β/β-helix structure and define the seed for tau aggregation. Paired helical and straight filaments differ in their interprotofilament packing, showing that they are ultrastructural polymorphs.40
Figure 5.
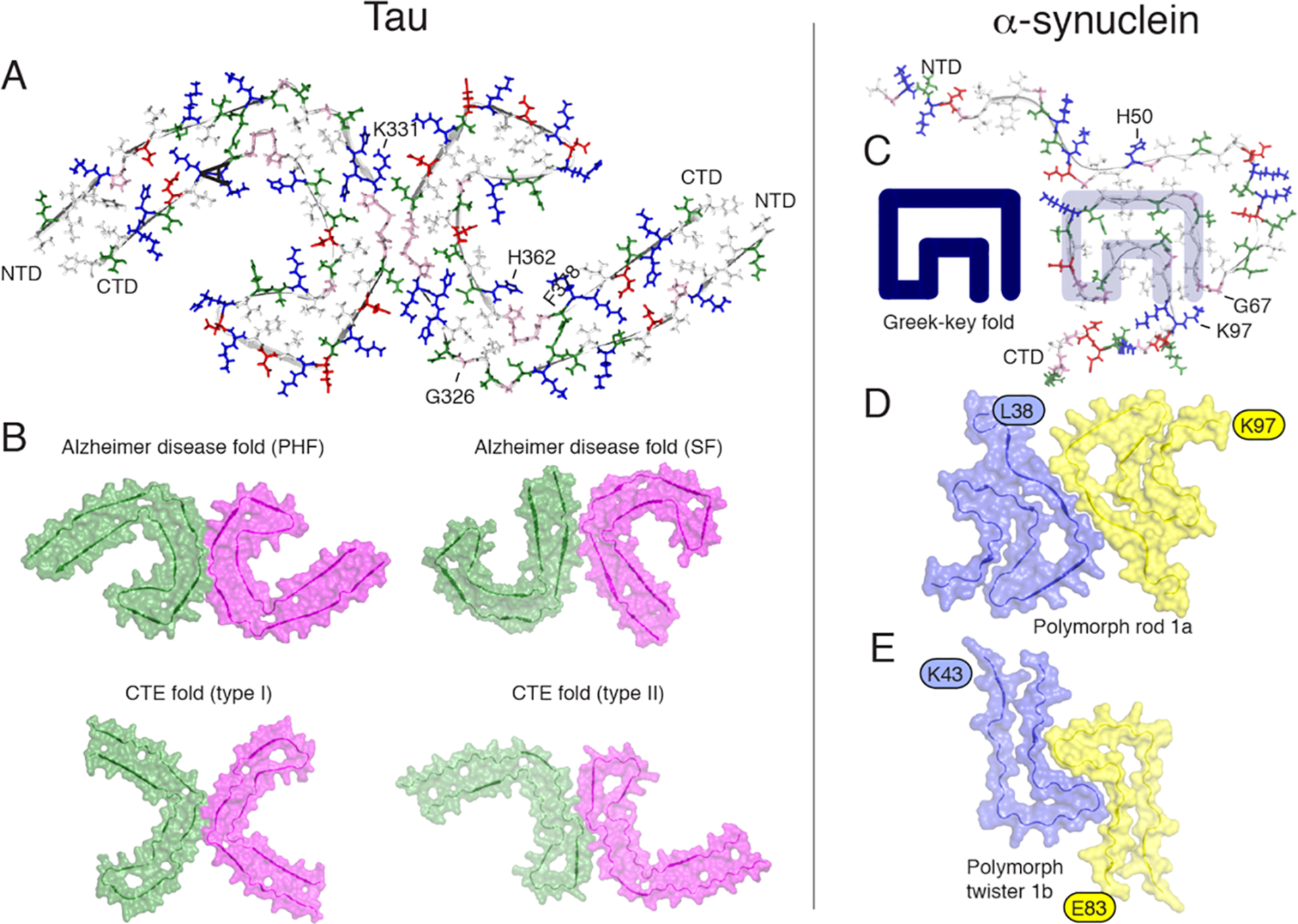
Orthogonal view of the three-dimensional structural models of tau filaments and α-synuclein amyloid fibrils. NTD and CTD are the N-terminal and C-terminal domains. (A) Solid-state tau paired PHF spanning residues 306–378 in the right protofilament. (B) Cryo-EM PHF and SF tau amyloid cores in AD. In chronic traumatic encephalopathy (CTE), tau filaments contain predominantly the type I (90%) and type II filaments. The interprotofilament interfaces are different compared to those in AD. (C) Amyloid core of human α-synuclein amyloid fibrils (PDB 2NOA), containing a Greek-key fold. (D and E) Amyloid core fibril structure of α-synuclein: polymorph rod 1a (PDB entry 6CU7) and polymorph twister 1b (PDB entry 6CU8). The authors prepared the figure with pymol.254
In Pick’s disease, the tau filament core comprises a distinct fold41 compared to the structures of AD’s PHF and SF. This suggests that a disease-specific amyloid fold might be the hallmark of clinical phenotypes, in analogy with structural strains observed in prion diseases.237 The structures of tau filaments from several brains of patients with chronic traumatic encephalopathy (CTE) confirmed this observation, revealing the presence of several structural polymorphs (Figure 5B) with a molecular fold and protofilament interfaces different to AD’s filaments.41 Additional density, not connected to tau, was observed in Pick’s disease tau filaments, suggesting a tight incorporation of biological cofactors into the structure. Structures of filaments from Pick’s disease consist of residues Lys254–Phe378 of 3R tau, explaining the selective incorporation of 3R tau in Pick bodies and the differences in phosphorylation relative to the tau filaments of AD. Interestingly, novel tau filament fold in chronic traumatic encephalopathy encloses hydrophobic molecules. Importantly, residues K274–R379 of 3R tau and S305–R379 of 4R tau form the ordered core of two identical C-shaped protofilaments, indicating common driving forces do exist in tau aggregation. The core of a CBD filament comprises residues lysine 274 to glutamate 380 of tau, spanning the last residue of the R1 repeat, the whole of the R2, R3, and R4 repeats, and 12 amino acids after R4.163 The core adopts a previously unseen four-layered fold, which encloses a large nonproteinaceous density. This density is surrounded by the side chains of lysine residues 290 and 294 from R2 and lysine 370 from the sequence after R4.
Finally, heparin-induced filaments of 2N4R tau have at least four different conformations. Cryo-EM structures of three of these conformations reveal a common, kinked hairpin fold, with differences in kink, helical twist, and offset distance of the ordered core from the helical axis. 2N3R tau filaments are structurally homogeneous and adopt a dimeric core, where the third repeats of two tau molecules pack in a parallel manner.173
3.2.2. α-Synuclein Fibrils.
Unlike tau, recombinant expression of αS led to the preparation of homogeneous fibrils amenable to structure determination. A first 3D structure was proposed by ssNMR,238 revealing a Greek-key topology (Figure 5C). This structure, later classified as the polymorph “1c”,225 contains a parallel in-register arrangement with hydrophobic side chain packing and a steric zipper. Cryo-EM studies have uncovered the high-resolution structures of various polymorphs, including the familial PD mutant H50Q.239 The interprotofibril arrangement is characterized by the presence of staggered β-strands, and familial mutations H50, G51, and A53 are localized at the protofilament interface and participate to its stability. Several wild type polymorphs have been solved (Figures 5D and E), revealing different protofilament interfaces. The familial mutations are localized at crucial positions that stabilize either the intramolecular fold or the inter protofilament interface.
The atomic structures of αS fibrils extracted from brains of individuals with multiple system atrophy (MSA) have been determined by cryo-EM.240 Two types of filaments were identified (named type I and II) each composed of two protofilaments. An astonishing feature of type I and type II αS filaments is the asymmetry of their protofilaments, leading to a different solvent exposure of the critical residues (e.g., K60). Post-translational modifications of only one protofilament have been proposed to explain the different conformations of the two protofilaments in the same fibril.240 Structures of brain-derived α-synuclein fibrils are distinct from fibrils obtained by recombinant expression and in vitro aggregation, as already observed for tau filaments.
3.2.3. Aβ40/42 Fibrils.
Because of its small size (compared to tau or α-synuclein proteins), its production by solid-phase peptide synthesis has offered a convenient way for numerous research groups to investigate its structure by biophysical techniques. To date, most methodological developments in structural biology of amyloid fibrils have been performed on Aβ peptides. Early studies using FTIR and CD have demonstrated the high propensity of the β-sheet structure of Aβ fragments.241 Pioneering work by the Tycko’s laboratory using solid-state242,243 determined the β-turn-β (U-shape) fold in an in-register, parallel arrangement for Aβ40 fibril with residues 12–24 and 30–40 in β-strand conformations based on distance restraints and torsion angle measurements.244
Several 3D models of AβA40 and Aβ42 (Figure 6A) have been proposed on the basis on ssNMR and cryo-EM,226,245–247 showing a diversity of intramolecular fold (U-shape or S-shape) and quaternary arrangement between Aβ40 and Aβ42. Small variations in the aggregation conditions can lead to distinct molecular conformation within the same peptide sequence and even different toxicity level.248 By combining cryo-EM and ssNMR, Schröder et al. presented a structure of Aβ42 fibrils assembled at low pH which is to date the highest resolution model of Aβ peptides.226 As already observed for other pathogenic amyloids, the staggering of nonplanar molecules (Figure 6B) constitutes a unique feature that has profound implications for fibril growth mechanisms, because the binding sites of the two fibril ends are different, implying a polarity of subunit growth. To date no high resolution of Aβ40 and Aβ42 fibrils extracted from brains has been solved. Tycko et al. have reported conformational studies at high resolution of Aβ40 and Aβ42 fibrils seeded from brain extracts.249,250 Studies of these human brain-derived fibrillar assemblies showed distinct molecular conformation by solid-state from each patient, suggesting the existence of structure-specific conformations depending on the patient AD clinical history. Comparison of Aβ40 and Aβ42 in this context revealed a predominant molecular conformation in Aβ40, while Aβ42 exhibited a larger degree of structural heterogeneity as revealed by ssNMR chemical shift analysis.250 It implies a greater structural susceptibility of Aβ42 brain seeds compared to those for Aβ40, as already suggested.251 As suggested for α-synuclein or tau, the existence of structure-based strains for Aβ40 and Aβ42, in analogy to conformational strains known for prion-like proteins, is still a matter of debate.
Figure 6.
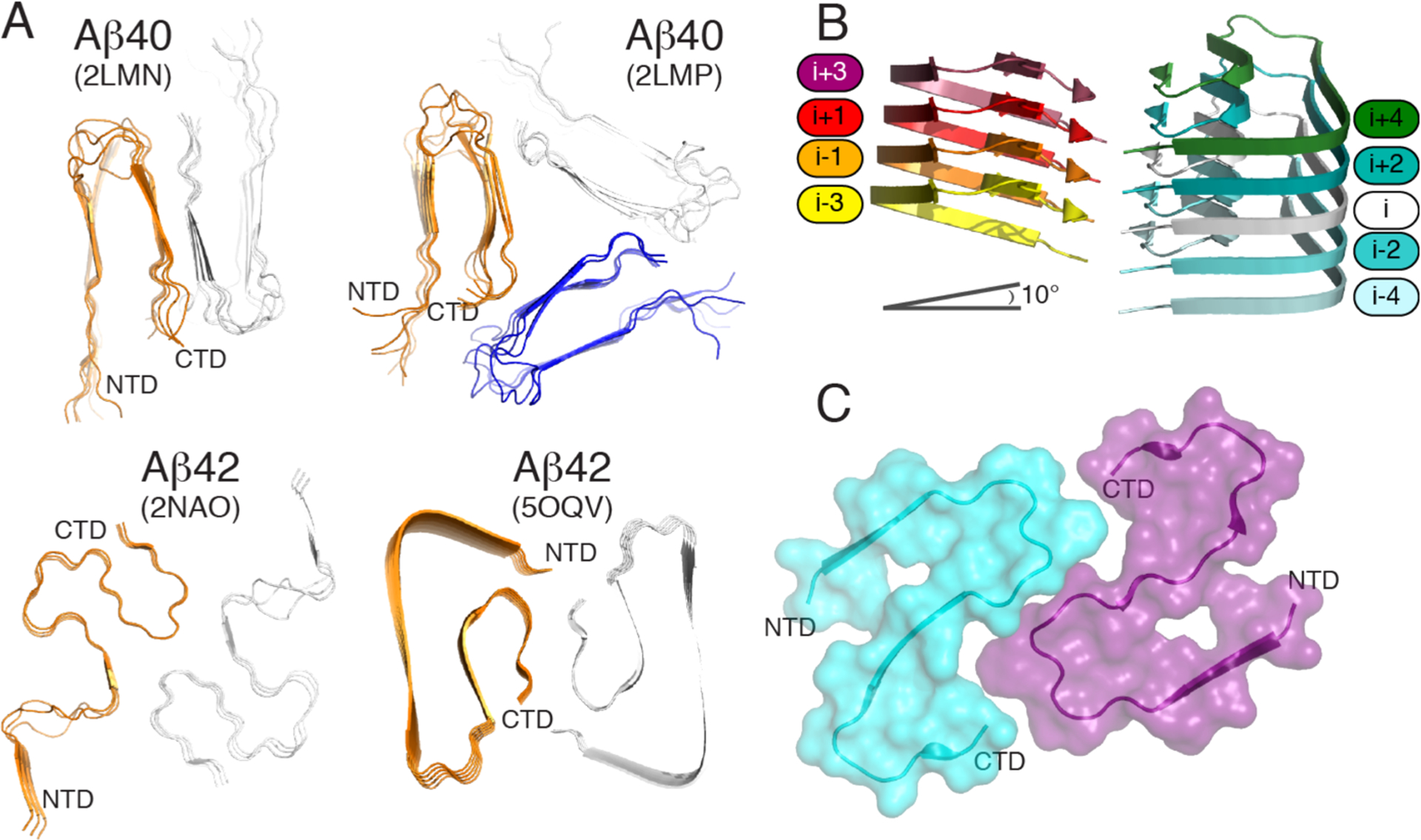
(A) Orthogonal view of Aβ40 structures (PDB entries 2LMN and 2LMP) spanning residues 9–40 and Aβ42 structures (PDB entries 2NAO and 5OQV) spanning residues 1–42. (B) Cryo-EM structure of Aβ42 showing the staggered arrangement of nonplanar Aβ42 subunits. (C) Cryo-EM structure of IAPP grown at physiological pH (PDB 6Y1A) spanning residues 13–37. The authors designed the figure with pymol.254
3.2.4. IAPP Fibrils.
Previous studies based on ss NMR of IAPP place the majority of the 37 residues into the fibril core and a U-shape conformation, with the N-terminus being at the periphery, but all models display substantial deviation.216,252 A very recent study by cryo-EM at physiological pH253 provides three polymorphs with the dominant one comprising residues 13–37 with two S-shaped, intertwined protofilaments (Figure 6C). The high similarity between this model and the Aβ42 model from Gremer is striking considering the link between T2D and AD.
3.3. Structures of Transient Oligomers
3.3.1. Aβ and IAPP.
The fibrillation of Aβ is preceded by a transition from a random-coil like structure to helical conformation where the latter facilitates the formation of β-sheet structured amyloid filaments.255 The helical conformation of Aβ1–40 monomer determined by NMR in a water-detergent solution adopts a helical structure in the K16–V24 and K28–V36 regions.256 The bihelical structure is shown to be disrupted in the oxidized Aβ1–40 and adopts a single helical region between residues K16 and V24.257 Another NMR study trapped a low population of an early intermediate of Aβ1–40 characterized by a 310 α-helix (Figure 7) spanning the core hydrophobic regions H13 to D23 in an aqueous medium containing no detergent.258 The partially folded 310-helical structure has also been observed in computations259 highlighting the identification of a potential target species from an on-pathway aggregation to design small-molecule Aβ inhibitors. The phenolic inhibitor EGCG is used to study the ligand–Aβ interaction using the 310 α-helical structure and shows a tendency to bind the hydrophilic N-terminus and the α-helical (H13 to D23) region.260
Figure 7.
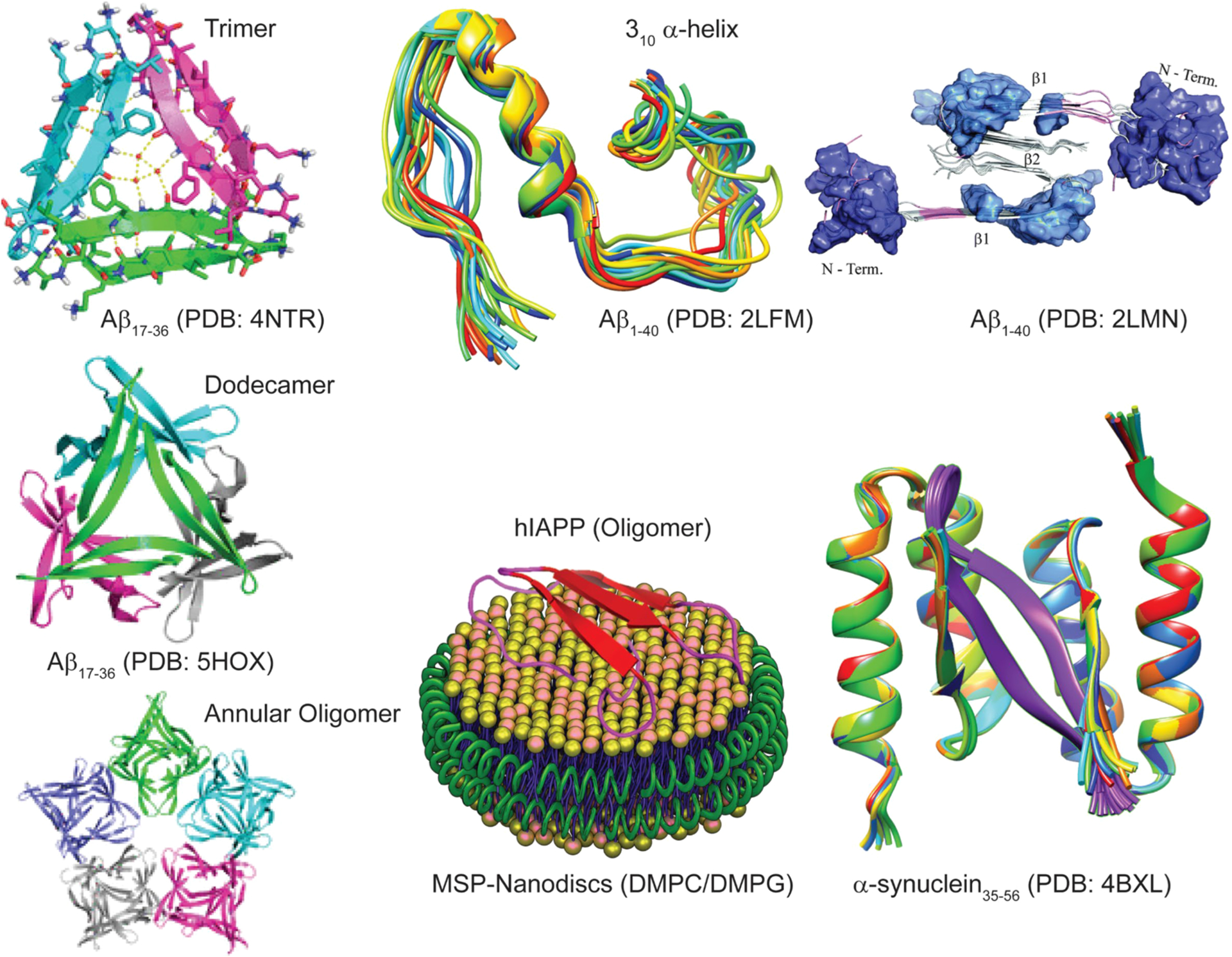
High-resolution structures of amyloid oligomers in solution or membrane-bound state solved using X-ray and NMR methods, with the exception being the model of Aβ40 assembly toxic surface (top, right).
Since high-resolution 3D model structure determination of pathologically relevant low- and high-ordered Aβ species is very challenging, a chemical cross-linking approach has been employed to homogenize the sample for structural determination. Macrocyclic β-sheet peptides that mimic β-hairpins of amyloid peptides and stabilize both low- and high-ordered oligomers have been developed for X-ray crystallography study.261 Using Aβ β-hairpin mimicking peptides,262 a twisted β-hairpin triangular Aβ trimer structure has been solved which also forms hexamers, dodecamers, and annular oligomers (Figure 7) through self-assembling that resembles annular structures reported for Aβ oligomers by EM.263,264 These Aβ oligomers present substantial neurotoxicity and thus are proposed to be useful molecular models of Aβ oligomer for structure-based inhibitor designing. Stable Aβ dimers and trimers characterized with parallel β-sheets and neurotoxicity have been developed by sequentially varying the position for cross-linking using chemical linkers or disulfide bridges.265–267 A combination of solution and ssNMR and AFM characterized highly disordered oligomers of Aβ that were formed in parallel to the fibril formation process.268 A recent study employed pressure-jump NMR to observe the oligomerization of Aβ.269 By combining solution NMR, DLS, EM, and wide-angle X-ray diffraction and cell viability, Melacini et al. provide an atomic resolution map of soluble Aβ toxic surfaces, with the exposure of a hydrophobic surface spanning residues 17–28 and the shielding of the N-terminus (Figure 7).270 While isolation and characterization of Aβ dimers, trimers, tetramers, etc. without chemical modification is challenging in experimental conditions, atomistic MD simulations complemented these limitations and provide high-resolution 3D structures. For example, simulations identified dimer, trimer, tetramer, and stable globular-like oligomers starting from disordered monomers with 13–23% of β-sheet content.271 While self-assembled Aβ oligomers are of interest, recent studies highlighted the formation of Aβ hetero-oligomers through cross seeding or binding to other proteins in the brain. Aβ1–40 and Aβ1–42 mixed oligomers are reported to have an intermediate structure between those of the self-assembled oligomers and proposed to have a large surface with antiparallel β-sheet structure.272 Aβ interaction with prion protein was reported to trap an antiparallel β-sheet oligomer but lacks a high-resolution structure; on the other hand, Aβ shows a random-coil structure when forming heterooligomers with apolipoprotein-E derived peptide fragments with enhanced neurotoxicity.273–275 Readers are referred to a review that discusses the high-resolution solution and ssNMR approaches to monitor the formation and characterization of Aβ oligomers.276
A number of biophysical techniques and different types of sample preparations have been used to investigate the aggregation mechanisms of IAPP. Helical intermediates of human-IAPP have been reported.277,278 NMR experiments have shown high-resolution structures for full-length human-IAPP,279 human-IAPP-1-19,280 and full-length rat-IAPP281 peptides both in solution as well as in a membrane environment. Investigation of the aggregation pathways revealed the formation intermediate species of IAPP,282–288 and Figure 7 shows a structure of membrane (nanodisc)-associated hIAPP oligomers. For additional details on the structure and kinetics of aggregation under various conditions, readers are referred to review articles on IAPP.289–291
3.3.2. α-Synuclein and Tau Proteins.
In PD, oligomerization of αS has been shown to increase in individuals with Lewy pathology. A low-resolution structural evolution of individual αS oligomers characterized by antiparallel β-sheet structure is detected using AFM-IR.292 Structural mapping of oligomers at all-stages of aggregation by AFM-IR highlighted the early oligomers (with 1 day incubation) are spherical in shape and dominated by a α-helix/random-coil secondary structure, but some of the oligomers showed mixed parallel and antiparallel β-sheet structures. The size of the spherical oligomers is observed to grow on day-2 with an increasing β-sheet and decreasing α-helix structures. Later on day-3, AFM-IR detected the presence of both spherical oligomers (α-helix/β-sheet rich structures) and protofibers with a predominant β-sheet structure.292 The structure of αS oligomers observed under cryo-EM is shown to have a cylinder-like appearance and be characterized with a predominant β-sheet structure (35 ± 5%).293 The formation of early aggregates of αS and interaction between monomers and stable bioengineered oligomers are studied using fluorescence spectroscopy by labeling αS species with differently labeled fluorophores.294 This study highlighted a comparatively higher binding affinity between oligomers as compared to oligomer–monomer/monomer–monomer interactions suggesting oligomeroligomer assembly is a major driving molecular process of aggregation in the early stages of the disease progression. Importantly, large oligomers are found to assemble to form aggregates sooner as compared to low-size oligomers (octamer > tetramer > dimer/monomer) but not competently assist nucleation and monomer recruitment.294 Another study showed αS oligomers do not seed the fibrillation reaction. This study underlined metastable spherical shape (10 nm), with a disordered conformation, for the αS oligomers by a small-angle neutron scattering method.295 Amyloid oligomers differ in their surface properties (in general hydrophobic residues are solvent exposed) which correlate to its competent membrane binding and toxicity. Hydrophobicity surface landscaping of individual αS oligomers by selective fluorescent molecules is probed using a super-resolution imaging technique.296 The hydrophobicity of the surface of the oligomers is found to be comparatively higher than that of aged fibers evidencing αS aggregation proceeds via generation of toxic intermediates characterized with hydrophobic heterogeneity like that which has been reported for amyloid proteins and peptides.296 A recent study showed that αS promotes formation of Aβ oligomers and inhibits fibrillation by stabilizing the oligomer structure.297 Notably, this effect is identified only by the soluble species of αS (monomers and oligomers) but not by fibers. EM studies revealed globular morphologies for the oligomer mixture of Aβ and αS. While structural information for the Aβ oligomers induced by αS has still not been reported, further investigation could provide more details to establish a correlation between AD and PD.297 The aggregation and folding of αS is shown to be modulated by the isomeric state of the five proline residues (P108, P117, P120, P128, and P138) located in the disordered C-terminus.298 Among the five-proline residues, isomerization of P128 is identified to be catalyzed by cyclophilin A (CypA), a protein belonging to the family of peptidylprolyl isomerases. CypA and αS are colocalized in cells, where CypA interacts with αS with a micromolar binding affinity.298 NMR titration experiments, combined with a crystal structure of the αS–CypA complex, showed CypA binds to the amyloid core preNAC hydrophobic domain (residues 47–56) of αS298 and also is known to bind membrane.299 The α-synuclein complexed by affibody displays a β-hairpin spanning residues 35–56 by NMR (Figure 7).261
Finally, we report on recent studies that provided structural and functional details for the tau oligomers. Antibody assay detected an increasing amount of soluble tau oligomers in the limbic regions of human brain with positive NFT, also referred to as Braak stages III and IV.300,301 Clusters of soluble tau multimers (globular) were observed in AFM with a measured height ranging from 10 to 30 nm.301 TEM imaging analysis reported a similar globular morphology of chemically stabilized tau oligomers prepared in vitro and detected by oligomer specific antibodies.302 Tau oligomer rich in β-sheet structure is shown to be influenced by both Aβ and αS oligomers.303 A recent NMR study reported tau monomers binding to the C-terminal disordered domain of αS monomer following the generation of toxic αS oligomers.304 Similarly, αS fibers and monomers are shown to induce tau oligomerization. Tau oligomerization is also shown to be triggered by its interaction with other molecules like β-arrestin and molecular chaperones ubiquitously expressed in cells.305,306 An increase in tau level shown to increase β-arrestin impairs tau clearance by stabilizing toxic tau species and promotes the aggregation of tau.305 Molecular chaperone Hsp90 complex with PPIase FKBP51 is shown to promote tau oligomerization. The ternary complex (Hsp90/FKBP51/tau) formation is proposed to have a synergistic effect where the chaperone and PPIase may influence tau proline isomerization leading to the formation of tau oligomers.306 Readers are referred to recent reviews on tau oligomers.307,308
3.4. Reproducibility Issue of In Vitro Experiments
The self-assembly of amyloidogenic peptides and proteins into amyloids is generally considered to work via a nucleation and elongation (a noncovalent polymerization) process. Depending on the amyloidogenic peptide/protein also other processes can be very important, such as secondary nucleation.309 This self-assembly is a stochastic process and susceptible to a lot of nonspecific interactions. Moreover, the process is autocatalytic and hence the addition of preformed aggregates can dramatically accelerate the self-assembly.
In vitro studies have shown that the self-assembly process can be influenced by numerous factors including peptide/protein concentration, salt, pH, temperature, metal ions, lipids, other molecules (such as small molecules as discussed in section 9), and cofactors such as for tau. Thus, for test tube experiments, reproducibility needs identical starting conditions. As the number and/or type of preaggregates are very difficult to control, the self-assembly process should be started with a sample of only monomeric species.193 Obtaining pure monomeric species can be very difficult but depends on the peptide/protein under investigation.310 Some are very strongly aggregating and hence difficult to monomerize.311 For instance αS is less aggregating than Aβ. Self-assembly of αS is often studied at higher concentration or stimulated by effectors, as it is slow.312 In contrast amyloid-β aggregates are very fast; particularly the Aβ42 aggregates are faster than the more abundant Aβ40 form. IAPP is another very fast aggregating peptide that is quite sensitive to pH and other conditions.
Amyloid-β (even Aβ40) and IAPP are very difficult to monomerize, and this is a reason why self-assembly is often not reproducible even when the same conditions were used. First, different batches of Aβ can vary in purity and in the preaggregation states, and therefore, reproducible results are more difficult to obtain between different batches. Even an extremely low amount of impurities (for example, metal ions) that cannot be detected by the regular analytical methods sometimes can have a significant influence on the reproducibility. In general, biologically (typically via expression from E. coli) obtained fresh peptides are higher in purity than synthetically obtained peptides and more likely to provide reproducible results. Then, a lot of other parameters, in addition to those described previously, influence the self-assembly, like ionic strength, surfaces (type and size), shaking, sheering forces, etc.).310,313 So, it is very important to report the complete details of the sample treatments including the monomerization steps and the use of several batches that are highly recommended, in particular for the very aggregation-prone amyloidogenic peptides (such as Aβ and IAPP).313,314
4. MULTISCALE SIMULATIONS OF THE EARLY AND FINAL AGGREGATION STEPS OF AMYLOID PEPTIDES
There are various sampling techniques to explore the configuration space of amyloids at different aggregation steps. Readers are referred to recent reviews describing their main features.12,14 Multiple protein representations are also used throughout this review and are shown in Figures 8 and 9.
Figure 8.
Protein representations from all-atom to coarse-grained models. (A) All-atom. (B) OPEP. (C) Geometric representation of the protein intermediate-resolution model, PRIME, for polyalanine. Covalent bonds are shown with thick gray lines connecting united atoms for N–H, C=O, Cα-H, and CH3 side chain. At least one of each type of pseudobond is shown with a colored line. Pseudobonds are used to maintain backbone bond angles, consecutive Cα distances, and l-isomerization. The united atoms are not shown full size for ease of viewing. (D) UNRES. (E) Shea’s model.
Figure 9.
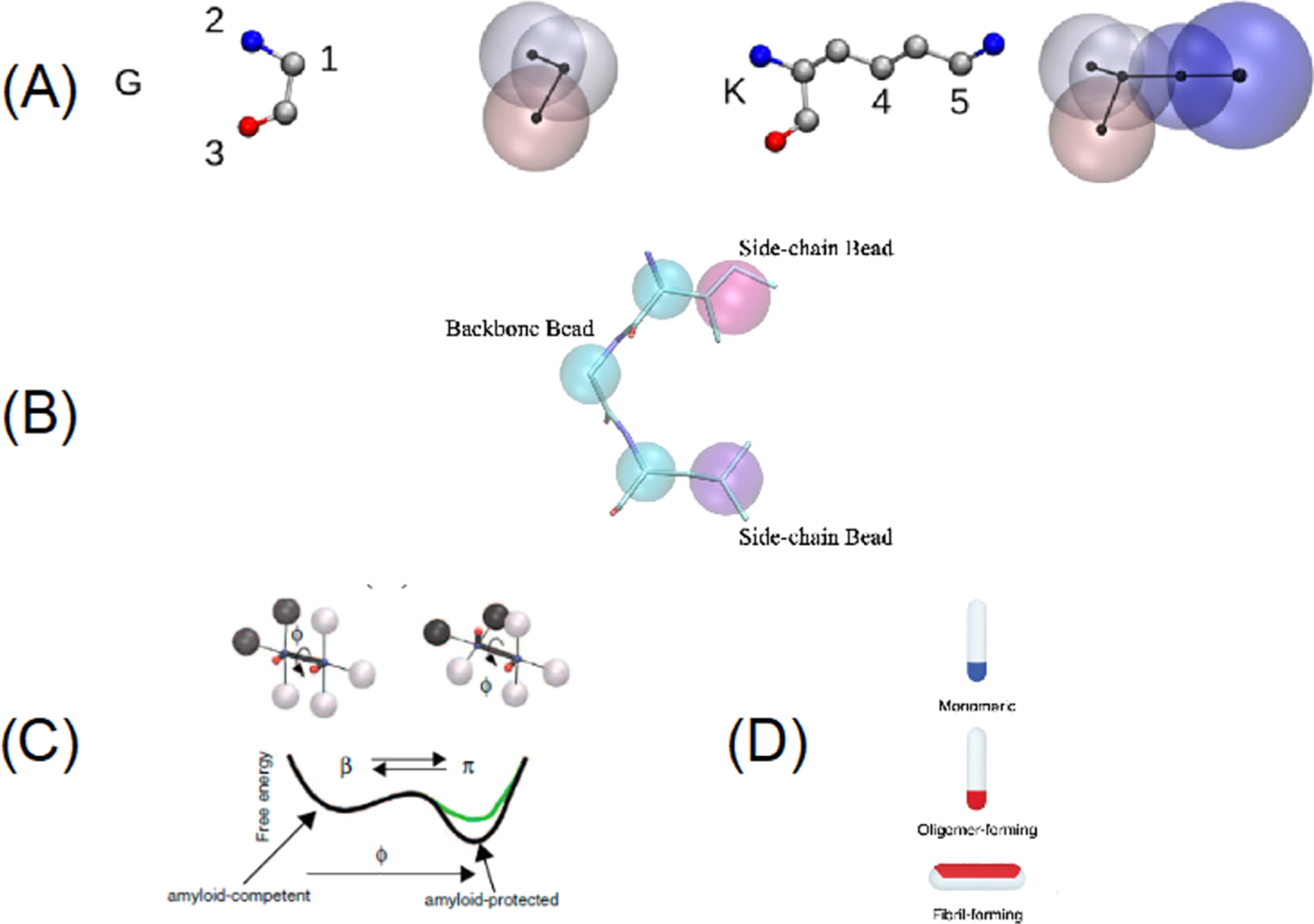
Protein representations from coarse-grained to mesoscopic models. (A) SIRAH. (B) SOP-IDP. (C) Caflisch’s model. (D) Frenkel’s model.
4.1. Thermodynamic Phase Diagram of Short Amyloid Peptide Aggregation from Simple to More Realistic Protein Models
Discontinuous molecular dynamics (DMD) is a fast event-driven alternative to conventional molecular dynamics in which particles interact with each other via discontinuous potentials including hard-sphere and square-well potentials.315 A moderately coarse-grained model, PRIME (PRotein Intermediate resolution Model), was developed for use with DMD simulation to model the folding behavior316–318 and fibril formation of homopolypeptides, in particular, polyalanine.319,320 In the PRIME model, as shown in Figure 8C, each amino acid is represented by three backbone spheres N–H, Cα-H, and C=O and one side chain sphere R. The two major nonbonded interactions captured in the PRIME model are the directional hydrogen bonding interactions between backbone N–H and C=O spheres and the nondirectional hydrophobic interaction between two side chain spheres. The other nonbonded excluded volume interactions between backbone spheres and side chain spheres are all modeled as hard-sphere potentials. The PRIME model was later extended to become the PRIME20 model which contains distinct square-well potential parameters to represent all possible side chain-side chain polar, electrostatic, and hydrophobic interactions between the 20 amino acids. This was achieved by Cheon et al.,321 who used a perceptron learning algorithm to derive knowledge-based side chain-side chain square-well potential parameters by fitting to the structures of 711 native-state globular proteins in the PDB and 2 million decoy structures. Later, Wang et al. related the reduced temperature and time scale to real units by matching a peptide helical folding profile and a self-diffusion coefficient from DMD/PRIME20 simulation with experimental measurements and atomistic simulations.322 The validity and efficiency of DMD simulations with the PRIME20 model for studying protein aggregation problems have been demonstrated by extensive investigation of self-assembly systems, including Aβ16–22;323,324 hexapeptides325 designed by Serrano et al.;326 tau fragment;327 Aβ17–36;328 and Aβ17–42;329 and coassembly systems of Aβ16–22 and Aβ40330 and the charge complementary peptide pair CATCH (±).331
Of note, researchers have adopted a number of different geometric and energetic representations of peptides to emphasis those aspects of protein aggregation that they consider most essential and to be compatible with their simulation engine. Examples of prominent CG models include the anisotropic rodlike model by Frenkel332 (Figure 9D), the lattice-based toy models by Thirumalai333 and by Frenkel,334 the midresolution off-lattice CG model of Shea335 (Figure 8E), the UNRES model by Scheraga336 (Figure 8D), the SOP-IDP model31 (Figure 9B), the OPEP model by Derreumaux337,338 (Figure 8B), the CG DMD model by Urbanc,339 the SIRAH CG model (Figure 9A) by Pantano,340 and the Caflisch’s model (Figure 9C).12,341
Although one might expect that DMD/PRIME20 and other MD simulations of CG protein models could be used to predict the equilibrium phase behavior of peptide systems and that by comparing their prediction to experimentally determined phase diagrams one could test each model’s validity, this turns out not to be the case. More precisely, the seemingly straightforward way to do this—simulate the fibril nucleation event and aggregation of a small system (e.g., <50) of randomly distributed peptides aggregates into a fibrillar structure on a short time scale (<5 μs) at each temperature (T), or equivalently, intermolecular energy (ε) and concentration (C), and then plot the boundary between the observed phases in the T-C or ε-C plane342–344—has two problems. The first problem is that the system will not necessarily equilibrate but will instead become kinetically trapped even if one simulates for a long time. The second problem is that since amyloid formation is a solid–liquid transition, the thermodynamic quantity to calculate is the solubility, the concentration of peptides in solution in equilibrium with the solid phase (the amyloid), not the total concentration as above. In other words, the microscopic “kinetics” phase diagram so derived cannot be used to calculate macroscopic thermodynamic quantities, such as solubility and latent heat, which can be directly measured from in vitro experiments. A more rigorous approach is described below.
From a thermodynamic point of view, amyloid formation is a spontaneous nucleation and growth process whose nucleation rate is determined by the driving force associated with the difference in the chemical potentials of the peptides in the fibril and in the solution phases. At solid–liquid phase equilibrium where the chemical potentials of the peptides in the fibril and in the solution phases equal each other, an amyloid fibril neither grows nor dissolves. The equilibrium protein composition in the solution phase is essentially the solubility of the protein, as mentioned before. In the light of classical nucleation theory (CNT), Kashchiev and Auer345 treated amyloid fibril nucleation as a classical two-dimensional nucleation problem in which fibrils are formed by “one step” direct polymerization. They derived a theoretical description of the work of nucleation, the critical nucleus size, and the nucleation rate for fibril formation as explicit functions of the concentration and temperature of a protein solution. Auer and Kashchiev346 first applied Monte Carlo (MC) simulation with the seeding approach347 to calculate the liquid–liquid coexistence line for helical-rich oligomers and the solid–liquid coexistence line for β-sheet-rich fibrils using a one-bead-per-residue homopolypeptide model.348 Later, Auer349 applied this amyloid nucleation theory approach and MC simulation to the same protein model to calculate the equilibrium solubility line for an infinite-layer β-sheet fibril which represents a macroscopic fibril.
We applied DMD/PRIME20 simulation and the nucleation theory approach of Auer to calculate solubilities, and hence the equilibrium phase diagram, of a real fibril-forming peptide, Aβ16–22 peptide, the archetypal amyloid former.350 To do this it was necessary to simulate many assembly and disassembly events for fibrillar aggregates which is difficult as it requires the breaking of many backbone hydrogen bonds. One reason for studying this peptide sequence is that the Aβ16–22 fibril structures predicted using DMD/PRIME20 simulation agree well with experiment.323 First, we applied constrained canonical ensemble DMD simulations and the seeding approach346,347 to determine the solubilities of a series of small aggregates (disordered oligomers and 2, 3, 4 β-sheet Aβ16–22 fibrils) formed by the Aβ16–22 peptides, as shown in Figures 10A–E. Specifically, the solubilities for the different aggregates at a given temperature were taken to be the equilibrium monomer concentration Ce at which the aggregate neither grows nor shrinks. During the simulation, the monomer peptides freely attach or detach from the two fibril ends but the creation of a new β-sheet on the preformed fibril is prevented. The latent heats of peptide aggregation into fibril phases from solution can be obtained by fitting the Ce(T) data to the van’t Hoff equation,
| (1) |
where Ce is the equilibrium fibril solubility, Cr is a temperature-independent reference concentration, kB is the Boltzmann constant, T is the simulation temperature, and L is the latent heat of monomer peptide aggregation into the oligomer or fibril. As shown in Figure 10F, at a given temperature, the solubilities for the 2, 3, and 4 β-sheet fibrils decrease with increasing thickness (number of β-sheet layers) of the fibril. This indicates that the stability of the fibril increases with increasing fibril thickness. However, the solubilities directly measured for 2, 3, and 4 β-sheet fibrils from simulation are not necessarily consistent with the experimental solubility measurements since the macroscopic fibril formed by short peptides may contain on the order of ten β-sheets.19,351
Figure 10.
(A–E) Simulation snapshots of non-HB oligomer; HB oligomer; and two, three, and four β-sheet fibrils, respectively. The peptides in the five aggregates and in solution are shown in green and magenta, respectively. (F) Phase diagram for Aβ16–22 peptide. Solubility, Ce, versus temperature data for the oligomer and fibril. The fibril and solution phases are colored yellow and cyan, respectively. (G) Summary of both the simulation-predicted temperature-dependent solubility line, Ce(T), for Aβ16–22 peptide (black curve) and the fibrillation experiments performed under given conditions. Red dots and red circles indicate conditions at which fibrils have been found to form and not to form, respectively, via TEM. Blue dots indicate that fibrils have been reported in the literature352–357 to form under these conditions. (H) Six selected TEM images (i–vi) showing that Aβ16–22 forms fibrils under conditions that correspond to the six red dots labeled i–vi in part A. (Scale bar: 200 nm.) Reprinted with permission from ref 350. Copyright 2019 National Academy of Science.
To estimate the solubility line for a macroscopic (i.e., thick) fibril, we adopt Auer’s approach346 and applied a linear fit of the simulation data (InCiβ vs 1/i) to eq 2 to extrapolate the solubility C∞β for an infinitely thick fibril. The dependence of the solubility of a fibril with i layers (i = 1, 2, 3, etc.) of a β-sheet, Ciβ, on fibril thickness (i) at fixed temperature was derived by Kashchiev and Auer:345
| (2) |
where C∞β is the solubility of an infinite thick fibril, Ψh = ahσh is the fibril surface energy parallel to the fibril thickening axis, σh is the specific surface energy of the face perpendicular to the fibril axis, and ah is the lateral surface area occupied by each peptide within one β-sheet.349
The predicted phase diagram is presented in Figure 10F, which shows that the solution phase and the macroscopic fibrillar phase are thermodynamically stable phases and that there also exists a hierarchy of metastable phases. To validate the in silico prediction of Aβ16–22 solubility at biophysically relevant temperatures, TEM was used to determine whether fibrils had formed after a predetermined time (2-wk incubation). Importantly, our prediction of Aβ16–22 solubility over temperatures from 277 to 330 K agrees well with experimental measurements (Figures 10G and 10H).
It has traditionally been challenging to create accurate thermodynamic phase diagrams for complex biomolecules such as polypeptides due to the lack of appropriate force fields and limited computational resources. Our work represents a significant milestone in efforts to overcome this challenge. By applying an advanced computational technique to a relatively realistic peptide model, we predicted an equilibrium concentration and temperature phase diagram for a short amyloid β peptide that is in remarkably good quantitative agreement with experiment.
Finally, it is important to point out that the solubility measurement approach adopted here assumes that peptides self-assemble through direct polymerization or “one-step” nucleation, which is mainly applicable for short polypeptides (e.g., <15 amino acids). In contrast, Aβ42, amylin, and αS undergo a two-step nucleation process, first forming oligomers (due to the non-negligible intrachain interactions) that eventually merge, rearrange, and nucleate to form a fibrillar structure. Thus, the nucleation process may involve oligomer formation358 which may be accompanied by liquid–liquid phase separation.359 In this case, a nonclassical nucleation theory needs to be developed for in silico prediction of protein solubility and the comparison with experimental measurements.360–362
4.2. Hydrodynamics, Shear, and Crowding Effects on Amyloid Formation
Even at in vitro conditions where proteins/peptides concentration is at least 3 orders of magnitude higher than in vivo, the formation of amyloid fibrils occurs in hours/days. Therefore, in order to get molecular insights on this process via computer simulation, it is necessary to make compromises. The first and more important is the use of simplified models representing the proteins and the aqueous environment by simplified representations where CG models and implicit solvent are used.363 The second compromise concerns the effective concentration that is used in simulations364 and that can be tuned so to favor the encounter of the molecules when the kinetics of aggregation and elongation is investigated. Finally, even with these simplifications in hand the size of the simulated systems is generally limited, generally with less than 103 monomers.365,366
The two principal aspects that have been studied via CG models are the early steps of aggregation and the mechanism of fibril elongation.367 Peptide aggregation can be described as a nucleation process, but whether the primary nucleation is a one-step or two-step process clearly depends on the amino acid sequence and the experimental condition (Figure 11A). As for many proteins, however, and notably the prion and Aβ proteins, it has been reported experimentally that the proteins first collapse into disordered aggregates that associate and dissociate, eventually producing fibrils that elongate and fragment,12,368–370 with all events involving very long-time scales for each event (Figure 11B).
Figure 11.
(A) Schematic representation of two extreme scenarios, the one-step and the two-steps nucleation process for early steps of amyloid aggregation.366 (B) Characteristic steps and kinetics of Aβ42 amyloid proliferation determined experimentally, with the reaction rates at a concentration of 5 microM.370 (C) Description of the dock and lock mechanism, left chart schematically reproduces the results,375 where the evolution of the β-structure of a monomer is followed in time during the dock and lock phases. (D) Effect of hydrodynamic interactions on the aggregation process of Aβ16–22 peptides (left) and protofibril elongation due to oligomer fusion (right) as observed in LBMD simulations.366,394
Simulations based on simplified molecular models have addressed these issues. In seminal works attention has been posed on how the molecular propensity of the amyloid peptide to sample the aggregation β-prone state influences the path of the initial aggregation. Pellarin and Caflish371 and Bellasia and Shea372 explored the aggregation mechanism by tuning the conformational stability of the “fibrillar-like” state of model peptides. Both studies clearly showed that when the peptide is able to access conformations different from that of fibrillar structures, the aggregation passes through disordered micelles. On the contrary for peptides having strong propensity for the β-configuration, the aggregation is funnelled directly toward structured aggregates. These, as other studies, highlighted the competition among thermodynamic and kinetic selection during aggregation, and the effect of the monomer configurational energetic landscape.371–374 A nucleation diagram as a function of solution concentration and intermonomer interaction strength has been reported on the basis of MC simulations of simple stick-like molecules, stressing the important role of the intermediate disordered aggregates and the predominant contribution of the two-steps primary nucleation process.365
Computational studies focused also on the behavior of a monomer when aggregating. Following the study by Massi and Straub,375 two idealized pictures can be drawn (Figure 11C). In the first one, the amylogenic protein/peptide acquires very rapidly its monomeric prone-aggregation configuration that drives fast adhesion with other monomers for the creation of a critical structured nuclei or with an existing fibril. In the second scenario, the adhesion and structuring processes of the monomer occur in two distinct phases, referred to as dock and lock. There is now a consensus from experiments and simulations that the dock–lock mechanism is the dominant elongation path in many conditions. There is a clear kinetic separation among the time scales of the dock and lock phases. The first one is limited by the diffusion and the energetics of desolvation, while the second one is controlled by the free energy barriers that separate peptide conformations and that can be affected by the local packing in the aggregate. According to several estimates, a difference of at least 2 orders of magnitude for the characteristic kinetics exists, τlock/τdock ≥ 100.376–379 Experimentally the elongation process results to be entropically driven with an entropy gain associated with the release of interfacial water into the bulk. Clearly in CG simulations such a contribution cannot be accounted explicitly. However, critical information about the change in conformational flexibility of the proteins/peptides upon aggregation has been obtained. The steps from the dock toward the multiple lock states have been related to the size of the proteins and their conformational space and described by monitoring the shift from intra- vs intermolecular interactions.77,380,381 A very detailed picture of the dock and lock mechanism has been recently drawn by combining atomistic simulations and the Markov state model. It was shown that for a simple amylogenic peptide, the fragment Aβ16–22, the locking phase requires a complex search in a multiminima landscape characterized by off-register configurations and with no evident funnel toward the fibrillar configuration.382
Simulations have also inquired the global process of elongation, and an asymmetric elongation has often been reported,367 with one cap of the fibril growing faster and involving local disorder. Thanks to the simulation of a very large system, a complementary view to the one monomer addition was explored. It was possible to probe that at large length-scale once the first oligomers are formed, multiple fusion events take place to elongate the protofibril with not clear dominant scheme365,366 and including lateral branching that uses the surface of the growing fibril as a seed.366,383 This finding confirms the theoretical intuition proposed to describe the kinetics of amyloid formation and that includes a key secondary nucleation process.309,384
When using implicit solvent CG models to explore protein aggregation two issues must alert the reader. The first concerns the estimate of thermodynamics quantities. As anticipated, solvation free energy is not explicitly accounted for, and empirical or approximated approaches are required to estimate relevant thermodynamic contributions such as the specific heat variation. Moreover, by reducing the degrees of freedom of a system the quantification of entropy is altered with respect to atomist models, with the risk to unbalance the entropy/enthalpy compensation effect. The simplification of the free energy landscape clearly impacts also the barriers separating states and therefore the resulting kinetics. However, a second aspect must be accounted for when focusing on the dynamic behavior. In an implicit solvent model there is not exchange of momenta between the particles and the solvent, and solvent mediated correlations are not accounted for. This is critical when dealing with many particles systems, and their aggregations since the hydrodynamics interactions (HI) might play an essential role. Several techniques can be used to cure this deficiency385–387 and have been applied in several contexts, such as protein folding388,389 or protein motion under crowding.390 More specifically, the Brownian dynamics including HI has been used to investigate lipid aggregation using a simplified dumbbell model.391 It was shown that HI speeds up the process since, as can be deduced by comparing the Zimm versus Rouse model for polymer motion,392 when the first oligomers are formed their diffusivity scales more favorably with the aggregate size than when HI is not included in the simulation. A similar result was obtained for amyloid aggregation by lattice Boltzmann MD simulations with the OPEP model for Aβ16–22 peptides,393,394 see Figure 11D. Notably the presence of HI coupled to the high-resolution and flexible OPEP model enhances size fluctuations of the formed oligomers. These size fluctuations generate solvent local flows at the nanoscale that contribute to the progress of the aggregation. This is especially important at a large length-scale when very extended protofibrils (each formed by hundreds of monomers) fuse together.366
The impact of fluid dynamics on amyloid aggregation is well documented by in vitro experiments where the action of stirring or shaking accelerates the kinetics of fibril elongation. For instance, the shearing of a protein solution in a Couette flow device favors the aggregation of beta-lactoglobulin by enhancing the formation of spheroidal seeds in the solution.395 It was also reported that the action of shear alters the morphology of the formed fibrils.396,397 The energetic landscape of β2-microglobulin fibrils was explored by combining isothermal titration calorimetry and stirring.398 The stirring force modifies the end-point of the aggregation process as measured by the solution enthalpy difference, and the kinetics of aggregation that results is enhanced. The action of shear may also perturb formed fibrils. For example, the capability of shear flow to break existing fibrils has been used to explore the different mechanisms coacting during fibril elongation. Namely shearing is used to enhance the kinetics of the fragmentation contribution over secondary and primary nucleation.384
The molecular mechanism of flow action on aggregation is still debated. In fact, for standard globular proteins the initial unfolding that should trigger aggregation can be forced only at very high shear rates,399–402 values much higher than what was found typically in physiological conditions that are <104–105 s−1. In a recent study elongational flow was used to perturb and trigger aggregation of several proteins, β2-microglobulin, bovine serum album (BSA), and monoclonal antibodies.16 It was shown that when exposed several times to an elongational flow at shear rate ~104–105 s−1 even a stable protein such as BSA can expose to solvent parts of the sequence generally screened in the folded state and can therefore induce aggregation. It must also be considered that in many neurodegenerative diseases the involved proteins are intrinsically disordered. For these latter ones even a weak fluid perturbation might alter the ensemble of conformational states accessed by the protein and therefore select the ones prone to aggregate or force preferential orientations that favor the fibrillar elongation. Moreover, shearing and interactions with a surface might act together and modify the structure of prefibrillar seeds. A possible impact of fluid flow in cerebrospinal and interstitial fluid on the amyloid aggregation has been proposed recently and discussed in detail.403
In order to complete the discussion on protein aggregation, we should address an extra problem. In fact, in vivo proteins generally move in a very crowded space such as the cytoplasm or the membranes. This crowding has multiple impacts on aggregation. The macromolecular crowding generates an excluded volume effect that locally alters the concentration of the aggregating species and thus could have a favorable impact on the aggregation with respect to a dilute reference solution. On the other hand, moving in a crowded space is more difficult, and the diffusion of a species is reduced as an effect of the reduced dimensionality and higher viscosity of the solution. The molecular collisions needed to start the nucleation, and the further elongation becomes rarer. Finally, the macromolecules surrounding the aggregating species are not inert; they might have specific interactions with them that could ease misfolding or compete with the aggregation.
Experimentally several studies have tackled these issues. In vitro, the crowding effect can be reproduced by adding polymers to a solution, e.g., Ficoll, dextran or PEG (polyethylene glycol), or proteins of different sizes, for example BSA proteins generate large excluded volumes. NMR studies reported that depending on the nature of crowders the αS monomer can adopt more or less compact configurations, indicating that the effect on protein extension is not merely an excluded volume effect but might depend on other features of the crowding agent.404 The ensemble of configurations explored by αS can also be tuned by two-dimensional crowding on a lipid surface.405 Interestingly, when focusing on aggregation it was reported that proteins linked to neurodegenerative diseases have indeed a propensity to aggregate more and faster under crowding conditions.406 Moreover, the morphology of the formed fibrils is also affected by crowding, for instance Ficoll induces a more packed state of β2-microglobulin fibrils.398
However, the heterogeneity of in vivo crowding cannot be easily mimicked in in vitro setups, and the effect on protein mobility, conformation, and stability is subtle. While the excluded volume effect predicts stabilization of proteins under crowding, a variety of studies reported a much more complex response, pointing to the importance of quinary interactions, the fifth degree of organization of a protein within its surrounding environment.407,408 For the B1 domain of protein G, the stability effect of a single amino-acid mutation was found to be amplified 10-fold by the crowded cytosol of Escherichia coli.407 On the other hand, mutating a single amino-acid residue in SOD1 sufficed to reverse the sign of the stability effect of the intracellular environment.408 Interestingly, for monomeric αS, it was shown that under crowding the conformation is flexible and disordered while one would have expected a compact state due to excluded volume.185
However, concerning the general picture, fibrillar aggregation of SOD1409 or Huntingtin exon 1 protein410 in cells has been characterized by means of different techniques, NMR and fluorescence spectroscopy, respectively, and the obtained results are similar to what was found in in vitro experiments. Of note when discussing the impact of aggregation on neurodegenerative diseases, one should also consider the aging of the cells, a difficult condition to reproduce in in vivo experiments.
A few simulations have tried to shed light on the mechanism of aggregation under crowding. A first set of studies has focused on the effect of crowding on the conformation sampled by IDPs and using different resolution models to compare with experiments such as SAXS or FRET.411–413 As an example, we refer to the work by Zhou et al.412 where by using a mixed resolution approach, atomistic for the IDP and CG for the crowders, three IDPs (for the N-terminal domain of HIV-1 integrase, the ACTR, and prothymosin α) were investigated in a PEG crowded solution. A good match with experimental FRET data was obtained only when a certain degree of attraction between the proteins and the crowders was incorporated in the model, while a pure repulsive interaction led to too compact states for the IDPs.412
In a second class of work the focus was placed on the aggregation kinetics. In the presence of inert crowders modeled as hard spheres it was shown that Aβ16–22 amyloid peptides simulated by the PRIME-DMD generally collapse quickly in disordered aggregates, and the structuring phase occurs as a second step.324 However, when the nature of the crowder is specified, e.g., being modeled as hydrophobic particles, the interactions with the peptides become competitive with the interpeptide interactions, causing the extension of the lag time of fibril elongation.324 The competitive effects of crowding, local concentration increase and diffusion slow down, were inspected for an ideal peptide considering the different propensity toward different aggregation paths.414 For peptides with low aggregation affinity, the formation of a fibril is rate limited by the creation of a first nuclei, and the crowders tend to stabilize the nuclei and therefore impact favorably aggregation; on the contrary for peptides owning high affinity, the elongation is clearly delayed as an effect of diffusion slowdown. All these studies used simplified or mixed molecular representations. However, CG simulations can be directly complemented by high-resolution atomistic approaches in a multiscale strategy as recently proposed for investigating the effect of crowding on the stability of the aggregation prone SOD1 protein;408,415 see Figure 12. Here, a large-scale lattice Boltzmann MD simulation served to identify states of local packing around SOD1 in a crowded protein solution (BSA), which was then closely examined using atomistic enhanced-sampling simulations. In line with experimental results, the presence of crowders was found to have only a minor effect on the overall thermal stability of SOD1, and crowding did not dramatically perturb the unfolding process. Nevertheless, the simulations identified a fragile region on the β barrel, susceptible to early unfolding and subsequently showing a strong propensity to interact with the crowder. This semi-unfolded intermediate state, appearing to be stabilized by the interactions with the crowded environment, was hypothesized to play a role in the aggregation of SOD1 in the crowded cellular conditions.
Figure 12.
Unfolding and stability of SOD1 in crowded conditions.408,415 (A) Snapshot from a 1 μs coarse-grain LBMD simulation of loop-truncated SOD1 monomers immersed in a 200 g/L BSA solution.408 (B) Representative states of local packing around SOD1 extracted from the coarse-grain simulation and converted into a fully all-atom representation. (C) Thermal stability—calculated using enhanced-sampling all-atom simulations—of SOD1 in the different states of local packing. The stability of the SOD1 monomer is expressed by means of the secondary-structure content of the protein. Comparison with a result obtained in dilute conditions reveals only a weak effect induced by crowding.415 (D) Semiunfolded intermediate state observed in the enhanced-sampling all-atom simulations. The blue clouds, representing the spatial distribution of BSA atoms in contact with SOD1, show that the denaturated region has an increased probability to interact with the crowder.415
5. EXTENSIVE SIMULATIONS ON MONOMERS AND SMALL OLIGOMERS
We discuss Aβ, α-synuclein and tau systems. Simulation results on IAPP can be found in ref 12.
5.1. Aβ40/42 in Solution
There have been numerous computational studies on monomers and oligomers of Aβ40 and Aβ42 (Figure 13A) to understand their links with the fibrils with intramolecular U- and S-shapes. We mainly discuss the works published during the last five years (Table 1).
Figure 13.
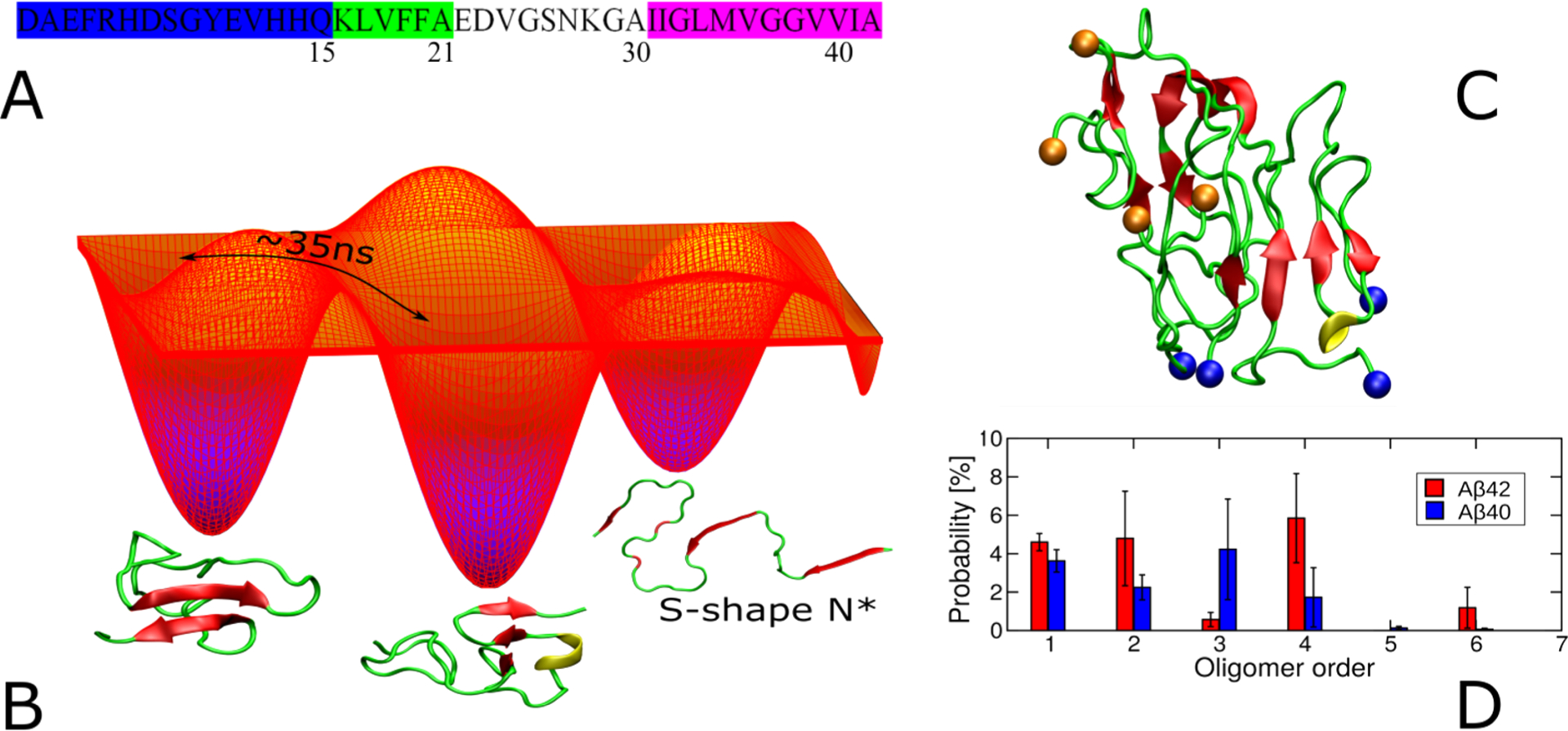
(A) N-terminus (blue, 1–15), CHC (green, 16–21), loop (gray, 22–29), and C-terminus (purple, 30–40/42). (B) Schematic free energy landscape of Aβ monomers. Switching between some conformations occurs within 35 ns, as reported by FRET data.428 The conformation of S-shape N* from the fibril structure with PDB ID 2NAO was also sampled in CG simulations.437 (C) Representative structure of the Aβ42 tetramer, obtained by using multiscale MD simulation.451 Blue and orange balls refer to the first and last residues, respectively, of monomer subunits. (D) Population of oligomer sizes obtained from simulations of 20-peptides.463
Table 1.
Computational Studies on Monomers and Oligomers of Aβ Alloforms in Solution after 2015
| ref | force field | Solvent model | Method | Time scale | Aβ alloform | Oligomer size |
|---|---|---|---|---|---|---|
| Rosenman419 | OPLS-AA/L | TIP3P-Ew | REMD | 52 μs | Aβ42 | 1 |
| AMBER99SB-ILDM | TIP4P | |||||
| CHARMM22* | TIP3P | |||||
| Weber416 | AMBER FF99SB | TIP3P | REMD | 5.28 μs | Aβ42 | 1 |
| CHARMM22/CMAP | ||||||
| Carballo-Pacheco.476 | OPLS, AMBER99SB, AMBER99SB-ILDN, AMBER99SBILDN-NMR, CHARMM22* | TIP3P | REMD | 6.4 μs | Aβ42 | 1 |
| TIP4P-Ew | ||||||
| Meng428 | AMBER99SB | TIP4P/2005 | REMD | 740 ns | Aβ42, Aβ40 | 1 |
| Krupa418 | AMBER14SB | TIP3P | REMD | 28.8 μs | Aβ42 | 1 |
| AMBER14SB_IDPS | ||||||
| AMBER99SB | ||||||
| CHARMM36 | ||||||
| CHARMM36m | ||||||
| Thu435 | OPLS | GB | REMD | 6 μs | Aβ42 and 19 variants | 1 |
| Frigori438 | Charmm22* | TIP4P/Ew | REMD | 19.2 μs | Aβ42, Aβ40 | 1 |
| Aggarwal434 | AMBER99SBILDN | SP C/E | MD | 100 ns | Aβ42 and 5 variants | 1 |
| Li468 | AMBER99SB | GB | REMD | 8 μs | Aβ40, Aβ40-A2 V and tautomer | 1 |
| Liu469 | AMBER99SB-ILDN | TIP3P | MD | 0.4–1.8 μs | Aβ42 | 1 |
| Bhattacharya426 | CHARMM36, CHARMM22*, CHARMM36m, AmberA03, AmberA03Ws | mTIP3P, TIP4P, TIP4P-D, TIP4P-Ew | MD | 33 μs | Aβ42 | 1 |
| Robustelli465 | AMBER99SB*-ILDN, CHARMM22* | TIP3P | MD | 30 μs | Aβ40 | 1 |
| CHARMM36m, AMBER03ws, AMBER99SB-ILDN | TIP4P-D | |||||
| AMBER99SB-disp | ||||||
| Zhang440 | AMBER99SB-ILDN | TIP3P | MD | 4 μs | Aβ42 | 2 |
| Man444 | AMBER99SB-ILDN | TIP3P | REMD | 25 μs | Aβ42 WT, S8C | 2 |
| Man445 | OPLS-AA, CHARMM22, AMBER99SB-ILDN, AMBERSB14 | TIP3P | REMD | 36 μs | Aβ42 | 2 |
| Nguyen448 | CHARMM22* | TIP3P | REMD | 24 μs | Aβ40, Aβ40-A2 V | 2 |
| Nguyen447 | CHARMM22* | TIP3P | REMD | 24 μs | Aβ40-A2T | 2 |
| Cao449 | PACE | MARTINI | REMD | 2.7 ms | Aβ40 | 2 |
| Hashemi470 | AMBER99SB-ILDN | TIP3P | MD | 4 μs | Aβ40 | 2 |
| Das443 | OPLS-AA | TIP3P | REMD | 51.2 μs | Aβ42, Aβ42-A2T | 2 |
| Sharma467 | CHARMM36 | TIP3P | MD+REMD | 1.5 μs | Aβ42-WT, Aβ42-A2 V, Aβ42-A2T | 2 |
| Mehrazmaal442 | GROMOS96–53a5 | SPC | MD | 9.5 μs | Aβ42 | 2 |
| AMBER99SB-ILDN | ||||||
| Press-Sandler471 | CHARMM36/CMAP | TIP3P | MD | 200 ns | Aβ42 | 2 |
| Nguyen451 | UNRES AMBER99SB-ILDN, OPLS-AA/L | TIP3P, TIP4P | MD+REMD | 50 μs | Aβ42 | 4 |
| Xi458 | CHARMM36 | TIP3P | MD | 800 ns | Aβ42 | 3–4 |
| Nguyen459 | AMBER99SB-ILDN, OPLS, AMBER99SB, CHARMM36m | TIP3P, TIP3P-modified, DISP | REMD | 101.9 μs | Aβ40, Aβ42 | 4 |
| Man464 | AMBER14SB | TIP3P | MD | 22.5 μs | Aβ42 | 2–4 |
| Zhang453 | DMD4B-HYDRA | DMD | 21.6 μs | Aβ40, Aβ42 | 32 | |
| Voelker456 | OPLS-AA | TIP3P, SPCE | MD | 24.35 μs | Aβ40, Aβ42 | 1–5 |
| Barz463 | OPLS-AA | GBSA | MD | 2.5 μs | Aβ42, Aβ40 | 20 |
| Zheng461 | AWSEM | MD, umbrella sampling | Aβ40 | 1–8 | ||
| Zheng462 | AWSEM | MD, umbrella sampling | 1 μs | Aβ42 | 1–8 |
5.1.1. Monomers.
Simulations showed that the conformational ensemble of both Aβ alloforms depends on the force field, water model, and sampling method, but a consensus has been reached.80,416–422 All-atom REMD simulations with both an implicit421,422 and explicit solvent80,419 confirmed that, in agreement with NMR relaxation data,423 the C-terminus of Aβ42 is more rigid and has a higher β-strand content than Aβ40. This fact is often invoked to explain the increased rate of Aβ42 aggregation. Regardless of the force field and simulation method, for both alloforms the α-helix content (≤10%) is lower than the β-strand content (10–27%), which is consistent with CD data showing the α-helix content is about 9% and the β-content is between 12 and 25%.33,424 NMR studies suggested the presence of an antiparallel β-sheet between CHC and residues 29–36 for monomeric Aβ42,80 and this was confirmed by REMD simulation using CHARMM36m, FF14SB-IDPs, FF14SB, and FF99SB force fields.418 A small β-hairpin, centered at residues 36–37, was populated in Aβ42 but not in Aβ40, and this may contribute to the difference between the two forms in the aggregation rates.425 Through MD simulations with ten combinations of distinct CHARMM, AMBER, and water models, it was shown that helical conformations at the N-terminus of Aβ42 are stable due primarily to hydrophobic interactions with CHC and due to salt bridges with other fragments playing a secondary role.426 The existence of such helices and the formation of α-sheets may promote the formation of an α-sheet in the lag phase.427
Meng et al. performed MD simulations and sm-FRET studies to explore the conformations of Aβ40 and Aβ42 at physiological conditions.428 They found that, similar to recent NMR data,255 both peptides adopt random coil conformations with Aβ42 being slightly more compact than Aβ40. Sm-FRET revealed435 that some conformations rapidly interconvert, and nanosecond fluorescence correlation spectroscopy provided the time scale of the transition of about 35 ns (Figure 13B).428 The same time scale was also obtained for other IPDs.429 The application of NMR-guided metadynamic sampling for Aβ40 monomer revealed that highly populated basins are separated by low barriers, and stability seems to increase with temperature, which contradicts the behavior of ordered proteins.430
Both experiment and simulation provided evidence that the aggregation rates of proteins are controlled by hydrophobicity, charge, and secondary structure.431–434 In a pioneering work by Chiti et al.,432 the latter factor was accessed as the propensity to convert from α-helical to β-sheet conformation in a monomeric state, and therefore, the direct relationship between aggregation rates and secondary structures remains unclear. In addition, the estimation of the free energy change for this conversion using empirical formulas is not sufficiently accurate.432–434 To solve this problem, Thu et al.435 calculated the β-content of 19 mutants of Aβ42 using REMD simulation with the OPLS force field and implicit water model and related it with the experimentally measured aggregation rate κ. They found κ ~ exp(cβ), where c = 0.071 and β is the percent of β-content, implying that the higher the β-structure in the monomeric state the faster the fibril formation. Thus, the propensity to aggregation is encoded in the conformations accessed by the monomers at equilibrium.
Li et al. introduced the concept of the fibril-prone states N*, which resemble monomers in the fibril structure.333 In lattice models N* is a single state,431 while in off-lattice models it comprises an ensemble of states in the same basin.436,437 Since N* can serve as a template for the nucleation step, it is rational to assume that the propensity to aggregate depends on the gap between N* and the ground state. Moreover, because the population of the fibril-prone state PN* is determined by this gap, the fibril formation time τfib is related to PN* as
| (3) |
where c is a constant and PN* is expressed as a percentage.431 Chakraborty et al.437 used eq 3 to understand why Aβ42 aggregates faster than Aβ40 and to shed light on the kinetics of fibril polymorphism. Using the SOP-IDP model31 (Figure 9B) for MD simulation and the striated fibril structure (PDB code 2M4J) as the reference state for N*, they estimated PN*(Aβ40) for Aβ40 monomer. For Aβ42 monomer, PN*U(Aβ42) and PN*S(Aβ42) were obtained using the U-bend (PDB code 2BEG) and the S-bend (PDB code 2NAO, Figure 13B) fibril structures, respectively. Overall, PN*(Aβ40) < PN*U(Aβ42) < PN*S(Aβ42), and using eq 3 with c = 1 and the estimated values of P N*, τfib of S-bend Aβ42 fibril is around 24 times smaller than Aβ40.437 This prediction is consistent with recent experiments showing that the rate of Aβ42 fibril formation is about an order of magnitude higher than Aβ40. The higher population of N* in Aβ42 than in Aβ40 was also confirmed by all-atom simulations.438 If the U-bend conformations are identified as N*, then the aggregation occurs only two times faster than Aβ40.437 Therefore, the N* theory can capture the fact that formation of various polymorphic structures such as U-bend and S-bend fibrils is time dependent or under kinetic control.371 Assuming that the Aβ aggregation obeys the Ostwald’s rule, which states that the least stable polymorph would form first, followed by a subsequent transition to a more stable form, one can predict that the S-bend Aβ42 fibril is more stable than the U-bend form, as the latter forms faster.437
5.1.2. Dimers.
The dimer has been subjected to many studies.439–450 Zhang et al.440 combined MD and MC pulling simulations with AFM experiments to obtain models for Aβ42 dimers. First, various structures were generated using MD and then an external force was applied to the Cα atom of the first Cys residue of each monomer to pull them away at a constant speed. The obtained patterns of the rupture force were compared with experimental data to select the best models. The Aβ42 dimer structures are mainly stabilized by the intermonomer interactions within the CHC regions and do not contain long β-strands that occur in the fibril state. An AFM-based force clamp was also applied for dissociating Aβ42 dimer and identified two transient states with lifetimes of 188 ± 52 and 317 ± 67 ms.441
By performing 9.5 μs MD with GROMOS96-53a5 and AMBER99SB-ILDN force fields in SPC solution, Mehrazma and Rauk reported442 that in addition to CHC, the hydrophobic C-terminal also plays a key role in Aβ42 dimer stabilization. Unlike other groups,440,443–445 their models are richer in β-structure than monomers, which may be due to the simulation setup, where the initial conformations were taken from predefined NMR structures. Using OPLS-AA, CHARMM22*, AMBER99sb-ildn, and AMBERsb14 with the TIP3P water model, Man et al. showed that the equilibrium ensembles of Aβ42 dimers are random coil stabilized by nonspecific interactions.453 Despite significant differences in the secondary structure, the cross collision sections and small-angle X-ray scattering profiles are independent of the force field and are consistent with experimental data.
Derreumaux et al. conducted REMD simulations to explore the equilibrium ensembles of the Aβ40 dimer and its mutants in solution.446–448 They found that the wild type dimer at 315 K is highly disordered, but the secondary structure of the β-strand and α-helix is richer than that of the monomer. Although conformations with an unstructured N-terminus and β-hairpins covering residues 17–21 and 30–36 are transiently populated, the antiparallel and perpendicular orientation of the peptides is preferable to parallel organization.
Combining a hybrid-resolution model and adaptive sampling techniques, Cao et al.449 carried out a 2.7 ms simulation in order to investigate the mechanisms of formation of Aβ40 dimers. They then developed a Markov state model (MSM) to characterize transition pathways and related kinetics, finding hairpin-containing and parallel in-register structures resembling fibrils (see recent review450 on the application of MSM to study the aggregation of amyloid peptides). Hairpin-like structures occur through one step nucleation, in which two preformed β-hairpins spanning residues 16–35 occasionally associate, preserving the β-hairpin conformation. This process occurs on a time scale of about 200 μs, which is ~100-fold faster than the formation of fibril-like dimers (25.8 ms). Transformation into fibril-like structures includes rapid hydrophobic collapse, followed by a slow configuration rearrangement, which proceeds via different routes but always requires transient unfolding of complexes. Interactions involving the N-terminal region play a crucial role in the dimerization kinetics.449
5.1.3. Oligomers.
The Aβ42 tetramers were explored by a multiscale approach involving REMD simulation with the UNRES force field (Figure 8D) followed by all-atom MD to refine the most representative CG structures.451 The models obtained are polymorphic and more compact than its fibril counterpart (Figure 13C). In both OPLS-AA/L AMBER99SB-ILDN force fields the calculated collision cross section (~2000 Å2), which falls into the experimental range,452 is lower than that of fibril (~2600 Å2). The high population of the β-structure in residues 9–14, 17–21, and 30–40 is in agreement with the experiment, and Aβ42 models are dominated by turn and coil, which is also consistent with experimental observations. Interaction with solvent promotes compactness while the interchain electrostatic interaction facilitates the formation of extended structures. As a consequence, when the number of chains becomes large the interplay between these two interactions should lead to fibrillar structures.451
Employing the DMD4B-HYDRA force field and DMD simulation, Zhang et al.453 investigated the role of cross-linking via tyrosines in Aβ self-assembly kinetics and morphology of aggregates. They found that cross-linking promotes aggregation, especially that of Aβ40, and significantly alters the shape of the oligomers by increasing the solvent exposure of hydrophobic residues, which leads to elongated oligomeric structures that differ from more globular structures of noncrosslinked partners. Recent experimental454 and simulation455 works confirmed that the oxidative reactivity of Cu-Aβ catalyzes the formation of Tyr–Tyr cross-links in peptide dimers. Voelker et al.456 used the structures obtained in the coarse-grained DMD4B-HYDRA simulations as initial conformations for multiple all-atom MD simulations of monomers and oligomers of 2–5 Aβ chains. They observed water-permeable pores in trimers, tetramers, and pentamers of both Aβ40 and Aβ42 and found that the tendency to form pores increases with increasing oligomer size.
Combining IM/MS, EM, AFM, and computational modeling, the Eisenberg’s group457 demonstrated that cylindrin-like barrels of tandem repeats of Aβ fragments with a length of 11 residues (residues 24–34, 25–35, 26–36) held together by two glycine residues are stable. To check whether a similar structure is possible for Aβ oligomers, Xi et al.458 built out-of-register Aβ42 assemblies and also discovered barrel-shaped structures consisting of β2-turn-β3 domains (residues 27–42). They occurred both in trimers and in tetramers, though the stability in the latter is higher than in the former after at least 200 ns of MD simulations with explicit water.
The stability of tetrameric Aβ40 and Aβ42 β-barrel structures, which are different from out-of-register barrels,458 was probed by REMD simulation with four atomistic force fields.459 In aqueous solution, due to a change in the CHC–CHC and C-end-C-end interfaces, a β-barrel structure, made of eight antiparallel β-strands covering residues 9–40/42 with two distinct β-hairpin types and an inner pore diameter of 0.7 nm, exists transiently and to a greater extent for Aβ42 than Aβ40.
Using the CG AWSEM460 (associative memory, water-mediated, structure, and energy model) force field, Zheng et al.461 studied the relative stabilities of Aβ40 monomer and oligomers up to an octamer. A transient hairpin structure populated in a monomer becomes increasingly more stable in oligomers, where hydrogen bonds between adjacent chains can form. Oligomers have either prefibrillar or fibrillar forms. Prefibrillar oligomers are polymorphic but typically have a cylindrin-like shape, consisting mainly of antiparallel β-strands, while fibrillar oligomers contain only parallel β-sheets.461 The aggregation free energy profile of Aβ42 is more downhill than Aβ40, and the two terminal residues stabilize the oligomeric structures for Aβ42 relative to Aβ40, which greatly facilitates the conversion from prefibrillar trimers to fibrillar tetramers.462
While Zheng et al. focused on the thermodynamics of Aβ aggregation, Barz et al. looked at the kinetic aspects by MD simulation of 20 Aβ peptides, which are initially disordered and randomly distributed, using the OPLS-AA force field and GBSA (generalized Born solvent area).463 They developed transition networks to show that both Aβ alloforms can form extended and compact oligomers, which play different roles; the former are apparently engaged in the formation of new aggregates, while the latter are likely metastable, off-pathway, and experimentally observable. Aβ40 and Aβ42 show distinct propensities for the formation of oligomers; that is, Aβ40 primarily populates dimers, trimers, and tetramers, while Aβ42 predominantly forms dimers, tetramers, and hexamers (Figure 13D), consistent with experiment452 apart from the absence of dodecamers for Aβ42. This may be due to either insufficient sampling or the small number of chains used in the simulation. Also, Aβ42 tetramers appear to be more involved in the formation of bigger oligomers than Aβ40 tetramers.463
Finally, Man et al.464 performed all-atom MD simulation for Aβ42 dimers, trimers, and tetramers and found that, in accordance with classical nucleation theory, the oligomerization time depends on the monomer concentration by a power of −2.4. Using this dependence and assuming a concentration of Aβ monomers in the human brain of 0.8 nM, they speculated that it will take 62 years for the formation of toxic Aβ42 oligomers, a time being equal to the age of AD onset.
Overall, we must recall that despite a constant improvement of atomistic force fields in explicit solvent,465 there are divergences between the simulation results on kinetics and thermodynamics,466 the impact of mutations,467,468 the population of critical states,469 and the shapes of oligomers.470,471
5.2. α-Synuclein in Solution
αS can form oligomers that are toxic to neuronal cells and cause cell death.472,473 To understand the aggregation mechanism and develop promising neuroprotective strategies, knowledge of the conformational characteristics of monomeric αS may be critical. The structural characterization of αS by traditional biophysical experiments or MD simulations has been challenging.474,475 Force fields developed for folded proteins in MD simulations tend to predict more compact structures of IDPs that contradict experimental measurement.476 To address these challenges, scientists modified force fields to improve the simulations of IDPs.477–481 Robustelli et al.465 performed MD simulations of 21 systems that contain folded and disordered proteins using six force fields and a 2.5 fs time step. Based on the results, they modified parameters and developed a force field, a99SB-disp, which is competent for accurate simulations of both ordered and disordered proteins. Other methods have been integrated with standard MD simulations to guide accurate modeling. Pietrek et al.45 used a hierarchical approach that explores all possible structures of IDPs by assembling protein fragments modeled by 100 ns all-atom simulations with the Amber99SB*-ILDN-q force field and the TIP3P water model. The generated ensemble of αS captured the local structures from NMR and the overall dimension from SAXS data.45 Experimentally constrained computational simulations have been widely applied to predict the αS atomistic structures (Table 2).482–488 Brodie et al. used a novel approach that integrates protein cross-linking constraints and discrete molecular dynamics (CL-DMD) simulations.488–491 Adding experimental data to DMD simulations reduces the conformational space and allows the simulations to achieve accurate protein folding on a reasonable time scale.489–494
Table 2.
Experimentally Guided Molecular Dynamics Simulations of α-Synuclein Monomer
| Simulation method | Force field | Water model | Experimental data used |
|---|---|---|---|
| Ensemble MD482 | CHARMM | Implicit solvent | Paramagnetic relaxation enhancement nuclear NMR |
| REMD483 | OPLS-AA | AGBNP implicit solvent | |
| REMD484 | CHARMM | EEF1 implicit solvent | NMR chemical shifts, NMR residual dipolar couplings (RDCs), and SAXS |
| Monte Carlo485 | Repulsive Lennard-Jones interactions and harmonic restraints derived from smFRET measurements | Not applicable | Single-molecule Förster resonance energy transfer (smFRET) |
| REMD486 | OPLS-AA | AGBNP implicit solvent | RDCs and PREs data |
| Monte Carlo487 | Rosetta score function and the constraint energy | Not applicable | Ensemble FRET data |
| Discrete molecular dynamics488 | Medusa force field | Implicit solvent | Short-distance cross-linking data by mass spectrometry |
Computationally predicted ensembles of αS conformations revealed the interactions that stabilize the monomer and the structures that drive the early stages of aggregation.495 The interaction between NAC and C-terminal was proposed to protect the highly hydrophobic NAC region from aggregation.482,483 However, the interaction between the N- and C-terminals places the NAC region in a solvent exposed orientation, which might trigger αS aggregation.484 Secondary structures, such as folded stable helices within the N-terminal and the NAC region,496,497 slow down the fibrillation rate by inhibiting the formation of partially structured helices.433,498 On the other hand, β-sheets and α-helix form frequently within the NAC domain, and exposure of these secondary structures in solvent may initiate the formation of toxic oligomers and promote fibril formation.488,499–503 Interestingly, transient trefoil knots were formed in an all-atom simulation with explicit solvent,504 and the authors deduced that the knots should increase accumulation of the protein and, hence, induce multimeric aggregation.
