

Abstract
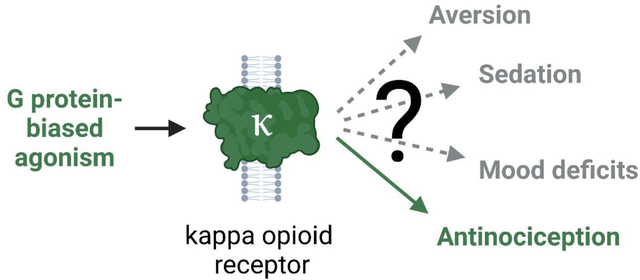
Kappa opioid receptor (κOR) agonists lack the abuse liability and respiratory depression effects of clinically used mu opioid receptor (μOR) analgesics and are hypothesized to be safer alternatives. However, κOR agonists have limiting adverse effects of their own, including aversion, sedation, and mood effects, that have hampered their clinical translation. Studies performed over the last 15 years have suggested that these adverse effects could result from activation of distinct intracellular signaling pathways that are dependent on β-arrestin, whereas signaling downstream of G protein activation produces antinociception. This led to the hypothesis that agonists biased away from β-arrestin signaling would have improved therapeutic windows over traditional unbiased agonists and allow for clinical development of analgesic G-protein-biased κOR agonists. Given a recent controversy regarding the benefits of G-protein-biased μOR agonists, it is timely to reassess the therapeutic promise of G-protein-biased κOR agonists. Here we review recent discoveries from preclinical κOR studies and critically evaluate the therapeutic windows of G-protein-biased κOR agonists in each of the adverse effects above. Overall, we find that G-protein-biased κOR agonists generally have improved therapeutic window relative to unbiased agonists, although frequently study design limits strong conclusions in this regard. However, a steady flow of newly developed biased κOR agonists paired with recently engineered behavioral and molecular tools puts the κOR field in a prime position to make major advances in our understanding of κOR function and fulfill the promise of translating a new generation of biased κOR agonists to the clinic.
Keywords: kappa opioid receptor, biased agonism, antinociception, aversion, sedation, depression
Graphical Abstract
1. Introduction to the kappa opioid receptor
The kappa opioid receptor (κOR) is a G protein-coupled receptor expressed throughout the central nervous system as well as in some peripheral tissues [1,2] and is activated by the endogenous opioid agonist peptide, dynorphin A, which was discovered in 1981 [3]. Dynorphin A is a cleavage product of its precursor protein, preprodynorphin, and is released from neurons through the merging of its resident large dense core vesicles to the plasma membrane [4]. When dynorphin A binds to the κOR, it initiates a signal transduction cascade involving the activation of the heterotrimeric G-protein complex, which for the κOR generally includes the inhibitory Gαi/o-proteins. Following activation, the κOR is rapidly phosphorylated which promotes recruitment of the β-arrestin class of scaffolding proteins [5]. Acutely, these events depress neuronal activity through G protein-mediated inhibition of voltage-gated Ca2+ channels, and the activation of hyperpolarizing channels such as the G protein-couple inwardly rectifying K+ [6], κOR desensitization, as well as β-arrestin-initiated trafficking of neurotransmitter re-uptake channels to the plasma membrane [7]. The longer-term effects of extended κOR activation include the engagement of kinases that lead to a variety of transcriptional changes, the downstream mediators of which we are only beginning to identify [8]. It is these intracellular events that ultimately allow endogenous and exogenous κOR agonists to impact behavior [6,9].
Like the mu opioid receptor (μOR), the κOR is appreciated for its role in nociception and its activation causes analgesia in mammals [10]. κOR activation does not share the same considerable adverse effects, commonly observed with clinical μOR agonists, like morphine and fentanyl, such as respiratory depression, constipation, and high abuse liability [6]. Yet, potent, efficacious κOR agonists have not translated into humans as alternative analgesic options as they are noteworthy for inducing aversion, dysphoria, sedation, and/or depressive and anxiety-related effects [10,11]. Nevertheless, analgesic drugs that modulate the κOR have been clinically approved, though often the drugs have low potency for κOR (e.g. tramadol) [12,13], are partial agonists (e.g. nalbuphine, butorphanol) [10], or even antagonists (buprenorphine) [14]. In the majority of these cases, the drug has strong(er) potency at μOR from which it derives most of the analgesic efficacy.
However, the concept of biased signaling, i.e. the idea that certain agonists can stabilize κOR conformations that preferentially engage one signaling pathway over another, has unveiled the opportunity for a fully efficacious κOR therapeutic agonist. This idea was driven by early studies in the Chavkin lab, which produced multiple papers suggesting that κOR agonist-induced dysphoria and aversion is dependent on β-arrestin-mediated p38 activation [7,15–17]. Thus, together with earlier in vivo studies that utilized the Gαi-protein inhibitor pertussis toxin to demonstrate that the analgesic effects of κOR agonists depended on G protein signaling [18–20], biasing κOR agonists towards G protein activation and away from β-arrestin signaling was predicted to be beneficial [5]. Later, the finding that κOR agonists remain antinociceptive in mice lacking β-arrestin 2 further confirmed that G protein signaling was necessary, whereas β-arrestin signaling was dispensable for therapeutic efficacy [21]. These studies, and others, some of which will be reviewed here, have fueled efforts to develop and discover novel G protein-biased κOR agonists for treatment of not only pain, but a variety of central and peripheral pathologies. In this review, we examine whether the initial promise of G-protein-biased κOR agonist as a path towards clinical relevance still holds true, needs refining or should be discarded. This is particularly important in light of a recent, and ongoing reassessment of the value of G protein-biased μOR agonists as safer analgesics [22–25].
2. Definition and calculation of bias
As discussed in Section 1, ligand “bias” refers to an agonist preferentially signaling through one pathway over another, relative to a reference agonist. Signaling bias at GPCRs can extend to cover any type of signal output produced following receptor activation, and can include, amongst others, different preferences in G protein isoform recruitment, receptor internalization, phosphorylation by GRK isoforms or recruitment of β-arrestin isoforms. Two pathways can be compared to generate a single scalar measure of bias, or “bias factor”, or several pathways compared simultaneously in spiderweb/radial plots [26] or forest plots [8]. Biased signaling at the κOR has been reviewed recently [27–29] and for this review we mostly limit our discussion to studies investigating bias for G protein signaling over β-arrestin recruitment.
As stated above, bias factors are always calculated with respect to a reference agonist, which ideally taken to be the endogenous agonist of the receptor, or an unbiased synthetic compound that activates and recruits both G protein and β-arrestin pathways to the same degree as the endogenous ligand. The advantage of choosing the endogenous agonist as the reference is that a comparison is made to native biology. A disadvantage of using an endogenous agonist is that endogenous κOR agonists are peptidic with poor central penetration, limiting ‘easy’ systemic administration in preclinical studies. Additional challenges for using dynorphins are that the endogenous opioid peptides have poor selectivity (relative to synthetic agonists designed to be more selective) [30,31] and that it is difficult to measure which endogenous peptide is the most relevant or dominant in a given circuit [32]. In contrast, synthetic small molecule κOR agonists can enable studies where the same reference compound is used to draw comparisons between cellular and behavioral pharmacology of the test compounds, without significant concerns over off-target signaling.
One parameter frequently used to measure and rank signal bias is the bias factor, defined as the 10^ΔΔlog(τ/KA), which compares the dissociation constant, KA, and τ, which estimates the efficacy of the receptor-agonist complex, for a test compound relative to the reference compound between two assays [33]. A downside of this method is that it conflates potency and efficacy; this is problematic as some κOR agonists are deemed G-protein-biased by nature of their potency to activate G proteins despite being able to recruit β-arrestin with 100% efficacy. These types of G-protein-biased agonists are sometimes referred to as affinity-dominant and are in contrast with efficacy-dominant agonists that are biased because the compound has minimal β-arrestin recruitment efficacy [27,34]. In this review, we discuss nalfurafine, the salvinorin A analogues RB-64, mesyl salvinorin B (Mesyl Sal B), ethoxymethyl ether salvinorin B (EOM Sal B), 16-Bromo salvinorin A (16-Bromo Sal A), and triazole 1.1 which are affinity-dominant G-protein-biased agonists (Table 1). We also discuss salvinorin A (Sal A) as a G-protein-biased agonist relative to the early-discovered U50,488 and U69,593 [35,36], though due to its weak bias this compound is often utilized as the unbiased reference compound [21]. In the efficacy-dominant category, we discuss BPHA, 6’-GNTI, HS665, HS666, as well as recently developed compounds LOR17, “compound 4a” and the new peptide agonist Helianorphin19 (Table 1).
Table 1. Signaling bias and antinociceptive potency of κOR agonists.
Overview of the bias profile (unbiased, G-protein-biased or arrestin-biased as well as affinity- or efficacy-dominant) for what are generally considered G-protein-biased κOR agonists. Antinociceptive ED50 values are provided where possible, otherwise the reported dose or dose range in mg/kg that was significantly antinociceptive in the study is provided in parentheses. Listed are the pain model and animal species/strain used in the study. Doses are systemic unless otherwise indicated. Select studies for the classic agonist U50,488 are included at the top as reference.
| Agonist name | Bias Direction | Bias Type | Bias Reference | Antinociceptive ED50, mg/kg | Antinociception Assay | Organism | In vivo Reference |
|---|---|---|---|---|---|---|---|
| U50,488 | Unbiased | NA | [144–146] | 7.2 nmol i.c.v. | Tail withdrawal | C57BL/6 mice | [65] |
| Arrestin (mκOR) | Affinity | [145] | 6.3–6.7 | Tail withdrawal | B6-SJL mice | [50,147] | |
| 5.2 | Tail flick | ddY mice | [48] | ||||
| 8.7 | Low temperature hot plate | ddY mice | [69] | ||||
| 1.2 | Acetic acid writhe | ddY mice | [48] | ||||
| 1.5 | Acetic acid writhe | CD-1 mice | [111] | ||||
| 0.58 | Formalin | CD-1 mice | [58] | ||||
| 11 | Paw pressure | Wistar rats | [49] | ||||
| (5 – 30) | Tail flick | C57BL/6J mice | [73] | ||||
| Nalfurafine | G protein | Affinity | [36,72,104] | (0.05 – 0.15) | Tail withdrawal | C57BL/6 mice | [104] |
| Unbiased | NA | [58] | (0.015 – 0.06) | Tail withdrawal | C57BL/6J mice | [36] | |
| Arrestin | Affinity | [144] | 0.062 | Tail flick | ddY mice | [48] | |
| 0.129 | Low temperature hot plate | ddY mice | [69] | ||||
| 0.0033 | Acetic acid | ddY mice | [48] | ||||
| 0.0058 | Formalin | CD-1 mice | [58] | ||||
| 0.0096 | Formalin | Wistar rats | [49] | ||||
| 0.009 | Tail pressure | ddY mice | [69] | ||||
| 0.035 | Tail pinch | ddY mice | [69] | ||||
| 0.064 | Paw pressure | Wistar rats | [49] | ||||
| Salvinorin A | G protein | Affinity | [35,36] | 1.4 | Tail withdrawal | Sprague-Dawley rats | [59] |
| Unbiased | NA | [144,148] | 2.1 | Tail withdrawal | B6-SJL/ptprca mice | [35] | |
| (1 – 2) | Formalin | B6-SJL/ptprca mice | [35] | ||||
| (2) | Hot plate | B6-SJL mice | [50] | ||||
| (2) | Formalin | B6-SJL mice | [50] | ||||
| (1) | Tail withdrawal | B6-SJL mice | [54] | ||||
| 2.1 | Tail withdrawal | B6-SJL mice | [147] | ||||
| RB-64 | G protein | Affinity | [21,148] | (3) | Hot plate | C57BL/6 mice | [21] |
| Mesyl Sal B | G protein | Affinity | [35,144] | 3.0 | Tail withdrawal | B6-SJL/ptprca mice | [35] |
| (1 – 2) | Formalin | B6-SJL/ptprca mice | [35] | ||||
| (1) | Tail withdrawal | B6-SJL mice | [54] | ||||
| EOM Sal B | G protein | Affinity | [36] | 0.83 | Tail withdrawal | Sprague-Dawley rats | [59] |
| (1) | Tail withdrawal | C57BL/6J mice | [36] | ||||
| 16-Bromo Sal A | G protein | Affinity | [50] | 2.1 | Tail withdrawal | B6-SJL mice | [50] |
| (2) | Hot plate | B6-SJL mice | [50] | ||||
| (1 – 2) | Formalin | B6-SJL mice | [50] | ||||
| Triazole 1.1 | G protein | Affinity | [36,73] | (15 – 30) | Tail flick | C57BL/6J mice | [73] |
| HS665 | G protein | Both | [65,144] | 3.7 nmol i.c.v. | Tail withdrawal | C57BL/6J mice | [65] |
| 1.9 | Acetic acid writhe | CD-1 mice | [111] | ||||
| HS666 | G protein | Both | [65] | 6 nmol i.c.v. | Tail withdrawal | C57BL/6J mice | [65] |
| G protein | Efficacy | [144] | 3.2 | Acetic acid writhe | CD-1 mice | [77] | |
| BPHA | G protein | Efficacy | [110,144] | Unknown | |||
| 6’-GNTI | G protein | Efficacy (may prefer heterodimers) | [66,149] | (10 nmol i.c.v) | Tail flick | ICR-CD1 mice | [66] |
| 0.36 nmol i.t. | Tail flick | 129S6 mice | [150] | ||||
| LOR17 | G protein | Efficacy | [52] | 10 | Tail withdrawal | CD-1 mice | [52] |
| 5.7 | Acetic acid writhe | CD-1 mice | [52] | ||||
| (1 – 20) | Oxaliplatin cold plate | CD-1 mice | [52] | ||||
| Compound 4a | G protein | Efficacy | [78] | (5) | Tail flick | C57BL/6J mice | [78] |
| (5) | Hot plate | C57BL/6J mice | [78] | ||||
| Helianorphin 19 | G protein | Both | [116] | (1 nmol i.c.) | Colorectal distension | C57BL/6J mice | [116] |
3. G biased agonism in analgesia
The κOR is expressed in dorsal root ganglia neurons and in the spinal cord, where they directly modulate nociception [37,38]. The κOR is also expressed throughout the basal ganglia and the mesolimbic pathway, putting it in key positions to regulate reward, motivation, mood and general motor behavior [9,39,40]. Generally, the κOR suppresses each of these behaviors. Thus, the κOR system is commonly summarized as analgesic in the periphery, with the undesirable side effects of κOR activation (aversion, sedation, anxiety, et cetera) in the central nervous system [6,41]. Supporting this, classic κOR agonists that penetrate the brain such as U50,488 [42] and U69,593 [43] produce antinociception but then are hampered by small therapeutic windows due to their activation of these alternative processes, giving them a negative side effect profile.
