

Summary
Pan-cancer studies sketched the genomic landscape of the tumor types spectrum. We delineated the purity- and ploidy-adjusted allele-specific profiles of 4,950 patients across 27 tumor types from the Cancer Genome Atlas (TCGA). Leveraging allele-specific data, we reclassified as loss of heterozygosity (LOH) 9% and 7% of apparent copy-number wild-type and gain calls, respectively, and overall observed more than 18 million allelic imbalance somatic events at the gene level. Reclassification of copy-number events revealed associations between driver mutations and LOH, pointing out the timings between the occurrence of point mutations and copy-number events. Integrating allele-specific genomics and matched transcriptomics, we observed that allele-specific gene status is relevant in the regulation of TP53 and its targets. Further, we disclosed the role of copy-neutral LOH in the impairment of tumor suppressor genes and in disease progression. Our results highlight the role of LOH in cancer and contribute to the understanding of tumor progression.
Keywords: cancer, allele specific, ploidy, genomic instability, somatic copy number, TP53, loss of heterozygosity
Graphical abstract
Highlights
-
•
Allele-specific analysis of TCGA collection identifies 18 million allelic imbalance events
-
•
Wild-type and copy-number gain calls are reclassified as LOH
-
•
LOH states associate with tumor suppressors reduced expression and prognosis
-
•
Activation of TP53 downstream targets reflects its allele-specific genomic status
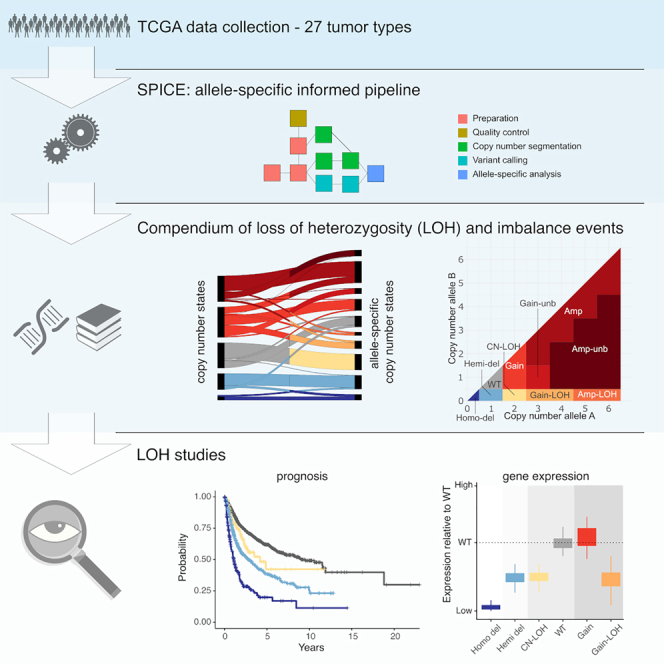
Ciani et al. delineated the purity and ploidy-adjusted allele-specific profiles across TCGA tumor types and identified 18 million allelic imbalance events. This led to the reclassification of wild-type and copy gain calls as loss of heterozygosity (LOH) and to an allele-specific genomic events catalog. The authors showed that the activation of p53 downstream targets is reflective of the allele-specific genomic status of TP53 and highlighted the pervasiveness of LOH and its association with prognosis and tumor suppressor genes expression.
Introduction
Pan-cancer genomic studies, pioneered by the Cancer Genome Atlas (TCGA), uncovered both tissue-specific and shared features of human tumors (Berger et al., 2018), enabled the characterization of the immune response to cancer (Thorsson et al., 2018), and detected at least one driver mutation in 91% of 2,658 analyzed whole-cancer genomes, mainly in coding regions (ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium, 2020).
In addition to mutations and changes in the tumor cell ploidy (aneuploidy) (Bielski et al., 2018b; Pfister et al., 2018) (Zack et al., 2013), an important class of events in cancer cells is the loss of heterozygosity (LOH). LOH occurs via heterozygous deletion of one allele. These events can be simple deletions or be accompanied by duplications of the remaining allele, giving rise to copy-neutral LOH (CN-LOH) or even to copy gain-LOH events. LOH has been interrogated in search for actionable vulnerabilities, since the lack of one allele and the subsequent reduced genomic redundancy can be exploited to specifically target cancer cells, for instance using allele-specific gene editing technology to target the remaining allele of essential or haploinsufficient genes (Nichols et al., 2020). Despite the interest as putative targets, to our knowledge, no study has systematically shown the relevance of LOH events in cancer-related processes at pan-cancer level.
The accurate measurement of tumor cells ploidy and the use of methods that can discriminate between alleles (Prandi and Demichelis, 2019; Shen and Seshan, 2016; Taylor et al., 2018) is essential for the comprehensive characterization of somatic copy-number aberrations (SCNA) and the ultimate delineation of allele-specific informed events. This is particularly relevant for identifying genes in CN-LOH status otherwise classified as wild type; in fact, CN-LOH can in principle lead to the duplication of a mutated allele in an oncogene or in a tumor suppressor gene, and duplication or loss of a methylated allele thus impacting on gene expression (Hagenkord et al., 2010; Yeung et al., 2018). Allele-specific informed data have been considered in tumor-type-specific studies (Buchwald et al., 2020; Ged et al., 2020; Hoff et al., 2020; Wilkinson et al., 2020), in the setting of haploinsufficiency detection for tumor suppressor genes (TSGs) (Davoli et al., 2013), for DNA repair genes in breast tissues (Karaayvaz-Yildirim et al., 2020), and also for the understanding of cancer aneuploidy (Taylor et al., 2018).
We hypothesized that a uniform harmonized characterization of tumor-allele-specific informed genomic landscape would deepen our understanding of the cancer genomes and of the role of previously unappreciated LOH events, such as CN-LOH and copy gain-LOH events, in cancer-related processes. This characterization can lead to the identification of molecular vulnerabilities (Nichols et al., 2020) and provide additional discovery tools for the assessment of biomarkers for patients’ enrollment into clinical trials. Therefore, here we present a framework for the analysis of allele-specific genomic features, a uniform harmonized characterization of the genomes of 4,950 patients from 27 TCGA datasets, and evidence that single-allele data provide an orthogonal component of information to the landscape of primary tumors whereby LOH is a common trait of impaired tumor-suppressive processes.
Results
A framework for allele-specific informed genomic features analysis
To comprehensively characterize the genomic landscape of human tumors at the single-allele level and define the spectrum of LOH events (including CN-LOH, copy gain (i.e., the allele presents 3 or 4 copies) and amplification LOH (i.e., the allele presents 5 or more copies) events, here referred to as Gain-LOH and Amp-LOH, respectively), we designed a framework that integrates a set of widely used tools (STAR Methods; Figures S1 and S2) to seamlessly process matched tumor and normal samples profiled by next-generation sequencing technologies and extract allele-dependent genomic information from segmented data upon tumor ploidy and tumor purity correction. The pipeline implements the computation of allele-specific copy-number (asCN) data (Prandi and Demichelis, 2019) that broaden the spectrum of assessable copy-number states. As any DNA copy number higher than one can be explained by more than one-allele-based combination (STAR Methods; Table S3), the set of possibilities also includes multiple LOH states such as CN-LOH, Gain-LOH, Amp-LOH, in addition to the most commonly studied hemizygous deletion (Hemi-del). We applied the pipeline to 8,183 primary cancer and matched normal samples data from 27 tumor types profiled with whole-exome sequencing (WES) from TCGA (Grossman et al., 2016) and identified 4,950 cases with overall high purity (pan-cancer median of 69%, interquartile range [IQR] [54% and 82%]) amenable to downstream analyses (Figures 1A and S3; Table S1). Overall, we observed more than 18 million events of allelic imbalance at the gene level (corresponding to a total of 177,650 genomic segments, corresponding to 1/3 of all the segments), 56.4% of which are contributed by apparent wild-type events that are in fact CN-LOH (Figure 1B). The full set of the study cohort asCN data are represented in Figure 1C where for each gene allele A and allele B correspond to the allele with the higher and the lower number of copies, respectively. Overall, the analysis revealed that 9.2% of wild-type gene calls according to conventional methods (i.e., not using allele-specific informed processing) are CN-LOH (2-0) and that 7.1% of Gains are Gain-LOH (3-0 or 4-0) and that allelic imbalance is widely present in both diploid and non-diploid genomes, as graphically represented in the asCN space (Figure 1C). Illustrative patient sample data are shown in Figures 1D–1G: the panels show the reclassification of log2 ratios to asCN call and detailed allelic fraction of patient’s heterozygous SNPs across genomic segments along a chromosome 8 (additional exemplificative sample in Figure S7).
Figure 1.

Allele-specific characterization of TCGA data across 27 tumor types upon tumor purity and ploidy adjustment
(A) Distribution of copy-number (CN)-based tumor purity (left) and of allele-specific informed ploidy (asP) values for each TCGA study (right). asP values are color-coded as high asP: > 2.5 and low asP: ≤ 1.5. Tumor types are sorted by decreasing percentage of high asP samples. Complete data in Table S1.
(B) Sankey diagram linking adjusted log2 ratios to allele-specific copy-number (asCN) states.
(C) Distribution of asCN events based on number of copies of allele A and B (allele with more and fewer copies, respectively). Complete table with number of events at gene levels is included in Table S3.
(D) Sankey diagram showing the classification of segments based on discretized raw log2 ratios (left), purity- and ploidy-adjusted discretized log2 ratios (middle) and allele-specific copy number (asCN, right) of an exemplificative study patient. For this high asP sample, purity and ploidy correction (middle) reclassifies apparent Hemi-del segments as wild type or as Gains. Additionally, asCN classification identifies wild-type segments as CN-LOH, and gain segments as Gain-LOH or as Gain-Unb. Detailed visualization of segments for the same patient is shown in (E, F, and G).
(E) Scatterplot showing adjusted log2 ratio and beta values of segments: each point represents a segment (details of labeled segments are shown in F and G). Beta values are estimations of the fraction of reads, equally representing the two parental alleles.
(F) Allelic fraction of informative SNPs on chr8 in the normal sample (top panel) and matched tumor sample (low panels).