The conformational ensemble of monomeric αS predicted by CL-DMD revealed interaction between the NAC region and the C-terminal portion and secondary structures, such as α-helix near the N-terminus and β-sheet in the NAC domain, which are in agreement with the predictions from the other simulations.488 The contents of α-helix (~2.4%), β-structure (~29.1%), and other secondary structures (~68.5%) are consistent with the data from other simulations and CD studies of the monomer (α-helix 3 ± 1%, β-sheet 23 ± 8%, and random coil 74 ± 10%).477,488,505,506 The structures of the cluster centroids (Figure 14), however, are more compact than those determined by simulations in combination with PRE-NMR, FRET, or SAXS data.482,485,488 This might be due to the short-range cross-linking data used in the study. A total of 44 cross-links were used for CL-DMD simulations, including the cross-linkers EDC, TATA, SDA, and ABAS with spacer lengths of 0, 5, 5, and 7 Å, respectively. The maximal sequence separation by cross-linkers is from the N-terminus to E126, and the minimal sequence separation is from D121 to Y125 or E28 to K32. The structures with lowest 10% of the energies selected for clustering, which eliminated the unfolded conformations, might also lead to the compact conformation of α-synuclein.488 Although the radii of gyration (Rg, 14.1 Å) of the centroids are relatively small, the Rg determined by different techniques (NMR, PRE, SAXS, and smFRET) display significant disagreement ranging from 22.6 to 50 Å.476,477,485,507,508 This inconsistency and the large Rg values might be attributed to the multimeric states of αS under the experimental conditions. Moreover, the lower energy conformations are verified by the long-distance cross-linking, hydrogen–deuterium exchange, surface modification, and CD data, suggesting that the predicted compact structural ensemble by CL-DMD is convincing.
Figure 14.
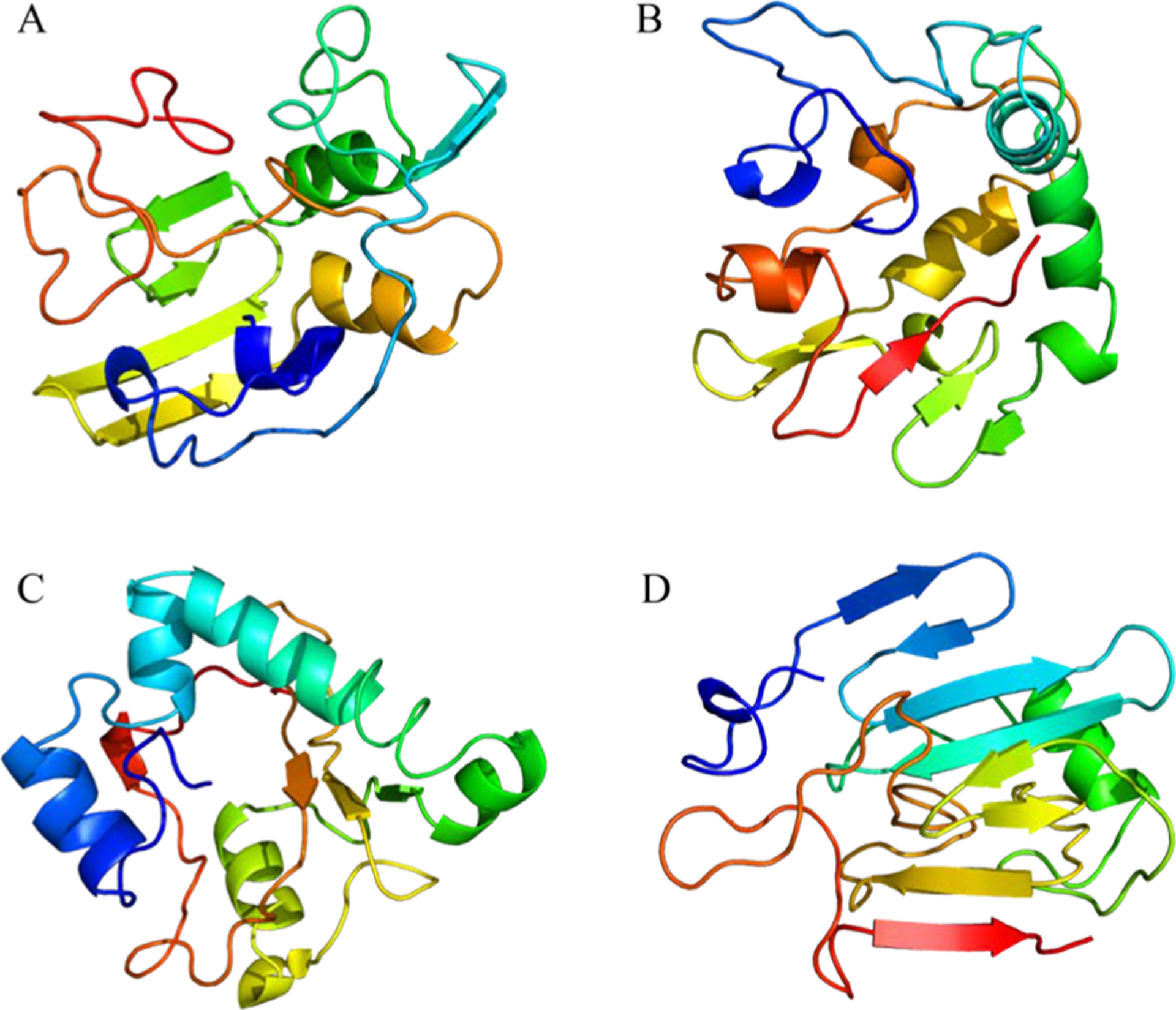
Representative structures of the αS conformational ensemble obtained by short-distance cross-linking constraint-guided DMD simulations.488 Conformers A, B, C, and D represent the first four clusters. Structures are colored from blue (N-terminus) to red (C-terminus). The figure was created using Pymol.254
Apart from the monomeric state of αS, MD simulations have been used to study dimeric and tetrameric conformations. In dimeric conformations, stable parallel or antiparallel β-sheets are formed within the NAC region through hydrophobic interactions.495,496,509 Dimer structures with β-sheets could generate stronger interactions between the monomers driving the oligomerization process toward fibrillar aggregation.510 Experiments revealed that αS can form a tetramer within the native cell environment.187 Simulations exhibit that the tetramer has less stable interchain β-sheets than the dimers, while the helices are more stable in the tetramer.509 The tetramer was stabilized by hydrophobic interactions mediated by helices in the N-terminal and NAC region and the salt bridge along with KTKEGV repeat motifs.484,511
Overall, simulations of αS predicted its heterogeneous conformational ensemble comprising metastable states.503 Analysis of key structural elements revealed the possible mechanisms that trigger oligomerization. The proposed mechanisms can be validated by future experiments, and the essential structural elements can be targeted to develop new therapies for neurodegenerative diseases.
5.3. Tau in All Its States
5.3.1. Impact of Phosphorylation and Other PTM on Tau Aggregation.
Phosphorylation and acetylation change tau aggregation and toxicity (Figure 15).159,160,512 While each kinase can phosphorylate 10–28 sites in tau, the combination of the four kinases can produce 32 (out of total of 52 phosphorylation sites when using the kinases separately).161 Furthermore, AD P-tau seeds hyperphosphorylated tau to form aggregates, which resist the dephosphorylation by PP2A, resulting in hyperphosphorylation and pathology of tau.513 To understand the self-acetylation activity and aggregation propensity of tau, Luo et al. investigated the conformational ensembles of K18 and K19 using REMD simulations.514 The simulation results revealed dynamically ordered conformations with close lysine–cysteine distances essential for tau self-acetylation. The “order in disorder” property in conformations provides the structural basis for tau self-acetylation. Interestingly, an acetylation–phosphorylation switch can regulate tau aggregation propensity and function.515 The acetylation on Lys-321 (within a KCGS motif) is both essential for acetylation-mediated inhibition of tau aggregation in vitro and a molecular tactic for preventing phosphorylation on the downstream Ser-324 residue. Phosphorylation of Ser-324 (pSer-324) has not previously been evaluated in the context of tauopathy, and increased deposition of pSer-324-positive tau has been observed in both mouse models of tauopathy and AD patients. These findings uncover a novel acetylation–phosphorylation switch at Lys-321/Ser-324 that coordinately regulates tau polymerization and function.515
Figure 15.
(A) Sequence alignment of the R1–R4 repeats and the sequence after R4 that are part of the CBD and AD fibril cores. The positions of filamentous β strands in both diseases are shown. PTMs detected by MS in tau fibrils from CBD case 1 and AD. The cryo-EM structures are shown with acetylation, ubiquitination, trimethylation, and phosphorylation sites marked with blue, orange, red, and green balls, respectively. Side chains with multiple PTMs detected are shown with two colors.518 (B and C) PTMs mapped onto schematics of the protofilament structures from (B) CBD case 1 and (C) AD. The same color scheme as described above is used to depict PTMs.518 (D) cis pT231-tau is highly neurotoxic and acts as an early driver of tauopathy, with bipolar illustrated here.525 (E) Snapshot from the simulation showing the α-helix and two salt bridge interactions (pThr231-Arg242 and pSer235-Arg242) of peptide htau225–250.521
Determining the functional relationship between tau phosphorylation and aggregation has proven a challenge owing to the multiple potential phosphorylation sites and their clustering in the tau sequence. For this reason, actual phosphorylation is often mimicked by mutating the selected amino acid into glutamate or aspartate. It has been shown that the two methods may produce a similar ensemble of conformations, even though the kinetic and chemical details that lead to it are quite different.516 Heparin-induced tau and in vitro phosphorylated tau have different conformations, properties, and activities.517 Decades of studies using the traditional methods and recent approaches of cryo-EM, specific kinases, and simulations provided unprecedented insights into phosphorylation effects on tau aggregation and toxicities.518–523
Cryo-EM and mass spectrometry of tau filaments from corticobasal degeneration (CBD) reveal that the CBD conformer is heavily decorated with PTMs, making it possible to map PTMs directly onto the structures (Figures 15A–C).518 By comparing the structures and PTMs of tau filaments from CBD and AD, it is found that phosphorylation occurs largely in the fuzzy coat. Previously, Xu et al. explored the conformational consequences of hyperphosphorylation on tau and also primarily in the fuzzy coat region.183 The presence of the phosphorylated terminal domains alters the relative stabilities of conformations in the K18 ensemble. The hyperphosphorylation of the two terminal domains decreases the attractive interactions among the N- and C-terminus and repeat domains. However, the structure with the straight repeats in the core region is still the most stable, and the exposure of the repeat domains upon hyperphosphorylation could enhance tau filament aggregation.183
The effects of phosphorylation are coupled with conformational and electrostatic contributions. In vitro kinase assays can generate well-characterized phosphorylated tau samples with combined phosphorylation at the Ser202/Thr205/Ser208 sites, together with the absence of pSer262. This phosphorylated tau readily forms fibers. Based on analysis of synthetic phosphorylated peptides, it was found that aggregation correlates with destabilization of the turn-like structure defined by phosphorylation of Ser202/Thr205.519 Using the time-resolved FRET method, Chin et al. studied the property of tau173–183 (AKTPPAPKTPP)524 and found that phosphorylation extends the end-to-end distance and increases the effective persistence length of this tau-derived peptide. However, the peptide extension is independent of salt concentration, indicative of a nonelectrostatic origin. These data indicate that geometric extension and stiffening at the peptide scale may be an important conformational consequence of phosphorylation in disordered proteins.
Two independent simulations found that the effect of phosphorylation on proline-rich domains of tau is mostly electrostatic.520,531 Lyons et al. modeled the phosphorylation induced conformational change on a peptide of htau225–250 with different phosphorylation patterns of the peptide (pThr231 and/or pSer235). All patterns were found to disrupt a nascent terminal β-sheet pattern (226VAVVR230 and 244QTAPVP249). The double pThr231/pSer235 phosphorylation pattern at experimental ionic strength resulted in the best agreement with NMR structural characterization, with the observation of a transient α-helix (239AKSRLQT245). They found that pThr231/pSer235 forms a salt bridge with Arg242 (Figure 15E).520 Another study provided a similar picture. Metadynamics simulations have been used to investigate the phosphorylation-induced conformational effects on a Tau segment (Tau225–246) from the proline-rich domain, 4 residues less than the first study.521 Two different phosphorylation patterns were investigated: group 1 with phosphorylation at Thr231 and Ser235 and group 2 with Thr231, Ser235, Ser237, and Ser238 phosphorylated. Phosphorylation leads to the formation of strong salt-bridge contacts with adjacent lysine and arginine residues, which disrupts the native β-sheet structure observed in Tau225–246. They also observed the formation of a transient α-helix (238SAKSRLQ244) when Tau225–246 is phosphorylated at four sites.521
The phosphorylation site Thr231 discussed above is followed by Pro232. Thr231 can be phosphorylated by various proline-directed kinases. Since that proline can exist in either trans- or cis-conformation defined by the prolyl bond, the coupling of the Thr231 phosphorylation and Pro232 isomerization generated a unique phenomenon and terminology in tauopathy: trans-tau and cis-tau (Figure 15D). Therefore, depending on the relative conformation of pThr231, the trans-tau and cis-tau may have different functions. A study found that cis pT231-tau is highly neurotoxic and acts as an early driver of tauopathy in several neurodegenerative diseases. Examination of bipolar and healthy human brain samples also detected cis p-tau in the patients’ brains.525
Antibodies have been developed to specifically recognize this unique phosphor-epitope in tau. The Fab fragment forms a complex with the tau peptide 224KKVAVVR(pT231)PPK-(pS235)PSSAKC241.526 In the Fab-peptide cocrystal structure, 10 amino acids (225KVAVVR(pT)PPK234) are visible, of which six (225KVAVVR(pT231)) interact directly with the Fab fragment, and the remaining eight residues of the peptide are disordered. The segment 224–241 is in the N-terminal side of the tau repeat domains, which starts at residue 243. Since the antibody recognizes the phosphorylated epitope in the intact molecule,526 the segment 224–241 must be exposed in the full-length tau. The critical phosphorylation site (pThr-231) is exclusively recognized by CDR-H2, which forms a positively charged pocket to accommodate the phosphate. The highly specific phosphate recognition explains why the antibody does not bind to nonphosphorylated peptides with the same sequence.526
Configuration specific antibodies were developed to distinguish cis- and trans-tau.527,528 It was found that cis, but not trans, p-tau appears early in mild cognitive impairment neurons and further accumulates in neurofibrillary degenerated neurons as AD progresses, localizing to the dystrophic neurites, an early hallmark change that correlates with synaptic and cognitive deficits. Unlike trans p-tau, the cis not only cannot promote microtubule assembly but also is more resistant to dephosphorylation and degradation and prone to aggregation.527
While the trans proline is thermodynamically more stable, the cis proline may form presumably by the help of peptidylprolyl isomerase. In order to examine the conformational preference of the Pro232, NMR was used to examine the conformation of all prolines in a functional tau fragment, Tau208–324.529 Although they detect and identify some minor conformers in the cis form, all prolines are for over 90% in the trans conformation. Phosphorylation by Thr231 specific kinase does not change preference of trans-configuration. The results hence disagree with the notion that specific prolyl bonds in tau would adopt preferentially the cis conformation.529 Other conflicting results were also observed in the proneness of aggregation. One study suggested that the trans isomer of tau peptide is prone to aggregate, and the WW domain of Pin1 drastically decreases its aggregation.530 It could be possible that both trans-tau and cis-tau are able to aggregate, depends in the environment in the experimental conditions to change their energy landscape. Accelerated MD were used to explore the conformational landscape of the tau segment containing the phosphorylated-Thr(231)-Pro(232) motif.531 The results show that intramolecular electrostatic interactions are coupled to the isomeric state of the peptidyl prolyl bond. Intramolecular electrostatic interactions are better formed in the trans isomer; however, the loss of intramolecular interactions and the more restricted conformational ensemble of the cis isomer could favor self-aggregation.532
Some phosphorylation sites in tau inhibit, rather than enhance, tau aggregation. It is within expectation when a phosphorylation site is in MBD. The effects of phosphorylation of two unique residues within MBD, Ser305 and Ser320, were examined in the context of established aggregation and seeding models.532 It was found that the S305E phosphomimetic significantly inhibited both tau seeding and tau aggregation in this model, while S320E did not. To further explore Ser305 phosphorylation in vivo, a monoclonal antibody (2G2) specific for tau phosphorylated at Ser305 was generated and characterized. Consistent with inhibition of tau aggregation, pSer305 was not detected in pathological tau inclusions in AD brain tissue.532 As can be seen in Figure 15, the Ser305 can be buried inside the fibril core (as in the case of CBD) or exposed on the fibril surface in AD fold. Therefore, depending on the diseases, we expected pSer305 may have a different outcome. Ser305 is not phosphorylated in CBD, since pSer305 would clearly disrupt the fibril core. In the AD fold, Ser305 could be phosphorylated since it is on the outside of the fibril core. However, the N-terminal or C-terminal fuzzy coat may prevent its binding with antibody 2G2. The second example of phosphorylation inhibition of tau aggregation also happens for the MBD sites. A total chemical synthetic approach to site-specifically phosphorylate the MBD of tau (K18) at single (pSer356) or multiple (pSer356/pSer262 and pSer356/pSer262/pSer258) residues was used to show that hyperphosphorylation of K18 inhibits (1) its aggregation in vitro, (2) its seeding activity in cells, (3) its binding to microtubules, and (4) its ability to promote microtubule polymerization.522 The inhibition increased with increasing the number of phosphorylated sites, with pSer262 having the strongest effect.522 The third example of phosphorylation effects is indirect. Proteolytic truncation of microtubule associated human (h) Tau protein by caspase-3 at the carboxy (C) terminus has been linked to the pathogenesis of AD. Ser422 phosphorylation blocks human tau cleavage by caspase-3 and suggests that the kinase associated with this Ser-phosphorylation may protect tau from aggregation.533 While the majority of phosphorylation sites in tau are threonine and serine, there are five tyrosine residues in Tau (Tyr-18, -29, -197, -310, and -394). As expected, phosphorylation of Tyr-310 would prevent tau aggregation, it is interesting to see that phosphorylation at multiple N-terminal tyrosine residues (Tyr-18, -29, and -197) also abolishes tau aggregation and inhibits its microtubule- and lipid-binding properties.534
Both Aβ monomer and oligomers are able to affect tau phosphorylation.523,535–537 Distinct Aβ assemblies activate neuronal signaling pathways in a selective manner and thus trigger tau phosphorylation.535 Soluble phosphorylated tau can happen even in initial increases of aggregate Aβ as early as two decades before the development of aggregated tau pathology.523 Aβ monomer can affect tau phosphorylation either through changing signaling pathways or directly interacting with tau protein. Aβ monomer induced phosphorylation of tau at Ser214 through both β2AR-cAMP/PKA-JNK and β2ARGRK signaling pathways.536 Intracellular binding of soluble Aβ to soluble nonphosphorylated tau promoted tau phosphorylation and Aβ nucleation.537 However, phosphorylation of Thr212, Ser214, Ser356, and Ser396 completely blocks Aβ42 binding.537 Ser356 phosphorylation also contributes to tau stabilization when PAR-1/MARK activity is elevated.538 Using 5 μs MD simulation of tau R3–R4 dimer with and without Ser356 phosphorylated, pSer356 not only perturbs the population of the β-helix motif spanning residues 336–354 but also induces distinct heterogeneous interfaces between the two chains compared to its WT counterpart. Also, pSer356 modulates the population of distinct architectures by increasing the number of globular shapes.539
5.3.2. Aggregation-Prone Conformation of Tau in Solution and Coacervation of Tau under Cellular Conditions.
In addition to forming fibrillar aggregates, tau has the ability to phase separate in the presence of RNA into liquid droplets. This process of liquid–liquid phase separation (also known as coacervation) into a polymer rich and a polymer depleted phase is observed in vivo for a number of IDPs (for example in the formation of membraneless organelles such as cajal bodies), and this process can be readily recapitulated in in vitro experiments. The role of tau coacervation in vivo is hotly debated. Coacervation has been proposed as a means of concentrating tau to facilitate microtubule binding. Its role could be a protective one, in which tau uses the droplets to store free tau and prevent its aggregation into fibrillar species. This premise is supported by experiments that show reversible formation of tau droplets.540 However, evidence of droplet “aging” in the context of hyperphosphorylated or disease mutant peptides suggests that in these pathological constructs, droplets may transition to a toxic, fibrillar state, thereby playing a role in neurodegeneration.541 We review recent literature focusing on simulations of the aggregation and coacervation of tau, with a focus on the MBD region (Figure 16). This region which consists of 4 repeat regions (R1, R2, R3, R4) is of particular interest as it can not only form fibrillar aggregates but also form liquid droplets in the presence of RNA.
Figure 16.
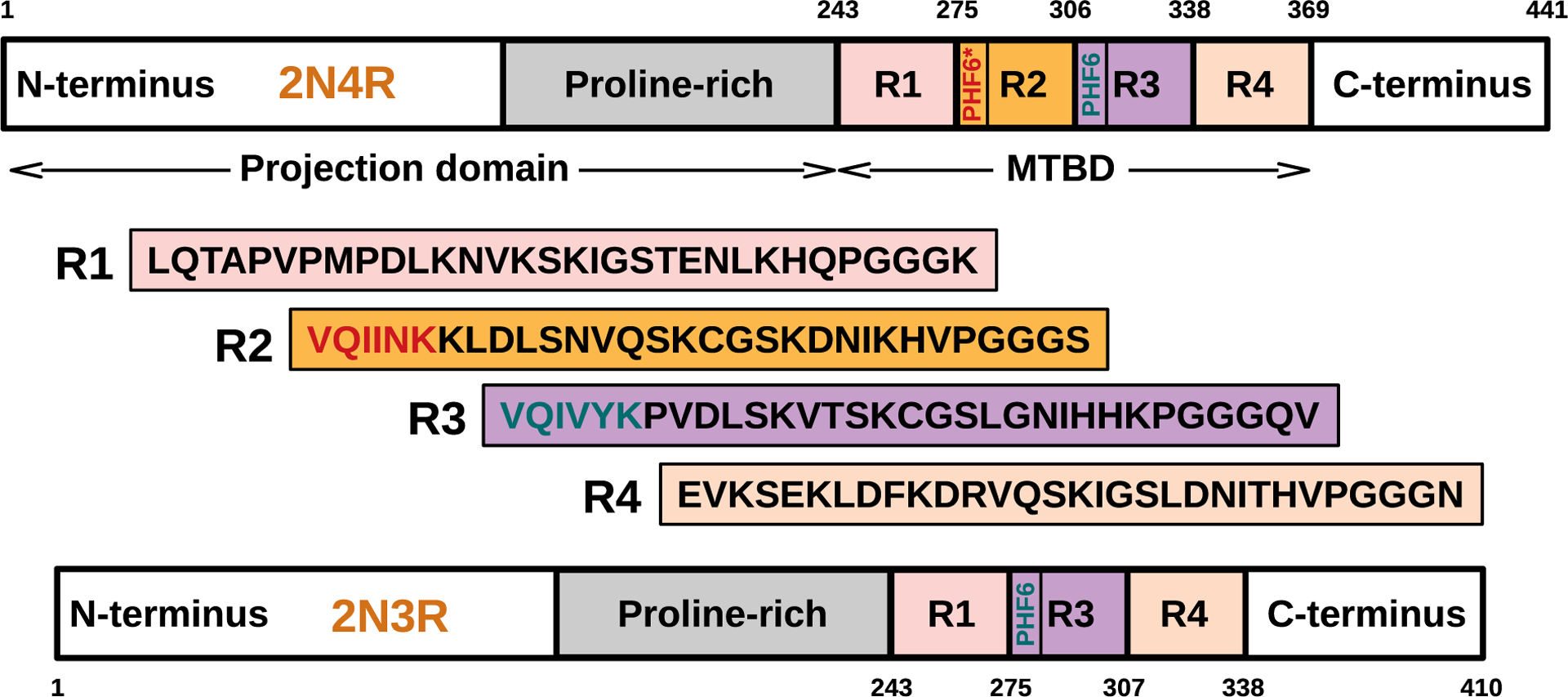
Schematic diagram of 4-repeat (2N4R) and 3-repeat (2N3R) human tau isoforms. The microtubules binding region (MBD) is highlighted, and the sequences for the repeat units are shown along with the hexapeptides PHF6* and PHF6.
5.3.2.1. Monomeric and Early Oligomeric Conformations of Tau in Solution.
In comparison to other IDPs such as Aβ and α-synuclein, the number of computational studies on tau is still fairly small. The primary reason is that full length tau is 441 amino acids in length, rendering its study computationally challenging. Hence much of the effort on the computational front has focused on studying fragments of tau rather than the full-length constructs. The smallest aggregating fragments of tau are PHF6* located near the R2 region and PHF6 located near the R3 region (Figure 16), and they serve as model systems for understanding the aggregation of this protein. The majority of tau simulation studies focus on PHF6/6* and on larger constructs encompassing these segments.
The earliest computational studies of tau involved MC485 and MD542 simulations aimed at understanding the conformational states sampled by tau in its monomeric form. An important outcome of these monomeric simulations has been to reveal that tau coexists between disordered structures with no detectable secondary structure along with structures that retain some elements of secondary structure. In addition, both extended and compact conformations were sampled in simulation. More recent simulations by Thirumalai and co-workers using the coarse-grained SOP-IDP model (Figure 9B) confirm that tau peptides do not behave simply as random coil chains but can adopt locally compact structures.31 The authors studied fragments of tau ranging from 99 to 441 amino acids in length and showed that the K25 fragment of tau populated an equilibrium ensemble of compact conformations determined not only by entropy but also from energetic interactions that are sequence dependent. Interestingly, a locally compact segment observed in the SOP-IDP simulations of the K25 fragment was also present in full length tau, highlighting the fact that the study of fragments can shed significant insights into the behavior of larger tau constructs.
Simulations support a picture in which monomers of small fragments can adopt a structure in the monomeric state that strongly resembles the structure adopted in the fibrillar state. For example, an aggregation-competent extended β-strand conformation was directly observed in simulations of the R2(273–284) tau fragment. In the case of the R2(273–284) tau fragment and its disease-implicated mutant R2(273–284, ΔK280), REMD simulations showed a net difference in the population of extended (aggregation-competent) structures in the case of the mutant over compact hairpin-like conformations, suggesting that the propensity to aggregate is encoded in the monomeric state.543–545 Simulations on dimers confirmed the hypothesis that structure encoded in the monomer could be transferred to oligomers. R2(273–284), R2(273–284, ΔK280), and the R3(306–317) homodimers were found to assume a mixture of compact and extended structures.543,546 However, the disease-related mutant showed a greater stability and a much greater probability of adopting extended homodimers than the less aggregation-prone variants of tau. Of interest is that the compact tau monomer conformations showed a gradual extension when the monomers formed dimers, higher order oligomers, and fibrils. Hence not only is fibril-structure encoded in the monomer, but oligomers can change the shapes of nonfibril competent conformations to further the aggregation process.547
Insights into the combined roles of the PHF6 and PHF6* peptides in promoting aggregation of tau can be gleaned from replica-exchange simulations of the longer fragment K18(244–372), which includes all the four repeat units. Simulations revealed a mixture of dynamically disordered and structured (α-helix and β-sheet) conformers514 and suggested that enhanced β-sheet conformations of the key aggregation-prone hexapeptides (PHF6 and PHF6*) and their relatively higher hydrophobic surface exposure could lead to nucleation and aggregation of tau. This conjecture about the role of the hydrophobic effect and secondary structure in driving aggregation of tau is supported by experimental studies that show that increased temperature induces compaction in tau and that furthermore, changes in secondary structure contribute to the thermal collapse (with entropic collapse as a driving factor).548,549
A number of simulations have focused not on the monomer structure but rather on elucidating the structures of stable oligomers and fibrils.327,543,546,550–554 Early MC study of the PHF6 domain predicted a fibril structure with mixed parallel and antiparallel β-strands, with an increase in the number of parallel strands for larger fibrils.550 Subsequent REMD simulations of the R3(306–317) domain (which contains PHF6) also predicted stable β-sheet structures for dimers with parallel orientation of the monomers.546 In contrast, the R2(273–284) (containing PHF6*) dimers were found to be consisting of antiparallel monomers. Using these antiparallel dimer structures as building blocks, fibrils were constructed using MD simulations by stacking two sheets made out of strands of R2(273–284).547 Antiparallel stacking of the sheets was shown to lead to more stable fibrils than parallel stacking, and this work provided a computational prediction of fibril structure of the R2(273–284) peptide. Comparing the stability of the dimers involving R2 (which includes PHF6*) and R3 (which includes PHF6), PHF6 was found to be significantly more aggregation-prone than PHF6*.543,546 Further REMD simulations by Bolhuis showed that PHF6 can form fibrils in a two-step process, while PHF6* remains as amorphous aggregates.344
Cryo-EM studies observed paired helical filaments of the R3-R4(306–378) domain of tau (extracted from the brain of an AD patient) and predicted C-shaped motifs for this birepeat.40 MD simulations of these filaments consisting of different birepeats of R1, R2, R3, and R4 showed that the C-shaped motif was only stable for R3–R4, while the R1–R2 repeat assumed a linear structure.555 However, a recent REMD study of an R3–R4 dimer did not identify C-shaped structures,539 which potentially indicates that the C-shaped motifs of this domain may only form in the context of the entire brain-derived filament, hence bearing the signature of pathogenesis.
All the simulations mentioned in this subsection were performed in water (with implicit or explicit description). The presence of small molecules and cosolvents in water can dramatically modulate the aggregation propensity of tau. The effect of the presence of osmolytic cosolvents, such as urea and methylamines in water, on the aggregation propensity of tau-fragments was studied by Shea and co-workers.556 ThT assay experiments showed that the protein-protective osmolyte trimethylamine N-oxide (TMAO) significantly enhances aggregation of R2(273–284), while protein-denaturant urea reduces it. Extensive REMD simulations showed that none of the osmolytes induces any new peptide conformation, otherwise absent without the osmolytes. However, urea was found to promote extended conformations of the peptide by directly binding to the peptide. In pure water, extension of the peptide would incur enhanced aggregation, as discussed above. Interestingly, since urea binds to the peptide, it reduces the number of available hydrogen bonding sites in the peptide which are crucial for the oligomerization process. Hence, urea inhibits the aggregation. In contrast to urea, TMAO was found to promote compact helical dimers of R2 that could further rearrange into β-rich structures. A TMAO-induced rearrangement of the water molecules around the amino acids was observed in simulations, and the importance of the dynamics of the surface water around Tau in the fibrillation process was further suggested in a combined neutron scattering and MD study by Weik and co-workers.557
5.3.2.2. Tau Coacervation.
The intrinsically disordered nature of tau, with its lack of a well-defined native state and of canonical secondary structure, predisposes it to exhibit a rich phase behavior.558 The balance between underlying entropic and molecular-level chemical interactions leads to the emergence of coherent mesoscale stable or metastable tangles with important biological implications. Tau liquid–liquid phase separation (LLPS) is a process in which the proteins, through a delicate interplay between solvation entropy and conformational energy, spontaneously assemble into a quasi-static dense liquid phase known as a coacervate, which is in coexistence with the surrounding supernatant-tau solution.559–561 The intramolecular energy scale of tau coacervation is typically of the order of the thermal energy fluctuation, and thus, the thermodynamic driving forces of LLPS can be affected by the conformational and compositional fluctuations.560 Favored by lower interfacial energy, the dense microphase of tau is in equilibrium, forming pseudospherical droplets.562 Recent in vitro experiments suggest that in some instances, coacervation, in the next stage of collective dynamics, might initiate and accommodate tau fibrillization.541,562–565 The mechanism of LLPS and the influence of various experimental parameters, namely, tau concentration, ionic strength, and temperature modulation, are largely unknown. Establishing the phase behavior of tau in physiological conditions is critical for understanding whether the thermodynamic state of tau coacervates in cellular environments is a stable mesophase or a transitional-intermediate state toward fibrillization.
In spite of recent advances in computational approaches and methodologies, many large-scale biological processes cannot be simulated with atomically detailed models. The computational limitations motivate the use of CG models that enable efficient simulations of complex systems. Mesoscopic physics models have been used to describe phase separation in the context of polymer models, and such approaches can be adapted to proteins.566–568 A promising computational approach, which can be used to understand sequence-dependent phase separation, is field-theoretic simulation (FTS) with complex-Langevin sampling. FTS is an approximation-free numerical method that fully accounts for inherent thermal fluctuations of the system. In FTS, by using the Hubbard–Stratonovich transformation, a particle-based model is exactly transformed to a statistical field theory through a self-consistent approach that decouples many-body interactions in the particle based model by introducing complex-valued auxiliary fields.540,569,570 Thus, by employing a CG model that properly describes the physics of tau polypeptide with its specific charge-sequence, the phase diagram and the relevant equilibrium properties can be computed accurately and efficiently. In this CG model, the solvent is treated implicitly, and the interactions are generalized into two types of potentials: long-range Coulomb interactions mediated by a solvent dielectric constant and a short-range repulsive interaction that reflects the solvent quality. Each amino acid of tau polypeptide is represented by a single monomeric unit, and the connectivity of the successive monomers of the chain is enforced by a simple harmonic potential. In this polymer physics model, the relevant thermodynamic state variables are an excluded volume parameter v, which parametrizes the strength of the repulsive potential at contact, and the Bjerrum length lB that parametrizes the strength of long-range electrostatic interactions.
Phase diagrams associated with biological phase separation have been the subject of a number of reviews, but to date, there has only been a single computational study of the phase diagram of tau, and we focus on this study below. A phase diagram maps out how the system behaves given a combination of state variables. Lin, McCarty, et al. employed FTS in conjunction with experiment to scrutinize the phase behavior of Tau-RNA LLPS and determine the conditions in which tau can undergo LLPS from a homogeneous phase.570 By performing an in vitro study of the N-terminus truncated isoform of human 4R, residues 255–441 (Figure 17a), they showed that tau-RNA LLPS is reversibly stable within a narrow range of biologically accessible conditions. The thermodynamic state and the phase diagram of tau-RNA solution were investigated under different solvent conditions (as described by the excluded volume parameter (v), ion concentration (that can implicitly be regulated by the Bjerrum length lB), and tau density. The results suggest that slight changes of the stimulus present inside the cellular environment (in other words, small changes in conditions inside the neuron) might be sufficient to induce LLPS. In Figure 17b, the instantaneous snapshots of the tau density profiles from FTS in two regimes are shown: (1) a homogeneous dilute solution phase in weak electrostatic strength and good solvent conditions; (2) a two-phase region in low salt concentration (or relatively large lB) and poor solvent quality, where a dilute supernatant and dense coacervate coexist. In both cases, the tau density is fixed and identical; however, due to the local density evolution, the uniformly distributed tau proteins throughout the solution gradually assemble and transform the conformational state from a single phase to a stable state in which tau phase separates into a dilute tau-depleted domain (white) and a condensed tau coacervate (red).
Figure 17.
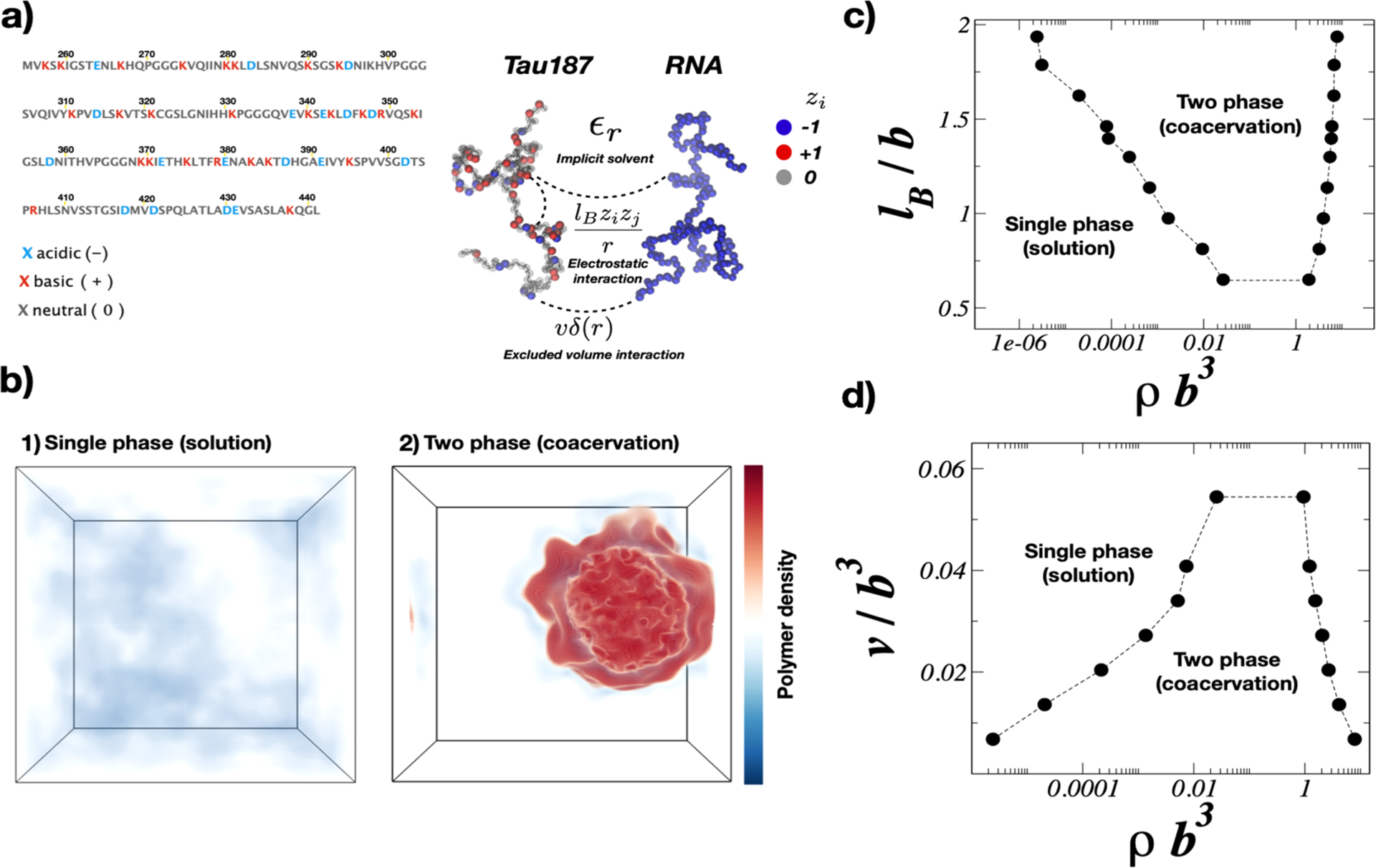
(a) Schematic representation of Tau and RNA models. Tau and RNA are modeled as chains of bonded monomeric units with size b in implicit solvent. The charge of each monomer is determined from the corresponding amino acid in the tau sequence at pH = 7. RNA is modeled as a uniformly charged polyanion. (b) Tau solution phases at fixed total density: (b-1) single phase solution in weak electrostatic strength and good solvent conditions (lB = 0.16 b, v = 0.02 b3); (b-2) two phase coacervate in relatively strong electrostatic interaction and low solvent quality (lB = 3.25 b, v = 0.0068 b3). (c) Coexistence phase boundary determined from FTS as a function of the Bjerrum length and tau density at fixed excluded volume of v = 0.0068 b3. (d) Binodal points as a function of the excluded volume at fixed Bjerrum length lB = 1.79 b.570
The first complete phase diagram of tau condensation studied in the literature is shown in Figure 17c, as a function of lB and tau density at fixed solvent conditions (constant v); similarly in Figure 17d, the phase behavior of tau solution is probed by varying the solvent quality at fixed lB. As is evident from Figures 17c and 17d, by increasing the solvent quality (or reducing v), the tendency of tau coacervation decreases. However, the increment of the electrostatic screening (or reduction of lB) results in inhibition of tau coacervation and promotes the appearance of a single homogeneous phase. The FTS phase diagrams shown in Figures 17c and 17d suggest that it is possible for cells to drive tau-RNA complex coacervation in vivo.
Experimentally, tau–RNA complex coacervation exhibits a lower critical solution temperature (LCST) phase diagram.540 By using the typical analytical polymer theories such as the Flory–Huggins–Voorn–Overbeek theory571 (with no charge sequence), one can adjust an effective temperature dependent χ parameter that enables an empirical fit to experiment; however, this approach provides limited insight due to the number of approximations involved. Instead, Lin, McCarty, et al. computed the phase diagram for tau–RNA complex coacervation using FTS with a linear temperature-dependent excluded volume.570 Using FTS with a proper polymer model that includes the polypeptide spatial charge-sequence (Figure 18) and by considering reasonable values of the model parameters to match the experimental conditions, one can provide direct approximation-free insights into the mechanism of tau LLPS in vivo. FTS simulations are emerging as a powerful tool to study the process of phase separation in vivo and hold the promise of not only guiding experimental studies but also shedding insight into the in vivo phase separation process.
Figure 18.
Coexistence points obtained from parametrized FTS at low salt (filled green circles) and at 120 mM NaCl (open green circles) as compared to experimental cloud point temperature as measured in turbidity experiments performed at 20 mM NaCl (filled red circles) and 120 mM (filled blue circles). The experimentally determined cloud point temperature corresponds to the low-density branch of the phase diagram. FTS simulations also predict the corresponding high-density branch of the binodal curve.570
6. INTERACTIONS OF AMYLOID PEPTIDES WITH CELLULAR MEMBRANES FROM SIMULATIONS AND EXPERIMENTS
Interactions of amyloid oligomers have been studied using different model membrane mimetic systems that include liposomes, bicelles, and nanodiscs by experiments and simulations.263,572–580 On one hand, amyloid proteins (e.g., Aβ, tau, hIAPP, α-synuclein, and prion protein), regardless of their sequences, structures, and normal functions, have a general ability to strongly interact with cell membranes. On the other hand, cell membranes serve as catalytic sites to facilitate the misfolding and formation of toxic amyloid oligomers, which in turn disrupt cell membranes via a two-step mechanism for Aβ, IAPP, and maybe others. A combination of ThT based fluorescence, dye-leakage fluorescence, AFM, solution NMR, and ssNMR experiments has been used to obtain insights into these mechanisms of membrane disruption, which is also shown to vary with the lipid composition and metal ions (such as Ca(II), Zn(II)). In what follows the following abbreviations are used: dipalmitoylphosphatidylcholine (DPPC), 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC), 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphatidylethanolamine (POPE), 1,2-dioleoyl-sn-glycero-3-phospho-l-serine (DOPS), 1,2-dihexanoyl-sn-glycero-3-phosphocholine (DHPC), dodecylphosphocholine (DPC), octyl glucoside (OG), dodecylmaltoside, decylmaltoside (DM), and lauryldimethylamine-N-oxide (LDAO).
6.1. Aβ40/42 and Its Mutants
Numerous studies have shown that Aβ can adsorb on, insert into, and destabilize cell membranes to induce cell membrane disruption via the direct and physical interactions between amyloid oligomers and cell membranes.
Experimental studies from AFM, electrophysiology, and cell calcium imaging have consistently shown that Aβ peptides can penetrate into the cell membrane to form ion-permeable, transmembrane amyloid pores.581–585 Aβ interaction with liposomes containing ganglioside (GM1), cholesterol, and sphingomyelin has been shown to form in-register parallel and two-residue-shifted antiparallel β-sheet structures that are tested with enhanced toxicity. By comparing the interaction of human and rodent Aβ with GM1 clusters in membrane, the formation of toxic fibers by human Aβ was identified, whereas rodent fibers are shown to be less toxic. Using MD simulations, it is shown that the side-chains of H13 and H14 residues of Aβ drive GM1 to clustering by interacting with its headgroup.573 Aβ is shown to interact and form oligomers at the membrane interface at nanomolar concentrations and is transient in nature as membrane-association and dissociation of oligomers are detected. An increase and decrease in the contents of β-sheet and α-helix, respectively, are observed for membrane bound preformed oligomers. Nanodiscs are shown to trap amyloid-intermediates in a near-native environment. Nanodisc trapped Aβ intermediates are shown to have a predominant β-sheet structure.263
With the exception of the very recent NMR structure of an Aβ42 oligomer formed in a membrane mimicking environment,586 namely an Aβ42 tetramer, which comprises a six stranded β-sheet core and reveals a mechanism of membrane disruption in which water permeation occurred through lipid-stabilized pores mediated by the hydrophilic residues located on the core β-sheets edges of the oligomer, a number of Aβ pores have been computationally constructed in different lipid bilayers using low-resolution structural data. Using NMR-determined β-strand-turn-β-strand amyloid monomers derived from Aβ fibrils as building block,587 Aβ pores were computationally constructed by preinserting annular Aβ oligomers with different numbers of peptides (12 to 36 monomers), pore sizes (2–4 nm inner diameter and 7–12 nm outer diameter), and pore topology (different β-strands facing the solvated pore or contacting with lipids) into different model lipid bilayers, including zwitterionic DOPC, DPPC, POPE, and mixed DPPC/DOPS bilayers.579,588–590 MD simulations showed some very interesting atomic details to describe pore structures/dynamics and ion permeability/selectivity. From a structural viewpoint, not all computationally constructed Aβ pores are structurally stable: (1) the lipid bilayers did not support too small 12-mer Aβ pores or too large 36-mer Aβ pores, and only 18- or 24-mer Aβ pores can retain their pore-like morphology across the lipid bilayers; (2) Aβ pores with CNpNC pore topology (i.e., hydrophobic C-terminal β-strands are in contact with lipids and hydrophilic N-terminal β-strands face the pore) are stable (Figure 19a), while Aβ pores with NCpCN are collapsed due to unfavorable peptide–lipid interactions.
Figure 19.
Different interaction models of full-length Aβ40/42 peptides with lipid membranes. (a) Aβ transmembrane pores with high Ca2+ permeability and selectivity.579 (b) Tetrameric Aβ42 β-barrel pores in PC/PS/cholesterol/sphingomyelin and DPPC bilayers.591 (c) Tetrameric Aβ42 α-helix-bundle pores in a DPPC bilayer with Ca2+ transport across the bilayer.596 (d) Aβ42 monomer with both α-helical and β-structure conformations being adsorbed on cholesterol-rich POPC bilayers, where increase of cholesterol promotes Aβ-membrane interactions and adsorption.603 (e) Aβ dimers on the GM1-clustering membrane, with C-terminal residues being inserted into the membrane.604 (f) Aβ tetramers with typical U-bent β-structure being preferentially adsorbed on and inserted into the POPE bilayer over the POPC bilayer, as driven by electrostatic interactions. (g) Aβ42 pentamer being adsorbed onto a POPC/POPG bilayer via Ca2+ ionic bridges between Glu22 and Asp23 and anionic headgroups of the lipid bilayer.610
Apart from the organization of Aβ strands parallel or antiparallel to lipids, another class of Aβ β-barrel pores were also proposed and modeled computationally with titled Aβ strands relative to lipids as assisted with structural data from experimental and theoretical results. Computational construction and MD simulations of tetrameric β-barrel transmembrane pores by Aβ42 or Aβ40 peptides in membrane bilayers of PC/PS/cholesterol/sphingomyelin and DPPC showed the higher structural stability of Aβ42β-barrels with different barrel topologies at the same diameter of 0.7 nm (Figure 19b), in sharp contrast to unstable Aβ40β-barrels.591 Mutations of D23N (more toxic mutant) or A2T (more protective mutant) did not largely affect the stability of Aβ42β-barrels in DPPC bilayers and thus appeared not to strongly correlate their structures with the known both mutant-induced toxicity.592 Computational Aβ42β-barrel pores, but not Aβ40 pores, also exhibited specific Zn2+ binding at the pore entrance, consistent with several experimental observations that Aβ42, not Aβ40, can (i) assemble into β-barrels in OG/DM/DHPC/LDAO/DPC membranes with varied pore diameters of 0.7 nm593 and 1.7–2.4 nm594 and (ii) induce ionic currents in planar lipid bilayers by electric recordings.593 Moreover, different from small Aβ β-barrel pores, they could also exist with the much larger sizes formed by six parallel Aβ hexamers (36 monomers) with N-terminus segments facing the pores in the POPE bilayer.588,589 A recent study from native ion mobility-mass spectrometry revealed that Aβ42 formed a diverse set of hexameric β-barrel pores in a membrane-mimicking environment, including tilted hexameric barrels, star shaped hexamers, dimers of trimers, and trimers of dimers,595 all of which shared the similar peptide organization by folding and assembling hydrophobic C-terminal ends of the Aβ-peptide into a pore center. In parallel to β-structure-based transmembrane pores of Aβ, α-helix bundles of the Aβ17–42 tetramer and trimer can also form transmembrane pore-like structures, which were stable enough to transport Ca2+ ions across the DPPC bilayer596 (Figure 19c). Ca2+ transport and binding sites were similar to a classical voltage-gated calcium pore but not an M2 proton pore. The α-helix pores of Aβ provide additional possible pathways for Ca2+ homeostasis.
While these membrane-supported Aβ pores have a wide variety of conformational heterogeneities and polymorphisms, they all adopt some common structural characteristics, including pore sizes, irregular shapes, and assembly patterns. Briefly, stable Aβ pores are made of four to six, small, dynamic oligomers (each oligomer contains 4–6 Aβ monomers), which are loosely associated with each other through some β-strand interactions to prevent pore dissociation. Such irregular and polymorphic Aβ pores suggest the possible pore formation pathways; that is, small Aβ aggregates (monomers or oligomers) first insert into the membranes, followed by the self-assembly of these inserted Aβ aggregates into different Aβ pores. From a membrane permeability viewpoint, these transmembrane Aβ pores indeed exhibited specific cation selectivity (particularly for Ca2+ and Zn2+) and characteristic stepwise ion permeability, presumably due to specific side chain orientations and organizations of Aβ.597 Ca2+-permeable Aβ pores are an attractive target to develop inhibitory compounds, and thus prevention of Aβ-induced Ca2+ influx is expected to restore the Ca2+ balance, cycling, and bioenergetics in neurons that would generate a potential therapeutic strategy for AD treatment. Taken together, Aβ pores were found in different lipid membranes, suggesting that the formation of a pathogenic, ion conducting pore by Aβ is considered as a general membranotropic mechanism of AD.593 In principle, amyloid polymorphism in solution and the cell membrane allows formation of a wide variety of amyloid pores, but only stable Aβ oligomers that adopted a specific structure and incorporated into membranes as pores are more biologically linked to neurotoxicity. On the other hand, it is known that membrane components such as anionic lipids, gangliosides, and cholesterol could influence Aβ pore formation by altering membrane structure and Aβ–membrane interactions.598–600
Apart from Aβ-induced ion permeable pores, numerous simulations have studied the interaction of Aβ monomers (Aβ40 or Aβ42) and oligomers of different sizes and conformations with different lipid bilayers and observed that the adsorption/insertion of Aβ peptides on/into cell membranes could also increase membrane conductance. On the other hand, some controversial results were also reported for membrane-induced Aβ conformations and Aβ-induced membrane thinning effects, probably due to different experimental/computational setting-up and conditions and different structural ensembles in a complex Aβ-membrane energy landscape. For instance, Aβ42 monomers with the disordered or β-hairpin structures can be adsorbed on both anionic DOPS and zwitterionic DPPC bilayers, but Aβ42 conformations were well retained on the DOPS bilayer as compared to the DPPC bilayer, due to the stronger electrostatic interactions between Aβ42 and DOPS.601 Aβ40 also behaved similarly to being strongly adsorbed on anionic DLPG liposomes, with a spontaneous structural transition from where it initially adopts mixtures of disordered and helical structures.602 Furthermore, increased cholesterol in POPC bilayer promoted Aβ42 monomer with different conformations (i.e., α-helical and β-hairpin) to be adsorbed on the POPC bilayer, which further facilitated Aβ aggregation and membrane insertion (Figure 19d).603 For a more realistic and complex POPC bilayer containing ganglioside (GM), sphingomyelin (SM), and cholesterol (Chol), Aβ42 monomer adsorption behavior was strongly dependent on the ratios of Aβ:membrane components;604 that is, Aβ42 monomers favored the α-helical conformation at a lower ratio of Aβ:GM but changed to the β-strand-rich conformation when this ratio was high (Figure 19e). At the same ratio, Aβ42 dimerization significantly promotes β-structure formation.605 Similar simulation results were presented where Aβ40 remained inserted in POPC, POPS, POPC/POPE, and raft membranes, and Aβ-GM1 interactions promote the structural conversion of Aβ from α-helix to β-strand.606 In parallel, numerous experiments from Z-scan fluorescence spectroscopy, cross-correlation spectroscopy, and fluorescence lifetime Förster resonance energy transfer have confirmed that sphingomyelin triggers oligomerization of Aβ40 and that GM1 is counteractive thus preventing oligomerization.607 These findings not only explain why Aβ is favorably bound to the GM1/SM/Chol membrane but also reveal the important role of Aβ concentration and initial oligomerization in promoting the transformation into a β-sheet structure in the presence of raft-like membrane in the brains of patients with AD.
Membrane-bound Aβ oligomers also showed preferential adsorption and insertion into the POPE bilayer over the POPC bilayer.608,609 While both POPE and POPC bilayers are electrostatically neutral, different structural morphologies between zwitterionic groups in both lipids lead to strong differences in electrostatic attraction between the proximal charged residues of Aβ oligomers and lipid headgroups (Figure 19f). Furthermore, comparison of the interactions between Aβ pentamer with neutral POPC and anionic POPC/POPG (3:1) bilayers revealed that Aβ pentamer had much stronger interactions with anionic POPC–POPG lipids than neutral POPC lipids, and such strong interaction with and adsorption on the anionic POPC/POPG bilayer were mainly driven by electrostatic interactions, consistent with experimental observation that Aβ adsorption and fibrillation are enhanced on anionic lipid bilayers.610 More importantly, Ca2+ ions are found to form ionic bridges to associate negatively charged residues of Aβ with anionic headgroups of the lipid bilayer, resulting in Aβ–Ca2+–PO4− complexes (Figure 19g). Further SPR and AFM data also confirmed that Ca2+ ions and the lipid bilayer concertedly accelerate the conformational change or misfolding of Aβ.611 Intensive Ca2+ bound to the lipid bilayer and Ca2+ ionic bridges may explain Ca2+ hemostasis responsible for neuronal dysfunction and death. Aβ oligomers also affect neuronal Ca2+ homeostasis by modulating the activity of NMDA receptors. Ca2+ bridges have also been shown to mediate the interaction between the membrane and the acidic tail of αS and hIAPP.612,613
While different and somewhat conflicting data have been reported due to notoriously more complex Aβ-membrane systems, some common observations regarding the interaction of the various Aβ species with membranes could still be achieved. First, upon adsorption of Aβ on different lipid membranes, Aβ aggregates usually undergo structural transition toward a β-sheet-rich structure, which appears to be a prerequisite step for facilitating Aβ aggregation and insertion to induce membrane damage. Second, electrostatic and hydrogen bonding interactions are often observed and considered as a crucial driving force for modulating the anchoring and insertion of Aβ peptides into lipid bilayers. Third, Ca2+ promotes membrane interactions of Aβ aggregates to facilitate their aggregation, adsorption, and insertion to form Ca2+-permeable pores in membranes or to increase membrane conductance. Blocking of Aβ pores by small-molecule compounds enabled reduction of the Ca2+ conductivity of Aβ pores and rescue of synaptic plasticity and memory function for amyloid pathology in a mouse.614 This may explain a possible link between calcium homeostasis and amyloid hypotheses. Finally, membrane components (e.g., lipids, cholesterol, sphingomyelin, ganglioside) with different structure, hydrophobicity, and charges play an important role in determining their interactions with Aβ (i.e., adsorption, orientation, and insertion) and the fate of cell membranes, but the exact mechanisms of membrane-induced Aβ aggregation and toxicity are still far more complex than what we expected. In a broader view, these types of membrane disruptions have been shown to be the molecular basis of cytotoxicity for other amyloid proteins and even antimicrobial peptides, including bacterial pore-forming toxins.615
6.2. Tau and IAPP
6.2.1. Interactions of hIAPP with Lipid Bilayers.
In general, hIAPP–membrane interactions have mutual effects on the structure and dynamics of both hIAPP and cell membrane. On one hand, hIAPP aggregates of different sizes and structures would disrupt the integrity, permeability, and functions of cell membranes, leading to cell death. On the other hand, the presence of cell membranes also modulates the aggregation kinetics, pathways, and structures of hIAPP; in most cases, cell membranes accelerate the structural transition to β-structure and aggregation kinetics toward amyloid fibrils.
hIAPP and Aβ have very similar properties with regard to membrane interactions, and both peptides adopt an α-helical conformation when interacting with lipid membranes prior to amyloid formation. Similar to other amyloid proteins, the interaction of hIAPP monomer with the lipid membrane is considered as the first step to initiate hIAPP aggregation on the cell membrane. A number of MD simulations have been performed to study the interaction of hIAPP monomers with different conformations (e.g., α-helix, β-hairpin, and coil structure) with lipid membranes (e.g., pure POPC, POPG, and DOPS and mixed DOPC/DOPS, POPC/POPE, DPPC/DPPS, and DVPC/DVPS). These MD simulations consistently showed that independent of initial hIAPP monomer conformations and membrane components, hIAPP monomers were always adsorbed on these lipid membranes.613,616–619 Such adsorption was largely driven by electrostatic interactions between positively charged residues from the N-terminal region of hIAPP and negatively charged headgroups of lipid membranes613,620–622 (Figure 20a). Upon initial adsorption, specific hIAPP–lipid interaction would drive hIAPP monomers to adopt α-helical623 or coil616 structure and orientations for better establishing stable and strong contacts with lipid membranes624 (Figure 20b), followed by subsequent full or partial insertion of hIAPP monomers into lipid membranes618 (Figure 20c). During this adsorption–insertion process, structural transition of hIAPP peptides toward β-sheet-rich aggregates would occur at any stage of the hIAPP aggregation process both in solution and on the cell membrane.624 All-atom MD simulations of hIAPP monomer with α-helical structure on DOPC, DOPS, and a mixed DOPC/DOPS (7:3) showed that the helicity of hIAPP monomers was reduced on DOPS bilayers, rearranged on DOPC bilayers, and enhanced on DOPC/DOPS bilayers.618 Moreover, hIAPP monomers on anionic POPG bilayers experienced the faster dynamics to orient their N-terminal toward the membranes than those on neutral POPC bilayers.620,621,625
Figure 20.
Typical interaction model of hIAPP monomer with lipid bilayers via a three-step approaching–adsorption–insertion process. The hIAPP monomers with the disordered structures approach the cell membrane to establish initial contacts with their N-terminal residues via electrostatic interactions, then adjust their orientation parallel to membrane surfaces with hydrophobic residues facing toward hydrophobic tails of lipids, and finally tend to insert partially or fully into cell membranes. During the whole process, hIAPP monomers always involve the conformational changes from the bulk phase to the membrane surface to the membrane interior for inducing their potential membrane-disruption activity.
Moreover, different lipid membranes lead to different insertion dynamics of hIAPP monomers. Unstructured hIAPP monomer tended to insert into POPC bilayers by establishing an initial contact of amphiphilic F23–F25, followed by a subsequent insertion of T4–T9. Differently, α-helical hIAPP monomer used its N-terminal residues (C7–Q10) to insert into POPG bilayers, and further insertion of F15–H18 led to the conformational change of hIAPP monomer to β-structure.616 Another simulation strategy was applied to preinsert hIAPP monomer into POPG lipid bilayer, in which hIAPP monomer liked to bind to POPG bilayer with α-helical structure. Moreover, REMD simulation of a hIAPP1–19 fragment with a POPG bilayer confirmed that hIAPP1–19 favored to adopt random coils with a parallel orientation to the POPG surface at the adsorption state but changed to helical structure inside the POPG bilayer at the insertion state. In addition, the presence of cholesterol in DPPC/DPPS bilayers promoted hIAPP1–19 aggregation, while the absence of cholesterol alleviated the insertion tendency of hIAPP1–19 into in DPPC/DPPS bilayer.617 This indicates that cholesterol may play a double-edged sword to accelerate hIAPP aggregation and prevent membrane damage from hIAPP insertion.
From a computational viewpoint, spontaneous insertion of any protein or peptide into the cell membrane encounters a substantial energy barrier, which is typically beyond the time scale explored by MD and REMD simulations. To overcome this issue, a hybrid simulation method combining the adaptive biasing force (ABF) method with the MARTINI CG force field is often used to probe a complete membrane insertion process of proteins or peptides with different insertion pathways. Potential mean force (PMF) is calculated, using the umbrella sampling protocol, to directly measure the free energy barrier required to transfer the peptides from the bulk water phase to the water–membrane interface to the bilayer interior. PMF profiles from the ABF-based simulations of hIAPP monomer insertion into a 7:3 DOPS/DOPC bilayer showed new observation that hIAPP monomers were more energetically favorable to insert their tails into the lipid membranes than their turn regions at a lower energy penalty by ~11 kcal/mol.613 Different from PC, PG, and PE-based bilayers that do not present in the outer leaflet of islet β-cell membranes, anionic ganglioside lipids (G-lipids) are the main component of the pancreas. A combination of CG and all-atom MD simulations were applied to study the interactions of hIAPP monomers with GM3-enriched DOPC (GM3/DOPC) bilayers.626 Simulations showed that GM3 lipids tended to induce conformational changes of hIAPP from α-helix to β-hairpin structure, in contrast to DOPC bilayers to retain the α-helix of hIAPP. The ratio of GM3/DOPC components determines not only the conformational population of hIAPP but also the aggregation kinetics (acceleration or inhibition) of hIAPP. Membrane binding was driven by electrostatic interactions between positively charged N-terminal residues and the anionic elements of the membrane. It is also interesting to observe from different simulations of hIAPP monomers with lipid membranes that the adsorbed hIAPP monomers, regardless of different conformations, often adopted highly populated α-helical conformations and oriented their hydrophilic residues toward lipid head groups, but their hydrophobic residues toward lipid tail groups, and such orientation would facilitate hIAPP–hIAPP interaction to promote their oligomerization and fibrillization on lipid membranes.
Numerous computational studies of the interaction of hIAPP oligomers with lipid membranes have also been reported, which are more biologically relevant to hIAPP oligomer-induced cell toxicity, particularly to the membrane disruption mechanism.617,627,628 hIAPP dimers as the smallest oligomers and building blocks have been computationally modeled with initial α-helical structures to interact with and penetrate into POPG bilayers.621 It was found that the interaction of hIAPP dimers with the POPG bilayer is mostly driven by the N-terminal positively charged residues via electrostatic interactions, and upon adsorption/insertion of hIAPP on/into the POPG bilayer, hIAPP dimers had a larger disturbance on the ordering of head groups of the POPG bilayer than hIAPP monomer, but both hIAPP dimers and monomers still largely retained their α-helical structures (Figure 21a). Later, a relatively large size of hIAPP pentamer with β-sheet conformation was selected to interact with both POPC and POPC/POPE (3:1) bilayers to examine the β-structure effect of hIAPP pentamer on the adsorption, orientation, and interactions of hIAPP on both bilayers.613 hIAPP pentamer requires a specific orientation to be adsorbed on both lipid bilayers via N-terminal residues, as driven by electrostatic interactions. Because dominant driving forces mainly stem from electrostatic interactions, hIAPP pentamer showed stronger interactions with mixed POPC/POPE lipids than pure POPC lipids, but no membrane disruption effect was observed (Figure 21c). Differently, when partially inserting hIAPP oligomers (e.g., trimer, tetramer, and pentamer) into DPPG or mixed DPPC/DPPG (7:3) bilayers, while all hIAPP oligomers disturbed the lipid ordering and the local bilayer thickness around hIAPP,629 hIAPP pentamer had a shallow insertion depth of ~1.62 nm and a small insertion title angle ~52° into DPPG bilayer, as compared to a deeper insertion of 1.75 nm and a larger title angle of ~77° into the DPPC/DPPG bilayer (Figure 21b).629
Figure 21.
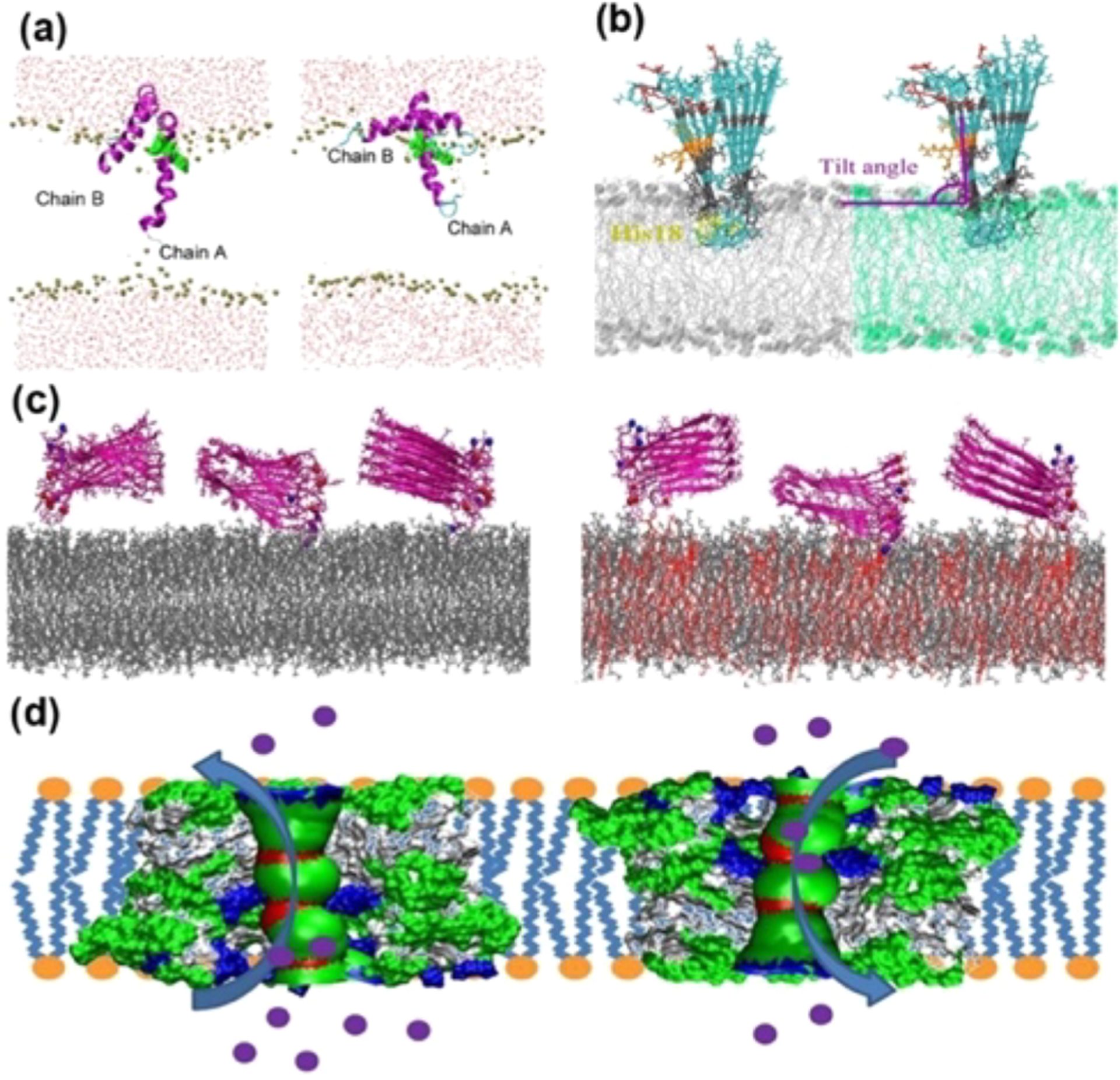
Different interaction models of full-length hIAPP1–37 oligomers with lipid membranes. (a) hIAPP dimers with α-helical structure insert into POPG bilayers with different insertion depths, strongly depending on interactive contacts of hIAPP dimers with POPG bilayers.621 (b) hIAPP pentamer with U-bent β-structures to be partially inserted into (left) DPPG and (right) DPPC/DPPG bilayers via the turn region.629 (c) hIAPP pentamer with U-bent β-structures to be adsorbed on (left) POPC and (right) POPG/POPE bilayers with different hIAPP orientations.613 (d) Double hIAPP pores with different (left) “turn-to-tail” and (right) “tail-to-turn” orientations in DOPC bilayers to show nonselective, ion-permeable activity.632
Besides adsorption and insertion models, the hIAPP pore model was also proposed and computationally studied in the presence of different lipid membranes, with special focus on ion selectivity and permeability of hIAPP pores. Using NMR-determined β-strand–turn–β-strand hIAPP monomers de rived from amyloid fibrillar structure as a building block, a series of hIAPP pore models with different sizes and orientations were constructed and preinserted in the presence of a DOPC bilayer.630,631 It was found that all individual hIAPP pores displayed high selectivity for Cl− ions over other cations (Na+, K+, and Ca2+), but the coexistence of “turn-to-tail” and “tail-to-turn” double hIAPP pores resulted in the nonionic selectivity. Double hIAPP pore simulations explain experimental observation that hIAPP forms nonselective, ion-permeable channels in planar phospholipid bilayer membranes, due to the coexistence of “turn-to-tail” and “tail-to-turn” channels in the membrane (Figure 21d).632 Computationally modeled hIAPP pores showed similar structural morphologies to AFM-identified hIAPP pores,581 including inner diameters of 1–2 nm and outer diameters of 8–12, irregular pore shapes assembled by multiple, small oligomers, and U-turn region orientation. Despite different simulations of hIAPP with lipid membranes, some general interaction modes between hIAPP and cell membranes are observed at the molecular level. Regardless of hIAPP sizes/conformations and lipid components, hIAPP peptides have high probability for its N-terminal residues to be adsorbed on and inserted into cell membranes as driven by electrostatic interactions. Consistently, hIAPP also favors to interact with anionic cell membranes over neutral ones, further highlighting the importance of electrostatic interactions in hIAPP–membrane interactions and potential membrane disruption mechanisms, consistent with experimental observation that hIAPP adsorption and fibrililation are enhanced on anionic lipid bilayers. A high-resolution structure of membrane-associated hIAPP was solved using NMR distance constraints and MD simulations. The structural model shows that each monomer subunit in the hIAPP oligomer is characterized with three antiparallel β-strands in the regions A8–L12, F15–H18, and I26–S29.285
6.2.2. Interaction of Tau with Lipid Bilayers.
Figure 22 illustrates the pathological process of tau production, hyperphosphorylation, oligomerization, fibrillization, and interaction with cell membranes. Several lines of evidence have shown that tau is able to directly interact with plasma membrane via multiple binding modes, including involving the N-terminal projection domain and R1–R4 repeats.633–637 Purified PHFs were found to contain different cell membrane components, including phosphatidylcholine (PC), cholesterol (CH), galactocerebrosides (GC), and sphingomyelin (SM), supporting the pathology mediated by tau–membrane interactions in AD.638 Tau exhibited the stronger interactions with anionic DMPG bilayers than neutral DPPC bilayer, leading to complete structural disruption of DMPG bilayers in sharp contrast to intact DPPC bilayer.639 In addition, tau also interacts with other ionic vesicles and micelles to promote tau oligomerization and PHFs formation.635,636,639–642 Tau proteins have the tendency to form porelike amyloid structures in brain tissue from patients with progressive supranuclear palsy (PSP) and dementia with Lewy bodies (DLB), as well as in the P301L mouse model,643 which mimic the membrane-disrupting properties of pore-forming protein toxins, consistent with Aβ, hIAPP, and αS pores.644 These studies from different aspects suggest that tau–membrane interactions not only mediate conformational change of tau but also accelerate nucleated seed formation, both of which modulate the aberrant aggregation of tau into mature fibrils and induce adverse effects on the structural integrity and biological functions of cell membranes.639,645,646 Considering its large size and its disorder, no molecular simulations have been conducted to study the interaction of full-length tau with lipid membranes. Thus, this remains as a major challenge to be overcome by continuous and fast progress in computational hardware technologies, simulation algorithms, and even machine learning methods.
Figure 22.
Pathological process of tau production, hyperphosphorylation, oligomerization, fibrillization, and interaction with cell membranes.
6.3. α-Synuclein Membrane Interaction
The large majority of the putative functions of αS are based on a fundamental step requiring the binding to biological membranes.189,647 The interaction with the membrane is indeed a signature of the biological active form of αS in vivo206 and plays a central role under pathological conditions by influencing the kinetics of αS aggregation, with enhancement or inhibition being observed depending on the experimental conditions648,649 and the toxicity of its oligomers.650
The key membrane interaction of αS in vivo is established with synaptic vesicles.194,200,201,204 In addition αS has been shown to bind a variety of other cellular membranes, including mitochondrial membranes and the presynaptic membrane,189 as well as nonphysiological interfaces such as detergent micelles651 and the water–air interface.652 In general, the membrane-binding affinity of αS has been shown to be favored by negative charges and high curvature of the membrane.653
Structural studies have clarified the topology of the amphipathic helical segments forming in αS upon membrane binding, indicating that these adopt a parallel orientation with respect to the membrane,299 with most of the protein remaining bound at the membrane surface while the first 12 residues of αS experience a mild insertion into the hydrophobic region of the bilayer.654 In the micelle-bound state, a structure of a “broken helix” with two helical segments (residues 3–37 and 45–92) followed by a disordered C-terminal region was refined,651 whereas structural studies of the membrane-bound state indicated mostly a single helix ranging from residues 1 to 97.655 But the key feature governing the biological properties of the membrane-bound state of αS appears to be the dynamical content enabling a multiplicity of binding modes to occur by the same protein.656 Conformational dynamics in the membrane-bound state differ significantly across the αS sequence, with a rigid N-terminal region (residues 1–25) serving as a primary binding site that anchors the protein to the membrane, a central region featuring an equilibrium between bound and unbound states (residues 26–97), and a disordered-unbound C-terminal region (residues 98–140)299 (Figure 23). Of these regions, the most enigmatic is certainly the central that, in addition to hosting the amyloidogenic NAC segment, has been shown to influence the binding affinity to acidic lipid bilayers as well as the ability of αS to bridge multiple membranes in a so-called “double-anchor” mechanism.199,203 The equilibrium between membrane-bound and detached states of the region 65 to 97, however, may also lead to aberrant aggregation of αS at the membrane surface as it determines the level of exposure of the NAC region. A view is emerging, therefore, that the balance between functional and pathological roles of αS are inextricably connected to the equilibrium between ordered and disordered conformations of the region 65 to 97 at the surface of biological membranes.647 This balance can be influenced by external factors such as pathological mutations, particularly A30P, G51D, and E56K having significant effects on the membrane binding, and PTMs such as phosphorylation of residues Ser 87,657 Ser 129,658 and Tyr 39659 that also regulate the equilibrium between membrane-bound and detached states of αS (Figure 23). A key posttranslational modification for the membrane binding of αS is the N-terminal acetylation,650,660 which is believed to define the predominant form of the protein in vivo and that enhances the α-helical propensity of the N-terminal residues to increase the membrane affinity.661
Figure 23.
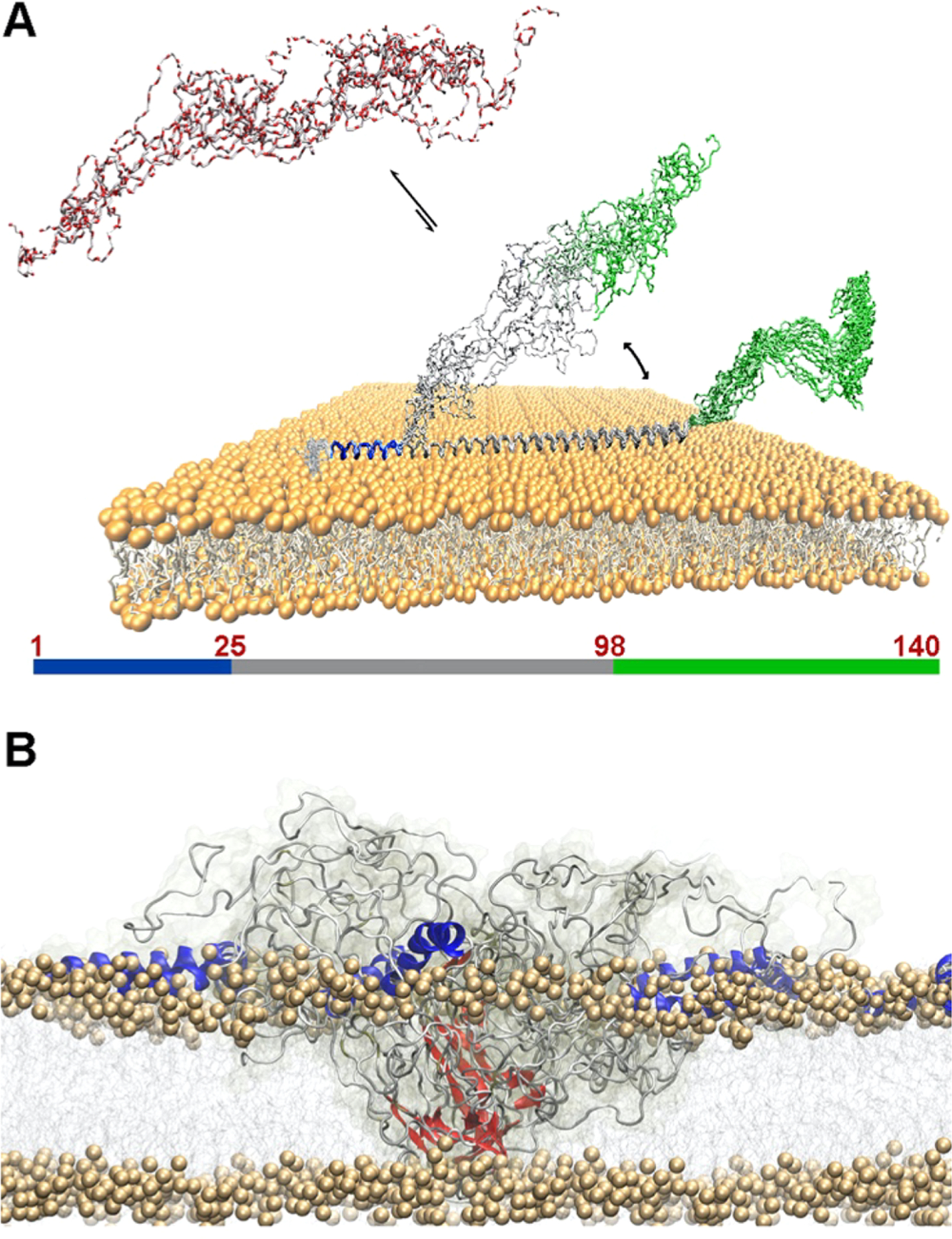
Membrane binding by monomeric and oligomeric αS. (A) Monomeric αS is disordered in solution (red) and binds the membrane with three regions having distinct structural and dynamical properties. The N-terminal region (blue) acts as a rigid membrane anchor. The central region (gray) is in conformational exchange between membrane-bound (helical) and detached (disordered) conformations. The C-terminal region (green) remains essentially unbound from the membrane.299 (B) The membrane binding by toxic αS oligomers involves N-terminal regions (blue) of αS molecules from the oligomer, strongly anchoring the oligomers to the membrane surface in a cooperative manner, and the prefibrilar β-sheeted rigid core (red), inserting into the lipid bilayer and disrupting its integrity.650
As expected, the binding modes of the physiological monomers of αS were found to differ significantly from those of its neurotoxic oligomers. In particular it was found that prefibrillar toxic αS oligomers bind neuronal membranes via a lipophilic element, which is composed of several copies of the N-terminal region of αS molecules from the oligomers, and insert their structured hydrophobic core into the lipid bilayer by disrupting its integrity.650 This key mechanism of membrane interaction and disruption is followed by imbalance in calcium and metal ions, leading to downstream processes of cellular toxicity such as the generation of reactive oxygen species and disruption of the mitochondrial function.650
7. ROLE OF METAL IONS IN AMYLOID DISEASES
Metal ions such as Cu, Zn, Ca, and Fe have been thought to play important roles in the development of a variety of amyloid diseases including AD and T2D. For example, high concentrations of metal ions have been found to be colocalized along with amyloid fibrillary aggregates in the senile plaques and with amyloid-β in blood vessels from the brains of AD patients.662 Specifically, zinc with its highest content in islets (millimolar)663 stored with insulin and IAPP is shown to have a direct correlation with the progression and severity of T2D. To this end, the effect of metal ions in modulating the aggregation kinetics and morphological and pathological phenotypes of several amyloidogenic peptides/proteins has been tested in vitro.664 Here, we cover the reported studies on the effects of metal ions on the amyloid aggregation and toxicity of amyloid-β, αS, tau, and IAPP.
7.1. Biophysical Probing the Effects of Metal Ions on Amyloid Aggregation
In general, the effects of metal ions on Aβ, αS, IAPP, and tau are very difficult to reconcile. The self-catalyzing aggregation process is very difficult to control, and hence, the effect of metal ions is very condition dependent, ranging from aggregation promoting effects, inhibition effects, change in structure of aggregates, more toxic, less toxic, etc. One robust result obtained is that Zn(II) and Cu(II) modulate the aggregation behavior, and this always in a metal-specific way. For recent reviews, see refs 15, 313, and 665–669.
The metal ions that were mostly investigated are the essential metal ions, in particular Cu and Zn ions, but also ions of Fe, Ca, etc. Here we concentrate on the most active research on Cu and Zn. In the case of Cu it is important to realize that its main redox state is different in the cytosol compared to that in the extracellular environment. In the thiol-rich cytosol and nucleus Cu is mainly present in its reduced form, Cu(I). In contrast, in extracellular spaces Cu(II) is predominant.670,671 So depending where the amyloidogenic peptide occurs, one or the other redox state is more relevant.
To better understand the potential roles of these metal ions in the pathology of amyloid diseases, a multitude of in vitro studies investigated the effects of metal ions on the structures of amyloidogenic peptides/proteins, the kinetics of amyloid aggregation, the mechanisms of the formation of toxic intermediates, off-pathway intermediates, and the final fibrillary assemblies of Aβ, αS, tau, etc. To overcome the many challenges posed by the complexities of the effects of metal ions on amyloid aggregation, a combination of various biophysical and biochemical approaches is employed to systematically dissect the complicated molecular processes and structural changes underlying the metal-induced effects on amyloid aggregation. The kinetics of amyloid aggregation is most commonly measured using ThT based fluorescence experiments, while the structural changes during the course of amyloid aggregation are typically investigated by using a combination of CD and NMR as well as MD simulations, which yield atomic-resolution structures of amyloid species. The morphologies of metal–amyloid–protein aggregates are usually obtained from TEM and AFM images. Recent studies have highlighted the value of HS (high speed)-AFM, which is able to monitor the real-time aggregation process of amyloid proteins as reported in a review.672 Other techniques such as EPR, ITC, and FTIR have also been used to study the interactions between metal ions and amyloidogenic peptides/proteins. Finally, chemical tools have been applied to control the effects of metal–protein interactions on amyloid aggregation and toxicity and also to probe the aggregation pathways.
7.2. Structure of Copper– and Zinc–Peptide Complexes
The coordination sites of Cu(I/II) and Zn(II) have been studied for several amyloidogenic peptides and their variants, but not to the same degree. The most intensely studied peptide is Aβ, followed by αS and IAPP, with tau being the amyloid protein with the least information on its interactions with metal ions. The so far best supported models of the binding modes between these peptides/proteins and Cu(I/II) or Zn(II) are summarized in Figure 24. For the more comprehensive and recent reviews on these interactions, see the reviews668,669,673 and references cited therein. The elucidation of these binding modes was often realized with the relevant fragments of the respective amyloid peptide/protein harboring the metal-binding domain, such as on Aβ1–16. However, it should be noted that for the shorter peptides Aβ and IAPP, a lot of studies have been performed not only on the metal-binding domain but also on the full-length peptides. Similarly, for the longer αS protein, several studies were also performed on the full-length form. This is, however, not the case for tau as it is much longer and occurs under different phosphorylated states. Phosphates are potential coordinating groups and could be very important in interactions with the harder (according to Pearson’s) metal ions, such as Fe(III). This is more likely restricted to the extracellular space, as in the thiol rich intracellular environment, Cu and Fe are mainly present in the reduced forms, Cu(I) and Fe(II). These metal ions are soft or intermediate and hence have less relative affinity compared to the oxidized Cu(II) and Fe(III), respectively.
Figure 24.
