

Abstract
ZFTA (C11orf95)–a gene of unknown function–partners with a variety of transcriptional co-activators in translocations that drive supratentorial ependymoma, a frequently lethal brain tumor. Understanding the function of ZFTA is key to developing therapies that inhibit these fusion proteins. Here, using a combination of transcriptomics, chromatin immunoprecipitation-sequencing, and proteomics, we interrogated a series of deletion-mutant genes to identify a tri-partite transformation mechanism of ZFTA-containing fusions, including: spontaneous nuclear translocation, extensive chromatin binding, and SWI/SNF, SAGA and NuA4/Tip60 HAT chromatin modifier complex recruitment. Thereby, ZFTA tethers fusion proteins across the genome, modifying chromatin to an active state, and enabling its partner transcriptional co-activators to promote promiscuous expression of a transforming transcriptome. Using mouse models, we validate further those elements of ZFTA-fusion proteins that are critical for transformation–including ZFTA zinc fingers and partner gene transactivation domains–thereby unmasking vulnerabilities for therapeutic targeting.
Keywords: Ependymoma, RELA, YAP1, ZFTA, translocation
INTRODUCTION
Brain tumors are the most common and lethal childhood cancers (1). Although these tumors frequently resist conventional therapies, they often contain genetic alterations that might serve as targets for novel treatments (2). But these genetic alterations are complex and poorly understood, impeding efforts to target them pharmacologically. Drugging brain tumor drivers effectively will require a better understanding of how they transform neural lineages.
Ependymomas are tumors of the brain and spinal cord that are incurable in up to 40% of patients (3). While histologically similar, ependymomas from the different regions of the central nervous system are distinct entities, with different lineage origins, transcriptomes, genetic alterations and clinical outcomes (4–9). Over 70% of supratentorial ependymomas (ST-EPs) contain translocations between ZFTA and RELA (ZFTA-RELA), or more rarely, between ZFTA and YAP1 (ZFTA-YAP1) or MAML2 (ZFTA-MAML2) (6,8): we term this subgroup of ependymomas, ST-EP-ZFTAFUS. Recurrent translocations between ZFTA and MRTFB have also been described in chondroid lipomas (10,11) and ZFTA-RELA or ZFTA-YAP1 transgenes drive ST-EP-ZFTAFUS tumors in mice (6,12).
While ZFTA fusion proteins are transforming, how this gene cooperates with its various translocation partners to drive tumorigenesis remains to be determined. Here, we identify a tri-partite transformation mechanism of ZFTA-containing fusions that includes: active nuclear trafficking; zinc finger (ZF)-dependent chromatin binding and remodeling; and promiscuous activation of gene expression.
RESULTS
ZFTA-ZFs promote nuclear trafficking
ZFTA contains four evenly distributed ZF domains: between one and four of these are incorporated into fusion proteins with RELA or YAP1 (Figure 1A) (6). To understand how these fusion proteins transform cells, we studied ZFTA-RELAFUS1, ZFTA-RELAFUS2 and ZFTA-YAP1FUS that are observed in 56%, 33% and 10% of ST-EP-ZFTAFUS ependymomas, respectively (6,8). Since protein-protein and protein-DNA interactions can be species and cell context specific, we studied fusion proteins in both human (HEK293) and mouse (mNSC) cells: the latter are proven cells of origin of ependymoma in mice (5,6,12).
Figure 1. ZFTA-fusions translocate to the nucleus.
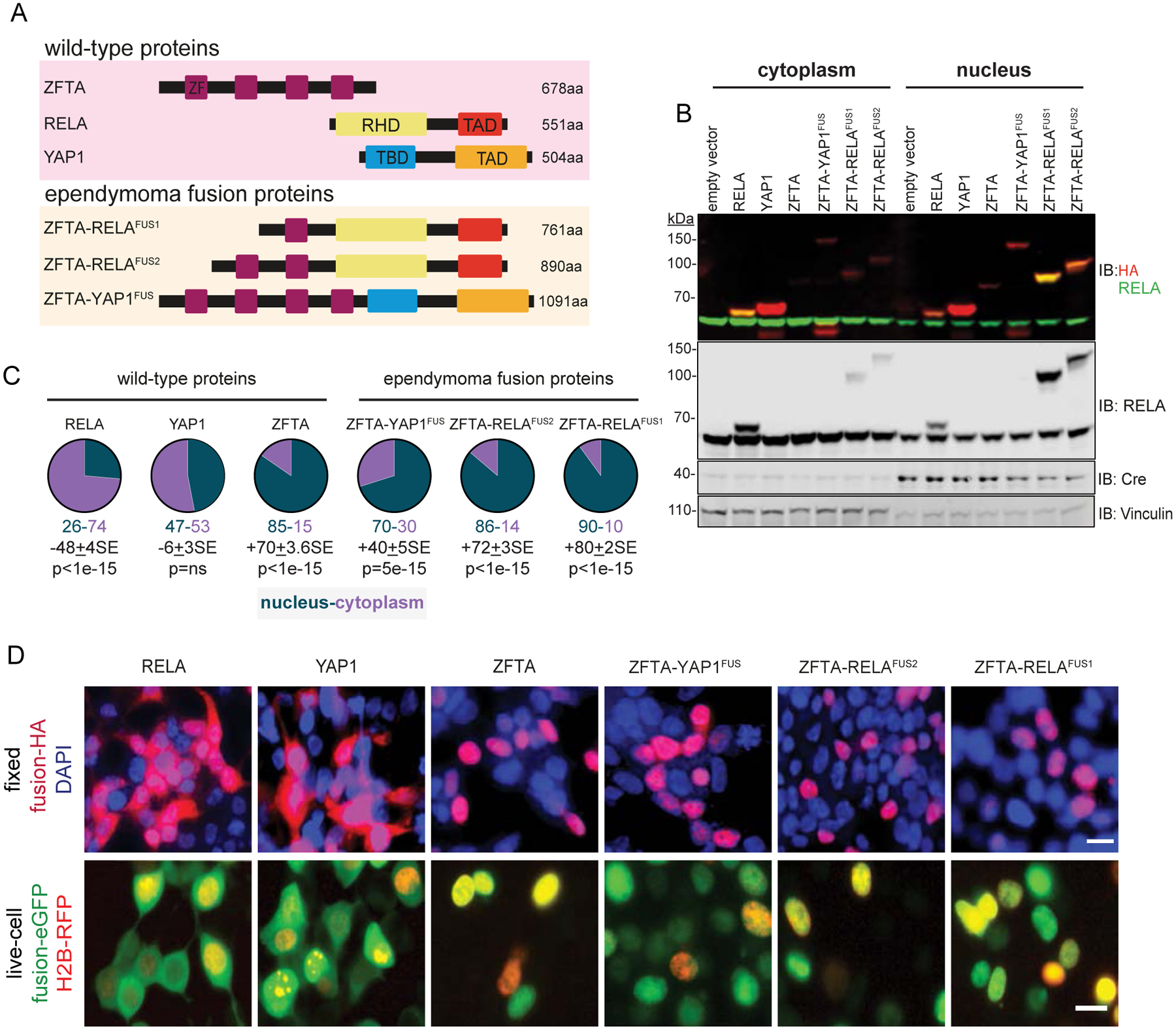
A. Schematics of wild-type and fusion proteins. Zinc Finger (ZF), REL homology (RHD), trans-activation (TAD) and TEAD-binding (TBD) domains. B. Immunoblots (IB) with indicated antibodies of cytoplasmic and nuclear fractions of HA-tagged proteins in HEK293 cells. Vinculin (cytoplasmic) and Cre (nuclear) are loading controls. C. Percent of proteins localized in cytoplasm or nucleus. Average difference + Standard Error (SE) and Mann-Whitney p-value (n=5), below. D. Top, localization of indicated HA-tagged proteins in DAPI counterstained and fixed HEK293 cells, or bottom, enhanced (e)-GFP tagged proteins in live, HEK293 cells expressing red fluorescence protein (RFP)-tagged Histone 2B (scale bar, 10μm).
Immunoblotting of subfractions of HEK293 cells transduced with hemagglutinin (3xHA)-tagged proteins, identified 74%±3.3SE (n=5) of RELA and 53%±2.7SE (n=5) of YAP1 in the cytoplasm, while 85%±3.4SE (n=5) of ZFTA was located in the nucleus (Figures 1B and C). Fusion of RELA or YAP1 to ZFTA reversed this cellular distribution: 90%±1.7SE of ZFTA-RELAFUS1 (n=5), 86%1.7±SE of ZFTA-RELAFUS2 (n=5) and 70%±1.8SE) of ZFTA-YAP1FUS (n=5) protein was located in the nucleus (Figure 1B and C). Similar distributions of fusion and wildtype partner proteins were observed by immunoblotting in mNSCs, and by immunofluorescence of fixed and living HEK293 cells (Figure 1D and Supplemental Figure S1A–C).
Deletion of the entire ZFTA-ZF–or just its core C2-H2 or bi-partite nuclear localization sequence–blocked ZFTA-RELAFUS1 nuclear trafficking in HEK293 and mNSCs (Figure 2A–E and Supplemental Figure S1B). Only one of the four ZFTA-ZFs within ZFTA-YAPFUS was required for nuclear trafficking (Figure 2D, E and Supplemental Figure S1C). Thus, a single ZFTA-ZF, and particularly its C2H2 and NLS motifs, is both necessary and sufficient for spontaneous nuclear trafficking of fusion proteins.
Figure 2. Domain mapping of ZFTA-fusion driven nuclear translocation.
