

Abstract
Bromodomain-containing protein 4 (BRD4), as the most studied member of the bromodomain and extra-terminal (BET) family, is a chromatin reader protein interpreting epigenetic codes through binding to acetylated histones and non-histone proteins, thereby regulating diverse cellular processes including cell cycle, cell differentiation, and cell proliferation. As a promising drug target, BRD4 function is closely related to cancer, inflammation, cardiovascular disease, and liver fibrosis. Currently, clinical resistance to BET inhibitors has limited their applications but synergistic antitumor effects have been observed when used in combination with other tumor inhibitors targeting additional cellular components such as PLK1, HDAC, CDK, and PARP1. Therefore, designing dual-target inhibitors of BET bromodomains is a rational strategy in cancer treatment to increase potency and reduce drug resistance. This review summarizes the protein structures and biological functions of BRD4 and discusses recent advances of dual BET inhibitors from a medicinal chemistry perspective. We also discuss the current design and discovery strategies for dual BET inhibitors, providing insight into potential discovery of additional dual-target BET inhibitors.
Keywords: bromodomain and extra-terminal (BET), bromodomain-containing protein 4, dual-target inhibitors, medicinal chemistry, structure-activity relationship
1 |. INTRODUCTION
Bromodomain (BRD) proteins are epigenetic regulators with highly conserved functional domains.1–5 They specifically recognize acetylated lysine (Kac), participate in gene regulation, and are closely associated with tumor formation, inflammation, cardiovascular disease, and liver fibrosis.6–11 According to their structural differences, BRD proteins can be divided into eight families, among which the bromodomain and extra-terminal (BET) family has been widely studied. The BET family has four members, including bromodomain-containing protein 2 (BRD2), 3 (BRD3), 4 (BRD4), and a testis-specific protein (BRDT).12–14 BRD4 is widely expressed in humans as an important functional protein of the BET family and has become one of the most investigated epigenetic targets in recent years.
As a scaffolding protein and a mitotic “bookmarking” factor, BRD4 plays an important role in cellular functions. It can interpret the epigenetic code, recognize acetylated histones including H3 lysine 9 (K9), H3K14, H4K5/8/12 or nonhistone proteins, control DNA replication and regulate gene transcription.15–21 However, abnormal acetylation levels and dysfunction of the BET proteins result in transcription disorders, which is associated with the development of many diseases, especially cancers, where the BET proteins regulate the expression of key oncogenes and antiapoptotic proteins.10,22,23 Zuber et al.24 found that BRD4 is a key factor required to maintain acute myeloid leukemia. Inactivation of BRD4 abolishes the expression of MYC in leukemia and limits its self-renewal, leading to myeloid differentiation and elimination of leukemia stem cells. Inhibiting BRD4 downregulates the expression of Kirsten rat sarcoma viral oncogene homolog (KRAS), fos-like 1 (FOSL1), and B-cell lymphoma 2 (BCL2), which play an important role respectively in glioblastoma, non-small cell lung cancer, and hematological malignancy.25 Therefore, targeting BET proteins is a promising strategy that helps promote the development of BET inhibitors in cancer therapy.
Currently, many pan-BET inhibitors with positive antitumor effects have entered clinical trials (Table 1). However, as a monotherapy, pan-BET inhibitors show moderate efficacy in preclinical models due to potential toxicity and side effects.42–44 Moreover, as the effectiveness of targeted therapies is often limited by the development of drug resistance, it is essential to explore resistance mechanisms for optimizing the clinical efficacy of these inhibitors. Restoring MYC transcription is one of the mechanisms of resistance to BET inhibitors. The increase of WNT/β-catenin signaling is a key driving factor for drug resistance in leukemia models, and it can maintain MYC expression independently of BRD4.45 Inhibiting the PRC2 complex can restore the transcriptional regulation of MYC.46 In castration-resistant prostate cancer (CRPC), maintaining MYC expression promotes resistance to BET inhibition.47 In addition, GLI2-dependent upregulation of c-MYC can mediate the resistance of pancreatic cancer cells.48 Kinome reprogramming is also related to resistance to BET inhibitors. For example, activation of receptor tyrosine kinase (RTK) overcomes BET inhibition and leads to resistance. The RTK network can promote downstream PI3K/ERK signaling to prevent JQ1-mediated apoptosis.49 Hyper-phosphorylation of BRD4 also has an impact on drug resistance. In the resistant triple-negative breast cancer cells, BRD4 is hyperphosphorylated (caused by decreased PP2A activity), and binds to MED1 more strongly, which promotes the BRD-independent chromatin recruitment mechanism.50 AMPK/ULK1-mediated autophagy is also one of the resistance mechanisms in AML leukemia stem cells.51 In prostate cancer cells with SPOP mutation, the molecular mechanism of drug resistance to BET inhibitors may be that mutation leads to stabilization of BET proteins. However, some WT SPOP samples also show high BRD4 protein levels, indicating that other mechanisms remain to be further investigated.52 Stabilization of BRD4 correlates with activation of AKT-mTORC1 signaling.53 In addition, targeting DUB3 can overcome drug resistance to BET inhibitors in tumors with NCOR2-HDAC10-DUB3-BRD4 signaling imbalance or SPOP mutation.54 Notably, the drug resistance problem involves multiple mechanisms and limits treatment potential with BET inhibitors. Thus, overcoming BET inhibitor resistance is indeed a significant challenge.
TABLE 1.
Summary of BET small-molecule inhibitors and dual-target inhibitors
| Type | Inhibitors | Crystal structure of BET protein | PDB code of other targets | Clinical phase | NCT identifiera | |||
|---|---|---|---|---|---|---|---|---|
| PDB code | Positions | Released date | Resolution (Å) | |||||
| BET small-molecule inhibitors | JQ1 | 3MXF15 | 42–168 | 2010.10.06 | 1.60 | NA | NA | NA |
| I-BET762 | 3P5O26 | 44–168 | 2010.11.17 | 1.60 | NA | II | NCT01943851 | |
| I-BET151 | 3ZYU6 | 44–168 | 2011.11.02 | 1.50 | NA | NA | NA | |
| CPI-0610 | 5HLS27 | 42–168 | 2016.02.10 | 2.18 | NA | I | NCT02157636 NCT01949883 | |
| PFI-1 | 4E9628 | 44–168 | 2012.04.18 | 1.92 | NA | NA | NA | |
| I-BET726 | 4BJX29 | 44–168 | 2013.10.02 | 1.59 | NA | NA | NA | |
| RVX-208 | 4MR630 | 346–455 | 2013.11.27 | 1.67 | NA | III | NCT02586155 | |
| ABBV-744 | 6E6J31 | 348–455 | 2019.07.31 | 2.44 | NA | I | NCT03360006 | |
| BET dual-target inhibitors | Dinaciclib | 4O7032 | 44–168 | 2014.03.05 | 1.55 | 4KD133 | III | NCT01580228 |
| Flavopiridol | 4O7132 | 44–168 | 2014.03.05 | 1.36 | 3BLR34 | II | NCT00023894, NCT00464633, NCT00003256 | |
| BI-2536 | 4O7435 | 44–168 | 2014.02.26 | 1.73 | 2RKU36 | II | NCT00701766, NCT00376623, NCT00706498, NCT00710710 | |
| TG101348 | 4OGJ35 | 44–168 | 2014.02.26 | 1.65 | NA | II | NCT04282187 | |
| TG101209 | 4O7632 | 44–168 | 2014-03-05 | 1.70 | 4JI937 | NA | NA | |
| SF2523 | 5U2838 | 44–180 | 2017.02.08 | 1.80 | NA | NA | NA | |
| SF2558HA | 5U2C38 | 342–460 | 2017.02.08 | 3.30 | NA | NA | NA | |
| SB610251B | 4O7E32 | 44–168 | 2014.03.05 | 1.85 | NA | NA | NA | |
| SB202190 | 4O7732 | 44–168 | 2014.03.05 | 2.00 | NA | NA | NA | |
| JWG-047 | 6CIS39 | 44–166 | 2018.08.29 | 1.51 | NA | NA | NA | |
| GW612286X | 4O7832 | 44–168 | 2014.03.05 | 1.34 | 3CJF40 | NA | NA | |
| NU7441 | 4O7232 | 44–168 | 2014.03.05 | 1.40 | NA | NA | NA | |
| SB409514 | 4O7A32 | 44–168 | 2014.03.05 | 1.34 | NA | NA | NA | |
| TW9 | 6YQN41 | 44–168 | 2020.05.06 | 1.05 | NA | NA | NA | |
Abbreviation: NA, not available.
http://clinicaltrials.gov; January 2021.
Multitarget drugs can produce synergistic effects on individual targets and ensure the effectiveness and durability of antitumor effects, providing a new scope for clinical research on BET inhibitors. Based on the latest progress of epigenetic drugs, BET and other antitumor inhibitors can enhance the antitumor effect by synergistically targeting abnormal signaling pathways. The combination of BET and other antitumor inhibitors can reduce the possibility of acquired resistance. Considering the obvious advantages and feasibility of combining BET and other antitumor inhibitors, considerable research is devoted to the development of dual-target BET inhibitors, as illustrated by the recent development of dual-target drugs targeting BET proteins (Table 1).
In this review, we will discuss the domain structures and biological functions of BRD4 and highlight the strategies for drug design/discovery and structure–activity relationships (SARs) of dual-target inhibitors from the medicinal chemistry perspective, in hopes of providing insight into clinical drug research of BET proteins and leading to discovery of new dual BET inhibitors.
2 |. DOMAIN FEATURES AND BIOLOGICAL FUNCTIONS OF BRD4 AND DUAL-TARGET DESIGN/DISCOVERY OF BET BRD INHIBITORS
2.1 |. Domain features of BRD4
Due to alternative 3′ mRNA splicing, BRD4 exists in three major protein isoforms differing at their C-terminal amino acid residues: BRD4-L (long form), BRD4-S(a) (short form a), and BRD4-S(b) (short form b; Figure 1A).55 All three BRD4 protein isoforms contain two tandem BRDs, namely bromodomain 1 (BD1) and bromodomain 2 (BD2), as well as the N-terminal cluster of phosphorylation sites (NPS) and the basic residue-enriched interaction domain (BID) and an extra-terminal (ET) domain (Figure 1A).56,57 BRD4-L has a C-terminal motif interacting with positive transcription elongation factor b (P-TEFb), whereas BRD4-S(b) contains a specific C-terminus associated with condensation protein complex II to regulate DNA damage response.58 BRD4-S(a) has three unique C-terminal residues (GPA) not present in BRD4-L and BRD4-S(b). Intramolecular interaction between NPS and BD2 is disrupted by casein kinase II (CK2)-mediated NPS phosphorylation that switches NPS contact with upstream BD2 to downstream BID, thereby allowing BD2 binding to acetylated chromatin.57 Importantly, the Chiang lab55 recently demonstrated that BRD4-S(a) has oncogenic activity and BRD4-L has tumor-inhibitory effects on breast cancer initiation and progression. Therefore, targeted therapy for oncogenic rather than tumor-suppressive activity of BRD4 is of vital importance.
FIGURE 1.
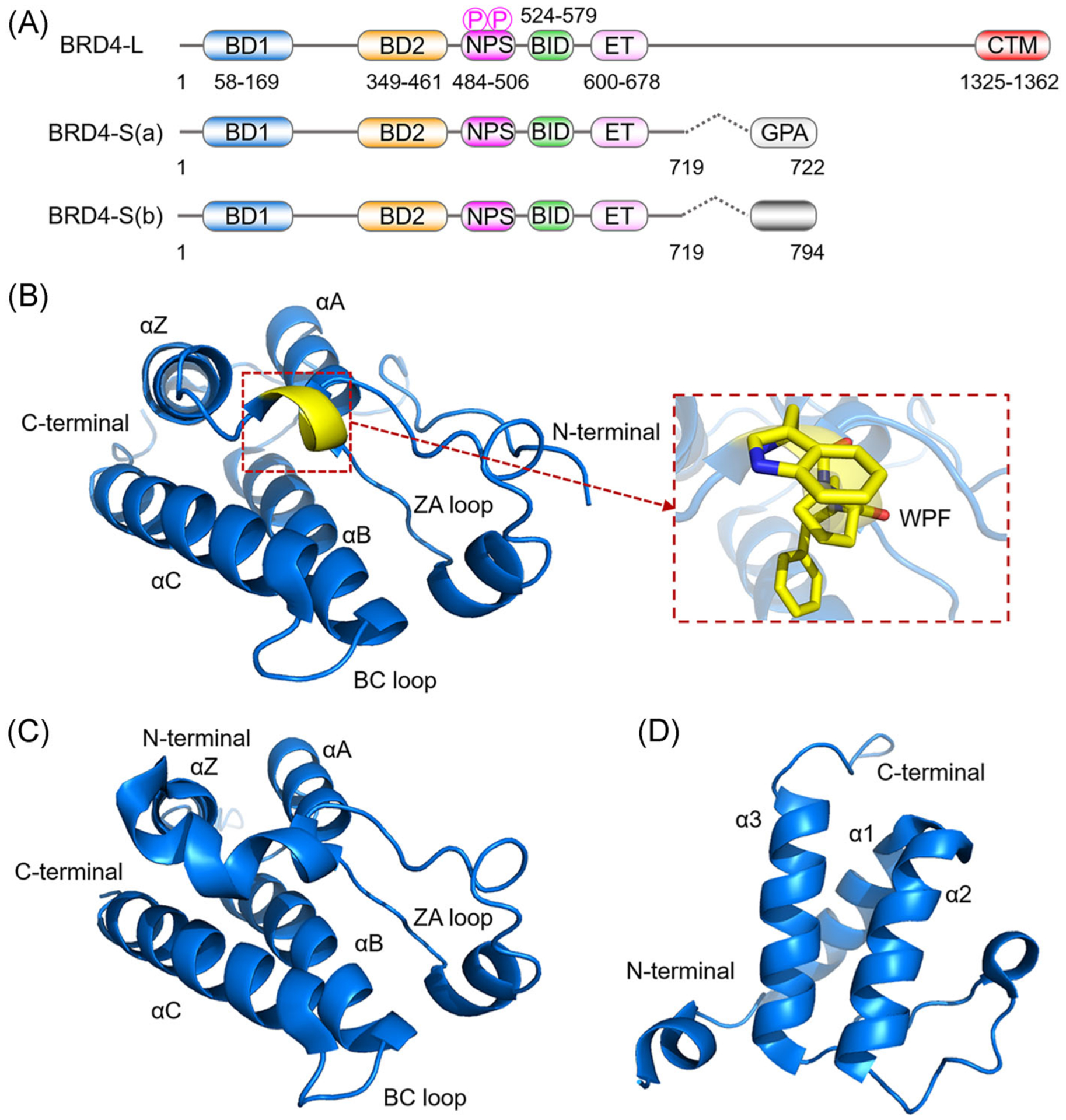
Schematic diagram of BRD4 domain features and crystal structures. (A) Domain architecture of three BRD4 protein isoforms. (B) Crystal structure of BRD4(1) (PDB ID: 3MXF). (C) Crystal structure of BRD4(2) (PDB ID: 5U2C). (D) NMR structure of the BRD4 ET domain (PDB ID: 6BNH). Protein is represented by blue ribbons with WPF shelf residues indicated by yellow sticks
BD1 and BD2 of BRD4, abbreviated as BRD4(1) and BRD4(2), are highly conserved and consist of ~110 amino acids with four antiparallel α helices (αZ, αA, αB, and αC), and two hydrophobic (ZA and BC) loops (Figure 1B,C).59 Co-crystal structures show that the ZA loop and BC loop form a hydrophobic cavity contacting specifically Kac, which is the key feature for BRD4 inhibitor development. In addition to the Kac-binding pocket, the WPF shelf and the ZA channel also contribute to the structural feature of the BET BRD. WPF, a hydrophobic region illustrated by Trp81, Pro82 and Phe83 of BRD4(1), forms a shelf to improve the binding affinity of BRD4 (Figure 1B).60,61 The ZA channel, a hydrophobic pocket formed by a portion of the ZA loop, is also an effective site for achieving small molecule inhibitor selectivity.62,63 Furthermore, the ET domain can regulate protein-protein interactions and also recruit chromatin regulators (such as NSD3, ATAD5, CHD4, GLTSCR1, and JMJD6) to mediate transcriptional activation (Figure 1D).64
Although BD1 and BD2 both bind to Kac, they are not functionally equivalent. Recently, researchers from GlaxoSmithKline have developed selective BD1 and BD2 inhibitors to explore respective functions of BD1 and BD2 in cancer and immunoinflammation.65 They found BD1 inhibitors have effects similar to pan-BET inhibitors in cancer and inflammation, whereas BD2 inhibitors are effective mainly in inflammatory and autoimmune disease models. However, ABBV-744 (a pan-BD2 inhibitor) is also active in prostate cancer and acute myeloid leukemia cell lines and has now entered the clinical trial of acute myeloid leukemia (NCT03360006).31,66 These findings will help optimize the strategies of targeted BET inhibition in different diseases in the future. In addition, differences in the loop regions of BD1 and BD2 have been selectively targeted for anticancer drug development.67
2.2 |. Biological functions of BRD4
BRD4 is a pivotal factor in gene-regulatory networks and plays a significant role in malignant tumor progression (Figure 2A). As a general coactivator, BRD4 can interact with P-TEFb, a complex formed by cyclin T1 (CCNT1) and cyclin-dependent kinase 9 (CDK9), and promote serine 2 phosphorylation of the C-terminal domain (CTD) of RNA polymerase II, thereby enhancing transcription elongation.68–70 BRD4 also upregulates the expression of oncogenic transcription factors such as myelocytomatosis viral oncogene cellular homolog (C-MYC), myelocytomatosis viral oncogene neuroblastoma-derived homolog (MYCN), FOSL1 and BCL2.56,71–73 In DNA synthesis, BRD4 promotes cell cycle progression in part by regulating the expression of Aurora B kinase and association with early 2 factor (E2F).74,75 In inflammatory response, BRD4 promotes the expression of interleukin-2 (IL-2), IL-8, and C-C motif chemokine ligand 2 (CCL2).76,77 BRD4 also associates with estrogen receptor (ER) and androgen receptor (AR) to regulate their target genes including growth regulating estrogen receptor binding 1 (GREB1), trefoil factor 1 (TFF1), puromycin-sensitive aminopeptidase, and calcium/calmodulin-dependent protein kinase kinase 2 (CAMKK2).78,79 BRD4 interacts with nucleophosmin1 (NPM1) to inhibit some transcription programs in acute myeloid leukemia.80 In addition, BRD4 phosphorylation is very important and represents the active form of BRD4 in binding to acetylated chromatin.57,81 Upon CK2 phosphorylation, BRD4 can recruit p53 and mediate p53 target gene transcription.57 In DNA damage response, BRD4 can recruit Condensin II, reshape chromatin structure, and inhibit DNA damage.58 BRD4 is a signal transducer of cellular response to oxidative stress. Downregulation of BRD4 can cause imbalance of the Kelch-like ECH-associated protein 1 (KEAP1) pathway, leading to disorder of heme oxygenase 1 (HMOX1) and reactive oxygen species.82
FIGURE 2.