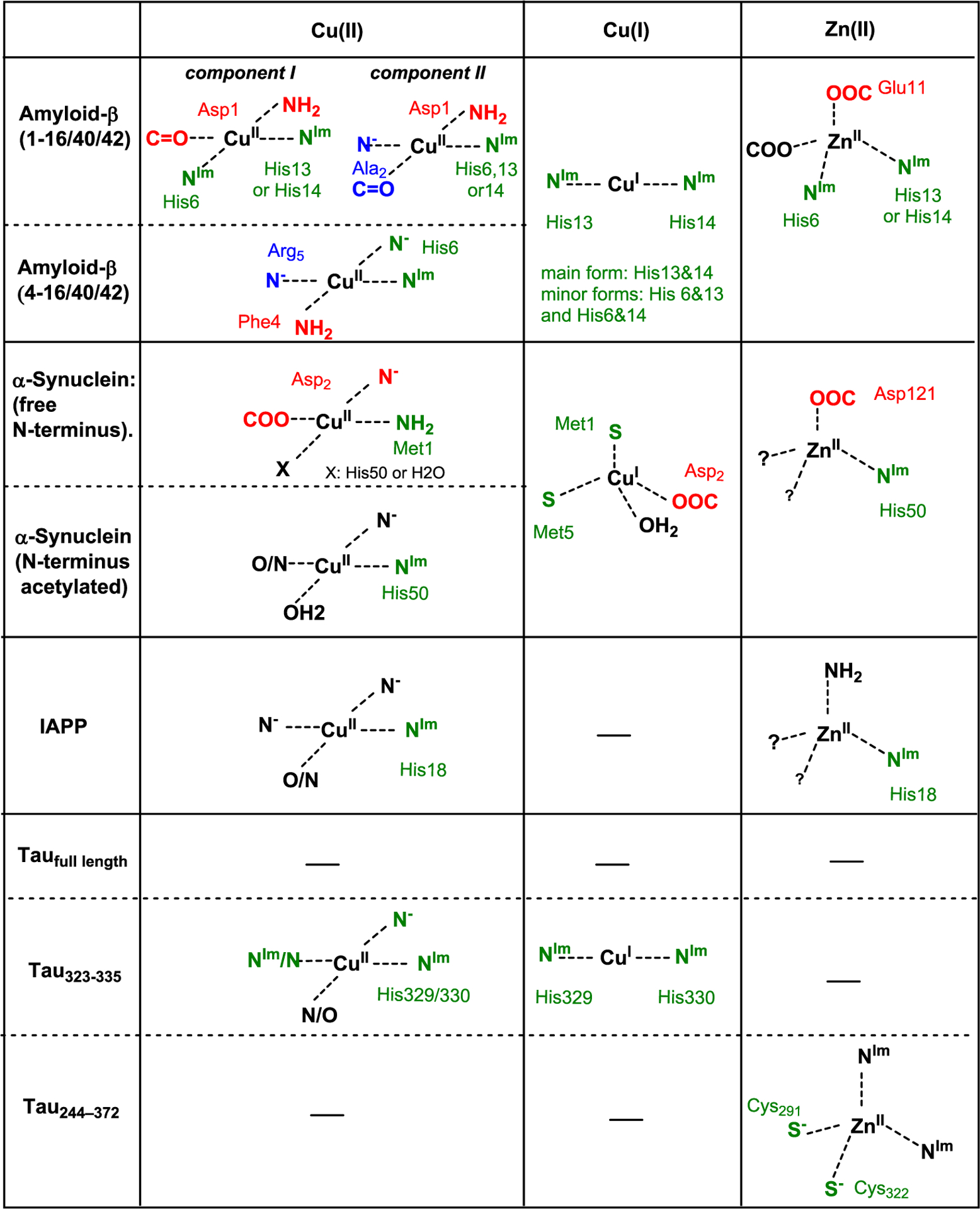
Coordination spheres of Aβ, αS, IAPP, and tau peptides and some variants with Cu(I), Cu(III), and Zn(II). Generally, the most populated states at pH 7.4 are shown. Often several coordination spheres (states) are present and differently populated. They are in fast equilibrium. An exception is Cu(II)-Aβ4–40/2 which has a well-defined coordination sphere. Abbreviations: NH2, N-terminal amine; NIm, nitrogen of imidazole ring of a His; N−, nitrogen of an amidate; N, any nitrogen of unknown ligand. Review668 and references.678–680
7.2.1. General Features of Cu(I/II) and Zn(II) Binding to Aβ, αSyn, IAPP, and Tau.
The binding of metal ions, especially Cu(I/II) and Zn(II) to the various amyloid peptides/proteins is characterized by certain similarities, which can be summarized as the following:
Importance of His: In most of the cases one or more His residues are involved in the binding of metal ions to amyloid peptides/proteins. Histidine is one of the most common metal coordinating ligands among the amino acids. They are intermediate bases (according to Pearson’s hard and soft acid base concept) and hence have a certain selectivity for intermediate metal ions, like Cu(II) and Zn(II). Though soft Cu(I) has also been found bound to His, but this is likely only relevant in the absence of or in combination with cysteines. Thus, Cu(I) binding to His only is unlikely to occur intracellularly.
Moderate affinities: In the cases where the metal ion binding affinities have been measured, they were found to be moderate and generally lower than affinities to metalloproteins. For Cu(II) and Cu(I) the log K values at pH 7.4 are classically between 7 and 10, and for Zn(II) they are even lower with values in the range of 4–7.673–675 This can be explained by the fact that the amyloid peptides/proteins are mainly intrinsically disordered and hence binding metal ions with different ligands bears an entropic penalty. This is an indication that the interaction of these peptides with metal ions is not physiological but rather pathophysiological. It further implies that an accumulation of peptides and/or metal ions, possibly originating from dysregulation in the respective disease, is needed before the interaction can occur.
Dynamic coordination: Again unlike metalloproteins, which can be usually characterized by binding of one or more metal ions to specific sites in the protein, there are no rigid, very well-defined metal-binding sites in amyloid peptides. In general, each metal ion can bind with different coordination spheres to the peptides as reported in the review.673 In Figure 24, only the most populated coordination sphere for each of the metal ion-amyloid peptide/protein combinations is shown, but in general several coordination modes exist, which are pH dependent and in fast exchange with each other. The existence of multiple metal binding modes is in line with the IDP signature, which is not inhibited by the binding of metal ions.673
An exception to this feature is the Cu(II)-binding to an abundant form of Aβ, where the first 3 amino acids are truncated (called Aβ4–40/2). Here, Cu(II) is bound to the amino acids 4 to 6 (Phe-Arg-His) by the N-terminal NH2, the two N− of the two amidates (from peptide bonds between Phe-Arg and Arg-His), and the Nπ of the His. This binding site belongs to a well-known N-terminal binding motif with the general sequence NH2-Xxx-Zzz-His (Xxx-Zzz are any amino acid, but Pro for Zzz) with high affinities of log K 13–15 at pH 7.4 and more inert Cu(II)-binding compared to other peptides.676,677
7.3. Reactivity of Cu-Amyloid Peptides with O2 and Reducing Agent to Produce ROS
Oxidative stress is a well-recognized factor in several amyloid-related diseases, although a cause or a consequence is not clear as reported in the review.681 Oxidative stress is an imbalance between ROS (reactive oxygen species) production and scavenging, leading to an accumulation of ROS, which can damage all types of biomolecules. Cu can be very potent in catalyzing the production of ROS from dioxygen and a reductant, such as ascorbate. In particular “loosely” or nonspecifically bound Cu is often more prone to ROS production. Cu coordinated to proteins and enzymes is normally strongly bound and in a well-controlled protein environment, which allows control of the reactivity of Cu.670
Thus, it is possible that Cu bound to amyloidogenic peptides (Aβ, αS, etc.) can contribute to oxidative stress via its catalytic redox activity and formation of ROS (Figure 25). The importance of this ROS production in vivo is not clear, but test tube experiments showed that these Cu–peptide complexes are quite competent in the catalysis of ROS production, at least more than the other tested Cu–peptide/protein complexes.682 An exception here is again the truncated Aβ4–40/42,676 in which Cu(II) is rigidly bound to the N-terminal motif Xxx-Zzz-His and is thus inefficient in ROS production.683
Figure 25.
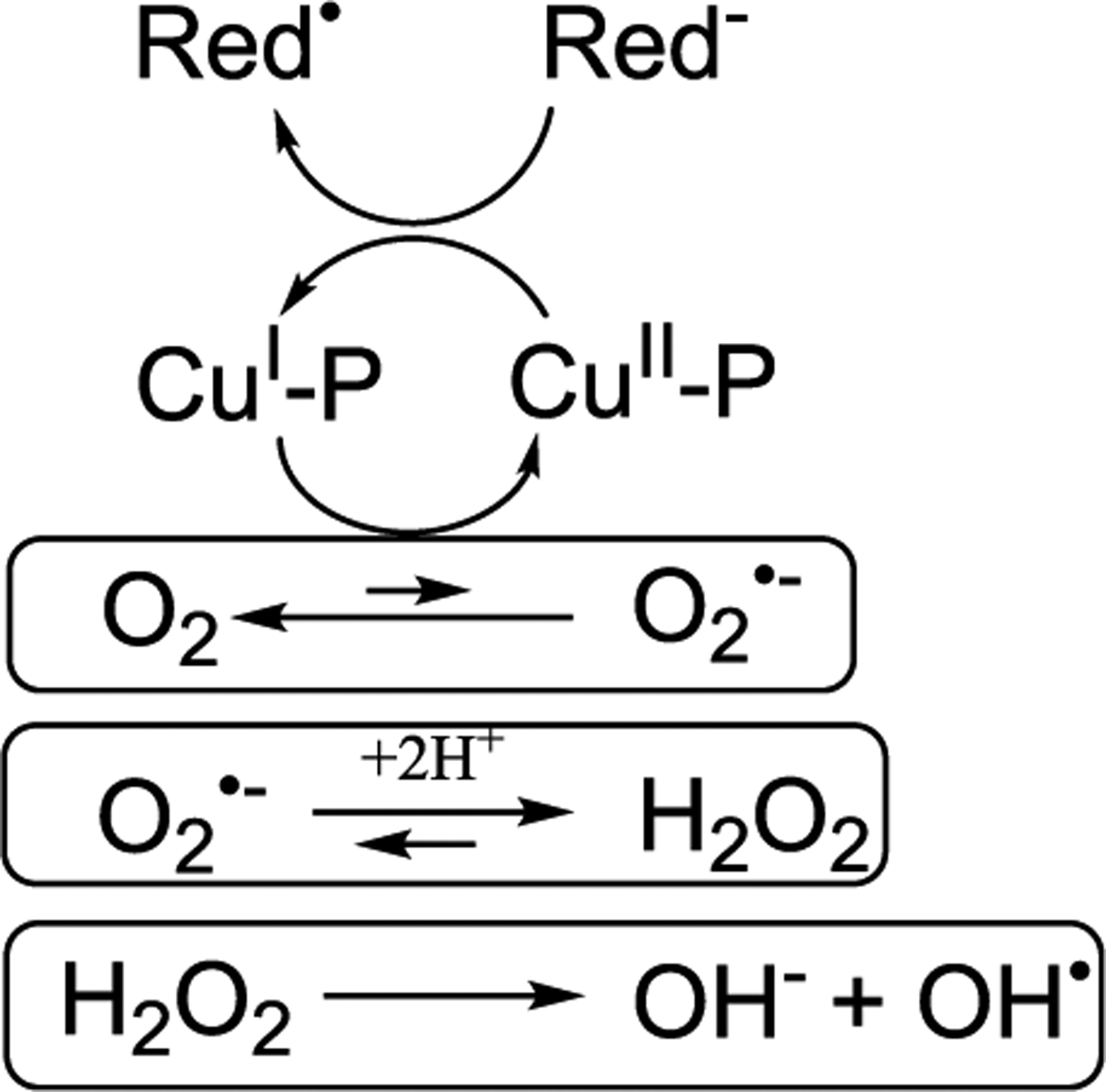
Proposed mechanism of ROS production by a Cu–peptide (Cu-P) in the presence of a reducing agent (Red−) and dioxygen.
As outlined above, a general feature of these peptides (apart from Cu(II)-Aβ4–40/2) is the moderate affinity for Cu(II) and Cu(I) and the quite different ligands in the coordination sphere between the two redox states. This can be explained by the conformational flexibility of the peptide that adapts to the very different coordination preferences of Cu(II) and Cu(I). This flexibility enables the redox reaction between Cu(I) and Cu(II), but the structural rearrangement between the two states makes it slow. Very detailed investigation on Cu-Aβ including both experimental and theoretical work (e.g.,684,685) suggested that the redox chemistry and ROS production is performed by a low populated state(s), which is different from the highest populated states shown in Figure 24 (see review668). These low populated states (also called in-between states), which are accessible due to the flexibility of Aβ and are responsible for the redox chemistry, likely also exist in the other amyloid peptides/proteins IAPP, αS, and the so far studied portions of tau, as the key features are also present there.678,686
The high structural flexibility and the presence of several metal binding modes with different structures speak against a controlled reactivity. First, the peptide is too flexible for selective substrate binding. Second, a tight control of structure is necessary to control the reaction. Thus, the flexibility is more in line with unspecific and not controlled reactions and Cu bound to these peptides seems potentially dangerous and not for physiological purposes.
In test tube studies of ROS production ascorbate was mostly used as reducing agent. Ascorbate is of physiological relevance and occurs at concentrations of around 10−4 M and 10−3 M extra- and intracellularly, respectively, as reported in a review.687 Other reducing agents have also been tested but are often much less potent than ascorbate, such as catechols. The intracellularly most abundant reducing agent, glutathione (GSH), at relevant concentrations is a stronger Cu(I)-chelator than Aβ, αS, etc. GSH first reduces Cu(II) to Cu(I) by forming GSSG (oxidized GSH), and second GSH (present in large excess) withdraws the Cu(I) from the amyloid peptides/proteins, which even occurs for the strongest complex, Cu(II)-Aβ4–40/42.688 Consequences are that, first, GSH cannot serve as a reducing agent to fuel the ROS production by Cu-peptide like ascorbate (as GSH withdraws Cu(I) from amyloids), and second, Aβ, αS, IAPP, and tau (at least to so far studied peptides with Cu) are most likely not strong enough Cu-binders to exist in a mM GSH environment like in the cytosol or nucleus.
7.4. Structural Studies Probing Metal Ion Interactions with Aβ
High-resolution structural information on amyloid proteins under various conditions is important to better understand the underlying molecular mechanism of the pathological progression of amyloid diseases as explained in other sections of this review. Such structural details would also provide insights into the roles of metal ions and other molecules on amyloid aggregation and also aid in the rational design of potential therapeutics to treat amyloid diseases. Therefore, the high-resolution metal bound structures of Aβ under various conditions would not only shed light on the mechanism of conversion of nontoxic to toxic amyloid species but also be helpful for the rational designing of compounds to suppress the metal induced toxicity. To this end, the interaction of metal ions (such as Zn(II), Cu(I/II), Cd(II), Ag(I), etc.) with Aβ peptides and their influence on the structure have been investigated to obtain atomic-resolution information using NMR techniques and MD simulations. In the following subsections, we limit the focus to briefly cover the structural interactions of Zn(II) and copper ions with Aβ.
7.4.1. NMR Studies.
The three major histidine residues (H6, H13, and H14) in Aβ are shown to coordinate with zinc based on 2D heteronuclear (1H/13C and 1H/15N) chemical shift correlation (HSQC) and 15N relaxation NMR experiments.689 A second, but lower affinity Zn(II) binding site is with D23 and K28 that disrupts the D23–K28 salt bridge, which otherwise is known to favor the nucleation of the Aβ aggregation process.690,691 Zinc binding has exhibited no significant effects on the longitudinal (or spin–lattice, T1) relaxation time and the diffusion rate, suggesting that the Zn(II)–Aβ complex is present as monomers. Diffusion NMR and size-exclusion chromatography have identified an increase in the compactness of monomeric Aβ bound to Zn(II) with a hydrodynamic radius of 1.5 nm compared to 1.7 nm for Aβ alone.692 This was later confirmed by obtaining a contraction upon Zn binding from 1.66 to 1.59 nm using pulsed field gradient diffusion measurements.693,694 Transverse relaxation rate analysis further highlighted that the Zn(II) binding increased the structural order in the N-terminal region while the C-terminus remained flexible.689 This observation is supported by a thermodynamic analysis model that suggested chaperonin activity of Zn(II) and induction of a short-lived folded Aβ conformation around the Zn(II)-binding site at the N-terminus.693
Zn(II) interaction with Aβ has been shown to generate nonfibrillary Aβ aggregates, and the underlying molecular mechanism has been probed using NMR experiments. Zn(II)-binding induces a rigid turn-like structure in the V24–K28 region of Aβ allowing the C-terminus to be highly dynamic.696 Investigation of the Zn–Aβ complex is limited by (i) structural plasticity of Aβ, (ii) multiple zinc coordination sites in Aβ, (iii) precipitation of sample at high Zn/Aβ concentration required for NMR detection, and (iv) inactive spectroscopic nature of zinc (d10 electronic configuration). Alies et al. addressed these limitations using a combination of approaches that include 1H NMR, X-ray absorption spectroscopy, and affinity measurements. They reported Zn(II) ions are tetrahedrally bound to Aβ with the Zn coordination sphere comprised of two histidine residues and two carboxylate side chains (Figure 26).695 The binding of zinc to Aβ is pH and concentration dependent as revealed by NMR.696–698 At substoichiometric concentration of Zn(II) (i.e., 0.2 equiv), line-broadening was observed for H6, S8, V12, H13, and H14. Further increase in Zn(II) concentration (i.e., above 0.5 equiv) broadened the signals from many N-terminal residues (E3, F4, R5, D7, S8, E11, V12, Q15, and K16) including all histidine residues beyond detection by solution NMR.697,698
Figure 26.
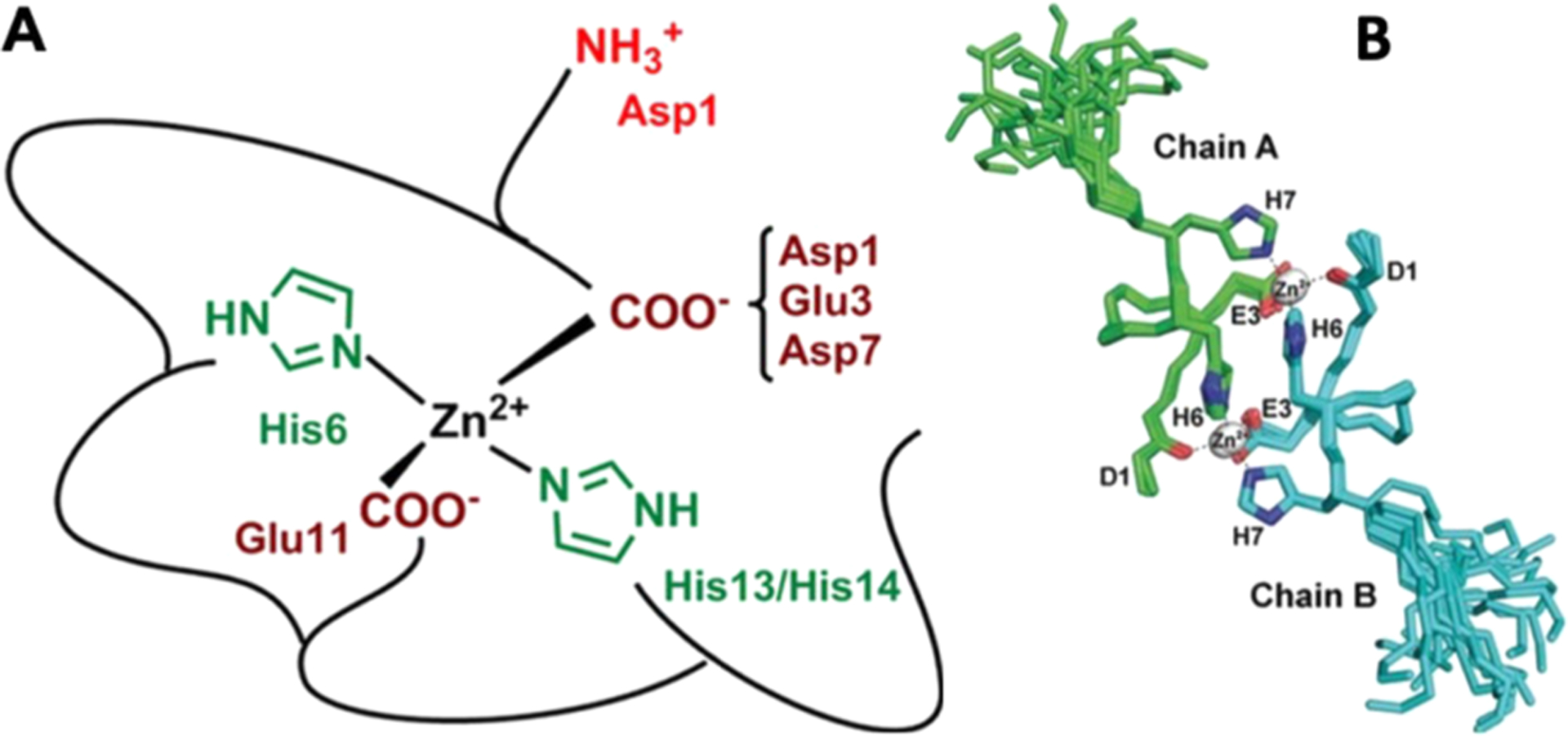
High-resolution structural characterization of zinc binding sites to Alzheimer’s Aβ. (A) At physiological pH (7.4), zinc was proposed to tetrahedrally bind with Aβ where the coordination sphere is contributed from two of three histidine (H6, H13, and H14) residues and two carboxylate side chains (E11 and one from D1, E3, or D7). (B) Superimposition of 20 NMR model structures of Taiwanese mutant Aβ fragment (D7H-Aβ1–10) showing zinc (gray sphere) induced formation of a homodimer. The dimer is stabilized by two zinc ions where one coordinates to D1 and H6 of one Aβ subunit and the other to H7 and E3 orienting the H6 residue of both subunits to form stacking interactions.695,700
Zinc binding has been shown to induce Aβ oligomerization and its pathological phenotype. NMR experiments have been used to probe the high-resolution structural details of zinc induced Aβ oligomers by using an Aβ fragment involving residues 1–16.699 Aβ1–16 forms a dimer in the presence of zinc and E11–H14 was identified as the key interface region to stabilize the dimer. Similarly, a binuclear zinc interaction is shown to form dimers for Aβ fragments derived from the Taiwanese mutation D7H. A dimer model structure shows the coordination of Zn(II) with residues D1 and H6 of one peptide and E3 and H7 from another peptide in the coordination sphere, bringing the H6 residues of both peptides to a close proximity for stacking interactions (Figure 26).700 In another study, zinc binding to phosphorylated Aβ (at S8), as detected in AD brain, showed the H6 imidazole ring and phosphate group of S8 to form three of the four Zn(II) coordination bonds promoting Aβ dimerization.699,701 Lee et al. reported Zn(II) induced Aβ oligomers are spherical with a diameter of ~12–14 nm and used homonuclear (13C/13C) magic angle spinning (MAS) solid-state NMR to study the Zn-Aβ aggregates where Aβ was selectively labeled with 13C at V24, A30, I31, G33, and L34.702 This study identified A30 in a rigid β-sheet that differed from its conformation in aged fibers, whereas L34 showed no conformational change highlighting the Zn-Aβ aggregates adopting a β-sheet structure but is relatively disordered when compared with aged Aβ fibers.702 In another MAS NMR study, Mithu et al. revealed a structural distortion in Zn–Aβ aggregates due to the disruption of the D23–K28 salt bridge but maintained a cross-β structure.691 In aged fibers, they observed two different chemical shifts for D23, K28, A30, and V36 indicating the presence of a heterogeneous structure. But in Zn–Aβ aggregates they observed a single chemical shift for D23 and K28 and two distinct chemical shifts for A30 and V36691 suggesting the Zn–Aβ aggregates are morphologically distinct from aged fibers.
Other metal ions such as Cu(II) share similar but not identical binding sites and modes with Aβ. Cu(II) binds in a pH-dependent fashion to Aβ, with two main forms present at pH 6–8 as shown by EPR, X-ray absorption, CD, and potentiometry (Figure 24) (reviewed in refs 668, 674, and 703). These two forms are often called components I and II. In contrast Cu(I) seems to be less pH dependent, at least from pH 6.5 to 9, and bound in a diagonal fashion to two His residues. Cu(I) bound to H13 and H14 is the dominant form but is in rapid exchange with Cu(I) bound to H6/H13 and H6/H14.703 NMR spectra of Cu(II)–Aβ (1:1) showed significant line-broadening for the N-terminal residues spanning the region E3–V18.704 It should be noted that both Cu(I) and Cu(II) are involved in the interaction with Aβ. 2D TOCSY NMR experiments identified Cu(I) binding to all three histidine residues in Aβ1–16. Cu(I) has been shown to bind to the imidazole nitrogen (H6–N, and H13–Nε, H14–Nε) that induced significant chemical shift perturbation for the side chain protons from all His residues.705 Proton NMR experiments revealed that Zn(II) does not alter the copper binding environment in Aβ in line with the known stronger binding of Cu(II) when compared to Zn(II). This is in contrast to EPR and XAS measurements, which showed that Zn slightly pushes the Cu(II) from component I to II.706,707 In addition, it is also shown that unlike the formation of amorphous aggregates upon zinc binding, copper binding does not alter the fibrillary state of Aβ.708 The paramagnetic effect of copper measured using MAS experiments has been used to identify the selective quenching of NMR peaks from the N-terminal residues (E3, E11, H13, and H14) and C-terminal V40 in Aβ40 fibrils mixed with copper, but no change in the structure was identified.708 Relaxation experiments under MAS further identified that the paramagnetic Cu(II) binding to Aβ changed to a diamagnetic Cu(I) which remained attached to the fiber. Interaction of other metal ions such as Ag(I) and Cd(II) with Aβ has also been studied using NMR spectroscopy. Cd(II) showed interaction with the N-terminal histidine residues, similar to Zn(II), with a binding stoichiometry of 1:1.698 Similarly, Ag(I) interaction studies with Aβ, and an Aβ mutant having no histidine, revealed interaction with the N-terminal residues reducing the NMR signal intensities of all histidine residues by ~75–80%.709
7.4.2. Molecular Simulations.
Molecular simulations complement the experimental studies of the interactions between amyloid peptides and metal ions as they provide atomic-level details of the metal binding and the resulting effects on the structure and dynamics of the peptide with femtosecond resolution. Different simulations ranging from quantum mechanical (QM) calculations and ab initio MD simulations to quantum mechanics/molecular mechanics (QM/MM) and pure classical MD simulations have been conducted that studied the binding of Zn(II) and Cu(I) or Cu(II) to Aβ. A comprehensive review summarizing the findings from a large set of these different simulation approaches is provided in ref 710. The aim of the simulations involving a QM approach is to shed light on the coordination spheres around the metal ions bound to Aβ. The results of these QM calculations are usually compared to experimental data and help to decide which of the possible binding modes best agrees with experimental signatures. Moreover, the stability of the different binding modes can be estimated based on the calculated molecular energies. To give an example, DFT calculations helped in the interpretation of extended X-ray absorption fine structure (EXAFS) spectra of Cu(II) in complex with Aβ. Different coordination spheres were constructed and their geometry optimized using DFT calculations. The optimized structures were then utilized in the EXAFS spectral data refinements, based on which a distorted six-coordinated arrangement with three histidine (H6, H13, and H14) residues and a carboxylate oxygen (from either E11 or D1) in an approximately equatorial planar arrangement and a water molecule and another carboxylate oxygen also from E11 or D1 as axial ligands was reported.711 Moreover, a pentacoordinated structure with Y10 involved712,713 could be ruled out. Several other DFT studies that also assessed the likelihood of Y10 as Cu(II) ligand found a preference of N-terminal E and D residues over Y10,714,715 in agreement with the findings from NMR, potentiometry, and electrochemistry that never identified Y10 as a main ligand for Aβ.
Studies on metal–Aβ complexes using ab initio MD simulations usually employed the Car–Parrinello molecular dynamics (CPMD) method. They also aimed at making predictions regarding the prevalence of different coordination spheres by testing their stability in short CPMD simulations. For example, different binding modes of Zn(II) in complex with Aβ1–16 were probed by CPMD simulations,716 reporting H6, E11, H13, and H14 as most likely ligands. Possible Zn(II)-bridged Aβ dimer structures were also suggested from CPMD simulations.717 Here, the most stable complex was found to be one in which two peptides are bridged by a Zn(II) ion in a four-histidine coordination mode involving H13 and H14 of both peptides. Another QM/MM study718 that assessed possible dimerization scenarios of Aβ involving Zn(II) concluded that the Zn(II) ion is likely chelated by E11 and H14, which is in agreement with the NMR results mentioned above699 and was also supported by ITC experiments of the sequence Aβ11–14.718 When replacing H13 with an arginine, the mutated peptide binds with the same affinity Zn(II) as the original peptide EVHH, indicating that H13 is not involved in Zn(II) binding.718 CPMD was also employed to probe the binding of Cu(I) to Aβ, testing whether two or three His residues coordinate with Cu(I).719 The coordination sphere with three His residues turned out not to be stable as one of them detached from Cu(I) during the CPMD simulation. This result is in accord with findings from XAS and NMR experiments. It was further found that H6 binding to Cu(I) is possible, but the preferred coordination involves H13 and H14.719
Classical MD simulations allow studying the impact of metal ion binding on the structural preferences of amyloid peptides. Here, special care has to be taken when modeling transition metal ions at the classical level. The most common approach is to describe the metal ion as a sphere with a partial charge and van der Waals parameters. However, this simplistic approach often fails to model the specific interactions between transition metals and the coordinating ligands so that the coordination site geometry is not retained during an MD simulation, especially if the protein is as flexible as Aβ. An approach to overcome this problem is provided by so-called bonded models, where bonds between the transition metal ion and ligated amino acid residues are explicitly modeled, enabling a stable coordination sphere geometry during MD simulations. To elucidate the effects of metal ion binding on amyloidogenic peptides, MD simulations on the microsecond time scale or enhanced sampling, such as REMD simulations, is needed to observe relevant conformational transitions in these peptides. From the multitude of MD simulations that studied conformational changes in monomeric or oligomeric Aβ upon metal ion binding (as comprehensively reviewed in ref 710) only those that used a bonded model for the coordination site and performed sufficient MD sampling are discussed here. As binding modes for Zn(II) and Cu(II), those previously reported from NMR spectroscopy and/or DFT calculations were generally adopted. In the case of monomeric Aβ, most of the simulations used a coordination sphere including H6, H13, and H14 plus D1 or E11 as fourth ligand. In one study, which employed REMD simulations, the most populated coordination sphere constituted by H6 and H13 as well as the amine and carbonyl groups of D1 (Figure 24) was considered,720 using previous QM/MM calculations and experimental findings as input.721 In general, similar observations were made by most of these simulation studies. Many of them found that both Zn(II)722,723 and Cu(II)720,724 binding increases the formation of β-sheets in the C-terminal residues ~30–40/42 of monomeric Aβ. In the N-terminal residues, on the other hand, where the metal ion binds, the amount of helix and β-sheets is reduced as the coordination geometry counteracts the formation of these secondary structures. Moreover, metal ion binding causes a compaction of the N-terminal half of Aβ, explaining the reduction of the hydrodynamic radius of Aβ upon Zn(II) binding as observed experimentally,692,693 which in turn interacts less with the C-terminal half of the peptide. The C-terminal residues instead interact with each other, leading to increased β-sheet formation. An REMD study of the Cu(II)-bridged Aβ42 dimer, where the metal ion was coordinated by a pair of H13 and H14 residues from the two peptides, also found a considerable increase in β-sheet in the C-terminal residues, along with a gain in the exposed hydrophobic surface area, which is expected to increase the aggregation tendency as compared to the metal-free peptide.725 It was suggested that the increased aggregation tendency will rather lead to amorphous aggregates as Cu(II) binding enhances the probability of closed Aβ structures with more intramolecular hydrogen bonds, including intramolecular β-sheets, which are different from the extended monomeric structures prone to fibril formation.726
7.5. Structural Studies on Metal Induced IAPP Aggregation
7.5.1. Roles of Zinc on IAPP Aggregation.
Human IAPP has been shown to aggregate to form amyloid fibrils at nanomolar concentrations under in vitro conditions. While the formation of both the amyloid fibers and smaller oligomeric species by hIAPP has been linked to β-cell destruction, it is safely stored in the secretory granule at millimolar concentrations. Given that hIAPP in isolation spontaneously aggregates at concentrations 2 to 3 orders of magnitude lower than those present in the secretory granule where it is stored, it is reasonable to look for other factors that act as chaperones to stabilize hIAPP in a nontoxic form in normal individuals. In contrast to other amyloid proteins for which high-affinity metal binding sites have been identified, the influence of metal binding on hIAPP has been almost entirely unexplored. Zinc is of particular interest as the zinc content of pancreatic β-cells is among the highest in the body and several clinical and epidemiological studies suggest zinc deficiency is a common symptom of type II diabetes. The recent discovery between the genetic linkage between the SLC30A8 gene, which transports zinc into the secretory granule where insulin and hIAPP are stored, and type II diabetes suggests zinc could have an impact on hIAPP cytotoxicity toward β-cells.
7.5.2. NMR and MD Simulation Studies on Zn-IAPP Complexes.
Structural characterization of zinc-bound IAPP complex is important to delineate the biological activity of IAPP and the role of zinc in type-2 diabetes. Poor understanding and the lack of high-resolution structural knowledge of the zinc bound IAPP species have been a major roadblock to designing structure-based therapeutics. Here we briefly present the advances in studying Zn–IAPP complex at a molecular level using NMR. Solution NMR experiments, 2D TOCSY and 2D NOESY, have been carried out to probe the interaction of zinc with human-IAPP. Based on the NMR results, a structural model reported for the Zn–IAPP complex identified His-18 to be the key residue in zinc binding.727 Zinc binding showed a significant chemical shift perturbation for Hε (0.208 ppm) and Hδ (0.106 ppm) from His18’s imidazole side chain. The binding of zinc (10 equiv) to human-IAPP neutralized the overall peptide charge and induces a local disruption of the secondary structure in the vicinity of His-18 spanning two short α-helices (Figure 27).727 On the other hand, under identical NMR conditions, a regular kinked helix-spanning region Arg11-Thr30 was found for IAPP in the absence of zinc, while the rest of the peptide regions were found to be disordered similar to that observed from the Zn-IAPP complex.727 Taken together, the comparative structural model analysis revealed the major structural difference between IAPP and Zn-IAPP was found to be centered at the His18 residue which has been shown to induce a profound kink in the Zn–IAPP complex with a pattern of helix–kink–helix conformation (Figure 27). Atomic insights further showed that a loss of Phe15-His18 and Leu16-Ser19 nuclear Overhauser enhancements (NOEs) due to zinc binding to IAPP is the reason for the conformational unwinding of IAPP helical structure.727
Figure 27.
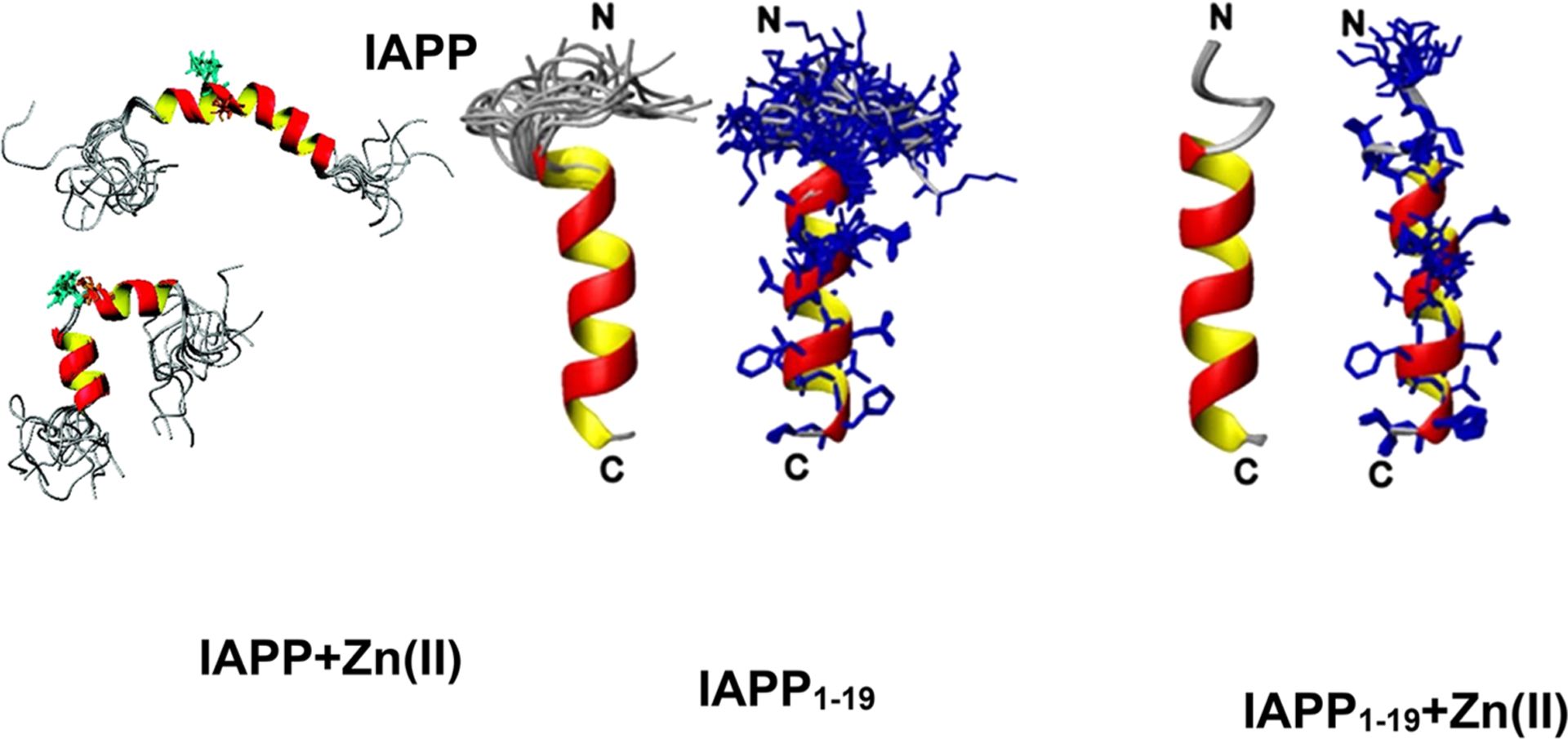
High-resolution NMR model structure of IAPP in the absence or presence of zinc. Full-length NMR structure of IAPP (top) and Zn-IAPP complex (bottom). Zinc binding to His18 (green sticks) induces helical perturbation and formation of a helix–kink–helix conformation. NMR model structures of IAPP1–19 and IAPP1–19 obtained from 2D 1H/1H TOCSY and NOESY experiments. Zinc binding transforms the disordered N-terminus (left) to an ordered structure (right).727,728
The intermediates of IAPP (oligomers) have the most pathological relevance, and zinc plays a crucial role in the formation of IAPP intermediates. In a follow-up study, Salamekh et al. showed the inhibition of dimers or fibers but the formation of IAPP hexamers upon zinc binding.728 Importantly, a second zinc-binding site in human-IAPP has been observed only at a high concentration. To obtain high-resolution structural details, 2D heteronuclear (1H/15N) SOFAST-HMQC and homonuclear (1H/1H) were carried out on the full-length (human-IAPP-1-37) and the N-terminal truncated (human-IAPP-1-19) peptides.728 These experiments showed a significant chemical shift change for His18 upon zinc binding in the 1H/15N spectrum that correlates to the previous observation and also chemical shift perturbation for the N-terminal residues including the disulfide bonded Cys residues that are disordered in the absence of zinc suggesting a structural transition. These studies further verified the structural change by comparing the 1H/1H correlation spectrum of IAPP(1–19) in the absence and presence of zinc.728 The observed results highlighted the zinc binding to be inducing an ordered conformation in the N-terminal as compared to the zinc free IAPP peptide (Figure 27). Another study probed the changes in the IAPP monomer–oligomer equilibrium upon zinc binding by analyzing the depletion in 1H signal intensities within the limit of NMR detection.729 Zinc binding to IAPP monomers did not show appearance of new peaks or substantial line broadening of the His18 imidazole peak. Time-lapse measurements and comparison of lag-times of aggregation between IAPP and Zn–IAPP complex using 2D SOFAST-HMQC (1H/15N) showed zinc to affect the monomer–oligomer equilibrium and generate prefibrillary IAPP aggregates that are not visible in NMR.729
The binding of Zn(II) to IAPP fibrils was probed by MD simulations.730,731 In these simulations IAPP was modeled in a preassembled fibril state and Zn(II) was placed at different potential binding sites including H18 but also C2 and C7 which were modeled without a disulfide bridge between them. The most stable binding was observed for Zn(II) being coordinated to two C2 and two C7 of two neighboring IAPP peptides in the fibrillary structure. In general it was found that Zn(II) ions increase the number of polymorphic states of the fibrillary IAPP by inducing conformational changes. However, these changes are overall minor and the fibrillary models are still very similar to each other so that they are unlikely to be captured as being different from each other by experimental means.
7.6. α-Synuclein and Metal Ions
Like Aβ and IAPP, αS can bind metal ions. A seminal study by Uversky et al. showed that metal ions, in particular Al(III) and Cu(II), can impact the fibrillation rate of αS, possibly by leading to the formation of a partially folded state that is more prone to aggregation than the monomeric protein in solution.732 However, the metal concentration used in this pioneering study was in the millimolar range, i.e. greater than those normally occurring in cerebral tissues. Subsequent studies investigated the impact of divalent metal ions (Fe(II), Cu(II), Zn(II), Mn(II), Co(II), and Ni(II)) using physiologically relevant μM concentrations. The major finding of these studies was that under these conditions only Cu(II) is able to accelerate the formation of amyloid fibrils.733 Further characterizations of the metal binding revealed that the metal ions generally have a high tendency to interact with αS through its C-terminal domain, which contains several Asp and Glu residues that are strong attractors of cations and positively charged molecules.733 The DPDNEA sequence, located at the C-terminal domain (Figure 28), is a nonspecific binding site (site 3) for many di- and trivalent metal ions. This site shows conditional dissociation constants in the order of ~10−6 M, which is indicative of a low binding affinity for different cations.734 For Cu(II) three binding sites in αS have been elucidated (Figure 28).734 Copper coordination in site 3, which has the lowest Cu(II) affinity, is mediated by the backbone carbonyl groups of D122 and by the carboxylate groups of D119, D121, and D123. Cu(II) binding to H50 (site 2) is pH-dependent, and at physiological pH the metal ion is anchored via the imidazole ring of H50, via the deprotonated backbone amides of H50 and V49 (or by the carbonyl group of V48), and by one water molecule. In site 1, which has the highest affinity for Cu(II), the cation is coordinated by the N-terminal NH2 group of M1, the deprotonated backbone amide, the carboxylic groups of D2, and a water molecule. However, it can also involve H50 depending on pH and concentration.735,736 Copper has been shown to be one of the most potent modulators of αS structure as well as a very effective accelerator of αS fibrillation in vitro.732 AFM-based experiments revealed that the presence of Cu(II) significantly enhanced the relative abundance of the β-sheetlike structure in the otherwise conformationally heterogeneous monomeric αS.737 Even at physiologically relevant concentrations, Cu(II) ions were effective in the acceleration of αS aggregation without affecting the resultant fibrillary structures.738
Figure 28.
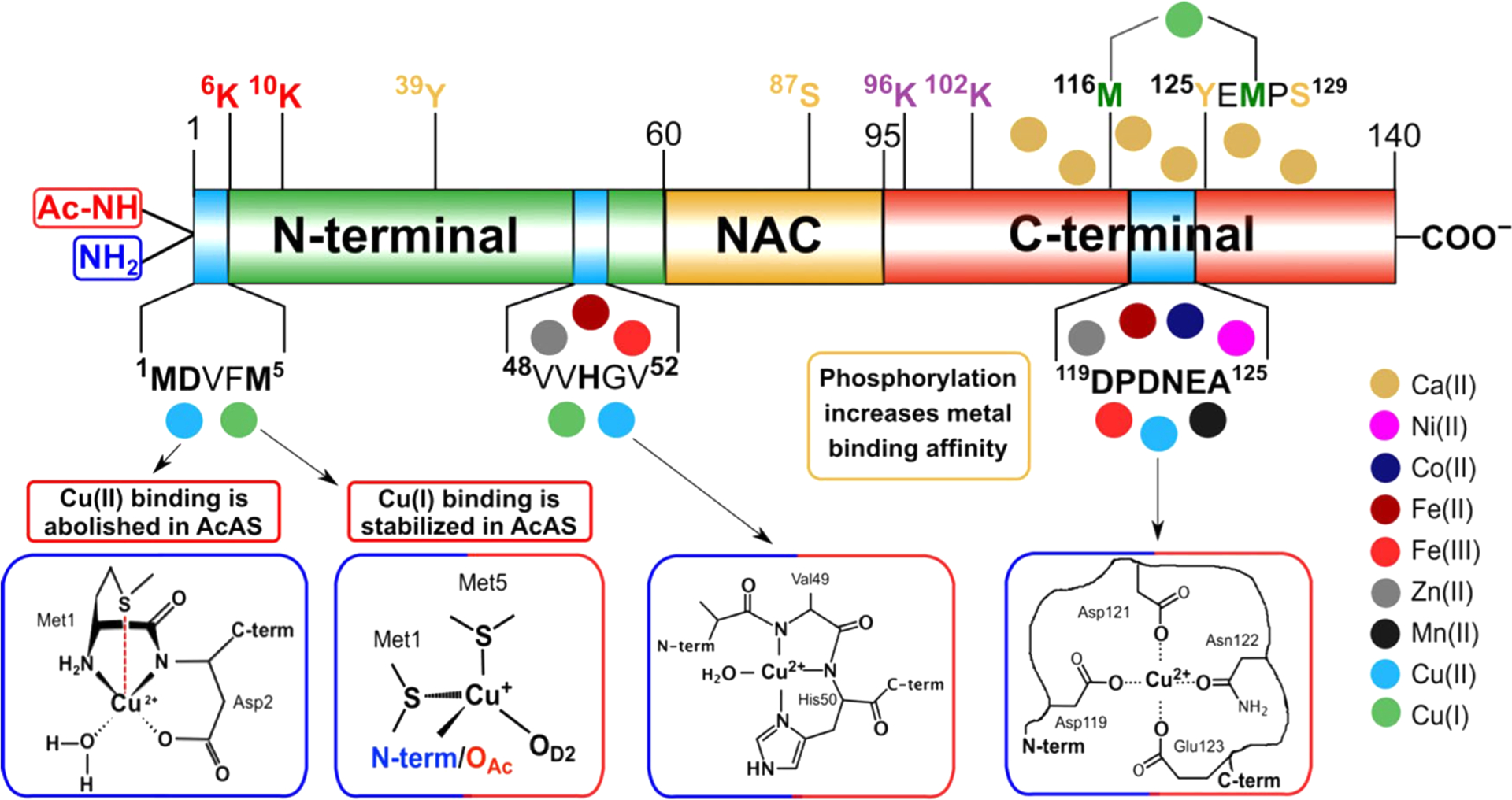
Metal binding sites and post-translational modifications of αS. αS harbors three binding sites for metal ions (displayed with colored circles): the low affinity, nonspecific metal binding site at the acidic DPDNEA segment in the C-terminal, the His50 site, and the first five residues at the N-terminal. His50 can anchor transition-metal ions such as Zn(II), Fe(III), and Fe(II), but the highest affinity is found for Cu(II) and Cu(I) ions. The N-terminus of αS is a high affinity and highly specific site for Cu ions. Reprinted with permission from ref 734. Copyright 2021 John Wiley and Sons.
We would like to conclude this section by indicating that lysozomes are responsible for degradation of biomolecules and stores for metal ions, and several forms of lysosomal dependent cell death have been identified in diseases, including cancers and neurodegenerative diseases.739 Rebalancing metal dyshomeostasis could be a feasible therapeutic strategy for AD.740
8. WHAT DO WE KNOW ABOUT ALS ETIOLOGY?
ALS causes motor neuron death leading to paralysis and eventual respiratory failure. Symptoms typically begin later in life with disease onset typically beginning around ages 40–60. The majority of patients die within 3 to 5 years of their first symptoms. More than 5,600 people worldwide are newly diagnosed with ALS each year, and the disease accounts for 2 deaths per 100,000 people.741,742 5–10% of cases are considered familial (fALS), i.e., genetic mutations can be traced back to relatives, while 90–95% of cases are considered sporadic (sALS).743 Despite a lack of family history in sALS, a pattern of genetic mutations is beginning to emerge across all cases.
Since 1993 over 46 different genetic mutations have been linked to ALS and more continue to be discovered each year. Most of the mutations fall into four main functional categories: RNA processing, protein trafficking/degradation, cytoskeletal/axonal dynamics, and mitochondria function.744–746 All these mutations could increase cell’s vulnerability to an overarching toxic effect.
Cytosolic antioxidant enzyme Cu, Zn superoxide dismutase (SOD1) is a dimeric protein responsible for the conversion of superoxide radicals into oxygen and hydrogen peroxide. Mutations in SOD1 lead to destabilization of the dimeric form and accumulation of toxic aggregates.747 SOD1 mutations are the second most commonly occurring fALS mutations, found in 10–20% of patients, and have also been found in 1–2% of sALS cases.748 Unlike the majority of other genetic mutations tied to ALS, normal SOD1 function does not fit into any of the previously mentioned functional categories. It was originally thought that SOD1 mutations prevented the protein from functioning as it should, leading to a buildup of superoxide radicals in cells; this hypothesis was disproved in studies comparing SOD1 deficient mice to SOD1 glycine-93 to alanine (G93A) mutant mice that showed a loss of motor function in the SOD1G93A mice but not in the SOD1 deficient mice.749,750 These findings led to the theory that SOD1 mutations cause a toxic gain of function.
Similar to tau, amyloid β, and αS, misfolded SOD1 aggregates are seen in large inclusion bodies in cells both in vivo and in vitro.751,752 Despite this, in 2016 Proctor et al. showed that trimeric SOD1 was the most toxic species in NSC-34 cells, and in 2018 Zhu et al. further showed that large aggregates of SOD1 were not only nontoxic to cells but actually protective.753,754 Other proteins associated with ALS, mainly TAR DNA-binding protein 43 kDa (TDP-43) and Fused in Sarcoma (FUS), are also found in large inclusion bodies during disease progression.755 TDP-43 and FUS are both important for RNA transport and are natively found in the nucleus, and mutations cause both proteins to mislocalize to the cytoplasm and aggregate into stress granules (large inclusion bodies).756–758 Yet, overexpression of either mutant is not sufficient to trigger the appearance of stress granules in healthy cells without a stress stimulus.759 Overexpression of misfolded SOD1 has been shown to cause mislocalization and aggregation of both mutant and nonmutant TDP-43 and FUS,760,761 and mutant TDP-43 and FUS, in a stressed environment, have been shown to coexist with misfolded wild-type SOD1 in cells.762–764 Stress granules containing TDP-43 and FUS aggregates can lead to cell death in two main ways; either through the unfolded protein response from the cell’s inability to break down the large insoluble aggregates or through loss of function and RNA dysfunction through a lack of correctly folded TDP-43 and FUS in the nucleus.765–767 Multiple other genetic mutations in the RNA processing and protein trafficking/degradation categories could act similarly to TDP-43 and FUS or contribute to these cell death pathways.768
Motor neurons are unique from other cells due to the length of their axons. Due to how large an area a neuron can span, the cytoskeleton system is especially crucial to deliver organelles and materials throughout the entire cell.769,770 Mutant SOD1 has been shown to alter cytoskeleton transport in two main areas: inhibiting anterograde trafficking between the ER and Golgi and affecting microtubule transport.771 Coat protein complex II (COPII) vesicles transport proteins between the ER and Golgi; loss of transport leads to ER stress and Golgi fragmentation.772,773 Expression of COPII with G93A or alanine-4 to valine substitution (A4 V) mutant SOD1 prevents stress granules and cell death, suggesting mutant SOD1 affects ER to Golgi transport by interfering with COPII vesicles.774,775 Misfolded SOD1 has been shown to interact with p38 MAP kinase (p38 MAPK).776 P38 MAPK phosphorylates kinase-1 preventing it from moving along microtubules.777 Disrupted functioning of p38 MAPK could cause excessive kinesin-1 phosphorylation which would inhibit microtubule transport; inhibition of p38 MAPK in mutant SOD1G93A cell models reduced cell death.778 HDAC6 expression is also affected in mutant SOD1 models; HDAC6 is a histone deacetylase which can destabilize microtubules. Deletion of HDAC6 slowed disease progression in SOD1G93A mouse models.779 Another pathological hallmark of ALS is the accumulation of neurofilaments;780,781 any defects in kinesin or microtubule stability could cause this accumulation.782,783 Genetic mutations in the cytoskeletal/axonal dynamics functional category have been linked to either ER-Golgi transport or microtubule transport, making it likely that these two systems are crucial to ALS pathology.
Kinesin-1 and microtubules are also responsible for the transport of mitochondria throughout the cell.784,785 In vitro ALS models expressing SOD1G93A showed a significant reduction in the number of axonal mitochondria, and the mitochondria present were spaced further apart than in the controls; the same was seen in SOD1G93A transgenic mice and rats.786,787 SOD1G93A has also been shown to interfere with the activity of voltage-dependent anion channel 1 (VDAC1) on the outer mitochondrial membrane.788 VDAC1 is necessary for the exchange of ATP and ADP across the mitochondrial membrane; in SOD1G93A mouse models depressed mitochondrial respiration rates and impaired ATP synthesis were seen in the brain and spinal cord well before disease onset.789,790 Bcl-2 proteins regulate the release of apoptotic cytochromes by VDAC1 and have also been shown to interact with misfolded SOD1.791
Dysfunction at multiple points throughout the cell can lead to problems with calcium signaling, ER stress can lead to depletion of calcium stores, and VDAC1 controls calcium signaling in mitochondria.792,793 Motor neurons trigger calcium release through glutamate signaling. Excess glutamate in the synaptic cleft is taken up by excitatory amino acid transporter 2 (EAAT2) which is expressed on glial cells.794–796 Misfolded SOD1 has been shown to reduce expression of EAAT2 leading to glutamate induced excitotoxicity.797–799 Riluzole, the first FDA approved drug to treat ALS, slows disease progression by increasing EAAT2 activity.800,801 Since Riluozole only slows disease progression and there are so many different mechanisms that can alter calcium homeostasis within neurons, it is likely that EAAT2 dysfunction is not a direct cause of cell death in ALS.
Although SOD1 is the most prevalent ALS genetic mutation that spans all the other categories of mutations, the role of misfolded SOD1 in sALS is debated. Studies using patient tissue samples conflict on whether misfolded SOD1 is present, even when using the same antibodies.802–804 One of the limitations of these patient studies is that most only look for misfolded SOD1 in stress granules instead of investigating its presence throughout the cell.805 For toxic SOD1 trimers to form, the stable dimer must dissociate into monomers and lose its metals. In 2004 Khare et al. showed SOD1A4V dimers have a 10,000 fold increase in dimer dissociation equilibrium constant compared to SOD1WT.806 SOD1A4V is only one out of more than 100 structurally diverse point mutations tied to SOD1 aggregation in ALS, many of which are not located in the dimer interface (Figure 29). The full database of mutant SOD1 variants tied to ALS can be found on the ALS online database (alsod.ac.uk).807 In 2005 Khare and Dokholyan showed that the monomers of SOD1 have coupled motion when a dimer is formed, specifically in the movement of the distal metal-binding loops.808,809 Mutations within the dimer interface and on the outside of the monomers shared the effect of disrupting the coupled motion of the dimer and destabilizing it. Beyond destabilization of the dimer, loss of metals in monomeric form is also crucial to SOD1 aggregation. Metal ions stabilize the glutamic acid-49 to asparagine-53 region of the zinc loop, protecting the β-sheet region of the monomer. Loss of metals exposes the β-sheet region, destabilizing the monomer and making it prone to aggregation.810–813 Simulations of the SOD1 barrel, without metals, found that the β-strands β5 and β6 are the weak spots for thermal unfolding. In simulations in a crowded environment, oscillations of the VII electrostatic loop region expose the same β-strands correlating with increased interactions with the crowder. Mutations of histidine 46 stabilized the VII loop in the compact state, thus protecting the β5 and β6 strands similar to how metal ions protect the barrel.415
Figure 29.
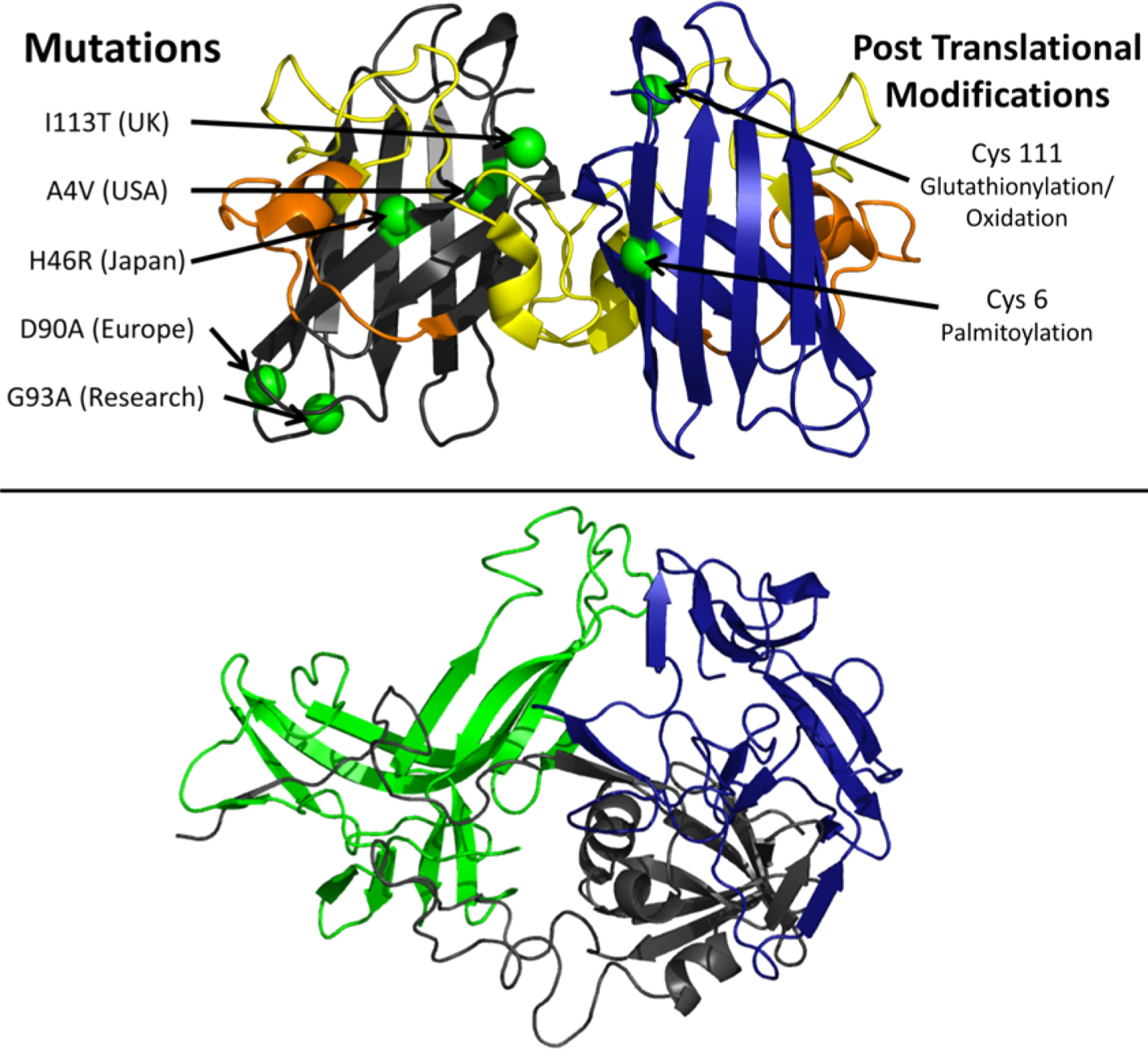
Mutations, modifications, and structural elements of SOD1. The crystal structure of SOD1WT is shown in the top panel (pdb: 2V0A)830 with the zinc binding loop, loop IV, spanning residues 49–84 (yellow) and the electrostatic loop, loop VII, spanning residues 122–143 (orange). The most prevalent ALS associated mutations are shown on the left monomer (green),829 and three SOD1 destabilizing post-translational modifications are shown on the right monomer (green).814 The bottom panel shows a possible trimeric structure of SOD1 modeled by Procter et al.753 The authors designed the figure with Pymol.254
Multiple studies have shown that PTMs can destabilize the dimer interface, increasing the chance of misfolding.814,815 In 2011 Redler showed that glutathionylation of dimeric SOD1WT increased the equilibrium dissociation constant 100,000 fold. Glutathionylation is a natural protective measure in response to oxidative stress and could propagate the SOD1 misfolding as neighboring cells are stressed.816,817 Oxidation of Cysteine 111 to sulfonic acid in SOD1WT in neurons led to similar phenotypes of familial ALS, misfolded SOD1 aggregates, and inhibition of kinesin-based fast axonal transport.818 SAPH-ire, a machine learning program that can predict post translational modifications to a protein, indicated that the 31 amino acids between Serine-98 and Lysine-128 had the highest likelihood to affect SOD1 protein function.814,819 That region of residues encompasses part of both the zinc binding loop VI and electrostatic loop VII (Figure 29), providing further evidence that alterations in these loops may affect the stability of dimeric SOD1. Palmitoylation of SOD1 at cysteine-6 impairs nuclear localization leading to mislocalized SOD1 accumulating in the cytoplasm.820 Antinone et al. found that misfolded SOD1 in SOD1G93A cell culture and mice spinal cords showed increased palmytoylation compared to SOD1WT.821 Other PTMs have been shown to protect SOD1. Cysteinylation of SOD1 could prevent oxidation of cysteine 111; the crystal structure of cysteinylated SOD1 showed a slight change at loops VI and VII.822 Fay et al. found that a phosphor-mimicking aspartic acid substitution at Threonine-2 stabilizes SOD1 A4 V dimers, suggesting that phosphorylation of SOD1 could be an innate cellular protection against SOD1 destabilization.823
Mutations from environmental toxins have also been shown to destabilize the dimer interface of SOD1.824–826 Most notably, exposure to β-methylamino-l-alanine (BMAA) is linked to 100 times increase in ALS incidence in the indigenous Chamorro population on Guam. A new study shows that substituting any serine for BMAA in SOD1 has the potential to destabilize the dimer interface and promote SOD1 aggregation.827 Post translational modifications and mutations from environmental toxins could explain why there is such a large amount of sALS cases with no known genetic cause.828 The toxic effects of mutant SOD1 on RNA processing, protein trafficking/degradation, cytoskeletal/axonal dynamics, and mitochondria function, along with the decrease in dimer stability from modifications and mutations, show the aggregation of SOD1 into toxic trimers leads to a toxic gain of function in both sporadic and familial ALS.
9. AD, PD, AND T2D THERAPIES
9.1. Small Molecules, Polymer-Based Molecules, and Carbon Nanoparticles
Understanding the mechanistic determinants of amyloid–inhibitor interaction is a prerequisite for developing new drugs, as discussed in recent reviews,193,831 and designing new strategies to reduce the toxicities by targeting Aβ, tau, αS, and IAPP.302,832–846
9.1.1. Small Molecules.
While a plethora of small molecules has been shown to be efficient in solution, the lipid membrane has been shown to catalyze the formation of toxic amyloid intermediates and influence amyloid aggregation. The amphipathic lipid membrane not only complicates the amyloid aggregation process but will also modulate the action of amyloid inhibitors. Therefore, to efficiently suppress amyloid aggregation, the action of small-molecule compounds must be examined in the presence of the lipid membrane. In a recent study, Cox et al. developed an approach for high-throughput screening of a library of >1800 molecules that inhibited Aβ aggregation in a membrane environment.847 This study reported five small molecules as efficient inhibitors of Aβ aggregation both in solution and in the presence of membrane. Using a combination of fluorescence and dot-blot assay, they validated the inhibitory efficacy of these five molecules (AQ-4, THQ-1, BF-3, DHQ-1, and DHQ-2, Figure 30) that generated nonfibrillary Aβ species in the presence of large unilamellar vesicles composed of mixed (zwitterionic/anionic) lipids.847 The mechanism of the action of these compounds to inhibit Aβ fibrillation in the membrane interface, as compared to widely studied natural phenolic compounds (EGCG, curcumin, etc.), needs to be explored. However, a suggested mechanism of action is the ability of the screened compounds to interact and adsorb on the membrane without affecting its interaction with Aβ. Another approach that promotes Aβ aggregation and reduces the formation of toxic intermediates has also been considered to treat AD. In this category, the major players are the cellular polyamines (Figure 30), present intracellularly with millimolar concentration, which have been shown to promote Aβ aggregation by suppressing the amount of toxic Aβ species.848,849 Recently, Dobson et al. discovered a natural product aminosterol trodusquemine (Figure 30) isolated from shark that enhanced Aβ aggregation following the suppression of neurotoxicity.850 Promotion of Aβ aggregation has also been observed in the presence of poly amino acids.852 Nonsteroidal anti-inflammatory drug (NSAID) sulindac sulfide is another small molecule that forms colloid particles and catalyzes Aβ fibrillation (Figure 30). Solid-state NMR results probing the action of NSAID have identified a homogeneous distribution of amyloid-fibers characterized by a canonical β-strand–turn–β-strand with reduced neurotoxicity.853
Figure 30.
Chemical structures of a few selected small molecules that are shown to inhibit amyloid aggregation, remodel aged fibers, and/or promote Aβ or IAPP fibrillation.
Small-molecule inhibitors of IAPP have been reviewed in ref 854. We report on recent studies on a few of these small molecules that modulate IAPP aggregation, morphology, and pathological phenotypes. Natural polyphenols such as EGCG present in green tea, curcumin present in turmeric, and resveratrol present in red wine inhibit IAPP amyloidogenesis (Figure 30).855 Curcumin is a planar biphenolic compound that delays IAPP aggregation when mixed with peptide monomers. The mechanism of action is proposed to be mediated by curcumin assisted disassembly of an α-helical intermediate of IAPP, that has been reported to nucleate the aggregation.856 Daval et al. highlighted a less likely therapeutic use of curcumin due to its toxicity to cells and proposed chemical modifications. This study showed curcumin to be toxic at micromolar concentration to β-cells and partially protect cells from IAPP induced apoptosis.857 Moreover, the poor water solubility of curcumin is a major limitation for its potential use to protect cells from amyloid toxicity. A chemically modified curcumin derivative (CurDac) was recently shown to significantly modulate IAPP aggregation (Figure 30).846 Pithadia et al. tested the effect of CurDac in rescuing the IAPP induced membrane damage using a variety of biophysical techniques.858 Using fluorescence and NMR, they observed a significant delay in IAPP aggregation when peptide monomers are mixed with CurDac. In addition, they identified the interaction of CurDac with aged IAPP fibers to be mediated by the aromatic ring atoms. More interestingly, they demonstrated that CurDac inhibits the membrane catalyzed IAPP fibrillation.858 A very recent study on CurDac interaction with IAPP fibers by Cox et al. presented some surprising results highlighting the limitations of the use of CurDac as a therapeutic compound. This study reported the ability of CurDac to disaggregate amyloid fibers as tested on three different amyloidogenic peptides (Aβ, IAPP, and human calcitonin).846 Toxicity measurements of the disintegrated peptide species from aged fibers, or monomers, incubated with CurDac were found to enhance the cytotoxicity of IAPP.846 Together, these studies’ direct small-molecule intervention of IAPP aggregation could be lethal and need further attention and chemical optimization. Like CurDac, EGCG and its stereoisomer (GCG) have been shown to be an effective inhibitor of monomeric Aβ and IAPP aggregation, remodeling toxicity of oligomers and disaggregate aged fibers in solution (Figure 30).859,860 In addition, a seeding reaction identified that EGCG bound IAPP species do not bind IAPP monomers and thus restrict fibrillation.859,861 Unlike curcumin, EGCG is shown to rescue cells from IAPP induced toxicity. Another recent study reported the combined effects of EGCG and zinc to suppress IAPP aggregation and toxicity.862 Morin hydrate is another good example of a small-molecule compound that is also shown to inhibit IAPP aggregation and disaggregates the preformed IAPP fibers (Figure 30).863 IAPP oligomers are also targeted by small molecules to reduce their cytotoxicity. For example, sulfonated triphenyl methane derivative acid fuchsin864 and EGCG859 are shown to effectively inhibit IAPP aggregation and suppress toxicity when added to the peptide during the lag-phase of growth where toxic intermediates are formed. In addition to in vitro observation, the effectiveness of EGCG was tested on IAPP transgenic mice.865 Saravanan et al. reported a diphenylpyrazole derived inhibitor anle145c to effectively bind and generate nontoxic IAPP oligomers (Figure 30).842 Computational study of an analogous compound anle145c shows remodeling of preformed Aβ and IAPP oligomers.866 Polyphenols and their effect on IAPP aggregation in the membrane interface have also been tested. Derivatives of resveratrol with improved solubility (Figure 30) are shown to abolish IAPP fibrillation in the presence of liposomes.867 In addition, suppression of IAPP induced membrane damage is revealed by conjugating resveratrol with lipids containing a dimyristoylphosphatidyl moiety.867 Lolicato et al. provided mechanistic insights into rescue of IAPP induced membrane damage. The simulation shows resveratrol disrupts IAPP interaction with an anionic membrane by binding to the hydrophobic region (23–25 and 32–34) of the peptide.868
Many compounds were synthesized to prevent amyloid aggregation.842,869 Of note, the compound anle138b also ameliorates tau pathology.869 Studies have also attempted to determine potential inhibitors from natural compounds870,871 since they normally show low side effects, well-known pharmacological properties, and high availability. Several compounds with high antioxidant and anti-inflammatory properties were found to be able to remodel or inhibit the neurotoxic conformations of amyloidogenic peptides via various pathways.872 The inhibition probably activates through direct binding with the peptides873 and/or interfering with π–π stacking interaction between aromatic rings of amyloidogenic residues according to the observation via microscopic and quantum chemical analyses.874 Interestingly, some natural compounds such as taxanthin,875–879 brazilin,880–882 curcumin,883–887 dopamine,888–894 EGCG,283,860,895–900 resveratrol,328,901–904 and rosmarinic acid905–910 can simultaneously target the proteins involved in AD, PD, T2D, and ALS where all experimental and computational studies are reported in Table 3. Interestingly, although the inhibitory mechanisms of these ligands to the diseases are probably different, they all adopt a similar binding pose to Aβ peptides, α-synuclein, hIAPP, insulin, and SOD1 via molecular docking simulations (Figure 31). As seen in Table 3, the knowledge of the interaction of some drugs with the amyloid proteins is still lacking.
Table 3.
List of Seven Inhibitors for AD, PD, T2D, and ALS
| N0 | Compound | Alzheimer | Parkinson | Type II Diabetes | ALS |
|---|---|---|---|---|---|
| 1 | Astaxanthin (Experiments/Computations) | Inhibits Aβ (cell viability assay, ThT, TEM, neurite outgrowth assay/docking, MD, REMD, linear interaction energy) | Inhibits αS (cell viability assay, indirect immunofluorescence microscopy, mitochondrial membrane potential, immunohistochemistry, immunoblot analysis/N/A) | Increases insulin concentration (Immunodetection, 2-deoxy-glucose uptake, ROS detection, /N/A) | Inhibits SOD1 (immunocytochemistry, neurite outgrowth assay, photomicrographs/N/A) |
| 2 | Brazilin (Experiments/Computations) | Remodels Aβ fibrils (cell viability assay, CD, ThT assay, TEM/docking, MD) | Inhibits αS (AFM, cell viability assay, fluorescein–propidium iodide double staining assay, ThT assay/MD, binding energy) | Inhibits hIAPP (cell viability assay, CD, ThT assay, TEM, /MD) | N/A |
| 3 | Curcumin (Experiments/Computations) | Blocks Aβ oligomers (cell viability assay, free radical assay, TEM,/docking, MD, free energy perturbation) | Prevents formation of αS oligomers and fibrils (aggregation assay, apoptosis study, cell uptake study, cell viability assay, in vivo bioavailability study, immunofluorescence, immunohistochemistry, Western blot analysis, /N/A) | Inhibits hIAPP (DLS/DMD) | Inhibits SOD1 (AFM, ATR-FTIR, DLS, ThT assay, TEM, MTT metabolic assay, Rayleigh scattering/docking, steered MD simulations) |
| 4 | Dopamine (Experiments/Computations) | Inhibits Aβ fibrils (AFM, light scattering, TEM, size-exclusion HPLC, MS/MD, REMD) | Inhibits Aβ and αS (AFM, light scattering, TEM, size-exclusion HPLC, MS, /MD) | Inhibits hIAPP protofilbrils (ESI-IMS-MS, ThT assay, TEM,/MD, REMD) | Inhibits SOD1 (aggregation assay, X-ray crystallography, small-angle X-ray scattering,/docking, MD) |
| 5 | EGCG (Experiments/Computations) | Remodels Aβ oligomers (aggregation assay, cell viability assay, CD, dot blot assay, DLS, nitroblue tetrazolium staining, size-exclusion chromatography, ThT, EM, /MD, REMD) | Inhibits αS (aggregation assay, cell viability assay, CD spectroscopy, dot blot assay, DLS, nitroblue tetrazolium staining, EM, size-exclusion chromatography, NMR, ThT/docking, REMD) | Remodels hIAPP fibrils (ESI-IM-MS, fibril depolymerization, ThT assay, TEM, /MD, REMD) | Inhibits SOD1 aggregation (ANS assay, collision induced unfolding, ESI-IM-MS, ThT assay, VC50 experiment, /docking, steered MD simulations) |
| 6 | Resveratrol (Experiments/Computations) | Reduces Aβ aggregation (aggregation assay, Bio-Rad protein assay/DMD) | Inhibits αS (cell viability assay, fibril formation assay, size-exclusion chromatography, CD, TEM/MD) | Inhibits hIAPP (DLS/DMD) | Inhibit SOD1 (aggregation assay, ESI-MS, MS, ThT assay/docking, DMD simulations, quantum mechanics calculation) |
| 7 | Rosmarinic Acid (Experiments/Computations) | Blocks Aβ oligomerization (AFM, cell viability assay, oligomer size distributions, electrophysiology, NMR, size-exclusion chromatography/docking) | Inhibits αS oligomerrization (AFM, CD, chemical cross-linking, size-exclusion, ThT, EM, chromatography, NMR, electrophysiology/MD) | Inhibits insulin (CD, NMR, ThT assay, TEM/docking) | Extends survival of rats having SOD1 (G93A) |
Figure 31.
Chemical structures and the binding poses of the seven natural ligands to (A) 4Aβ11–42 peptides (PDB 2MXU),911, (B) αS (2X6M),912 (C) hIAPP (6Y1A),253 (D) insulin (1GUJ),913 and (E) SOD1 (6FLH).914 Binding poses were obtained by Autodock vina915 with exhaustiveness of 8 and a grid of 60 × 60 × 60 Å covering the whole receptor.916 Astaxanthin, brazilin, curcumin, dopamine, EGCG, resveratrol, and rosmarinic acid are highlighted in red, green, light blue, blue, yellow, magenta, and orange, respectively.
9.1.2. Polymer-Based Molecules.
Recently bioinspired peptides containing natural and unnatural amino acids, synthetic polymers, and polymer-based particles have been developed to control amyloid aggregation. Selectivity, specificity, toxicity, rational designing, membrane permeability, and proteolytic stability are the advantages of these molecules to effectively target amyloidogenic proteins/peptides. Sahoo et al. developed a cationic PMAQA polymer and proposed its multifunctional use against AD and T2D.851 It is shown that PMAQA917 accelerates the fibrillation (~1 min) of Aβ and delays IAPP aggregation (~3 days). In another study, star-shaped poly(2-hydroxyethyl acrylate) polymers are shown to accelerate the fibrillation of IAPP thus reducing the formation of toxic intermediates.918 Recently, short anionic and cationic styrene-based copolymers (~2.2 kDa) are shown to catalyze IAPP aggregation by quickly inducing the formation of nontoxic fibers, whereas cationic styrene copolymers inhibit IAPP aggregation and induce generation of nontoxic globulomers.919 Smith et al. recently developed polymer–peptide conjugates to control the molecular ordering of Aβ structure and stability, but the pathological phenotypes of these nanostructure are yet to be tested.920 Thermoresponsive polymers varying in hydrophobicity/hydrophilicity ratio are shown to selectively modulate Aβ aggregation kinetics.921 Poly(p-phenylenevinylene) derived polymer is shown to degrade Aβ fibers that are nontoxic and to clear the Aβ plaques from the brain tested ex vivo.922 Polymer nanocomposites have been shown to eliminate toxic amyloid species under in vitro and in vivo conditions. Zhao et al. developed a nanocomposite that wrapped a protein with a cross-linked Aβ fragment “KLVFF” polymer layer. The nanocomposite is shown to reduce toxic oligomers by trapping Aβ following the formation of coassembled nanoclusters, and its efficacy is measured in vivo using Alzheimer’s mice.923 Dendrimers designed using a poly(propyleneimine) core and a maltose-histidine shell have been shown to enhance its ability to cross the blood–brain-barrier (BBB) and protect from the progression of AD in transgenic mice.924 Poly(amidoamine) dendrimer and N-isopropylacrylamide:N-tert-butylacrylamide copolymeric nanoparticles are shown to effectively abolish IAPP aggregation and its toxicity.925,926 Lipid-nanodiscs prepared by conjugating synthetic polymers or peptides with natural phospholipids were recently tested on Aβ.263,274 Polymer nanodiscs are shown to strongly bind Aβ following a quick induction of fibers that tested non-neurotoxic.263 Protein and peptide lipid-nanodiscs show promising application in trapping amyloid intermediates and delaying aggregation of Aβ or IAPP.274,285 Apolipoprotein J conjugated nanoparticles have been shown to reduce the amount of cerebral Aβ in transgenic mice.927 Cationic fluorescent conjugated polymer nanoparticles are shown to inhibit Aβ aggregation.928 Nanoliposomes decorated with Aβ binding surface containing benzothiazolyl are proposed to be an effective therapeutic systems for AD treatment.929 For other peptide-based inhibitors, readers are referred to a recent review930 and recent articles.931–933
9.1.3. Carbon Nanoparticles.
Carbon nanoparticles (CNPs, including fullerene, carbon nanotube, graphene, and their derivatives) have received great interest due to their exceptional physicochemical properties (such as large surface-to-volume ratio and capability of translocation across the BBB), as discussed in two recent reviews.836,838 As early as 1997, it was reported by Dugan et al. that C60 derivatives (i.e., carboxyfullerenes) can function as neuroprotective drugs in vivo.934 In the following years, a few research groups investigated the influence of carbon nanoparticles on the aggregation of amyloidogenic proteins. For example, in 2003, a ThT study by Kim et al. showed that fullerenes inhibit strongly the amyloid aggregation of Aβ1–40 at the early stage.935 In 2007, TEM and in vivo investigations by Podolski et al. demonstrated that hydrated fullerenes impede the fibrillization of Aβ25–35 peptide and improve the performance of the cognitive task in control rats.936 In the same year, Linse et al. reported that carbon nanotubes (CNTs) enhance the fibril formation of human β2-microglobulin by accelerating the nucleation process.937 Those early experimental studies revealed the distinct roles of CNPs on the aggregation of different proteins and inspired more research in this direction. In order to determine the physicochemical determinants of CNPs in modulating protein aggregation and to find more effective inhibitors against protein aggregation, a growing number of computational and experimental studies have been carried out over the past decade. In what follows, we mostly describe the most recent simulation studies aimed at understanding the inhibitory mechanisms of CNPs against the fibrillization of Aβ fragments, full-lenth Aβ, hIAPP, and αS fragment (Table 4).
Table 4.
Summary of Atomistic Simulation of the Inhibition of Carbon Nanoparticles (CNPs) on the Fibrillization of Amyloid Peptides, Starting from Oligomers or Protofibrilsa
| ref | Peptide | Carbon nanoparticle | Force field | Solvent model | Method | T (K) | Time (ns) | Oligomer or Protofibril |
|---|---|---|---|---|---|---|---|---|
| 938 | Aβ16–22 | CNT | Gromos96 43a1 | SPC | REMD | 310–420 | 110 ns × 40 | 8mer |
| 939 | Aβ16–22 | CNT-(OH)30 | Gromos96 43a1 | SPC | REMD | 310–420 | 200 ns × 40 | 8mer, fibrillar β-sheet |
| 940 | Aβ16–22 | C60, C180 | Gromos96 43a1 | SPC | REMD | 310–420 | 200 ns × 40 | 8mer |
| 942 | Aβ16–21 | Graphene nanosheet | OPLS/AA | SPC | MD | 310 | 1000 ns × 5 | Protofibril |
| 945 | Aβ42 | C60 | Gromos96 53a6 | SPC | REMD | 306–418 | 340 ns × 54 | 2mer |
| 946 | Aβ42 | GO60 | Gromos96 53a6 | SPC | REMD | 306–418 | 410 ns × 54 | 2mer |
| 308–419 | 500 ns × 54 | |||||||
| 950 | Aβ11–42 | CNT-(OH)30 | OPLS/AA | TIP4P | MD | 310 | 100 ns × 3 | Protofibril |
| 953 | Aβ33–42 | GO60, GO120 | Amber99sb-ILDN | TIP3P | REMD | 307–437 | 400 ns × 54 | 4mer |
| 954 | hIAPP | GO nanosheet | Medusa | Implicit solvent EEF1 | DMD | 290 | 50 ns × 10 | 2mer, 4mer, 6mer |
| 955 | hIAPP | GQD | Medusa | Implicit solvent EEF1 | DMD | 300 | 400 ns × 2 | 2mer |
| 957 | hIAPP | CNT-(OH)30 | OPLS/AA | TIP4P | REMD | 306–409 | 400 ns × 48 | 2mer, Protofibril |
| MD | 305–445 | 400 ns × 48 | ||||||
| 310 | 200 ns × 3 | |||||||
| 958, 959 | hIAPP | C60, C60(OH)8 | OPLS/AA | TIP4P | REMD | 306–409 | 360 ns × 48 | 2mer, Protofibril |
| C60(OH)24 | MD | 310 | 300 ns × 6 | |||||
| 960 | αS68–78 | C60(OH)4n (n = 0–5, 10) | Medusa | Implicit solvent EEF1 | DMD | 300 | 200 ns × 20 | 10mer |
Abbreviations: GO, graphene oxide; CNT, carbon nanotube; GQD, graphene quantum dot.
9.1.3.1. Inhibition of CNPs against Fibrillization of Aβ/Aβ-Fragment and Shape/Surface Curvature Effects of CNPs.
In 2011, Li et al. investigated the effects of carbon nanotubes on the oligomerization of Aβ16–22 peptide by atomistic REMD simulations.938 Their simulations demonstrated that CNTs not only inhibit the formation of β-sheet-rich octamers (with an average β-sheet content of 7.9% in the presence of CNT relative to 44.5% in the absence of CNT) but also destabilize fibrillar β-sheets through hydrophobic and π–π stacking interactions. The authors proposed that CNT is likely to impede the fibrillation of Aβ16–22 and of the full-length Aβ, as β-sheet is the dominant secondary structure content of Aβ fibril and Aβ16–22 is the central hydrophobic core of Aβ. Two years later, Xie et al. examined the influence of a hydroxylated carbon nanotube (CNT-OH) on Aβ16–22 aggregation using a combined simulation and experimental method.939 REMD simulations revealed that a hydroxylated CNT, with the same diameter and length as the pristine CNT used in ref 978, can also prevent the formation of ordered β-sheet-rich octamers (with a β-sheet content of 17.5% in the presence of CNT-OH). Hydrophobic, π–π stacking and electrostatic interactions between CNT-OH and Aβ16–22 play important roles in inhibiting β-sheet formation. Their AFM and thioflavin ThT fluorescence experiments confirmed the inhibitory effect of both CNT and CNT-OH on Aβ16–22 fibrillization, in support of the REMD simulation predictions.938,939 Next, Xie et al. investigated the effect of fullerenes with different surface curvatures (C60 and C180) on the β-sheet formation of Aβ16–22 octamers by REMD simulations.940 They found that C60 significantly reduces the β-sheet probability (25.7% with C60 relative to 44.5% without C60) and increases the coil propensity, thus retarding the fibril formation of Aβ16–22. The simulation-predicted inhibitory effect of C60 on Aβ16–22 fibrillization was confirmed by AFM and ThT experiments.940 Interestingly, it was found that C180 (with the same total mass as C60) exhibits a stronger inhibitory effect on the β-sheet formation (with a β-sheet content of 18.1%) than C60. This observation can be explained by the strong hydrophobic and aromatic stacking interactions between fullerene hexagonal rings and the Phe rings of Aβ16–22 as C180 contains more hexagonal rings than C60. This result highlights the significant role of fullerene hexagonal rings in the inhibition of Aβ16–22 fibrillation. By comparing the β-sheet probability of Aβ16–22 oligomers in the presence of CNT, C60, and C180 (with almost the same total mass) obtained by the two REMD studies,938,940 it was found that CNT displays the strongest inhibitory effect on the β-sheet formation, followed by C180 and then C60. The different inhibitory abilities are attributed to the delicate balance of hydrophobic and aromatic stacking interactions dictated by the shape and/or surface curvature of CNPs. Consistent with REMD results, AFM experiments by Wang et al. demonstrated that GO nanosheets have the strongest inhibitory effect on Aβ33–42 aggregation, followed by nanotubes and nanodots.941 These results reveal the important role of shape and surface curvature of CNP in inhibiting Aβ16–22/Aβ33–42 fibrillation. Meanwhile, Yang et al. examined the influence of graphene nanosheets on preformed Aβ16–21 fibrils by performing MD simulations. They found that graphene nanosheets can penetrate amyloid fibrils, extract a large number of peptides from them, and destruct Aβ16–21 fibrils.942
In order to reveal at the atomistic level the molecular mechanism behind the experimental observations showing that both C60 and graphene oxide (GO) nanosheet can inhibit the fibrillization of full-length Aβ peptide,935,942–944 Sun et al. and Jin et al. performed REMD simulations on Aβ1–42 dimer in the absence and presence of four C60 molecules or four GO nanosheets.945,946 The GO nanosheet contains the same number of carbon atoms as C60, which is denoted as GO60. These REMD simulations showed that Aβ1–42 peptides form diverse β-hairpin-containing dimeric conformations, whereas those conformations are significantly reduced in the absence of C60 or GO60. C60 suppresses the β-sheet formation of Aβ1–42 mostly by binding to the central hydrophobic motif LVFFA and the C-terminal hydrophobic region I31 ~ V40, while GO60 inhibits the β-sheet formation by mainly binding to charged residues and hydrophobic residues. GO60 displays a better inhibitory effect on the β-sheet formation of Aβ1–42 dimer than C60,946 indicating that the shape of CNP may play a role in the inhibition of Aβ42 aggregation. In silico and in vitro experiments showed that water-soluble fullerenol C60(OH)16 tightly binds to negatively charged residues of Aβ1–40 monomers and effectively reduces the formation of amyloid fibrils.947 The interactions of C60/C60OH with U-/LS-shaped Aβ protofibrils were studied by explicit-solvent MD simulations.948,949 It was found that C60 and C60OH exhibit different binding mechanisms and the binding of C60 to Aβ protofibril results in disruption of the D23–K28 salt bridge. More recently, a combined MD simulation and experimental study by Liu et al.950 reported that hydroxylated carbon nanotubes (CNT-OHs) also inhibit Aβ42 fibrillization, disaggregate mature fibrils, and reduce Aβ42-induced cytotoxicity. The six residues H13, H14, Q15, V36, G37, and G38 contribute most to the interactions between CNT-OH and Aβ11–42 protofibril.
