

Abstract
Monoclonal antibodies (mAbs) are one of the most rapidly growing drug classes used for the treatment of cancer, infectious and autoimmune diseases. Complement-dependent cytotoxicity (CDC) is one of the effector functions for antibodies to deplete target cells. We report here an efficient chemoenzymatic synthesis of structurally well-defined conjugates of a monoclonal antibody with a rhamnose- and an αGal trisaccharide-cluster to recruit natural anti-rhamnose and anti-αGal antibodies, respectively, to enhance the CDC-dependent targeted cell killing. The synthesis was achieved by using a modular antibody Fc-glycan remodeling method that includes site-specific chemoenzymatic Fc-glycan functionalization and subsequent click conjugation of synthetic rhamnose- and αGal trisaccharide-cluster to provide the respective homogeneous antibody conjugates. Cell-based assays indicated that the antibody-rhamnose cluster conjugates could mediate potent CDC activity for targeted cancer cell killing and showed much more potent efficacy than the antibody-αGal trisaccharide cluster conjugates for CDC effects.
Keywords: anti-Rha antibody, antibody conjugates, chemoenzymatic synthesis, glycoengineering, immunotherapy
Graphical Abstract
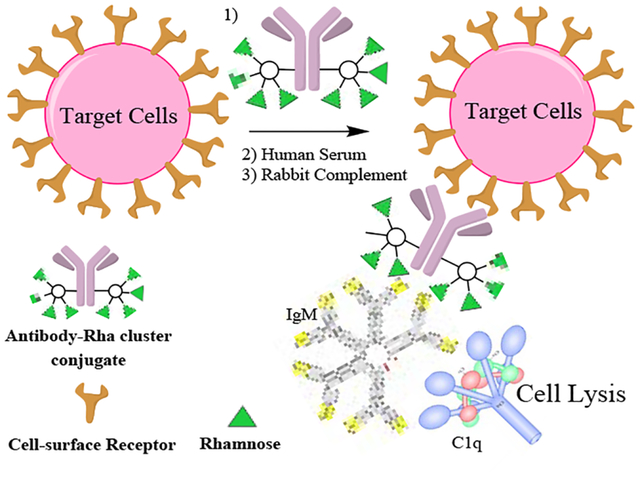
A highly efficient chemoenzymatic synthesis of homogeneous antibody-rhamnose and antibody-αGal cluster conjugates is described. The synthesis was achieved through a modular antibody Fc-glycan remodeling that includes site-specific chemoenzymatic Fc-glycan functionalization and subsequent click conjugation of synthetic rhamnose- and αGal trisaccharide clusters. A comparative cell-based assay indicated that the antibody-rhamnose cluster conjugates showed potent complement-dependent targeted cancer cell killing by recruiting natural anti-rhamnose antibodies.
Introduction
Complement-dependent cytotoxicity (CDC) is one of the major mechanisms for antibody-mediated killing of target cells.[1] Nevertheless, many therapeutic antibodies are limited by their low potency in stimulating a strong complement-dependent cytotoxicity. One strategy to achieve complement-dependent targeted cell killing is to explore novel bi-functional molecules consisting of a target-binding motif and a specific antigenic structure to recruit naturally abundant antibodies, such as the anti-αGal and anti-rhamnose (anti-Rha) antibodies to the target cells.[2] The α-Gal epitope, Galα1–3Galβ1–4GlcNAc-R, is expressed abundantly on glycolipids and glycoproteins in non-human primates and New World monkeys, but it is not present on human cells. As a result, humans produce large quantities of anti-αGal antibodies in circulation: up to 1–2% of total serum IgG and 3–8% of total IgM natural antibodies are anti-αGal antibodies.[3] The interaction between αGal and anti-αGal antibodies are largely responsible for the rejection of the transplanted tissues following xenotransplantation, mainly due to activation of the complement systems.[4] On the other hand, anti-Rha antibodies are also abundant in human sera, which have been found even at higher concentrations than the anti-αGal antibodies in human serum samples.[2b, 5] Moreover, the majority of anti-Rha antibodies are of IgM type, while most of the anti-αGal antibodies are of IgG type.[2b, 6] It has been shown that the IgM type antibody is much more efficient than the monomeric IgG type antibody to initiate complement-dependent cytotoxicity, as the IgMs is inherently a pentamer that can efficiently bind to the multi-subunit C1q complement protein in a multivalent fashion to trigger the downstream effects, while the IgG antibody must form hexamer complex to engage an efficient multivalent interactions with the multi-subunit C1q complement protein.[7]
To explore the potential of natural anti-αGal or anti-Rha antibodies for targeting cancer, bacterial and/or virus-infected cells, several groups have previously designed and synthesized bi-functional small molecules, by conjugating a target-binding ligand to an αGal oligosaccharide or a rhamnose moiety, to recruit natural anti-αGal or anti-Rha antibodies for complement-dependent cell killing.[2] For example, Kiessling and co-workers have reported the synthesis of αGal-integrin ligand conjugates for targeting cancer;[2a, 2b] Yi and co-workers have assembled bi-functional liposomes incorporating rhamnose and folic acid to recruit anti-Rha antibodies to target folate receptor-overexpressing tumor cells.[2e] Recently, Fukase and co-workers have reported the first synthesis of αGal oligosaccharide-antibody conjugates, and the cell-based assays have shown that the presentation of multiple copies of the αGal epitopes (multi-valency) was important for an efficient cancer cell killing.[8] While this study provides proof-of-concept data indicating the feasibility of using antibody-αGal conjugates for targeted cancer cell killing, the lysine-based antibody-αGal conjugation and the reduction of antibodies into monomeric antibodies for thiol-maleimide ligation both lead to mixtures of heterogeneous conjugates. We report in this paper a highly efficient chemoenzymatic synthesis and comparative study of structurally well-defined, homogeneous conjugates of an antibody with rhamnose and αGal oligosaccharide clusters, respectively. Trastuzumab, a therapeutic antibody targeting HER2 over-expressing cancer cells, was selected as a model antibody, and the carbohydrate antigens were introduced specifically at the Fc glycosylation site by a chemoenzymatic Fc glycan remodeling approach. The resulting glycoengineered antibodies carrying the rhamnose and αGal oligosaccharide clusters were designed to recruit natural anti-Rha and anti-αGal antibodies, respectively, for targeted cell killing (Figure 1). It was found that the antibody-rhamnose cluster conjugates were much more efficient than the antibody-αGal cluster conjugate for the complement-dependent cell killing. During the submission of this manuscript, Wu and co-workers have reported rituximab-rhamnose conjugates by reaction of partially reduced rituximab with a maleimide-functionalized rhamnose derivative, and the resulting antibody-rhamnose conjugates demonstrate antitumor efficacy in a xenograft model.[9]
Figure 1.
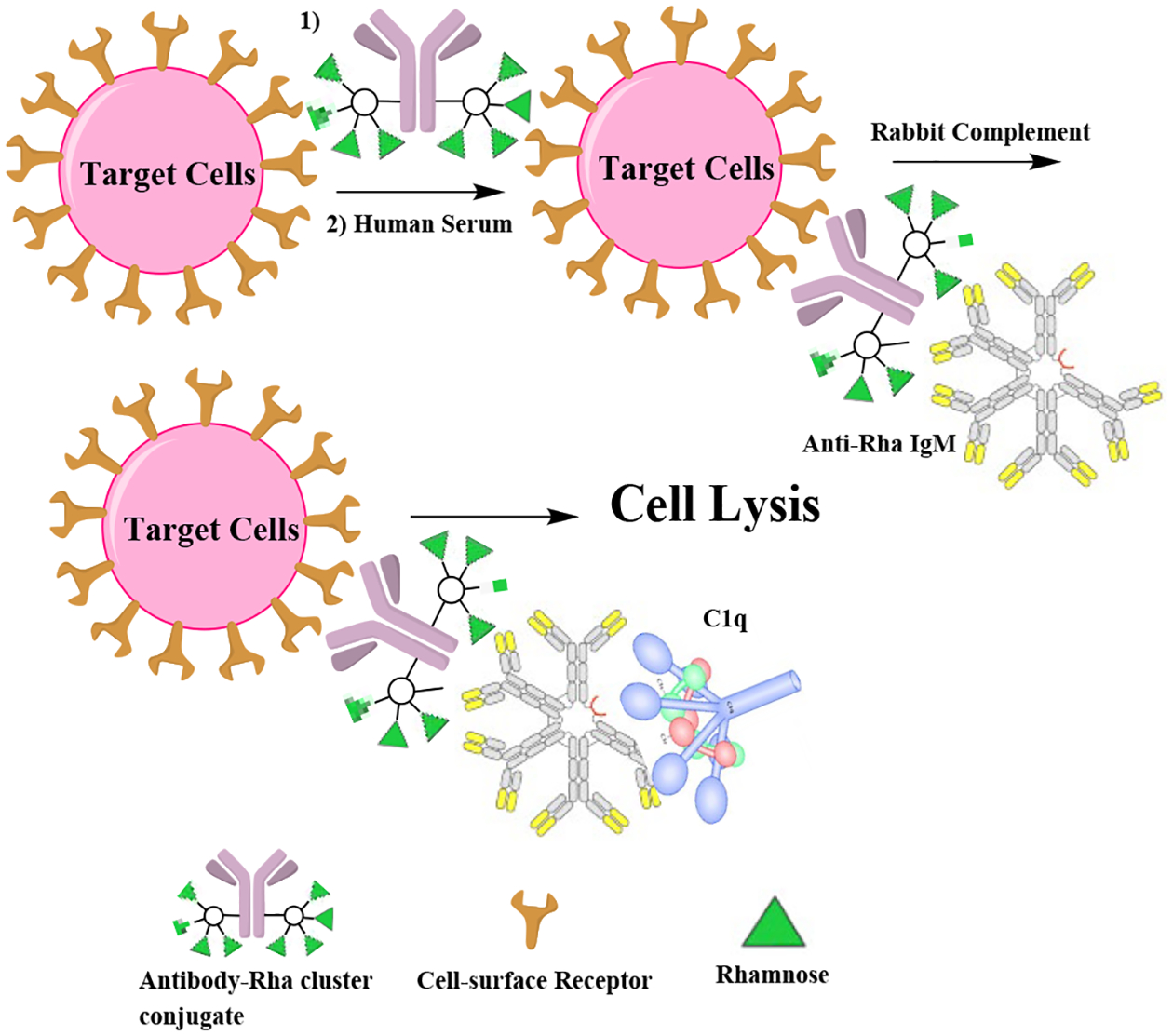
Homogenous antibody conjugates that recruit serum antibodies and initiate CDC.
