

Abstract
The genus Styrax L. consists of approximately 130 species distributed in the Americas, eastern Asia, and the Mediterranean region. The phylogeny and evolutionary history of this genus are not clear. Knowledge of the phylogenetic relationships and the method for species identification will be critical for the evolution of this genus. In this study, we sequenced the chloroplast genome of 17 Styrax samples and added 17 additional chloroplast genome sequences from GenBank. The data were used to investigate chloroplast genome evolution, infer phylogenetic relationships, and access the species identification rate within Styrax. The Styrax chloroplast genome contains typical quadripartite structures, ranging from 157,641 bp to 159,333 bp. The chloroplast genome contains 114 unique genes. The P distance among the Styrax species ranged from 0.0003 to 0.00611. Seventeen small inversions and SSR sites were discovered in the Styrax chloroplast genome. By comparing with the chloroplast genome sequences, six mutation hotspots were identified, and the markers ycf1b and trnT-trnL were identified as the best Styrax-specific DNA barcodes. The specific barcodes and superbarcode exhibited higher discriminatory power than universal barcodes. Chloroplast phylogenomic results improved the resolution of the phylogenetic relationships of Styrax compared to previous analyses.
1. Introduction
Chloroplasts are involved in photosynthesis and energy transformation in plants [1, 2]. Its own genome is known as the chloroplast genome, plastid gene, or plastome, which commonly occurs in multiple copies within the organelle. The important role of the chloroplast genome is functioning of the photosynthesis and other metabolic processes. The chloroplast genome is 120-160 kb in length [2, 3] and has a highly conserved quadripartite circular organization. This organization contains two single-copy regions (LSC and SSC) separated by two copies of inverted repeat (IR) regions [4]. The chloroplast genome encodes approximately 80 protein-coding genes, four rRNAs, and 30 tRNA genes [1, 2].
Advances in DNA sequencing technology have provided scientists with high efficiency and low cost to obtain complete chloroplast genome sequences. The chloroplast genomes are mostly inherited uniparentally, lack recombination, have compact size; thus, they effectively expand genetic information. Although the genome structure is conserved, mutational events, including indels, SSRs, and single-nucleotide substitutions (SNPs), are frequently occurring even in related species [5, 6]. These mutational resources provide rich information to infer evolutionary patterns [7], establish relationships among the plants [8–10], and provide effective genetic markers to resolve complex evolutionary histories [11, 12]. Moreover, plant DNA barcodes rely heavily on chloroplast genome sequences. Chloroplast genome markers, including rbcL, matK, and trnH-psbA, have been used as core DNA barcodes for plants [13]. Comparison of complete chloroplast genome sequences also provides an opportunity to identify specific plant DNA barcodes [14, 15]. Whole chloroplast genome sequences have been used as superbarcodes for plants in recent years [16, 17].
The genus Styrax L. consists of approximately 130 species distributed in the Americas, eastern Asia, and the Mediterranean region [18]. The Styrax species have important medicinal, ornamental, and economic values. The seed oil or resin of several species is a valuable medicinal ingredient and raw material for the manufacture of aromatic oils.
The most taxonomic treatment of Styrax is from Fritsch [18] who conducted a phylogenetic analysis based on 34 morphological characters. In this treatment, Styrax was divided into section Valvatae (predominantly tropical evergreen species) and section Styrax (north-temperate deciduous species). The section Styrax was divided into two series: Cyrta and Styrax. Series Cyrta were distributed in eastern Asia and eastern North America with serrated leaf margins and included 31 species. Series Styrax is distributed in western North America and western Eurasia with entire leaf margins and included three species. The Valvatae section also included two series. Series Valvatae was a strictly neotropical clade including about 77 species, and the series Benzoin was a strictly paleotropical clade including nine species.
The taxonomy of Styrax species remains incomplete, and several new species have been published based on their morphological characteristics [19–22]. Several studies have used molecular data to infer the phylogeny of Styrax species, such as the nuclear ribosomal DNA ITS [23] and chloroplast markers ndhF-rpl32-trnL, trnK, trnL-trnF, trnS-trnG, trnV-ndhC, rpoC1, and rpoC2 [23–25]. However, these results showed that those markers had low divergence. Therefore, sampling more genetic characters, such as the chloroplast genome sequences, may enhance the species identification.
To better understand the chloroplast genome evolution in Styrax and identify the variable markers to species identification within Styrax, we sequenced the chloroplast genome of 17 samples of Styrax and added published data from GenBank. Specifically, we attempted to (1) elucidate the chloroplast genome evolution and (2) determine whether the variable chloroplast markers or whole chloroplast genome data can be effective for Styrax species identification.
2. Materials and Methods
2.1. Plant Materials and DNA Extraction
A total of 17 species of the genus Styrax were obtained from the field and the DNA Bank of China, Institute of Botany, Chinese Academy of Sciences, and the DNA Bank of China has been permitted obtaining from the materials of the specimens in PE (Institute of Botany, Chinese Academy of Sciences). The details of the 17 species are shown in Table S1. Fresh leaf tissues from each accession were immediately dried with a silica gel before DNA extraction. Total DNA was extracted using a modified CTAB DNA extraction protocol (mCTAB) [26]. In addition to the newly collected material for DNA sequencing, publicly available complete chloroplast genome sequences (17 accessions, Table S1) of Styrax were also included. In total, the dataset of sequenced samples and GenBank accessions consisted of 34 individuals representing 29 Styrax species.
2.2. Chloroplast Genome Sequencing, Assembly, and Annotation
Chloroplast genome sequencing was performed at Novogene (Beijing) using the Illumina HiSeq X-ten platform. Total DNA was sheared to 350 bp fragments using an ultrasonicator. A rapid library was prepared using the NEBNext® Ultra™ DNA Library Prep Kit. Each sample yielded approximately 4 GB of data.
Illumina data were filtered using Trimmomatic v0.36 [27] to remove the adaptors and low-quality reads with Q − value ≤ 20. The parameters were set as follows: leading: 20; trailing: 20; sliding window: 4 : 15; MIN LEN: 36; and AVG QUAL: 20. The clean data were used to assemble the chloroplast genome using GetOrganelle [28], and the k-mer length was set to 85, 95, and 105. Complete chloroplast genomes were annotated using Plann [29], and the published chloroplast genome sequences of S. obassis (GenBank Accession number: MN560143) was used as the reference. Circular chloroplast genome maps were visualized using OGDRAW [30]. The final annotated chloroplast genomes were deposited in GenBank under accession numbers MZ285733 to MZ285749.
2.3. Repeat Analysis and Whole Genome Comparison
SSRs in the chloroplast genome were identified using the Perl script microsatellite identification (MISA) software. The parameters implemented in MISA are as follows: repeat units ≥ 10 for mononucleotides, repeat units ≥ 5 for dinucleotides, repeat units ≥ 4 for trinucleotides, and repeat units ≥ 3 for tetranucleotides, pentanucleotides, and hexanucleotides.
Small inversions were identified based on the aligned chloroplast genome sequence matrix, according to Dong et al. [7]. Inversions form a stem-loop structure, including inversion sequences and inverted repeats at the opposite flanking end [7].
The mVISTA program was used to analyze the variation in the Styrax chloroplast genomes [31], for which sequence annotation of S. agrestis was used as the reference. The 47 Styrax chloroplast genomes were aligned using MAFFT v7.0 and then adjusted manually using Se-Al v2.0 [32]. To explore the sequence divergence with the whole chloroplast genome in the 27 Styrax species, genetic P distances were calculated with MEGA X [33].