However, there is evidence that some of the antinociceptive components of κOR activation occur centrally [44,45]. Additionally, pain is multidimensional: though the detection of noxious stimuli occurs in the periphery, the aversive component of chronic pain is centrally modulated via κORs in the nucleus accumbens [46]. Thus, attempts to generate successful, peripherally restricted κOR analgesics have so far failed, leaving the idea that biased agonism can biochemically separate analgesic from undesired effects as the most promising avenue for bringing κOR analgesics into the clinic [47]. G-protein-biased κOR agonists have been tested in a variety of pain models – both acute and chronic – including thermal (e.g. hot plate/tail flick), mechanical (e.g. formalin/Von Frey), visceral (e.g. acetic acid writhe test) and neuropathic (e.g. nerve constriction test). From these studies it can be concluded that both balanced and G-protein-biased κOR agonists generally are antinociceptive (Table 1), supporting the idea that analgesia is driven through G protein-associated pathways at the κOR [6,27].
Though all G-protein-biased κOR agonists tested to date cause some form of antinociception, certain agonists appear more effective against certain types of pain than others. For example, nalfurafine (also known as TRK-820) was nearly 20 times more potent in the acetic acid writhe test than the tail flick assay in ddY mice [48]. When evaluated in Wistar rats, nalfurafine was approximately seven times more potent in the formalin (aqueous formaldehyde) test than a paw pressure test [49]. Mesyl Sal B [35] and 16-Bromo Sal B [50] demonstrated an opposite pattern: each showed antinociception in the warm water tail withdrawal model of thermal pain but were much less effective in a formalin model of pain in mice. In contrast, the reference compounds in these studies, Sal A [35] and the novel balanced agonist 16-ethynyl salvinorin A [50] were effective in both pain assays. Likewise, the newly developed SLL039/SLL1206 exhibited similar potencies in the hot plate, formalin, and acetic acid writhe tests [51]. The efficacy-dominant G-protein-biased κOR agonist LOR17 also displayed similar potencies in the hot plate and acetic acid writhe tests [52].
Thus, there is more nuance to antinociception by the κOR than can be captured by merely G protein bias vs β-arrestin. The data also cannot be explained by relative selectivity for the κOR. For example, though nalfurafine is only moderately selective for the κOR over the μOR [53], both Mesyl Sal B and Sal A are extremely selective for the κOR [54], and yet show different patterns of potency in the nociceptive assays. Gaining pharmacokinetic data, including information on the relative tissue distribution of these compounds, may help explain the differences in their performance. Additionally, applying newer techniques such as phosphoproteomic approaches that evaluate the impact of an agonist on many signaling partners simultaneously may reveal specific biochemical pathways that are responsible [8]. As discussed in the following sections, these compounds also differ in their potencies in generating adverse effects, which can potentially influence behavior in antinociceptive assays.
4. Aversion
If “rewarding” is defined as the quality that makes an organism repeat a behavior or desire an outcome, then “punishment” is the quality that makes an organism avoid or dislike a behavioral outcome [55]. The rewarding or punishing nature of a drug, when paired with neutral environmental stimuli (e.g. different patterning in a testing chamber), will generate a preference or an aversion, respectively, of those environmental stimuli that is readily evaluated in a conditioned place preference test [56,57]. Aversion stands as a serious hurdle to patient compliance with κOR-mediated drugs. Thus, it is a key effect that researchers have sought to engineer out of κOR agonists by introducing G protein bias.
It was discovered early that classic, unbiased κOR agonists such as U50,488 [42] and U69,593 [43] are aversive, significantly restricting their therapeutic window. For example, U50,488 has an ED50 in the formalin test of 0.58 mg/kg in male CD-1 mice but causes aversion at doses as low as 0.25 mg/kg [58]. In C57BL/6J mice, U50,488 has an ED50 of 5.0 mg/kg in the tail withdrawal assay [59] but is aversive at a 2.5 mg/kg dose [15]. The same holds true in Sprague-Dawley rats, which show a 12.7 mg/kg ED50 for U50,488 in the hot plate assay [60], with aversion occurring at just 10 mg/kg [59].
Nalfurafine is a biased κOR agonist that is affinity-dominant (Table 1) and has very strong potency in the formalin pain model; nalfurafine consistently has ED50 values below 10 μg/kg [49,58]. However, nalfurafine does not produce aversion up to a dose of 40 μg/kg in Sprague-Dawley rats [61], 30 μg/kg in male ddY mice [62], and up to 20 μg/kg in CD-1 mice [58] and 15 μg/kg in C57BL/6J mice [36]. Thus, nalfurafine has a clear therapeutic window in rodents, supporting its clinical approval in Japan for uremic pruritis [63,64].
Salvinorin A and its analogue RB-64, which is significantly more G-protein-biased than salvinorin A, were both aversive at antinociceptive doses tested (3 mg/kg) in the hot plate assay [21]. Like nalfurafine, RB-64 is an affinity-dominant G-protein-biased agonist that efficaciously recruits β-arrestin 2, yet lacks an apparent therapeutic window for aversion [21]. It may be that RB-64 is antinociceptive at lower doses than 3 mg/kg, and the data underestimates its potency. Another possible reason for this discrepancy is that nalfurafine was more potent in formalin and acetic acid pain tests than in thermal pain models (Table 1) [48,58], and perhaps RB-64 might similarly be more potent in those pain assays, resulting potentially in a more significant therapeutic window.
Another salvinorin A analogue, EOM Sal B, did not produce aversion at 0.1 mg/kg, a dose at which it reduced cocaine-primed reinstatement in Sprague-Dawley rats [59]. EOM Sal B displayed some antinociceptive potency (EC50 = 0.8 mg/kg in the warm water tail withdrawal assay), but aversion was not tested at higher doses so it is unclear if there is a therapeutic window for analgesia. Another study found that a 1 mg/kg dose of EOM Sal B was aversive in C57BL/6J mice [36]. Similar gaps are present for Mesyl Sal B, which has an antinociceptive ED50 of 3 mg/kg in the tail withdrawal assay and is at least not aversive at a dose of 0.3 mg/kg, but higher doses were not evaluated [35].
With regard to efficacy-dominant G-protein-biased κOR agonists, studies suggest that limited β-arrestin 2 recruitment efficacy correlates with limited conditioned place aversion. Intriguingly, the diphenylethylamines HS665 and HS666 both displayed potent single digit nanomole ED50 values in the tail withdrawal assay, however HS665 which recruits β-arrestin 2 recruitment more efficaciously than HS666, displayed conditioned place aversion at 30 nmol (i.c.v.) whereas HS666 did not, even at 150 nmol [65]. Similarly, the efficacy-dominant G protein-biased agonist 6’-GNTI, which is antinociceptive at 10 nmol i.c.v. in the tail flick assay in ICR-CD1 mice [66] was not aversive in C57BL/6N mice up to 30 nmol i.c.v. [67].
Thus, nearly all explored affinity-dominant G-protein-biased agonists produce aversion at a large enough dose, and agonists biased in efficacy or both efficacy and affinity may have higher therapeutic windows for aversion. Still, a limitation for reaching such a conclusion is that many studies utilized different species, strains and pain assays, making proper comparisons difficult. Moreover, despite early studies linking aversion to arrestin-dependent signaling in cells [7,15–17], RB-64 and salvinorin A remained aversive in β-arrestin 2 knockout mice, suggesting this response was not dependent on β-arrestin 2 [21]. Given the diverse phosphorylation patterns in κOR agonists, it may be that reducing κOR signaling to simply “G protein” or “arrestin” may not be sufficient for dissecting the plethora of behavioral effects of the κOR [8].
5. Sedation
Beyond aversion, the promise of G-protein-biased κOR agonism comes from studies suggesting that avoiding β-arrestin may reduce the sedative or hypolocomotive properties of κOR agonists. Sedation is typically measured through either measuring an animal’s ambulatory locomotor behavior (hypolocomotion), sometimes after the “novel stimulus” of entering the testing chamber, or through a rotarod experiment that measures the length of time a rodent is able to maintain walking on a rotating beam [68]. These two assays have some nuances in that the locomotion is motivated differently (exploring environment versus fear of falling from rod) and in that the rotarod is also a measure of muscle coordination. These nuances can sometimes be detected by κOR agonists. For example, whereas 20 μg/kg nalfurafine does not cause hypolocomotion it does show impairment on the rotarod at that dose [58].
Generally, nalfurafine has an ED50 of 27 μg/kg or higher on the rotarod [69,70], which is above its antinociceptive ED50 in several pain assays (Table 1). Similarly, nalfurafine’s therapeutic antipruritic effects require a 10-fold lower dose (0.1 μg/kg) than the 1 μg/kg nalfurafine in rhesus monkeys that reduces overall activity [71]. Thus, nalfurafine has a therapeutic window both for antinociception and antipruritic activity over sedative adverse effects. The importance of bias with regards to improving therapeutic window is exemplified by comparing nalfurafine with its analog nalfurafine 42B. Nalfurafine 42B showed significant hypolocomotor effects at just two times its antinociceptive midpoints and produced muscle incoordination on the rotarod at sub-antinociceptive doses [72]. The difference between nalfurafine and nalfurafine 42B is that the former has a G protein bias factor of 4.49, whereas the latter’s is 2.85 [72]. Similarly, the κOR agonist U50,488, which is generally considered to have limited bias relative to dynorphin, also has poor resolution between doses causing antinociception and doses causing sedation. For example, 5 mg/kg U50,488 produced only modest antinociception in a tail flick assay in C57BL/6 mice, a dose that caused significant deficits in novelty-induced locomotion [73]. Another study reported ED50 values for hot plate and acetic acid writhe to be 4.42 and 0.89 mg/kg, respectively, with an ED50 of 3.32 mg/kg for U50,488 in the rotarod test, confirming a negligible therapeutic window for U50,488 [74].
Similarly to U50,488, salvinorin A produces hypolocomotive effects in rodents [21,50] and non-human primates [71,75]. However, derivatives of salvinorin A with greater G protein-bias can have higher thresholds for sedation. For example, 10 and 3 mg/kg RB-64 did not have sedative effects in either the rotarod or novelty-induced locomotion tests, respectively, compared to a 3 mg/kg dose producing antinociception in a hot plate assay in C57BL/6J mice [21].
Unfortunately, the locomotor effects of the salvinorin analogue Mesyl Sal B were evaluated at doses lower than those for antinociception, making it difficult to draw conclusions with regard to its therapeutic. Specifically, two studies found Mesyl Sal B to be effective in the 1 – 3 mg/kg range in the tail withdrawal assay but tested for locomotor deficits at 0.3 – 2 mg/kg range in rodents [35,54]. Since no hypolocomotive effects were seen, this data neither rules out nor demonstrates a therapeutic window for Mesyl Sal B.
While Mesyl Sal B may yet have its therapeutic window demonstrated, two other salvinorin A analogues, EOM Sal B and 16-Bromo Sal A, look less promising. For example, one study measured the ED50 of EOM Sal B in the warm water tail withdrawal test to be 0.83 mg/kg, and found that 0.3 mg/kg EOM Sal B did not show decreased ambulatory activity in Sprague-Dawley rats [59]. However, another study found that a 1 mg/kg dose of EOM Sal B that was antinociceptive in a tail flick assay in C57BL/6 mice also inhibited novelty-induced locomotor activity, arguing against the pursuit of EOM Sal B as a clinical candidate [36]. Likewise, 16-Bromo Sal A showed mild muscle incoordination on the rotarod assay at 1 mg/kg, compared to an ED50 of 2.1 mg/kg in the tail withdrawal test [50]. Though 16-Bromo Sal A did not impact ambulatory locomotor activity at the 1 mg/kg dose [76], 16-Bromo Sal A is unlikely to have a strong therapeutic window for sedation. This data is another example of the rotarod assay being more sensitive to κOR effects than ambulatory locomotion.
The affinity-dominant G-protein-biased κOR agonist Triazole 1.1 did not show alterations in locomotor activity in male C57BL/6 mice up to 30 mg/kg but elicited antipruritic and antinociceptive responses at doses as low as 1 and 15 mg/kg, respectively [73]. This held true across species as doses of triazole 1.1 up to 0.32 mg/kg i.v. did not promote sedative behaviors in rhesus monkeys but blocked oxycodone-induced scratching [71]. Two κOR agonists biased in both efficacy and affinity, HS665 and HS666, also showed no motor impairment in the rotarod assay when injected at 30 nmol i.c.v., much higher than the minimal antinociceptive dose of 3 nmol in C57BL/6 mice [65]. This result repeated when the agonists were injected subcutaneously in CD-1 mice, with HS665 and HS666 not causing muscle incoordination at up to 10 and 20 mg/kg, respectively, compared with ED50 values of 1.91 and 3.23 mg/kg for antinociception in an acetic acid writhe test [77]. Thirdly, 6’GNTI, an efficacy-dominant agonist, did not modulate ambulatory locomotor activity of C57BL/6N mice at doses up to 30 nmol (i.c.v.) [67].