9.1.3.2. Size Effect of Carbon Nanoparticles on the Inhibition of Aβ33–42 Aggregation.
CD spectra and AFM experiments by Dong’s group reported that GO interferes with the aggregation pathway of Aβ33–42 peptide, leading to a reduced β-sheet content and considerably short fibrils, and large-size GO displays a strong capacity in impeding Aβ33–42 aggregation.951,952 In order to unravel the mechanistic basis of the inhibition and size effect of GO nanosheets on the fibrillization of Aβ33–42 peptide, Chen et al.953 investigated the oligomerization of Aβ33–42 peptide in the absence and presence of four GO60 nanosheets or two GO120 nanosheets by performing all-atom REMD simulations starting from four extended Aβ33–42 peptide chains. Their simulations showed that in the absence of GO nanosheets, four Aβ33–42 chains can form β-barrel-like structures and antiparallel β-sheets. Inter-peptide hydrophobic interactions among residues M35, V36, V39, and V40 and backbone hydrogen bonding interactions play crucial roles in the formation of β-barrels/β-sheets. GO retards Aβ33–42 oligomerization by interfering with peptide–peptide interactions. Consisting of the same total carbon atoms and oxidation groups as GO60, GO120 exhibits a stronger capacity in inhibiting Aβ33–42 aggregation than GO60. The oxidation groups of GO120, especially the hydroxyl groups, can form more hydrogen bonds with Aβ33–42 than those of GO60. Moreover, GO120 has a larger hydrophobic contact surface area with Aβ33–42 than GO60. These results reveal that GO120 displays stronger hydrogen-bonding and hydrophobic interactions with Aβ33–42 than GO60, thus leading to a better inhibitory effect on Aβ33–42 aggregation. These simulation results provide mechanistic insights into the inhibition and size effects of GO nanosheets on the aggregation of Aβ33–42 peptide.953
9.1.3.3. Inhibition of CNPs against hIAPP/αS Aggregation and Effect of Surface Hydroxylation Extents of CNPs.
The influence of zero-, one-, and two-dimensional carbon nanoparticles on the amyloid fibrillization of other amyloid proteins has also been reported in the last five years. Nedumpully-Govindan et al. investigated the effect of GO nanosheets on hIAPP fibrillization and cytotoxicity by combining computational modeling, biophysical characterization, and cell toxicity measurements.954 DMD simulations showed a strong binding of hIAPP monomers/oligomers on the surface of GO nanosheets. TEM images and cell toxicity assays indicated that small GO nanosheets inhibit hIAPP aggregation and reduce hIAPP toxicity. Following that work, Wang et al.955 demonstrated the use of graphene quantum dots (GQDs: a single- or few-layer graphene with a tiny size less than 100 nm) as inhibitors against the aggregation and toxicity of hIAPP using DMD simulations, in vitro ThT and β-cell viability assays, and an embryonic zebrafish model. Their recent study956 reported that GQDs can rescue protein dysregulation of pancreatic β-cells exposed to hIAPP, pointing to the potential of using GQDs for in vivo mitigation of T2D amyloidosis.
By combining all-atom REMD and MD simulations, AFM and TEM images, ThT, and cell toxicity assays, Mo et al.957–959 examined the influences of hydroxylated carbon nanotubes and fullerenes on the fibrillization of hIAPP. REMD and MD simulations demonstrated that both CNT-OHs and C60OHs not only suppress the formation of β-sheet and β-hairpin-containing amyloidogenic precursors of hIAPP but also remodel or disassemble preformed hIAPP protofibrils/fibrils. Hydrogen-bonding, hydrophobic, cation–π, and π–π stacking interactions between hIAPP and CNT-OH/C60OH are the dominant driving forces in inhibiting hIAPP aggregation. Turbidity analyses, ThT fluorescence assays, CD microscopies, and TEM and AFM images provide direct evidence for the simulation-predicted β-sheet inhibition and protofibril-disruption by hydroxylated CNTs/fullerenes. SH-SY5Y cell viability assays demonstrated the rescue effect of C60OHs on hIAPP-induced cytotoxicity.958 The REMD simulations by Bai et al.958,959 showed that C60, C60(OH)8, and C60(OH)24 possess different inhibitory effects on the β-sheet formation of hIAPP: the β-sheet content of hIAPP dimer in the presence of C60, C60(OH)8, and C60(OH)24 is respectively 1.8, 4.2, and 5.6% (the β-sheet content of hIAPP dimer alone is 10.8%). These data reveal that hydroxylation extents modulate the inhibition capacity of C60OHs against hIAPP aggregation. Recently, Sun et al.960 explored the effects of hydroxylation extents of C60 on β-sheet formation of the nonamyloidogenic core (NAC) fragment (residues 68–78) of αS by DMD simulations. Their simulation data showed that the αS68–78 peptide can assemble into cross-β aggregates and β-barrel intermediates. Hydroxylated C60 (C60(OH)4n with n = 1–5) significantly inhibits β-sheet formation and aggregation of αS68–78 peptide, while C60(OH)40 displays very weak inhibitory impact. These results suggest the necessity of the amphiphilic surface chemistry of hydroxylated fullerenes in hindering the amyloid aggregation of α-Syn68–78 peptide. The influence of GQDs on the aggregation of α-synuclein (α-Syn) protein was investigated by Kim et al. using a combination of in silico, in vitro, and in vivo methods.961 It was shown that GQDs not only inhibit α-Syn fibrillization and disaggregate fibrils but also penetrate the BBB and possess unique neuroprotective effects against the neuropathological αS aggregates/fibrils.961 A recent study demonstrated the antiaggregation effect of fullerenols (C60(OH)30 and C70(OH)30) on αS protein and the neuroprotective activity in Drosophila PD models.962 The observed neuroprotective effects of CNPs point to carbon-based nanomedicine as a new frontier against human amyloid diseases although challenges of CNPs in the application of biomedicine still exist, as discussed in a recent review.963
Taken together, all studies demonstrate that CNPs/small molecules can effectively interfere with the fibrillization process and reduce the amyloid-induced toxicity, providing new clues for de novo design of antiamyloid inhibitors. The schematic diagram in Figure 32 illustrates the inhibitory mechanisms of CNPs/small molecules against protein aggregation.
Figure 32.
Schematic representation of the molecular mechanisms by which carbon nanoparticles (CNPs) and small molecules inhibit the aggregation of proteins/peptides discussed in this review. Amyloid proteins can self-assemble into toxic β-sheet-rich oligomers (such as β-barrels) and protofibrils/fibrils. CNPs/small molecules can prevent β-sheet formation and disassemble preformed protofibrils/fibrils through physical interactions and finally lead to the inhibition of protein amyloid formation. CNPs include graphene, fullerene, carbon nanotube (CNT), and their derivatives (such as hydroxylated CNT/fullerene, graphene oxide, and graphene quantum dot). Many small-molecule inhibitors have been reported in the literature, such as dopamine, EGCG, curcumin, and resveratrol. Due to space limitations, only fullerene, carbon nanotube, dopamine, and EGCG are illustrated in the diagram.
9.2. Active and Passive Immunizations as Alzheimer’s and Parkinson’s Therapies
9.2.1. Active Immunization as AD Therapy.
Active immunization is where the exposure of the body to an antigen generates an adaptive immune response. While the response can take days or weeks to fully develop, immunization is long-lasting and potentially even lifelong. Vaccines that work in this way are a triumph of medicine, providing protection against diphtheria, tetanus, whooping cough, smallpox, polio, measles, mumps, rubella, and numerous other major diseases. Vaccine antigens are either live attenuated, killed inactivated, toxin mimics, or virus subunits. Subunits are made either using recombinant DNA technology or normal bacteriological growth processes. Vaccines also contain excipients, present to stabilize the vaccine, help with delivery to the right part of the body, or improve the immune response (adjuvants).964,965
The success of vaccines in general suggested the tantalizing prospect of developing a vaccine for AD. The pioneering study in this field was AN-1792: synthetic Aβ(1–42) coupled with a glycosidic adjuvant called QS-21.966 While it might seem paradoxical to use Aβ therapeutically if excess Aβ leads to AD, the idea was that Aβ might beneficially stimulate antibody production, rather than neurodegeneration, if given early. In a mouse model for AD that overexpressed mutant human APP, administration of AN-1792 to young mice prevented the later development of Aβ plaques, neuritic dystrophy, and astrogliosis. Even older animals benefitted from treatment, showing reduced AD pathology.966 These encouraging results led to a Phase 2a trial in 273 patients with mild to moderate AD. The trial had to be halted after 6% of patients developed brain inflammation. Follow up studies of recipients showed mixed results: Post-mortems showed that while plaque could be cleared, neurofibrillary tangles were not.967 A minority of patients developed antibodies against the N-terminal region of Aβ.968 Some patients showed functional benefit several years later.969 Phospho-tau was reduced.970
The AN-1792 results prompted various studies aimed at developing an AD vaccine. Most designs use a fragment of Aβ or repeats of a fragment, enough to stimulate an immune response while avoiding any potential problems from using the full-length, aggregation-prone sequence. The N-terminal region of Aβ is most often used, as most antibodies target this region of the peptide. Various other excipients are included to help with delivery and stability or to help stimulate an immune response. Most active AD vaccine candidates have not progressed beyond Phase 1 trials or have been discontinued, presumably due to lack of efficacy. A few trials in AD patients are ongoing, however:
AB-vac40 consists of multiple repeats of a short C-terminal fragment of Aβ40 conjugated to the keyhole limpet cyanine carrier protein and formulated with the adjuvant alum hydroxide.971 Using the C-terminal end of Aβ is designed to minimize binding to the parent APP when APP is within a membrane. It will also be unaffected by N-terminal reactions of Aβ. Coupling to the keyhole limpet protein boosts the immune response. A pilot study in 12 participants found that 11 successfully developed antibodies after three injections of ABvac40. There were no safety issues. A Phase 2 trial of AB-vac40 is currently underway, with 120 participants at the earliest stage of AD or MCI, monitored over 2 years using amyloid PET (position emission tomography) scans, bio-markers, and cognitive and quality-of-life measures.
UB-311 is two Aβ(1–14) peptides linked to T-cell peptide epitopes, formulated in a proprietary delivery system, biased to Th2 cells.972 It is safe and well-tolerated and successfully generates an immune response in almost all patients. After encouraging pilot data, Phase 2 study continues.
Naturally, there is considerable interest in targeting tau as well as Aβ in AD.973 Since tau aggregates are intracellular, it seemed doubtful whether antibody administration could be beneficial. However, promising results in tau transgenic mice974,975 gave some encouragement. The AADvac1 vaccine is amino acids 294 to 305 of tau (KDNIKHVPGGGS), coupled to keyhole limpet hemocyanin with the aluminum hydroxide adjuvant. In transgenic rats it reduced tau pathology and improved function; it reduced AD-type hyperphosphorylation of tau by approximately 95%.976 In patients, 98% generated antitau antibodies and showed reduced hyperphosphorylation.977 Patterns of tau phosphorylation are distinct markers of disease progression over decades.523
Tau-targeting antibodies may enter neurons using Fcγ receptors or endocytosis, after which they can promote intracellular clearance of pathological tau.978 Alternatively, clearing extracellular tau may be all that is needed, as this could stop transmission between cells.979 Intracellular tau oligomers are known to be secreted and taken up by nearby cells.980
GV001 is a 16 amino acid peptide with a sequence from the human enzyme telomerase reverse transcriptase, an enzyme commonly upregulated in cancer. It has anti-inflammatory and antioxidative stress properties. It was first developed as an unsuccessful immunotherapy for pancreatic cancer. However, in rat neuronal stem cells, GV1001 reduced Aβ oligomer-induced toxicity and protected against oxidative stress,981,982 so it is being repurposed as an AD vaccine. A small phase 2 study showed promising effects at halting cognitive decline.983
9.2.2. Passive Immunization against AD.
Elderly people typically show a weak response to antigens, making active immunization a challenge. Alternatively, passive immunization involves creating antibodies to AD targets outside the body and using them as a therapy. Antibodies can be generated in an animal either against antigens of Aβ or tau fragments, or against aggregates to achieve specificity against pathogenic forms of Aβ. The monoclonal antibodies are collected and given to a patient with AD. Their own immune system then clears the antigens bound to the antibodies.984 A big problem for this approach is that few immunoglobulins dosed into blood will enter the brain. Crossing the BBB might not be necessary, however. The peripheral sink hypothesis holds that Aβ can slowly leave the brain, down a concentration gradient, if it is mopped up in the blood.985 Alternatively, passive immunization may work by antibodies crossing the BBB, activating microglia, and triggering phagocytosis of antibody-bound antigens, perhaps using strategies to facilitate this process.984
A second concern with passive immunization is initiating unwanted inflammation in the brain, already recognized as a pathological feature of AD. Using IgG1 and IgG4 immunoglobulins and avoiding proinflammatory subclasses such as IgG3 can reduce this risk.986 Examples of promising antibodies currently in clinical trials for AD include the following:
Aducanumab shows a strong preference for binding to aggregated forms of Aβ over monomers, a highly desirable property given that oligomers are likely to be the most toxic form of the peptide. It has more than 10,000-fold selectivity for aggregates over monomers,987,988 binding to Aβ residues 3–7 in an extended conformation.989 In a mouse model, the antibody reduced Aβ deposits by 70%, mostly likely by microglia-mediated phagocytosis.987 A phase 1b trial of 165 prodromal or mild AD patients showed that aducanumab could clear Aβ plaques with no toxicity, and there were hints of cognitive benefits and decreased inflammation.987 Large phase 2 trials, called ENGAGE and EMERGE were therefore initiated.
In March 2019, both trials were stopped early, due to an apparent inability to slow cognitive decline, the primary end point of the trial.990,991 Most unusually, however, in October 2019, Biogen announced that this conclusion was premature: later analysis of a larger EMERGE data set showed that patients on the highest dose of aducanumab (10 mg/kg), given to ApoE4 carriers, had a significant reduction in cognitive decline; the low dose group also showed benefit, but this was not statistically significant. Similarly, the ENGAGE trial showed that a subgroup of people receiving the higher dose declined more slowly, though without meeting its primary end point. In addition, patients showed dose-dependent reduction in brain amyloid and phosphor-tau in CSF.992 Biogen claims that this is the first time that a phase 3 study has shown that clearing Aβ deposits can reduce cognitive decline in AD. As such, this would be a great boost to the amyloid hypothesis of AD, the immunotherapy approach, and AD drug discovery in general. Caution is needed however: the data is not yet published in detail in a peer-reviewed journal. Differences between the ENGAGE and EMERGE outcomes also need further investigation. In addition, positive claims based on retrospective analyses of subsets of participants are notoriously unreliable. Release of complete data and analyses is needed.993
Similarly to aducanumab, the IgG1 antibody BAN2401 shows a strong preference for binding to soluble protofibrils of Aβ, rather than the monomeric peptide, binding to the N-terminal region. In transgenic mice, a murine version of the antibody reduced Aβ protofibrils, while leaving monomers and insoluble plaques untouched.994 In phase 1 trials, the humanized antibody was nontoxic.995 An 18-month phase 2 study of 856 early AD patients showed statistically significant and dose-dependent slowing of amyloid deposition in the brain and cognitive function. Larger trials are underway.996
Solanezumab is a humanized monoclonal IgG1 antibody that binds the central region of monomeric Aβ. The IgG1 antibody Gantenerumab binds to N-terminal and central amino acids of Aβ, preferentially interacting with Aβ aggregates.997 Both reached phase 3 trials for mild AD, though they were unsuccessful.998,999 Nevertheless, as both have good safety records and some promising activity, they are now being tested in asymptomatic carriers of dominant mutations in APP, PSEN1, and PSEN2. If their phase 3 failures are because they are given too late in the disease progression, they may still work if given at an earlier stage to people sure to be on the path to AD. This long-term trial will first track AD biomarkers before adding the earliest cognitive effects.1000
A common form of Aβ in plaques is Aβ(p3–42), which has pyroglutamate at its N-terminus.1001 Donanemab is a humanized IgG1 monoclonal antibody that binds to this plaque-specific form. It is effective at clearing plaques from transgenic mice1002 and in patients with AD and a positive amyloid-PET scan.1003 A phase 2 trial is underway.
Work on antibodies that target tau antigens979 is less advanced than those that target Aβ, though phase 2 trials are in progress for a few. Gosuranemab is a humanized IgG4 monoclonal antibody that binds to extracellular N-terminal fragments of tau. These secreted forms of tau may cause neuronal hyperactivity, which leads to increased Aβ production,1004 one of many positive feedback loops in AD.1005 The antibody was tested in patients with progressive supranuclear palsy and showed >90% reduction in secreted tau fragments, while CSF total tau and phosphorylated tau were unchanged.1006 Semorinemab is an antitau IgG4 antibody that targets extracellular human tau, binding to the N-terminal region in both monomeric and oligomeric conformations, for all six tau isoforms and independent of phosphorylation.1007 Zagotenemab is a humanized antibody that is selective for soluble tau aggregates, binding to the N-terminal region of tau.1008 Phase 2 trials for participants with MCI or mild AD are ongoing for all three of these antibodies.
9.2.3. Antibody Therapies for Synucleinopathies.
To date, work on antibodies that targets αS is still little explored. PRX002 is a humanized IgG1 monoclonal antibody that binds to epitopes near the C-terminus of αS. In mouse models of PD and DLB, it reduces αS accumulation and counteracts behavioral deterioration.1009 In humans, PRX002 had a good safety profile. It nearly entirely removed free α-synuclein in the blood, though it did not reduce αS levels in CSF.1010 BIIB054 is a human IgG1 monoclonal antibody directed at an epitope near the N-terminus of αS, selective for aggregated forms.1011 In a pilot study in PD patients published in 2019, BIIB054 formed plasma complexes with αS. Phase 2 trials for both antibodies are underway. Recently, antibodies targeting the N-terminal domain of αS to neutralize the toxicity of oligomers have been attempted under in vitro and in vivo conditions. A primary antibody designed from the highly lipophilic region present in the N-terminus of αS (residues 1–25) showed promising activity in rescuing the neuronal cell damage induced by αS oligomers.1012
9.3. Infrared Laser, Ultrasound, and Electromagnetic and Electric Fields
9.3.1. Infrared Laser.
The low-level near-infrared laser irradiation with wavelength 600–1000 nm is widely used because it is able to deliver low energy, nonheating infrared light into tissues and nerves deeply.1013 So far, the first clinical trial with near-infrared at 810 nm wavelength pulsed at 10 Hz and power of 14.2 mW/cm2 was performed on five patients with mild to moderate AD for 12 weeks of active treatment and no treatment in the four follow-up weeks. Significant improvements were obtained in cognition after 12 weeks of active treatment. Also, increased function, better sleep, fewer angry outbursts, and less anxiety and wandering were also reported. There were no side effects such as diarrhea, nausea, vomiting, anorexia, or dizziness.1014
By in vivo studies, De Taboada et al. performed a near-infrared (808 nm) laser experiment to determine the effect of laser irradiation in an Aβ protein precursor transgenic mouse model. After six months, the numbers of Aβ plaques were significantly reduced in the brain as indicated by reduction in the plasma Aβ peptide levels of 17.7–39.8%, depending on the laser parameters.1015 Grillo et al. used near-infrared laser at 1072 nm wavelength to treat female TASTPM mice—an AD mouse model, and after 7 months, significant reduction in Aβ42 plaques (approximate 15%) was observed in the cerebral cortex.1016 Near-infrared laser was also applied to APP/PS1 transgenic mouse brain as well. Results show that Aβ plaques were reduced by more than 30%, neurofibrillary tangles and the hyperphosphorylation of tau were attenuated in the neocortex, hippocampus, and cerebellum as compared to that of nontreated mice.1017,1018 The authors suggested that the reduction in amyloid plaques is likely due to the enhanced mitochondrial function. It is unclear whether the reduction is also due to the direct interaction between the laser and amyloid fibrils.
By in vitro studies, several approaches using infrared laser have been developed to dissociate directly Aβ fibrils and oligomers. Li et al. used graphene oxide covalently connected to thioflavin-S, which can bind to Aβ aggregates.1019 The graphene oxide plays a role as a local heater, which absorbs strongly the near-infrared laser energy, to dissociate Aβ fibrils. Recently, Kawasaki and colleagues have demonstrated that the mid-infrared laser can be used to disassembly amyloid fibrils.1020–1023 They have developed a free-electron laser having specific oscillation characteristics of a picosecond pulse structure, a tunable wavelength within infrared frequencies, and a high photon density. Tuning the laser frequency to that of the amide I bands, they were able to dissociate various amyloid fibrils, including amyloid fibrils of lysozyme and of short peptide of the thyroid hormone, into soluble monomers. Because the amide I frequency of amyloid fibrils is shifted compared to single proteins, the laser-irradiation targeted to the amide I bands of fibrils should minimize the damage to surrounding molecules.1024 In addition to near- and mid-infrared techniques, Kawasaki et al. also employed the far-infrared laser to dissociate a fibril formed by calcitonin hormone peptide.1025 The far-infrared laser exhibits high penetration against the biological substance even under the low radiation energy. The results showed that the far-infrared irradiation changes the fibril structure more remarkably than the mid-infrared laser, suggesting that deep penetration of the far-infrared laser could disrupt the hydrogen-bond network inside of fibril more effectively.1025
At the computational level, we developed a laser-induced nonequilibrium MD simulation method and applied it to study the dissociation process of various fibrils, made of 5-mer U-shape Aβ17–42, 5-mer β-solenoid HET-s, and 200-mer GNNQQNY.1026,1027 Samples containing amyloid fibrils, DNA duplexes, and globular proteins are also considered, and the simulations show that the surrounding protein and DNA molecules are not affected, demonstrating therefore laser frequency selectivity for dissociation.1026 Simulations showed that the primary step in the dissociation process is due to the strong resonance between the fibril amide I vibrations and the tuned laser frequency and not just the deposited infrared thermal energy.
Figure 33 shows the results obtained from a joint experimental/simulation study of the dissociation of the GNNQQNY amyloid fibril.1027 We first constructed the fibril sample so that its 2D and 3D structures are similar to those of experiment before FEL irradiation. By tuning the laser frequency to the amide I band of the fibril, the resonance takes place and dissociation occurs. The calculated and observed wide-angle X-ray scattering profiles and secondary structures before and after laser irradiation being identical, we proposed a dissociation mechanism with high confidence from our simulations. We find that dissociation starts in the core of the fibrils by fragmenting the intermolecular H-bonds and separating the peptides and then propagates to the fibril extremities leading to the formation of unstructured expanded oligomers. This should be a generic mechanism of the laser-induced dissociation of amyloid fibrils.1027
Figure 33.
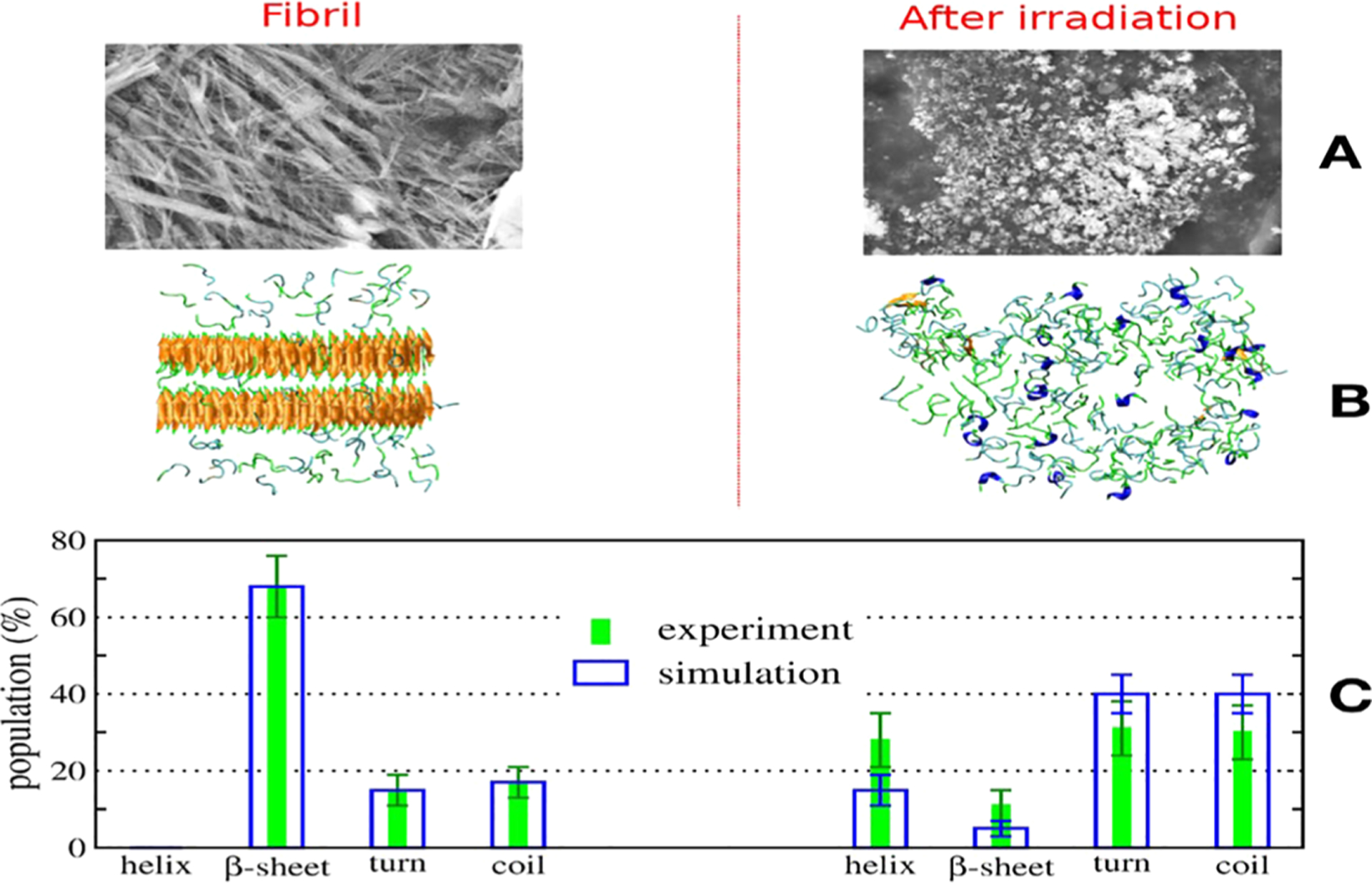
Images of the GNNQQNY fibril sample prior to and after laser irradiation of the GNNQQNY obtained from scanning EM experiment (A) and simulation (B). Their secondary structures are in (C).1027
We find that the effects of the laser irradiation are determined by a balance between fibril formation and dissociation. Understanding the factors that are responsible for this balance is important. The method was also employed to study the relative stability of polyQ and PolyN fibrils. The results show polyQ is more stable than polyN because the enthalpic contributions to the free energy favor polyQ over PolyN.1028
9.3.2. Ultrasound.
Ultrasound waves are mechanical waves which create oscillations with frequency between 20 kHz and 10 THz in the media through which they pass, generating thermal and nonthermal effects which are the basis of various therapeutic applications. The thermal effect arises from the absorption of ultrasound energy that creates heat in the system if the rate of heat production is greater than the rate of heat removal.1029 The nonthermal effects include acoustic radiation force, acoustic streaming, shock waves, and bubble cavitation.1030 Ultrasound waves are already used to treat certain types of cancer and glaucoma.
By in vivo studies, most works use focused ultrasound to open temporarily the blood–brain-barrier and thus facilitate the transport of molecules from the blood to the brain.1031 However, several studies using ultrasound alone also show positive results. Jordao et al. demonstrate that transcranial focused ultrasound (frequency of 0.5 MHz, pressure of 0.3 MPa) application leads to a significant reduction (20%) in mean plaque size 4 days after a single treatment in the TgCRND8 mouse model of AD. This is presumably due to the treatment facilitating endogenous antibodies to reach the brain and/or activation of microglial cells.1032 Leinenga et al. utilized the repeated scanning ultrasound (0.7-MPa peak rarefactional pressure, 10-Hz pulse repetition frequency, 10% duty cycle, and 6-s sonication time per spot) in combination with microbubble injection to treat APP23 mice for several weeks.1033 The histological and biochemistry analysis of brain tissues after treatment reveals a reduction of about 50% in the dense core amyloid plaque and soluble species of Aβ peptides, and no evidence of neuronal death, edema, erythrocyte extravasation, or ischemic changes. The memory deficit is successfully recovered. The authors postulate that the ultrasound somehow triggers microglial activation and enhanced Aβ phagocytosis.1033 However, it could also be possible that ultrasound increases the activation of Aβ-degrading enzymes, such as neprilysin, insulin degrading enzyme, and plasmin. It is unclear whether the ultrasound has any direct impact on the amyloid plaques. With a study on dogs with AD, O’Reilly et al. showed, however, that there is no significant reduction in Aβ load.1034 In the first clinical trial, Beisteiner et al. developed a technique using single ultrashort (3 μs) ultrasound pulses with typical energy levels of 0.2–0.3 mJ mm−2 and pulse frequencies of 1–5 Hz to stimulate the human brain. The treatment of 35 patients with AD shows neuropsychological scores are improved significantly, last up to three months, and have no major side effects.1035
By in vitro studies, several experiments have been carried out to study the direct interaction of ultrasound with amyloid fibrils. Sato et al. showed that ultrasound with frequency of 1 MHz and power of 2.5 W/cm2 can dissociate soluble Aβ peptides from fibrils.1036 The Aβ42 is more resistant to dissociation from fibrils to monomers and/or low molecular weight soluble oligomers than Aβ40. This is consistent with the fact that Aβ42 peptides aggregate faster than Aβ40 counterparts. Because of low ultrasound power, the thermal effect is excluded in the interpretation of results. The authors suggested that the microstreaming generated by bubble cavitation may change the structure of fibrils.1036 Goto et al. studied the effects of low frequency ultrasound (7–20 kHz) irradiation on both aggregation and dissociation of Aβ40 peptide. They determined a critical peptide concentration of 0.7–0.9 μM, below which the ultrasound accelerates the dissociation of fibrils into monomers.1037 Above this critical concentration, ultrasound can promote nucleation of fibrillation by lowering the energy barrier agitating effects. However, the newly formed fibrils can also be broken. These two processes lead to the production of minimal and mono-dispersed fibrils.1038,1039 The small homogeneous fibrils will be useful for characterizing the structure and dynamics of amyloid fibrils. Again, the shearing forces of liquid flows induced by ultrasound are believed to be the causes of fibril formation and dissociation. Similar to the ultrasound, Foguel et al. used a cyclic high-pressure technique to dissociate the fibrils of αS and transthyretin.1040 Here, the fibrils are compressed at a pressure of 3000 bar for 30 min at 37 °C at pH 5 and then released. When the changes in the light scattering leveled off, pressure is applied again to evaluate its effects on the fibril structure. The authors observe that fibrils undergo rapid disassembly upon compression and aggregation after decompression. The mechanism could be due to the pressure induced disruption of hydrophobic interactions and eliminate water-excluded cavities.1040
A few nonequilibrium MD simulations have been carried out to understand the molecular mechanism of the ultrasound induced dissociation of amyloid fibrils. Okumura et al. carry out simulations of the Aβ17–42 fibril of various sizes under high ultrasound pressure of 200 MPa and very fast period of 1 ns.1041 The simulation shows that the inertial cavitation of bubbles, which are formed during the ultrasound rarefaction phase, is responsible for the fibril disruption. This mechanism could support the in vitro results reviewed above. However, in many in vivo applications, the stable cavitation of bubbles is more preferable, because the inertial cavitation could damage biomolecules. To simulate such bubble stable oscillation, we developed a new nonequilibrium MD method,1042,1043 where a bubble is represented by a particle with low mass and no charge and interacts with surrounding waters by a time-dependent Lennard-Jones potential. The time-dependent potential changes harmonically during the simulation, pushing and pulling waters back and forth, respectively, mimicking the bubble stable cavitation. The method was applied to study the effect of the stable cavitation on the pentamer Aβ17–42 amyloid fibril. The simulation shows that after 5 ns of excitation by 100 stable bubble cavitation periods, the β-structure in the initial fibril structure is reduced to 12%, and significant amounts of turn (30%) and coil (57%), with ≤1% of α-helices being formed. The simulation shows that the harmonic fluctuation in the water pressure induced by the stable bubble cavitation is the origin of the fibril dissociation.1042,1043 The method has also been used to study the stability of fibrils having U-shaped and S-shaped motifs exposed to ultrasound stable cavitation. It is shown that there is a marked difference in the kinetics of destabilization of fibrils having different shapes.1044
9.3.3. Electromagnetic and Electric Fields.
The transcranial electromagnetic treatment (TEMT) has emerged as a promising and safe approach to treat AD.1045 This method uses electromagnetic field to provide stimulatory/inhibitory effects on neuronal activity. With the frequencies in the radio frequency range (around 1 GHz), TEMT easily penetrates deep human brain areas and all neurons to impact intraneuronal pathologic processes, such as Aβ and tau. The method has been used in a number of preclinical studies involving AD mice, and the prevention as well as reversal of cognitive impairment at multiple ages of mice was obtained. It has been shown that TEMT can prevent/reverse Aβ oligomers/fibrils both inside and outside neurons, disaggregate tau oligomers, enhance mitochondrial function, and increase neuronal activity.1046–1048 So far, a first clinical trial of TEMT has been carried out on eight patients with mild to moderate AD, and the results show that TEMT enhances the cognitive performance of AD patients without behavioral/physiological side effects or brain abnormalities.1049
By in vitro study, Saikia et al. investigated the influence of the external electric field and magnetic field of varying strengths on the fibrillogenesis of the Aβ16–22 and the Aβ42 peptides.1050 Using electric field strengths of 150–300 V/cm, Aβ-elicited toxicity of EF-treated samples in two neuroblastoma cell lines and human embryonic kidney cell line was found to be 15–38% less toxic than the electric field untreated ones under identical conditions. However, the magnetic field around 0.8 T has little ability to induce a conformational switch. A magnetic-field-based therapy may be difficult to implement and, hence, has minimal therapeutic value.1050
Nonequilibrium all-atom MD simulations have been carried out. The simulation by English et al. showed that under external static electric field the total dipole moment of the hen egg white lysozyme aligns with the field, and this induces changes in the protein secondary structure relative to the zero-field state.1051 Zerbetto et al. simulated the interaction of Aβ peptide with electric field of varying strengths and showed that the electric field can switch the Aβ peptide from helical to β-sheet conformation.1052 For the amyloidogenic apoC-II(60–70) peptide, the simulation shows that high strength electromagnetic field can align the peptide dipole, resulting in the disruption of the inherent β-hairpin conformation known to be the intermediate state for fibril formation. Weaker field-strength can accelerate dynamics which leads to the increased population of structures with fibril-inhibiting characteristics.1053,1054
10. CROSSTALK AND CROSS-SEEDING BETWEEN AMYLOID PROTEINS
10.1. What Do We Know from In Vitro and In Vivo Conditions?
Despite intrinsic differences among the protein misfolding disordered diseases, notably the underlying neural circuit affected by pathology, they share a common molecular pathological mechanism: the misfolding, aggregation, tissue accumulation of a protein whose protein sequences differ greatly, and cell-to-cell propagation.193,1055–1061 In principle, these shared mechanistic and pathological characteristics also suggest that protein misfolding processes occurring simultaneously might synergistically interact among each other thereby accelerating disease pathogenesis. Recent studies are indeed providing new evidence for prevalent mixed proteinopathies across neurodegenerative diseases, with aging and APOE ε4 status constituting risk factors.1062 These observations are reminiscent of prior studies documenting that mixed neuropathologies are the most common cause of the clinical syndrome of dementia and are also common among persons with mild cognitive impairment or cognitive decline.1063–1066 Furthermore, these findings support earlier observations indicating that a more rapid rate of cognitive decline is observed in 30–40% of AD cases presenting with aS inclusions known as Lewy bodies (LB) and Lewy neurites (LN) compared to subjects with AD without αS pathology.51,1067 The co-occurrence of these proteinopathies thus provides support to accumulating observations documenting potential molecular cross-talks among amyloid proteins (Figure 34).
Figure 34.
Co-occurring pathologies across common neurodegenerative diseases. Schematic illustrating the relative proportions (numbers within colored disks) of amyloid pathologies formed of Aβ (blue), tau (pink), and αS (green) within each disease class. Question marks indicate unknown proportions for hybrid assemblies (i.e., Aβ/tau, Aβ/αS, αS/tau, and Aβ/αS/tau). The numbers inserted within the colored disks are taken from ref 1062.
Here, we define molecular cross-talk by molecular interactions and cross-seeding between aggregates of amyloid proteins with functional consequences for disease pathogenesis or disease progression (Figure 35). Cross-seeding can occur within the same cell or through cell-to-cell transmission of pathological amyloid aggregates, which is intrinsically part of spreading. Many excellent reviews on this subject are available.193,1055–1059,1061,1068
Figure 35.
Cross-seeding and coaggregation of amyloid proteins. Schematic illustrating the in vitro formation of amyloids (depicted by different shapes) including oligomers, protofibrils, and fibrils from tau (pink), αS (green), Aβ (blue), IAPP (orange), and PrP (gray) proteins. This scheme also summarizes the in vitro formation of coaggregates (αS:tau, Aβ:αS, Aβ:IAPP, Aβ:PrP, tau:PrP) by cross-seeding/coaggregation mechanism demonstrated in various studies. Different shapes, colors, and their numbers used here arbitrarily represent amyloid aggregates; they do not indicate aggregates of any specific size or shape.
10.1.1. Aβ and αS.
Cross seeding between aggregated Aβ and αS has been observed in vitro,1069 along with hybrid oligomer formation.1070 Many studies in fact have suggested that Aβ and αS are capable of direct and indirect cross-talks in the brain, along with hybrid oligomer formation.1069–1075 Similar findings were shown in a recent study, where Aβ42 and pyroglutamate Aβ3–42 (pGlu-Aβ3–42) peptides accelerated αS aggregation in vitro. Colocalization of the two Aβ species with αS was also shown in the APP-transgenic Tg2576 mouse brain sections, suggesting an in vivo occurrence of the Aβ and αS coaggregates.1076 To assess a potential crosstalk between fibrillar αS and fibrillar Aβ, Bassil and co-workers in 2020 injected mouse αS preformed fibrils (αS-PFFs) into young adult 5XFAD mice harboring amyloid plaques. The presence of Aβ deposits enhanced αS pathology and spreading throughout the brain. These findings have led the authors to suggest a “feed-forward” mechanism whereby Aβ plaques potentiate αS seeding and spreading over time.1077
10.1.2. αSyn and Tau.
Lately, the relationship between αS and tau has received much attention, which has also been supported by GWAS studies showing strong association between the genes encoding αS and tau proteins (SNCA and MAPT, respectively) in PD and dementia with Lewy body (DLB) pathologies.1078,1079 The in vivo association of the two proteins was reported in several studies from post-mortem brain tissues showing their co-occurrence and even coaggregation in PD, DLB, ALS/parkinsonism-dementia complex pathologies.1080–1083 Robinson and co-workers recently characterized the presence of co-occurring pathologies of four major amyloid proteins, i.e. Aβ, tau, αS, and TDP-43, in several neurodegenerative diseases.1062 Nearly all synucleinopathies including DLB and multiple system atrophy defined by αS inclusions invariably exhibited tau proteinopathy (92–100% of cases). By contrast, Aβ deposits were found in 38–80% of cases while TDP-43 copathology was identified in 0–22% of cases. It is tempting to hypothesize that this quasi-universal co-occurrence of αS and tau pathologies observed in synucleinopathies might result from a potential cross-seeding between these proteins.1062
Several groups have shown in vitro cross-seeding among αS and tau, which occasionally was a synergistic effect on both the proteins forming aggregates.1084–1089 In vitro aggregation studies indicate that monomeric αS directly interacts with two synthetic forms of tau including the K19 construct formed from the aggregation-prone repeat domain of the microtubule-binding domain due to its ability to aggregate faster in the absence of the flanking regions,1090 thereby promoting tau fibrillization.1089 This interaction is mediated by the C-terminus of αS and is exacerbated by phosphorylation of αS at serine 129, a characteristic PTM of αS pathology.1089 Additional work is needed to examine the possible heterologous nature of the assemblies formed and to establish the relevance of this in vitro study using artificial recombinant proteins.
Abundant tau pathology was shown in transgenic mouse models of PD expressing either A53T mutant αSyn (TgA53T, M83 line) or E46K mutant αS (TgE46K, M47 line).1085,1091 It is interesting to note that while TgE46K mice displayed a greater number of tau inclusions compared to TgA53T mice, in vitro studies demonstrated that mutant αSE46K is less efficient than wild-type αS (αSWT) at promoting tau inclusions in cultured QBI293 cells.1091 The reason for the accumulation of hyperphosphorylated tau inclusions in TgE46K mice is unclear and could result from environment, host, and cell type differences between the in vitro and in vivo paradigms used. Additionally, co-occurring aggregates of tau and αS were not only reported in neurons from M83 TgA53T mice but also exacerbated in oligodendrocytes from a bigenic CNP-TauP301L/αSWT mouse model overexpressing P301L mutant tau and human αSWT driven by the murine 2′,3′-cyclic nucleotide 3′-phosphodiesterase (CNP) promoter, in which inclusions were positive to Thioflavin S.1085 Because these seminal studies focused on fibril formation as opposed to oligomerization per se, the functional role of αS/tau cross-talks was further investigated where passive immunotherapy against tau oligomers prevented brain protein pathology and cognitive and motor deficits in M83 TgA53T mice.1092 By contrast, work from Singh and colleagues suggests that the role of tau in young and middle-aged adult G2.3 TgA53T mice is independent from αS/tau crosstalks because no differences in o-αSyn nor o-Tau were found in the absence or presence of MAPT deletion.1093 With aging, it is likely that this molecular crosstalk would strengthen and have an impact on the phenotype of this PD mouse model.
Several studies have also documented the existence of a functional coupling between αS and tau. Most notably, αS was shown to regulate a key kinase for tau phosphorylation and glycogen synthase kinase-3β (GSK3β), in several mouse models of PD.1094–1098 Another comprehensive study from the Sidhu group has shown the colocalization of active GSK3β (p-GSK3β-Y216) with phosphorylated tau (p-tau) or αS (p-αS) in the brain tissues of a transgenic mouse model expressing human GSK3β with a point mutation (S9A). Furthermore, the authors have shown that in addition to tau, GSK3β also phosphorylates αS in vitro and that both tau and αS cooperate with each other to increase the extent of their phosphorylation in vitro.1099 Exogenously added αS was shown to modulate the GSK3β-mediated hyperphosphorylation of tau causing microtubule destabilization in rat pheochromocytoma (PC12) cells.1100 These observational studies suggest an indirect association between tau and αS.
10.1.3. Aβ and IAPP.
Accumulating evidence of a potential link between T2D and AD pathologies has led to the investigation of the molecular interaction between Aβ and IAPP. Studies have shown in vitro that IAPP promotes the aggregation of Aβ42.1101 Additionally, cross-seeding between IAPP and Aβ42 resulted in hybrid aggregate formation, which could associate with an artificial lipid membrane, thus reducing its fluidity.1102–1105 Earlier work had reported IAPP deposition in the brain tissues of AD patients without any diabetes history, indicating the presence of brain insulin resistance.1106 Plaques of IAPP were found individually or embedded in the Aβ plaques in these AD brain tissues to the extent that they were almost indistinguishable by the staining procedures specific for these amyloids. The findings from this study are supportive of a co-occurrence of these pathologies in AD lesions and suggestive of a potential in vivo cross-talk between Aβ and IAPP.1103 More recently, colocalization of IAPP and Aβ was also confirmed by proximity ligation assay in both cerebral and vascular Aβ deposits in AD brain tissues.1107 Work from the Soto group reported that intracerebral injection of IAPP aggregates derived pancreatic homogenates into APP transgenic mice exacerbated Aβ deposition compared to the nontransgenic animals injected with the same inoculum, suggesting an in vivo cross-seeding mechanism between the two proteins.1108
10.1.4. αSyn and IAPP.
The in vitro cross-seeding of IAPP has been shown to promote αS aggregation. The two proteins formed coaggregates when incubated together in their monomeric forms, suggesting a possible reason for T2D to be a risk factor for PD.1109
10.1.5. PrP, Aβ, and Tau.
Cellular prion protein (PrPC) was shown to be a receptor for Aβ oligomers with high affinity, thus facilitating oligomer-induced synaptic dysfunction in mice.1110,1111 Several groups have reported the coimmunoprecipitation of human PrPC with Aβ from the brain tissues of AD patients.1112,1113 In another study, intraperitoneal inoculation of misfolded PrP aggregates (PrPSc) in the Tg2576 AD mouse model resulted in the accumulation of both Aβ and pathogenic PrPSc with concomitant histopathological features of prion disease.1114 The same authors also showed that protein misfolding can be enhanced by a cross-seeding mechanism in vitro.1114
Overexpression of the longest human tau isoform was shown to regulate cellular trafficking of PrPC and reduce its expression on the cell surface bound with concomitant accumulation of insoluble PrPC in primary cortical neurons.1115 Additionally, abundant tau pathology was reported in inherited prion diseases.1116,1117 Finally, oligomeric assemblies of Aβ, PrP, αS, and TDP-43 proteins were shown to colocalize in AD pathology,1118 indicating a possible cross-talk between these amyloid proteins.
10.2. What Have We Learned from Simulations?
10.2.1. IAPP/Aβ.
The attempt to investigate the coaggregation between amyloids via computational tools requires the structures of the amyloid aggregates that were solved by ssNMR or crystal structures. The cross-seeding between disordered monomers of Aβ (PDB ID: 1Z0Q) and IAPP (PDB ID: 2L86) using REMD simulations and DMD simulations with CHARMM force field (FF) and TIP3P water has been investigated. It was found that IAPP promotes Aβ aggregation via interactions of IAPP with residues16–22 of Aβ. In addition, two IAPP fibrils with U-shape structures were solved, by ssNMR216 and by X-ray crystallography.1119 Applying these IAPP fibril structures, further polymorphic IAPP fibrils were investigated via computational tools.1120 The interactions of each polymorphic IAPP fibril with Aβ fibril were investigated using MD simulations with CHARMM FF and TIP3P water.1121 Different interactions and orientations between polymorphic IAPP fibrils and Aβ fibril were studied for single-layer and double-layer conformations.1105,1121
It has been suggested that in water solution the cross-seeding between these two amyloid fibrils is preferred to form polymorphic single-layer conformations (in-register interactions) rather than double-layer conformations. It is thus expected that when two amyloid fibrils will interact in-register to form a single layer conformation, a synergistic effect will be produced and a promotion of the coaggregation will be presented. In some double-layer conformations, this phenomenon does not occur or is less preferred to occur.1121 The mechanisms that lead to coaggregation between these two amyloids indicate a strong tendency to form single-layer conformations.
It has been shown experimentally that the toxicity of Aβ oligomers to neuronal cells has been demonstrated to occur via a two-step mechanism of membrane disruption.1122 Yet, the cross-seeding between Aβ and IAPP aggregates has not been investigated at the atomic resolution in the membrane environment via experimental tools. The interactions between the U-shape structures of IAPP fibrils and Aβ fibrils associated with different types of membranes were investigated using MD simulations with CHARMM FF and TIP3P water by Zheng’s group.1105 It has been shown that the cross-seeding fibrils more strongly interact with the POPC/POPG bilayer than the POPC bilayer. It was therefore concluded that the electrostatic interactions between the cross-seeding fibrils and the membrane are the crucial interactions that stabilize peptide–lipid interactions. In such interactions, the N-termini of Aβ fibrils associate with the membrane and stabilize the contacts between the cross-seeding fibrils and the membrane. Finally, it has been shown that the cross-seeding of IAPP-Aβ induces the disruption of the cell membrane via altering calcium homeostasis and the cell membrane phase.
10.2.2. Aβ/NAC(αS).
The study of the cross-seeding between Aβ fibrils and αS fibrils is more challenging, due to the relatively large number of amino acids in αS. The short time scale of MD simulations with CHARMM FF and TIP3P water was performed to investigate the interactions between Aβ monomer and two αS monomers associated with a membrane.1070 The interactions of these two amyloids in the membrane indicated a promotion of the formation of stable ringlike oligomers. These oligomers were found to be composed of both Aβ and αS that are docked in the membrane. To simplify the study of the interactions between Aβ fibrils and αS fibrils, the interactions between Aβ fibrils and NAC fibrils were investigated using MD simulations in water solution. While the structures of Aβ fibrils were solved experimentally, the NAC fibrils were not solved. The NAC fibril S-shape structure was solved for the first time by molecular modeling tools.1123 Then, the interactions between the NAC fibril and the U-shape Aβ fibril structure were studied by MD simulations.1124 Various associations between NAC fibril and Aβ were investigated to form polymorphic single and double-layer conformations in the cross-seeding states.
The simulations demonstrated that the polymorphic Aβ fibrils prefer to interact with NAC fibril to form double-layer than single-layer conformations. The hydrophobic and electrostatic interactions are the driving forces that stabilize the cross-seeding double-layer conformations. In the single layer conformations, the NAC fibril affects the structure of Aβ fibrils. It is well-known that Aβ fibrils consist of two β-strands connected by one U-turn. Interestingly, the NAC fibrils in the cross-seeding aggregates induce the formation of a third β-strand and further U-turn in the Aβ fibril. The NAC fibril consists of three β-strands connected by two U-turn domains. Therefore, the cross-seeding between this NAC fibril and Aβ fibrils initiates the formation of a similar fibrillary structure for Aβ with three β-strands connected by two U-turn domains. A further effect of the NAC fibril on Aβ fibrils is related to the stabilization of the cross-β structure. While the inner core of NAC in the single-layer conformations of the cross-seeding aggregates does not change, the inner core of Aβ fibrils is decreased, i.e. forming a more compact stable cross-β structure due to the strong hydrophobic interactions in the inner core domain.1124
One can conclude that there is a lack of a synergistic effect in the cross-seeding NAC-Aβ fibrils. While the NAC fibril strongly affects the polymorphic Aβ fibrils, these polymorphic Aβ fibrils do not affect the structure of NAC fibril. This event is probably due to the compact structure of polymorphic NAC that consists of a relatively large number of hydrophobic interactions in the inner core, compared to the inner core of the polymorphic Aβ fibrils.
10.2.3. NAC(αS)/IAPP.
The cross-seeding between the U-shaped structure of IAPP aggregates and the S-shaped structure of NAC aggregates was investigated by MD simulations using the CHARMM FF and TIP3P water in Miller’s group.1125 In the case of the NAC fibril interacting with polymorphic Aβ fibrils, the double-layer conformations of the cross-seeding states are more preferred. Here, in the case where NAC fibril interacts with polymorphic IAPP fibrils, the single-layer conformations of the cross-seeding states are more preferred. Extensive structural analyses have shown that the NAC fibril stabilizes the structural properties of the polymorphic IAPP fibrils. For instance, the percentage of the hydrogen bond interactions between the β-sheets in the fibrillary states of IAPP is increased, compared to the separated IAPP fibrils. Moreover, the distance in the inner core of the cross-β structure of IAPP fibrils is decreased, indicating a compact cross-β structure.
Finally, for both the case of the cross-seeding between NAC and Aβ and the case of the cross-seeding between NAC and IAPP fibrils, the IAPP and Aβ in the cross-seeding states do not affect the structural features of NAC. Therefore, there is a lack of synergism between these amyloids. The ability of NAC fibril to enhance the stability of amyloids, such as IAPP and Aβ is due to its hydrophobic properties of the sequence in NAC.
10.2.4. Tau/Tau Isoforms.
The coaggregation between K18 and K19 was investigated by MD simulations.552 Simulations showed that coaggregation of some of the K18 or K19 with the tau R3 repeat exhibited a stable L-shaped fibril. The coaggregation of other K18 or K19 with tau R3 repeat demonstrated straight-line shaped fibrils. The aggregation of K18 is strongly initiated by both R2 and R3 repeats, while the aggregation of K19 is induced by only a R3 repeat. Therefore, these tau repeats are critical for aggregation of K18 and K19. Interestingly, it was proposed that the polymorphic core units of K18 and K19 yield to a cross-seeding barrier for K18 to trigger K19 fibril growth, because R2 is not available in K19. The polymorphic nature in amyloids may impede or initiate fibril formation and may affect the differences in barriers between K18 and K19.
10.2.5. Aβ-Tau/Mutated Tau.
There is a synergy between the tau and Aβ pathology in the mitochondria,1126–1130 and it was suggested that Aβ may accelerate tau neurofibrillary tangles.1131–1133 Experimental studies proposed that coaggregation of tau with Aβ occurs via interactions between the β strands of Aβ and β-stands of tau.1134–1138 Yet, there is a challenge to investigate the interactions between the full-length Aβ aggregates and the full-length tau aggregates or mutated tau aggregates at the atomic resolution both by experimental techniques and by computational modeling tools. The interactions between the Aβ25–35 fragment and the Tau273–284 fragment were investigated using REMD simulations and the OPLS-AA FF and TIP3P water.1139 It was shown that these interactions initiate cross-seeding between these fragments.
The tau repeats R2, R3, and R4 are known to form β-strands. The fibrillary structures of these U-shape tau repeat fibrils were studied by MD with CHARMM FF and TIP3P water molecules.1140 The interactions between each one of these tau repeat fibrils with the full-length Aβ U-shaped fibrils were studied also by simulations with CHARMM FF and TIP3P water.1140 Simulations showed that there are fewer hydrogen bond interactions between a neighboring monomer of Aβ fibril and a monomer of tau repeat R3 or R4 fibrils. Furthermore, conformational energy analyses illustrated that the “reaction coordinate” of Aβ fibrils with tau repeat R2 fibril presents exothermic reaction. The “reaction coordinate” of Aβ fibrils with tau repeats R3 or R4 fibrils demonstrated endothermic reaction. Therefore, it was proposed that tau repeats R3 and R4 are less preferred to interact with Aβ and tau repeat R2 has a strong tendency to coaggregate with Aβ. Furthermore, it was suggested that there is a synergistic effect between tau R2 repeat and Aβ.
One of the main hallmarks of the fronto-temporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) is the accumulation of neurofibrillary tangles in the brain as an outcome of the aggregation of mutated tau protein. This process occurs due to a number of genetic mutations in the MAPT gene. One of these mutations is the ΔK280 mutation in the tau R2 repeat domain, which promotes the aggregation vis-à-vis that for the wild-type tau. Experimental studies have shown that Aβ forms aggregates both with itself and with wild-type tau. By analogy, in FTDP-17, it is likely that there are interactions between Aβ and mutated tau. Yet, it is challenging to investigate the interactions between Aβ and mutated tau at the atomic resolution via both experimental techniques and computational modeling tools. The molecular mechanisms underlying such interactions between U-shaped Aβ fibrils and mutated tau ΔK280 R2 repeat U-shaped fibrils were studied by MD simulations with CHARMM FF and TIP3P water molecules.1141
Two predicted polymorphic mutated tau ΔK280 R2 repeat fibrils were investigated. Each one of these two mutated tau fibrils were interacted with Aβ fibril, producing single- and double-layer conformations. The interactions of one of the mutated tau polymorph fibrils with Aβ illustrated that thermodynamically the double-layer conformations are more preferred than the single-layer conformations. Interestingly, the polymorphic cross-seeding mutated tau-Aβ fibrils are more preferred than the separated mutated tau fibrils and Aβ fibrils. The mutated tau fibrils and Aβ fibrils synergistically stabilize each other, leading to polymorphic states. The synergistic stabilization was confirmed by the formation of well-defined β-sheet structures for both Aβ fibrils and mutated tau fibrils along the fibril axis. Finally, interactions between trimers of Aβ alternating with trimers of mutated tau in the cross-seeding fibrils form complexes that are thermodynamically less preferred but structurally Aβ trimers and mutated tau trimers stabilize each other to form similar intersheet distances in the core domain along the fibril axis.1141
11. CONCLUSIONS AND PERSPECTIVES
This review overviews the state of the art in computer, in vitro, in vivo, and pharmacological (small molecule and antibody) experiments related to AD, PD, T2D, and ALS. We have reported on the most recent experimental and computational findings on the monomers, oligomeric intermediates, and fibrils of the key amyloid proteins implicated in these diseases ranging from aqueous solution (free or with metal ions and inhibitors) to membrane mimetic systems to in vivo. The crosstalk and cross-seeding between the Aβ, tau, αS, and IAPP proteins and the failed and ongoing therapy assays and developments are also discussed. Over the last two decades, a huge amount of data has been gathered from different standpoints due to the investments and continued efforts by researchers from various disciplines, and this is very important to achieve a superior understanding.
Which are the next steps and promising directions that can potentially be handled by experiments? We will list some of them here: (a) The application of cryo-EM analysis, in combination with solid-state NMR and other biophysical techniques, on Aβ, tau, and αS fibrils isolated from the brain extracts of hundreds of patients would be an important step toward the atomic structure and dynamics determination of these proteins in the context of the disease propagation. (b) Determination of the high-resolution structures of toxic oligomers resulting from the coaggregation of human brain-derived Aβ, pyroglutamate Aβs, and αS proteins interacting with mitochondrial membranes would be useful as mitochondria are playing a pivotal role in AD and PD.1142,1143 Probing the many roles of the cell membrane by utilizing novel membrane mimetics such as lipid-nanodiscs that offer several advantages such as stability, solubility, monodispersity, size tunability, and lipid heterogeneity would also be useful. (c) Knowledge of the structures of SOD1 oligomers upon mutations and PTMs could be a real breakthrough to better understand the molecular events underlying the ALS pathology. Of note a strong challenge is imposed by the limitation for isolating amyloid aggregates in their native conditions, which will accurately mimic the species in human brains1144 and availability of reagents and tools to study these protein aggregates in their most stable forms. (d) Consider how propagation might proceed in the context of co-occurring proteinopathies, how multiple cell populations or circuits might be affected, and how spreading occurs through multiple pathways (e.g., exosome or prion-like seeding). Of note there is an urgent need for novel experimental approaches to resolve amyloid cross-toxicity, at the neuro-histochemical and organismal levels, to make progress in understanding the cross-seeding.
Which are the next steps and promising directions that can potentially be handled by both experiments and simulations? Some of them are (a) investigate the role of multibody interactions involving oligomers of different amyloid proteins, cellular proteins, and membranes, (b) elucidate the role played by cholesterol in direct interactions with APP and gamma-secretase as well as indirect influences through changes in the membrane phase, (c) understand the role of membrane microdomains in partitioning and colocalizing secretases and substrate during Aβ genesis, (d) determine whether and how liquid–liquid phase and other biomolecules can induce pathogenic tau conformations in vivo,1145,1146 (e) understand how the balance between functional and pathological interactions is regulated in both αS and IAPP and how the exact function of αS is connected with its interaction at the surface of synaptic membranes, and (f) investigate the formation of amyloid oligomer/phospholipid complexes.1147,1148
What are the next steps and promising directions that can potentially be handled by simulations? We know that PMTs mediate the structural diversity of tauopathy strains. The similarities and differences among amyloid formation of different tau isoforms therefore provide excellent opportunities to understand the driving forces leading to different pathological diseases. Computational simulations are fully appropriate to explore the initial nucleation of aggregation, the energy landscape connecting different fibril structures and possible related oligomers, and conformational effects of phosphorylation and other PTMs. One of the particularly interesting problems is the phosphorylation effects in cis- and trans-tau, which is difficult to examine experimentally. Another interesting problem involves phosphorylation at residue 217, a tau site closely linked to the first stage of AD, and at residue 181,523 as advanced studies on blood sample analysis reported that detection of this phosphorylated tau could differentiate healthy participants from those with AD.1149,1150 What are the effects of these two phosphorylations on the tau energy landscape? Simulations could also investigate the circulation of Aβ or αS proteins in a crowded environment and in a constricted geometry with AD and PD aging conditions. Indeed, it is known that shear flows in microfluidic devices accelerate aggregation kinetics,16 synucleopathies alter nanoscale organization and diffusion in the brain extracellular space,1151 and the geometry of the brain interstitial system undergoes alterations with aging and the accumulation of Aβ and tau.1152–1154
Of note, we are only at the beginning of the road to adapt the computational methods that generate confidence in the field of globular protein science to IDPs.44,1155 Similarly, it is not clear yet which atomistic force field is the most reliable for describing the monomeric and oligomeric structures of Aβ, tau, and αS in and their aggregation pathways. Despite extensive research toward better atomistic force fields, the aggregation pathways and the dominant microstates in bulk solution are still more sensitive to the choice of the force field than a change in the IDP sequence.445,1156–1158 The same limitation holds for coarse-grained simulations and mesoscopic approaches, but it is important to recall that these simulations are designed to capture qualitative rather quantitative properties of the aggregation process. Coarse-grained simulations and mesoscopic approaches, due to the elimination of many unimportant degrees of freedom and no explicit solvent interactions, are appropriate to study the dependence of fibril formation and the primary and secondary nucleation mechanisms as a function of amino acid sequence, temperature, protein concentration, the ratio between intramolecular and intermolecular interactions, and the presence of simplified crowders or cell membranes for instance.324,365,371,462,1159 Coarse-grained simulations, which allow longer time- and length-scales at the expense of a reduced energy accuracy, are continuously improving.31,321,337,1160 They are complementary to all-atom simulations, and in some cases, their results are further explored by all-atom simulations, referred to as multiscale approaches.111,451,1161 We emphasize that there is no emerging state-of-the art simulation protocol for the aggregation of IDPs, even though coupling all-atom simulations with Markov state chain modeling approaches is becoming popular at least on small amyloid peptides and aggregate sizes.450,1162 Overall, we face multiple key issues such as the force field accuracy and the sampling and concentration bottleneck over a wide range of time scales toward efficient elucidation of IDPs aggregation kinetics and thermodynamics and accurate predictions from atomistic and coarse-grained simulations. Whether combining deep learning and statistical mechanics through Boltzmann generators can help solve the sampling issue for amyloid aggregation remains to be determined.1163
Developing disease-modifying therapies for AD presents an immense challenge, with little success despite intense work over decades.314,1164–1167 Some of the reasons why drug development for AD continues to fail include the following: (a) in vitro experiments poorly mimic in vivo conditions, (b) Aβ and tau exist in multiple covalent and aggregated forms of uncertain relevance to pathology and we have a poor knowledge of their structures, and (c) animal models are limited.314 Among animal models, genetically modified mouse lines are invaluable and widely used models for understanding genes, proteins, and biological pathways, evaluating disease progression, and assessing safety of molecules for therapeutic purposes in many diseases including neurodegenerative diseases, cancer, and diabetes. However, there are several significant intrinsic limitations in using in vivo animal studies to replicate the pathological environment occurring in the human brain undergoing neurodegeneration.1168 First, there is an obvious cross-species comparability factor between human and rodents, importantly including aging for which aging in mice does not accurately reflect aging in humans.1169,1170 Second, human and mice brains differ in neuroanatomy, in brain biochemistry, and in blood–brain-barrier (BBB) permeability.1169,1171–1173 Considering the differences in respective life spans for humans and mice, it is naive to take at face value the development of phenotypes in young mice for symptoms that are usually developed in elderly human subjects.1169 The push to generate models that rapidly progressed disease phenotype often resulted in either worsening of the pathology to nonphysiological levels or inducing aberrant early lethality in mice.1174 Third, the reliance on inbred mouse models has been widely documented as a translational issue whereby specific alleles can predispose the animals to certain phenotypes prior to genetic manipulation.1175 Fourth, the phenotypes observed in mice can also be influenced by potential artifacts resulting from the overexpression of transgenes, from the location of the transgene insertion or from environmental factors.1169,1176 Finally, to observe the desired pathology and phenotypes, the commonly used transgenic mouse models carry multiple mutations, such as in both Aβ and tau, which rarely happens in human brains.1176 Thus, challenges are faced while attempting to recapitulate the features of human diseases in mice.
After more than 20 years, we do not know whether the accumulation of Aβ oligomers is the cause or consequence of AD.1177,1178 Other researchers in the field place tau aggregates upstream of Aβ in AD. As a matter of fact, all promising drugs and antibodies have failed in phase III clinical, with the majority of these compounds even unable to reach such a step. Caution is needed until the full analyses from the phase III aducanumab trials are released. A few weeks ago, the results from the TAURIEL phase II clinical trial of semorinemab in early AD were announced.1179 This antibody, which binds the N-terminus of all six isoforms of human tau independently of the phosphorylation status, did not show any benefit over placebo. Should we move away from the amyloid cascade hypothesis or the series of events starting from tau for efficient AD treatment? Undoubtedly, the answer is no. As quoted by Karran and De Strooper, “it is one thing to test a drug and quite another to test a hypothesis”1177 for several reasons.
First, clinical trials take place too late in the disease process.314,1180,1181 However, diagnosing AD before the onset of symptoms, where drugs might have a beneficial effect, is not easy, though substantial progress is being made using biomarkers and imaging. Alternatively, drugs can be tested on people with dominant mutations for AD while they are asymptomatic. We do not know much however about the asymptomatic phase of AD, and there is a lot of heterogeneity in AD in terms of its presymptomatic and symptomatic phases and pathological features.251,1182
Second, approaches such as genome wide association studies have found over 50 loci significantly associated with AD onset. Pathway and functional genomic analyses show that in addition to Aβ and tau processing, immunity, endocytosis, cholesterol transport, and ubiquitination are involved in AD. Inflammation, oxidative stress, infection, diabetes, loss of protein degradation, and metal deposition are also likely to play roles in AD.1183 Many proteins play a significant role in developing AD, such as the prion protein,1184 APOE4,1185,1186 and the lipoprotein receptor related protein 1 (LRP1), with the latter discovered recently to be a master regular of tau uptake and spread.1187 There is also increasing evidence that cerebral vascular disease, the BBB, and the cerebrospinal fluid environment play an integral role in the development of AD.1178,1188,1189 Notably, the LRP1 protein, a component of the BBB, contributes to the clearance of Aβ from the central nervous system. The pivotal role of mitochondria and their dynamics in the brain1142 and changes in microtubule structure and dynamics1190 have also been discussed. It is very interesting that Lashuel et al. recently demonstrated that the process of Lewy body formation involves a complex interplay between αS aggregates and membrane organelles involving mitochondria and the autophagosome, inducing mitochondrial damage and deficits and synaptic dysfunctions.1147 Improving our understanding of all these crucial topics should aid drug development.
Finally, there is accumulating pathological evidence that many neurodegenerative diseases are mixed proteinopathies. Copathologies and cross-talks are likely a common feature of aging and neurological disorders, whereby the prevalence of these pathologies ultimately determines the type of disorder development in the aging brain. Therefore, the amyloid hypothesis in AD needs to be modified and to integrate cross-talk with other significant pathologies. This realization of copathologies and the failure of all previous monotherapies suggest that multitherapies, targeting simultaneously Aβ, tau, and αS, could become the norm, as discussed already in 2000.1067 Clearly, the creation of the 2019 EU-US clinical trials on Alzheimer’s disease is good news for AD, PD, T2D, and ALS.1191
ACKNOWLEDGMENTS
Laura Dominguez gratefully acknowledges the support of PAIP 5000-9155, LANCAD-UNAM-DGTIC-306, and CONACyT Ciencia Básica A1-S-8866. John E. Straub gratefully acknowledges the generous support of the National Science Foundation (Grant No. CHE-1900416) and the National Institutes of Health (Grant No. R01 GM107703). Alfonso De Simone acknowledges funding from the European Research Council (ERC), Consolidator Grant (CoG) “BioDisOrder” (819644). Yiming Wang and Carol K. Hall acknowledge the support of a Cheney Visiting Scholar Fellowship from the University of Leeds. The work was also supported by NSF Division of Chemical, Bioengineering, Environmental, and Transport Systems Grants 1743432 and 1512059. Antoine Loquet thanks the ERC starting Grant no. 639020. For Buyong Ma and Ruth Nussinov, this project has been funded in whole or in part with federal funds from the National Cancer Institute, National Institutes of Health, under contract HHSN26120080001E. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does mention of trade names, commercial products, or organizations imply endorsement by the U.S. Government. This Research was supported [in part] by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. Birgit Strodel acknowledges funding by a Helmholtz ERC Recognition Award. Jie Zheng acknowledges funding from NSF (1806138 and 1825122). Stepan Timr acknowledges funding from the European Union’s Horizon 2020 research and innovation programme under the Marie Sklodowska-Curie grant agreement No 840395. Fabio Sterpone acknowledges funding from the ERC (FP7/2007-2013) Grant Agreement no. 258748. Nikolay Dokholyan acknowledges support from the National Institutes for Health grants 1R35 GM134864 and UL1 TR002014 and the Passan Foundation. Joan-Emma Shea acknowledges computational support from the Extreme Science and Engineering Discovery Environment (XSEDE) through the National Science Foundation (NSF) grant number TG-MCA05S027. J.-E. Shea acknowledges the support from the National Science Foundation (NSF Grant MCB-1716956). The funding from the National Institutes of Health (NIH grant R01-GM118560-01A) and partial support from the National Science Foundation MRSEC grant No. DMR 1720256 is also acknowledged. She thanks the Center for Scientific Computing at the California Nanosystems Institute (NSF Grant CNS-1725797). The work of Sylvain Lesné was supported by grants from the National Institutes of Health (NIH) to (RF1-AG044342, R21-AG065693, R01-NS092918, R01-AG062135, and R56-NS113549). Additional support included start-up funds from the University of Minnesota Foundation and bridge funds from the Institute of Translational Neuroscience to S.L. Rakez Kayed was supported by National Institute of Health grants R01AG054025 and R01NS094557. Mai Suan Li was supported by Narodowe Centrum Nauki in Poland (grant 2019/35/B/ST4/02086) and the Department of Science and Technology, Ho Chi Minh City, Vietnam (grant 07/2019/HĐ-KHCNTT). Yifat Miller thanks the Israel Science Foundation, grant no. 532/15 and FP7-PEOPLE-2011-CIG, research grant no. 303741. Son Tung Ngo was supported by Vietnam National Foundation for Science & Technology Development (NAFOSTED) grant #104.99-2019.57. Research in the Ramamoorthy lab is supported by NIH (AG048934). Guanghong Wei acknowledges the financial support from the National Science Foundation of China (Grant Nos. 11674065 and 11274075) and National Key Research and Development Program of China (2016YFA0501702). Philippe Derreumaux acknowledges the support of the Université de Paris, ANR SIMI7 GRAL 12-BS07-0017, “Initiative d’Excellence” program from the French State (Grant “DYNAMO”, ANR-11-LABX-0011-01) and the CNRS Institute of Chemistry (INC) for two years of délégation in 2017 and 2018. We thank the reviewers for their insightful comments and suggestions.
Biographies
Phuong H. Nguyen is a CNRS researcher at the Laboratory of Theoretical Biochemistry, France. He received his M.Sc. from ICTP, Italy, and his Ph.D. from the University of Bielefeld, Germany, in Theoretical Condensed Matter Physics. He was then a Postdoctoral Fellow at the University of Frankfurt. His current research focuses on the development and application of theoretical methods for studying equilibrium and nonequilibrium structure, dynamics, and thermodynamics of single proteins, amyloids, and membranes at all-atom and coarse-grained levels.
Ayyalusamy Ramamoorthy joined the faculty at the University of Michigan, Ann Arbor, after getting his Ph.D. in Chemistry from the Indian Institute of Technology (Kanpur, India), working in the Central Leather Research Institute (Chennai, India) and JEOL Ltd (Tokyo, Japan), and completing postdoctoral training at the University of Pennsylvania, Philadelphia. His current research focuses on the structural biophysics of amyloid proteins and membrane proteins and on the development and applications of solid-state NMR spectroscopy.
Bikash R. Sahoo obtained a B.Sc. degree in physics from the Fakir Mohan University and an M.Sc. degree from Orissa University of Agriculture and Technology, India. He received Ph.D. from Osaka University in Molecular Biophysics studying the design of novel anticancer peptides in September 2016. He then joined the University of Michigan as a postdoctoral research fellow and is currently working with Professor Ayyalusamy Ramamoorthy. His research focuses on the structural and functional investigation of toxic amyloid intermediates associated with Alzheimer’s disease and Type II Diabetes.
Jie Zheng is a Professor of the Department of Chemical, Biomolecular, and Corrosion Engineering at the University of Akron. He received his B.S. and Ph.D. degrees in Chemical Engineering from Zhejiang University, China, in 1995 and from University of Washington in 2005. Zheng is the recipient of the NSF CAREER Award (2010), 3M Non-Tenure Faculty Award (2008), and Anton Award from National Resource for Biomedical Super-computing (2010). His main research interest focuses on design and study of better bioinspired, biofunctional, and biomimetic soft materials for engineering and biomedical applications.
Peter Faller is a Professor of Chemistry at the University of Strasbourg (F) and a group leader at the Institute of Chemistry (UMR 7177). He studied at the University of Zürich (CH) earning a Ph.D. in biochemistry on metallothioneins with M. Vasak and did postdoctoral studies on photosystem II at the CEA near Paris and in Freiburg (D). He was a professor and group leader at the “Laboratoire de Chimie de Coordination” in Toulouse (F) for more than a decade, where his group focused on bioinorganic chemistry of amyloidogenic peptides. In 2015 he moved to Strasbourg (F). Ongoing research projects of his group concern the role of copper and other 1st row d-block metals in biology and medicine.
John E. Straub is a Professor of Chemistry at Boston University. He received his B.S. in Chemistry in 1982 from the University of Maryland at College Park, where he worked with Millard Alexander on quantum scattering theory, and his Ph.D. in Chemical Physics in 1987 from Columbia University, where he worked with Bruce Berne on chemical research rate theory and simulation. He was an NIH Postdoctoral Fellow at Harvard University, where he worked with Martin Karplus on theory and simulation of protein dynamics, before joining the faculty of Boston University in 1990. His research explores the dynamics and thermodynamics of protein, membrane, and complex molecular assemblies, as well as algorithmic development for optimization, enhanced sampling, and long-time dynamics. He has served as President of the Telluride Science Research Center (TSRC), Phi Beta Kappa National Visiting Scholar, and is an Associate Editor of The Journal of Chemical Physics. He is the author of the textbook Mathematical Methods for Molecular Science (2020).
Laura Dominguez is a Professor at the School of Chemistry of the National Autonomous University of Mexico (UNAM). She received her Chemistry degree in 2004 from UNAM, where she worked on traffic dynamics and game theory. She earned a master’s degree in 2008 in Biochemistry from UNAM, working in protein dynamics with Lorenzo Segovia and Matthew Jacobson at UCSF, and a doctoral degree from the same program in 2011, working in Alejandro Sosa’s lab in protein simulation methodologies. She was a Schlumberger Faculty for the Future Postdoctoral Fellow at Boston University, where she worked with John Straub on simulation of protein dynamics and enhanced sampling techniques. She joined the faculty at UNAM’s School of Chemistry in 2014 at the Physical Chemistry department working on complex simulations of protein dynamics, interactions, and function.
Joan-Emma Shea received a B.Sc. in Chemistry from McGill University and a Ph.D. in physical chemistry from the Massachusetts Institute of Technology. Following postdoctoral studies at the Scripps Research Institute, she joined the faculty at the University of California, Santa Barbara where she is currently a full professor in the department of Chemistry and Biochemistry and in the Department of Physics. Her research involves developing and applying methods of computational chemistry and physics to biological systems.
Nikolay Dokholyan received his Ph.D. in Physics in 1999 at Boston University and completed postdoctoral training at Harvard University in the Department of Chemistry and Chemical Biology as a NIH NRSA Fellow. Dr. Dokholyan joined the Department of Biochemistry and Biophysics at the University of North Carolina at Chapel Hill School of Medicine as an Assistant Professor in 2002 and was promoted to Full Professor in 2011. Dr. Dokholyan has served as the Director of the Center for Computational and Systems Biology and the Graduate Director of the Program in Molecular and Cellular Biophysics at UNC. In 2014, Dr. Dokholyan was named the Michael Hooker Distinguished Professor. In 2018, he assumed the position of the G. Thomas Passananti Professor and Vice Chair for Research in the Department of Pharmacology and the Director of the Center for Translational Systems Research a position at the Penn State University Hershey Medical Center. Dr. Dokholyan was elected to be a Fellow of the American Physical Society (2012) and American Association for Advancements in Science (2019).
Alfonso De Simone obtained his Ph.D. at the University of Padova in 2007 and subsequently joined the lab of Chris Dobson at the University of Cambridge to work on the underlying molecular mechanisms of protein misfolding diseases. In 2011 he joined the Imperial College London, where he is currently professor of biological NMR spectroscopy. Since 2020 he has also been a professor of molecular biology at the University of Naples, Federico II. His current research interests are in the molecular properties and interactions of disordered proteins at the surface of membranes in the context of neuronal communication and neurodegeneration.
Buyong Ma is a Professor in the School of Pharmacy, Shanghai Jiaotong University. In 1995, he received his Ph.D. in physical chemistry from the Center for Computational Quantum Chemistry, The University of Georgia at Athens. From 1995 to 1998, he was a postdoc with Professor Allinger, focusing on development and application of molecular mechanics. In 1998, he joined the NCI-NIH and started his research in computational biology. After a brief one-year of work at Locus Pharmaceuticals, he accepted a Senior Scientist position in Leidos Biomedical Research Inc, National Cancer Institute of NIH in 2003. He has authored over 180 scientific papers. His current research area covers computational immunology and computational antibody design related to cancer and protein aggregation diseases.
Ruth Nussinov received her Ph.D. in 1977 from Rutgers University and did postdoctoral work in the Structural Chemistry Department of the Weizmann Institute. Subsequently she was at the Chemistry Department at Berkeley, the Biochemistry Department at Harvard, and a visiting scientist at the NIH. In 1984 she joined the Department of Human Genetics, at the Medical School at Tel Aviv University. In 1985, she accepted a concurrent position at the National Cancer Institute of the NIH, Leidos Biomedical Research, where she is a Senior Principal Scientist and Principle Investigator heading the Computational Structural Biology Section at the NCI. She has authored over 650 scientific papers. She is the Editor-in-Chief of Current Opinion in Structural Biology, was the Editor-in-Chief of PLoS Computational Biology, and is Associate Editor and on the Editorial Boards of a number of journals. She is a frequent speaker at Domestic and International meetings, symposia, and academic institutions, has won several awards, and has been elected a fellow of several societies. Her National Cancer Institute website gives further details. https://ccr.cancer.gov/ruth-nussinov.
Saeed Najafi received his Ph.D. in Physics from the Max Planck Institute for Polymer Research in 2017. Under the supervision of Prof. Potestio in the Theory Group, he conducted theoretical and computational study of topologically constrained biopolymers. He is currently a Postdoctoral Fellow in the Chemistry Department at UCSB. Under the guidance of Prof. Shea and Prof. Fredrickson, his research primarily focuses on elucidating the phase behavior of biological and artificial polymers.
Son Tung Ngo received his B.Sc. and M.Sc. degrees in Theoretical Physics at the Ho Chi Minh University of Science, followed by his Ph.D. in Physics at the Institute of Physics Polish Academy of Science. He currently is head of the Laboratory of Theoretical and Computational Biophysics at Ton Duc Thang University. He has been applying computational studies to understand the binding mechanism of protein–ligand interactions and the misfolding of amyloidogenic peptides in various environments. The structural change of metalloproteins, including polysaccharide and deoxyhypu-sine monooxygenases, is also evaluated by using rigorous computational methods.
Antoine Loquet studied chemistry at the Ecole Normale Superieure de Lyon and the University of Lyon. He obtained a Ph.D. in biophysics with A. Bockmann at the IBCP in Lyon and was an EMBO postdoc with A. Lange at the Max Planck Institute for Biophysical Chemistry, Gottingen. He is a CNRS research director at the IECB and at the CBMN in Bordeaux.
Mara Chiricotto is a postdoctoral researcher associate at the University of Manchester. She received her Ph.D. from Université Sorbonne Paris Cité under the supervision of F. Sterpone and P. Derreumaux. After completion of her Ph.D., she moved to University of Massachusetts Amherst, as a postdoctoral fellow. Her research interests are mainly focused on the hydrodynamic effects of amyloid aggregation using multiscale simulation methods, GPU acceleration of molecular dynamics algorithms, and the interfacial properties between liquids and solid surfaces.
Pritam Ganguly is an assistant project scientist in the department of Chemistry and Biochemistry at the University of California Santa Barbara (PI: Prof. Joan-Emma Shea). He completed his Ph.D. in Chemistry from the Technische Universität Darmstadt, Germany, in 2013 (supervisor: Prof. Nico van der Vegt). His research focuses on the solvation and aggregation of proteins.
James McCarty, Ph.D., is an Assistant Professor of Chemistry at Western Washington University where his research applies theoretical and computational approaches to study the conformational dynamics of biological macromolecules. After obtaining his Ph.D. in Chemistry at the University of Oregon under the supervision of Prof. Marina Guenza, he worked as a postdoctoral fellow in the group of Prof. Michele Parrinello in the Department of Chemistry and Applied Biosciences at ETH Zürich, Switzerland. He then moved to the University of California Santa Barbara as a postdoctoral fellow working in the groups of Prof. Joan-Emma Shea in the Department of Chemistry and Glenn H. Fredrickson in the Materials Research Laboratory. He joined the Chemistry Department at Western Washington in 2019.
Mai Suan Li is Professor of Physics at the Institute of Physics, Polish Academy of Sciences. He obtained his Ph.D. in theoretical physics from Chisinau State University, Moldova, and his habilitation degree from the Institute of Physics, Polish Academy of Sciences. His research interests are (a) development of coarse-grained models for studying protein folding and aggregation, (b) factors governing protein aggregation, (c) cotranslational folding on ribosomes, (d) protein unfolding under an external mechanical force, and (e) computer-aided drug design for Alzheimer’s disease, influenza A virus, breast cancer, and Covid-19.
Carol K. Hall is the Camille Dreyfus Distinguished University Professor of Chemical and Biomolecular Engineering at North Carolina State University. Originally trained in physics, she was one of the first women to join a chemical engineering faculty in the United States. Her research focuses on applying statistical thermodynamics and molecular-level computer simulation to topics of chemical, biological, or engineering interest involving macromolecules or complex fluids. Hall first turned her attention to protein aggregation and amyloid formation in the late 90s, motivated by her father’s death from Pick’s disease. Hall and her student, Hung Nguyen, were among the first to tackle the protein aggregation problem computationally by applying the very-fast discontinuous molecular dynamics simulation technique to an intermediate-resolution model of protein geometry and energetics, orders of magnitude quicker than atomic-level simulations. That work helped to set the stage for a new era in which “in silico” exploration of amyloid formation in various human diseases is added to the arsenal of experimental techniques used to study this important problem.
Yiming Wang received his B.S. degree in Chemical Engineering from East China University of Science and Technology in 2013. He obtained his Ph.D. degree in Chemical Engineering from North Carolina State University under the supervision of Dr. Carol K. Hall in 2018. He is currently a postdoctoral researcher in Dr. Pablo G. Debenedetti’s group in the Department of Chemical and Biological Engineering at Princeton University. His research involves using molecular dynamics simulation to investigate the thermodynamics and kinetics underlying various biomolecule self-assembly and phase transition processes, including amyloidogenic peptide aggregation, chiral molecule liquid–liquid phase separation, and criticality phenomena.
Yifat Miller received her Ph.D. in Physical Chemistry from the Hebrew University of Jerusalem. During 2008–2011 she was a postdoctoral fellow in the group of Ruth Nussinov at the National institute of Health, Maryland, USA. In 2011, she was offered an assistant professorship position at the Department of Chemistry of the Ben-Gurion University of the Negev, and she was promoted to professor in 2016. Her research group has extensively investigated the molecular mechanisms of cross-seeding amyloid interactions using classical molecular dynamics simulations. The cross-amyloid studies led her group to investigate the effect of insulin on amyloid aggregation. Since 2018, Miller’s group work focuses on understanding the initial steps of amyloid seeding and cross-amyloid aggregation at the atomic resolution and on designing novel compounds for amyloid and cross-amyloid aggregation.