2.4. Mutation Hotspots Identified and DNA Barcoding Analysis
Three factors, including nucleotide diversity (π), mean distance (D), and the proportion of zero pairwise genetic distances (Z) for each species in the matrix, were used to explore the mutation hotspots in the Styrax chloroplast genome. Nucleotide diversity was calculated using the software DnaSP v6 [34]. Mean window distance and the proportion of zero pairwise genetic distances for each species in the matrix were calculated using the slideAnalyses function of the SPIDER package [35] in R. The window length was set to 600 bp, with a 50 bp step size.
Nucleotide diversity and variable and parsimony-informative sites were used to evaluate marker variability. The three universal chloroplast DNA barcodes, rbcL, matK, and trnH-psbA, were also used in this analysis. Nucleotide diversity was determined using the DnaSP v6 software [34], and variable and parsimony-informative sites were calculated using MEGA v7 software [36].
Distance and tree-building methods were used to assess the marker discriminatory power. The distance method uses the nearneighbor function of SPIDER. The tree-based method was applied using ML. ML analysis was conducted using RAxML nonparametric bootstrapping and 1000 ML pseudoreplicates. The best-fitting models were selected using ModelFinder [37].
2.5. Phylogenetic Analyses
Phylogenetic analysis was conducted to elucidate the interspecific phylogenetic relationships within Styrax. Two datasets were created to infer the Styrax phylogeny. The first data were whole chloroplast genome sequences of 34 Styrax samples with Huodendron tibeticum and H. biaristatum used as the outgroup. The second dataset was the concatenation of the 80 coding genes. Maximum likelihood (ML) and Bayesian inference (BI) methods were used to infer phylogenetic relationships. All phylogenetic analyses used the best-fitting models of nucleotide substitution selected in ModelFinder [37] under the Bayesian information criterion. Maximum likelihood (ML) analyses were performed in RAxML-NG [38] with 500 bootstrap replicates. The BI tree was inferred to be MrBayes v3.2 [39]. The BI analysis was run with two independent chains and prior for 20 million generations, with sampling every 1000 generations. The initial 25% of the sampled trees were discarded as burn-ins. Stationarity was considered to have been reached when the average standard deviation of the split frequencies remained below 0.01.
3. Results
3.1. Structural Characteristics of the Styrax Chloroplast Genome
Illumina paired-end sequencing produced between 11,971,102 (S. japonicus) and 40,957,798 (S. rugosus) paired-end clean reads per samples. After screening these paired-end reads through mapping with Styrax chloroplast genome using Geneious V9, 67,059 to 1,702,907 chloroplast genome reads were extracted with 64 × (S.americanus) to 1,618 × (S.roseus) coverage (Table S2).
All 17 newly sequenced chloroplast genomes were assembled entirely, and their sequence lengths and structures were very similar (Table 1, Figure 1). The chloroplast genome length ranged from 157,641 bp (S. japonicus) to 159,333 bp (S. suberifolius). The chloroplast genome has a quadripartite structure typical of angiosperms composed of an LSC region (87,250–88,656 bp), SSC region (17,993–18,412 bp), and two IR copies (26,017–26,352 bp). The overall G/C content was approximately 37%. The Styrax chloroplast genome encodes 114 genes, including 80 protein-coding genes, 30 transfer RNA (tRNA) genes, and four ribosomal RNA (rRNA) genes. The mVISTA results revealed collineation, no rearrangement, and high sequence similarity across the Styrax chloroplast genomes (Figure S1).
Table 1.
Summary statistics for the assembly of 17 Styrax chloroplast genomes.
| LSC | IR | SSC | Total | GC% | Number of genes | Protein-coding genes | tRNA | rRNA | |
|---|---|---|---|---|---|---|---|---|---|
| Styrax agrestis | 87,495 | 26,048 | 18,279 | 157,870 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax americanus | 87,448 | 26,053 | 18,272 | 157,826 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax argenteus | 87,898 | 26,017 | 18,343 | 158,275 | 36.9 | 114 | 80 | 30 | 4 |
| Styrax casearifolius | 87,526 | 26,048 | 18,310 | 157,932 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax formosanus | 87,664 | 26,047 | 18,295 | 158,053 | 36.9 | 114 | 80 | 30 | 4 |
| Styrax hemsleyanus | 87,586 | 26,048 | 18,293 | 157,975 | 36.9 | 114 | 80 | 30 | 4 |
| Styrax huanus | 87,507 | 26,060 | 18,290 | 157,917 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax japonicus | 87,250 | 26,047 | 18,297 | 157,641 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax obassia | 87,528 | 26,051 | 18,279 | 157,909 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax perkinsiae | 87,444 | 26,047 | 18,272 | 157,810 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax roseus | 87,546 | 26,034 | 18,292 | 157,906 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax rugosus | 87,755 | 26,041 | 18,412 | 158,249 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax serrulatus | 87,489 | 26,049 | 18,313 | 157,900 | 37.0 | 114 | 80 | 30 | 4 |
| Styrax suberifolius | 88,656 | 26,342 | 17,993 | 159,333 | 36.9 | 114 | 80 | 30 | 4 |
| Styrax tonkinensis | 87,642 | 26,049 | 18,318, | 158,058 | 36.9 | 114 | 80 | 30 | 4 |
| Styrax tonkinensis | 87,553 | 26,049 | 18,317 | 157,968 | 36.9 | 114 | 80 | 30 | 4 |
| Styrax tonkinensis | 87,622 | 26,352 | 18,027 | 158,353 | 36.9 | 114 | 80 | 30 | 4 |
Figure 1.
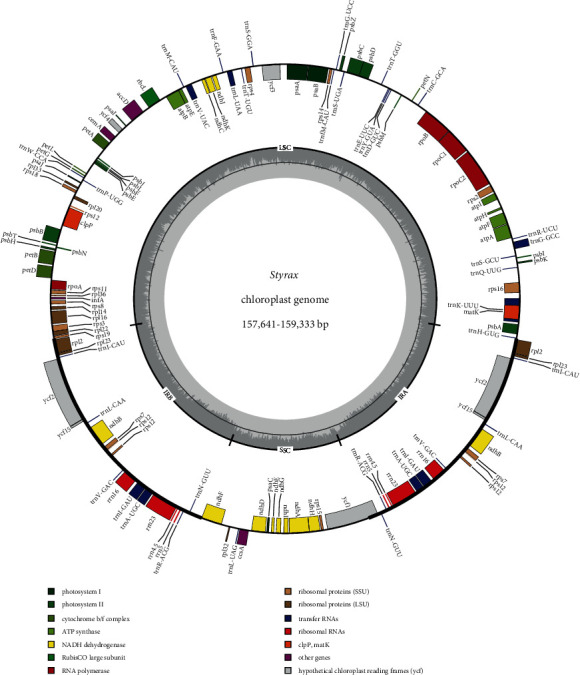
Chloroplast genome map of Styrax. Genes shown inside a circle are transcribed counterclockwise; genes outside are transcribed clockwise. Difference functional groups of genes are shown in different colors.
