

Abstract
In a recent report in Science Signaling (Gillis, A., et al. Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signaling 2020, 13, eaaz3140 10.1126/scisignal.aaz3140), it was suggested that low intrinsic agonism, and not biased agonism, leads to an improvement in the separation of potency in opioid-induced respiratory suppression versus antinociception. Although many of the compounds that were tested have been shown to display G protein signaling bias in prior publications, the authors conclude that because they cannot detect biased agonism in their cellular signaling studies the compounds are therefore not biased agonists. Rather, they conclude that it is low intrinsic efficacy that leads to the therapeutic window improvement. Intrinsic efficacy is the extent to which an agonist can stimulate a G protein-coupled receptor response in a system, while biased agonism takes into consideration not only the intrinsic efficacy but also the potency of an agonist in an assay. Herein, we have reanalyzed the data presented in the published work (10.1126/scisignal.aaz3140) [including the recent Erratum (10.1126/scisignal.abf9803)] to derive intrinsic efficacy and bias factors as ΔΔlog(τ/KA) and ΔΔlog(Emax/EC50), respectively. On the basis of this reanalysis, the data support the conclusion that biased agonism, favoring G protein signaling, was observed. Moreover, a conservation of rank order intrinsic efficacy was not observed upon comparing responses in each assay, further suggesting that multiple active receptor states were present. These observations agree with prior studies in which oliceridine, PZM21, and SR-17018 were first described as biased agonists with improvement in antinociception over respiratory suppression in mice. Therefore, the data in the Science Signaling paper provide strong corroborating evidence that G protein signaling bias may be a means of improving opioid analgesia while avoiding certain undesirable side effects.
Graphical Abstract
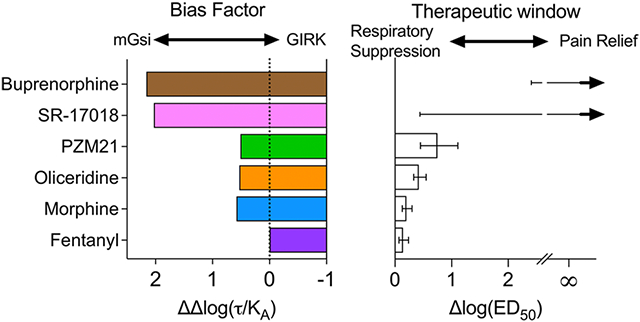
In the March 31, 2020, issue of Science Signaling, a research article by Gillis et al.1,2 (hereafter termed the SS-manuscript) investigated a series of mu opioid agonists for activity in a compendium of in vitro cell-based bioluminescence resonance energy transfer (BRET) studies (summarized in Table 1) to evaluate the pharmacological basis of functional selectivity. In particular, the authors focused on two discrete avenues of response: those medicated by G protein signaling or βarrestin2 recruitment. The SS-manuscript also included studies in mice that were intended to compare the therapeutic window of the selected compounds by determining potency in agonist-induced antinociception (hot-plate latency) and respiratory suppression (respiratory frequency) measures. These physiological responses are relevant as they reflect a similar relationship to the therapeutic window observed in clinical use of known opioids. While the study confirmed that the selected biased agonists showed an improved therapeutic window, the authors failed to detect biased agonism in their signaling assays and concluded that intrinsic efficacy and not biased agonism was responsible for the improved therapeutic window.1
Table 1.
Assay Abbreviations from the SS-Manuscript1
| assay | measure | effector | MOR | cells | indicator |
|---|---|---|---|---|---|
| Nb33 | Giα nanobody recruitment | Nb33-venus | flag-mMOR-NLuc | transient HEK293 | BRET |
| mGsi | mini-soluble Gi protein recruitment | NES-venus-mGsi | flag-mMOR-NLuc | transient HEK293 | BRET |
| Gai2 | association of Gβ with GRK3ct and dissociation from Gαi2 | Gαi2 venus-155–239-Gβ1 venus-1–155-Gγ2 |
flag-mMOR-NLuc | transient HEK293 | BRET |
| cAMP | cAMP biosensor | CAMYEL YFP-Epac-Rluc | flag-mMOR | transient HEK293 | BRET |
| βarr2+GRK2 | βarrestin2 recruitment with overexpressed GRK2 | βarr2-YFP (bovine) | flag-MOR-NLuc | transient HEK293 | BRET |
The pursuit of agonists that display G protein signaling over βarrestin2 recruitment was inspired by early observations of mice lacking βarrestin2, in which antinociception was enhanced and respiratory suppression was diminished in response to morphine.3-5 Several laboratories have developed such agonists, including PZM21,6 a series of Scripps Research “SR” compounds,7,8 a fungal natural product,9 derivatives of mitragynine,10 and, importantly, the first G protein signaling-biased MOR agonist, oliceridine (TRV-130, Olinvyk11), that has progressed to the clinic.12 Moreover, an improvement in producing antinociception with fewer signs of respiratory suppression has been seen for these compounds in rodents (with the exception of the fungal agonist that was not tested in vivo). Importantly, with the emerging clinical studies demonstrating an improvement in analgesic efficacy over respiratory suppression for oliceridine,13,14 it would seem that the early mouse studies were predictive of a useful strategy for improving opioid therapeutics by developing G protein signaling-biased agonists.
Determining whether a compound shows a preference for signaling in one pathway over another has proven to be less straightforward than anticipated. These measures are complicated by the use of overexpression systems in different cellular backgrounds, varying receptor levels, tags and modifications to the receptors and effectors to amplify the signal, and the assumption that a plethora of assays designed to detect G protein-mediated signaling are equal surrogates for that response. This leads to the assumption that the determination of potency (EC50) and efficacy (Emax) across a diverse array of “G protein signaling” assays will define the agonist’s ability to stimulate G proteins. In reality, the receptor number, signal amplification, and sensitivity of the assay will greatly affect the determination of potency and efficacy; however, if all agonists have an equivalent propensity to activate different pathways, then their rank order efficacy should be preserved (the most potent and efficacious agonist should remain so in all assays).15-17
In the SS-manuscript,1 it is proposed that ligand rank order intrinsic efficacy and not ligand bias is the driving factor behind the separation of antinociception from respiratory suppression of PZM21, oliceridine, buprenorphine, and SR-17018. The authors quantified “biased agonism” and agonist intrinsic efficacy across cellular signaling assays using the operational model originally described by Black and Leff.18 Notably, the authors reanalyzed efficacy (Emax) values from several recent papers (including PZM216 and SR-170188) to support the proposal that it is intrinsic efficacy, and not biased agonism, that leads to improvements in the therapeutic window.1,19,20 The conclusions of this report1 and the value of partial agonist efficacy have been the subject of a number of recent review papers.19-21
The operational model has become commonplace in studying agonist performance across a variety of experimental systems and serves as a means of quantifying biased agonism. The key feature that makes this model appealing is its capacity to produce a simplified correction for nonlinear occupancy–response relationships. The model employs a hyperbolic occupancy–response relationship and assumes that, at a partial level of agonist occupancy, some agonists are able to fully activate their effector proteins. The products of this analysis are two unique parameters, tau (τ) and KA. KA is an equilibrium affinity constant that describes the avidity by which an agonist–receptor complex is formed, while τ is an (intrinsic) efficacy parameter that describes how well the agonist–receptor complex can convey a signal to the effector protein. An easy way to understand the scale of τ is that when an agonist has an Emax of 50%, it will have a τ of 1.22 This can be realized by substitution of τ = 1 into eq 1, from Black and Leff:18
| (1) |
As the Emax of the agonist approaches 0, the value of τ will approach 0. For most experimental systems, however, τ will be a positive value, and because τ is positive, it can be expressed as log(τ), which will then extend across positive and negative numbers. Concern about this assumption could be warranted if any of the agonists inhibited the response below the baseline, but this was not the case in any assay herein.