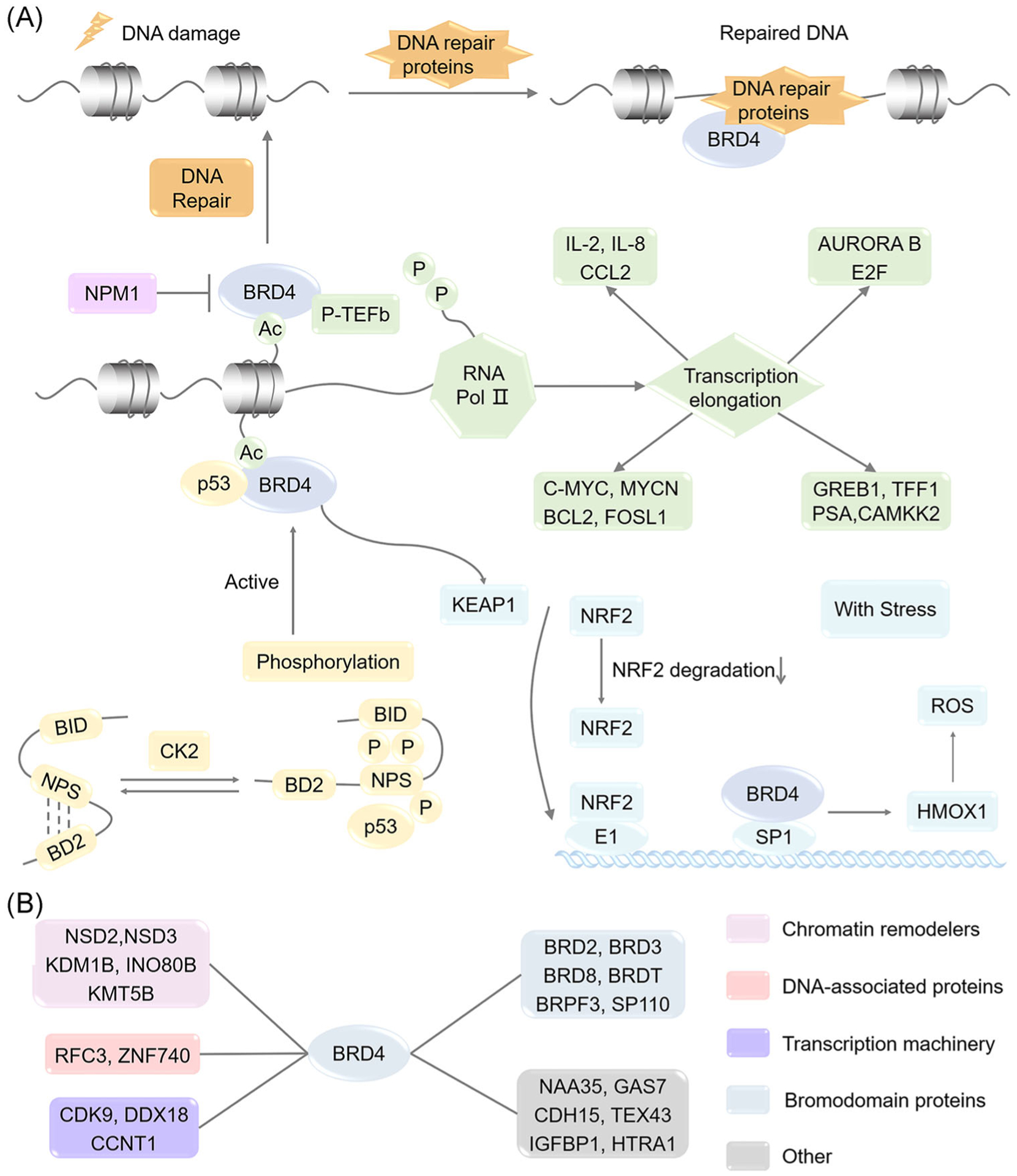
Schematic diagram of gene regulation and protein interactome networks of BRD4. (A) Schematic diagram of BRD4 gene-regulatory networks. (B) Schematic diagram of BRD4 interactome. BCL2, B-cell lymphoma 2; BD2, bromodomain 2; BID, basic residue-enriched interaction domain; BRD2, bromodomain-containing protein 2; BRD3, bromodomain-containing protein 3; BRD4, bromodomain-containing protein 4; BRD8, bromodomain-containing protein 8; BRDT, bromodomain testis-specific protein; BRPF3, bromodomain and PHD finger-containing protein 3; CAMKK2, calcium/calmodulin-dependent protein kinase kinase 2; CCL2, C-C motif chemokine ligand 2; CCNT1, cyclin T1; CDH15, cadherin 15; C-MYC, myelocytomatosis viral oncogene cellular homolog; CDK9, cyclin-dependent kinase 9; CK2, casein kinase II; DDX18, DEAD-box helicase 18; E2F, early 2 factor; FOSL1, fos like 1; GAS7, growth arrest specific 7; GREB1, growth regulating estrogen receptor binding 1; HMOX1, heme oxygenase 1; HTRA1, HtrA serine peptidase 1; IGFBP1, insulin-like growth factor-binding protein 1; IL-2, interleukin-2; IL-8, interleukin-8; INO80B, INO80 complex subunit B; KDM1B, lysine demethylase 1B; KEAP1, Kelch-like ECH-associated protein 1; KMT5B, lysine methyltransferase 5B; MYCN, myelocytomatosis viral oncogene neuroblastoma-derived homolog; NAA35, N-alpha-acetyltransferase 35; NPM1, nucleophosmin1; NPS, N-terminal cluster of CK2 phosphorylation sites; NRF2, nuclear factor erythroid 2-related factor 2; NSD2, nuclear receptor binding SET domain protein 2; NSD3, nuclear receptor binding SET domain protein 3; PSA, puromycin-sensitive aminopeptidase; P-TEFb, positive transcription elongation factor b; RFC3, replication factor C subunit 3; ROS, reactive oxygen species; SP110, SP110 nuclear body protein; TEX43, testis-expressed 43; TFF1, trefoil factor 1; ZNF740, zinc finger protein 740
At the cellular level, BRD4 activity can be disrupted by inhibiting BRD4-interacting complexes.83 Through systematic proteomics-based BRD4-interacting network analysis, Kim et al.84 identified chromatin remodelers, DNA-associated proteins, transcription-related factors, and BRD proteins that interact with BRD4 with crucial roles in gene transcription, genomic maintenance, and DNA repair (Figure 2B).
2.3 |. Synergistic effects of BET inhibitors and other co-targeted inhibitors
BET proteins are the most distinctive epigenetic readers and have become attractive therapeutic targets for cancer treatment. However, in early clinical trials, BET inhibitors exhibit toxic effects including severe thrombocytopenia, fatigue, nausea, and gastrointestinal side effects.85 Research shows that BET and other antitumor inhibitors have synergistic cancer-suppressing effects. JQ1, when combined with rapamycin, a mammalian target of rapamycin (mTOR) inhibitor, can synergistically inhibit the growth and survival of human osteosarcoma cells in vitro and in vivo.86 Combination of JQ1 and THZ1 (a CDK7 inhibitor) has a synergistic effect on inhibiting pancreatic ductal adenocarcinoma.87 Furthermore, BET inhibitors also show synergy with inhibitors of other cancer-related proteins such as BCL2 and Aurora A.73,88,89
Epigenetic synergy is one of the most promising areas of cancer research. BET and histone deacetylase (HDAC) inhibitors can induce similar genes with comparable biological effects, synergistically suppressing MYC-driven mouse lymphoma cell growth and inducing apoptosis.90 Moreover, combined SP-2509 and JQ1, as respective histone lysine demethylase 1 (LSD1) and BET-regulatory agents, alter the growth and invasion of prostate cancer.91
Undoubtedly, synthetic lethal interaction is also a promising new strategy for cancer treatment.92 Synthetic lethality means that a defect in one of the two genes or proteins in a cell or organism has little effect, but combined defects in both genes/proteins can cause significant impairment.93 Inhibiting BRD4 downregulates the expression of C-terminal binding protein-interacting protein (CtIP) and induces homologous recombination deficiency, making cells with homologous recombination deficiency sensitive to poly ADP-ribose polymerase (PARP) inhibitors.94 Therefore, BET inhibitors and PARP inhibitors show synthetic lethality in inducing homologous recombination defects. Although synthetic lethal interactions are promising, identifying such interaction pairs is challenging. Methods for identifying synthetic lethal pairs include: (1) high-throughput screening by chemical library screening, genome-wide RNAi analysis and CRISPR–Cas9 mutagenesis; (2) analysis of conservative connections using genetically engineered lower eukaryotes, such as yeast models; and (3) bioinformatics prediction strategy.93,95
BET inhibitors together with immune checkpoint inhibitors also enhance anticancer effects. BRD4 binds directly to the promoter of the programmed death ligand 1 (PD-L1) gene and activates PD-L1 transcription.96 In MYC-induced lymphoma, JQ1 and programmed death 1 (PD-1) monoclonal antibodies can significantly reduce tumor burden and extend the overall survival time.97
2.4 |. Discovery and design of dual-target inhibitors of BET BRDs
Considering the synergistic effect and synthetic lethality of BET inhibitors and various target inhibitors, developing multitarget drugs that concurrently inhibit BET and other targets is a promising strategy to overcome tumor drug resistance and prolong clinical treatment outcome. Drug discovery and design strategies for dual inhibitors of BET and other targets are reviewed here, including medicinal chemistry and computational approaches.98 Medicinal chemistry design strategies primarily include drug repurposing, skeleton modification of lead compounds, and pharmacophore fusion.
Drugs approved or under investigation may involve more than one pathology and trigger unpredictable targets. Drug repurposing can explore new therapeutic options. It is worth mentioning that screening compound libraries can also discover other potential and unpredictable targets for small-molecule inhibitors. In 2014, Schönbrunn and Knapp’s groups32,35 used kinase inhibitor libraries to identify that BET could be a potential target for multiple kinase inhibitors through robotic co-crystallization screening and AlphaScreen analysis. Additionally, Urick et al.99 established protein-observed fluorine NMR to screen the kinase inhibitor library and discover dual kinase-BRD inhibitors. It is worth noting that some dual kinase-BRD inhibitors have entered clinical trials. Since BET proteins are potential targets for many kinase inhibitors, it is feasible to design dual kinase-BRD inhibitors by expanding the repertoire of these kinase inhibitors.
Skeleton modification of lead compounds is also an effective and commonly used strategy to obtain dual-target inhibitors. By modifying the structure of BI-2536 (a dual kinase-BRD inhibitor), dual-target inhibitors with balanced selectivity can be obtained.100,101 Skeleton modification can also improve drug absorption/solubility, reduce toxic and side effects, improve metabolic stability, and thus improve drug specificity. But it is only suitable for certain scaffold types of small-molecule compounds.
Pharmacophore fusion combines different pharmacophores in different target inhibitors, which is conducive to the rational design of dual-target or even multitarget drugs with good inhibitory activity. The pharmacophore of HDAC inhibitors consists of a cap, a linker and a zinc-binding group (ZBG), in which the cap can be replaced by a BET inhibitor. In addition, the linker is usually located in a solvent-exposed region or the ZA channel of the BET proteins, providing more possibilities for the development of dual BET/HDAC inhibitors with improved drug properties.
At present, more and more evidence show that multipharmacological methods based on BET targeting are promising, so ligand- and structure-based computational methods would be helpful to identify dual-target BET inhibitors. Allen et al.102 developed a computational screen to identify dual BRD/kinase inhibitors. Their virtual screening method combines highly predicted ligands and structure-based models constructed from small molecule kinase inhibition datasets, BRD4 protein structures, and predictors of physical and chemical properties to successfully identify novel BRD4 inhibitors and dual EGFR/BRD4 inhibitors from more than 6 million commercial compounds. In 2016, Carlino et al.103 also introduced computational methods that can be used to identify dual kinase/BRD inhibitors, and generate ad hoc pharmacophores and docking models. This computational method is applicable to identify dual BET and other target inhibitors with potential to treat multiple cancers.
These strategies, as mentioned above, will provide new perspectives for future design of dual BET inhibitors to facilitate the discovery of new cancer therapeutics.
3 |. DUAL-TARGET INHIBITORS OF BET BRDS AND OTHER CANCER-RELATED TARGETS
3.1 |. Dual inhibitors of BET BRDs and kinases
3.1.1 |. Dual inhibitors of BET BRDs and CDKs
In 2012, Devaiah et al.104 reported that in addition to recognizing Kac, BRD4 is an atypical kinase that phosphorylates serine 2 of the RNA polymerase II CTD. However, the potential of BRD4 BRDs to interact with kinase inhibitors is still unclear. In 2013, Martin et al.33 found that dinaciclib (1, SCH-727965; Figure 3A), a potent CDK inhibitor, interacts with the Kac recognition pocket of BRDT, providing evidence for possible interaction of kinase inhibitors with a BET BRD. In 2014, the Schönbrunn group32 identified the binding mode of Dinaciclib in BRD4(1) with cocrystal structures showing the pyrazolo-pyrimidine moiety of Dinaciclib forms two hydrogen bonds with Asn 140 of BD1. Additional hydrogen bonds are formed by pyridine oxide with Asp 145 and Ile 146. Simultaneously, hydroxyl and the ZA channel form additional hydrogen bonds through water molecules (Figure 3B). Structural analysis of dinaciclib binding to CDK2 is shown in Figure 3C. Dinaciclib inhibits CDK1, CDK2, CDK5, CDK9, and BRD4 with IC50 values of 3, 1, 1, 4 nM, and 18.7 μM, respectively.32,105 It has entered Phase III clinical trials for treating chronic lymphocytic leukemia (NCT01580228).
FIGURE 3.
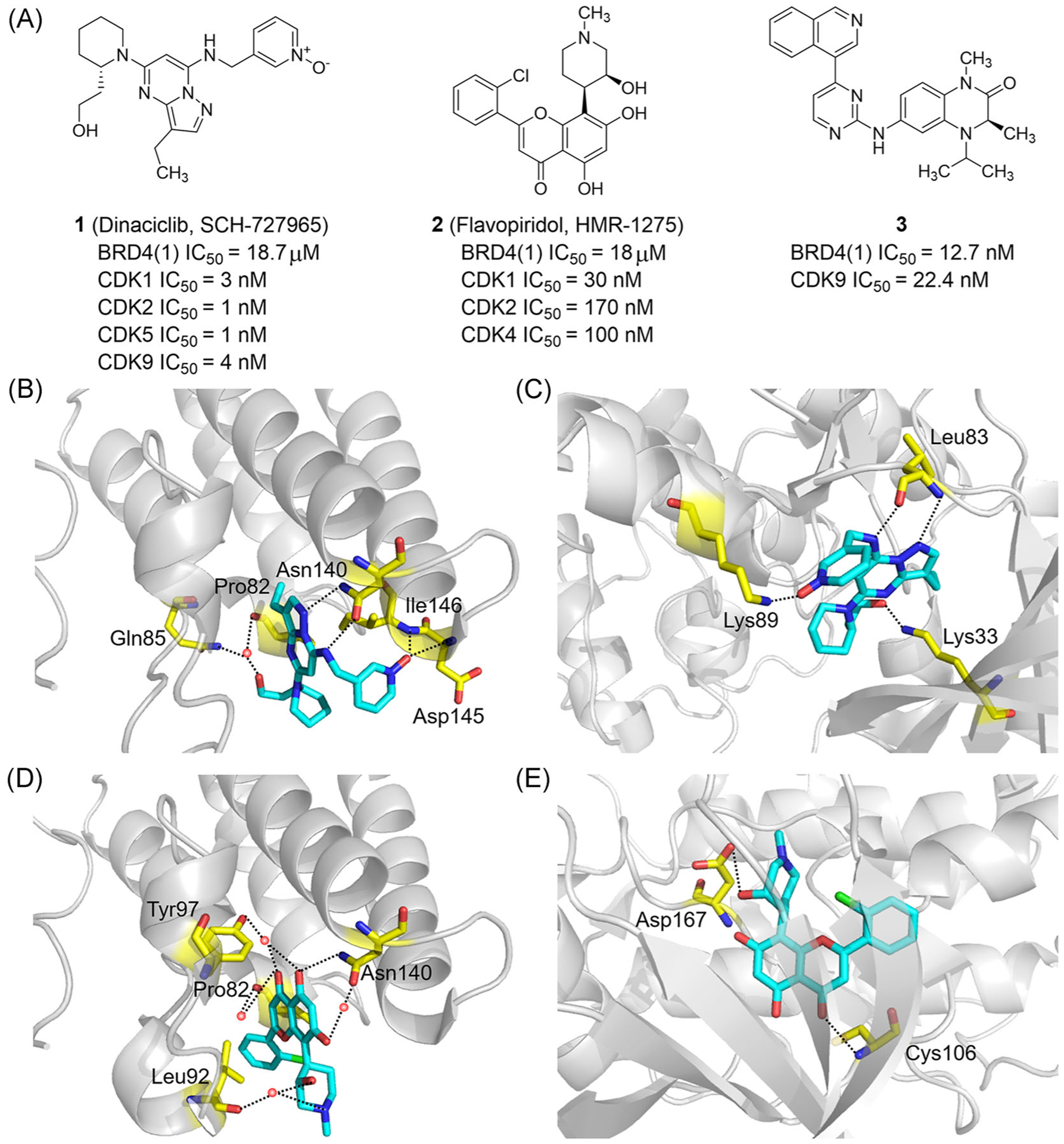
Dual-target inhibitors of BET bromodomains and CDKs. (A) Chemical structures of dinaciclib, flavopiridol and compound 3. (B) X ray cocrystal structure of Dinaciclib bound to BRD4(1) (PDB ID: 4O70). (C) X ray cocrystal structure of dinaciclib bound to CDK2 (PDB ID: 4KD1). (D) X ray cocrystal structure of Flavopiridol bound to BRD4 (PDB ID: 4O71). (E) X ray cocrystal structure of flavopiridol bound to CDK9/cyclin T1 (PDB ID: 3BLR). Protein is represented by gray ribbons, with key compound-contacting residues indicated by yellow sticks. The compound is shown in sticks and colored according to the atom type (C, light blue; N, blue; O, red; Cl, green). Black dashed line = hydrogen bond; orange sphere = water molecule
Flavopiridol (2, HMR-1275; Figure 3A) is a broad-spectrum CDK inhibitor and the first CDK inhibitor used in clinical trials with IC50 values of 30, 170, and 100 nM for CDK1, CDK2, and CDK4, respectively.106 In 2013, Flavopiridol completed Phase II clinical trials for treating endometrial cancer, chronic lymphocytic leukemia, and recurrent prostate cancer (NCT00023894, NCT00464633, and NCT00003256). Flavopiridol is also a ligand for the Kac-binding pocket, with an IC50 value of 18 μM for BRD4(1). Flavopiridol forms hydrogen bonds with Asn140 of BD1 through chromenone hydroxyl, while carbonyl oxygen interacts with the ZA channel (Figure 3D).32 Flavopiridol forms two hydrogen bonds with Cys106 and Asp167 of CDK9 (Figure 3E).
In 2020, through pharmacophore fusion, Lv et al.107 found that compound 3 (Figure 3A) has balanced activities of BRD4(1) (IC50 = 12.7 nM) and CDK9 (IC50 = 22.4 nM). Moreover, compound 3 shows good antiproliferative activity on a small cancer cell panel, indicating a synergistic effect in the cellular context. In HCT116 cell line, compound 3 demonstrates good inhibitory activity in vitro and large doses could block the activities of CDK9 and BRD4 in a short time.
Collectively, these results provide strong evidence for the potential of kinase inhibitors to inhibit BET proteins, and provide a conceptual framework for the design of new BET inhibitors and dual kinase and BET inhibitors.
3.1.2 |. Dual inhibitors of BET BRDs and polo-like kinase 1 (PLK1)
PLK1, a crucial cell cycle regulator, is important in DNA damage response, cell proliferation, and cell cycle control.108,109 It is closely linked to the development of malignant tumors.110 Importantly, PLK1 is related to the expression of C-MYC.111 PLK1 inhibitors not only significantly inhibit PLK1, but also reduce downstream C-MYC phosphorylation, and weaken the binding of BRD4 to C-MYC, thereby inhibiting C-MYC function. Therefore, dual-target inhibitors of BET and PLK1 have the potential to increase the efficacy of both targets and reduce toxicity.
The Schönbrunn group32 found the PLK1 inhibitor BI-2536 (4) was the most effective BET inhibitor in the Selleck library by co-crystallization screening of the kinase libraries, with an IC50 value of 25 nM for BRD4(1). BI-2536 interacts with the Kac site of BRD4(1) through a complex network of hydrogen bonding and hydrophobic interaction (Figure 4A). The carbonyl group of BI-2536 forms a hydrogen bond with Asn140 of BD1. Meanwhile, the aminopyrimidine moiety binds to Pro82 and water molecules of the ZA channel. Key hydrogen bonds between BI-2536 and PLK1 are shown in Figure 4B, with the hinge-binding region of BI-2536 pointing to the ZA channel and Pro82 of BRD4(1). Significantly, the Knapp group35 also found that BI-2536 is an effective dual BET/kinase inhibitor, which displaces BRD4 from chromatin and inhibits the expression of C-MYC. Kinome-wide profiling confirmed the interaction of BI-2536 in the whole BRD proteins is mainly limited to BET BRDs, while its activity against non-BET BRDs such as BRD7, BRPF1B, and BRWD3(2) was weak. Most noteworthy is the methoxy group in BI-2536 pointing to the solvent region of BRD4(1) is very important for PLK selectivity. Thus, it is possible to modify this position to keep the BRD activity unchanged while regulating the ability to recognize different kinases.