3.2. Repeats and Small Inversions
A total of 61–74 SSRs were found in the Styrax chloroplast genomes. Mono-, di-, tri-, tetra-, penta-, and hexanucleotide SSRs were identified (Figure 2). The majority of SSRs were mononucleotide repeats in all Styrax species, followed by trinucleotide repeats. Pentanucleotide repeats were limited to one occurrence in S. ramirezii. Most mononucleotide repeats were composed of A/T with minimal G/C. The LSC region contained the most significant SSRs (76.91%), with 14.49% identified in the SSC region and 8.61% in the IR region.
Figure 2.
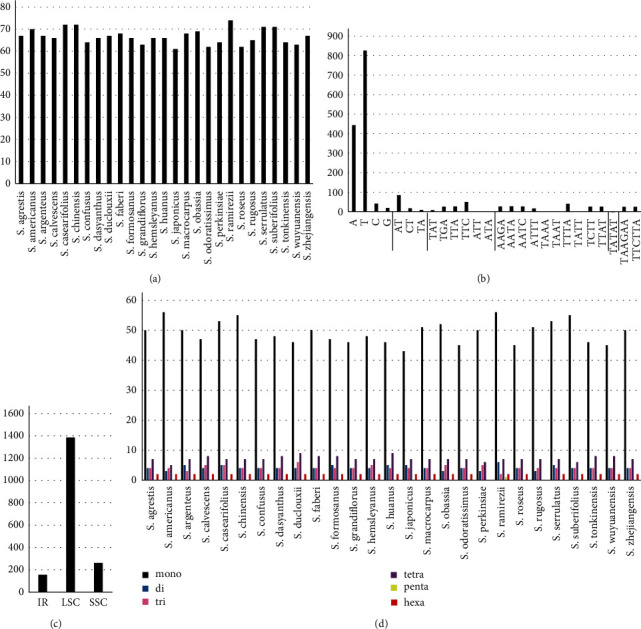
Frequency of SSRs in the Styrax chloroplast genomes: (a) the number of SSRs detected in the different Styrax species; (b) the number of SSR motifs in different repeat class types; (c) the frequency of SSRs in LSC, IR, and SSC regions; (d) the number of SSR types in different Styrax species.
Seventeen small inversions were identified in the Styrax chloroplast genome (Table 2). All inversions and their inverted repeating flanking sequences formed stem-loop structures. The inversion length was 4 to 164 bp, and the flanking repeats ranged from 6 bp to 28 bp. The longest inversion occurred in the trnSUGA –psbZ region. Except for the two inversions, the others were all located in the LSC region. All inversions were located in noncoding regions, including 14 in space and three in intron regions. The trnFGAA –ndhJ region included three inversions, and ycf3–trnSGGA had two inversions. Seven inversions (trnCGCA–petN, ycf3–trnSGGA 01, trnTUGU–trnLUAA, trnFGAA–ndhJ 01, trnFGAA–ndhJ 02, clpP, and trnRACG–trnNGUU) were specific to one species (Table S3). For example, the inversion in trnCGCA–petN was specific to S. duclouxii. The inversion in trnSGCU–trnGGCC and psaJ–rpl33 occurred in only one sample of S. agrestis, whereas inversions in trnSGCU–trnGGCC and petN–psbM occurred in some samples of S. tonkinensis. This suggests that these three inversions are polymorphic in one species.
Table 2.
The size and locations of small inversions in the Styrax chloroplast genomes.
| Region | Position | Location | Length (bp) | |
|---|---|---|---|---|
| Loop | Stem | |||
| LSC | trnSGCU–trnGGCC | Spacer | 4 | 12 |
| LSC | atpF–atpH | Spacer | 3 | 14 |
| LSC | rpoC1 | Intron | 4 | 10 |
| LSC | trnCGCA–petN | Spacer | 4 | 8 |
| LSC | petN–psbM | Spacer | 10 | 17 |
| LSC | trnSUGA–psbZ | Spacer | 164 | 11 |
| LSC | ycf3–trnSGGA 01 | Spacer | 4 | 12 |
| LSC | ycf3–trnSGGA 02 | Spacer | 4 | 7 |
| LSC | trnTUGU–trnLUAA | Spacer | 5 | 14 |
| LSC | trnFGAA–ndhJ 01 | Spacer | 6 | 6 |
| LSC | trnFGAA–ndhJ 02 | Spacer | 3 | 9 |
| LSC | trnFGAA–ndhJ 03 | Spacer | 10 | 15 |
| LSC | petA–psbJ | Spacer | 27 | 15 |
| LSC | psaJ–rpl33 | Spacer | 9 | 28 |
| LSC | clpP | Intron | 13 | 14 |
| IR | trnIGAU | Intron | 16 | 18 |
| IR | trnRACG–trnNGUU | Spacer | 8 | 11 |
3.3. Universal DNA Barcodes of Styrax
Three universal candidate DNA barcodes, rbcL, matK, and trnH-psbA, were analyzed to test the species discrimination power of Styrax (Table 3). The core barcode of rbcL had an aligned length of 695 bp, with 14 variable sites and nine parsimony-informative sites. Nucleotide diversity was 0.00297. The matK barcode was more variable than rbcL, with an aligned length of 878 bp and 25 variable sites in Styrax. The trnH-psbA barcode is an intergenic space region with an aligned length of 486 bp. It contained 25 variable sites and 14 parsimony-informative sites. According to the nucleotide diversity values, trnH-psbA was the most variable marker among the three chloroplast universal markers.
Table 3.
The variability of the three universal DNA barcodes and six variable markers in Styrax.
| Markers | Length | Variable sites | Parsimony-informative sites | Discrimination success (%) based on distance method | Nucleotide diversity | ||
|---|---|---|---|---|---|---|---|
| Numbers | % | Numbers | % | ||||
| rbcL | 695 | 14 | 2.01 | 9 | 1.29 | 18.52 | 0.00297 |
| matK | 878 | 25 | 2.85 | 13 | 1.48 | 44.44 | 0.00322 |
| trnH-psbA | 486 | 25 | 5.14 | 14 | 2.88 | 48.15 | 0.00888 |
| rbcL+matK | 1,573 | 39 | 2.48 | 22 | 1.40 | 48.15 | 0.00311 |
| rbcL+matK+trnH-psbA | 2,059 | 64 | 3.11 | 36 | 1.75 | 70.37 | 0.00412 |
| rps16-trnQ | 1,173 | 63 | 5.37 | 34 | 2.90 | 51.85 | 0.00800 |
| trnT-trnL | 1,055 | 46 | 4.36 | 17 | 1.61 | 70.37 | 0.00761 |
| ndhC-trnV | 701 | 54 | 7.70 | 27 | 3.85 | 59.26 | 0.00544 |
| petA-psbJ | 718 | 42 | 5.85 | 17 | 2.37 | 70.37 | 0.00807 |
| rpl32-trnL | 1,296 | 69 | 5.32 | 34 | 2.62 | 59.26 | 0.00766 |
| ycf1b | 1,350 | 73 | 5.41 | 42 | 3.11 | 92.59 | 0.00816 |
| petA-psbJ+ycf1b | 2,068 | 115 | 5.56 | 59 | 2.85 | 92.59 | 0.00813 |
| trnT-trnL+ycf1b | 2,405 | 119 | 4.95 | 59 | 2.45 | 100.00 | 0.00795 |
| Six-marker combination | 6,293 | 347 | 5.51 | 171 | 2.72 | 100.00 | 0.00760 |
Using the distance-based species identification methods, the three universal DNA barcodes had 18.4% (rbcL), 44.4% (matK), and 48.8% (trnH-psbA) discriminatory power in Styrax. Combining rbcL and matK, the success rate was 48.15%, and the success rate of the combined three barcodes was 70.37%. The results obtained using the tree-based method are shown in Figure 3(a). The phylogenetic tree had a lower resolution and lower support values. Four individuals of S. tonkinensis did not form a monophyletic clade.