In the SS-manuscript, the operational model is employed to calculate the intrinsic efficacy (τ) for several partial agonists to assess MOR engagement with G protein or other effectors relative to the 100% maximum system response produced by DAMGO (see Table 1 for the assay description; abbreviated names of the signaling assays will be used throughout). The resulting values for log(τ) (SS-Table 1), Emax (SS-Table 2), EC50 (SS-Table 3), and Δlog(τ/KA) (SS-Table S1) are presented in the SS-manuscript (for the sake of clarity, figures and tables from the SS-manuscript will be cited with the prefix SS, i.e., SS-Table 11). An Erratum2 included the following changes: SS-Table S1 [Δlog(τ/KA)] values for cAMP for all compounds were replaced, and buprenorphine values were added for βarr2 and βarr2 (+GRK2). SS-Table 3 (EC50) values for SR-17018 in βarr2 (+GRK2) were changed. No graphs or conclusions were changed in the Erratum. All analyses presented herein include the “Erratum” values.
ANALYSIS
Discrepancies between τ and Emax.
Because the primary conclusion of the manuscript is that intrinsic efficacy, and not agonist bias, correlates with an improved therapeutic window, it was reasonable to investigate these measures of τ and Emax. As noted, the τ value can be calculated from Emax, and vice versa (eq 11,18); therefore, the Emax values presented after conversion from SS-Table 1 (conversion using substitution into eq 1) were compared to the Emax values presented in SS-Table 2 in Figure 1. For this comparison, only partial agonists and only the assays that presented all of the values in both SS-Tables 1 and 2 were included. While morphine shows the expected consistency between the Emax derived from the two tables, there is considerably less agreement for other agonists.
Figure 1.
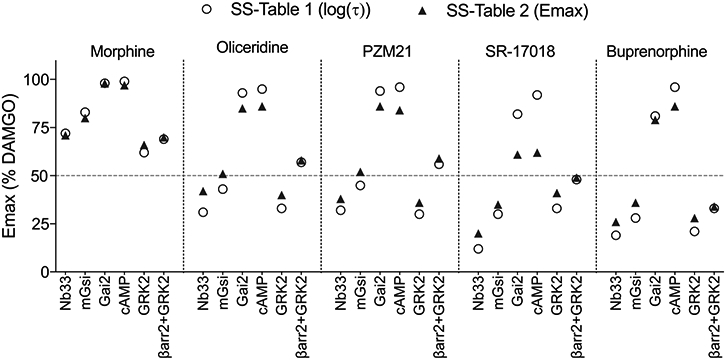
Emax is graphically presented from the values provided in SS-Table 2 in comparison to the Emax calculated from log(τ) (SS-Table 1) using eq 1.
Interestingly, the Emax is overestimated when derived from log(τ) and this is consistent for most of the amplified assays (Gαi2 and cAMP) while underestimated for the assays that most proximally detect G protein–receptor interaction (Nb33 and mGsi). The discrepancy is most pronounced for the data provided for SR-17018 where there is significant separation between the Emax prediction from τ (SS-Table 1) and the Emax estimate (SS-Table 2). Furthermore, it is remarkable that certain log(τ) values ± the standard error of the mean (SEM) in SS-Table 1 are negative. Specifically, while the Emax for oliceridine was reported as 51% for mGsi in SS-Table 2, a negative log(τ) value of −0.12 ± 0.05 was presented in SS-Table 1, which would require that the Emax be <50%, by definition. It should be mentioned that although the mean ± SEM does not overlap zero, this information is secondary to the primary Emax versus log(τ) comparison. In other words, a mean Emax of >50% cannot agree with a mean log(τ) of <0 regardless of any consideration of the standard error of the mean. Overall, these discrepancies raise questions about the utility of the determination of τ, as presented in SS-Table 1, for assigning rank order intrinsic efficacy.
The use of the operational model centrally assumes a nonlinear occupancy–response relationship. As such, an agonist response curve (potency) is expected to reside to the left of the agonist occupancy binding curve (affinity). That is, the agonist affinity is amplified by the system to produce the observed agonist EC50 in a given response. Concern is warranted when a hyperbolic occupancy–response function is employed to analyze what would be better described as a linear occupancy–response relationship, i.e., when there is minimal separation between the response and binding curve.23,20 Specifically, this can alter the way τ and KA relate to Emax and EC50 due to an inappropriate correction for system amplification. However, as τ and KA, and Emax and EC50, appear to result from systems with some amplification, this concern may not apply. As partial agonists were included in the analysis, an occupancy–response relationship (in a system with some amplification) is likely a close approximation; therefore, the calculations presented herein should be reasonable.
Potency Comparisons and Interpretations.
An agonist’s affinity for the receptor is expected to be a system-independent parameter and, by definition, constant. In systems in which the agonist is observed to produce a submaximal response, the agonist potency for the response is expected to be extremely close to that affinity constant. It is noteworthy that some partial agonists produced EC50 values that are orders of magnitude apart across assays (SS-Table 3 and Figure 2). In theory, the potency values should be much closer together. It is possible that ligand binding kinetic properties are leading to overestimates of potency in certain assays; this would also artificially affect the perception of bias.24 In the SS-manuscript, agonist association kinetics (on-rate) were measured as time-course studies of Nb33 and mGsi recruitment immediately after the addition of each agonist at a concentration of 10 μM (SS-Figure S2).1 In most cases, the agonist causes rapid recruitment of each BRET sensor. The one exception is SR-17018, which took 5–10 min to reach peak recruitment. Additionally, the authors measure the rate that agonist stimulation is lost in the same experiment in which, after treatment with 10 μM agonist, the cells were treated with 10 μM naloxone. The purpose of this experiment was to estimate the dissociation rate of the agonist as the high concentration of the antagonist would prevent any further agonist stimulation and loss of signal would reflect disintegration of the agonist–receptor complex.
Figure 2.

Graphic representation of potency values (pEC50) with the standard error of the mean from SS-Table 3. The red error bars indicate SEM values that are one-half log order or greater. The SS-manuscript states that means are the average of 3–14 experiments.
It is particularly interesting that naloxone reversal of agonist-stimulated mGsi was incomplete (compared to baseline), for all agonists tested, with the exception of SR-17018, which was completely reversed by naloxone (SS-Figure S2B). This would suggest that the dissociation of SR-17018 is extremely rapid and that association of SR-17018 plays a major role in reaching equilibrium. Furthermore, if the kinetics are delayed for the other agonists, this would lead to overestimates of potency for those compounds (including the reference agonist) for the mGsi assay, which was subsequently used for bias calculations and comparison to the therapeutic window. Indeed, if this were the case, then it is possible that all bias estimates are significantly underestimated due to lack of equilibrium and overestimation of agonist potency.24
Comparison of Efficacy (τ values) Derived from Provided Log(τ), Emax, and Log(τ/KA) Values.
For the purposes of bias analysis in the SS-manuscript, the authors focus on five specific responses [Nb33, mGsi, Gαi2 activation, cAMP, and βarrestin2 (+GRK2) recruitment] (see Table 1 for a description of the assays). βArrestin2 recruitment is measured in the presence of overexpressed GRK2 as the authors propose that GRK2 overexpression does not alter the analysis of bias (SS-Figure 9). The specific bias analysis that was employed is a reparameterization of the operational model that produces a transduction coefficient, log(τ/KA).25,26 When compared to a reference agonist, this efficacy and affinity integration can be used to normalize and consolidate agonist activity into a single composite parameter, namely the normalized transduction coefficient, Δlog(τ/KA). Many of the agonists investigated in the SS-manuscript were partial agonists, and both efficacy and affinity can be addressed by submaximal stimulation (<90%) at saturating agonist concentrations. In addition, the analysis used in the SS-manuscript assumes that the responses measured exhibit concentration–response relationships with a Hill slope constrained to one. As such, the authors state that the systems have minimal amplification due to the expected stoichiometric measurement of protein–protein interactions. Constraining the Hill slope to one can be appropriate as log(τ/KA) values have been shown to remain linear across a wide range of Hill slope values, including one.26 Simulated concentration–response curves using the parameter values listed in SS-Tables 2 and 3 reveal that the Hill slopes do not appear to be different from 1; therefore, the mathematical conversions we present should be generally correct (simulations are included for visual comparison to SS-Figures 1 and 2 in Figure S1).