Results and Discussion
Chemical synthesis of the dibenzocyclooctyne (DBCO)-functionalized rhamnose and αGal clusters
As a general synthetic design, we sought to introduce azide tags into the antibody through the chemoenzymatic Fc glycan remodeling method that we have recently described,[10] and then to click the synthetic oligosaccharide antigens to the antibody to construct the conjugates. For this purpose, we first synthesized the DBCO-functionalized rhamnose and rhamnose clusters (Scheme 1). The synthesis of DBCO-tagged rhamnose (3) was achieved by reaction of rhamnose derivative 1 with the NHS-active ester (2) (Scheme 1a). For constructing a tetravalent rhamnose cluster, we chose a tri-lysine core as the scaffold. The synthesis of the tri-lysine core (7) was shown in Scheme 1b. At the C-terminus of 4, a short N-methyl ethylenediamine spacer with an Fmoc protecting group was introduced to provide a handle for further functionalization. The N-methyl spacer (5) was specifically chosen to provide linker stability without premature release.[11] For the synthesis of the tetravalent rhamnose cluster (13), the tri-lysine core (7) was reacted with NHS-activated rhamnose derivative (10) to give the rhamnose cluster (11). After de-O-acetylation followed by removal of the Fmoc group, the resulting compound (12) was reacted with the NHS activated ester (2) to give the DBCO-tagged rhamnose cluster (13) after LH20 size exclusion chromatography (Scheme 1c).
Scheme 1.
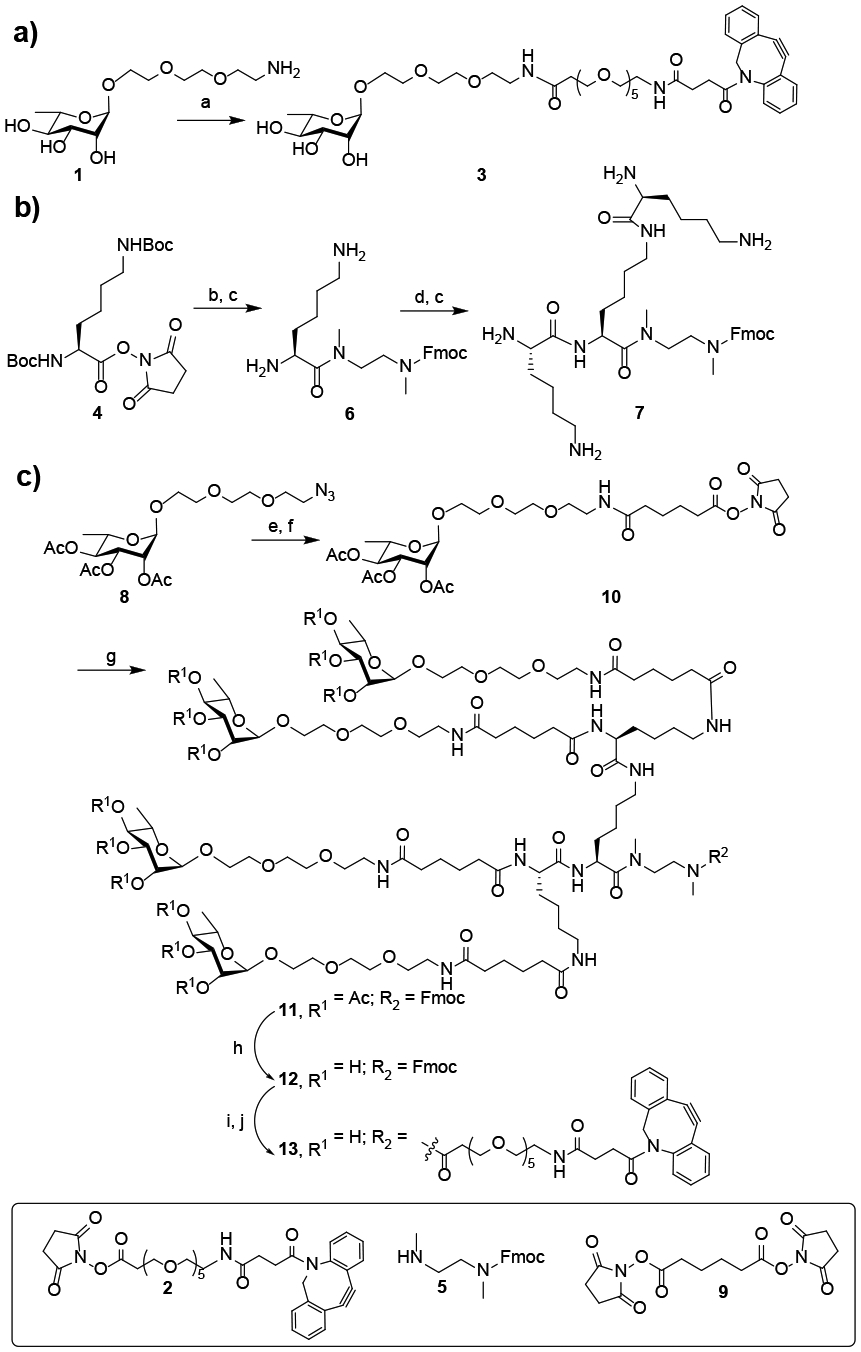
Chemical synthesis of the monovalent and multivalent rhamnose-based antibody recruiting molecules. a) 2, DMSO, Et3N, RT, 60%; b) 5, CH2Cl2, Et3N, RT; c) TFA/ CH2Cl2, (1:1, v/v) 0°C; d) 4, CH2Cl2, Et3N, RT; e) Pd/C, H2, MeOH; f) 9, CH2Cl2, Et3N, RT, 79% over two steps; g) 7, DMSO, Et3N, RT; h) hydrazine, H2O, RT; i) 20% piperidine, RT; j) 2, DMSO, Et3N, RT, 73% over four steps.
To construct the DBCO-functionalized αGal trisaccharide and its cluster, we first synthesized the αGal trisaccharide following a previously published procedure.[12] Then the amine-functionalized αGal was reacted with 2 to give the DBCO-functionalized αGal trisaccharide (15) (Scheme 2a). To synthesize a cluster of α-Gal trisaccharide, 14 was activated as an NHS ester, then it was reacted with the tri-lysine core (7) to give the tetravalent α-Gal oligosaccharide (17). Finally, deprotection of 17 followed by reaction with 2 gave the desired DBCO-functionalized α-Gal trisaccharide cluster (18) (Scheme 2b).
Scheme 2.
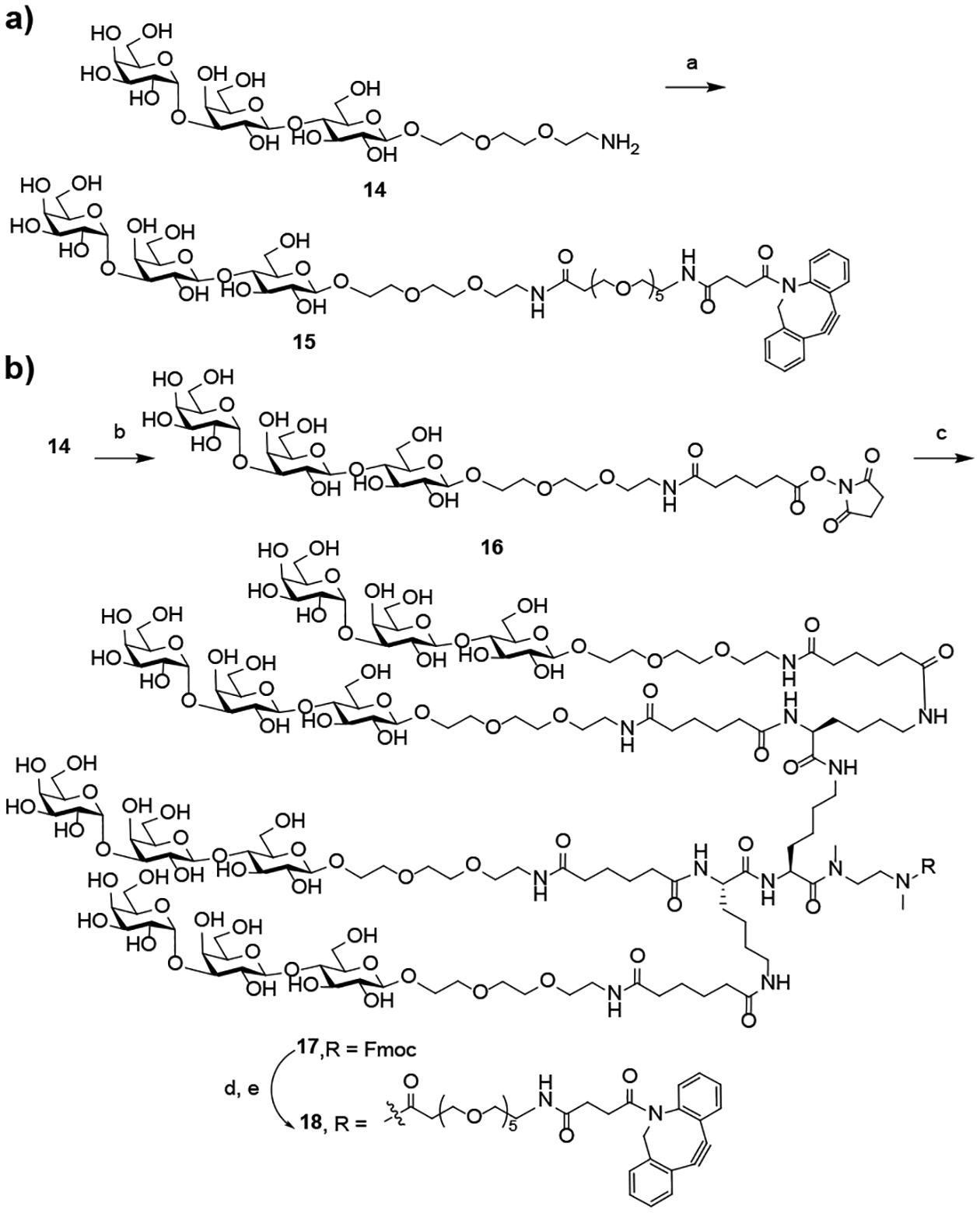
Chemical synthesis of the monovalent and multivalent α-Gal-based antibody recruiting molecules. a) 2, DMSO, Et3N, RT, 58%; b) 9, DMSO, Et3N, RT, 67%; c) 7, DMSO, Et3N, RT; d) piperidine, RT; e) 2, DMSO, Et3N, RT, 65% over three steps.