(G) Distribution of allelic fractions in chr8 in normal sample (top panel) and matched tumor sample (low panel), stratified by asCN (defined based on tumor sample).
Building on the asCN data of each tumor sample, we here defined a measure of the tumor cell nuclear DNA content, similar to the concept of DNA index from DNA cytometry (Danielsen et al., 2016), which we termed allele-specific informed ploidy (asP) index (Figures 1A and S4), as it is computed as the weighted mean of the asCN of homologous chromosomes. By definition, perfectly diploid cells have asP equal to 2. The obtained asP values are overall concordant with ABSOLUTE ploidy measures (Carter et al., 2012) (Figure S5A). Overall, 1,305 tumors (26.4%) in the study cohort show high asP (defined as asP > 2.5) with marked variability among tumor types, ranging from 81.2% (n = 101) in testicular germ cell tumors (TGCT) to none (n = 16) in acute myeloid leukemia (LAML). Of note, 46 tumors (1%) show low asP (defined as asP ≤ 1.5), the majority of which are adrenocortical carcinoma (ACC). When comparing asP values with other ploidy-related or genome instability measures (Figures S4B–S4E), we observed that the specific information captured by each genomic measure is also reflected by the diverse associations with overall survival (OS) and progression-free interval (PFI) (Figure S6A). Concerning PFI, asP is the only measure that detects significant association in ACC, a tumor type with frequent low asP (Figure 1A), with high asP demonstrating worse outcome.
To query for potential tumor fingerprints driven by allele-specific information, we applied a data dimensionality reduction method (McInnes et al., 2018) to asCN data of autosomal chromosomes. Upon removal of the tumor ploidy component (STAR Methods; Figures S9A and S9B), we observed a well-structured tumor samples organization (Figure S9C; Table S4) in which 20 clusters (tested for stability, median adjusted rand index = 0.9895, SD: 8.9e−3, calculated on n = 10 random samplings, Figure S8) were identified by the density-based procedure DBSCAN (Ester et al., 1996). Clusters characterization showed that they are not uniquely driven by specific tumor types (column margins in Figure S9D) but also by the distribution of recurrent genomic lesions in key TSG and oncogenes (OG) (row margins) (Tables S5 and S6). Altogether, this unsupervised analysis of allele-specific informed fingerprints identifies genomic commonalities among tumors that are not strictly defined by tumor type but rather by specific sets of oncogenic events.
The application of the framework for the analysis of allele-specific informed genomic features to the TCGA WES collection highlighted that LOH is a relatively common genomic status in primary tumors. This characterization can be beneficial to the study of cancer-related processes in pan-cancer studies by offering a more accurate assessment of the genome. The here proposed pipeline is an easy-to-use tool to directly obtain asCN data from matched tumor and normal samples; this study also makes available such data for 27 tumor types from TCGA.
Combined analysis of asCN and SNV allelic fractions facilitates considerations on the timing of cancer-driving events
Given the widespread presence of allelic imbalance, we next looked at the representation of asCN states in the presence of point mutation events. We observed depletion of wild-type and balanced states compared with unbalanced conditions for TSGs (Figure 2A), in line with the observation previously reported in advanced solid cancer patient samples (Bielski et al., 2018a).
Figure 2.

Association between loss of heterozygosity, SNVs incidence, and their allelic fraction
(A) Fraction of SNVs (N = 601,177) in each asCN state, stratified by class of gene (TSG, OG, other genes). Color code is shared for (A, C, D, E, and F).
(B) Top-ten ranked genes with uneven distributions of SNVs across asCN classes. Dot size is proportional to the number of samples with SNV. Oncogenes and TSG with FDR < 0.001 (chi-square test) and with at least 20 SNV events are shown.
(C) Distribution of allelic fraction of SNV (N = 6,657, Mann-Whitney tests) across asCN classes comparing tumor suppressor genes and oncogenes.
(D) Parallel sets plot showing the mapping of TP53 log2 discretized copy-number values to asCN status. Barplots on the sides of CN bars show the fraction of samples harboring TP53 deleterious SNVs in each class. Chi-square test indicates the non-independence between asCN status and presence of SNVs.
(E) Distribution of allelic fractions of TP53 LOF mutations (N = 384, Mann-Whitney tests).
(F) Barplots showing the TP53 asCN status in each TCGA study.
(G) Fraction of samples with TP53 loss-of-function SNVs that have retained or lost the wild-type copy. Each row represents a TCGA study as indicated in (F).
(H) Expression levels of CDKN1A and MDM2 in samples stratified based on presence of wild-type copies of TP53.
We next explored at gene level the relationship between the incidence of SNVs and asCN of both OG and TSG by stratifying tumors in three asCN classes, namely LOH, heterozygous, and unbalanced. We observed multiple genes with uneven distributions of SNVs across classes (chi-square test, FDR < 0.05, only keeping classes with at least 20 SNV events, Table S8), with enrichment of TSGs (Fisher’s exact test, p-value = 0.008) in the LOH asCN class, with TP53, PTEN, SMAD4, CIC, VHL, and RB1 as top-ranked (Figure 2B). Further, when querying their CN adjusted allelic fractions (AF) among asCN classes, we often observed AF close to 1 for SNVs in LOH states, thus indicating that deleterious SNVs within TSGs are driver events (Figure 2C). On the other hand, OG data demonstrate a bimodal distribution (Figure 2C) evidencing that point mutations occur upon the loss of one allele, in line with the notion that bi-allelic mutations are not required for oncogenic activation.
We then hypothesized that LOH asCN states of TSG are enriched for deleterious SNVs as a means of increasing cell fitness via full impairment of tumor-suppressive processes. We therefore tested whether in-depth allele-specific informed analysis of the recurrently mutated TSG TP53 (Hollstein et al., 1991; Olivier et al., 2010; Petitjean et al., 2007) could lead to new insights on cancer-related biological processes with respect to conventional CN analysis (Figure 2D) where CN-LOH are frequently misclassified as wild-type copy number. For instance, we detected a significantly lower proportion of deleterious SNVs when focusing on asCN-based wild-type segments as opposed to apparent wild-type segments (based on log2 ratio only analysis) (proportion test, p value = 1.6e−54; 7% versus 24%). Conversely, we observed an enrichment of SNVs in Gain-LOH asCN segments with respect to log2 ratio = 1 segments (proportion test, p value = 2.8e−11; 55.7% versus 16.9%). Taking all asCN statuses into account (WT, CN-LOH, Hemi-del, Gain-LOH, and Gain), TP53 SNV status is not independent of the allele-specific status (chi-square test, p value < 2.2e−16). Altogether, TP53 LOH tumors are enriched for TP53 deleterious SNVs (37.2%, 51.4%, and 55.7% in Hemi-dels, CN-LOH, and Gain-LOH, respectively). The distribution of AF of loss-of-function (LOF) SNVs of TP53 has higher values in CN-LOH and Gain-LOH events with respect to WT, supporting the concept that SNVs and LOH are driver events and that gene amplifications (leading to CN-LOH and Gain-LOH) occur at a later time (Figure 2E). Despite the variability of TP53 mutation frequency, this association is conserved across tumor types (Figures 2F and 2G). When TP53 is mutated, some tumors still retain a wild-type copy (Figure 2G, right). This can modulate the effects of TP53 mutations through a “dominant-positive mechanism,” where a wild-type copy can change the stoichiometry and function of p53 tetramers (Gogna et al., 2012; Walerych et al., 2018).
We next investigated the transcript levels of the p53 target genes CDKN1A (coding for p21) and MDM2 with respect to TP53-allele-specific status (Figure 2H). Independently of TP53 expression level (Figure S10), significantly lower levels of expression of CDKN1A and MDM2 (Mann-Whitney test, p value = 0.012 and p value = 0.0002, respectively) were observed in tumors harboring exclusively mutated copies of TP53 compared with tumors that retained at least one wild-type copy. These pan-cancer observations suggest a TP53-dependent transcriptional regulation mechanism for MDM2 that is coherent with previous publications (Midgley and Lane, 1997; Terzian et al., 2008; Vijayakumaran et al., 2015), whereby the lack of wild-type TP53 impairs the transcriptional activation of MDM2, which is responsible for the ubiquitination and translocation of p53 to the proteasome. RB1 expression is also reduced in absence of wild-type TP53 (Mann-Whitney test, p value = 0.007 considering only LOF mutations, p value = 6.8e−05 considering all deleterious SNVs). In line with previous work showing that TP53 mutational status is linked to ploidy and that aneuploid mammalian cells activate p53 (Hinchcliffe et al., 2016) (Li et al., 2010; Soto et al., 2017; Thompson and Compton, 2010), we observed that TP53 SNVs are enriched in high asP samples (chi-square test p value = 4.7e−21, Table S7). As TP53 is involved in cell-cycle regulation and, based on recent data, in aneuploidy-mediated activation of proteotoxic stress response in cells (Santaguida et al., 2015), in the impact of SCNA on the amount of proteins (Stingele et al., 2012), and in proteasome regulation (Walerych et al., 2016), we tested the activation of a set of selected proliferative (STAR Methods; Table S18) (Sheltzer, 2013) and proteasome-related pathways (Levin et al., 2018; Wang et al., 2017) in high asP samples compared with diploid samples (Figure S10). The results suggest the activation of opposite transcriptional programs upon the presence of high asP in different tumor types (Table S16) possibly related to different genetic and tissue-specific transcriptional backgrounds and their interactions with TP53 status, affecting proliferation and global protein homeostasis. Altogether, the observation in this pan-cancer setting of the allele-specific effect of TP53 on downstream targets expression and processes corroborates the hypothesis that the tumor genomic make-up contributes to shaping the proliferative response to aneuploidy by regulating both transcription and protein degradation.