FIGURE 4.
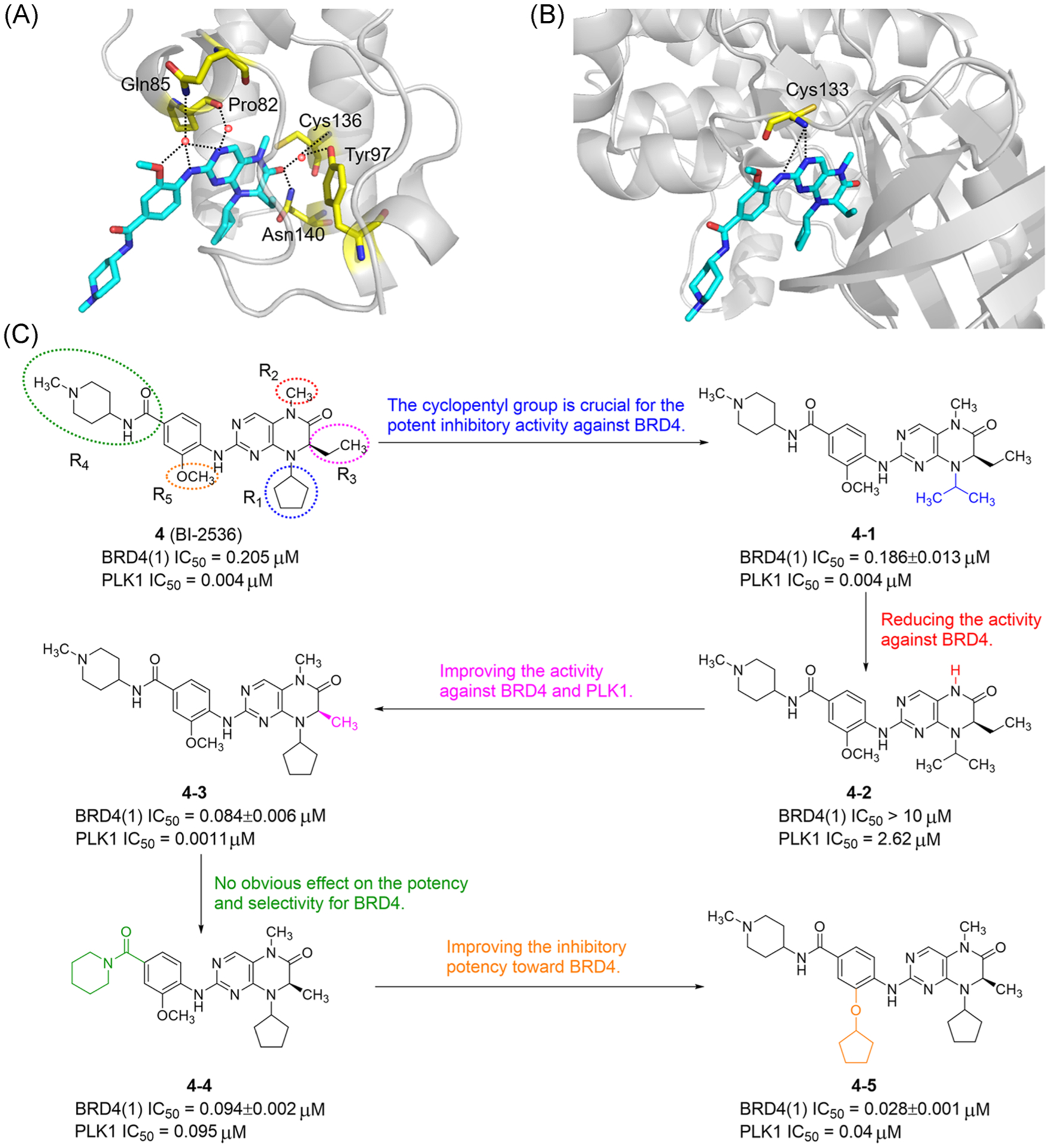
Development of dual-target inhibitors of BET bromodomains and PLK1. (A) X ray cocrystal structure of BI-2536 bound to BRD4(1) (PDB ID: 4O74). (B) X ray cocrystal structure of BI-2536 bound to PLK1 (PDB ID: 2RKU). (C) SARs of BI-2536. Protein is represented by gray ribbons with key compound-contacting residues highlighted in yellow sticks. The compound is shown in sticks and colored according to the atom type (C, light blue; N, blue; O, red). Black dashed line = hydrogen bond; orange sphere = water molecule
To obtain balanced dual-target inhibitors of PLK1 and BET BRDs, Liu et al.100 conducted a pharmacochemical study with BI-2536 as the core skeleton (Figure 4C). The results showed the cyclopentyl group at the R1 position and methylated amides at the R2 position are very important to inhibit BRD4. After the cyclopentyl group was replaced, compound 4–1 was less effective against BRD4 than BI-2536. The affinity of compound 4–2 to BRD4 decreased more than 1000 times. Compound 4–3 with methyl substituents on the asymmetric dihydropteridinone carbon atom showed good inhibitory activity against BRD4 and PLK1. Substitution of terminal cyclic amines with different aromatic amines at the R4 position did not seriously affect the performance and selectivity of compounds to BRD4, such as compound 4–4. Upon replacement of methoxy with cyclopentanoxyl at the R5 position, the effect of compound 4–5 on BRD4 was increased and the effect on PLK1 was decreased. In 2020, Timme et al.112 found that compound 4–5 (UMB160) had synergistic antitumor effects, induced apoptosis of pediatric tumor cells at low nanomolar concentrations, and had strong antitumor activity against neuroblastoma, medulloblastoma, and rhabdomyosarcoma cells.
Cyclopentyl modification can adjust the BRD activity, which for BI-6727 (5, Volasertib; Figure 5) leads to a decrease in the affinity for BRD4(1).35 BI-6727 has good pharmacokinetic properties and tolerability. The main toxicity in clinical studies is reversible myelosuppression.113 Due to its limited non-hematological toxicity, BI-6727 is also an attractive drug for combination therapy. Additionally, Kolosenko et al.114 reported that BI-6727 affects normal cells in PBMCs stimulated by CD3.
FIGURE 5.
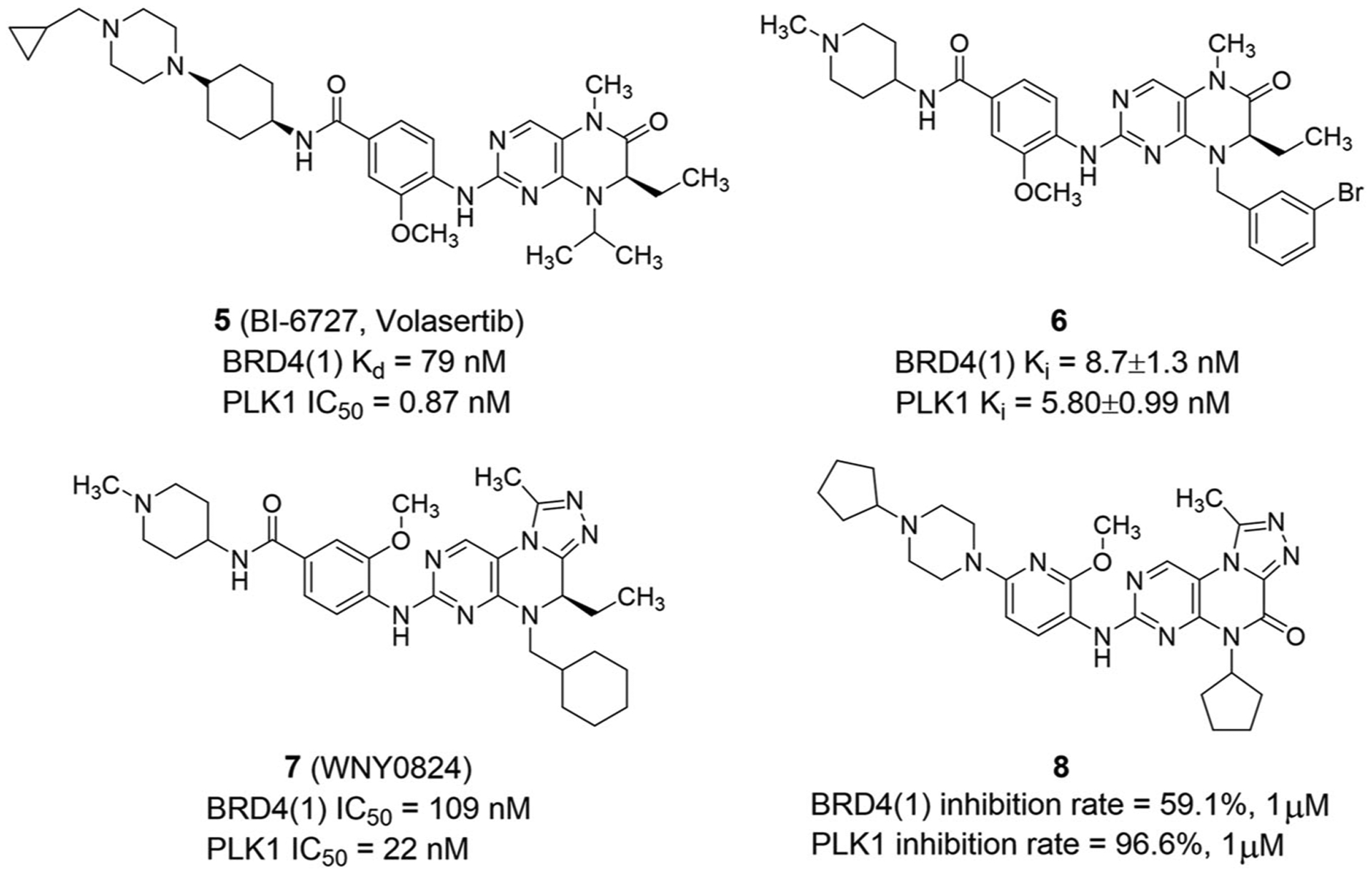
Other dual-target inhibitors of BET bromodomains and PLK1
Chen et al.101 reported that modifying the 3-methoxy group on the benzene ring is essential to increase BRD4(1) binding and maintain dual-target inhibition. But at the same time, pyrimidine NH, cyclopentyl, and R-stereochemistry of the ethyl group should be retained. Among them, compound 6 is most effective with a Ki value of 8.7 nM for BRD4(1), which is equivalent to that of PLK1 (Figure 5). The high affinity of compound 6 for BRD4 may be due to increased interaction with the WPF shelf. Besides, the pyrimidine NH was replaced by an oxygen atom, and the cyclopentyl group and the 3-methoxy group on the benzene ring were modified to obtain a selective sub-nanomolar BRD4 inhibitor.
In 2020, Wang et al.115 found that WNY0824 (7) had strong antiproliferative activity against cancer cells, with IC50 values of 109 and 22 nM for BRD4 and PLK1, respectively (Figure 5). WNY0824 exhibited antitumor activity in vivo at a dose of 60 mg/kg and the antitumor rate was 66% with no obvious toxicity in an MV4–11 mouse xenograft model. WNY0824 also inhibited the expression of C-MYC, MYCN, and BCL2. Moreover, WNY0824 could alter the AR-dependent transcription program and inhibit the ETS pathway, with excellent antiproliferative activity on AR-positive CRPC cells.116 Oral administration of WNY0824 inhibited the growth of CRPC xenograft tumors resistant to enzalutamide.
Qi et al.117 found that the inhibition rates of compound 8 (Figure 5) for BRD4(1) and PLK1 at 1 μM were 59.1% and 96.6%, respectively. Compound 8 exhibited cytotoxic activity against A549, HCT116, PC-3 and MCF-7 cell lines, with IC50 values of 1.27, 1.36, 3.85, and 4.06 μM, respectively. But its activity was slightly weaker than that of BI-2536. In HCT116 cell line, compound 8 significantly inhibited cell proliferation, induced apoptosis, and blocked the S phase. As a potential anticancer agent for further study, compound 8 can be modified to improve BRD4 inhibitory activity. These results provide the rationale for design of PLK1-BET inhibitors.
3.1.3 |. Dual inhibitors of BET BRDs and anaplastic lymphoma kinase (ALK)
ALK is a RTK belonging to the insulin receptor subfamily and is important in various cancers including neuroblastoma,118–120 in which ALK kinase is often mutated to ALKF1174L with concurrent MYCN gene amplification.121 Suppressing BRD4 causes downregulation of MYCN expression, providing an effective strategy for treating MYCN-dysregulated neuroblastoma.122 Therefore, dual-target inhibitors that synergistically inhibit two targets may provide an effective cancer therapy.
Starting with BI-2536, Watts et al.123 adopted a structure-based design to develop dual-target inhibitors of BET BRDs and ALK by regulating kinase selectivity (Figure 6A). They found the IC50 values of BI-2536 for ALK and ALKF1174L were 390 and 190 nM, respectively. As an analog of Kac, methyl amide plays an important role in binding BRD4. Aminopyrimidine interacts strongly with the hinge region of PLK1 kinase. The interaction between aminopyrimidine of BI-2536 and the hinge region of ALK is similar to that of PLK1. By maintaining the key groups on the core of dihydropteridinone that interact with ALK and BRD4, Watts et al. optimized BI-2536 to reduce the activity of PLK1, improve the activity of ALK, and maintain the activity of BRD4 (Figure 6B). After the methoxy group at the R1 position was substituted with an ethoxy group, the effect of compound 9–1 on ALKF1174L was decreased, while the effect on PLK1 was greater. For BRD4, the R1 position had tolerance for larger groups. The R-ethyl group at the R2 position was critical for the effectiveness of ALKF1174L, PLK1, and BRD4. After it was changed to a hydrogen atom, the activity of compound 9–2 decreased significantly. After removing the amide group at the R3 position, compound 9–3 showed a four-fold decrease in potency against PLK1. Hence, the amide group was very important to maintain the activity of PLK1, and had little effect on the activity of ALK and BRD4. Because the cyclopentyl group of BI-2536 had a great influence on the activity of PLK1, it was converted into benzyl at the R4 position in compound 9–4, whose activity for PLK1 was reduced by 30–40 fold. Compound 9–5 changed it to a thiophene ring. Although compound 9–5 improved IC50 and Kd values for ALKF1174L and BRD4, the selectivity for ALKF1174L and PLK1 were decreased. When the methyl group of the thiophene ring of compound 9–5 was changed to the 4-position, the IC50 and Kd values of compound 9–6 were further increased, and the selectivity for ALKF1174L and PLK1 was increased by seven times. The methyl amide part of compound 9–6 forms a key hydrogen bond with Asn 140 (Figure 6C). It is gratifying that 9–6 shows excellent BET family selectivity and extensive kinome selectivity. Both nanoBRET and ALK autophosphorylation assay proved that 9–6 simultaneously inhibited BRD4 and ALKF1174L in cells. In addition, 9–6 induced mitotic arrest.123
FIGURE 6.
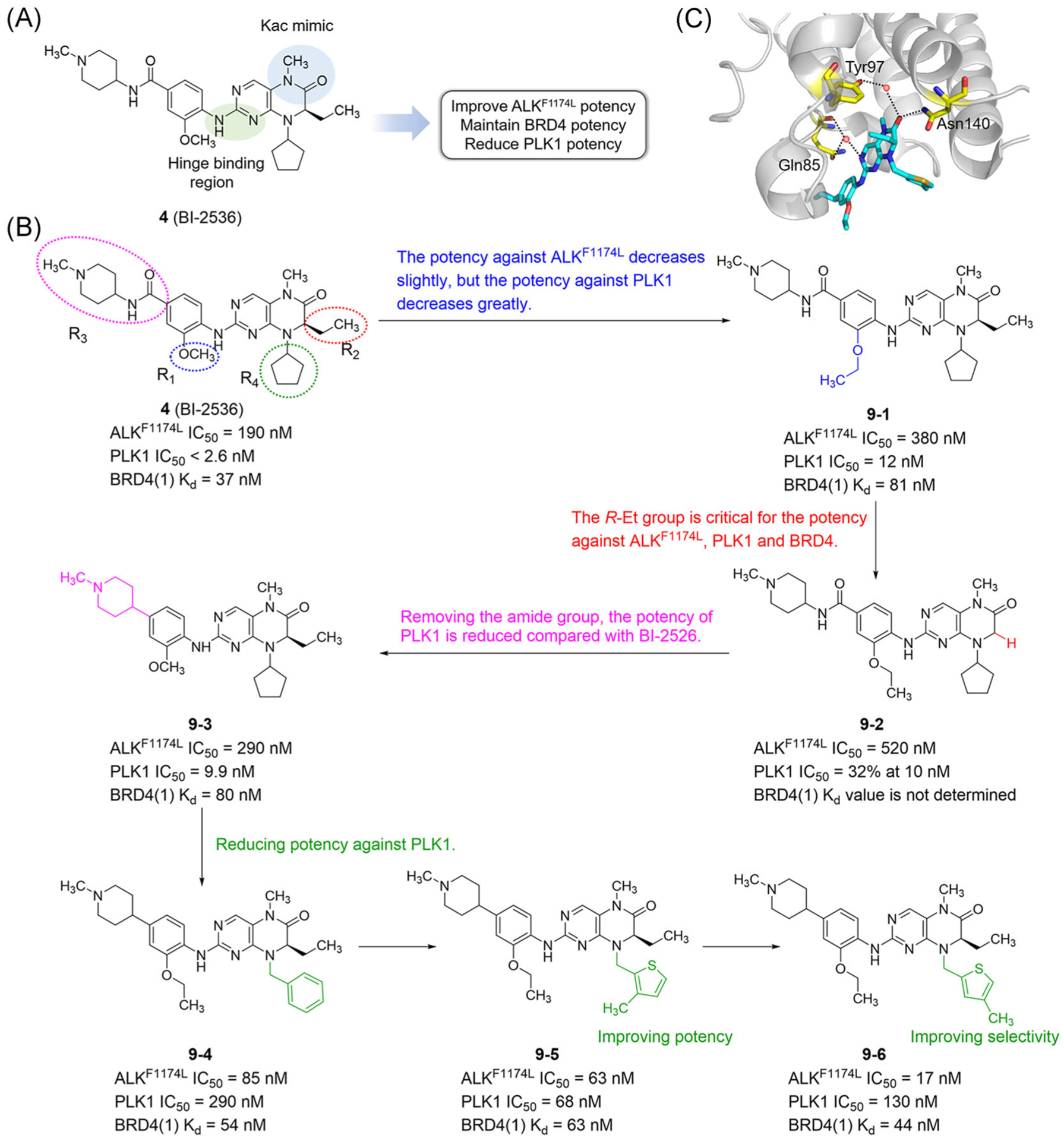
Design and SARs of dual-target inhibitors of BET bromodomains and ALK. (A) BI-2536 forms key interaction with BRD4 (blue) and PLK1 (green). (B) SARs of dual-target inhibitors of BET bromodomains and ALK. (C) X ray cocrystal structure of compound 9–6 bound to BRD4(1) (PDB ID: 6Q3Z). Protein is represented by gray ribbons with key compound-contacting residues highlighted in yellow sticks. The compound is shown in sticks and colored according to the atom type (C, light blue; N, blue; O, red; S, yellow). Black dashed line = hydrogen bond; orange sphere = water molecule
Currently, compound 9–6 is the first dual BRD4-ALK inhibitor reported. It is worth mentioning that the authors changed PLK1 kinase activity to ALK, taking an important step towards the design and discovery of new dual kinase/BRD inhibitors. Their work combines two different pharmacophores into a drug-like compound, which not only effectively inhibits two targets, but also has selectivity in two protein families. For the design of dual-target inhibitors, the starting point is very important, because the choice of chemical optimization tends to be more limited. Importantly, when both targets are tolerant to a range of different chemical inhibitors, the discovery of dual-target inhibitors will increase the chance of success.
