

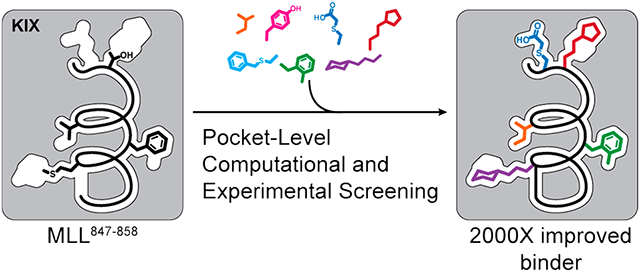
Abstract
Constrained peptides have proven to be a rich source of ligands for protein surfaces, but are often limited in their binding potency. Deployment of nonnatural side chains that access unoccupied crevices on the receptor surface offers a potential avenue to enhance binding affinity. We recently described a computational approach to create topographic maps of protein surfaces to guide the design of nonnatural side chains [J. Am. Chem. Soc. 2017, 139, 15560]. The computational method, AlphaSpace, was used to predict peptide ligands for the KIX domain of the p300/CBP coactivator. KIX has been the subject of numerous ligand discovery strategies, but potent inhibitors of its interaction with transcription factors remain difficult to access. Although the computational approach provided a significant enhancement in the binding affinity of the peptide, fine-tuning of nonnatural side chains required an experimental screening method. Here we implement a peptide-tethering strategy to screen fragments as nonnatural side chains on conformationally defined peptides. The combined computational–experimental approach offers a general framework for optimizing peptidomimetics as inhibitors of protein–protein interactions.
Graphical Abstract
INTRODUCTION
Rationally designed mimics of protein secondary and tertiary structures can serve as modulators of biomolecular interactions. The hypothesis governing the design of protein mimics is that elements of protein surfaces can be miniaturized to obtain cell permeable, proteolytically resistant ligands that retain the binding interactions of the native protein.1 However, these mimics often suffer from an inherent limitation: by definition, minimal mimics of proteins do not encompass all contact points available to native proteins, and the loss of contacts correlates with a reduction in binding affinity and specificity for their cognate receptors.2,3 Potential solutions to this challenge include enhancement of the synthetic mimics with electrophilic warheads to elicit covalent contacts with the receptor4 or inclusion of nonnatural side chains, which can compensate for the loss in binding affinity by noncovalent engagement of pockets not accessible to native residues.5
We developed a combined computational and experimental approach to engineer peptidomimetics with judiciously designed nonnatural side chains. In our preliminary efforts, we focused on developing ligands for the coactivator KIX.6 The KIX domain of transcriptional coactivators CBP and p300 serves as a docking site for transcription factors.7 Figure 1A shows an NMR model of transactivation domains from cMyb and MLL binding on two allosterically linked faces of KIX.8 Binding of one transcription factor on KIX enhances binding of the other transcription factor, highlighting KIX’s plasticity.9 Interactions of KIX with aberrant transcription factors lead to abnormal gene expression and disease.10 Several approaches, including structure-guided design,6,11 NMR-based screening,12 high-throughput screening,13 phage display,14 and disulfide protein tethering,15,16 have been employed to develop ligands for one or both binding surfaces of KIX to block the transcription factor–coactivator interactions. The conformational plasticity of KIX and a lack of contiguous binding pockets on its surface have made the discovery of potent ligands difficult—many small molecules and peptides designed or isolated from the above strategies have shown weak affinity in the high micromolar to millimolar range for KIX. A potent peptide ligand that links cMyb and MLL sequences was recently described; this study further illustrates the challenge in targeting an individual KIX cavity with low to medium molecular weight compounds.17
Figure 1.