Lastly, two new efficacy-dominant G-protein-biased κOR agonists show promise for antinociception without sedation. LOR17 did not alter rotarod performance at 10 mg/kg s.c., double its ED50 (5.74 mg/kg) in the acetic acid writhe test and equal to its ED50 in the hot plate test (10.07 mg/kg) [52]. Similarly, a novel beta-carboline (compound 4a) was antinociceptive in the hot plate assay in C57BL/6J mice at 5 mg/kg i.p. but did not decline rotarod performance at 10 mg/kg (although this dose was administered orally) [78].
Overall, G-protein-biased κOR agonists show a wide range of abilities to avoid sedation. Nalfurafine, RB-64, triazole 1.1, the diphenethylamines HS665 and HS666, and the efficacy-dominant agonists 6’-GNTI, LOR17, and compound 4a tend to not cause sedation at antinociceptive doses. In contrast, salvinorin A, EOM Sal B, and to a lesser extent 16-Bromo Sal A do cause hypolocomotion at antinociceptive doses. The results for Mesyl Sal B are promising but ambiguous until doses above the antinociceptive range are tested. Of note is that drugs tested in the rotarod tend to have lower therapeutic windows for sedation than those tested by ambulatory locomotor activity (e.g. nalfurafine, [58]; 16-Bromo Sal A, [50,76]). Thus, muscle coordination may be more sensitive to κOR activation than general sedation and could therefore be more important to assess in order to establish safety and maximal tolerable doses.
The above results would be meaningfully extended by studies evaluating the effects of biased κOR agonists on electrocortical activity. Large scale changes in brain activity patterns between sleep and wakefulness produce unique signatures that can be readily detected by electroencephalographic methods. These techniques can therefore give a more direct measure of whether a novel κOR agonist is altering movement due to sedation as opposed to other psychological effects [79]. These measurements can be made concurrently with electromyographs to give additional insights on whether immobility is due to sedation or incoordination. Though the invasive nature of these methods makes them less amenable to initial drug screening, they offer a strong complement to behavioral methods for establishing the sedative profile of candidate drugs.
6. Pro-anxiety and pro-depressive behaviors
A major hurdle for the translational development of κOR agonists has been their ability to induce anxiety-like (often measured by the elevated plus maze test [80]), depression-like (forced-swim test [81]) behaviors as well as anhedonia (intracranial self-stimulation (ICSS) [82]) and dysphoria. The classic and relatively unbiased/balanced κOR agonists U50,488 and U69,693 cause these undesirable effects at doses close to antinociceptive doses. For example, U69,593 increased immobility time in the forced swim test, i.e. increased depression-like behavior, at doses as low as 0.3 mg/kg i.p. in Sprague-Dawley rats [83]. As little as 0.25 mg/kg U69,593 can cause anhedonia in an ICSS test in Sprague-Dawley rats [84,85]. U50,488 promotes anhedonia in the ICSS test in Fisher 344 rats at a 6 mg/kg dose [73] and decreases sucrose responding (an alternative measure for anhedonia to ICSS) at 5 and 10 mg/kg in Long-Evans [86] and Sprague-Dawley [59] rats, respectively. In mice, doses as low as 0.5 and 1 mg/kg of U50,488 and U69,593, respectively, were sufficient to raise ICSS thresholds [21,58]. In contrast, the G-protein-biased nalfurafine did not alter ICSS in CD-1 mice up to a dose of 20 μg/kg, three times higher than the ED50 for reducing pain in the formalin test in this study [58].
Inconsistency in doses selected for assessing anxiety, depression, and anhedonia relative to the agonist’s therapeutic effect hampers evaluating the therapeutic window of several G-protein-biased agonists that have been studied in rodent models. For example, RB-64 is antinociceptive at a 3 mg/kg dose, but was tested in ICSS at 1 mg/kg [21]. Though RB-64 did not alter ICSS thresholds, a core measure of anhedonia, it did create a rightward shift in how mice responded to different frequencies of stimulation, an alternative measure of anhedonia. Thus, the results are ambiguous for RB-64 [21]. Mesyl Sal B produced depression-like effects at 0.3 mg/kg (well below the antinociceptive dose, Table 1), but was not anxiogenic at this dose in male Sprague-Dawley rats, nor did a 0.3 and 1 mg/kg Mesyl Sal B dose alter sucrose self-administration [35]. EOM Sal B and salvinorin A did not induce anhedonia in male Sprague-Dawley rats at 0.1 and 0.3 mg/kg doses, respectively, as measured by decreases in responding to sucrose [59]. Additionally, while 0.1 and 0.3 mg/kg EOM Sal B did not alter time spent in the open arm in the elevated maze test, 0.3 mg/kg salvinorin A decreased open arm time (promoted anxiety) [59]. These doses of EOM Sal B also did not alter mobility in the forced swim test [59]. However, similarly to the studies on RB-64 and Mesyl Sal B [21,35], the tested doses of EOM Sal B and salvinorin A are lower than their warm water tail flick ED50 values of 0.83 mg/kg and 1.4 mg/kg, respectively [59]. Thus, the no observed adverse effect level (NOAEL) of salvinorin A is lower than its antinociceptive dose, whereas the results for EOM Sal B are ambiguous as it lacks a Maximum Tolerated Dose, relative to its respective tail-flick ED50.
In contrast to the above studies, 16-Bromo Sal A was tested for anxiety measures at antinociceptive doses [50]. 16-Bromo Sal A up to 2 mg/kg did not alter time in open arm or arm entries in the elevated zero maze test, nor did it alter marble burying (a measure of anxiety/compulsivity) in C57BL/6J mice, though 1 and 2 mg/kg was sufficient for thermal antinociception in B6-SJL mice [50]. Triazole 1.1 also did not affect ICSS in Fisher 344 rats at doses up to 24 mg/kg, compared with antinociceptive responses at doses as low as 15 mg/kg in C57BL/6J [73]. the 24 mg/kg dose of Triazole 1.1 muted the ability of 1.8% lactic acid to reduce ICSS in the ventral tegmental area in C57BL/6J mice [73]. Thus, not only is triazole 1.1 not anhedonic on its own, but it can relieve anhedonia brought on by painful stimuli [11].
LOR17, a recently discovered G-protein-biased κOR agonist, is the only efficacy-dominant agonist in this review to have been tested for depression-like behavior. LOR17 did not alter mobility time in the forced swim paradigm at 10 mg/kg, double its ED50 (5.74 mg/kg) in the acetic acid writhing test and equal to its ED50 (10.07 mg/kg) in the hot plate test [52]. More work is needed to identify the therapeutic window in this assay and whether this extends to other efficacy-dominant G-protein-biased agonists at the κOR.
Despite the limitations of interpreting therapeutic windows for G-biased κOR agonists relative to mood-related adverse effects, it appears that multiple biased agonists, including nalfurafine, triazole 1.1, 16-Bromo Sal A, and LOR17 can avoid negative mood behavior at antinociceptive doses, and thus can differentiate themselves from unbiased κOR agonists.
7.1. Utility for G-protein-biased κOR agonists to reduce substance use disorders
The therapeutic potential of κOR agonists is not restricted to their use as alternatives to μOR analgesics. Two indications actively being explored are substance use disorders and pruritus (discussed in the following subsection). Agonism of the κOR can block the rewarding aspects of μOR agonists, supporting the idea that κOR agonists may serve as adjuvants to analgesic drugs to mute their abuse liability [36,87]. This idea remains true for G-protein-biased κOR agonists. For example, 15 μg/kg nalfurafine and 1 mg/kg EOM Sal B each reduced morphine-induced conditioned place preference in C57BL/6J mice [36]. Additionally, nalfurafine disrupted self-administration of fentanyl over food reward in male and female Sprague-Dawley rats [88]. This effect was specific to nalfurafine co-administered with fentanyl, as pretreatment with nalfurafine, even at doses that decreased overall responding (presumably through sedation), had no effect [88]. Likewise, nalfurafine and triazole 1.1 decreased oxycodone (56 μg/kg/injection) self-administration in Sprague-Dawley rats when co-administered at doses as low as 3.2 and 1800 μg/kg/injection, respectively [60]. Notably, nalfurafine and the relatively unbiased agonist salvinorin A also prevented increased responding for oxycodone (25 – 100 μg/kg/injection) in male rhesus monkeys when co-administered at doses as low as 0.18 and 6 μg/kg/injection, respectively [89].
Thus, G-protein-biased agonists can block the place preference of μOR analgesics and limit their self-administration, suggesting co-administration of these drugs can limit clinical abuse liability. In addition, when co-administered, κOR agonists often potentiate the analgesia of traditional pharmaceuticals, which could help lower the amount used and prevent the development of a substance use disorder. For example, 15 μg/kg nalfurafine potentiated the antinociceptive effect of 5 mg/kg morphine in both the tail immersion and hot plate assays [36]. Additionally, both nalfurafine and triazole 1.1 co-administered with oxycodone significantly increased the antinociceptive potency of oxycodone in Sprague-Dawley rats [60]. 10–15 μg/kg nalfurafine can also block morphine-induced hyperlocomotion in rodents, an effect that is blocked by the κOR antagonist norBNI [36,62]. Note that it is important to assess whether κOR agonists can block μOR agonist reward and hyperlocomotion at doses where the κOR agonists are not sedative on their own. For example, one study found mild hypolocomotive effects at mid-range doses of nalfurafine (15 μg/kg), an effect that passed within 30 minutes [36]. 1 mg/kg EOM Sal B showed more pronounced hypolocomotive effects in C57BL/6J mice [36].
Note that many of the findings on κOR agonists blocking morphine reward also generalize to other substances of abuse. For example, doses of 3–20 μg/kg nalfurafine can block cocaine place preference [61,70]. Thus, the utility of biased κOR agonists may extend beyond preventing development of opioid use disorders to reducing relapse/reinstatement following abstinence of other substances of abuse. Historically, κOR antagonists have been pursued to block drug reinstatement and prevent relapse [90,91]. A number of studies have suggested that part of the mechanism by which unbiased κOR agonists can promote reinstatement involves β-arrestin signaling [7,92,93]. As such it can be hypothesized that G-protein-biased agonism may be able to inhibit relapse as it could serve as a functional antagonist of dynorphin-mediated β-arrestin signaling.
Supporting this, pre-injection of either EOM Sal B (0.1 or 0.3 mg/kg), salvinorin A (0.3 mg/kg) [59], or Mesyl Sal A (0.3 or 1.0, but not 0.1 mg/kg) [54] prevented cocaine-induced cocaine self-administration in Sprague-Dawley rats, a model of re-instatement. 16-Bromo Sal A at 0.3 and 1 mg/kg attenuated cocaine reinstatement primed by a 20 mg/kg injection of cocaine in Sprague-Dawley rats [76]. 0.3 mg/kg Mesyl Sal B also blocked sensitization to cocaine-induced hyperlocomotion in Sprague-Dawley rats previously receiving cocaine [35]. Similarly, 0.1 mg/kg EOM Sal B [59] and 0.3 mg/kg Mesyl Sal B [35] reduced cocaine-induced hyperlocomotion in Sprague-Dawley rats. 0.3 and 1 mg/kg Mesyl Sal B co-administered with 1 mg/kg of the FDA-approved alcohol use disorder treatment naltrexone also decreased ethanol consumption in an intermittent access drinking model in male and female C57BL/6J mice [94]. A 10x higher dose of Mesyl Sal B was required to lower ethanol consumption when administered without naltrexone. These effects didn’t repeat in a lower-consumption drinking-in-the-dark model, suggesting this result may be specific to animals with a history of high ethanol consumption [94].
However, pretreatment with 10 μg/kg, but not 3, nalfurafine increased self-administration of cocaine in C57BL/6 mice, an effect that was blocked by the reversible κOR agonist LY2444296 [70]. Indeed, studies with the unbiased κOR agonist U50,488 find that effects on drug reward and self-administration are time and dose-dependent, with U50,488 able to either block or potentiate both cocaine/ethanol place preference [95–97] and cocaine self-administration [98]. Therefore, since the field is new enough that few results have been published with multiple time points for G-protein-biased agonists, caution should be used when interpreting the above findings.
Overall, co-administration of G-protein-biased κOR agonists consistently lowers the place preference and self-administration of μOR analgesics while improving their antinociceptive potency. However, especially considering the potential confounding effects of timing and dose seen with unbiased κOR agonists, this field is still in its infancy and it is too soon to ascertain a therapeutic window relative to hypolocomotion or aversion for the presented biased κOR agonists. With regard to treating substance use disorders that are already developed, the data is even more complex. Nonetheless, triazole 1.1 and, to a lesser extent, nalfurafine, show promise in this area. Note that of the fourteen G-protein-biased agonists in Table 1, only six have data in the substance use disorder field. Diversifying these studies to include a wider array of G-protein-biased agonists will also help develop our understanding of the potential of these agonists in this area.
7.2. Utility for G-protein-biased κOR agonists to treat itch and inflammation
κORs are expressed in human mast cells and κOR stimulation reduces mast cell numbers [99] which may attribute to the established antipruritic efficacy of κOR agonists [100,101]. As our understanding of pruritus and the role of κOR therein continues to expand, G-protein-biased κOR agonists continue to show promise in this area. An early study looking at orally administered nalfurafine found that 100 μg/kg nalfurafine reduced scratching in substance P-induced itch in ICR mice, an effect that was blocked by norBNI [102]. This same study found that both 30 and 100 μg/kg nalfurafine p.o. inhibited scratching in histamine-induced (10 μg/site) itch [102]. These early studies helped nalfurafine to become the first κOR agonist approved for use in humans in Japan for uremic pruritus [63,64]. Nalfurafine continues to be a standard bearer in testing in this area. Nalfurafine decreased scratching behavior induced by the MRGPRX2 agonist compound 48/80 with an ED50 of 8.0 μg/kg [58]. Nalfurafine also reduced scratching behavior in an ethynylestradiol (2 mg/kg/day for 14 days) cholestatic liver disease model in Sprague-Dawley rats with an ED50 of 13 μg/kg [103]. Recently, doses as low as 0.1 μg/kg nalfurafine were found to reduce spontaneous scratching behavior in rhesus monkeys [71]. Pretreatment with 50 μg/kg nalfurafine also inhibited scratching induced by the κOR antagonist 5’-GNTI in C57BL/6 mice (30 μg/kg) [104].
