

Abstract
Prions are a self-propagating misfolded conformation of a cellular protein. Prions are found in several eukaryotic organisms with mammalian prion diseases encompassing a wide range of disorders. The first recognized prion disease, the transmissible spongiform encephalopathies (TSEs), affect several species including humans. Alzheimer’s disease, synucleinopathies, and tauopathies share a similar mechanism of self-propagation of the prion form of the disease-specific protein reminiscent of the infection process of TSEs. Strain diversity in prion disease is characterized by differences in the phenotype of disease that is hypothesized to be encoded by strain-specific conformations of the prion form of the disease-specific protein. Prion therapeutics that target the prion form of the disease-specific protein can lead to the emergence of drug-resistant strains of prions, consistent with the hypothesis that prion strains exist as a dynamic mixture of a dominant strain in combination with minor substrains. To overcome this obstacle, therapies that reduce or eliminate the template of conversion are efficacious, may reverse neuropathology, and do not result in the emergence of drug resistance. Recent advancements in preclinical diagnosis of prion infection may allow for a combinational approach that treats the prion form and the precursor protein to effectively treat prion diseases.
1. Prion diseases
Prions are a self-propagating misfolded conformation of a cellular protein. The conversion of the normal isoform to the misfolded prion form occurs in a stepwise manner, but the exact mechanism is unknown.1–4 Prions are found in several eukaryotic organisms. Yeast prions are a dominant, non-Mendelian form of epigenetic inheritance that causes a detectable phenotype. For example, the [URE3], [SWI+], and [GAR+] prions in Saccharomyces cerevisiae enhance growth on nutrient-poor sources, while the [PSI+] prion enables translational readthrough.5–9 The prion state in yeast is dependent on a chromosomal gene and transient overproduction of the normal cellular protein promotes the prion state which can be reversibly cured.8,10,11 The [Het-s] prion of Podospora anserina is infectious and has a necessary role in heterokaryon incompatibility, a process that restricts fusion of hyphae to genetically similar partners.12–14 Prions found in yeast, fungi, and mammals can have advantageous roles. Functional prion-like proteins involved in memory, RNA metabolism, and immunity have been identified.15–19 However, prions in mammals are most notably associated with neurodegenerative diseases.
Mammalian prion diseases encompass a wide range of disorders. The first recognized prion disease, the transmissible spongiform encephalopathies (TSEs), are characterized by long incubation periods followed by a relatively short duration of clinical signs that ultimately leads to death of the host.1,20 TSEs occur in both domesticated populations of sheep and goats (scrapie), cattle (bovine spongiform encephalopathy, BSE), and mink (transmissible mink encephalopathy, TME). Chronic wasting disease (CWD) is unique among TSEs as it can affect both wild and domestic populations of cervids.21–23 Camel prion disease (CPD) in domesticated dromedary camels was recently identified, but the geographic distribution and potential of transmission to wild populations is unknown.24 Human TSEs can have infectious (Kuru), inherited (Gerstmann-Sträussler-Scheinker, GSS), or sporadic (Creutzfeldt-Jakob disease, CJD) etiologies, with sporadic being the most prevalent.25 Iatrogenic transmission of PrP prions can occur via cadaveric human growth hormone (c-hGH), dura mater grafts, or corneal transplants.26–29 TSE agent replication involves the misfolded infectious prion conformation of the prion protein, PrPSc, binding to the normal, cellular conformation, PrPC, initiating further seeded conversion.
Alzheimer’s disease, synucleinopathies, and tauopathies are characterized by the misfolding and aggregation of amyloid-β (Aβ; a peptide derived from the host cellular amyloid precursor protein (APP)), α-synuclein protein (α-syn), and tau protein, respectively. Similar to TSEs, these protein misfolding diseases share a similar mechanism of self-propagation of the prion form of the disease-specific protein and cell to cell spreading of the host protein in the prion state.30–39 Experimental injection of the prion form of the protein into animals that express the respective precursor protein can accelerate the pathogenesis of disease, reminiscent of the infection process of TSEs (or PrP prions).34,40–46 Although these non-prion protein disorders have both inherited and sporadic etiologies, conflicting evidence for infectious transmission has been reported. Epidemiological studies concluded that a history of one or more blood transfusions was not associated with a higher risk of developing AD compared to age- and gender-matched controls for each patient case.47,48 Injection of nonhuman primates with over 600 patient samples from individuals diagnosed with nonspongiform diseases (i.e., AD, PD, etc.) failed to transmit disease.49 No cases of AD or PD were identified among recipients of c-hGH in the United States despite finding mild amounts of pathological Aβ, tau, and α-synuclein by immunohistochemical analysis of pituitary glands from neurodegenerative disease and control patients.50 However, a recent study reported that recipients of c-hGH contaminated with PrP prions subsequently developed CJD with concurrent Aβ deposition in the brain, suggesting iatrogenic transmission of Aβ.51 Retrospective studies on the archived c-hGH samples identified significant amounts of tau, Aβ42 and Aβ40.52 Intracerebral injection of these c-hGH samples to transgenic mice expressing a human mutant of the amyloid precursor protein resulted in the development of brain pathology similar to the patients injected with the same material.52 This finding suggests, under certain conditions, iatrogenic transmission of AD is possible. Overall, the aforementioned neurodegenerative disorders share profound similarities at the biochemical and cellular level.
2. Prion strain diversity
Prion strains are operationally defined by heritable differences in the biological properties of disease under controlled agent and host parameters.53–56 In yeast, prion strains are classified based on several traits that differ depending on the prion state (i.e., [PSI+] or [URE3]) but were initially distinguished by strength of phenotype (i.e., “strong” versus “weak”).57–59 TSE strains are classified by differences in incubation period and neuropathology but can differ in their clinical signs and biochemical features of PrPSc.60,61 Similar to TSEs, Aβ, tau, and ɑ-synuclein strains are defined by differences in abnormal protein deposition and pathology in the brain as well as protein conformation or seeding activity of the prion form of the respective protein.38,44,62–64 The relationship between the strain-specific biochemical features of the prion form of the protein and the phenotype of disease is poorly understood.
2.1. Prion strain diversity in yeast
Studies of yeast prion strain diversity have added much to the knowledge of prion strain diversity. Yeast prion strains differ in strength of phenotype, stability of propagation, toxicity/lethality, ability to overcome inter- or intraspecies transmission barriers, and biochemical and physical properties.57–59,65–67 Nuclear magnetic resonance (NMR) spectroscopy identified conformational differences between [PSI+] yeast prion strains, and atomic force microscopy (AFM) reveals strain-specific differences in yeast prion fibril morphology.68 There are strong correlations between biochemical properties of the prion form of the protein and strain phenotype in yeast. The strength of the yeast prion phenotype corresponds with an increased fragility of protein aggregates.69 Mechanistically, increased fragility of prion aggregates results in rapid generation of new free ends, accelerating prion formation (Fig. 1, step 5).69
Fig. 1.
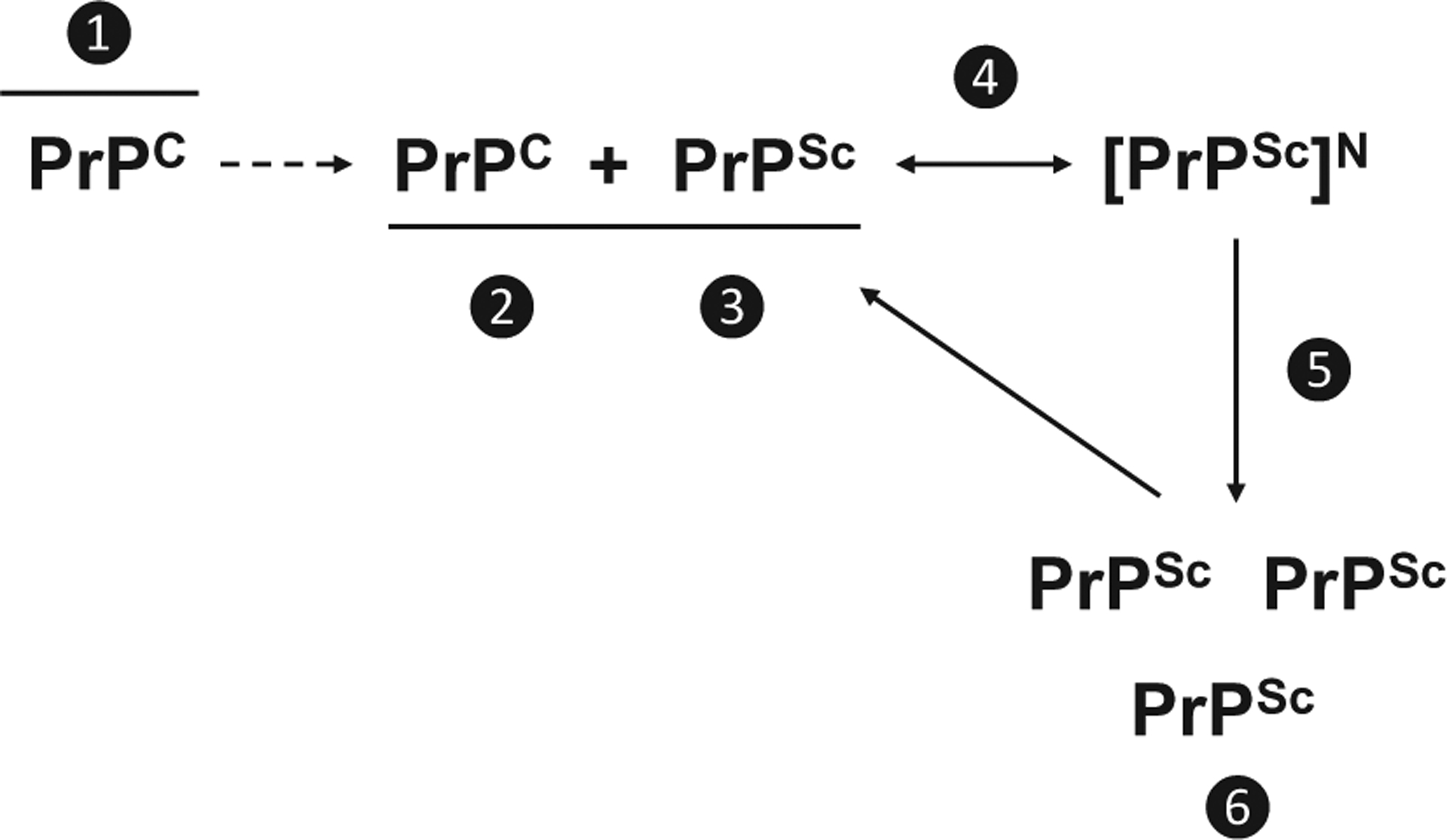
Mechanistic location of therapeutic targets for PrP prion diseases. Therapeutic targets: ❶ PrPC post-translational processing, metabolism, cellular trafficking, or localization; ❷ PrPC by binding or inducing conformational change that prevents PrPC interaction; ❸ PrPSc by stabilization, redistribution; ❹ conversion process by blocking binding site on PrPC, capping growth, inhibition of cofactor interactions, or ❺ promoting fragmentation or ❻ clearance from the host.
2.2. TSE strain diversity
Strain diversity is hypothesized to be encoded by strain-specific conformations of PrPSc.63,64,68,70–72 Strain-specific differences in the biochemical properties of PrPSc were first identified in mice.73 These differences were later observed in hamsters infected with the hyper (HY) or drowsy (DY) strains of hamster-adapted transmissible mink encephalopathy (TME).74 Detergent extraction and protease digestion of HY and DY PrPSc revealed strain-specific Western blot migration profiles suggesting strain-specific differences in the PK digestion site on PrPSc.75 Edman protein sequencing of PrPSc from HY or DY TME-infected hamsters confirmed that PrPSc from these two strains had different PK cleavage sites.70 Based on these observations, the authors proposed the hypothesis that strain-specific conformations of PrPSc encoded prion strain diversity.70,75,76 Consistent with this hypothesis, the molecular weight of the proteinase K-resistant fragment of PrPSc after deglycosylation in fatal familial insomnia is 19 kDa while the same fragment was 21 kDa in sporadic and familial Creutzfeldt Jakob disease.72 When inoculated into mice, the strain-specific migration pattern of PrPSc from human prion strains was preserved supporting the hypothesis that strain-specific conformations of PrPSc is the basis of strain diversity.72
The presence of cellular cofactors during prion conversion may be responsible for the maintenance of strain-specific, infectious conformations of PrPSc. Development of synthetic, in vitro-generated prions able to successfully infect wild-type rodents relied on cofactors such as polyanions and lipids.77,78 Propagation of a recombinant prion strain in PMCA in the presence of both recombinant murine PrP and the lipid phosphatidylethanolamine (PE) retained infectivity.79 Withdrawal of the cofactor (PE) led to the emergence of a protein-only strain, named OSU, that did not cause disease in mice suggesting cellular cofactors are required for infectivity. Additionally, in vitro PMCA propagation of three distinct prion strains with PE as the sole cofactor resulted in the three strains evolving into a single strain.79 This work on the directed evolution of prions suggests that cellular cofactors are involved in prion strain diversity. Recent work has clarified the role of prion cofactors. Using a novel in vitro system where protein only PrPSc preparations, in the absence of PMCA adaptation, are directly infectious to animals, but only in the presence of co-factors.80 Importantly, protein-only and cofactor generated PrPSc had identical strain properties indicating that the conformation of PrPSc is sufficient to encode prion strain information and that cellular co-factors are needed for prion propagation.80 Overall, these data provide a unified model of prion infectivity where strain information is encoded by the structure of PrPSc and host cellular cofactors are necessary for prion conversion and subsequent infectivity.