Simone Melchionna is a researcher at the Institute for Complex Systems of the Consiglio Nazionale delle Ricerche (CNR) and founder of startup Lexma Technology specialized in biofluidics. He has a Ph.D. in chemistry from the University of Rome La Sapienza. During his Ph.D. study, he developed techniques in molecular dynamics of biological systems, such as constrained mechanics, enhanced sampling, and isothermal–isobaric dynamical approaches. At Cambridge University he worked on confined fluids and water via density functional theory and other theoretical approaches. Subsequently, he developed lattice Boltzmann and multiscale simulation numerical methods at Harvard University and at CNR, with applications to DNA translocation and physiological fluid transport.
Birgit Habenstein studied biotechnology at the TU Berlin. She obtained her Ph.D. in biophysics with A. Bockmann at the IBCP in Lyon. She then joined the group of B. H. Meier at the ETH Zurich for a short postdoctoral stay. She was an EMBO postdoctoral fellow with A. Lange at the Max Planck Institute for Biophysical Chemistry, Gottingen. Since 2015 she has been a CNRS researcher at the IECB and at the CBMN in Bordeaux.
Stepan Timr received his Ph.D. in Biophysics from Charles University (Czech Republic) under the supervision of Pavel Jungwirth (2017). Since 2017, he has been working as a postdoctoral associate and, starting from 2019, as a Marie Skłodowska-Curie fellow with Fabio Sterpone at the Laboratory of Theoretical Biochemistry of the CNRS in Paris, where he has been investigating computationally protein stability and diffusion in crowded environments.
Jiaxing Chen received his master’s degree in Pharmacognosy from Peking University in China, 2015. He is now a Ph.D. candidate in the Bioinformatics and Genomics program and a research assistant of Dr. Dokholyan’s lab in the Department of Pharmacology at the Pennsylvania State University.
Brianna Hnath received her Bachelor’s degree in Neuroscience from the University of Scranton in 2017. She is now a research technologist and the lab manager for Dr. Nikolay Dokholyan’s lab in the Department of Pharmacology at the Pennsylvania State University College of Medicine.
Birgit Strodel is head of the Computational Biochemistry Group at the Jülich Research Centre (D) and was appointed Professor at Heinrich Heine University Düsseldorf (D) in 2011. She studied chemistry at Heinrich Heine University Düsseldorf and the University of North Carolina at Chapel Hill (USA), received her Ph.D. in Theoretical Chemistry from the University of Frankfurt/Main (D), and did postdoctoral studies at the University of Cambridge (UK). Her research interests primarily involve the development and application of molecular simulation techniques to understand protein aggregation.
Rakez Kayed, Ph.D., is the John Sealy Distinguished Chair in Parkinson Disease Research, Professor at the Departments of Neurology and Neuroscience & Cell Biology & Anatomy, and a member of George & Cynthia Mitchell Center for Neurodegenerative Diseases at University of Texas Medical Branch, Galveston, Texas. Rakez received his Ph.D. in Medicinal Chemistry from university of Tubingen and Post-Doctoral training in Alzheimer’s disease and Protein Misfolding Diseases, University of California, Irvine, CA, in Prof. Charles Glabe’s laboratory. His research focuses on toxic amyloid oligomers formed by the disease-associated proteins tau, α-synuclein, and amyloid in neurodegenerative disorders.
Sylvain E. Lesné received his B.Sc. in Cell Biology, Immunology & Microbiology and his M.Sc. in Biochemistry and Neuroscience. He was a graduate student with Drs. Alain Buisson and Denis Vivien and received his Ph.D. in Molecular & Cellular Biology from the Université de Caen-Basse-Normandie, Caen (France). He was a Postdoctoral Fellow in the laboratory of Karen H. Ashe at the University of Minnesota. He is currently an Associate Professor in Neuroscience at the University of Minnesota School of Medicine and a Scholar at the Institute of Translational Neuroscience. His research is focused on molecular and cellular mechanisms induced by soluble forms of aggregation-prone proteins in neurodegenerative diseases.
Guanghong Wei received her Ph.D. degree in Physics from Fudan University in 1998 and joined Fudan University as an assistant professor in the same year. From 2001 to 2004, she was a postdoctoral researcher with Normand Mousseau at the University of Montreal. Then, she went for a second postdoctoral fellowship in the group of Joan-Emma Shea at University of California, Santa Barbara. In 2005, she was offered an associate professorship position in the Department of Physics at Fudan University, and she was promoted to full professor in 2009. Her current research focuses on mechanistic understanding of peptide self-assembly, amyloidogenic protein aggregation, and its inhibition by nanoparticles/small molecules.
Fabio Sterpone is currently a research director at the CNRS, France, and works at the Laboratoire de Biochimie Théeorique at IBPC in Paris. He graduated from the University of Paris UPMC (biophysics). He has a broad experience in the application and development of computational methods to soft-matter systems. Presently, he is mainly interested in the study of protein stability and aggregation by applying and developing multiscale simulation methodologies.
Andrew J. Doig is Professor of Biochemistry and Biochemistry Programme Director at the University of Manchester. He studied Natural Science and Chemistry at the University of Cambridge and Biochemistry at Stanford University Medical School. He became a lecturer in Manchester in 1994, where he has been ever since. His research is on computational biology, neuroscience, dementia, bioinformatics, statistical thermodynamics, developmental biology, and protein structure. He has a major interest in amyloidosis, specifically Alzheimer’s Disease, Parkinson’s Disease, and diabetes. He founded two spin-out biotechnology companies from his research on Alzheimer’s drug discovery: Senexis developed new drugs for Alzheimer’s, while PharmaKure repositions existing drugs to work on Alzheimer’s patients.
Philippe Derreumaux is a Professor at Université de Paris, is a senior member of the Institut Universitaire de France (IUF), and has been the director of UPR9080 CNRS at IBPC from 2007 to 2016. He has been working on the development of coarse-grained protein and RNA/DNA models and the self-assembly of amyloid proteins by computer simulations.
Footnotes
Complete contact information is available at: https://pubs.acs.org/10.1021/acs.chemrev.0c01122
The authors declare no competing financial interest.
Contributor Information
Phuong H. Nguyen, CNRS, UPR9080, Université de Paris, Laboratory of Theoretical Biochemistry, IBPC, Fondation Edmond de Rothschild, PSL Research University, Paris 75005, France;.
Ayyalusamy Ramamoorthy, Biophysics and Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109-1055, United States;.
Bikash R. Sahoo, Biophysics and Department of Chemistry, University of Michigan, Ann Arbor, Michigan 48109-1055, United States;.
Jie Zheng, Department of Chemical & Biomolecular Engineering, The University of Akron, Akron, Ohio 44325, United States;.
Peter Faller, Institut de Chimie, UMR 7177, CNRS-Université de Strasbourg, 67000 Strasbourg, France;.
John E. Straub, Department of Chemistry, Boston University, Boston, Massachusetts 02215, United States;.
Laura Dominguez, Facultad de Química, Departamento de Fisicoquímica, Universidad Nacional Autónoma de México, Mexico City 04510, Mexico;.
Joan-Emma Shea, Department of Chemistry and Biochemistry, and Department of Physics, University of California, Santa Barbara, California 93106, United States;.
Nikolay V. Dokholyan, Department of Pharmacology and Biochemistry & Molecular Biology, Penn State University College of Medicine, Hershey, Pennsylvania 17033, United States; Department of Chemistry, and Biomedical Engineering, Pennsylvania State University, University Park, Pennsylvania 16802, United States;.
Alfonso De Simone, Department of Life Sciences, Imperial College London, London SW7 2AZ, U.K.; Molecular Biology, University of Naples Federico II, Naples 80138, Italy.
Buyong Ma, Basic Science Program, Leidos Biomedical Research, Inc., Cancer and Inflammation Program, National Cancer Institute, Frederick, Maryland 21702, United States; School of Pharmacy, Shanghai Jiao Tong University, Shanghai, China;.
Ruth Nussinov, Basic Science Program, Leidos Biomedical Research, Inc., Cancer and Inflammation Program, National Cancer Institute, Frederick, Maryland 21702, United States; Sackler Institute of Molecular Medicine, Department of Human Genetics and Molecular Medicine Sackler School of Medicine, Tel Aviv University, Tel Aviv, Israel.
Saeed Najafi, Department of Chemistry and Biochemistry, and Department of Physics, University of California, Santa Barbara, California 93106, United States.
Son Tung Ngo, Laboratory of Theoretical and Computational Biophysics & Faculty of Applied Sciences, Ton Duc Thang University, 33000 Ho Chi Minh City, Vietnam;.
Antoine Loquet, Institute of Chemistry & Biology of Membranes & Nanoobjects, (UMR5248 CBMN), CNRS, Université Bordeaux, Institut Européen de Chimie et Biologie, 33600 Pessac, France;.
Mara Chiricotto, Department of Chemical Engineering and Analytical Science, University of Manchester, Manchester M13 9PL, U.K..
Pritam Ganguly, Department of Chemistry and Biochemistry, and Department of Physics, University of California, Santa Barbara, California 93106, United States;.
James McCarty, Chemistry Department, Western Washington University, Bellingham, Washington 98225, United States;.
Mai Suan Li, Institute for Computational Science and Technology, Ho Chi Minh City 700000, Vietnam; Institute of Physics, Polish Academy of Sciences, 02-668 Warsaw, Poland;.
Carol Hall, Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, North Carolina 27695-7905, United States;.
Yiming Wang, Department of Chemical and Biomolecular Engineering, North Carolina State University, Raleigh, North Carolina 27695-7905, United States;.
Yifat Miller, Department of Chemistry and The Ilse Katz Institute for Nanoscale Science & Technology, Ben-Gurion University of the Negev, Be’er Sheva 84105, Israel;.
Simone Melchionna, ISC-CNR, 00185 Rome, Italy.
Birgit Habenstein, Institute of Chemistry & Biology of Membranes & Nanoobjects, (UMR5248 CBMN), CNRS, Université Bordeaux, Institut Européen de Chimie et Biologie, 33600 Pessac, France.
Stepan Timr, CNRS, UPR9080, Université de Paris, Laboratory of Theoretical Biochemistry, IBPC, Fondation Edmond de Rothschild, PSL Research University, Paris 75005, France.
Jiaxing Chen, Department of Pharmacology and Biochemistry & Molecular Biology, Penn State University College of Medicine, Hershey, Pennsylvania 17033, United States.
Brianna Hnath, Department of Pharmacology and Biochemistry & Molecular Biology, Penn State University College of Medicine, Hershey, Pennsylvania 17033, United States.
Birgit Strodel, Institute of Complex Systems: Structural Biochemistry (ICS-6), Forschungszentrum Jülich 52425, Jülich, Germany;.
Rakez Kayed, Mitchell Center for Neurodegenerative Diseases, and Departments of Neurology, Neuroscience and Cell Biology, University of Texas Medical Branch, Galveston, Texas 77555, United States;.
Sylvain Lesné, Department of Neuroscience, University of Minnesota, Minneapolis, Minnesota 55455, United States.
Guanghong Wei, Department of Physics, State Key Laboratory of Surface Physics, and Key Laboratory for Computational Physical Science, Multiscale Research Institute of Complex Systems, Fudan University, Shanghai 200438, China;.
Fabio Sterpone, CNRS, UPR9080, Université de Paris, Laboratory of Theoretical Biochemistry, IBPC, Fondation Edmond de Rothschild, PSL Research University, Paris 75005, France;.
Andrew J. Doig, Division of Neuroscience and Experimental Psychology, School of Biological Sciences, Faculty of Biology, Medicine and Health, University of Manchester, Manchester M13 9PT, U.K.;.
Philippe Derreumaux, CNRS, UPR9080, Université de Paris, Laboratory of Theoretical Biochemistry, IBPC, Fondation Edmond de Rothschild, PSL Research University, Paris 75005, France; Laboratory of Theoretical Chemistry and Faculty of Pharmacy, Ton Duc Thang University, 33000 Ho Chi Minh City, Vietnam;.
REFERENCES
- (1).Benson MD; Buxbaum JN; Eisenberg DS; Merlini G; Saraiva MJM; Sekijima Y; Sipe JD; Westermark P Amyloid Nomenclature 2018: Recommendations by the International Society of Amyloidosis (ISA) Nomenclature Committee. Amyloid 2018, 25, 215–219. [DOI] [PubMed] [Google Scholar]
- (2).Edler MK; Sherwood CC; Meindl RS; Hopkins WD; Ely JJ; Erwin JM; Mufson EJ; Hof PR; Raghanti MA Aged Chimpanzees Exhibit Pathologic Hallmarks of Alzheimer’s Disease. Neurobiol. Aging 2017, 59, 107–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Berchtold NC; Cotman CW Evolution in the Conceptualization of Dementia and Alzheimer’s Disease: Greco-Roman Period to the 1960s. Neurobiol. Aging 1998, 19, 173–189. [DOI] [PubMed] [Google Scholar]
- (4).Glenner GG; Wong CW Alzheimer’s Disease and Down’s Syndrome: Sharing of a Unique Cerebrovascular Amyloid Fibril Protein. Biochem. Biophys. Res. Commun 1984, 122, 1131–1135. [DOI] [PubMed] [Google Scholar]
- (5).Goldberg MS; Lansbury PT Jr Is there a Cause-and-Effect Relationship Between Alpha-synuclein Fibrillization and Parkinson’s Disease? Nat. Cell Biol 2000, 2, E115–E119. [DOI] [PubMed] [Google Scholar]
- (6).Hardy J; Selkoe DJ The Amyloid Hypothesis of Alzheimer’s Disease: Progress and Problems on the Road to Therapeutics. Science 2002, 297, 353–356. [DOI] [PubMed] [Google Scholar]
- (7).Koo EH; Lansbury PT Jr; Kelly JW Amyloid Diseases: Abnormal Protein Aggregation in Neurodegeneration. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 9989–9990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Dobson CM Protein Misfolding, Evolution and Disease. Trends Biochem. Sci 1999, 24, 329–332. [DOI] [PubMed] [Google Scholar]
- (9).Forman MS; Trojanowski JQ; Lee VM TDP-43: A Novel Neurodegenerative Proteinopathy. Curr. Opin. Neurobiol 2007, 17, 548–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Khan MAI; Respondek M; Kjellström S; Deep S; Linse S; Akke M Cu/Zn Superoxide Dismutase Forms Amyloid Fibrils under Near-Physiological Quiescent Conditions: The Roles of Disulfide Bonds and Effects of Denaturant. ACS Chem. Neurosci 2017, 8, 2019–2026. [DOI] [PubMed] [Google Scholar]
- (11).Meisl G; Michaels TCT; Linse S; Knowles TPJ Kinetic Analysis of Amyloid Formation. Methods Mol. Biol 2018, 1779, 181–196. [DOI] [PubMed] [Google Scholar]
- (12).Ilie IM; Caflisch A Simulation Studies of Amyloidogenic Polypeptides and Their Aggregates. Chem. Rev 2019, 119, 6956–6993. [DOI] [PubMed] [Google Scholar]
- (13).Dobson M; Knowles TPJ; Vendruscolo M The Amyloid Phenomenon and its Significance in Biology and Medicine. Cold Spring Harbor Perspect. Biol 2020, 12, No. a033878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Nasica-Labouze J; Nguyen PH; Sterpone F; Berthoumieu O; Buchete NV; Coté S; De Simone A; Doig AJ; Faller P; Garcia A; et al. Amyloid beta Protein and Alzheimer’s Disease: When Computer Simulations Complement Experimental Studies. Chem. Rev 2015, 115, 3518–3563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Owen MC; Gnutt D; Gao M; Wärmländer SKTS; Jarvet J; Gräslund A; Winter R; Ebbinghaus S; Strodel B Effects of in vivo Conditions on Amyloid Aggregation. Chem. Soc. Rev 2019, 48, 3946–3996. [DOI] [PubMed] [Google Scholar]
- (16).Dobson J; Kumar A; Willis LF; Tuma R; Higazi DR; Turner R; Lowe DC; Ashcroft AE; Radford SE; Kapur N; et al. Inducing Protein Aggregation by Extensional Flow. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 4673–4678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Chiti F; Webster P; Taddei N; Clark A; Stefani M; Ramponi G; Dobson CM Designing Conditions for in vitro Formation of Amyloid Protofilaments and Fibrils. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 3590–3594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Adler-Abramovich L; Vaks L; Carny O; Trudler D; Magno A; Caflisch A; Frenkel D; Gazit E Phenylalanine Assembly into Toxic Fibrils Suggests Amyloid Etiology in Phenylketonuria. Nat. Chem. Biol 2012, 8, 701–706. [DOI] [PubMed] [Google Scholar]
- (19).Lu K; Jacob J; Thiyagarajan P; Conticello VP; Lynn DG Exploiting Amyloid Fibril Lamination for Nanotube Self-assembly. J. Am. Chem. Soc 2003, 125, 6391–6393. [DOI] [PubMed] [Google Scholar]
- (20).Cherny I; Gazit E Amyloids: Not only Pathological Agents but also Ordered Nanomaterials. Angew. Chem., Int. Ed 2008, 47, 4062–4069. [DOI] [PubMed] [Google Scholar]
- (21).Monsellier E; Chiti F Prevention of Amyloid-like Aggregation as a Driving Force of Protein Evolution. EMBO Rep 2007, 8, 737–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Loquet A; Saupe SJ; Romero D Functional Amyloids in Health and Disease. J. Mol. Biol 2018, 430, 3629–3630. [DOI] [PubMed] [Google Scholar]
- (23).Rufo CM; Moroz YS; Moroz OV; Stöhr J; Smith TA; Hu X; DeGrado WF; Korendovych IV Short Peptides Self-assemble to Produce Catalytic Amyloids. Nat. Chem 2014, 6, 303–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Omosun TO; Hsieh MC; Childers WS; Das D; Mehta AK; Anthony NR; Pan T; Grover MA; Berland KM; Lynn DG Catalytic Diversity in Self-propagating Peptide Assemblies. Nat. Chem 2017, 9, 805–809. [DOI] [PubMed] [Google Scholar]
- (25).Al-Garawi ZS; McIntosh BA; Neill-Hall D; Hatimy AA; Sweet SM; Bagley MC; Serpell LC The Amyloid Architecture Provides a Scaffold for Enzyme-like Catalysts. Nanoscale 2017, 9, 10773–10783. [DOI] [PubMed] [Google Scholar]
- (26).Greenwald J; Friedmann MP; Riek R Amyloid Aggregates Arise from Amino Acid Condensations under Prebiotic Conditions. Angew. Chem., Int. Ed 2016, 55, 11609–11613. [DOI] [PubMed] [Google Scholar]
- (27).Brack A; Orgel LE Beta Structures of Alternating Polypeptides and their Possible Prebiotic Significance. Nature 1975, 256, 383–387. [DOI] [PubMed] [Google Scholar]
- (28).Langenberg T; Gallardo R; van der Kant R; Louros N; Michiels E; Duran-Romaña R; Houben B; Cassio R; Wilkinson H; Garcia T; et al. Thermodynamic and Evolutionary Coupling between the Native and Amyloid State of Globular Proteins. Cell Rep 2020, 31, 107512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Arai M; Sugase K; Dyson HJ; Wright PE Conformational Propensities of Intrinsically Disordered Proteins Influence the Mechanism of Binding and Folding. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 9614–9619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Dunker AK; Babu MM; Barbar E; Blackledge M; Bondos SE; Dosztányi Z; Dyson HJ; Forman-Kay J; Fuxreiter M; Gsponer J; et al. What’s in a name? Why These Proteins are Intrinsically Disordered: Why these Proteins are Intrinsically Disordered. Intrinsically Disord. Proteins 2013, 1, No. e24157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Baul U; Chakraborty D; Mugnai ML; Straub JE; Thirumalai D Sequence Effects on Size, Shape, and Structural Heterogeneity in Intrinsically Disordered Proteins. J. Phys. Chem. B 2019, 123, 3462–3474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Aliyan A; Cook NP; Martí AA Interrogating Amyloid Aggregates using Fluorescent Probes. Chem. Rev 2019, 119, 11819–11856. [DOI] [PubMed] [Google Scholar]
- (33).Ono K; Condron MM; Teplow DB Structure-neurotoxicity Relationships of Amyloid beta-protein Oligomers. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 14745–14750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gralle M; Botelho MM; de Oliveira CL; Torriani I; Ferreira ST Solution Studies and Structural Model of the Extracellular Domain of the Human Amyloid Precursor Protein. Biophys. J 2002, 83, 3513–3524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Fraser PE; Nguyen JT; Inouye H; Surewicz WK; Selkoe DJ; Podlisny MB; Kirschner DA Fibril Formation by Primate, Rodent, and Dutch-hemorrhagic Analogues of Alzheimer amyloid beta protein. Biochemistry 1992, 31, 10716–10723. [DOI] [PubMed] [Google Scholar]
- (36).Garzon-Rodriguez W; Sepulveda-Becerra M; Milton S; Glabe CG Soluble Amyloid Abeta-(1–40) Exists as a Stable Dimer at Low Concentrations. J. Biol. Chem 1997, 272, 21037–21044. [DOI] [PubMed] [Google Scholar]
- (37).Tang C; Ghirlando R; Clore GM Visualization of Transient Ultra-weak Protein Self-association in Solution using Paramagnetic Relaxation Enhancement. J. Am. Chem. Soc 2008, 130, 4048–4056. [DOI] [PubMed] [Google Scholar]
- (38).Safar J; Roller PP; Gajdusek DC; Gibbs CJ Jr Scrapie Amyloid (prion) Protein has the Conformational Characteristics of an Aggregated Molten Globule Folding Intermediate. Biochemistry 1994, 33, 8375–8383. [DOI] [PubMed] [Google Scholar]
- (39).Wasmer C; Lange A; Van Melckebeke H; Siemer AB; Riek R; Meier BH Amyloid Fibrils of the HET-s(218–289) Prion Form a beta Solenoid with a Triangular Hydrophobic Core. Science 2008, 319, 1523–1526. [DOI] [PubMed] [Google Scholar]
- (40).Fitzpatrick AWP; Falcon B; He S; Murzin AG; Murshudov G; Garringer HJ; Crowther RA; Ghetti B; Goedert M; Scheres SHW Cryo-EM Structures of Tau Filaments from Alzheimer’s Disease. Nature 2017, 547, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Falcon B; Zhang W; Murzin AG; Murshudov G; Garringer HJ; Vidal R; Crowther RA; Ghetti B; Scheres SHW; Goedert M Structures of Filaments from Pick’s Disease Reveal a Novel Tau Protein Fold. Nature 2018, 561, 137–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Nelson R; Sawaya MR; Balbirnie M; Madsen A; Riekel C; Grothe R; Eisenberg D Structure of the Cross-beta Spine of Amyloid-like Fibrils. Nature 2005, 435, 773–778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (43).Shrestha UR; Juneja P; Zhang Q; Gurumoorthy V; Borreguero JM; Urban V; Cheng X; Pingali SV; Smith JC; O’Neill HM; et al. Generation of the Configurational Ensemble of an Intrinsically Disordered Protein from Unbiased Molecular Dynamics Simulation. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 20446–20452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (44).Nguyen PH; Derreumaux P Structures of the Intrinsically Disordered Aβ, tau and α-synuclein Proteins in Aqueous Solution from Computer Simulations. Biophys. Chem 2020, 264, 106421. [DOI] [PubMed] [Google Scholar]
- (45).Pietrek LM; Stelzl LS; Hummer G Hierarchical Ensembles of Intrinsically Disordered Proteins at Atomic Resolution in Molecular Dynamics Simulations. J. Chem. Theory Comput 2020, 16, 725–737. [DOI] [PubMed] [Google Scholar]
- (46).Lindorff-Larsen K; Maragakis P; Piana S; Shaw DE Picosecond to Millisecond Structural Dynamics in Human Ubiquitin. J. Phys. Chem. B 2016, 120, 8313–8320. [DOI] [PubMed] [Google Scholar]
- (47).Selkoe DG; Hardy J The Amyloid Hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med 2016, 8, 595–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).Choi ML; Gandhi S Crucial Role of Protein Oligomerization in the Pathogenesis of Alzheimer’s and Parkinson’s Diseases. FEBS J 2018, 285, 3631–3644. [DOI] [PubMed] [Google Scholar]
- (49).Haataja L; Gurlo T; Huang CJ; Butler PC Islet Amyloid in Type 2 Diabetes, and the Toxic Oligomer Hypothesis. Endocr. Rev 2008, 29, 303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Wright GSA; Antonyuk SV; Hasnain SS The Biophysics of Superoxide dismutase-1 and Amyotrophic Lateral Sclerosis. Q. Rev. Biophys 2019, 52, No. e12. [DOI] [PubMed] [Google Scholar]
- (51).Hamilton RL Lewy Bodies in Alzheimer’s Disease: a Neuropathological Review of 145 Cases using alpha-synuclein Immunohistochemistry. Brain Pathol 2000, 10, 378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Chaudhuri P; Prajapati KP; Anand BG; Dubey K; Kar K Amyloid Cross-seeding Raises New Dimensions to Understanding of Amyloidogenesis Mechanism. Ageing Res. Rev 2019, 56, 100937. [DOI] [PubMed] [Google Scholar]
- (53).Ren B; Zhang Y; Zhang M; Liu Y; Zhang D; Gong X; Feng Z; Tang J; Chang Y; Zheng J Fundamentals of Cross-seeding of Amyloid Proteins: An Introduction. J. Mater. Chem. B 2019, 7, 7267–7282. [DOI] [PubMed] [Google Scholar]
- (54).Krotee P; Griner SL; Sawaya MR; Cascio D; Rodriguez JA; Shi D; Philipp S; Murray K; Saelices L; Lee J; et al. Common Fibrillar Spines of Amyloid-β and Human Islet Amyloid Polypeptide Revealed by Microelectron Diffraction and Structure-based Inhibitors. J. Biol. Chem 2018, 293, 2888–2902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (55).Laferla FM; Tinkle BT; Bieberich CJ; Haudenschild CC; Jay G The Alzheimer’s Aβ Peptide Induces Neurodegeneration and Apoptotic Cell Death in Transgenic Mice. Nat. Genet 1995, 9, 21–30. [DOI] [PubMed] [Google Scholar]
- (56).Olson MI; Shaw CM Presenile Dementia and Alzheimer’s Disease in Mongolism. Brain 1969, 92, 147–56. [DOI] [PubMed] [Google Scholar]
- (57).Brouwers N; Sleegers K; Engelborghs S; Maurer-Stroh S; Gijselinck I; Van Der Zee J; Pickut BA; Van Den Broeck M; Mattheijssens M; Peeters.; et al. Genetic Variability in Progranulin Contributes to Risk for Clinically Diagnosed Alzheimer Disease. Neurology 2008, 71, 656–664. [DOI] [PubMed] [Google Scholar]
- (58).Langosch D; Scharnagl C; Steiner H; Lemberg MK Understanding Intramembrane Proteolysis: From Protein Dynamics to Reaction Kinetics. Trends Biochem. Sci 2015, 40, 318–227. [DOI] [PubMed] [Google Scholar]
- (59).Winkler E; Julius A; Steiner H; Langosch D Homodimerization Protects the Amyloid Precursor Protein C99 Fragment from Cleavage by Secretase. Biochemistry 2015, 54, 6149–6152. [DOI] [PubMed] [Google Scholar]
- (60).Sanders CR How γ-Secretase Hits a Moving Target. eLife 2016, 5, No. e20043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Yagishita S; Morishima-Kawashima M; Ishiura S; Ihara Y Aβ46 Is Processed to Aβ40 and Aβ43, but Not to Aβ42, in the Low Density Membrane Domains. J. Biol. Chem 2008, 283, 733–738. [DOI] [PubMed] [Google Scholar]
- (62).Matsumura N; Takami M; Okochi M; Wada-Kakuda S; Fujiwara H; Tagami S; Funamoto S; Ihara Y; Morishima-Kawashima M γ-Secretase Associated with Lipid Rafts: Multiple Interactive Pathways in the Stepwise Processing Of β-Carboxylterminal Fragment. J. Biol. Chem 2014, 289, 5109–5121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Beel AJ; Mobley CK; Kim HJ; Tian F; Hadziselimovic A; Jap B; Prestegard JH; Sanders CR Structural Studies of the Transmembrane {C}-Terminal Domain of the Amyloid Precursor Protein (APP): Does APP Function as a Cholesterol Sensor? Biochemistry 2008, 47, 9428–9446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Haapasalo A; Kovacs DM The Many Substrates of Presenilin/γ-Secretase. J. Alzheimer’s Dis 2011, 25, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Jurisch-Yaksi N; Sannerud R; Annaert W A Fast Growing Spectrum of Biological Functions of γ-Secretase in Development and Disease. Biochim. Biophys. Acta, Biomembr 2013, 1828, 2815–2827. [DOI] [PubMed] [Google Scholar]
- (66).Struhl G; Adachi A Requirements for Presenilin-Dependent Cleavage of Notch and Other Transmembrane Proteins. Mol. Cell 2000, 6, 625–636. [DOI] [PubMed] [Google Scholar]
- (67).Chen GF; Xu TH; Yan Y; Zhou YR; Jiang Y; Melcher K; Xu HE Defining the Minimum Substrate and Charge Recognition Model of Gamma-Secretase. Acta Pharmacol. Sin 2017, 38, 1205–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (68).Xu TH; Yan Y; Kang Y; Jiang Y; Melcher K; Xu HE Alzheimer’s Disease-Associated Mutations Increase Amyloid Precursor Protein Resistance to γ-Secretase Cleavage and the Aβ42/Aβ40 Ratio. Cell Discovery 2016, 2, 16026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).Hitzenberger M; Zacharias M γ-Secretase Studied by Atomistic Molecular Dynamics Simulations: Global Dynamics, Enzyme Activation, Water Distribution and Lipid Binding. Front. Chem 2019, 6, 640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (70).Shah S; Lee SF; Tabuchi K; Hao YH; Yu C; LaPlant Q; Ball H; Dann CE; Südhof T; Yu G Nicastrin Functions as a γ-Secretase-Substrate Receptor. Cell 2005, 122, 435–447. [DOI] [PubMed] [Google Scholar]
- (71).Vetrivel KS; Cheng H; Lin W; Sakurai T; Li T; Nukina N; Wong PC; Xu H; Thinakaran G Association of γ-Secretase with Lipid Rafts in Post-Golgi and Endosome Membranes. J. Biol. Chem 2004, 279, 44945–44954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Chyung JH; Raper DM; Selkoe DJ γ-Secretase Exists on the Plasma Membrane as an Intact Complex That Accepts Substrates and Effects Intramembrane Cleavage. J. Biol. Chem 2005, 280, 4383–4392. [DOI] [PubMed] [Google Scholar]
- (73).Frykman S; Hur JY; Frènberg J; Aoki M; Winblad B; Nahalkova J; Behbahani H; Tjernberg LO Synaptic and Endosomal Localization of Active γ-Secretase in Rat Brain. PLoS One 2010, 5, No. e8948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (74).Winkler E; Kamp F; Scheuring J; Ebke A; Fukumori A; Steiner H Generation of Alzheimer Disease-Associated Amyloid β 42/43 Peptide by γ-Secretase Can Be Inhibited Directly by Modulation of Membrane Thickness. J. Biol. Chem 2012, 287, 21326–21334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Benilova I; Karran E; De Strooper B The Toxic Aβ Oligomer and Alzheimer’s Disease: An Emperor in Need of Clothes. Nat. Neurosci 2012, 15, 349–357. [DOI] [PubMed] [Google Scholar]
- (76).Kayed R; Lasagna-Reeves CA Molecular Mechanisms of Amyloid Oligomers Toxicity. J. Alzheimer’s Dis 2012, 33 (1), S67–S78. [DOI] [PubMed] [Google Scholar]
- (77).Gurry T; Stultz CM Mechanism of Amyloid-β Fibril Elongation. Biochemistry 2014, 53, 6981–6991. [DOI] [PubMed] [Google Scholar]
- (78).Sengupta U; Nilson AN; Kayed R The Role of Amyloid-β Oligomers in Toxicity, Propagation, and Immunotherapy. EBioMedicine 2016, 6, 42–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (79).Lee SJC; Nam E; Lee HJ; Savelieff MG; Lim MH Towards an Understanding of Amyloid-β Oligomers: Characterization, Toxicity Mechanisms, and Inhibitors. Chem. Soc. Rev 2017, 46, 310–323. [DOI] [PubMed] [Google Scholar]
- (80).Rosenman DJ; Connors CR; Chen W; Wang C; García AE Aβ Monomers Transiently Sample Oligomer and Fibril-like Configurations: Ensemble Characterization Using a Combined MD/NMR Approach. J. Mol. Biol 2013, 425, 3338–3359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Ball KA; Phillips AH; Wemmer DE; Head-Gordon T Differences in β-Strand Populations of Monomeric Aβ40 and Aβ42. Biophys. J 2013, 104, 2714–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Straub JE; Thirumalai D Principles Governing Oligomer Formation in Amyloidogenic Peptides. Curr. Opin. Struct. Biol 2010, 20, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (83).Mengel D; Hong W; Corbett GT; Liu W; DeSousa A; Solforosi L; Fang C; Frosch MP; Collinge J; Harris DA; et al. PrP-Grafted Antibodies Bind Certain Amyloid β-Protein Aggregates, but Do Not Prevent Toxicity. Brain Res 2019, 1710, 125–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (84).Weidemann A; König G; Bunke D; Fischer P; Salbaum JM; Masters CL; Beyreuther K Identification, Biogenesis, and Localization of Precursors of Alzheimer’s Disease A4 Amyloid Protein. Cell 1989, 57, 115–126. [DOI] [PubMed] [Google Scholar]
- (85).Goldgaber D; Lerman MI; McBride OW; Saffiotti U; Gajdusek DC Characterization and Chromosomal Localization of a CDNA Encoding Brain Amyloid of Alzheimer’s Disease. Science 1987, 235, 877–880. [DOI] [PubMed] [Google Scholar]
- (86).Goldgaber D; Lerman MI; McBride WO; Saffiotti U; Gajdusek DC Isolation, Characterization, and Chromosomal Localization of Human Brain CDNA Clones Coding for the Precursor of the Amyloid of Brain in Alzheimer’s Disease, Down’s Syndrome and Aging. J. Neural Transm. Suppl 1987, 24, 23–28. [PubMed] [Google Scholar]
- (87).Goate A; Chartier-Harlin MC; Mullan M; Brown J; Crawford F; Fidani L; Giuffra L; Haynes A; Irving N; James L; et al. ; Segregation of a Missense Mutation in the Amyloid Precursor Protein Gene with Familial Alzheimer’s Disease. Nature 1991, 349, 704–706. [DOI] [PubMed] [Google Scholar]
- (88).Kimberly WT; LaVoie MJ; Ostaszewski BL; Ye W; Wolfe MS; Selkoe DJ γ-Secretase Is a Membrane Protein Complex Comprised of Presenilin, Nicastrin, Aph-1, and Pen-2. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Edbauer D; Winkler E; Regula JT; Pesold B; Steiner H; Haass C Reconstitution of γ- Secretase Activity. Nat. Cell Biol 2003, 5, 486–488. [DOI] [PubMed] [Google Scholar]
- (90).Takasugi N; Tomita T; Hayashi I; Tsuruoka M; Niimura M; Takahashi Y; Thinakaran G; Iwatsubo T The Role of Presenilin Cofactors in the γ-Secratase Complex. Nature 2003, 422, 438–441. [DOI] [PubMed] [Google Scholar]
- (91).Bolduc DM; Montagna DR; Gu Y; Selkoe DJ; Wolfe MS Nicastrin Functions to Sterically Hinder γ-Secretase-Substrate Interactions Driven by Substrate Transmembrane Domain. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E509–E518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Aguayo-Ortiz R; Dominguez L APH-1A Component of γ-Secretase Forms an Internal Water and Ion-Containing Cavity. ACS Chem. Neurosci 2019, 10, 2931–2938. [DOI] [PubMed] [Google Scholar]
- (93).De Strooper B; Iwatsubo T; Wolfe MS Presenilins and γ-Secretase: Structure, Function, and Role in Alzheimer Disease. Cold Spring Harbor Perspect. Med 2012, 2, No. a006304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (94).Wolfe MS; Xia W; Ostaszewski BL; Diehl TS; Kimberly WT; Selkoe DJ Two Transmembrane Aspartates in Presenilin-1 Required for Presenilin Endoproteolysis and γ-Secretase Activity. Nature 1999, 398, 513–517. [DOI] [PubMed] [Google Scholar]
- (95).Zhou R; Yang G; Guo X; Zhou Q; Lei J; Shi Y Recognition of the Amyloid Precursor Protein by Human G-Secretase. Science 2019, 363, No. eaaw0930. [DOI] [PubMed] [Google Scholar]
- (96).Szaruga M; Munteanu B; Lismont S; Veugelen S; Horré K; Mercken M; Saido TC; Ryan NS; De Vos T; Savvides SN; et al. Alzheimer’s-Causing Mutations Shift Aβ Length by Destabilizing γ-Secretase-Aβn Interactions. Cell 2017, 170, 443–456 (e14). [DOI] [PubMed] [Google Scholar]
- (97).De Strooper B; Karran E The Cellular Phase of Alzheimer’s Disease. Cell 2016, 164, 603–615. [DOI] [PubMed] [Google Scholar]
- (98).Chávez-Gutiérrez L; Bammens L; Benilova I; Vandersteen A; Benurwar M; Borgers M; Lismont S; Zhou L; Van Cleynenbreugel S; Esselmann H; et al. The Mechanism of γ-Secretase Dysfunction in Familial Alzheimer Disease. EMBO J 2012, 31, 2261–2274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (99).Kuperstein I; Broersen K; Benilova I; Rozenski J; Jonckheere W; Debulpaep M; Vandersteen A; Segers-Nolten I; Van Der Werf K; Subramaniam V; et al. Neurotoxicity of Alzheimer’s Disease Aβ Peptides Is Induced by Small Changes in the Aβ42 to Aβ40 Ratio. EMBO J 2010, 29, 3408–3020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Miyashita N; Straub JE; Thirumalai D Structures of β-Amyloid Peptide 1–40, 1–42, and 1–55-the 672–726 Fragment of APP-in a Membrane Environment with Implications for Interactions with γ-Secretase. J. Am. Chem. Soc 2009, 131, 17843–17852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Lu JX; Yau WM; Tycko R Evidence from Solid-State NMR for Nonhelical Conformations in the Transmembrane Domain of the Amyloid Precursor Protein. Biophys. J 2011, 100, 711–719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (102).Tang T-C; Hu Y; Kienlen-Campard P; El Haylani L; Decock M; Van Hees J; Fu Z; Octave J-N; Constantinescu SN; Smith SO Conformational Changes Induced by the A21G Flemish Mutation in the Amyloid Precursor Protein lead to Increased Aβ Production. Structure 2014, 22, 387–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (103).Pantelopulos GA; Straub JE; Thirumalai D; Sugita Y Structure of APP-C991-99 and Implications for Role of Extra-Membrane Domains in Function and Oligomerization. Biochim. Biophys. Acta Biomembr 2018, S0005–2736(18)30112–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Sato T; Tang TC; Reubins G; Fei JZ; Fujimoto T; Kienlen-Campard P; Constantinescu SN; Octave JN; Aimoto S; Smith SO A Helix-to-Coil Transition at the Σ-Cut Site in the Transmembrane Dimer of the Amyloid Precursor Protein Is Required for Proteolysis. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 1421–1426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Czirr E; Cottrell BA; Leuchtenberger S; Kukar T; Ladd TB; Esselmann H; Paul S; Schubenel R; Torpey JW; Pietrzik CU; et al. Independent Generation of Aβ42 and Aβ38 Peptide Species by γ-Secretase. J. Biol. Chem 2008, 283, 17049–17054. [DOI] [PubMed] [Google Scholar]
- (106).Munter LM; Voigt P; Harmeier A; Kaden D; Gottschalk KE; Weise C; Pipkorn R; Schaefer M; Langosch D; Multhaup G GxxxG Motifs within the Amyloid Precursor Protein Transmembrane Sequence Are Critical for the Etiology of Aβ42. EMBO J 2007, 26, 1702–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Kienlen-Campard P; Tasiaux B; Van Hees J; Li M; Huysseune S; Sato T; Fei JZ; Aimoto S; Courtoy PJ; Smith SO; Constantinescu SN; Octave JN Amyloidogenic Processing but Not Amyloid Precursor Protein (APP) Intracellular C-Terminal Domain Production Requires a Precisely Oriented APP Dimer Assembled by Transmembrane GXXXG Motifs. J. Biol. Chem 2008, 283, 7733–7734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (108).Khalifa N; Ben Van Hees J; Tasiaux B; Huysseune S; Smith SO; Constantinescu SN; Octave JN; Kienlen-Campard P What Is the Role of Amyloid Precursor Protein Dimerization? Cell Adhes. Migr 2010, 4, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Song Y; Hustedt EJ; Brandon S; Sanders CR Competition between Homodimerization and Cholesterol Binding to the C99 Domain of the Amyloid Precursor Protein. Biochemistry 2013, 52, 5051–5064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (110).Dominguez L; Foster L; Meredith SC; Straub JE; Thirumalai D Structural Heterogeneity in Transmembrane Amyloid Precursor Protein Homodimer Is a Consequence of Environmental Selection. J. Am. Chem. Soc 2014, 136, 9619–9626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Dominguez L; Foster L; Straub JE; Thirumalai D Impact of Membrane Lipid Composition on the Structure and Stability of the Transmembrane Domain of Amyloid Precursor Protein. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E5281–E5287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Capone RF; Tiwari A; Fricke N; Hadziselimovic A; Kenworthy AK; Sanders CR Use of Giant Plasma Membrane Vesicles (GPMV) to Examine the Lo/Ld Phase Preference of the C99 Domain of the Amyloid Precursor Protein. Biophys. J 2020, 118 (1), 392A. [Google Scholar]
- (113).Miyashita N; Straub JE; Thirumalai D; Sugita Y Transmembrane Structures of Amyloid Precursor Protein Dimer Predicted by Replica-Exchange Molecular Dynamics Simulations. J. Am. Chem. Soc 2009, 131, 3438–3439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Dominguez L; Meredith SC; Straub JE; Thirumalai D Transmembrane Fragment Structures of Amyloid Precursor Protein Depend on Membrane Surface Curvature. J. Am. Chem. Soc 2014, 136, 854–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Cutler RG; Kelly J; Storie K; Pedersen WA; Tammara A; Hatanpaa K; Troncoso JC; Mattson MP Involvement of Oxidative Stress-Induced Abnormalities in Ceramide and Cholesterol Metabolism in Brain Aging and Alzheimer’s Disease. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 2070–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Shobab LA; Hsiung GYR; Feldman HH Cholesterol in Alzheimer’s Disease. Lancet Neurol 2005, 4, P841–P852. [DOI] [PubMed] [Google Scholar]
- (117).Puglielli L; Ellis BC; Saunders AJ; Kovacs DM Ceramide Stabilizes β-Site Amyloid Precursor Protein-Cleaving Enzyme 1 and Promotes Amyloid β-Peptide Biogenesis. J. Biol. Chem 2003, 278, 19777–19783. [DOI] [PubMed] [Google Scholar]
- (118).Kojro E; Gimpl G; Lammich S; März W; Fahrenholz F Low Cholesterol Stimulates the Nonamyloidogenic Pathway by Its Effect on the α-Secretase ADAM 10. Proc. Natl. Acad. Sci. U. S. A 2001, 98, 5815–5820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).De Meyer F; Smit B Effect of Cholesterol on the Structure of a Phospholipid Bilayer. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 3654–3658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).Ehehalt R; Keller P; Haass C; Thiele C; Simons K Amyloidogenic Processing of the Alzheimer β-Amyloid Precursor Protein Depends on Lipid Rafts. J. Cell Biol 2003, 160, 113–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Hicks DA; Nalivaeva NN; Turner AJ Lipid Rafts and Alzheimer’s Disease: Protein-Lipid Interactions and Perturbation of Signaling. Front. Physiol 2012, 3, 189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Panahi A; Bandara A; Pantelopulos GA; Dominguez L; Straub JE Specific Binding of Cholesterol to C99 Domain of Amyloid Precursor Protein Depends Critically on Charge State of Protein. J. Phys. Chem. Lett 2016, 7, 3535–3541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Cordy JM; Hussain I; Dingwall C; Hooper NM; Turner AJ Exclusively Targeting β-Secretase to Lipid Rafts by GPI-Anchor Addition up-Regulates β-Site Processing of the Amyloid Precursor Protein. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 11735–11740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (124).Kim S Il; Yi JS; Ko YG Amyloid β Oligomerization Is Induced by Brain Lipid Rafts. J. Cell. Biochem 2006, 99, 878–889. [DOI] [PubMed] [Google Scholar]
- (125).Kosicek M; Malnar M; Goate A; Hecimovic S Cholesterol Accumulation in Niemann Pick Type C (NPC) Model Cells Causes a Shift in APP Localization to Lipid Rafts. Biochem. Biophys. Res. Commun 2010, 393, 404–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (126).Bhattacharyya R; Barren C; Kovacs DM Palmitoylation of Amyloid Precursor Protein Regulates Amyloidogenic Processing in Lipid Rafts. J. Neurosci 2013, 33, 11169–11183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (127).Vetrivel KS; Thinakaran G Membrane Rafts in Alzheimer’s Disease Beta-Amyloid Production. Biochim. Biophys. Acta, Mol. Cell Biol. Lipids 2010, 1801, 860–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (128).Liu L; Ding L; Rovere M; Wolfe MS; Selkoe DJ A Cellular Complex of BACE1 and γ-Secretase Sequentially Generates Aβ from Its Full-Length Precursor. J. Cell Biol 2019, 218, 644–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (129).De Strooper B; Saftig P; Craessaerts K; Vanderstichele H; Guhde G; Annaert W; Von Figura K; Van Leuven F Deficiency of Presenilin-1 Inhibits the Normal Cleavage of Amyloid Precursor Protein. Nature 1998, 391, 387–390. [DOI] [PubMed] [Google Scholar]
- (130).Fabelo N; Martín V; Marín R; Moreno D; Ferrer I; Díaz M Altered Lipid Composition in Cortical Lipid Rafts Occurs at Early Stages of Sporadic Alzheimer’s Disease and Facilitates APP/BACE1 Interactions. Neurobiol. Aging 2014, 35, 1801–1812. [DOI] [PubMed] [Google Scholar]
- (131).Osenkowski P; Ye W; Wang R; Wolfe MS; Selkoe DJ Direct and Potent Regulation of γ-Secretase by Its Lipid Microenvironment. J. Biol. Chem 2008, 283, 22529–22540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (132).Holmes O; Paturi S; Ye W; Wolfe MS; Selkoe DJ Effects of Membrane Lipids on the Activity and Processivity of Purified γ-Secretase. Biochemistry 2012, 51, 3565–3575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Aguayo-Ortiz R; Straub JE; Dominguez L Influence of Membrane Lipid Composition on the Structure and Activity of γ-Secretase. Phys. Chem. Chem. Phys 2018, 20, 27294–27304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (134).Hitzenberger M; Götz A; Menig S; Brunschweiger B; Zacharias M; Scharnagl C Seminars in Cell & Developmental Biology The Dynamics of γ-Secretase and Its Substrates. Semin. Cell Dev. Biol 2020, 105, 86–101. [DOI] [PubMed] [Google Scholar]
- (135).Kong R; Chang S; Xia W; Wong STC Molecular Dynamics Simulation Study Reveals Potential Substrate Entry Path into γ-Secretase/Presenilin-1. J. Struct. Biol 2015, 191, 120–129. [DOI] [PubMed] [Google Scholar]
- (136).Audagnotto M; Kengo Lorkowski A; Dal Peraro M Recruitment of the Amyloid Precursor Protein by γ-Secretase at the Synaptic Plasma Membrane. Biochem. Biophys. Res. Commun 2018, 498, 334–341. [DOI] [PubMed] [Google Scholar]
- (137).MartíDel Moral A; Fortique F Omega-3 fatty Acids and Cognitive Decline: a Systematic Review. Nutr. Hosp 2019, 36, 939–949. [DOI] [PubMed] [Google Scholar]
- (138).Loef M; Walach H The omega-6/omega-3 Ratio and Dementia or Cognitive Decline: a Systematic Review on Human Studies and Biological Evidence. J. Nutr. Gerontol. Geriatr 2013, 32, 1–23. [DOI] [PubMed] [Google Scholar]
- (139). https://www.alzforum.org/mutations.
- (140).Humphrey W; Dalke A; Schulten K VMD - Visual Molecular Dynamics. J. Mol. Graphics 1996, 14, 33–38. [DOI] [PubMed] [Google Scholar]
- (141).Nizynski B; Dzwolak W; Nieznanski K Amyloidogenesis of Tau Protein. Protein Sci 2017, 26, 2126–2150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Kellogg EH; Hejab NMA; Poepsel S; Downing KH; DiMaio F; Nogales E Near-atomic Model of Microtubule-tau Interactions. Science 2018, 360, 1242–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Novak P; Cehlar O; Skrabana R; Novak M Tau Conformation as a Target for Disease-Modifying Therapy: The Role of Truncation. J. Alzheimer’s Dis 2018, 64, S535–S546. [DOI] [PubMed] [Google Scholar]
- (144).Quinn JP; Corbett NJ; Kellett KAB; Hooper NM Tau Proteolysis in the Pathogenesis of Tauopathies: Neurotoxic Fragments and Novel Biomarkers. J. Alzheimer’s Dis 2018, 63, 13–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (145).Al-Hilaly YK; Foster BE; Biasetti L; Lutter L; Pollack SJ; Rickard JE; Storey JMD; Harrington CR; Xue WF; Wischik CM; et al. Tau (297–391) Forms Filaments that Structurally Mimic the Core of Paired Helical Filaments in Alzheimer’s Disease Brain. FEBS Lett 2020, 594, 944–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (146).Schlegel K; Awwad K; Heym RG; Holzinger D; Doell A; Barghorn S; Jahn TR; Klein C; Mordashova Y; Schulz M; et al. N368-Tau fragments Generated by Legumain are Detected Only in Trace Amount in the Insoluble Tau Aggregates Isolated from AD Brain. Acta Neuropathol. Commun 2019, 7, 177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (147).Mohamed T; Gujral SS; Rao PPN Tau Derived Hexapeptide AcPHF6 Promotes Beta-Amyloid (Abeta) Fibrillogenesis. ACS Chem. Neurosci 2018, 9, 773–782. [DOI] [PubMed] [Google Scholar]
- (148).Chen D; Drombosky KW; Hou Z; Sari L; Kashmer OM; Ryder BD; Perez VA; Woodard DR; Lin MM; Diamond MI; et al. Tau Local Structure shields an Amyloid-forming Motif and Controls Aggregation Propensity. Nat. Commun 2019, 10, 2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Qi R; Luo Y; Wei G; Nussinov R; Ma B Aβ “Stretching-and-Packing” Cross-Seeding Mechanism Can Trigger Tau Protein Aggregation. J. Phys. Chem. Lett 2015, 6, 3276–3282. [Google Scholar]
- (150).Stohr J; Wu H; Nick M; Wu Y; Bhate M; Condello C; Johnson N; Rodgers J; Lemmin T; Acharya S; et al. A 31-residue Peptide Induces Aggregation of Tau’s Microtubule-binding Region in Cells. Nat. Chem 2017, 9, 874–881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Liu H; Zhong H; Liu X; Zhou S; Tan S; Liu H; Yao X Disclosing the Mechanism of Spontaneous Aggregation and Template-Induced Misfolding of the Key Hexapeptide (PHF6) of Tau Protein Based on Molecular Dynamics Simulation. ACS Chem. Neurosci 2019, 10, 4810–4823. [DOI] [PubMed] [Google Scholar]
- (152).Chemerovski-Glikman M; Frenkel-Pinter M; Mdah R; Abu-Mokh A; Gazit E; Segal D Inhibition of the Aggregation and Toxicity of the Minimal Amyloidogenic Fragment of Tau by Its Pro-Substituted Analogues. Chem. - Eur. J 2017, 23, 9618. [DOI] [PubMed] [Google Scholar]
- (153).Seidler PM; Boyer DR; Murray KA; Yang TP; Bentzel M; Sawaya MR; Rosenberg G; Cascio D; Williams CK; Newell KL; et al. Structure-based Inhibitors halt Prion-like Seeding by Alzheimer’s Disease-and Tauopathy-derived Brain Tissue Samples. J. Biol. Chem 2019, 294, 16451–16464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Seidler PM; Boyer DR; Rodriguez JA; Sawaya MR; Cascio D; Murray K; Gonen T; Eisenberg DS Structure-based Inhibitors of Tau Aggregation. Nat. Chem 2018, 10, 170–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (155).Kuret J; Congdon EE; Li G; Yin H; Yu X; Zhong Q Evaluating Triggers and Enhancers of Tau Fibrillization. Microsc. Res. Tech 2005, 67, 141–155. [DOI] [PubMed] [Google Scholar]
- (156).Lippens G; Sillen A; Landrieu I; Amniai L; Sibille N; Barbier P; Leroy A; Hanoulle X; Wieruszeski JM Tau Aggregation in Alzheimer’s Disease: What Role for Phosphorylation? Prion 2007, 1, 21–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (157).Johnson GV; Stoothoff WH Tau Phosphorylation in Neuronal Cell Function and Dysfunction. J. Cell Sci 2004, 117, 5721–5729. [DOI] [PubMed] [Google Scholar]
- (158).Martin L; Latypova X; Terro F Post-translational Modifications of Tau Protein: Implications for Alzheimer’s Disease. Neurochem. Int 2011, 58, 458–471. [DOI] [PubMed] [Google Scholar]
- (159).Funk KE; Thomas SN; Schafer KN; Cooper GL; Liao Z; Clark DJ; Yang AJ; Kuret J Lysine Methylation is an Endogenous Post-translational Modification of Tau Protein in Human Brain and a Modulator of Aggregation Propensity. Biochem. J 2014, 462, 77–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Cohen TJ; Guo JL; Hurtado DE; Kwong LK; Mills IP; Trojanowski JQ; Lee VMY The Acetylation of Tau Inhibits its Function and Promotes Pathological Tau Aggregation. Nat. Commun 2011, 2, 252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (161).Hanger DP; Byers HL; Wray S; Leung KY; Saxton MJ; Seereeram A; Reynolds CH; Ward MA; Anderton BH Novel Phosphorylation Sites in Tau from Alzheimer Brain Support a Role for Casein Kinase 1 in Disease Pathogenesis. J. Biol. Chem 2007, 282, 23645–23654. [DOI] [PubMed] [Google Scholar]
- (162).Lippens G; Gigant B Elucidating Tau Function and Dysfunction in the Era of Cryo-EM. J. Biol. Chem 2019, 294, 9316–9325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Zhang W; Tarutani A; Newell KL; Murzin AG; Matsubara T; Falcon B; Vidal R; Garringer HJ; Shi Y; Ikeuchi T; et al. Novel Tau Filament Fold in Corticobasal Degeneration. Nature 2020, 580, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).Falcon B; Zivanov J; Zhang W; Murzin AG; Garringer HJ; Vidal R; Crowther RA; Newell KL; Ghetti B; Goedert M; et al. Novel Tau Filament Fold in Chronic Traumatic Encephalopathy Encloses Hydrophobic Molecules. Nature 2019, 568, 420–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Luo Y; Dinkel P; Yu X; Margittai M; Zheng J; Nussinov R; Wei G; Ma B Molecular Insights into the Reversible Formation of Tau Protein Fibrils. Chem. Commun (Cambridge, U. K.) 2013, 49, 3582–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (166).Adamcik J; Sánchez-Ferrer A; Ait-Bouziad N; Reynolds NP; Lashuel HA; Mezzenga R Microtubule-Binding R3 Fragment from Tau Self-Assembles into Giant Multistranded Amyloid Ribbons. Angew. Chem., Int. Ed 2016, 55, 618–622. [DOI] [PubMed] [Google Scholar]
- (167).Holmes BB; DeVos SL; Kfoury N; Li M; Jacks R; Yanamandra K; Ouidja MO; Brodsky FM; Marasa J; Bagchi DP; et al. Heparan Sulfate Proteoglycans Mediate Internalization and Propagation of Specific Proteopathic Seeds. Proc. Natl. Acad. Sci. U. S. A 2013, 110, E3138–E31147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Wickramasinghe SP; Lempart J; Merens HE; Murphy J; Huettemann P; Jakob U; Rhoades E Polyphosphate Initiates Tau Aggregation through Intra- and Intermolecular Scaffolding. Biophys. J 2019, 117, 717–728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Mukrasch MD; Biernat J; von Bergen M; Griesinger C; Mandelkow E; Zweckstetter M Sites of Tau Important for Aggregation Populate β-Structure and Bind to Microtubules and Polyanions. J. Biol. Chem 2005, 280, 24978–24986. [DOI] [PubMed] [Google Scholar]
- (170).Akoury E; Mukrasch MD; Biernat J; Tepper K; Ozenne V; Mandelkow E; Blackledge M; Zweckstetter M Remodeling of the Conformational Ensemble of the Repeat Domain of Tau by an Aggregation Enhancer. Protein Sci 2016, 25, 1010–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (171).Zhao J; Huvent I; Lippens G; Eliezer D; Zhang A; Li Q; Tessier P; Linhardt RJ; Zhang F; Wang C Glycan Determinants of Heparin-Tau Interaction. Biophys. J 2017, 112, 921–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (172).Fichou Y; Vigers M; Goring AK; Eschmann NA; Han S Heparin-induced Tau Filaments are Structurally Heterogeneous and Differ from Alzheimer’s Disease Filaments. Chem. Commun (Cambridge, U. K.) 2018, 54, 4573–4576. [DOI] [PubMed] [Google Scholar]
- (173).Zhang W; Falcon B; Murzin AG; Fan J; Crowther RA; Goedert M; Scheres SH Heparin-induced Tau Filaments are Polymorphic and Differ from those in Alzheimer’s and Pick’s Diseases. eLife 2019, 8, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (174).Chen X; Chen M; Schafer NP; Wolynes PG Exploring the Interplay between Fibrillization and Amorphous Aggregation Channels on the Energy Landscapes of Tau Repeat Isoforms. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 4125–4130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (175).Kjaergaard M; Dear AJ; Kundel F; Qamar S; Meisl G; Knowles TPJ; Klenerman D Oligomer Diversity during the Aggregation of the Repeat Region of Tau. ACS Chem. Neurosci 2018, 9, 3060–3071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (176).Dregni AJ; Mandala VS; Wu H; Elkins MR; Wang HK; Hung I; DeGrado WF; Hong M In vitro 0N4R Tau Fibrils Contain a Monomorphic beta-sheet Core Enclosed by Dynamically Heterogeneous Fuzzy Coat Segments. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 16357–16366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Brion JP; Hanger DP; Bruce MT; Couck AM; Flament-Durand J; Anderton BH Tau in Alzheimer Neurofibrillary Tangles. N- and C-terminal Regions are Differentially Associated with Paired Helical Filaments and the Location of a Putative Abnormal Phosphorylation Site. Biochem. J 1991, 273, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (178).Geng J; Xia L; Li W; Dou F The C-Terminus of Tau Protein Plays an Important Role in Its Stability and Toxicity. J. Mol. Neurosci 2015, 55, 251–259. [DOI] [PubMed] [Google Scholar]
- (179).Gamblin TC; Berry RW; Binder LI Tau Polymerization: Role of the Amino Terminus. Biochemistry 2003, 42, 2252–2257. [DOI] [PubMed] [Google Scholar]
- (180).Horowitz PM; LaPointe N; Guillozet-Bongaarts AL; Berry RW; Binder LI N-Terminal Fragments of Tau Inhibit Full-Length Tau Polymerization in Vitro. Biochemistry 2006, 45, 12859–12866. [DOI] [PubMed] [Google Scholar]
- (181).Yanagawa H; Chung S-H; Ogawa Y; Sato K; Shibata-Seki T; Masai J; Ishiguro K Protein Anatomy: C-Tail Region of Human Tau Protein as a Crucial Structural Element in Alzheimer’s Paired Helical Filament Formation in Vitro. Biochemistry 1998, 37, 1979–1988. [DOI] [PubMed] [Google Scholar]
- (182).Weismiller HA; Murphy R; Wei G; Ma B; Nussinov R; Margittai M Structural Disorder in Four-repeat Tau Fibrils Reveals a New Mechanism for Barriers to Cross-seeding of Tau isoforms. J. Biol. Chem 2018, 293, 17336–17348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (183).Xu L; Zheng J; Margittai M; Nussinov R; Ma B How Does Hyperphopsphorylation Promote Tau Aggregation and Modulate Filament Structure and Stability? ACS Chem. Neurosci 2016, 7, 565–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (184).Lashuel HA; Overk CR; Oueslati A; Masliah E The Many Faces of alpha-synuclein: From Structure and Toxicity to Therapeutic Target. Nat. Rev. Neurosci 2013, 14, 38–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (185).Theillet FX; Binolfi A; Bekei B; Martorana A; Rose HM; Stuiver M; Verzini S; Lorenz D; van Rossum M; Goldfarb D; et al. Structural Disorder of Monomeric alpha-synuclein Persists in Mammalian Cells. Nature 2016, 530, 45–50. [DOI] [PubMed] [Google Scholar]
- (186).Fauvet B; Mbefo MK; Fares MB; Desobry C; Michael S; Ardah MT; Tsika E; Coune P; Prudent M; Lion N; et al. alpha-Synuclein in Central Nervous System and From Erythrocytes, Mammalian Cells, and Escherichia coli Exists Predominantly as Disordered Monomer. J. Biol. Chem 2012, 287, 15345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (187).Bartels T; Choi JG; Selkoe DJ Alpha-Synuclein Occurs Physiologically as a Helically Folded Tetramer that Resists Aggregation. Nature 2011, 477, 107–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (188).Uversky VN; Eliezer D Biophysics of Parkinson’s Disease: Structure and Aggregation of alpha-synuclein. Curr. Protein Pept. Sci 2009, 10, 483–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (189).Snead D; Eliezer D Alpha-synuclein Function and Dysfunction on Cellular Membranes. Exp. Neurobiol 2014, 23, 292–313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (190).Ueda K; Fukushima H; Masliah E; Xia Y; Iwai A; Yoshimoto M; Otero DA; Kondo J; Ihara Y; Saitoh T Molecular Cloning of cDNA Encoding an Unrecognized Component of Amyloid in Alzheimer Disease. Proc. Natl. Acad. Sci. U. S. A 1993, 90, 11282–11286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Galvin JE; Uryu K; Lee VM; Trojanowski JQ Axon Pathology in Parkinson’s Disease and Lewy Body Dementia Hippocampus Contains alpha-, beta-, and gamma-synuclein. Proc. Natl. Acad. Sci. U. S. A 1999, 96, 13450–13455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (192).Spillantini MG; Crowther RA; Jakes R; Cairns NJ; Lantos PL; Goedert M Filamentous alpha-synuclein Inclusions Link Multiple System Atrophy with Parkinson’s Disease and Dementia with Lewy Bodies. Neurosci. Lett 1998, 251, 205–208. [DOI] [PubMed] [Google Scholar]
- (193).Chiti F; Dobson CM Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu. Rev. Biochem 2017, 86, 27–68. [DOI] [PubMed] [Google Scholar]
- (194).Burre J The Synaptic Function of alpha-Synuclein. J. Parkinson’s Dis 2015, 5, 699–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (195).Murphy DD; Rueter SM; Trojanowski JQ; Lee VM Synucleins are Developmentally Expressed, and alpha-synuclein Regulates the Size of the Presynaptic Vesicular Pool in Primary Hippocampal Neurons. J. Neurosci 2000, 20, 3214–3220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (196).George JM; Jin H; Woods WS; Clayton DF Characterization of a Novel protein Regulated during the Critical Period for Song Learning in the Zebra Finch. Neuron 1995, 15, 361–372. [DOI] [PubMed] [Google Scholar]
- (197).Burre J; Sharma M; Sudhof TC Alpha-synuclein Assembles into Higher-order Multimers upon Membrane Binding to Promote SNARE Complex Formation. Proc. Natl. Acad. Sci. U. S. A 2014, 111, E4274–E4283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (198).Auluck PK; Caraveo G; Lindquist S Alpha-Synuclein: Membrane Interactions and Toxicity in Parkinson’s Disease. Annu. Rev. Cell Dev. Biol 2010, 26, 211–233. [DOI] [PubMed] [Google Scholar]
- (199).Lautenschlager J; Stephens AD; Fusco G; Strohl F; Curry N; Zacharopoulou M; Michel CH; Laine R; Nespovitaya N; Fantham M; et al. C-terminal Calcium Binding of alpha-synuclein Modulates Synaptic Vesicle Interaction. Nat. Commun 2018, 9, 712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (200).Burre J; Sharma M; Tsetsenis T; Buchman V; Etherton MR; Sudhof TC Alpha-synuclein Promotes SNARE-complex Assembly in vivo and in vitro. Science 2010, 329, 1663–1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (201).Chandra S; Gallardo G; Fernandez-Chacon R; Schluter OM; Sudhof TC Alpha-synuclein Cooperates with CSPalpha in Preventing Neurodegeneration. Cell 2005, 123, 383–396. [DOI] [PubMed] [Google Scholar]
- (202).Pelkonen A; Yavich L Neuromuscular Pathology in Mice Lacking Alpha-synuclein. Neurosci. Lett 2011, 487, 350–353. [DOI] [PubMed] [Google Scholar]
- (203).Fusco G; Pape T; Stephens AD; Mahou P; Costa AR; Kaminski CF; Kaminski Schierle GS; Vendruscolo M; Veglia G; Dobson CM; et al. Structural Basis of Synaptic Vesicle Assembly Promoted by a-synuclein. Nat. Commun 2016, 7, 12563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Diao J; Burre J; Vivona S; Cipriano DJ; Sharma M; Kyoung M; Sudhof TC; Brunger AT Native alpha-synuclein Induces Clustering of Synaptic-vesicle Mimics via Binding to Phospholipids and Synaptobrevin-2/VAMP2. eLife 2013, 2, No. e00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (205).Cooper AA; Gitler AD; Cashikar A; Haynes CM; Hill KJ; Bhullar B; Liu K; Xu K; Strathearn KE; Liu F; et al. Alpha-synuclein Blocks ER-Golgi Traffic and Rab1 Rescues Neuron Loss in Parkinson’s Models. Science 2006, 313, 324–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (206).Maltsev AS; Chen J; Levine RL; Bax A Site-specific Interaction between Alpha-synuclein and Membranes Probed by NMR-observed Methionine Oxidation Rates. J. Am. Chem. Soc 2013, 135, 2943–2946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (207).Newberry RW; Leong JT; Chow ED; Kampmann M; DeGrado WF Deep Mutational Scanning Reveals the Structural Basis for alpha-synuclein Activity. Nat. Chem. Biol 2020, 16, 653–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (208).Eliezer D; Kutluay E; Bussell R, Jr; Browne, G. Conformational Properties of alpha-synuclein in its Free and Lipid-associated States. J. Mol. Biol 2001, 307, 1061–1073. [DOI] [PubMed] [Google Scholar]
- (209).Cookson MR The Biochemistry of Parkinson’s disease. Annu. Rev. Biochem 2005, 74, 29–52. [DOI] [PubMed] [Google Scholar]
- (210).Hoppener JW; Ahren B; Lips CJ Islet Amyloid and Type 2 Diabetes Mellitus. N. Engl. J. Med 2000, 343, 411–419. [DOI] [PubMed] [Google Scholar]
- (211).Higham CE; Hull RL; Lawrie L; Shennan KI; Morris JF; Birch NP; Docherty K; Clark A Processing of Synthetic Pro-islet Amyloid Polypeptide (proIAPP) ‘amylin’ by Recombinant Prohormone Convertase Enzymes, PC2 and PC3, in vitro. Eur. J. Biochem 2000, 267, 4998–5004. [DOI] [PubMed] [Google Scholar]
- (212).Lorenzo A; Razzaboni B; Weir GC; Yankner BA Pancreatic Islet Cell Toxicity of Amylin associated with Type-2 Diabetes Mellitus. Nature 1994, 368, 756–760. [DOI] [PubMed] [Google Scholar]
- (213).Janciauskiene S; Ahren B Different Sensitivity to the Cytotoxic Action of IAPP Fibrils in two Insulin-producing Cell Lines, HIT-T15 and RINm5F Cells. Biochem. Biophys. Res. Commun 1998, 251, 888–893. [DOI] [PubMed] [Google Scholar]
- (214).Narita R; Toshimori H; Nakazato M; Kuribayashi T; Toshimori T; Kawabata K; Takahashi K; Masukura S Islet Amyloid Polypeptide (IAPP) and Pancreatic Islet Amyloid Deposition in Diabetic and Non-diabetic Patients. Diabetes Res. Clin. Pract 1992, 15, 3–14. [DOI] [PubMed] [Google Scholar]
- (215).Zhang J; Tan J; Pei R; Ye S Acidic Environment Significantly Alters Aggregation Pathway of Human Islet Amyloid Polypeptide at Negative Lipid Membrane. Langmuir 2020, 36, 1530–1537. [DOI] [PubMed] [Google Scholar]
- (216).Luca S; Yau WM; Leapman R; Tycko R Peptide Conformation and Supramolecular Organization in Amylin Fibrils: Constraints from Solid-state NMR. Biochemistry 2007, 46, 13505–13522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (217).Tycko R; Wickner RB Molecular Structures of Amyloid and Prion Fibrils: Consensus versus Controversy. Acc. Chem. Res 2013, 46, 1487–1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (218).Berman HM; Bhat TN; Bourne PE; Feng Z; Gilliland G; Weissig H; Westbrook J The Protein Data Bank and the Challenge of Structural Genomics. Nat. Struct. Biol 2000, 7, 957–959. [DOI] [PubMed] [Google Scholar]
- (219).Serpell LC; Berriman J; Jakes R; Goedert M; Crowther RA Fiber Diffraction of Synthetic Alpha-synuclein Filaments Shows Amyloid-like Cross-beta Conformation. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 4897–4902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (220).Bronsoms S; Trejo SA Applications of Mass Spectrometry to the Study of Protein Aggregation. Methods Mol. Biol 2015, 1258, 331–345. [DOI] [PubMed] [Google Scholar]
- (221).Lee YH; Goto Y Kinetic Intermediates of Amyloid Fibrillation Studied by Hydrogen Exchange Methods with Nuclear Magnetic Resonance. Biochim. Biophys. Acta, Proteins Proteomics 2012, 1824, 1307–1323. [DOI] [PubMed] [Google Scholar]
- (222).Shen Y; Delaglio F; Cornilescu G; Bax A TALOS+: a Hybrid Method for Predicting Protein Backbone Torsion Angles from NMR Chemical Shifts. J. Biomol. NMR 2009, 44, 213–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (223).Luca S; Filippov DV; van Boom JH; Oschkinat H; de Groot HJ; Baldus M Secondary Chemical Shifts in Immobilized Peptides and Proteins: a Qualitative Basis for Structure Refinement under Magic Angle Spinning. J. Biomol. NMR 2001, 20, 325–331. [DOI] [PubMed] [Google Scholar]
- (224).Fitzpatrick AW; Saibil HR Cryo-EM of Amyloid Fibrils and Cellular Aggregates. Curr. Opin. Struct. Biol 2019, 58, 34–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (225).Guerrero-Ferreira R; Taylor NM; Arteni AA; et al. Two New Polymorphic Structures of Human Full-length alpha-synuclein Fibrils Solved by Cryo-electron Microscopy. eLife 2019, 8, No. e48907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (226).Gremer L; Schölzel D; Schenk C; Reinartz E; Labahn J; Ravelli RBG; Tusche M; Lopez-Iglesias C; Hoyer W; Heise H; et al. Fibril Structure of amyloid-β(1–42) by Cryo-electron Microscopy. Science 2017, 358, 116–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Sunde M; Serpell LC; Bartlam M; Fraser PE; Pepys MB; Blake CC Common Core Structure of Amyloid Fibrils by Synchrotron X-ray Diffraction. J. Mol. Biol 1997, 273, 729–739. [DOI] [PubMed] [Google Scholar]
- (228).Lashuel HA; Wall JS Molecular Electron Microscopy Approaches to Elucidating the Mechanisms of Protein Fibrillogenesis. Methods Mol. Biol 2004, 299, 81–101. [DOI] [PubMed] [Google Scholar]
- (229).Chen B; Thurber KR; Shewmaker F; Wickner RB; Tycko R Measurement of Amyloid Fibril Mass-per-length by Tilted-Beam Transmission Electron Microscopy. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 14339–14344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Castellani F; van Rossum B; Diehl A; Schubert M; Rehbein K; Oschkinat H Structure of a Protein Determined by Solid-state Magic-angle-spinning NMR Spectroscopy. Nature 2002, 420, 99–102. [DOI] [PubMed] [Google Scholar]
- (231).Etzkorn M; Böckmann A; Lange A; Baldus M Probing Molecular Interfaces using 2D Magic-angle-spinning NMR on Protein Mixtures with Different Uniform Labeling. J. Am. Chem. Soc 2004, 126, 14746–14751. [DOI] [PubMed] [Google Scholar]
- (232).Meyer V; Margittai M Spin Labeling and Characterization of Tau Fibrils Using Electron Paramagnetic Resonance (EPR). Methods Mol. Biol 2016, 1345, 185–199. [DOI] [PubMed] [Google Scholar]
- (233).Rodriguez JA; Eisenberg DS; Gonen T Taking the Measure of MicroED. Curr. Opin. Struct. Biol 2017, 46, 79–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Kidd M Paired Helical Filaments in Electron Microscopy of Alzheimer’s Disease. Nature 1963, 197, 192–193. [DOI] [PubMed] [Google Scholar]
- (235).Yagishita S; Itoh Y; Nan W; Amano N Reappraisal of the Fine Structure of Alzheimer’s Neurofibrillary Tangles. Acta Neuropathol 1981, 54, 239–246. [DOI] [PubMed] [Google Scholar]
- (236).Fichou Y; Lin Y; Rauch JN; Vigers M; Zeng Z; Srivastava M; Keller TJ; Freed JH; Kosik KS; Han S Cofactors are Essential Constituents of Stable and Seeding-active Tau Fibrils. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 13234–13239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Prusiner SB Biology and Genetics of Prions Causing Neurodegeneration. Annu. Rev. Genet 2013, 47, 601–623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Tuttle MD; Comellas G; Nieuwkoop AJ; Covell DJ; Berthold DA; Kloepper KD; Courtney JM; Kim JK; Barclay AM; Kendall A; et al. Solid-state NMR Structure of a Pathogenic Fibril of Full-length Human α-synuclein. Nat. Struct. Mol. Biol 2016, 23, 409–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Boyer DR; Li B; Sun C; Fan W; Zhou K; Hughes MP; Sawaya MR; Jiang L; Eisenberg DS Structures of Fibrils formed by α-synuclein Hereditary Disease Mutant H50Q Reveal New Polymorphs. Nat. Struct. Mol. Biol 2019, 26, 1044–1052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Schweighauser M; Shi Y; Tarutani A; Kametani F; Murzin AG; Ghetti B; Matsubara T; Tomita T; Ando T; Hasegawa K; et al. Structures of α-synuclein Filaments from Multiple System Atrophy. Nature 2020, 585, 464–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Lansbury PTJ; Costa PR; Griffiths JM; Simon EJ; Auger M; Halverson KJ; Kocisko DA; Hendsch ZS; Ashburn TT; Spencer RG; et al. Structural Model for the beta-amyloid Fibril Based on Interstrand Alignment of an Antiparallel-sheet Comprising a C-terminal Peptide. Nat. Struct. Mol. Biol 1995, 2, 990–998. [DOI] [PubMed] [Google Scholar]
- (242).Antzutkin ON; Balbach JJ; Leapman RD; Rizzo NW; Reed J; Tycko R Multiple Quantum solid-state NMR Indicates a Parallel, not Antiparallel, Organization of beta-sheets in Alzheimer’s beta-amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 13045–13050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Petkova AT; Ishii Y; Balbach JJ; Antzutkin ON; Leapman RD; Delaglio F; Tycko R A Structural Model for Alzheimer’s beta -amyloid Fibrils Based on Experimental Constraints from Solid state NMR. Proc. Natl. Acad. Sci. U. S. A 2002, 99, 16742–16747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (244).Petkova AT; Yau WM; Tycko R Experimental Constraints on Quaternary Structure in Alzheimer’s beta-amyloid Fibrils. Biochemistry 2006, 45, 498–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Paravastu AK; Leapman RD; Yau WM; Tycko R Molecular Structural Basis for Polymorphism in Alzheimer’s beta-amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 18349–18354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (246).Zhang R; Hu X; Khant H; Ludtke SJ; Chiu W; Schmid MF; Frieden C; Lee JM Interprotofilament Interactions between Alzheimer’s Abeta1–42 Peptides in Amyloid Fibrils Revealed by CryoEM. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 4653–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Wälti MA; Ravotti F; Arai H; Glabe CG; Wall JS; Böckmann A; Güntert P; Meier BH; Riek R Atomic-resolution Structure of a Disease-relevant Aβ(1–42) Amyloid Fibril. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E4976–E4984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (248).Petkova AT; Leapman RD; Guo Z; Yau WM; Mattson MP; Tycko R Self-propagating, Molecular-level Polymorphism in Alzheimer’s beta-amyloid Fibrils. Science 2005, 307, 262–265. [DOI] [PubMed] [Google Scholar]
- (249).Lu JX; Qiang W; Yau WM; Schwieters CD; Meredith SC; Tycko R Molecular Structure of β-amyloid Fibrils in Alzheimer’s Disease Brain Tissue. Cell 2013, 154, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Qiang W; Yau WM; Lu JX; Collinge J; Tycko R Structural Variation in Amyloid-β Fibrils from Alzheimer’s Disease Clinical Subtypes. Nature 2017, 541, 217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Cohen ML; Kim C; Haldiman T; ElHag M; Mehndiratta P; Pichet T; Lissemore F; Shea M; Cohen Y; Chen W; et al. Rapidly Progressive Alzheimer’s Disease Features Distinct Structures of Amyloid-β. Brain 2015, 138, 1009–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Bedrood S; Li Y; Isas JM; Hegde BG; Baxa U; Haworth IS; Langen R Fibril Structure of Human Islet Amyloid Polypeptide. J. Biol. Chem 2012, 287, 5235–5241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Röder C; Kupreichyk T; Gremer L; Schäfer LU; Pothula KR; Ravelli RBG; Willbold D; Hoyer W; Schröder GF Cryo-EM Structure of Islet Amyloid Polypeptide Fibrils Reveals Similarities with Amyloid-β Fibrils. Nat. Struct. Mol. Biol 2020, 27, 660–667. [DOI] [PubMed] [Google Scholar]
- (254).DeLano WL PyMOL; DeLano Scientific: San Carlos, CA, 2002; p 700. [Google Scholar]
- (255).Roche J; Shen Y; Lee JH; Ying J; Bax A Monomeric Aβ1–40 and Aβ1–42 Peptides in Solution Adopt Very Similar Ramachandran Map Distributions That Closely Resemble Random Coil. Biochemistry 2016, 55, 762–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Coles M; Bicknell W; Watson RA; Fairlie DP; Craik DJ Solution Structure of Amyloid β-Peptide(1–40) in a Water-Micelle Environment. Is the Membrane-Spanning Domain Where We Think It Is? Biochemistry 1998, 37, 11064–11077. [DOI] [PubMed] [Google Scholar]
- (257).Watson AA; Fairlie DP; Craik DJ Solution Structure of Methionine-Oxidized Amyloid β-Peptide (1–40). Does Oxidation Affect Conformational Switching? Biochemistry 1998, 37, 12700–12706. [DOI] [PubMed] [Google Scholar]
- (258).Vivekanandan S; Brender JR; Lee SY; Ramamoorthy A A Partially Folded Structure of Amyloid-Beta(1–40) in an Aqueous Environment. Biochem. Biophys. Res. Commun 2011, 411, 312–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Bossis F; Palese LL Amyloid Beta(1–42) in Aqueous Environments: Effects of Ionic Strength and E22Q (Dutch) Mutation. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 2486–2493. [DOI] [PubMed] [Google Scholar]
- (260).Hyung SJ; Detoma AS; Brender JR; Lee S; Vivekanandan S; Kochi A; Choi JS; Ramamoorthy A; Ruotolo BT; Lim MH Insights into Antiamyloidogenic Properties of the Green Tea Extract (−)-Epigallocatechin-3-Gallate toward Metal-Associated Amyloid-β Species. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 3743–3748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (261).Kreutzer AG; Nowick JS Elucidating the Structures of Amyloid Oligomers with Macrocyclic β-Hairpin Peptides: Insights into Alzheimer’s Disease and Other Amyloid Diseases. Acc. Chem. Res 2018, 51, 706–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Pham JD; Spencer RK; Chen KH; Nowick JS A Fibril-like Assembly of Oligomers of a Peptide Derived from β-Amyloid. J. Am. Chem. Soc 2014, 136, 12682–12690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Sahoo BR; Genjo T; Bekier M; Cox SJ; Stoddard AK; Ivanova M; Yasuhara K; Fierke CA; Wang Y; Ramamoorthy A Alzheimer’s Amyloid-Beta Intermediates Generated Using Polymer-Nanodiscs. Chem. Commun 2018, 54, 12883–12886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Lashuel HA; Hartley D; Petre BM; Walz T; Lansbury PT Neurodegenerative Disease: Amyloid Pores from Pathogenic Mutations. Nature 2002, 418, 291. [DOI] [PubMed] [Google Scholar]
- (265).Irie Y; Murakami K; Hanaki M; Hanaki Y; Suzuki T; Monobe Y; Takai T; Akagi KI; Kawase T; Hirose K; Irie K Synthetic Models of Quasi-Stable Amyloid B40 Oligomers with Significant Neurotoxicity. ACS Chem. Neurosci 2017, 8, 807–816. [DOI] [PubMed] [Google Scholar]
- (266).Matsushima Y; Yanagita RC; Irie K Control of the Toxic Conformation of Amyloid B42 by Intramolecular Disulfide Bond Formation. Chem. Commun 2020, 56, 4118–4121. [DOI] [PubMed] [Google Scholar]
- (267).Irie Y; Hanaki M; Murakami K; Imamoto T; Furuta T; Kawabata T; Kawase T; Hirose K; Monobe Y; Akagi K; Ichi Yanagita RC; Irie K Synthesis and Biochemical Characterization of Quasi-Stable Trimer Models of Full-Length Amyloid B40 with a Toxic Conformation. Chem. Commun 2019, 55, 182–185. [DOI] [PubMed] [Google Scholar]
- (268).Kotler SA; Brender JR; Vivekanandan S; Suzuki Y; Yamamoto K; Monette M; Krishnamoorthy J; Walsh P; Cauble M; Holl MMB; Marsh ENG; Ramamoorthy A High-Resolution NMR Characterization of Low Abundance Oligomers of Amyloid-β without Purification. Sci. Rep 2015, 5, 11811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Barnes CA; Robertson AJ; Louis JM; Anfinrud P; Bax A Observation of β-Amyloid Peptide Oligomerization by Pressure-Jump NMR Spectroscopy. J. Am. Chem. Soc 2019, 141, 13762–13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Ahmed R; Akcan M; Khondker A; Rheinstädter MC; Bozelli JC Jr; Epand RM; Huynh V; Wylie RG; Boulton S; Huang J; et al. Atomic Resolution Map of the Soluble Amyloid beta Assembly Toxic Surfaces. Chem. Sci 2019, 10, 6072–6082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).Dorosh L; Stepanova M Probing Oligomerization of Amyloid Beta Peptide in Silico. Mol. BioSyst 2017, 13, 165–182. [DOI] [PubMed] [Google Scholar]
- (272).Baldassarre M; Baronio CM; Morozova-Roche LA; Barth A Amyloid β-Peptides 1–40 and 1–42 Form Oligomers with Mixed β-Sheets. Chem. Sci 2017, 8, 8247–8254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Sahoo BR; Bekier ME; Liu Z; Kocman V; Stoddard AK; Anantharamaiah GM; Nowick J; Fierke CA; Wang Y; Ramamoorthy A Structural Interaction of Apolipoprotein A-I Mimetic Peptide with Amyloid-β Generates Toxic Hetero-Oligomers. J. Mol. Biol 2020, 432, 1020–1034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Sahoo BR; Genjo T; Cox SJ; Stoddard AK; Anantharamaiah GM; Fierke C; Ramamoorthy A Nanodisc-Forming Scaffold Protein Promoted Retardation of Amyloid-Beta Aggregation. J. Mol. Biol 2018, 430, 4230–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Younan ND; Sarell CJ; Davies P; Brown DR; Viles JH The Cellular Prion Protein Traps Alzheimer’s Aβ in an Oligomeric Form and Disassembles Amyloid Fibers. FASEB J 2013, 27, 1847–1858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Sahoo BR; Cox SJ; Ramamoorthy A High-Resolution Probing of Early Events in Amyloid-β Aggregation Related to Alzheimer’s Disease. Chem. Commun (Cambridge, U. K.) 2020, 56, 4627–4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Williamson JA; Miranker AD Direct Detection of Transient α-Helical States in Islet Amyloid Polypeptide. Protein Sci 2007, 16, 110–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Patel HR; Pithadia AS; Brender JR; Fierke CA; Ramamoorthy A In Search of Aggregation Pathways of IAPP and Other Amyloidogenic Proteins: Finding Answers through NMR Spectroscopy. J. Phys. Chem. Lett 2014, 5, 1864–1870. [DOI] [PubMed] [Google Scholar]
- (279).Nanga RPR; Brender JR; Vivekanandan S; Ramamoorthy A Structure and Membrane Orientation of IAPP in Its Natively Amidated Form at Physiological pH in a Membrane Environment. Biochim. Biophys. Acta, Biomembr 2011, 1808, 2337–2342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (280).Nanga RPR; Brender JR; Xu J; Veglia G; Ramamoorthy A Structures of Rat and Human Islet Amyloid Polypeptide IAPP1–19 in Micelles by NMR Spectroscopy. Biochemistry 2008, 47, 12689–12697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Nanga RPR; Brender JR; Xu J; Hartman K; Subramanian V; Ramamoorthy A Three-Dimensional Structure and Orientation of Rat Islet Amyloid Polypeptide Protein in a Membrane Environment by Solution NMR Spectroscopy. J. Am. Chem. Soc 2009, 131, 8252–8261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Abedini A; Plesner A; Cao P; Ridgway Z; Zhang J; Tu LH; Middleton CT; Chao B; Sartori DJ; Meng F; et al. Time-Resolved Studies Define the Nature of Toxic IAPP Intermediates, Providing Insight for Anti-Amyloidosis Therapeutics. eLife 2016, 5, No. e12977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Young LM; Cao P; Raleigh DP; Ashcroft AE; Radford SE Ion Mobility Spectrometry-Mass Spectrometry Defines the Oligomeric Intermediates in Amylin Amyloid Formation and the Mode of Action of Inhibitors. J. Am. Chem. Soc 2014, 136, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (284).Bram Y; Frydman-Marom A; Yanai I; Gilead S; Shaltiel-Karyo R; Amdursky N; Gazit E Apoptosis Induced by Islet Amyloid Polypeptide Soluble Oligomers Is Neutralized by Diabetes-Associated Specific Antibodies. Sci. Rep 2014, 4, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Rodriguez Camargo DC; Korshavn KJ; Jussupow A; Raltchev K; Goricanec D; Fleisch M; Sarkar R; Xue K; Aichler M; Mettenleiter G; et al. Stabilization and Structural Analysis of a Membrane-Associated HIAPP Aggregation Intermediate. eLife 2017, 6, No. e31226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Rodriguez Camargo DC; Tripsianes K; Buday K; Franko A; Göbl C; Hartlmüller C; Sarkar R; Aichler M; Mettenleiter G; Schulz M; et al. The Redox Environment Triggers Conformational Changes and Aggregation of HIAPP in Type II Diabetes. Sci. Rep 2017, 7, 44041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Buchanan LE; Dunkelberger EB; Tran HQ; Cheng PN; Chiu CC; Cao P; Raleigh DP; De Pablo JJ; Nowick JS; Zanni MT Mechanism of IAPP Amyloid Fibril Formation Involves an Intermediate with a Transient β-Sheet. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 19285–19290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Brender JR; Krishnamoorthy J; Sciacca MFM; Vivekanandan S; D’Urso L; Chen J; La Rosa C; Ramamoorthy A Probing the Sources of the Apparent Irreproducibility of Amyloid Formation: Drastic Changes in Kinetics and a Switch in Mechanism Due to Micellelike Oligomer Formation at Critical Concentrations of IAPP. J. Phys. Chem. B 2015, 119, 2886–2896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Akter R; Cao P; Noor H; Ridgway Z; Tu LH; Wang H; Wong AG; Zhang X; Abedini A; Schmidt AM; Raleigh DP Islet Amyloid Polypeptide: Structure, Function, and Pathophysiology. J. Diabetes Res 2016, 2016, 2798269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (290).Pithadia A; Brender JR; Fierke CA; Ramamoorthy A Inhibition of IAPP Aggregation and Toxicity by Natural Products and Derivatives. J. Diabetes Res 2016, 2016, 2046327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (291).Khemtemourian L; Gazit E; Miranker A Recent Insight in Islet Amyloid Polypeptide Morphology, Structure, Membrane Interaction, and Toxicity in Type 2 Diabetes. J. Diabetes Res 2016, 2016, 2535878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Zhou L; Kurouski D Structural Characterization of Individual α-Synuclein Oligomers Formed at Different Stages of Protein Aggregation by Atomic Force Microscopy-Infrared Spectroscopy. Anal. Chem 2020, 92, 6806–6810. [DOI] [PubMed] [Google Scholar]