Figure 3.
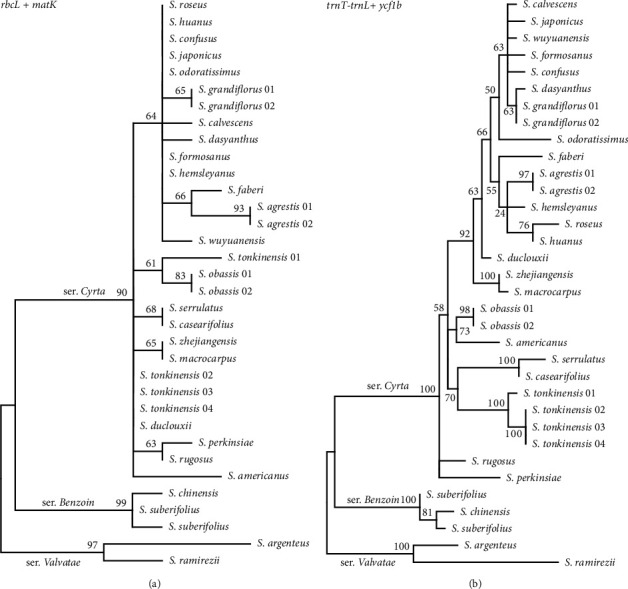
Phylogenetic tree of Styrax based on gene markers: (a) standard DNA barcodes (rbcL+matK); (b) specific DNA barcodes (trnT-trnL+ycf1b).
3.4. Identification of Specific DNA Barcodes of Styrax
Using the slide window method, π values ranged from 0 to 0.01113 in a 600 bp window size, the D values ranged from 0 to 0.0202, and Z values ranged from 0.2059 to 1. We considered variable regions with π values > 0.008, D values > 0.01, and Z values < 0.3. Six variable regions (rps16-trnQ, trnT-trnL, ndhC-trnV, petA-psbJ, rpl32-trnL, and ycf1b) were identified in the Styrax chloroplast genome (Figure 4). These regions included five intergenic regions (rps16-trnQ, trnT-trnL, ndhC-trnV, petA-psbJ, and rpl32-trnL), and one was the coding region of ycf1 (ycf1b). Four intergenic regions (rps16-trnQ, trnT-trnL, ndhC-trnV, and petA-psbJ) were located in the LSC region, and rpl32-trnL and ycf1b were located in the SSC region.
Figure 4.
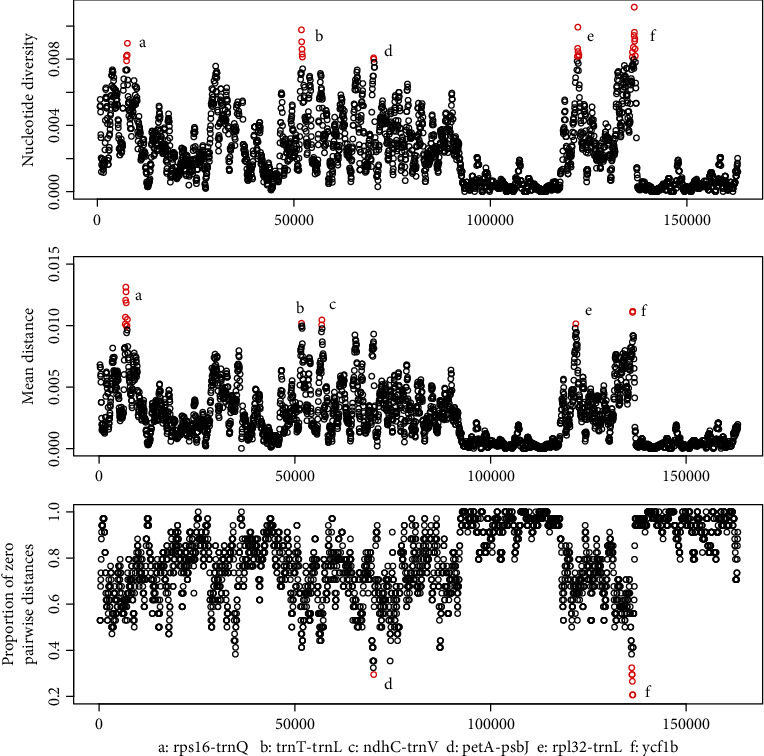
Specific DNA barcode development. Window length: 600 bp; step size: 50 bp; x-axis: position of the midpoint of a window. The three figures are the nucleotide diversity (π), mean sequence distance (D), and the proportion of zero pairwise distances (Z) among the species.
The percentage of variable sites among these six regions ranged from 4.36 to 5.85, and the parsimony-informative sites ranged from 1.61 to 3.85. According to the π values, ycf1b showed the highest variability in Styrax, followed by petA-psbJ, rps16-trnQ, rpl32-trnL, trnT-trnL, and ndhC-trnV. Using the distance methods, ycf1b had 92.56% discriminatory power, followed by trnT-trnL and petA-psbJ. Combined with ycf1b and trnT-trnL, all Styrax species were successfully distinguished. The tree-based results are presented in Figure 3(b). Compared to universal DNA barcodes, the combination of ycf1b and trnT-trnL had a higher resolution. Thus, ycf1b and trnT-trnL were chosen as Styrax-specific chloroplast DNA barcodes. The primers designed for the two regions are listed in Table S4, and the primers were tested to work well.
3.5. Superbarcode of Styrax
The 47 whole Styrax chloroplast genomes had an aligned length of 163,099 sites with 3,160 variable sites (1.94%) and 1,481 parsimony-informative sites (0.91%). The mean nucleotide diversity was found to be 0.00231 (Table 4). The genetic P distance of the Styrax species is shown in Figure 5. The mean genetic distance was 0.00244, the lowest divergence (0.0003) was between S. macrocarpus and S. zhejiangensis, and the largest sequence divergence (0.00611) was between S. casearifolia and S. ramirezii. The discriminatory power of the whole chloroplast genome as a DNA barcode was assessed using distance- and tree-based methods. Compared to the universal DNA barcodes or the six newly specific DNA barcodes, the whole chloroplast genome data exhibited the highest discriminatory power (Table 4 and Figure 6).
Table 4.
Chloroplast genome sequence variable in the Styrax.
| Regions | Length | Variable sites | Information sites | Nucleotide diversity | ||
|---|---|---|---|---|---|---|
| Numbers | % | Numbers | % | |||
| LSC | 91,693 | 2,301 | 2.51 | 1,088 | 1.19 | 0.0031 |
| SSC | 19,066 | 649 | 3.40 | 302 | 1.58 | 0.00399 |
| IR | 26,172 | 106 | 0.41 | 46 | 0.18 | 0.00045 |
| Complete cp genome | 163,099 | 3,160 | 1.94 | 1,481 | 0.91 | 0.00231 |
Figure 5.