Lesion profiles, cellular assays, and the biochemical properties of PrPSc are used to differentiate prion strains. The scrapie cell assay (SCA) and the cell panel assay (CPA) assess the relative susceptibility or resistance of murine neuroblastoma cell lines to infection by different prion strains.81–83 The Western blot migration profile, conformational stability, and sensitivity to proteolytic digestion of PrPSc can be strain specific. It is unclear which, if any, of the strain-specific biochemical differences of PrPSc directly contributes the strain-specific phenotype of disease. For example, in murine PrP prions, decreased PrPSc conformational stability is positively correlated with a shorter incubation period, however, in humans and hamsters the inverse relationship between PrPSc conformational stability and incubation period was identified.71,84–87
2.3. Aβ strain diversity
The heterogeneous clinical presentation of AD may be attributed to strain diversity.88 The Arctic and Swedish APP mutations in humans result in distinct AD pathologies.89–92 It was hypothesized that the two AD clinical disease outcomes were a result of two distinct Aβ strains. Conformational stability studies of Aβ from patient brains with Arctic AD and Swedish AD mutations established that aggregates of Aβ found in the Arctic AD patients were significantly more sensitive to denaturation with guanidine hydrochloride compared to Swedish AD.44 Further, injecting transgenic mice expressing human APP containing the Swedish mutation with the Arctic or Swedish AD brain material resulted in two distinct phenotypes of disease that were maintained upon serial passage consistent with the predicted behavior of prion strains.44 Luminescent conjugated oligothiophenes, a class of amyloid binding dyes, to investigate how the Aβ amyloid structure in familial AD differs from sporadic AD.93 The emission spectra of luminescent conjugated oligothiophenes changes when bound to amyloid of differing structure. The emission spectra of luminescent conjugated oligothiophenes bound to Aβ some of the subtypes of familial and sporadic AD varied significantly, suggesting a variety of Aβ structures exist in different AD cases.93 Solid state nuclear magnetic resonance imaging analysis of Aβ from patient tissue samples from PCA-AD or rp-AD indicated that Aβ from PCA-AD resembled typical AD with predominant Aβ40 fibril structure while rp-AD samples were composed of a cloud of variant Aβ40 structures.94 Overall, this data is consistent with the hypothesis that Aβ structural variance contribute to the clinical and pathological heterogeneity of AD.
2.4. Tau strain diversity
Alzheimer’s disease (AD), corticobasal degeneration (CBD), progressive supranuclear palsy (PSP), Pick’s disease (PiD), and argyrophilic grain disease (AGD) are unique tauopathies defined by the pattern of tau deposition in the brain along with distinct clinical phenotypes.95 Transmission studies in wild-type and transgenic mouse models have established that tau extracts from various tauopathies transmit distinct tau neuropathology.38,64,96–98 For example, AD-tau variants can differentially seed tau pathology after intracranial injection of wild-type mice; the tau variants were determined to be structurally different by trypsin digests that resulted in three distinct trypsin resistant fragments.38 Injecting wild-type mice with tau from AD, CBD, or PSP resulted in differences in tau conversion efficiency and cellular tropism in the CNS suggesting the three tau isolates are distinct prion strains.64 Consistent with this hypothesis, tau fibrils from the tau strains showed varying resistance to Gdn-HCl denaturation with CBD-tau, AD-tau, and PSP-tau having the lowest to highest stability, respectively.64 Two recombinant tau strains in HEK293 cells expressing the tau repeat domain that propagated conformationally distinct forms of tau that could be serially passaged in PS19 transgenic tau mice that retained the strain-specific conformation of tau.99 Further, the HEK293 cell model was used to examine AD, CBD, and AGD patient samples, and each tauopathy induced unique tau inclusion morphology.99 Consistent with this finding, 18 tau strains were identified based on differences in limited proteolysis and in vitro seeding activity of tau in combination with hippocampal tau pathology when transmitted to PS19 transgenic tau mice.100 Taken together, these data support that tau structural variance may explain the distinct pathologies and clinical presentation of the tauopathies.
2.5. ɑ-synuclein strain diversity
Strain-specific prion forms of α-synuclein have been identified in synucleinopathies including Parkinson’s disease (PD), dementia with Lewy bodies, and multiple system atrophy (MSA). Synthetic α-synuclein fibrils generated under varying salt concentrations form distinct conformations with differences in seeding ability and toxicity in vivo and in vitro.62,101,102 Recombinant fibrillar α-synuclein created under different laboratory conditions varied in ability to cross-seed tau aggregation in cell culture and in vivo.63 Consistent with this observation, proteinase K digestion revealed differences in protein cleavage between the synthetic α-synuclein as well as two distinct α-synuclein digestion products isolated from Parkinson’s disease with dementia (PDD) patient brains.63 Distinct digestion products are suggestive of two α-synuclein conformations reminiscent of prior observations in PrP prions.72,75,103–105 In vitro seeding assays utilizing a cell line that readily propagates MSA α-synuclein was not susceptible to α-synuclein from PD patient brains suggesting that PD contains a different strain of α-synuclein.106 Brain material from PD and MSA patients has distinct seeding activity and cellular inclusion pathology in HEK293T cells, suggestive of distinct strain of α-synuclein.107 Overall, α-synuclein strains share many features of PrP prion strains that include differences in biochemical features, neuropathology, incubation period, and clinical manifestation of disease.38,62,63
3. Prion strains exist as a mixture
Multiple prion strains can co-exist in an individual host. In cases of sporadic Creutzfeldt-Jakob disease (sCJD), the polymorphism at codon 129 and different proteinase K cleavage patterns via Western blot classify disease as Type 1 (21 kDa) or 2 sCJD (19 kDa).103 Type 1 and Type 2 sCJD PrPSc can co-exist in the CNS of the same patient.108–116 It is suggested that up to 40% of sCJD cases contain more than one strain, however, the prevalence rate is controversial since methodological differences in strain typing and the region of the brain that is evaluated can both affect the outcome.108–116 It is not well understood if strains of Aβ can co-exist in a single host, however, distinct strains of Aβ have been identified raising the possibility that sporadic AD may be a mixture of strains.44 Mapping phospho-tau patterns of deposition in the hippocampal regions of individuals with various tauopathies established that the pattern of pathology is disease-specific with regional selectivity reminiscent of strain targeting in TSE’s.117 This same study determined that AD-related tau hippocampal deposition is greatly influenced by simultaneous argyrophilic grain disease (AGD), another tauopathy, suggesting that coexisting tau strains can create heterogeneous brain pathologies and overlapping clinical features highlighting the need for discriminatory methods of post-mortem diagnosis.117 Antibodies that distinguish AD tau from non-AD tau deposits of CBD, PSP, and Pick’s Disease (PiD) can discern the differential deposition in co-occurring pathologies.118 While ample experimental evidence suggests that different α-synucleinopathy strains exist, the coexistence of α-synucleinopathy strains has not been described.
Coexisting prion strains can interfere with each other. Strain interference is a phenomenon in which a slowly-converting blocking strain can delay or prevent the emergence of a quickly-converting strain. A number of parameters govern prion strain interference. Increasing the titer of the blocking strain results in a corresponding increase in the interference effect.119–122 Importantly, prion conversion in a common population of cells is required for interference to occur as the strains compete for a limiting cellular resource, PrPC.123,124 Consistent with this hypothesis, co-infection of two low conversion efficiency prion strains results in each strain converting independently without interference that is due to PrPC not becoming limiting.125 Overall, strain interference is an important parameter in the emergence of a dominant strain from a mixture.
Currently, strain interference has not been reported for mammalian non-PrP prions, and further investigation is required to determine if this phenomenon is a common property of mammalian prions. In yeast, there is evidence that strain variants compete within a cell during and after mating, with “strong” variants outcompeting “weak” variants for the template of conversion.126 Mechanistically, “weak” variants are characterized by quick growth but low frangibility, whereas “strong” variants grow slowly but are more easily fragmented, leading to increased free ends for growth and therefore a competitive advantage over “weak” variants.69
4. Prion therapeutics
Therapeutics that target prion conversion can work via several mechanisms. Drugs can target PrPC prior to its interaction with PrPSc by affecting the post-translational processing, metabolism, cellular trafficking, or localization of PrPC (Fig. 1, step 1). Drugs that bind to or induce a conformational change in PrPC can hinder the interaction between PrPC and PrPSc (Fig. 1, step 2). Drugs can target PrPSc by stabilizing or redistributing PrPSc, interfering with the interaction between PrPC and PrPSc (Fig. 1, step 3). The conversion of PrPC to PrPSc can be targeted by drugs that block the PrPSc binding site on PrPC, cap PrPSc aggregate growth, or inhibit cofactors that enable conversion (Fig. 1, step 4). Therapeutics that target clearance of PrPSc either increase fragmentation of PrPSc aggregates (Fig. 1, step 5) or promote clearance of PrPSc from cells (Fig. 1, step 6).
4.1. Treatments that target PrPSc
Several drugs that directly interact with PrPSc have anti-prion effects (Table 1 and Fig. 1). Congo red, an amyloid binding dye, was one of the first compounds identified to extend the incubation period of prion-infected animals and can reduce the formation of PrPSc in scrapie-infected mouse neuroblastoma cells (Sc+ MNB).127 Congo red is hypothesized to directly bind and over-stabilize PrPSc, preventing further prion formation.127–129 Oral administration of Compound B, an amyloidophilic compound, extends the incubation period of TSEs and is hypothesized to inhibit formation of PrPSc by directly binding PrPSc or by interacting with cofactors/chaperones necessary for prion conversion.134,135 The diphenyl-pyrazole anle138b inhibits PrPSc amplification and reduces neurotoxicity by shifting PrPSc oligomers to a smaller size by reducing intermolecular hydrogen bonding and obstructing the formation of higher order oligomers, modulating the formation of toxic PrPSc aggregates.136,137 Anle138b has anti-prion activity both in vitro and in vivo and substantially prolonged survival even when treatment began after onset of signs of prion disease.137
Table 1.
Anti-prion therapeutics.
| Drug | Type | Model | Efficacy | Suggested mechanism | Strain Specificity | References |
|---|---|---|---|---|---|---|
| PrPsc target | ||||||
| Congo Red | Sulfonated amyloid dye | Neuronal cell culture, hamster | Decreases PrPSc accumulation and inhibits propagation in culture, prolongs mean incubation period in vivo | Stabilizes PrPSc, inhibits GAG-PrP binding and potentially PrPC-PrPSc binding | 127S, Sc237, PrP (27–30), 263K, 139H | 127–131 |
| LIN5001, LIN7002, LIN5044 | Luminescent conjugated polythiophenes (LCPs) | Mouse | IV infusion prolongs survival, reduces infectivity | Aggregate hyperstabilization | RML6, 263K | 132,133 |
| Compound B | 4-pyridinecarboxaldehyde, 2-[4-(5-oxazolyl)phenyl] hydrazone | Neuronal cell culture, mouse | Eliminates PrPSc in culture; prolongs incubation period/life, reduces PrP deposition, large quantity needed for efficacy | Binds to abnormal PrP, inhibits new formation of abnormal PrP | RML, 22L (marginally), Fukuoka-1 GSS, 263K (barely) | 134,135 |
| anle138b | 3,5-diphenyl-pyrazole (DPP) | Neuronal cell culture, mouse | Antiprion activity in vitro and in vivo, substantial prolongation of survival (txt after clinical onset), good bioavailability, BBB penetration | Directly blocks PrPSc amplification, causes shift towards smaller PrPSc oligomer size, inhibits pathological aggregation (PrPSc specific) | RML, ME7, 301c, sCJD, vCJD, a-syn | 136–138 |
| U18666A | Cholesterol synthesis inhibitor | Neuronal cell culture | Significantly decreased PrP-res levels | Redistributes PrPSc to late endosome-lysosome, degradation of PrPSc in secondary lysosomes | 22L | 139 |
| Pentosan Polysulfate (PPS) | Cysteine protease inhibitor | Neuronal cell culture, mouse, human | IV infusion prolonged survival even when administered after abnormal PrP deposition, rapidly and significantly decreased PrPSc levels, various efficacy in humans | May act as a competitive coreceptor (with endogenous GAG or other proteoglycans) for PrP at cell surface or causes fragmentation of PrPSc at the cell surface | 263K, RML, Fukuoka-1 GSS, 22L, iCJD, vCJD | 139–147 |
| Heterologous protein | Hamster PrP | Neuronal cell culture, mouse | Decreases PrP-res in culture, slows disease progression, increases survival, significantly delays onset of clinical signs, decreases PrPSc accumulation in brain/spleen | Either binds to PrPSc creating a functionally impotent aggregate unable to produce additional PrPSc or binds and blocks the conversion site from PrPC | RML-Chandler | 148–151 |
| IND24, IND81, IND114338, IND125, IND126461 | 2-aminothiazoles | Neuronal cell culture, mouse | Strain-specific and administration time-dependent extension in survival, decreased & altered distribution of PrPSc in brain, can lead to drug resistance (e.g. IND24-resistant RML) | Inhibits formation of new PrPSc, reduces PrPSc load via drug-like mechanism, might promote clearance of PrPSc | RML, ME7, 22L, CWD | 138,152–156 |
| Amphotericin B | Fungizone (polyene antibiotic) | Neuronal cell culture, mouse, hamster, human | Administration time-dependent prolongation of incubation period, delays onset of clinical signs without improvement of neurological symptoms or prolongation of clinical disease, delays accumulation ofPrPSc in the brain | Potentially could: interact with PrPC/PrPSc, interfere with uptake of PrPSc into cells, target detergent-resistant microdomains (modify cell surface distribution and/or trafficking of PrP), regulate microglial/glial activation, directly bind and either “cap” amyloid growth or overstabilize PrPSc | 263K, CJD | 140,157–164 |
| PrPC Target | ||||||
| 771, active ASO 1, active ASO 2 | Antisense oligonucleotide | Neuronal cell culture, mouse | Reduces PrPC/PrPSc in cell culture and in vivo, delays onset of clinical signs, slows disease progression, toxicity issues when administered after prion inoculation | Decreased PrPC levels slow accumulation of PrPSc | RML, potentially more since PrPC is the target | 165,166 |
| J1, J8, J20, J35 | Chalcones | Neuronal cell culture | Decreases PrPSc levels, completely blocks PrP109–149 aggregation | Directly binds to PrPC (C-terminal), decreases amount of PrPC on cell surface (arrest in ER), may stabilize PrPC or bind seeding surface | RML, 22L, CWD, 263K, CJD | 167,168 |
| Y13, Y17 | Oxadiazole | Neuronal cell culture | Could not totally block conversion but Increased lag phase for conversion; minor decrease in PrP-res formed in RT-QuIC | Direct binding to PrPC (stabilize PrPC or bind at seeding surface), potentially interacts with PrPC/PrPSc N-terminal region, decrease amount of PrPC on cell surface causing arrest in ER | RML, 263K, CJD | 167,168 |