- (293).Chen SW; Drakulic S; Deas E; Ouberai M; Aprile FA; Arranz R; Ness S; Roodveldt C; Guilliams T; De-Genst EJ; et al. Structural Characterization of Toxic Oligomers That Are Kinetically Trapped during α-Synuclein Fibril Formation. Proc. Natl. Acad. Sci. U. S. A 2015, 112, E1994–E2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Li X; Dong C; Hoffmann M; Garen CR; Cortez LM; Petersen NO; Woodside MT Early Stages of Aggregation of Engineered α-Synuclein Monomers and Oligomers in Solution. Sci. Rep 2019, 9, 1734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (295).Bhak G; Lee S; Kim TH; Lee JH; Yang JE; Joo K; Lee J; Char K; Paik SR Morphological Evaluation of Meta-Stable Oligomers of α-Synuclein with Small-Angle Neutron Scattering. Sci. Rep 2018, 8, 14295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (296).Lee JE; Sang JC; Rodrigues M; Carr AR; Horrocks MH; De S; Bongiovanni MN; Flagmeier P; Dobson CM; Wales DJ; et al. Mapping Surface Hydrophobicity of α-Synuclein Oligomers at the Nanoscale. Nano Lett 2018, 18, 7494–7501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (297).Candreva J; Chau E; Rice ME; Kim JR Interactions between Soluble Species of β-Amyloid and α-Synuclein Promote Oligomerization While Inhibiting Fibrillization. Biochemistry 2020, 59, 425–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (298).Favretto F; Baker JD; Strohäker T; Andreas LB; Blair LJ; Becker S; Zweckstetter M The Molecular Basis of the Interaction of Cyclophilin A with α-Synuclein. Angew. Chem., Int. Ed 2020, 59, 5643–5646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (299).Fusco G; De Simone A; Gopinath T; Vostrikov V; Vendruscolo M; Dobson CM; Veglia G Direct Observation of the Three Regions in α-Synuclein That Determine Its Membrane-Bound Behaviour. Nat. Commun 2014, 5, 3827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Braak H; Braak E Neuropathological Stageing of Alzheimer-Related Changes. Acta Neuropathol 1991, 82, 239–259. [DOI] [PubMed] [Google Scholar]
- (301).Ercan-Herbst E; Ehrig J; Schöndorf DC; Behrendt A; Klaus B; Gomez Ramos B; Prat Oriol N; Weber C; Ehrnhoefer DE A Post-Translational Modification Signature Defines Changes in Soluble Tau Correlating with Oligomerization in Early Stage Alzheimer’s Disease Brain. Acta Neuropathol. Commun 2019, 7, 192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (302).Karikari TK; Nagel DA; Grainger A; Clarke-Bland C; Hill EJ; Moffat KG Preparation of Stable Tau Oligomers for Cellular and Biochemical Studies. Anal. Biochem 2019, 566, 67–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Lasagna-Reeves CA; Castillo-Carranza DL; Guerrero-Muñoz MJ; Jackson GR; Kayed R Preparation and Characterization of Neurotoxic Tau Oligomers. Biochemistry 2010, 49, 10039–10041. [DOI] [PubMed] [Google Scholar]
- (304).Dasari AKR; Kayed R; Wi S; Lim KH Tau Interacts with the C-Terminal Region of α-Synuclein, Promoting Formation of Toxic Aggregates with Distinct Molecular Conformations. Biochemistry 2019, 58, 2814–2821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (305).Woo JAA; Liu T; Fang CC; Castaño MA; Kee T; Yrigoin K; Yan Y; Cazzaro S; Matlack J; Wang X; et al. β-Arrestin2 Oligomers Impair the Clearance of Pathological Tau and Increase Tau Aggregates. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 5006–5015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Oroz J; Chang BJ; Wysoczanski P; Lee CT; Pérez-Lara Á; Chakraborty P; Hofele RV; Baker JD; Blair LJ; Biernat J; et al. Structure and Pro-Toxic Mechanism of the Human Hsp90/PPIase/Tau Complex. Nat. Commun 2018, 9, 4532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (307).Skachokova Z; Martinisi A; Flach M; Sprenger F; Naegelin Y; Steiner-Monard V; Sollberger M; Monsch AU; Goedert M; Tolnay M; et al. Cerebrospinal Fluid from Alzheimer’s Disease Patients Promotes Tau Aggregation in Transgenic Mice. Acta Neuropathol. Commun 2019, 7, 72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (308).Maeda S; Takashima A Tau Oligomers. Adv. Exp. Med. Biol 2019, 1184, 373–380. [DOI] [PubMed] [Google Scholar]
- (309).Törnquist M; Michaels TCT; Sanagavarapu K; Yang X; Meisl G; Cohen SIA; Knowles TPJ; Linse S Secondary Nucleation in Amyloid Formation. Chem. Commun 2018, 54, 8667–8684. [DOI] [PubMed] [Google Scholar]
- (310).Hellstrand E; Boland B; Walsh DM; Linse S Amyloid β-protein Aggregation Produces highly Reproducible Kinetic Data and Occurs by a Two-phase Process. ACS Chem. Neurosci 2010, 1, 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Jan A; Hartley DM; Lashuel HA Preparation and Characterization of Toxic Abeta Aggregates for Structural and Functional Studies in Alzheimer’s Disease Research. Nat. Protoc 2010, 5, 1186–1209. [DOI] [PubMed] [Google Scholar]
- (312).Marvian AT; Koss DJ; Aliakbari F; Morshedi D; Outeiro TF In vitro Models of Synucleinopathies: Informing on Molecular mechanisms and protective strategies. J. Neurochem 2019, 150, 535–565. [DOI] [PubMed] [Google Scholar]
- (313).Weibull MGM; Simonsen S; Oksbjerg CR; Tiwari MK; Hemmingsen L Effects of Cu(II) on the Aggregation of Amyloid-β. JBIC, J. Biol. Inorg. Chem 2019, 24, 1197–1215. [DOI] [PubMed] [Google Scholar]
- (314).Doig AJ; Del Castillo-Frias MP; Berthoumieu O; Tarus B; Nasica-Labouze J; Sterpone F; Nguyen PH; Hooper NM; Faller P; Derreumaux P Why Is Research on Amyloid-β Failing to Give New Drugs for Alzheimer’s Disease? ACS Chem. Neurosci 2017, 8, 1435–1437. [DOI] [PubMed] [Google Scholar]
- (315).Alder BJ; Wainwright TE Studies in Molecular Dynamics.1. General Method. J. Chem. Phys 1959, 31, 459–466. [Google Scholar]
- (316).Smith AV; Hall CK Alpha-helix Formation: Discontinuous Molecular Dynamics on an Intermediate-resolution Protein Model. Proteins: Struct., Funct., Genet 2001, 44, 344–360. [DOI] [PubMed] [Google Scholar]
- (317).Smith AV; Hall CK Assembly of a Tetrameric Alpha-helical Bundle: Computer Simulations on an Intermediate-resolution Protein Model. Proteins: Struct., Funct., Genet 2001, 44, 376–391. [DOI] [PubMed] [Google Scholar]
- (318).Smith AV; Hall CK Protein Refolding versus Aggregation: Computer Simulations on an Intermediate-resolution Protein Model. J. Mol. Biol 2001, 312, 187–202. [DOI] [PubMed] [Google Scholar]
- (319).Nguyen HD; Hall CK Molecular Dynamics Simulations of Spontaneous Fibril Formation by Random-coil Peptides. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 16180–16185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (320).Nguyen HD; Hall CK Spontaneous Fibril Formation by Polyalanines; Discontinuous Molecular Dynamics Simulations. J. Am. Chem. Soc 2006, 128, 1890–1901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (321).Cheon M; Chang I; Hall CK Extending the PRIME Model for Protein Aggregation to all 20 Amino Acids. Proteins: Struct., Funct., Genet 2010, 78, 2950–2960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Wang Y; Shao Q; Hall CK N-terminal Prion Protein Peptides (PrP(120–144)) Form Parallel In-register β-Sheets via Multiple Nucleation-dependent Pathways. J. Biol. Chem 2016, 291, 22093–22105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Cheon M; Chang I; Hall CK Spontaneous Formation of Twisted A beta(16–22) Fibrils in Large-Scale Molecular-Dynamics Simulations. Biophys. J 2011, 101, 2493–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (324).Latshaw DC; Cheon M; Hall CK Effects of Macromolecular Crowding on Amyloid Beta (16–22) Aggregation Using Coarse-Grained Simulations. J. Phys. Chem. B 2014, 118, 13513–13526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (325).Wagoner VA; Cheon M; Chang I; Hall CK Fibrillization Propensity for Short Designed Hexapeptides Predicted by Computer Simulation. J. Mol. Biol 2012, 416, 598–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (326).de la Paz ML; Serrano L Sequence Determinants of Amyloid Fibril Formation. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 87–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (327).Cheon M; Chang I; Hall CK Influence of Temperature on Formation of Perfect Tau Fragment Fibrils using PRIME20/DMD Simulations. Protein Sci 2012, 21, 1514–1527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (328).Wang Y; Latshaw DC; Hall CK Aggregation of Aβ(17–36) in the Presence of Naturally Occurring Phenolic Inhibitors Using Coarse-Grained Simulations. J. Mol. Biol 2017, 429, 3893–3908. [DOI] [PubMed] [Google Scholar]
- (329).Cheon M; Hall CK; Chang I Structural Conversion of Abeta(17–42) Peptides from Disordered Oligomers to U-Shape Protofilaments via Multiple Kinetic Pathways. PLoS Comput. Biol 2015, 11, No. e1004258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (330).Bunce SJ; Wang Y; Stewart KL; Ashcroft AE; Radford SE; Hall CK; Wilson AJ Molecular Insights into the Surface Catalysed Secondary Nucleation of amyloid-β (Aβ40) by the Peptide Fragment Aβ16–22. Sci. Adv 2019, 5, No. eaav8216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Shao Q; Wong KM; Seroski DT; Wang Y; Liu R; Paravastu AK; Hudalla GA; Hall CK Anatomy of a Selectively Coassembled β-sheet Peptide Nanofiber. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 4710–4717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Vácha R; Frenkel D Relation between Molecular Shape and the Morphology of Self-assembling Aggregates: a Simulation Study. Biophys. J 2011, 101, 1432–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (333).Li MS; Klimov DK; Straub JE; Thirumalai D Probing the Mechanisms of Fibril Formation using Lattice Models. J. Chem. Phys 2008, 129, 175101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (334).Abeln S; Frenkel D Accounting for Protein-solvent Contacts Facilitates Design of Nonaggregating Lattice Proteins. Biophys. J 2011, 100, 693–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Bellesia G; Shea J-E Self-assembly of β-sheet Forming Peptides into Chiral Fibrillar Aggregates. J. Chem. Phys 2007, 126, 245104. [DOI] [PubMed] [Google Scholar]
- (336).Rojas AV; Maisuradze GG; Scheraga HA Dependence of the Formation of Tau and Aβ Peptide Mixed Aggregates on the Secondary Structure of the N-terminal of Aβ. J. Phys. Chem. B 2018, 122, 7049–7056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (337).Sterpone F; Melchionna S; Tuffery P; Pasquali S; Mousseau N; Cragnolini T; Chebaro Y; St-Pierre JF; Kalimeri M; Barducci A; et al. , The OPEP Protein Model: from Single Molecules, Amyloid Formation, Crowding and Hydrodynamics to DNA/RNA Systems. Chem. Soc. Rev 2014, 43, 4871–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (338).Santini S; Wei G; Mousseau N; Derreumaux P Pathway Complexity of Alzheimer’s β-amyloid Aβ16–22 Peptide Assembly. Structure 2004, 12, 1245–1255. [DOI] [PubMed] [Google Scholar]
- (339).Urbanc B; Borreguero JM; Cruz L; Stanley HE Ab initio Discrete Molecular Dynamics Approach to Protein Folding and Aggregation. Methods Enzymol 2006, 412, 314–338. [DOI] [PubMed] [Google Scholar]
- (340).Darré L; Machado MR; Brandner AF; González HC; Ferreira S; Pantano S SIRAH: A Structurally Unbiased Coarse-grained Force Field for Proteins with Aqueous Solvation and Long-range Electrostatics. J. Chem. Theory Comput 2015, 11, 723–739. [DOI] [PubMed] [Google Scholar]
- (341).Morriss-Andrews A; Shea JE Simulations of Protein Aggregation: Insights from Atomistic and Coarse-grained Models. J. Phys. Chem. Lett 2014, 5, 1899–1908. [DOI] [PubMed] [Google Scholar]
- (342).Nguyen HD; Hall CK Phase Diagrams Describing Fibrillization by Polyalanine Peptides. Biophys. J 2004, 87, 4122–4134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Ranganathan S; Maji SK; Padinhateeri R Defining a Physical Basis for Diversity in Protein Self-Assemblies Using a Minimal Model. J. Am. Chem. Soc 2016, 138, 13911–13922. [DOI] [PubMed] [Google Scholar]
- (344).Ni R; Abeln S; Schor M; Cohen Stuart MA; Bolhuis PG Interplay between Folding and Assembly of Fibril Forming Polypeptides. Phys. Rev. Lett 2013, 111, 058101. [DOI] [PubMed] [Google Scholar]
- (345).Kashchiev D; Auer S Nucleation of amyloid fibrils. J. Chem. Phys 2010, 132, 215101. [DOI] [PubMed] [Google Scholar]
- (346).Auer S; Kashchiev D Phase Diagram of α-helical and β-sheet Forming Peptides. Phys. Rev. Lett 2010, 104, 168105. [DOI] [PubMed] [Google Scholar]
- (347).Bai XM; Li M Calculation of Solid-liquid Interfacial Free Energy: A Classical Nucleation Theory Based Approach. J. Chem. Phys 2006, 124, 124707. [DOI] [PubMed] [Google Scholar]
- (348).Hoang TX; Trovato A; Seno F; Banavar JR; Maritan A Geometry and Symmetry Presculpt the Free-energy Landscape of Proteins. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 7960–7964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Auer S Phase Diagram of Polypeptide Chains. J. Chem. Phys 2011, 135, 175103. [DOI] [PubMed] [Google Scholar]
- (350).Wang Y; Bunce SJ; Radford SE; Wilson AJ; Auer S; Hall CK Thermodynamic Phase Diagram of Amyloid-β (16–22) Peptide. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 2091–2096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Kirschner DA; Inouye H; Duffy LK; Sinclair A; Lind M; Selkoe DJ Synthetic Peptide Homologous to beta Protein from Alzheimer Disease Forms Amyloid-like Fibrils in vitro. Proc. Natl. Acad. Sci. U. S. A 1987, 84, 6953–6957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Hsieh MC; Liang C; Mehta AK; Lynn DG; Grover MA Multistep Conformation Selection in Amyloid Assembly. J. Am. Chem. Soc 2017, 139, 17007–17010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Childers WS; Anthony NR; Mehta AK; Berland KM; Lynn DG Phase Networks of Cross-β Peptide Assemblies. Langmuir 2012, 28, 6386–6395. [DOI] [PubMed] [Google Scholar]
- (354).Petty SA; Decatur SM Experimental Evidence for the Reorganization of β-strands within Aggregates of the Aβ(16–22) Peptide. J. Am. Chem. Soc 2005, 127, 13488–13489. [DOI] [PubMed] [Google Scholar]
- (355).Preston GW Analysis of Biomolecular Interactions using Photo-induced Covalent Cross-linking. PhD thesis, University of Leeds, 2012. [DOI] [PubMed] [Google Scholar]
- (356).Balbach JJ; Ishii Y; Antzutkin ON; Leapman RD; Rizzo NW; Dyda F; Reed J; Tycko R Amyloid Fibril Formation by Abeta(16–22), a seven-residue Fragment of the Alzheimer’s beta-amyloid Peptide, and Structural Characterization by Solid state NMR. Biochemistry 2000, 39, 13748–13759. [DOI] [PubMed] [Google Scholar]
- (357).Senguen FT; Lee NR; Gu X; Ryan DM; Doran TM; Anderson EA; Nilsson BL Probing Aromatic, Hydrophobic, and Steric Effects on the Self-assembly of an Amyloid-β Fragment Peptide. Mol. BioSyst 2011, 7, 486–496. [DOI] [PubMed] [Google Scholar]
- (358).Auer S; Ricchiuto P; Kashchiev D Two-step Nucleation of Amyloid Fibrils: Omnipresent or Not? J. Mol. Biol 2012, 422, 723–730. [DOI] [PubMed] [Google Scholar]
- (359).Conicella AE; Dignon GL; Zerze GH; Schmidt HB; D’Ordine AM; Kim YC; Rohatgi R; Ayala YM; Mittal J; Fawzi NL TDP-43 α-helical Structure Tunes Liquid-liquid Phase Separation and Function. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 5883–5894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (360).Adachi M; Noji M; So M; Sasahara K; Kardos J; Naiki H; Goto Y Aggregation Phase Diagram of β2 Microglobulin Reveal Temperature and Salt Effects on Competitive Formation of Amyloids versus Amorphous Aggregates. J. Biol. Chem 2018, 293, 14775–14785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (361).Adachi M; So M; Sakurai K; Kardos J; Goto Y Supersaturation-Limited and Unlimited Phase Transitions Compete to Produce the Pathway Complexity in Amyloid Formation. J. Biol. Chem 2015, 290, 18134–18145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Sawada M; Yamaguchi K; Hirano M; Noji M; So M; Otzen DE; Kawata Y; Goto Y Amyloid Formation of α-synculein Based on the Solubility and Supersaturation Dependent Mechanism. Langmuir 2020, 36, 4671–4681. [DOI] [PubMed] [Google Scholar]
- (363).Wu C; Shea JE Coarse-Grained Models for Protein Aggregation. Curr. Opin. Struct. Biol 2011, 21, 209–220. [DOI] [PubMed] [Google Scholar]
- (364).Carballo-Pacheco M; Strodel B Advances in the Simulation of Protein Aggregation at the Atomistic Scale. J. Phys. Chem. B 2016, 120, 2991–2999. [DOI] [PubMed] [Google Scholar]
- (365).Saric A; Chebaro YC; Knowles TPJ; Frenkel D Crucial Role of Nonspecific Interactions in Amyloid Nucleation. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 17869–17874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (366).Chiricotto M; Melchionna S; Derreumaux P; Sterpone F Multiscale Aggregation of the Amyloid Aβ16–22 Peptide: From Disordered Coagulation and Lateral Branching to Amorphous Prefibrils. J. Phys. Chem. Lett 2019, 10, 1594–1599. [DOI] [PubMed] [Google Scholar]
- (367).Cao Y; Tang X; Yuan M; Han W Computational Studies of Protein Aggregation Mediated by Amyloid: Fibril Elongation and Secondary Nucleation. Prog. Mol. Biol. Transl. Sci 2020, 170, 461–504. [DOI] [PubMed] [Google Scholar]
- (368).Serio TR; Cashikar AG; Kowal AS; Sawicki GJ; Moslehi JJ; Serpell L; Arnsdorf M; Lindquist SL Nucleated Conformational Conversion and the Replication of Conformational Information by a Prion Determinant. Science 2000, 289, 1317–1321. [DOI] [PubMed] [Google Scholar]
- (369).Lee J; Culyba EK; Powers ET; Kelly JW Amyloid-β Forms Fibrils by Nucleated Conformational Conversion of Oligomers. Nat. Chem. Biol 2011, 7, 602–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (370).Michaels TCT; Ćarić A; Curk S; Bernfur K; Arosio P; Meisl G; Dear AJ; Cohen SIA; Dobson CM; Vendruscolo M; et al. Dynamics of Oligomer Populations Formed during the Aggregation of Alzheimer’s Aβ42 Peptide. Nat. Chem 2020, 12, 445–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (371).Pellarin R; Schuetz P; Guarnera E; Caflisch A Amyloid Fibril Polymorphism is under Kinetic Control. J. Am. Chem. Soc 2010, 132, 14960–14970. [DOI] [PubMed] [Google Scholar]
- (372).Bellesia G; Shea JE Effect of β-Sheet Propensity on Peptide Aggregation. J. Chem. Phys 2009, 130, 145103. [DOI] [PubMed] [Google Scholar]
- (373).Straub JE; Thirumalai D Toward a Molecular Theory of Early and Late Events in Monomer to Amyloid Fibril Formation. Annu. Rev. Phys. Chem 2011, 62, 437–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (374).Tran TT; Nguyen PH; Derreumaux P Lattice Model for Amyloid Peptides: OPEP Force Field Parametrization and Applications to the Nucleus Size of Alzheimer’s Peptides. J. Chem. Phys 2016, 144, 205103. [DOI] [PubMed] [Google Scholar]
- (375).Massi F; Straub J Energy Landscape Theory for Alzheimer’s Amyloid β-Peptide Fibril Elongation. Proteins: Struct., Funct., Genet 2001, 42, 217–229. [DOI] [PubMed] [Google Scholar]
- (376).Brown S; Fawzi NJ; Head-Gordon T Coarse-Grained Sequences for Protein Folding and Design. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 10712–10717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (377).Han W; Schulten K Fibril Elongation by Aβ(17–42): Kinetic Network Analysis of Hybrid-Resolution Molecular Dynamics Simulations. J. Am. Chem. Soc 2014, 136, 12450–12460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (378).Reddy G; Straub JE; Thirumalai D Dynamics of Locking of Peptides onto Growing Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 11948–11953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Schwierz N; Frost CV; Geissler PL; Zacharias M Dynamics of Seeded Aβ40-Fibril Growth from Atomistic Molecular Dynamics Simulations: Kinetic Trapping and Reduced Water Mobility in the Locking Step. J. Am. Chem. Soc 2016, 138, 527–539. [DOI] [PubMed] [Google Scholar]
- (380).Rojas AV; Liwo A; Scheraga HA Molecular Dynamics with the United-Residue Force Field: Ab Initio Folding Simulations of Multichain Proteins. J. Phys. Chem. B 2007, 111, 293–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Roder K; Wales DJ Energy Landscapes for the Aggregation of Aβ17–42. J. Am. Chem. Soc 2018, 140, 4018–4027. [DOI] [PubMed] [Google Scholar]
- (382).Jia Z; Schmit JD; Chen J Amyloid Assembly is Dominated by Misregistered Kinetic Traps on an Unbiased Energy Landscape. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 10322–10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (383).Saric A.đe.; Buell AK; Meisl G; Michaels TCT; Dobson CM; Linse S; Knowles TPJ; Frenkel D Physical Determinants of the Self-Replication of Protein Fibrils. Nat. Phys 2016, 12, 874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (384).Cohen SIA; Linse S; Luheshi LM; Hellstrand E; White DA; Rajah L; Otzen DE; Vendruscolo M; Dobson CM; Knowles TPJ Proliferation of Amyloid-β42 Aggregates Occurs through a Secondary Nucleation Mechanism. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 9758–9763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Ermak DL; McCammon JA Brownian Dynamics with Hydrodynamic Interactions. J. Chem. Phys 1978, 69, 1352. [Google Scholar]
- (386).Malevanets A; Kapral R Mesoscopic Model for Solvent Dynamics. J. Chem. Phys 1999, 110, 8605–8613. [Google Scholar]
- (387).Ahlrichs P; Dünweg B Simulation of a Single Polymer Chain in Solution by Combining Lattice Boltzmann and Molecular Dynamics. J. Chem. Phys 1999, 111, 8225–8239. [Google Scholar]
- (388).Frembgen-Kesner T; Elcock AH Striking Effects of Hydrodynamic Interactions on the Simulated Diffusion and Folding of Proteins. J. Chem. Theory Comput 2009, 5, 242–256. [DOI] [PubMed] [Google Scholar]
- (389).Zegarra FC; Homouz D; Eliaz Y; Gasic AG; Cheung MS The Impact of Hydrodynamic Interactions on Protein Folding Rates Depends on Temperature. Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top 2018, 97, 032402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (390).Ando T; Skolnick J Crowding and Hydrodynamic Interactions Likely Dominate In Vivo Macromolecular Motion. Proc. Natl. Acad. Sci. U. S. A 2010, 107, 18457–18462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (391).Ando T; Skolnick J On the Importance of Hydrodynamic Interactions in Lipid Membrane Formation. Biophys. J 2013, 104, 96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (392).Zimm BH Dynamics of Polymer Molecules in Dilute Solution: Viscoelasticity, Flow Birefringence and Dielectric Loss. J. Chem. Phys 1956, 24, 269–278. [Google Scholar]
- (393).Sterpone F; Derreumaux P; Melchionna S Protein Simulations in Fluids: Coupling the OPEP Coarse-Grained Force Field with Hydrodynamics. J. Chem. Theory Comput 2015, 11, 1843–1853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (394).Chiricotto M; Melchionna S; Derreumaux P; Sterpone F Hydrodynamic Effects on β-Amyloid (16–22) Peptide Aggregation. J. Chem. Phys 2016, 145, 035102. [DOI] [PubMed] [Google Scholar]
- (395).Hill EK; Krebs B; Goodall DG; Howlett GJ; Dunstan DE Shear Flow Induces Amyloid Fibril Formation. Biomacromolecules 2006, 7, 10–13. [DOI] [PubMed] [Google Scholar]
- (396).Collins SR; Douglass A; Vale RD; Weissman JS Mechanism of Prion Propagation: Amyloid Growth Occurs by Monomer Addition. PLoS Biol 2004, 2, No. e321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Bekard IB; Dunstan DE Chapter 44 - The Effect of Shear Flow on Amyloid Fibril Formation and Morphology. Bio-nanoimaging. Protein Misfolding and Aggregation 2014, 503–513. [Google Scholar]
- (398).Kinoshita M; Lin Y; Dai I; Okumura M; Markova N; Ladbury JE; Sterpone F; Lee Y-H Energy Landscape of Polymorphic Amyloid Generation of β2-Microglobulin Revealed by Calorimetry. Chem. Commun 2018, 54, 7995–7998. [DOI] [PubMed] [Google Scholar]
- (399).Jaspe J; Hagen SJ Do Protein Molecules Unfold in a Simple Shear Flow? Biophys. J 2006, 91, 3415–3424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Ashton L; Dusting J; Imomoh E; Balabani S; Blanch EW Susceptibility of Different Proteins to Flow-Induced Conformational Changes Monitored with Raman Spectroscopy. Biophys. J 2010, 98, 707–714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).Sterpone F; Derreumaux P; Melchionna S Molecular Mechanism of Protein Unfolding under Shear: A Lattice Boltzmann Molecular Dynamics Study. J. Phys. Chem. B 2018, 122, 1573–1579. [DOI] [PubMed] [Google Scholar]
- (402).Languin-Cattoën O; Melchionna S; Derreumaux P; Stirnemann G; Sterpone F Three Weaknesses for Three Perturbations: Comparing Protein Unfolding Under Shear, Force, and Thermal Stresses. J. Phys. Chem. B 2018, 122, 11922–11930. [DOI] [PubMed] [Google Scholar]
- (403).Trumbore CN Shear-Induced Amyloid Formation of IDPs in the Brain. Prog. Mol. Biol. Transl. Sci 2019, 166, 225–309. [DOI] [PubMed] [Google Scholar]
- (404).Bai J; Liu M; Pielak GJ; Li C Macromolecular and Small Molecular Crowding Have Similar Effects on α-Synuclein Structure. ChemPhysChem 2017, 18, 55–58. [DOI] [PubMed] [Google Scholar]
- (405).Banerjee PR; Moosa MM; Deniz AA Two-Dimensional Crowding Uncovers a Hidden Conformation of α-Synuclein. Angew. Chem., Int. Ed 2016, 55, 12789–12792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (406).Zhou Z; Fan JB; Zhu HL; Shewmaker F; Yan X; Chen X; Chen J; Xiao GF; Guo L; Liang Y Crowded Cell-like Environment Accelerates the Nucleation Step of Amyloidogenic Protein Misfolding. J. Biol. Chem 2009, 284, 30148–30158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Monteith WB; Cohen RD; Smith AE; Guzman-Cisneros E; Pielak GJ Quinary Structure Modulates Protein Stability in Cells. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 1739–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (408).Gnutt D; Timr S; Ahlers J; Ko B; Manderfeld E; Heyden M; Sterpone F; Ebbinghaus S Stability Effect of Quinary Interactions Reversed by Single Point Mutations. J. Am. Chem. Soc 2019, 141, 4660–4669. [DOI] [PubMed] [Google Scholar]
- (409).Lang L; Zetterstrom P; Brannstrom T; Marklund SL; Danielsson J; Oliveberg M SOD1 Aggregation in ALS Mice Shows Simplistic Test Tube Behavior. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 9878–9883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (410).Büning S; Sharma A; Vachharajani S; Newcombe E; Ormsby A; Gao M; Gnutt D; Vöpel T; Hatters DM; Ebbinghaus S Conformational Dynamics and Self-Association of Intrinsically Disordered Huntingtin Exon 1 in Cells. Phys. Chem. Chem. Phys 2017, 19, 10738–10747. [DOI] [PubMed] [Google Scholar]
- (411).Li X; Mehler EL Simulation of Molecular Crowding Effects on an Alzheimer’s α-Amyloid Peptide. Cell Biochem. Biophys 2006, 46, 123–141. [DOI] [PubMed] [Google Scholar]
- (412).Nguemaha V; Qin S; Zhou HX Atomistic Modeling of Intrinsically Disordered Proteins Under Polyethylene Glycol Crowding: Quantitative Comparison with Experimental Data and Implication of Protein-Crowder Attraction. J. Phys. Chem. B 2018, 122, 11262–11270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (413).Fagerberg E; Lenton S; Skepö M Evaluating Models of Varying Complexity of Crowded Intrinsically Disordered Protein Solutions Against SAXS. J. Chem. Theory Comput 2019, 15, 6968–6983. [DOI] [PubMed] [Google Scholar]
- (414).Magno A; Caflisch A; Pellarin R Crowding Effects on Amyloid Aggregation Kinetics. J. Phys. Chem. Lett 2010, 1, 3027–3032. [Google Scholar]
- (415).Timr S; Gnutt D; Ebbinghaus S; Sterpone F The Unfolding Journey of Superoxide Dismutase 1 Barrels under Crowding: Atomistic Simulations Shed Light on Intermediate States and Their Interactions with Crowders. J. Phys. Chem. Lett 2020, 11, 4206–4212. [DOI] [PubMed] [Google Scholar]
- (416).Weber OC; Uversky VN How Accurate are your Simulations? Effects of Confined Aqueous Volume and AMBER FF99SB and CHARMM22/CMAP Force Field Parameters on Structural Ensembles of Intrinsically Disordered Proteins: Amyloid-β42 in water. Intrinsically Disord. Proteins 2017, 5, No. e1377813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (417).Carballo-Pacheco M; Strodel B Comparison of Force Fields for Alzheimer’s: A Case Study for Intrinsically Disordered Proteins. Protein Sci 2017, 26, 174–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Krupa P; Huy PDQ; Li MS Properties of Monomeric Aβ42 Probed by Different Sampling Methods and Force Fields: Role of Energy Components. J. Chem. Phys 2019, 151, 055101. [Google Scholar]
- (419).Rosenman DJ; Wang C; García AE Characterization of Aβ Monomers through the Convergence of Ensemble Properties among Simulations with Multiple Force Fields. J. Phys. Chem. B 2016, 120, 259–277. [DOI] [PubMed] [Google Scholar]
- (420).Lincoff J; Sasmal S; Head-Gordon T The Combined Force Field-sampling Problem in Simulations of Disordered Amyloid-β Peptides. J. Chem. Phys 2019, 150, 104108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (421).Linh NH; Minh Thu TT; Tu L; Hu C-K; Li MS Impact of Mutations at C-Terminus on Structures and Dynamics of Aβ40 and Aβ42: A Molecular Simulation Study. J. Phys. Chem. B 2017, 121, 4341–4354. [DOI] [PubMed] [Google Scholar]
- (422).Nguyen HL; Thi Minh Thu T; Truong PM; Lan PD; Man VH; Nguyen PH; Tu LA; Chen Y-C; Li MS Aβ41 Aggregates More Like Aβ40 than Like Aβ42: In Silico and in Vitro Study. J. Phys. Chem. B 2016, 120, 7371–7379. [DOI] [PubMed] [Google Scholar]
- (423).Yan Y; Wang C Aβ42 is More Rigid than Aβ40 at the C Terminus: Implications for Aβ Aggregation and Toxicity. J. Mol. Biol 2006, 364, 853–862. [DOI] [PubMed] [Google Scholar]
- (424).Kirkitadze MD; Condron MM; Teplow DB Identification and Characterization of Key Kinetic Intermediates in amyloid β-protein Fibrillogenesis. J. Mol. Biol 2001, 312, 1103–1119. [DOI] [PubMed] [Google Scholar]
- (425).Roychaudhuri R; Yang M; Deshpande A; Cole GM; Frautschy S; Lomakin A; Benedek GB; Teplow DB C-Terminal Turn Stability Determines Assembly Differences between Aβ40 and Aβ42. J. Mol. Biol 2013, 425, 292–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (426).Bhattacharya S; Xu L; Thompson D Molecular Simulations Reveal Terminal Group Mediated Stabilization of Helical Conformers in Both Amyloid-β42 and α-Synuclein. ACS Chem. Neurosci 2019, 10, 2830–2842. [DOI] [PubMed] [Google Scholar]
- (427).Shea D; Hsu C-C; Bi TM; Paranjapye N; Childers MC; Cochran J; Tomberlin CP; Wang L; Paris D; Zonderman J; et al. α-Sheet Secondary Structure in amyloid β-peptide Drives Aggregation and Toxicity in Alzheimer’s Disease. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 8895–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Meng F; Bellaiche MMJ; Kim J-Y; Zerze GH; Best RB; Chung HS Highly Disordered Amyloid-β Monomer Probed by Single-Molecule FRET and MD Simulation. Biophys. J 2018, 114, 870–884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Schuler B; Soranno A; Hofmann H; Nettels D Single-Molecule FRET Spectroscopy and the Polymer Physics of Unfolded and Intrinsically Disordered Proteins. Annu. Rev. Biophys 2016, 45, 207–231. [DOI] [PubMed] [Google Scholar]
- (430).Granata D; Baftizadeh F; Habchi J; Galvagnion C; De Simone A; Camilloni C; Laio A; Vendruscolo M The Inverted Free Energy Landscape of an Intrinsically Disordered Peptide by Simulations and Experiments. Sci. Rep 2015, 5, 15449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (431).Li MS; Co NT; Reddy G; Hu CK; Straub JE; Thirumalai D Factors Governing Fibrillogenesis of Polypeptide Chains Revealed by Lattice Models. Phys. Rev. Lett 2010, 105, 218101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (432).Chiti F; Stefani M; Taddei N; Ramponi G; Dobson CM Rationalization of the Effects of Mutations on Peptide and Protein Aggregation Rates. Nature 2003, 424, 805. [DOI] [PubMed] [Google Scholar]
- (433).Chong SH; Ham S Interaction with the Surrounding Water Plays a Key role in Determining the Aggregation Propensity of Proteins. Angew. Chem 2014, 126, 4042–4045. [DOI] [PubMed] [Google Scholar]
- (434).Aggarwal L; Biswas P Effect of Alzheimer’s Disease Causative and Protective Mutations on the Hydration Environment of Amyloid-β. J. Phys. Chem. B 2020, 124, 2311–2322. [DOI] [PubMed] [Google Scholar]
- (435).Thu TTM; Co NT; Tu LA; Li MS Aggregation Rate of Amyloid beta Peptide is Controlled by beta-content in Monomeric State. J. Chem. Phys 2019, 150, 225101. [DOI] [PubMed] [Google Scholar]
- (436).Nam HB; Kouza M; Zung H; Li MS Relationship between Population of the Fibril-prone Conformation in the Monomeric State and Oligomer Formation Times of Peptides: Insights from all-atom Simulations. J. Chem. Phys 2010, 132, 165104. [DOI] [PubMed] [Google Scholar]
- (437).Chakraborty D; Straub JE; Thirumalai D Differences in the Free Energies between the Excited States of Aβ40 and Aβ42 Monomers Encode their Distinct Aggregation Propensities. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 19926–19937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Frigori RB; Barroso da Silva FL; Carvalho PPD; Alves NA Occurrence of Biased Conformations as Precursors of Assembly States in Fibril Elongation of Amyloid-β Fibril Variants: An In Silico Study. J. Phys. Chem. B 2020, 124, 2798–2805. [DOI] [PubMed] [Google Scholar]
- (439).Nagel-Steger L; Owen MC; Strodel B An Account of Amyloid Oligomers: Facts and Figures Obtained from Experiments and Simulations. ChemBioChem 2016, 17, 657–676. [DOI] [PubMed] [Google Scholar]
- (440).Zhang Y; Hashemi M; Lv Z; Lyubchenko YL Self-assembly of the Full-length Amyloid Aβ42 Protein in Dimers. Nanoscale 2016, 8, 18928–18937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Maity S; Lyubchenko YL Force Clamp Approach for Characterization of Nano-assembly in Amyloid beta 42 Dimer. Nanoscale 2019, 11, 12259–12265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Mehrazma B; Rauk A Exploring Amyloid-β Dimer Structure Using Molecular Dynamics Simulations. J. Phys. Chem. A 2019, 123, 4658–4670. [DOI] [PubMed] [Google Scholar]
- (443).Das P; Chacko AR; Belfort G Alzheimer’s Protective Cross-Interaction between Wild-Type and A2T Variants Alters Aβ42 Dimer Structure. ACS Chem. Neurosci 2017, 8, 606–618. [DOI] [PubMed] [Google Scholar]
- (444).Man VH; Nguyen PH; Derreumaux P Conformational Ensembles of the Wild-Type and S8C Aβ1–42 Dimers. J. Phys. Chem. B 2017, 121, 2434–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (445).Man VH; Nguyen PH; Derreumaux P High-Resolution Structures of the Amyloid-β 1–42 Dimers from the Comparison of Four Atomistic Force Fields. J. Phys. Chem. B 2017, 121, 5977–5987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (446).Tarus B; Tran TT; Nasica-Labouze J; Sterpone F; Nguyen PH; Derreumaux P Structures of the Alzheimer’s Wild-Type Aβ1–40 Dimer from Atomistic Simulations. J. Phys. Chem. B 2015, 119, 10478–10487. [DOI] [PubMed] [Google Scholar]
- (447).Nguyen PH; Sterpone F; Pouplana R; Derreumaux P; Campanera JM Dimerization Mechanism of Alzheimer Aβ40 Peptides: The High Content of Intrapeptide-Stabilized Conformations in A2V and A2T Heterozygous Dimers Retards Amyloid Fibril Formation. J. Phys. Chem. B 2016, 120, 12111–12126. [DOI] [PubMed] [Google Scholar]
- (448).Nguyen PH; Sterpone F; Campanera JM; Nasica-Labouze J; Derreumaux P Impact of the A2V Mutation on the Heterozygous and Homozygous Aβ1–40 Dimer Structures from Atomistic Simulations. ACS Chem. Neurosci 2016, 7, 823–832. [DOI] [PubMed] [Google Scholar]
- (449).Cao Y; Jiang X; Han W Self-Assembly Pathways of β-Sheet-Rich Amyloid-β(1–40) Dimers: Markov State Model Analysis on Millisecond Hybrid-Resolution Simulations. J. Chem. Theory Comput 2017, 13, 5731–5744. [DOI] [PubMed] [Google Scholar]
- (450).Narayan B; Yuan Y; Fathizadeh A; Elber R; Buchete N-V Chapter Five - Long-time methods for molecular dynamics simulations: Markov State Models and Milestoning. In Progress in Molecular Biology and Translational Science; Strodel B; Barz B, Eds.; Academic Press: 2020, Vol. 170, pp 215–237. [DOI] [PubMed] [Google Scholar]
- (451).Nguyen HL; Krupa P; Hai NM; Linh HQ; Li MS Structure and Physicochemical Properties of the Aβ42 Tetramer: Multiscale Molecular Dynamics Simulations. J. Phys. Chem. B 2019, 123, 7253–7269. [DOI] [PubMed] [Google Scholar]
- (452).Bernstein SL; Dupuis NF; Lazo ND; Wyttenbach T; Condron MM; Bitan G; Teplow DB; Shea J-E; Ruotolo BT; Robinson CV; et al. Amyloid-β protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer’s Disease. Nat. Chem 2009, 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Zhang S; Fox DM; Urbanc B Insights into Formation and Structure of Aβ Oligomers Cross-Linked via Tyrosines. J. Phys. Chem. B 2017, 121, 5523–5535. [DOI] [PubMed] [Google Scholar]
- (454).Gu M; Bode DC; Viles JH Copper Redox Cycling Inhibits Aβ Fibre Formation and Promotes Fibre Fragmentation, while Generating a Dityrosine Aβ Dimer. Sci. Rep 2018, 8, 16190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).La Penna G; Li M S Computational Models Explain how Copper Binding to amyloid-β Peptide Oligomers Enhances Oxidative Pathways. Phys. Chem. Chem. Phys 2019, 21, 8774–8784. [DOI] [PubMed] [Google Scholar]
- (456).Voelker MJ; Barz B; Urbanc B Fully Atomistic Aβ40 and Aβ42 Oligomers in Water: Observation of Porelike Conformations. J. Chem. Theory Comput 2017, 13, 4567–4583. [DOI] [PubMed] [Google Scholar]
- (457).Do TD; LaPointe NE; Nelson R; Krotee P; Hayden EY; Ulrich B; Quan S; Feinstein SC; Teplow DB; Eisenberg D; et al. Amyloid β-Protein C-Terminal Fragments: Formation of Cylindrins and β-Barrels. J. Am. Chem. Soc 2016, 138, 549–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (458).Xi W; Vanderford EK; Hansmann UH E. Out-of-Register Aβ42 Assemblies as Models for Neurotoxic Oligomers and Fibrils. J. Chem. Theory Comput 2018, 14, 1099–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (459).Nguyen PH; Campanera JM; Ngo ST; Loquet A; Derreumaux P Tetrameric Aβ40 and Aβ42 β-Barrel Structures by Extensive Atomistic Simulations. II. In Aqueous Solution. J. Phys. Chem. B 2019, 123, 6750–6756. [DOI] [PubMed] [Google Scholar]
- (460).Davtyan A; Schafer NP; Zheng W; Clementi C; Wolynes PG; Papoian GA AWSEM-MD: Protein Structure Prediction Using Coarse-Grained Physical Potentials and Bioinformatically Based Local Structure Biasing. J. Phys. Chem. B 2012, 116, 8494–8503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (461).Zheng W; Tsai M-Y; Chen M; Wolynes PG Exploring the Aggregation Free Energy Landscape of the amyloid-β Protein (1–40). Proc. Natl. Acad. Sci. U. S. A 2016, 113, 11835–11840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Zheng W; Tsai M-Y; Wolynes PG Comparing the Aggregation Free Energy Landscapes of Amyloid Beta(1–42) and Amyloid Beta(1–40). J. Am. Chem. Soc 2017, 139, 16666–16676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (463).Barz B; Liao Q; Strodel B Pathways of Amyloid-β Aggregation Depend on Oligomer Shape. J. Am. Chem. Soc 2018, 140, 319–327. [DOI] [PubMed] [Google Scholar]
- (464).Man VH; He X; Ji B; Liu S; Xie X-Q; Wang J Molecular Mechanism and Kinetics of Amyloid-β42 Aggregate Formation: A Simulation Study. ACS Chem. Neurosci 2019, 10, 4643–4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Robustelli P; Piana S; Shaw DE Developing a Molecular Dynamics Force Field for both Folded and Disordered Protein States. Proc. Natl. Acad. Sci. U. S. A 2018, 115, E4758–E4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Saha S; Deep S Protein Aggregation: Elucidation of the Mechanism and Determination of Associated Thermodynamic and Kinetic Parameters. Curr. Phys. Chem 2014, 4, 114–136. [Google Scholar]
- (467).Sharma B; Ranganathan SV; Belfort G Weaker N-Terminal Interactions for the Protective over the Causative Aβ Peptide Dimer Mutants. ACS Chem. Neurosci 2018, 9, 1247–125. [DOI] [PubMed] [Google Scholar]
- (468).Li H; Salimi A; Lee JY Intrinsic Origin of Amyloid Aggregation: Collective Effects of the Mutation and Tautomerism of Histidine. ACS Chem. Neurosci 2019, 10, 4729–4734. [DOI] [PubMed] [Google Scholar]
- (469).Liu C; Zhao W; Xing X; Shi H; Kang B; Liu H; Li P; Ai H An Original Monomer Sampling from a Ready-Made Aβ42 NMR Fibril Suggests a Turn-β-Strand Synergetic Seeding Mechanism. ChemPhysChem 2019, 20, 1649–1660. [DOI] [PubMed] [Google Scholar]
- (470).Hashemi M; Zhang Y; Lv Z; Lyubchenko YL Spontaneous self-assembly of Amyloid β (1–40) into Dimers. Nanoscale Adv 2019, 1, 3892–3899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (471).Press-Sandler O; Miller Y Distinct Primary Nucleation of Polymorphic Aβ Dimers Yields to Distinguished Fibrillation Pathways. ACS Chem. Neurosci 2019, 10, 4407–4413. [DOI] [PubMed] [Google Scholar]
- (472).Cremades N; Chen SW; Dobson CM Structural Characteristics of α-Synuclein Oligomers. Int. Rev. Cell Mol. Biol 2017, 329, 79–143. [DOI] [PubMed] [Google Scholar]
- (473).Ilie IM; Den Otter WK; Briels WJ A Coarse Grained Protein Model with Internal Degrees of Freedom. Application to α-synuclein Aggregation. J. Chem. Phys 2016, 144, 085103. [DOI] [PubMed] [Google Scholar]
- (474).Eliezer D Biophysical Characterization of Intrinsically Disordered Proteins David. Curr. Opin. Struct. Biol 2009, 19, 23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Dokholyan NV Experimentally-driven Protein Structure Modeling. J. Proteomics 2020, 220, 103777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (476).Piana S; Donchev AG; Robustelli P; Shaw DE Water Dispersion Interactions strongly Influence Simulated Structural Properties of Disordered Protein States. J. Phys. Chem. B 2015, 119, 5113–5123. [DOI] [PubMed] [Google Scholar]
- (477).Ramis R; Ortega-Castro J; Casasnovas R; Mariño L; Vilanova B; Adrover M; Frau J A Coarse-Grained Molecular Dynamics Approach to the Study of the Intrinsically Disordered Protein α-Synuclein. J. Chem. Inf. Model 2019, 59, 1458–1471. [DOI] [PubMed] [Google Scholar]
- (478).Zhang Y; Liu H; Yang S; Luo R; Chen HF Well-Balanced Force Field ff03 CMAP for Folded and Disordered Proteins. J. Chem. Theory Comput 2019, 15, 6769–6780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (479).Latham AP; Zhang B Maximum Entropy Optimized Force Field for Intrinsically Disordered Proteins. J. Chem. Theory Comput 2020, 16, 773–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (480).Song D; Liu H; Luo R; Chen H-F Environment-Specific Force Field for Intrinsically Disordered and Ordered Proteins. J. Chem. Inf. Model 2020, 60, 2257–2267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Yu L; Li DW; Brüschweiler R Balanced Amino-Acid-Specific Molecular Dynamics Force Field for the Realistic Simulation of Both Folded and Disordered Proteins. J. Chem. Theory Comput 2020, 16, 1311–1318. [DOI] [PubMed] [Google Scholar]
- (482).Dedmon MM; Lindorff-Larsen K; Christodoulou J; Vendruscolo M; Dobson CM Mapping Long-range Interactions in α-synuclein Using Spin-label NMR and Ensemble Molecular Dynamics Simulations. J. Am. Chem. Soc 2005, 127, 476–477. [DOI] [PubMed] [Google Scholar]
- (483).Wu K-H; Weinstock DS; Narayanan C; Levy RM; Baum J Structural Reorganization of α-synuclein at Low pH Observed by NMR and REMD Simulations. J. Mol. Biol 2009, 391, 784–796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (484).Ullman O; Fisher CK; Stultz CM Explaining the Structural Plasticity of α-synuclein. J. Am. Chem. Soc 2011, 133, 19536–19546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (485).Nath A; Sammalkorpi M; DeWitt DC; Trexler AJ; Elbaum-Garfinkle S; O’Hern CS; Rhoades E The Conformational Ensembles of α-synuclein and tau: Combining Single-molecule FRET and Simulations. Biophys. J 2012, 103, 1940–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (486).Narayanan C; Weinstock DS; Wu KP; Baum J; Levy RM Investigation of the Polymeric Properties of α-Synuclein and Comparison with NMR Experiments: A Replica Exchange Molecular Dynamics Study. J. Chem. Theory Comput 2012, 8, 3929–3942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Ferrie JJ; Haney CM; Yoon J; Pan B; Lin YC; Fakhraai Z; Rhoades E; Nath A; Petersson EJ Using a FRET Library with Multiple Probe Pairs To Drive Monte Carlo Simulations of α-Synuclein. Biophys. J 2018, 114, 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (488).Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH Conformational Ensemble of Native α-synuclein in Solution as Determined by Short-distance Crosslinking Constraint-guided Discrete Molecular Dynamics Simulations. PLoS Comput. Biol 2019, 15, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (489).Proctor EA; Ding F; Dokholyan NV Discrete Molecular Dynamics. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2011, 1, 80–92. [Google Scholar]
- (490).Dokholyan NV; Buldyrev SV; Stanley HE; Shakhnovich EI Discrete Molecular Dynamics Studies of the Folding of a Protein-like Model. Folding Des 1998, 3, 577–587. [DOI] [PubMed] [Google Scholar]
- (491).Shirvanyants D; Ding F; Tsao D; Ramachandran S; Dokholyan NV Discrete Molecular Dynamics: An Efficient and Versatile Simulation Method for Fine Protein Characterization. J. Phys. Chem. B 2012, 116, 8375–8382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (492).Brodie NI; Popov KI; Petrotchenko EV; Dokholyan NV; Borchers CH Solving Protein Structures using Short-distance Cross-linking Constraints as a Guide for Discrete Molecular Dynamics Simulations. Sci. Adv 2017, 3, No. e1700479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Ding F; Jha RK Scaling Behavior and Structure of Denatured Proteins. Structure 2005, 13, 1047–1054. [DOI] [PubMed] [Google Scholar]
- (494).Chen Y; Campbell SL; Dokholyan NV Deciphering Protein Dynamics from NMR data Using Explicit Structure Sampling and Selection. Biophys. J 2007, 93, 2300–2306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Churchill CDM; Healey MA; Preto J; Tuszynski JA; Woodside MT Probing the Basis of α-Synuclein Aggregation by Comparing Simulations to Single-Molecule Experiments. Biophys. J 2019, 117, 1125–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (496).Zhang Y; Hashemi M; Lv Z; Williams B; Popov KI; Dokholyan NV; Lyubchenko YL High-speed Atomic Force Microscopy Reveals Structural Dynamics of α-synuclein Monomers and Dimers. J. Chem. Phys 2018, 148, 123322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (497).Dułak D; Gadzała M; Banach M; Konieczny L; Roterman I Alternative Structures of α-synuclein. Molecules 2020, 25, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (498).Rossetti G; Musiani F; Abad E; Dibenedetto D; Mouhib H; Fernandez CO; Carloni P Conformational Ensemble of Human α-synuclein Physiological Form Predicted by Molecular Simulations. Phys. Chem. Chem. Phys 2016, 18, 5702–5706. [DOI] [PubMed] [Google Scholar]
- (499).Graen T; Klement R; Grupi A; Haas E; Grubmuüller H Transient Secondary and Tertiary Structure Formation Kinetics in the Intrinsically Disordered State of α-Synuclein from Atomistic Simulations. ChemPhysChem 2018, 19, 2507–2511. [DOI] [PubMed] [Google Scholar]
- (500).Balupuri A; Choi KE; Kang NS Computational Insights into the Role of α-strand/sheet in Aggregation of α-synuclein. Sci. Rep 2019, 9, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Healey MA; Woodside MT; Tuszynski JA Phase Transitions and Structure Analysis in wild-type, A30P, E46K, and A53T Mutants of α-synuclein. Eur. Biophys. J 2016, 45, 355–364. [DOI] [PubMed] [Google Scholar]
- (502).Jónsson SA; Mohanty S; Irbäck A Distinct Phases of Free α-synuclein - A Monte Carlo Study. Proteins: Struct., Funct., Genet 2012, 80, 2169–2177. [DOI] [PubMed] [Google Scholar]
- (503).Yu H; Han W; Ma W; Schulten K Transient β-hairpin Formation in alpha-synuclein Monomer Revealed by Coarse-grained Molecular Dynamics Simulation. J. Chem. Phys 2015, 143, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Chwastyk M; Cieplak M Conformational Biases of α-Synuclein and Formation of Transient Knots. J. Phys. Chem. B 2020, 124, 11–19. [DOI] [PubMed] [Google Scholar]
- (505).Weinreb PH; Zhen W; Poon AW; Conway KA; Lansbury PT NACP, a Protein Implicated in Alzheimer’s Disease and Learning, is Natively Unfolded. Biochemistry 1996, 35, 13709–13715. [DOI] [PubMed] [Google Scholar]
- (506).Davidson WS; Jonas A; Clayton DF; George JM Stabilization of α-Synuclein Secondary Structure upon Binding to Synthetic Membranes. J. Biol. Chem 1998, 273, 9443–9449. [DOI] [PubMed] [Google Scholar]
- (507).Schwalbe M; Ozenne V; Bibow S; Jaremko M; Jaremko L; Gajda M; Jensen MR; Biernat J; Becker S; Mandelkow E; et al. Predictive Atomic Resolution Descriptions of Intrinsically Disordered hTau40 and α-synuclein in solution from NMR and Small Angle Scattering. Structure 2014, 22, 238–249. [DOI] [PubMed] [Google Scholar]
- (508).Dibenedetto D; Rossetti G; Caliandro R; Carloni P A Molecular Dynamics Simulation-based Interpretation of Nuclear Magnetic Resonance Multidimensional Heteronuclear Spectra of α-synuclein·dopamine Adducts. Biochemistry 2013, 52, 6672–6683. [DOI] [PubMed] [Google Scholar]
- (509).Mane JY; Stepanova M Understanding the Dynamics of Monomeric, Dimeric, and Tetrameric α-synuclein Structures in Water. FEBS Open Bio 2016, 6, 666–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Sahu KK; Woodside MT; Tuszynski JA A-Synuclein Dimer Structures Found From Computational Simulations. Biochimie 2015, 116, 133–140. [DOI] [PubMed] [Google Scholar]
- (511).Cote Y; Delarue P; Scheraga HA; Senet P; Maisuradze GG From a Highly Disordered to a Metastable State: Uncovering Insights of α-Synuclein. ACS Chem. Neurosci 2018, 9, 1051–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (512).Schaffert LN; Carter WG Do Post-Translational Modifications Influence Protein Aggregation in Neurodegenerative Diseases: A Systematic Review. Brain Sci 2020, 10, 232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (513).Miao J; Shi R; Li L; Chen F; Zhou Y; Tung YC; Hu W; Gong CX; Iqbal K; Liu F Pathological Tau From Alzheimer’s Brain Induces Site-Specific Hyperphosphorylation and SDS- and Reducing Agent-Resistant Aggregation of Tau in vivo. Front. Aging Neurosci 2019, 11, 34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (514).Luo Y; Ma B; Nussinov R; Wei G Structural Insight into Tau Protein’s Paradox of Intrinsically Disordered Behavior, Self-Acetylation Activity, and Aggregation. J. Phys. Chem. Lett 2014, 5, 3026–3021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Carlomagno Y; Chung DC; Yue M; Castanedes-Casey M; Madden BJ; Dunmore J; Tong J; DeTure M; Dickson DW; Petrucelli L; et al. An Acetylation-phosphorylation Switch that Regulates Tau Aggregation Propensity and Function. J. Biol. Chem 2017, 292, 15277–15286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (516).Prokopovich DV; Whittaker JW; Muthee MM; Ahmed A; Larini L Impact of Phosphorylation and Pseudophosphorylation on the Early Stages of Aggregation of the Microtubule-Associated Protein Tau. J. Phys. Chem. B 2017, 121, 2095–2103. [DOI] [PubMed] [Google Scholar]
- (517).Despres C; Di J; Cantrelle FX; Li Z; Huvent I; Chambraud B; Zhao J; Chen J; Chen S; Lippens G; et al. Major Differences between the Self-Assembly and Seeding Behavior of Heparin-Induced and in Vitro Phosphorylated Tau and Their Modulation by Potential Inhibitors. ACS Chem. Biol 2019, 14, 1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (518).Arakhamia T; Lee CE; Carlomagno Y; Duong DM; Kundinger SR; Wang K; Williams D; DeTure M; Dickson DW; Cook CN; et al. Posttranslational Modifications Mediate the Structural Diversity of Tauopathy Strains. Cell 2020, 180, 633–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (519).Despres C; Byrne C; Qi H; Cantrelle FX; Huvent I; Chambraud B; Baulieu EE; Jacquot Y; Landrieu I; Lippens G; et al. Identification of the Tau Phosphorylation Pattern that Drives its Aggregation. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 9080–9085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (520).Lyons AJ; Gandhi NS; Mancera RL Molecular Dynamics Simulation of the Phosphorylation-induced Conformational Changes of a Tau Peptide Fragment. Proteins: Struct., Funct., Genet 2014, 82, 1907–1918. [DOI] [PubMed] [Google Scholar]
- (521).Rani L; Mittal J; Mallajosyula SS Effect of Phosphorylation and O-GlcNAcylation on Proline-Rich Domains of Tau. J. Phys. Chem. B 2020, 124, 1909–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Haj-Yahya M; Gopinath P; Rajasekhar K; Mirbaha H; Diamond MI; Lashuel HA Site-Specific Hyperphosphorylation Inhibits, Rather than Promotes, Tau Fibrillization, Seeding Capacity, and Its Microtubule Binding. Angew. Chem., Int. Ed 2020, 59, 4059–4067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Barthelemy NR; Li Y; Joseph-Mathurin N; Gordon BA; Hassenstab J; Benzinger TLS; Buckles V; Fagan AM; Perrin RJ; Goate AM; et al. A Soluble Phosphorylated Tau Signature Links Tau, Amyloid and the Evolution of Stages of Dominantly Inherited Alzheimer’s Disease. Nat. Med 2020, 26, 398–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Chin AF; Toptygin D; Elam WA; Schrank TP; Hilser VJ Phosphorylation Increases Persistence Length and End-to-End Distance of a Segment of Tau Protein. Biophys. J 2016, 110, 362–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Naserkhaki R; Zamanzadeh S; Baharvand H; Nabavi SM; Pakdaman H; Shahbazi S; Vosough M; Ghaedi G; Barzegar A; Mirtorabi D; et al. cis pT231-Tau Drives Neurodegeneration in Bipolar Disorder. ACS Chem. Neurosci 2019, 10, 1214–1221. [DOI] [PubMed] [Google Scholar]
- (526).Shih HH; Tu C; Cao W; Klein A; Ramsey R; Fennell BJ; Lambert M; Shúilleabháin DN; Autin B; Kouranova E An Ultra-specific Avian Antibody to Phosphorylated Tau Protein Reveals a unique Mechanism for Phosphoepitope Recognition. J. Biol. Chem 2012, 287, 44425–44434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (527).Nakamura K; Zhou XZ; Lu KP Distinct Functions of Cis and Trans Phosphorylated Tau in Alzheimer’s Disease and their Therapeutic Implications. Curr. Mol. Med 2013, 13, 1098–1109. [DOI] [PubMed] [Google Scholar]
- (528).Wang JZ; Zhang Y Configuration-specific Immunotherapy Targeting cis pThr231-Pro232 Tau for Alzheimer Disease. J. Neurol. Sci 2015, 348, 253–255. [DOI] [PubMed] [Google Scholar]
- (529).Ahuja P; Cantrelle FX; Huvent I; Hanoulle X; Lopez J; Smet C; Wieruszeski JM; Landrieu I; Lippens G Proline Conformation in a Functional Tau Fragment. J. Mol. Biol 2016, 428, 79–91. [DOI] [PubMed] [Google Scholar]
- (530).Ikura T; Tochio N; Kawasaki R; Matsuzaki M; Narita A; Kikumoto M; Utsunomiya-Tate N; Tate SI; Ito N The Trans Ssomer of Tau Peptide is Prone to Aggregate, and the WW Domain of Pin1 Drastically Decreases its Aggregation. FEBS Lett 2018, 592, 3082–3091. [DOI] [PubMed] [Google Scholar]
- (531).Barman A; Hamelberg D Loss of Intramolecular Electrostatic Interactions and Limited Conformational Ensemble May Promote Self-association of cis-tau Peptide. Proteins: Struct., Funct., Genet 2015, 83, 436–444. [DOI] [PubMed] [Google Scholar]
- (532).Strang KH; Sorrentino ZA; Riffe CJ; Gorion KM; Vijayaraghavan N; Golde TE; Giasson BI Phosphorylation of Serine 305 in Tau Inhibits Aggregation. Neurosci. Lett 2019, 692, 187–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (533).Sandhu P; Naeem MM; Lu C; Kumarathasan P; Gomes J; Basak A Ser(422) Phosphorylation Blocks Human Tau Cleavage by Caspase-3: Biochemical Implications to Alzheimer’s Disease. Bioorg. Med. Chem. Lett 2017, 27, 642–653. [DOI] [PubMed] [Google Scholar]
- (534).Ait-Bouziad N; Chiki A; Limorenko G; Xiao S; Eliezer D; Lashuel HA Phosphorylation of the Overlooked Tyrosine 310 Regulates the Structure, Aggregation, and Microtubule- and Lipid-binding Properties of Tau. J. Biol. Chem 2020, 295, 7905–7922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Amar F; Sherman MA; Rush T; Larson M; Boyle G; Chang L; Gotz J; Buisson A; Lesne SE The amyloid-beta Aligomer Abeta*56 Induces Specific Alterations in Neuronal Signaling that lead to Tau Phosphorylation and Aggregation. Sci. Signaling 2017, 10, No. eaal2021. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (536).Wu H; Wei S; Huang Y; Chen L; Wang Y; Wu X; Zhang Z; Pei Y; Wang D Abeta Monomer Induces Phosphorylation of Tau at Ser-214 through beta2AR-PKA-JNK Signaling Pathway. FASEB J 2020, 34, 5092–5105. [DOI] [PubMed] [Google Scholar]
- (537).Guo JP; Arai T; Miklossy J; McGeer PL Abeta and Tau Form Soluble Complexes that May Promote Self Aggregation of both into the Insoluble Forms Observed in Alzheimer’s Disease. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 1953–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Ando K; Oka M; Ohtake Y; Hayashishita M; Shimizu S; Hisanaga S; Iijima KM Tau Phosphorylation at Alzheimer’s Disease-related Ser356 Contributes to tau stabilization when PAR-1/MARK Activity is Elevated. Biochem. Biophys. Res. Commun 2016, 478, 929–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (539).Derreumaux P; Man VH; Wang J; Nguyen PH Tau R3-R4 Domain Dimer of the Wild Type and Phosphorylated Ser356 Sequences. I. In Solution by Atomistic Simulations. J. Phys. Chem. B 2020, 124, 2975–2983. [DOI] [PubMed] [Google Scholar]
- (540).Lin YX; McCarty J; Rauch JN; Delaney KT; Kosik KS; Fredrickson GH; Shea JE; Han S Narrow Equilibrium Window for Complex Coacervation of Tau and RNA under Cellular Conditions. eLife 2019, 8, No. e42571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Wegmann S; Eftekharzadeh B; Tepper K; Zoltowska KM; Bennett RE; Dujardin S; Laskowski PR; MacKenzie D; Kamath T; Commins C; et al. Tau Protein liquid-liquid Phase Separation can Initiate Tau Aggregation. EMBO J 2018, 37, No. e98049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (542).Battisti A; Ciasca G; Grottesi A; Bianconi A; Tenenbaum A Temporary Secondary Structures in Tau, an Intrinsically Disordered Protein. Mol. Simul 2012, 38, 525–533. [Google Scholar]
- (543).Larini L; Gessel MM; LaPointe NE; Do TD; Bowers MT; Feinstein SC; Shea JE Initiation of Assembly of tau(273–284) and its Delta K280 Mutant: an Experimental and Computational study. Phys. Chem. Chem. Phys 2013, 15, 8916–8928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (544).Thirumalai D; Klimov DK; Dima RI Emerging ideas on the Molecular Basis of Protein and Peptide Aggregation. Curr. Opin. Struct. Biol 2003, 13, 146–159. [DOI] [PubMed] [Google Scholar]
- (545).Zhuravlev PI; Reddy G; Straub JE; Thirumalai D Propensity to Form Amyloid Fibrils Is Encoded as Excitations in the Free Energy Landscape of Monomeric Proteins. J. Mol. Biol 2014, 426, 2653–2666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Ganguly P; Do TD; Larini L; LaPointe NE; Sercel AJ; Shade MF; Feinstein SC; Bowers MT; Shea JE Tau Assembly: The Dominant Role of PHF6 (VQIVYK) in Microtubule Binding Region Repeat R3. J. Phys. Chem. B 2015, 119, 4582–4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (547).Eschmann NA; Georgieva ER; Ganguly P; Borbat PP; Rappaport MD; Akdogan Y; Freed JH; Shea JE; Han S Signature of an Aggregation-prone Conformation of Tau. Sci. Rep 2017, 7, 44739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Battisti A; Ciasca G; Grottesi A; Tenenbaum A Thermal Compaction of the Intrinsically Disordered Protein Tau: Entropic, Structural, and Hydrophobic Factors. Phys. Chem. Chem. Phys 2017, 19, 8435–8446. [DOI] [PubMed] [Google Scholar]
- (549).Ciasca G; Campi G; Battisti A; Rea G; Rodio M; Papi M; Pernot P; Tenenbaum A; Bianconi A Continuous Thermal Collapse of the Intrinsically Disordered Protein Tau Is Driven by Its Entropic Flexible Domain. Langmuir 2012, 28, 13405–13410. [DOI] [PubMed] [Google Scholar]
- (550).Li DW; Mohanty S; Irback A; Huo SH Formation and Growth of Oligomers: A Monte Carlo Study of an Amyloid Tau Fragment. PLoS Comput. Biol 2008, 4, No. e1000238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (551).Siddiqua A; Luo Y; Meyer V; Swanson MA; Yu X; Wei GH; Zheng J; Eaton GR; Ma BY; Nussinov R; Eaton SS; Margittai M Conformational Basis for Asymmetric Seeding Barrier in Filaments of Three- and Four-Repeat Tau. J. Am. Chem. Soc 2012, 134, 10271–10278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (552).Yu X; Luo Y; Dinkel P; Zheng J; Wei GH; Margittai M; Nussinov R; Ma BY Cross-seeding and Conformational Selection between Three- and Four-repeat Human Tau Proteins. J. Biol. Chem 2012, 287, 14950–14959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (553).Meyer V; Dinkel PD; Luo Y; Yu X; Wei GH; Zheng J; Eaton GR; Ma BY; Nussinov R; Eaton SS; Margittai M Single Mutations in Tau Modulate the Populations of Fibril Conformers through Seed Selection. Angew. Chem., Int. Ed 2014, 53, 1590–1593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (554).Matthes D; Gapsys V; Daebel V; de Groot BL Mapping the Conformational Dynamics and Pathways of Spontaneous Steric Zipper Peptide Oligomerization. PLoS One 2011, 6, No. e19129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Li XH; Dong XW; Wei GH; Margittai M; Nussinov R; Ma BY The Distinct Structural Preferences of tau Protein Repeat Domains. Chem. Commun 2018, 54, 5700–5703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (556).Levine ZA; Larini L; LaPointe NE; Feinstein SC; Shea JE Regulation and Aggregation of Intrinsically Disordered Peptides. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 2758–2763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (557).Fichou Y; Schiro G; Gallat FX; Laguri C; Moulin M; Combete J; Zamponi M; Hartlein M; Picart C; Mossou E; Lortat-Jacob H; Colletier JP; Tobias DJ; Weik M Hydration Water Mobility is Enhanced around Tau Amyloid Fibers. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 6365–6370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Ambadipudi S; Biernat J; Riedel D; Mandelkow E; Zweckstetter M Liquid-liquid Phase Separation of the Microtubule-binding Repeats of the Alzheimer-related protein Tau. Nat. Commun 2017, 8, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).Anderson P; Kedersha N RNA granules. J. Cell Biol 2006, 172, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (560).Wippich F; Bodenmiller B; Trajkovska MG; Wanka S; Aebersold R; Pelkmans L Dual Specificity Kinase DYRK3 Couples Stress Granule Condensation/Dissolution to mTORC1 Signaling. Cell 2013, 152, 791–805. [DOI] [PubMed] [Google Scholar]
- (561).Brangwynne CP; Eckmann CR; Courson DS; Rybarska A; Hoege C; Gharakhani J; Julicher F; Hyman AA Germline P Granules Are Liquid Droplets That Localize by Controlled Dissolution/Condensation. Science 2009, 324, 1729–1732. [DOI] [PubMed] [Google Scholar]
- (562).Molliex A; Temirov J; Lee J; Coughlin M; Kanagaraj AP; Kim HJ; Mittag T; Taylor JP Phase Separation by Low Complexity Domains Promotes Stress Granule Assembly and Drives Pathological Fibrillization. Cell 2015, 163, 123–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (563).Hyman AA; Weber CA; Juelicher F Liquid-Liquid Phase Separation in Biology. Annu. Rev. Cell Dev. Biol 2014, 30, 39–58. [DOI] [PubMed] [Google Scholar]
- (564).Murakami T; Qamar S; Lin JQ; Schierle GSK; Rees E; Miyashita A; Costa AR; Dodd RB; Chan FTS; Michel CH; et al. ALS/FTD Mutation-Induced Phase Transition of FUS Liquid Droplets and Reversible Hydrogels into Irreversible Hydrogels Impairs RNP Granule Function. Neuron 2015, 88, 678–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (565).Patel A; Lee HO; Jawerth L; Maharana S; Jahnel M; Hein MY; Stoynov S; Mahamid J; Saha S; Franzmann TM; et al. A Liquid-to-Solid Phase Transition of the ALS Protein FUS Accelerated by Disease Mutation. Cell 2015, 162, 1066–1077. [DOI] [PubMed] [Google Scholar]
- (566).Andreev M; Prabhu VM; Douglas JF; Tirrell M; de Pablo JJ Complex Coacervation in Polyelectrolytes from a Coarse-Grained Model. Macromolecules 2018, 51, 6717–6723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (567).Dignon GL; Zheng WW; Kim YC; Best RB; Mittal J Sequence Determinants of Protein Phase Behavior from a Coarse-grained Lodel. PLoS Comput. Biol 2018, 14, No. e1005941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (568).Feric M; Vaidya N; Harmon TS; Mitrea DM; Zhu L; Richardson TM; Kriwacki RW; Pappu RV; Brangwynne CP Coexisting Liquid Phases Underlie Nucleolar Subcompartments. Cell 2016, 165, 1686–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (569).Delaney KT; Fredrickson GH Recent Developments in Fully Fluctuating Field-Theoretic Simulations of Polymer Melts and Solutions. J. Phys. Chem. B 2016, 120, 7615–7634. [DOI] [PubMed] [Google Scholar]
- (570).McCarty J; Delaney KT; Danielsen SPO; Fredrickson GH; Shea JE Complete Phase Diagram for Liquid-Liquid Phase Separation of Intrinsically Disordered Proteins. J. Phys. Chem. Lett 2019, 10, 1644–1652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Voorn MJ Complex Coacervation.5. Experiments on the Distribution of Salts - Comparison of Theory and Experiment. Recl. Trav. Chim. Pay B 1956, 75, 1021–1030. [Google Scholar]
- (572).Kotler SA; Walsh P; Brender JR; Ramamoorthy A Differences between Amyloid-β Aggregation in Solution and on the Membrane: Insights into Elucidation of the Mechanistic Details of Alzheimer’s Disease. Chem. Soc. Rev 2014, 43, 6692–6700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (573).Butterfield SM; Lashuel HA Amyloidogenic Protein-membrane Interactions: Mechanistic Insight from Model Systems. Angew. Chem., Int. Ed 2010, 49, 5628–5654. [DOI] [PubMed] [Google Scholar]
- (574).Liu Y; Ren B; Zhang Y; Sun Y; Chang Y; Liang G; Xu L; Zheng J Molecular Simulation Aspects of Amyloid Peptides at Membrane Interface. Biochim. Biophys. Acta, Biomembr 2018, 1860, 1906–1916. [DOI] [PubMed] [Google Scholar]
- (575).Lee S; Zheng XY; Krishnamoorthy J; Savelieff MG; Park HM; Brender JR; Kim JH; Derrick JS; Kochi A; Lee HJ; et al. Rational Design of a Structural Framework with Potential Use to Develop Chemical Reagents That Target and Modulate Multiple Facets of Alzheimer’s Disease. J. Am. Chem. Soc 2014, 136, 299–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (576).Brender JR; Salamekh S; Ramamoorthy A Membrane Disruption and Early Events in the Aggregation of the Diabetes Related Peptide IAPP from a Molecular Perspective. Acc. Chem. Res 2012, 45, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (577).Zhang MZ; Zhao J; Zheng J Molecular Understanding of a Potential Functional Link between Antimicrobial and Amyloid Peptides. Soft Matter 2014, 10, 7425–7451. [DOI] [PubMed] [Google Scholar]
- (578).Murray IV; Liu L; Komatsu H; Uryu K; Xiao G; Lawson JA; Axelsen PH Membrane-mediated Amyloidogenesis and the Promotion of Oxidative Lipid Damage by Amyloid beta Proteins. J. Biol. Chem 2007, 282, 9335–9345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Jang H; Zheng J; Nussinov R Models of beta-amyloid Ion Channels in the Membrane Suggest that Channel Formation in the Bilayer is a Dynamic Process. Biophys. J 2007, 93, 1938–1949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (580).Jang H; Connelly L; Arce FT; Ramachandran S; Kagan BL; Lal R; Nussinov R Mechanisms for the Insertion of Toxic, Fibril-like beta-Amyloid Oligomers into the Membrane. J. Chem. Theory Comput 2013, 9, 822–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (581).Quist A; Doudevski I; Lin H; Azimova R; Ng D; Frangione B; Kagan B; Ghiso J; Lal R Amyloid Ion Channels: A Common Structural Link for Protein-misfolding Disease. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 10427–10432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (582).Lin H; Bhatia R; Lal R Amyloid-beta Protein Forms Ion Channels: Implications for Alzheimer’s Disease Pathophysiology. FASEB J 2001, 15, 2433–2444. [DOI] [PubMed] [Google Scholar]
- (583).Connelly L; Jang H; Teran Arce F; Capone R; Kotler SA; Ramachandran S; Kagan BL; Nussinov R; Lal R Atomic Force Microscopy and MD Simulations Reveal Pore-like Structures of all-d-enantiomer of Alzheimer’s β-amyloid Peptide: Relevance to the Ion Channel Mechanism of AD Pathology. J. Phys. Chem. B 2012, 116, 1728–1735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (584).Jang H; Arce FT; Ramachandran S; Capone R; Lal R; Nussinov R beta-Barrel Topology of Alzheimer’s beta-amyloid Ion Channels. J. Mol. Biol 2010, 404, 917–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (585).Korshavn KJ; Satriano C; Lin Y; Zhang R; Dulchavsky M; Bhunia A; Ivanova MI; Lee Y-H; La Rosa C; Lim MH; et al. Reduced Lipid Bilayer Thickness Regulates the Aggregation and Cytotoxicity of Amyloid-β. J. Biol. Chem 2017, 292, 4638–4650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (586).Ciudad S; Puig E; Botzanowski T; Meigooni M; Arango AS; Do J; Mayzel M; Bayoumi M; Chaignepain S; Maglia G; et al. Aβ(1–42) Tetramer and Octamer Structures Reveal Edge Conductivity Pores as a Mechanism for Membrane Damage. Nat. Commun 2020, 11, 3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Luhrs T; Ritter C; Adrian M; Riek-Loher D; Bohrmann B; Dobeli H; Schubert D; Riek R 3D Structure of Alzheimer’s amyloid-beta(1–42) Fibrils. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 17342–17347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (588).Shafrir Y; Durell S; Arispe N; Guy HR Models of Membrane-bound Alzheimer’s Abeta Peptide Assemblies. Proteins: Struct., Funct., Genet 2010, 78, 3473–3487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (589).Jang H; Arce FT; Ramachandran S; Capone R; Lal R; Nussinov R β-barrel Topology of Alzheimer’s β-Amyloid Ion Channels. J. Mol. Biol 2010, 404, 917–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (590).Jang H; Teran Arce F; Ramachandran S; Capone R; Lal R; Nussinov R Structural Convergence among Diverse, Toxic β-sheet Ion Channels. J. Phys. Chem. B 2010, 114, 9445–9451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (591).Nguyen PH; Campanera JM; Ngo ST; Loquet A; Derreumaux P Tetrameric Aβ40 and Aβ42 β-Barrel Structures by Extensive Atomistic Simulations. I. In a Bilayer Mimicking a Neuronal Membrane. J. Phys. Chem. B 2019, 123, 3643–3648. [DOI] [PubMed] [Google Scholar]
- (592).Ngo ST; Nguyen PH; Derreumaux P Impact of A2T and D23N Mutations on Tetrameric Aβ42 Barrel within a Dipalmitoylphosphatidylcholine Lipid Bilayer Membrane by Replica Exchange Molecular Dynamics. J. Phys. Chem. B 2020, 124, 1175–1182. [DOI] [PubMed] [Google Scholar]
- (593).Serra-Batiste M; Ninot-Pedrosa M; Bayoumi M; Gairí M; Maglia G; Carulla N Aβ42 Assembles into Specific β-barrel Pore-forming Oligomers in Membrane-mimicking Environments. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 10866–10871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (594).Bode DC; Baker MD; Viles JH Ion Channel Formation by Amyloid-β42 Oligomers but Not Amyloid-β40 in Cellular Membranes. J. Biol. Chem 2017, 292, 1404–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Österlund N; Moons R; Ilag LL; Sobott F; Gräslund A Native Ion Mobility-Mass Spectrometry Reveals the Formation of β-Barrel Shaped Amyloid-β Hexamers in a Membrane-Mimicking Environment. J. Am. Chem. Soc 2019, 141, 10440–10450. [DOI] [PubMed] [Google Scholar]
- (596).Ngo ST; Derreumaux P; Vu VV Probable Transmembrane Amyloid α-Helix Bundles Capable of Conducting Ca2+ Ions. J. Phys. Chem. B 2019, 123, 2645–2653. [DOI] [PubMed] [Google Scholar]
- (597).Fantini J; Di Scala C; Yahi N; Troadec J-D; Sadelli K; Chahinian H; Garmy N Bexarotene Blocks Calcium-Permeable Ion Channels Formed by Neurotoxic Alzheimer’s β-Amyloid Peptides. ACS Chem. Neurosci 2014, 5, 216–224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (598).Kakio A; Nishimoto S-I; Yanagisawa K; Kozutsumi Y; Matsuzaki K Cholesterol-dependent Formation of GM1 Ganglio-side-bound Amyloid β-Protein, an Endogenous Seed for Alzheimer Amyloid. J. Biol. Chem 2001, 276, 24985–24990. [DOI] [PubMed] [Google Scholar]
- (599).Fantini J; Yahi N Molecular Insights into Amyloid Regulation by Membrane Cholesterol and Sphingolipids: Common Mechanisms in Neurodegenerative Diseases. Expert Rev. Mol. Med 2010, 12, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Kandel N; Matos JO; Tatulian SA Structure of amyloid β(25–35) in Lipid Environment and Cholesterol-dependent Membrane Pore Formation. Sci. Rep 2019, 9, 2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (601).Davis CH; Berkowitz ML Structure of the amyloid-beta (1–42) Monomer Absorbed to Model Phospholipid Bilayers: a Molecular Dynamics Study. J. Phys. Chem. B 2009, 113, 14480–14486. [DOI] [PubMed] [Google Scholar]
- (602).Xiong J; Roach CA; Oshokoya OO; Schroell RP; Yakubu RA; Eagleburger MK; Cooley JW; JiJi RD Role of Bilayer Characteristics on the Structural Fate of Aβ(1–40) and Aβ(25–40). Biochemistry 2014, 53, 3004–3011. [DOI] [PubMed] [Google Scholar]
- (603).Yu X; Zheng J Cholesterol Promotes the Interaction of Alzheimer β-amyloid Monomer with Lipid bilayer. J. Mol. Biol 2012, 421, 561–571. [DOI] [PubMed] [Google Scholar]
- (604).Hoshino T; Mahmood MI; Mori K; Matsuzaki K Binding and Aggregation Mechanism of Amyloid β-Peptides onto the GM1 Ganglioside-Containing Lipid Membrane. J. Phys. Chem. B 2013, 117, 8085–8094. [DOI] [PubMed] [Google Scholar]
- (605).Manna M; Mukhopadhyay C Binding, Conformational Transition and Dimerization of amyloid-β peptide on GM1-containing Ternary Membrane: Insights from Molecular Dynamics Simulation. PLoS One 2013, 8, No. e71308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (606).Lemkul JA; Bevan DR Lipid Composition Influences the Release of Alzheimer’s amyloid β-peptide from Membranes. Protein Sci 2011, 20, 1530–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (607).Amaro M; Šachl R; Aydogan G; Mikhalyov II; Vácha R; Hof M GM1 Ganglioside Inhibits β-Amyloid Oligomerization Induced by Sphingomyelin. Angew. Chem., Int. Ed 2016, 55, 9411–9415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (608).Tofoleanu F; Buchete N-V Molecular Interactions of Alzheimer’s Aβ Protofilaments with Lipid Membranes. J. Mol. Biol 2012, 421, 572–586. [DOI] [PubMed] [Google Scholar]
- (609).Tofoleanu F; Brooks BR; Buchete N-V Modulation of Alzheimer’s Aβ Protofilament-Membrane Interactions by Lipid Headgroups. ACS Chem. Neurosci 2015, 6, 446–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (610).Yu X; Wang Q; Pan Q; Zhou F; Zheng J Molecular Interactions of Alzheimer amyloid-beta Oligomers with Neutral and Negatively Charged lipid Bilayers. Phys. Chem. Chem. Phys 2013, 15, 8878–8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (611).Yi X; Zhang Y; Gong M; Yu X; Darabedian N; Zheng J; Zhou F Ca2+ Interacts with Glu-22 of Aβ(1–42) and Phospholipid Bilayers to Accelerate the Aβ(1–42) Aggregation below the Critical Micelle Concentration. Biochemistry 2015, 54, 6323–6332. [DOI] [PubMed] [Google Scholar]
- (612).Tamamizu-Kato S; Kosaraju MG; Kato H; Raussens V; Ruysschaert J-M; Narayanaswami V Calcium-Triggered Membrane Interaction of the α-Synuclein Acidic Tail. Biochemistry 2006, 45, 10947–10956. [DOI] [PubMed] [Google Scholar]
- (613).Zhang M; Ren B; Liu Y; Liang G; Sun Y; Xu L; Zheng J Membrane interactions of hIAPP Monomer and Oligomer with Lipid Membranes by Molecular Dynamics Simulations. ACS Chem. Neurosci 2017, 8, 1789–1800. [DOI] [PubMed] [Google Scholar]
- (614).Martinez Hernandez A; Urbanke H; Gillman AL; Lee J; Ryazanov S; Agbemenyah HY; Benito E; Jain G; Kaurani L; Grigorian G; et al. The Diphenylpyrazole Compound anle138b Blocks Aβ Channels and Rescues Disease Phenotypes in a Mouse Model for amyloid pathology. EMBO Mol. Med 2018, 10, 32–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (615).Last NB; Miranker AD Common Mechanism Unites Membrane Poration by Amyloid and Antimicrobial Peptides. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 6382–6387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (616).Qiao Q; Wei G; Yao D; Song Z Formation of α-helical and β-sheet Structures in Membrane-bound Human IAPP Monomer and the Resulting Membrane Deformation. Phys. Chem. Chem. Phys 2019, 21, 20239–20251. [DOI] [PubMed] [Google Scholar]
- (617).Xu WX; Wei GH; Su HB; Nordenskiold L; Mu YG Effects of Cholesterol on Pore Formation in Lipid Bilayers induced by Human Islet Amyloid Polypeptide Fragments: A Coarse-grained Molecular Dynamics Study. Phys. Rev. E 2011, 84, 051922. [DOI] [PubMed] [Google Scholar]
- (618).Dignon GL; Zerze GH; Mittal J Interplay Between Membrane Composition and Structural Stability of Membrane-Bound hIAPP. J. Phys. Chem. B 2017, 121, 8661–8668. [DOI] [PubMed] [Google Scholar]
- (619).Guo C; Cote S; Mousseau N; Wei GH Distinct Helix Propensities and Membrane Interactions of Human and Rat IAPP(1–19) Monomers in Anionic Lipid Bilayers. J. Phys. Chem. B 2015, 119, 3366–3376. [DOI] [PubMed] [Google Scholar]
- (620).Jia Y; Qian ZY; Zhang Y; Wei GH Adsorption and Orientation of Human Islet Amyloid Polypeptide (hIAPP) Monomer at Anionic Lipid Bilayers: Implications for Membrane-Mediated Aggregation. Int. J. Mol. Sci 2013, 14, 6241–6258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (621).Zhang Y; Luo Y; Deng YH; Mu YG; Wei GH Lipid Interaction and Membrane Perturbation of Human Islet Amyloid Polypeptide Monomer and Dimer by Molecular Dynamics Simulations. PLoS One 2012, 7, No. e38191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (622).Skeby KK; Andersen OJ; Pogorelov TV; Tajkhorshid E; Schiott B Conformational Dynamics of the Human Islet Amyloid Polypeptide in a Membrane Environment: Toward the Aggregation Prone Form. Biochemistry 2016, 55, 2031–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (623).Duan M; Fan J; Huo S Conformations of Islet Amyloid Polypeptide Monomers in a Membrane Environment: Implications for Fibril Formation. PLoS One 2012, 7, No. e47150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (624).Sun Y; Kakinen A; Xing Y; Faridi P; Nandakumar A; Purcell AW; Davis TP; Ke PC; Ding F Amyloid Self-Assembly of hIAPP8–20 via the Accumulation of Helical Oligomers, α-Helix to β-Sheet Transition, and Formation of β-Barrel Intermediates. Small 2019, 15, 1805166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (625).Qian ZJ; Yan Wei G Binding Orientations and Lipid Interactions of Human Amylin at Zwitterionic and Anionic Lipid Bilayers. J. Diabetes Res 2016, 2016, 1749196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (626).Christensen M; Schiott B Revealing a Dual Role of Ganglioside Lipids in the Aggregation of Membrane-Associated Islet Amyloid Polypeptide. J. Membr. Biol 2019, 252, 343–356. [DOI] [PubMed] [Google Scholar]
- (627).Kayed R; Head E; Thompson JL; McIntire TM; Milton SC; Cotman CW; Glabe CG Common Structure of Soluble amyloid Oligomers Implies Common Mechanism of Pathogenesis. Science 2003, 300, 486–489. [DOI] [PubMed] [Google Scholar]
- (628).Jiang P; Xu WX; Mu YG Amyloidogenesis Abolished by Proline Substitutions but Enhanced by Lipid Binding. PLoS Comput. Biol 2009, 5, No. e1000357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (629).Qian ZY; Zou Y; Zhang QW; Chen PJ; Ma BY; Wei GH; Nussinov R Atomistic-level Study of the Interactions between hIAPP Protofibrils and Membranes: Influence of pH and lipid Composition. Biochim. Biophys. Acta, Biomembr 2018, 1860, 1818–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (630).Zhao J; Luo Y; Jang HB; Yu X; Wei GH; Nussinov R; Zheng J Probing Ion Channel Activity of Human islet Amyloid Polypeptide (amylin). Biochim. Biophys. Acta, Biomembr 2012, 1818, 3121–3130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (631).Zhao J; Hu RD; Sciacca MFM; Brender JR; Chen H; Ramamoorthy A; Zheng J Non-selective Ion Channel Activity of Polymorphic Human islet Amyloid Polypeptide (amylin) Double Channels. Phys. Chem. Chem. Phys 2014, 16, 2368–2377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (632).Mirzabekov TA; Lin M-C; Kagan BL Pore Formation by the Cytotoxic Islet Amyloid Peptide Amylin. J. Biol. Chem 1996, 271, 1988–1992. [DOI] [PubMed] [Google Scholar]
- (633).Arrasate M; Perez M; Avila J Tau Dephosphorylation at tau-1 Site Correlates with its Association to Cell Membrane. Neurochem. Res 2000, 25, 43–50. [DOI] [PubMed] [Google Scholar]
- (634).Shea TB Phospholipids alter Tau Conformation, Phosphorylation, Proteolysis, and Association with Microtubules: Implication for tau Function under Normal and Degenerative Conditions. J. Neurosci. Res 1997, 50, 114–122. [DOI] [PubMed] [Google Scholar]
- (635).Georgieva ER; Xiao SF; Borbat PP; Freed JH; Eliezer D Tau Binds to Lipid Membrane Surfaces via Short Amphipathic Helices Located in Its Microtubule-Binding Repeats. Biophys. J 2014, 107, 1441–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (636).Barre P; Eliezer D Folding of the Repeat Domain of Tau upon Binding to Lipid Surfaces. J. Mol. Biol 2006, 362, 312–326. [DOI] [PubMed] [Google Scholar]
- (637).Brandt R; Léger J; Lee G Interaction of Tau with the Neural Plasma Membrane Mediated by Tau’s amino-terminal Projection Domain. J. Cell Biol 1995, 131, 1327–1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (638).Press-Sandler O; Miller Y Molecular Mechanisms of Membrane-associated Amyloid Aggregation: Computational Perspective and Challenges. Biochim. Biophys. Acta, Biomembr 2018, 1860, 1889–1905. [DOI] [PubMed] [Google Scholar]
- (639).Jones EM; Dubey M; Camp PJ; Vernon BC; Biernat J; Mandelkow E; Majewski J; Chi EY Interaction of Tau Protein with Model Lipid Membranes Induces Tau Structural Compaction and Membrane Disruption. Biochemistry 2012, 51, 2539–2550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (640).Barré P; Eliezer D Structural Transitions in Tau k18 on Micelle Binding Suggest a Hierarchy in the Efficacy of Individual Microtubule-binding Repeats in Filament Nucleation. Protein Sci 2013, 22, 1037–1048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (641).Chirita CN; Necula M; Kuret J Anionic Micelles and Vesicles Induce Tau Fibrillization in vitro. J. Biol. Chem 2003, 278, 25644–25650. [DOI] [PubMed] [Google Scholar]
- (642).Kunze G; Barre P; Scheidt HA; Thomas L; Eliezer D; Huster D Binding of the Three-repeat Domain of Tau to Phospholipid Membranes Induces an Aggregated-like State of the Protein. Biochim. Biophys. Acta, Biomembr 2012, 1818, 2302–2313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (643).Lasagna-Reeves CA; Sengupta U; Castillo-Carranza D; Gerson JE; Guerrero-Munoz M; Troncoso JC; Jackson GR; Kayed R The formation of tau pore-like structures is prevalent and cell specific: possible implications for the disease phenotypes. Acta Neuropathol. Commun 2014, 2, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (644).Ghio S; Camilleri A; Caruana M; Ruf VC; Schmidt F; Leonov A; Ryazanov S; Griesinger C; Cauchi RJ; Kamp F; et al. Cardiolipin Promotes Pore-Forming Activity of Alpha-Synuclein Oligomers in Mitochondrial Membranes. ACS Chem. Neurosci 2019, 10, 3815–3829. [DOI] [PubMed] [Google Scholar]
- (645).Fanni AM; Van der Zanden CM; Majewska PV; Majewski J; Chi EY Membrane-mediated Fibrillation and Toxicity of the tau Hexapeptide PHF6. J. Biol. Chem 2019, 294, 15304–15317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (646).Li K; Arikan MC; Andreadis A Modulation of the Membrane-binding Domain of Tau Protein: Splicing Regulation of Exon 2. Mol. Brain Res 2003, 116, 94–105. [DOI] [PubMed] [Google Scholar]
- (647).Fusco G; Sanz-Hernandez M; De Simone A Order and Disorder in the Physiological Membrane Binding of alpha-synuclein. Curr. Opin. Struct. Biol 2018, 48, 49. [DOI] [PubMed] [Google Scholar]