CN-LOH events are frequent and associate with prognosis
While LOH events due to monoallelic deletions are commonly reported, CN-LOH events most often remain hidden in large genomic studies, either incorrectly classified as wild-type segments or not explored for their functional relevance. Here, we estimated an overall pan-cancer median CN-LOH burden per sample of 2% (IQR [0%, 9%]), with significant increase in high asP samples (p value < 0.001, Wilcoxon rank sum test with continuity correction) (Figure S11A) with median values of 14% (IQR [8%, 22%]) compared with 0.4% (IQR [0%, 4%]) and 0.2% (IQR [0%, 4%]) in diploid and low asP tumors, respectively. Remarkably, 30% of the high asP samples have at least 20% of the genome with CN-LOH signal (Figure S11B), while the percentage drops below 3% for diploid and low asP samples. When considering tumor-specific data (Figure S11C), few exceptions emerged as TGCT and colon adenocarcinoma (COAD) (Table S9). Albeit less frequent, LOH events also include Gain-LOH and Amp-LOH, observed in 36.5% of the study samples and enriched in high asP samples (Figure S11D; Table S10). Since LOH events can involve entire chromosomal arms (Figure S11E), a great number of genes can be affected by Hemi-del and CN-LOH in each sample (Figure S12A). We observed that Hemi-dels are underrepresented in high asP samples (p value < 0.001, Wilcoxon rank sum test with continuity correction), which are enriched for CN-LOH events instead (Figure S12A, inset). In a recent work, Nichols et al. (Nichols et al., 2020) focused on LOH events on essential genes to identify putative cancer vulnerabilities: we estimated frequencies of LOH in essential genes obtaining results concordant with previously published data (Figure S12B). With respect to what was reported by Nichols et al., we here add the characterization of Gain-LOH and Amp-LOH events in essential genes, potentially expanding the panel of putative cancer vulnerabilities to genes with copy-number gains.
When studying the extension of LOH genomic events at the level of chromosomal arms (arm-wide versus partial) across tumor types, we observed that TSGs such as TP53, BCL2, and CDKN2A are included in chromosomal arms which show extremely widespread LOH regions (Figures 3A and S11E). We estimated that almost 40% and 15% of samples have at least two TSG with CN-LOH and Gain-LOH, respectively (Figure 3B). Patterns of Hemi-dels are not random, preferentially encompassing TSGs and antiproliferative genes (Solimini et al., 2012), and cumulative haploinsufficiency may contribute to tumor evolution and maintenance (Davoli et al., 2013). Given these premises, we tested whether the asCN status of TSGs (Table S20) has prognostic value (considering all TSGs with at least 10 events per asCN status at pan-cancer level). We performed pan-cancer multivariate cox hazard analysis in diploid samples using PFI as readout, with asCN gene status combined with the presence of SNVs as predictor and tumor type (study) as covariate. A total of 20 TSGs resulted as significant predictors of risk when comparing Hemi-del versus WT (N = 11) or CN-LOH versus WT (N = 12), in the absence of deleterious SNVs (Table S11, results for all samples, independently of ploidy, are shown in Table S12). Of note, 12 genes showed statistical significance when considering CN-LOH status versus WT (in absence of deleterious SNVs), thus supporting the relevance of explicitly assessing the asCN status of TSG. Among the top significant genes of the multivariate cox hazard models, the analysis unveiled the associations between disease progression and events in CDKN2A and in the multiple endocrine neoplasia type I gene, MEN1, already reported as a haploinsufficient tumor suppressor (Lejonklou et al., 2012) (Figures 3C–3E). Despite the limitations stemming from the correlative nature of the analysis, we here observed clear evidence for TSGs CN-LOH association with tumor progression.
Figure 3.

Loss of heterozygosity events and their impact on prognosis
(A) Chromosomal arm spread of LOH burden distinguishes two patterns, arm-wide spread, and heterogeneous spread. Gene density does not associate with LOH burden spread.
(B) Distribution of number of TSGs in LOH status, stratified by asCN. CN-LOH is the second most common event of LOH for TSGs (after Hemi-del), while Gain-LOH and Amp-LOH, albeit less common, are present in a significant portion of samples.
(C) Volcano plot showing coefficient and significance of Cox proportional hazards models testing asCN and SNV status of tumor suppressor genes using PFI as endpoint. Analysis performed on diploid samples with tumor type as covariate.
(D and E) PFI survival curves, corresponding to (C) analysis, of selected tumor suppressor genes CDKN2A and MEN1. SNV related curves are omitted due to low numerosity. p-values from log rank tests and Cox proportional hazards models.
LOH associates with TSG expression
It has been observed that deletions of TSGs are early events (Deng et al., 1996) and, therefore, the most plausible scenario for the origin of CN-LOH involving TSGs is likely the loss of one allele followed by the duplication of the remaining allele. This is supported by our observation of the AF of SNVs in TSGs which present CN-LOH or Gain-LOH status (Figure 2C). On this premise, we next tested whether CN-LOH events, despite restoring two copies of the involved genes, demonstrated reduced gene expression with respect to the WT counterpart as opposed to rescuing the basal levels. Specifically, we hypothesize that genes that lose one allele and then regain additional copies (CN-LOH, Gain-LOH, or Amp-LOH) cannot rescue their basal expression level if that is at a disadvantage to the cancer cell, such as in the case of TSGs.
To test this hypothesis, we built a linear model to predict the expression level of a gene based on two variables derived by asCN information: the total number of copies (CN tot) and the presence of LOH (Figure 4A). In the specific scenario of a TSG, we can test whether CN-LOH events associate with reduced expression level with respect to the WT level and further if Gain-LOH and Amp-LOH also show a reduction in expression, independently of the total number of copies.
Figure 4.

Loss of heterozygosity events and their impact on gene expression
(A) Synthetic example showing the expression levels, stratified by asCN, of a gene for which LOH has negative effect on expression.
(B) Density of association coefficients for parameters a α (CN tot) and β (LOH) of the linear model. Negative values indicate a negative impact on gene expression. Sets of genes included are in Table S20.
(C) Proportion of genes showing a significant decrease of gene expression upon LOH (p-values are computed with the proportion test).
(D) Distribution of ratios of aberrant asCN to WT expression levels. Dashed line indicates half expression with respect to WT. Ticks on the x axis indicate single events (N = 37).
This is opposed to a model that uses classical CN information instead of asCN and considers the level of expression dependent solely on the total number of copies (Figure S13A). We tested the model (STAR Methods) on all genes at study (cancer type) specific level and observed that, globally, the total number of copies positively impacts on expression while LOH has a negative impact (Figure 4B, left panel): this effect is even more pronounced in specific studies such as BRCA (Figure 4B, right panel) and it is stronger for TSGs with respect to OGs and all other genes (Figure 4B) (tables with full of results are available at https://github.com/demichelislab/SPICE-pipeline). We selected all the genes whose LOH is significantly associated with gene expression reduction ( < 0, FDR < 0.1) in at least two tumor types: enrichment analysis showed that this phenomenon is significantly more present in TSGs with respect to OGs or all other genes (Figure 4C). This supports our hypothesis that TSGs are subject to selective pressure when in CN-LOH status. A significant proportion of essential genes showed reduced expression upon LOH as well. Even if potentially counterintuitive, this can be related to the higher stability of essential proteins compared with non-essential ones (Yen et al., 2008), thus reducing the negative impact of reduced expression. We next nominated pathways and biological functions that are possibly impaired by LOH dependent expression reduction (Figure S13B; Tables S13 and S14). The most relevant terms are related to RNA metabolic processes as well as to molecular localization and transport. Other enriched molecular functions, such as “vacuolar transport,” “autophagy,” and “oligosaccharide-lipid intermediate biosynthetic process” are relevant for tumorigenesis and cancer progression (Mulcahy Levy and Thorburn, 2020).
In total, we identified 18 TSGs under putative selective pressure for reduced expression upon CN-LOH in at least two studies (Figure S13C). When comparing the magnitude of the expression reduction with respect to WT, we observed that Hemi-del and CN-LOH have a similar impact, with a median ratio (see STAR Methods) of 0.54 and 0.66, respectively. Globally, we observed that CN-LOH, Gain-LOH, and Amp-LOH can all be associated with reduced expression compared with the wild-type state for specific TSGs in a tumor-specific manner (Figures S13D–S13G).
Discussion
The quantitative assessment of tumor ploidy and asCN alterations that we here proposed broadens the characterization of primary tumor genomes. For instance, low ploidy tumors are naturally distinguished from high ploidy by asP as in the case of ACC, an observation that would have been missed by other genomic indices (Beroukhim et al., 2010; Burrell et al., 2013; Carter et al., 2012; Mouliere et al., 2018; Taylor et al., 2018). The combined utilization of multiple genomic stability indices and ploidy assessment could help distinguish between markedly diverse cancer genomes states, from chaotic disruptions to whole-genome duplications, in turn pointing to the study of specific molecular targets.
Allele-specific informed analysis, applied via our framework, allows for precise detection of a variety of LOH states such as CN-LOH, Amp-LOH, and Gain-LOH. This is particularly relevant for CN-LOH events, which are otherwise incorrectly classified as WT, potentially leading to incorrect biological interpretations. In fact, we observed that CN-LOH results in the enrichment of loss and gain-of-function mutations in TSGs. Although we did not explicitly study in detail the time dependency between imbalance and point mutations, it recently emerged that whereas allelic imbalance is not driven by the occurrence of a mutant allele, a mutant allele dosage increase favors the fitness of a malignant clone (“exaptation” phenomenon) (Bielski et al., 2018a). In the context of copy-neutral and Gain-LOH events of cancer genes, the analysis of CN adjusted AF of deleterious SNVs suggests that point mutations rarely occur after the relevant amplification event (i.e., concomitant or prior events) as opposed to what is observed in the context of heterozygous states. Unbalanced status of point mutations could be an additional feature, beyond the position and the type of substitution, by which OG might achieve the ideal level of signaling (“sweet spot”), as recently suggested for KRAS (Li et al., 2018).
This study also quantified the widespread presence of allelic imbalance events, more pronounced in high asP tumors. These events could be a result of the processing of DNA double-strand breaks occurring via breakage-induced replication (Elango et al., 2017), particularly for the cases where arm-wide events are prevalent, and, which may also contribute to the high asP phenotype. Our results suggest that these events are more widespread than expected, possibly related to alterations in DNA-damage checkpoint and DNA repair capacity or underlying the acquisition of an alternative lengthening of telomeres (ALT) phenotype (Heaphy et al., 2011). It is possible that p53 status, which impacts on DNA replication and repair mechanisms (Klusmann et al., 2016) (Janic et al., 2018), can also impact on the occurrence of CN-LOH events.