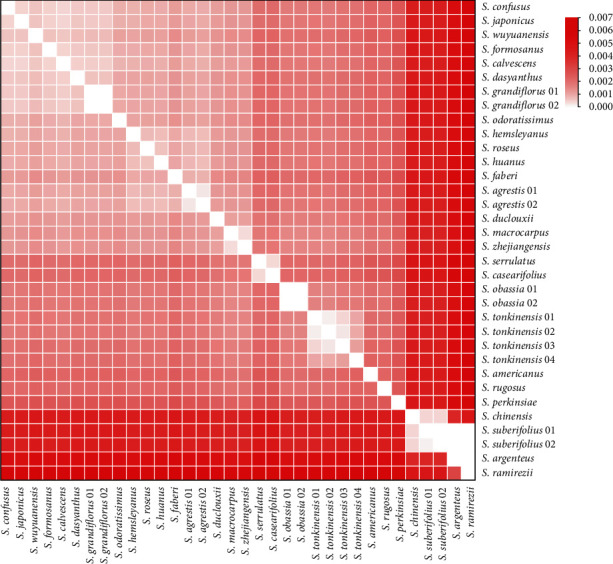
Pairwise genetic distances among Styrax samples.
Figure 6.
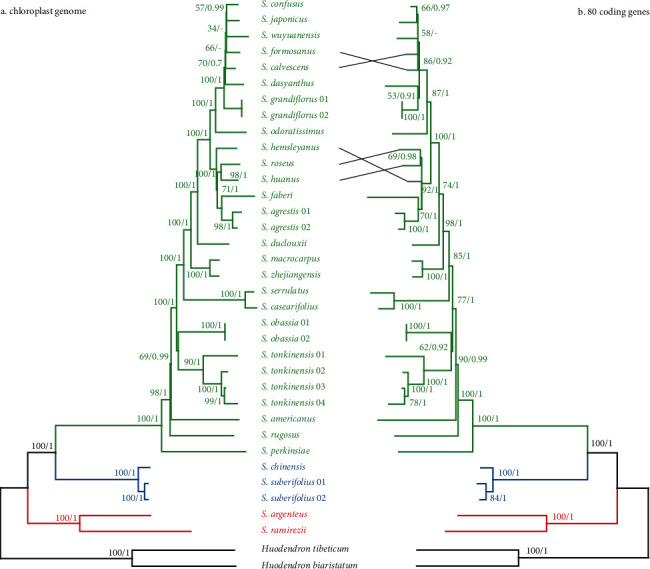
Phylogenetic trees of Styrax: (a) ML tree based on the whole chloroplast genome; (b) ML tree using 80 coding genes. ML bootstrap support value/Bayesian posterior probability presented at each node.
3.6. Phylogenetic Inference
The phylogenetic tree inferred from the chloroplast genome and 80 coding gene datasets was similar to the phylogenetic relationships of Styrax species (Figure 6). The best-fit model GTR+G from ModelFinder was used for the ML and BI analyses. The topologies of the ML and BI trees and the two datasets were nearly identical. All Styrax species formed a monophyletic clade (BS = 100/PP = 1), and some notes had shortened branches, indicating low divergence among some Styrax species. Three lineages were formed in the phylogenetic tree. Styrax ramirezii and S. argenteus were the first diverging branches (series Valvatae) and were sisters to the remaining species. Styrax chinensis and S. suberifolius formed the second lineage (series Benzoin). The remaining species formed the third lineage (series Cyrta) with a 100% bootstrap value.
4. Discussion
4.1. Styrax Chloroplast Genome Evolution
The Styrax chloroplast genomes were similar to other angiosperms, indicating that the chloroplast genome was a quadripartite structure, including a large single-copy, a small single-copy, and a pair of inverted repeats. The Styrax chloroplast genomes have highly similar genome structures, genome sizes, and gene contents (Figure 1), and the single-copy regions and noncoding regions are more variable than the IRs and coding regions (Figure S1).
SSRs, which consist of tandemly repeated motifs of six base pairs (bp) or less, are important markers for population genetics and germplasm management [5, 40, 41]. In the chloroplast genome, SSRs are dominated by mono- and dinucleotide repeats and A/T bases are the most common [7, 42, 43]. This was consistent with previous findings that the chloroplast genome is usually composed of polyA and polyT repeats [44]. A total of 61–74 SSRs were found in the Styrax chloroplast genomes (Figure 2), which were more abundant than other species of Styracaceae [45].
Small inversions have been found in most related species [46–48]. All of the inversions formed stem-loop structures, and there was no correlation between the lengths of inversions and inverted repeats [7]. Many small inversions are generated by parallel or back mutation events during chloroplast genome evolution [49, 50]. The inversion in trnSGCU–trnGGCC and petN–psbM occurred in some samples of S. tonkinensis, and trnSGCU–trnGGCC and psaJ–rpl33 occurred in only one sample of S. agrestis. These inversions did not show phylogenetic signals (Table S3). Recent studies suggest that some small inversions are valuable for phylogenetic relationships in some groups [51–54].
4.2. Species Identification from Genes to Genomes
Rapid and accurate species delimitation is very important in biology. Morphological characteristics are the key methods used to identify the samples [55]. DNA barcoding is a new effective tool widely used in species identification since 2003 [56]. Selecting a DNA marker as a universal DNA barcode is essential for the diversity of organisms [57]. However, the selection of universal barcode(s) in plants is more complex than other organisms. The CBOL Working Group recommended three chloroplast markers (rbcL, matK, and trnH-psbA) and nuclear ITS as universal DNA barcodes for higher plants [58]. More evidence has shown that these markers have lower variability and discrimination power [15, 59–61]. This study assessed the three chloroplast markers in Styrax to evaluate their suitability for species resolution. Using the distance- and tree-based methods, their discrimination power was barely satisfactory (Table 3). Combining the two core DNA barcodes (rbcL and matK) had a resolution rate of less than 50%. ITS is regarded as a powerful phylogenetic marker at the species level, showing high interspecific divergence [62]. However, phylogenetic resolution using ITS data was also limited in Styrax [23].
The chloroplast genome sequence mutations (SNPs and indels) were not random and clustered into mutation hotspot regions, and these regions were selected as specific DNA barcodes [5, 59, 60, 63]. Specific DNA barcodes revealed a higher resolution rate than universal DNA barcodes. For example, Oryza chloroplast genomes were compared and five or six specific DNA barcodes for Oryza were identified [14, 64]. Using comparison of oak species, two intergenic regions matK-trnK-rps16 and trnR-atpA and two coding regions ndhF and ycf1b were developed as specific DNA barcodes [15].
This study identified six variable markers (rps16-trnQ, trnT-trnL, ndhC-trnV, petA-psbJ, rpl32-trnL, and ycf1b). These markers had higher variable and species resolutions than the universal DNA barcodes (Table 3). According to the success discrimination rate, two markers (trnT-trnL and ycf1b) were selected as Styrax-specific chloroplast DNA barcodes. TrnT-trnL is an intergenic spacer region and has been frequently used in plant phylogeny [65, 66]. Dong et al. were the first to report ycf1a and ycf1b markers, located in the second-longest gene ycf1 [60, 61]. ycf1b was more variable than the two core DNA barcodes, rbcL and matK [61]. Combining ycf1b and trnT-trnL significantly improved the identification success rate, and these two markers were chosen as the Styrax-specific DNA barcodes.