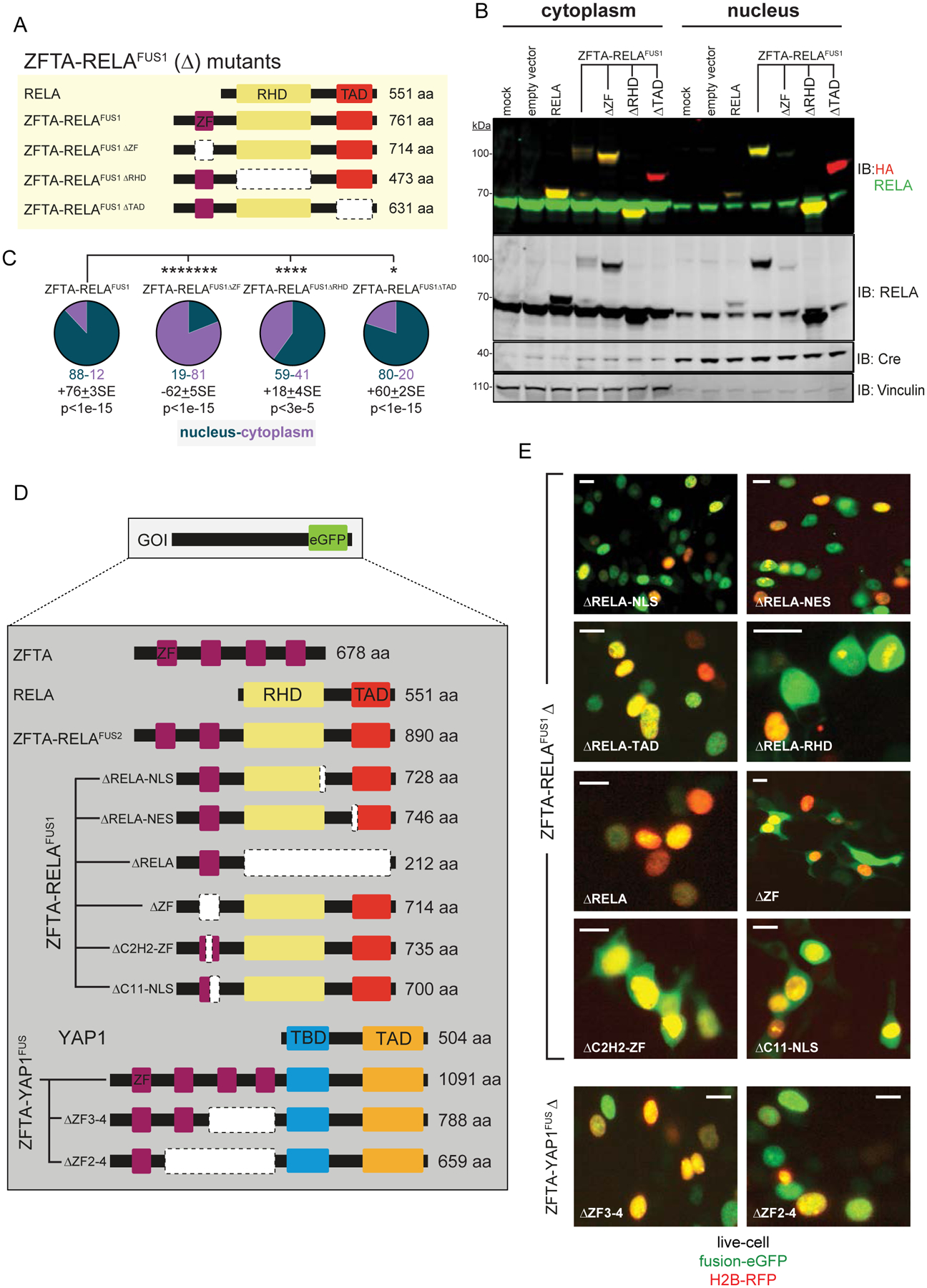
A. Schematics of deletion (Δ)-mutant fusion proteins. B. Immunoblots probed with indicated antibodies of cytoplasmic and nuclear fractions of HA-tagged proteins in HEK293 cells. Vinculin (cytoplasmic) and Cre (nuclear) are loading controls. C. Percent of proteins localized in cytoplasm or nucleus. Average difference ± Standard Error (SE) and Mann-Whitney p-value (n=5), below. D. Schematics of deletion-mutant fusion proteins. E. Localization of enhanced (e)-GFP tagged proteins in live HEK293 cells (scale bar, 10μm).
Deletion of the REL homology domain (RHD) reduced nuclear fusion protein content by ~30%, suggesting this region might mediate nuclear retention of fusion proteins, possibly through DNA or protein binding (Figure 2A–E and Supplemental Figure S1B). Conversely, the RELA transactivation domain (TAD) and its nuclear export sequence (NES) were dispensable for nuclear trafficking.
ZFTA-fusions upregulate ependymoma signature genes
Since RELA and YAP1 are transcription factors (13,14) and ZFTA-fusion proteins drive their translocation to the nucleus, then we reasoned that these fusion proteins might promote aberrant transcription. As a first step to test this, we looked for similarities among the transcriptomes of human ST-EP-ZFTAFUS ependymomas and mouse models of these tumors. Twenty-six percent (93/359) of genes that were upregulated specifically in two independent cohorts of human ST-EP-ZFTAFUS relative to other ependymomas (hereon, ‘ZFTAFUS-sig’; Supplementary Table S1) (6,8), were also upregulated in at least two out of four mouse models of these tumors (overlap, p=1.55e−08; Supplemental Figure S2A–C) (6,12,15,16). These included the validated ependymoma oncogene EPHB2 (5) and NES, IGF2 and JAG1 that regulate NSCs and ependymoma progenitor-like cells (4,7,17). ZFTAFUS-sig also included GLI2, which together with data presented in two accompanying manuscripts, implicates this Hedgehog signaling effector as a critical regulator of ependymoma tumorigenesis (15,16).
To test more directly if ZFTA-fusions drive aberrant gene expression, we performed total RNA sequencing of mNSCs and HEK293 cells transduced with wild-type partner genes, ZFTA-fusions or empty vector. In keeping with its superior transforming potency (6), ZFTA-RELAFUS1 drove the greatest transcriptomic change relative to ZFTA-RELAFUS2, ZFTA-YAP1FUS or wild-type partner proteins (Figure 3A and B). Seventy-five percent (70/93, overlap p=2.26e−39) and 45% (42/93, overlap p=2.11e−41) of ZFTAFUS-sig genes were upregulated by ZFTA-RELAFUS1 and ZFTA-RELAFUS2 in HEK293, respectively (Figures 3C). Furthermore, genes upregulated by ZFTA-RELAFUS1 or ZFTA-RELAFUS2 in HEK293 cells were enriched for the ZFTAFUS-sig and ‘human ST-EP-ZFTAFUS ependymoma specific’ genesets more than any other (18,225 gene sets tested; Supplemental Figure S3A to D). Less significant enrichment of genes involved in development and neurogenesis was also observed.
Figure 3. ZFTA-fusion proteins drive an aberrant transcriptome.
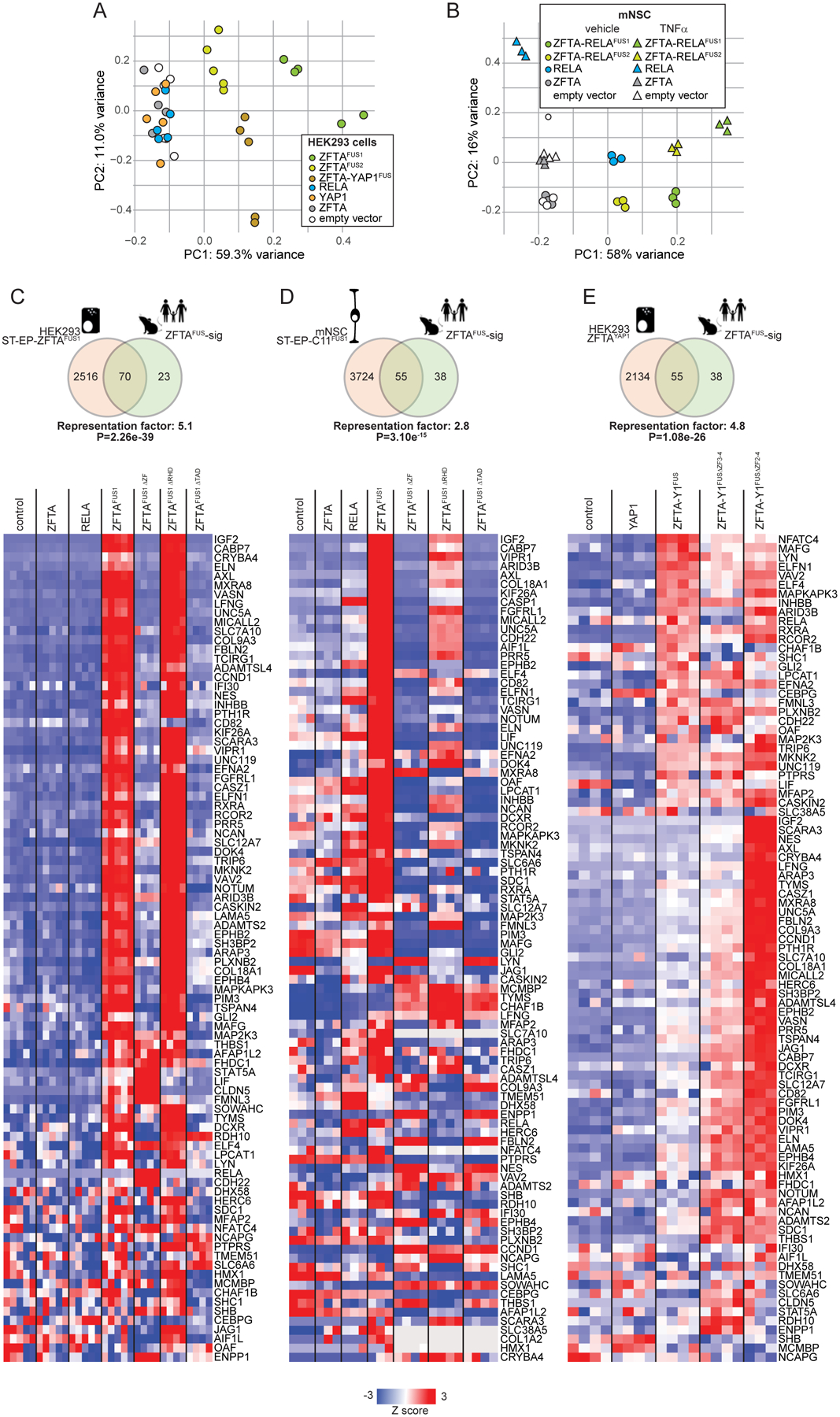
Principal components analysis of RNAseq profiles of HEK293 (A) and mNSCs (B) transduced with the indicated genes. mNSCs were also treated with TNFα (2 hours) or control (phosphate buffered saline). C-E, top in each, Venn diagram of overlap in genes upregulated (FDR<0.05) by the indicated ZFTA-fusion protein in the indicated cell type, with the ZFTAFUS-sig gene set (representation factor and p-value for overlap are shown). Below in each, heatmaps reporting expression of ZFTAFUS-sig genes in corresponding cells harboring the indicated gene.