Chemoenzymatic synthesis of the antibody-rhamnose and αGal cluster conjugates
With the successful synthesis of DBCO-functionalized αGal and rhamnose clusters, we sought to construct the antibody conjugates utilizing the Fc glycan-mediated chemoenzymatic modification approach, which is validated to achieve antibody conjugates with high homogeneity and great efficiency.[10, 13] Thus, trastuzumab was trimmed with immobilized Endo-S2, followed by efficient Fc modification with glycosynthase EndoS2-D184M and the di-N3-SCT-oxazoline to yield the azide functionalized antibody (20).[10] Then, the homogenous antibody-rhamnose (21 and 22) and antibody-αGal-conjugates (23 and 24) were generated by incubating 20 at 5 mg/mL with the DBCO derivatives (3, 13, 15, and 18, respectively, 12 molar equivalents per antibody) at room temperature (Scheme 3). The reactions were monitored by LC-MS analysis. It was found that the click reaction with DBCO derivatives (3 and 15) were completed within 10 h, while the reactions of DBCO derivatives 13 and 18 took a much longer time, which required about 24 h to complete, probably due to the bulkiness of the clickable oligosaccharide clusters. The final products were purified by affinity chromatography on a protein A column in an excellent isolated yield. The identity and homogeneity were confirmed by LC-ESI-MS analysis of both the intact antibodies (Figure 2a–2d) and the Fc domains after IdeS treatment (Figure 2e–2h). The observed molecular mass (deconvolution data) of 21, 22, 23 and 24 was 154521 Da, 161718 Da, 155882 Da and 167156 Da, which matched well with their calculated value of 154518 Da, 161714 Da, 155879 Da and 167159 Da, respectively. In addition to the intact antibody analysis, LC-ESI-MS analysis of the monomeric Fc domain of 21–24 released from the IdeS treatment of the antibody conjugates further confirmed the site-selectivity and homogeneity of the final products (Figures 2e–2h). The raw LC-ESI-MS data were shown in Figures S1–S8 (Supporting information).
Scheme 3.
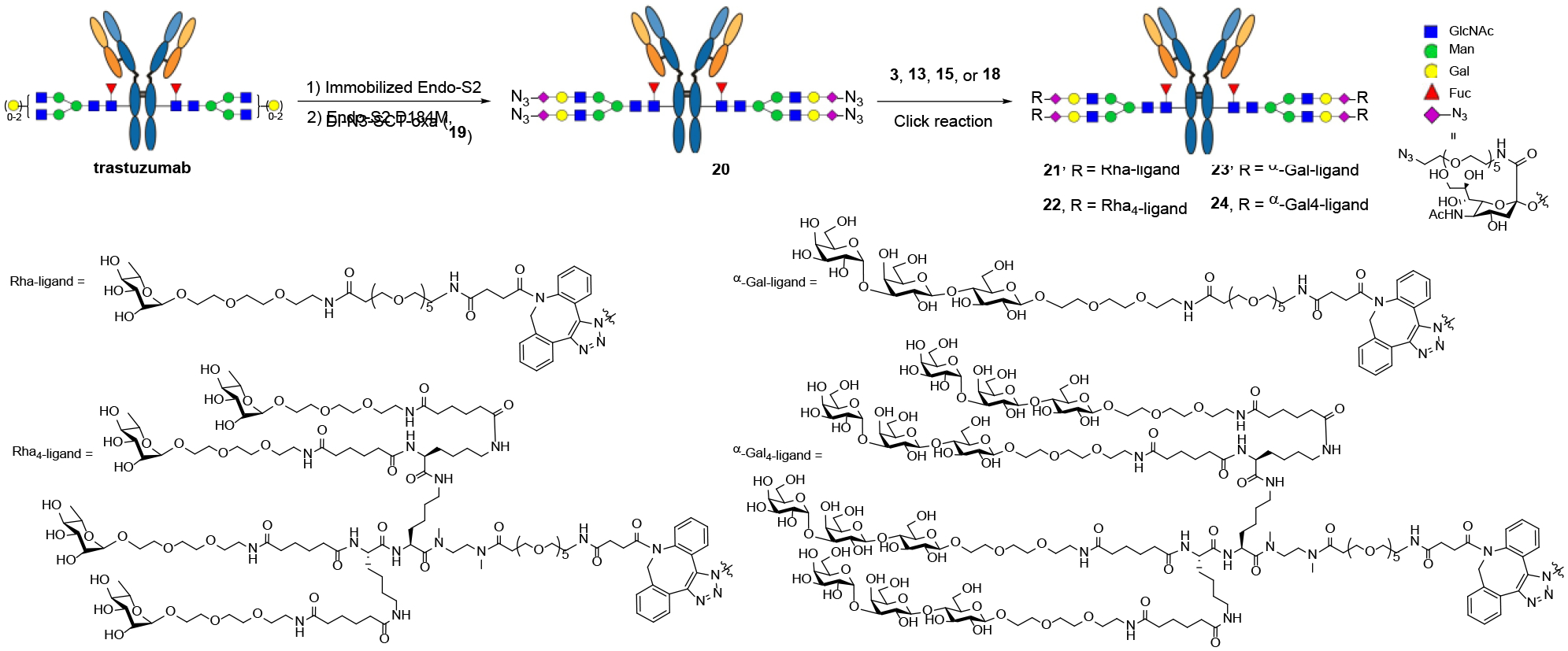
Synthesis of the antibody conjugates via chemoenzymatic Fc glycan remodeling followed by click reaction
Figure 2.
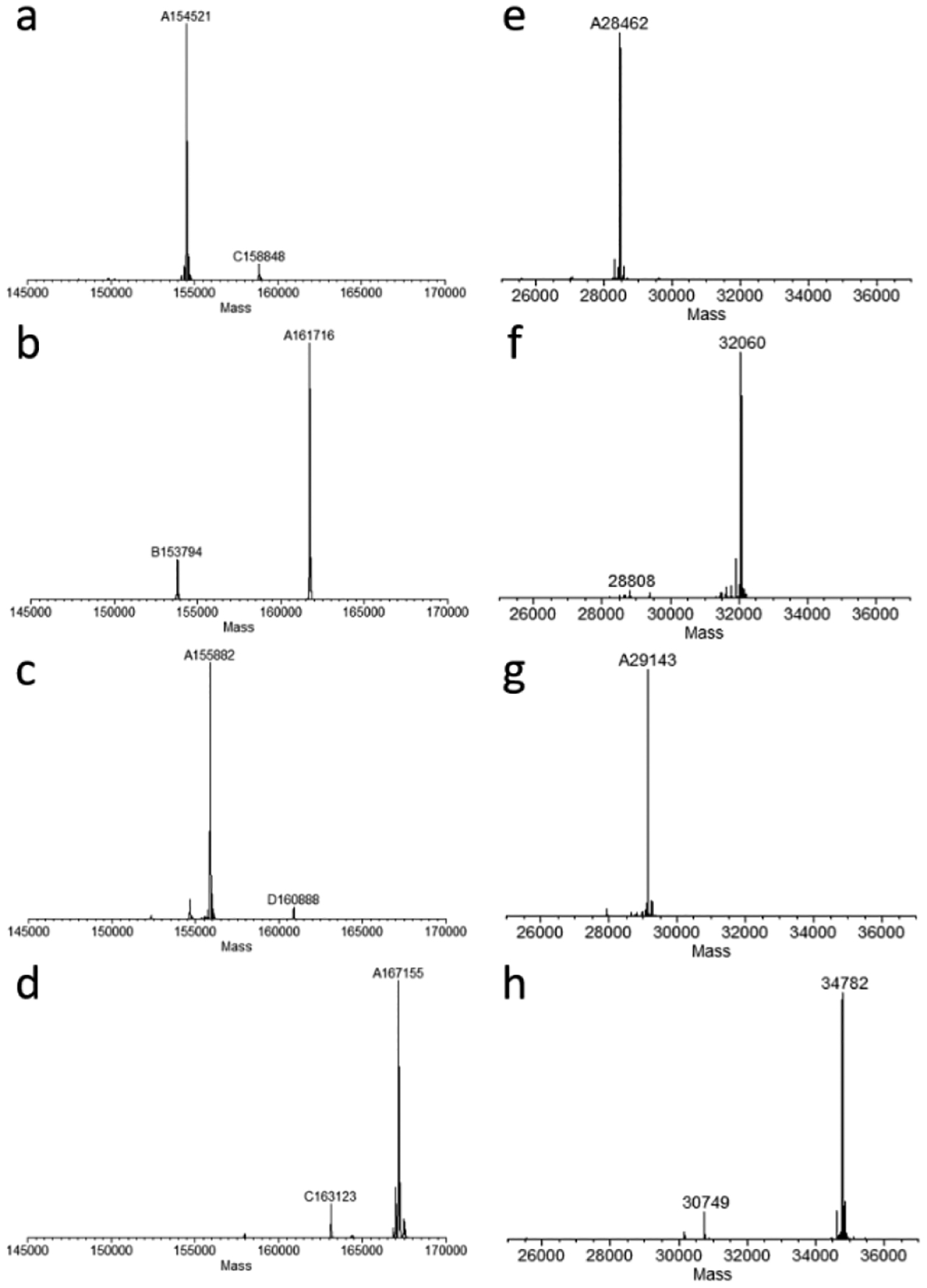
LC-ESI-MS analysis of the intact conjugates (21-24) and the Fc domains released by IdeS treatment. a) the deconvoluted mass of intact rhamnose conjugate (21); b) the deconvoluted mass of intact rhamnose cluster conjugate (22); c) the deconvoluted mass of intact α-Gal conjugate (23); d) the deconvoluted mass of intact α-Gal cluster conjugate (24); e) the deconvoluted mass of the Fc domain of rhamnose conjugate (21); f) the deconvoluted mass of the Fc domain of rhamnose cluster conjugate (22); g) the deconvoluted mass of the Fc domain of α-Gal conjugate (23); h) the deconvoluted mass of the Fc domain of α-Gal cluster conjugate (24).