The advent of next-generation sequencing (NGS) technologies has led to a decrease in the cost of genome sequencing. Genomic data have extended the concept of the DNA barcoding approach, referred to as “superbarcoding” [14, 16], “ultrabarcoding” [67], or “plant barcoding 2.0” [17]. Compared to the nuclear and mitochondrial genomes, the chloroplast genomes were easily sequenced using genomic skimming [68–70], and the chloroplast genome has sufficient sequence variation in closely related species [6]. More studies showed that the chloroplast genome had a sufficiently high mutation rate which enables species identification and it may be best suited as superbarcodes for plants [70]. For example, using the chloroplast genome sequences, all 20 sampled Olea species had been successfully distinguished and even some subspecies of O. europaea can be identified [70]. Wu et al. also indicated that the chloroplast genome can be used to effectively differentiate Fritillaria species [71]. Moreover, chloroplast genome data have been widely used in plant phylogenetics at different taxonomic levels [8, 72, 73]. In this study, the chloroplast genome showed sufficient information for Styrax species identification (Table 4 and Figure 6). In addition to assembling the chloroplast genomes, clean reads from NGS could further be used to retrieve nuclear genome sequences, giving the possibilities for accurate species identification and phylogenetic relationship reconstruction.
5. Conclusions
The analyzed 34 Styrax chloroplast genomes have a similar structure, gene number, and gene order. SSR sites and small inversions were also identified. Comparisons of the Styrax chloroplast genome sequence divergences revealed that rps16-trnQ, trnT-trnL, ndhC-trnV, petA-psbJ, rpl32-trnL, and ycf1b were variable markers. Furthermore, ycf1b and trnT-trnL were suggested as Styrax-specific DNA barcodes. The whole chloroplast genome is potentially available as a superbarcode for Styrax species. This study demonstrated the potential of chloroplast genome data to improve the phylogenetic resolution.
Acknowledgments
This work was supported by 2020 Research Program of Sanya Yazhou Bay Science and Technology City (Grant No. SKJC-2020-02-015). We thank the DNA Bank of China for providing materials.
Contributor Information
Yun Song, Email: sydef1016@163.com.
MingFu Li, Email: limf9@sina.com.
Data Availability
The 17 Styrax chloroplast genomes are available in the GenBank database (accession numbers: MZ285733 to MZ285749).
Conflicts of Interest
The authors declare that there are no conflicts of interest regarding the publication of this paper.
Authors' Contributions
Yun Song, Wenjun Zhao, and Mingfu Li designed the experiment and drafted and made revisions to the manuscript. Jin Xu collected samples and performed the experiment. Yun Song and Jin Xu analyzed the data. Mingfu Li and Yongjiang Zhang contributed reagents and analysis tools. All of the authors have read and approved the final manuscript.
Supplementary Materials
Table S1: taxa included in this study with locality, voucher, and GenBank accession numbers. The bold font samples were sequenced in this study. Table S2: summary of the sequencing data for 17 Styrax samples. Table S3: the information of small inversions. Table S4: the primers used for amplification the specific Styrax DNA barcodes. Figure S1: visualization of genome alignment of the Styrax chloroplast genomes. The x-axis showed the coordinate between the chloroplast genomes.
References
- 1.Wicke S., Schneeweiss G. M., Depamphilis C. W., Muller K. F., Quandt D. The evolution of the plastid chromosome in land plants: gene content, gene order, gene function. Plant Molecular Biology . 2011;76(3-5):273–297. doi: 10.1007/s11103-011-9762-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daniell H., Lin C.-S., Yu M., Chang W.-J. Chloroplast genomes: diversity, evolution, and applications in genetic engineering. Genome Biology . 2016;17(1):1–29. doi: 10.1186/s13059-016-1004-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Olejniczak S. A., Lojewska E., Kowalczyk T., Sakowicz T. Chloroplasts: state of research and practical applications of plastome sequencing. Planta . 2016;244(3):517–527. doi: 10.1007/s00425-016-2551-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhao F., Chen Y.-P., Salmaki Y., et al. An updated tribal classification of Lamiaceae based on plastome phylogenomics. BMC Biology . 2021;19(1):p. 2. doi: 10.1186/s12915-020-00931-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Dong W., Liu Y., Xu C., et al. Chloroplast phylogenomic insights into the evolution of Distylium (Hamamelidaceae) BMC Genomics . 2021;22(1):p. 293. doi: 10.1186/s12864-021-07590-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Dong W., Xu C., Liu Y., Shi J., Li W., Suo Z. Chloroplast phylogenomics and divergence times of Lagerstroemia (Lythraceae) BMC Genomics . 2021;22(1):p. 434. doi: 10.1186/s12864-021-07769-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dong W., Xu C., Wen J., Zhou S. Evolutionary directions of single nucleotide substitutions and structural mutations in the chloroplast genomes of the family Calycanthaceae. BMC Evolutionary Biology . 2020;20(1):p. 96. doi: 10.1186/s12862-020-01661-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Xu L.-S., Herrando-Moraira S., Susanna A., Galbany-Casals M., Chen Y.-S. Phylogeny, origin and dispersal of Saussurea (Asteraceae) based on chloroplast genome data. Molecular Phylogenetics and Evolution . 2019;141, article ??? doi: 10.1016/j.ympev.2019.106613. [DOI] [PubMed] [Google Scholar]
- 9.Zhang X., Deng T., Moore M. J., et al. Plastome phylogenomics of Saussurea (Asteraceae: Cardueae) BMC Plant Biology . 2019;19(1):p. 290. doi: 10.1186/s12870-019-1896-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang X., Zhang H. J., Landis J. B., et al. Plastome phylogenomic analysis ofTorreya(Taxaceae) Journal of Systematics and Evolution . 2019;57(6):607–615. doi: 10.1111/jse.12482. [DOI] [Google Scholar]
- 11.Mohamoud Y. A., Mathew L. S., Torres M. F., et al. Novel subpopulations in date palm (Phoenix dactylifera) identified by population-wide organellar genome sequencing. BMC Genomics . 2019;20(1):p. 498. doi: 10.1186/s12864-019-5834-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Qiao J., Zhang X., Chen B., et al. Comparison of the cytoplastic genomes by resequencing: insights into the genetic diversity and the phylogeny of the agriculturally important genus Brassica. BMC Genomics . 2020;21(1):p. 480. doi: 10.1186/s12864-020-06889-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chase M. W., Cowan R. S., Hollingsworth P. M., et al. A proposal for a standardised protocol to barcode all land plants. Taxon . 2007;56(2):295–299. doi: 10.1002/tax.562004. [DOI] [Google Scholar]
- 14.Zhang W., Sun Y., Liu J., et al. DNA barcoding of Oryza: conventional, specific, and super barcodes. Plant Molecular Biology . 2021;105(3):215–228. doi: 10.1007/s11103-020-01054-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pang X., Liu H., Wu S., et al. Species identification of oaks (Quercus L., Fagaceae) from gene to genome. International Journal of Molecular Sciences . 2019;20(23):p. 5940. doi: 10.3390/ijms20235940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li X., Yang Y., Henry R. J., Rossetto M., Wang Y., Chen S. Plant DNA barcoding: from gene to genome. Biological Reviews . 2015;90(1):157–166. doi: 10.1111/brv.12104. [DOI] [PubMed] [Google Scholar]
- 17.Hollingsworth P. M., Li D. Z., M van der Bank, Twyford A. D. Telling plant species apart with DNA: from barcodes to genomes. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences . 2016;371(1702):p. 20150338. doi: 10.1098/rstb.2015.0338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Fritsch P. W. Phylogeny of Styrax based on morphological characters, with implications for biogeography and infrageneric classification. Systematic Botany . 1999;24(3):356–378. doi: 10.2307/2419695. [DOI] [Google Scholar]
- 19.Fritsch P. W. New species and taxonomic changes in Styrax (Styracaceae) from South America. Novon . 2004;14(1):43–57. [Google Scholar]
- 20.Fritsch P. W. Two new species ofStyrax(Styracaceae) from South America. Novon: A Journal for Botanical Nomenclature . 2015;24(1):9–13. doi: 10.3417/2014030. [DOI] [Google Scholar]
- 21.Zhou H., Yang W., Li M., Yu X.-L. Styrax rhytidocarpus (Styracaceae), a new species from Hunan, China. Phytotaxa . 2015;230(1):87–91. doi: 10.11646/phytotaxa.230.1.9. [DOI] [Google Scholar]
- 22.Xu W., Yang P., Huang Y., Zhang R., Tang M. <strong><em>Styrax hwangiae</em> (Styracaceae), a new species endemic to limestone areas of Guangxi, China</strong>. Phytotaxa . 2021;478(1):171–178. doi: 10.11646/phytotaxa.478.1.13. [DOI] [Google Scholar]
- 23.Fritsch P. W. Phylogeny and biogeography of the flowering plant genus Styrax (Styracaceae) based on chloroplast DNA restriction sites and DNA sequences of the internal transcribed spacer region. Molecular Phylogenetics and Evolution . 2001;19(3):387–408. doi: 10.1006/mpev.2001.0933. [DOI] [PubMed] [Google Scholar]
- 24.Susilowati A., Rachmat H. H., Fadilah W. N., Kusuma Y. S. Phylogeny of kemenyan toba (Styrax sumatrana) Inferred from trnl-trnf Chloroplast DNA Sequence. Proceedings of the International Conference on Natural Resources and Technology (ICONART 2019) 2019. pp. 26–29.