The affinity-dominant G-protein-biased triazole 1.1 inhibited chloroquine phosphate (40 mg/kg)-induced scratching at doses as low as 1 mg/kg in C57BL/6J mice [73]. Additionally, doses up to 0.32 mg/kg i.v. did not promote sedative behaviors in rhesus monkeys but blocked scratching induced by 0.1 mg/kg oxycodone [71].
Overall, the G-protein-biased κOR agonists nalfurafine and triazole 1.1. consistently mitigate scratching behaviors in animal models. In contrast, 6’-GNTI was found to induce scratching behavior, though it should be noted that 6’-GNTI-induced scratching behavior in C57BL/6J mice was sustained in κOR KO mice, suggesting this is a unique off-target effect [105]. The peripherally restricted κOR agonist CR845 (difelikefalin) recently received approval in the United States for treatment of pruritus associated with chronic kidney disease in hemodialysis patients [106]. With both CR845 and nalfurafine approved in humans for pruritus, it is likely work in this area will continue to expand. In particular, as the mechanisms behind pruritus continue to be elucidated, it will be interesting to see whether some of the benefits of G-protein-biased agonists can be generalized to other inflammatory diseases, such as multiple sclerosis [107]. Indeed, the κOR is expressed on numerous immune cells and its full therapeutic potential in this area has not yet been realized [108].
8.1. Overview of recently developed κOR agonists with known G protein bias
Only a handful of biased κOR agonists have been investigated extensively across multiple studies and in some cases multiple research teams. Yet there are numerous signal-biased κOR agonists that have been characterized solely in the paper where they were first described. In the following section we review some of the more recently developed biased κOR agonists and other agonists recently shown to be biased in order to highlight the structural diversity that exists amongst signal-biased κOR agonists. Additionally, this section may promote follow-up studies that could aid in understanding the promise and limitations associated with affinity- and efficacy-dominant G-protein-biased κOR agonists.
Extracts from the kratom plant Mitragyna speciosa have been found to have opioidergic activities via a collection of β-carboline compounds [109]. These compounds are generally non-specific between the opioid receptors, but in a recent collaboration between the Yadav and Batra labs researchers developed a series of fused ring β-carbolines that were highly specific for the κOR [78]. Compound 4a was the most potent (46 nM, Glosensor) and showed bias toward G protein signaling (cAMP) and away from β-arrestin 2 (Tango) and ERK1/2 signaling (SRE-luc). A dose of 5 mg/kg compound 4a was antinociceptive in the tail-flick and hot plate tests – an effect that was blocked by norBNI – while no locomotor deficits were observed even at 10 mg/kg [78].
Two other agonists, N-n-butyl- N-phenylethyl-N-3-hydroxyphenylethyl-amine (BPHA [110], or compound 5 in [111]) and 16-Bromo Sal A [50,76] are derived from other G-protein-biased scaffolds. BPHA is a structural derivative of HS665 and HS666 with 10-fold lower affinity than HS665 [111]. BPHA has strong G protein activation with almost zero observed efficacy for β-arrestin recruitment and did not show sedation at doses up to 30 mg/kg in C57BL/6 mice [110]. 16-Bromo Sal A is a derivative of salvinorin A and has 7.7x bias for G protein signaling over arrestin signaling at the κOR [50]. 16-Bromo Sal A was significantly antinociceptive in a warm water tail withdrawal assay and to a lesser extent in a formalin model of pain in B6-SJL mice [50]. 16-Bromo Sal A was not anxiogenic in the elevated zero maze or the marble burying tests in C57Bl/6J mice, and only had mild incoordination in the rotarod test in B6-SJL mice. The specificity of 16-Bromo Sal A for the κOR over other opioid receptors is unknown.
While biased signaling may be one way to ameliorate the adverse effects associated with κOR agonism, another approach is to avoid engaging centrally expressed κORs in the first place. This is a potentially relevant strategy given that peripheral κORs are of therapeutic interest, as can be gleaned from difelikefalin. Difelikefalin is a synthetic κOR agonists that is peripherally restricted by nature of its peptidic structure and is devoid of aversion and dysphoria [112,113]. Unfortunately, we have been unable to find any reports on the signal-bias of difelikefalin or characterization of the tetrapeptide in an arrestin-recruitment assay, limiting our ability to discuss it in light of biased signaling. Regardless, one does not need to rely on exogenous peptides to discover signal-biased κOR peptides; one recent study characterized 20 endogenous opioid peptides at μOR, δOR and κOR and identified intriguing profiles for several of them at the κOR [31]. Using GTPγS and β-arrestin 2 pathHunter assays to calculate bias factors, the study found that DynorphinA1–13 and metorphamide are β-arrestin-biased peptides at the κOR. Additionally, Stefanucci et al computationally screened 6 million tripeptides and found two sequences, H-D-Tyr-Val-Val-O-(3-Br)-Bz and H-D-Tyr-Val-Trp-OBz, that they then tested in vivo [114]. Both peptides show antinociception in the tail flick (i.c.v) and formalin (i.p.) tests in CD-1 mice, with the effect in the tail flick experiment lasting for up to 90 minutes. More work is needed to understand the level of central penetration of these peptides, as well as their signaling mechanisms and in vivo effects.
Cyclized peptides can overcome some of the metabolic liabilities of natural peptides and have been successfully produced for μOR and δOR. Building on previous work looking at cyclized δOR and μOR tetrapeptides [115], a novel cyclotetrapeptide (c[Phe-Gly-(b-Ala)-D- Trp]) named LOR17 that has ≥105-fold selectivity for κOR over μOR or δOR was recently discovered [52]. Additionally, the authors calculated a bias factor for G protein (cAMP EIA kit) over β-arrestin 2 (PathHunter) signaling of 853 for LOR17. The study also utilized two human cell lines, U87-MG (ATCC HTB-14) and normal human astrocytes (Lonza CC-2565) that endogenously express κORs to cross-validate their in vitro results. LOR17 was found to be antinociceptive in a panel of assays, including tests of thermal (warm water tail withdrawal), visceral (acetic acid writhing), and neuropathic pain (oxaliplatin cold plate test). The same dose (10 mg/kg) was found to have no sedative effects in a small battery of locomotor and coordination tests [52].
Another group, inspired by the antinociceptive cyclopeptides in sunflower, cyclized dynorphin and added two cysteine residues to bridge the ring across the middle of the sequence [116]. Their top hit, named helianorphin-19 after the species name for sunflower, Helianthus annuus, has ~200-fold selectivity for κOR over μOR and δOR. Helianorphin-19 is a full agonist in the cAMP assay (CisBio HTRF) but only a partial agonist for β-arrestin 2 in a BRET assay, with similar shifts in potency between helianorphin-19 and dynorphin. Thus, it likely has some degree of bias toward G protein signaling over β-arrestin 2. Helianorphin-19 ablated hypersensitivity in a model of chronic visceral pain (dinitrobenzene sulfonic acid – induced colitis). This effect seems to be due to a lowered response in nociceptor firing rate to mechanical stimuli, an effect that was blocked by norBNI. Moreover, a 5 mg/kg dose of helianorphin-19 did not cause incoordination in a rotarod assay. As the peptide was delivered in different routes, it is difficult to infer a therapeutic window from these tests. Nonetheless, the relative dosing suggests helianorphin-19 is a promising candidate for visceral pain.
8.2. Summary of newer κOR agonists with unknown signaling properties
A number of recent studies have described preclinical efficacy of novel κOR agonists for which bias is unknown but may be inferred based on the known profile of the compounds they are modelled after. For example, The Shao lab has recently found a series of 6,14-endoethanotetrahydronorthebaines, chemical structures related to the G-protein-biased nalfurafine, to be potent, κOR-selective agonists [117,118]. Both SLL-039 [117] and SLL-1206 [118] showed antinociception in hot plate, abdominal constriction, and formalin tests [51,117,118]. These two compounds have long-lasting antinociception, each peaking at 4–6 hours post-injection. Moreover, each have 3–10x therapeutic windows for analgesia over hypolocomotor and aversion However, the hypolocomotor and aversive effects of SLL-039 and SLL-1206 were evaluated at 2 to 3 hours post-injection, which matches the timing for the antinociception assays but misses early timepoints where κOR agonists can have acute effects (e.g. [21,35]). Further investigating these compounds in this acute time range will help establish their clinical potential.
The Abels lab recently synthesized two new series of perhydroquinoxalines with excellent potency and selectivity for the κOR [119,120]. They selected molecules for restriction to the periphery and found candidates with anti-inflammatory activity. When tested in human skin organ culture, they found that compound 5a [119] decreased the number of mast cells, supporting its use as an antipruritic [99]. While the bias of the agonists in their 2017 series has not yet been evaluated, the agonists in the 2019 series are arrestin biased relative to their reference compound salvinorin A. Since salvinorin A has itself been found to have G protein bias relative to dynorphin [35,36], these calculations should be followed up on. Nonetheless, it would be interesting to see these compounds characterized in models of pain and pruritus.
The Wunsch lab recently engineered novel potent κOR agonists building off earlier work [121] that investigated compounds wherein the N-atom of the dichlorophenylacetamide group of the κOR agonist U50,488 is incorporated into a piperidine ring [122]. Their approach was to further modify the piperidine ring into bicyclic moieties. Their top hits have excellent selectivity for κOR over μOR, δOR, and σ2 receptors, with moderate (8 – 22x) selectivity over σ1 receptors.
The Tao group recently designed a series of peptides modifying the termini of the peripheral κOR agonist difelikefalin/CR845 [123]. Their top hit, SHR0687, is peripherally restricted, highly selective κOR agonist showing antinociception in a carrageenan-induced allodynia model (paw withdrawal threshold). Notably, in this model SHR0687 produced antinociception with similar potency as difelikefalin.
9. Conclusions, recommendations, and future directions
Despite being proposed over a decade ago [5] G-protein-biased agonism at the κOR is still in its infancy with regard to understanding the pharmacological basis of its adverse effect profile (Table 2). Given the availability of a diverse array of efficacy- and affinity-dominant G-protein-biased agonists, this is an excellent time to begin evaluating the pharmacology of these compounds in more stringent preclinical models for pain and substance use disorders, as well as testing their adverse effects in multiple models, at multiple time points, and at multiple doses. Additionally, assessment of tachyphylaxis upon repeated administration of efficacy- and affinity-dominant G protein-biased agonists to either the analgesic or adverse effects is not typically performed, but would be of great value for drug lead development [124]. It is also critical to carry out pharmacokinetic evaluations and perform absorption, distribution, metabolism, and excretion (ADME) screening to determine how much of the biased κOR agonists enter the brain, and for how long. Additionally, the use of female subjects should be part of the study design as sex-differences have been reported for κOR ligands [125–127].
Table 2. Therapeutic windows of G protein-biased κOR agonists in antinociception versus various adverse effects.
Green indicates that the minimum dose producing an adverse effect is higher than the antinociceptive dose or the antinociceptive ED50. Red indicates that the adverse effect occurs at or below the antinociceptive dose/ED50. Yellow indicates that the doses tested were below the antinociceptive dose/ED50 and the results are therefore ambiguous. A “u” means this adverse effect is untested for this agonist. Design credit: [47].
| Aversion | Sedation | Pro-anxiety/ depression | |
|---|---|---|---|
| Nalfurafine | |||
| Sal A | |||
| RB-64 | |||
| EOM Sal B | |||
| Mesyl Sal B | |||
| 16-Bromo Sal A | u | ||
| Triazole 1.1 | u | ||
| HS665 | u | u | |
| HS666 | u | ||
| BPHA | u | u# | u |
| 6’-GNTI | u | ||
| LOR17 | u | ||
| Compound 4a | u | u | |
| Helianorphin19 | u | u | u |
This property was studied but there is no antinociceptive data for BPHA
In particular, no efficacy-dominant κOR agonist has been fully characterized in all three of the adverse effect areas in this review. Moreover, among the affinity-dominant agonists, the evaluation of the therapeutic window for the salvinorin A derivative RB-64 is based only on single- and two-point tests. For certain κOR agonists, such as EOM Sal B and Mesyl Sal B, adverse effects were measured at doses that were lower than the antinociceptive doses, making these results ambiguous [35,59]. We recommend testing for adverse effects at least at 1x and 3x (roughly a half log unit above) the antinociceptive dose tested (or the ED50 value). Among the adverse effects, we recommend evaluating sedation first, since this can potentially confound nearly every other behavioral assay (including antinociception itself). Even if rotarod/ambulatory activity cannot be quantified, having some qualitative measure of locomotor effects adds significant confidence to the interpretation of any other behavioral tests performed. After screening in rodents, the establishment of a non-sedative profile in nonhuman primates for promising κOR agonists can be performed using qualitative measurements of behavior [71], and is particularly important for motivating future translational studies of the drug.
The evaluation of many of the agonists are also based on only one type of nociception, most commonly thermal pain, which κOR agonists are commonly not as potent for as chemical (formalin) and visceral pain (Section 3) [48,52]. In addition to more broadly characterizing agonists with the pain models discussed above, a host of new pain models have been developed that have been validated with traditional analgesics [47]. These include the evaluation of restoration of depressed behaviors following pain stimuli as well as operant measures, such as the restoration of ICSS responding following a painful stimulus [47,73]. Classic κOR agonists typically fail in these antinociceptive assays, making them a more stringent test of effectiveness for new G-protein-biased κOR agonists [11,47]. It should be noted that although the G-protein-biased agonist nalfurafine failed to progress in clinical trials for analgesia, it is still debated whether nalfurafine is G protein- or arrestin-biased (Table 1) [11]. Broader characterization of κOR agonists with a diverse range in bias will help better prune candidates and define the profile of a successful κOR analgesic [11,47].