Although ZFTAFUS-sig genes were also upregulated by ZFTA-RELAFUS1 or ZFTA-RELAFUS2 in mNSC, this occurred to a much lesser extent than in HEK293 cells and neither fusion upregulated the ‘human ST-EP-ZFTAFUS ependymoma specific’ geneset in mNSCs (Figure 3D and Supplementary Figure S3A to D). Thus, ZFTA-fusions drive ZFTAFUS-sig gene expression. This is modeled more faithfully in HEK293 cells than in mNSCs, presumably because of species-specific protein-protein or protein-DNA interactions. Indeed, in an accompanying manuscript we show that ZFTA-fusion proteins display species-specific selectivity for certain transcription factor binding sites (15).
In keeping with its less frequent selection in human ST-EP-ZFTAFUS and reduced transforming potency in mice (6), ZFTA-YAP1FUS upregulated fewer ZFTAFUS-sig genes in HEK293 than did ZFTA-RELAFUS1 (55/93, overlap p=1.08e−26; Figure 3E). However, comparison of all genes upregulated by ZFTA-RELAFUS1, ZFTA-RELAFUS2 or ZFTA-YAP1FUS in HEK293 identified a common set of 273 genes that was highly enriched for ZFTAFUS-sig genes (adjusted enrichment p=2.26e−45; Supplementary Figure S3E and F). Thus, ZFTA-fusion proteins drive a core set of genes, highly-enriched for the ZFTAFUS-sig transcriptome and regulators of development and neurogenesis, that is likely important for ependymoma tumorigenesis.
Deletion of either the ZFTA-ZF that is required for fusion protein nuclear trafficking (Figure 2 and Supplemental Figure S1) or the RELA-TAD that recruits transcriptional co-regulators, blocked ZFTAFUS-sig gene expression in ZFTA-RELAFUS1 transduced HEK293 and mNSCs (Figure 3C and D). Conversely, the RELA-RHD that mediates DNA binding and dimerization of NF-κB proteins was dispensable for ZFTAFUS-sig expression. Interestingly, deletion of all but one ZF from ZFTA-YAP1 enhanced ZFTAFUS-sig expression (Figure 3E), potentially explaining why ZFTA-RELAFUS1 that also contains a single ZFTA-ZF is the most frequently observed fusion in human ST-EP-ZFTAFUS (6).
ZFTA-RELA fusions are chromatin-bound transcription factors.
To understand how ZFTA-fusion proteins drive aberrant transcription, we used ChIP-seq to map ZFTA, RELA, and ZFTA-RELAFUS1 binding sites across the genome. Since the ZFTAFUS-sig was driven most faithfully in HEK293 cells, then we used these cells for ChIP-seq experiments. Both ZFTA (n=29,477 sites) and ZFTA-RELAFUS1 (n=32,447 sites) displayed extensive genome-wide binding that was distinct from that of RELA (n=1,156; Figure 4A to C). Remarkably, 75% (n=24,497/32,447) of ZFTA binding sites overlapped with those of ZFTA-RELAFUS1, suggesting ZFTA governs fusion protein chromatin binding (overlap, p<1.0e−250; Figure 4B and C). Overlapping binding sites included 91% (85/93) of ZFTAFUS-sig genes, including EPHB2 and GLI2 as well as L1CAM that is used as a diagnostic marker of human ST-EP-ZFTAFUS ependymoma (6); however, only ZFTA-RELAFUS1 binding was associated with increased gene expression (Figure 4D and Supplemental Figure S4A). In stark contrast, RELA bound only 11 ZFTAFUS-sig genes, with no detectable gene induction. Together, these data support the notion that ZFTA tethers ZFTA-RELAFUS1 to the genome, allowing the RELA-TAD to promote aberrant gene transcription.
Figure 4. ZFTA-RELAFUS1 remodels the chromatin landscape.
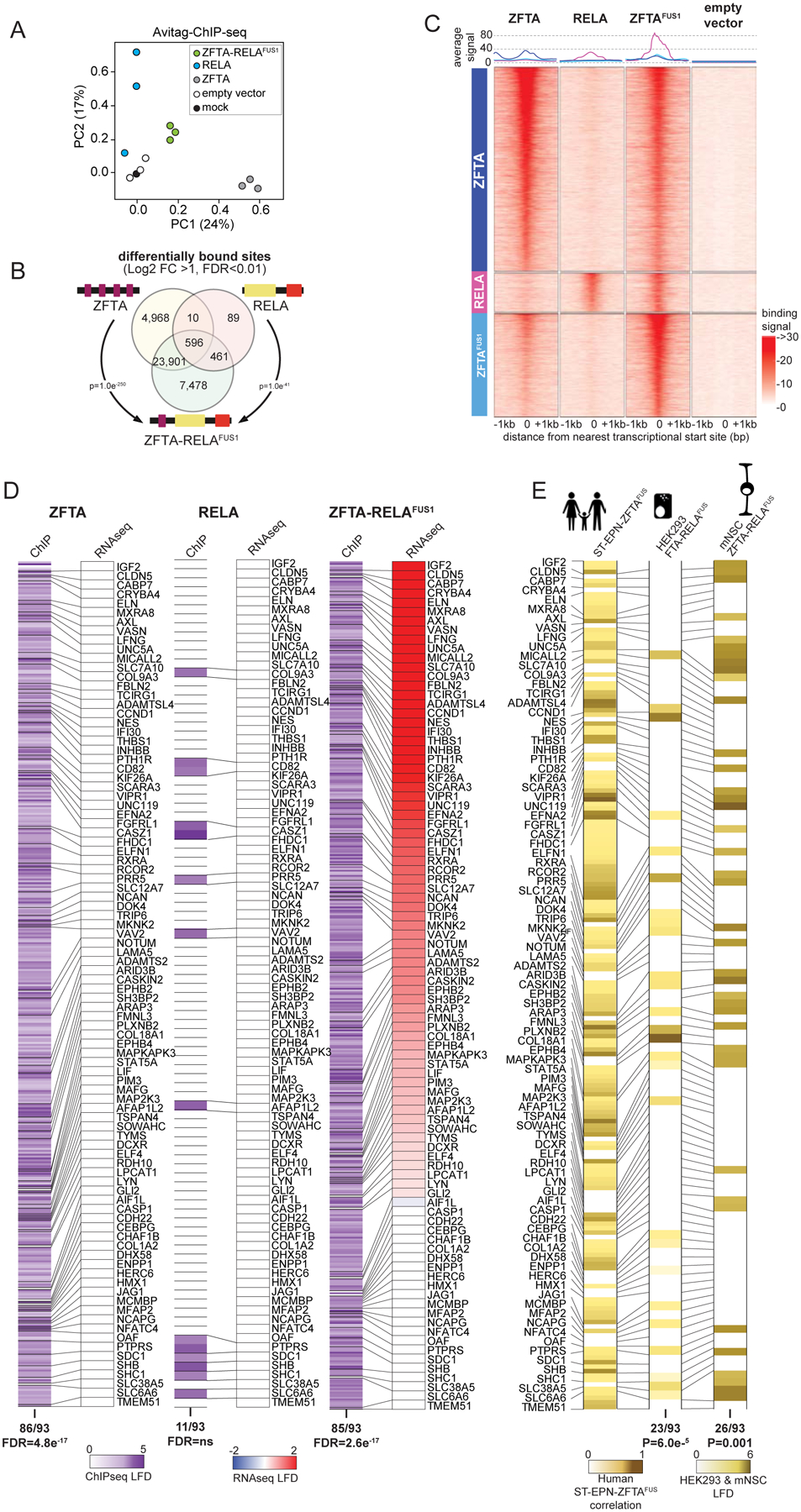
A. Principal components analysis of ChIP-seq profiles of chromatin binding by the indicated proteins in HEK293 cells. B. Venn diagram of overlap in ChIP-seq detected DNA binding sites of the indicated proteins (DESeq2 detected consensus peaks in ≥2 ChIP-seq samples). P-values report representation factor for overlap of indicated binding sites. C. Signal profile heatmaps of representative ChIP-seq samples for ZFTA, RELA, ZFTA-RELAFUS1 and empty vector transduced HEK293 cells showing distance to nearest translational start site (+/− 1Mbp). Three groups of binding sites (left column), sorted by decreasing number of overlapping reads, are used: top, 5000 ZFTA differentially bound sites, middle, 958 RELA differentially bound sites not also bound by ZFTA, bottom, ZFTA-RELAFUS1 top 2500 sites differentially bound, not also bound by ZFTA or RELA; all vs. empty vector. Above, average signal for each binding site group. D. Heatmaps of ChIP seq DNA binding (left in each) and RNAseq expression (right in each) level of ZFTAFUS-sig genes in HEK293 cells transduced with the indicated fusion. E. Heat map of H3K27ac marks in ZFTAFUS-sig genes in ST-EP-ZFTAFUS human tumors (left), or ZFTA-RELAFUS1 transduced HEK293 cells (middle) or mNSC (right).