- 25.Fritsch P. W., Cruz B. C., Simison W. B., Campbell A. J., Harris J. K. Early phylogenetic divergence of gynodioecious species warrants the recognition of subseries in <I>Styrax</I> series <I>Valvatae</I>. Systematic Botany . 2016;40(4):1081–1092. doi: 10.1600/036364415X690120. [DOI] [Google Scholar]
- 26.Li J., Wang S., Jing Y., Wang L., Zhou S. A modified CTAB protocol for plant DNA extraction. Chinese Bulletin of Botany . 2013;48(1):72–78. doi: 10.3724/SP.J.1259.2013.00072. [DOI] [Google Scholar]
- 27.Bolger A. M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics . 2014;30(15):2114–2120. doi: 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jin J.-J., Yu W.-B., Yang J.-B., et al. GetOrganelle: a fast and versatile toolkit for accurate de novo assembly of organelle genomes. Genome Biology . 2020;21(1):p. 241. doi: 10.1186/s13059-020-02154-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Huang D. I., Cronk Q. C. B. Plann: a command-line application for annotating plastome sequences. Applications in Plant Sciences . 2015;3(8):p. 1500026. doi: 10.3732/apps.1500026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Greiner S., Lehwark P., Bock R. OrganellarGenomeDRAW (OGDRAW) version 1.3.1: expanded toolkit for the graphical visualization of organellar genomes. Nucleic Acids Research . 2019;47(W1):W59–W64. doi: 10.1093/nar/gkz238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Frazer K. A., Pachter L., Poliakov A., Rubin E. M., Dubchak I. VISTA: computational tools for comparative genomics. Nucleic Acids Research . 2004;32(Web Server):W273–W279. doi: 10.1093/nar/gkh458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rambaut A. Se-Al: sequence alignment editor. version 2.0. 1996. http://tree.bio.ed.ac.uk/software/seal/
- 33.Kumar S., Stecher G., Li M., Knyaz C., Tamura K. MEGA X: molecular evolutionary genetics analysis across computing platforms. Molecular Biology and Evolution . 2018;35(6):1547–1549. doi: 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rozas J., Ferrer-Mata A., Sanchez-DelBarrio J. C., et al. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Molecular Biology and Evolution . 2017;34(12):3299–3302. doi: 10.1093/molbev/msx248. [DOI] [PubMed] [Google Scholar]
- 35.Brown S. D., Collins R. A., Boyer S., et al. Spider: an R package for the analysis of species identity and evolution, with particular reference to DNA barcoding. Molecular Ecology Resources . 2012;12(3):562–565. doi: 10.1111/j.1755-0998.2011.03108.x. [DOI] [PubMed] [Google Scholar]
- 36.Kumar S., Stecher G., Tamura K. MEGA7: molecular evolutionary genetics analysis version 7.0 for bigger datasets. Molecular Biology and Evolution . 2016;33(7):1870–1874. doi: 10.1093/molbev/msw054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Kalyaanamoorthy S., Minh B. Q., Wong T. K. F., von Haeseler A., Jermiin L. S. ModelFinder: fast model selection for accurate phylogenetic estimates. Nature Methods . 2017;14(6):587–589. doi: 10.1038/nmeth.4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kozlov A. M., Darriba D., Flouri T., Morel B., Stamatakis A. RAxML-NG: a fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics . 2019;35(21):4453–4455. doi: 10.1093/bioinformatics/btz305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ronquist F., Teslenko M., van der Mark P., et al. MrBayes 3.2: efficient Bayesian phylogenetic inference and model choice across a large model space. Systematic Biology . 2012;61(3):539–542. doi: 10.1093/sysbio/sys029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Li B., Lin F., Huang P., Guo W., Zheng Y. Development of nuclear SSR and chloroplast genome markers in diverse Liriodendron chinense germplasm based on low-coverage whole genome sequencing. Biological Research . 2020;53(1):p. 21. doi: 10.1186/s40659-020-00289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cao J., Jiang D., Zhao Z., et al. Development of chloroplast genomic resources in Chinese yam (Dioscorea polystachya) BioMed Research International . 2018;2018:11. doi: 10.1155/2018/6293847.6293847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ping J., Feng P., Li J., Zhang R., Su Y., Wang T. Molecular evolution and SSRs analysis based on the chloroplast genome ofCallitropsis funebris. Ecology and Evolution . 2021;11(9):4786–4802. doi: 10.1002/ece3.7381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wang M., Wang X., Sun J., et al. Phylogenomic and evolutionary dynamics of inverted repeats across Angelica plastomes. BMC Plant Biology . 2021;21(1):p. 26. doi: 10.1186/s12870-020-02801-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sablok G., Mudunuri S. B., Patnana S., Popova M., Fares M. A., Porta N. L. ChloroMitoSSRDB: open source repository of perfect and imperfect repeats in organelle genomes for evolutionary genomics. DNA Research . 2013;20(2):127–133. doi: 10.1093/dnares/dss038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yan M., Moore M. J., Meng A., Yao X., Wang H. The first complete plastome sequence of the basal asterid family Styracaceae (Ericales) reveals a large inversion. Plant Systematics and Evolution . 2017;303(1):61–70. doi: 10.1007/s00606-016-1352-0. [DOI] [Google Scholar]
- 46.Dong W., Xu C., Li D., et al. Comparative analysis of the complete chloroplast genome sequences in psammophyticHaloxylonspecies (Amaranthaceae) PeerJ . 2016;4, article e2699 doi: 10.7717/peerj.2699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dong W., Cheng T., Xu C., Wen J., Zhou S. Patterns and rates of nucleotide substitutions, microstructural mutations in plastid genomes of Calycanthaceae. BMC Evolutionary Biology . 2019:p. e2699. [Google Scholar]
- 48.Whitlock B. A., Hale A. M., Groff P. A. Intraspecific inversions pose a challenge for the trnH-psbA plant DNA barcode. PLoS One . 2010;5(7, article e11533) doi: 10.1371/journal.pone.0011533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim K. J., Lee H. L. Widespread occurrence of small inversions in the chloroplast genomes of land plants. Molecules and Cells . 2005;19(1):104–113. [PubMed] [Google Scholar]
- 50.Catalano S. A., Saidman B. O., Vilardi J. C. Evolution of small inversions in chloroplast genome: a case study from a recurrent inversion in angiosperms. Cladistics . 2009;25(1):93–104. doi: 10.1111/j.1096-0031.2008.00236.x. [DOI] [PubMed] [Google Scholar]