Despite challenges in translating κOR analgesics to humans, κOR agonists have had some success in the clinic. Thus far, one of the most promising areas of translation of κOR agonists is in treating pruritus. Nalfurafine was the first κOR-selective agonist to receive clinical authorization, for drug-resistant itch in uremic patients in Japan [64]. Peripherally restricted κOR agonists, which are designed with the mindset that most unwanted side-effects are centrally mediated, have also failed to translate to the clinic as analgesic option [47]. Nonetheless, the peripherally restricted κOR agonist CR845 (difelikefalin) was recently approved in the United States under the brand name KORSUVA for pruritus in chronic kidney disease [106]. It is also currently in clinical trials for pruritus in patients with primary biliary cholangitis, notalgia paresthetica, and atopic dermatitis [128]. As discussed above in Section 7.2, G-protein-biased agonists tend to mitigate scratching behavior in animal models. Despite the clinical success of κOR agonists in treating pruritus, of the sixteen studies summarized in Section 8 on new and understudied κOR agonists, eleven of them used some measure of pain, while only one [51] used a measure of pruritus. Incorporating antipruritic behavioral tests would greatly enhance evaluations the clinical potential of new κOR agonists.
Advances in tool development may further aid in probing in vivo effects of G-protein-biased κOR agonists. For example, optodialysis probes [129] and the fluorescent, genetically encoded dynorphin sensor kLight[130] can be used to study to what degree G-protein-biased κOR agonists compete for dynorphin binding or modulate dynorphin release. Efforts could be expended to create G-protein-biased agonists for chemogenetic tools such as κORdi [131], a κOR that has been engineered to respond to Sal B but not to κOR ligands. Similarly, G-protein-biased photoactivable dynorphin analogues [132] could be developed to help study biased κOR signaling in vivo. Advanced mass spectrometry methods are also useful for characterizing divergent downstream signaling cascades, for example those that include p38 [15] and mTOR [58]. Finally, the use of specific nanobodies that stabilize signal-biased conformations will be useful both in cellular assays and in generating Cryo-EM or X-ray crystallographic structural information of κOR bound to biased agonists [133–136]. These efforts together will help refine what the ideal signaling profile of a κOR agonist looks like, in an iterative process that will improve translational efforts at this receptor.
While much of the above has focused on G protein signaling, further tool development is needed to understand the role of β-arrestins at the κOR. Though early studies suggested a link between β-arrestin 2-dependent p38 signaling and adverse effects at the κOR [7,15–17], new data in β-arrestin 2 KO mice suggests this protein is not essential for aversive components of κOR signaling [21]. Following up with conditional β-arrestin 2 KO mice [137,138] could reduce any confounding compensatory changes in congenic β-arrestin 2 KO mice. Additionally, nearly all of the work summarized to date uses β-arrestin 2 recruitment as a proxy for arrestin signaling at the κOR. However, when exploring biased signaling, β-arrestin 1 should not be discounted, as increasing evidence suggests the two β-arrestin isoforms fulfill unique and even opposing roles [139–143]. New tools to study these isoforms in detail will further our understanding of how β-arrestins contribute to the diverse behavioral profiles of κOR agonists.
In summary, despite a decade of effort, there is much work to do in understanding the potential of G protein-biased κOR agonists for clinical use in humans (Table 2), however we would argue their potential remains. With two κOR agonists now approved for pruritus [64,106], testing in this area should be a major consideration when demonstrating initial therapeutic potential of new ligands, even among research teams searching for novel κOR analgesics. Lastly, the plethora of new tools developed for studying the κOR, including the ligands reviewed here, offer exciting opportunities to expand our understanding of how biased agonism translates into altered brain function, and the multifaceted roles of the κOR in health and disease.
Acknowledgements
Research reported in this publication was supported by the NIAAA of the NIH under Award Number R01AA025368 (RMvR).
Abbreviations:
- κOR
kappa opioid receptor
- μOR
mu opioid receptor
- δOR
delta opioid receptor
- 6’-GNTI
6’-guanidinonaltrindol
- 16-Bromo Sal A
16-Bromo salvinorin A
- BRET
bioluminescence resonance energy transfer
- cAMP
cyclic adenosine monophosphate
- EOM Sal B
ethoxymethyl ether salvinorin B
- GPCR
G protein-coupled receptor
- HS665
3-[2-(Cyclobutylmethyl-phenethyl-amino)-ethyl]-phenol Hydrochloride
- HS666
3-(2-((Cyclopropylmethyl)(phenethyl)amino)ethyl)phenol hydrochloride
- KO
knockout
- LOR17
c[Phe-Gly-(b-Ala)-D-Trp]
- Mesyl Sal B
mesyl salvinorin B
- nalfurafine
(2E)-N-[(5α,6β)-17-(cyclopropylmethyl)- 3,14-dihydroxy- 4,5-epoxymorphinan- 6-yl]- 3-(3-furyl)-N-methylacrylamide hydrochloride
- norBNI
norbinaltorphimine
- U50,488
2-(3,4-dichlorophenyl)-N-methyl-N-[(1R,2R)-2-pyrrolidin-1-ylcyclohexyl]acetamide
- U69,593
a,7a,8b)-N-methyl-N-(7-[1-pyrrolidinyl]-1-oxaspiro[4.5]dec8-yl)-benzenacetamide
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of competing interests
Authors have no competing interests to disclose.
CRediT Author Statement
Alex French: Writing - Original draft, Writing – Review and editing, conceptualization; Richard van Rijn:. Writing – Review and editing, conceptualization, supervision, funding acquisition
References
- [1].Chen C, Willhouse AH, Huang P, Ko N, Wang Y, Xu B, Huang LHM, Kieffer B, Barbe MF, Liu-Chen LY, Characterization of a knock-in mouse line expressing a fusion protein of k opioid receptor conjugated with tdtomato: 3-dimensional brain imaging via clarity, ENeuro. 7 (2020) 1–18. 10.1523/ENEURO.0028-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Peng J, Sarkar S, Chang SL, Opioid receptor expression in human brain and peripheral tissues using absolute quantitative real-time RT-PCR, Drug and Alcohol Dependence. 124 (2012) 223–228. 10.1016/j.drugalcdep.2012.01.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Goldstein A, Fischli W, Lowney LI, Hunkapillert M, Hoodt L, Porcine pituitary dynorphin : Complete amino acid sequence of the biologically active heptadecapeptide, 78 (1981) 7219–7223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Chavkin C, Dynorphin-Still an Extraordinarily Potent Opioid Peptide, Molecular Pharmacology. 83 (2012) 729–736. 10.1124/mol.112.083337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Bruchas MR, Chavkin C, Kinase cascades and ligand-directed signaling at the kappa opioid receptor, Psychopharmacology. 209 (2010) 137–147. 10.1007/s00213-009-1768-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Machelska H, Celik MÖ, Advances in Achieving Opioid Analgesia Without Side Effects., Frontiers in Pharmacology. 9 (2018) 1388. 10.3389/fphar.2018.01388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Bruchas MR, Schindler AG, Shankar H, Messinger DI, Miyatake M, Land BB, Lemos JC, Hagan CE, Neumaier JF, Quintana A, Palmiter RD, Chavkin C, Selective p38α MAPK deletion in serotonergic neurons produces stress resilience in models of depression and addiction, Neuron. 71 (2011) 498–511. 10.1016/j.neuron.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Liu JJ, Sharma K, Zangrandi L, Chen C, Humphrey SJ, Chiu YT, Spetea M, Liu-Chen LY, Schwarzer C, Mann M, In vivo brain GPCR signaling elucidated by phosphoproteomics, Science. 360 (2018). 10.1126/science.aao4927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Cahill C, Tejeda HA, Spetea M, Chen C, Liu-Chen L-Y, Fundamentals of the Dynorphins/Kappa Opioid Receptor System: From Distribution to Signaling and Function, (2021). 10.1007/164_2021_433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Paton KF, Atigari D. v., Kaska S, Prisinzano T, Kivell BM, Strategies for developing k opioid receptor agonists for the treatment of pain with fewer side effects, Journal of Pharmacology and Experimental Therapeutics. 375 (2020) 332–348. 10.1124/JPET.120.000134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lazenka MF, Antinociceptive Effects of Kappa-Opioid Receptor Agonists, (2021). 10.1007/164_2020_430. [DOI] [PubMed] [Google Scholar]
- [12].Raffa RB, Friderichs E, Reimann W, Shank RP, Codd EE, Vaught JL, Opioid and nonopioid components independently contribute to the mechanism of action of tramadol, an “atypical” opioid analgesic, Journal of Pharmacology and Experimental Therapeutics. 260 (1992) 275–285. [PubMed] [Google Scholar]
- [13].Sun HL, Zheng JW, Wang K, Liu RK, Liang JH, Tramadol reduces the 5-HTP-induced head-twitch response in mice via the activation of μ and κ opioid receptors, Life Sciences. 72 (2003) 1221–1230. 10.1016/S0024-3205(02)02345-7. [DOI] [PubMed] [Google Scholar]
- [14].Lutfy K, Cowan A, Buprenorphine: A Unique Drug with Complex Pharmacology, Current Neuropharmacology. 2 (2004) 395–402. 10.2174/1570159043359477.Buprenorphine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bruchas MR, Land BB, Aita M, Xu M, Barot SK, Li S, Chavkin C, Stress-induced p38 mitogen-activated protein kinase activation mediates κ-opioid-dependent dysphoria, Journal of Neuroscience. 27 (2007) 11614–11623. 10.1523/JNEUROSCI.3769-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Ehrich JM, Messinger DI, Knakal CR, Kuhar JR, Schattauer SS, Bruchas MR, Zweifel LS, Kieffer BL, Phillips PEM, Chavkin C, Kappa Opioid Receptor-Induced Aversion Requires p38 MAPK Activation in VTA Dopamine Neurons, Journal of Neuroscience. 35 (2015) 12917–12931. 10.1523/jneurosci.2444-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Land BB, Bruchas MR, Lemos JC, Xu M, Melief EJ, Chavkin C, The dysphoric component of stress is encoded by activation of the dynorphin κ-opioid system, Journal of Neuroscience. 28 (2008) 407–414. 10.1523/JNEUROSCI.4458-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Hernandez A, Soto-Moyano R, Mestre C, Eschalier A, Pelissier T, Paeile C, Contreras E, Intrathecal pertussis toxin but not cyclic AMP blocks kappa opioid-induced antinociception in rat, International Journal of Neuroscience. 81 (1995) 193–197. 10.3109/00207459509004886. [DOI] [PubMed] [Google Scholar]
- [19].Gullapalli S, Ramarao P, Role of L-type Ca2+ channels in pertussis toxin induced antagonism of U50,488H analgesia and hypothermia, Brain Research. 946 (2002) 191–197. 10.1016/S0006-8993(02)02880-9. [DOI] [PubMed] [Google Scholar]
- [20].Goicoechea C, Ormazábal MJ, Abalo R, Alfaro MJ, Martín MI, Calcitonin reverts pertussis toxin blockade of the opioid analgesia in mice, Neuroscience Letters. 273 (1999) 175–178. 10.1016/S0304-3940(99)00640-0. [DOI] [PubMed] [Google Scholar]
- [21].White KL, Robinson JE, Zhu H, DiBerto JF, Polepally PR, Zjawiony JK, Nichols DE, Malanga CJ, Roth BL, The G Protein–Biased κ -Opioid Receptor Agonist RB-64 Is Analgesic with a Unique Spectrum of Activities In Vivo, Journal of Pharmacology and Experimental Therapeutics. 352 (2015) 98–109. 10.1124/jpet.114.216820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kliewer A, Gillis A, Hill R, Schmiedel F, Bailey C, Kelly E, Henderson G, Christie MJ, Schulz S, Morphine-induced respiratory depression is independent of β -arrestin2 signalling, (2020) 2923–2931. 10.1111/bph.15004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Gillis A, Gondin AB, Kliewer A, Sanchez J, Lim HD, Alamein C, Manandhar P, Santiago M, Fritzwanker S, Schmiedel F, Katte TA, Reekie T, Grimsey NL, Kassiou M, Kellam B, Krasel C, Halls ML, Connor M, Lane JR, Schulz S, Christie MJ, Canals M, Low intrinsic efficacy for G protein activation can explain the improved side effect profiles of new opioid agonists, Science Signaling. 13 (2020). 10.1126/scisignal.aaz3140. [DOI] [PubMed] [Google Scholar]