The use of the selectively azide-tagged bi-antennary N-glycan for Fc glycan remodeling and subsequent conjugation has several advantages, including the site-specific conjugation and the preservation of the natural Fc N-glycan core after remodeling, which could be important for maintaining antibody stability and favorable pharmacokinetic property. Our previous studies have demonstrated that glycosynthase EndoS2-D184M possesses quite relaxed substrate specificity in transferring different natural and selectively modified glycan oxazolines,[10, 14] Recently, we have shown that both the wild type and the EndoS2-D184M mutant of Endo-S2 can efficiently transfer selectively azide-functionalized Manβ1,4-GlcNAc oxazoline to the deglycosylated antibody for constructing homogeneous antibody-drug conjugates.[14a] This method could provide an alternative approach for making the antibody-rhamnose or αGal cluster conjugates.
Given the broad substrate specificity of the Endo-S2 mutant, we reasoned that the enzyme might be able to transfer ligand-preloaded glycan oxazolines to make the final antibody conjugates in a single step. To test this hypothesis, we first conjugated the monomeric and tetrameric rhamnose to the azide-functionalized N-glycan using the ring-strain-promoted cycloaddition reaction to give 26a and 26b, respectively. The ligand-functionalized sialylated glycan was then converted to oxazolines 27a and 27b using the single-step glyan oxazoline formation reaction with dehydrating agent 3-chloro-1,3-dimethylimidazolinium chloride (DMC) in water.[15] We found that EndoS2-D184M could tolerate the rhamnose modification on the glycan oxazoline and could smoothly transfer the antigen-loaded glycan to the deglycosylated trastuzumab (28) to give the corresponding antibody conjugate (21) (Scheme 4). Using 30 equivalents of the glycan oxazoline (27a), the reaction completed within 40 min (Figure S9). However, we found that the larger Rha cluster-containing glycan oxazoline (27b) was transferred much more slowly than the simpler Rha-loaded glycan oxazoline (27a). (Scheme 4) Interestingly, the major product was a monosubstituted antibody when the reaction was carried out for 60 min (Figure S10). The results suggest that the Endo-S2 mutant could tolerate the modifications on the glycan oxazolines, but a modification with a larger moiety such as the Rha clusters slows down the transglycosylation reaction, which would require optimization of the conditions to drive the reaction to completion.
Scheme 4.
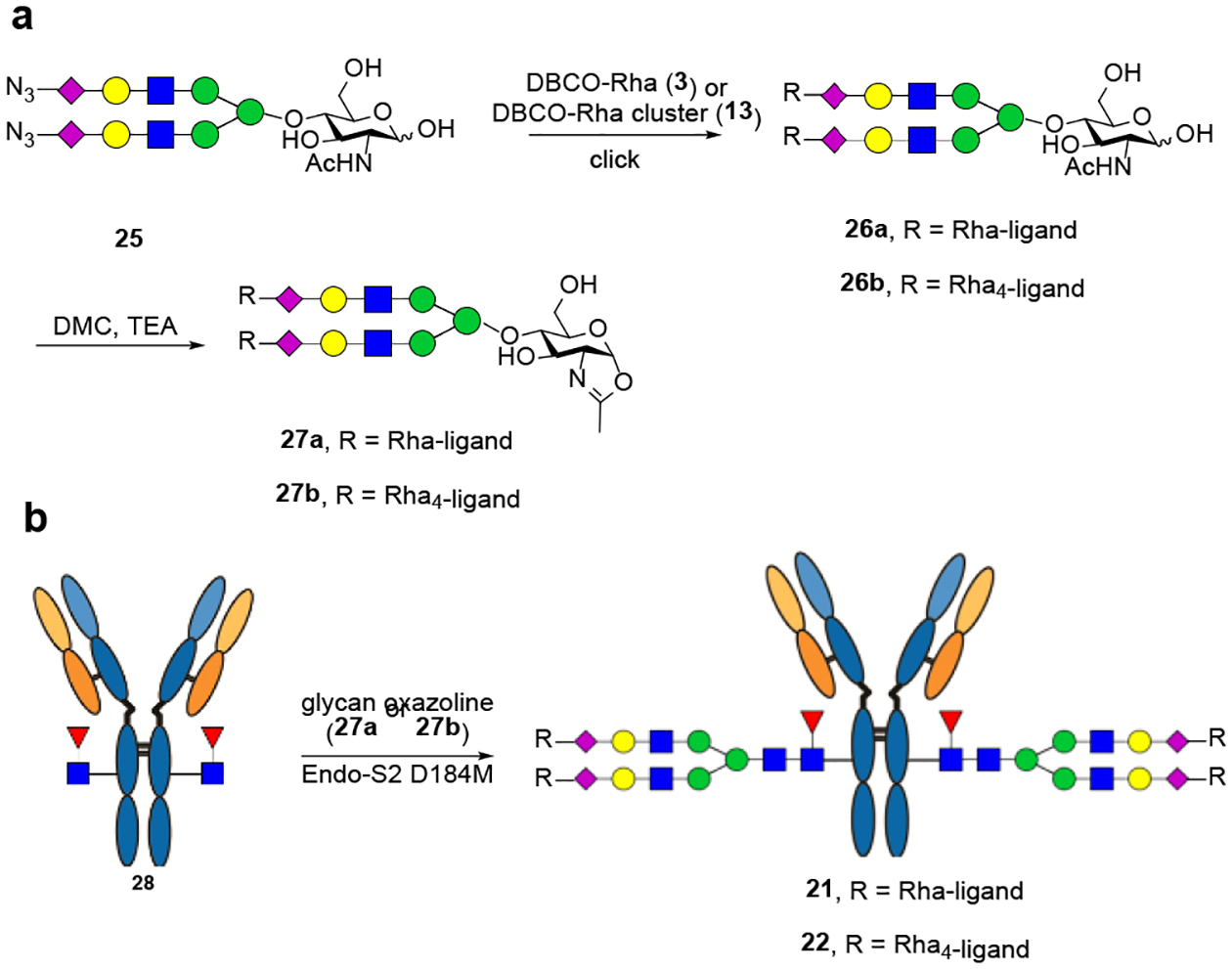
Direct enzymatic transfer of Rha ligand-loaded glycans to antibodies using the Endo-S2 D184M.