- 51.Graham S. W., Reeves P. A., Burns A. C. E., Olmstead R. G. Microstructural changes in noncoding chloroplast DNA: interpretation, evolution, and utility of indels and inversions in basal angiosperm phylogenetic inference. International Journal of Plant Sciences . 2000;161(S6):S83–S96. doi: 10.1086/317583. [DOI] [Google Scholar]
- 52.Lehtonen S., Myllys L., Huttunen S. Phylogenetic analysis of non-coding plastid DNA in the presence of short inversions. Phytotaxa . 2009;1(1):3–20. doi: 10.11646/phytotaxa.1.1.2. [DOI] [Google Scholar]
- 53.Wolf P. G., Duffy A. M., Roper J. M. Phylogenetic use of inversions in fern chloroplast genomes. American Fern Journal . 2009;99(2):132–134. [Google Scholar]
- 54.Liao Y. Y., Liu Y., Liu X., et al. The complete chloroplast genome ofMyriophyllum spicatumreveals a 4-kb inversion and new insights regarding plastome evolution in Haloragaceae. Ecology and Evolution . 2020;10(6):3090–3102. doi: 10.1002/ece3.6125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cowan R. S., Fay M. F. Challenges in the DNA barcoding of plant material. Methods in Molecular Biology . 2012;862:23–33. doi: 10.1007/978-1-61779-609-8_3. [DOI] [PubMed] [Google Scholar]
- 56.Hebert P. D. N., Cywinska A., Ball S. L., DeWaard J. R. Biological identifications through DNA barcodes. Proceedings of the Royal Society of London, Series B: Biological Sciences . 2003;270(1512):313–321. doi: 10.1098/rspb.2002.2218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hollingsworth P. M., Graham S. W., Little D. P. Choosing and using a plant DNA barcode. PLoS One . 2011;6(5, article e19254) doi: 10.1371/journal.pone.0019254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Kress W. J., Wurdack K. J., Zimmer E. A., Weigt L. A., Janzen D. H. Use of DNA barcodes to identify flowering plants. Proceedings of the National Academy of Sciences of the United States of America . 2005;102(23):8369–8374. doi: 10.1073/pnas.0503123102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Song Y., Wang S., Ding Y., et al. Chloroplast genomic resource of Paris for species discrimination. Scientific Reports . 2017;7(1):p. 3427. doi: 10.1038/s41598-017-02083-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dong W., Liu J., Yu J., Wang L., Zhou S. Highly variable chloroplast markers for evaluating plant phylogeny at low taxonomic levels and for DNA barcoding. PLoS One . 2012;7(4, article e35071) doi: 10.1371/journal.pone.0035071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dong W., Xu C., Li C., et al. ycf1, the most promising plastid DNA barcode of land plants. Scientific Reports . 2015;5(1):p. ???. doi: 10.1038/srep08348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.B O L G China Plant, Li D. Z., Gao L. M., et al. Comparative analysis of a large dataset indicates that internal transcribed spacer (ITS) should be incorporated into the core barcode for seed plants. Proceedings of the National Academy of Sciences of the United States of America . 2011;108(49):19641–19646. doi: 10.1073/pnas.1104551108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Song Y., Zhang Y., Xu J., Li W., Li M. Characterization of the complete chloroplast genome sequence of Dalbergia species and its phylogenetic implications. Scientific Reports . 2019;9(1):p. 20401. doi: 10.1038/s41598-019-56727-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Song Y., Chen Y., Lv J., et al. Development of chloroplast genomic resources for Oryza species discrimination. Frontiers in Plant Science . 2017;8:p. 1854. doi: 10.3389/fpls.2017.01854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hamzeh M., Dayanandan S. Phylogeny of Populus (Salicaceae) based on nucleotide sequences of chloroplast TRNT-TRNF region and nuclear rDNA. American Journal of Botany . 2004;91(9):1398–1408. doi: 10.3732/ajb.91.9.1398. [DOI] [PubMed] [Google Scholar]
- 66.Achigan-Dako E. G., Degbey H., Hale I., Blattner F. R. Georeferenced phylogenetic analysis of a global collection of wild and cultivatedCitrullusspecies. Ecology and Evolution . 2021;11(4):1918–1936. doi: 10.1002/ece3.7189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kane N., Sveinsson S., Dempewolf H., et al. Ultra-barcoding in cacao (Theobroma spp.; Malvaceae) using whole chloroplast genomes and nuclear ribosomal DNA. American Journal of Botany . 2012;99(2):320–329. doi: 10.3732/ajb.1100570. [DOI] [PubMed] [Google Scholar]
- 68.Liu L., Wang Y., He P., et al. Chloroplast genome analyses and genomic resource development for epilithic sister genera Oresitrophe and Mukdenia (Saxifragaceae), using genome skimming data. BMC Genomics . 2018;19(1):p. 235. doi: 10.1186/s12864-018-4633-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang Y. B., Liu B. B., Nie Z. L., et al. Major clades and a revised classification ofMagnoliaand Magnoliaceae based on whole plastid genome sequences via genome skimming. Journal of Systematics and Evolution . 2020;58(5):673–695. doi: 10.1111/jse.12588. [DOI] [Google Scholar]
- 70.Dong W., Sun J., Liu Y., et al. Phylogenomic relationships and species identification of the olive genusOlea(Oleaceae) Journal of Systematics and Evolution . 2021 doi: 10.1111/jse.12802. [DOI] [Google Scholar]
- 71.Wu L., Wu M., Cui N., et al. Plant super-barcode: a case study on genome-based identification for closely related species of Fritillaria. Chinese Medicine . 2021;16(1):p. 52. doi: 10.1186/s13020-021-00460-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Burke S. V., Ungerer M. C., Duvall M. R. Investigation of mitochondrial-derived plastome sequences in the Paspalum lineage (Panicoideae; Poaceae) BMC Plant Biology . 2018;18(1):p. 152. doi: 10.1186/s12870-018-1379-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sun Y., Moore M. J., Landis J. B., et al. Plastome phylogenomics of the early-diverging eudicot family Berberidaceae. Molecular Phylogenetics and Evolution . 2018;128:203–211. doi: 10.1016/j.ympev.2018.07.021. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: taxa included in this study with locality, voucher, and GenBank accession numbers. The bold font samples were sequenced in this study. Table S2: summary of the sequencing data for 17 Styrax samples. Table S3: the information of small inversions. Table S4: the primers used for amplification the specific Styrax DNA barcodes. Figure S1: visualization of genome alignment of the Styrax chloroplast genomes. The x-axis showed the coordinate between the chloroplast genomes.
Data Availability Statement
The 17 Styrax chloroplast genomes are available in the GenBank database (accession numbers: MZ285733 to MZ285749).