Following chromatin binding, transcription factors recruit chromatin modifying complexes that create a permissive or suppressive transcriptional environment (18). Review of published ChIP-seq profiles (19) revealed that 59% (38/93 overlap, p=4.61e−49) of ZFTAFUS-sig genes in human ST-EP-ZFTAFUS ependymomas are marked as ‘active’ with H3K27ac, including EPHB2, GLI2, CCND1 and L1CAM (Figures 4E and Supplementary Figures S4A and B). Furthermore, H3K27ac marking of genes in ZFTA-RELAFUS1 transduced HEK293 and mNSCs were significantly enriched for ZFTAFUS-sig genes (Figure 4D), and our mouse model of ST-EP-ZFTAFUS, as well as a patient derived xenograft of ST-EP-ZFTAFUS, expressed much greater levels of H3K27ac than did a non-fusion driven ST-EP ependymoma (Supplementary Figure S4C). Together, these data are compatible with the notion that fusion proteins bind and remodel chromatin to an active state, promoting the transcription of ZFTAFUS-sig genes to drive ependymoma tumorigenesis. Indeed, an accompanying study also identified ZFTA-RELAFUS1 binding to, and activation of, ZFTAFUS-sig genes (15).
To assess further the extent to which ZFTA-RELAFUS1 binding modifies chromatin, we performed ChIP-seq of H3K4me1 (poised/active enhancers), H3K36me3 (gene elongation), H3K27me3 (down-regulated genes) and H2AK119ub (repressed developmental genes) in HEK293 and mNSCs (Supplementary Figure S4D and E). Robust ChIP-seq profiles of these marks could not be obtained in HEK293 cells, presumably because they did not tolerate prolonged expression of ZFTA-RELAFUS1. However, the histone landscape of mNSCs that did tolerate longer exposure to fusion proteins was extensively remodeled by ZFTA-RELAFUS1, resulting in overwhelming gains of H3K4me1 (2,614/3,016 significant peaks) and H3K36me3 (639/1,071) and loss of H3K27me3 (n=407/459). Thus, ZFTA-RELAFUS1 appears to function as an aberrant transcription factor, binding and remodeling chromatin to drive expression of a transforming transcriptome.
ZFTA-RELAFUS1 recruits chromatin remodeling complexes and transcriptional activators
To characterize how ZFTA-RELAFUS1 modifies chromatin to drive gene transcription, we quantified its protein-protein interactions on chromatin using a cross-linking immunoprecipitation and mass spectrometry technique termed qPLEX-RIME (20). We again focused our studies in HEK293 cells in which ZFTA-RELAFUS1 bound and activated ZFTAFUS-sig genes most faithfully. Principal component analysis positioned the ZFTA-RELAFUS1 protein interactome between those of ZFTA and RELA, suggesting these proteins share binding partners (Figure 5A). In keeping with this notion and their widespread chromatin-binding, proteins interacting with ZFTA and ZFTA-RELAFUS1 were highly enriched for nuclear proteins and/or those involved in chromatin binding, remodeling and transcription (Figure 5B and Supplementary Figure S5A). Conversely, RELA and ZFTA-RELAFUS1 but not ZFTA bound components of the NF-κB interactome, including NFκB2 (Figure 5B and Supplementary Figure S5A). This observation was confirmed by independent co-immunoprecipitation studies without cross-linking (Figure 5C). Conversely, SMARCA4, SMARCC1, SMARCD1, SMARCD2 and SMARCE1 that are key components of the SWI/SNF (SWItch/Sucrose Non-Fermentable) complex that regulates nucleosome positioning (21) and the histone acetylator CREBBP, were unique to the ZFTA-RELAFUS1 interactome.
Figure 5. Protein interactomes of ZFTA, RELA, and ZFTA-RELAFUS1.
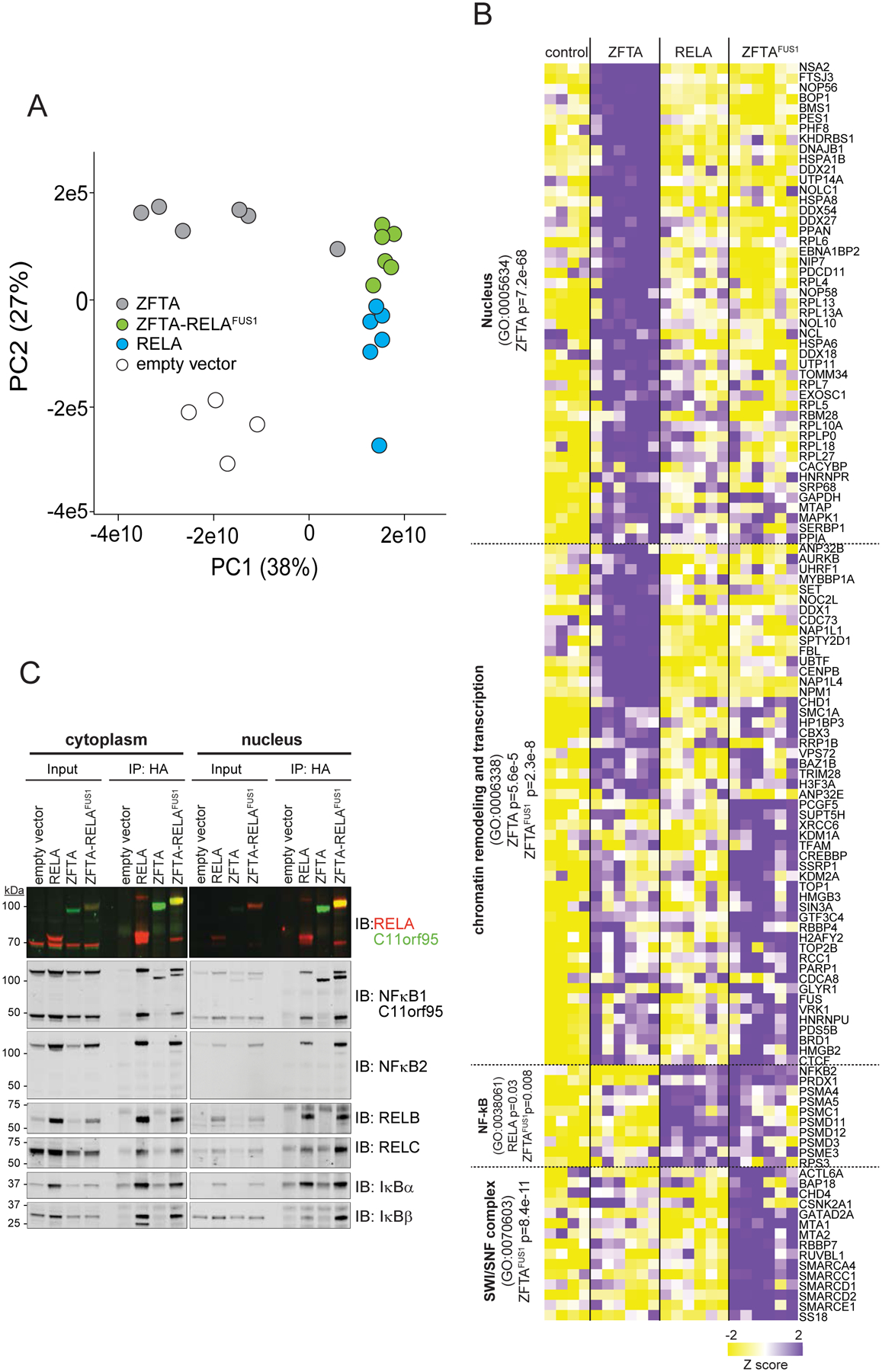
A. PCA of qPLEX-RIME protein interactome profiles of the indicated proteins in HEK293 cells. B. Heat map of significant protein interactors (right) with the indicated proteins (top). Pathways significantly enriched among binding partners are shown left. C. Immunoblot following co-immunoprecipitation from cytoplasmic and nuclear lysates of HEK293 cells expressing the indicated 3xHA-tagged cDNAs.