Cell-based assay of the complement-dependent cytotoxicity of the antibody conjugates
To compare the potency of the conjugates 21-24, we studied the in vitro cytotoxicity of the antibody conjugates with BT474 (high HER-2 expressing) and T47D (low HER-2 expressing) cells. In this assay, the anti-αGal and anti-rhamnose antibodies were provided by commercially available human serum. Serum-free RPMI media was used to carry out the assays to avoid the possible αGal contamination from the FBS. The complement required to lysate the cells was provided by standard rabbit complement. Since rabbit complement itself could lysate cells even at relatively low concentration, we screened for the best combination amount of human serum and rabbit complement to minimize the effect of non-specific CDC. We found that 10 μL of human serum (Millipore Sigma) and 4 μL of rabbit complement (Cedarlane Laboratories) per well gave the best balance between non-specific killing caused by the complement (about 10–15%) and the efficacy of the conjugates. Under this condition, we found that conjugate 22, with sixteen rhamnose antigen, was the most potent antibody conjugate, which killed more than 70% of the HER2 high-expression cancer cells within 2 h (Figure 3a). The calculated EC50 value for 22 was 0.57 nM (92 ng/mL). To our amazement, the conjugate 21, with only four rhamnose antigen, could also lysate up to 50% of BT474 cells in relatively short period of time (Figure 3a). The reason that the best antibody-rhamnose conjugates did not reach to 100% target cell killings may be due to the varied levels of HER2 expression of BT474 and/or the unstable nature of the complements during the incubation of the assays. It is expected that these results could be more efficient when the complement is persistent as in the case of in vivo situation. In contrast, the killing of the αGal conjugates (23 and 24) were much weaker than the antibody-rhamnose cluster conjugates (21 and 22). It was found that conjugate 24, with sixteen αGal epitope managed to lysate up to 20% of the BT474, while conjugate 23, with four αGal epitope could lysate only about 10% of the BT474 at 25 μg/mL. (Figure 3b) To eliminate the possibility that the difference between the rhamnose conjugates and the αGal conjugates was caused by the specific batch of the human serum that was used for the assay, the cytotoxicity assay was also carried out by using the human serum obtained from a different source (Cosmo Bio USA). The results were almost identical (Figure S11). Given the fact that the average anti-rhamnose antibodies in human blood is usually higher than the anti-αGal antibodies,[2b, 5a] with a higher percentage of the anti-rhamnose antibodies being of the IgM type,[2b, 6a, 6c] These results suggest that recruiting the more potent anti-Rha IgM antibodies could be a more efficient strategy than targeting the anti-αGal antibodies. For the HER-2 low-expressing cell line, none of these four antibody conjugates showed cytotoxicity in a dose-dependent manner up to 50 μg/mL (Figure 3c–3d). This study demonstrates that the antibody-rhamnose cluster conjugates are a promising construct for augmenting potent complement-dependent cytotoxicity for targeted cell killing.
Figure 3.
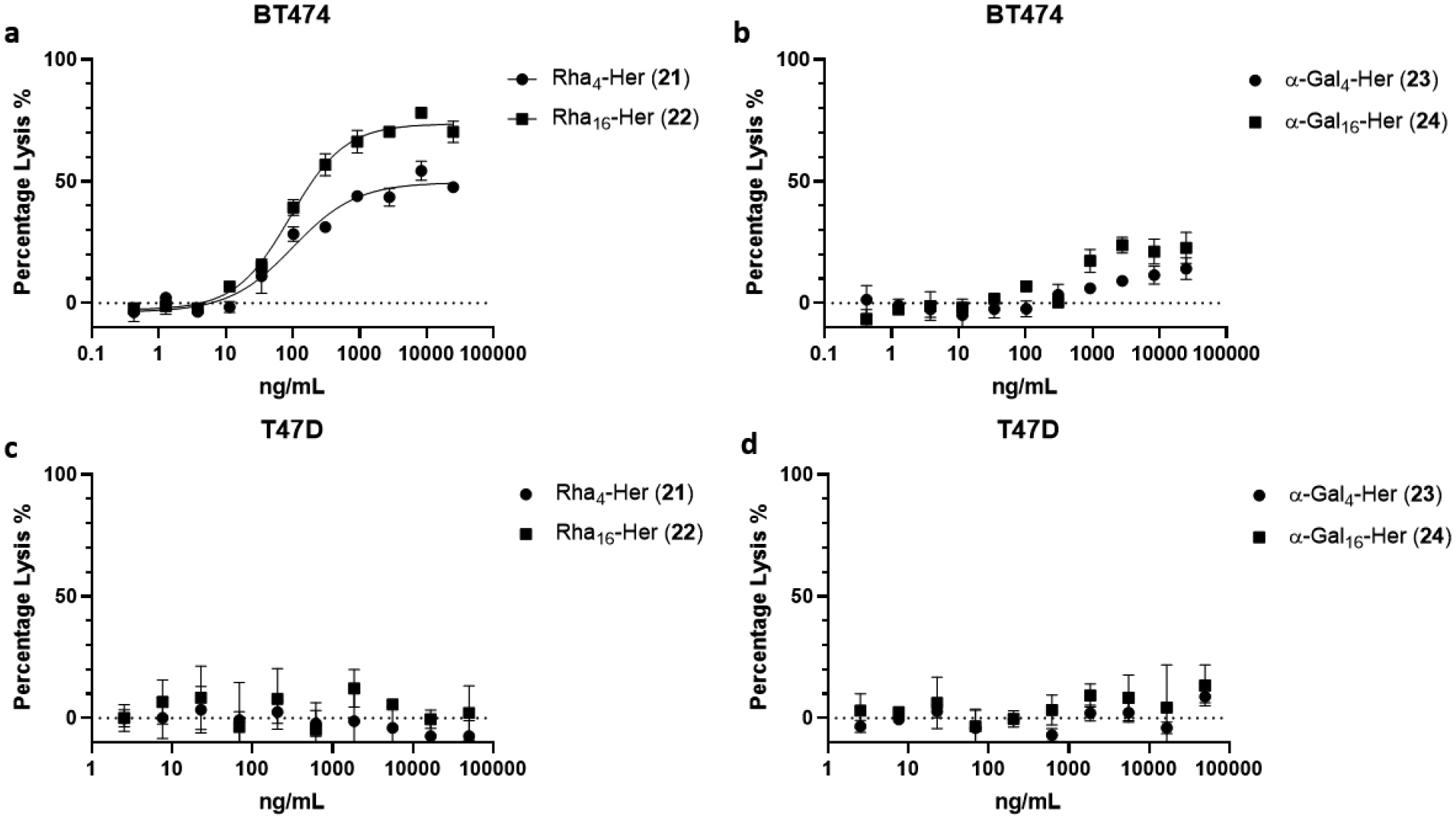
Cell killing assays for breast cancer cell lines a) rhamnose conjugates with BT-474 (HER2 overexpression); b) α-Gal conjugates with BT-474 (HER2 overexpression); c) rhamnose conjugates with T47D (HER2 low expression); d) α-Gal conjugates with T47D (HER2 low expression). All assays were performed in triplicate.
Conclusion
A highly efficient chemoenzymatic synthesis of homogeneous antibody-rhamnose cluster and antibody-αGal cluster conjugates is described. The construction of the antibody conjugates was achieved by site-specific chemoenzymatic Fc glycan remodeling followed by a click reaction. Alternatively, the antibody-rhamnose conjugate could also be achieved by direct Fc glycan remodeling with a rhamnose-preloaded glycan oxazoline. A comparative study reveals that the antibody-rhamnose cluster conjugates are more potent than the corresponding antibody-αGal oligosaccharide conjugates in recruiting natural antibodies for targeted cancer cell killing. These results suggest that antibody-rhamnose conjugates represent a promising strategy for augmenting complement-dependent targeted cell killing by recruiting the more effective natural anti-Rha IgM antibodies. Future studies should be directed to evaluation of the cytotoxicity of the antibody conjugates in animal models.
Experimental Section
Materials and Methods.
Chemicals, reagents, and solvents were purchased from Sigma–Aldrich and/or TCI and used as received unless otherwise specified. Monoclonal antibody Hercptin was purchased from Premium Health Services Inc. (Columbia, MD). All moisture sensitive reactions were carried out under argon atmosphere, using standard Schlenk techniques. All dry solvents were prepared according to standard procedures. Thin-layer chromatography was performed on silica gel 60-F254 on glass plates (Merck) and revealed with p-anisaldehyde stain. Silica gel (200–425 mesh) used in flash chromatography for large-scale reactions was purchased from Sigma-Aldrich. Columns for flash chromatography for small-scale reactions were performed on Isolera One system with ZIP KP-Sil columns (Biotage) with elution condition specified for each target compound. Solvent gradients were given refer to stepped gradients and concentrations are reported as % v/v. Preparative HPLC was performed with Waters 1525 Binary HPLC pump coupled with 2489 UV/Vis Detector under UV 214 nm and 280 nm with a Waters Symmetry C18 column (7 μm, 19 × 300 mm) using water containing 0.1% trifluoracetic acid as phase A, MeCN containing 0.1% trifluoracetic acid as phase B. Semi-preparative HPLC for the toxic payloads was performed on the same instrument with an Aglient Eclipse XDB-C18 column (5 μm, 9.4× 250 mm) using water containing 0.1% formic acid as phase A, MeCN containing 0.1% formic acid as phase B.
Purification of antibody and antibody conjugates using AKTA prime plus FPLC system.
The FPLC system (GE Healthcare) was used for purification of the functionalized antibodies equipped with 1mL HiTrap protein A column (GE Heatlthcare). Concentration of antibodies was determined by NanoDrap 200c (Thermo Scientific).
LC-ESI-MS analysis of antigen-DBCO payloads
LC-MS for glycans, glycopeptides and payload derivatives were performed on HPLC-SQ2 detector (Waters) with a Waters XBridge C18 column (3.5 μm, 2.1× 50 mm) using water containing 0.1% formic acid as phase A, MeCN containing 0.1% formic acid as phase B. The analytical HPLC for payload derivatives was analyzed with an Aglient Eclipse SDB-C18 column (5 μm, 3.0× 250 mm) under UV 214 nm and 280 nm with methods specialized for each compound. HR-ESI-MS was performed with Exactive Plus Orbitrap Mass Spectrometer (Thermo Scientific) equipped with a Waters XBridge C18 column (3.5 μm, 2.1× 50 mm).
LC-ESI-MS analysis of intact antibody derivatives.
LC-ESI-MS analysis of intact tagged antibodies and antibody-drug conjugates was performed with Exactive Plus Orbitrap Mass Spectrometer (Thermo Scientific) equipped with a Waters XBridge BEH300 C-4 column (3.5 μm, 2.1 × 50 mm) with gradient elution of water containing 0.1% formic acid as phase A, MeCN containing 0.1% formic acid as phase B. Mass spectra were deconvoluted using MagTran (ver 1.03 b2).
LC-ESI-MS analysis of Fc domains released by IdeS treatment.