- (648).Zhu M; Fink AL Lipid Binding Inhibits alpha-synuclein Fibril Formation. J. Biol. Chem 2003, 278, 16873–16877. [DOI] [PubMed] [Google Scholar]
- (649).Perrin RJ; Woods WS; Clayton DF; George JM Exposure to Long Chain Polyunsaturated Fatty Acids Triggers Rapid Multimerization of Synucleins. J. Biol. Chem 2001, 276, 41958–41962. [DOI] [PubMed] [Google Scholar]
- (650).Fusco G; Chen SW; Williamson PTF; Cascella R; Perni M; Jarvis JA; Cecchi C; Vendruscolo M; Chiti F; Cremades N; et al. Structural Basis of Membrane Disruption and Cellular Toxicity by alpha-synuclein Oligomers. Science 2017, 358, 1440–1443. [DOI] [PubMed] [Google Scholar]
- (651).Ulmer TS; Bax A; Cole NB; Nussbaum RL Structure and Dynamics of Micelle-bound Human alpha-synuclein. J. Biol. Chem 2005, 280, 9595–9603. [DOI] [PubMed] [Google Scholar]
- (652).Campioni S; Carret G; Jordens S; Nicoud L; Mezzenga R; Riek R The Presence of an air-water Interface Affects Formation and Elongation of alpha-Synuclein Fibrils. J. Am. Chem. Soc 2014, 136, 2866–2875. [DOI] [PubMed] [Google Scholar]
- (653).Middleton ER; Rhoades E Effects of Curvature and Composition on alpha-synuclein Binding to Lipid Vesicles. Biophys. J 2010, 99, 2279–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (654).Fusco G; De Simone A; Arosio P; Vendruscolo M; Veglia G; Dobson CM Structural Ensembles of Membrane-bound alpha-Synuclein Reveal the Molecular Determinants of Synaptic Vesicle Affinity. Sci. Rep 2016, 6, 27125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (655).Georgieva ER; Ramlall TF; Borbat PP; Freed JH; Eliezer D Membrane-bound alpha-synuclein Forms an Extended Helix: Long-distance Pulsed ESR Measurements using Vesicles, Bicelles, and Rodlike Micelles. J. Am. Chem. Soc 2008, 130, 12856–12857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (656).Bodner CR; Dobson CM; Bax A Multiple Tight Phospholipid-binding Modes of alpha-synuclein Revealed by Solution NMR spectroscopy. J. Mol. Biol 2009, 390, 775–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (657).Oueslati A; Paleologou KE; Schneider BL; Aebischer P; Lashuel HA Mimicking Phosphorylation at serine 87 Inhibits the Aggregation of Human alpha-synuclein and Protects against its Toxicity in a Rat model of Parkinson’s Disease. J. Neurosci 2012, 32, 1536–1544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (658).Stephens AD; Zacharopoulou M; Moons R; Fusco G; Seetaloo N; Chiki A; Hooper PJ; Mela I; Lashuel H; Philips JJ; et al. Extent of N-terminus Exposure by Altered Long-range Interactions of Monomeric alpha-synuclein Determines its Aggregation Propensity. Nat. Commun 2020, 11, 2820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (659).Dikiy I; Fauvet B; Jovicic A; Mahul-Mellier AL; Desobry C; El-Turk F; Gitler AD; Lashuel HA; Eliezer D Semisynthetic and in Vitro Phosphorylation of Alpha-Synuclein at Y39 Promotes Functional Partly Helical Membrane-Bound States Resembling Those Induced by PD Mutations. ACS Chem. Biol 2016, 11, 2428–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (660).Runfola M; De Simone A; Vendruscolo M; Dobson CM; Fusco G The N-terminal Acetylation of alpha-Synuclein Changes the Affinity for Lipid Membranes but not the Structural Properties of the Bound State. Sci. Rep 2020, 10, 204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (661).Maltsev AS; Ying J; Bax A Impact of N-terminal Acetylation of alpha-Synuclein on its Random Coil and Lipid Binding Properties. Biochemistry 2012, 51, 5004–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (662).Lovell MA; Robertson JD; Teesdale WJ; Campbell JL; Markesbery WR Copper, Iron and Zinc in Alzheimer’s Disease Senile Plaques. J. Neurol. Sci 1998, 158, 47–52. [DOI] [PubMed] [Google Scholar]
- (663).Chimienti F; Devergnas S; Favier A; Seve M Identification and Cloning of a β-Cell-Specific Zinc Transporter, ZnT-8, Localized into Insulin Secretory Granules. Diabetes 2004, 53, 2330–2337. [DOI] [PubMed] [Google Scholar]
- (664).Alghrably M; Czaban I; Jaremko Ł; Jaremko M Interaction of Amylin Species with Transition Metals and Membranes. J. Inorg. Biochem 2019, 191, 69–76. [DOI] [PubMed] [Google Scholar]
- (665).Wärmländer SKTS; Österlund N; Wallin C; Wu J; Luo J; Tiiman A; Jarvet J; Gräslund A Metal Binding to the Amyloid-β Peptides in the Presence of Biomembranes: Potential Mechanisms of Cell Toxicity. JBIC, J. Biol. Inorg. Chem 2019, 24, 1189–1196. [DOI] [PubMed] [Google Scholar]
- (666).Faller P; Hureau C; Berthoumieu O Role of Metal Ions in the Self-Assembly of the Alzheimer’s Amyloid-β Peptide. Inorg. Chem 2013, 52, 12193–12206. [DOI] [PubMed] [Google Scholar]
- (667).Rana M; Sharma AK Consequence of Coordination on Aggregation and Formation of Neurotoxic Soluble Aβ Oligomers. Metallomics 2019, 11, 64–84. [DOI] [PubMed] [Google Scholar]
- (668).Atrián-Blasco E; González P; Santoro A; Alies B; Faller P; Hureau C Cu and Zn Coordination to Amyloid Peptides: From Fascinating Chemistry to Debated Pathological Relevance. Coord. Chem. Rev 2018, 371, 38–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (669).Barnham KJ; Bush AI Biological Metals and Metal-Targeting Compounds in Major Neurodegenerative Diseases. Chem. Soc. Rev 2014, 43, 6727–6749. [DOI] [PubMed] [Google Scholar]
- (670).Kim B-E; Nevitt T; Thiele DJ Mechanisms for Copper Acquisition, Distribution and Regulation. Nat. Chem. Biol 2008, 4, 176–185. [DOI] [PubMed] [Google Scholar]
- (671).Lutsenko S; Bhattacharjee A; Hubbard AL Copper Handling Machinery of the Brain. Metallomics 2010, 2, 596. [DOI] [PubMed] [Google Scholar]
- (672).Watanabe-Nakayama T; Ono K High-Speed Atomic Force Microscopy of Individual Amyloidogenic Protein Assemblies. In Nanoscale Imaging. Methods in Molecular Biology; Lyubchenko Y, Eds.; Humana Press: New York, NY, 2018; p 1814. [DOI] [PubMed] [Google Scholar]
- (673).Faller P; Hureau C; La Penna G Metal Ions and Intrinsically Disordered Proteins and Peptides: From Cu/Zn Amyloid-Beta to General Principles. Acc. Chem. Res 2014, 47, 2252–2259. [DOI] [PubMed] [Google Scholar]
- (674).Viles JH Metal Ions and Amyloid Fiber Formation in Neurodegenerative Diseases. Copper, Zinc and Iron in Alzheimer’s, Parkinson’s and Prion Diseases. Coord. Chem. Rev 2012, 256, 2271–2284. [Google Scholar]
- (675).Xiao Z; Gottschlich L; van der Meulen R; Udagedara SR; Wedd AG Evaluation of Quantitative Probes for Weaker Cu(i) Binding Sites Completes a Set of Four Capable of Detecting Cu(i) Affinities from Nanomolar to Attomolar. Metallomics 2013, 5, 501–513. [DOI] [PubMed] [Google Scholar]
- (676).Stefaniak E; Bal W CuII Binding Properties of N-Truncated Aβ Peptides: In Search of Biological Function. Inorg. Chem 2019, 58, 13561–13577. [DOI] [PubMed] [Google Scholar]
- (677).Gonzalez P; Bossak K; Stefaniak E; Hureau C; Raibaut L; Bal W; Faller P N-Terminal Cu-Binding Motifs (Xxx-Zzz-His, Xxx-His) and Their Derivatives: Chemistry, Biology and Medicinal Applications. Chem. - Eur. J 2018, 24, 8029–8041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (678).Bacchella C; Gentili S; Bellotti D; Quartieri E; Draghi S; Baratto MC; Remelli M; Valensin D; Monzani E; Nicolis S; et al. Binding and Reactivity of Copper to R1 and R3 Fragments of Tau Protein. Inorg. Chem 2020, 59, 274–286. [DOI] [PubMed] [Google Scholar]
- (679).Shin BK; Saxena S Insight into Potential Cu(II)-Binding Motifs in the Four Pseudorepeats of Tau Protein. J. Phys. Chem. B 2011, 115, 15067–15078. [DOI] [PubMed] [Google Scholar]
- (680).Mo ZY; Zhu YZ; Zhu HL; Fan JB; Chen J; Liang Y Low Micromolar Zinc Accelerates the Fibrillization of Human Tau via Bridging of Cys-291 and Cys-322. J. Biol. Chem 2009, 284, 34648–34657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (681).Singh A; Kukreti R; Saso L; Kukreti S A Key Modulator in Neurodegenerative Diseases. Molecules 2019, 24, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (682).Guilloreau L; Combalbert S; Sournia-Saquet A; Mazarguil H; Faller P Redox Chemistry of Copper-Amyloid-β: The Generation of Hydroxyl Radical in the Presence of Ascorbate Is Linked to Redox-Potentials and Aggregation State. ChemBioChem 2007, 8, 1317–1325. [DOI] [PubMed] [Google Scholar]
- (683).Santoro A; Walke G; Vileno B; Kulkarni PP; Raibaut L; Faller P Low Catalytic Activity of the Cu(II)-Binding Motif (Xxx-Zzz-His; ATCUN) in Reactive Oxygen Species Production and Inhibition by the Cu(i)-Chelator BCS. Chem. Commun 2018, 54, 11945–11948. [DOI] [PubMed] [Google Scholar]
- (684).Cheignon C; Jones M; Atrián-Blasco E; Kieffer I; Faller P; Collin F; Hureau C Identification of Key Structural Features of the Elusive Cu-Aβ Complex That Generates ROS in Alzheimer’s Disease. Chem. Sci 2017, 8, 5107–5118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (685).Prosdocimi T; De Gioia L; Zampella G; Bertini L On the Generation of OH· Radical Species from H2O2 by Cu(I) Amyloid Beta Peptide Model Complexes: A DFT Investigation. JBIC, J. Biol. Inorg. Chem 2016, 21, 197–212. [DOI] [PubMed] [Google Scholar]
- (686).Rodríguez EE; Arcos-López T; Trujano-Ortiz LG; Fernández CO; González FJ; Vela A; Quintanar L Role of N-Terminal Methionine Residues in the Redox Activity of Copper Bound to Alpha-Synuclein. JBIC, J. Biol. Inorg. Chem 2016, 21, 691–702. [DOI] [PubMed] [Google Scholar]
- (687).Rice ME Ascorbate Regulation and Its Neuroprotective Role in the Brain. Trends Neurosci 2000, 23, 209–216. [DOI] [PubMed] [Google Scholar]
- (688).Santoro A; Wezynfeld N; Vasak M; Bal W; Faller P Cysteine and Glutathione Trigger the Cu-Zn Swap between Cu(II)-Amyloid-B4–16 Peptide and Zn7-Metallothionein-3. Chem. Commun 2017, 53, 11634–11637. [DOI] [PubMed] [Google Scholar]
- (689).Danielsson J; Pierattelli R; Banci L; Gräslund A High-Resolution NMR Studies of the Zinc-Binding Site of the Alzheimer’s Amyloid β-Peptide. FEBS J 2007, 274, 46–59. [DOI] [PubMed] [Google Scholar]
- (690).Sciarretta KL; Gordon DJ; Petkova AT; Tycko R; Meredith SC Aβ40-Lactam(D23/K28) Models a Conformation Highly Favorable for Nucleation of Amyloid. Biochemistry 2005, 44, 6003–6014. [DOI] [PubMed] [Google Scholar]
- (691).Mithu VS; Sarkar B; Bhowmik D; Chandrakesan M; Maiti S; Madhu PK Zn+2 Binding Disrupts the Asp 23-Lys 28 Salt Bridge without Altering the Hairpin-Shaped Cross-β Structure of Aβ 42 Amyloid Aggregates. Biophys. J 2011, 101, 2825–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (692).Talmard C; Guilloreau L; Coppel Y; Mazarguil H; Faller P Amyloid-Beta Peptide Forms Monomeric Complexes with CuII and ZnII Prior to Aggregation. ChemBioChem 2007, 8, 163–165. [DOI] [PubMed] [Google Scholar]
- (693).Abelein A; Gräslund A; Danielsson J Zinc as Chaperone-Mimicking Agent for Retardation of Amyloid β Peptide Fibril Formation. Proc. Natl. Acad. Sci. U. S. A 2015, 112, 5407–5412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (694).Rezaei-Ghaleh N; Giller K; Becker S; Zweckstetter M Effect of Zinc Binding on β-Amyloid Structure and Dynamics: Implications for Aβ Aggregation. Biophys. J 2011, 101, 1202–1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (695).Alies B; Conte-Daban A; Sayen S; Collin F; Kieffer I; Guillon E; Faller P; Hureau C Zinc(II) Binding Site to the Amyloid-β Peptide: Insights from Spectroscopic Studies with a Wide Series of Modified Peptides. Inorg. Chem 2016, 55, 10499–10509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (696).Ghalebani L; Wahlström A; Danielsson J; Wärmländer SKTS; Gräslund A PH-Dependence of the Specific Binding of Cu(II) and Zn(II) Ions to the Amyloid-β Peptide. Biochem. Biophys. Res. Commun 2012, 421, 554–560. [DOI] [PubMed] [Google Scholar]
- (697).Valiente-Gabioud AA; Torres-Monserrat V; Molina-Rubino L; Binolfi A; Griesinger C; Fernández CO Structural Basis behind the Interaction of Zn2+ with the Protein α-Synuclein and the Aβ Peptide: A Comparative Analysis. J. Inorg. Biochem 2012, 117, 334–341. [DOI] [PubMed] [Google Scholar]
- (698).Syme CD; Viles JH Solution 1H NMR Investigation of Zn2+ and Cd2+ Binding to Amyloid-Beta Peptide (Aβ) of Alzheimer’s Disease. Biochim. Biophys. Acta, Proteins Proteomics 2006, 1764, 246–256. [DOI] [PubMed] [Google Scholar]
- (699).Istrate AN; Kozin SA; Zhokhov SS; Mantsyzov AB; Kechko OI; Pastore A; Makarov AA; Polshakov VI Interplay of Histidine Residues of the Alzheimer’s Disease Aβ Peptide Governs Its Zn-Induced Oligomerization. Sci. Rep 2016, 6, 21734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (700).Polshakov VI; Mantsyzov AB; Kozin SA; Adzhubei AA; Zhokhov SS; van Beek W; Kulikova AA; Indeykina MI; Mitkevich VA; Makarov AA A Binuclear Zinc Interaction Fold Discovered in the Homodimer of Alzheimer’s Amyloid-β Fragment with Taiwanese Mutation D7H. Angew. Chem., Int. Ed 2017, 56, 11734–11739. [DOI] [PubMed] [Google Scholar]
- (701).Kulikova AA; Tsvetkov PO; Indeykina MI; Popov IA; Zhokhov SS; Golovin AV; Polshakov VI; Kozin SA; Nudler E; Makarov AA Phosphorylation of Ser8 Promotes Zinc-Induced Dimerization of the Amyloid-β Metal-Binding Domain. Mol. BioSyst 2014, 10, 2590–2596. [DOI] [PubMed] [Google Scholar]
- (702).Lee MC; Yu WC; Shih YH; Chen CY; Guo ZH; Huang SJ; Chan JCC; Chen YR Zinc Ion Rapidly Induces Toxic, off-Pathway Amyloid-β Oligomers Distinct from Amyloid-β Derived Diffusible Ligands in Alzheimer’s Disease. Sci. Rep 2018, 8, 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (703).Hureau C Coordination of Redox Active Metal Ions to the Amyloid Precursor Protein and to Amyloid-β Peptides Involved in Alzheimer Disease. Part 1: An Overview. Coord. Chem. Rev 2012, 256, 2164–2174. [Google Scholar]
- (704).Hou L; Zagorski MG NMR Reveals Anomalous Copper(II) Binding to the Amyloid Aβ Peptide of Alzheimer’s Disease. J. Am. Chem. Soc 2006, 128, 9260–9261. [DOI] [PubMed] [Google Scholar]
- (705).De Gregorio G; Biasotto F; Hecel A; Luczkowski M; Kozlowski H; Valensin D Structural Analysis of Copper(I) Interaction with Amyloid β Peptide. J. Inorg. Biochem 2019, 195, 31–38. [DOI] [PubMed] [Google Scholar]
- (706).Alies B; Sasaki I; Proux O; Sayen S; Guillon E; Faller P; Hureau C Zn Impacts Cu Coordination to Amyloid-β, the Alzheimer’s Peptide, but Not the ROS Production and the Associated Cell Toxicity. Chem. Commun 2013, 49, 1214–1216. [DOI] [PubMed] [Google Scholar]
- (707).Silva KI; Saxena S Zn(II) Ions Substantially Perturb Cu(II) Ion Coordination in Amyloid-β at Physiological pH. J. Phys. Chem. B 2013, 117, 9386–9394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (708).Karr JW; Akintoye H; Kaupp LJ; Szalai VA N-Terminal Deletions Modify the Cu2+ Binding Site in Amyloid-β. Biochemistry 2005, 44, 5478–5487. [DOI] [PubMed] [Google Scholar]
- (709).Wallin C; Jarvet J; Biverstèl H; Wärmländer S; Danielsson J; Gräslund A; Abelein A Metal Ion Coordination Delays Amyloid-β Peptide Self-Assembly by Forming an Aggregation-Inert Complex. J. Biol. Chem 2020, 295, 7224–7234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (710).Strodel B; Coskuner-Weber O Transition Metal Ion Interactions with Disordered Amyloid-β Peptides in the Pathogenesis of Alzheimer’s Disease: Insights from Computational Chemistry Studies. J. Chem. Inf. Model 2019, 59, 1782–1805. [DOI] [PubMed] [Google Scholar]
- (711).Streltsov VA; Titmuss SJ; Epa VC; Barnham KJ; Masters CL; Varghese JN The Structure of the Amyloid-β Peptide High-Affinity Copper II Binding Site in Alzheimer Disease. Biophys. J 2008, 95, 3447–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (712).Stellato F; Menestrina G; Serra MD; Potrich C; Tomazzolli R; Meyer-Klaucke W; Morante S Metal Binding in Amyloid β-peptides Shows Intra- and Inter-peptide Coordination Modes. Eur. Biophys. J 2006, 35, 340–351. [DOI] [PubMed] [Google Scholar]
- (713).Minicozzi V; Stellato F; Comai M; Serra MD; Potrich C; Meyer-Klaucke W; Morante S Identifying the Minimal Copper- and Zinc-binding Site Sequence in Amyloid-β Peptides. J. Biol. Chem 2008, 283, 10784–10792. [DOI] [PubMed] [Google Scholar]
- (714).Marino T; Russo N; Toscano M; Pavelka M On the metal ion (Zn(2+), Cu(2+)) coordination with beta-amyloid peptide: DFT computational study. Interdiscip. Sci.: Comput. Life Sci 2010, 2, 57. [DOI] [PubMed] [Google Scholar]
- (715).Coskuner-Weber O Revisiting Cu(II) Bound Amyloid-β40 and Amyloid-β42 Peptides: Varying Coordination Chemistries. J. Turkish Chem. Soc., Section A: Chemistry 2018, 5, 981–1008. [Google Scholar]
- (716).Furlan S; La Penna G Modeling of the Zn2+ binding in the 1–16 region of the amyloid β peptide involved in Alzheimer’s disease. Phys. Chem. Chem. Phys 2009, 11, 6468–6481. [DOI] [PubMed] [Google Scholar]
- (717).Giannozzi P; Jansen K; La Penna G; Minicozzi V; Morante S; Rossi G; Stellato F Zn induced structural aggregation patterns of β-amyloid peptides by first-principle simulations and XAS measurements. Metallomics 2012, 4, 156–165. [DOI] [PubMed] [Google Scholar]
- (718).Kozin SA; Mezentsev YV; Kulikova AA; Indeykina MA; Golovin AV; Ivanov AS; Tsvetkov PO; Makarov AA Zinc-induced dimerization of the amyloid-β metal-binding domain 1–16 is mediated by residues 11–14. Mol. BioSyst 2011, 7, 1053–1055. [DOI] [PubMed] [Google Scholar]
- (719).Furlan S; Hureau C; Faller P; La Penna G Modeling the Cu+ Binding in the 1–16 Region of the Amyloid-β Peptide Involved in Alzheimer’s Disease. J. Phys. Chem. B 2010, 114, 15119–15133. [DOI] [PubMed] [Google Scholar]
- (720).Liao Q; Owen MC; Olubiyi OO; Barz B; Strodel B Conformational Transitions of the Amyloid-β Peptide Upon Copper(II) Binding and pH Changes. Isr. J. Chem 2017, 57, 771–784. [Google Scholar]
- (721).Ali-Torres J; Marechal J-D; Rodriguez-Santiago L; Sodupe M Three Dimensional Models of Cu2+-Aβ(1–16) Complexes from Computational Approaches. J. Am. Chem. Soc 2011, 133, 15008–15014. [DOI] [PubMed] [Google Scholar]
- (722).Wise-Scira O; Xu L; Perry G; Coskuner O Structures and Free Energy Landscapes of Aqueous Zinc(II)-bound Amyloid-β(1–40) and Zinc(II)-bound Amyloid-β(1–42) With Dynamics. JBIC, J. Biol. Inorg. Chem 2012, 17, 927–938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (723).Pan L; Patterson JC Molecular Dynamics Study of Zn(Ab) and Zn(Ab)2. PLoS One 2013, 8, No. e70681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (724).Coskuner O Divalent copper ion bound amyloid-β(40) and amyloid-β(42) alloforms are less preferred than divalent zinc ion bound amyloid-β(40) and amyloid-β(42) alloforms. JBIC, J. Biol. Inorg. Chem 2016, 21, 957–973. [DOI] [PubMed] [Google Scholar]
- (725).Liao Q; Owen MC; Bali S; Barz B; Strodel B Aβ under stress: the effects of acidosis, Cu2+-binding, and oxidation on amyloid β-peptide dimers. Chem. Commun 2018, 54, 7766–7769. [DOI] [PubMed] [Google Scholar]
- (726).Pham DQH; Li MS; La Penna G Copper Binding Induces Polymorphism in Amyloid-β Peptide: Results of Computational Models. J. Phys. Chem. B 2018, 122, 7243–7252. [DOI] [PubMed] [Google Scholar]
- (727).Brender JR; Hartman K; Nanga RPR; Popovych N; de La Salud Bea R; Vivekanandan S; Marsh ENG; Ramamoorthy A Role of Zinc in Human Islet Amyloid Polypeptide Aggregation. J. Am. Chem. Soc 2010, 132, 8973–8983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (728).Salamekh S; Brender JR; Hyung SJ; Nanga RPR; Vivekanandan S; Ruotolo BT; Ramamoorthy A A Two-Site Mechanism for the Inhibition of IAPP Amyloidogenesis by Zinc. J. Mol. Biol 2011, 410, 294–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (729).Brender JR; Krishnamoorthy J; Messina GML; Deb A; Vivekanandan S; La Rosa C; Penner-Hahn JE; Ramamoorthy A Zinc Stabilization of Prefibrillar Oligomers of Human Islet Amyloid Polypeptide. Chem. Commun 2013, 49, 3339–3341. [DOI] [PubMed] [Google Scholar]
- (730).Wineman-Fisher V; Miller Y Effect of Zn2+ Ions on the Assembly of Amylin Oligomers: Insight Into the Molecular Mechanisms. Phys. Chem. Chem. Phys 2016, 18, 21590–21599. [DOI] [PubMed] [Google Scholar]
- (731).Wineman-Fisher V; Miller Y Insight Into a New Binding Site of Zinc Ions in Fibrillar Amylin. ACS Chem. Neurosci 2017, 8, 2078–2087. [DOI] [PubMed] [Google Scholar]
- (732).Uversky VN; Li J; Fink AL Metal-triggered Structural Transformations, Aggregation, and Fibrillation of Human Alpha-Synuclein. A Possible Molecular NK Between Parkinson’s Disease and Heavy Metal Exposure. J. Biol. Chem 2001, 276, 44284–44296. [DOI] [PubMed] [Google Scholar]
- (733).Binolfi A; Rasia RM; Bertoncini CW; Ceolin M; Zweckstetter M; Griesinger C; Jovin TM; Fernández CO Interaction of Alpha-Synuclein With Divalent Metal Ions Reveals Key Differences: A Link Between Structure, Binding Specificity and Fibrillation Enhancement. J. Am. Chem. Soc 2006, 128, 9893–9901. [DOI] [PubMed] [Google Scholar]
- (734).Gonzalez N; Arcos-López T; König A; Quintanar L; Menacho Marquez M; Outeiro TF; Fernández CO Effects of Alpha-Synuclein Post-Translational Modifications on Metal Binding. J. Neurochem 2019, 150, 507–521. [DOI] [PubMed] [Google Scholar]
- (735).De Ricco R; Valensin D; Dell’Acqua S; Casella L; Dorlet P; Faller P; Hureau C Remote His50 Acts as a Coordination Switch in the High-Affinity N-Terminal Centered Copper(II) Site of alpha-Synuclein. Inorg. Chem 2015, 54, 4744–4751. [DOI] [PubMed] [Google Scholar]
- (736).Tian Y; Stanyon HF; Barritt JD; Mayet U; Patel P; Karamani E; Fusco G; Viles JH Copper2+ Binding to α-Synuclein. Histidine50 Can Form a Ternary Complex With Cu2+ at the N-Terminus but Not a Macrochelate. Inorg. Chem 2019, 58, 15580–15589. [DOI] [PubMed] [Google Scholar]
- (737).Sandal M; Valle F; Tessari I; Mammi S; Bergantino E; Musiani F; Brucale M; Bubacco L; Samorì B Conformational Equilibria in Monomeric α-Synuclein at the Single-Molecule Level. PLoS Biol 2008, 6, No. e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (738).Rasia RM; Bertoncini CW; Marsh D; Hoyer W; Cherny D; Zweckstetter M; Griesinger C; Jovin TM; Fernández CO Structural Characterization of copper(II) Binding to Alpha-Synuclein: Insights Into the Bioinorganic Chemistry of Parkinson’s Disease. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 4294–4299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (739).Nagakannan P; Tabeshmehr P; Eftekharpour E Oxidative Damage of Lysosomes in Regulated Cell Death Systems: Pathophysiology and Pharmacologic Interventions. Free Radic. Biol. Med 2020, S0891–5849(20)30319–1. [DOI] [PubMed] [Google Scholar]
- (740).Yang GJ; Liu H; Ma DL; Leung CH Rebalancing Metal Dyshomeostasis for Alzheimer’s Disease Therapy. JBIC, J. Biol. Inorg. Chem 2019, 24, 1159–1170. [DOI] [PubMed] [Google Scholar]
- (741).van Es MA; Hardiman O; Chio A; Al-Chalabi A; Pasterkamp RJ; Veldink JH; Van den Berg LH Amyotrophic Lateral Sclerosis. Lancet 2017, 390, 2084–2098. [DOI] [PubMed] [Google Scholar]
- (742).Redler RL; Dokholyan NV The Complex Molecular Biology of Amyotrophic Lateral Sclerosis (ALS). Prog. Mol. Biol. Transl. Sci 2012, 107, 215–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (743).Al-Chalabi A; Hardiman O The Epidemiology of ALS: A Conspiracy of genes, Environment and Time. Nat. Rev. Neurol 2013, 9, 617–628. [DOI] [PubMed] [Google Scholar]
- (744).Mejzini R; Flynn LL; Pitout IL; Fletcher S; Wilton SD; Akkari PA ALS Genetics, Mechanisms, and Therapeutics: Where are we now? Front. Neurosci 2019, 13, 1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (745).Alsultan AA; Waller R; Heath PR; Kirby J The Genetics of Amyotrophic Lateral Sclerosis: Current Insights. Degener. Neurol. Neuromuscular Dis 2016, 6, 49–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (746).Chen S; Sayana P; Zhang X; Le W Genetics of Amyotrophic Lateral Sclerosis: an update. Mol. Neurodegener 2013, 8, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (747).Rosen DR; Siddique T; Patterson D; Figlewicz DA; Sapp P; Hentati A; Donaldson D; Goto J; O’Regan JP; Deng HX; et al. Mutations in Cu/Zn Superoxide Dismutase Gene are associated with Familial Amyotrophic Lateral Sclerosis. Nature 1993, 362, 59–62. [DOI] [PubMed] [Google Scholar]
- (748).Hayashi Y; Homma K; Ichijo H SOD1 in Neurotoxicity and its Controversial Roles in SOD1Mutation-negative ALS. Adv. Biol. Regul 2016, 60, 95–104. [DOI] [PubMed] [Google Scholar]
- (749).Gurney ME; Pu H; Chiu AY; Dal Canto MC; Polchow CY; Alexander DD; Caliendo J; Hentati A; Kwon YW; Deng HX; et al. Motor Neuron Degeneration in Mice that Express a Human Cu, Zn Superoxide Dismutase Mutation. Science 1994, 264, 1772–1775. [DOI] [PubMed] [Google Scholar]
- (750).Reaume AG; Elliott JL; Hoffman EK; Kowall NW; Ferrante RJ; Siwek DR; Wilcox HM; Flood DG; Beal MF; Brown RH Jr; et al. Motor Neurons in Cu/Zn Superoxide Dismutase-deficient Mice Develop Normally but Exhibit Enhanced Cell Death after Axonal Injury. Nat. Genet 1996, 13, 43–47. [DOI] [PubMed] [Google Scholar]
- (751).Ayers JI; Fromholt SE; O’Neal VM; Diamond JH; Borchelt DR Prion-like Propagation of Mutant SOD1Misfolding and Motor Neuron Disease Spread along Neuroanatomical Pathways. Acta Neuropathol 2016, 131, 103–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (752).Forsberg K; Graffmo K; Pakkenberg B; Weber M; Nielsen M; Marklund S; Brännström T; Andersen PM Misfolded SOD1 Inclusions in Patients with Mutations in C9orf72 and other ALS/FTD-associated Genes. J. Neurol., Neurosurg. Psychiatry 2019, 90, 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (753).Proctor EA; Fee L; Tao Y; Redler RL; Fay JM; Zhang Y; Lv Z; Mercer IP; Deshmukh M; Lyubchenko YL; et al. Nonnative SOD1 Trimer is Toxic to Motor Neurons in a Model of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 614–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (754).Zhu C; Beck MV; Griffith JD; Deshmukh M; Dokholyan NV Large SOD1 Aggregates, unlike Trimeric SOD1, Do not Impact Cell Viability in a Model of Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 4661–4665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (755).Farrawell NE; Lambert-Smith IA; Warraich ST; Blair IP; Saunders DN; Hatters DM; Yerbury JJ Distinct Partitioning of ALS Associated TDP-43, FUS and SOD1Mutants into Cellular Inclusions. Sci. Rep 2015, 5, 13416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (756).Kim HJ; Taylor JP Lost in Transportation: Nucleocytoplasmic Transport Defects in ALS and other Neurodegenerative Diseases. Neuron 2017, 96, 285–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (757).Gautam M; Jara JH; Kocak N; Rylaarsdam LE; Kim KD; Bigio EH; Özdinler PH Mitochondria, ER, and Nuclear Membrane Defects Reveal Early Mechanisms for upper Motor Neuron Vulnerability with respect to TDP-43 Pathology. Acta Neuropathol 2019, 137, 47–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (758).Chou CC; Zhang Y; Umoh ME; Vaughan SW; Lorenzini I; Liu F; Sayegh M; Donlin-Asp PG; Chen YH; Duong DM; et al. TDP-43 pathology Disrupts Nuclear Pore Complexes and Nucleocytoplasmic Transport in ALS/FTD. Nat. Neurosci 2018, 21, 228–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (759).Aulas A; Vande Velde C Alterations in Stress Granule Dynamics driven by TDP-43 and FUS: a Link to Pathological Inclusions in ALS? Front. Cell. Neurosci 2015, 9, 423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (760).Zeineddine R; Farrawell NE; Lambert-Smith IA; Yerbury JJ Addition of Exogenous SOD1 Aggregates Causes TDP-43 Mislocalisation and Aggregation. Cell Stress Chaperones 2017, 22, 893–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (761).Vildan C; Sule D; Turker B; Hilmi U; Sibel KB Genetic Alterations of C9orf72, SOD1, TARDBP, FUS, and UBQLN2 Genes in Patients with Amyotrophic Lateral Sclerosis. Cogent Medicine 2019, 6, 1582400. [Google Scholar]
- (762).Jeon GS; Shim YM; Lee DY; Kim JS; Kang M; Ahn SH; Shin JY; Geum D; Hong YH; Sung JJ Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1Mutations. Mol. Neurobiol 2019, 56, 2007–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (763).Higashi S; Tsuchiya Y; Araki T; Wada K; Kabuta T TDP-43 physically Interacts with Amyotrophic Lateral Sclerosis-linked Mutant CuZn Superoxide Dismutase. Neurochem. Int 2010, 57, 906–913. [DOI] [PubMed] [Google Scholar]
- (764).Pokrishevsky E; Grad LI; Yousefi M; Wang J; Mackenzie IR; Cashman NR Aberrant Localization of FUS and TDP43 is Associated with Misfolding of SOD1 in Amyotrophic Lateral Sclerosis. PLoS One 2012, 7, No. e35050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (765).Liu EY; Cali CP; Lee EB RNA Metabolism in Neurodegenerative Disease. Dis. Model Mech 2017, 10, 509–518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (766).Dewey CM; Cenik B; Sephton CF; Johnson BA; Herz J; Yu G TDP-43 Aggregation in Neurodegeneration: are Stress Granules the key? Brain Res 2012, 1462, 16–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (767).Buratti E; Baralle FE TDP-43: Gumming up Neurons through protein-protein and protein-RNA Interactions. Trends Biochem. Sci 2012, 37, 237–247. [DOI] [PubMed] [Google Scholar]
- (768).Blokhuis AM; Groen EJ; Koppers M; van den Berg LH; Pasterkamp RJ Protein Aggregation in Amyotrophic Lateral Sclerosis. Acta Neuropathol 2013, 125, 777–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (769).De Vos KJ; Hafezparast M Neurobiology of Axonal Transport Defects in Motor Neuron Diseases: Opportunities for Translational Research? Neurobiol. Dis 2017, 105, 283–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (770).Spaulding EL; Burgess RW Accumulating Evidence for Axonal Translation in Neuronal Homeostasis. Front. Neurosci 2017, 11, 312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (771).Burk K; Pasterkamp RJ Disrupted Neuronal Trafficking in Amyotrophic Lateral Sclerosis. Acta Neuropathol 2019, 137, 859–877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (772).Brandizzi F; Barlowe C Organization of the ER-Golgi Interface for Membrane Traffic Control. Nat. Rev. Mol. Cell Biol 2013, 14, 382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (773).Heffernan LF; Simpson JC The Trials and Tubule-ations of Rab6 Involvement in Golgi-to-ER Retrograde Transport. Biochem. Soc. Trans 2014, 42, 1453–1459. [DOI] [PubMed] [Google Scholar]
- (774).Atkin JD; Farg MA; Soo KY; Walker AK; Halloran M; Turner BJ; Nagley P; Horne MK Mutant SOD 1 Inhibits ER-Golgi Transport in Amyotrophic Lateral Sclerosis. J. Neurochem 2014, 129, 190–204. [DOI] [PubMed] [Google Scholar]
- (775).Soo KY; Halloran M; Sundaramoorthy V; Parakh S; Toth RP; Southam KA; McLean CA; Lock P; King A; Farg MA; et al. Rab1-dependent ER-Golgi Transport Dysfunction is a Common Pathogenic Mechanism in SOD1, TDP-43 and FUS-associated ALS. Acta Neuropathol 2015, 130, 679–697. [DOI] [PubMed] [Google Scholar]
- (776).Bosco DA; Morfini G; Karabacak NM; Song Y; Gros-Louis F; Pasinelli P; Goolsby H; Fontaine B; Lemay N; McKenna-Yasek D; et al. Wild-type and Mutant SOD1 Share an Aberrant Conformation and a Common Pathogenic Pathway in ALS. Nat. Neurosci 2010, 13, 1396–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (777).Morfini GA; Bosco DA; Brown H; Gatto R; Kaminska A; Song Y; Molla L; Baker L; Marangoni MN; Berth S; et al. Inhibition of Fast Axonal Transport by Pathogenic SOD1 Involves Activation of p38 MAP Kinase. PLoS One 2013, 8, No. e65235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (778).Dewil M; dela Cruz VF; Van Den Bosch L; Robberecht W Inhibition of p38 Mitogen Activated Protein Kinase Activation and Mutant SOD1G93A-induced Motor Neuron Death. Neurobiol. Dis 2007, 26, 332–341. [DOI] [PubMed] [Google Scholar]
- (779).Taes I; Timmers M; Hersmus N; Bento-Abreu A; Van Den Bosch L; Van Damme P; Auwerx J; Robberecht W Hdac6 Deletion Delays Disease Progression in the SOD1G93A Mouse Model of ALS. Hum. Mol. Genet 2013, 22, 1783–1790. [DOI] [PubMed] [Google Scholar]
- (780).Munoz DG; Greene C; Perl DP; Selkoe DJ Accumulation of Phosphorylated Neurofilaments in Anterior Horn Motoneurons of Amyotrophic Lateral Sclerosis Patients. J. Neuropathol. Exp. Neurol 1988, 47, 9–18. [DOI] [PubMed] [Google Scholar]
- (781).Williamson TL; Bruijn LI; Zhu Q; Anderson KL; Anderson SD; Julien JP; Cleveland DW Absence of Neurofilaments Reduces the Selective Vulnerability of Motor Neurons and Slows Disease caused by a Familial Amyotrophic Lateral Sclerosis-linked Superoxide Dismutase 1 Mutant. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 9631–9636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (782).Prahlad V; Helfand BT; Langford GM; Vale RD; Goldman RD Fast Transport of Neurofilament Protein along Microtubules in Squid Axoplasm. J. Cell Sci 2000, 113, 3939–3946. [DOI] [PubMed] [Google Scholar]
- (783).Sunil N; Lee S; Shea TB Interference with Kinesin-based Anterograde Neurofilament Axonal Transport Increases Neurofilament-neurofilament Bundling. Cytoskeleton 2012, 69, 371–379. [DOI] [PubMed] [Google Scholar]
- (784).Pilling AD; Horiuchi D; Lively CM; Saxton WM Kinesin-1 and Dynein are the Primary Motors for Fast Transport of Mitochondria in Drosophila Motor Axons. Mol. Biol. Cell 2006, 17, 2057–2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (785).Chen S; Owens GC; Makarenkova H; Edelman DB HDAC6 Regulates Mitochondrial Transport in Hippocampal Neurons. PLoS One 2010, 5, No. e10848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (786).De Vos KJ; Chapman AL; Tennant ME; Manser C; Tudor EL; Lau KF; Brownlees J; Ackerley S; Shaw PJ; McLoughlin DM; et al. Familial Amyotrophic Lateral Sclerosis-linked SOD1Mutants Perturb Fast Axonal Transport to Reduce Axonal Mitochondria Content. Hum. Mol. Genet 2007, 16, 2720–2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (787).Xie Y; Zhou B; Lin MY; Wang S; Foust KD; Sheng ZH Endolysosomal Deficits Augment Mitochondria Pathology in Spinal Motor Neurons of Asymptomatic fALS Mice. Neuron 2015, 87, 355–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (788).Israelson A; Arbel N; Da Cruz S; Ilieva H; Yamanaka K; Shoshan-Barmatz V; Cleveland DW Misfolded Mutant SOD1 Directly Inhibits VDAC1 Conductance in a Mouse Model of Inherited ALS. Neuron 2010, 67, 575–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (789).Kirkinezos IG; Bacman SR; Hernandez D; Oca-Cossio J; Arias LJ; Perez-Pinzon MA; Bradley WG; Moraes CT Cytochrome c Association with the Inner Mitochondrial Membrane is Impaired in the CNS of G93A-SOD1 mice. J. Neurosci 2005, 25, 164–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (790).Mattiazzi M; D’Aurelio M; Gajewski CD; Martushova K; Kiaei M; Beal MF; Manfredi G Mutated Human SOD1 Causes Dysfunction of Oxidative Phosphorylation in Mitochondria of Transgenic Mice. J. Biol. Chem 2002, 277, 29626–29633. [DOI] [PubMed] [Google Scholar]
- (791).Pasinelli P; Belford ME; Lennon N; Bacskai BJ; Hyman BT; Trotti D; Brown RH Jr, Amyotrophic Lateral Sclerosis-associated SOD1Mutant Proteins Bind and Aggregate with Bcl-2 in Spinal Cord Mitochondria. Neuron 2004, 43, 19–30. [DOI] [PubMed] [Google Scholar]
- (792).Edens BM; Miller N; Ma YC Impaired Autophagy and Defective Mitochondrial Function: Converging Paths on the Road to Motor Neuron Degeneration. Front. Cell. Neurosci 2016, 10, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (793).Tadic V; Prell T; Lautenschlaeger J; Grosskreutz J The ER Mitochondria Calcium Cycle and ER Stress Response as Therapeutic Targets in Amyotrophic Lateral Sclerosis. Front. Cell. Neurosci 2014, 8, 147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (794).Yuan S; Zhang ZW; Li ZL Cell Death-autophagy Loop and Glutamate-glutamine Cycle in Amyotrophic Lateral Sclerosis. Front. Mol. Neurosci 2017, 10, 231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (795).Foran E; Trotti D Glutamate Transporters and the Excitotoxic Path to Motor Neuron Degeneration in Amyotrophic Lateral Sclerosis. Antioxid. Redox Signaling 2009, 11, 1587–1602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (796).Kim K; Lee S-G; Kegelman TP; Su ZZ; Das SK; Dash R; Dasgupta S; Barral PM; Hedvat M; Diaz P; et al. Role of Excitatory Amino Acid Transporter-2 (EAAT2) and Glutamate in Neurodegeneration: Opportunities for Developing Novel Therapeutics. J. Cell. Physiol 2011, 226, 2484–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (797).Tortarolo M; Crossthwaite AJ; Conforti L; Spencer JP; Williams RJ; Bendotti C; Rattray M Expression of SOD1 G93A or wild-type SOD1 in Primary Cultures of Astrocytes down-regulates the Glutamate Transporter GLT-1: Lack of Involvement of Oxidative Stress. J. Neurochem 2004, 88, 481–493. [DOI] [PubMed] [Google Scholar]
- (798).Roy J; Minotti S; Dong L; Figlewicz DA; Durham HD Glutamate Potentiates the Toxicity of Mutant Cu/Zn-Superoxide Dismutase in Motor Neurons by Postsynaptic calcium-dependent Mechanisms. J. Neurosci 1998, 18, 9673–9684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (799).Bendotti C; Tortarolo M; Suchak SK; Calvaresi N; Carvelli L; Bastone A; Rizzi M; Rattray M; Mennini T Transgenic SOD1 G93A Mice Develop Reduced GLT-1 in Spinal Cord without Alterations in Cerebrospinal Fluid Glutamate Levels. J. Neurochem 2001, 79, 737–746. [DOI] [PubMed] [Google Scholar]
- (800).Lin CLG; Kong Q; Cuny GD; Glicksman MA Glutamate Transporter EAAT2: a New Target for the Treatment of Neurodegenerative Diseases. Future Med. Chem 2012, 4, 1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (801).Doble A The Pharmacology and Mechanism of Action of Riluzole. Neurology 1996, 47, 233S–241S. [DOI] [PubMed] [Google Scholar]
- (802).Fujimori K; Ishikawa M; Otomo A; Atsuta N; Nakamura R; Akiyama T; Hadano S; Aoki M; Saya H; Sobue G; et al. Modeling Sporadic ALS in iPSC-derived Motor Neurons Identifies a Potential Therapeutic Agent. Nat. Med 2018, 24, 1579–1589. [DOI] [PubMed] [Google Scholar]
- (803).Da Cruz S; Bui A; Saberi S; Lee SK; Stauffer J; McAlonis-Downes M; Schulte D; Pizzo DP; Parone PA; Cleveland D; et al. Misfolded SOD1 is not a Primary Component of Sporadic ALS. Acta Neuropathol 2017, 134, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (804).Atlasi RS; Malik R; Corrales CI; Tzeplaeff L; Whitelegge JP; Cashman NR; Bitan G Investigation of Anti-SOD1 Antibodies Yields New Structural Insight into SOD1Misfolding and Surprising Behavior of the Antibodies Themselves. ACS Chem. Biol 2018, 13, 2794–2807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (805).Mackenzie IR; Bigio EH; Ince PG; Geser F; Neumann M; Cairns NJ; Kwong LK; Forman MS; Ravits J; Stewart H; et al. Pathological TDP-43 Distinguishes Sporadic Amyotrophic Lateral Sclerosis from Amyotrophic Lateral Sclerosis with SOD1Mutations. Ann. Neurol 2007, 61, 427–434. [DOI] [PubMed] [Google Scholar]
- (806).Khare SD; Caplow M; Dokholyan NV The Rate and Equilibrium Constants for a Multistep reaction Sequence for the Aggregation of Superoxide Dismutase in Amyotrophic Lateral Sclerosis. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 15094–15099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (807).Abel O; Powell JF; Andersen PM; Al-Chalabi A ALSoD: A user-friendly Online Bioinformatics Tool for Amyotrophic Lateral Sclerosis Genetics. Hum. Mutat 2012, 33, 1345–1351. [DOI] [PubMed] [Google Scholar]
- (808).Khare SD; Ding F; Dokholyan NV Folding of Cu, Zn Superoxide Dismutase and Familial Amyotrophic Lateral Sclerosis. J. Mol. Biol 2003, 334, 515–525. [DOI] [PubMed] [Google Scholar]
- (809).Khare SD; Dokholyan NV Common Dynamical Signatures of Familial Amyotrophic Lateral Sclerosis-associated Structurally Diverse Cu, Zn Superoxide Dismutase Mutants. Proc. Natl. Acad. Sci. U. S. A 2006, 103, 3147–3152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (810).Khare SD; Wilcox KC; Gong P; Dokholyan NV Sequence and Structural Determinants of Cu, Zn Superoxide Dismutase Aggregation. Proteins: Struct., Funct., Genet 2005, 61 (3), 617–632. [DOI] [PubMed] [Google Scholar]
- (811).Khare SD; Caplow M; Dokholyan NV FALS Mutations in Cu, Zn Superoxide Dismutase Destabilize the Dimer and Increase Dimer Dissociation Propensity: A Large-scale Thermodynamic Analysis. Amyloid 2006, 13, 226–235. [DOI] [PubMed] [Google Scholar]
- (812).Ding F; Dokholyan NV Dynamical Roles of Metal Ions and the Disulfide Bond in Cu, Zn Superoxide Dismutase Folding and Aggregation. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 19696–19701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (813).Redler RL; Shirvanyants D; Dagliyan O; Ding F; Kim DN; Kota P; Proctor EA; Ramachandran S; Tandon A; Dokholyan NV Computational Approaches to Understanding Protein Aggregation in Neurodegeneration. J. Mol. Cell Biol 2014, 6, 104–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (814).Banks CJ; Andersen JL Mechanisms of SOD1 Regulation by Post-translational Modifications. Redox Biol 2019, 26, 101270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (815).Wilcox KC; Zhou L; Jordon JK; Huang Y; Yu Y; Redler RL; Dokholyan NV Modifications of Superoxide Dismutase (sod1) in Human Erythrocytes a Possible Role in Amyotrophic Lateral Sclerosis. J. Biol. Chem 2009, 284, 13940–13947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (816).Redler RL; Fee L; Fay JM; Caplow M; Dokholyan NV Non-native Soluble Oligomers of Cu/Zn Superoxide Dismutase (SOD1) Contain a Conformational Epitope Linked to Cytotoxicity in Amyotrophic Lateral Sclerosis (ALS). Biochemistry 2014, 53, 2423–2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (817).Redler RL; Wilcox KC; Proctor EA; Fee L; Caplow M; Dokholyan NV Glutathionylation at Cys-111 Induces Dissociation of wild type and FALS Mutant SOD1 Dimers. Biochemistry 2011, 50, 7057–7066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (818).Fujiwara N; Nakano M; Kato S; Yoshihara D; Ookawara T; Eguchi H; Taniguchi N; Suzuki K Oxidative Modification to Cysteine Sulfonic Acid of Cys111 in Human copper-zinc Superoxide Dismutase. J. Biol. Chem 2007, 282, 35933–35944. [DOI] [PubMed] [Google Scholar]
- (819).Dewhurst HM; Choudhury S; Torres MP Structural Analysis of PTM Hotspots (SAPH-ire)-a Quantitative Informatics Method Enabling the Discovery of Novel Regulatory Elements in Protein Families. Mol. Cell. Proteomics 2015, 14, 2285–2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (820).Marin EP; Derakhshan B; Lam TT; Davalos A; Sessa WC Endothelial Cell Palmitoylproteomic Identifies Novel lipid-modified Targets and Potential Substrates for Protein acyl transferases. Circ. Res 2012, 110, 1336–1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (821).Antinone SE; Ghadge GD; Lam TT; Wang L; Roos RP; Green WN Palmitoylation of Superoxide Dismutase 1 (SOD1) is Increased for Familial Amyotrophic Lateral Sclerosis-linked SOD1Mutants. J. Biol. Chem 2013, 288, 21606–21617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (822).Auclair JR; Brodkin HR; D’Aquino JA; Petsko GA; Ringe D; Agar JN Structural Consequences of Cysteinylation of Cu/Zn-superoxide Dismutase. Biochemistry 2013, 52, 6145–6150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (823).Fay JM; Zhu C; Proctor EA; Tao Y; Cui W; Ke H; Dokholyan NV A Phosphomimetic Mutation Stabilizes SOD1 and Rescues Cell Viability in the Context of an ALS-associated Mutation. Structure 2016, 24, 1898–1906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (824).Xu WC; Liang JZ; Li C; He ZX; Yuan HY; Huang BY; Liu XL; Tang B; Pang DW; Du HN; et al. Pathological Hydrogen Peroxide Triggers the Fibrillization of wild-type SOD1 via sulfenic acid Modification of Cys-111. Cell Death Dis 2018, 9, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (825).Hjørnevik LV; Fismen L; Young FM; Solstad T; Fladmark KE Nodularin Exposure Induces SOD1 Phosphorylation and Disrupts SOD1 Co-localization with Actin Filaments. Toxins 2012, 4, 1482–1499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (826).Sutedja NA; Veldink JH; Fischer K; Kromhout H; Heederik D; Huisman MH; Wokke JH; van den Berg LH Exposure to Chemicals and Metals and Risk of Amyotrophic Lateral Sclerosis: a Systematic Review. Amyotrophic Lateral Scler 2009, 10, 302–309. [DOI] [PubMed] [Google Scholar]
- (827).Proctor EA; Mowrey DD; Dokholyan NV β-Methylamino-L-alanine Substitution of Serine in SOD1 Suggests a Direct Role in ALS etiology. PLoS Comput. Biol 2019, 15, No. e1007225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (828).Zarei S; Carr K; Reiley L; Diaz K; Guerra O; Altamirano PF; Pagani W; Lodin D; Orozco G; Chinea A A Comprehensive Review of Amyotrophic Lateral Sclerosis. Surgical neurology international 2015, 6, 171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (829).Kaur SJ; McKeown SR; Rashid S Mutant SOD1Mediated Pathogenesis of Amyotrophic Lateral Sclerosis. Gene 2016, 577, 109–118. [DOI] [PubMed] [Google Scholar]
- (830).Strange RW; Yong CW; Smith W; Hasnain SS Molecular Dynamics using Atomic-resolution Structure Reveal Structural Fluctuations that May Lead to Polymerization of Human Cu-Zn Superoxide Dismutase. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 10040–10044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (831).Nguyen P; Derreumaux P Understanding Amyloid Fibril Nucleation and Aβ Oligomer/Drug Interactions from Computer Simulations. Acc. Chem. Res 2014, 47, 603–611. [DOI] [PubMed] [Google Scholar]
- (832).Armiento V; Spanopoulou A; Kapurniotu A Peptide-Based Molecular Strategies To Interfere with Protein Misfolding, Aggregation, and Cell Degeneration. Angew. Chem., Int. Ed 2020, 59, 3372–3384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (833).Panza F; Lozupone M; Logroscino G; Imbimbo BP A Critical Appraisal of Amyloid-β-Targeting Therapies for Alzheimer Disease. Nat. Rev. Neurol 2019, 15, 73–88. [DOI] [PubMed] [Google Scholar]
- (834).Doig AJ; Derreumaux P Inhibition of Protein Aggregation and Amyloid Formation by Small Molecules. Curr. Opin. Struct. Biol 2015, 30, 50–56. [DOI] [PubMed] [Google Scholar]
- (835).Pujols J; Peña-Díaz S; Pallarès I; Ventura S Chemical Chaperones as Novel Drugs for Parkinson’s Disease. Trends Mol. Med 2020, 26, 408–421. [DOI] [PubMed] [Google Scholar]
- (836).Mahmoudi M; Kalhor HR; Laurent S; Lynch I Protein Fibrillation and Nanoparticle Interactions: Opportunities and Challenges. Nanoscale 2013, 5, 2570–2588. [DOI] [PubMed] [Google Scholar]
- (837).Zhang M; Mao X; Yu Y; Wang C-X; Yang Y-L; Wang C Nanomaterials for Reducing Amyloid Cytotoxicity. Adv. Mater 2013, 25, 3780–3801. [DOI] [PubMed] [Google Scholar]
- (838).Ke PC; Pilkington EH; Sun Y; Javed I; Kakinen A; Peng G; Ding F; Davis TP Mitigation of Amyloidosis with Nanomaterials. Adv. Mater 2020, 32, 1901690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (839).Viswanathan GK; Shwartz D; Losev Y; Arad E; Shemesh C; Pichinuk E; Engel H; Raveh A; Jelinek R; Cooper I; et al. Purpurin Modulates Tau-Derived VQIVYK Fibrillization and Ameliorates Alzheimer’s Disease-Like Symptoms in Animal Model. Cell. Mol. Life Sci 2020, 77, 2795–2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (840).Baggett DW; Nath A The Rational Discovery of a Tau Aggregation Inhibitor. Biochemistry 2018, 57, 6099–6107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (841).Afitska K; Priss A; Yushchenko DA; Shvadchak VV Structural Optimization of Inhibitors of α-Synuclein Fibril Growth: Affinity to the Fibril End as a Crucial Factor. J. Mol. Biol 2020, 432, 967–977. [DOI] [PubMed] [Google Scholar]
- (842).Saravanan MS; Ryazanov S; Leonov A; Nicolai J; Praest P; Giese A; Winter R; Khemtemourian L; Griesinger C; Killian JA The Small Molecule Inhibitor Anle145c Thermodynamically Traps Human Islet Amyloid Peptide in the Form of Non-Cytotoxic Oligomers. Sci. Rep 2019, 9, 19023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (843).Boulton S; Selvaratnam R; Ahmed R; Van K; Cheng X; Melacini G Mechanisms of Specific versus Nonspecific Interactions of Aggregation-Prone Inhibitors and Attenuators. J. Med. Chem 2019, 62, 5063–5079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (844).Araújo AR; Camero S; Taboada P; Reis RL; Pires RA Vescalagin and Castalagin Reduce the Toxicity of Amyloid-Beta42 Oligomers Through the Remodelling of its Secondary Structure. Chem. Commun 2020, 56, 3187–3190. [DOI] [PubMed] [Google Scholar]
- (845).Li G; Yang W-Y; Li W-H; Luo Y-Y; Lim Y-J; Li Y; Paul A; Segal D; Hong L; Li Y-M Rational Design of a Cocktail of Inhibitors against Aβ Aggregation. Chem. - Eur. J 2020, 26, 3499–3503. [DOI] [PubMed] [Google Scholar]
- (846).Cox SJ; Rodriguez Camargo DC; Lee Y-H; Ivanova MI; Padmini V; Reif B; Ramamoorthy A Small Molecule Induced Toxic Human-IAPP Species Characterized by NMR. Chem. Commun 2020, 56, 13129–13132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (847).Cox SJ; Lam B; Prasad A; Marietta HA; Stander NV; Joel JG; Sahoo BR; Guo F; Stoddard AK; Ivanova MI; Ramamoorthy A High-Throughput Screening at the Membrane Interface Reveals Inhibitors of Amyloid-β. Biochemistry 2020, 59, 2249–2258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (848).Luo J; Yu C-H; Yu H; Borstnar R; Kamerlin SCL; Gräslund A; Abrahams JP; Wärmländer SKTS Cellular Polyamines Promote Amyloid-Beta (Aβ) Peptide Fibrillation and Modulate the Aggregation Pathways. ACS Chem. Neurosci 2013, 4, 454–462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (849).Luo J; Mohammed I; Wärmländer SKTS; Hiruma Y; Gräslund A; Abrahams JP Endogenous Polyamines Reduce the Toxicity of Soluble Aβ Peptide Aggregates Associated with Alzheimer’s Disease. Biomacromolecules 2014, 15, 1985–1991. [DOI] [PubMed] [Google Scholar]
- (850).Limbocker R; Chia S; Ruggeri FS; Perni M; Cascella R; Heller GT; Meisl G; Mannini B; Habchi J; Michaels TCT; et al. Trodusquemine Enhances Aβ42 Aggregation but Suppresses its Toxicity by Displacing Oligomers from Cell Membranes. Nat. Commun 2019, 10, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (851).Sahoo BR; Genjo T; Nakayama TW; Stoddard AK; Ando T; Yasuhara K; Fierke CA; Ramamoorthy A A Cationic Polymethacrylate-Copolymer Acts as an Agonist for β-Amyloid and an Antagonist for Amylin Fibrillation. Chem. Sci 2019, 10, 3976–3986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (852).Assarsson A; Linse S; Cabaleiro-Lago C Effects of Polyamino Acids and Polyelectrolytes on Amyloid β Fibril Formation. Langmuir 2014, 30, 8812–8818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (853).Prade E; Barucker C; Sarkar R; Althoff-Ospelt G; Lopez del Amo JM; Hossain S; Zhong Y; Multhaup G; Reif B Sulindac Sulfide Induces the Formation of Large Oligomeric Aggregates of the Alzheimer’s Disease Amyloid-β Peptide Which Exhibit Reduced Neurotoxicity. Biochemistry 2016, 55, 1839–1849. [DOI] [PubMed] [Google Scholar]
- (854).Young LM; Ashcroft AE; Radford SE Small Molecule Probes of Protein Aggregation. Curr. Opin. Chem. Biol 2017, 39, 90–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (855).Davinelli S; Sapere N; Zella D; Bracale R; Intrieri M; Scapagnini G Pleiotropic Protective Effects of Phytochemicals in Alzheimer’s Disease. Oxid. Med. Cell. Longevity 2012, 2012, 386527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (856).Sparks S; Liu G; Robbins KJ; Lazo ND Curcumin Modulates the Self-Assembly of the Islet Amyloid Polypeptide by Disassembling α-Helix. Biochem. Biophys. Res. Commun 2012, 422, 551–555. [DOI] [PubMed] [Google Scholar]
- (857).Daval M; Bedrood S; Gurlo T; Huang C-J; Costes S; Butler PC; Langen R The Effect of Curcumin on Human Islet Amyloid Polypeptide Misfolding and Toxicity. Amyloid 2010, 17, 118–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (858).Pithadia AS; Bhunia A; Sribalan R; Padmini V; Fierke CA; Ramamoorthy A Influence of a Curcumin Derivative on hIAPP Aggregation in the Absence and Presence of Lipid Membranes. Chem. Commun 2016, 52, 942–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (859).Cao P; Raleigh DP Analysis of the Inhibition and Remodeling of Islet Amyloid Polypeptide Amyloid Fibers by Flavanols. Biochemistry 2012, 51, 2670–2683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (860).Ahmed R; VanSchouwen B; Jafari N; Ni X; Ortega J; Melacini G Molecular Mechanism for the (−)-Epigallocatechin Gallate-Induced Toxic to Nontoxic Remodeling of Aβ Oligomers. J. Am. Chem. Soc 2017, 139, 13720–13734. [DOI] [PubMed] [Google Scholar]
- (861).Meng F; Abedini A; Plesner A; Verchere CB; Raleigh DP The Flavanol (−)-Epigallocatechin 3-Gallate Inhibits Amyloid Formation by Islet Amyloid Polypeptide, Disaggregates Amyloid Fibrils, and Protects Cultured Cells against IAPP-Induced Toxicity. Biochemistry 2010, 49, 8127–8133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (862).Lee Y-H; Lin Y; Cox SJ; Kinoshita M; Sahoo BR; Ivanova M; Ramamoorthy A Zinc Boosts EGCG’s hIAPP Amyloid Inhibition both in Solution and Membrane. Biochim. Biophys. Acta, Proteins Proteomics 2019, 1867, 529–536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (863).Noor H; Cao P; Raleigh DP Morin Hydrate Inhibits Amyloid Formation by Islet Amyloid Polypeptide and Disaggregates Amyloid Fibers. Protein Sci 2012, 21, 373–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (864).Meng F; Abedini A; Plesner A; Middleton CT; Potter KJ; Zanni MT; Verchere CB; Raleigh DP The Sulfated Triphenyl Methane Derivative Acid Fuchsin Is a Potent Inhibitor of Amyloid Formation by Human Islet Amyloid Polypeptide and Protects against the Toxic Effects of Amyloid Formation. J. Mol. Biol 2010, 400, 555–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (865).Franko A; Rodriguez Camargo DC; Böddrich A; Garg D; Rodriguez Camargo A; Rathkolb B; Janik D; Aichler M; Feuchtinger A; Neff F; et al. Epigallocatechin Gallate (EGCG) Reduces the Intensity of Pancreatic Amyloid Fibrils in Human Islet Amyloid Polypeptide (hIAPP) Transgenic Mice. Sci. Rep 2018, 8, 1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (866).Matthes D; Gapsys V; Griesinger C; de Groot BL Resolving the Atomistic Modes of Anle138b Inhibitory Action on Peptide Oligomer Formation. ACS Chem. Neurosci 2017, 8, 2791–2808. [DOI] [PubMed] [Google Scholar]
- (867).Sciacca MFM; Chillemi R; Sciuto S; Greco V; Messineo C; Kotler SA; Lee D-K; Brender JR; Ramamoorthy A; La Rosa C; et al. A Blend of two Resveratrol Derivatives Abolishes hIAPP Amyloid Growth and Membrane Damage. Biochim. Biophys. Acta, Biomembr 2018, 1860, 1793–1802. [DOI] [PubMed] [Google Scholar]
- (868).Lolicato F; Raudino A; Milardi D; La Rosa C Resveratrol Interferes with the Aggregation of Membrane-Bound Human-IAPP: A Molecular Dynamics Study. Eur. J. Med. Chem 2015, 92, 876–881. [DOI] [PubMed] [Google Scholar]
- (869).Brendel M; Deussing M; Blume T; Kaiser L; Probst F; Overhoff F; Peters F; von Ungern-Sternberg B; Ryazanov S; Leonov A; et al. Late-Stage Anle138b Treatment Ameliorates Tau Pathology and Metabolic Decline in a Mouse Model of Human Alzheimer’s Disease Tau. Alzheimer’s Res. Ther 2019, 11, 67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (870).Ambure P; Bhat J; Puzyn T; Roy K Identifying Natural Compounds as Multi-Target-Directed Ligands Against Alzheimer’s Disease: an in silico Approach. J. Biomol. Struct. Dyn 2019, 37, 1282–1306. [DOI] [PubMed] [Google Scholar]
- (871).Perni M; Galvagnion C; Maltsev A; Meisl G; Muller MBD; Challa PK; Kirkegaard JB; Flagmeier P; Cohen SIA; Cascella R; et al. A Natural Product Inhibits the Initiation of alpha-synuclein Aggregation and Suppresses its Toxicity. Proc. Natl. Acad. Sci. U. S. A 2017, 114, E1009–E1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (872).Stoilova T; Colombo L; Forloni G; Tagliavini F; Salmona M A New Face for Old Antibiotics: Tetracyclines in Treatment of Amyloidoses. J. Med. Chem 2013, 56, 5987–6006. [DOI] [PubMed] [Google Scholar]
- (873).Young LM; Saunders JC; Mahood RA; Revill CH; Foster RJ; Tu L-H; Raleigh DP; Radford SE; Ashcroft AE Screening and Classifying Small-Molecule Inhibitors of Amyloid Formation using Ion Mobility Spectrometry-Mass Spectrometry. Nat. Chem 2015, 7, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (874).Ladiwala ARA; Dordick JS; Tessier PM Aromatic Small Molecules Remodel Toxic Soluble Oligomers of Amyloid β through Three Independent Pathways. J. Biol. Chem 2011, 286, 3209–3218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (875).Alghazwi M; Smid S; Musgrave I; Zhang W In vitro Studies of the Neuroprotective Activities of Astaxanthin and Fucoxanthin Against Amyloid Beta (Aβ1–42) Toxicity and Aggregation. Neurochem. Int 2019, 124, 215–224. [DOI] [PubMed] [Google Scholar]
- (876).Minh Hung H; Nguyen MT; Tran P-T; Truong VK; Chapman J; Quynh Anh LH; Derreumaux P; Vu VV; Ngo ST Impact of the Astaxanthin, Betanin and EGCG Compounds on Small Oligomers of the Amyloid Aβ40 Peptide. J. Chem. Inf. Model 2020, 60, 1399–1408. [DOI] [PubMed] [Google Scholar]
- (877).Lee D-H; Kim C-S; Lee YJ Astaxanthin Protects Against MPTP/MPP+-Induced Mitochondrial Dysfunction and ROS Production in vivo and in vitro. Food Chem. Toxicol 2011, 49, 271–280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (878).Ishiki M; Nishida Y; Ishibashi H; Wada T; Fujisaka S; Takikawa A; Urakaze M; Sasaoka T; Usui I; Tobe K Impact of Divergent Effects of Astaxanthin on Insulin Signaling in L6 Cells. Endocrinology 2013, 154, 2600–2612. [DOI] [PubMed] [Google Scholar]
- (879).Isonaka R; Hiruma H; Katakura T; Kawakami T Inhibition of Superoxide Dismutase Selectively Suppresses Growth of Rat Spinal Motor Neurons: Comparison with Phosphorylated Neurofilament-Containing Spinal Neurons. Brain Res 2011, 1425, 13–19. [DOI] [PubMed] [Google Scholar]
- (880).Du W-J; Guo J-J; Gao M-T; Hu S-Q; Dong X-Y; Han Y-F; Liu F-F; Jiang S; Sun Y Brazilin Inhibits Amyloid β-Protein Fibrillogenesis, Remodels Amyloid Fibrils and Reduces Amyloid Cytotoxicity. Sci. Rep 2015, 5, 7992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (881).Liu FF; Wang Y; Sang JC; Wei W; Zhao WP; Chen BB; Zhao F; Jia LG; Lu FP Brazilin Inhibits alpha-Synuclein Fibrillogenesis, Disrupts Mature Fibrils, and Protects against Amyloid-Induced Cytotoxicity. J. Agric. Food Chem 2019, 67, 11769–11777. [DOI] [PubMed] [Google Scholar]
- (882).Guo J; Sun W; Li L; Liu F; Lu W Brazilin Inhibits Fibrillogenesis of Human Islet Amyloid Polypeptide, Disassembles Mature Fibrils, and Alleviates Cytotoxicity. RSC Adv 2017, 7, 43491–43501. [Google Scholar]
- (883).Ngo ST; Fang S-T; Huang S-H; Chou C-L; Huy PDQ; Li MS; Chen Y-C Anti-Arrhythmic Medication Propafenone a Potential Drug for Alzheimer’s Disease Inhibiting Aggregation of Aβ: In Silico and In Vitro Studies. J. Chem. Inf. Model 2016, 56, 1344–1356. [DOI] [PubMed] [Google Scholar]
- (884).Kundu P; Das M; Tripathy K; Sahoo SK Delivery of Dual Drug Loaded Lipid Based Nanoparticles across the Blood-Brain Barrier Impart Enhanced Neuroprotection in a Rotenone Induced Mouse Model of Parkinson’s Disease. ACS Chem. Neurosci 2016, 7, 1658–1670. [DOI] [PubMed] [Google Scholar]
- (885).Nedumpully-Govindan P; Kakinen A; Pilkington EH; Davis TP; Chun Ke P; Ding F Stabilizing Off-pathway Oligomers by Polyphenol Nanoassemblies for IAPP Aggregation Inhibition. Sci. Rep 2016, 6, 19463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (886).Bhatia NK; Srivastava A; Katyal N; Jain N; Khan MAI; Kundu B; Deep S Curcumin Binds to the Pre-Fibrillar Aggregates of Cu/Zn Superoxide Dismutase (SOD1) and Alters its Amyloidogenic Pathway Resulting in Reduced Cytotoxicity. Biochim. Biophys. Acta, Proteins Proteomics 2015, 1854, 426–436. [DOI] [PubMed] [Google Scholar]
- (887).Srinivasan E; Rajasekaran R Computational Investigation of Curcumin, a Natural Polyphenol that Inhibits the Destabilization and the Aggregation of Human SOD1Mutant (Ala4Val). RSC Adv 2016, 6, 102744–102753. [Google Scholar]
- (888).Li J; Zhu M; Manning-Bog AB; Monte DAD; Fink AL Dopamine and L-dopa Disaggregate Amyloid Fibrils: Implications for Parkinson’s and Alzheimer’s Disease. FASEB J 2004, 18, 962–964. [DOI] [PubMed] [Google Scholar]
- (889).Tavanti F; Pedone A; Menziani MC Computational Insight into the Effect of Natural Compounds on the Destabilization of Preformed Amyloid-β(1–40) Fibrils. Molecules 2018, 23, 1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (890).Zou Y; Qian Z; Gong Y; Tang Y; Wei G; Zhang Q Critical Nucleus of Greek-key-like Core of α-synuclein Protofibril and its Disruption by Dopamine and Norepinephrine. Phys. Chem. Chem. Phys 2020, 22, 203–211. [DOI] [PubMed] [Google Scholar]
- (891).Lao Z; Chen Y; Tang Y; Wei G Molecular Dynamics Simulations Reveal the Inhibitory Mechanism of Dopamine against Human Islet Amyloid Polypeptide (hIAPP) Aggregation and Its Destabilization Effect on hIAPP Protofibrils. ACS Chem. Neurosci 2019, 10, 4151–4159. [DOI] [PubMed] [Google Scholar]
- (892).Saunders JC; Young LM; Mahood RA; Jackson MP; Revill CH; Foster RJ; Smith DA; Ashcroft AE; Brockwell DJ; Radford SE An in vivo Platform for Identifying Inhibitors of Protein Aggregation. Nat. Chem. Biol 2016, 12, 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (893).Wright GSA; Antonyuk SV; Kershaw NM; Strange RW; Samar Hasnain S Ligand Binding and Aggregation of Pathogenic SOD1. Nat. Commun 2013, 4, 1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (894).Huang H-J; Chang T-T; Chen H-Y; Chen CY-C Finding Inhibitors of Mutant Superoxide Dismutase-1 for Amyotrophic Lateral Sclerosis Therapy from Traditional Chinese Medicine. Evid. Based Complement Alternat. Med 2014, 2014, 156276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (895).Ehrnhoefer DE; Bieschke J; Boeddrich A; Herbst M; Masino L; Lurz R; Engemann S; Pastore A; Wanker EE EGCG Redirects Amyloidogenic Polypeptides into Unstructured, Off-Pathway Oligomers. Nat. Struct. Mol. Biol 2008, 15, 558–566. [DOI] [PubMed] [Google Scholar]
- (896).Zhang T; Zhang J; Derreumaux P; Mu Y Molecular Mechanism of the Inhibition of EGCG on the Alzheimer Aβ1–42 Dimer. J. Phys. Chem. B 2013, 117, 3993–4002. [DOI] [PubMed] [Google Scholar]
- (897).Pujols J; Peña-Díaz S; Lázaro DF; Peccati F; Pinheiro F; González D; Carija A; Navarro S; Conde-Giménez M; García J; et al. Small Molecule Inhibits α-Synuclein Aggregation, Disrupts Amyloid Fibrils, and Prevents Degeneration of Dopaminergic Neurons. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 10481–10486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (898).Mo Y; Lei J; Sun Y; Zhang Q; Wei G Conformational Ensemble of hIAPP Dimer: Insight into the Molecular Mechanism by which a Green Tea Extract inhibits hIAPP Aggregation. Sci. Rep 2016, 6, 33076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (899).Zhao B; Zhuang X; Pi Z; Liu S; Liu Z; Song F Determining the Effect of Catechins on SOD1 Conformation and Aggregation by Ion Mobility Mass Spectrometry Combined with Optical Spectroscopy. J. Am. Soc. Mass Spectrom 2018, 29, 734–741. [DOI] [PubMed] [Google Scholar]
- (900).Srinivasan E; Rajasekaran R Probing the Inhibitory Activity of Epigallocatechin-Gallate on Toxic Aggregates of Mutant (L84F) SOD1 Protein Through Geometry Based Sampling and Steered Molecular Dynamics. J. Mol. Graphics Modell 2017, 74, 288–295. [DOI] [PubMed] [Google Scholar]
- (901).Regitz C; Fitzenberger E; Mahn FL; Dussling LM; Wenzel U Resveratrol Reduces Amyloid-Beta (A Beta(1–42))-Induced Paralysis Through Targeting Proteostasis in an Alzheimer Model of Caenorhabditis Elegans. Eur. J. Nutr 2016, 55, 741–747. [DOI] [PubMed] [Google Scholar]
- (902).Gautam S; Karmakar S; Batra R; Sharma P; Pradhan P; Singh J; Kundu B; Chowdhury PK Polyphenols in Combination with β-Cyclodextrin can Inhibit and disaggregate α-Synuclein Amyloids under Cell Mimicking Conditions: A Promising Therapeutic Alternative. Biochim. Biophys. Acta, Proteins Proteomics 2017, 1865, 589–603. [DOI] [PubMed] [Google Scholar]
- (903).Zhuang XY; Li XX; Zhao B; Liu ZQ; Song FR; Lu JZ Native Mass Spectrometry Based Method for Studying the Interactions between Superoxide Dismutase 1 and Stilbenoids. ACS Chem. Neurosci 2020, 11, 184–190. [DOI] [PubMed] [Google Scholar]
- (904).Srinivasan E; Rajasekaran R Quantum Chemical and Molecular Mechanics Studies on the Assessment of Interactions Between Resveratrol and Mutant SOD1 (G93A) Protein. J. Comput.-Aided Mol. Des 2018, 32, 1347–1361. [DOI] [PubMed] [Google Scholar]
- (905).Ono K; Li L; Takamura Y; Yoshiike Y; Zhu LJ; Han F; Mao X; Ikeda T; Takasaki J; Nishijo H; et al. Phenolic Compounds Prevent Amyloid beta-Protein Oligomerization and Synaptic Dysfunction by Site-specific Binding. J. Biol. Chem 2012, 287, 14631–14643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (906).Taguchi R; Hatayama K; Takahashi T; Hayashi T; Sato Y; Sato D; Ohta K; Nakano H; Seki C; Endo Y; Tokuraku K; Uwai K Structure-activity Relations of Rosmarinic Acid Derivatives for the amyloid beta Aggregation Inhibition and Antioxidant Properties. Eur. J. Med. Chem 2017, 138, 1066–1075. [DOI] [PubMed] [Google Scholar]
- (907).Takahashi R; Ono K; Takamura Y; Mizuguchi M; Ikeda T; Nishijo H; Yamada M Phenolic Compounds Prevent the Oligomerization of α-Synuclein and Reduce Synaptic Toxicity. J. Neurochem 2015, 134, 943–955. [DOI] [PubMed] [Google Scholar]
- (908).Kamelabad MR; Sardroodi JJ; Ebrahimzadeh AR The Interaction of Curcumin and Rosmarinic Acid with Non-Amyloid-Component Domain of Alpha-Synuclein: A Molecular Dynamics Study. Chemistry Select 2020, 5, 3312–3320. [Google Scholar]
- (909).Zheng Q; Lazo ND Mechanistic Studies of the Inhibition of Insulin Fibril Formation by Rosmarinic Acid. J. Phys. Chem. B 2018, 122, 2323–2331. [DOI] [PubMed] [Google Scholar]
- (910).Shimojo Y; Kosaka K; Noda Y; Shimizu T; Shirasawa T Effect of Rosmarinic Acid in Motor Dysfunction and Life Span in a Mouse Model of Familial Amyotrophic Lateral Sclerosis. J. Neurosci. Res 2010, 88, 896–904. [DOI] [PubMed] [Google Scholar]
- (911).Xiao Y; Ma B; McElheny D; Parthasarathy S; Long F; Hoshi M; Nussinov R; Ishii Y Aβ(1–42) Fibril Structure Illuminates Self-Recognition and Replication of Amyloid in Alzheimer’s Disease. Nat. Struct. Mol. Biol 2015, 22, 499–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (912).De Genst EJ; Guilliams T; Wellens J; O’Day EM; Waudby CA; Meehan S; Dumoulin M; Hsu S-TD; Cremades N; Verschueren KHG; et al. Structure and Properties of a Complex of α-Synuclein and a Single-Domain Camelid Antibody. J. Mol. Biol 2010, 402, 326–343. [DOI] [PubMed] [Google Scholar]
- (913).Whittingham JL; Scott DJ; Chance K; Wilson A; Finch J; Brange J; Dodson G Insulin at pH 2: Structural Analysis of the Conditions Promoting Insulin Fibre Formation. J. Mol. Biol 2002, 318, 479–490. [DOI] [PubMed] [Google Scholar]
- (914).Yang F; Wang H; Logan DT; Mu X; Danielsson J; Oliveberg M The Cost of Long Catalytic Loops in Folding and Stability of the ALS-Associated Protein SOD1. J. Am. Chem. Soc 2018, 140, 16570–16579. [DOI] [PubMed] [Google Scholar]
- (915).Trott O; Olson AJ Improving the Speed and Accuracy of Docking with a New Scoring Function, Efficient Optimization, and Multithreading. J. Comput. Chem 2010, 31, 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (916).Nguyen NT; Nguyen TH; Pham TNH; Huy NT; Bay MV; Pham MQ; Nam PC; Vu VV; Ngo ST Autodock Vina Adopts More Accurate Binding Poses but Autodock4 Forms Better Binding Affinity. J. Chem. Inf. Model 2020, 60, 204–211. [DOI] [PubMed] [Google Scholar]
- (917).Yasuhara K; Arakida J; Ravula T; Ramadugu SK; Sahoo B; Kikuchi J-I; Ramamoorthy A Spontaneous Lipid Nanodisc Fomation by Amphiphilic Polymethacrylate Copolymers. J. Am. Chem. Soc 2017, 139, 18657–18663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (918).Pilkington EH; Lai M; Ge X; Stanley WJ; Wang B; Wang M; Kakinen A; Sani M-A; Whittaker MR; Gurzov EN; et al. Star Polymers Reduce Islet Amyloid Polypeptide Toxicity via Accelerated Amyloid Aggregation. Biomacromolecules 2017, 18, 4249–4260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (919).Sahoo BR; Souders CL; Ivanova M; Deng Z; Nakayama TW; Suladze S; Reif B; Ando T; Martyniuk CJ; Ramamoorthy A Conformational Tuning of Amylin by Charged SMA Copolymers. bioRxiv 2020, 2020.04.23.057547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (920).Smith JW; Jiang X; An H; Barclay AM; Licari G; Tajkhorshid E; Moore EG; Rienstra CM; Moore JS; Chen Q Polymer-Peptide Conjugates Convert Amyloid into Protein Nano-bundles through Fragmentation and Lateral Association. ACS Appl. Nano Mater 2020, 3, 937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (921).Evgrafova Z; Voigt B; Roos AH; Hause G; Hinderberger D; Balbach J; Binder WH Modulation of Amyloid β Peptide Aggregation by Hydrophilic Polymers. Phys. Chem. Chem. Phys 2019, 21, 20999–21006. [DOI] [PubMed] [Google Scholar]
- (922).Sun H; Liu J; Li S; Zhou L; Wang J; Liu L; Lv F; Gu Q; Hu B; Ma Y; Wang S Reactive Amphiphilic Conjugated Polymers for Inhibiting Amyloid β Assembly. Angew. Chem., Int. Ed 2019, 58, 5988–5993. [DOI] [PubMed] [Google Scholar]
- (923).Zhao Y; Cai J; Liu Z; Li Y; Zheng C; Zheng Y; Chen Q; Chen H; Ma F; An Y; Xiao L; Jiang C; Shi L; Kang C; Liu Y Nanocomposites Inhibit the Formation, Mitigate the Neurotoxicity, and Facilitate the Removal of β-Amyloid Aggregates in Alzheimer’s Disease Mice. Nano Lett 2019, 19, 674–683. [DOI] [PubMed] [Google Scholar]
- (924).Aso E; Martinsson I; Appelhans D; Effenberg C; Benseny-Cases N; Cladera J; Gouras G; Ferrer I; Klementieva O Poly(Propylene Imine) Dendrimers with Histidine-Maltose Shell as Novel Type of Nanoparticles for Synapse and Memory Protection. Nanomedicine 2019, 17, 198–209. [DOI] [PubMed] [Google Scholar]
- (925).Gurzov EN; Wang B; Pilkington EH; Chen P; Kakinen A; Stanley WJ; Litwak SA; Hanssen EG; Davis TP; Ding F; et al. Inhibition of hIAPP Amyloid Aggregation and Pancreatic β-Cell Toxicity by OH-Terminated PAMAM Dendrimer. Small 2016, 12, 1615–1626. [DOI] [PubMed] [Google Scholar]
- (926).Cabaleiro-Lago C; Lynch I; Dawson KA; Linse S Inhibition of IAPP and IAPP(20–29) Fibrillation by Polymeric Nanoparticles. Langmuir 2010, 26, 3453–3461. [DOI] [PubMed] [Google Scholar]
- (927).Fernández-De-Retana S; Cano-Sarabia M; Marazuela P; Sánchez-Quesada JL; Garcia-Leon A; Montañola A; Montaner J; Maspoch D; Hernández-Guillamon M Characterization of ApoJ-Reconstituted High-Density Lipoprotein (rHDL) Nanodisc for the Potential Treatment of Cerebral β-Amyloidosis. Sci. Rep 2017, 7, 14637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (928).Ye Z; Wei L; Li Y; Xiao L Efficient Modulation of β-Amyloid Peptide Fibrillation with Polymer Nanoparticles Revealed by Super-Resolution Optical Microscopy. Anal. Chem 2019, 91, 8582–8590. [DOI] [PubMed] [Google Scholar]
- (929).Mourtas S; Christodoulou P; Klepetsanis P; Gatos D; Barlos K; Antimisiaris SG Preparation of Benzothiazolyl-Decorated Nanoliposomes. Molecules 2019, 24, 1540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (930).Goyal D; Shuaib S; Mann S; Goyal B Rationally Designed Peptides and Peptidomimetics as Inhibitors of Amyloid-β (Aβ) Aggregation: Potential Therapeutics of Alzheimer’s Disease. ACS Comb. Sci 2017, 19, 55–80. [DOI] [PubMed] [Google Scholar]
- (931).Niu Z; Prade E; Malideli E; Hille K; Jussupow A; Mideksa YG; Yan L-M; Qian C; Fleisch M; Messias AC; et al. Structural Insight into IAPP-Derived Amyloid Inhibitors and Their Mechanism of Action. Angew. Chem., Int. Ed 2020, 59, 5771–5781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (932).Griner S; Seidler P; Bowler J; Murray K; Yang T; Sahay S; Sawaya M; Cascio D; Rodriguez J; Philipp S; et al. Structure-Based Inhibitors of Amyloid Beta Core Suggest a Common Interface with Tau. eLife 2019, 8, No. e46924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (933).Elfgen A; Hupert M; Bochinsky K; Tusche M; González de San Román Martin E; Gering I; Sacchi S; Pollegioni L; Huesgen PF; Hartmann R; et al. Metabolic Resistance of the D-peptide RD2 Developed for Direct Elimination of Amyloid-β Oligomers. Sci. Rep 2019, 9, 5715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (934).Dugan LL; Turetsky DM; Du C; Lobner D; Wheeler M; Almli CR; Shen CK-F; Luh T-Y; Choi DW; Lin T-S Carboxyfullerenes as Neuroprotective Agents. Proc. Natl. Acad. Sci. U. S. A 1997, 94, 9434–9439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (935).Kim JE; Lee M Fullerene inhibits β-Amyloid Peptide Aggregation. Biochem. Biophys. Res. Commun 2003, 303, 576–579. [DOI] [PubMed] [Google Scholar]
- (936).Podolski IY; Podlubnaya ZA; Kosenko EA; Mugantseva EA; Makarova EG; Marsagishvili LG; Shpagina MD; Kaminsky YG; Andrievsky GV; Klochkov VK Effects of Hydrated Forms of C60 Fullerene on Amyloid 1-Peptide Fibrillization in vitro and Performance of the Cognitive Task. J. Nanosci. Nanotechnol 2007, 7, 1479–1485. [DOI] [PubMed] [Google Scholar]
- (937).Linse S; Cabaleiro-Lago C; Xue W-F; Lynch I; Lindman S; Thulin E; Radford SE; Dawson KA Nucleation of Protein Fibrillation by Nanoparticles. Proc. Natl. Acad. Sci. U. S. A 2007, 104, 8691–8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (938).Li H; Luo Y; Derreumaux P; Wei G Carbon Nanotube Inhibits the Formation of β-Sheet-Rich Oligomers of the Alzheimer’s Amyloid-β(16–22) Peptide. Biophys. J 2011, 101, 2267–2276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (939).Xie L; Lin D; Luo Y; Li H; Yang X; Wei G Effects of Hydroxylated Carbon Nanotubes on the Aggregation of Aβ16–22 Peptides: A Combined Simulation and Experimental Study. Biophys. J 2014, 107, 1930–1938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (940).Xie L; Luo Y; Lin D; Xi W; Yang X; Wei G The Molecular Mechanism of Fullerene-Inhibited Aggregation of Alzheimer’s β-Amyloid Peptide Fragment. Nanoscale 2014, 6, 9752–9762. [DOI] [PubMed] [Google Scholar]
- (941).Wang J; Zhu Z; Bortolini C; Hoffmann SV; Amari A; Zhang HX; Liu L; Dong MD Dimensionality of Carbon Nanomaterial Impacting on the Modulation of Amyloid Peptide Assembly. Nanotechnology 2016, 27, 304001. [DOI] [PubMed] [Google Scholar]
- (942).Yang Z; Ge C; Liu J; Chong Y; Gu Z; Jimenez-Cruz CA; Chai Z; Zhou R Destruction of Amyloid Fibrils by Graphene Through Penetration and Extraction of Peptides. Nanoscale 2015, 7, 18725–18737. [DOI] [PubMed] [Google Scholar]
- (943).Mahmoudi M; Akhavan O; Ghavami M; Rezaee F; Ghiasi SMA Graphene Oxide Strongly inhibits Amyloid Beta Fibrillation. Nanoscale 2012, 4, 7322–7325. [DOI] [PubMed] [Google Scholar]
- (944).Liu Y; Xu L-P; Dai W; Dong H; Wen Y; Zhang X Graphene Quantum Dots for the Inhibition of β Amyloid Aggregation. Nanoscale 2015, 7, 19060–19065. [DOI] [PubMed] [Google Scholar]
- (945).Sun Y; Qian Z; Wei G The Inhibitory Mechanism of a Fullerene Derivative Against Amyloid-β Peptide Aggregation: an Atomistic Simulation Study. Phys. Chem. Chem. Phys 2016, 18, 12582–12591. [DOI] [PubMed] [Google Scholar]
- (946).Jin Y; Sun Y; Chen Y; Lei J; Wei G Molecular Dynamics Simulations Reveal the Mechanism of Graphene Oxide Nanosheet Inhibition of Aβ1–42 Peptide Aggregation. Phys. Chem. Chem. Phys 2019, 21, 10981–10991. [DOI] [PubMed] [Google Scholar]
- (947).Bednarikova Z; Huy PDQ; Mocanu M-M; Fedunova D; Li MS; Gazova Z Fullerenol C60(OH)16 Prevents Amyloid Fibrillization of Aβ40 - in vitro and in silico Approach. Phys. Chem. Chem. Phys 2016, 18, 18855–18867. [DOI] [PubMed] [Google Scholar]
- (948).Zhou X; Xi W; Luo Y; Cao S; Wei G Interactions of a Water-Soluble Fullerene Derivative with Amyloid-β Protofibrils: Dynamics, Binding Mechanism, and the Resulting Salt-Bridge Disruption. J. Phys. Chem. B 2014, 118, 6733–6741. [DOI] [PubMed] [Google Scholar]
- (949).Liu Z; Zou Y; Zhang Q; Chen P; Liu Y; Qian Z Distinct Binding Dynamics, Sites and Interactions of Fullerene and Fullerenols with Amyloid-β Peptides Revealed by Molecular Dynamics Simulations. Int. J. Mol. Sci 2019, 20, 2048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (950).Liu F; Wang W; Sang J; Jia L; Lu F Hydroxylated Single-Walled Carbon Nanotubes Inhibit Aβ42 Fibrillogenesis, Disaggregate Mature Fibrils, and Protect against Aβ42-Induced Cytotoxicity. ACS Chem. Neurosci 2019, 10, 588–598. [DOI] [PubMed] [Google Scholar]
- (951).Wang J; Cao Y; Li Q; Liu L; Dong M Size Effect of Graphene Oxide on Modulating Amyloid Peptide Assembly. Chem. - Eur. J 2015, 21, 9632–9637. [DOI] [PubMed] [Google Scholar]
- (952).Li Q; Liu L; Zhang S; Xu M; Wang X; Wang C; Besenbacher F; Dong M Modulating Aβ33–42 Peptide Assembly by Graphene Oxide. Chem. - Eur. J 2014, 20, 7236–7240. [DOI] [PubMed] [Google Scholar]
- (953).Chen Y; Chen Z; Sun Y; Lei J; Wei G Mechanistic Insights into the Inhibition and Size Effects of Graphene Oxide Nanosheets on the Aggregation of an Amyloid-β Peptide Fragment. Nanoscale 2018, 10, 8989–8997. [DOI] [PubMed] [Google Scholar]
- (954).Nedumpully-Govindan P; Gurzov EN; Chen P; Pilkington EH; Stanley WJ; Litwak SA; Davis TP; Ke PC; Ding F Graphene Oxide Inhibits hIAPP Amyloid Fibrillation and Toxicity in Insulin-Producing NIT-1 Cells. Phys. Chem. Chem. Phys 2016, 18, 94–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (955).Wang M; Sun Y; Cao X; Peng G; Javed I; Kakinen A; Davis TP; Lin S; Liu J; Ding F; Ke PC Graphene Quantum Dots Against Human IAPP Aggregation and Toxicity in vivo. Nanoscale 2018, 10, 19995–20006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (956).Faridi A; Sun Y; Mortimer M; Aranha RR; Nandakumar A; Li Y; Javed I; Kakinen A; Fan Q; Purcell AW; et al. Graphene Quantum Dots Rescue Protein Dysregulation of Pancreatic β-Cells Exposed to Human Islet Amyloid Polypeptide. Nano Res 2019, 12, 2827–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (957).Mo Y; Brahmachari S; Lei J; Gilead S; Tang Y; Gazit E; Wei G The Inhibitory Effect of Hydroxylated Carbon Nanotubes on the Aggregation of Human Islet Amyloid Polypeptide Revealed by a Combined Computational and Experimental Study. ACS Chem. Neurosci 2018, 9, 2741–2752. [DOI] [PubMed] [Google Scholar]
- (958).Bai C; Lin D; Mo Y; Lei J; Sun Y; Xie L; Yang X; Wei G Influence of Fullerenol on hIAPP Aggregation: Amyloid Inhibition and Mechanistic Aspects. Phys. Chem. Chem. Phys 2019, 21, 4022–4031. [DOI] [PubMed] [Google Scholar]
- (959).Bai C; Lao Z; Chen Y; Tang Y; Wei G Pristine and Hydroxylated Fullerenes Prevent the Aggregation of Human Islet Amyloid Polypeptide and Display Different Inhibitory Mechanisms. Front. Chem 2020, 8, 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (960).Sun Y; Kakinen A; Zhang C; Yang Y; Faridi A; Davis TP; Cao W; Ke PC; Ding F Amphiphilic Surface Chemistry of Fullerenols is Necessary for Inhibiting the Amyloid Aggregation of Alpha-Synuclein NACore. Nanoscale 2019, 11, 11933–11945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (961).Kim D; Yoo JM; Hwang H; Lee J; Lee SH; Yun SP; Park MJ; Lee M; Choi S; Kwon SH; et al. Graphene Quantum Dots Prevent α-Synucleinopathy in Parkinson’s Disease. Nat. Nanotechnol 2018, 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (962).Golomidov I; Bolshakova O; Komissarov A; Sharoyko V; Slepneva E; Slobodina A; Latypova E; Zherebyateva O; Tennikova T; Sarantseva S The Neuroprotective Effect of Fullerenols on a Model of Parkinson’s Disease in Drosophila Melanogaster. Biochem. Biophys. Res. Commun 2020, 523, 446–451. [DOI] [PubMed] [Google Scholar]
- (963).Reina G; González-Domínguez JM; Criado A; Vázquez E; Bianco A; Prato M Promises, Facts and Challenges for Graphene in Biomedical Applications. Chem. Soc. Rev 2017, 46, 4400–4416. [DOI] [PubMed] [Google Scholar]
- (964).Cacabelos R How Plausible is an Alzheimer’s Disease Vaccine? Expert Opin. Drug Discovery 2020, 15, 6. [DOI] [PubMed] [Google Scholar]
- (965).Baxter D Active and Passive Immunity, Vaccine Types, Excipients and Licensing. Occupational Medicine-Oxford 2007, 57, 552–556. [DOI] [PubMed] [Google Scholar]
- (966).Schenk D; Barbour R; Dunn W; Gordon G; Grajeda H; Guido T; Hu K; Huang J; Johnson-Wood K; Khan K; et al. Immunization with amyloid-beta Attenuates Alzheimer-disease-like Pathology in the PDAPP Mouse. Nature 1999, 400, 173–177. [DOI] [PubMed] [Google Scholar]
- (967).Nicoll JAR; Wilkinson D; Holmes C; Steart P; Markham H; Weller RO Neuropathology of Human Alzheimer Disease after Immunization with amyloid-β Peptide: A Case Report. Nat. Med 2003, 9, 448–452. [DOI] [PubMed] [Google Scholar]
- (968).Lee M; Bard F; Johnson-Wood K; Lee C; Hu K; Griffith SG; Black RS; Schenk D; Seubert P Abeta42 Immunization in Alzheimer’s Disease Generates Abeta N-terminal Antibodies. Ann. Neurol 2005, 58, 430–435. [DOI] [PubMed] [Google Scholar]
- (969).Vellas B; Black R; Thal LJ; Fox NC; Daniels M; McLennan G; Tompkins C; Leibman C; Pomfret M; Grundman M; Team AQ-S Long-Term Follow-Up of Patients Immunized with AN1792: Reduced Functional Decline in Antibody Responders. Curr. Alzheimer Res 2009, 6, 144–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (970).Boche D; Donald J; Love S; Harris S; Neal JW; Holmes C; Nicoll JA Reduction of Aggregated Tau in Neuronal Processes but not in the Cell Bodies after Abeta42 Immunisation in Alzheimer’s Disease. Acta Neuropathol 2010, 120, 13–20. [DOI] [PubMed] [Google Scholar]
- (971).Lacosta AM; Pascual-Lucas M; Pesini P; Casabona D; Perez-Grijalba V; Marcos-Campos I; Sarasa L; Canudas J; Badi H; Monleon I; et al. Safety, Tolerability and Immunogenicity of an Active Anti-A beta(40) Vaccine (ABvac40) in Patients with Alzheimer’s Disease: a Randomised, Double-blind, Placebo-controlled, Phase I Trial. Alzheimer’s Res. Ther 2018, 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (972).Wang CY; Wang PN; Chiu MJ; Finstad CL; Lin F; Lynn S; Tai YH; De Fang X; Zhao K; Hung CH; et al. UB-311, a Novel UBITh amyloid β peptide Vaccine for Mild Alzheimer’s Disease. Alzheimers Dement. (N Y) 2017, 3, 262–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (973).Congdon EE; Sigurdsson EM Tau-targeting therapies for Alzheimer disease. Nat. Rev. Neurol 2018, 14, 399–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (974).Asuni AA; Boutajangout A; Quartermain D; Sigurdsson EM Immunotherapy Targeting Pathological Tau Conformers in a Tangle Mouse Model Reduces Brain Pathology with Associated Functional Improvements. J. Neurosci 2007, 27, 9115–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (975).Boutajangout A; Ingadottir J; Davies JP; Sigurdsson EM P3–427: Passive Tau Immunotherapy Diminishes Functional Decline and Clears Tau aggregates in a Mouse Model of Tauopathy. Alzheimer’s Dementia 2010, 6, S578–S578. [Google Scholar]
- (976).Kontsekova E; Zilka N; Kovacech B; Novak P; Novak M First-in-man Tau Vaccine Targeting Structural Determinants Essential for Pathological tau-tau interaction Reduces Tau Oligomerisation and Neurofibrillary Degeneration in an Alzheimer’s Disease Model. Alzheimer’s Res. Ther 2014, 6, 44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (977).Axon_Neuroscience, Axon announces Positive Results from Phase II ADAMANT Trial for AADvac1 in Alzheimer’s Disease 2019. https://www.axon-neurosci.eu.