3.1.4 |. Dual inhibitors of BET BRDs and JAK2
Janus kinase 2 (JAK2) inhibitors, TG101348 (10; Figure 7A) and TG101209 (11; Figure 7A), bind BET BRDs and can be classified as dual kinase-BRD inhibitors.35 The IC50 values of TG101348 and TG101209 for BRD4(1), measured by AlphaScreen assay, are 290 and 130 nM, respectively. Importantly, TG101348 also effectively inhibits C-MYC expression in MM.1S multiple myeloma cells. It is worth mentioning that the secondary amine nitrogen and 5-methyl-pyrimidine ring of TG101348 form two hydrogen bonds with Asn140 of BD1 and also interact with the kinase hinge region. Additional hydrogen bonds are simultaneously formed by sulfonamide oxygen and pyrrolidine nitrogen with Leu92 through water molecules (Figure 7B). In 2020, TG101348 and decitabine entered a Phase II clinical trial in treating myeloproliferative tumors (NCT04282187).
FIGURE 7.
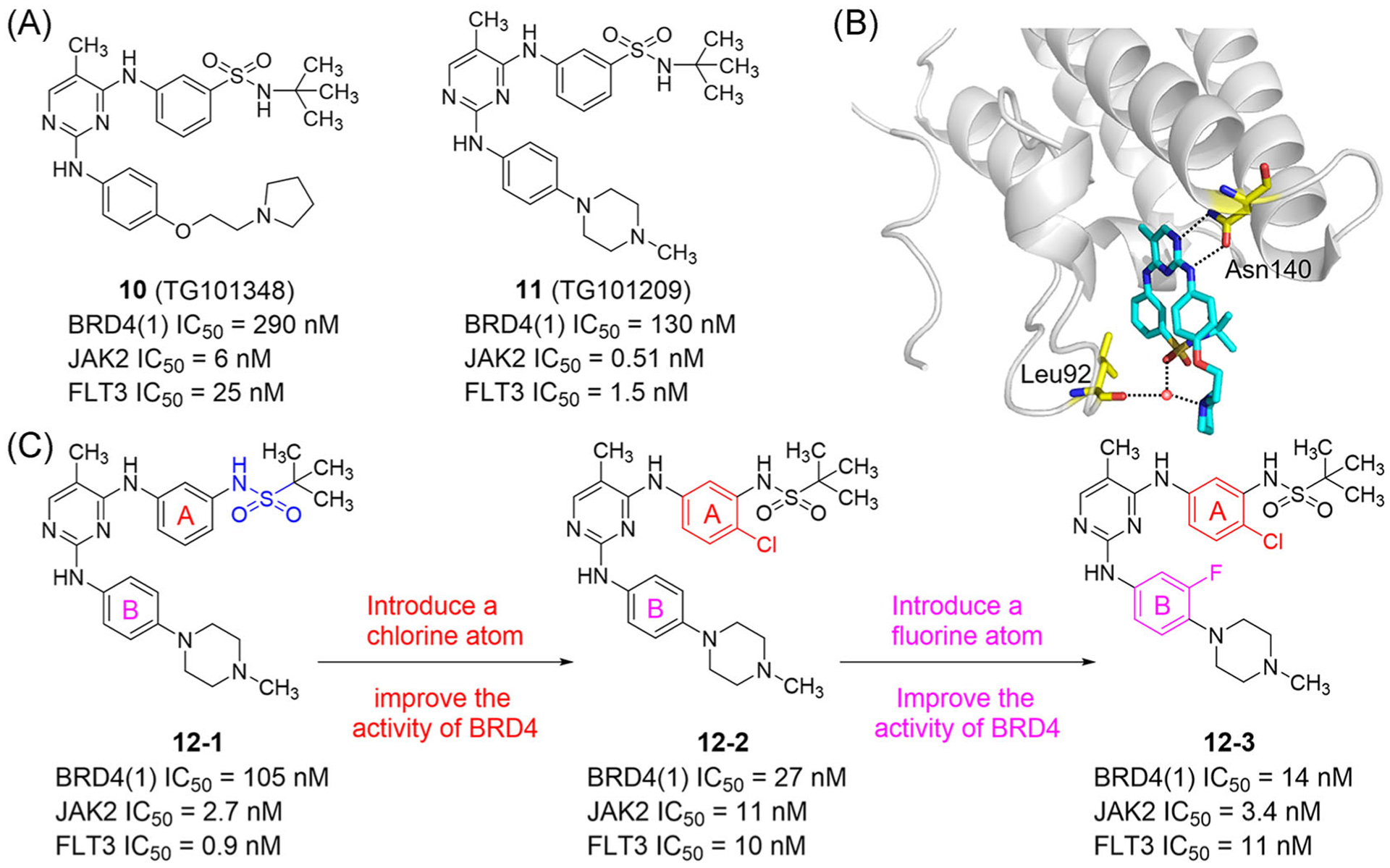
Development of dual inhibitors of BET bromodomains and JAK2. (A) Chemical structures of TG101348 and TG101209. (B) X ray cocrystal structure of TG101348 bound to BRD4(1) (PDB ID: 4OGJ). (C) SARs of dual inhibitors of BET bromodomains and JAK2. Protein is represented by gray ribbons, with key compound-contacting residues indicated by yellow sticks. The compound is shown in sticks and colored according to the atom type (C, light blue; N, blue; O, red; S, yellow). Black dashed line = hydrogen bond; orange sphere = water molecule
In 2017, Ember et al.124 designed effective dual BET/kinase inhibitors using the dianilinopyrimidine framework of TG101348 and TG101209. Upon reversal of the sulfonamide group at the 3′ position of the A ring, the binding potential of compound 12–1 to BRD4, JAK2, and FLT3 was similar to that of TG101209. Introducing a chlorine atom at the 4′ position of the A ring increased the activity of compound 12–2 for BRD4 by four times, but decreased the activities for JAK2 and FLT3. After introducing a fluorine atom at the 3″ position of the B ring, the binding activity of compound 12–3 to BRD4 increased, likely due to formation of hydrogen bonds between the NH group and Asn140 and Pro82 promoted by the fluorine atom as an electron absorber. Compound 12–3 has low activity against kinases, and compounds 12–2 and 12–3 show little difference in inhibition of BET, JAK2, and FLT3 (Figure 7C).
3.1.5 |. Dual inhibitors of BET BRDs and phosphoinositide 3-kinases (PI3Ks)
PI3Ks are a specific type of phosphatidylinositol kinases. They regulate malignant progression of tumors and are one of the research focuses in tumor targeting.125–127 Research shows that downregulation of BRD4 inhibits PI3K/AKT pathway, proliferation, and metastasis of gallbladder cancer cells, and induces apoptosis.128 LY294002 (13; Figure 8A), a small-molecule probe commonly used in cell-signaling research, belongs to the PI3K inhibitors (Figure 8A). In 2014, Dittmann et al.,129 through chemoproteomic target profiling, found that LY294002 inhibited Kac binding of BD1 but not BD2 of BET proteins. Therefore, LY294002 is a dual inhibitor of PI3K and BET proteins and its chromen-4-one scaffold can be used as a new BET pharmacophore.
FIGURE 8.
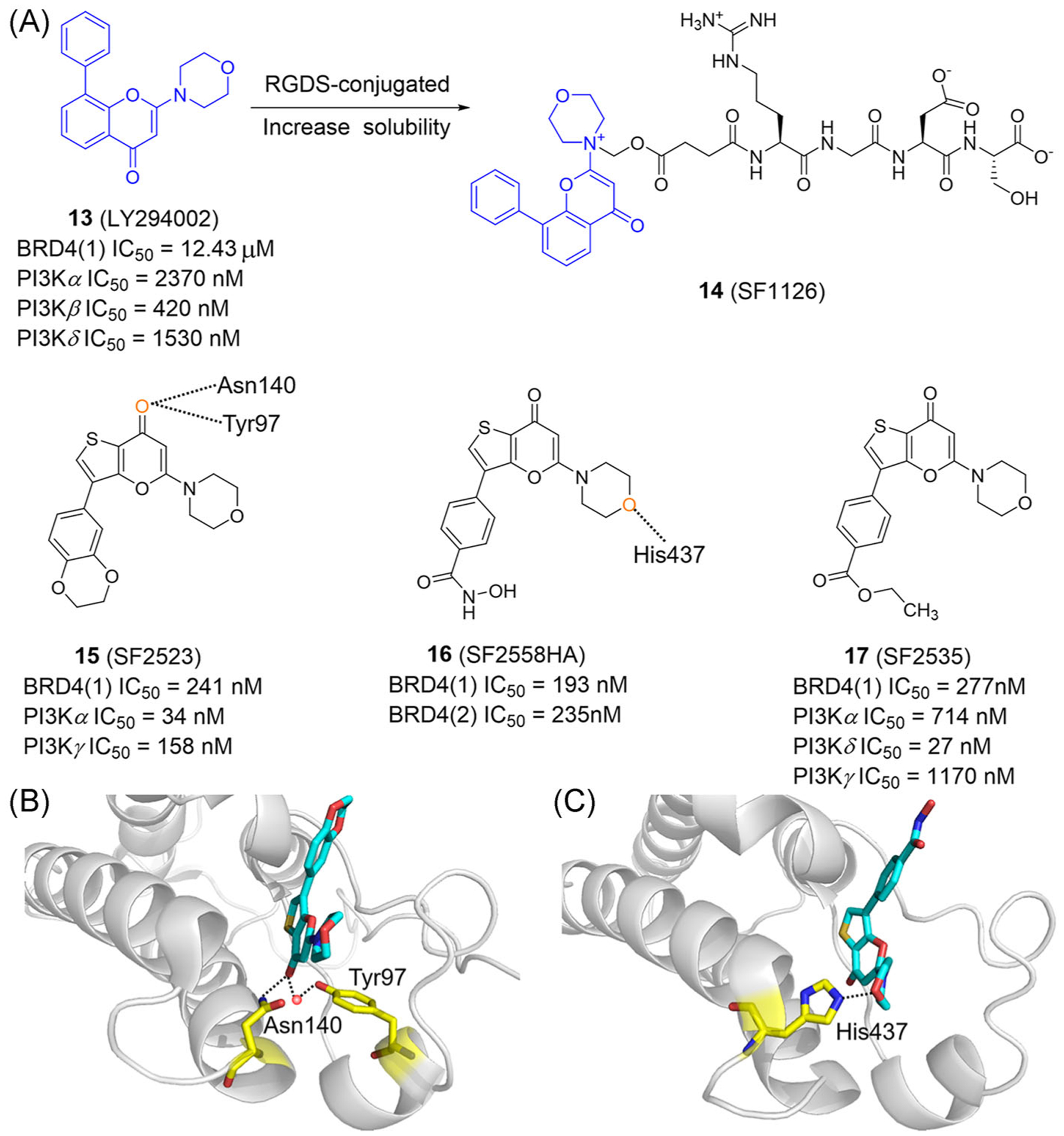
Dual-target inhibitors of BET bromodomains and PI3K. (A) Chemical structures of dual inhibitors of BET bromodomains and PI3K. (B) X ray cocrystal structure of SF2523 bound to BRD4(1) (PDB ID: 5U28). (C) X ray cocrystal structure of SF2558HA bound to BRD4(2) (PDB ID: 5U2C). Protein is represented by gray ribbons, with compound-contacting residues indicated by yellow sticks. The compound is shown in sticks and colored according to the atom type (C, light blue; N, blue; O, red; S, yellow). Black dashed line = hydrogen bond; orange sphere = water molecule
However, LY294002 has poor solubility and a short half-life. SF1126 (14) is an Arg-Gly-Asp-Ser (RGDS)-conjugated LY294002 prodrug with favorable solubility and pharmacokinetics (Figure 8A).130 In 2016, Singh et al.131 confirmed that SF1126 is a potent BRD4/PI3K inhibitor with IC50 values of 5.05, 6.89, 3.14, and 2.14 μM in Hep3B, HepG2, SK-Hep1, and Huh7 cells, respectively. SF1126 can inhibit tumor angiogenesis and down-regulate the expression of proangiogenic factors, such as VEGF, secreted by macrophages. Moreover, SF1126 can inhibit the growth of colorectal cancer cells in vivo and in vitro, primarily through PI3K-AKT-mTOR, BRD4, and p38 signaling pathways.132 Obviously, the selectivity of LY294002 (the active ingredients of SF1126) for PI3K is better than BET proteins.
In 2019, Joshi et al.133 found that SF2523 (15; Figure 8A) is an effective BRD4 and PI3K inhibitor, which downregulates MYC expression, inhibits tumor growth and metastasis, restores CD8+ T cell activity, and stimulates antitumor immune responses. Cocrystal structures show a key hydrogen bond is formed by the carbonyl oxygen in the thienopyrano group of SF2523 and Asn140 of BRD4(1). Simultaneously, the carbonyl oxygen forms a water-mediated hydrogen bond with Tyr97 (Figure 8B).38 SF2558HA (16; Figure 8A) can inhibit PI3K/BRD4 signaling both in vivo and in vitro, and significantly down-regulate MYC to inhibit the growth and metastasis of cancer cells.38 Importantly, SF2558HA has a stronger inhibitory activity for BRD4 than SF2523, with IC50 values of 193 and 235 nM for BRD4(1) and BRD4(2), respectively. The morpholino oxygen of SF2558HA forms a hydrogen bond with His437 of BRD4(2) (Figure 8C). In addition, SF2535 (17) is also a dual inhibitor of BRD4 and PI3K, inhibiting BRD4, PI3Kα, PI3Kδ, and PI3Kγ with IC50 values of 277, 714, 27, and 1170 nM, respectively.38,103 Notably, the morpholinothienopyrano skeleton is important for binding BRD4.
3.1.6 |. Dual inhibitors of BET BRDs and p38 kinase
p38 kinase inhibitors have low micromolar inhibitory activity against BET BRDs, and can also be used as dual kinase/BRD inhibitors, such as SB610251B (18), SB614067R (19), SB202190 (20), SB203580 (21), SB251527 (22), SB284847BT (23), and SB284851BT (24) (Figure 9).32,36,99 This provides a good starting point for designing representative ligands, researching multipharmacology, and exploring potential synergy between p38 and BET proteins. In 2018, Divakaran et al.134 synthesized a series of SB284851BT analogs as dual kinase-BRD inhibitors and evaluated their selectivity for the BET BRD family. The IC50 value of compound 25 against BRD4(1) is 1.8 μM (Figure 9). The selectivity of compound 25 to BD1 exceeds BD2. Besides, the selectivity of compound 25 to BRD4 is 6- and 16-fold better than that of BRD2 and BRD3, respectively. These results provide a valuable scaffold for the development of BD1-selective inhibitors of the BET BRDs. Importantly, the authors found that the observed selectivity was due to displacement of ordered water and conformational flexibility of Lys141 in BRD4(1). Compound 25 inhibits the expression of C-MYC oncoprotein in MM.1S cells and reduces the phosphorylation level of MSK1, a downstream phosphorylation target of p38α kinase. Such 1,4,5-trisubstituted imidazoles may be valuable starting points for dual kinase-BRD inhibition.
FIGURE 9.
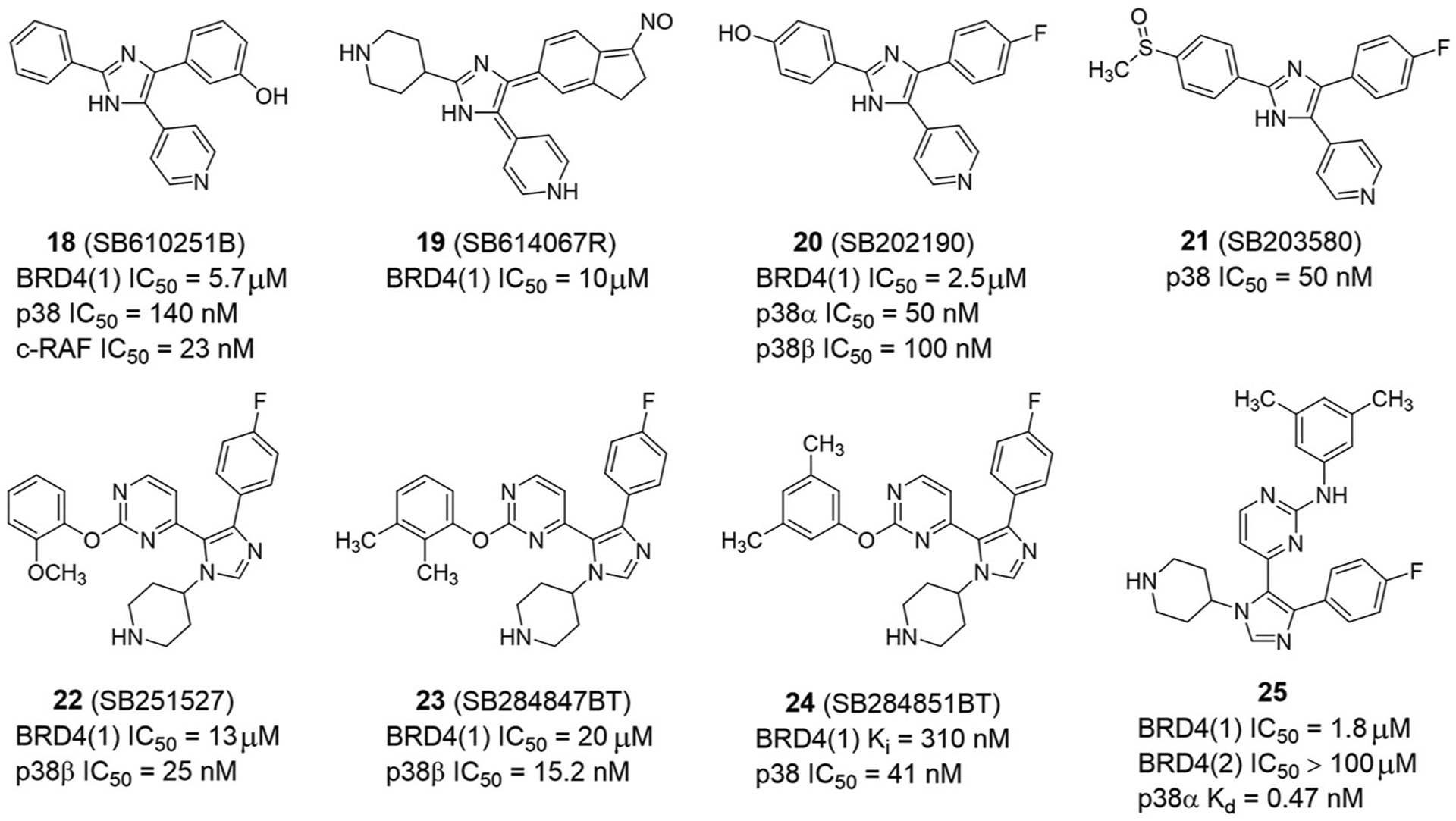
Dual-target inhibitors of BET bromodomains and p38 kinase
3.1.7 |. Dual inhibitors of BET BRDs and other kinases
Extracellular signal-regulated kinase 5 (ERK5) inhibitors also have potent BRD4-dependent pharmacology.39 structure–activity studies revealed that compounds JWG-069 (26; Figure 10) and XMD17–26 (27; Figure 10) with cyclobutyl and cyclopentyl substitutions not only show good binding to BRD4, but are also well tolerated by ERK5 and LRRK2, indicating that they are rational designs of BET/kinase multipharmacology. Compound JWG-047 (28; Figure 10) slightly enhances the binding to BRD4, while reduces the inhibition of ERK5 and LRRK2. These molecules with the benzo [e]pyrimido-[5,4-b]diazepine-6(11H)-one framework represent a new class of dual kinase and BRD inhibitors.
FIGURE 10.