In-depth characterization of TP53 genomics through an unsupervised integrated pan-cancer analysis highlighted the relationship between mutations and aneuploidy state, providing a link between TP53 status, aneuploidy, cell proliferation, and proteasome activation. Further, TP53 LOH associates with the presence of TP53 SNVs, exacerbating the deregulation of tumor-suppressive pathways. On one hand, the complete loss of wild-type copies impairs the ability of TP53 to regulate its targets, on the other hand, the presence of mutated copies allows for new interactions and oncogenic processes such as the establishment of a mutant-p53 proteasome axis (Walerych et al., 2016).
Integration of allele-specific genomics and matched transcriptomic data pointed to processes of RNA metabolisms, known to be quantitatively fine-tuned in the maintenance of normal cells, as perturbed upon LOH. We further confirmed a more prominent association of LOH and TSGs expression, as opposed to the whole transcriptome (Davoli et al., 2013), and showed that this is heavily contributed by CN-LOH events often undisclosed in genome-wide studies. In-depth analysis of LOH can shed the light on previously unexplored selective pressure mechanisms involving specific genes or pathways. Although we made the effort to study asCN genomic features and their functional impact by considering the specific contribution of diverse tumor types, our results are intrinsically limited by the modest frequencies of specific event subclasses in the study cohorts. Further, we omitted to consider additional type of events that could provide compensatory mechanisms in tumor evolution and disease progression (Persi et al., 2021).
Broadly, we showed that the detailed characterization of cancer genomic alterations benefits the study of oncogenic and tumor progression events while empowering cancer mechanisms investigations. To acknowledge the importance of reproducibility and easy access to asCN-based future studies, we implemented the study pipeline also using the common workflow language (CWL) and provide the full set of allele-specific informed genomic data for the studied cohort. Altogether, we envision that this orthogonal genomic feature will eventually allow the refined charting of tumor evolution paths, the detection of synthetic lethality combinations, and the accurate assessment of genomic biomarkers for patients’ enrollment in clinical trials.
STAR★Methods
Key resources table
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Yari Ciani (yari.ciani@unitn.it).
Materials availability
This study did not generate new materials.
Method details
TCGA studies inclusion criteria
Via the GDC Data Transfer Tool Client provided by the Genomic Data Commons (GDC) (Grossman et al., 2016), all Whole Exome Sequencing (WES) BAM files of the TCGA collection available on January 2018 were downloaded (N BAM=22,196). Figure S2D reports inclusion criteria -and relevant numbers- of the current study. Briefly, samples for which either kit annotation was unavailable or multiple kits were specified were excluded from downstream processing (N samples excluded=2,885). All the possible pairs of normal-tumor samples were identified per patient (N pairs=10,581). Tumor-normal pairs were excluded if gender information was not available (gender required for log2 ratio inference) (N pairs excluded=75) or MuTect2 SNV calls from GDC were not available (N pairs excluded=1353). The SPIA genetic distance based tool (Demichelis et al., 2008) applied to verify the correct annotation of paired samples nominated 14 alleged pairs with incompatible genotypes (11 of which from the same tumor type study (DLBC)), which we excluded from subsequent analyses (N patients excluded=14). When more than one pair was available for the same patient, we included the one with the highest tumor purity. Only patients with a primary tumor available were retained (i.e. metastases or recurrent tumors were excluded). Last, studies with less than 60 patients, prior to ploidy and purity correction, were excluded. Based on the above requirements, a total number of 8,183 patients (i.e. normal-tumor pairs) across 27 tumor types was selected (see Table S1; Figure S2D). Further quality filters for allele-specific genomic analysis then nominated 4,950 pairs as adequate for downstream study investigations. (reliable copy number based clonality by CLONETv2: 4,950; reliable SNV based clonality by TPES: 4,299, of which 1,246, not included in the CLONETv2 set).
The allele-specific informed pipeline
The pipeline was designed to generate a comprehensive analysis of matched normal and tumor next-generation sequencing aligned data, including whole-exome, whole-genome, and targeted panels. The peculiarity of the pipeline is the heavy use of individual-specific germline information, from quality check steps to the assessment of somatic lesion clonality and allele-specific events. The pipeline named SPICE is composed of several modules each designed to handle a different part of the analysis (Figure S1A). Each module is self-contained and, by maintaining the input/output interface, can be replaced with custom modules. The pipeline is structured so that each normal/tumor pair is analyzed independently. All tools used in the pipeline modules have been previously published either by our group or by others and custom scripts were created for their integration. The bash version of the pipeline, including integration scripts, is available at https://github.com/demichelislab/SPICE-pipeline. A CWL version of the pipeline is available at https://github.com/demichelislab/SPICE-CWL-pipeline. A list of the pipeline modules with relevant references is available below and at https://github.com/demichelislab/SPICE-CWL-pipeline.
Resources organization
Preparation:
The pipeline configuration file includes the parameters needed to perform all analyses, such as the reference genome build, the dbSNP version, the identifier of the sequencing kit, the gender of the individual, and the BAM files paths. During this phase, the folder structure used by each tool during the analysis is created. After consistency checks related to the configuration setup, the pipeline verifies if the indices of the BAM files are present otherwise BAM indexing is run. The last step of the preparation phase is the computation of the SNP pileups (Valentini et al., 2019) confined to regions covered by the sequencing kit, as utilized multiple times throughout the pipeline.
Quality Control (QC):
As part of this module the following steps are run; collection of statistics of the sequencing data (picard HsMetrics, URL http://broadinstitute.github.io/picard/); inference of individual’s ethnicity (EthSEQ (Romanel et al., 2017)); normal-tumor match check by genetic distance based on a set of relevant SNPs (SPIA (Demichelis et al., 2008)). Number of SNPs used for each sample spans from 223 to 497, median 460.
Segmentation:
After the QC phase is completed the pipeline proceeds with the “Copy number segmentation phase,” where the copy number profile of the tumor is computed by CNVKit (Talevich et al., 2016) CBS segmentation that returns a log2 ratio of tumor against control for each segment; given the low computational cost (Figures S2A–S2C), two additional segmentation methods are run for ancillary analyses, i.e. CNVKit with SLM based segmentation (Orlandini et al., 2017) and FACETS (Shen and Seshan, 2016). By default, the CNVKit CBS-based segmentation is used; the user can select other segmentation outputs via a parameter (i.e. configuration file).
Variant calling:
In this phase, SNVs and indels are called using MuTect2 (https://doi.org/10.1101/861054) and annotated with Variant Effect Predictor (VEP) (McLaren et al., 2016). The information about the coverage of the SNV sites is integrated with the annotation produced by VEP.
CLONETv2:
As last phase, the pipeline runs tools to assess copy number data, allele-specific copy number data, and SNV features upon tumor ploidy and purity correction. First, the copy number based tool CLONETv2 (Prandi and Demichelis, 2019) is applied. CLONET corrects the effects that tumor ploidy and admixture have on the copy number of the tumor and determines the level of SCNA clonality. Second, an SNV based tool, TPES (Locallo et al., 2019), is applied to ensure tumor purity assessment of tumors with quiet genomes. Last, the pipeline combines the information about clonality that is generated by CLONETv2 with the SNVs to estimate the clonality of each SNV.
An option to compute the MuTect2 panel of normal is available and that is reported as phase “Other” in the figure.
Performance
The pipeline collects the runtime of each step. The execution time on a single core machine is moderate (Figure S2A) and allows to scale to large parallel machines. All tools were run on all the study samples, with the exception of MuTect2; in-house MuTect2 calls for 2,000 randomly selected patients were compared to those generated by the Genomic Data Commons (GDC), resulting in high concordance. The more time-consuming steps are those which process the entire BAMs namely PaCBAM (Valentini et al., 2019) (SNP pileup, SNV pileup), Picard HSMetrics, and CNVKit.
The median execution times of the entire pipeline for a tumor/normal pair with and without MuTect2 computations on a single core are ∼21 hours and 5.5 hours, respectively. We analyzed the entire set of selected patients in ∼20 days of computing time on 3 HPC machines with 40 cores (for a total of 120 cores) and 256 Gb of RAM each.
The pipeline analyzes each sample on a single core thus allowing the easy implementation of external parallelization strategies. The pipeline uses GNU parallel (DOI http://doi.org/10.5281/zenodo.1146014) as parallelization mechanism. CPU and memory usage of a test run on 158 normal/tumor pairs were assessed (Figures S2B and S2C). The test was conducted on a machine with 40 physical cores and 256 Gb of RAM. By default, each sample is configured so to use a single core in order to have a predictable behavior both in terms of CPU and memory usage. This single-threaded nature enables us to maximize the load on the machines as visible in Figure S2C. The pipeline peak memory usage was of ∼130 GB. If we consider the memory usage per-core, the pipeline used less than 3.5GB throughout the whole execution. The entire per-core memory usage is shown as a gray line in the bottom part of Figure Figure S2C.
Pipeline output tables
Table S21 lists the steps that are included in the bash version of the pipeline and the output files produced by each step. Figure S1 shows the dependencies between all the steps that are part of the pipeline using a flowchart.
CWL implementation of the SPICE pipeline
The SPICE pipeline is also available using the Common Workflow Language (Amstutz et al., 2016), a standard specification for the description of computational workflows that enables easily portable and scalable pipelines. Using one of the many available CWL implementations, it is possible to run SPICE on a variety of architectures (from single machines to clusters or cloud services) to easily scale up as needed. In order to enable ease of use and reproducible analyses, the tools that are used in the pipeline are ready on Docker Hub as containers. To run the pipeline, it is sufficient to create a single configuration file per tumor/normal pair, where the user provides the required options (e.g. BAM files, reference genome) (https://github.com/demichelislab/SPICE-CWL-pipeline).