(A) KIX domain of coactivators p300/CBP interacts with a multitude of transcription factors. An NMR-derived model of KIX in complex with MLL and cMyb is shown (PDB code 2AGH). (B) Helical domain of MLL847–858 provides a template for the development of synthetic ligands for KIX. The topographical map of KIX suggests that several pockets on its surface are not optimally occupied by native MLL residues, and nonnatural residues may be designed to provide enhanced affinity. The figure shows AlphaSpace analysis of the KIX/MLL complex. The yellow spheres depict the centroid of potential pockets near the MLL helix. (C) In published studies, we showed that a computationally designed peptide with noncanonical amino acids replacing M850, F852, V854, and T857 makes superior contacts with KIX. Here we build on the computational method and describe an experimental fragment screening approach to identify synthetic side chains to engage KIX.
In our previous studies, we used the computational algorithm AlphaSpace18 to topographically map protein surfaces and design natural and nonnatural side chains on peptide mimics. AlphaSpace analysis suggested that several MLL residues do not optimally occupy the closest lying pockets on KIX (Figure 1B) and that nonnatural residues may be designed to capture cryptic pockets. Application of AlphaSpace led to a significantly improved minimal MLL motif; the optimized sequence featured several nonnatural residues and bound KIX with low micromolar affinity as compared to the millimolar binding affinity observed for the short native sequence. Specifically, computational design predicted that S-benzylcysteine (in place of M850), 2-methyl-phenylalanine (in place of F852), isoleucine (in place of V853), and tyrosine (in place of T857) could successfully occupy pockets on KIX (Figure 1B). Here, we merge this computational fragment design approach with an experimental side-chain screening method, which allows fragments to be individually screened at the pocket level within the context of a constrained helix (Figure 1C).
Our experimental strategy builds on a combination of protein tethering19,20 and fragment linking approaches.21 In protein tethering, or site-directed ligand discovery, an engineered or native cysteine residue is employed to form covalent linkages with fragments from a library and guide individual fragments into a neighboring protein pocket (Figure 2A).20,22 Tethering increases the local concentration of the fragment, allowing for screening of fragments at lower concentrations than if they were not tethered. Protein tethering has been successfully utilized to develop inhibitors and stabilizers of protein–protein interactions (PPIs),23,24 and to probe for ligandability.25 Fragment linking has become a useful approach to generate potent multivalent ligands from weak-binding fragments.26,27 We envisioned peptide tethering to involve judicious placement of a reactive group on a rationally designed peptidomimetic as opposed to the protein itself (Figure 2B). The fragments are covalently linked to a peptide containing a fluorophore before incubation of the fragment-modified peptide with the protein. The relative impact of fragments on binding can then be quantified by using a fluorescence polarization assay (Figure 2C). For this strategy to succeed, residues to be screened must be directed into nearby pockets; we rationalized that if the peptide retains several native critical binding residues, its native orientation would be retained to allow screening at individual sites. For example, in Figure 2B,C, the green hexagon is expected to anchor the peptide into the correct position on the receptor and allow side-chain fragment screening at the chosen site.
Figure 2.

(A) Protein tethering versus (B) peptide tethering. In protein tethering, native or nonnative reactive moieties such as cysteine residues direct fragments into nearby pockets. Fragment screening is often performed to target multiple pockets on the protein; multiple fragments are then linked together. In peptide tethering, the fragments are tethered to cysteine or another reactive group on the peptidomimetic. (C) Experimental setup: the peptide has a fluorophore, and fragments are covalently linked before individually incubating with the protein and determining fluorescence polarization (FP). The process can be repeated at multiple sites on the peptide.
We envisioned different strategies for modifying the peptide with fragments, including disulfide bridges, thiol alkylation, and potentially various metal-mediated reactions. Our computational analysis suggested that aromatic or aliphatic fragments would lead to enhanced binding because the pockets being investigated are largely hydrophobic, with the exception of a KIX pocket near the MLL C-terminus (Supporting Information, Figure S1). Based on this hypothesis, the cysteine group on the peptide was modified with a library of fragments largely consisting of aliphatic and aromatic groups. We chose three different positions on the MLL peptide for optimization. Successive implementation of peptide tethering at the individual sites led to a submicromolar ligand for KIX, which corresponds to a >2000-fold improvement for the combined computational–experimental side-chain fragment identification strategy.
RESULTS AND DISCUSSION
The KIX/MLL complex serves as a model system to explore the potential of the combined computational–experimental side-chain fragment screening method to predict optimal nonnatural side-chain groups.6 The AlphaSpace topographical mapping of the protein complex reveals that the MLL helical domain does not optimally engage the pockets on the KIX surface (Figure 1A). On the basis of this analysis, we first pursued a rational design approach to endow the MLL peptide with noncanonical side-chain groups. These efforts produced Peptide I (Ac-SDI-Bcs-D-FMe-ILKNYP-NH2, Ki = 22 ± 8 μM, Bcs = benzylcysteine), which inhibited the interaction between KIX and fluorescently labeled MLL peptide with >50-fold higher potency than the wild-type sequence MLL847–858 (Ac-SDIMDFVLKNTP-NH2, Ki > 1000 μM; Table S1).
We pursued the peptide tethering strategy on the MLL sequence to further improve upon the computational campaign. We began the experimental screening studies by converting the lead linear peptide into covalently constrained helix mimics. We utilized the hydrogen bond surrogate (HBS) strategy to stabilize the helix conformation (Figure 3A). HBS α-helices have previously been extensively evaluated in biophysical, biochemical, cell culture, and in vivo assays.28-31 AlphaSpace analysis suggests native residues M850, F852, V853, T857, and P858 may be optimized. Substitution of F852 with methylphenylalanine and V853 with isoleucine leads to near full occupation of the cognate pockets, but an experimental fragment screen is required to identify optimal residues for positions 850, 857, and 858. We focused the side-chain fragment screening efforts on the computationally optimized sequence and sought analogues for benzylcysteine850, Y857, and P858 (Figure 3B). We synthesized three parent HBS helices with cysteine residues at positions 850, 857, and 858 for peptide tethering screens (Figure S2). As part of these studies, we also extended the peptide with a tryptophan residue at the C-terminus to potentially engage a cationic patch on the KIX through cation–π interactions. Addition of the tryptophan residue enhanced the binding affinity by roughly 2-fold (Table S1).
Figure 3.