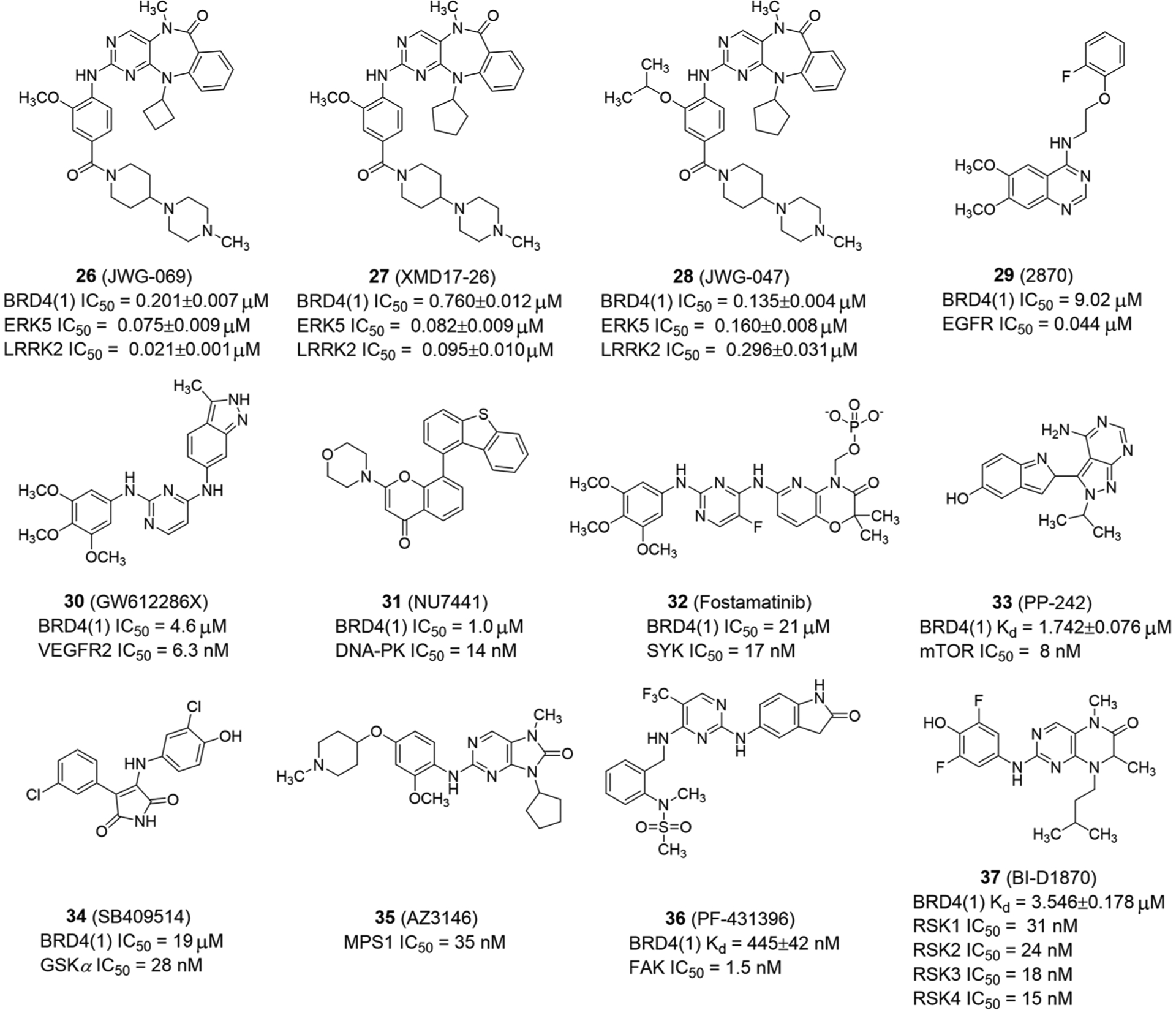
Dual-target inhibitors of BET bromodomains and other kinases
In 2015, Allen et al.102 identified a dual epidermal growth factor receptor (EGFR) and BRD4 inhibitor, namely 2870 (29), through computational screening, with IC50 values of 0.044 and 9.02 μM for EGFR and BRD4(1), respectively (Figure 10). Simulation analysis shows pyrimidine nitrogen at the 6 position of 2870 forms a conserved interaction with Asn140, Pro82, Leu 92, and Ile146 of BRD4. The 4-amino-quinazoline group interacts with the EGFR hinge region. GW612286X (30), as a VEGFR inhibitor, is also a moderate inhibitor of BET proteins with an IC50 value of 4.6 μM for BRD4(1) (Figure 10).
In addition, Schönbrunn’s and Knapp’s groups also identified other dual inhibitors of BET/kinase, such as NU7441 (31, DNA-PK inhibitor), Fostamatinib (32, SYK inhibitor), PP-242 (33, mTOR inhibitors), SB409514 (34, GSK inhibitor), AZ-3146 (35, MPS1 inhibitor), PF-431396 (36, FAK inhibitor), and BI-D1870 (37, RSK inhibitor) (Figure 10).
3.2 |. Dual inhibitors of BET BRDs and non-kinases
3.2.1 |. Dual inhibitors of BET BRDs and HDACs
HDACs are epigenetic erasers that deacetylate histones and regulate physiological processes such as cell differentiation, proliferation, and apoptosis.135 HDACs are frequently overexpressed in cancer cells, leading to abnormal acetylation of histones and perturbed gene expression and malignant tumor development.136,137 Therefore, HDACs are promising targets in the research and development of antitumor drugs. HDAC inhibitors (HDACi), such as SAHA (38), CI994 (39), and MS275 (40), usually consist of a cap group, a linker, and a ZBG group (Figure 11A). Importantly, BET and HDAC inhibitors work synergistically in MYC-induced mouse lymphoma, and they have similar target genes and biological phenotypes.90,138,139 Therefore, designing dual-target inhibitors of BET and HDAC, as a new treatment strategy, has great potential in cancer therapy.
FIGURE 11.
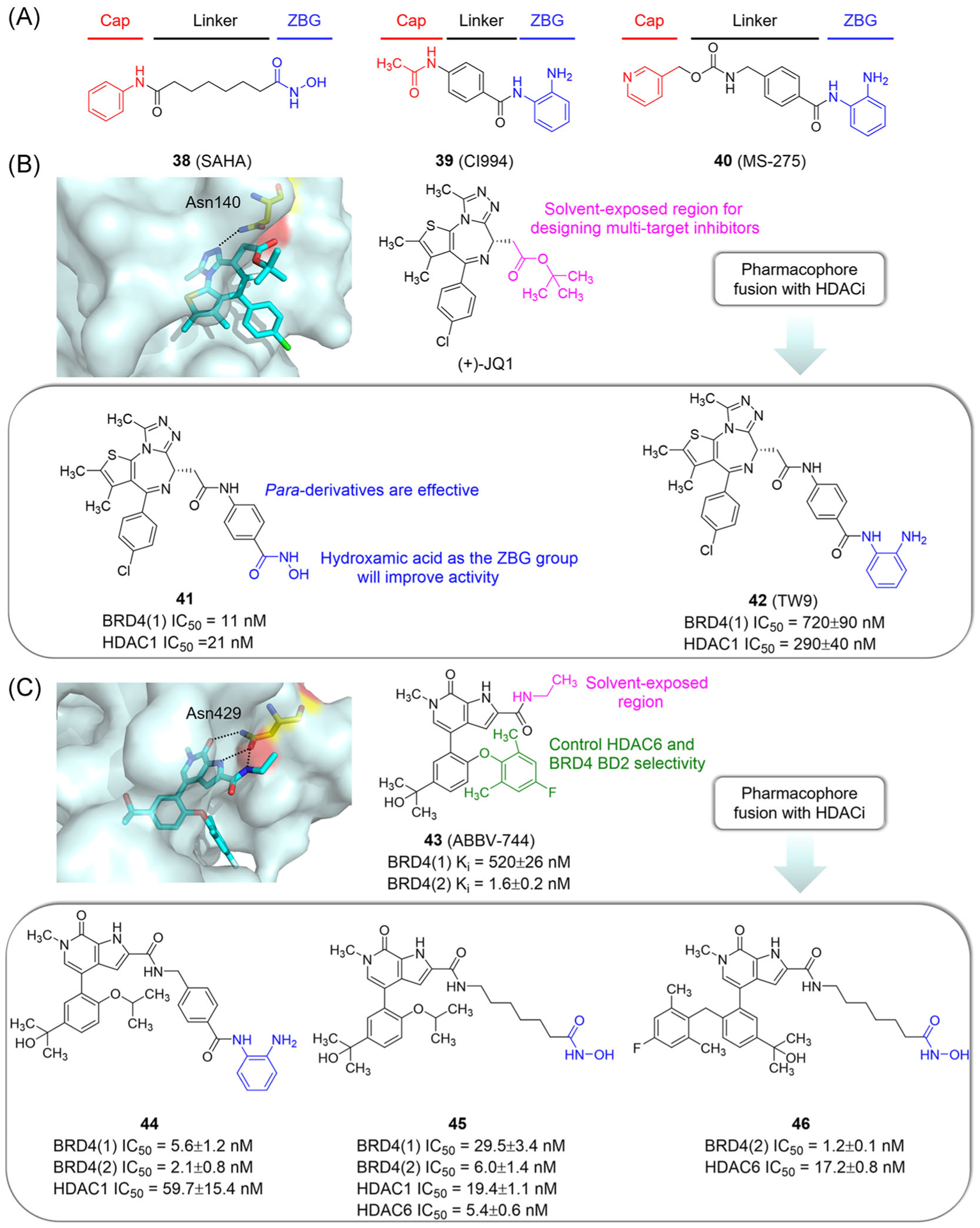
Design of dual-target inhibitors of BET and HDAC based on a pharmacophore fusion strategy. (A) The pharmacophore model of HDACi. (B) Design of compounds 41 and 42. X ray cocrystal structure of JQ1 bound to BRD4(1) (PDB ID: 3MXF). (C) Design of compounds 44, 45, and 46. X ray cocrystal structure of ABBV-744 bound to BRD2(2) (PDB ID: 6E6J). Protein is represented by light blue surface, with compound-contacting residues indicated by yellow sticks. The compound is shown in sticks and colored according to the atom type (C, light blue; N, blue; O, red; Cl, green; F, light cyan). Black dashed line = hydrogen bond
In 2020, He et al.140 reported a high-efficiency pan-BET/HDAC dual inhibitor, compound 41, with balanced activity against BRD4 BD1 (IC50 = 11 nM) and HDAC1 (IC50 = 21 nM) (Figure 11B). It was found that JQ1 had a complementary shape with the binding cavity of Kac and formed a key hydrogen bond with Asn140. The t-butyloxycarboryl group, exposed to the solvent, provides a feasible site for multitarget design. The ZBG is a crucial group for inhibiting HDAC activity. Inspired by these findings, He et al. designed a dual inhibitor of BET and HDAC through a pharmacophore fusion strategy. The structure–activity study showed that when the ZBG group was hydroxamic acid instead of o-aminoanilide, the activity against HDAC1 was improved, along with enhanced anti-tumor ability. Para-substituted derivatives are more effective than the corresponding meta derivatives. Compound 41 is a potent pan-BET and pan-HDAC inhibitor with excellent antitumor effect in vitro. In Capan-1 cells, it significantly inhibits the expression of C-MYC and CDC25B, and induces apoptosis at a concentration of 0.5 μM. It also has good metabolic stability, with a T1/2 value of 44.29 min. The antitumor effect of compound 41 in vivo is significantly better than BET inhibitors and HDAC inhibitors used alone or combined, and the toxicity is lower. These findings indicate that this novel BET/HDAC dual inhibitor has the potential to treat pancreatic cancer and is expected to help treat pancreatic cancer in the future.
Based on JQ1 and CI994, Zhang et al.41 designed a new dual inhibitor of BET and HDAC, TW9 (42; Figure 11B). The binding mode of TW9 and BRD4 (1) is the same as that of JQ1, and its triazole moiety forms a key hydrogen bond with Asn140. Crucially, the HDAC-inhibiting group of TW9 is solvent-exposed, indicating that TW9 could interact with HDAC even in the BET-bound state. TW9 is more effective than CI994 (an HDAC inhibitor) and has the best BRD4-inhibitory activity among all the dual HDAC/BET inhibitors reviewed in this article. TW9 downregulates C-MYC but upregulates histone H3 acetylation. It also inhibits the growth of pancreatic cancer cells better than its parent molecules. In 2020, Laszig et al.141 reported that TW9 induces mitochondrial apoptosis and has strong antitumor activity in rhabdomyosarcoma cells.
ABBV-744 (43) has an ethyl amide group extending toward the solvent region to connect the pharmacophore of HDAC inhibitors.31 To maintain the pyrrolopyridone core in BET pharmacophore, Chen et al.142 used a pharmacophore fusion strategy to identify three new HDAC/BRD4 dual inhibitors: compounds 44 (HDAC1 and BRD4), 45 (BRD4 and pan-HDAC) and 46 (BRD4-BD2 and HDAC6) (Figure 11C). The structure–activity study showed the pyrrolopyridone 2-site in the solvent region is suitable for connecting the ZBG without interfering with key binding interactions with BRD4. In MV4–11 cells, compound 44, as an HDAC1-selective inhibitor, mainly increases H3 acetylation and strongly induces G0/G1 cell cycle arrest and apoptosis. As a pan-HDAC inhibitor, compound 45 upregulates the acetylation levels of histone H3 and tubulin. Compound 46 is the first dual inhibitor of HDAC6/BRD4(2), which increases the level of acetylated tubulin but reduces C-MYC expression in a dose-dependent manner.
In 2014, Atkinson et al.143 designed a series of BET/HDAC inhibitors with the tetrahydroquinoline skeleton through pharmacophore fusion. I-BET295 (47) is highly complementary to BRD4 BRDs in shape (Figure 12A). The carbonyl group of I-BET295 forms a hydrogen bond with Asn140 and a water-mediated hydrogen bond with Tyr97. Importantly, the para-position of a 6-aryl substituent of I-BET295 provides a vector protruding toward the solvent and is at a suitable location for modification. DUAL946 (48), as a representive compound, inhibits BRD4 (IC50 = 50 nM) and downregulates the expression of C-MYC. In NUT midline carcinoma and acute myeloid leukemia cells, DUAL946 inhibits cell growth but does not significantly improve its efficacy compared with I-BET295 and SAHA.
FIGURE 12.
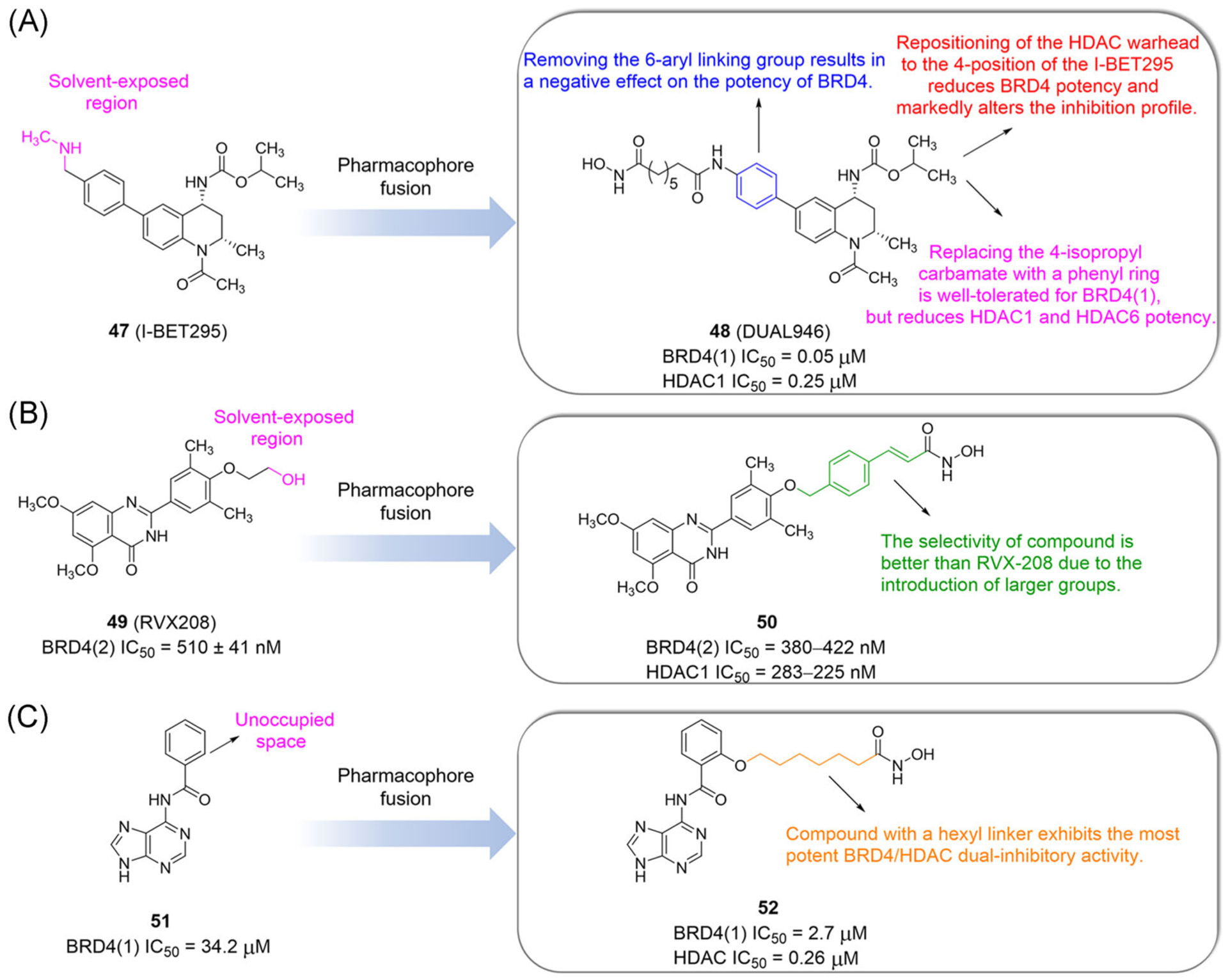
Design and SARs of dual-target inhibitors of BET and HDAC based on a pharmacophore fusion strategy (tetrahydroquinoline skeleton, quinolinone skeleton, and N6-benzoyladenine skeleton)
In 2017, Shao et al.144 found the hydroxy-ethylether part of RVX-208 (49) is located outside the Kac-binding pocket and rarely contacts the surface of the BRD, providing a site for the design of dual BET and HDAC inhibitors. They used various types of linkers to connect the free hydroxyl group of RVX208 with the ZBG group, and obtained a dual inhibitor with a quinolinone skeleton (Figure 12B). Compound 50 is an effective BRD4/HDAC inhibitor with inhibitory activity against BRD4(2), and its quinazolinone and Asn433 of BRD4(2) form a hydrogen bond. In vitro, compound 50 decreases the expression of MYC and inhibits the proliferation of human acute myeloid leukemia cell lines.
Compound 51 with an N6-benzoyladenine skeleton has inhibitory activity against BRD4 with an unoccupied space at the 2-position of the benzoyl group (Figure 12C).145,146 In 2017, Amemiya et al.147 synthesized compound 52 based on the idea of multitarget drugs by fusing N6-benzoyladenine and SAHA. Compound 52 suppresses the growth of HL-60 cells, inhibits the production of C-MYC, induces apoptosis, and intriguingly resensitizes BRD4 inhibitor-resistant cells to growth inhibition.
Compound 53, with the skeleton of dimethylisoxazole, is an effective inhibitor of BET BRDs (Figure 13A).148 The 3,5-dimethylisoxazole moiety of compound 53 occupies the Kac-binding pocket. The hydroxyl group of compound 53 does not completely occupy the ZA channel, and the flexible chain group may be more advantageous in the ZA channel. Considering the considerable similarity between the ZA channel of BRD4 and the tubular channel of HDAC, Zhang et al.149 introduced the SAHA linker and the ZBG group into hydroxyl position of compound 53 to design a series of 3,5-dimethylisoxazole-based BET/HDAC inhibitors (Figure 13A). Representative compound 54 has antiproliferative activity in K562 and MV4–11 cells (IC50 = 1.86 and 0.91 μM). The cocrystal structures show a hydrogen bond formed by the oxygen atom of isoxazole in compound 54 and Asn140 of BRD4(1). Another hydrogen bond is formed by the nitrogen atom and Tyr97 of BRD4(1) through a water molecule. Moreover, the hydroxamate of compound 54 forms hydrogen bonds with His131, His132, and Tyr297 of HDAC1.
FIGURE 13.