- [24].Stahl EL, Bohn LM, Low Intrinsic Efficacy Alone Cannot Explain the Improved Side Effect Profiles of New Opioid Agonists, Biochemistry. (2021). 10.1021/acs.biochem.1c00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].He L, Gooding SW, Lewis E, Felth LC, Gaur A, Whistler JL, Pharmacological and genetic manipulations at the μ-opioid receptor reveal arrestin-3 engagement limits analgesic tolerance and does not exacerbate respiratory depression in mice, Neuropsychopharmacology. 46 (2021) 2241–2249. 10.1038/s41386-021-01054-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zhou L, Lovell KM, Frankowski KJ, Slauson SR, Phillips AM, Streicher JM, Stahl E, Schmid CL, Hodde P, Madoux F, Cameron MD, Prisinzano TE, Aubé J, Bohn LM, Development of functionally selective, small molecule agonists at kappa opioid receptors, Journal of Biological Chemistry. 288 (2013) 36703–36716. 10.1074/jbc.M113.504381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Mores KL, Cummins BR, Cassell RJ, van Rijn RM, A Review of the Therapeutic Potential of Recently Developed G Protein-Biased Kappa Agonists, Frontiers in Pharmacology. 10 (2019) 1–14. 10.3389/fphar.2019.00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].de Neve J, Barlow TMA, Tourwé D, Bihel F, Simonin F, Ballet S, Comprehensive overview of biased pharmacology at the opioid receptors: biased ligands and bias factors, RSC Medicinal Chemistry. 12 (2021) 828–870. 10.1039/d1md00041a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Dogra S, Yadav PN, Biased agonism at kappa opioid receptors: Implication in pain and mood disorders, European Journal of Pharmacology. 763 (2015) 184–190. 10.1016/j.ejphar.2015.07.018. [DOI] [PubMed] [Google Scholar]
- [30].Corbett AD, Paterson SJ, Kosterlitz HW, Selectivity of Ligands for Opioid Receptors, in: Opioids. Handbook of Experimental Pharmacology, 1993: pp. 645–679. 10.1007/978-3-642-77460-7_26. [DOI] [Google Scholar]
- [31].Gomes I, Sierra S, Lueptow L, Gupta A, Gouty S, Margolis EB, Cox BM, Devi LA, Biased signaling by endogenous opioid peptides., Proceedings of the National Academy of Sciences of the United States of America. (2020) 1–9. 10.1073/pnas.2000712117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Karkhanis AN, Al-Hasani R, Dynorphin and its role in alcohol use disorder, Brain Research. 1735 (2020) 146742. 10.1016/j.brainres.2020.146742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Kenakin T, Watson C, Muniz-Medina V, Christopoulos A, Novick S, A simple method for quantifying functional selectivity and agonist bias, ACS Chemical Neuroscience. 3 (2012) 193–203. 10.1021/cn200111m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Kenakin T, The effective application of biased signaling to new drug discovery, Molecular Pharmacology. 88 (2015) 1055–1061. 10.1124/mol.115.099770. [DOI] [PubMed] [Google Scholar]
- [35].Kivell BM, Paton KF, Kumar N, Morani AS, Culverhouse A, Shepherd A, Welsh SA, Biggerstaff A, Crowley RS, Prisinzano TE, Kappa opioid receptor agonist mesyl sal B attenuates behavioral sensitization to cocaine with fewer aversive side-effects than salvinorin a in rodents, Molecules. 23 (2018) 1–25. 10.3390/molecules23102602. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Kaski SW, White AN, Gross JD, Trexler KR, Wix K, Harland AA, Prisinzano TE, Aubé J, Kinsey SG, Kenakin T, Siderovski DP, Setola V, Preclinical Testing of Nalfurafine as an Opioid-sparing Adjuvant that Potentiates Analgesia by the Mu Opioid Receptor-targeting Agonist Morphine, Journal of Pharmacology and Experimental Therapeutics. 371 (2019) 487–499. 10.1124/jpet.118.255661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Snyder LM, Chiang MC, Loeza-Alcocer E, Omori Y, Hachisuka J, Sheahan TD, Gale JR, Adelman PC, Sypek EI, Fulton SA, Friedman RL, Wright MC, Duque MG, Lee YS, Hu Z, Huang H, Cai X, Meerschaert KA, Nagarajan V, Hirai T, Scherrer G, Kaplan DH, Porreca F, Davis BM, Gold MS, Koerber HR, Ross SE, Kappa Opioid Receptor Distribution and Function in Primary Afferents, Neuron. 99 (2018) 1274–1288.e6. 10.1016/j.neuron.2018.08.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Jhamandas K, Sutak M, Lemaire S, Comparative spinal analgesic action of dynorphin 1–8, dynorphin 1–13, and a kappa-receptor agonist U50,488, Canadian Journal of Physiology and Pharmacology. 64 (1986) 263–268. 10.1139/y86-042. [DOI] [PubMed] [Google Scholar]
- [39].Ji MJ, Yang J, Gao ZQ, Zhang L, Liu C, The Role of the Kappa Opioid System in Comorbid Pain and Psychiatric Disorders: Function and Implications, Frontiers in Neuroscience. 15 (2021) 1–10. 10.3389/fnins.2021.642493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Estave PM, Spodnick MB, Karkhanis AN, KOR Control over Addiction Processing: An Exploration of the Mesolimbic Dopamine Pathway, (2020) 1–27. 10.1007/164_2020_421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Ko M-C, Husbands SM, Pleiotropic Effects of Kappa Opioid Receptor-Related Ligands in Non-human Primates, Handbook of Experimental Pharmacology. (2020) 435–452. 10.1007/164_2020_419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Vonvoigtlander PF, Lahti RA, Ludens JH, U-50,488: a selective and structurally novel non-Mu (kappa) opioid agonist., The Journal of Pharmacology and Experimental Therapeutics. 224 (1983) 7–12. http://www.ncbi.nlm.nih.gov/pubmed/6129321. [PubMed] [Google Scholar]
- [43].Lahti RA, Mickelson MM, McCall JM, von Voigtlander PF, [3H]U-69593 a highly selective ligand for the opioid kappa receptor., European Journal of Pharmacology. 109 (1985) 281–4. 10.1016/0014-2999(85)90431-5. [DOI] [PubMed] [Google Scholar]
- [44].Ge Y, Lundeberg T, Yu L, Blockade effect of mu and kappa opioid antagonists on the anti-nociception induced by intra-periaqueductal grey injection of oxytocin in rats, 927 (2002) 204–207. [DOI] [PubMed] [Google Scholar]
- [45].Ji G, Neugebauer V, Kappa opioid receptors in the central amygdala modulate spinal nociceptive processing through an action on amygdala CRF neurons, Molecular Brain. 13 (2020) 128. 10.1186/s13041-020-00669-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Massaly N, Copits BA, Wilson-Poe AR, Hipólito L, Markovic T, Yoon HJ, Liu S, Walicki MC, Bhatti DL, Sirohi S, Klaas A, Walker BM, Neve R, Cahill CM, Shoghi KI, Gereau RW, McCall JG, Al-Hasani R, Bruchas MR, Morón JA, Pain-Induced Negative Affect Is Mediated via Recruitment of The Nucleus Accumbens Kappa Opioid System, Neuron. 102 (2019) 564–573.e6. 10.1016/j.neuron.2019.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Negus SS, Core Outcome Measures in Preclinical Assessment of Candidate Analgesics., Pharmacological Reviews. 71 (2019) 225–266. 10.1124/pr.118.017210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nagase H, Hayakawa J, Kawamura K, Kawai K, Takezawa Y, Matsuura H, Tajima C, Endo T, Discovery of a structurally novel opioid kappa-agonist derived from 4,5-epoxymorphinan., Chemical & Pharmaceutical Bulletin. 46 (1998) 366–9. 10.1248/cpb.46.366. [DOI] [PubMed] [Google Scholar]
- [49].Endoh T, Tajima A, Suzuki T, Kamei J, Suzuki T, Narita M, Tseng L, Nagase H, Characterization of the antinociceptive effects of TRK-820 in the rat, European Journal of Pharmacology. 387 (2000) 133–140. 10.1016/S0014-2999(99)00815-8. [DOI] [PubMed] [Google Scholar]
- [50].Paton KF, Biggerstaff A, Kaska S, Crowley RS, la Flamme AC, Prisinzano TE, Kivell BM, Evaluation of Biased and Balanced Salvinorin A Analogs in Preclinical Models of Pain, Frontiers in Neuroscience. 14 (2020) 1–13. 10.3389/fnins.2020.00765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].yuan Wei Y, Ma Y, yu Yao S, hui Kong L, Liu X, rui Chai J, Chen J, Li W, jun Wang Y, ming Shao L, gen Liu J, Novel selective κ agonists SLL-039 and SLL-1206 produce potent antinociception with fewer sedation and aversion, Acta Pharmacologica Sinica. (2021) 1–11. 10.1038/s41401-021-00761-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Bedini A, di Cesare Mannelli L, Micheli L, Baiula M, Vaca G, de Marco R, Gentilucci L, Ghelardini C, Spampinato S, Functional Selectivity and Antinociceptive Effects of a Novel KOPr Agonist, Frontiers in Pharmacology. 11 (2020) 1–22. 10.3389/fphar.2020.00188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Seki T, Awamura S, Kimura C, Ide S, Sakano K, Minami M, Nagase H, Satoh M, Pharmacological properties of TRK-820 on cloned μ-, δ- and κ-opioid receptors and nociceptin receptor, European Journal of Pharmacology. 376 (1999) 159–167. 10.1016/S0014-2999(99)00369-6. [DOI] [PubMed] [Google Scholar]
- [54].Simonson B, Morani AS, Ewald AWM, Walker L, Kumar N, Simpson D, Miller JH, Prisinzano TE, Kivell BM, Pharmacology and anti-addiction effects of the novel κ opioid receptor agonist Mesyl Sal B, a potent and long-acting analogue of salvinorin A, British Journal of Pharmacology. 172 (2015) 515–531. 10.1111/bph.12692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Huffman DC, The use of place conditioning in studying the neuropharmacology of drug reinforcement, Brain Research Bulletin. 23 (1989) 373–387. 10.1016/0361-9230(89)90224-4. [DOI] [PubMed] [Google Scholar]
- [56].Cunningham CL, Gremel CM, Groblewski PA, Drug-induced conditioned place preference and aversion in mice, Nature Protocols. 1 (2006) 1662–1670. 10.1038/nprot.2006.279. [DOI] [PubMed] [Google Scholar]
- [57].Hillhouse T, Prus A, Conditioned Place Preference Test for Assessing the Rewarding Effects of, in: The Brain Reward System, Neuromethods, 2021: pp. 263–278. 10.1007/978-1-0716-1146-3_13. [DOI] [Google Scholar]
- [58].Liu JJ, Chiu YT, DiMattio KM, Chen C, Huang P, Gentile TA, Muschamp JW, Cowan A, Mann M, Liu-Chen LY, Phosphoproteomic approach for agonist-specific signaling in mouse brains: mTOR pathway is involved in κ opioid aversion, Neuropsychopharmacology. 44 (2019) 939–949. 10.1038/s41386-018-0155-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Ewald AWM, Bosch PJ, Culverhouse A, Crowley RS, Neuenswander B, Prisinzano TE, Kivell BM, The C-2 derivatives of salvinorin A, ethoxymethyl ether Sal B and β-tetrahydropyran Sal B, have anti-cocaine properties with minimal side effects, Psychopharmacology. 234 (2017) 2499–2514. 10.1007/s00213-017-4637-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Zamarripa CA, Pareek T, Schrock HM, Prisinzano TE, Blough BE, Sufka KJ, Freeman KB, The kappa-opioid receptor agonist, triazole 1.1, reduces oxycodone self-administration and enhances oxycodone-induced thermal antinociception in male rats, Psychopharmacology. (2021). 10.1007/s00213-021-05965-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Mori T, Nomura M, Nagase H, Narita M, Suzuki T, Effects of a newly synthesized κ-opioid receptor agonist, TRK-820, on the discriminative stimulus and rewarding effects of cocaine in rats, Psychopharmacology. 161 (2002) 17–22. 10.1007/s00213-002-1028-z. [DOI] [PubMed] [Google Scholar]
- [62].Tsuji M, Takeda H, Matsumiya T, Nagase H, Narita M, Suzuki T, The novel κ-opioid receptor agonist TRK-820 suppresses the rewarding and locomotor-enhancing effects of morphine in mice, Life Sciences. 68 (2001) 1717–1725. 10.1016/S0024-3205(01)00957-2. [DOI] [PubMed] [Google Scholar]
- [63].Kumagai H, Ebata T, Takamori K, Muramatsu T, Nakamoto H, Suzuki H, Effect of a novel kappa-receptor agonist, nalfurafine hydrochloride, on severe itch in 337 haemodialysis patients: A Phase III, randomized, double-blind, placebo-controlled study, Nephrology Dialysis Transplantation. 25 (2010) 1251–1257. 10.1093/ndt/gfp588. [DOI] [PubMed] [Google Scholar]
- [64].Kumagai H, Ebata T, Takamori K, Miyasato K, Muramatsu T, Nakamoto H, Kurihara M, Yanagita T, Suzuki H, Efficacy and safety of a novel κ-agonist for managing intractable pruritus in dialysis patients, American Journal of Nephrology. 36 (2012) 175–183. 10.1159/000341268. [DOI] [PubMed] [Google Scholar]
- [65].Spetea M, Eans SO, Ganno ML, Lantero A, Mairegger M, Toll L, Schmidhammer H, McLaughlin JP, Selective κ receptor partial agonist HS666 produces potent antinociception without inducing aversion after i.c.v. administration in mice, British Journal of Pharmacology. 174 (2017) 2444–2456. 10.1111/bph.13854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Waldhoer M, Fong J, Jones RM, Lunzer MM, Sharma SK, Kostenis E, Portoghese PS, Whistler JL, A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers, Proceedings of the National Academy of Sciences. 102 (2005) 9050–9055. 10.1073/pnas.0501112102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Zangrandi L, Burtscher J, Mackay JP, Colmers WF, Schwarzer C, The G-protein biased partial κ opioid receptor agonist 6′-GNTI blocks hippocampal paroxysmal discharges without inducing aversion, British Journal of Pharmacology. 173 (2016) 1756–1767. 10.1111/bph.13474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Jones BJ, Roberts DJ, The quantitative measurement of motor inco-ordination in naive mice using an accelerating rotarod, Journal of Pharmacy and Pharmacology. 20 (1968) 302–304. 10.1111/j.2042-7158.1968.tb09743.x. [DOI] [PubMed] [Google Scholar]
- [69].Endoh T, Matsuura H, Tajima A, Izumimoto N, Tajima C, Suzuki T, Saitoh A, Suzuki T, Narita M, Tseng L, Nagase H, Potent antinociceptive effects of TRK-820, a novel κ-opioid receptor agonist, Life Sciences. 65 (1999) 1685–1694. 10.1016/S0024-3205(99)00417-8. [DOI] [PubMed] [Google Scholar]