The antibody samples in PBS were incubated with IdeS at 37 °C for 2 h. The samples were analyzed by with Exactive Plus Orbitrap Mass Spectrometer (Thermo Scientific) equipped with an Agilent Poroshell 300SB C8 column (5 μm, 1.0 × 75 mm) with gradient elution of water containing 0.1% formic acid as phase A, MeCN containing 0.1% formic acid as phase B. Mass spectra were deconvoluted using MagTran (ver 1.03 b2).
NMR analysis.
1H, 13C, and 1H–1H COSY NMR spectra were recorded on 400 MHz or 600 MHz spectrometer (Bruker) with CDCl3, MeOD-d4, D2O or DMSO-d6 as the solvent (solvent residue peak 7.26, 3.31, 4.79, 2.50 ppm). All 13C NMR spectra were performed with proton decoupling, and all chemical shifts are reported in part per million (ppm) and referenced to residual solvent. 1H-NMR chemical shifts were recorded relative to the solvent residual peak (CDCl3 at 7.26 ppm, MeOD-d4 at 3.31ppm, D2O at 4.79 ppm, DMSO-d6 at 2.50 ppm). 13C NMR chemical shifts are reported relative to the solvent residual peak (CDCl3 at 77.00 ppm, MeOD-d4 at 49.00 ppm, DMSO-d6 at 39.51 ppm). The number of protons (n) corresponding to a resonance signal was indicated by nH and spin-spin coupling constants (J value) recorded in Hz.
Synthesis of the Antibody recruiting molecules
Synthesis of Rhamnose-PEG-DBCO (3).
The rhamnose-PEG-NH2 1 (3.5 mg, 11.9 μmol) was weighed into a 1.5 mL centrifuge tube. A solution of the DBCO-PEG-NHS (2) (8 mg, 11.5 μmol, 50 mg/mL) was added, followed by 1 μL of TEA. The mixture reacted at room temperature for 2 hours before HPLC-SQ2 analysis showed completion of the reaction. The mixture was diluted with water and purified by semi prep-HPLC to afford 3 (6 mg, 60%). HR-ESI-MS: [M+H]+ calcd for C44H64N3O15+, 874.4332; found (m/z), 874.4304. HPLC (0.4 mL/min, 25–60%B, 30min, tR = 14.2 min)
Synthesis of compound 6.
To a solution of 4 (857 mg, 1.5 mmol) and 5 (400 mg, 1.3 mmol) in DCM (10 mL), TEA (303 mg, 3 mmol) was added at room temperature. After the mixture was stirred at room temperature for 16 hours, the solvent was removed in vacuo and the intermediate was purified by flash silica gel column chromatography by hexane and acetone (9:1 to 2:1, v/v). After it was concentrated under vacuo, the intermediate was treated with TFA/DCM (1:1, v/v) on ice for 30 minutes. The product in TFA salt form was concentrated and used without further purification (700 mg).
Synthesis of compound 7.
In a 25 mL round-bottom flask, 4 (350 mg, 0.52 mmol) and 6 (850 mg, 1.92 mmol) were dissolved in DCM (10 mL). The mixture was stirred for 3 hours after TEA (255 mg, 2.5 mmol) was added. The solvent was removed in vacuo and the intermediate was purified by flash silica gel column chromatography by hexane and acetone (9:1 to 1:1, v/v). After it was concentrated under vacuo, the intermediate was treated with TFA/DCM (1:1, v/v) on ice for 30 minutes. The product as a TFA salt was concentrated and used without further purification (614 mg).
Synthesis of compound 10.
2-(2-(2-azido)ethoxy)ethyl-2,3,4-tri-O-acetyl-α-L-Rhamnose [16] (1.7 g, 3.8 mmol) was dissolved in MeOH (20 mL) with 1M HCl (200 μL). The solution was stirred with 5% Pd/C (78 mg) under H2 atmosphere at room temperature for 1 hour. After filtration and concentration under vacuo, the resulting amine was used for next step without further purification. To a solution of 9 (700 mg, 2.05 mmol) in DCM (20 mL), a solution of pre-OAc-Rha amine (290 mg, 0.69 mmol) in DCM (10 mL) was added dropwise. The reaction mixture was stirred for another 2 hours at room temperature before the solvent was removed in vacuo and the intermediate was purified by flash silica gel column chromatography by hexane and EtOAc (9:1 to 1:1 with 0.5% AcOH, v/v). The product was isolated as a colorless oil (284 mg, 79%, over two steps). 1H NMR (400 MHz, DMSO-d6): δ = 1.12 (3H, d, J = 6.24 Hz), 1.57 (4H, m), 1.93 (3H, s), 2.03 (3H, s), 2.10 (5H, m), 2.66 (2H, t, J = 6.60 Hz), 2.81 (4H, br), 3.18 (2H, m), 3.40 (2H, t, J = 5.88 Hz), 3.45–3.65 (8H, m), 3.70 (1H, m), 3.86 (1H, m), 4.82 (1H, d, J = 1.24 Hz), 4.88 (1H, t, J = 9.94 Hz), 5.05–5.10 (2H, m), 7.87 (1H, m). 13C NMR (100 MHz, DMSO-d6): δ = 17.68, 20.91, 20.95, 21.07, 24.26, 24.78, 25.90, 30.37, 35.09, 38.90, 33.15, 66.94, 69.10, 69.43, 69.65, 69.74, 70.02, 70.21, 70.54, 97.06, 169.37, 170.18, 170.72, 172.23. ESI-MS: [M+H]+ calcd for C28H43N2O15+, 647.2658; found (m/z), 647.4511.
Synthesis of Rha4-dendrimer-DBCO (13).
To a solution of 7 (6.5 mg, 7.7 μmol) in DMSO (100 μL), a solution of 10 (30 mg, 46.4 μmol) in DMSO (300 μL) was added followed by TEA (1.17 mg, 11.4 μmol) at room temperature. After HPLC-SQ2 analysis suggested the reaction was completed, the reaction mixture was diluted with 2 mL water. Hydrazine was added so the final hydrazine concentration was 3%. The mixture was left at room temperature overnight before piperidine (200 μL) was added to the mixture to remove the Fmoc protection group at room temperature. Fmoc removal was completed within 30 minutes. The mixture was lyophilized and purified by LH-20 size exclusion chromatography. A solution of 2 (6.9 mg, 10 μmol) in DMSO (140 μL) was added to the intermediate. The mixture reacted at room temperature for 6 hours before HPLC-SQ2 analysis showed completion of the reaction. The mixture was diluted with water and purified by semi prep-HPLC to afford 13 (15 mg, 73%, over four steps). HR-ESI-MS: [M+H]+ calcd for C126H211N14O47+, 2673.4549 (100%); found (m/z), 2673.4546. HPLC (0.4 mL/min, 10–50%B, 30min, tR = 20.8 min)
Synthesis of compound α-Gal-PEG-DBCO (15).
To a solution of 14 [2c] (2 mg, 3.15 μmol) in DMSO (100 μL), the DBCO-PEG-NHS (2) (4 mg, 5.8 μmol) in DMSO (80 μL) was added, followed by 0.5 μL of TEA. The mixture reacted at room temperature for 2 hours before HPLC-SQ2 analysis showed completion of the reaction. The mixture was diluted with water and purified by semi prep-HPLC to afford 15 (2.2 mg, 58%). ESI-MS: [M+H]+ calcd for C56H84N3O26+, 1214.5338; found (m/z), 1214.8262. HPLC (0.4 mL/min, 10–50%B, 30min, tR = 21.6 min)
Synthesis of compound 16.
To a stirring solution of 9 (21.4 mg, 63 μmol) in DMSO (0.6 mL) the solution of 14 (10 mg, 15.7 μmol) was added dropwise over 30 minutes at room temperature. The reaction was stirred at room temperature for another 2 hours before HPLC-SQ2 analysis showed completion of the reaction. The mixture was diluted with water, lyophilized, and then purified by semi-prep HPLC to afford 16 (8.5 mg, 67%) as a white power. 1H NMR (600 MHz, D2O): δ = 1.69 (4H, m), 2.28 (2H, t, J = 6.99 Hz), 2.72 (2H, t, J = 6.96 Hz), 2.91 (4H, br), 3.32 (1H, m), 3.36 (2H, t, J = 5.25 Hz), 3.60–3.85 (21H, m), 3.92 (1H, m), 3.95 (1H, m), 3.99 (1H, d, J = 2.94 Hz), 4.03 (1H, m), 4.14–4.18 (2H, m), 4.49 (2H, dd, J1 = 7.98 Hz J2 = 2.22 Hz), 5.11 (1H, d, J = 3.84 Hz). 13C NMR (150 MHz, D2O): δ = 22.74, 23.99, 24.67, 25.02, 29.57, 34.69, 38.39, 59.67, 60.42, 60.48, 64.31, 67.71, 68.24, 68.37, 68.63, 68.80, 68.87, 69.08, 69.14, 69.19, 70.34, 72.29, 73.90, 74.26, 74.56, 76.71, 78.17, 94.94, 101.61, 102.38, 170.09, 172.92, 175.99. HR-ESI-MS: [M+H]+ calcd for C34H57N2O23+, 861.3347; found (m/z), 861.3313.
Synthesis of DBCO functionalized α-Gal dendrimer (18).