| Fe(III)-TMPyP | Cationic tetrapyrrole | Neuronal cell culture, mouse | Inhibits replication in vitro/cell culture, prolongs survival, inhibits amplification of PrPSc, inhibits cytotoxic effects of delta CR PrP | Interacts with C-terminal domain of PrPC (Helix-3), acts as pharmacological chaperone for PrPC (decreases prion-induced misfolding) | RML, 22L, bank vole strains (Italian, UK (SCR1), and CH1641) | 169,170 |
| D18 | Anti-recombinant PrP (29–231) antibody (residues 132–156; helix A) Fabs | Neuronal cell culture, mouse | Abolishes prion replication and clears existing PrPSc in cell culture (2 week txt), prolongs incubation period and decreased titer in mice | Binds to PrPC on cell surface and hinders docking of PrPSc/a cofactor needed for conversion | Scrapie | 171 |
| 6H4 | Anti-murine PrPC antibody (Mab and expressed as transgene) | Neuronal cell | Prevents infection of susceptible cells, cures chronically infected cultures, conferred anti-PrP titers (Prnp0/0 mice) w/o induction of autoimmune disease, no infectivity detected (Prnp+/0-6H4u mice) when challenged, prevents or drastically delays pathogenesis | Occludes PrPC at prion replication sites, captures and degrades (immune-mediated) incoming PrPSc inoculum, steric competition with template-directed refolding, interference with seeded PrPSc nucleation reaction | RML | 172,173 |
| ICSM 18 | Isotype IgG1, anti-murine alpha-PrP antibody (residues 146–159) | Neuronal cell culture, mouse | Reduces splenic PrPSc levels, increases survival, passive transfer of Abs had no effect late in incubation period | Direct inhibition of PrPSc production, binding may bury “active residues” and stabilize helix 1 | RML | 174,175 |
| 110 | Monoclonal anti-PrP antibody (PHGGGWG at aa 59–65 and aa 83–89; octarepeat region) | Neuronal cell culture | Reduced PrPSc accumulation (dose-dependent) in cell culture, long term txt increased PrPC levels | Retains PrPC on cell surface (may interfere with PrPC metabolism), mAb-PrPC binding inhibits PrPC-PrPSc interaction by occupying binding domain or through steric interference | Chandler (139A), RML | 176 |
| 6D11 | Monoclonal antibody, recognizes PrPC, PrPSc, and recPrP (epitope between 93–109) | Neuronal cell culture | Complete abrogation of PrPSc, no re-emergence for at least 14 days, (following removal of txt), pre-incubation prevents infection | May prevent PrPC-PrPSc interaction or interfere with binding auxiliary molecules needed for prion propagation | 22L | 177 |
| 7H6 | Monoclonal Ab, recognizes residues 130–140 | Neuronal cell culture | Complete abrogation of PrPSc, no re-emergence for at least 14 days, (following removal of txt) | Similar to 6D11? | 22L | 177 |
| 7A12 | Monoclonal Ab, recognizes residues 143–155 (alpha-helix A) | Neuronal cell culture | Complete abrogation of PrPSc, no re-emergence for at least 14 days, (following removal of txt), weakest preventative effect | Similar to 6D11? | 22L | 177 |
| SAF34 | Monoclonal Ab, recognizes residues 59–89 (HuPrP) | Neuronal cell culture | Dose-dependent decrease of PrPSc levels, increased inhibition when used in conjunction with SAF61, re-emergence of PrPSc following removal of txt | Decreases half-life of PrPC, antibodies act on PrPC or PrPSc isoforms, antibodies prevent PrPC-PrPSc interaction | 22L | 178 |
| SAF61 | Monoclonal Ab, recognizes residues 144–152 (HuPrP) | Neuronal cell culture | Dose-dependent decrease of PrPSc levels, increased inhibition of PrPSc when used in conjunction with SAF34, no re-emergence ofPrPSc following removal oftxt | Decreases half-life of PrPC, antibodies act on PrPC or PrPSc isoforms, antibodies prevent PrPC-PrPSc interaction, increase clearance/degradation of PrPC, modulate cellular trafficking of PrP, induce conformational change exposing PrP to protease attacks | 22L | 178 |
| PrioV3 | Camelid anti-PrP antibody (residues between 171 and 190, YYR motif) | Neuronal cell culture, endo-thelial cells, mouse | Crosses BBB in vitro and in vivo, abrogates PrPSc replication in cells, permanent depletion of PrPSc in cells, marked inhibition ofPrPSc accumulation in spleen | Alters PrPC expression (direct neutralizing effect on PrPC/PrPSc), could block PrPC incorporation into infectious PrPSc | RML | 179,180 |
| 44B1 | Monoclonal antibody | Neuronal cell culture | Rapidly and significantly decreased PrPSc levels | Retains PrPC on cell surface as an antigen-antibody complex, interferes with internalization and trafficking of PrPC to endocytic compartments | 22L | 139 |
| POM2 | Anti-PrP antibody to flexible tail of PrPC | Mouse COCS | Prevented prion-mediated neurodegeneration but prion titer was not decreased | Induces shift in distribution ofPrPSc moieties without affecting overall quantity | RML | 181 |
| LD7, JZ107 | Phenethyl piperidine | Neuronal cell culture | Reduced PrPSc by 50%, permanently cured RML-infected cells (1 month exposure), protects against dendritic spine loss in presence of PrPSc | Might interact with PrPC in a cellular context, perhaps in conjunction with other cell-surface receptors, may interact with PrPC substrate | RML, 22L | 182 |
| Alprenolol hydrochloride | B-adrenergic blocker | Neuronal cell culture, mouse | Reduced levels of PrPSc in cells and brains of infected mice, inhibited PrPSc accumulation and spongiform changes but did not prolong survival | May inhibit via interaction with PrPC | Fukuoka-1 | 183 |
| R12, R24, R12-A-R12 | RNA aptamer | Neuronal cell culture | Significantly reduces PrPSc levels in cell culture | Tightly binds to and stabilizes PrPC, blocking conversion to PrPSc | Fukuoka-1 | 184,185 |
| DE10, DC2, EB8, EF2 | Monoclonal antibodies to N-terminal region of PrPSc | Neuronal cell culture | Promoted complete clearance ofPrPSc in cell culture | Binds to a region on PrPC that PrPSc also binds, blocking PrPC-PrPSc interaction | RML | 186 |
| PrP C /PrP Sc | ||||||
| Chlorpromazine (CPZ) | Phenothiazine | Neuronal cell culture | No to modest effect on PrPSc accumulation, inhibits prion replication in cells but not in vitro | Decreases PrPC at cell surface by inhibiting clathrin-mediated endocytosis, redistributes PrPSc to late endosome-lysosome | sheep scrapie 127S, Sc237, Type 1 CJD, RML, 22L | 129,139,187 |
| Quinacrine | Lysosomotropic agent | Neuronal cell culture, mouse, humans | Inhibits cell growth at concentrations greater than 2.0 μM, no prolongation effect in mice and humans, may lead to drug resistance | Competitive inhibition of GAG-PrP interaction, unfolding of PrP-res, destablilization of PrP-res conformation, induces conformational change that disfavors PrPSc conformation | Scrapie, 263K, vCJD, sCJD, iatrogenic CJD, genetic CJD | 140,188–193 |
| E-64d | Cysteine protease inhibitor | Neuronal cell culture, mouse | No toxicity to cell growth at [ ] up to 100 μM, no prolongation effect | Competitive inhibition of GAG-PrP interaction, unfolding of PrP-res, destabilization of PrP-res conformation | Scrapie, 263K | 140,190 |
| MS-8209 | Amphotericin B derivative | Neuronal cell culture, mouse, hamster | Prolongs incubation period (dose- and timing dependent), delays onset of PrP-res/GFAP accumulation, vacuolation, spongiosis, and astrogliosis in brain, variable efficacy across prion species | Potentially affects conversion, may interact with astrocyte lysosomal system and limit propagation of PrP at inoculation site | Sheep scrapie 127S, 139A, Sc237 (weaker with 139H), type 1 CJD, C506M3 (similar to ME7), 263K | 129,194–200 |
| 31C6 | Monoclonal anti-PrP antibody (residues 143–149), IgG1 | Neuronal cell culture, mouse | Reduces PrPSc in cell culture (dose-dependent), no re-emergence following txt, mAb txt at 120 dpi increased survival (not statistically significant), slowed weight loss, disease progression, and accumulation ofPrPSc in brain | Direct inhibition of PrPC-PrPSc interaction by occupying binding domains, could interfere with PrPC metabolism by retaining PrPC on cell surface | Chandler (139A), RML | 176,201 |
| Doxycycline | Antibiotic | Neuronal cell culture, human | Variably affected PrP-res accumulation in culture, variable prolongation of survival, reduction in widespread & severe lesions (early txt) | Destabilize abnormal PrP, could operate at cell level by modulating formation of PrP aggregates | CJD (sporadic and genetic–E200K or V210I), sCJDMM1, sCJDVV2a, vCJDMM2b, iCJDMM1 | 202–206 |
| Other | ||||||
| GSK2606414 | Protein kinase RNA-like ER kinase (PERK) inhibitor | Mouse | Oral treatment reversed cognitive deficits and prevented clinical disease, effective pre-and post-symptomatic, halted progression in spongiform degeneration, protected from neuronal loss | Prevents activation of UPR branch that mediates prion neurotoxicity by inhibiting PERK | RML | 207 |
Cellular redistribution of PrPSc can inhibit prion formation. Chlorpromazine (CPZ), an antipsychotic, redistributes PrPSc from the early endosomal/endocytic recycling pathway to the late endosomal/lysosomal pathway resulting in an inhibition of prion conversion in prion-infected N2a cells; however, CPZ was ineffective in vivo.139 A cholesterol synthesis inhibitor, U18666A, can redistribute PrPSc from the early endosomal/endocytic recycling pathway to the late endosomal/lysosomal pathway, causing an increase in PrPSc degradation that corresponds to a decrease in total PrPSc in prion-infected N2a cells.139
Increasing degradation of PrPSc can ameliorate prion infection (Fig. 1, step 6). The tyrosine kinase inhibitor STI571 decreases the half-life of PrPSc in prion-infected ScN2a cells via interaction with the tyrosine kinase c-Abl without affecting the cellular location or trafficking of PrPSc.208 Several drugs increase degradation of PrPSc by inducing autophagy. A-12 and A-14, derivatives of the antitumor drug celecoxib, can reduce or eliminate PrPSc in prion-infected neuronal cells lines by stimulating autophagy.209 Lithium, which is used to treat depression, reduces PrPSc in prion-infected neuronal and non-neuronal cells cultures by inducing autophagy.210
Pentosan polysulfate (PPS), a cysteine protease inhibitor, induces a reduction in prion conversion by causing fragmentation of PrPSc at the cell surface or utilizing endogenous glycosaminoglycans (GAGs) to competitively interfere with the binding of PrPC to PrPSc.139,140 Treatment of prion-infected mice with PPS decreased PrPSc accumulation and prolonged survival compared to untreated controls even when administered after the onset of PrPSc deposition in the CNS.139,140 While effective in rodents, treatment of human prion diseases with PPS has varied results. In some patients treated with intravenous PPS, there is a significant increase in survival compared with untreated patients141–143; however, there are several cases indicating treatment is not effective at extending survival or ameliorating clinical features of disease.144–146
Treatment of prion disease with heterologous prion proteins (HetPrP) can interfere with the formation of PrPSc in both Sc+MNB cells and in vivo.148–151 Treatment of RML-infected mice with hamster HetPrP significantly delayed onset of clinical symptoms and significantly decreased PrPSc accumulation in brain and spleen.150 Mechanistically, it is hypothesized that HetPrP either incorporates itself into a growing PrPSc aggregate and, unable to be converted to PrPSc by the host species PrPSc, hinders conversion or binds directly to the conversion site on PrPSc blocking the PrPC conversion site.148 Overall, drugs that target PrPSc that are effective in prion-infected cell cultures and animals are ineffective in human clinical trials. The lack of efficacy in human clinical trials could be partly due to timing of treatment initiation. In prion-infected rodents, anti-prion drugs administered prior to or shortly after prion infection have the highest efficacy, with administration at the onset of clinical disease being less effective. In human prion disease, treatment is typically not begun until after onset of clinical signs, decreasing treatment efficacy.211 In an attempt to overcome this, an ongoing study is treating subjects at risk for developing fatal familial insomnia with doxycycline.212
4.2. Strain-specific efficacy of anti-prion treatments
Treatment of prion-infected animals or cell cultures with anti-prion drugs can lead to the emergence of drug resistant prion strains. Swainsonine inhibits the processing of asparagine-linked glycoproteins by impeding the action of Golgi α-mannosidase II as well as lysosomal mannosidase.213–215 Treatment of prion-infected neuroblastoma-derived R33 cells with swainsonine can result in the emergence of swainsonine-resistant prions.216 Importantly, PrPSc from swainsonine-resistant and swainsonine-sensitive prions from cell culture have different PrPSc conformational stabilities suggesting they are distinct prion strains.216,217 Treatment of prion-infected cells with swainsonine selected for a drug resistant substrain, but when the inhibitor is removed, the susceptible substrain reverted to a drug sensitive population in cell culture.218 Additionally, passage of swainsonine-resistant RML prions from AMO10 to PK1 cells in the presence of swainsonine resulted in the selection of swainsonine-dependent prion variants with an increased efficiency to propagate in the presence of the drug.219 However, passaging the swainsonine-dependent prions in the absence of drug resulted in reversion to a swainsonine-sensitive prion population in cells.219
RML-infected ScN2a cells treated with the 2-aminothiazole IND24 resulted in the emergence of an IND24-resistant prion strain (RML [IND24]) that had a different host cell range, biochemical and neurological features compared to RML-infected mice treated with vehicle (RML [V]).152 In addition to IND24, this drug-resistant prion strain was also resistant to the anti-prion drugs Compound B and quinacrine.152 This suggests that the mechanism of resistance between the three anti-prion compounds is similar. Susceptibility of the RML[IND24] strain to IND24 could be restored when passaged in the absence of IND24 resulting in the reversion of the RML[IND24] phenotype to the RML[V] phenotype in mice.152
The mechanism underlying the emergence of drug-resistant prion strains is not known. Anti-prion treatments can be strain specific.152,157 One hypothesis is that compounds effective against the most predominant, but not all, strains in a mixture can allow for the emergence of a drug resistant strain. Prion strains are thought to exist as a dynamic mixture of substrains, or mutant spectra.220,221 In this scenario, the dominant strain suppresses the emergence of substrains and, once the dominant strain is removed by the anti-prion drug, the suppressive effect is diminished allowing for emergence of a preexisting drug resistant strain. Studies of strain interference have established the conditions and mechanism of how a dominant strain can suppress the emergence of a minor strain consistent with this hypothesis.121,123,221–223 Selective removal of a dominant strain from a mixture using strain-specific differences in the stability of PrPSc can allow for the emergence of a highly pathogenic substrain from a mixture, providing additional support for this hypothesis.224 Alternatively, it is possible that anti-prion compounds directly interact with PrPSc altering its conformation (i.e., mutation) and therefore strain properties. Anti-prion compounds can bind to PrPSc consistent with this hypothesis, however, RML[IND24] remained resistant to IND24 for up to 20 passages in CAD5 cells in the absence of IND24 suggesting that IND24 interaction with PrPSc is not required for the resistant phenotype.152 Overall, it is unclear if the emergence of drug resistant prion strains is due to selection of a preexisting mutant strain from the mutant spectra and/or if the anti-prion drug acts as a prion mutagen to generate the drug resistant strain.