To further define the domains of ZFTA-RELAFUS1 that recruit protein complexes, we repeated our qPLEX-RIME and immunoprecipitation studies in HEK293 cells expressing either ZFTA-RELAFUS1ΔZF, ZFTA-RELAFUS1ΔRHD, ZFTA-RELAFUS1ΔTAD or the full-length fusion (Figure 6A and B). Loss of the ZFTA-ZF markedly impaired fusion protein binding of three major chromatin remodeling complexes (enrichment relative to the CORUM database (22); Figure 6A and Supplementary Figure S5B): SWI/SNF (SMARCA4, SMARCB1, SMARCC2, SMARCD2, SMARCD3, ACTL6A, ARID1A; FDR=5.89e−16); SAGA (Spt-Ada-Gcn5 acetyltransferase) that acetylates histones (TADA3, TAF5L, TAF6L, TADA2B, TTRAP, SUPT20H, USP22, SUPT3H, SGF29, TADA1, ATXN7L2; FDR=2.60e−23) (23); and NuA4/Tip60-HAT (NuA4/Tip60 histone acetyltransferase) that acetylates histone H4 and H2A (YEATS4, EP400, EPC1, TRRAP, VPS72; FDR=1.57e−15) (24). Deletion of this domain also blocked binding of other key chromatin remodeling proteins including BRD4 and ATRX. These data were validated by independent co-immunoprecipitation studies of selected proteins in the reverse direction (Figure 6C). Notably, Cut-n-Run-Sequencing studies in an accompanying manuscript also identify co-recruitment of Brd4, Ep300, Crebbp, and RNA polymerase 2 to most ZFTA-RELAFUS1 binding sites (15).
Figure 6. Protein complexes recruited by ZFTA-RELAFUS1.
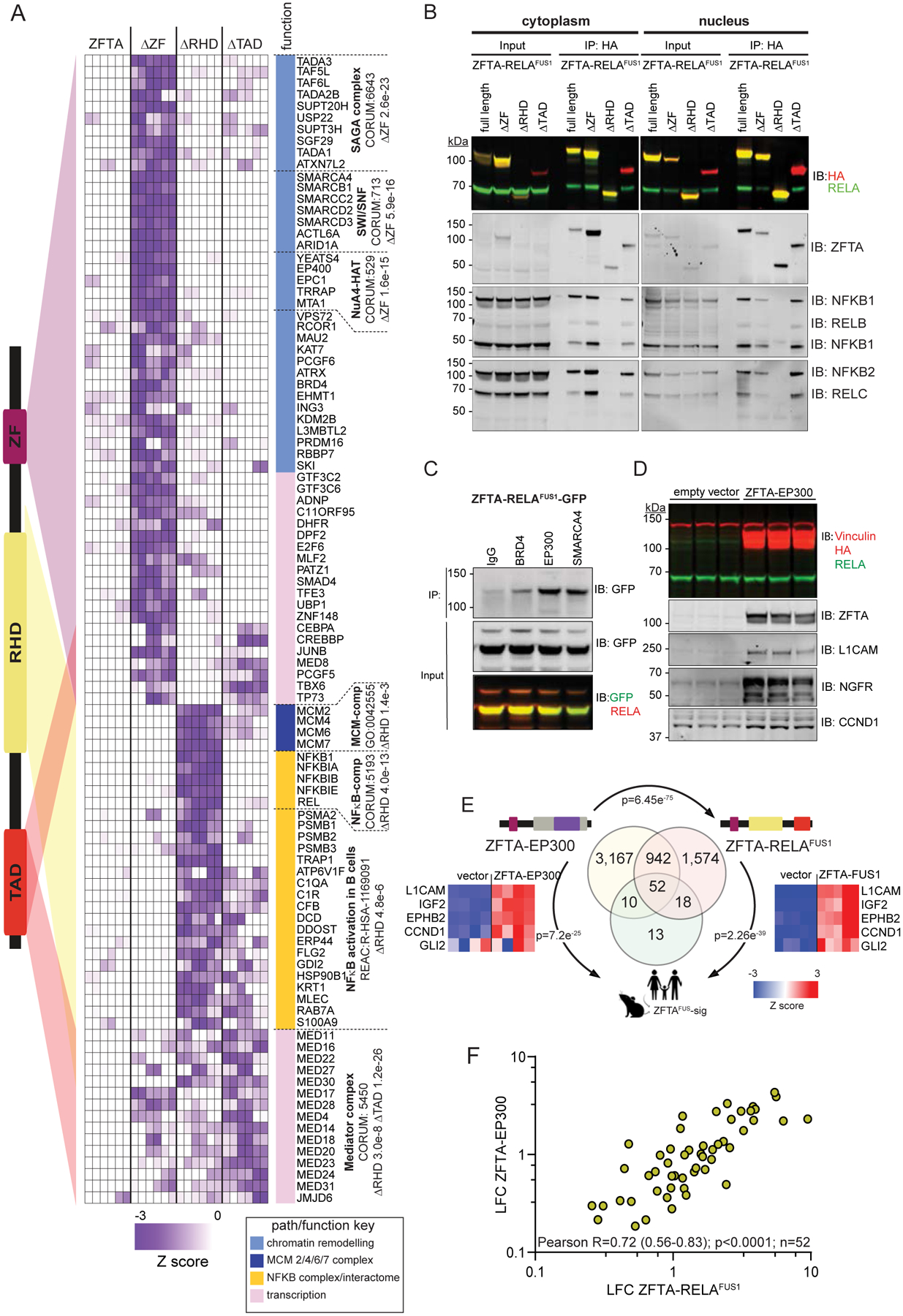
A. Left, schematic of ZFTA-RELAFUS1. Colored triangles indicate proteins in heatmap (right) that are lost from the ZFTA-RELAFUS1 interactome when the associated protein domain is deleted. Pathways enriched with significant interacting proteins are shown right. Western blot of protein lysates of HEK293 expressing ZFTA-RELAFUS1 (B and C) or ZFTA-EP300 (D) tagged proteins that were immunoprecipitated (IP) and then probed (IB) with the indicated antibodies. E. Venn diagram of overlap in genes upregulated (FDR<0.05) by the indicated fusion protein in HEK293 cells with the ZFTAFUS-sig gene set (p-value for overlap). Heatmaps report representative ZFTAFUS-sig gene expression including GLI2. F. Correlation of 52 ZFTAFUS-sig genes upregulated by the ZFTA-EP300 and ZFTA-RELAFUS1 in HEK293 cells.
While failure of ZFTA-RELAFUS1ΔZF to access the nucleus very likely impeded its binding of chromatin remodeling complexes (Figure 2A–C), neither the RELA-RHD (ZFTA-RELAFUS1ΔRHD) or TAD (ZFTA-RELAFUS1ΔTAD) were required to recruit SWI/SNF, SAGA or NuA4/Tip60 HAT complexes (Figures 6A and Supplementary Figure S5B). Thus, we propose that the ZFTA-ZF coordinates both chromatin binding and recruitment of chromatin remodeling complexes to fusion proteins.
As expected, NF-κB complex binding (NFKB1, NFKBIA, NFKBIB, NFKBIE, REL; FDR=3.98e−13) was confined largely to the RELA-RHD (Figures 6A and Supplementary Figure S5B). Deletion of the RELA-RHD also ablated binding of the minichromosome maintenance (MCM) 2/4/6/7 complex that is required for both DNA replication initiation and elongation (25). Interaction between the NF-κB and MCM2/4/6/7 complexes has not been reported, suggesting this is a novel property of the ZFTA-RELAFUS1 protein. Deletion of the RELA-RHD also disrupted fusion protein recruitment of a several other proteins involved in the NF-κB interactome, including components of the 20S core proteasome complex responsible for proteolytic processing of NFKB1 (Figures 5B and 6A) (26).
In keeping with the property of TADs to provide a protein scaffold for transcription coregulators (27), deletion of the RELA-TAD ablated ZFTA-RELAFUS1 interaction with the Mediator complex (MED11, MED16, MED22, MED27, MED30, MED4, MED14, MED18, MED20, MED23, MED24, MED31, JMJD6; FDR=1.24e−26; Figure 6A and Supplementary Figure S5B). This transcription coregulator communicates between active enhancers and promoters by interacting with proteins that bind to either of these two classes of regulatory DNA elements (28). Thus, the RELA-TAD that is critical for driving ZFTA-RELAFUS1-induced aberrant gene expression (Figure 3C and D), likely mediates the recruitment of key transcriptional activators. Proteins within the Mediator complex were enriched to a lesser extent within the ZFTA-ZF and RELA-RHD interactomes, suggesting these domains might also coordinate ZFTA-RELAFUS1-Mediator complex binding (Figure 6A and Supplementary Figure S5B).
To compare the interactomes of different ZFTA-fusion proteins, we also catalogued protein binding partners of ZFTA-YAP1 using qPLEX-RIME (Supplementary Figure 6). The interactome of ZFTA-YAP1 was remarkably similar to that of ZFTA-RELAFUS1 (overlap, p=3.61e−40; Supplementary Figure S6A and B) and included chromatin binding proteins (FDR=8.2e−9), SWI/SNF (FDR=8.2e−9), MCM (FDR=2.3e−3) and SAGA complexes (FDR=0.0004), as well as members of the Mediator and NuA4/Tip60 HAT complexes. As expected, ZFTA-YAP1 did not recruit the NF-κB complex but did bind known interactors of YAP1 including CTNNB1, TEAD1 and TEAD3. The interactomes of ZFTA-YAP1 and the ZFTA-YAP1ΔZF2–3 deletion mutant were almost identical, supporting further the notion that a single ZF is both necessary and sufficient for fusion-driven transformation (overlap, <1.0e−300; Supplementary Figure S6A). Thus, ZFTA-fusion proteins recruit similar interactomes regardless of binding partners, explaining why different fusions drive similar transcriptomes.