- [70].Dunn A, Windisch K, Ben-Ezra A, Pikus P, Morochnik M, Erazo J, Reed B, Kreek MJ, Modulation of cocaine-related behaviors by low doses of the potent KOR agonist nalfurafine in male C57BL6 mice, Psychopharmacology. 237 (2020) 2405–2418. 10.1007/s00213-020-05543-7. [DOI] [PubMed] [Google Scholar]
- [71].Huskinson SL, Platt DM, Brasfield M, Follett ME, Prisinzano TE, Blough BE, Freeman KB, Quantification of observable behaviors induced by typical and atypical kappa-opioid receptor agonists in male rhesus monkeys, Psychopharmacology. (2020). 10.1007/s00213-020-05519-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Cao D, Huang P, Chiu YT, Chen C, Wang H, Li M, Zheng Y, Ehlert FJ, Zhang Y, Liu-Chen LY, Comparison of pharmacological properties between the kappa opioid receptor agonist nalfurafine and 42b, its 3-dehydroxy analogue: Disconnect between in vitro agonist bias and in vivo pharmacological effects, ACS Chemical Neuroscience. 11 (2020) 3036–3050. 10.1021/acschemneuro.0c00407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Brust TF, Morgenweck J, Kim SA, Rose JH, Locke JL, Schmid CL, Zhou L, Stahl EL, Cameron MD, Scarry SM, Aubé J, Jones SR, Martin TJ, Bohn LM, Biased agonists of the kappa opioid receptor suppress pain and itch without causing sedation or dysphoria., Science Signaling. 9 (2016) ra117. 10.1126/scisignal.aai8441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Wang YJ, Tao YM, Li FY, Wang YH, Xu EJ, Chen J, Cao YL, Chi ZQ, Neumeyer JL, Zhang A, Liu JG, Pharmacological characterization of ATPM [(−)-3-amino- Thiazolo[5,4-b]-N-cyclopropylmethylmorphinan hydrochloride], a novel mixed κ-agonist and μ-agonist/-antagonist that attenuates morphine antinociceptive tolerance and heroin self-administration behavi, Journal of Pharmacology and Experimental Therapeutics. 329 (2009) 306–313. 10.1124/jpet.108.142802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Butelman ER, Prisinzano TE, Deng H, Rus S, Kreek MJ, Unconditioned behavioral effects of the powerful κ-opioid hallucinogen salvinorin A in nonhuman primates: Fast onset and entry into cerebrospinal fluid, Journal of Pharmacology and Experimental Therapeutics. 328 (2009) 588–597. 10.1124/jpet.108.145342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Riley AP, Groer CE, Young D, Ewald AW, Kivell BM, Prisinzano TE, Synthesis and κ-opioid receptor activity of furan-substituted salvinorin A analogues, Journal of Medicinal Chemistry. 57 (2014) 10464–10475. 10.1021/jm501521d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Erli F, Guerrieri E, ben Haddou T, Lantero A, Mairegger M, Schmidhammer H, Spetea M, Highly Potent and Selective New Diphenethylamines Interacting with the κ-Opioid Receptor: Synthesis, Pharmacology, and Structure–Activity Relationships, Journal of Medicinal Chemistry. 60 (2017) 7579–7590. 10.1021/acs.jmedchem.7b00981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Yadav VD, Kumar L, Kumari P, Kumar S, Singh M, Siddiqi MI, Yadav PN, Batra S, Synthesis and Assessment of Fused β-Carboline Derivatives as Kappa Opioid Receptor Agonists, ChemMedChem. 16 (2021) 1917–1926. 10.1002/cmdc.202100029. [DOI] [PubMed] [Google Scholar]
- [79].Montandon G, Horner RL, Electrocortical changes associating sedation and respiratory depression by the opioid analgesic fentanyl, Scientific Reports. 9 (2019) 1–11. 10.1038/s41598-019-50613-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Walf AA, Frye CA, The use of the elevated plus maze as an assay of anxiety-related behavior in rodents, Nature Protocols. 2 (2007) 322–328. 10.1038/nprot.2007.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Slattery DA, Cryan JF, Using the rat forced swim test to assess antidepressant-like activity in rodents, Nature Protocols. 7 (2012) 1009–1014. 10.1038/nprot.2012.044. [DOI] [PubMed] [Google Scholar]
- [82].Carlezon WA, Chartoff EH, Intracranial self-stimulation (ICSS) in rodents to study the neurobiology of motivation, Nature Protocols. 2 (2007) 2987–2995. 10.1038/nprot.2007.441. [DOI] [PubMed] [Google Scholar]
- [83].Mague SD, Pliakas AM, Todtenkopf MS, Tomasiewicz HC, Zhang Y, Stevens WC, Jones RM, Portoghese PS, Carlezon WA, Antidepressant-Like Effects of κ-Opioid Receptor Antagonists in the Forced Swim Test in Rats, Journal of Pharmacology and Experimental Therapeutics. 305 (2003) 323–330. 10.1124/jpet.102.046433. [DOI] [PubMed] [Google Scholar]
- [84].Tomasiewicz HC, Todtenkopf MS, Chartoff EH, Cohen BM, Carlezon WA, The Kappa-Opioid Agonist U69,593 Blocks Cocaine-Induced Enhancement of Brain Stimulation Reward, Biological Psychiatry. 64 (2008) 982–988. 10.1016/j.biopsych.2008.05.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Todtenkopf MS, Marcus JF, Portoghese PS, Carlezon WA, Effects of κ-opioid receptor ligands on intracranial self-stimulation in rats, Psychopharmacology. 172 (2004) 463–470. 10.1007/s00213-003-1680-y. [DOI] [PubMed] [Google Scholar]
- [86].Henderson-Redmond A, Czachowski C, Effects of systemic opioid receptor ligands on ethanol- and sucrose seeking and drinking in alcohol-preferring (P) and Long Evans rats, Psychopharmacology. 231 (2014) 4309–4321. 10.1007/s00213-014-3571-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Bruijnzeel AW, kappa-Opioid receptor signaling and brain reward function, Brain Research Reviews. 62 (2009) 127–146. 10.1016/j.brainresrev.2009.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Townsend EA, Effects of kappa opioid receptor agonists on fentanyl vs. food choice in male and female rats: contingent vs. non-contingent administration, Psychopharmacology. 238 (2021) 1017–1028. 10.1007/s00213-020-05749-9. [DOI] [PubMed] [Google Scholar]
- [89].Zamarripa CA, Naylor JE, Huskinson SL, Townsend EA, Prisinzano TE, Freeman KB, Kappa opioid agonists reduce oxycodone self-administration in male rhesus monkeys, Psychopharmacology. 237 (2020) 1471–1480. 10.1007/s00213-020-05473-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Wise RA, Koob GF, The development and maintenance of drug addiction, Neuropsychopharmacology. 39 (2014) 254–262. 10.1038/npp.2013.261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Koob GF, Buck CL, Cohen A, Edwards S, Park PE, Schlosburg JE, Schmeichel B, Vendruscolo LF, Wade CL, Whitfield TW, George O, Addiction as a stress surfeit disorder, Neuropharmacology. 76 (2014) 370–382. 10.1016/j.neuropharm.2013.05.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Schindler AG, Messinger DI, Smith JS, Shankar H, Gustin RM, Schattauer SS, Lemos JC, Chavkin NW, Hagan CE, Neumaier JF, Chavkin C, Stress produces aversion and potentiates cocaine reward by releasing endogenous dynorphins in the ventral striatum to locally stimulate serotonin reuptake, Journal of Neuroscience. 32 (2012) 17582–17596. 10.1523/JNEUROSCI.3220-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Land BB, Bruchas MR, Schattauer S, Giardino WJ, Aita M, Messinger D, Hnasko TS, Palmiter RD, Chavkin C, Activation of the kappa opioid receptor in the dorsal raphe nucleus mediates the aversive effects of stress and reinstates drug seeking, Proceedings of the National Academy of Sciences of the United States of America. 106 (2009) 19168–19173. 10.1073/pnas.0910705106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Zhou Y, Crowley RS, Ben K, Prisinzano TE, Kreek MJ, Synergistic blockade of alcohol escalation drinking in mice by a combination of novel kappa opioid receptor agonist Mesyl Salvinorin B and naltrexone, Brain Research. 1662 (2017) 75–86. 10.1016/j.brainres.2017.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Sperling RE, Gomes SM, Sypek EI, Carey AN, McLaughlin JP, Endogenous kappa-opioid mediation of stress-induced potentiation of ethanol-conditioned place preference and self-administration, Psychopharmacology. 209 (2010) 199–209. 10.1007/s00213-010-1844-5. [DOI] [PubMed] [Google Scholar]
- [96].Logrip ML, Janak PH, Ron D, Blockade of ethanol reward by the kappa opioid receptor agonist U50,488H, Alcohol. 43 (2009) 359–365. 10.1016/j.alcohol.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].McLaughlin JP, Land BB, Li S, Pintar JE, Chavkin C, Prior activation of kappa opioid receptors by U50,488 mimics repeated forced swim stress to potentiate cocaine place preference conditioning, Neuropsychopharmacology. 31 (2006) 787–794. 10.1038/sj.npp.1300860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Kuzmin A. v., Semenova S, Gerrits MAFM, Zvartau EE, van Ree JM, κ-Opioid receptor agonist U50,488H modulates cocaine and morphine self-administration in drug-naive rats and mice, European Journal of Pharmacology. 321 (1997) 265–271. 10.1016/S0014-2999(96)00961-2. [DOI] [PubMed] [Google Scholar]
- [99].Chéret J, Gherardini J, Soeberdt M, Hundt JE, Abels C, Bertolini M, Paus R, Non-neuronal kappa-opioid receptor activation enhances epidermal keratinocyte proliferation, and modulates mast cell functions in human skin ex vivo, Journal of Dermatology. 47 (2020) 917–921. 10.1111/1346-8138.15407. [DOI] [PubMed] [Google Scholar]
- [100].Soeberdt M, Kilic A, Abels C, Small molecule drugs for the treatment of pruritus in patients with atopic dermatitis, European Journal of Pharmacology. 881 (2020) 173242. 10.1016/j.ejphar.2020.173242. [DOI] [PubMed] [Google Scholar]
- [101].Reszke R, Krajewski P, Szepietowski JC, Emerging Therapeutic Options for Chronic Pruritus, American Journal of Clinical Dermatology. 21 (2020) 601–618. 10.1007/s40257-020-00534-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Togashi Y, Umeuchi H, Okano K, Ando N, Yoshizawa Y, Honda T, Kawamura K, Endoh T, Utsumi J, Kamei J, Tanaka T, Nagase H, Antipruritic activity of the κ-opioid receptor agonist, TRK-820, European Journal of Pharmacology. 435 (2002) 259–264. 10.1016/S0014-2999(01)01588-6. [DOI] [PubMed] [Google Scholar]
- [103].Inan S, Cowan A, Nalfurafine, a kappa opioid receptor agonist, inhibits scratching behavior secondary to cholestasis induced by chronic ethynylestradiol injections in rats, Pharmacology Biochemistry and Behavior. 85 (2006) 39–43. 10.1016/j.pbb.2006.07.004. [DOI] [PubMed] [Google Scholar]
- [104].Schattauer SS, Kuhar JR, Song A, Chavkin C, Nalfurafine is a G-protein biased agonist having significantly greater bias at the human than rodent form of the kappa opioid receptor, Cellular Signalling. 32 (2017) 59–65. 10.1016/j.cellsig.2017.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Cowan A, Liu-Chen LY, Inan S, Itching-like behavior: A common effect of the kappa opioid receptor antagonist 5′-guanidinonaltrindole and the biased kappa opioid receptor agonist 6′-guanidinonaltrindole in mice, Medicine in Drug Discovery. 11 (2021) 100097. 10.1016/j.medidd.2021.100097. [DOI] [Google Scholar]
- [106].Fishbane S, Jamal A, Munera C, Wen W, Menzaghi F, A phase 3 trial of difelikefalin in hemodialysis patients with pruritus, New England Journal of Medicine. 382 (2020) 222–232. 10.1056/NEJMoa1912770. [DOI] [PubMed] [Google Scholar]
- [107].Denny L, al Abadey A, Robichon K, Templeton N, Prisinzano TE, Kivell BM, la Flamme AC, Nalfurafine reduces neuroinflammation and drives remyelination in models of CNS demyelinating disease, Clinical and Translational Immunology. 10 (2021) 1–19. 10.1002/cti2.1234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Rogers TJ, Kappa Opioid Receptor Expression and Function in Cells of the Immune System, in: Liu-Chen L, Inan S (Eds.), Handbook of Experimental Pharmacology, 2021: pp. 419–433. 10.1007/164_2021_441. [DOI] [PubMed] [Google Scholar]
- [109].Jansen KLR, Prast CJ, Ethnopharmacology of kratom and the Mitragyna alkaloids, Journal of Ethnopharmacology. 23 (1988) 115–119. 10.1016/0378-8741(88)90121-3. [DOI] [PubMed] [Google Scholar]
- [110].Dunn AD, Reed B, Guariglia C, Dunn AM, Hillman JM, Kreek MJ, Structurally related kappa opioid receptor agonists with substantial differential signaling bias: Neuroendocrine and behavioral effects in C57BL6 Mice, International Journal of Neuropsychopharmacology. 21 (2018) 847–857. 10.1093/ijnp/pyy034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Spetea M, Berzetei-Gurske IP, Guerrieri E, Schmidhammer H, Discovery and pharmacological evaluation of a diphenethylamine derivative (HS665), a highly potent and selective κ opioid receptor agonist, Journal of Medicinal Chemistry. 55 (2012) 10302–10306. 10.1021/jm301258w. [DOI] [PubMed] [Google Scholar]