Maintenance of a drug resistant phenotype may have a high fitness cost. Passage of drug resistant prion strains in the absence of the anti-prion drug can result in rapid reversion to the original drug sensitive strain. This suggests that the drug resistant prion strain has relatively poor fitness for the host. In the absence of drug, the mutant spectra that arises from prion conversion produces mutants that have increased fitness for the host. This observation raises several questions. First, reversion of the drug resistant strain in the drug-free environment leads to the emergence of the original drug sensitive strain. It is unclear why the drug resistant strain would revert to the parent strain as opposed to another strain if generation of the mutant spectra is random. This suggests that the drug resistant strain may contain a “memory” that favors reversion to the original strain. Second, treatment of a host with an anti-prion drug can extend the incubation period of disease without the emergence of a drug resistant strain. The mechanism behind this observation is unknown. It is possible that certain strains can still retain sufficient suppressive effect of substrains during anti-prion treatment. The relative distribution of the mutant spectra in any given isolate may differ depending on its passage history. For example, biologically cloned strains may have a more limited mutant spectra compared to uncloned or field isolates. Finally, highly stable class I strains may have a more limited mutant spectra compared to unstable class III strains.225 Overall, very little is known about how the interaction between the host and the strain influences prion strain fitness.
The emergence of drug resistant prions is a significant barrier to the development of effective treatments of prion diseases that target the prion form of the protein. Treatments that reduce or eliminate the abundance or the ability of the conversion template to form the prion form of the protein may offer an alternative strategy for anti-prion therapies.
4.3. Treatments that target PrPC
Prion conversion is dependent on the presence of the template of conversion (Fig. 1, step 2). Genetic ablation of PrP in mice results in complete resistance to prion infection with little known detriment to development or function.226,227 Similarly, ablation of PrPC in cattle does not result in major functional or immunological deficits and tissue from these animals does not support prion conversion.228 In a conditional PrPC knockout model, transgenic mice express PrPC until approximately 12 weeks of age before undergoing gene-directed depletion of PrPC.229 When these mice are challenged with RML prions at 3–4 weeks of age, prion infection was established, but the depletion of PrPC at 12 weeks halted progression to the clinical phase of prion disease and reversed spongiosis.229 Overall, these studies provide evidence that elimination of PrPC, even after the establishment of prion infection, has potential as a therapeutic strategy.
Antisense oligonucleotides (ASOs) can modulate expression of PrPC. Sequence-specific ASOs lower levels of PrPC and PrPSc in ScN2a cells.165 When sequence specific ASOs were administered to mice via either i.p. or intracerebroventricular (ICV) routes, ASOs were well tolerated and resulted in a decrease in Prnp mRNA levels in the brain compared to PBS negative controls.165,166 When ASOs were administered prophylactically or at time of inoculation with the RML prion strain, there was diminished PrPC expression and a corresponding reduction in PrPSc in the brains of ASO-treated mice compared to PBS controls. Reduction of PrPSc levels correlated to a prolongation of the incubation period of disease, but animals ultimately succumbed to prion disease.165,166 When ASOs are administered just prior to the onset of clinical signs of disease, this resulted in many of the animals rapidly succumbing to the toxic effect of ASO administration.166 Encouragingly, the mice that survived the ASO treatment had a delayed onset of clinical disease as well as an extended clinical duration of disease that was three times longer than saline treated controls.166 Overall, ASOs can delay the onset of prion disease whether given prophylactically or after the establishment of infection and, one issues of toxicity are addressed, may be an attractive method of depressing PrPC expression to treat prion diseases.
Treatments that alter the expression of PrPC on the cell surface can reduce prion conversion. For example, a group of chalcones and oxadiazoles can directly bind to PrPC and decrease its cell surface expression arresting PrPC trafficking to the endoplasmic reticulum resulting in a decrease in PrPSc abundance and aggregation in prion-infected N2a cells.167,168 Alternatively, some of these compounds prevent conversion of PrPC to PrPSc by binding to and stabilizing PrPC or directly binding to PrPC at the seeding surface.168,184,185
Alteration of post translational processing of PrPC can inhibit prion formation. PrPC can undergo either α- or β-endoproteolytic cleavage resulting in C1 and C2 fragments, respectively.230 The C2 amino acid sequence approximately aligns with that of the PrPSc protease resistant core, and has been found in abundance in prion infected brains.231–237 The production of proteolytic cleavage products with varying pathogenic propensities is similar to what is observed of amyloid precursor protein, where APP cleavage is central to the development of AD.238 Processing of APP by the enzymes β-secretase and γ-secretase leads to production of the peptides Aβ-40 and Aβ-42. While Aβ-40 has neuroprotective qualities,239,240 Aβ-42 has been implicated in AD pathogenesis.241,242 Endopeptidase cleavage of α-synuclein results in aggregation and toxicity that ultimately causes PD.243 Pharmacological alteration of cellular proteolytic cleavage of the precursor proteins is an attractive druggable target. For example, a drug that preferentially inhibits the production of the C2 fragment would be a viable anti-prion treatment as the unfavorable PrPC cleavage product (C2) would be sequestered while the C1 fragment would be preserved and able to continue to perform a protective function.244 The metalloprotease, ADAM10, plays a role PrPC shedding from the cell surface, but has also been suggested to contribute the α-cleavage of PrPC.232,245–249 Inhibition of ADAM10 resulted in an earlier onset of prion disease and slower spread to other brain regions in mice as a result of PrPC sequestration, suggesting ADAM10 may cleave newly formed PrPSc from the surface of infected cells.247,250 These data suggest that a therapeutic that increases PrPC shedding, instead of inhibiting post-translational processing of PrPC, may be more effective at dampening disease progression.
Monoclonal antibodies (mAb) directed against PrPC can inhibit prion conversion. Several mAbs (D18, 6H4, ICSM 18, 31C6, etc.) are able to abrogate prion conversion and cure prion-infected N2a cells.171,172,176–180,251 In prion-infected N2a cells, the inhibition of prion conversion depends on the binding affinity between the mAb and PrPC at the cell surface, with the most potent inhibitors often binding the ɑ-helical domain of PrP.174,176,177 Inhibition of PrPSc formation by antibodies in cell culture has been proposed to occur via several mechanisms. The antibodies D18 and 6D11 can abolish prion conversion by binding to PrPC at the cell surface and hindering PrPSc-templated conversion.171,177 Similarly, treatment of prion-infected N2a cells with mAb 44B1 retains PrPC at the cell surface in an antigen–antibody complex rendering it unavailable for conversion resulting in a significant decrease of PrPSc levels.139 A group of N-terminal monoclonal antibodies (DE10, DC2, EB8, and EF2) are hypothesized to directly bind to a region on PrPC that has been shown to tightly bind PrPSc, effectively blocking the PrPC-PrPSc interaction.186 Antibodies that disrupt PrPC metabolism or trafficking either by preventing internalization of PrPC or reducing the half-life of PrPC, result in a decrease in PrPSc in cell culture models.176,178,251 Although the aforementioned antibodies were reported to have no effect on cell growth or induce cellular toxicity, there are reports of anti-PrP antibodies (e.g., POM1) that are neurotoxic.181,252–255 Anti-PrP antibodies that are toxic tend to bind epitopes in the globular domain of PrP and may trigger similar pathogenic cell signaling pathways as prion disease.181,254
Monoclonal antibodies directed against PrPC can alter prion pathogenesis and delay the onset of clinical signs of prion infection. ICSM 18, when administered to mice either intraperitoneal (i.p.) or i.c. 7 or 30 days after i.p. inoculation with RML was found to decrease PrPSc levels in the spleen and delay the onset of clinical signs of prion infection compared to untreated controls.175 However, treatment was ineffective when administered at the onset of clinical signs or if mice were i.c. inoculated with RML.175 Treatment of mice infected with the Chandler strain of murine-adapted scrapie with mAb 31C6 120 dpi led to an increase in the incubation period, a decrease in PrPSc levels, and delayed progression of neuropathological lesions in the cerebellum.201 Creating PrP0/0 mice expressing the 6H4 antibody as a transgene (Prnp0/0-6H4μ) led to these mice producing anti-PrP titers in sera.173 Crossbreeding of Prnp0/0-6H4μ mice with C57BL/6 mice led to re-introduction of one or two PrP alleles, generating Prnp+/0-6H4μ and Prnp+/+-6H4μ mice, respectively, which developed anti-PrP serum levels.173 Following intraperitoneal inoculation of the RML strain of scrapie, Prnp+/0 6H4μ mice lacked deposition of PrPSc in the spleen or the brain, whereas Prnp+/0 mice had deposition in both tissues.173 Expression of the 6H4 antibody as a transgene drastically delayed prion pathogenesis in mice.173 Overall, anti-PrP antibodies can reduce or eliminate PrPSc formation in cells and animals and provides proof of principle of the efficacy of this approach.
Vaccines that result in host induced production of antibodies that recognize PrPSc or PrPC have been attempted. Vaccination of mice or white-tailed deer with attenuated Salmonella expressing PrP resulted in significant prolongation of onset of clinical disease in the vaccinated group with complete protection in a portion of the animals as determined by the absence of PrPSc deposition; however, these studies involved a relatively small sample size.256,257 Vaccines consisting of peptides derived from the primary amino acid sequence of the prion protein resulted in a humoral immune response to all peptides and delayed the onset of clinical signs of prion infection compared to control prion-infected mice.258 Other studies have focused on protein epitopes specific to PrPSc with strong induction of antibody responses after vaccination of mice, sheep, and white-tailed deer, strengthening the possibility of an effective PrPSc-specific vaccine.259–262 In contrast to the previous studies, vaccinating elk to induce a antibody response against PrPSc resulted in a significant shortening of incubation period of CWD.263 The acceleration in disease onset could be attributed to antibody-mediated misfolding of PrPC or by promoting the uptake of PrPSc in the gut.263 Since PrPC is a host-encoded protein, development of an effective vaccine directed immune response is impeded by host tolerance. While many approaches have aimed to overcome tolerance, there are negative consequences of inducing an antibody response to a host-encoded cellular protein. The effects of antibodies directed at the globular domain of PrPC results in varying degrees of neurotoxicity while other PrPC-specific antibodies lead to apoptosis and improper signaling cascades.181,253,264 Vaccination strategies have successfully induced strong humoral and mucosal immune responses in multiple animal models; however, more work is required to evaluate the most effective vaccination strategy to prevent disease onset while avoiding toxicity.
5. Future prospects
Development of ultra-sensitive methods of PrPSc detection have greatly improved and may lead to routine detection of prion infection in asymptomatic individuals. Protein misfolding cyclic amplification (PMCA) and real time quaking induced conversion (RT-QuIC) can detect a single infectious prion particle.265,266 Both in vitro methods can detect PrPSc in urine, feces, blood, saliva, and cerebrospinal fluid (CSF).267–272 PMCA detects PrPSc from vCJD patient blood and urine samples, while RT-QuIC is currently being used for the diagnosis of sCJD from CSF and nasal mucosa swab samples.267,269–271,273 Advancements in in vitro techniques have enabled reliable preclinical detection in experimental prion disease. PMCA can detect prions in the blood or skin from pre-clinical prion-infected mice and hamsters.274–276 The sensitivity of RT-QuIC can detect prions in the blood of prion-infected preclinical hamsters and deer at timepoints as early as 5 days and 1 month post inoculation, respectively.277,278 Like PrPSc, Aβ, tau, and α-synuclein are present in biological fluids making these in vitro techniques promising for preclinical diagnosis.279–283 Overall, preclinical diagnosis of prion diseases will provide a larger window for therapeutic treatments and consequently a greater likelihood of success. Challenging this is the presence of multiple prion strains. Strain-specific efficacy of anti-prion drugs or vaccines can result in emergence of resistant strains that may be more pathogenic or have a different host range compared to the original dominant strain. These challenges are reminiscent of those faced by bacterial, fungal, and viral drug development and a successful anti-prion therapy may require a multidrug approach. The unique nature of prions, however, provides an additional therapeutic target, namely the precursor protein. Treatments that target the prion precursor protein can inhibit prion formation and may reverse pathological changes. A combinational approach that treats the prion form and the precursor protein will likely be needed to effectively treat the broad spectrum of prion diseases.