To test more directly if the fusion partners of ZFTA recruit transcriptional coregulators, we created an artificial fusion protein between ZFTA and the histone acetyltransferase EP300 that interacts with ZFTA-RELAFUS1 (Figure 6C) and was reported recently to co-locate with the Mediator complex at super enhancers in NSCs (29). ZFTA-EP300 upregulated 4,171 genes in HEK293 cells including 67% (62/93) of ZFTAFUS-sig genes (overlap p=7.21e-25; Figure 6E). Thirty-seven percent of genes upregulated by ZFTA-RELAFUS1 in HEK293 cells were also upregulated by ZFTA-EP300 (overlap p=6.45e−75; Figure 6E), including 56% (52/93) of ZFTAFUS-sig genes. Expression levels of ZFTAFUS-sig genes correlated closely in ZFTA-EP300 and ZFTA-RELAFUS1 transduced HEK293 cells (R=0.72, P<0.0001; Figure 6F). Despite these similarities, ZFTA-EP300 failed to upregulate GLI2 that is shown in an accompanying manuscript to be crucial for ZFTA-fusion driven transformation (Figure 6E; Ref. 15). This paper further shows that ZFTA-EP300 does not drive brain tumorigenesis in mice, supporting the notion that aberrant hedgehog signaling is an important mediator of fusion-driven ependymoma tumorigenesis.
Together, these data suggest a model in which the ZFTA-ZF promotes genome-wide chromatin binding of ZFTA-fusion proteins and the recruitment of key chromatin remodelling proteins including the SWI/SNF, SAGA and NuA4/Tip60 complexes (Figure 7A). Aberrant transcription is then driven by fusion partners including RELA and YAP1 that involves transcriptional coactivators and the Mediator complex. Although the RELA-RHD recruits the NF-κB and the MCM complex, this is not required for fusion-driven gene expression.
Figure 7. Molecular and cancer phenotype characteristics of ZFTA-RELAFUS1 protein domains.
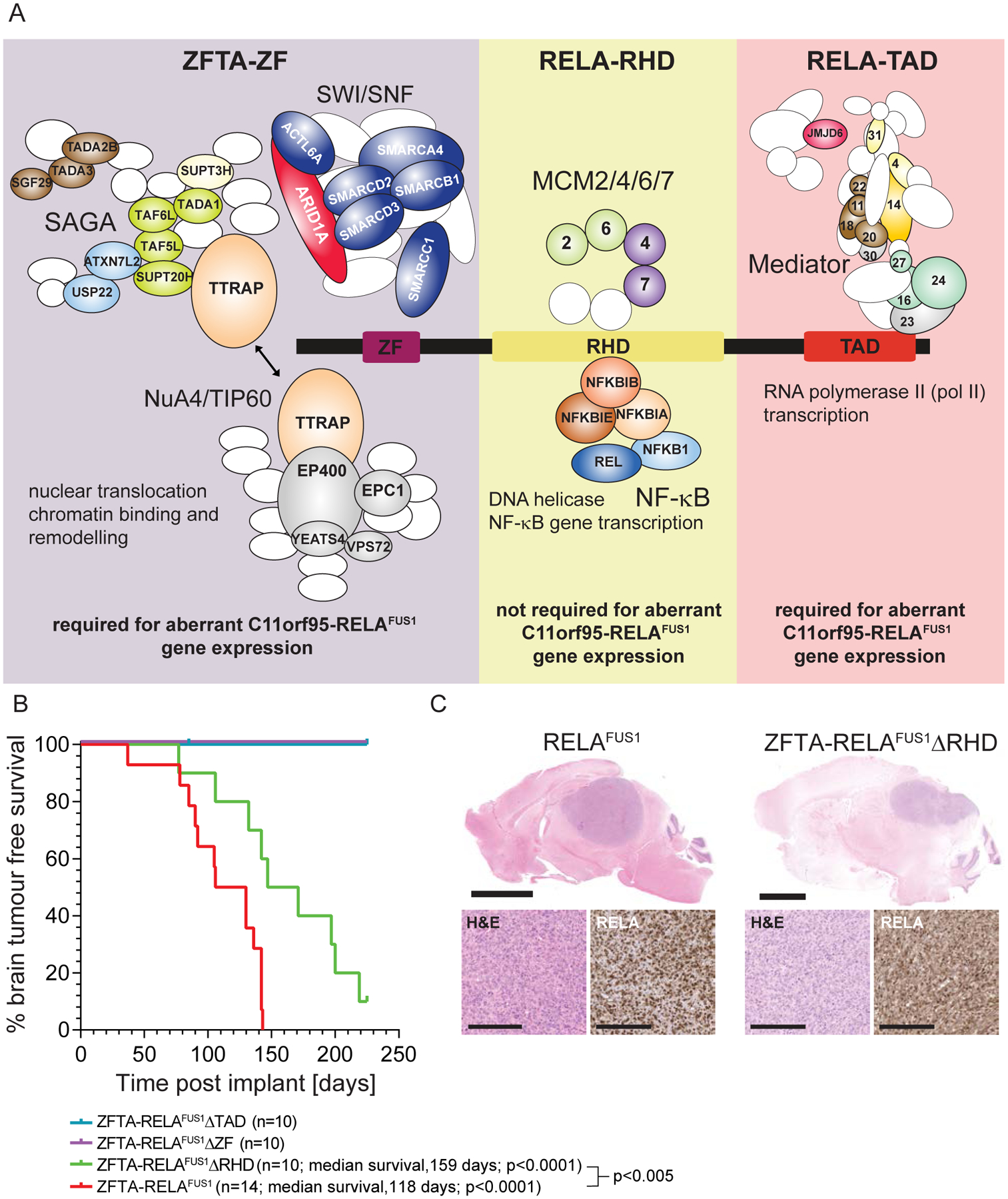
A. Schematic depicting the protein complexes that bind each ZFTA-RELAFUS1 domain. Protein subunits identified in each domain interactome by qPLEX-RIME are colored and labelled in complexes. Complex components not identified are white ovals/circles. B. Kaplan-Meier brain tumor-free survival curves of mice allografted with mNSCs transduced with the indicate cDNAs. P values report the Log-Rank statistic. C. Top (low power, scale=3mm) and bottom left (high power, scale=10μm) in each figure, photomicrographs of hematoxylin and eosin stained tumors in the brains of mice allografted with mNSCs transduced with the indicated cDNA. Bottom right in each figure are p65-RELA immunostains of tumor sections.
ZFTA-RELAFUS1 ZF and TAD, but not RHD, are required for tumorigenesis
We showed previously that mNSCs transduced with either ZFTA-RELAFUS1, ZFTA-RELAFUS2 or ZFTA-YAP1FUS, but not wild-type ZFTA, RELA or YAP1, form brain tumors in mice (6,12). Therefore, to test directly which motifs within ZFTA-RELAFUS1 are required for transformation, we transduced mNSCs with either full length ZFTA-RELAFUS1, ZFTA-RELAFUS1ΔZF, ZFTA-RELAFUS1ΔRHD or ZFTA-RELAFUS1ΔTAD and injected suspensions of 1×106 of these cells, separately into the brains of four week-old immunocompromised mice exactly as described previously (6).
As expected, all mice harboring ZFTA-RELAFUS1-transduced mNSCs succumbed to brain tumors (n=14, median survival, 118 days). In stark contrast, neither cells transduced with ZFTA-RELAFUS1ΔZF (n=10) or ZFTA-RELAFUS1ΔTAD (n=10) formed tumors (surveillance, 225 days post-implant; Figure 7B). This requirement of ZFTA-ZF to drive ependymomas in mice was also confirmed in an accompanying study (16). Thus, the chromatin binding and remodeling functions of ZFTA-ZF, and the transcription co-activator properties of the RELA-TAD, are indispensable for fusion-driven tumorigenesis.
In stark contrast, all mice injected with ZFTA-RELAFUS1ΔRHD-transduced mNSCs developed brain tumors, in keeping with its redundancy in fusion chromatin remodeling complex recruitment and aberrant gene expression. These tumors were histologically indistinguishable from those driven by full-length ZFTA-RELAFUS1, but took significantly longer to form (p<0.005; Figure 7B and C). Thus, while not required for tumorigenesis, recruitment of the NF-κB and MCM complexes, and/or nuclear localization of ZFTA-RELAFUS1 by the RHD, might contribute to tumor development (Figures 2C and 7A).
DISCUSSION
We identify three functions conferred by ZFTA on oncogenic fusion proteins: nuclear translocation, chromatin binding, and the recruitment of chromatin modifiers. ZFTA thereby tethers fusion proteins across the genome, modifying chromatin to an active state, and enabling its partner transcriptional co-activators (RELA, YAP1, MAML2 or MRTFB) to promote promiscuous expression of a transforming transcriptome.
ZF domains promote nuclear translocation, in part by binding the nuclear import protein KPNB1 via their C2H2 and/or NLS motifs (30). A single ZFTA-ZF (its C2H2 or NLS motifs in particular) were necessary and sufficient to traffic fusion proteins to the nucleus. Neither the RELA-RHD nor RELA-TAD were required for nuclear translocation, although deletion of the RELA-RHD reduced nuclear trafficking by ~30%. Thus, ZFTA-RELAFUS may be maintained in the nucleus by RELA-RHD-mediated protein-protein or protein-DNA interactions: these may include the NF-κB and/or MCM complexes that bound the RELA-RHD. The MCM complex forms a DNA helicase important in DNA replication, potentially explaining why RELA-RHD deletion prolonged fusion-driven tumor latency.