To a solution of 7 (1 mg, 1.14 μmol) in DMSO (100 μL), a solution of 16 (4.8 mg, 5.7 μmol) in DMSO (100 μL) was added followed by TEA (1.17 mg, 11.4 μmol) at room temperature. After HPLC-SQ2 analysis suggested the reaction was completed, the reaction mixture was diluted with 1 mL water. Piperidine (120 μL) was added to the mixture to remove the Fmoc protection group at room temperature. Deprotection was completed within 30 minutes. The mixture was lyophilized, before a solution of 2 (1 mg, 1.4 μmol) in DMSO (100 μL) was added. The mixture reacted at room temperature for 6 hours before HPLC-SQ2 analysis showed completion of the reaction. The mixture was diluted with water and purified by semi prep-HPLC to afford 18 (3.1 mg, 65%, over three steps). HR-ESI-MS: [M+2H]2+ calcd for C174H292N14O912+, 2017.4337 (100%); found (m/z), 2017.3817. HPLC (0.4 mL/min, 10–50%B, 30min, tR = 16.5 min)
Synthesis of Rhamose functionalized SCT-oxa (27a).
A solution of Di-N3-SCT (25) (5 mg, 1.92 μmol) and 3 (4.2 mg, 4.8 μmol) in DI water (400 μL) was incubated at room temperature for 4 h when LC-MS indicated the completion of the reaction. The solution was then cooled on ice, and TEA (11.7 mg, 115 μmol) and 2-chloro-1,3-dimethylimidazolinium chloride (DMC) (8.1 mg, 45.5 μmol) were added, following the previously described procedure.[15] The reaction mixture was kept on ice for 45 min, then it was diluted with 0.1 % ammonium hydroxide, and the glycan oxaozline was purified by semi-prep HPLC with gradient elution (10%–70%B, 40min) of water containing 0.1% ammonium hydroxide as phase A, MeCN containing 0.1% ammonium hydroxide as phase B. 27a was isolated as a white powder after lyophilization (6 mg, 72%, over two steps). 1H NMR (600 MHz, D2O): δ = 7.92 – 7.83 (m, 1H), 7.82 – 7.74 (m, 1H), 7.72 – 7.65 (m, 2H), 7.64 –7.49 (m, 4H), 7.44 – 7.21 (m, 8H), 6.04 (d, J = 7.2 Hz, H-1-GlcNAc-Ox), 5.91 – 5.80 (m, 3H), 5.61 – 5.53 (m, 2H), 5.18 – 5.16 (m, 1H), 5.12 – 5.00 (m, 3H), 4.95 – 4.86 (m, 4H), 4.78 – 3.02 (m, 183H), 2.71 – 2.60 (m, 4H), 2.51 – 2.38 (m, 8H), 2.05 – 1.97 (m, 15H), 1.85 – 1.75 (m, 3H), 1.71 – 1.54 (m, 4H), 1.28 – 1.21 (m, 6H). HR-ESI-MS [M+2H]2+ calcd for C188H299N19O942+, 2164.4628 (100%); found (m/z), 2164.4403.
Synthesis of rhamose cluster functionalized SCT-oxa (27b).
A solution of Di-N3-SCT (25) (1 mg, 0.385 μmol) and 13 (2.5 mg, 0.962 μmol) in DI water (200 μL) was incubated at room temperature for 4 h. Then, the solution was cooled at 0°C, and TEA (2.6 mg, 25 μmol) and DMC (1.6 mg, 9.62 μmol) were added, and the reaction mixture was incubated at 0 °C for 45 min. The glycan oxazoline was purified by semi-prep HPLC with gradient elution (10%–70%B, 40min) of water containing 0.1% ammonium hydroxide as phase A, MeCN containing 0.1% ammonium hydroxide as phase B. 27b was isolated as a white powder after lyophilization (1.8 mg, 59%, over two steps). 1H NMR (600 MHz, D2O): δ = 7.90 – 7.85 (m, 1H), 7.82 – 7.74 (m, 1H), 7.72 – 7.65 (m, 4H), 7.64 –7.49 (m, 4H), 7.42 – 7.25 (m, 8H), 6.04 (d, J = 7.3 Hz, H-1-GlcNAc-Ox), 5.91 – 5.80 (m, 3H), 5.61 – 5.53 (m, 2H), 5.18 – 5.16 (m, 1H), 5.12 – 5.00 (m, 3H). 4.94 – 4.88 (m, 2H), 1.28 – 1.21 (m, 24H). HR-ESI-MS [M+4H]4+ calcd for C352H595N41O1584+, 1981.9966 (100%); found (m/z), 1981.9896.
Preparation of Antibody Conjugates
Synthesis of Rha4-Herceptin conjugate (21).
A solution of azide-tagged antibody 20 (2 mg, 13 nmol) and the Rha-PEG-DBCO (3) (113 μg, 0.13 μmol, 10 eq) in a phosphate buffer (50 mM, pH 7.2) (final volume, 500 μL) was incubated at ambient temperature (23 °C). The reaction mixture was shielded from light and gently vortexed. The reaction was monitored by LC-ESI-MS analysis. After 8 h, the click reaction was complete as indicated by LC-ESI-MS. The mixture was then diluted with phosphate buffer (5 mL, 50 mM, pH 7.2) and filtered by 0.22 μm syringe filter. The conjugate product in the filtrate was purified by protein A chromatography to give 21 (1.8 mg, 87%). ESI-MS of 21: calcd. M = 154518 Da; found (m/z), 2810.50 [M + 55H]55+, 2862.53 [M + 54H]54+, 2916.46 [M + 53H]53+, 2972.55 [M + 52H]52+, deconvolution of the ESI-MS, M = 154521 Da. Fc analysis: calcd, M = 28463 Da; found (m/z), 2034.03 [M + 14H]14+, 2090.38 [M + 13H]13+, 2372.80 [M + 12H]12+, deconvolution data, M = 28462 Da.
Synthesis of Rha16-Herceptin conjugate (22).
A solution of azide-tagged antibody 20 (2 mg, 13 nmol) and the Rha4-PEG-DBCO (13) (721 μg, 0.27 μmol, 20 eq) in a phosphate buffer (50 mM, pH 7.2) (final volume, 500 μL) was incubated at ambient temperature (23 °C). The reaction mixture was shielded from light and gently vortexed. The reaction was monitored by LC-ESI-MS analysis. After 16 h, the click reaction was complete as indicated by LC-ESI-MS. The mixture was then diluted with phosphate buffer (5 mL, 50 mM, pH 7.2) and filtered by 0.22 μm syringe filter. The conjugate product in the filtrate was purified by protein A chromatography to give 22 (1.9 mg, 88%). ESI-MS of 22: calcd. M = 161714 Da; found (m/z), 2941.30 [M + 55H]55+, 3052.25 [M + 53H]53+, 3110.84 [M + 52H]52+, 3171.81 [M + 51H]51+, deconvolution of the ESI-MS, M = 161718 Da. Fc analysis: calcd, M = 32061 Da; found (m/z), 2291.05 [M + 14H]14+, 2467.16 [M + 13H]13+, 2672.64 [M + 12H]12+, deconvolution data, M = 32060 Da.
Synthesis of α-Gal4-Herceptin conjugate (23).
A solution of azide-tagged antibody 20 (2 mg, 13 nmol) and the α-Gal-PEG-DBCO (15) (157 μg, 0.13 μmol, 10 eq) in a phosphate buffer (50 mM, pH 7.2) (final volume, 500 μL) was incubated at ambient temperature (23 °C). The reaction mixture was shielded from light and gently vortexed. The reaction was monitored by LC-ESI-MS analysis. After 8 h, the click reaction was complete as indicated by LC-ESI-MS. The mixture was then diluted with phosphate buffer (5 mL, 50 mM, pH 7.2) and filtered by 0.22 μm syringe filter. The conjugate product in the filtrate was purified by protein A chromatography to give 23 (1.9 mg, 87%). ESI-MS of 23: calcd. M = 155879 Da; found (m/z), 2835.26 [M + 55H]55+, 2887.68 [M + 54H]54+, 2942.18 [M + 53H]53+, 2998.70 [M + 52H]52+, deconvolution of the ESI-MS, M = 155882 Da. Fc analysis: calcd, M = 29143 Da; found (m/z), 2082.71 [M + 14H]14+, 2242.71 [M + 13H]13+, 2429.57 [M + 12H]12+, deconvolution data, M = 29143 Da.
Synthesis of α-Gal16-Herceptin conjugate (24).
A solution of azide-tagged antibody 20 (2 mg, 13 nmol) and the α-Gal-PEG-DBCO (15) (1.09 mg, 0.27 μmol, 20 eq) in a phosphate buffer (50 mM, pH 7.2) (final volume, 500 μL) was incubated at ambient temperature (23 °C). The reaction mixture was shielded from light and gently vortexed. The reaction was monitored by LC-ESI-MS analysis. After 40 h, the click reaction was complete as indicated by LC-ESI-MS. The mixture was then diluted with phosphate buffer (5 mL, 50 mM, pH 7.2) and filtered by 0.22 μm syringe filter. The conjugate product in the filtrate was purified by protein A chromatography to give 24 (2.0 mg, 90%). ESI-MS of 24: calcd. M = 167159 Da; found (m/z), 3040.18 [M + 55H]55+, 3096.46 [M + 54H]54+, 3154.81 [M + 53H]53+, 3215.53 [M + 52H]52+, deconvolution of the ESI-MS, M = 167158 Da. Fc analysis: calcd, M = 34783 Da; found (m/z), 2319.77 [M + 15H]15+, 2485.47 [M + 14H]14+, 2676.52 [M + 13H]13+, deconvolution data, M = 34782 Da.
Synthesis of Rha4-Herceptin conjugate (21) via transglycosylation.
To the solution of 28 (500 μg, 25 mg/ml in 100 mM Tris buffer), glycan oxazoline 27a (941 μg, 30 eq, 100 mg/mL in 100 mM Tris buffer) was added. After adjusting the pH to 7.4, Endo-S2 D184M mutant (13 mg/mL) was added to the solution. The final enzyme concentration was 0.1 mg/mL. The reaction was carried out at 30 °C and monitored by LC-MS every 20 minutes. The reaction reached completion in 60 min. The product was purified by protein A chromatography to yield 21 (420 μg, 82%). ESI-MS of 21: calcd. M = 154518 Da; found (m/z), 2810.48 [M + 55H]55+, 2862.51 [M + 54H]54+, 2916.44 [M + 53H]53+, 2972.53 [M + 52H]52+, deconvolution of the ESI-MS, M = 154519 Da.