References
- 1.Prusiner SB. Novel proteinaceous infectious particles cause scrapie. Science. 1982;216 (4542):136–144. [DOI] [PubMed] [Google Scholar]
- 2.Horiuchi M, Caughey B. Specific binding of normal prion protein to the scrapie form via a localized domain initiates its conversion to the protease-resistant state. EMBO J. 1999;18(12):3193–3203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bessen RA, Kocisko DA, Raymond GJ, Nandan S, Lansbury PT, Caughey B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature. 1995;375(6533):698–700. [DOI] [PubMed] [Google Scholar]
- 4.Kocisko DA, Come JH, Priola SA, et al. Cell-free formation of protease-resistant prion protein. Nature. 1994;370(6489):471–474. [DOI] [PubMed] [Google Scholar]
- 5.Du Z, Park KW, Yu H, Fan Q, Li L. Newly identified prion linked to the chromatin-remodeling factor Swi1 in Saccharomyces cerevisiae. Nat Genet. 2008;40(4):460–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jarosz DF, Lancaster AK, Brown JCS, Lindquist S. An evolutionarily conserved prion-like element converts wild fungi from metabolic specialists to generalists. Cell. 2014;158(5):1072–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Masison DC, Wickner RB. Prion-inducing domain of yeast Ure2p and protease resistance of Ure2p in prion-containing cells. Science. 1995;270(5233):93–95. [DOI] [PubMed] [Google Scholar]
- 8.Wickner RB. [URE3] as an altered URE2 protein: evidence for a prion analog in Saccharomyces cerevisiae. Science. 1994;264(5158):566–569. [DOI] [PubMed] [Google Scholar]
- 9.Uptain SM, Sawicki GJ, Caughey B, Lindquist S. Strains of [PSI(+)] are distinguished by their efficiencies of prion-mediated conformational conversion. EMBO J. 2001;20(22):6236–6245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chernoff Y, Lindquist S, Ono B, Inge-Vechtomov S, Liebman S. Role of the chaperone protein Hsp104 in propagation of the yeast prion-like factor [psi+]. Science. 1995;268(5212):880–884. [DOI] [PubMed] [Google Scholar]
- 11.Wickner RB, Bezsonov E, Bateman DA. Normal levels of the antiprion proteins Btn2 and Cur1 cure most newly formed [URE3] prion variants. Proc Natl Acad Sci U S A. 2014;111(26):E2711–E2720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Coustou V, Deleu C, Saupe S, Begueret J. The protein product of the het-s heterokaryon incompatibility gene of the fungus Podospora anserina behaves as a prion analog. Proc Natl Acad Sci U S A. 1997;94(18):9773–9778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Maddelein ML, Dos Reis S, Duvezin-Caubet S, Coulary-Salin B, Saupe SJ. Amyloid aggregates of the HET-s prion protein are infectious. Proc Natl Acad Sci U S A. 2002;99(11):7402–7407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Saupe SJ. The [Het-s] prion of Podospora anserina and its role in heterokaryon incompatibility. Semin Cell Dev Biol. 2011;22(5):460–468. [DOI] [PubMed] [Google Scholar]
- 15.Fioriti L, Myers C, Huang YY, et al. The persistence of hippocampal-based memory requires protein synthesis mediated by the prion-like protein CPEB3. Neuron. 2015;86(6):1433–1448. [DOI] [PubMed] [Google Scholar]
- 16.Gilks N, Kedersha N, Ayodele M, et al. Stress granule assembly is mediated by prion-like aggregation of TIA-1. Mol Biol Cell. 2004;15(12):5383–5398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hou F, Sun L, Zheng H, Skaug B, Jiang QX, Chen ZJ. MAVS forms functional prion-like aggregates to activate and propagate antiviral innate immune response. Cell. 2011;146(3):448–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Li X, Rayman JB, Kandel ER, Derkatch IL. Functional role of Tia1/Pub1 and Sup35 prion domains: directing protein synthesis machinery to the tubulin cytoskeleton. Mol Cell. 2014;55(2):305–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stephan JS, Fioriti L, Lamba N, et al. The CPEB3 protein is a functional prion that interacts with the actin cytoskeleton. Cell Rep. 2015;11(11):1772–1785. [DOI] [PubMed] [Google Scholar]
- 20.Bolton DC, Mckinley MP, Prusiner SB. Identification of a protein that purifies with the scrapie prion. Science. 1982;218(4579):1309–1311. [DOI] [PubMed] [Google Scholar]
- 21.Baeten LA, Powers BE, Jewell JE, Spraker TR, Miller MW. A natural case of chronic wasting disease in a free-ranging moose (Alces alces shirasi). J Wildl Dis. 2007; 43(2):309–314. [DOI] [PubMed] [Google Scholar]
- 22.Benestad SL, Mitchell G, Simmons M, Ytrehus B, Vikoren T. First case of chronic wasting disease in Europe in a Norwegian free-ranging reindeer. Vet Res. 2016; 47(1):88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Williams ES, Young S. Chronic wasting disease of captive mule deer: a spongiform encephalopathy. J Wildl Dis. 1980;16(1):89–98. [DOI] [PubMed] [Google Scholar]
- 24.Babelhadj B, Di Bari MA, Pirisinu L, et al. Prion disease in dromedary camels Algeria. Emerg Infect Dis. 2018;24(6):1029–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Imran M, Mahmood S. An overview of human prion diseases. Virol J. 2011;8:559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Duffy P, Wolf J, Collins G, DeVoe AG, Streeten B, Cowen D. Letter: possible person-to-person transmission of Creutzfeldt-Jakob disease. N Engl J Med. 1974;290(12):692–693. [PubMed] [Google Scholar]
- 27.Heckmann JG, Lang CJ, Petruch F, et al. Transmission of Creutzfeldt-Jakob disease via a corneal transplant. J Neurol Neurosurg Psychiatry. 1997;63(3):388–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koch TK, Berg BO, De Armond SJ, Gravina RF. Creutzfeldt-Jakob disease in a young adult with idiopathic hypopituitarism. Possible relation to the administration of cadaveric human growth hormone. N Engl J Med. 1985;313(12):731–733. [DOI] [PubMed] [Google Scholar]
- 29.Thadani V, Penar PL, Partington J, et al. Creutzfeldt-Jakob disease probably acquired from a cadaveric dura mater graft. Case report. J Neurosurg. 1988;69(5):766–769. [DOI] [PubMed] [Google Scholar]
- 30.Frost B, Jacks RL, Diamond MI. Propagation of tau misfolding from the outside to the inside of a cell. J Biol Chem. 2009;284(19):12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kfoury N, Holmes BB, Jiang H, Holtzman DM, Diamond MI. Trans-cellular propagation of Tau aggregation by fibrillar species. J Biol Chem. 2012;287(23):19440–19451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Langer F, Eisele YS, Fritschi SK, Staufenbiel M, Walker LC, Jucker M. Soluble Aβ seeds are potent inducers of cerebral β-amyloid deposition. J Neurosci. 2011;31:14488–14495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Eisele YS, Obermuller U, Heilbronner G, et al. Peripherally applied Abeta-containing inoculates induce cerebral beta-amyloidosis. Science. 2010;330(6006):980–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Clavaguera F, Bolmont T, Crowther RA, et al. Transmission and spreading of tauopathy in transgenic mouse brain. Nat Cell Biol. 2009;11(7):909–913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Desplats P, Lee HJ, Bae EJ, et al. Inclusion formation and neuronal cell death through neuron-to-neuron transmission of alpha-synuclein. Proc Natl Acad Sci U S A. 2009; 106(31):13010–13015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fehlinger A, Wolf H, Hossinger A, et al. Prion strains depend on different endocytic routes for productive infection. Sci Rep. 2017;7(1):6923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Freundt EC, Maynard N, Clancy EK, et al. Neuron-to-neuron transmission of alpha-synuclein fibrils through axonal transport. Ann Neurol. 2012;72(4):517–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Guo JL, Narasimhan S, Changolkar L, et al. Unique pathological tau conformers from Alzheimer’s brains transmit tau pathology in nontransgenic mice. J Exp Med. 2016;213(12):2635–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holmes BB, DeVos SL, Kfoury N, et al. Heparan sulfate proteoglycans mediate internalization and propagation of specific proteopathic seeds. Proc Natl Acad Sci U S A. 2013;110(33):E3138–E3147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chandler RL. Encephalopathy in mice produced by inoculation with scrapie brain material. Lancet. 1961;1(7191):1378–1379. [DOI] [PubMed] [Google Scholar]
- 41.Bolmont T, Clavaguera F, Meyer-Luehmann M, et al. Induction of tau pathology by intracerebral infusion of amyloid-beta -containing brain extract and by amyloid-beta deposition in APP × Tau transgenic mice. Am J Pathol. 2007;171(6):2012–2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Luk KC, Kehm VM, Zhang B, O’Brien P, Trojanowski JQ, Lee VM. Intracerebral inoculation of pathological alpha-synuclein initiates a rapidly progressive neurodegenerative alpha-synucleinopathy in mice. J Exp Med. 2012;209(5):975–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Watts JC, Giles K, Oehler A, et al. Transmission of multiple system atrophy prions to transgenic mice. Proc Natl Acad Sci U S A. 2013;110(48):19555–19560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Watts JC, Condello C, Stöhr J, et al. Serial propagation of distinct strains of Aβ prions from Alzheimer’s disease patients. Proc Natl Acad Sci U S A. 2014;111: 10323–10328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.He Z,Guo JL,McBride JD, et al. Amyloid-β plaques enhanceAlzheimer’s braintau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med. 2018;24:29–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Woerman AL, Oehler A, Kazmi SA, et al. Multiple system atrophy prions retain strain specificity after serial propagation in two different Tg(SNCA*A53T) mouse lines. Acta Neuropathol. 2019;137(3):437–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.O’Meara ES, Kukull WA, Schellenberg GD, et al. Alzheimer’s disease and history of blood transfusion by apolipoprotein-E genotype. Neuroepidemiology. 1997;16(2):86–93. [DOI] [PubMed] [Google Scholar]
- 48.Bohnen NI, Warner MA, Kokmen E, Beard CM, Kurland LT. Prior blood transfusions and Alzheimer’s disease. Neurology. 1994;44(6):1159. [DOI] [PubMed] [Google Scholar]
- 49.Brown P, Gibbs CJ Jr, Rodgers-Johnson P, et al. Human spongiform encephalopathy: the National Institutes of Health series of 300 cases of experimentally transmitted disease. Ann Neurol. 1994;35(5):513–529. [DOI] [PubMed] [Google Scholar]
- 50.Irwin DJ, Abrams JY, Schonberger LB, et al. Evaluation of potential infectivity of Alzheimer and Parkinson disease proteins in recipients of cadaver-derived human growth hormone. JAMA Neurol. 2013;70(4):462–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jaunmuktane Z, Mead S, Ellis M, et al. Evidence for human transmission of amyloid-beta pathology and cerebral amyloid angiopathy. Nature. 2015;525(7568):247–250. [DOI] [PubMed] [Google Scholar]
- 52.Purro SA, Farrow MA, Linehan J, et al. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature. 2018;564:415–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bartz JC. Prion strain diversity. Cold Spring Harb Perspect Med. 2016;6(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Chien P, Weissman JS. Conformational diversity in a yeast prion dictates its seeding specificity. Nature. 2001;410(6825):223–227. [DOI] [PubMed] [Google Scholar]
- 55.Verges KJ, Smith MH, Toyama BH, Weissman JS. Strain conformation, primary structure and the propagation of the yeast prion [PSI+]. Nat Struct Mol Biol. 2011;18(4): 493–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fraser H, Dickinson AG. Distribution of experimentally induced scrapie lesions in the brain. Nature. 1967;216(5122):1310–1311. [DOI] [PubMed] [Google Scholar]
- 57.Brachmann A, Baxa U, Wickner RB. Prion generation in vitro: amyloid of Ure2p is infectious. EMBO J. 2005;24(17):3082–3092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Derkatch IL, Chernoff YO, Kushnirov VV, Inge-Vechtomov SG, Liebman SW. Genesis and variability of [PSI] prion factors in Saccharomyces cerevisiae. Genetics. 1996;144(4):1375–1386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Schlumpberger M, Prusiner SB, Herskowitz I. Induction of distinct [URE3] yeast prion strains. Mol Cell Biol. 2001;21(20):7035–7046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dickinson AG, Meikle VM, Fraser H. Identification of a gene which controls the incubation period of some strains of scrapie agent in mice. J Comp Pathol. 1968;78(3): 293–299. [DOI] [PubMed] [Google Scholar]
- 61.Fraser H, Dickinson AG. The sequential development of the brain lesions of scrapie in three strains of mice. J Comp Pathol. 1968;78:301–311. [DOI] [PubMed] [Google Scholar]
- 62.Bousset L, Pieri L, Ruiz-Arlandis G, et al. Structural and functional characterization of two alpha-synuclein strains. Nat Commun. 2013;4:2575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Guo JL, Covell DJ, Daniels JP, et al. Distinct α-synuclein strains differentially promote tau inclusions in neurons. Cell. 2013;154:103–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Narasimhan S, Guo JL, Changolkar L, et al. Pathological tau strains from human brains recapitulate the diversity of tauopathies in nontransgenic mouse brain. J Neurosci. 2017;37(47):11406–11423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Tanaka M, Chien P, Naber N, Cooke R, Weissman JS. Conformational variations in an infectious protein determine prion strain differences. Nature. 2004;428(6980):323–328. [DOI] [PubMed] [Google Scholar]
- 66.McGlinchey RP, Kryndushkin D, Wickner RB. Suicidal [PSI+] is a lethal yeast prion. Proc Natl Acad Sci U S A. 2011;108(13):5337–5341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Edskes HK, McCann LM, Hebert AM, Wickner RB. Prion variants and species barriers among Saccharomyces Ure2 proteins. Genetics. 2009;181(3):1159–1167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Ohhashi Y, Yamaguchi Y, Kurahashi H, et al. Molecular basis for diversification of yeast prion strain conformation. Proc Natl Acad Sci U S A. 2018;115(10):2389–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tanaka M, Collins SR, Toyama BH, Weissman JS. The physical basis of how prion conformations determine strain phenotypes. Nature. 2006;442(7102):585–589. [DOI] [PubMed] [Google Scholar]
- 70.Bessen RA, Marsh RF. Distinct PrP properties suggest the molecular basis of strain variation in transmissible mink encephalopathy. J Virol. 1994;68(12):7859–7868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Safar J, Wille H, Itri V, et al. Eight prion strains have PrP(Sc) molecules with different conformations. Nat Med. 1998;4(10):1157–1165. [DOI] [PubMed] [Google Scholar]
- 72.Telling GC, Parchi P, DeArmond SJ, et al. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science. 1996;274(5295):2079–2082. [DOI] [PubMed] [Google Scholar]
- 73.Kascsak RJ, Rubenstein R, Merz PA, Carp RI, Wisniewski HM, Diringer H. Biochemical differences among scrapie-associated fibrils support the biological diversity of scrapie agents. J Gen Virol. 1985;66(pt 8):1715–1722. [DOI] [PubMed] [Google Scholar]
- 74.Bessen RA, Marsh RF. Identification of two biologically distinct strains of transmissible mink encephalopathy in hamsters. J Gen Virol. 1992;73(pt 2):329–334. [DOI] [PubMed] [Google Scholar]