Peptide tethering to screen side-chain fragments for MLL helix. (A) The computationally optimized sequence was stabilized into the helical conformation with the hydrogen bond surrogate strategy. (B) The constrained helix was iteratively screened at three positions to identify improved side-chain groups at these sites.
We created a focused library of fragments consisting of aromatic and aliphatic fragments for this initial analysis of the potential of peptide tethering to afford optimized ligands (Figure 4A). The library components were chosen from commercially available aryl bromides, aliphatic thiols, and iodoacetates and acetamides. We chose aryl bromides and iodoacetates for their potential to efficiently react with cysteine residues (Supporting Information). Conversely, alkyl bromides or iodides did not provide high yields of the alkylated cysteine products, and therefore alkyl fragments 14–22 were incorporated via disulfide linkages. The alkyl thiol groups were preactivated as 5-nitropyridyl mixed disulfides in a one-pot synthesis (Figure 4B and Supporting Information). In brief, fragments were incubated with the fluorescently tagged peptide, confirmed by mass spectrometry, diluted, and individually incubated with protein before analyzing the fluorescence polarization (Figure 2C and Supporting Information). The fluorescence polarization assay provides a direct readout of the potential of the alkylated fragment to engage KIX.
Figure 4.

(A) Fragments utilized in this study. (B) Side-chain fragments were incorporated into HBS peptides by cysteine alkylation or as disulfide linkages. Screening of fragment results at sites (C) 850, (D) 857, and (E) 858.
Results of the fragment screen at sites 850, 857, and 858 are depicted in Figures 4C-E and the Supporting Information. The bar graphs show fluorescence polarization intensity for 15 nM peptide library and 40 μM KIX; these conditions were chosen based on our prior analysis of binding of MLL derivatives and found to offer the requisite dynamic range for differentiating the impact of different fragments. Screening results for site 850 validate the computational results—notably, we find that hydrophobic residues at this position are favorable, in keeping with the prior AlphaSpace prediction of a benzylcysteine fragment at position 850. (The results between an HBS-constrained helix and unconstrained peptide vary slightly for the N-terminus Cys-850 residue because the constraint places a hydrophobic bridge next to this residue in addition to rigidifying the backbone.) The screen detected that polar residues (fragments 12, 13, 18, and 19) are not optimal for the hydrophobic pocket and selected nonpolar aliphatic and aromatic fragments that complement this pocket. In addition, the experimental screen suggested that cyclohexyl disulfide fragment 20 is a better candidate at this position than the aromatic ring (Figure 4C). This result highlights the key advantage of the experimental screen to build on the computational platform as the computational analysis is unable to discriminate between the aromatic and aliphatic six-membered rings because of their nearly identical physicochemical attributes and binding profiles. Encouraged by the screening results at site 850, we next evaluated optimal fragments for sites 857 and 858. Results suggest that a cyclopentyl fragment 21 is the top candidate in our screening library for site 857 (Figure 4D) while the iodoacetic acid fragment 13 emerged as the top hit for site 858 (Figure 4E). The emergence of a carboxylate fragment at this position can be rationalized and highlights the potential of the screening strategy: position 858 is surrounded by a cationic patch on KIX C-terminus (residues 667–671)7 (Figure S1).
We resynthesized an HBS helix that incorporates the lead hit fragments from each of the three screens to assess the success of the peptide tethering approach to yield high affinity binders. We reasoned that the limited stability of surface-exposed disulfide bonds may restrict the potential of peptidomimetics in advanced biological assays and replaced the disulfide bridges employed for fragment screens with hydrocarbon linkages to obtain HBS II (Figure 5A). The conformational stability of the constrained helix was investigated by circular dichroism spectroscopy—we observed a CD signature expected of helical peptides (Figure S7). We next evaluated the potential of HBS II to inhibit the KIX–MLL interaction in a competition fluorescence polarization assay. HBS II showed a submicromolar inhibitory constant (Ki = 480 ± 130 nM), which equates to an ~2000-fold binding affinity enhancement over the wild type MLL sequence (MLL847–858) and a roughly 50-fold binding affinity increase over computationally designed Peptide I (Figure 5B and Table S1). We utilized a direct binding fluorescence polarization assay during the screen but measured the affinity of the lead sequences with the hydrocarbon linked fragments using a competition assay. The binding affinities obtained from the direct binding and competition binding assays are slightly different, potentially reflecting the impact of the fluorophore; however, the fold-improvement observed is similar with and without the fluorophore in these assays.
Figure 5.