When the normalized transduction coefficient is determined in the SS-manuscript, an estimate for agonist affinity (KA) is also produced. With the values of log(τ/KA) and KA known, τ and log(τ) can subsequently be determined by eqs 2 and 3:
| (2) |
| (3) |
A recent review article highlighted the relationship between the three (or four)-parameter equation and the values that result from the operational analysis.20 This follows from the parameter definition originally described by Black and Leff,18 in eq 1 (shown above) and eq 4:
| (4) |
It is possible to estimate KA using the EC50 values from SS-Table 3 by applying eq 5:
| (5) |
More interestingly, because log(τ/KA) was produced for a number of agonists in SS-Table S1, it is possible to solve for τ directly by using eq 6:
| (6) |
It should be pointed out that these equations are most appropriate for partial agonists as the Emax of a partial agonist is defined at 100% occupancy. Therefore, τ was estimated for only agonists that produced <90% efficacy wherein the maximum response could be achieved only at levels that saturate the available receptor population. Using the log(τ/KA) for the partial agonists (SS-Table S1), Emax (SS-Table 2), pEC50 (SS-Table 3), and τ can be independently determined; a comparison of these derivations of τ is shown in Figure 3. Immediately, it is evident that there is poor agreement among τ values derived from the three sources for the Gαi2 and cAMP measures, and this was least consistent for SR-17018.
Figure 3.
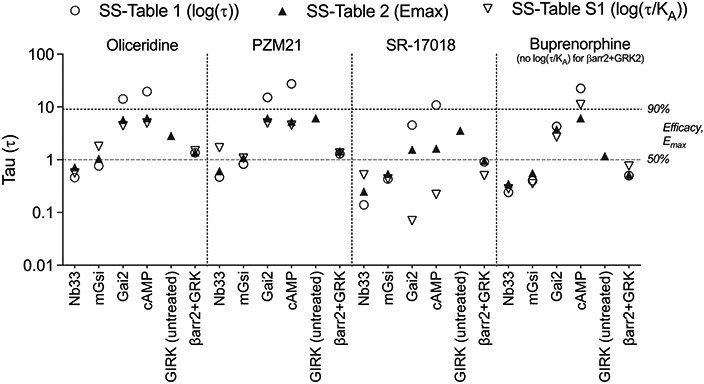
Graphic presentation of τ after its derivation from log(τ) (SS-Table 1), Emax (SS-Table 2), or log(τ/KA) (SS-Table S1) using eqs 1-6.
As the focus of the SS-manuscript was the correlation of agonist efficacy or bias with the therapeutic window, it was reasonable to produce Δlog(τ/KA) values that are used to evaluate functional selectivity (bias). As such, an estimate of log(τ/KA) can be derived from the three-parameter curve fit using eq 7:
| (7) |
This equation is easily adapted to Microsoft Excel for both parameter conversion and the further bias analysis that is the principal end point. For the sake of completeness, all Microsoft Excel spreadsheets for all figures used for the analyses, herein, are provided for examination in the Supporting Information.
Comparison of ΔLog(τ/KA) Derived from EC50 and Emax Compared to ΔLog(τ/KA) Values Presented.
In bias analysis, the Δlog(τ/KA) values specifically provide a discrete measure of each agonists activity in each experimental system. The Δ in the name comes from the correction to the activity of the reference agonist (usually a full agonist for which τ cannot be explicitly determined; in the SS-manuscript, DAMGO was used) within each system. The log(τ/KA)Reference is defined as the pEC50 of the reference agonist in each system, and the Δlog(τ/KA) for each test agonist is expressed in eq 8 as
| (8) |
In Figure 4, the Δlog(τ/KA) for each agonist in each response is plotted on the X-axis (calculated from SS-Table S1). In cases in which Δlog(τ/KA) was determined for full agonists, the comparison is presented as ΔpEC50(Test-Reference). The Y-axis presents the Δlog(τ/KA) that was produced using eqs 7 and 8 with EC50, Emaxsystem, and Emax values from SS-Tables 2 and 3. Emaxsystem is defined as the response produced by DAMGO in each system (100%). The coefficient of determination (R2) when the slope is constrained to one is also presented in each panel to demonstrate the strength of the agreement between Δlog(τ/KA) presented in SS-Table S1 and Δlog(τ/KA) derived from SS-Tables 2 and 3. Notably, the values do not agree for the Gai2 (R2 = 0.3638) and cAMP (R2 = 0.7182) measures; values for SR-17018 are in disagreement in most assays.
Figure 4.

Linear regression of the correlation between the Δlog(τ/KA) values derived from potency and efficacy values, presented in SS-Tables 2 and 3, and the Δlog(τ/KA) values derived from the log(τ/KA) provided in SS-Table S1. The red line indicates a perfect match of the data points (slope = 1), and the black line is the linear fit of the data points with a 95% confidence interval shown as dashed gray lines.
Comparison of ΔΔLog(τ/KA) Values Presented in Tables and Graphs and Derived from Emax and EC50 Values.
Having normalized the performance of each agonist in each assay to the performance of the reference agonist DAMGO [mean Δlog(τ/KA)], we can further compare an agonist’s relative performance between two assays using eq 9:
| (9) |
As written in eq 9, a positive number would indicate a preference for assay 1 and a negative number would indicate a preference for assay 2. When ΔΔlog(τ/KA) and its 95% confidence intervals do not include zero, the value is indicative of an agonist that exhibits selectivity between one response and another (i.e., functional selectivity, biased agonism).26 More broadly stated, ΔΔlog(τ/KA) ≠ 0 indicates that the agonist promotes a discrete selection of an active state that prefers one effector over another; as such, ΔΔlog(τ/KA) is used to quantify “biased agonism”. The ΔΔlog(τ/KA) is frequently expressed as extending toward or away from one response or another, depending on the perspective of preferring or not preferring a receptor active state. This concept was first proposed mechanistically by a number of studies both in brain and recombinant cell systems.17,27
Therefore, using the process described above, the bias factor [as ΔΔlog(τ/KA)] can be calculated from the data included in SS-Tables 2, 3, and S1. First, using SS-Table S1, the Δlog(τ/KA) value of βarr2+GRK2 can be subtracted from the Δlog(τ/KA) for each of the G protein signaling pathways to generate the ΔΔlog(τ/KA) (eq 9) to determine the preference over βarr2+GRK2 recruitment. These calculated parameters [ΔΔlog(τ/KA)] are presented in Figure 5A and are compared to values graphed in SS-Figure 5 [as estimated from the graphic because the ΔΔlog(τ/KA) values were not included in the SS-manuscript]. Although the error of these calculations is not readily accessible, the error propagation does not directly inform or affect the comparison of the means, as discussed above.
Figure 5.

Bias factor analysis presented by calculated ΔΔlog(τ/KA) from (A) SS-Table 1 [Δlog(τ/KA)] or (B) Δlog(τ/KA) values calculated using the potency and efficacy presented in SS-Tables 2 and 3. The four-point star represents the values published in SS-Figure 5 as determined by graphical overlay trace estimation. The figure legend is the same for panels A and B.