Genotype based analyses, SPIA and EthSeq
A genotype-based tool (SPIA) and an ethnicity caller (EthSeq) were applied to all study samples (Demichelis et al., 2008) (Romanel et al., 2017). Briefly, SPIA measures the similarity between two samples using a set of high-MAF selected SNPs (N_SNP median = 460; range: 223:497), whereby matched normal and tumor samples are expected to have high similarity. Figure S3A reports the results of the analysis on all possible pairings of the study samples. The vast majority of the samples were correctly paired with few exceptions (red dots), where samples annotated as related demonstrate high genotype distance (N=13) and samples annotated as unrelated demonstrate distances compatible with a match (N=15). Most of the unexpected results involve samples from the TCGA-DLBC (Diffuse Large B-cell Lymphoma) project (complete list of samples in Table S15). Those samples were excluded from the study (Figure S2). The total numbers of samples included in the test is 18,309 (i.e. 167,600,582, pairs tested) with number of pairs expected to match equal to 12,383 (504 SNPs used; Probability mismatch match=0.1; Probability mismatch non-match=0.6; Standard deviations match=2; Standard deviations mismatch=4; Similar maximum threshold=0.13; Different minimum threshold=0.50.
As somatic copy number aberrations might affect the genotype of variants within the genomic stretch, we plotted the genotype distance of matching pairs against the genomic burden; Figure S2B shows the relationship between genomic burden and the distance. Figure S3C shows the distribution of the ethnicities inferred by EthSEQ (Romanel et al., 2017) (Carrot-Zhang et al., 2020) for the whole study cohort (European ethnicity (84%), African (9%), and East Asian (5%)) and for each tumor type (Table S1). Finally, as SNPs Minor Allele Frequencies might be different in different ethnicities and this could reflect in genotype distances, we stratified the distance of correctly paired samples by ethnicity (Figure S2D) and did not observe significant changes. Further, distances of non-matching samples are stratified by the ethnicity of the pair components (Figures S2D and S2E); the heatmap shows the median distance within each group. Intra-ethnicity comparisons have a lower median distance compared to inter-ethnicity ones.
Purity and ploidy correction of log2 ratios
Purity and ploidy estimation and log2 ratios correction have been performed using CLONETv2 (Prandi and Demichelis, 2019). Briefly, for each segment spanning a set of SNPs that are heterozygous in the individual under study (informative SNPs), a Beta value (i.e. an estimation of the fraction of reads equally representing the two parental alleles) is computed by comparing the observed distribution of the SNPs allelic fractions against a set of expected distributions that assume diploid genomes for non-transformed cells in the tumor sample. The tumor sample purity and ploidy are then inferred from the log2 ratios and the Beta values with error minimization approaches and segmented data are adjusted for tumor ploidy and purity. Finally, allele-specific copy number (asCN) are assigned to each segment. Complex and ambiguous samples have been manually inspected in the log2 ratio/Beta space and adjusted accordingly.
Allele-specific ploidy (asP) and other indices
Segmentation algorithms assign to each identified genomic segment s few values, including the log2 ratio (tumor over normal), rs∈, and the segment coordinates, therefore the length ws∈. The total copy number of s is defined as cn(s)=; further, the asCN of the genomic segment s is represented as a pair of real values (cnA(s), cnB(s)), where cn(s) = (cnA(s) + cnB(s)), with cnA(s) ≥ cnB(s)) by definition. We here implemented a measure that is proportional to the average amount of DNA per cell. Given a genome G defined by a set of segments s∈G, the allele-specific ploidy (asP) is defined as the weighted mean of the allele-specific copy number of the segments s in G, that is
Discretized asP identifies three classes: low asP when (asP(G)<1.5), for instance when at least half of the genome G retains only one allele; high asP when (asP(G) ≥2.5), for instance when half of the genome presents at least three copies; diploid otherwise. By definition, asP(G) range is [0,∞). A diploid cell without any CN aberration by definition has asP = 2.
Hereafter, we report the definition of six indexes related to the genomic status of tumor cells. In addition to allele-specific ploidy introduced in this study, the other indexes were introduced and/or used in recent landscape studies exploiting next-generation sequencing or high-density array data from human tumor samples. A set of examples to highlight the behavior of each measure in different genomic contexts is listed in Figure S4C. A direct comparison between asP and ABSOLUTE ploidy is shown in Figure S5. Despite the overall concordance between the measures, there is a fundamental difference in the calculation: whereby ABSOLUTE relies on modeling of karyotypes and, for ambiguous samples, it relies on the most common study cohort karyotypes, asP is instead calculated independently for each sample and the cohort composition does not contribute to the calculation.
-
1.
Median absolute deviation (MAD): MAD has been calculated as in (Mouliere et al., 2018). The median absolute deviation (MAD) statistics quantifies the spread of a distribution. In genomics, MAD is conventionally the median absolute deviation from copy number neutrality, computed as . MAD has been used extensively to normalize and improve the quality of genotype calling in array data (Mouliere et al., 2018). MAD ranges in the interval is [0,∞).
-
2.
Genomic burden (GB): The genomic burden (GB) has been calculated as in (Beroukhim et al., 2010). It is a measure of the quietness of the genome; it is defined as the percentage of a genome G that is not wild-type (i.e., number of alleles different from two). GB is equal to 0 when no SCNA is detected and equal to 1 when no wild type genomic segment is present. Triploid and tetraploid cells have genomic burden equal to 1. The Genomic burden range is [0,1].
-
3.
Whole genome doubling (WGD): Whole genome doubling (WGD) is computed with ABSOLUTE (Carter et al., 2012) from Affymetrix SNP 6.0 array data of tumor samples (Carter et al., 2012). Briefly, ABSOLUTE estimates sample purity and ploidy from segmented copy number data and pre-computed models of cancer karyotypes. WGD assumes values 0, 1, and 2, corresponding to no duplication event, one duplication event, and more than one duplication event, respectively.
-
4.
Aneuploidy score (Taylor et al., 2018) of a tumor sample is defined as the number of chromosome arms with “large” somatic copy number alterations (SCNA). For each chromosome arm, the size of the SCNA is computed by first applying Gaussian mixture model to cluster SCNAs with similar length and genomic location. Then, three classes were identified based on the percentage of chromosome arm covered by the nominated SCNA cluster: more than 80% (value +1), less than the 20% (value 0), and intermediate length (no call). The Aneuploidy score is the sum of the arm level values returned by the described procedure. As not all chromosomal arms are typically sequenced, the Aneuploidy score range is [0,39].
-
5.
Weighted genomic instability index (GII): The genomic instability index GII (Chin et al., 2007) was originally defined for Affymetrix SNP 6.0 array assay as the percentage of SNPs within aberrant copy number segments. Weighted GII (wGII) (Burrell et al., 2013) improves over GII to account for different chromosome sizes: GII computed for each chromosome and wGII is the mean over the 22 chromosomes.
We define weighted ploidy as:
Following (Bakhoum et al., 2018), we adapted wGII to whole exome sequencing data by:
computing the weighted ploidy plw (A) for each chromosome arm A
extracting the set of segments T⊆A such that t∈T iff |cn(t) - plw(A)|>0.5
calculating the GII of chromosome arm A as the fraction of A that differs from plw(A) as
The wGII of a genome G is the mean for each chromosome arm A in genome G. The wGII range is [0,1].
-
6.
Microsatellite instability (MSI): Microsatellites are short repeated DNA sequences. Microsatellite instability (MSI) implies a change in the length of the inherited microsatellites and it is usually associated to defects in the mismatch repair mechanism. The extent of MSI has been recently characterized in 39 TCGA cancer types (Bonneville et al., 2017). The MSI detection tool MANTIS (Kautto et al., 2017) distinguishes MSI positive (named MSI-high or MSI-H) from microsatellite stable tumor samples (MSI-stable or MSS).
In summary, asP is proportional to the total amount of DNA (e.g. equals to 2, 3, and 4 for diploid, triploid and tetraploid genomes, respectively), can measure the DNA quantity resulting from catastrophic events as chromothripsis, and reflects the difference between monoallelic gain or monoallelic loss of the same genomic fraction, as opposed to other genomic indexes (see Figure S4C).
Allele-specific copy number and SNV analyses
Somatic copy number levels are conventionally grouped into five classes (Cerami et al., 2012; Gao et al., 2013), deep or homozygous deletions, shallow or hemizygous deletions, wild type, gain (3 and 4 copies), and amplification (5+ copies), based on log2 ratio values from tumor over matched normal signals. However, this abstraction masks relevant allele-specific copy number features as diverse combinations of allele counts result in the same group; for instance, for all autosomal chromosomes, both wild type copy number (one copy per allele; cnA=1, cnB=1) and copy-neutral loss of heterozygosity (one allele lost and one allele duplicated; cnA=2, cnB=0) result in the conventional wild type class. To solve these ambiguities and to study the landscape of allele-specific signal in primary tumors, we extended the five-level classification to a ten-level allele-specific copy number classification (Figure 1B, Table S3). Specifically, in addition to CN-LOH (2,0), we introduced Gain-LOH (3,0;4,0), Amp-LOH (5+,0), Gain- Umb (3,1), Amp-Umb (4,1;4,2;5,1;5,2;5,3;…). Applied de facto thresholds for discretized log2 ratio (rs) upon CN adjustment for ploidy and purity were as follows: if if if ; if ; if
Similarly, the contribution of SNVs to tumor evolution needs to account for underlying allelic imbalance whereby for an SNV at position p, allele-specific information about the genomic segment spanning p informs the number of genomic alleles that harbor the alternative base (Prandi and Demichelis, 2019). SNVs annotated in OnkoKB (Chakravarty et al., 2017) (version 1.19 patch 1) have been used to classify mutations as loss-of-function (LOF) and gain-of-function (GOF). For TP53 SNVs the “Transactivation Class” annotation of the IARC TP53 database (R20, (Bouaoun et al., 2016)) has been used. The current study uses TCGA WES data and does not utilize the matched SNP arrays. On one hand, this choice allows for the study expansion to other WES data cohorts and exploits the higher coverage with respect to WGS data. Allele-specific copy number and SNVs statuses of genes are annotated using the gene model reported in Table S17.