- [112].Shram MJ, Spencer RH, Qian J, Munera CL, Lewis ME, Henningfield JE, Webster L, Menzaghi F, Evaluation of the abuse potential of difelikefalin, a selective kappa-opioid receptor agonist, in recreational polydrug users, Clinical and Translational Science. (2021) 1–13. 10.1111/cts.13173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [113].Vanderah TW, Schteingart CD, Trojnar J, Junien JL, Lai J, Riviere PJM, FE200041 (D-Phe-D-Phe-D-Nle-D-Arg-NH2): A peripheral efficacious κ opioid agonist with unprecedented selectivity, Journal of Pharmacology and Experimental Therapeutics. 310 (2004) 326–333. 10.1124/jpet.104.065391. [DOI] [PubMed] [Google Scholar]
- [114].Stefanucci A, Iobbi V, della Valle A, Scioli G, Pieretti S, Minosi P, Mirzaie S, Novellino E, Mollica A, In silico identification of tripeptides as lead compounds for the design of kor ligands, Molecules. 26 (2021). 10.3390/molecules26164767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].de Marco R, Bedini A, Spampinato S, Cavina L, Pirazzoli E, Gentilucci L, Versatile Picklocks to Access All Opioid Receptors: Tuning the Selectivity and Functional Profile of the Cyclotetrapeptide c[Phe- d -Pro-Phe-Trp] (CJ-15,208), Journal of Medicinal Chemistry. 59 (2016) 9255–9261. 10.1021/acs.jmedchem.6b00420. [DOI] [PubMed] [Google Scholar]
- [116].Muratspahić E, Tomašević N, Koehbach J, Duerrauer L, Hadžić S, Castro J, Schober G, Sideromenos S, Clark RJ, Brierley SM, Craik DJ, Gruber CW, Design of a Stable Cyclic Peptide Analgesic Derived from Sunflower Seeds that Targets the κ-Opioid Receptor for the Treatment of Chronic Abdominal Pain, Journal of Medicinal Chemistry. 64 (2021) 9042–9055. 10.1021/acs.jmedchem.1c00158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Xiao L, Wang Y, Zhang M, Wu W, Kong L, Ma Y, Xu X, Liu X, He Q, Qian Y, Sun H, Wu H, Lin C, Huang H, Ye R, Jiang S, Ye RF, Yuan C, Fang S, Xue D, Yang X, Chen H, Zheng Y, Yu L, Xie Q, Zheng L, Fu W, Li W, Qiu Z, Liu J, Shao L, Discovery of a Highly Selective and Potent κ Opioid Receptor Agonist from N-Cyclopropylmethyl-7α-phenyl-6,14-endoethanotetrahydronorthebaines with Reduced Central Nervous System (CNS) Side Effects Navigated by the Message-Address Concept, Journal of Medicinal Chemistry. 62 (2019) 11054–11070. 10.1021/acs.jmedchem.9b00857. [DOI] [PubMed] [Google Scholar]
- [118].He Q, Wei Y, Liu X, Ye R, Kong L, Li Z, Jiang S, Yu L, Chai J, Xie Q, Fu W, Wang Y, Li W, Qiu Z, Liu J, Shao L, Discovery of an M-Substituted N-Cyclopropylmethyl-7α-phenyl-6,14-endoethanotetrahydronorthebaine as a Selective, Potent, and Orally Active κ-Opioid Receptor Agonist with an Improved Central Nervous System Safety Profile, Journal of Medicinal Chemistry. 64 (2021) 12414–12433. 10.1021/acs.jmedchem.1c01082. [DOI] [PubMed] [Google Scholar]
- [119].Soeberdt M, Molenveld P, Storcken RPM, Bouzanne Des Mazery R, Sterk GJ, Autar R, Bolster MG, Wagner C, Aerts SNH, van Holst FR, Wegert A, Tangherlini G, Frehland B, Schepmann D, Metze D, Lotts T, Knie U, Lin KY, Huang TY, Lai CC, Ständer S, Wünsch B, Abels C, Design and Synthesis of Enantiomerically Pure Decahydroquinoxalines as Potent and Selective κ-Opioid Receptor Agonists with Anti-Inflammatory Activity in Vivo, Journal of Medicinal Chemistry. 60 (2017) 2526–2551. 10.1021/acs.jmedchem.6b01868. [DOI] [PubMed] [Google Scholar]
- [120].Tangherlini G, Kalinin D. v., Schepmann D, Che T, Mykicki N, Ständer S, Loser K, Wünsch B, Development of Novel Quinoxaline-Based κ-Opioid Receptor Agonists for the Treatment of Neuroinflammation, Journal of Medicinal Chemistry. 62 (2019) 893–907. 10.1021/acs.jmedchem.8b01609. [DOI] [PubMed] [Google Scholar]
- [121].Vecchietti V, Giordani A, Giardina G, Colle R, Clarke GD, (2S)-1-(Arylacetyl)-2-(aminomethyl)piperidine derivatives: novel, highly selective .kappa. opioid analgesics, Journal of Medicinal Chemistry. 34 (1991) 397–403. 10.1021/jm00105a061. [DOI] [PubMed] [Google Scholar]
- [122].Jonas H, Aiello D, Frehland B, Lehmkuhl K, Schepmann D, Köhler J, Diana P, Wünsch B, Synthesis and pharmacological evaluation of enantiomerically pureendo-configured KOR agonists with 2-azabicyclo[3.2.1]octane scaffold, Organic and Biomolecular Chemistry. 19 (2021) 8384–8396. 10.1039/d1ob01498f. [DOI] [PubMed] [Google Scholar]
- [123].Li X, Wan H, Dong P, Wang B, Zhang L, Hu Q, Zhang T, Feng J, He F, Bai C, Zhang L, Tao W, Discovery of SHR0687, a Highly Potent and Peripheral Nervous System-Restricted KOR Agonist, ACS Medicinal Chemistry Letters. 11 (2020) 2151–2155. 10.1021/acsmedchemlett.0c00287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Liu-Chen L-Y, Agonist-induced regulation and trafficking of $kappa; opioid receptors, Life Sciences. 75 (2004) 511–536. 10.1016/j.lfs.2003.10.041. [DOI] [PubMed] [Google Scholar]
- [125].Chartoff EH, Mavrikaki M, Sex differences in kappa opioid receptor function and their potential impact on addiction, Frontiers in Neuroscience. 9 (2015). 10.3389/fnins.2015.00466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Huang P, Chen C, Cao D, Huang M, Liu-Chen L-Y, Agonist-promoted kappa opioid receptor (KOR) phosphorylation has behavioral endpoint-dependent and sex-specific effects, Neuropharmacology. 202 (2021) 108860. 10.1016/j.neuropharm.2021.108860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Vijay A, Wang S, Worhunsky P, Zheng M-Q, Nabulsi N, Ropchan J, Krishnan-Sarin S, Huang Y, Morris ED, PET imaging reveals sex differences in kappa opioid receptor availability in humans, in vivo., American Journal of Nuclear Medicine and Molecular Imaging. 6 (2016) 205–14. http://www.ncbi.nlm.nih.gov/pubmed/27648372. [PMC free article] [PubMed] [Google Scholar]
- [128].US National Library of Medicine, ClinicalTrials.gov, (2021). https://clinicaltrials.gov/ct2/results?cond=&term=difelikefalin+OR+CR845&cntry=&state=&city=&dist= (accessed December 15, 2021).
- [129].Al-Hasani R, Wong JMT, Mabrouk OS, McCall JG, Schmitz GP, Porter-Stransky KA, Aragona BJ, Kennedy RT, Bruchas MR, In vivo detection of optically-evoked opioid peptide release, ELife. 7 (2018) 1–13. 10.7554/eLife.36520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Patriarchi T, Cho JR, Merten K, Howe MW, Marley A, Xiong WH, Folk RW, Broussard GJ, Liang R, Jang MJ, Zhong H, Dombeck D, von Zastrow M, Nimmerjahn A, Gradinaru V, Williams JT, Tian L, Ultrafast neuronal imaging of dopamine dynamics with designed genetically encoded sensors, Science. 360 (2018). 10.1126/science.aat4422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Vardy E, Robinson JE, Li C, Olsen RHJ, DiBerto JF, Giguere PM, Sassano FM, Huang XP, Zhu H, Urban DJ, White KL, Rittiner JE, Crowley NA, Pleil KE, Mazzone CM, Mosier PD, Song J, Kash TL, Malanga CJ, Krashes MJ, Roth BL, A New DREADD Facilitates the Multiplexed Chemogenetic Interrogation of Behavior, Neuron. 86 (2015) 936–946. 10.1016/j.neuron.2015.03.065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Banghart MR, Sabatini BL, Photoactivatable Neuropeptides for Spatiotemporally Precise Delivery of Opioids in Neural Tissue, Neuron. 73 (2012) 249–259. 10.1016/j.neuron.2011.11.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Manglik A, Kobilka BK, Steyaert J, Nanobodies to Study G Protein-Coupled Receptor Structure and Function, Annual Review of Pharmacology and Toxicology. 57 (2017) 19–37. 10.1146/annurev-pharmtox-010716-104710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Staus DP, Strachan RT, Manglik A, Pani B, Kahsai AW, Kim TH, Wingler LM, Ahn S, Chatterjee A, Masoudi A, Kruse AC, Pardon E, Steyaert J, Weis WI, Prosser RS, Kobilka BK, Costa T, Lefkowitz RJ, Allosteric nanobodies reveal the dynamic range and diverse mechanisms of G-protein-coupled receptor activation, Nature. 535 (2016) 448–452. 10.1038/nature18636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Che T, Majumdar S, Zaidi SA, Ondachi P, McCorvy JD, Wang S, Mosier PD, Uprety R, Vardy E, Krumm BE, Han GW, Lee M-Y, Pardon E, Steyaert J, Huang X-P, Strachan RT, Tribo AR, Pasternak GW, Carroll FI, Stevens RC, Cherezov V, Katritch V, Wacker D, Roth BL, Structure of the Nanobody-Stabilized Active State of the Kappa Opioid Receptor, Cell. 172 (2018) 55–67.e15. 10.1016/j.cell.2017.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Che T, English J, Krumm BE, Kim K, Pardon E, Olsen RHJ, Wang S, Zhang S, Diberto JF, Sciaky N, Carroll FI, Steyaert J, Wacker D, Roth BL, Nanobody-enabled monitoring of kappa opioid receptor states, Nature Communications. 11 (2020) 1145. 10.1038/s41467-020-14889-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Urs NM, Gee SM, Pack TF, McCorvy JD, Evron T, Snyder JC, Yang X, Rodriguiz RM, Borrelli E, Wetsel WC, Jin J, Roth BL, O’Donnell P, Caron MG, Snyder SH, Distinct cortical and striatal actions of a β-arrestin-biased dopamine D2 receptor ligand reveal unique antipsychotic-like properties, Proceedings of the National Academy of Sciences of the United States of America. 113 (2016) E8178–E8186. 10.1073/pnas.1614347113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Huang B, Li Y, Cheng D, He G, Liu X, Ma L, β-Arrestin-biased β-adrenergic signaling promotes extinction learning of cocaine reward memory, Science Signaling. 11 (2018) 1–11. 10.1126/scisignal.aam5402. [DOI] [PubMed] [Google Scholar]
- [139].Zurkovsky L, Sedaghat K, Ahmed MR, Gurevich V. v., Gurevich E. v., Arrestin-2 and arrestin-3 differentially modulate locomotor responses and sensitization to amphetamine, Neuropharmacology. 121 (2017) 20–29. 10.1016/j.neuropharm.2017.04.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Fang Y, Jiang Q, Li S, Zhu H, Xu R, Song N, Ding X, Liu J, Chen M, Song M, Ding J, Lu M, Wu G, Hu G, Opposing functions of β-arrestin 1 and 2 in Parkinson’s disease via microglia inflammation and Nprl3, Cell Death and Differentiation. 28 (2021) 1822–1836. 10.1038/s41418-020-00704-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Pradhan AA, Perroy J, Walwyn WM, Smith ML, Vicente-Sanchez A, Segura L, Bana A, Kieffer BL, Evans CJ, Agonist-specific recruitment of arrestin isoforms differentially modify delta opioid receptor function, Journal of Neuroscience. 36 (2016) 3541–3551. 10.1523/JNEUROSCI.4124-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Chiang T, Sansuk K, van Rijn RM, β-Arrestin 2 dependence of δ opioid receptor agonists is correlated with alcohol intake, British Journal of Pharmacology. 173 (2016) 332–343. 10.1111/bph.13374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Robins MT, Chiang T, Berry JN, Ko MJ, Ha JE, van Rijn RM, Behavioral characterization of β-arrestin 1 knockout mice in anxiety-like and alcohol behaviors, Frontiers in Behavioral Neuroscience. 12 (2018) 1–11. 10.3389/fnbeh.2018.00054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Dunn AD, Reed B, Erazo J, Ben-Ezra A, Kreek MJ, Signaling Properties of Structurally Diverse Kappa Opioid Receptor Ligands: Toward in Vitro Models of in Vivo Responses, ACS Chemical Neuroscience. 10 (2019) 3590–3600. 10.1021/acschemneuro.9b00195. [DOI] [PubMed] [Google Scholar]
- [145].DiMattio KM, Ehlert FJ, Liu-Chen LY, Intrinsic relative activities of κ opioid agonists in activating Gα proteins and internalizing receptor: Differences between human and mouse receptors, European Journal of Pharmacology. 761 (2015) 235–244. 10.1016/j.ejphar.2015.05.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Li J-G, Zhang F, Jin X-L, Liu-Chen L-Y, Differential Regulation of the Human κ Opioid Receptor by Agonists: Etorphine and Levorphanol Reduced Dynorphin A- and U50,488H-Induced Internalization and Phosphorylation, Journal of Pharmacology and Experimental Therapeutics. 305 (2003) 531–540. 10.1124/jpet.102.045559. [DOI] [PubMed] [Google Scholar]
- [147].Paton KF, Kumar N, Crowley RS, Harper JL, Prisinzano TE, Kivell BM, The analgesic and anti-inflammatory effects of Salvinorin A analogue b -tetrahydropyran Salvinorin B in mice, 1 (2017) 1039–1050. 10.1002/ejp.1002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].White KL, Scopton AP, Rives M-L, Bikbulatov R. v., Polepally PR, Brown PJ, Kenakin T, Javitch JA, Zjawiony JK, Roth BL, Identification of Novel Functionally Selective κ-Opioid Receptor Scaffolds, Molecular Pharmacology. 85 (2014) 83–90. 10.1124/mol.113.089649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Rives ML, Rossillo M, Liu-Chen LY, Javitch JA, 6′-Guanidinonaltrindole (6′-GNTI) is a G protein-biased κ-opioid receptor agonist that inhibits arrestin recruitment, Journal of Biological Chemistry. 287 (2012) 27050–27054. 10.1074/jbc.C112.387332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Ansonoff MA, Portoghese PS, Pintar JE, Consequences of opioid receptor mutation on actions of univalent and bivalent kappa and delta ligands, (2010) 161–168. 10.1007/s00213-010-1826-7. [DOI] [PubMed] [Google Scholar]