Synthesis of Rha16-Herceptin conjugate (22) via transglycosylation.
To the solution of 28 (105 μg, 35 mg/ml in 100 mM Tris buffer), glycan oxazoline 27b (220 μg, 40 eq, 100 mg/mL in 100 mM Tris buffer) was added. After adjusting the pH to 7.4, Endo-S2 D184M mutant (13 mg/mL) was added to the solution. The final enzyme concentration was 0.4 mg/mL. The reaction was carried out at 30 °C and monitored by LC-MS every 20 minutes.
In Vitro Cytotoxicity Assay
BT474 cells (ATCC HTB-20) were cultured in HybriCare medium (ATCC 46-X) supplemented with 10% fetal bovine serum (FBS), 100 U/mL penicillin, and 100 μg/mL streptomycin in T-75 flasks (CELLTREAT). T47D cells (ATCC HTB-133) were maintained in RPMI-1640 medium (ATCC 30– 2001) containing FBS, 4 mg/L insulin, 100 U/mL penicillin, and 100 μg/mL streptomycin in T-75 flasks (CELLTREAT).
For the cytotoxicity assays, cells were seeded into 96-well plates with 25,000 cells per well and grown overnight at 37 °C and 5% CO2. The FBS containing media was removed. The serum free RPMI media with the antibody conjugates (starting at a concentration of 25 μg/mL for BT474 and 50 μg/mL for T47D and serially diluted 1:3) was added. Each compound was assessed in triplicate wells, the cells without compound served as control. After incubation for 2 hours, the antibody conjugates solution was removed. 70μL serum free RPMI and 10 μL human serum was added to each well and incubated for 15 minutes. Then 20 μL of 20% rabbit complement diluted by serum free RPMI was added to each well and incubated for 2 hours. The cells were then washed with PBS before the incubation with RPMI media containing 10% cell counting kit-8 (Dojindo). The absorbance of formazan released by viable cells was measured at 450 nm using a spectrophotometer after incubation for 2–3 h at 37 °C and 5% CO2. Finally, the cell viability curve and the EC50 values were calculated using GraphPad Prism software.
Supplementary Material
Acknowledgements
This work was supported by the US National Institutes of Health (NIH grant R01 AI155716).
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Tsao LC, Force J, Hartman ZC, Cancer Res 2021, 81, 4641–4651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].a) Owen RM, Carlson CB, Xu J, Mowery P, Fasella E, Kiessling LL, Chembiochem 2007, 8, 68–82; [DOI] [PubMed] [Google Scholar]; b) Sheridan RT, Hudon J, Hank JA, Sondel PM, Kiessling LL, Chembiochem 2014, 15, 1393–1398; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Naicker KP, Li H, Heredia A, Song H, Wang LX, Org. Biomol. Chem 2004, 2, 660–664; [DOI] [PubMed] [Google Scholar]; d) Perdomo MF, Levi M, Sallberg M, Vahlne A, Proc. Natl. Acad. Sci. U. S. A 2008, 105, 12515–12520; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Li X, Rao X, Cai L, Liu X, Wang H, Wu W, Zhu C, Chen M, Wang PG, Yi W, ACS Chem. Biol 2016, 11, 1205–1209. [DOI] [PubMed] [Google Scholar]
- [3].a) Galili U, Rachmilewitz EA, Peleg A, Flechner I, J. Exp. Med 1984, 160, 1519–1531; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Galili U, Wang L, LaTemple DC, Radic MZ, Subcell. Biochem 1999, 32, 79–106; [DOI] [PubMed] [Google Scholar]; c) Parker W, Bruno D, Holzknecht ZE, Platt JL, J. Immunol 1994, 153, 3791–3803. [PubMed] [Google Scholar]
- [4].Galili U, Immunol. Today 1993, 14, 480–482. [DOI] [PubMed] [Google Scholar]
- [5].a) Oyelaran O, McShane LM, Dodd L, Gildersleeve JC, J. Proteome Res 2009, 8, 4301–4310; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Huflejt ME, Vuskovic M, Vasiliu D, Xu H, Obukhova P, Shilova N, Tuzikov A, Galanina O, Arun B, Lu K, Bovin N, Mol. Immunol 2009, 46, 3037–3049. [DOI] [PubMed] [Google Scholar]
- [6].a) Hossain MK, Vartak A, Karmakar P, Sucheck SJ, Wall KA, ACS Chem. Biol 2018, 13, 2130–2142; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Jakobsche CE, Parker CG, Tao RN, Kolesnikova MD, Douglass EF Jr., Spiegel DA, ACS Chem. Biol 2013, 8, 2404–2411; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Chen W, Gu L, Zhang W, Motari E, Cai L, Styslinger TJ, Wang PG, ACS Chem. Biol 2011, 6, 185–191. [DOI] [PubMed] [Google Scholar]
- [7].a) Sharp TH, Boyle AL, Diebolder CA, Kros A, Koster AJ, Gros P, Proc. Natl. Acad. Sci. U. S. A 2019, 116, 11900–11905; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Ugurlar D, Howes SC, de Kreuk BJ, Koning RI, de Jong RN, Beurskens FJ, Schuurman J, Koster AJ, Sharp TH, Parren P, Gros P, Science 2018, 359, 794–797; [DOI] [PubMed] [Google Scholar]; c) Diebolder CA, Beurskens FJ, de Jong RN, Koning RI, Strumane K, Lindorfer MA, Voorhorst M, Ugurlar D, Rosati S, Heck AJ, van de Winkel JG, Wilson IA, Koster AJ, Taylor RP, Saphire EO, Burton DR, Schuurman J, Gros P, Parren PW, Science 2014, 343, 1260–1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Sianturi J, Manabe Y, Li HS, Chiu LT, Chang TC, Tokunaga K, Kabayama K, Tanemura M, Takamatsu S, Miyoshi E, Hung SC, Fukase K, Angew. Chem. Int. Ed 2019, 58, 4526–4530. [DOI] [PubMed] [Google Scholar]
- [9].Zhou K, Hong H, Lin H, Gong L, Li D, Shi J, Zhou Z, Xu F, Wu Z, J. Med. Chem 2022, 65, 323–332 [DOI] [PubMed] [Google Scholar]
- [10].Ou C, Li C, Zhang R, Yang Q, Zong G, Dai Y, Francis RL, Bournazos S, Ravetch JV, Wang LX, Bioconjug. Chem 2021, 32, 1888–1897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].a) Elgersma RC, Coumans RG, Huijbregts T, Menge WM, Joosten JA, Spijker HJ, de Groot FM, van der Lee MM, Ubink R, van den Dobbelsteen DJ, Egging DF, Dokter WH, Verheijden GF, Lemmens JM, Timmers CM, Beusker PH, Mol. Pharm 2015, 12, 1813–1835; [DOI] [PubMed] [Google Scholar]; b) Nicolaou KC, Li R, Chen Q, Lu Z, Pitsinos EN, Schammel A, Lin B, Gu C, Sarvaiya H, Tchelepi R, Valdiosera A, Clubb J, Barbour N, Sisodiya V, Sandoval J, Lee C, Aujay M, Gavrilyuk J, J. Am. Chem. Soc 2020, 142, 12890–12899. [DOI] [PubMed] [Google Scholar]
- [12].Zhang W, Wang J, Li J, Yu L, Wang PG, J. Carbohydr. Chem 2008, 18, 1009–1017. [Google Scholar]
- [13].a) Huang W, Giddens J, Fan SQ, Toonstra C, Wang LX, J. Am. Chem. Soc 2012, 134, 12308–12318; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Parsons TB, Struwe WB, Gault J, Yamamoto K, Taylor TA, Raj R, Wals K, Mohammed S, Robinson CV, Benesch JL, Davis BG, Angew. Chem. Int. Ed 2016, 55, 2361–2367; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tang F, Yang Y, Tang Y, Tang S, Yang L, Sun B, Jiang B, Dong J, Liu H, Huang M, Geng MY, Huang W, Org. Biomol. Chem 2016, 14, 9501–9518; [DOI] [PubMed] [Google Scholar]; d) Manabe S, Yamaguchi Y, Matsumoto K, Fuchigami H, Kawase T, Hirose K, Mitani A, Sumiyoshi W, Kinoshita T, Abe J, Yasunaga M, Matsumura Y, Ito Y, Bioconjug. Chem 2019, 30, 1343–1355. [DOI] [PubMed] [Google Scholar]
- [14].a) Zhang X, Ou C, Liu H, Prabhu SK, Li C, Yang Q, Wang LX, ACS Chem. Biol 2021, 16, 2502–2514; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li T, Tong X, Yang Q, Giddens JP, Wang LX, J. Biol. Chem 2016, 291, 16508–16518; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Li T, DiLillo DJ, Bournazos S, Giddens JP, Ravetch JV, Wang LX, Proc. Natl. Acad. Sci. U. S. A 2017, 114, 3485–3490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].a) Noguchi M, Tanaka T, Gyakushi H, Kobayashi A, Shoda S.-i., J. Org. Chem 2009, 74, 2210–2212; [DOI] [PubMed] [Google Scholar]; b) Huang W, Yang Q, Umekawa M, Yamamoto K, Wang LX, ChemBioChem 2010, 11, 1350–1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Lin H, Hong H, Wang J, Li C, Zhou Z, Wu Z, Chem. Commun. (Camb) 2020, 56, 13959–13962. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.