Given the discrepancy between calculated ΔΔlog(τ/KA) values and those graphed in SS-Figure 5, and a concern that perhaps the data in SS-Table S1 were incorrectly derived in the published paper, Δlog(τ/KA) values were then calculated from the Emax and EC50 values presented in SS-Tables 2 and 3 as described in eq 7. The bias factors [ΔΔlog(τ/KA)] were again produced by simple subtraction (eqs 8 and 9) and are presented in Figure 5B. The calculations that produced all values determined in Figure 5 are provided in the Supporting Information.
The comparison of ΔΔlog(τ/KA) (Figure 5) taken from the three different sources in the manuscript [SS-Figure 5 (simulation), SS-Table S1 (subtraction), and SS-Tables 2 and 3 (eq 7 using Emax and EC50)] reveals that values are fairly consistent but with some striking exceptions. The values presented in SS-Figure 5 consistently underestimate the G protein signaling preference for SR-17018 across all assays compared to the data obtained from SS-Table S1 (Figure 5A,B). The SS-manuscript concludes that SR-17018 “showed no statistically significant bias toward or away from any G protein activation measure”. If the same error in SS-Figure 5 were applied to the newly calculated ΔΔlog(τ/KA), the 95% confidence interval (which spans more than 2 orders of magnitude) would overlap zero even though the new mean calculated ΔΔlog(τ/KA) value changes substantially (by an order of magnitude, in some cases). As the 95% confidence intervals for ΔΔlog(τ/KA) values are not symmetrical (SS-Figures 5 and S6), how these intervals were determined is not clearly described. However, the standard method proposed26 was not used, indicating that a direct reassignment to these newly calculated mean values would not be possible or useful. This would lead to the general conclusion that the presence of functional selectivity cannot be proven or disproven, based solely on this analysis, as the error is so large as to preclude any meaningful interpretation.
Comparison of Efficacy and the Therapeutic Window.
The primary goal of the SS-manuscript was to demonstrate that intrinsic efficacy, and not bias, was responsible for the improvement in the therapeutic window of the biased MOR agonists. To that end, a series of in vivo experiments were produced to determine the therapeutic window as measured by the ΔlogED50 between antinociception (hot-plate latency) and respiratory suppression (respiratory frequency). These therapeutic window values were then correlated with ligand efficacy, log(τ), as well as functional selectivity [bias, ΔΔlog(τ/KA)], between different responses.
For this comparison, the authors focused on intrinsic efficacy in the mGsi assays and bias comparison between mGsi and βarr2+GRK2 assays in SS-Figure 8. Figure 6 presents a representation of the intrinsic efficacy, log(τ), taken from SS-Table 1 or derived from SS-Table 2 (Emax) graphed for mGsi (Figure 6A) and βarr2+GRK2 (Figure 6B); this is compared with the therapeutic window (Figure 6C). It should be noted that it was not possible to derive the log(τ) value for fentanyl in the mGsi assay as the maximum response of fentanyl intersects with the maximal response of the system. As such, the log(τ) value for fentanyl, in assays in which it approaches or exceeds 100%, will approach infinity. The Emax for the βarr2+GRK2 assay (Emax = 92% in SS-Table 2) was used to estimate log(τ) in Figure 6B; no value was provided in SS-Table 1. As the authors point out in SS-Figure 3 of the SS-manuscript, the partial agonists are partial in both G protein signaling assays and βarrestin2 recruitment. We can therefore interpret that decreasing the intrinsic efficacy for recruiting βarrestin2 as well as G protein correlates with an improved therapeutic window (Figure 6A-C). However, in both cases, there is not much refinement in the assessment, as PZM21 shows a wider therapeutic window, yet the intrinsic efficacy is the same as that observed for oliceridine in both assays.
Figure 6.
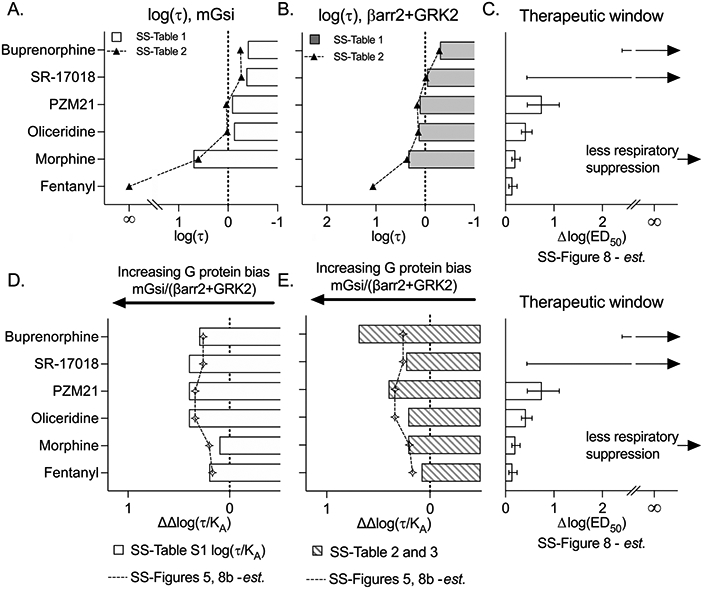
Graphical representation of agonist log(τ) and bias factors [ΔΔlog(τ/KA)] in comparison to the therapeutic index as presented in SS-Figure 8. Log(τ) values were plotted from SS-Table 1 or derived from SS-Table 2 and plotted for (A) mGsi and (B) βarr2+GRK2 assays and aligned for comparison with the (C) therapeutic window and error values estimated from SS-Figure 8. Infinity (∞) indicates an inability to estimate the mean, and error bars are provided as an indication of the lower confidence interval with an arrow pointing toward infinity according to SS-Figure 8D. The ΔΔlog(τ/KA) values calculated from the Δlog(τ/KA) values provided in SS-Table S1 are compared to the values presented in SS-Figures 5 and 8B (estimated). (E) ΔΔLog(τ/KA) values derived from EC50 (SS-Table 3) and Emax (SS-Table 2) values as described in the text. The therapeutic window plot is shown again for visual alignment.
Because therapeutic window values were not included in the manuscript, the plot in Figure 6C was created by tracing and estimating values that correspond with SS-Figure 8B; the error is also included on the basis of this same method. Additionally, the mean therapeutic window values for SR-17108 and buprenorphine were not included as an estimate of ΔED50 could not be calculated (as these agonists did not produce 50% maximum respiratory suppression). Therefore, it is not possible to estimate either the mean or the error for the ΔED50 for SR-17018 or buprenorphine as their means approach infinity. The therapeutic window for these compounds was not measured; any representation of therapeutic windows that does define either of these parameters is, at minimum, an overinterpretation of the data. With this recognition, the therapeutic window graph in Figure 6C presents the lower confidence intervals for means that extend to infinity as depicted in SS-Figure 8.
Panels D and E of Figure 6 are from Figure 5 and are plotted, as presented in SS-Figure 8, for comparison to the therapeutic window plot (shown again within the figure). Figure 6D shows the Δlog(τ/KA) values calculated from SS-Table S1 [log(τ/KA)], and these are shown in comparison to the values that were presented in graphical form in SS-Figures 5 and 8B. Here one can clearly see, using the Emax and EC50 values to derive log(τ/KA), that the biased ligands show an improvement in their therapeutic window; moreover, the differences in bias between PZM21 and oliceridine now reflect the differences observed in the therapeutic window.
Comparison of the Relative Intrinsic Activity and Therapeutic Window.