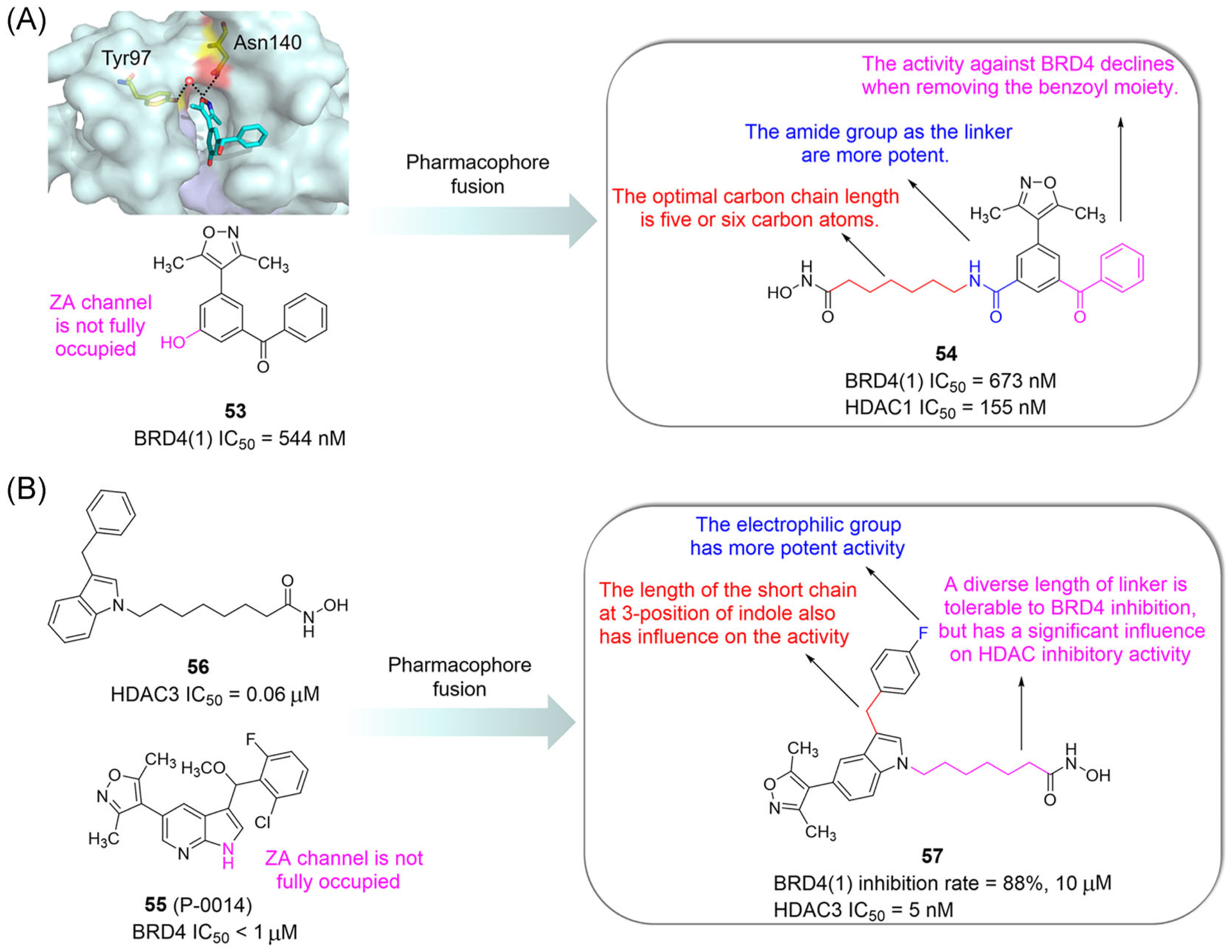
Design and SARs of dual-target inhibitors of BET and HDAC based on the pharmacophore fusion strategy (3,5-dimethylisoxazole skeleton and indole skeleton). Protein is represented by light blue surface, with compound-contacting residues indicated by yellow sticks. The compound is shown in sticks and colored according to the atom type (C, light blue; N, blue; O, red). Black dashed line = hydrogen bond
The binding mode of P-0014 (a BET inhibitor, 55) and BRD4 indicates that the ZA channel is not fully occupied. Cheng et al.150 speculated that the HDACi linker may be more beneficial to the ZA channel. P-0014 is structurally similar to compound 56 with an indole skeleton present in an HDAC inhibitor, thus allowing the use of pharmacophore fusion to design and synthesize BET/HDAC inhibitors (Figure 13B). Compound 57 inhibits HDAC3 (IC50 = 5 nM) with an 88% inhibition rate of BRD4 at 10 μM and decreases the expression of C-MYC while upregulates the expression of acetylated H3.
In 2020, Pan et al.151 synthesized a series of BET/HDAC inhibitors based on hydroxamic acid and thieno[2,3-d] pyrimidine (Figure 14). They first designed a novel BRD4 inhibitor skeleton based on fragment design using the ZINC database. The 2-position phenyl substituents of thieno[2,3-b] pyrimidin-4-one of compound 58 are crucial in inhibiting the activity of BRD4. The activity of compound 59 on BRD4 was comparable to that of compound 58. They took compounds 58 and 59 as the cap group and connected various ZBG groups to design a novel dual-target inhibitor of BRD4 and HDAC (Figure 14). SAR analysis suggests that the HDAC-inhibitory activity of the ZBG based on hydroxamic acid is better than that of o-phenylenediamine. Importantly, the inhibitory activity of BRD4 was completely lost due to the linkage of the ZBG group on the piperidine ring. The ZBG and the linker had little effect on the inhibition of BRD4. Representative compound 60 inhibits the expression of C-MYC and increases the acetylation level of histone H3. It also inhibits IL6-JAK-STAT pathway, induces cell cycle arrest and apoptosis, and effectively suppresses tumor growth.
FIGURE 14.
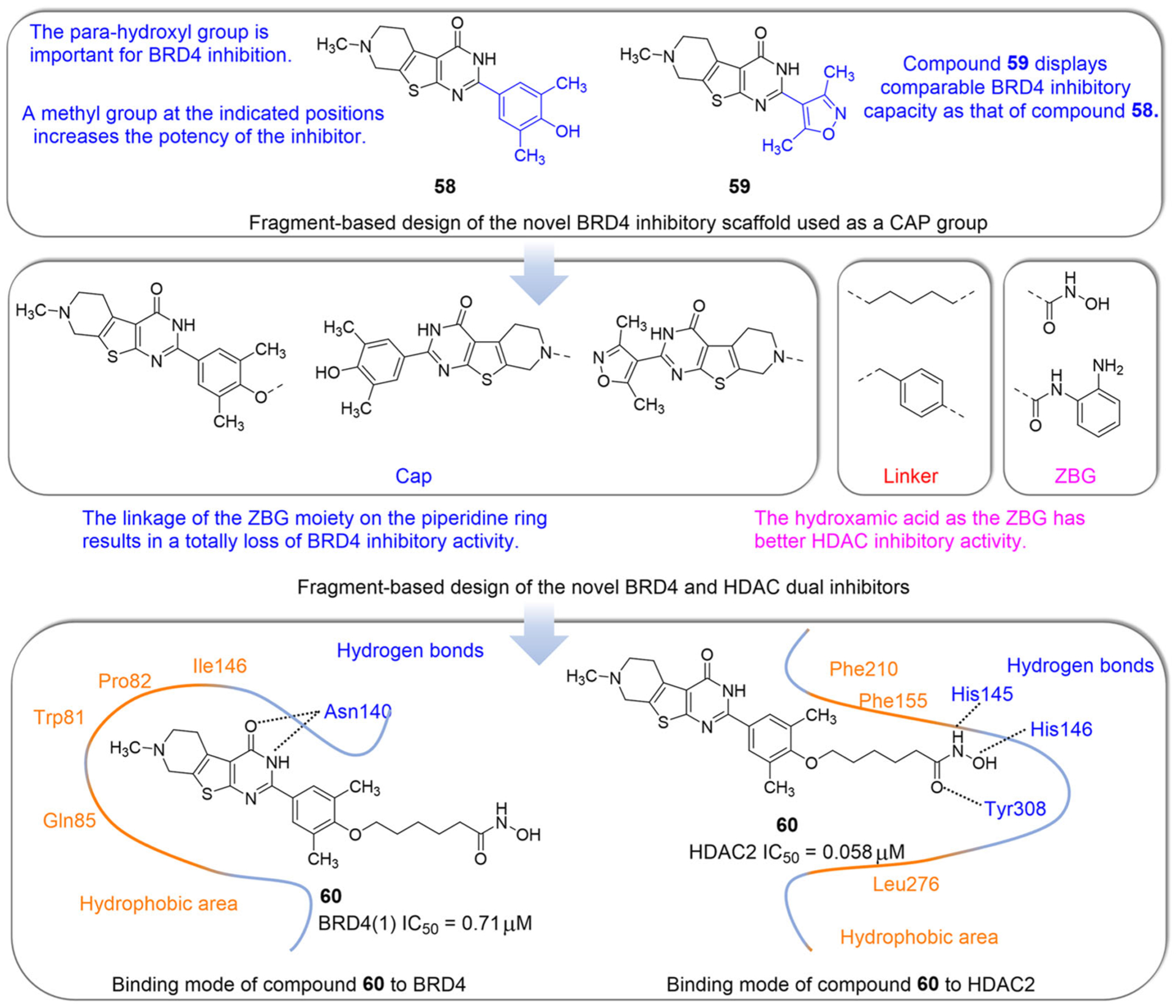
Design and SARs of dual-target inhibitors of BET/HDAC based on pharmacophore fusion of the thieno[2,3-d] pyrimidine skeleton
3.2.2 |. Dual inhibitors of BET BRDs and other BRD inhibitors
Cyclic adenosine monophosphate response element-binding protein (CBP) and p300 histone acetyltransferase (HAT) play important roles in lysine acetylation of histones and nonhistone proteins, and both are BRD-containing transcriptional coactivators.152 With the homology of BRDs present in CBP/p300 and BET proteins, small molecules with dual inhibition of CBP/p300 and BET proteins can be designed. In 2020, Morrison-Smith et al.153 found that inhibiting p300/CBP and BET BRDs synergistically depletes MYC and inhibits NUT-midline carcinoma (NMC) growth. NEO2734 (61; Figure 15) and NEO1132 (62; Figure 15), as dual inhibitors of BET and CBP/p300, can induce differentiation and cell cycle arrest in G1-phase. In preclinical xenograft mouse models, NEO2734 is capable of inhibiting NMC cell growth and prolonging survival significantly better than BET BRD inhibition or “standard” chemotherapy. Prostate cancer (PCa) carrying speckle-type POZ protein (SPOP) hotspot mutation, such as F133V, is typically resistant to BET inhibitors. Unlike F133V hotspot mutant tumors, patient-derived xenografts (PDXs) and organoids with Q165P mutation are modestly sensitive to BET inhibitor JQ1.154 Importantly, NEO2734 is active in PCa cells containing either F133V or Q165P SPOP mutation in vivo and in vitro, which provides the basis for clinical research on the anticancer effect of NEO2734 in SPOP-mutated PCa patients. In 2020, Spriano et al.155 demonstrated that NEO2734 has preclinical antitumor activity, especially in lymphoma and leukemia. Both NEO2734 and NEO1132 have strong antitumor activity against multiple myeloma.156
FIGURE 15.
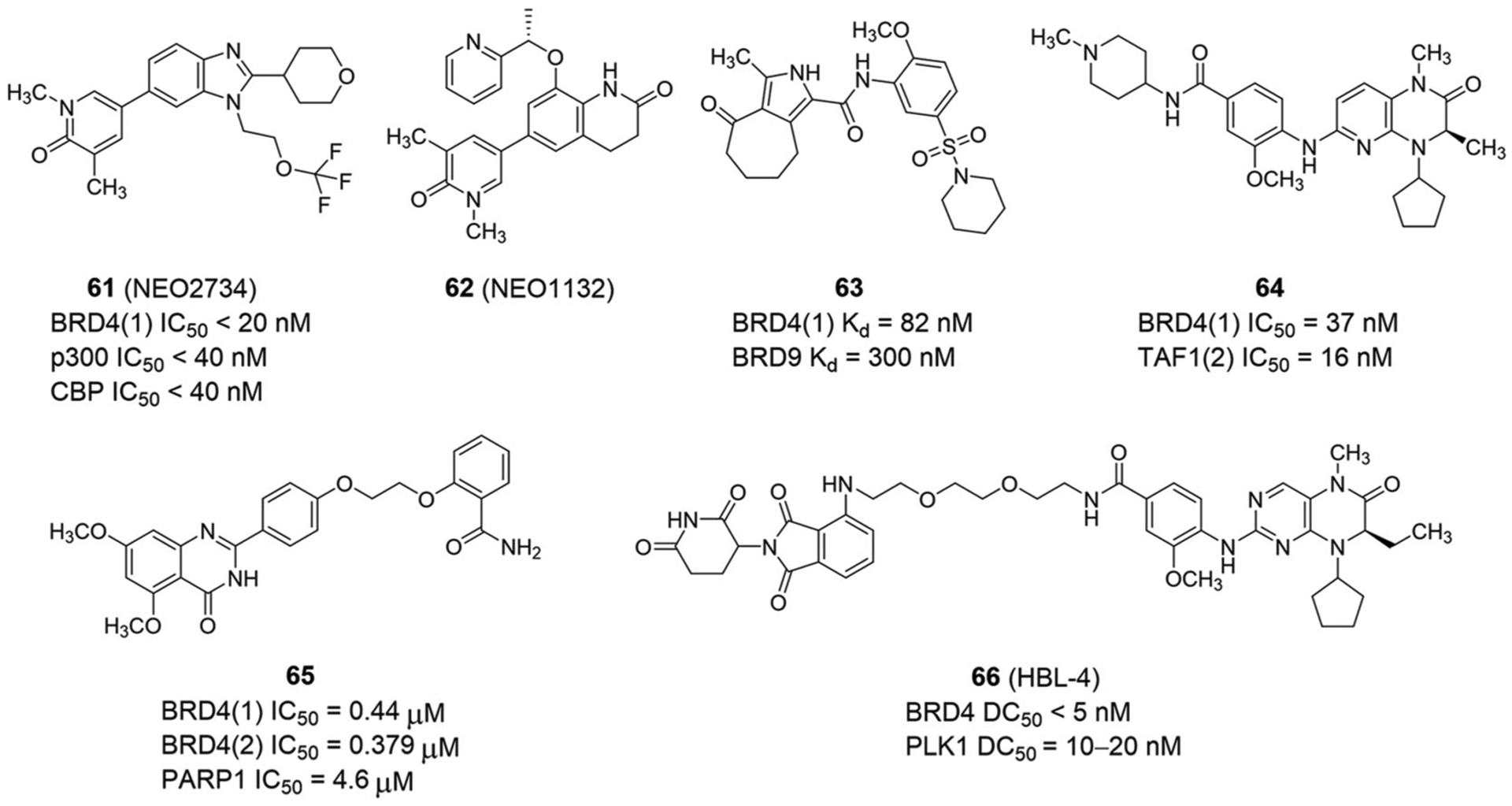
Dual-target inhibitors of BET bromodomains and other non-kinases and dual PROTAC of BET and PLK1
In 2020, Hügle et al.157 reported a group of dual BET-BRD7/9 BRD inhibitors with a 4-acyl pyrrole scaffold had strong antiproliferative effects on diverse cancer cell lines, such as MCF-7 breast cancer cells and SK-MEL-5 melanoma cells. The activity of representative compound 63 (Figure 15) against MCF-7 and SK-MEL-5 cells is synergized by BET-BRD7/9 dual inhibition. This indicates that BRD7/9 inhibitors have the potential to enhance the effect of BET inhibitors on cancer cell lines that are less sensitive to BET inhibition.
TBP-associated factor 1 (TAF1) is a component of the TFIID core promoter-binding factor with an important role in transcription initiation and cell cycle progression.158 Through epigenetic compound library screening performed in a human haploid reporter cell line, a small compound inhibitor with high affinity to the second BRD of TAF1 showed a comparable relief of heterochromatin silencing and MYC transcription inhibition as seen with JQ1 treatment or knockdown of BRD4 but not other BET proteins,159 indicating a functional association between TAF1 and BRD4 in transcriptional regulation159 and providing a proof-of-concept for developing dual-target inhibitors against TAF1 and BRD4. With the BI-2536 chemical scaffold, Remillard et al.160 used a structure-guided design and identified compound 64 as a highly effective dual inhibitor of TAF1(2) and BRD4(1), with IC50 values of 16 and 37 nM for TAF1(2) and BRD4(1), respectively (Figure 15).
3.2.3 |. Dual inhibitors of BET BRDs and PARP1
In 2020, Chang et al.161 rationally designed the first dual-target inhibitor of BET and PARP, compound 65, based on fragment combinatorial screening, with IC50 values of 0.44, 0.379, and 4.6 μM for BRD4(1), BRD4(2), and PARP1, respectively (Figure 15). The quinazoline ring of compound 65 forms key hydrogen bonds with Asn140 of BD1 and Asn433 of BD2. In PARP1, the benzamide of compound 65 forms pivotal hydrogen bonds with Ser904 and Gly863. Also, quinazoline substituted by dimethoxy forms a hydrogen bond with Arg878.
Conceivably, dual-target BET inhibitors have synergistic effects to inhibit BET and other biological targets, potentially increasing cell-killing capacity and reducing the risk of developing drug resistance. Although the design and development of dual-target inhibitors are challenging, especially with structurally distinct targets that do not belong to the same protein family, dual-target inhibitors are an attractive therapeutic approach expected to provide new chemical probes for cancer treatment.
3.3 |. Dual proteolysis targeting chimera of BET proteins and PLK1
In 2001, Crews and colleagues162 proposed the concept of proteolysis-targeting chimera (PROTAC) as a new strategy for drug discovery. PROTAC is widely used to induce the degradation of BRD4 and has become one of the research hotspots in cancer treatment.163 Theoretically, depleting key oncogenic proteins in cancer cells is usually better than simply inhibiting the activity or binding of the same protein.
In 2020, Mu et al.164 reported a dual BRD4 and PLK1 degrader HBL-4 (66; Figure 15) created by linking a small molecule recruiting the E3 ubiquitin ligase Cereblon (CRBN) to the dual BET/PLK1 inhibitor BI-2536. Based on the design of dual BET/PLK1 inhibitor and PROTAC technology, HBL-4 quickly and effectively degrades BRD4 and PLK1 proteins in MV4–11 cells. It also significantly inhibits C-MYC expression, induces apoptosis, and inhibits cell proliferation more effectively and lastingly compared to BI-2536, thus reducing the dosage and time of administration and side effects. At well-tolerated dosage, HBL-4 achieved complete and sustained tumor regression in MV4–11 acute leukemia xenograft mouse models with excellent pharmacokinetics in vivo. HBL-4 is the first reported dual degrader of kinase and BRDs, representing an important advance in PROTAC-based cancer treatment.
In general, BRD4 PROTACs can selectively degrade target protein BRD4, showing better preclinical antitumor activity and therapeutic effects than their corresponding inhibitors. The development of dual-target protein degraders such as HBL-4 provides a proof-of-concept for future design of PROTACs targeting BRD4 and other kinases. Although BRD4 PROTACs have not yet entered clinical trials, they have great potential as powerful biological and pharmacological agents to significantly improve clinical efficacy and drug resistance of BRD4- and BET-targeting small-molecule inhibitors.
4 |. CONCLUSIONS AND PERSPECTIVES
The development and progression of malignant tumors are complex, involving perturbation of multiple related targets. Target-selective inhibitors frequently affect only one dominant oncogene in the tumor, which may lead to rapid drug resistance in responsive patients. Dysfunctioning of BET proteins is closely related to cancer development. Unfortunately, BET inhibitors are less successful in recent clinical trials, not only because of the toxicity and side effects but also due to drug resistance-altered signaling and transcription pathways. Encouragingly, multitarget drugs have been increasingly recognized in emerging cancer therapy. The synergy between BET and multiple tumor-related target inhibitors provides a conceptual framework for development of powerful dual-target BET inhibitors.
At present, commonly used strategies for dual BET inhibitor development focus mainly on drug repurposing, skeleton modification of lead compounds, pharmacophore fusion, and computational approaches. In this review, we summarize the research progress on dual inhibitors of BET proteins with kinase and non-kinase targets, paving the road for future design of dual BET inhibitors. Likewise, dual-target protein degraders, such as HBL-4, also provide a new strategy to design PROTACs targeting BET and other targets. Dual BET inhibitors have not been widely applied to new targets, such as BCL2 and Aurora A that show functional synergy with BET, and direct BET-interacting proteins. Although research on dual-target inhibitors of BET proteins has brought great expectations for tumor treatment, few dual-target inhibitors have successfully entered clinical trials. Given the increase in relative molecular mass and structural complexity, the physicochemical properties and pharmacokinetics of dual-target drugs may not be ideal and require significant optimization considering the involvement of multiple targets with sometimes unforeseeable functions and synergy in the cell. The current challenge is to balance the activities of dual targets and obtain inhibitors with desirable pharmacological properties and safety.