- 75.Bessen RA, Marsh RF. Biochemical and physical properties of the prion protein from two strains of the transmissible mink encephalopathy agent. J Virol. 1992;66(4): 2096–2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Marsh RF, Bessen RA. Physicochemical and biological characterizations of distinct strains of the transmissible mink encephalopathy agent. Philos Trans R Soc Lond B Biol Sci. 1994;343(1306):413–414. [DOI] [PubMed] [Google Scholar]
- 77.Deleault NR, Harris BT, Rees JR, Supattapone S. Formation of native prions from minimal components in vitro. Proc Natl Acad Sci U S A. 2007;104(23):9741–9746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Wang F, Wang X, Yuan CG, Ma J. Generating a prion with bacterially expressed recombinant prion protein. Science. 2010;327(5969):1132–1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Deleault NR, Walsh DJ, Piro JR, et al. Cofactor molecules maintain infectious conformation and restrict strain properties in purified prions. Proc Natl Acad Sci U S A. 2012;109(28):E1938–E1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burke CM, Walsh DJ, Steele AD, et al. Full restoration of specific infectivity and strain properties from pure mammalian prion protein. PLoS Pathog. 2019;15(3):e1007662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Mahal SP, Baker CA, Demczyk CA, Smith EW, Julius C, Weissmann C. Prion strain discrimination in cell culture: the cell panel assay. Proc Natl Acad Sci U S A. 2007;104(52):20908–20913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Oelschlegel AM, Fallahi M, Ortiz-Umpierre S, Weissmann C. The extended cell panel assay characterizes the relationship of prion strains RML, 79A, and 139A and reveals conversion of 139A to 79A-like prions in cell culture. J Virol. 2012;86(9):5297–5303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Karapetyan YE, Saa P, Mahal SP, et al. Prion strain discrimination based on rapid in vivo amplification and analysis by the cell panel assay. PLoS One. 2009;4(5):e5730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peretz D, Scott MR, Groth D, et al. Strain-specified relative conformational stability of the scrapie prion protein. Protein Sci. 2001;10(4):854–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Cescatti M, Saverioni D, Capellari S, et al. Analysis of conformational stability of abnormal prion protein aggregates across the spectrum of Creutzfeldt-Jakob disease prions. J Virol. 2016;90(14):6244–6254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ayers JI, Schutt CR, Shikiya RA, Aguzzi A, Kincaid AE, Bartz JC. The strain-encoded relationship between PrP replication, stability and processing in neurons is predictive of the incubation period of disease. PLoS Pathog. 2011;7(3):e1001317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Legname G, Nguyen HO, Peretz D, Cohen FE, DeArmond SJ, Prusiner SB. Continuum of prion protein structures enciphers a multitude of prion isolate-specified phenotypes. Proc Natl Acad Sci U S A. 2006;103(50):19105–19110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Condello C, Lemmin T, Stöhr J, et al. Structural heterogeneity and intersubject variability of Aβ in familial and sporadic Alzheimer’s disease. Proc Natl Acad Sci U S A. 2018;115:E782–E791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nilsberth C, Westlind-Danielsson A, Eckman CB, et al. The ‘Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat Neurosci. 2001;4:887–893. [DOI] [PubMed] [Google Scholar]
- 90.Lannfelt L, Bogdanovic N, Appelgren H, et al. Amyloid precursor protein mutation causes Alzheimer’s disease in a Swedish family. Neurosci Lett. 1994;168(1–2):254–256. [DOI] [PubMed] [Google Scholar]
- 91.Mullan M, Crawford F, Axelman K, et al. A pathogenic mutation for probable Alzheimer’s disease in the APP gene at the N-terminus of beta-amyloid. Nat Genet. 1992;1(5):345–347. [DOI] [PubMed] [Google Scholar]
- 92.Philipson O, Lord A, Lalowski M, et al. The Arctic amyloid-β precursor protein (AβPP) mutation results in distinct plaques and accumulation of N- and C-truncated Aβ. Neurobiol Aging. 2012;33:1010.e1011–1010.e1013. [DOI] [PubMed] [Google Scholar]
- 93.Rasmussen J, Mahler J, Beschorner N, et al. Amyloid polymorphisms constitute distinct clouds of conformational variants in different etiological subtypes of Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017;114(49):13018–13023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Qiang W, Yau WM, Lu JX, Collinge J, Tycko R. Structural variation in amyloid-β fibrils from Alzheimer’s disease clinical subtypes. Nature. 2017;541:217–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee VM, Goedert M, Trojanowski JQ. Neurodegenerative tauopathies. Annu Rev Neurosci. 2001;24:1121–1159. [DOI] [PubMed] [Google Scholar]
- 96.Clavaguera F, Akatsu H, Fraser G, et al. Brain homogenates from human Tauopathies induce tau inclusions in mouse brain. Proc Natl Acad Sci U S A. 2013;110(23): 9535–9540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Boluda S, Iba M, Zhang B, Raible KM, Lee VM, Trojanowski JQ. Differential induction and spread of tau pathology in young PS19 tau transgenic mice following intracerebral injections of pathological tau from Alzheimer’s disease or corticobasal degeneration brains. Acta Neuropathol. 2015;129(2):221–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gibbons GS, Banks RA, Kim B, et al. GFP-mutant human tau transgenic mice develop Tauopathy following CNS injections of Alzheimer’s brain-derived pathological tau or synthetic mutant human tau fibrils. J Neurosci. 2017;37(47):11485–11494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Sanders DW, Kaufman SK, DeVos SL, et al. Distinct tau prion strains propagate in cells and mice and define different tauopathies. Neuron. 2014;82(6):1271–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kaufman SK, Sanders DW, Thomas TL, et al. Tau prion strains dictate patterns of cell pathology, progression rate, and regional vulnerability in vivo. Neuron. 2016;92(4): 796–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Peelaerts W, Bousset L, Van der Perren A, et al. α-Synuclein strains cause distinct synucleinopathies after local and systemic administration. Nature. 2015;522:340–344. [DOI] [PubMed] [Google Scholar]
- 102.Peelaerts W, Baekelandt V. α-Synuclein strains and the variable pathologies of synucleinopathies. J Neurochem. 2016;139(suppl 1):256–274. [DOI] [PubMed] [Google Scholar]
- 103.Parchi P, Castellani R, Capellari S, et al. Molecular basis of phenotypic variability in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 1996;39:767–778. [DOI] [PubMed] [Google Scholar]
- 104.Benestad SL, Sarradin P, Thu B, Schonheit J, Tranulis MA, Bratberg B. Cases of scrapie with unusual features in Norway and designation of a new type, Nor98. Vet Rec. 2003;153(7):202–208. [DOI] [PubMed] [Google Scholar]
- 105.Biacabe AG, Laplanche JL, Ryder S, Baron T. Distinct molecular phenotypes in bovine prion diseases. EMBO Rep. 2004;5(1):110–115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Woerman AL, Stohr J, Aoyagi A, et al. Propagation of prions causing synucleinopathies in cultured cells. Proc Natl Acad Sci U S A. 2015;112(35):E4949–E4958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Yamasaki TR, Holmes BB, Furman JL, et al. Parkinson’s disease and multiple system atrophy have distinct alpha-synuclein seed characteristics. J Biol Chem. 2019;294(3): 1045–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Cali I, Castellani R, Alshekhlee A, et al. Co-existence of scrapie prion protein types 1 and 2 in sporadic Creutzfeldt-Jakob disease: Its effect on the phenotype and prion-type characteristics. Brain. 2009;132(pt 10):2643–2658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Uro-Coste E, Cassard H, Simon S, et al. Beyond PrPres type 1/type 2 dichotomy in Creutzfeldt-Jakob disease. PLoS Pathog. 2008;4:e1000029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Parchi P, Giese A, Capellari S, et al. Classification of sporadic Creutzfeldt-Jakob disease based on molecular and phenotypic analysis of 300 subjects. Ann Neurol. 1999;46(2): 224–233. [PubMed] [Google Scholar]
- 111.Puoti G, Giaccone G, Rossi G, Canciani B, Bugiani O, Tagliavini F. Sporadic Creutzfeldt-Jakob disease: co-occurrence of different types of PrP(Sc) in the same brain. Neurology. 1999;53(9):2173–2176. [DOI] [PubMed] [Google Scholar]
- 112.Kovács GG, Head MW, Hegyi I, et al. Immunohistochemistry for the prion protein: comparison of different monoclonal antibodies in human prion disease subtypes. Brain Pathol. 2006;12(1):1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Haik S, Peoc’h K, Brandel JP, et al. Striking PrPsc heterogeneity in inherited prion diseases with the D178N mutation. Ann Neurol. 2004;56(6):909–910. [author reply 910–901]. [DOI] [PubMed] [Google Scholar]
- 114.Head MW, Bunn TJ, Bishop MT, et al. Prion protein heterogeneity in sporadic but not variant Creutzfeldt-Jakob disease: UK cases 1991–2002. Ann Neurol. 2004;55(6): 851–859. [DOI] [PubMed] [Google Scholar]
- 115.Lewis V, Hill AF, Klug GM, Boyd A, Masters CL, Collins SJ. Australian sporadic CJD analysis supports endogenous determinants of molecular-clinical profiles. Neurology. 2005;65(1):113–118. [DOI] [PubMed] [Google Scholar]
- 116.Schoch G, Seeger H, Bogousslavsky J, et al. Analysis of prion strains by PrPSc profiling in sporadic Creutzfeldt-Jakob disease. PLoS Med. 2006;3(2):e14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Milenkovic I, Petrov T, Kovacs GG. Patterns of hippocampal tau pathology differentiate neurodegenerative dementias. Dement Geriatr Cogn Disord. 2014;38(5–6):375–388. [DOI] [PubMed] [Google Scholar]
- 118.Gibbons GS, Banks RA, Kim B, et al. Detection of Alzheimer disease (AD)-specific tau pathology in AD and NonAD tauopathies by immunohistochemistry with novel conformation-selective tau antibodies. J Neuropathol Exp Neurol. 2018;77(3):216–228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Bartz JC, Bessen RA, McKenzie D, Marsh RF, Aiken JM. Adaptation and selection of prion protein strain conformations following interspecies transmission of transmissible mink encephalopathy. J Virol. 2000;74(12):5542–5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Dickinson AG, Outram GW. The scrapie replication-site hypothesis and its implications for pathogenesis. In: Hadlow WJ, Prusiner SB, eds. Slow transmissible diseases of the nervous system. New York: Academic Press, Inc; 1979:13–31. vol 2. [Google Scholar]
- 121.Kimberlin RH, Walker CA. Competition between strains of scrapie depends on the blocking agent being infectious. Intervirology. 1985;23(2):74–81. [DOI] [PubMed] [Google Scholar]
- 122.Taylor DM, Dickinson AG, Fraser H, Marsh RF. Evidence that transmissible mink encephalopathy agent is biologically inactive in mice. Neuropathol Appl Neurobiol. 1986;12(2):207–215. [DOI] [PubMed] [Google Scholar]
- 123.Bartz JC, Kramer ML, Sheehan MH, et al. Prion interference is due to a reduction in strain-specific PrPSc levels. J Virol. 2007;81(2):689–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Shikiya RA, Ayers JI, Schutt CR, Kincaid AE, Bartz JC. Coinfecting prion strains compete for a limiting cellular resource. J Virol. 2010;84(11):5706–5714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Eckland TE, Shikiya RA, Bartz JC. Independent amplification of co-infected long incubation period low conversion efficiency prion strains. PLoS Pathog. 2018;14(10): e1007323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Bradley ME, Edskes HK, Hong JY, Wickner RB, Liebman SW. Interactions among prions and prion “strains” in yeast. Proc Natl Acad Sci U S A. 2002;99(suppl 4):16392–16399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Caughey B, Ernst D, Race RE. Congo red inhibition of scrapie agent replication. J Virol. 1993;67(10):6270–6272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Caspi S, Halimi M, Yanai A, Sasson SB, Taraboulos A, Gabizon R. The anti-prion activity of Congo red. Putative mechanism. J Biol Chem. 1998;273(6):3484–3489. [DOI] [PubMed] [Google Scholar]
- 129.Cronier S, Beringue V, Bellon A, Peyrin JM, Laude H. Prion strain- and species-dependent effects of antiprion molecules in primary neuronal cultures. J Virol. 2007;81(24):13794–13800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Ingrosso L, Ladogana A, Pocchiari M. Congo red prolongs the incubation period in scrapie-infected hamsters. J Virol. 1995;69(1):506–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Milhavet O, Mange A, Casanova D, Lehmann S. Effect of Congo red on wild-type and mutated prion proteins in cultured cells. J Neurochem. 2000;74(1):222–230. [DOI] [PubMed] [Google Scholar]
- 132.Herrmann US, Schutz AK, Shirani H, et al. Structure-based drug design identifies polythiophenes as antiprion compounds. Sci Transl Med. 2015;7(299):299ra123. [DOI] [PubMed] [Google Scholar]
- 133.Margalith I, Suter C, Ballmer B, et al. Polythiophenes inhibit prion propagation by stabilizing prion protein (PrP) aggregates. J Biol Chem. 2012;287(23):18872–18887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Kawasaki Y, Kawagoe K, Chen CJ, Teruya K, Sakasegawa Y, Doh-ura K. Orally administered amyloidophilic compound is effective in prolonging the incubation periods of animals cerebrally infected with prion diseases in a prion strain-dependent manner. J Virol. 2007;81(23):12889–12898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lu D, Giles K, Li Z, et al. Biaryl amides and hydrazones as therapeutics for prion disease in transgenic mice. J Pharmacol Exp Ther. 2013;347(2):325–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Matthes D, Gapsys V, Griesinger C, de Groot BL. Resolving the atomistic modes of Anle138b inhibitory action on peptide oligomer formation. ACS Chem Nerosci. 2017; 8(12):2791–2808. [DOI] [PubMed] [Google Scholar]
- 137.Wagner J, Ryazanov S, Leonov A, et al. Anle138b: a novel oligomer modulator for disease-modifying therapy of neurodegenerative diseases such as prion and Parkinson’s disease. Acta Neuropathol. 2013;125(6):795–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Giles K, Berry DB, Condello C, et al. Different 2-aminothiazole therapeutics produce distinct patterns of Scrapie prion neuropathology in mouse brains. J Pharmacol Exp Ther. 2015;355(1):2–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Yamasaki T, Suzuki A, Hasebe R, Horiuchi M. Comparison of the anti-prion mechanism of four different anti-prion compounds, anti-PrP monoclonal antibody 44B1, pentosan polysulfate, chlorpromazine, and U18666A, in prion-infected mouse neuroblastoma cells. PLoS One. 2014;9(9):e106516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Doh-ura K, Ishikawa K, Murakami-Kubo I, et al. Treatment of transmissible spongiform encephalopathy by intraventricular drug infusion in animal models. J Virol. 2004;78(10):4999–5006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Bone I, Belton L, Walker AS, Darbyshire J. Intraventricular pentosan polysulphate in human prion diseases: an observational study in the UK. Eur J Neurol. 2008;15(5): 458–464. [DOI] [PubMed] [Google Scholar]