In this reanalysis of the data presented in the SS-manuscript, efforts were made to rederive τ and KA from Emax and EC50, presented in the paper, to arrive at the ΔΔlog(τ/KA) bias factors (Figure 5). However, given that these are partial agonists, it is also possible to use the values directly produced by the three-parameter concentration–response function to determine relative intrinsic activity25 in eq 10:
| (10) |
The relative intrinsic activity provides a means of interpreting the activity of the agonist in different systems without necessarily fully characterizing the stimulus–response relationship. As such, it is an empirical method of identifying agonists that show a preference for a subset of responses compared to other agonists that remain impartial between different responses. The results of this analysis can be visualized in Figure 7A by comparing ΔΔlog(Emax/EC50), where a preference for G protein signaling over βarr2 (+GRK) is observed across the four platforms used to assess G protein signaling. This analysis is in direct agreement with the ΔΔlog(τ/KA) values derived from analysis of Emax and EC50 to generate τ and KA in Figure 5B.
Figure 7.

Bias factors determined as ΔΔlog(Emax/EC50) from values provided in SS-Tables 2 and 3 comparing responses between (A) the indicated assays and βarr2+GRK2 as ΔΔlog(Emax/EC50)[G protein assay–βarr2+GRK2 assay] and (B) βarr2 vs βarr2+GRK2 as ΔΔlog(Emax/EC50)[βarr2 assay–βarr2+GRK2 assay] and (C) the indicated assays and GIRK (untreated) as ΔΔlog(Emax/EC50)[G protein assay–GIRK untreated assay]. (D) Comparison of ΔΔlog(Emax/EC50)[mGsi assay–GIRK assay] and ΔΔlog(τ/KA) [mGsi assay–GIRK untreated assay] to the therapeutic window from SS-Figure 8 as also shown in Figure 6.
The authors chose to focus on comparing responses to βarr2 only in the presence of GRK2. GRK2 overexpression is included in the βarr2 assay to improve system sensitivity, as GRK2 phosphorylates MOR and facilitates βarr2 recruitment.1,28 Therefore, it is expected that while GRK2 overexpression will enhance agonist potency and efficacy, the degree of enhancement should be consistent between the agonists. The preference for βarr2 recruitment in the absence of GRK, compared to GRK overexpression, is presented in Figure 7B. There is divergence from unity [ΔΔlog(Emax/EC50) ≠ 0], and it is readily apparent that a number of agonists exhibit a preference for βarr2 recruitment in the absence of GRK2 relative to their activity in the presence of GRK2 overexpression. This would suggest that GRK2 overexpression could be acting to decrease system sensitivity and diminish the extent of βarr2 recruitment produced by the agonist, which is in direct contrast to the expected role of GRK2 in this system (to facilitate βarr2 recruitment). Overall, it is unusual that GRK overexpression worsens an agonist’s ability to recruit βarr2 to the receptor.
We also note that most of the agonists show a preference away from GIRK recruitment and toward βarr2+GRK2; therefore, we asked whether the agonists would demonstrate functional selectivity among the G protein signaling assays. While GIRK was considered an orthogonal assay for G protein-mediated signaling, the data demonstrate robust signaling bias between the ability to activate the other G protein pathways over GIRK (Figure 7C). This is particularly intriguing as activation of GIRK has been implicated in MOR-mediated respiratory suppression;29 indeed, as presented in Figure 7D, the bias away from GIRK activation aligns well with the therapeutic window spectra presented in Figure 6.
Active State Affinity and Rank Order Efficacy Are Not Preserved.
Because the conclusions of the SS-manuscript are that intrinsic activity is directly related to therapeutic index, directly comparing the intrinsic efficacy observed across the different assays was relevant. Moreover, because most of the ligands of interest generally act as partial agonists, it was reasonable to generate the active state selectivity predictions that would result from analysis with the two-state model.17,30 The active state selectivity ratio (α) can be calculated15 as shown in eq 11:
| (11) |
where K* is the active state affinity and K is the inactive state affinity. Emax is the maximum response of the full agonist, and E is the maximum response of the agonist of interest (from SS-Table 2). L is the system intrinsic isomerization ratio (R/R*) and was constrained to a value of 100 for all responses.15,17 This value was originally chosen to describe a system with minimal constitutive activity, and we used this value (L = 100) in this analysis based on the same assumption.17 With the calculated active state selectivity ratio (α), it is possible to use the EC50 to calculate the active state affinity constant of the agonist for the receptor as shown in eq 12:17
| (12) |
where K* is the agonist affinity constant for the active state in each response using the EC50 that is produced from each response. The K*/K is calculated in eq 11, where, again, L is set equal to 100. These calculations are related to the operational fit but can be determined independent of full agonist response curves.31
Conceptually, the potency and efficacy of full agonists are highly sensitive to receptor reserve and system sensitivity. That is, a small number of receptors saturated with a full agonist can lead to a full response of the system at a concentration that is lower than the active state affinity constant. Partial agonists are less subject to this type of system-dependent amplification as their activity is directly proportional to their occupancy. For this reason, it would be expected that the active state affinity (K*) of a partial agonist would be generally conserved across multiple assays, despite differences in signal amplification. However, Figure 8A shows that the active state affinity (K*) of several of the partial agonists studied spans two orders of magnitude. This suggests that the differences in affinity may be due to multiple active states of the receptor as previously proposed.17
Figure 8.

Comparison of the intrinsic efficacy of each agonist in the indicated assays as determined by (A) calculated pK* (potency values were not included for morphine at Gai and cAMP and buprenorphine at βarr2), (B) Emax with SEM from SS-Table 2, and (C) τ as shown in Figure 2 as derived from log(τ) (SS-Table 1). Missing values in panel A for morphine are indicated by interrupted lines. Changes in rank order efficacy are easily visualized by the intersection of lines.
However, even if partial agonists could exhibit different affinities for the active state of the receptor (due to system-dependent parameters, including receptor-reserve or threshold sensitivity), the rank order of agonist efficacy should remain constant in different systems if a single active state receptor is responsible for mediating all responses.16,17 Panels B and C of Figure 8 present the comparison of efficacy as Emax and τ (derived from SS-Table S1), where it is clear that the relative intrinsic activity (also known as rank order efficacy) is not conserved across the assays. It should be noted that estimates of intrinsic efficacy are not subject to agonist binding kinetics as these values are determined at saturation. These findings distinctly and explicitly implicate more than one active state of the receptor in being responsible for stimulating different responses; moreover, the differences in active state selectivity are directly related to the distinction of biased agonists.
DISCUSSION
The comparison of agonist-induced activity across multiple assays to determine functional selectivity can be a daunting task. While many assays are developed to enhance signal-to-noise ratios, this can come at a cost of signal amplification. Moreover, certain assays are not amenable to developing stable cells lines and necessitate repeated transient transfections, which can in turn alter receptor number and assay sensitivity. Therefore, it is imperative that comparisons are always made to a reference agonist’s performance (or more than one reference agonist) and that assays are performed in parallel (to avoid changes in the systems over time). The most important requirement for determining functional selectivity is actually twofold: high-quality consistent data and the appropriate application of mechanistic pharmacology analysis. In this evaluation of existing data, we have presented several known approaches for analyzing data to identify a divergence in receptor signaling across assays and have presented an additional analysis adapted from the two-state model of receptor activation (K*). The benefit of determining K* is that it produces a microaffinity constant for the active state preference over the inactive state of the receptor. This allows for normalization of efficacy and potency to produce affinity estimates. If there is no functional selectivity, it would be expected that the rank order of these affinity estimates would be conserved across all systems that are being investigated. Divergence from this conservation will indicate a preference for an assay-dependent active state. In addition, the utilization of mechanistic analysis to underpin more empirical analysis can lend a degree of confidence to the general conclusion that ligand bias is observed within a set of data.