Our finding that both ZFTA and ZFTA-RELAFUS1 display widespread, overlapping chromatin binding provides strong evidence that the ZFTA-ZF also mediates DNA binding of fusion proteins. This was associated with the recruitment of the SWI/SNF, SAGA and NuA4/TIP60 complexes and profound remodeling of the chromatin landscape, converting thousands of genes–including ZFTAFUS-sig genes–to a transcriptionally-active state. Thus, we propose that ZF domains that are known to recruit chromatin modifiers, confer this property on fusion proteins (31–33). But important caveats warrant further investigation. Although nuclear trafficking and DNA binding of ZFTA and ZFTA-RELAFUS1 were similar, only the later was detected in association with chromatin remodeling proteins. And, SWI/SNF, SAGA and NuA4/TIP60 complex binding to ZFTA-RELAFUS1 was only revealed by comparison with the ZFTA-RELAFUS1ΔZF mutant that does not access the nucleus. Nevertheless, both ZFTA-RELAFUS1ΔRHD and ZFTA-RELAFUS1ΔTAD that do translocate to the nucleus as well as ZFTA-YAP1, readily recruited SWI/SNF, SAGA and NuA4/Tip60 HAT complexes. Furthermore, our artificial ZFTA-EP300 fusion as well as the array of disparate partner genes in ZFTA-containing fusions, drive expression of ZFTAFUS-sig genes. Thus, we propose that the ZFTA-ZF mediates SWI/SNF, SAGA and NuA4/TIP60 complex recruitment as well as nuclear translocation and chromatin binding.
The observation that HEK293 cells recapitulate the ZFTAFUS-sig more faithfully than mNSCs that drive ependymoma in mice was surprising, and suggests important species-specific differences in ZFTA-fusion activity. Further, since ZFTA-fusions possess the capacity to remodel diverse genomes, then the almost exclusive restriction of these fusions to human supratentorial ependymoma suggests NSCs are uniquely susceptible to the 11q chromothripsis that drive ZFTA translocations, rather than to fusion-driven aberrant gene transcription.
The notion that ZFTA confers critical chromatin binding and remodeling functions on fusion proteins is supported further by the observation that it is the common partner in an array if oncogenic fusions. Fusion partners with ZFTA include RELA, YAP1, MAML2 and MRTFB which are all transcriptional co-regulators. The conjoining of their transcription-promoting activity with the DNA binding and chromatin remodeling of ZFTA likely accounts for the potent induction of gene transcription by fusion proteins. The ‘selection’ of RELA, YAP1 and MAML2 as partners of ZFTA likely represents topological ‘marriages of convenience’: in human ST-SP-ZFTAFUS, extensive chromothripsis of 11q brings ZFTA (11q13.1) into close proximity with RELA (11q13.1), YAP1 (11q22.1) or MAML2 (11q21) (6). ZFTA-partner genes also share an ability to recruit the Mediator complex. Since the Mediator complex organizes the transcriptional network that determines NSC identity (29) and genes upregulated by ZFTA-fusion included regulators of neurogenesis, then perturbation of neural cell fate is likely fundamental to fusion-driven tumor initiation.
Finally, together with complementary data in two accompanying studies, we unmask novel approaches for treating fusion-driven ependymoma (15,16). Zheng et al., (16) identify GLI2 as an important target of fusion-driven transformation that we show is bound and upregulated by ZFTA-RELAFUS1. Notably, our artificial fusion ZFTA-EP300 that drove expression of most ZFTAFUS-sig genes, did not upregulate GLI2 and lacked transforming capacity in vivo. Thus, aberrant Hedgehog signaling might prove a useful therapeutic target. Similar to Arabzade et al., (15), we show also that ZFTA-RELAFUS1 localizes to enhancers and promoters and recruits transcriptional activation complexes. Thus, thalidomide analogs that cause the rapid ubiquitination and proteasomal degradation of ZF proteins (34) are a second potential therapeutic approach worthy of further study.
MATERIAL AND METHODS
Cell culture, western blot and fluorescence microscopy
Embryonic day (E) 14.5 Cdkn2a−/− mouse neural stem cells (mNSCs) were harvested from mice (under project license approved by the University of Cambridge Animal Welfare and Ethical Review Body) and expanded as neurospheres as described previously (5) in Neurobasal media supplemented with B27, N2, l-Glutamine and Pen-Strep, in the presence of 20 ng ml−1 EGF and FGF2 (all reagents are described in Table S2). Human embryonic kidney (HEK) 293 cells were obtained from the American Type Culture Collection and maintained in standard DMEM with 10% fetal calf serum. ELISA tests were performed monthly to ensure mycoplasma-free cell status and cell identity was authenticated prior to each set of in vitro and in vivo experiments using Single Tandem Repeat genotyping. mNSCs and HEK293 were plated on mouse laminin or poly-d-Lysine coated plates, respectively. Nuclear and cytoplasmic extractions of cells were prepared using the NE-PER kit (ThermoFisher Sci). Western blotting and fluorescence microscopy of cultured cells was conducted exactly as described previously (4,5) using antibodies described in Supplementary Table S2.
Generation of ZFTA fusion mutants
Deletion mutants of ZFTA-RELAFUS1 were generated using the In-Fusion system (Clontech) by deleting the first ZF (ΔZF, W109-W155), C2-H2 ZF (ΔC2-H2, M124-H149), REL homology domain (ΔRHD, P229-Y516), RELA transactivation domain (ΔTAD, P625-A754), RELA nuclear localization signal (ΔRELA-NLS, L499-P531), or RELA nuclear export signal (ΔRELA-NES, L646-L660). ZFTA-YAP1 mutants deleted ZFs 3–4 (W344-R649) or ZFs 2–4 (K214-R649). All mutants were sub-cloned into retrovirus carrying the indicated protein tag.
Virus production
Retro- and lenti-viruses were generated in 293T cells (American Type Culture Collection) by Lipofectamine 2000 transfection with viral packaging plasmids encoding pmd2.G, psPAX2 (Addgene), and the transfer vector expressing the construct of interest. Virus-containing supernatant was collected at 48 and 72-hours post-transfection, filtered and concentrated with PEG-it virus precipitation solution (SBI) overnight at 4° C.
RNA sequencing
Total RNA libraries were generated using the TruSeq mRNA library prep kit (Illumina), 50 base-pair single-end reads acquired using the HiSeq 4000, and aligned to the human (hg38) or mouse (mm10) genome using TopHat (35). Read counts were normalised and differential gene expression determined using DESeq2 (36).
Chromatin immunoprecipitation and sequencing (ChIP-seq)
Cells transfected with Avi-tagged proteins (Genecopoeia) were cross-linked after 48 hours in 1% formaldehyde and nuclei sonicated. For histone ChIP-seq, cells were cross-linked and sonicated in ChIP Lysis Buffer before primary antibody-bead immunoprecipitation. Sheared chromatin was incubated with magnetic streptavidin beads (Thermo Fisher Sci) to generate ChIP-seq and input libraries using the ThruPlex sample prep kit (Illumina). ChIP-seq sequences were aligned to hg19 or mm10 and peaks analyzed using MACS and DiffBind (37). Profileplyr was used to count overlapping reads, annotate genomic features, and generate plots. Reads were counted in 25bp bins covering 1000bp up- and down-stream of each 500bp differentially bound site (total width of each region: 2500bp).
qPLEX-RIME
qPLEX-RIME assays were performed as described previously (20). Briefly, cells expressing the appropriate biotinylated protein were cross-linked, nuclei sonicated and incubated with HA-coated magnetic beads (ThermoFisher Sci). Beads were subjected to protein digestion and peptides dried (via speedvac), reconstituted in 0.1M triethylammonium bicarbonate and labelled using TMT-10plex reagents (Thermo Fisher Sci). The peptide mixture was fractionated with Reversed-Phase cartridges at high pH (Pierce) and reconstituted peptides analyzed using a Dionex Ultimate 3000 UHPLC system with nano-ESI Fusion Lumos (Thermo Fisher Sci). CID tandem mass spectra were processed using SequestHT on Proteome Discoverer 2.1 and analyzed using qPLEXanalyzer, R-bioconductor package (20).
Mouse tumor studies
All experiments involving animals were carried out under a project license approved by the University of Cambridge Animal Welfare and Ethical Review Body (AWERB). Orthotopic allografts of ST-EP in mice were generated in accordance with UK Home Office regulations under project license exactly as described previously (5,6). Formalin-fixed paraffin embedded (FFPE) sections of tumors in mice were analysed using hematoxylin and eosin, RELA and H3K27acetyl mark immunostaining as described (6).
Supplementary Material
SIGNIFICANCE.
Ependymomas are hard to treat brain tumors driven by translocations between ZFTA and a variety of transcriptional co-activators. We dissect the transforming mechanism of these fusion proteins and identify protein domains indispensable for tumorigenesis, thereby providing insights into the molecular basis of ependymoma tumorigenesis and vulnerabilities for therapeutic targeting.