- (978).Congdon EE; Gu J; Sait HB; Sigurdsson EM Antibody Uptake into Neurons occurs primarily via Clathrin-dependent Fcgamma Receptor Endocytosis and is a Prerequisite for acute Tau Protein Clearance. J. Biol. Chem 2013, 288, 35452–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (979).Lee HE; Lim D; Lee JY; Lim SM; Pae AN Recent Tau-targeted Clinical Strategies for the Treatment of Alzheimer’s Disease. Future Med. Chem 2019, 11, 1845–1848. [DOI] [PubMed] [Google Scholar]
- (980).Kfoury N; Holmes BB; Jiang H; Holtzman DM; Diamond MI Trans-cellular Propagation of Tau Aggregation by Fibrillar Species. J. Biol. Chem 2012, 287, 19440–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (981).Park HH; Lee KY; Kim S; Lee JW; Choi NY; Lee EH; Lee YJ; Lee SH; Koh SH Novel Vaccine Peptide GV1001 effectively Blocks beta-amyloid Toxicity by Mimicking the Extra-telomeric Functions of Human Telomerase Reverse Transcriptase. Neurobiol. Aging 2014, 35, 1255–74. [DOI] [PubMed] [Google Scholar]
- (982).Park HH; Yu HJ; Kim S; Kim G; Choi NY; Lee EH; Lee YJ; Yoon MY; Lee KY; Koh SH Neural stem Cells Injured by Oxidative Stress can be Rejuvenated by GV1001, a Novel Peptide, through Scavenging Free Radicals and enhancing survival Signals. NeuroToxicology 2016, 55, 131–141. [DOI] [PubMed] [Google Scholar]
- (983).Alzforum, At CTAD, Early Failures and Hints of Success, from Small Trials 2019. https://www.alzforum.org/news/conference-coverage/ctad-early-failures-and-hints-success-small-trials#GV1001.
- (984).Vander Zanden CM; Chi EY Passive Immunotherapies Targeting Amyloid Beta and Tau Oligomers in Alzheimer’s Disease. J. Pharm. Sci 2020, 109, 68–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (985).Mangialasche F; Solomon A; Winblad B; Mecocci P; Kivipelto M Alzheimer’s Disease: Clinical trials and Drug development. Lancet Neurol 2010, 9, 702–716. [DOI] [PubMed] [Google Scholar]
- (986).Vidarsson G; Dekkers G; Rispens T IgG Subclasses and Allotypes: from Structure to effector Functions. Front. Immunol 2014, 5, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (987).Sevigny J; Chiao P; Bussière T; Weinreb PH; Williams L; Maier M; Dunstan R; Salloway S; Chen T; Ling Y; et al. The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2016, 537, 50–56. [DOI] [PubMed] [Google Scholar]
- (988).Sevigny J; Chiao P; Bussière T; Weinreb PH; Williams L; Maier M; Dunstan R; Salloway S; Chen T; Ling Y; et al. Addendum: The Antibody Aducanumab Reduces Aβ Plaques in Alzheimer’s Disease. Nature 2017, 546, 564–564. [DOI] [PubMed] [Google Scholar]
- (989).Arndt JW; Qian F; Smith BA; Quan C; Kilambi KP; Bush MW; Walz T; Pepinsky RB; Bussiere T; Hamann S; Cameron TO; Weinreb PH Structural and Kinetic Basis for the Selectivity of Aducanumab for Aggregated Forms of amyloid-beta. Sci. Rep 2018, 8, 6412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (990).Alzforum, Biogen/Eisai Halt Phase 3 Aducanumab Trials https://www.alzforum.org/news/research-news/biogeneisai-halt-phase-3-aducanumab-trials, 2019.
- (991).Selkoe DJ Alzheimer Disease and Aducanumab: Adjusting our approach. Nat. Rev. Neurol 2019, 15, 365–366. [DOI] [PubMed] [Google Scholar]
- (992).Biogen, Biogen Plans Regulatory Filing for Aducanumab in Alzheimer’s Disease Based on New Analysis of Larger Dataset from Phase 3 Studies https://investors.biogen.com/news-releases/news-release-details/biogen-plans-regulatory-filing-aducanumab-alzheimers-disease, 2019.
- (993).Howard R; Liu KY Questions EMERGE as Biogen Claims Aducanumab Turnaround. Nat. Rev. Neurol 2020, 16, 63–64. [DOI] [PubMed] [Google Scholar]
- (994).Lord A; Gumucio A; Englund H; Sehlin D; Sundquist VS; Soderberg L; Möller C; Gellerfors P; Lannfelt L; Pettersson FE; Nilsson LNG An Amyloid-beta Protofibril-selective Antibody Prevents Amyloid Formation in a Mouse Model of Alzheimer’s Disease. Neurobiol. Dis 2009, 36, 425–434. [DOI] [PubMed] [Google Scholar]
- (995).Logovinsky V; Satlin A; Lai R; Swanson C; Kaplow J; Osswald G; Basun H; Lannfelt L Safety and Tolerability of BAN2401-a Clinical Study in Alzheimer’s Disease with a Protofibril Selective Abeta Antibody. Alzheimer’s Res. Ther 2016, 8, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (996).Satlin A; Wang J; Logovinsky V; Berry S; Swanson C; Dhadda S; Berry DA Design of a Bayesian Adaptive Phase 2 Proof-of-concept Trial for BAN2401, a Putative Disease-modifying Monoclonal Antibody for the Treatment of Alzheimer’s Disease. Alzheimers Dement (NY: ) 2016, 2, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (997).Bohrmann B; Baumann K; Benz J; Gerber F; Huber W; Knoflach F; Messer J; Oroszlan K; Rauchenberger R; Richter WF; et al. Gantenerumab: A Novel Human Anti-A beta Antibody Demonstrates Sustained Cerebral Amyloid-b Binding and Elicits Cell-Mediated Removal of Human Amyloid-beta. J. Alzheimer’s Dis 2012, 28, 49–69. [DOI] [PubMed] [Google Scholar]
- (998).Honig LS; Vellas B; Woodward M; Boada M; Bullock R; Borrie M; Hager K; Andreasen N; Scarpini E; Liu-Seifert H; et al. Trial of Solanezumab for Mild Dementia Due to Alzheimer’s Disease. N. Engl. J. Med 2018, 378, 321–330. [DOI] [PubMed] [Google Scholar]
- (999).Ostrowitzki S; Lasser RA; Dorflinger E; Scheltens P; Barkhof F; Nikolcheva T; Ashford E; Retout S; Hofmann C; Delmar P; et al. A phase III Randomized Trial of Gantenerumab in Prodromal Alzheimer’s Disease. Alzheimer’s Res. Ther 2017, 9, 95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1000).Alzforum, Solanezumab https://www.alzforum.org/therapeutics/solanezumab, 2019.
- (1001).Jawhar S; Wirths O; Bayer TA Pyroglutamate Amyloid-beta (A beta): A Hatchet Man in Alzheimer Disease. J. Biol. Chem 2011, 286, 38825–38832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1002).Demattos RB; Lu J; Tang Y; Racke MM; Delong CA; Tzaferis JA; Hole JT; Forster BM; McDonnell PC; Liu F; et al. A Plaque-specific Antibody Clears Existing beta-amyloid Plaques in Alzheimer’s Disease mice. Neuron 2012, 76, 908–20. [DOI] [PubMed] [Google Scholar]
- (1003).Alzforum, Four Immunotherapies Now Banish Amyloid From the Brain https://www.alzforum.org/news/conference-coverage/four-immunotherapies-now-banish-amyloid-brain, 2018.
- (1004).Bright J; Hussain S; Dang V; Wright S; Cooper B; Byun T; Ramos C; Singh A; Parry G; Stagliano N; Griswold-Prenner I Human Secreted Tau Increases amyloid-beta Production. Neurobiol. Aging 2015, 36, 693–709. [DOI] [PubMed] [Google Scholar]
- (1005).Doig AJ Positive Feedback Loops in Alzheimer’s Disease: The Alzheimer’s Feedback Hypothesis. J. Alzheimer’s Dis 2018, 66, 25–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1006).Boxer AL; Qureshi I; Ahlijanian M; Grundman M; Golbe LI; Litvan I; Honig LS; Tuite P; McFarland NR; O’Suilleabhain P; et al. Safety of the Tau-directed Monoclonal Antibody BIIB092 in Progressive Supranuclear Palsy: a Randomised, Placebo-controlled, Multiple Ascending Dose Phase 1b Trial. Lancet Neurol 2019, 18, 549–558. [DOI] [PubMed] [Google Scholar]
- (1007).Lee SH; Le Pichon CE; Adolfsson O; Gafner V; Pihlgren M; Lin H; Solanoy H; Brendza R; Ngu H; Foreman O; et al. Antibody-Mediated Targeting of Tau In Vivo Does Not Require Effector Function and Microglial Engagement. Cell Rep 2016, 16, 1690–1700. [DOI] [PubMed] [Google Scholar]
- (1008).Alzforum, Zagotenemab 2018, https://www.alzforum.org/therapeutics/zagotenemab.
- (1009).Games D; Seubert P; Rockenstein E; Patrick C; Trejo M; Ubhi K; Ettle B; Ghassemiam M; Barbour R; Schenk D; et al. Axonopathy in an alpha-Synuclein Transgenic Model of Lewy Body Disease Is Associated with Extensive Accumulation of C-Terminal Truncated alpha-Synuclein. Am. J. Pathol 2013, 182, 940–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1010).Jankovic J; Goodman I; Safirstein B; Schenk D; Kinney G; Koller M; Ness D; Griffith S; Grundman M; Soto J; et al. Results from a Phase 1b Multiple Ascending-dose study of prx002, an Anti-alpha-synuclein Monoclonal Antibody, in Patients with Parkinson’s Disease. Mov. Disord 2017, 32, 1.28124430 [Google Scholar]
- (1011).Weihofen A; Liu YT; Arndt JW; Huy C; Quan C; Smith BA; Baeriswyl JL; Cavegn N; Senn L; Su LH; et al. Development of an Aggregate-selective, Human-derived alpha-synuclein Antibody BIIB054 that Ameliorates Disease Phenotypes in Parkinson’s Disease Models. Neurobiol. Dis 2019, 124, 276–288. [DOI] [PubMed] [Google Scholar]
- (1012).Cascella R; Perni M; Chen SW; Fusco G; Cecchi C; Vendruscolo M; Chiti F; Dobson CM; De Simone A Probing the Origin of the Toxicity of Oligomeric Aggregates of α-Synuclein with Antibodies. ACS Chem. Biol 2019, 14, 1352–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1013).Hamblin MR Photobiomodulation for Alzheimer’s Disease: Has the Light Dawned? Photonics 2019, 6, 77–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1014).Saltmarche AE; Naeser MA; Ho KF; Hamblin MR; Lim L Significant Improvement in Cognition in Mild to Moderately Severe Dementia Cases Treated with Transcranial Plus Intranasal Photobiomodulation: Case Series Report. Photomed. Laser Surg 2017, 35, 432–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1015).De Taboada L; Yu J; Gattoni-Celli S; Richieri S; McCarthy T; Streeter J; Kindy MS Transcranial Laser Therapy Attenuates Amyloid-β Peptide Neuropathology in Amyloid-β Protein Precursor Transgenic Mice. J. Alzheimer’s Dis 2011, 23, 521–535. [DOI] [PubMed] [Google Scholar]
- (1016).Grillo SL; Duggett NA; Ennaceur A; Chazot PL Non-invasive Infra-red Therapy (1072 nm) Reduces β-amyloid Protein Levels in the Brain of an Alzheimer’s Disease Mouse Model, TASTPM. J. Photochem. Photobiol., B 2013, 123, 13–22. [DOI] [PubMed] [Google Scholar]
- (1017).Purushothuman S; et al. Near Infrared Light Mitigates Cerebellar Pathology in Transgenic Mouse Models of Dementia. Neurosci. Lett 2015, 591, 155–159. [DOI] [PubMed] [Google Scholar]
- (1018).Purushothuman S; Johnstone DM; Nandasena C; Mitrofanis J; Stone J Photobiomodulation With Near Infrared Light Mitigates Alzheimer’s Disease-related Pathology in Cerebral Cortex - Evidence from Two Transgenic Mouse Models. Alzheimer’s Res. Ther 2014, 6, 2–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1019).Li M; Yang X; Ren J; Qu K; Qu X Using Graphene Oxide High Near-infrared Absorbance for Photothermal Treatment of Alzheimer’s Disease. Adv. Mater 2012, 24, 1722–1728. [DOI] [PubMed] [Google Scholar]
- (1020).Kawasaki T; Fujioka J; Imai T; Torigoe K; Tsukiyama K Mid-infrared Free-electron Laser Tuned to the Amide I Band for Converting Insoluble Amyloid-like Protein Fibrils Into the Soluble Monomeric Form. Lasers Med. Sci 2014, 29, 1701–1707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1021).Kawasaki T; Fujioka J; Imai T; Tsukiyama K Effect of Mid-infrared Free-Electron Laser Irradiation on Refolding of Amyloid-Like Fibrils of Lysozyme into Native Form. Protein J 2012, 31, 710–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1022).Kawasaki T; Imai T; Tsukiyama K Use of a Mid-Infrared Free-Electron Laser (MIR-FEL) for Dissociation of the Amyloid Fibril Aggregates of a Peptide. J. Anal. Sci., Methods Instrum 2014, 4, 9–18. [Google Scholar]
- (1023).Kawasaki T; Yaji T; Imai T; Ohta T; Tsukiyama K Synchrotron-Infrared Microscopy Analysis of Amyloid Fibrils Irradiated by Mid-Infrared Free-Electron Laser. Am. J. Anal. Chem 2014, 5, 384–394. [Google Scholar]
- (1024).Shivu B; Seshadri S; Li J; Oberg KA; Uversky VN; Fink AL Distinct β-sheet Structure in Protein Aggregates Determined by ATR-FTIR Spectroscopy. Biochemistry 2013, 52, 5176–5183. [DOI] [PubMed] [Google Scholar]
- (1025).Kawasaki T; Tsukiyama K; Irizawa A Dissolution of a Fibrous Peptide by Terahertz Free Electron Laser. Sci. Rep 2019, 9, 10636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1026).Viet MH; Derreumaux P; Li MS; Christopher R; Sagui C; Nguyen PH Picosecond Dissociation of Amyloid Fibrils With Infrared Laser: a Nonequilibrium Simulation Study. J. Chem. Phys 2015, 143, 155101. [DOI] [PubMed] [Google Scholar]
- (1027).Kawasaki T; Man HV; Sugimoto Y; Sugiyama N; Yamamoto H; Tsukiyama K; Wang J; Derreumaux P; Nguyen PH Infrared Laser Induced Amyloid Fibril Dissociation: A Joint experimental/theoretical Study on the GNNQQNY Peptide. J. Phys. Chem. B 2020, 124, 6266–6277. [DOI] [PubMed] [Google Scholar]
- (1028).Zhang Y; Viet MH; Christopher R; Sagui C Amyloid Properties of Asparagine and Glutamine in Prion-like Proteins. ACS Chem. Neurosci 2016, 7, 576–587. [DOI] [PubMed] [Google Scholar]
- (1029).Dalecki D Mechanical bioeffects of ultrasound. Annu. Rev. Biomed. Eng 2004, 6, 229–248. [DOI] [PubMed] [Google Scholar]
- (1030).Brennen CE Cavitation and Bubble Dynamics; University Press: Oxford, 1995. [Google Scholar]
- (1031).Souza RMDCE; da Silva ICS; Delgado ABT; da Silva PHV; Costa VRX Focused Ultrasound and Alzheimer’s Disease A systematic Review. Dement. Neuropsychol 2018, 12, 353–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1032).Jordáo JF; Thévenot E; Markham-Coultes K; Scarcelli T; Weng YQ; Xhima K; O’Reilly M; Huang Y; McLaurin J; Hynynen K; et al. Amyloid-β Plaque Reduction, Endogenous Antibody Delivery and Glial Activation by Brain-targeted, Transcranial Focused Ultrasound . Exp. Neurol 2013, 248, 16–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1033).Leinenga G; Gotz J Scanning Ultrasound Removes Amyloid-β and Restores Memory in an Alzheimer’s Disease Mouse Model. Sci. Transl. Med 2015, 7, 278ra33. [DOI] [PubMed] [Google Scholar]
- (1034).O’Reilly MA; Jones RM; Barrett E; Schwab A; Head E; Hynynen K Investigation of the Safety of Focused Ultrasound-Induced Blood-Brain Barrier Opening in a Natural Canine Model of Aging. Theranostics 2017, 7, 3573–3584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1035).Beisteiner R; Matt E; Fan C; Baldysiak H; Schönfeld M; Philippi Novak T; Amini A; Aslan T; Reinecke R; Lehrner J; et al. Transcranial Pulse Stimulation with Ultrasound in Alzheimer’s Disease - A New Navigated Focal Brain. Therapy. Adv. Sci 2020, 7, 1902583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1036).Sato N; Okochi M; Taniyama Y; Kurinami H; Shimamura M; Takeuchi D; Hamada H; Fukumori A; Kiyosue K; Taguchi T; et al. Development of New Screening System for Alzheimer Disease, in vitro Abeta Sink Assay, to Identify the Dissociation of Soluble Abeta From Fibrils. Neurobiol. Dis 2006, 22, 487–495. [DOI] [PubMed] [Google Scholar]
- (1037).Yagi H; Hasegawa K; Yoshimura Y; Goto Y Acceleration of the Depolymerization of Amyloid β Fibrils by Ultrasonication. Biochim. Biophys. Acta, Proteins Proteomics 2013, 1834, 2480–2485. [DOI] [PubMed] [Google Scholar]
- (1038).Chatani E; Lee YH; Yagi H; Yoshimura Y; Naiki H; Goto Y Ultrasonication-Dependent Production and Breakdown Lead to Minimum-Sized Amyloid Fibrils. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 11119–11124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1039).Yoshimura Y; Sakurai K; Lee YH; Ikegami T; Chatani E; Naiki H; Goto Y Direct Observation of Minimum-Sized Amyloid Fibrils Using Solution NMR Spectroscopy. Protein Sci 2010, 19, 2347–2355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1040).Foguel D; Suarez MC; Ferréo-Gonzales AD; Porto TC; Palmieri L; Einsiedler CM; Andrade LR; Lashuel HA; Lansbury PT; Kelly J; et al. Dissociation of Amyloid Fibrils of alpha-Synuclein and Transthyretin by Pressure Reveals Their Reversible Nature and the Formation of Water-Excluded Cavities. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 9831–9836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1041).Okumura H; Itoh SG Amyloid Fibril Disruption by Ultrasonic Cavitation: Nonequilibrium Molecular Dynamics Simulations. J. Am. Chem. Soc 2014, 136, 10549–10552. [DOI] [PubMed] [Google Scholar]
- (1042).Viet MH; Derreumaux P; Nguyen PH Nonequilibrium All-Atom Molecular Dynamics Simulation of the Ultrasound Induced Bubble Cavitation and Application to Dissociate Amyloid Fibril. J. Chem. Phys 2016, 145, 174113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1043).Viet MH; Li MS; Derreumaux P; Nguyen PH Rayleigh-Plesset Equation of the Bubble Stable Cavitation in Water: A Nonequilibrium All-Atom Molecular Dynamics Simulation Study. J. Chem. Phys 2018, 148, 094505. [Google Scholar]
- (1044).Miceli M; Muscat S; Morbiducci U; Cavaglià M; Deriu MA Ultrasonic Waves Effect on S-Shaped β-Amyloids Conformational Dynamics by Non-equilibrium Molecular Dynamics. J. Mol. Graphics Modell 2020, 96, 107518. [DOI] [PubMed] [Google Scholar]
- (1045).Arendash GW Review of the Evidence that Transcranial Electromagnetic Treatment will be a Safe and Effective Therapeutic Against Alzheimer’s Disease. J. Alzheimer’s Dis 2016, 53, 753–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1046).Dragicevic N; Bradshaw P; Mamcartz M; Lin X; Wang L; Cao C; Arendash GW Long-term Electromagnetic Field Treatment Enhances Brain Mitochondrial Function of both Alzheimer’s Transgenic Mice and Normal Mice: A Mechanism for Electromagnetic Field-induced Cognitive Benefit? Neuroscience 2011, 185, 135–149. [DOI] [PubMed] [Google Scholar]
- (1047).Arendash GW; Sanchez-Ramos J; Mori T; Mamcarz M; Lin X; Runfeldt M; Wang L; Zhang G; Sava V; Tan J; Cao C Electromagnetic Field Treatment Protects Against and Reverses Cognitive Impairment in Alzheimer’s Transgenic Mice. J. Alzheimer’s Dis 2010, 19, 191. [DOI] [PubMed] [Google Scholar]
- (1048).Arendash GW; Mori T; Dorsey M; Gonzalez R; Tajiri N; Borlongan C Electromagnetic Treatment to Old Alzheimer’s Mice Reverses β-amyloid Deposition, Modifies Cerebral Blood Flow, and Provides Selected Cognitive Benefit. PLoS One 2012, 7, No. e35751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1049).Arendash GW; Cao C; Abulaban H; Baranowski R; Wisniewski G; Becerra L; Andel R; Lin X; Zhang X; Wittwer D; et al. A Clinical Trial of Transcranial Electromagnetic Treatment in Alzheimer’s Disease: Cognitive Enhancement and Associated Changes in Cerebrospinal Fluid, Blood, and Brain Imaging. J. Alzheimer’s Dis 2019, 71, 57–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1050).Saikia J; Pandey G; Sasidharan S; Antony F; Nemade HB; Kumar S; Chaudhary N; Ramakrishnan V Electric Field Disruption of Amyloid Aggregation: Potential Noninvasive Therapy for Alzheimer’s Disease. ACS Chem. Neurosci 2019, 10, 2250–2262. [DOI] [PubMed] [Google Scholar]
- (1051).English N; Solomentsev G; O’Brien P Non-equilibrium Molecular Dynamics Study of Electric and Low-frequency Microwave Fields on Hen Egg White Lysozyme. J. Chem. Phys 2009, 131, 035106. [DOI] [PubMed] [Google Scholar]
- (1052).Toschi F; Lugli F; Biscarini F; Zerbetto F Effects of Electric Field Stress on a β-Amyloid Peptide. J. Phys. Chem. B 2009, 113, 369–376. [DOI] [PubMed] [Google Scholar]
- (1053).Todorova N; Bentveizen A; English N; Yarovsky I Electromagnetic-Field Effects on Structure and Dynamics of Amyloidogenic Peptides. J. Chem. Phys 2016, 144, 085101. [DOI] [PubMed] [Google Scholar]
- (1054).Todorova N; Bentveizen A; English N; Yarovsky I Electromagnetic Field Modulates Aggregation Propensity of Amyloid Peptides. J. Chem. Phys 2020, 152, 035104. [DOI] [PubMed] [Google Scholar]
- (1055).Brettschneider J; Del Tredici K; Lee VM; Trojanowski JQ Spreading of Pathology in Neurodegenerative Diseases: a Focus on Human studies. Nat. Rev. Neurosci 2015, 16, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1056).Goedert M; Eisenberg DS; Crowther RA Propagation of Tau Aggregates and Neurodegeneration. Annu. Rev. Neurosci 2017, 40, 189–240. [DOI] [PubMed] [Google Scholar]
- (1057).Jucker M; Walker LC Self-propagation of Pathogenic Protein Aggregates in Neurodegenerative Diseases. Nature 2013, 501, 45–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1058).Jucker M; Walker LC Propagation and Spread of Pathogenic Protein Assemblies in Neurodegenerative Diseases. Nat. Neurosci 2018, 21, 1341–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1059).Peng C; Trojanowski JQ; Lee VM Protein Transmission in Neurodegenerative Disease. Nat. Rev. Neurol 2020, 16, 199–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1060).Uemura N; Uemura MT; Luk KC; Lee VM; Trojanowski JQ Cell-to-Cell Transmission of Tau and α-Synuclein. Trends Mol. Med 2020, 26, 936–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1061).Vasili E; Dominguez-Meijide A; Outeiro TF Spreading of alpha-Synuclein and Tau: A Systematic Comparison of the Mechanisms Involved. Front. Mol. Neurosci 2019, 12, 107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1062).Robinson JL; Lee EB; Xie SX; Rennert L; Suh E; Bredenberg C; Caswell C; Van Deerlin VM; Yan N; Yousef A; et al. Neurodegenerative Disease Concomitant Proteinopathies are Prevalent, Age-related and APOE4-associated. Brain 2018, 141, 2181–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1063).Boyle PA; Wilson RS; Yu L; Barr AM; Honer WG; Schneider JA; Bennett DA Much of Late Life Cognitive Decline is not due to Common Neurodegenerative Pathologies. Ann. Neurol 2013, 74, 478–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1064).Boyle PA; Yu L; Wilson RS; Leurgans SE; Schneider JA; Bennett DA Person-specific Contribution of Neuropathologies to Cognitive Loss in Old Age. Ann. Neurol 2018, 83, 74–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1065).Kapasi A; DeCarli C; Schneider JA Impact of Multiple Pathologies on the Threshold for Clinically overt Dementia. Acta Neuropathol 2017, 134, 171–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1066).Schneider JA; Arvanitakis Z; Bang W; Bennett DA Mixed Brain Pathologies Account for Most Dementia Cases in Community-dwelling Older Persons. Neurology 2007, 69, 2197–2204. [DOI] [PubMed] [Google Scholar]
- (1067).Trojanowski JQ; Lee VM Fatal attractions” of Proteins. A Comprehensive Hypothetical Mechanism underlying Alzheimer’s Disease and other Neurodegenerative Disorders. Ann. N. Y. Acad. Sci 2000, 924, 62–67. [DOI] [PubMed] [Google Scholar]
- (1068).Uemura N; Yagi H; Uemura MT; Hatanaka Y; Yamakado H; Takahashi R Inoculation of alpha-synuclein Preformed Fibrils into the Mouse Gastrointestinal Tract Induces Lewy body-like Aggregates in the Brainstem via the Vagus Nerve. Mol. Neurodegener 2018, 13, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1069).Ono K; Takahashi R; Ikeda T; Yamada M Cross-seeding Effects of amyloid beta-protein and alpha-synuclein. J. Neurochem 2012, 122, 883–890. [DOI] [PubMed] [Google Scholar]
- (1070).Tsigelny IF; Crews L; Desplats P; Shaked GM; Sharikov Y; Mizuno H; Spencer B; Rockenstein E; Trejo M; Platoshyn O; et al. Mechanisms of Hybrid Oligomer Formation in the Pathogenesis of Combined Alzheimer’s and Parkinson’s Diseases. PLoS One 2008, 3, No. e3135. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (1071).Chia S; Flagmeier P; Habchi J; Lattanzi V; Linse S; Dobson CM; Knowles TPJ; Vendruscolo M Monomeric and Fibrillar alpha-synuclein Exert Opposite Effects on the Catalytic Cycle that Promotes the Proliferation of Abeta42 Aggregates. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 8005–8010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1072).Mandal PK; Pettegrew JW; Masliah E; Hamilton RL; Mandal R Interaction between Abeta Peptide and alpha synuclein: Molecular Mechanisms in Overlapping Pathology of Alzheimer’s and Parkinson’s in Dementia with Lewy body Disease. Neurochem. Res 2006, 31, 1153–1662. [DOI] [PubMed] [Google Scholar]
- (1073).Marsh SE; Blurton-Jones M Examining the Mechanisms that Link beta-amyloid and alpha-synuclein Pathologies. Alzheimer’s Res. Ther 2012, 4, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1074).Masliah E; Mallory M; Alford M; DeTeresa R; Hansen LA; McKeel DW Jr; Morris JC Altered Expression of Synaptic Proteins Occurs early during Progression of Alzheimer’s Disease. Neurology 2001, 56, 127–129. [DOI] [PubMed] [Google Scholar]
- (1075).Roberts HL; Schneider BL; Brown DR alpha-Synuclein Increases beta-amyloid Secretion by Promoting beta-/gamma-secretase processing of APP. PLoS One 2017, 12, No. e0171925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1076).Koppen J; Schulze A; Machner L; Wermann M; Eichentopf R; Guthardt M; Hahnel A; Klehm J; Kriegeskorte MC; Hartlage-Rubsamen M; et al. Amyloid-Beta Peptides Trigger Aggregation of Alpha-Synuclein In Vitro. Molecules 2020, 25, 580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1077).Bassil F; Brown HJ; Pattabhiraman S; Iwasyk JE; Maghames CM; Meymand ES; Cox TO; Riddle DM; Zhang B; Trojanowski JQ; et al. Amyloid-Beta (Abeta) Plaques Promote Seeding and Spreading of Alpha-Synuclein and Tau in a Mouse Model of Lewy Body Disorders with Abeta Pathology. Neuron 2020, 105, 260–275 (e6). [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1078).Colom-Cadena M; Gelpi E; Marti MJ; Charif S; Dols-Icardo O; Blesa R; Clarimon J; Lleo A MAPT H1 Haplotype is Associated with Enhanced alpha-synuclein Deposition in Dementia with Lewy Bodies. Neurobiol. Aging 2013, 34, 936–942. [DOI] [PubMed] [Google Scholar]
- (1079).Simon-Sanchez J; Schulte C; Bras JM; Sharma M; Gibbs JR; Berg D; Paisan-Ruiz C; Lichtner P; Scholz SW; Hernandez DG; et al. Genome-wide Association Study Reveals Genetic Risk Underlying Parkinson’s Disease. Nat. Genet 2009, 41, 1308–1312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1080).Arima K; Hirai S; Sunohara N; Aoto K; Izumiyama Y; Ueda K; Ikeda K; Kawai M Cellular Co-localization of Phosphorylated tau- and NACP/alpha-synuclein-epitopes in Lewy Bodies in Sporadic Parkinson’s Disease and in Dementia with Lewy Bodies. Brain Res 1999, 843, 53–61. [DOI] [PubMed] [Google Scholar]
- (1081).Forman MS; Schmidt ML; Kasturi S; Perl DP; Lee VM; Trojanowski JQ Tau and alpha-synuclein Pathology in Amygdala of Parkinsonism-dementia Complex Patients of Guam. Am. J. Pathol 2002, 160, 1725–1731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1082).Iseki E; Takayama N; Marui W; Ueda K; Kosaka K Relationship in the Formation Process between Neurofibrillary Tangles and Lewy Bodies in the Hippocampus of Dementia with Lewy Bodies Brains. J. Neurol. Sci 2002, 195, 85–91. [DOI] [PubMed] [Google Scholar]
- (1083).Sengupta U; Guerrero-Munoz MJ; Castillo-Carranza DL; Lasagna-Reeves CA; Gerson JE; Paulucci-Holthauzen AA; Krishnamurthy S; Farhed M; Jackson GR; Kayed R Pathological Interface between Oligomeric alpha-synuclein and tau in Synucleinopathies. Biol. Psychiatry 2015, 78, 672–683. [DOI] [PubMed] [Google Scholar]
- (1084).Castillo-Carranza DL; Guerrero-Munoz MJ; Sengupta U; Gerson JE; Kayed R alpha-Synuclein Oligomers Induce a Unique Toxic Tau Strain. Biol. Psychiatry 2018, 84, 499–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1085).Giasson BI; Forman MS; Higuchi M; Golbe LI; Graves CL; Kotzbauer PT; Trojanowski JQ; Lee VM Initiation and Synergistic Fibrillization of tau and alpha-synuclein. Science 2003, 300, 636–640. [DOI] [PubMed] [Google Scholar]
- (1086).Guo JL; Covell DJ; Daniels JP; Iba M; Stieber A; Zhang B; Riddle DM; Kwong LK; Xu Y; Trojanowski JQ; et al. Distinct alpha-synuclein Strains differentially Promote Tau Inclusions in Neurons. Cell 2013, 154, 103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1087).Nubling G; Bader B; Levin J; Hildebrandt J; Kretzschmar H; Giese A Synergistic Influence of Phosphorylation and Metal Ions on Tau Oligomer Formation and Coaggregation with alpha-synuclein at the Single Molecule Level. Mol. Neurodegener 2012, 7, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1088).Waxman EA; Giasson BI Induction of Intracellular Tau Aggregation is Promoted by alpha-synuclein Qeeds and Provides Novel Insights into the Hyperphosphorylation of Tau. J. Neurosci 2011, 31, 7604–7618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1089).Lu J; Zhang S; Ma X; Jia C; Liu Z; Huang C; Liu C; Li D Structural Basis of the Interplay between alpha-synuclein and Tau in Regulating Pathological Amyloid Aggregation. J. Biol. Chem 2020, 295, 7470–7480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1090).Gustke N; Trinczek B; Biernat J; Mandelkow EM; Mandelkow E Domains of Tau Protein and Interactions with Microtubules. Biochemistry 1994, 33, 9511–9522. [DOI] [PubMed] [Google Scholar]
- (1091).Emmer KL; Waxman EA; Covy JP; Giasson BI E46K Human alpha-synuclein Transgenic Mice Develop Lewy-like and Tau Pathology associated with Age-dependent, Detrimental Motor Impairment. J. Biol. Chem 2011, 286, 35104–35118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1092).Gerson JE; Farmer KM; Henson N; Castillo-Carranza DL; Carretero Murillo M; Sengupta U; Barrett A; Kayed R Tau Oligomers Mediate alpha-synuclein Toxicity and can be Targeted by Immunotherapy. Mol. Neurodegener 2018, 13, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1093).Singh B; Covelo A; Martell-Martinez H; Nanclares C; Sherman MA; Okematti E; Meints J; Teravskis PJ; Gallardo C; Savonenko AV; et al. Tau is Required for Progressive Synaptic and Memory Deficits in a Transgenic Mouse Model of alpha-synucleinopathy. Acta Neuropathol 2019, 138, 551–574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1094).Duka T; Duka V; Joyce JN; Sidhu A Alpha-Synuclein Contributes to GSK-3beta-catalyzed Tau Phosphorylation in Parkinson’s Disease Models. FASEB J 2009, 23, 2820–2830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1095).Duka T; Rusnak M; Drolet RE; Duka V; Wersinger C; Goudreau JL; Sidhu A Alpha-synuclein Induces Hyperphosphorylation of Tau in the MPTP Model of Parkinsonism. FASEB J 2006, 20, 2302–2312. [DOI] [PubMed] [Google Scholar]
- (1096).Haggerty T; Credle J; Rodriguez O; Wills J; Oaks AW; Masliah E; Sidhu A Hyperphosphorylated Tau in an alpha-synuclein-overexpressing Transgenic Model of Parkinson’s Disease. Eur. J. Neurosci 2011, 33, 1598–1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1097).Khan SS; LaCroix M; Boyle G; Sherman MA; Brown JL; Amar F; Aldaco J; Lee MK; Bloom GS; Lesne SE Bidirectional Modulation of Alzheimer Phenotype by alpha-synuclein in Mice and Primary Neurons. Acta Neuropathol 2018, 136, 589–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1098).Teravskis PJ; Covelo A; Miller EC; Singh B; Martell-Martinez HA; Benneyworth MA; Gallardo C; Oxnard BR; Araque A; Lee MK; et al. A53T Mutant Alpha-Synuclein Induces Tau-Dependent Postsynaptic Impairment Independently of Neurodegenerative Changes. J. Neurosci 2018, 38, 9754–9767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1099).Credle JJ; George JL; Wills J; Duka V; Shah K; Lee YC; Rodriguez O; Simkins T; Winter M; Moechars D; et al. GSK-3beta Dysregulation Contributes to Parkinson’s-like Pathophysiology with associated Region-specific Phosphorylation and Accumulation of tau and alpha-synuclein. Cell Death Differ 2015, 22, 838–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1100).Gassowska M; Czapski GA; Pajak B; Cieslik M; Lenkiewicz AM; Adamczyk A Extracellular alpha-synuclein Leads to Microtubule Destabilization via GSK-3beta-dependent Tau Phosphorylation in PC12 Cells. PLoS One 2014, 9, No. e94259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1101).Ge X; Yang Y; Sun Y; Cao W; Ding F Islet Amyloid Polypeptide Promotes Amyloid-Beta Aggregation by Binding-Induced Helix-Unfolding of the Amyloidogenic Core. ACS Chem. Neurosci 2018, 9, 967–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1102).Andreetto E; Yan LM; Tatarek-Nossol M; Velkova A; Frank R; Kapurniotu A Identification of Hot Regions of the Abeta-IAPP Interaction Interface as High-affinity Binding Sites in both Cross- and Self-association. Angew. Chem., Int. Ed 2010, 49, 3081–3085. [DOI] [PubMed] [Google Scholar]
- (1103).Seeliger J; Weise K; Opitz N; Winter R The Effect of Abeta on IAPP Aggregation in the Presence of an Isolated beta-cell Lembrane. J. Mol. Biol 2012, 421, 348–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1104).Zhang M; Hu R; Chen H; Gong X; Zhou F; Zhang L; Zheng J Polymorphic Associations and Structures of the Cross-Seeding of Abeta1–42 and hIAPP1–37 Polypeptides. J. Chem. Inf. Model 2015, 55, 1628–1639. [DOI] [PubMed] [Google Scholar]
- (1105).Zhang M; Hu R; Ren B; Chen H; Jiang B; Ma J; Zheng J Molecular Understanding of Abeta-hIAPP Cross-Seeding Assemblies on Lipid Membranes. ACS Chem. Neurosci 2017, 8, 524–537. [DOI] [PubMed] [Google Scholar]
- (1106).Jackson K; Barisone GA; Diaz E; Jin LW; DeCarli C; Despa F Amylin Deposition in the Brain: A second Amyloid in Alzheimer Disease? Ann. Neurol 2013, 74, 517–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1107).Oskarsson ME; Paulsson JF; Schultz SW; Ingelsson M; Westermark P; Westermark GT In vivo Seeding and Cross-seeding of Localized Amyloidosis: a Molecular Link between Type 2 Diabetes and Alzheimer Disease. Am. J. Pathol 2015, 185, 834–846. [DOI] [PubMed] [Google Scholar]
- (1108).Moreno-Gonzalez I; Edwards G Iii; Salvadores N; Shahnawaz M; Diaz-Espinoza R; Soto C Molecular Interaction between Type 2 Diabetes and Alzheimer’s Disease through Cross-seeding of Protein Misfolding. Mol. Psychiatry 2017, 22, 1327–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1109).Horvath I; Wittung-Stafshede P Cross-talk between Amyloidogenic proteins in Type-2 Diabetes and Parkinson’s Disease. Proc. Natl. Acad. Sci. U. S. A 2016, 113, 12473–12477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1110).Lauren J; Gimbel DA; Nygaard HB; Gilbert JW; Strittmatter SM Cellular Prion Protein Mediates Impairment of Synaptic Plasticity by amyloid-beta Oligomers. Nature 2009, 457, 1128–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1111).Um JW; Nygaard HB; Heiss JK; Kostylev MA; Stagi M; Vortmeyer A; Wisniewski T; Gunther EC; Strittmatter SM Alzheimer amyloid-beta Oligomer Bound to Postsynaptic Prion Protein Activates Fyn to Impair Neurons. Nat. Neurosci 2012, 15, 1227–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1112).Larson ME; Sherman MA; Greimel S; Kuskowski M; Schneider JA; Bennett DA; Lesne SE Soluble alpha-synuclein is a Novel Modulator of Alzheimer’s Disease Pathophysiology. J. Neurosci 2012, 32, 10253–10266. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- (1113).Zou WQ; Xiao X; Yuan J; Puoti G; Fujioka H; Wang X; Richardson S; Zhou X; Zou R; Li S; et al. Amyloid-beta42 Interacts Mainly with Insoluble Prion Protein in the Alzheimer Brain. J. Biol. Chem 2011, 286, 15095–15105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1114).Morales R; Estrada LD; Diaz-Espinoza R; Morales-Scheihing D; Jara MC; Castilla J; Soto C Molecular Cross Talk between Misfolded Proteins in Animal Models of Alzheimer’s and Prion Diseases. J. Neurosci 2010, 30, 4528–4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1115).Canu N; Filesi I; Pristera A; Ciotti MT; Biocca S Altered Intracellular Distribution of PrPC and Impairment of Proteasome Activity in tau overexpressing Cortical Neurons. J. Alzheimer’s Dis 2011, 27, 603–613. [DOI] [PubMed] [Google Scholar]
- (1116).Ishizawa K; Komori T; Shimazu T; Yamamoto T; Kitamoto T; Shimazu K; Hirose T Hyperphosphorylated tau Deposition Parallels Prion Protein Burden in a Case of Gerstmann-Straussler-Scheinker Syndrome P102L Mutation Complicated with Dementia. Acta Neuropathol 2002, 104, 342–350. [DOI] [PubMed] [Google Scholar]
- (1117).Reiniger L; Lukic A; Linehan J; Rudge P; Collinge J; Mead S; Brandner S Tau, Prions and Abeta: the Triad of Neurodegeneration. Acta Neuropathol 2011, 121, 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1118).Guerrero-Munoz MJ; Castillo-Carranza DL; Krishnamurthy S; Paulucci-Holthauzen AA; Sengupta U; Lasagna-Reeves CA; Ahmad Y; Jackson GR; Kayed R Amyloid-beta oligomers as a template for secondary amyloidosis in Alzheimer’s disease. Neurobiol. Dis 2014, 71, 14–23. [DOI] [PubMed] [Google Scholar]
- (1119).Wiltzius JJ; Sievers SA; Sawaya MR; Cascio D; Popov D; Riekel C; Eisenberg D Atomic Structure of the Cross-beta Spine of Islet Amyloid Polypeptide (IAPP). Protein Sci 2008, 17, 1467–1474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1120).Wineman-Fisher V; Atsmon-Raz Y; Miller Y Orientations of Residues along the beta-arch of Self-assembled IAPP Fibril-like Structures lead to Polymorphism. Biomacromolecules 2015, 16, 156–165. [DOI] [PubMed] [Google Scholar]
- (1121).Baram M; Atsmon-Raz Y; Ma B; Nussinov R; Miller Y IAPP-Abeta Oligomers at Atomic Resolution using Molecular Dynamics Simulations: a Link between Type 2 Diabetes and Alzheimer’s Disease. Phys. Chem. Chem. Phys 2016, 18, 2330–2338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1122).Sciacca MF; Milardi D; Messina GM; Marletta G; Brender JR; Ramamoorthy A; La Rosa C Cations as Switches of Amyloid-mediated Membrane Disruption Mechanisms: Calcium and IAPP. Biophys. J 2013, 104, 173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1123).Atsmon-Raz Y; Miller Y A Proposed Atomic Structure of the Self-Assembly of the Non-Amyloid-beta Component of Human alpha-Synuclein As Derived by Computational Tools. J. Phys. Chem. B 2015, 119, 10005–10015. [DOI] [PubMed] [Google Scholar]
- (1124).Atsmon-Raz Y; Miller Y Non-Amyloid-beta Component of Human alpha-synuclein Oligomers Induces Formation of new Abeta Oligomers: Insight into the Mechanisms that Link Parkinson’s and Alzheimer’s Diseases. ACS Chem. Neurosci 2016, 7, 46–55. [DOI] [PubMed] [Google Scholar]
- (1125).Atsmon-Raz Y; Miller Y Molecular Mechanisms of the Bindings between Non-amyloid beta Component Oligomers and IAPP Oligomers. J. Phys. Chem. B 2016, 120, 10649–10659. [DOI] [PubMed] [Google Scholar]
- (1126).Rhein V; Song X; Wiesner A; Ittner LM; Baysang G; Meier F; Ozmen L; Bluethmann H; Drose S; Brandt U; et al. Amyloid-beta and Tau Synergistically Impair the Oxidative Phosphorylation System in Triple Transgenic Alzheimer’s Disease Mice. Proc. Natl. Acad. Sci. U. S. A 2009, 106, 20057–20062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1127).David DC; Hauptmann S; Scherping I; Schuessel K; Keil U; Rizzu P; Ravid R; Drose S; Brandt U; Muller WE; et al. Proteomic and Functional Analyses Reveal a Mitochondrial Dysfunction in P301L Tau Transgenic Mice. J. Biol. Chem 2005, 280, 23802–23814. [DOI] [PubMed] [Google Scholar]
- (1128).Hauptmann S; Keil U; Scherping I; Bonert A; Eckert A; Muller WE Mitochondrial Dysfunction in Sporadic and Genetic Alzheimer’s Disease. Exp. Gerontol 2006, 41, 668–673. [DOI] [PubMed] [Google Scholar]
- (1129).Eckert A; Hauptmann S; Scherping I; Rhein V; Muller-Spahn F; Gotz J; Muller WE Soluble beta-amyloid Leads to Mitochondrial Defects in Amyloid Precursor Protein and Tau Transgenic Mice. Neurodegener. Dis 2008, 5, 157–9. [DOI] [PubMed] [Google Scholar]
- (1130).Gotz J; Lim YA; Ke YD; Eckert A; Ittner LM Dissecting Toxicity of tau and beta-amyloid. Neurodegener. Dis 2010, 7, 10–12. [DOI] [PubMed] [Google Scholar]
- (1131).Lewis J; Dickson DW; Lin WL; Chisholm L; Corral A; Jones G; Yen SH; Sahara N; Skipper L; Yager D; et al. Enhanced Neurofibrillary Degeneration in Transgenic Mice Expressing Mutant Tau and APP. Science 2001, 293, 1487–1491. [DOI] [PubMed] [Google Scholar]
- (1132).Gotz J; Chen F; van Dorpe J; Nitsch RM Formation of Neurofibrillary Tangles in P301l Tau Transgenic Mice Induced by Abeta42 Fibrils. Science 2001, 293, 1491–1495. [DOI] [PubMed] [Google Scholar]
- (1133).Oddo S; Caccamo A; Shepherd JD; Murphy MP; Golde TE; Kayed R; Metherate R; Mattson MP; Akbari Y; LaFerla FM Triple-transgenic Model of Alzheimer’s Disease with Plaques and Tangles: Intracellular Abeta and Synaptic Dysfunction. Neuron 2003, 39, 409–421. [DOI] [PubMed] [Google Scholar]
- (1134).von Bergen M; Friedhoff P; Biernat J; Heberle J; Mandelkow EM; Mandelkow E Assembly of tau Protein into Alzheimer Paired Helical Filaments Depends on a Local Sequence Motif ((306)VQIVYK(311)) Forming beta Structure. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5129–5134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1135).Goux WJ; Kopplin L; Nguyen AD; Leak K; Rutkofsky M; Shanmuganandam VD; Sharma D; Inouye H; Kirschner DA Formation of Straight and Twisted Filaments from Short tau Peptides. J. Biol. Chem 2004, 279, 26868–26875. [DOI] [PubMed] [Google Scholar]
- (1136).Barrantes A; Sotres J; Hernando-Perez M; Benitez MJ; de Pablo PJ; Baro AM; Avila J; Jimenez JS Tau Aggregation Followed by Atomic Force Microscopy and Surface Plasmon Resonance, and Single Molecule tau-tau Interaction Probed by Atomic Force Spectroscopy. J. Alzheimer’s Dis 2009, 18, 141–51. [DOI] [PubMed] [Google Scholar]
- (1137).Peterson DW; Zhou H; Dahlquist FW; Lew J A soluble Oligomer of Tau associated with Fiber Formation Analyzed by NMR. Biochemistry 2008, 47, 7393–7404. [DOI] [PubMed] [Google Scholar]
- (1138).Margittai M; Langen R Side Chain-dependent Stacking Modulates Tau Filament Structure. J. Biol. Chem 2006, 281, 37820–37827. [DOI] [PubMed] [Google Scholar]
- (1139).Do TD; Economou NJ; Chamas A; Buratto SK; Shea JE; Bowers MT Interactions between amyloid-β and Tau Fragments Promote Aberrant Aggregates: Implications for amyloid Toxicity. J. Phys. Chem. B 2014, 118, 11220–11230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1140).Miller Y; Ma B; Nussinov R Synergistic Interactions between Repeats in Tau Protein and Abeta amyloids May be Responsible for Accelerated Aggregation via Polymorphic States. Biochemistry 2011, 50, 5172–5181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1141).Raz Y; Miller Y Interactions between Abeta and Mutated Tau Lead to Polymorphism and Induce Aggregation of Abeta-mutated Tau Oligomeric Complexes. PLoS One 2013, 8, No. e73303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1142).Grimm A; Eckert A Brain Aging and Neurodegeneration: from a Mitochondrial Point of View. J. Neurochem 2017, 143, 418–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1143).Mahul-Mellier AL; Burtscher J; Maharjan N; Weerens L; Croisier M; Kuttler F; Leleu M; Knott GW; Lashuel HA The process of Lewy Body Formation, rather than simply α-synuclein Fibrillization, is one of the Major Drivers of Neurodegeneration. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 4971–4982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1144).Esparza TJ; Wildburger NC; Jiang H; Gangolli M; Cairns NJ; Bateman RJ; Brody DL Soluble Amyloid-beta Aggregates from Human Alzheimer’s Disease Brains. Sci. Rep 2016, 6, 38187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1145).Kanaan NM; Hamel C; Grabinski T; Combs B Liquid-liquid Phase Separation Induces Pathogenic Tau Conformations in vitro. Nat. Commun 2020, 11, 2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1146).Tetz G; Pinho M; Pritzkow S; Mendez N; Soto C; Tetz V Bacterial DNA Promotes Tau aggregation. Sci. Rep 2020, 10, 2369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1147).Ait-Bouziad N; Lv G; Mahul-Mellier AL; Xiao S; Zorludemir G; Eliezer D; Walz T; Lashuel HA Discovery and Characterization of Stable and Toxic Tau/phospholipid Oligomeric Complexes. Nat. Commun 2017, 8, 1678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1148).Habchi J; Chia S; Galvagnion C; Michaels TCT; Bellaiche MMJ; Ruggeri FS; Sanguanini M; Idini I; Kumita JR; Sparr E; et al. Cholesterol Catalyses Aβ42 Aggregation through a Heterogeneous Nucleation Pathway in the Presence of Lipid Membranes. Nat. Chem 2018, 10, 673–683. [DOI] [PubMed] [Google Scholar]
- (1149).Thijssen EH; La Joie R; Wolf A; Strom A; Wang P; Iaccarino L; Bourakova V; Cobigo Y; Heuer H; Spina S; et al. Diagnostic Value of Plasma Phosphorylated tau181 in Alzheimer’s Disease and Frontotemporal Lobar Degeneration. Nat. Med 2020, 26, 387–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1150).Karikari TK; Pascoal TA; Ashton NJ; Janelidze S; Benedet AL; Rodriguez JL; Chamoun M; Savard M; Kang MS; Therriault J; et al. Blood Phosphorylated tau 181 as a Biomarker for Alzheimer’s Disease: a Diagnostic Performance and Prediction Modelling Study using Data from four Prospective Cohorts. Lancet Neurol 2020, 19, 422–433. [DOI] [PubMed] [Google Scholar]
- (1151).Soria FN; Paviolo C; Doudnikoff E; Arotcarena ML; Lee A; Danné N; Mandal AK; Gosset P; Dehay B; Groc L; et al. Synucleinopathy Alters Nanoscale Organization and Diffusion in the Brain Extracellular Space through Hyaluronan Remodeling. Nat. Commun 2020, 11, 3440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1152).Kingsmore KM; Vaccari A; Abler D; Cui SX; Epstein FH; Rockne RC; Acton ST; et al. MRI Analysis to Map Interstitial Flow in the Brain Tumor Microenvironment. APL Bioeng 2018, 2, 031905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1153).Chatterjee K; Carman-Esparza CM; Munson JM Methods to Measure, Model and Manipulate Fluid Flow in Brain. J. Neurosci. Methods 2020, 333, 108541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1154).Lei Y; Han H; Yuan F; Javeed A; Zhao Y The Brain Interstitial System: Anatomy, Modeling, in vivo Measurement, and Applications. Prog. Neurobiol 2017, 157, 230–246. [DOI] [PubMed] [Google Scholar]
- (1155).Brini E; Simmerling C; Dill K Protein Storytelling through Physics. Science 2020, 370 (6520), No. eaaz3041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1156).Man VH; He X; Derreumaux P; Ji B; Xie XQ; Nguyen PH; Wang J Effects of All-Atom Molecular Mechanics Force Fields on Amyloid Peptide Assembly: The Case of Aβ16–22 Dimer. J. Chem. Theory Comput 2019, 15, 1440–1452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1157).Nguyen PH; Sterpone F; Derreumaux P Aggregation of Disease-related Peptides. Prog. Mol. Biol. Transl. Sci 2020, 170, 435–460. [DOI] [PubMed] [Google Scholar]
- (1158).Strodel B Amyloid aggregation Simulations: Challenges, Advances and Perspectives. Curr. Opin. Struct. Biol 2021, 67, 145–152. [DOI] [PubMed] [Google Scholar]
- (1159).Krausser J; Knowles TPJ; Šarić A Physical Mechanisms of Amyloid Nucleation on Fluid Membranes. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 33090–33098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1160).Wang J; Olsson S; Wehmeyer C; Pérez A; Charron NE; de Fabritiis G; Noé F; Clementi C Machine Learning of Coarse-Grained Molecular Dynamics Force Fields. ACS Cent. Sci 2019, 5, 755–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1161).Chebaro Y; Jiang P; Zang T; Mu Y; Nguyen PH; Mousseau N; Derreumaux P Structures of Aβ17–42 Trimers in Isolation and with Five Small-molecule Drugs using a Hierarchical Computational Procedure. J. Phys. Chem. B 2012, 116, 8412–8422. [DOI] [PubMed] [Google Scholar]
- (1162).Sengupta U; Carballo-Pacheco M; Strodel B Automated Markov State Models for Molecular Dynamics Simulations of Aggregation and Self-assembly. J. Chem. Phys 2019, 150, 115101. [DOI] [PubMed] [Google Scholar]
- (1163).Noé F; Olsson S; Köhler J; Wu H Boltzmann Generators: Sampling Equilibrium States of Many-body Systems with Deep Learning. Science 2019, 365, No. eaaw1147. [DOI] [PubMed] [Google Scholar]
- (1164).Cummings J; Lee G; Ritter A; Sabbagh M; Zhong K Alzheimer’s Disease Drug Development Pipeline. Alzheimer’s Dement (N.Y.) 2019, 5, 272–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1165).Bullain S; Doody RJ What Works and What doesn’t Work in Alzheimer’s Disease? From Interventions on Risk Factors to Anti-amyloid Trials. J. Neurochem 2020, 155, 120–136. [DOI] [PubMed] [Google Scholar]
- (1166).Lao K; Ji N; Qiao W; Tang Z; Gou X Drug Development for Alzheimer’s Disease: Review. J. Drug Target 2019, 27, 164–173. [DOI] [PubMed] [Google Scholar]
- (1167).Ballard C; Aarsland D; Cummings J; O’Brien J; Mills R; Molinuevo JL; Fladby T; Williams G; Doherty P; Corbett A; Sultana J Drug Repositioning and Repurposing for Alzheimer Disease. Nat. Rev. Neurol 2020, 16, 661–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1168).Jucker M The Benefits and Limitations of Animal Models for Translational Research in Neurodegenerative Diseases. Nat. Med 2010, 16, 1210–1214. [DOI] [PubMed] [Google Scholar]
- (1169).Fisher EMC; Bannerman DM Mouse Models of Neurodegeneration: Know your Question, Know your Mouse. Sci. Transl. Med 2019, 11, No. eaaq1818. [DOI] [PubMed] [Google Scholar]
- (1170).Jackson SJ; Andrews N; Ball D; Bellantuono I; Gray J; Hachoumi L; Holmes A; Latcham J; Petrie A; Potter P; et al. Does Age Matter? The Impact of Rodent Age on Study Outcomes. Lab. Anim 2017, 51, 160–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1171).Lui JH; Hansen DV; Kriegstein AR Development and Evolution of the Human Neocortex. Cell 2011, 146, 18–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1172).Oberheim NA; Wang X; Goldman S; Nedergaard M Astrocytic Complexity Distinguishes the Human Brain. Trends Neurosci 2006, 29, 547–543. [DOI] [PubMed] [Google Scholar]
- (1173).Stevens JC; Banks GT; Festing MF; Fisher EM Quiet Mutations in Inbred Strains of Mice. Trends Mol. Med 2007, 13, 512–519. [DOI] [PubMed] [Google Scholar]
- (1174).Jankowsky JL; Zheng H Practical Considerations for Choosing a Mouse Model of Alzheimer’s Disease. Mol. Neurodegener 2017, 12, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1175).Gamache J; Benzow K; Forster C; Kemper L; Hlynialuk C; Furrow E; Ashe KH; Koob MD Factors other than hTau Overexpression that Contribute to Tauopathy-like Phenotype in rTg4510 Mice. Nat. Commun 2019, 10, 2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1176).Lin JH Applications and Limitations of Genetically Modified Mouse Models in Drug Discovery and Development. Curr. Drug Metab 2008, 9, 419–438. [DOI] [PubMed] [Google Scholar]
- (1177).Karran E; De Strooper B The Amyloid Cascade Hypothesis: Are we Poised for Success or Failure? J. Neurochem 2016, 139 (2), 237–252. [DOI] [PubMed] [Google Scholar]
- (1178).Wang S; Mims PN; Roman RJ; Fan F Accumulation a Cause or Consequence of Alzheimer’s Disease? J. Alzheimers Parkinsonism Dement 2016, 1, 7. [PMC free article] [PubMed] [Google Scholar]
- (1179).Semorinemab. https://www.alzforum.org/.
- (1180).Abbott A Neuroscience: The Plaque Plan. Nature 2008, 456, 161–4. [DOI] [PubMed] [Google Scholar]
- (1181).Dubois B The Emergence of a New Conceptual Framework for Alzheimer’s Disease. J. Alzheimer’s Dis 2018, 62, 1059–1066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1182).Scialò C; De Cecco E; Manganotti P; Legname G Prion and Prion-Like Protein Strains: Deciphering the Molecular Basis of Heterogeneity in Neurodegeneration. Viruses 2019, 11, 261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1183).Sims R; Hill M; Williams J The Multiplex Model of the Genetics of Alzheimer’s Disease. Nat. Neurosci 2020, 23, 311–322. [DOI] [PubMed] [Google Scholar]
- (1184).De Cecco E; Celauro L; Vanni S; Grandolfo M; Bistaffa E; Moda F; Aguzzi A; Legname G The Uptake of tau Amyloid Fibrils is Facilitated by the Cellular Prion Protein and Hampers Prion Propagation in Cultured Cells. J. Neurochem 2020, 155, 577–591. [DOI] [PubMed] [Google Scholar]
- (1185).Therriault J; Benedet AL; Pascoal TA; Mathotaarachchi S; Savard M; Chamoun M; Thomas E; Kang MS; Lussier F; Tissot C et al. APOEε4 Potentiates the Relationship between amyloid-β and tau Pathologies. Mol. Psychiatry 2020, DOI: 10.1038/s41380-020-0688-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1186).Sala Frigerio C; Wolfs L; Fattorelli N; Thrupp N; Voytyuk I; Schmidt I; Mancuso R; Chen WT; Woodbury ME; Srivastava G; et al. The Major Risk Factors for Alzheimer’s Disease: Age, Sex, and Genes Modulate the Microglia Response to Aβ Plaques. Cell Rep 2019, 27, 1293–1306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1187).Rauch JN; Luna G; Guzman E; Audouard M; Challis C; Sibih YE; Leshuk C; Hernandez I; Wegmann S; Hyman BT; et al. LRP1 is a Master Regulator of tau Uptake and Spread. Nature 2020, 580, 381–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1188).Castellano JM; Deane R; Gottesdiener AJ; Verghese PB; Stewart FR; West T; Paoletti AC; Kasper TR; DeMattos RB; Zlokovic BV; et al. Low-density Lipoprotein Receptor Overexpression Enhances the Rate of Brain-to-Blood Aβ Clearance in a Mouse Model of β-amyloidosis. Proc. Natl. Acad. Sci. U. S. A 2012, 109, 15502–15507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (1189).Ikeda T; Ono K; Elashoff D; Condron MM; Noguchi-Shinohara M; Yoshita M; Teplow DB; Yamada M Cerebrospinal Fluid from Alzheimer’s Disease Patients Promotes amyloid beta-protein Oligomerization. J. Alzheimer’s Dis 2010, 21, 81–86. [DOI] [PubMed] [Google Scholar]
- (1190).Brandt R; Bakota L Microtubule Dynamics and the Neurodegenerative Triad of Alzheimer’s Disease: The Hidden Connection. J. Neurochem 2017, 143, 409–417. [DOI] [PubMed] [Google Scholar]
- (1191).Aisen PS; Cummings J; Doody R; Kramer L; Sallloway S; Selkoe DJ; Sims J; Sperling RA; Vellas B The Future of Anti-Amyloid Trials. J. Prev. Alzheimers Dis 2020, 7, 146–151. [DOI] [PubMed] [Google Scholar]