In the recalculation of bias from the SS-manuscript, it was not possible to interpret the error for the data set as there was only limited information provided by the authors; for example, the number of experiments was provided as a range (3–14 replications), and the SEM was presented in the tables. However, because the derivation of τ and KA is dependent on the potency (EC50) and efficacy (Emax) within each assay, the ability to produce a “bias factor” with any reasonable error of certainty will require that the individual assays be, first, reproducible and, second, adequately powered by replicates. There is reasonable concern that the experimental measures presented herein exhibit large errors in the estimation of potency (pEC50 in SS-Table 3) and efficacy (Emax in SS-Table 2). This can be visualized in Figure 2, where the error in potency exceeds a half-log order in several assays. Because the authors report running 3–14 experiments per data set, it would seem that perhaps, in cases in which the error is very high, the studies were underpowered. This initial error propagates throughout additional analyses, leading to 95% confidence intervals that are greater than the mean (SS-Figures 5 and S6). Therefore, it is apparent that several assays reported in the SS-manuscript are underpowered and do not support the conclusion that there is no “significant” bias observed for any of the compounds. This concern with reproducibility is further supported by the degree of error present upon averaging the parameters derived from DAMGO response curves. DAMGO, as the reference agonist, would be included in every experiment (presumably accounting for the upper n = 14 described in SS-Tables 1–3), yet the margin of error in DAMGO’s potency was wider than expected for several of the signaling assays (Figure 2 and SS-Table 3).
The reproducibility issues may stem from the use of transient transfection systems with variable receptor density and effector expression levels. Moreover, there was no indication that protein and receptor expression levels were monitored for consistency between experiments. Differences in receptor expression and receptor–effector coupling efficiency may induce the system to exhibit activity that does not reflect the true activity of the agonist. In effect, the experimental system may be primed to favor one response over another due to system-dependent properties. Interestingly, in this case, it appears that the agonist activity is muted, or disfavored, and system sensitivity is acting as a barrier to some responses. This system bias has been previously discussed as a confounding effect in experimental systems that requires appropriate vetting of experimental systems.22,32 One example of this is presented in Figure 7B for agonist activity in the absence and presence of GRK2 overexpression. In all cases, overexpression of GRK2 would be expected to increase agonist potency for βarrestin2 recruitment; however, this is not the case in the SS-manuscript, where GRK2 acts to mute the agonist activity in a way not previously observed.33,34 In addition, the high potency observed for PZM-21 and SR-17018 agonists in the transiently transfected MOR:βarr2 BRET assay1 does not agree with the prior reports for these compounds wherein stable, commercially available, cell lines were used.6,8
In many ways, the SS-manuscript reproduces observations from several investigators that have proposed that improving G protein signaling while avoiding βarrestin2 recruitment may be a useful approach to limiting respiratory suppression while preserving antinociception.3-6,8,11 Specifically, the authors reproduce in vivo observations originally published for SR-17018,8 oliceridine,11 and PZM21.6 Moreover, the demonstration that PZM21, at doses of ≤100 mg/kg, produces less than half of the respiratory suppression induced by 10 mg/kg morphine is very encouraging as it had been tested only up to 40 mg/kg in the first study, where it showed no respiratory suppression compared to the vehicle.6 It should be noted that these observations are in direct contrast to those of another study that showed PZM21 and morphine producing equivalent respiratory suppression at 10 mg/kg.35
The idea that a decreasing intrinsic efficacy may serve to improve the therapeutic utility of a mu opioid receptor agonist to manage pain and limit side effects has been pursued for some time, resulting in compounds such as buprenorphine.36 However, caution should be maintained in translating efficacy observed in cultured systems to expectations observed in clinical use. In particular, buprenorphine is fully efficacious in the treatment of some pain conditions,37 as is morphine, even though both perform as partial agonists across many assay systems, as demonstrated in the SS-manuscript. As such, there has not been a direct correlation between partial agonism observed in different cell-based assays and the effects of a drug in vivo; therefore, it may not be fully predictive that a partial agonist may be safer regarding respiratory suppression. Indeed, buprenorphine produces respiratory suppression in humans,38 with the additional complication that it is difficult to reverse with naloxone39 as buprenorphine is a long-lasting, high-affinity agonist.36 In rats, buprenorphine produces respiratory suppression to the same extent as fentanyl, which would also seem to contradict the efficacy hypothesis as well as the biased agonism hypothesis.40 However, studies with buprenorphine are further complicated by the fact that it has activity at other opioid receptors and also has nonselective active metabolites that can suppress respiration.41-43
The conclusion of the SS-manuscript is that partial agonism is responsible for the avoidance of respiratory suppression, and this is supported by the choice of agonists and how those agonists performed in the G protein signaling assays selected. However, the same could be said for the efficacy in the βarrestin2 recruitment assays, as the biased agonists act as partial agonists there, as well, in a manner that also aligns with an improved therapeutic window. The data presented here may support the role of agonist efficacy in determining the extent of respiratory suppression in mice; however, no counter argument was purposefully explored (i.e., testing of a full agonist with less respiratory suppression or a partial agonist with more respiratory suppression).
The study by Schmid et al. addressed this question by generating a series of structurally related MOR agonists that spanned a spectrum of both bias and intrinsic efficacy.8 One example from that study is SR-11501, a partial agonist that has potency and efficacy similar to those of morphine in G protein signaling and in antinociception studies. However, SR-11501 displays bias toward βarrestin2 recruitment over G protein signaling and produces fentanyl-like respiratory suppression, resulting in a very narrow therapeutic window. Moreover, examples of two full agonists that display bias toward G protein signaling, SR-14968 and SR-14969, were shown to produce a therapeutic window that was wider than that of morphine (less respiratory suppression vs antinociception).8 Schmid et al. also showed that fentanyl, which has a narrow therapeutic window, acts as a partial agonist in mouse brainstem membranes using 35S-GTPγS binding assays.8 Fentanyl has also been shown to act as a partial agonist in spinal cord44 and in rat thalamus45 in studies assessing 35S-GTPγS binding. Therefore, intrinsic efficacy can be uncoupled from biased agonism, and within the SR series of agonists, G protein signaling bias was shown to correlate with a decreased level of respiratory suppression, independent of intrinsic efficacy.8
Regardless of the distinction of bias, the comparison of active state affinity constants and the observation that rank order efficacy was not conserved across the spectrum of assays investigated provide compelling support for the idea that these experimental compounds present interesting and substantially novel pharmacology. Ultimately, the determination of biased agonism, or intrinsic efficacy for that matter, will be dependent on the cellular signaling systems used (i.e., receptor density, effector expression, and amplification of signal) as well as reference agonist used to determine the maximum possible signaling response in the system. It is not unexpected that perceptions of bias and efficacy will vary between studies.22,46 In a recent example, similar BRET-based studies were elegantly employed for similar analysis by Ehrlich et al.,47 investigating ligand bias at the mu opioid receptor where they demonstrate diverse signaling profiles of oliceridine, PZM21, and buprenorphine in neurons. In time, the value of these cellular readouts as predictors of agonist function may be supported by detecting state-dependent structural conformations of MOR induced by biased ligands binding to the receptor.48-51 This does not make their pharmacology less remarkable, nor does it disprove the hypothesis that ligands may demonstrate active state selectivity. On the contrary, as more studies present interesting and novel findings regarding the spectrum of activity these compounds demonstrate, it may be possible to more definitively establish these differences. In particular, the demonstration of changes in rank order efficacy is a compelling finding that further substantiates and validates the value of these compounds. From a practical standpoint, however, the correlation between the pharmacological properties and the improvement in therapeutic efficacy while limiting side effects in human studies should drive further investigations. Indeed, oliceridine, which is a partial agonist in both G protein signaling and βarrestin2 recruitment, as well as a biased agonist for G protein signaling, is producing superior analgesia/respiratory suppression in post-operative pain.11,13,14 Therefore, while partial agonism could be a desirable pharmacological property, biased agonism may serve as a means to further improve the therapeutic window.