- 142.Newman PK, Todd NV, Scoones D, et al. Postmortem findings in a case of variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulfate. J Neurol Neurosurg Psychiatry. 2014;85(8):921–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Parry A, Baker I, Stacey R, Wimalaratna S. Long term survival in a patient with variant Creutzfeldt-Jakob disease treated with intraventricular pentosan polysulphate. J Neurol Neurosurg Psychiatry. 2007;78(7):733–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Honda H, Sasaki K, Minaki H, et al. Protease-resistant PrP and PrP oligomers in the brain in human prion diseases after intraventricular pentosan polysulfate infusion. Neuropathology. 2012;32(2):124–132. [DOI] [PubMed] [Google Scholar]
- 145.Terada T, Tsuboi Y, Obi T, et al. Less protease-resistant PrP in a patient with sporadic CJD treated with intraventricular pentosan polysulphate. Acta Neurol Scand. 2010;121(2):127–130. [DOI] [PubMed] [Google Scholar]
- 146.Tsuboi Y, Doh-Ura K, Yamada T. Continuous intraventricular infusion of pentosan polysulfate: clinical trial against prion diseases. Neuropathology. 2009;29(5):632–636. [DOI] [PubMed] [Google Scholar]
- 147.Larramendy-Gozalo C, Barret A, Daudigeos E, et al. Comparison of CR36, a new heparan mimetic, and pentosan polysulfate in the treatment of prion diseases. J Gen Virol. 2007;88(pt 3):1062–1067. [DOI] [PubMed] [Google Scholar]
- 148.Horiuchi M, Priola SA, Chabry J, Caughey B. Interactions between heterologous forms of prion protein: binding, inhibition of conversion, and species barriers. Proc Natl Acad Sci U S A. 2000;97(11):5836–5841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Priola SA, Caughey B, Race RE, Chesebro B. Heterologous PrP molecules interfere with accumulation of protease-resistant PrP in scrapie-infected murine neuroblastoma cells. J Virol. 1994;68(8):4873–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Skinner PJ, Kim HO, Bryant D, et al. Treatment of prion disease with heterologous prion proteins. PLoS One. 2015;10(7):e0131993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Vorberg I, Groschup MH, Pfaff E, Priola SA. Multiple amino acid residues within the rabbit prion protein inhibit formation of its abnormal isoform. J Virol. 2003;77(3):2003–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Berry DB, Lu D, Geva M, et al. Drug resistance confounding prion therapeutics. Proc Natl Acad Sci U S A. 2013;110(44):E4160–E4169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Berry D, Giles K, Oehler A, Bhardwaj S, DeArmond SJ, Prusiner SB. Use of a 2-aminothiazole to treat chronic wasting disease in transgenic mice. J Infect Dis. 2015;212(suppl 1):S17–S25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Gallardo-Godoy A, Gever J, Fife KL, Silber BM, Prusiner SB, Renslo AR. 2-Aminothiazoles as therapeutic leads for prion diseases. J Med Chem. 2011;54(4): 1010–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Ghaemmaghami S, May BC, Renslo AR, Prusiner SB. Discovery of 2-aminothiazoles as potent antiprion compounds. J Virol. 2010;84(7):3408–3412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Silber BM, Rao S, Fife KL, et al. Pharmacokinetics and metabolism of 2-aminothiazoles with antiprion activity in mice. Pharm Res. 2013;30(4):932–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.McKenzie D, Kaczkowski J, Marsh R, Aiken J. Amphotericin B delays both scrapie agent replication and PrP-res accumulation early in infection. J Virol. 1994;68(11): 7534–7536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Hartsel SC, Weiland TR. Amphotericin B binds to amyloid fibrils and delays their formation: a therapeutic mechanism? Biochemistry. 2003;42(20):6228–6233. [DOI] [PubMed] [Google Scholar]
- 159.Mange A, Milhavet O, McMahon HE, Casanova D, Lehmann S. Effect of amphotericin B on wild-type and mutated prion proteins in cultured cells: putative mechanism of action in transmissible spongiform encephalopathies. J Neurochem. 2000;74(2):754–762. [DOI] [PubMed] [Google Scholar]
- 160.Masullo C, Macchi G, Xi YG, Pocchiari M. Failure to ameliorate Creutzfeldt-Jakob disease with amphotericin B therapy. J Infect Dis. 1992;165(4):784–785. [DOI] [PubMed] [Google Scholar]
- 161.Motoyoshi-Yamashiro A, Takano K, Kawabe K, et al. Amphotericin B induces glial cell line-derived neurotrophic factor in the rat brain. J Vet Med Sci. 2014;76(10): 1353–1358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Motoyoshi A, Nakajima H, Takano K, Moriyama M, Kannan Y, Nakamura Y. Effects of Amphotericin B on the expression of neurotoxic and neurotrophic factors in cultured microglia. Neurochem Int. 2008;52(6):1290–1296. [DOI] [PubMed] [Google Scholar]
- 163.Pocchiari M, Schmittinger S, Masullo C. Amphotericin B delays the incubation period of scrapie in intracerebrally inoculated hamsters. J Gen Virol. 1987;68(pt 1):219–223. [DOI] [PubMed] [Google Scholar]
- 164.Xi YG, Ingrosso L, Ladogana A, Masullo C, Pocchiari M. Amphotericin B treatment dissociates in vivo replication of the scrapie agent from PrP accumulation. Nature. 1992;356(6370):598–601. [DOI] [PubMed] [Google Scholar]
- 165.Nazor Friberg K, Hung G, Wancewicz E, et al. Intracerebral infusion of antisense oligonucleotides into prion-infected mice. Mol Ther Nucleic Acids. 2012;1:e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Raymond GJ, Zhao HT, Race B, et al. Antisense oligonucleotides extend survival of prion-infected mice. JCI Insight. 2019;5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ferreira NC, Ascari LM, Hughson AG, et al. A promising antiprion trimethoxychalcone binds to the globular domain of the cellular prion protein and changes its cellular location. Antimicrob Agents Chemother. 2018;62(2):e01441–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Ferreira NC, Marques IA, Conceicao WA, et al. Anti-prion activity of a panel of aromatic chemical compounds: in vitro and in silico approaches. PLoS One. 2014; 9(1):e84531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Massignan T, Cimini S, Stincardini C, et al. A cationic tetrapyrrole inhibits toxic activities of the cellular prion protein. Sci Rep. 2016;6:23180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Nicoll AJ, Trevitt CR, Tattum MH, et al. Pharmacological chaperone for the structured domain of human prion protein. Proc Natl Acad Sci U S A. 2010;107(41): 17610–17615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Peretz D, Williamson RA, Kaneko K, et al. Antibodies inhibit prion propagation and clear cell cultures of prion infectivity. Nature. 2001;412(6848):739–743. [DOI] [PubMed] [Google Scholar]
- 172.Enari M, Flechsig E, Weissmann C. Scrapie prion protein accumulation by scrapie-infected neuroblastoma cells abrogated by exposure to a prion protein antibody. Proc Natl Acad Sci U S A. 2001;98(16):9295–9299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Heppner FL, Musahl C, Arrighi I, et al. Prevention of scrapie pathogenesis by transgenic expression of anti-prion protein antibodies. Science. 2001;294(5540):178–182. [DOI] [PubMed] [Google Scholar]
- 174.Antonyuk SV, Trevitt CR, Strange RW, et al. Crystal structure of human prion protein bound to a therapeutic antibody. Proc Natl Acad Sci U S A. 2009;106(8):2554–2558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.White AR, Enever P, Tayebi M, et al. Monoclonal antibodies inhibit prion replication and delay the development of prion disease. Nature. 2003;422(6927):80–83. [DOI] [PubMed] [Google Scholar]
- 176.Kim CL, Karino A, Ishiguro N, Shinagawa M, Sato M, Horiuchi M. Cell-surface retention of PrPC by anti-PrP antibody prevents protease-resistant PrP formation. J Gen Virol. 2004;85(pt 11):3473–3482. [DOI] [PubMed] [Google Scholar]
- 177.Pankiewicz J, Prelli F, Sy MS, et al. Clearance and prevention of prion infection in cell culture by anti-PrP antibodies. Eur J Neurosci. 2006;23(10):2635–2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Perrier V, Solassol J, Crozet C, et al. Anti-PrP antibodies block PrPSc replication in prion-infected cell cultures by accelerating PrPC degradation. J Neurochem. 2004;89(2):454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.David MA, Jones DR, Tayebi M. Potential candidate camelid antibodies for the treatment of protein-misfolding diseases. J Neuroimmunol. 2014;272(1–2):76–85. [DOI] [PubMed] [Google Scholar]
- 180.Jones DR, Taylor WA, Bate C, David M, Tayebi M. A camelid anti-PrP antibody abrogates PrP replication in prion-permissive neuroblastoma cell lines. PLoS One. 2010;5(3):e9804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Herrmann US, Sonati T, Falsig J, et al. Prion infections and anti-PrP antibodies trigger converging neurotoxic pathways. PLoS Pathog. 2015;11(2):e1004662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Imberdis T, Heeres JT, Yueh H, et al. Identification of anti-prion compounds using a novel cellular assay. J Biol Chem. 2016;291(50):26164–26176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Miyazaki Y, Ishikawa T, Kamatari YO, et al. Identification of alprenolol hydrochloride as an anti-prion compound using surface plasmon resonance imaging. Mol Neurobiol. 2019;56(1):367–377. [DOI] [PubMed] [Google Scholar]
- 184.Mashima T, Lee JH, Kamatari YO, et al. Development and structural determination of an anti-PrP(C) aptamer that blocks pathological conformational conversion of prion protein. Sci Rep. 2020;10(1):4934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Mashima T, Nishikawa F, Kamatari YO, et al. Anti-prion activity of an RNA aptamer and its structural basis. Nucleic Acids Res. 2013;41(2):1355–1362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Didonna A, Venturini AC, Hartman K, Vranac T, Curin Serbec V, Legname G. Characterization of four new monoclonal antibodies against the distal N-terminal region of PrP(c). PeerJ. 2015;3:e811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Stincardini C, Massignan T, Biggi S, et al. An antipsychotic drug exerts anti-prion effects by altering the localization of the cellular prion protein. PLoS One. 2017;12(8): e0182589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Barret A, Tagliavini F, Forloni G, et al. Evaluation of quinacrine treatment for prion diseases. J Virol. 2003;77(15):8462–8469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Collinge J, Gorham M, Hudson F, et al. Safety and efficacy of quinacrine in human prion disease (PRION-1 study): a patient-preference trial. Lancet Neurol. 2009;8(4):334–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Doh-Ura K, Iwaki T, Caughey B. Lysosomotropic agents and cysteine protease inhibitors inhibit scrapie-associated prion protein accumulation. J Virol. 2000;74(10): 4894–4897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Georgieva D, Schwark D, von Bergen M, Redecke L, Genov N, Betzel C. Interactions of recombinant prions with compounds of therapeutical significance. Biochem Biophys Res Commun. 2006;344(2):463–470. [DOI] [PubMed] [Google Scholar]
- 192.Geschwind MD, Kuo AL, Wong KS, et al. Quinacrine treatment trial for sporadic Creutzfeldt-Jakob disease. Neurology. 2013;81:2015–2023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Ghaemmaghami S, Ahn M, Lessard P, et al. Continuous quinacrine treatment results in the formation of drug-resistant prions. PLoS Pathog. 2009;5(11):e1000673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Adjou KT, Demaimay R, Deslys JP, et al. MS-8209, a water-soluble amphotericin B derivative, affects both scrapie agent replication and PrPres accumulation in Syrian hamster scrapie. J Gen Virol. 1999;80(pt 4):1079–1085. [DOI] [PubMed] [Google Scholar]
- 195.Adjou KT, Privat N, Demart S, et al. MS-8209, an amphotericin B analogue, delays the appearance of spongiosis, astrogliosis and PrPres accumulation in the brain of scrapie-infected hamsters. J Comp Pathol. 2000;122(1):3–8. [DOI] [PubMed] [Google Scholar]
- 196.Adjou KT, Demaimay R, Lasmezas C, Deslys JP, Seman M, Dormont D. MS-8209, a new amphotericin B derivative, provides enhanced efficacy in delaying hamster scrapie. Antimicrob Agents Chemother. 1995;39(12):2810–2812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Adjou KT, Demaimay R, Lasmezas CI, Seman M, Deslys JP, Dormont D. Differential effects of a new amphotericin B derivative, MS-8209, on mouse BSE and scrapie: implications for the mechanism of action of polyene antibiotics. Res Virol. 1996;147(4): 213–218. [DOI] [PubMed] [Google Scholar]
- 198.Beringue V, Adjou KT, Lamoury F, et al. Opposite effects of dextran sulfate 500, the polyene antibiotic MS-8209, and Congo red on accumulation of the protease-resistant isoform of PrP in the spleens of mice inoculated intraperitoneally with the scrapie agent. J Virol. 2000;74(12):5432–5440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Demaimay R, Adjou K, Lasmezas C, et al. Pharmacological studies of a new derivative of amphotericin B, MS-8209, in mouse and hamster scrapie. J Gen Virol. 1994; 75(pt 9):2499–2503. [DOI] [PubMed] [Google Scholar]
- 200.Grigoriev VB, Adjou KT, Sales N, et al. Effects of the polyene antibiotic derivative MS-8209 on the astrocyte lysosomal system of scrapie-infected hamsters. J Mol Neurosci. 2002;18(3):271–281. [DOI] [PubMed] [Google Scholar]
- 201.Ohsawa N, Song CH, Suzuki A, Furuoka H, Hasebe R, Horiuchi M. Therapeutic effect of peripheral administration of an anti-prion protein antibody on mice infected with prions. Microbiol Immunol. 2013;57(4):288–297. [DOI] [PubMed] [Google Scholar]
- 202.Forloni G, Iussich S, Awan T, et al. Tetracyclines affect prion infectivity. Proc Natl Acad Sci U S A. 2002;99(16):10849–10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Gu Y, Singh N. Doxycycline and protein folding agents rescue the abnormal phenotype of familial CJD H187R in a cell model. Brain Res Mol Brain Res. 2004;123(1–2): 37–44. [DOI] [PubMed] [Google Scholar]
- 204.Haïk S, Marcon G, Mallet A, et al. Doxycycline in Creutzfeldt-Jakob disease: A phase 2, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2014;13(2): 150–158. [DOI] [PubMed] [Google Scholar]
- 205.Hannaoui S, Gougerot A, Privat N, et al. Cycline efficacy on the propagation of human prions in primary cultured neurons is strain-specific. J Infect Dis. 2014;209(7): 1144–1148. [DOI] [PubMed] [Google Scholar]
- 206.Pocchiari M, Ladogana A. Rethinking of doxycycline therapy in Creutzfeldt-Jakob disease. J Neurol Neurosurg Psychiatry. 2015;86(7):705. [DOI] [PubMed] [Google Scholar]
- 207.Moreno JA, Halliday M, Molloy C, et al. Oral treatment targeting the unfolded protein response prevents neurodegeneration and clinical disease in prion-infected mice. Sci Transl Med. 2013;5(206):206ra138. [DOI] [PubMed] [Google Scholar]
- 208.Ertmer A, Gilch S, Yun SW, et al. The tyrosine kinase inhibitor STI571 induces cellular clearance of PrPSc in prion-infected cells. J Biol Chem. 2004;279(40):41918–41927. [DOI] [PubMed] [Google Scholar]
- 209.Abdulrahman BA, Abdelaziz D, Thapa S, et al. The celecoxib derivatives AR-12 and AR-14 induce autophagy and clear prion-infected cells from prions. Sci Rep. 2017; 7(1):17565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Heiseke A, Aguib Y, Riemer C, Baier M, Schatzl HM. Lithium induces clearance of protease resistant prion protein in prion-infected cells by induction of autophagy. J Neurochem. 2009;109(1):25–34. [DOI] [PubMed] [Google Scholar]