Dimensionality reduction and clustering
Uniform Manifold Approximation and Projection (UMAP) dimensionality reduction algorithm (McInnes and Healy, 2018) and fast Principal Component Analysis (fPCA) (Zheng et al., 2017) were applied on copy number allele-specific data to look for similarities within and across tumor types (Figure S9; Table S4). Genomic segments that lacked allele-specific cnA and cnB status (due to coverage and/or informative SNPs restrictions) were first assigned a proxy value via interpolation. Briefly, given a genomic segment g with undefined allele-specific copy number, we identified the nearest 3’ and 5’, g3 and g5, with defined allele-specific copy number and assigned to g the mean of the allele-specific copy number of g3 and g5, weighed by the length of g3 and g5. To remove the asP effect from UMAP analysis and to characterize the allele-specific profile of each tumor sample (Figures S9A and S9B), we first applied fPCA to allele-specific copy number data and then we applied UMAP to all but the first fPCA component allele-specific data. As input for UMAP analysis we used continuous allele A and B (allele A and B corresponding to the allele present with more and less copies, respectively) copy values at gene level. Continuous values from bulk DNA analysis allow for subclonal events signal (together with uncertainty around the estimates). Finally, we identified clusters of tumor samples with similar allele-specific copy number profiles utilizing the Density-Based Spatial Clustering of Applications with Noise (DBSCAN) algorithm (Ester et al., 1996). DBSCAN groups together samples that are close (reachability distance and reachability minimum number of points, set to 0.24 and 15) in the UMAP space, while isolated samples result as outliers. Aberration enrichment analysis in DBSCAN clusters is performed with two tailed Fisher's exact test.
To measure the stability of clusters, after dimensionality reduction (UMAP), we randomly subdivided diploid samples into ten groups. We then ran the clustering algorithm (DBSCAN) while excluding each group alternatively for a total of ten runs. As a measure of stability, we used the Adjusted Rand Index (ARI) (https://doi.org/10.2307/2284239), a rescaled version of the Rand Index. Figure S8 shows the Adjusted Rand Index computed on each subset of samples. To calculate the measure, we excluded, from the original clustering, the points that were removed in each fold. As clearly visible from the figure, the values are very high (median: 0.9895, SD: 8.9e-3), suggesting that our original clustering is indeed stable. Moreover, if we compare the original clustering to a version where the labels are randomly shuffled, we obtain very low ARI values (median: 0.02175, SD: 1.6e-3; Figure S8B).
Association of LOH with gene expression
The matched expression data was downloaded from recount2 project (Collado-Torres et al., 2017). Genomic and transcriptomic data were matched using the case_id provided by the NCI Genomic Data Commons (GDC) (Grossman et al., 2016) (Table S1). Recount2 expression data were normalized with function scale_counts of R package recount using default parameters. To estimate the impact of LOH on gene expression we built a linear model for each gene in each TCGA study , using the copy number (CN, defined as the sum of copies of alleles A and B) and the presence of LOH as variables. We retained genes for which the model returned β < 0 and statistical significance for the LOH variable (fdr <0.05). Coefficient of association for CN and LOH are calculated by dividing β by the standard error.
Only genes with mean expression 20 and at least 10 events of LOH in each specific cancer type were tested. To calculate enrichment for LOH impact on expression in classes of genes (TSG, OG, ESSENTIAL and OTHER) we considered genes with < 0 and significance for LOH variable (fdr < 0.05) in at least two tumor types and performed independence test (Chi-squared test).
In order to take into account expression due to non-cancer cells (1-purity), the ratio of aberrant asCN (Hemi del, CN-LOH, Gain-LOH and Amp-LOH) with respect to WT asCN, was calculated for each gene as follows:
Functional annotation analysis was performed using ClusterProfiler (Yu et al., 2012)(Table S13). To reduce the redundancy and improve visualization of GO biological terms, we clustered the significant ones (fdr<0.1) in at least two tumor types in the semantic space, based on the Resnick distance (arXiv:cmp-lg/9511007), using ReviGO (Supek et al., 2011). Only terms with dispensability < 0.2 are labelled with text in Figure S13B.
Synthetic data used in Figures 4A and S13A are generated as follows: for each asCN class we generated a vector of n=100 normally distributed random numbers. The mean of each distribution is defined based on the expected level for that asCN, with mean of WT = 1. For instance, for Hemi-del we expect half the expression in respect to WT, so mean of Hemi-del = 0.5. These data have been used exclusively to generate Figures 4A and S13A and have no impact on the calculation of the linear models or any other result.
Gene signatures analysis
The selection of the gene signatures was hypothesis-driven; gene signatures were obtained from the literature (Table S18). Each signature was tested in each study comparing high asP and diploid samples using Mann-Whitney test (p<0.05, one-sided based on biological hypothesis). Hierarchical clustering was performed using Pearson’s correlation as distance applying hierarchical clustering algorithm with complete linkage. In this context, focal copy number events were defined spanning genomic sizes shorter than 25% of the chromosomal arm and with CN deviation of at least 0.5 from the average arm CN signal.
Tumor suppressor genes and Oncogenes lists
Lists of TSGs and OG were obtained from Futreal and Zhao publications (Futreal et al., 2004; Zhao et al., 2016). For TSGs, only genes present in both lists were kept (Table S20).
TP53 status analysis
asCN calls of TP53 were obtained through the SPICE pipeline. Proportions of asCN and SNV states were calculated and plotted using the mosaic function from the vcd package (Meyer D, Zeileis A, Hornik K (2020). vcd: Visualizing Categorical Data. R package version 1.4-7) and graphically adapted for the figure.
Survival Analysis
Univariate and multivariate analysis were performed using the survival (http://CRAN.R-project.org/package=survival) and survminer (https://CRAN.R-project.org/package=survminer) packages for R. Proportional hazard regression models were calculated using type of tumor (study, reference=BRCA) and genes genomic status (reference as wt_0, 0=SNV absent, 1=SNV present) as predictor variables and progression free interval (PFI) as response. For TSG and OG, for each gene, genomic status values were considered only if in the number of events is at least 10 in the cohort.
Significant variables (fdr<0.05) in the univariate analysis were used in the multivariate analysis. Forest plot, survival curves and Kaplan-Meier estimator were calculated and plotted using the survminer package.
Quantification and statistical analysis
Statistical tests applied throughout the study are specified in results, figure legends, and in the methods accordingly.
Acknowledgments
We thank current and previous members of the Demichelis laboratory and Mark A Rubin for fruitful discussions. This project received funding from the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (grant agreement no. 648670; SPICE) to F.D., from the Italian Ministry of University and Research (FARE Program) to F.D., from the National Cancer Institute (SPORE P50-CA211024) to F.D., from the AIRC Foundation under MFAG 2019 - ID. 23560, and Giovanni Armenise-Harvard Foundation (CDA 2017) to L.L.F.
Author contributions
Y.C., T.F., D.P., and F.D. conceived the study. Y.C., T.F., D.P., A.L., F.L., and P.G. designed and/or performed data analysis. G.M.F., M.B., O.E., L.L.F., and A.I. contributed to data discussion and interpretation. All authors participated in editing or reviewing of the manuscript, and all authors approved the submitted manuscript. F.D. supervised the work.
Declaration of interests
The authors declare no competing interests. Co-authors D.P. and A.L. are currently at Fondazione Bruno Kessler, Trento, Italy and Biotechnology Research and Innovation Center, University of Copenhagen, Denmark.
Published: November 2, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.cels.2021.10.001.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. Processed data have been deposited at 10.5281/zenodo.5266542 and are publicly available as of the date of publication. Accession numbers of analyzed data and DOI of processed data are listed in the key resources table.
-
•
Original source code for data processing is publicly available; DOIs are listed in the key resources table. The code implemented to generate the figures is available from the lead contact upon request.
-
•
Any additional information required to reproduce this work is available from the lead contact.