ACKNOWLEDGEMENTS
This work was supported by grants to: R. Gilbertson (Major Centre Core and Children’s Brain Tumour Centre from Cancer Research UK; The Brain Tumour Charity; and P01CA96832 and R01CA129541 from the US National Cancer Research); S. Mack (RR170023, Cancer Prevention and Research Institute of Texas) and K. Pajtler (Collaborative Ependymoma Research Network). We thank the Cambridge Institute genomics, proteomics, biorepository unit (BRU), and histology core facilities for technical assistance.
Financial support:
Cancer Research UK, NCI (R01CA129541 and U54CA243125), SOHN foundation, Mathile foundation.
Footnotes
Conflict of interest disclosures: The authors declare no conflict of interest.
REFERENCES
- 1.Jones DTW, Banito A, Grünewald TGP, Haber M, Jäger N, Kool M, et al. Molecular characteristics and therapeutic vulnerabilities across paediatric solid tumours. Nature Reviews Cancer 2019;19(8):420–38 doi 10.1038/s41568-019-0169-x. [DOI] [PubMed] [Google Scholar]
- 2.Aldape K, Brindle KM, Chesler L, Chopra R, Gajjar A, Gilbert MR, et al. Challenges to curing primary brain tumours. Nat Rev Clin Oncol 2019;16(8):509–20 doi 10.1038/s41571-019-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pajtler KW, Mack SC, Ramaswamy V, Smith CA, Witt H, Smith A, et al. The current consensus on the clinical management of intracranial ependymoma and its distinct molecular variants. Acta Neuropathol 2017;133(1):5–12 doi 10.1007/s00401-016-1643-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor MD, Poppleton H, Fuller C, Su X, Liu Y, Jensen P, et al. Radial glia cells are candidate stem cells of ependymoma. Cancer Cell 2005;8(4):323–35 doi 10.1016/j.ccr.2005.09.001. [DOI] [PubMed] [Google Scholar]
- 5.Johnson RA, Wright KD, Poppleton H, Mohankumar KM, Finkelstein D, Pounds SB, et al. Cross-species genomics matches driver mutations and cell compartments to model ependymoma. Nature 2010;466(7306):632–6 doi 10.1038/nature09173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Parker M, Mohankumar KM, Punchihewa C, Weinlich R, Dalton JD, Li Y, et al. C11orf95-RELA fusions drive oncogenic NF-kappaB signalling in ependymoma. Nature 2014;506(7489):451–5 doi 10.1038/nature13109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mohankumar KM, Currle DS, White E, Boulos N, Dapper J, Eden C, et al. An in vivo screen identifies ependymoma oncogenes and tumor-suppressor genes. Nature genetics 2015;47(8):878–87 doi 10.1038/ng.3323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pajtler KW, Witt H, Sill M, Jones DT, Hovestadt V, Kratochwil F, et al. Molecular Classification of Ependymal Tumors across All CNS Compartments, Histopathological Grades, and Age Groups. Cancer Cell 2015;27(5):728–43 doi 10.1016/j.ccell.2015.04.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mack SC, Witt H, Piro RM, Gu L, Zuyderduyn S, Stutz AM, et al. Epigenomic alterations define lethal CIMP-positive ependymomas of infancy. Nature 2014;506(7489):445–50 doi 10.1038/nature13108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Huang D, Sumegi J, Dal Cin P, Reith JD, Yasuda T, Nelson M, et al. C11orf95-MKL2 is the resulting fusion oncogene of t(11;16)(q13;p13) in chondroid lipoma. Genes Chromosomes Cancer 2010;49(9):810–8 doi 10.1002/gcc.20788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Flucke U, Tops BB, Saint Aubain Somerhausen N, Bras J, Creytens DH, Kusters B, et al. Presence of C11orf95-MKL2 fusion is a consistent finding in chondroid lipomas: a study of eight cases. Histopathology 2013;62(6):925–30 doi 10.1111/his.12100. [DOI] [PubMed] [Google Scholar]
- 12.Ozawa T, Arora S, Szulzewsky F, Juric-Sekhar G, Miyajima Y, Bolouri H, et al. A De Novo Mouse Model of C11orf95-RELA Fusion-Driven Ependymoma Identifies Driver Functions in Addition to NF-kappaB. Cell Rep 2018;23(13):3787–97 doi 10.1016/j.celrep.2018.04.099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manning SA, Kroeger B, Harvey KF. The regulation of Yorkie, YAP and TAZ: new insights into the Hippo pathway. Development 2020;147(8) doi 10.1242/dev.179069. [DOI] [PubMed] [Google Scholar]
- 14.Zhang Q, Lenardo MJ, Baltimore D. 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 2017;168(1–2):37–57 doi 10.1016/j.cell.2016.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Arabzade A C11ORF95-RELA Directs Oncogenic Transcription and Informs Therapeutic Targets in Ependymoma. Cancer Discovery 2020. [Google Scholar]
- 16.Zheng T C11orf95 rearrangements dictate oncogenic dependencies in supratentorial brain tumors with various histologies. Cancer Discovery 2020. [Google Scholar]
- 17.Calabrese C, Poppleton H, Kocak M, Hogg TL, Fuller C, Hamner B, et al. A perivascular niche for brain tumor stem cells. Cancer Cell 2007;11(1):69–82 doi 10.1016/j.ccr.2006.11.020. [DOI] [PubMed] [Google Scholar]
- 18.Spitz F, Furlong EE. Transcription factors: from enhancer binding to developmental control. Nat Rev Genet 2012;13(9):613–26 doi 10.1038/nrg3207. [DOI] [PubMed] [Google Scholar]
- 19.Mack SC, Pajtler KW, Chavez L, Okonechnikov K, Bertrand KC, Wang X, et al. Therapeutic targeting of ependymoma as informed by oncogenic enhancer profiling. Nature 2018;553(7686):101–5 doi 10.1038/nature25169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Papachristou EK, Kishore K, Holding AN, Harvey K, Roumeliotis TI, Chilamakuri CSR, et al. A quantitative mass spectrometry-based approach to monitor the dynamics of endogenous chromatin-associated protein complexes. Nat Commun 2018;9(1):2311 doi 10.1038/s41467-018-04619-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mittal P, Roberts CWM. The SWI/SNF complex in cancer - biology, biomarkers and therapy. Nat Rev Clin Oncol 2020;17(7):435–48 doi 10.1038/s41571-020-0357-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, et al. CORUM: the comprehensive resource of mammalian protein complexes-2019. Nucleic Acids Res 2019;47(D1):D559–d63 doi 10.1093/nar/gky973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmlinger D, Tora L. Sharing the SAGA. Trends Biochem Sci 2017;42(11):850–61 doi 10.1016/j.tibs.2017.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Doyon Y, Côté J. The highly conserved and multifunctional NuA4 HAT complex. Current opinion in genetics & development 2004;14(2):147–54 doi 10.1016/j.gde.2004.02.009. [DOI] [PubMed] [Google Scholar]
- 25.Bell SD, Botchan MR. The minichromosome maintenance replicative helicase. Cold Spring Harb Perspect Biol 2013;5(11):a012807 doi 10.1101/cshperspect.a012807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moorthy AK, Savinova OV, Ho JQ, Wang VY-F, Vu D, Ghosh G. The 20S proteasome processes NF-kappaB1 p105 into p50 in a translation-independent manner. EMBO J 2006;25(9):1945–56 doi 10.1038/sj.emboj.7601081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Mitchell PJ, Tjian R. Transcriptional regulation in mammalian cells by sequence-specific DNA binding proteins. Science 1989;245(4916):371–8 doi 10.1126/science.2667136. [DOI] [PubMed] [Google Scholar]
- 28.Soutourina J Transcription regulation by the Mediator complex. Nature reviews Molecular cell biology 2018;19(4):262–74 doi 10.1038/nrm.2017.115. [DOI] [PubMed] [Google Scholar]
- 29.Quevedo M, Meert L, Dekker MR, Dekkers DHW, Brandsma JH, van den Berg DLC, et al. Mediator complex interaction partners organize the transcriptional network that defines neural stem cells. Nat Commun 2019;10(1):2669 doi 10.1038/s41467-019-10502-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mingot J-M, Vega S, Maestro B, Sanz JM, Nieto MA. Characterization of Snail nuclear import pathways as representatives of C2H2 zinc finger transcription factors. J Cell Sci 2009;122(9):1452–60 doi 10.1242/jcs.041749. [DOI] [PubMed] [Google Scholar]
- 31.Schmitges FW, Radovani E, Najafabadi HS, Barazandeh M, Campitelli LF, Yin Y, et al. Multiparameter functional diversity of human C2H2 zinc finger proteins. Genome Res 2016;26(12):1742–52 doi 10.1101/gr.209643.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Barazandeh M, Lambert SA, Albu M, Hughes TR. Comparison of ChIP-Seq Data and a Reference Motif Set for Human KRAB C2H2 Zinc Finger Proteins. G3 (Bethesda, Md) 2018;8(1):219–29 doi 10.1534/g3.117.300296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Imbeault M, Helleboid PY, Trono D. KRAB zinc-finger proteins contribute to the evolution of gene regulatory networks. Nature 2017;543(7646):550–4 doi 10.1038/nature21683. [DOI] [PubMed] [Google Scholar]
- 34.Sievers QL, Petzold G, Bunker RD, Renneville A, Slabicki M, Liddicoat BJ, et al. Defining the human C2H2 zinc finger degrome targeted by thalidomide analogs through CRBN. Science 2018;362(6414) doi 10.1126/science.aat0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biology 2013;14(4):R36 doi 10.1186/gb-2013-14-4-r36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love MI, Huber W, Anders S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biology 2014;15(12):550 doi 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481(7381):389–93 doi 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.