- 211.Mead S, Tagliavini F. Clinical trials. Handb Clin Neurol. 2018;153:431–444. [DOI] [PubMed] [Google Scholar]
- 212.Forloni G, Tettamanti M, Lucca U, et al. Preventive study in subjects at risk of fatal familial insomnia: Innovative approach to rare diseases. Prion. 2015;9(2):75–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Elbein AD, Solf R, Dorling PR, Vosbeck K. Swainsonine: An inhibitor of glycoprotein processing. Proc Natl Acad Sci U S A. 1981;78(12):7393–7397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Tulsiani DR, Harris TM, Touster O. Swainsonine inhibits the biosynthesis of complex glycoproteins by inhibition of Golgi mannosidase II. J Biol Chem. 1982;257(14): 7936–7939. [PubMed] [Google Scholar]
- 215.Tulsiani DRP, Touster O. Swainsonine, a potent mannosidase inhibitor, elevates rat liver and brain lysosomal α-d-mannosidase, decreases Golgi α-d-mannosidase II, and increases the plasma levels of several acid hydrolases. Arch Biochem Biophys. 1983; 224(2):594–600. [DOI] [PubMed] [Google Scholar]
- 216.Mahal SP, Browning S, Li J, Suponitsky-Kroyter I, Weissmann C. Transfer of a prion strain to different hosts leads to emergence of strain variants. Proc Natl Acad Sci U S A. 2010;107(52):22653–22658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Li J, Mahal SP, Demczyk CA, Weissmann C. Mutability of prions. EMBO Rep. 2011;12(12):1243–1250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Li J, Browning S, Mahal SP, Oelschlegel AM, Weissmann C. Darwinian evolution of prions in cell culture. Science. 2010;327(5967):869–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Oelschlegel AM, Weissmann C. Acquisition of drug resistance and dependence by prions. PLoS Pathog. 2013;9(2):e1003158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Collinge J, Clarke AR. A general model of prion strains and their pathogenicity. Science. 2007;318(5852):930–936. [DOI] [PubMed] [Google Scholar]
- 221.Langenfeld KA, Shikiya RA, Kincaid AE, Bartz JC. Incongruity between prion conversion and incubation period following coinfection. J Virol. 2016;90(12):5715–5723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Bartz JC, Aiken JM, Bessen RA. Delay in onset of prion disease for the HY strain of transmissible mink encephalopathy as a result of prior peripheral inoculation with the replication-deficient DY strain. J Gen Virol. 2004;85(pt 1):265–273. [DOI] [PubMed] [Google Scholar]
- 223.Dickinson AG, Fraser H, Meikle VM, Outram GW. Competition between different scrapie agents in mice. Nat New Biol. 1972;237(77):244–245. [DOI] [PubMed] [Google Scholar]
- 224.Holec SAM, Yuan Q, Bartz JC. Alteration of prion strain emergence by nonhost factors. mSphere. 2019;4(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Bruce ME,Dickinson AG. Biological stability ofdifferent classesofscrapie agent.In: Slow Transmissible Disease of the Nervous System. Academic Press, Inc; 1979:71–86. vol. 2. [Google Scholar]
- 226.Büeler H, Fisher M, Lang Y, et al. Normal development and behaviour of mice lacking the neuronal cell-surface PrP protein. Nature. 1992;356:577–582. [DOI] [PubMed] [Google Scholar]
- 227.Büeler H, Aguzzi A, Sailer A, et al. Mice devoid of PrP are resistant to scrapie. Cell. 1993;73:1339–1347. [DOI] [PubMed] [Google Scholar]
- 228.Richt JA, Kasinathan P, Hamir AN, et al. Production of cattle lacking prion protein. Nat Biotechnol. 2007;25:132–138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Mallucci G, Dickinson A, Linehan J, Klohn PC, Brandner S, Collinge J. Depleting neuronal PrP in prion infection prevents disease and reverses spongiosis. Science. 2003;302(5646):871–874. [DOI] [PubMed] [Google Scholar]
- 230.Lewis V Proteolytic processing of the prion protein. In: Collins S, Lawson V, eds. The Cellular and Molecular Biology of Prion Disease. Kerala: Research Signpost; 2011:53–71. [Google Scholar]
- 231.Chen SG, Teplow DB, Parchi P, Teller JK, Gambetti P, Autilio-Gambetti L. Truncated forms of the human prion protein in normal brain and in prion diseases. J Biol Chem. 1995;270(32):19173–19180. [DOI] [PubMed] [Google Scholar]
- 232.Jiménez-Huete A, Lievens PMJ, Vidal R, et al. Endogenous proteolytic cleavage of normal and disease-associated isoforms of the human prion protein in neural and non-neural tissues. Am J Pathol. 1998;153(5):1561–1572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Zanusso G, Righetti PG, Ferrari S, et al. Two-dimensional mapping of three phenotype-associated isoforms of the prion protein in sporadic Creutzfeldt-Jakob disease. Electrophoresis. 2002;23(2):347–355. [DOI] [PubMed] [Google Scholar]
- 234.Owen JP, Rees HC, Maddison BC, et al. Molecular profiling of ovine prion diseases by using thermolysin-resistant PrPSc and endogenous C2 PrP fragments. J Virol. 2007;81(19):10532–10539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Hope J, Reekie LJD, Hunter N, et al. Fibrils from brains of cows with new cattle disease contain scrapie-associated protein. Nature. 1988;336:390–392. [DOI] [PubMed] [Google Scholar]
- 236.Hope J, Morton LJ, Farquhar CF, Multhaup G, Beyreuther K, Kimberlin RH. The major polypeptide of scrapie-associated fibrils (SAF) has the same size, charge distribution and N-terminal protein sequence as predicted for the normal brain protein (PrP). EMBO J. 1986;5(10):2591–2597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Pan T, Wong P, Chang B, et al. Biochemical fingerprints of prion infection: accumulations of aberrant full-length and N-terminally truncated PrP species are common features in mouse prion disease. J Virol. 2005;79(2):934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.O’Brien RJ, Wong PC. Amyloid precursor protein processing and Alzheimer’s disease. Annu Rev Neurosci. 2011;34:185–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Kim J, Onstead L, Randle S, et al. Abeta40 inhibits amyloid deposition in vivo. J Neurosci. 2007;27(3):627–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Deng Y, Tarassishin L, Kallhoff V, et al. Deletion of presenilin 1 hydrophilic loop sequence leads to impaired gamma-secretase activity and exacerbated amyloid pathology. J Neurosci. 2006;26(14):3845–3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Iijima K, Liu HP, Chiang AS, Hearn SA, Konsolaki M, Zhong Y. Dissecting the pathological effects of human Abeta40 and Abeta42 in drosophila: A potential model for Alzheimer’s disease. Proc Natl Acad Sci U S A. 2004;101(17):6623–6628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.McGowan E, Pickford F, Kim J, et al. Abeta42 is essential for parenchymal and vascular amyloid deposition in mice. Neuron. 2005;47(2):191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Zhang Z, Kang SS, Liu X, et al. Asparagine endopeptidase cleaves alpha-synuclein and mediates pathologic activities in Parkinson’s disease. Nat Struct Mol Biol. 2017;24(8): 632–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Bremer J, Baumann F, Tiberi C, et al. Axonal prion protein is required for peripheral myelin maintenance. Nat Neurosci. 2010;13(3):310–318. [DOI] [PubMed] [Google Scholar]
- 245.Vincent B, Paitel E, Saftig P, et al. The disintegrins ADAM10 and TACE contribute to the constitutive and phorbol ester-regulated normal cleavage of the cellular prion protein. J Biol Chem. 2001;276(41):37743–37746. [DOI] [PubMed] [Google Scholar]
- 246.Cisse MA, Sunyach C, Lefranc-Jullien S, Postina R, Vincent B, Checler F. The disintegrin ADAM9 indirectly contributes to the physiological processing of cellular prion by modulating ADAM10 activity. J Biol Chem. 2005;280(49):40624–40631. [DOI] [PubMed] [Google Scholar]
- 247.Linsenmeier L, Mohammadi B, Wetzel S, et al. Structural and mechanistic aspects influencing the ADAM10-mediated shedding of the prion protein. Mol Neurodegener. 2018;13(1):18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Taylor DR, Parkin ET, Cocklin SL, et al. Role of ADAMs in the ectodomain shedding and conformational conversion of the prion protein. J Biol Chem. 2009;284(34): 22590–22600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Altmeppen HC, Prox J, Puig B, et al. Lack of a-disintegrin-and-metalloproteinase ADAM10 leads to intracellular accumulation and loss of shedding of the cellular prion protein in vivo. Mol Neurodegener. 2011;6:36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Altmeppen HC, Prox J, Krasemann S, et al. The sheddase ADAM10 is a potent modulator of prion disease. Elife. 2015;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Feraudet C, Morel N, Simon S, et al. Screening of 145 anti-PrP monoclonal antibodies for their capacity to inhibit PrPSc replication in infected cells. J Biol Chem. 2005;280(12):11247–11258. [DOI] [PubMed] [Google Scholar]
- 252.Lefebvre-Roque M, Kremmer E, Gilch S, et al. Toxic effects of intracerebral PrP antibody administration during the course of BSE infection in mice. Prion. 2007;1(3): 198–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Solforosi L, Criado JR, McGavern DB, et al. Cross-linking cellular prion protein triggers neuronal apoptosis in vivo. Science. 2004;303(5663):1514–1516. [DOI] [PubMed] [Google Scholar]
- 254.Sonati T, Reimann RR, Falsig J, et al. The toxicity of antiprion antibodies is mediated by the flexible tail of the prion protein. Nature. 2013;501(7465):102–106. [DOI] [PubMed] [Google Scholar]
- 255.Tayebi M, David M, Bate C, et al. Epitope-specific anti-prion antibodies upregulate apolipoprotein E and disrupt membrane cholesterol homeostasis. J Gen Virol. 2010;91(pt 12):3105–3115. [DOI] [PubMed] [Google Scholar]
- 256.Goni F, Knudsen E, Schreiber F, et al. Mucosal vaccination delays or prevents prion infection via an oral route. Neuroscience. 2005;133(2):413–421. [DOI] [PubMed] [Google Scholar]
- 257.Goni F, Mathiason CK, Yim L, et al. Mucosal immunization with an attenuated Salmonella vaccine partially protects white-tailed deer from chronic wasting disease. Vaccine. 2015;33(5):726–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Pilon J, Loiacono C, Okeson D, et al. Anti-prion activity generated by a novel vaccine formulation. Neurosci Lett. 2007;429(2–3):161–164. [DOI] [PubMed] [Google Scholar]
- 259.Hedlin PD, Cashman NR, Li L, et al. Design and delivery of a cryptic PrP(C) epitope for induction of PrP(Sc)-specific antibody responses. Vaccine. 2010;28(4):981–988. [DOI] [PubMed] [Google Scholar]
- 260.Marciniuk K, Maattanen P, Taschuk R, et al. Development of a multivalent, PrP(Sc)-specific prion vaccine through rational optimization of three disease-specific epitopes. Vaccine. 2014;32(17):1988–1997. [DOI] [PubMed] [Google Scholar]
- 261.Taschuk R, Van der Merwe J, Marciniuk K, et al. In vitro neutralization of prions with PrP(Sc)-specific antibodies. Prion. 2015;9(4):292–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Taschuk R, Scruten E, Woodbury M, et al. Induction of PrP(Sc)-specific systemic and mucosal immune responses in white-tailed deer with an oral vaccine for chronic wasting disease. Prion. 2017;11(5):368–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Wood ME, Griebel P, Huizenga ML, et al. Accelerated onset of chronic wasting disease in elk (Cervus canadensis) vaccinated with a PrP(Sc)-specific vaccine and housed in a prion contaminated environment. Vaccine. 2018;36(50):7737–7743. [DOI] [PubMed] [Google Scholar]
- 264.Arsenault RJ, Li Y, Potter A, Griebel PJ, Kusalik A, Napper S. Induction of ligand-specific PrP (C) signaling in human neuronal cells. Prion. 2012;6(5):477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Saborio GP, Permanne B, Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature. 2001;411(6839):810–813. [DOI] [PubMed] [Google Scholar]
- 266.Wilham JM, Orru CD, Bessen RA, et al. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010;6(12): e1001217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Lacroux C, Comoy E, Moudjou M, et al. Preclinical detection of variant CJD and BSE prions in blood. PLoS Pathog. 2014;10(6):e1004202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Mathiason CK, Powers JG, Dahmes SJ, et al. Infectious prions in the saliva and blood of deer with chronic wasting disease. Science. 2006;314(5796):133–136. [DOI] [PubMed] [Google Scholar]
- 269.McGuire LI, Peden AH, Orru CD, et al. Real time quaking-induced conversion analysis of cerebrospinal fluid in sporadic Creutzfeldt-Jakob disease. Ann Neurol. 2012;72(2):278–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Moda F, Gambetti P, Notari S, et al. Prions in the urine of patients with variant Creutzfeldt-Jakob disease. N Engl J Med. 2014;371(6):530–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Orru CD, Groveman BR, Hughson AG, Zanusso G, Coulthart MB, Caughey B. Rapid and sensitive RT-QuIC detection of human Creutzfeldt-Jakob disease using cerebrospinal fluid. MBio. 2015;6(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Safar JG, Lessard P, Tamguney G, et al. Transmission and detection of prions in feces. J Infect Dis. 2008;198(1):81–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Orru CD, Bongianni M, Tonoli G, et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N Engl J Med. 2014;371(6):519–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Saa P, Castilla J, Soto C. Presymptomatic detection of prions in blood. Science. 2006;313(5783):92–94. [DOI] [PubMed] [Google Scholar]
- 275.Sawyer EB, Edgeworth JA, Thomas C, Collinge J, Jackson GS. Preclinical detection of infectivity and disease-specific PrP in blood throughout the incubation period of prion disease. Sci Rep. 2015;5:17742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wang Z, Manca M, Foutz A, et al. Early preclinical detection of prions in the skin of prion-infected animals. Nat Commun. 2019;10(1):247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Elder AM, Henderson DM, Nalls AV, et al. In vitro detection of prionemia in TSE-infected cervids and hamsters. PLoS One. 2013;8(11):e80203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Elder AM, Henderson DM, Nalls AV, et al. Immediate and ongoing detection of prions in the blood of hamsters and deer following oral, nasal, or blood inoculations. J Virol. 2015;89(14):7421–7424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Fairfoul G, McGuire LI, Pal S, et al. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol. 2016;3(10):812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Saijo E, Ghetti B, Zanusso G, et al. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017;133(5):751–765. [DOI] [PubMed] [Google Scholar]
- 281.Salvadores N, Shahnawaz M, Scarpini E, Tagliavini F, Soto C. Detection of misfolded Abeta oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell Rep. 2014;7(1):261–268. [DOI] [PubMed] [Google Scholar]
- 282.Shahnawaz M, Tokuda T, Waragai M, et al. Development of a biochemical diagnosis of Parkinson disease by detection of alpha-synuclein misfolded aggregates in cerebrospinal fluid. JAMA Neurol. 2017;74(2):163–172. [DOI] [PubMed] [Google Scholar]
- 283.Saijo E, Groveman BR, Kraus A, et al. Ultrasensitive RT-QuIC seed amplification assays for disease-associated tau, alpha-synuclein, and prion aggregates. Methods Mol Biol. 2019;1873:19–37. [DOI] [PubMed] [Google Scholar]