References
- Amstutz P., Crusoe M.R., Tijanić N., Chapman B., Chilton J., Heuer M., Kartashov A., Leehr D., Ménager H., Nedeljkovich M., et al. 2016. Common Workflow Language, v1.0. figshare. [DOI] [Google Scholar]
- Bakhoum S.F., Ngo B., Laughney A.M., Cavallo J.-A., Murphy C.J., Ly P., Shah P., Sriram R.K., Watkins T.B.K., Taunk N.K., et al. Chromosomal instability drives metastasis through a cytosolic DNA response. Nature. 2018;553:467–472. doi: 10.1038/nature25432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A.C., Korkut A., Kanchi R.S., Hegde A.M., Lenoir W., Liu W., Liu Y., Fan H., Shen H., Ravikumar V., et al. A comprehensive pan-cancer molecular study of gynecologic and breast cancers. Cancer Cell. 2018;33:690–705.e9. doi: 10.1016/j.ccell.2018.03.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroukhim R., Mermel C.H., Porter D., Wei G., Raychaudhuri S., Donovan J., Barretina J., Boehm J.S., Dobson J., Urashima M., et al. The landscape of somatic copy-number alteration across human cancers. Nature. 2010;463:899–905. doi: 10.1038/nature08822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielski C.M., Donoghue M.T.A., Gadiya M., Hanrahan A.J., Won H.H., Chang M.T., Jonsson P., Penson A.V., Gorelick A., Harris C., et al. Widespread selection for oncogenic mutant allele imbalance in cancer. Cancer Cell. 2018;34:852–862.e4. doi: 10.1016/j.ccell.2018.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bielski C.M., Zehir A., Penson A.V., Donoghue M.T.A., Chatila W., Armenia J., Chang M.T., Schram A.M., Jonsson P., Bandlamudi C., et al. Genome doubling shapes the evolution and prognosis of advanced cancers. Nat. Genet. 2018;50:1189–1195. doi: 10.1038/s41588-018-0165-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonneville R., Krook M.A., Kautto E.A., Miya J., Wing M.R., Chen H.-Z., Reeser J.W., Yu L., Roychowdhury S. Landscape of microsatellite instability across 39 cancer types. JCO Precis. Oncol. 2017;2017 doi: 10.1200/PO.17.00073. PO.17.00073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouaoun L., Sonkin D., Ardin M., Hollstein M., Byrnes G., Zavadil J., Olivier M. TP53 variations in human cancers: new lessons from the IARC TP53 database and genomics data. Hum. Mutat. 2016;37:865–876. doi: 10.1002/humu.23035. [DOI] [PubMed] [Google Scholar]
- Buchwald Z.S., Tian S., Rossi M., Smith G.H., Switchenko J., Hauenstein J.E., Moreno C.S., Press R.H., Prabhu R.S., Zhong J., et al. Genomic copy number variation correlates with survival outcomes in WHO grade IV glioma. Sci. Rep. 2020;10:7355. doi: 10.1038/s41598-020-63789-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell R.A., McClelland S.E., Endesfelder D., Groth P., Weller M.-C., Shaikh N., Domingo E., Kanu N., Dewhurst S.M., Gronroos E., et al. Replication stress links structural and numerical cancer chromosomal instability. Nature. 2013;494:492–496. doi: 10.1038/nature11935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrot-Zhang J., Chambwe N., Damrauer J.S., Knijnenburg T.A., Robertson A.G., Yau C., Zhou W., Berger A.C., Huang K.-L., Newberg J.Y., et al. Comprehensive analysis of genetic ancestry and its molecular correlates in cancer. Cancer Cell. 2020;37:639–654.e6. doi: 10.1016/j.ccell.2020.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carter S.L., Cibulskis K., Helman E., McKenna A., Shen H., Zack T., Laird P.W., Onofrio R.C., Winckler W., Weir B.A., et al. Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol. 2012;30:413–421. doi: 10.1038/nbt.2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami E., Gao J., Dogrusoz U., Gross B.E., Sumer S.O., Aksoy B.A., Jacobsen A., Byrne C.J., Heuer M.L., Larsson E., et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–404. doi: 10.1158/2159-8290.CD-12-0095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakravarty D., Gao J., Phillips S.M., Kundra R., Zhang H., Wang J., Rudolph J.E., Yaeger R., Soumerai T., Nissan M.H., et al. OncoKB: a precision oncology knowledge base. JCO Precis. Oncol. 2017;2017 doi: 10.1200/PO.17.00011. PO.17.00011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chin S.F., Teschendorff A.E., Marioni J.C., Wang Y., Barbosa-Morais N.L., Thorne N.P., Costa J.L., Pinder S.E., van de Wiel M.A., Green A.R., et al. High-resolution aCGH and expression profiling identifies a novel genomic subtype of ER negative breast cancer. Genome Biol. 2007;8:R215. doi: 10.1186/gb-2007-8-10-r215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collado-Torres L., Nellore A., Kammers K., Ellis S.E., Taub M.A., Hansen K.D., Jaffe A.E., Langmead B., Leek J.T. Reproducible RNA-seq analysis using recount2. Nat. Biotechnol. 2017;35:319–321. doi: 10.1038/nbt.3838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielsen H.E., Pradhan M., Novelli M. Revisiting tumour aneuploidy - the place of ploidy assessment in the molecular era. Nat. Rev. Clin. Oncol. 2016;13:291–304. doi: 10.1038/nrclinonc.2015.208. [DOI] [PubMed] [Google Scholar]
- Davoli T., Xu A.W., Mengwasser K.E., Sack L.M., Yoon J.C., Park P.J., Elledge S.J. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell. 2013;155:948–962. doi: 10.1016/j.cell.2013.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demichelis F., Greulich H., Macoska J.A., Beroukhim R., Sellers W.R., Garraway L., Rubin M.A. SNP panel identification assay (SPIA): a genetic-based assay for the identification of cell lines. Nucleic Acids Res. 2008;36:2446–2456. doi: 10.1093/nar/gkn089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng G., Lu Y., Zlotnikov G., Thor A.D., Smith H.S. Loss of heterozygosity in normal tissue adjacent to breast carcinomas. Science. 1996;274:2057–2059. doi: 10.1126/science.274.5295.2057. [DOI] [PubMed] [Google Scholar]
- Elango R., Sheng Z., Jackson J., DeCata J., Ibrahim Y., Pham N.T., Liang D.H., Sakofsky C.J., Vindigni A., Lobachev K.S., et al. Break-induced replication promotes formation of lethal joint molecules dissolved by Srs2. Nat. Commun. 2017;8:1790. doi: 10.1038/s41467-017-01987-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ester M., Kriegel H.-P., Sander J., Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. Kdd, Conference Proceedings. 1996;96:226–231. [Google Scholar]
- Futreal P.A., Coin L., Marshall M., Down T., Hubbard T., Wooster R., Rahman N., Stratton M.R. A census of human cancer genes. Nat. Rev. Cancer. 2004;4:177–183. doi: 10.1038/nrc1299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J., Aksoy B.A., Dogrusoz U., Dresdner G., Gross B., Sumer S.O., Sun Y., Jacobsen A., Sinha R., Larsson E., et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 2013;6:pl1. doi: 10.1126/scisignal.2004088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ged Y., Chaim J.L., DiNatale R.G., Knezevic A., Kotecha R.R., Carlo M.I., Lee C.-H., Foster A., Feldman D.R., Teo M.Y., et al. DNA damage repair pathway alterations in metastatic clear cell renal cell carcinoma and implications on systemic therapy. J. Immunother. Cancer. 2020;8:e000230. doi: 10.1136/jitc-2019-000230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogna R., Madan E., Kuppusamy P., Pati U. Chaperoning of mutant p53 protein by wild-type p53 protein causes hypoxic tumor regression. J. Biol. Chem. 2012;287:2907–2914. doi: 10.1074/jbc.M111.317354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A., Staudt L.M. Toward a shared vision for cancer genomic data. N. Engl. J. Med. 2016;375:1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagenkord J.M., Monzon F.A., Kash S.F., Lilleberg S., Xie Q., Kant J.A. Array-based karyotyping for prognostic assessment in chronic lymphocytic leukemia: performance comparison of Affymetrix 10K2.0, 250K nsp, and SNP6.0 arrays. J. Mol. Diagn. 2010;12:184–196. doi: 10.2353/jmoldx.2010.090118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heaphy C.M., Subhawong A.P., Hong S.-M., Goggins M.G., Montgomery E.A., Gabrielson E., Netto G.J., Epstein J.I., Lotan T.L., Westra W.H., et al. Prevalence of the alternative lengthening of telomeres telomere maintenance mechanism in human cancer subtypes. Am. J. Pathol. 2011;179:1608–1615. doi: 10.1016/j.ajpath.2011.06.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinchcliffe E.H., Day C.A., Karanjeet K.B., Fadness S., Langfald A., Vaughan K.T., Dong Z. Chromosome missegregation during anaphase triggers p53 cell cycle arrest through histone H3.3 Ser31 phosphorylation. Nat. Cell Biol. 2016;18:668–675. doi: 10.1038/ncb3348. [DOI] [PubMed] [Google Scholar]
- Hoff A.M., Kraggerud S.M., Alagaratnam S., Berg K.C.G., Johannessen B., Høland M., Nilsen G., Lingjærde O.C., Andrews P.W., Lothe R.A., et al. Frequent copy number gains of SLC2A3 and ETV1 in testicular embryonal carcinomas. Endocr. Relat. Cancer. 2020;27:457–468. doi: 10.1530/ERC-20-0064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollstein M., Sidransky D., Vogelstein B., Harris C.C. p53 mutations in human cancers. Science. 1991;253:49–53. doi: 10.1126/science.1905840. [DOI] [PubMed] [Google Scholar]
- ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium Pan-cancer analysis of whole genomes. Nature. 2020;578:82–93. doi: 10.1038/s41586-020-1969-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janic A., Valente L.J., Wakefield M.J., Di Stefano L., Milla L., Wilcox S., Yang H., Tai L., Vandenberg C.J., Kueh A.J., et al. DNA repair processes are critical mediators of p53-dependent tumor suppression. Nat. Med. 2018;24:947–953. doi: 10.1038/s41591-018-0043-5. [DOI] [PubMed] [Google Scholar]
- Karaayvaz-Yildirim M., Silberman R.E., Langenbucher A., Saladi S.V., Ross K.N., Zarcaro E., Desmond A., Yildirim M., Vivekanandan V., Ravichandran H., et al. Aneuploidy and a deregulated DNA damage response suggest haploinsufficiency in breast tissues of BRCA2 mutation carriers. Sci. Adv. 2020;6:eaay2611. doi: 10.1126/sciadv.aay2611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kautto E.A., Bonneville R., Miya J., Yu L., Krook M.A., Reeser J.W., Roychowdhury S. Performance evaluation for rapid detection of pan-cancer microsatellite instability with MANTIS. Oncotarget. 2017;8:7452–7463. doi: 10.18632/oncotarget.13918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klusmann I., Rodewald S., Müller L., Friedrich M., Wienken M., Li Y., Schulz-Heddergott R., Dobbelstein M. p53 activity results in DNA replication fork processivity. Cell Rep. 2016;17:1845–1857. doi: 10.1016/j.celrep.2016.10.036. [DOI] [PubMed] [Google Scholar]
- Lejonklou M.H., Barbu A., Stålberg P., Skogseid B. Accelerated proliferation and differential global gene expression in pancreatic islets of five-week-old heterozygous Men1 mice: Men1 is a haploinsufficient suppressor. Endocrinology. 2012;153:2588–2598. doi: 10.1210/en.2011-1924. [DOI] [PubMed] [Google Scholar]