In summary, dual-target inhibitors of BET proteins are encouraging, but still have room for improvement. With more understanding of the mechanisms underlying tumor initiation/progression and applications of pharmacophore fusion and 3D spatial structure similarity of binding sites, multiple-targeting remains one of the most promising directions in medicinal chemistry to design and develop dual-target inhibitors based on key targets that can produce synergistic, antagonistic, or compensatory mechanisms with BET proteins. We believe dual-target BET inhibitors will show great promise in future antitumor drug development.
ACKNOWLEDGEMENTS
This study was supported by grants from National Natural Science Foundation of China (Grant nos. 81922064, 81874290, 81803755, and 91853109), Project of science and Technology Department of Sichuan Province (Grant no. 20YYJC3921), National Clinical Research Center for Geriatrics, West China Hospital, Sichuan University (Grant no. Z20201004), US National Institutes of Health (NIH) (Grant nos. 1RO1CA251698-01) and the Cancer Prevention and Research Institute of Texas (CPRIT) (Grant nos. RP180349 and RP190077).
AUTHOR BIOGRAPHIES
Lu Feng is a master candidate at State Key Laboratory of Biotherapy and Cancer Center of West China Hospital at Sichuan University. Her research interest focuses on pharmaceutical chemistry.
Guan Wang is Assistant Professor at State Key Laboratory of Biotherapy and Cancer Center, Innovation Center of Nursing Research, Nursing Key Laboratory of Sichuan Province, National Clinical Research Center for Geriatrics, West China Hospital, and Collaborative Innovation Center of Biotherapy, Sichuan University China. His research interest focuses on bioinformatics, pharmaceutical chemistry, structure-based design of drugs and bioactive compound.
Yi Chen is Associate Professor at State Key Laboratory of Biotherapy and Cancer Center of West China Hospital at Sichuan University. His research interest focuses on target identification and structure-based drug design.
Gu He received his PhD degree in Medicinal Chemistry from West China School of Pharmacy, Sichuan University, in 2008. He is now working as Professor at State Key Laboratory of Biotherapy and Cancer Center of West China Hospital at Sichuan University. His research interests include the design and synthesis of small molecule drugs, mechanism of action, and drug targeted delivery systems.
Bo Liu received his PhD degree in Bioinformatics (drug design) from School of Life Sciences, Sichuan University, in 2010. In 2012, he joined the faculty of State Key Laboratory of Biotherapy and Cancer Center of West China Hospital at Sichuan University as Professor. His research interests include autophagic target identification and structure-based drug design.
Jie Liu received his PhD degree in Organic Chemistry from Department of Chemistry, Sichuan University, in 2009. He is now working as Professor at State Key Laboratory of Biotherapy and Cancer Center of West China Hospital at Sichuan University. He has long been engaging in green organic synthesis methodology, the design and synthesis of drugs based on various original targets, the basic research on the application of lead compound screening, and the development of related new drug target recognition methods and drug discovery.
Cheng-Ming Chiang received his PhD degree from Department of Biochemistry at University of Rochester in 1991. He has been Assistant Professor at University of Illinois at Urbana-Champaign (1995–2000), Associate Professor at Case Western Reserve University (2000–2007), and Professor at University of Texas Southwestern Medical Center since 2007. His research interests span transcription mechanisms, chromatin, epigenetics, HPV, post-translational modification, cancer biology, and BRD4 therapeutics.
Liang Ouyang received his PhD degree in Medicinal Chemistry from West China School of Pharmacy, Sichuan University, in 2010. He began his academic career in 2012 at State Key Laboratory of Biotherapy and Cancer Center, Innovation Center of Nursing Research, Nursing Key Laboratory of Sichuan Province, National Clinical Research Center for Geriatrics, West China Hospital, and Collaborative Innovation Center of Biotherapy, Sichuan University as Professor. His research interests include identification of novel drug targets in cell death and structure-based discovery of small-molecule and peptide drugs.
Footnotes
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
REFERENCES
- 1.Wu SY, Chiang CM. The double bromodomain-containing chromatin adaptor BRD4 and transcriptional regulation. j Biol Chem. 2007;282(18):13141–13145. [DOI] [PubMed] [Google Scholar]
- 2.Filippakopoulos P, Picaud S, Mangos M, et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell. 2012;149(1):214–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Arrowsmith CH, Bountra C, Fish PV, Lee K, Schapira M. Epigenetic protein families: a new frontier for drug discovery. Nat Rev Drug Discov. 2012;11(5):384–400. [DOI] [PubMed] [Google Scholar]
- 4.Romero FA, Taylor AM, Crawford TD, Tsui V, Côté A, Magnuson S. Disrupting acetyl-lysine recognition: progress in the development of bromodomain inhibitors. J Med Chem. 2016;59(4):1271–1298. [DOI] [PubMed] [Google Scholar]
- 5.Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-Myc. Cell. 2011;146(6):904–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dawson MA, Prinjha RK, Dittmann A, et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature. 2011;478(7370):529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Duan Q, McMahon S, Anand P, et al. BET bromodomain inhibition suppresses innate inflammatory and profibrotic transcriptional networks in heart failure. Sci Transl Med. 2017;9(390):eaah5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dey A, Yang W, Gegonne A, et al. BRD4 directs hematopoietic stem cell development and modulates macrophage inflammatory responses. EMBO J. 2019;38(7):e100293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Stratton MS, Bagchi RA, Felisbino MB, et al. Dynamic chromatin targeting of BRD4 stimulates cardiac fibroblast activation. Circ Res. 2019;125(7):662–677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Segura MF, Fontanals-Cirera B, Gaziel-Sovran A, et al. BRD4 sustains melanoma proliferation and represents a new target for epigenetic therapy. Cancer Res. 2013;73(20):6264–6276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ding N, Hah N, Yu RT, et al. BRD4 is a novel therapeutic target for liver fibrosis. Proc Natl Acad Sci USA. 2015; 112(51):15713–15718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kulikowski E, Rakai BD, Wong NCW. Inhibitors of bromodomain and extra-terminal proteins for treating multiple human diseases. Med Res Rev. 2021;41(1):223–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang CY, Filippakopoulos P. Beating the odds: BETs in disease. Trends Biochem Sci. 2015;40(8):468–479. [DOI] [PubMed] [Google Scholar]
- 14.Prinjha RK, Witherington J, Lee K. Place your BETs: the therapeutic potential of bromodomains. Trends Pharmacol Sci. 2012;33(3):146–153. [DOI] [PubMed] [Google Scholar]
- 15.Filippakopoulos P, Qi J, Picaud S, et al. Selective inhibition of BET bromodomains. Nature. 2010;468(7327): 1067–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Devaiah BN, Case-Borden C, Gegonne A, et al. BRD4 is a histone acetyltransferase that evicts nucleosomes from chromatin. Nat Struct Mol Biol. 2016;23(6):540–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wen X, Klionsky DJ. BRD4 is a newly characterized transcriptional regulator that represses autophagy and lysosomal function. Autophagy. 2017;13(11):1801–1803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Belkina AC, Denis GV. BET domain co-regulators in obesity, inflammation and cancer. Nat Rev Cancer. 2012;12(7): 465–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gaub A, Sheikh BN. Evolutionary conserved NSL complex/BRD4 axis controls transcription activation via histone acetylation. Nat Commun. 2020;11(1):2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li X, Baek G, Ramanand SG, et al. BRD4 promotes DNA repair and mediates the formation of TMPRSS2-ERG gene rearrangements in prostate cancer. Cell Rep. 2018;22(3):796–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chiang CM. BRD4 engagement from chromatin targeting to transcriptional regulation: selective contact with acetylated histone H3 and H4. F1000 Biol Rep. 2009;1(98):98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Feng Q, Zhang Z, Shea MJ, et al. An epigenomic approach to therapy for tamoxifen-resistant breast cancer. Cell Res. 2014;24(7):809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Filippakopoulos P, Knapp S. Targeting bromodomains: epigenetic readers of lysine acetylation. Nat Rev Drug Discov. 2014;13(5):337–356. [DOI] [PubMed] [Google Scholar]
- 24.Zuber J, Shi J, Wang E, et al. RNAi screen identifies BRD4 as a therapeutic target in acute myeloid leukaemia. Nature. 2011;478(7370):524–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Donati B, Lorenzini E, Ciarrocchi A. BRD4 and cancer: going beyond transcriptional regulation. Mol Cancer. 2018; 17(1):164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nicodeme E, Jeffrey KL, Schaefer U, et al. Suppression of inflammation by a synthetic histone mimic. Nature. 2010; 468(7327):1119–1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Albrecht BK, Gehling VS, Hewitt MC, et al. Identification of a benzoisoxazoloazepine inhibitor (CPI-0610) of the bromodomain and extra-terminal (BET) family as a candidate for human clinical trials. J Med Chem. 2016;59(4): 1330–1339. [DOI] [PubMed] [Google Scholar]
- 28.Fish PV, Filippakopoulos P, Bish G, et al. Identification of a chemical probe for bromo and extra C-terminal bromodomain inhibition through optimization of a fragment-derived hit. J Med Chem. 2012;55(22):9831–9837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wyce A, Ganji G, Smitheman KN, et al. BET inhibition silences expression of MYCN and BCL2 and induces cytotoxicity in neuroblastoma tumor models. PLOS One. 2013;8(8):e72967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Picaud S, Wells C, Felletar I, et al. RVX-208, an inhibitor of BET transcriptional regulators with selectivity for the second bromodomain. Proc Natl Acad Sci USA. 2013;110(49):19754–19759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Faivre EJ, McDaniel KF, Albert DH, et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nature. 2020;578(7794):306–310. [DOI] [PubMed] [Google Scholar]
- 32.Ember SW, Zhu JY, Olesen SH, et al. Acetyl-lysine binding site of bromodomain-containing protein 4 (BRD4) interacts with diverse kinase inhibitors. ACS Chem Biol. 2014;9(5):1160–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Martin MP, Olesen SH, Georg GI, Schönbrunn E. Cyclin-dependent kinase inhibitor Dinaciclib interacts with the acetyl-lysine recognition site of bromodomains. ACS Chem Biol. 2013;8(11):2360–2365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Baumli S, Lolli G, Lowe ED, et al. The structure of P-TEFb (CDK9/cyclin T1), its complex with flavopiridol and regulation by phosphorylation. EMBO J. 2008;27(13):1907–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ciceri P, Müller S, O’Mahony A, et al. Dual kinase-bromodomain inhibitors for rationally designed polypharmacology. Nat Chem Biol. 2014;10(4):305–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kothe M, Kohls D, Low S, et al. Research article: selectivity-determining residues in PLK1. Chem Biol Drug Des. 2007; 70(6):540–546. [DOI] [PubMed] [Google Scholar]
- 37.Siu M, Pastor R, Liu W, et al. 2-Amino-[1,2,4]triazolo[1,5-a]pyridines as JAK2 inhibitors. Bioorg Med Chem Lett. 2013; 23(17):5014–5021. [DOI] [PubMed] [Google Scholar]
- 38.Andrews FH, Singh AR, Joshi S, et al. Dual-activity PI3K-BRD4 inhibitor for the orthogonal inhibition of MYC to block tumor growth and metastasis. Proc Natl Acad Sci USA. 2017;114(7):E1072–E1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wang J, Erazo T, Ferguson FM, et al. Structural and atropisomeric factors governing the selectivity of pyrimido-benzodiazipinones as inhibitors of kinases and bromodomains. ACS Chem Biol. 2018;13(9): 2438–2448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Harris PA, Boloor A, Cheung M, et al. Discovery of 5-[[4-[(2,3-Dimethyl-2H-indazol-6-yl)methylamino]-2-pyrimidinyl]amino]-2-methyl-benzenesulfonamide (Pazopanib), a novel and potent vascular endothelial growth factor receptor inhibitor. J Med Chem. 2008;51(15):4632–4640. [DOI] [PubMed] [Google Scholar]
- 41.Zhang X, Zegar T, Weiser T, et al. Characterization of a dual BET/HDAC inhibitor for treatment of pancreatic ductal adenocarcinoma. Int J Cancer. 2020;147(10):2847–2861. [DOI] [PubMed] [Google Scholar]
- 42.Bechter O, Schöffski P. Make your best BET: the emerging role of BET inhibitor treatment in malignant tumors. Pharmacol Ther. 2020;208:107479. [DOI] [PubMed] [Google Scholar]
- 43.Bolden JE, Tasdemir N, Dow LE, et al. Inducible in vivo silencing of BRD4 identifies potential toxicities of sustained BET protein inhibition. Cell Rep. 2014;8(6):1919–1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tang P, Zhang J, Liu J, Chiang CM, Ouyang L. Targeting bromodomain and extraterminal proteins for drug discovery: from current progress to technological development. J Med Chem. 2021;64(5):2419–2435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fong CY, Gilan O, Lam EY, et al. BET inhibitor resistance emerges from leukaemia stem cells. Nature. 2015; 525(7570):538–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Rathert P, Roth M, Neumann T, et al. Transcriptional plasticity promotes primary and acquired resistance to BET inhibition. Nature. 2015;525(7570):543–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Coleman DJ, Gao L, Schwartzman J, et al. Maintenance of MYC expression promotes de novo resistance to BET bromodomain inhibition in castration-resistant prostate cancer. Sci Rep. 2019;9(1):3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kumar K, Raza SS, Knab LM, et al. GLI2-dependent c-MYC upregulation mediates resistance of pancreatic cancer cells to the BET bromodomain inhibitor JQ1. Sci Rep. 2015;5(1):9489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kurimchak AM, Shelton C, Duncan KE, et al. Resistance to BET bromodomain inhibitors is mediated by kinome reprogramming in ovarian cancer. Cell Rep. 2016;16(5):1273–1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shu S, Lin CY, He HH, et al. Response and resistance to BET bromodomain inhibitors in triple-negative breast cancer. Nature. 2016;529(7586):413–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jang JE, Eom JI, Jeung HK, et al. AMPK-ULK1-mediated autophagy confers resistance to BET inhibitor JQ1 in acute myeloid leukemia stem cells. Clin Cancer Res. 2017;23(11):2781–2794. [DOI] [PubMed] [Google Scholar]
- 52.Dai X, Gan W, Li X, et al. Prostate cancer–associated SPOP mutations confer resistance to BET inhibitors through stabilization of BRD4. Nat Med. 2017;23(9):1063–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang P, Wang D, Zhao Y, et al. Intrinsic BET inhibitor resistance in SPOP-mutated prostate cancer is mediated by BET protein stabilization and AKT–mTORC1 activation. Nat Med. 2017;23(9):1055–1062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jin X, Yan Y, Wang D, et al. DUB3 promotes BET inhibitor resistance and cancer progression by deubiquitinating BRD4. Mol Cell. 2018;71(4):592–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wu SY, Lee CF, Lai HT, et al. Opposing functions of BRD4 isoforms in breast cancer. Mol Cell. 2020;78(6): 1114–1132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shi J, Vakoc CR. The mechanisms behind the therapeutic activity of BET bromodomain inhibition. Mol Cell. 2014; 54(5):728–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wu SY, Lee AY, Lai HT, Zhang H, Chiang CM. Phospho switch triggers BRD4 chromatin binding and activator recruitment for gene-specific targeting. Mol Cell. 2013;49(5):843–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Floyd SR, Pacold ME, Huang Q, et al. The bromodomain protein BRD4 insulates chromatin from DNA damage signalling. Nature. 2013;498(7453):246–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Montserrat PS, Esteller M. Bromodomain inhibitors and cancer therapy: From structures to applications. Epigenetics. 2017;12(5):323–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang M, Zhang Y, Song M, et al. Structure-based discovery and optimization of benzo[d]isoxazole derivatives as potent and selective BET inhibitors for potential treatment of castration-resistant prostate cancer (CRPC). J Med Chem. 2018;61(7):3037–3058. [DOI] [PubMed] [Google Scholar]
- 61.Chung CW, Coste H, White JH, et al. Discovery and characterization of small molecule inhibitors of the BET family bromodomains. J Med Chem. 2011;54(11):3827–3838. [DOI] [PubMed] [Google Scholar]
- 62.Prieto-Martínez FD, Medina-Franco JL. Flavonoids as putative epi-modulators: insight into their binding mode with BRD4 bromodomains using molecular docking and dynamics. Biomolecules. 2018;8(3):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bharatham N, Slavish PJ, Shadrick WR, Young BM, Shelat AA. The role of ZA channel water-mediated interactions in the design of bromodomain-selective BET inhibitors. J Mol Graph Model. 2018;81:197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Konuma T, Yu D, Zhao C, Ju Y, Sharma R, Ren C. Structural mechanism of the oxygenase JMJD6 recognition by the extraterminal (ET) domain of BRD4. Sci Rep. 2017;7(1):16272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Gilan O, Rioja I, Knezevic K, et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science. 2020;368(6489):387–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Sheppard GS, Wang L, Fidanze SD, et al. Discovery of N-Ethyl-4-[2-(4-fluoro-2,6-dimethyl-phenoxy)-5-(1-hydroxy-1-methyl-ethyl)phenyl]-6-methyl-7-oxo-1H-pyrrolo[2,3-c]pyridine-2-carboxamide (ABBV-744), a BET bromodomain inhibitor with selectivity for the second bromodomain. J Med Chem. 2020;63(10):5585–5623. [DOI] [PubMed] [Google Scholar]
- 67.Vollmuth F, Blankenfeldt W, Geyer M. Structures of the dual bromodomains of the P-TEFb-activating protein BRD4 at atomic resolution. J Biol Chem. 2009;284(52):36547–36556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Itzen F, Greifenberg AK, Bösken CA, Geyer M. BRD4 activates P-TEFb for RNA polymerase II CTD phosphorylation. Nucleic Acids Res. 2014;42(12):7577–7590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Jang MK, Mochizuki K, Zhou M, Jeong HS, Brady JN, Ozato K. The bromodomain protein BRD4 is a positive regulatory component of P-TEFb and stimulates RNA polymerase II-dependent transcription. Mol Cell. 2005;19(4): 523–534. [DOI] [PubMed] [Google Scholar]
- 70.Rahl PB, Lin CY, Seila AC, et al. c-Myc regulates transcriptional pause release. Cell. 2010;141(3):432–445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li C, Bonazzoli E, Bellone S, et al. Mutational landscape of primary, metastatic, and recurrent ovarian cancer reveals c-MYC gains as potential target for BET inhibitors. Proc Natl Acad Sci USA. 2019;116(2):619–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Henssen A, Althoff K, Odersky A, et al. Targeting MYCN-driven transcription by BET-bromodomain inhibition. Clin Cancer Res. 2016;22(10):2470–2481. [DOI] [PubMed] [Google Scholar]
- 73.Peirs S, Frismantas V, Matthijssens F, et al. Targeting BET proteins improves the therapeutic efficacy of BCL-2 inhibition in T-cell acute lymphoblastic leukemia. Leukemia. 2017;31(10):2037–2047. [DOI] [PubMed] [Google Scholar]