Supplementary Material
Funding
This work is funded by National Institute on Drug Abuse Grants DA033073 and DA038964.
Footnotes
Special Issue Paper
Published as part of the Biochemistry virtual special issue “Biochemistry of Pain”.
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.biochem.1c00466.
Supplemental Figure 1 (PDF)
Graphic simulations of the parameters from the SS-manuscript1 (XLSX)
The authors declare no competing financial interest.
REFERENCES
- (1).Gillis A; Gondin AB; Kliewer A; Sanchez J; Lim HD; Alamein C; Manandhar P; Santiago M; Fritzwanker S; Schmiedel F; Katte TA; Reekie T; Grimsey NL; Kassiou M; Kellam B; Krasel C; Halls ML; Connor M; Lane JR; Schulz S; Christie MJ; Canals M Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signaling 2020, 13, eaaz3140. [DOI] [PubMed] [Google Scholar]
- (2).Gillis A; Gondin AB; Kliewer A; Sanchez J; Lim HD; Alamein C; Manandhar P; Santiago M; Fritzwanker S; Schmiedel F; Katte TA; Reekie T; Grimsey NL; Kassiou M; Kellam B; Krasel C; Halls ML; Connor M; Lane JR; Schulz S; Christie MJ; Canals M Erratum for the Research Article: “Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists. Sci. Signaling 2021, 14, eabf9803. [DOI] [PubMed] [Google Scholar]
- (3).Bohn LM; Lefkowitz RJ; Gainetdinov RR; Peppel K; Caron MG; Lin FT Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999, 286, 2495–2498. [DOI] [PubMed] [Google Scholar]
- (4).Bohn LM; Gainetdinov RR; Lin FT; Lefkowitz RJ; Caron MG Mu-opioid receptor desensitization by beta-arrestin-2 determines morphine tolerance but not dependence. Nature 2000, 408, 720–723. [DOI] [PubMed] [Google Scholar]
- (5).Raehal KM; Walker JK; Bohn LM Morphine side effects in beta-arrestin 2 knockout mice. J. Pharmacol. Exp. Ther 2005, 314, 1195–1201. [DOI] [PubMed] [Google Scholar]
- (6).Manglik A; Lin H; Aryal DK; McCorvy JD; Dengler D; Corder G; Levit A; Kling RC; Bernat V; Hubner H; Huang XP; Sassano MF; Giguere PM; Lober S; Da D; Scherrer G; Kobilka BK; Gmeiner P; Roth BL; Shoichet BK Structure-based discovery of opioid analgesics with reduced side effects. Nature 2016, 537, 185–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Kennedy NM; Schmid CL; Ross NC; Lovell KM; Yue Z; Chen YT; Cameron MD; Bohn LM; Bannister TD Optimization of a Series of Mu Opioid Receptor (MOR) Agonists with High G Protein Signaling Bias. J. Med. Chem 2018, 61, 8895–8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Schmid CL; Kennedy NM; Ross NC; Lovell KM; Yue Z; Morgenweck J; Cameron MD; Bannister TD; Bohn LM Bias Factor and Therapeutic Window Correlate to Predict Safer Opioid Analgesics. Cell 2017, 171, 1165–1175.e13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dekan Z; Sianati S; Yousuf A; Sutcliffe KJ; Gillis A; Mallet C; Singh P; Jin AH; Wang AM; Mohammadi SA; Stewart M; Ratnayake R; Fontaine F; Lacey E; Piggott AM; Du YP; Canals M; Sessions RB; Kelly E; Capon RJ; Alewood PF; Christie MJ A tetrapeptide class of biased analgesics from an Australian fungus targets the micro-opioid receptor. Proc. Natl. Acad. Sci. U. S. A 2019, 116, 22353–22358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Varadi A; Marrone GF; Palmer TC; Narayan A; Szabo MR; Le Rouzic V; Grinnell SG; Subrath JJ; Warner E; Kalra S; Hunkele A; Pagirsky J; Eans SO; Medina JM; Xu J; Pan YX; Borics A; Pasternak GW; McLaughlin JP; Majumdar S Mitragynine/Corynantheidine Pseudoindoxyls As Opioid Analgesics with Mu Agonism and Delta Antagonism, Which Do Not Recruit beta-Arrestin-2. J. Med. Chem 2016, 59, 8381–8397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).DeWire SM; Yamashita DS; Rominger DH; Liu G; Cowan CL; Graczyk TM; Chen XT; Pitis PM; Gotchev D; Yuan C; Koblish M; Lark MW; Violin JD A G protein-biased ligand at the mu-opioid receptor is potently analgesic with reduced gastrointestinal and respiratory dysfunction compared with morphine. J. Pharmacol. Exp. Ther 2013, 344, 708–717. [DOI] [PubMed] [Google Scholar]
- (12).Lambert D; Calo G Approval of oliceridine (TRV130) for intravenous use in moderate to severe pain in adults. Br. J. Anaesth 2020, 125, e473–e474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Ayad S; Demitrack MA; Burt DA; Michalsky C; Wase L; Fossler MJ; Khanna AK Evaluating the Incidence of Opioid-Induced Respiratory Depression Associated with Oliceridine and Morphine as Measured by the Frequency and Average Cumulative Duration of Dosing Interruption in Patients Treated for Acute Postoperative Pain. Clin. Drug Invest 2020, 40, 755–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bergese S; Berkowitz R; Rider P; Ladouceur M; Griffith S; Segura Vasi A; Cochrane K; Wase L; Demitrack MA; Habib AS Low Incidence of Postoperative Respiratory Depression with Oliceridine Compared to Morphine: A Retrospective Chart Analysis. Pain Research and Management 2020, 2020, 7492865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Leff P The two-state model of receptor activation. Trends Pharmacol. Sci 1995, 16, 89–97. [DOI] [PubMed] [Google Scholar]
- (16).Leff P; Scaramellini C; Law C; McKechnie K A three-state receptor model of agonist action. Trends Pharmacol. Sci 1997, 18, 355–362. [DOI] [PubMed] [Google Scholar]
- (17).Berg KA; Maayani S; Goldfarb J; Scaramellini C; Leff P; Clarke WP Effector pathway-dependent relative efficacy at serotonin type 2A and 2C receptors: evidence for agonist-directed trafficking of receptor stimulus. Mol. Pharmacol 1998, 54, 94–104. [PubMed] [Google Scholar]
- (18).Black JW; Leff P Operational models of pharmacological agonism. Proc. R. Soc. B 1983, 220, 141–162. [DOI] [PubMed] [Google Scholar]
- (19).Gillis A; Kliewer A; Kelly E; Henderson G; Christie MJ; Schulz S; Canals M Critical Assessment of G Protein-Biased Agonism at the mu-Opioid Receptor. Trends Pharmacol. Sci 2020, 41, 947–959. [DOI] [PubMed] [Google Scholar]
- (20).Gillis A; Sreenivasan V; Christie MJ Intrinsic Efficacy of Opioid Ligands and Its Importance for Apparent Bias, Operational Analysis, and Therapeutic Window. Mol. Pharmacol 2020, 98, 410–424. [DOI] [PubMed] [Google Scholar]
- (21).Faouzi A; Varga BR; Majumdar S Biased Opioid Ligands. Molecules 2020, 25, 4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kenakin T; Christopoulos A Signalling bias in new drug discovery: detection, quantification and therapeutic impact. Nat. Rev. Drug Discovery 2013, 12, 205–216. [DOI] [PubMed] [Google Scholar]