- Levin A., Minis A., Lalazar G., Rodriguez J., Steller H. PSMD5 inactivation promotes 26S proteasome assembly during colorectal tumor progression. Cancer Res. 2018;78:3458–3468. doi: 10.1158/0008-5472.CAN-17-2296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M., Fang X., Baker D.J., Guo L., Gao X., Wei Z., Han S., van Deursen J.M., Zhang P. The ATM-p53 pathway suppresses aneuploidy-induced tumorigenesis. Proc. Natl. Acad. Sci. USA. 2010;107:14188–14193. doi: 10.1073/pnas.1005960107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li S., Balmain A., Counter C.M. A model for RAS mutation patterns in cancers: finding the sweet spot. Nat. Rev. Cancer. 2018;18:767–777. doi: 10.1038/s41568-018-0076-6. [DOI] [PubMed] [Google Scholar]
- Locallo A., Prandi D., Fedrizzi T., Demichelis F. TPES: tumor purity estimation from SNVs. Bioinformatics. 2019;35:4433–4435. doi: 10.1093/bioinformatics/btz406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes L., Healy J. arXiv; 2018. UMAP: uniform manifold approximation and projection for dimension reduction.https://arxiv.org/abs/1802.03426 [Google Scholar]
- McInnes L., Healy J., Melville J. UMAP: uniform manifold approximation and projection for dimension reduction. arXiv. 2018 https://arxiv.org/abs/1802.03426 [Google Scholar]
- McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R., Thormann A., Flicek P., Cunningham F. The Ensembl variant effect predictor. Genome Biol. 2016;17:122. doi: 10.1186/s13059-016-0974-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midgley C.A., Lane D.P. p53 protein stability in tumour cells is not determined by mutation but is dependent on Mdm2 binding. Oncogene. 1997;15:1179–1189. doi: 10.1038/sj.onc.1201459. [DOI] [PubMed] [Google Scholar]
- Mouliere F., Chandrananda D., Piskorz A.M., Moore E.K., Morris J., Ahlborn L.B., Mair R., Goranova T., Marass F., Heider K., et al. Enhanced detection of circulating tumor DNA by fragment size analysis. Sci. Transl. Med. 2018;10:eaat4921. doi: 10.1126/scitranslmed.aat4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulcahy Levy J.M., Thorburn A. Autophagy in cancer: moving from understanding mechanism to improving therapy responses in patients. Cell Death Differ. 2020;27:843–857. doi: 10.1038/s41418-019-0474-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols C.A., Gibson W.J., Brown M.S., Kosmicki J.A., Busanovich J.P., Wei H., Urbanski L.M., Curimjee N., Berger A.C., Gao G.F., et al. Loss of heterozygosity of essential genes represents a widespread class of potential cancer vulnerabilities. Nat. Commun. 2020;11:2517. doi: 10.1038/s41467-020-16399-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivier M., Hollstein M., Hainaut P. TP53 mutations in human cancers: origins, consequences, and clinical use. Cold Spring Harb. Perspect. Biol. 2010;2:a001008. doi: 10.1101/cshperspect.a001008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlandini V., Provenzano A., Giglio S., Magi A. SLMSuite: a suite of algorithms for segmenting genomic profiles. BMC Bioinformatics. 2017;18:321. doi: 10.1186/s12859-017-1734-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Persi E., Wolf Y.I., Horn D., Ruppin E., Demichelis F., Gatenby R.A., Gillies R.J., Koonin E.V. Mutation-selection balance and compensatory mechanisms in tumour evolution. Nat. Rev. Genet. 2021;22:251–262. doi: 10.1038/s41576-020-00299-4. [DOI] [PubMed] [Google Scholar]
- Petitjean A., Achatz M.I., Borresen-Dale A.L., Hainaut P., Olivier M. TP53 mutations in human cancers: functional selection and impact on cancer prognosis and outcomes. Oncogene. 2007;26:2157–2165. doi: 10.1038/sj.onc.1210302. [DOI] [PubMed] [Google Scholar]
- Pfister K., Pipka J.L., Chiang C., Liu Y., Clark R.A., Keller R., Skoglund P., Guertin M.J., Hall I.M., Stukenberg P.T. Identification of drivers of aneuploidy in breast tumors. Cell Rep. 2018;23:2758–2769. doi: 10.1016/j.celrep.2018.04.102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prandi D., Demichelis F. Ploidy- and purity-adjusted allele-specific DNA analysis using CLONETv2. Curr. Protoc. Bioinformatics. 2019;67:e81. doi: 10.1002/cpbi.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanel A., Zhang T., Elemento O., Demichelis F. EthSEQ: ethnicity annotation from whole exome sequencing data. Bioinformatics. 2017;33:2402–2404. doi: 10.1093/bioinformatics/btx165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santaguida S., Vasile E., White E., Amon A. Aneuploidy-induced cellular stresses limit autophagic degradation. Genes Dev. 2015;29:2010–2021. doi: 10.1101/gad.269118.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheltzer J.M. A transcriptional and metabolic signature of primary aneuploidy is present in chromosomally unstable cancer cells and informs clinical prognosis. Cancer Res. 2013;73:6401–6412. doi: 10.1158/0008-5472.CAN-13-0749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen R., Seshan V.E. FACETS: allele-specific copy number and clonal heterogeneity analysis tool for high-throughput DNA sequencing. Nucleic Acids Res. 2016;44:e131. doi: 10.1093/nar/gkw520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solimini N.L., Xu Q., Mermel C.H., Liang A.C., Schlabach M.R., Luo J., Burrows A.E., Anselmo A.N., Bredemeyer A.L., Li M.Z., et al. Recurrent hemizygous deletions in cancers may optimize proliferative potential. Science. 2012;337:104–109. doi: 10.1126/science.1219580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto M., Raaijmakers J.A., Bakker B., Spierings D.C.J., Lansdorp P.M., Foijer F., Medema R.H. p53 prohibits propagation of chromosome segregation errors that produce structural aneuploidies. Cell Rep. 2017;19:2423–2431. doi: 10.1016/j.celrep.2017.05.055. [DOI] [PubMed] [Google Scholar]
- Stingele S., Stoehr G., Peplowska K., Cox J., Mann M., Storchova Z. Global analysis of genome, transcriptome and proteome reveals the response to aneuploidy in human cells. Mol. Syst. Biol. 2012;8:608. doi: 10.1038/msb.2012.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Supek F., Bošnjak M., Škunca N., Šmuc T. REVIGO summarizes and visualizes long lists of gene ontology terms. PLoS One. 2011;6:e21800. doi: 10.1371/journal.pone.0021800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talevich E., Shain A.H., Botton T., Bastian B.C. CNVkit: genome-wide copy number detection and visualization from targeted DNA sequencing. PLoS Comput. Biol. 2016;12:e1004873. doi: 10.1371/journal.pcbi.1004873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor A.M., Shih J., Ha G., Gao G.F., Zhang X., Berger A.C., Schumacher S.E., Wang C., Hu H., Liu J., et al. Genomic and functional approaches to understanding cancer aneuploidy. Cancer Cell. 2018;33:676–689.e3. doi: 10.1016/j.ccell.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terzian T., Suh Y.-A., Iwakuma T., Post S.M., Neumann M., Lang G.A., Van Pelt C.S., Lozano G. The inherent instability of mutant p53 is alleviated by Mdm2 or p16INK4a loss. Genes Dev. 2008;22:1337–1344. doi: 10.1101/gad.1662908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson S.L., Compton D.A. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J. Cell Biol. 2010;188:369–381. doi: 10.1083/jcb.200905057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorsson V., Gibbs D.L., Brown S.D., Wolf D., Bortone D.S., Ou Yang T.-H., Porta-Pardo E., Gao G.F., Plaisier C.L., Eddy J.A., et al. The immune landscape of cancer. Immunity. 2018;48:812–830.e14. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentini S., Fedrizzi T., Demichelis F., Romanel A. PaCBAM: fast and scalable processing of whole exome and targeted sequencing data. BMC Genomics. 2019;20:1018. doi: 10.1186/s12864-019-6386-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakumaran R., Tan K.H., Miranda P.J., Haupt S., Haupt Y. Regulation of mutant p53 protein expression. Front. Oncol. 2015;5:284. doi: 10.3389/fonc.2015.00284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walerych D., Lisek K., Sommaggio R., Piazza S., Ciani Y., Dalla E., Rajkowska K., Gaweda-Walerych K., Ingallina E., Tonelli C., et al. Proteasome machinery is instrumental in a common gain-of-function program of the p53 missense mutants in cancer. Nat. Cell Biol. 2016;18:897–909. doi: 10.1038/ncb3380. [DOI] [PubMed] [Google Scholar]
- Walerych D., Pruszko M., Zyla L., Wezyk M., Gaweda-Walerych K., Zylicz A. Wild-type p53 oligomerizes more efficiently than p53 hot-spot mutants and overcomes mutant p53 gain-of-function via a "dominant-positive" mechanism. Oncotarget. 2018;9:32063–32080. doi: 10.18632/oncotarget.25944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang X., Chemmama I.E., Yu C., Huszagh A., Xu Y., Viner R., Block S.A., Cimermancic P., Rychnovsky S.D., Ye Y., et al. The proteasome-interacting Ecm29 protein disassembles the 26S proteasome in response to oxidative stress. J. Biol. Chem. 2017;292:16310–16320. doi: 10.1074/jbc.M117.803619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson S., Harmon S.A., Terrigino N.T., Karzai F., Pinto P.A., Madan R.A., VanderWeele D.J., Lake R., Atway R., Bright J.R., et al. A case report of multiple primary prostate tumors with differential drug sensitivity. Nat. Commun. 2020;11:837. doi: 10.1038/s41467-020-14657-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yen H.-C.S., Xu Q., Chou D.M., Zhao Z., Elledge S.J. Global protein stability profiling in mammalian cells. Science. 2008;322:918–923. doi: 10.1126/science.1160489. [DOI] [PubMed] [Google Scholar]
- Yeung C.C.S., McElhone S., Chen X.Y., Ng D., Storer B.E., Deeg H.J., Fang M. Impact of copy neutral loss of heterozygosity and total genome aberrations on survival in myelodysplastic syndrome. Mod. Pathol. 2018;31:569–580. doi: 10.1038/modpathol.2017.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G., Wang L.-G., Han Y., He Q.-Y. clusterProfiler: an R package for comparing biological themes among gene clusters. Omics. 2012;16:284–287. doi: 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zack T.I., Schumacher S.E., Carter S.L., Cherniack A.D., Saksena G., Tabak B., Lawrence M.S., Zhsng C.-Z., Wala J., Mermel C.H., et al. Pan-cancer patterns of somatic copy number alteration. Nat. Genet. 2013;45:1134–1140. doi: 10.1038/ng.2760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M., Kim P., Mitra R., Zhao J., Zhao Z. TSGene 2.0: an updated literature-based KnowledgeBase for tumor suppressor genes. Nucleic Acids Res. 2016;44:D1023–D1031. doi: 10.1093/nar/gkv1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng G., Li S., Szekely G. Elsevier; 2017. Statistical Shape And Deformation Analysis: Methods Implementation and Applications; pp. 1–508. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. Processed data have been deposited at 10.5281/zenodo.5266542 and are publicly available as of the date of publication. Accession numbers of analyzed data and DOI of processed data are listed in the key resources table.
-
•
Original source code for data processing is publicly available; DOIs are listed in the key resources table. The code implemented to generate the figures is available from the lead contact upon request.
-
•
Any additional information required to reproduce this work is available from the lead contact.