- 74.Wang X, Helfer CM, Pancholi N, Bradner JE, You J. Recruitment of BRD4 to the human papillomavirus type 16 DNA replication complex is essential for replication of viral DNA. J Virol. 2013;87(7):3871–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dong X, Hu X, Chen J, Hu D, Chen LF. BRD4 regulates cellular senescence in gastric cancer cells via E2F/miR-106b/p21 axis. Cell Death Dis. 2018;9(2):203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zou Z, Huang B, Wu X, et al. BRD4 maintains constitutively active NF-κB in cancer cells by binding to acetylated RelA. Oncogene. 2014;33(18):2395–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Huang B, Yang XD, Zhou MM, Ozato K, Chen LF. BRD4 coactivates transcriptional activation of NF-kappaB via specific binding to acetylated RelA. Mol Cell Biol. 2009;29(5):1375–1387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Urbanucci A, Barfeld SJ, Kytölä V, et al. Androgen receptor deregulation drives bromodomain-mediated chromatin alterations in prostate cancer. Cell Rep. 2017;19(10):2045–2059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nagarajan S, Hossan T, Alawi M, et al. Bromodomain protein BRD4 is required for estrogen receptor-dependent enhancer activation and gene transcription. Cell Rep. 2014;8(2):460–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Dawson MA, Gudgin EJ, Horton SJ, et al. Recurrent mutations, including NPM1c, activate a BRD4-dependent core transcriptional program in acute myeloid leukemia. Leukemia. 2014;28(2):311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Chiang C-M. Phospho-BRD4: transcription plasticity and drug targeting. Drug Discov Today: Technol. 2016;19:17–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hussong M, Börno ST, Kerick M, et al. The bromodomain protein BRD4 regulates the KEAP1/NRF2-dependent oxidative stress response. Cell Death Dis. 2014;5(4):e1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Brasier AR, Zhou J. Validation of the epigenetic reader bromodomain-containing protein 4 (BRD4) as a therapeutic target for treatment of airway remodeling. Drug Discov Today. 2020;25(1):126–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Kim JJ, Lee SY, Gong F, et al. Systematic bromodomain protein screens identify homologous recombination and R-loop suppression pathways involved in genome integrity. Genes Dev. 2019;33(23–24):1751–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Doroshow DB, Eder JP, LoRusso PM. BET inhibitors: a novel epigenetic approach. Ann Oncol. 2017;28(8): 1776–1787. [DOI] [PubMed] [Google Scholar]
- 86.Lee DH, Qi J, Bradner JE, et al. Synergistic effect of JQ1 and rapamycin for treatment of human osteosarcoma. Int J Cancer. 2015;136(9):2055–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Huang CS, You X, Dai C, et al. Targeting super-enhancers via nanoparticle-facilitated BRD4 and CDK7 inhibitors synergistically suppresses pancreatic ductal adenocarcinoma. Adv Sci. 2020;7(7):1902926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Takimoto-Shimomura T, Tsukamoto T, Maegawa S, et al. Dual targeting of bromodomain-containing 4 by AZD5153 and BCL2 by AZD4320 against B-cell lymphomas concomitantly overexpressing c-MYC and BCL2. Invest New Drugs. 2019;37(2):210–222. [DOI] [PubMed] [Google Scholar]
- 89.Felgenhauer J, Tomino L, Selich-Anderson J, Bopp E, Shah N. Dual BRD4 and AURKA inhibition is synergistic against mycn-amplified and nonamplified neuroblastoma. Neoplasia. 2018;20(10):965–974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Bhadury J, Nilsson LM, Muralidharan SV, et al. BET and HDAC inhibitors induce similar genes and biological effects and synergize to kill in Myc-induced murine lymphoma. Proc Natl Acad Sci USA. 2014;111(26):E2721–E2730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang J, Yu Q, Qiu Z, et al. The combined effect of epigenetic inhibitors for LSD1 and BRD4 alters prostate cancer growth and invasion. Aging. 2020;12(1):397–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Shu S, Wu HJ, Ge JY, et al. Synthetic lethal and resistance interactions with BET bromodomain inhibitors in triple-negative breast cancer. Mol Cell. 2020;78(6):1096–1113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ashworth A, Lord CJ. Synthetic lethal therapies for cancer: what’s next after PARP inhibitors? Nat Rev Clin Oncol. 2018;15(9):564–576. [DOI] [PubMed] [Google Scholar]
- 94.Sun C, Yin J, Fang Y, et al. BRD4 inhibition is synthetic lethal with PARP inhibitors through the induction of homologous recombination deficiency. Cancer Cell. 2018;33(3):401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Jackson RA, Chen ES. Synthetic lethal approaches for assessing combinatorial efficacy of chemotherapeutic drugs. Pharmacol Ther. 2016;162:69–85. [DOI] [PubMed] [Google Scholar]
- 96.Zhu H, Bengsch F, Svoronos N, et al. BET bromodomain inhibition promotes anti-tumor immunity by suppressing PD-L1 expression. Cell Rep. 2016;16(11):2829–2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hogg SJ, Vervoort SJ, Deswal S, et al. BET-bromodomain inhibitors engage the host immune system and regulate expression of the immune checkpoint ligand PD-L1. Cell Rep. 2017;18(9):2162–2174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Sun D, Zhao Y, Zhang S, Zhang L, Liu B, Ouyang L. Dual-target kinase drug design: current strategies and future directions in cancer therapy. Eur J Med Chem. 2020;188:112025. [DOI] [PubMed] [Google Scholar]
- 99.Urick AK, Hawk LM, Cassel MK, et al. Dual screening of BPTF and BRD4 using protein-observed fluorine NMR uncovers new bromodomain probe molecules. ACS Chem Biol. 2015;10(10):2246–2256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Liu S, Yosief HO, Dai L, et al. Structure-guided design and development of potent and selective dual bromodomain 4 (BRD4)/polo-like Kinase 1 (PLK1) inhibitors. J Med Chem. 2018;61(17):7785–7795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chen L, Yap JL, Yoshioka M, et al. BRD4 structure-activity relationships of dual PLK1 kinase/BRD4 bromodomain inhibitor BI-2536. ACS Med Chem Lett. 2015;6(7):764–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Allen BK, Mehta S, Ember SWJ, Schonbrunn E, Ayad N, Schürer SC. Large-scale computational screening identifies first in class multitarget inhibitor of EGFR kinase and BRD4. Sci Rep. 2015;5(1):16924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Carlino L, Rastelli G. Dual kinase-bromodomain inhibitors in anticancer drug discovery: a structural and pharmacological perspective. J Med Chem. 2016;59(20):9305–9320. [DOI] [PubMed] [Google Scholar]
- 104.Devaiah BN, Lewis BA, Cherman N, et al. BRD4 is an atypical kinase that phosphorylates serine2 of the RNA polymerase II carboxy-terminal domain. Proc Natl Acad Sci USA. 2012;109(18):6927–6932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Parry D, Guzi T, Shanahan F, et al. Dinaciclib (SCH 727965), a novel and potent cyclin-dependent kinase inhibitor. Mol Cancer Ther. 2010;9(8):2344–2353. [DOI] [PubMed] [Google Scholar]
- 106.Misra RN, Xiao HY, Williams DK, et al. Synthesis and biological activity of N-aryl-2-aminothiazoles: potent pan inhibitors of cyclin-dependent kinases. Bioorg Med Chem Lett. 2004;14(11):2973–2977. [DOI] [PubMed] [Google Scholar]
- 107.Lv K, Chen W, Chen D, et al. Rational design and evaluation of 6-(pyrimidin-2-ylamino)-3,4-dihydroquinoxalin-2(1H)-ones as polypharmacological inhibitors of BET and kinases. J Med Chem. 2020;63(17):9787–9802. [DOI] [PubMed] [Google Scholar]
- 108.Liu Z, Sun Q, Wang X. PLK1, a potential target for cancer therapy. Transl Oncol. 2017;10(1):22–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gutteridge RE, Ndiaye MA, Liu X, Ahmad N. PLK1 inhibitors in cancer therapy: from laboratory to clinics. Mol Cancer Ther. 2016;15(7):1427–1435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hassan QN 2nd, Alinari L, Byrd JC. PLK1: a promising and previously unexplored target in double-hit lymphoma. J Clin Invest. 2018;128(12):5206–5208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Murga-Zamalloa C, Polk A, Hanel W, et al. Polo-like-kinase 1 (PLK-1) and c-myc inhibition with the dual kinase-bromodomain inhibitor volasertib in aggressive lymphomas. Oncotarget. 2017;8(70):114474–114480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Timme N, Han Y, Liu S, et al. Small-molecule dual PLK1 and BRD4 inhibitors are active against preclinical models of pediatric solid tumors. Transl Oncol. 2020;13(2):221–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Brandwein JM. Targeting polo-like kinase 1 in acute myeloid leukemia. Ther Adv Hematol. 2015;6(2):80–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Kolosenko I, Edsbäcker E, Björklund A-C, et al. RNAi prodrugs targeting PLK1 induce specific gene silencing in primary cells from pediatric T-acute lymphoblastic leukemia patients. J Controlled Release. 2017;261:199–206. [DOI] [PubMed] [Google Scholar]
- 115.Wang NY, Xu Y, Xiao KJ, et al. Design, synthesis, and biological evaluation of 4,5-dihydro-[1,2,4]triazolo[4,3-f] pteridine derivatives as novel dual-PLK1/BRD4 inhibitors. Eur J Med Chem. 2020;191:112152. [DOI] [PubMed] [Google Scholar]
- 116.Xu Y, Wang Q, Xiao K, et al. Novel dual BET and PLK1 inhibitor WNY0824 exerts potent antitumor effects in CRPC by inhibiting transcription factor function and inducing mitotic abnormality. Mol Cancer Ther. 2020;19(6):1221–1231. [DOI] [PubMed] [Google Scholar]
- 117.Qi Y, Xu L, Li Z, et al. Design, synthesis and biological evaluation of novel pteridinone derivatives as potent dual inhibitors of PLK1 and BRD4. New J Chem. 2020;44(38):16477–16490. [Google Scholar]
- 118.Kong X, Pan P, Sun H. Drug discovery targeting anaplastic lymphoma kinase (ALK). J Med Chem. 2019;62(24): 10927–10954. [DOI] [PubMed] [Google Scholar]
- 119.Mossé YP, Laudenslager M, Longo L, et al. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature. 2008;455(7215):930–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hallberg B, Palmer RH. The role of the ALK receptor in cancer biology. Ann Oncol. 2016;27:iii4–iii15. [DOI] [PubMed] [Google Scholar]
- 121.Berry T, Luther W, Bhatnagar N, et al. The ALK(F1174L) mutation potentiates the oncogenic activity of MYCN in neuroblastoma. Cancer Cell. 2012;22(1):117–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Puissant A, Frumm SM, Alexe G, et al. Targeting MYCN in neuroblastoma by BET bromodomain inhibition. Cancer Discov. 2013;3(3):308–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Watts E, Heidenreich D, Tucker E, et al. Designing dual inhibitors of anaplastic lymphoma kinase (ALK) and bromodomain-4 (BRD4) by tuning kinase selectivity. J Med Chem. 2019;62(5):2618–2637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Ember SW, Lambert QT, Berndt N, et al. Potent dual BET bromodomain-kinase inhibitors as value-added multi-targeted chemical probes and cancer therapeutics. Mol Cancer Ther. 2017;16(6):1054–1067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Fruman DA, Chiu H, Hopkins BD, Bagrodia S, Cantley LC, Abraham RT. The PI3K pathway in human disease. Cell. 2017;170(4):605–635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lien EC, Dibble CC, Toker A. PI3K signaling in cancer: beyond AKT. Curr Opin Cell Biol. 2017;45:62–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Yue Y, Hui K, Wu S, et al. MUC15 inhibits cancer metastasis via PI3K/AKT signaling in renal cell carcinoma. Cell Death Dis. 2020;11(5):336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hao J, Yang Z, Wang L, et al. Downregulation of BRD4 inhibits gallbladder cancer proliferation and metastasis and induces apoptosis via PI3K/AKT pathway. Int J Oncol. 2017;51(3):823–831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dittmann A, Werner T, Chung CW, et al. The commonly used PI3-kinase probe LY294002 is an inhibitor of BET bromodomains. ACS Chem Biol. 2014;9(2):495–502. [DOI] [PubMed] [Google Scholar]
- 130.Mahadevan D, Chiorean EG, Harris WB, et al. Phase I pharmacokinetic and pharmacodynamic study of the pan-PI3K/mTORC vascular targeted pro-drug SF1126 in patients with advanced solid tumours and B-cell malignancies. Eur J Cancer. 2012;48(18):3319–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Singh AR, Joshi S, Burgoyne AM, et al. Single agent and synergistic activity of the “first-in-class” dual PI3K/BRD4 inhibitor SF1126 with sorafenib in hepatocellular carcinoma. Mol Cancer Ther. 2016;15(11):2553–2562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Qin AC, Li Y, Zhou LN, Xing CG, Lu XS. Dual PI3K-BRD4 inhibitor SF1126 inhibits colorectal cancer cell growth in vitro and in vivo. Cell Physiol Biochem. 2019;52(4):758–768. [DOI] [PubMed] [Google Scholar]
- 133.Joshi S, Singh AR, Liu KX, et al. SF2523: dual PI3K/BRD4 inhibitor blocks tumor immunosuppression and promotes adaptive immune responses in cancer. Mol Cancer Ther. 2019;18(6):1036–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Divakaran A, Talluri SK, Ayoub AM, et al. Molecular basis for the N-terminal bromodomain-and-extra-terminal-family selectivity of a dual kinase–bromodomain inhibitor. J Med Chem. 2018;61(20):9316–9334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Fraga MF, Ballestar E, Villar-Garea A, et al. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat Genet. 2005;37(4):391–400. [DOI] [PubMed] [Google Scholar]
- 136.Groselj B, Sharma NL, Hamdy FC, Kerr M, Kiltie AE. Histone deacetylase inhibitors as radiosensitisers: effects on DNA damage signalling and repair. Br J Cancer. 2013;108(4):748–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Tng J, Lim J. Achiral derivatives of hydroxamate AR-42 potently inhibit class I HDAC enzymes and cancer cell proliferation. J Med Chem. 2020;63(11):5956–5971. [DOI] [PubMed] [Google Scholar]
- 138.Shahbazi J, Liu PY, Atmadibrata B, et al. The bromodomain inhibitor JQ1 and the histone deacetylase inhibitor panobinostat synergistically reduce n-myc expression and induce anticancer effects. Clin Cancer Res. 2016;22(10): 2534–2544. [DOI] [PubMed] [Google Scholar]
- 139.Liu T, Wan Y, Xiao Y, Xia C. Dual-target inhibitors based on HDACs: novel antitumor agents for cancer therapy. J Med Chem. 2020;63(17):8977–9002. [DOI] [PubMed] [Google Scholar]
- 140.He S, Dong G, Li Y, Wu S, Wang W, Sheng C. Potent dual BET/HDAC inhibitors for efficient treatment of pancreatic cancer. Angew Chem Int Ed. 2020;59(8):3028–3032. [DOI] [PubMed] [Google Scholar]
- 141.Laszig S, Boedicker C, Weiser T, Knapp S, Fulda S. The novel dual BET/HDAC inhibitor TW09 mediates cell death by mitochondrial apoptosis in rhabdomyosarcoma cells. Cancer Lett. 2020;486:46–57. [DOI] [PubMed] [Google Scholar]
- 142.Chen J, Li Y, Zhang J, et al. Discovery of selective HDAC/BRD4 dual inhibitors as epigenetic probes. Eur J Med Chem. 2021;209:112868. [DOI] [PubMed] [Google Scholar]
- 143.Atkinson SJ, Soden PE, Angell DC, et al. The structure based design of dual HDAC/BET inhibitors as novel epigenetic probes. MedChemComm. 2014;5(3):342–351. [Google Scholar]
- 144.Shao M, He L, Zheng L, et al. Structure-based design, synthesis and in vitro antiproliferative effects studies of novel dual BRD4/HDAC inhibitors. Bioorg Med Chem Lett. 2017;27(17):4051–4055. [DOI] [PubMed] [Google Scholar]
- 145.Noguchi-Yachide T, Sakai T, Hashimoto Y, Yamaguchi T. Discovery and structure–activity relationship studies of N6-benzoyladenine derivatives as novel BRD4 inhibitors. Biorg Med Chem. 2015;23(5):953–959. [DOI] [PubMed] [Google Scholar]
- 146.Amemiya S, Yamaguchi T, Sakai T, Hashimoto Y, Noguchi-Yachide T. Structure-activity relationship study of N(6)-benzoyladenine-type BRD4 inhibitors and their effects on cell differentiation and TNF-α production. Chem Pharm Bull. 2016;64(9):1378–1383. [DOI] [PubMed] [Google Scholar]
- 147.Amemiya S, Yamaguchi T, Hashimoto Y, Noguchi-Yachide T. Synthesis and evaluation of novel dual BRD4/HDAC inhibitors. Biorg Med Chem. 2017;25(14):3677–3684. [DOI] [PubMed] [Google Scholar]
- 148.Hewings DS, Wang M, Philpott M, et al. 3,5-Dimethylisoxazoles act as acetyl-lysine-mimetic bromodomain ligands. J Med Chem. 2011;54(19):6761–6770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhang Z, Hou S, Chen H, et al. Targeting epigenetic reader and eraser: rational design, synthesis and in vitro evaluation of dimethylisoxazoles derivatives as BRD4/HDAC dual inhibitors. Bioorg Med Chem Lett. 2016;26(12):2931–2935. [DOI] [PubMed] [Google Scholar]
- 150.Cheng G, Wang Z, Yang J, et al. Design, synthesis and biological evaluation of novel indole derivatives as potential HDAC/BRD4 dual inhibitors and anti-leukemia agents. Bioorg Chem. 2019;84:410–417. [DOI] [PubMed] [Google Scholar]
- 151.Pan Z, Li X, Wang Y, et al. Discovery of thieno[2,3-d]pyrimidine-based hydroxamic acid derivatives as bromodomain-containing protein 4/histone deacetylase dual inhibitors induce autophagic cell death in colorectal carcinoma cells. J Med Chem. 2020;63(7):3678–3700. [DOI] [PubMed] [Google Scholar]
- 152.Dancy BM, Cole PA. Protein lysine acetylation by p300/CBP. Chem Rev. 2015;115(6):2419–2452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Morrison-Smith CD, Knox TM, Filic I. Combined targeting of the BRD4-NUT-p300 axis in NUT midline carcinoma by dual selective bromodomain inhibitor, NEO2734. Mol Cancer Ther. 2020;19(7):1406–1414. [DOI] [PubMed] [Google Scholar]
- 154.Yan Y, Ma J, Wang D, et al. The novel BET-CBP/p300 dual inhibitor NEO2734 is active in SPOP mutant and wild-type prostate cancer. EMBO Mol Med. 2019;11(11):e10659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Spriano F, Gaudio E, Cascione L, et al. Antitumor activity of the dual BET and CBP/EP300 inhibitor NEO2734. Blood Adv. 2020;4(17):4124–4135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Ryan KR, Giles F, Morgan GJ. Targeting both BET and CBP/EP300 proteins with the novel dual inhibitors NEO2734 and NEO1132 leads to anti-tumor activity in multiple myeloma. Eur J Haematol. 2021;106(1):90–99. [DOI] [PubMed] [Google Scholar]
- 157.Hügle M, Regenass P, Warstat R, et al. 4-Acyl pyrroles as dual BET-BRD7/9 bromodomain inhibitors address BETi insensitive human cancer cell lines. J Med Chem. 2020;63(24):15603–15620. [DOI] [PubMed] [Google Scholar]
- 158.Thomas MC, Chiang CM. The general transcription machinery and general cofactors. Crit Rev Biochem Mol Biol. 2006; 41(3):105–178. [DOI] [PubMed] [Google Scholar]
- 159.Sdelci S, Lardeau CH, Tallant C, et al. Mapping the chemical chromatin reactivation landscape identifies BRD4-TAF1 cross-talk. Nat Chem Biol. 2016;12(7):504–510. [DOI] [PubMed] [Google Scholar]
- 160.Remillard D, Buckley DL, Seo HS, et al. Dual inhibition of TAF1 and BET bromodomains from the BI-2536 kinase inhibitor scaffold. ACS Med Chem Lett. 2019;10(10):1443–1449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Chang X, Sun D, Shi D, et al. Design, synthesis, and biological evaluation of quinazolin-4(3H)-one derivatives co-targeting poly(ADP-ribose) polymerase-1 and bromodomain containing protein 4 for breast cancer therapy. Acta Pharm Sin B. 2021;11(1):156–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Sakamoto KM, Kim KB, Kumagai A, Mercurio F, Crews CM, Deshaies RJ. PROTACS: chimeric molecules that target proteins to the Skp1-Cullin-F box complex for ubiquitination and degradation. Proc Natl Acad Sci U S A. 2001;98(15): 8554–8559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Bushweller JH. Targeting transcription factors in cancer—from undruggable to reality. Nat Rev Cancer. 2019;19(11): 611–624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Mu X, Bai L, Xu Y, Wang J, Lu H. Protein targeting chimeric molecules specific for dual bromodomain 4 (BRD4) and Polo-like kinase 1 (PLK1) proteins in acute myeloid leukemia cells. Biochem Biophys Res Commun. 2020;521(4): 833–839. [DOI] [PubMed] [Google Scholar]