- (23).Kelly E Efficacy and ligand bias at the mu-opioid receptor. Br. J. Pharmacol 2013, 169, 1430–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Klein Herenbrink C; Sykes DA; Donthamsetti P; Canals M; Coudrat T; Shonberg J; Scammells PJ; Capuano B; Sexton PM; Charlton SJ; Javitch JA; Christopoulos A; Lane JR The role of kinetic context in apparent biased agonism at GPCRs. Nat. Commun 2016, 7, 10842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Griffin MT; Figueroa KW; Liller S; Ehlert FJ Estimation of agonist activity at G protein-coupled receptors: analysis of M2 muscarinic receptor signaling through Gi/o,Gs, and G15. J. Pharmacol. Exp. Ther 2007, 321, 1193–1207. [DOI] [PubMed] [Google Scholar]
- (26).Kenakin T; Watson C; Muniz-Medina V; Christopoulos A; Novick S A simple method for quantifying functional selectivity and agonist bias. ACS Chem. Neurosci 2012, 3, 193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Mailman RB; Lawler CP; Lewis MM; Blake B; Nichols DE Functional Effects of Novel Dopamine Ligands: Dihydrexidine and Parkinson’s Disease as a First Step. In Dopamine Receptor Subtypes; Demirdamar P, Ed.; IOS Press, Inc.: Amsterdam, 1998; pp 64–83. [Google Scholar]
- (28).Krupnick JG; Benovic JL The role of receptor kinases and arrestins in G protein-coupled receptor regulation. Annu. Rev. Pharmacol. Toxicol 1998, 38, 289–319. [DOI] [PubMed] [Google Scholar]
- (29).Montandon G; Ren J; Victoria NC; Liu H; Wickman K; Greer JJ; Horner RL G-protein-gated Inwardly Rectifying Potassium Channels Modulate Respiratory Depression by Opioids. Anesthesiology 2016, 124, 641–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Ehlert FJ Functional studies cast light on receptor states. Trends Pharmacol Sci. 2015, 36, 596–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Tran JA; Chang A; Matsui M; Ehlert FJ Estimation of relative microscopic affinity constants of agonists for the active state of the receptor in functional studies on M2 and M3 muscarinic receptors. Mol Pharmacol. 2015, 75, 381–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Gundry J; Glenn R; Alagesan P; Rajagopal S A Practical Guide to Approaching Biased Agonism at G Protein Coupled Receptors. Front. Neurosci 2017, 11, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Miess E; Gondin AB; Yousuf A; Steinborn R; Mosslein N; Yang Y; Goldner M; Ruland JG; Bunemann M; Krasel C; Christie MJ; Halls ML; Schulz S; Canals M Multisite phosphorylation is required for sustained interaction with GRKs and arrestins during rapid mu-opioid receptor desensitization. Sci. Signaling 2018, 11, eaas9609. [DOI] [PubMed] [Google Scholar]
- (34).Zhang J; Ferguson SS; Barak LS; Bodduluri SR; Laporte SA; Law PY; Caron MG Role for G protein-coupled receptor kinase in agonist-specific regulation of mu-opioid receptor responsiveness. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 7157–7162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Hill R; Disney A; Conibear A; Sutcliffe K; Dewey W; Husbands S; Bailey C; Kelly E; Henderson G The novel mu-opioid receptor agonist PZM21 depresses respiration and induces tolerance to antinociception. Br. J. Pharmacol 2018, 175, 2653–2661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Lewis JW Buprenorphine. Drug Alcohol Depend. 1985, 14, 363–372. [DOI] [PubMed] [Google Scholar]
- (37).Raffa RB; Haidery M; Huang HM; Kalladeen K; Lockstein DE; Ono H; Shope MJ; Sowunmi OA; Tran JK; Pergolizzi JV Jr. The clinical analgesic efficacy of buprenorphine. J. Clin. Pharm. Ther 2014, 39, 577–583. [DOI] [PubMed] [Google Scholar]
- (38).Warner NS; Warner MA; Cunningham JL; Gazelka HM; Hooten WM; Kolla BP; Warner DO A Practical Approach for the Management of the Mixed Opioid Agonist-Antagonist Buprenorphine During Acute Pain and Surgery. Mayo Clin. Proc 2020, 95, 1253–1267. [DOI] [PubMed] [Google Scholar]
- (39).van Dorp E; Yassen A; Sarton E; Romberg R; Olofsen E; Teppema L; Danhof M; Dahan A Naloxone reversal of buprenorphine-induced respiratory depression. Anesthesiology 2006, 105, 51–57. [DOI] [PubMed] [Google Scholar]
- (40).Kuo A; Wyse BD; Meutermans W; Smith MT In vivo profiling of seven common opioids for antinociception, constipation and respiratory depression: no two opioids have the same profile. Br. J. Pharmacol 2015, 172, 532–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Brown SM; Campbell SD; Crafford A; Regina KJ; Holtzman MJ; Kharasch ED P-glycoprotein is a major determinant of norbuprenorphine brain exposure and antinociception. J. Pharmacol. Exp. Ther 2012, 343, 53–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Huang P; Kehner GB; Cowan A; Liu-Chen LY Comparison of pharmacological activities of buprenorphine and norbuprenorphine: norbuprenorphine is a potent opioid agonist. J. Pharmacol. Exp. Ther 2001, 297, 688–695. [PubMed] [Google Scholar]
- (43).Sadee W; Rosenbaum JS; Herz A Buprenorphine: differential interaction with opiate receptor subtypes in vivo. J. Pharmacol. Exp. Ther 1982, 223, 157–162. [PubMed] [Google Scholar]
- (44).Madia PA; Navani DM; Yoburn BC [(35)S]GTPgammaS binding and opioid tolerance and efficacy in mouse spinal cord. Pharmacol., Biochem. Behav 2012, 101, 155–165. [DOI] [PubMed] [Google Scholar]
- (45).Selley DE; Sim LJ; Xiao R; Liu Q; Childers SR mu-Opioid receptor-stimulated guanosine-5’-O-(gamma-thio)-triphosphate binding in rat thalamus and cultured cell lines: signal transduction mechanisms underlying agonist efficacy. Mol. Pharmacol 1997, 51, 87–96. [DOI] [PubMed] [Google Scholar]
- (46).Kenakin TP Pharmacological onomastics: what’s in a name? Br. J. Pharmacol 2008, 153, 432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Ehrlich AT; Semache M; Gross F; Da Fonte DF; Runtz L; Colley C; Mezni A; Le Gouill C; Lukasheva V; Hogue M; Darcq E; Bouvier M; Kieffer BL Biased Signaling of the Mu Opioid Receptor Revealed in Native Neurons. iScience 2019, 14, 47–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).de Waal PW; Shi J; You E; Wang X; Melcher K; Jiang Y; Xu HE; Dickson BM Molecular mechanisms of fentanyl mediated beta-arrestin biased signaling. PLoS Comput. Biol 2020, 16, e1007394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Podlewska S; Bugno R; Kudla L; Bojarski AJ; Przewlocki R Molecular Modeling of micro Opioid Receptor Ligands with Various Functional Properties: PZM21, SR-17018, Morphine, and Fentanyl-Simulated Interaction Patterns Confronted with Experimental Data. Molecules 2020, 25, 4636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Schneider S; Provasi D; Filizola M How Oliceridine (TRV-130) Binds and Stabilizes a mu-Opioid Receptor Conformational State That Selectively Triggers G Protein Signaling Pathways. Biochemistry 2016, 55, 6456–6466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Cong X; Maurel D; Demene H; Vasiliauskaite-Brooks I; Hagelberger J; Peysson F; Saint-Paul J; Golebiowski J; Granier S; Sounier R Molecular insights into the biased signaling mechanism of the mu-opioid receptor. Mol. Cell 2021, DOI: 10.1016/j.mol-cel.2021.07.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.