Analysis of the impact of nonnatural side chains on the binding affinities of MLL peptides. (A) The fragment screen yielded optimized peptide HBS II from HBS I as a starting point. (B) HBS II binds KIX with submicromolar affinity, which corresponds to a 2000-fold improvement over MLL847–858 and a 50-fold improvement over the computationally optimized Peptide I. (C) HBS helices III*–VI* with single and double side-chain fragment hits were evaluated. Each nonnatural appendage offers cumulative enhancement to peptide affinity. *Denotes fluorescently labeled peptide. The binding constants for the peptides are listed in Table S2.
We assessed the impact of the individual fragments on binding of HBS II by individually substituting the top fragment hits with native residues (Figure S3). The goal of this analysis was to determine if the screened fragments are providing a synergistic increase to the binding affinity of HBS II or if one fragment is dominating the binding profile. We incorporated a single experimentally screened fragment in HBS II at sites 850, 857 and 858 and tested direct binding (Figure 5C and Table S2). The results suggest that each fragment offers a marginal (~2–4-fold) improvement consistent with multivalent systems where each contact point provides a small gain in binding energy. The binding profile supports the hypothesis that the additive effect of multiple fragments can provide a high affinity ligand.
Peptides that engage the MLL face of KIX, including the current optimized peptide, include a ϕ–x–x–ϕ–ϕ motif, where ϕ is a hydrophobic residue.10 Such helical motifs and hydrophobic residues, in general, are commonly found at protein–protein interfaces.32-34 To test if the designed helix with several hydrophobic modifications binds nonspecifically to other receptors that are known to accommodate the ϕ–x–x–ϕ–ϕ helical partners, we incubated the fluorescently labeled HBS II (termed HBS II*) with MDM2, which is a well-known binding partner of the p53 activation domain. We did not observe significant binding of the fragment-optimized HBS II* with MDM2 suggesting that this compound retains specificity for KIX (Figure S4).
Lastly, we utilized titration HSQC 15N NMR spectroscopy to evaluate the binding site profile of HBS II on KIX. Addition of 0.5 and 1 mol equivalent of HBS II to 15N labeled produced changes in chemical shifts of KIX backbone amide protons located on the MLL and cMyb faces of KIX (Figure 6A, Figure S5 and Table S3). Figure 6B depicts the changes chemical shifts of backbone amide mapped onto the MLL/KIX/cMyb protein complex (PDB 2AGH). Chemical shift changes were observed on both MLL and cMyb faces of KIX, which is not surprising because the two faces are allosterically connected.9,35 The largest shift was observed for K659, which is located near the strand portion of MLL and predicted to contact position 850 of HBS II. K659 has been shown to participate in the allosteric communication between different transcription factor binding sites on KIX.36 It is likely that the lead peptide is binding both faces of KIX, as is observed for native MLL.37 The exact mechanism of HBS II binding will require further explorations.
Figure 6.

(A) Bar graph shows mean chemical shift changes observed for the 15N-labeled KIX upon addition of HBS II. (B) Chemical shift changes are mapped on to a model from PDB 2AGH. Shifts are observed at both Myb and MLL faces, indicating that the compound is potentially modulating these allosterically connected faces of KIX.
CONCLUSION
Miniature mimics of proteins can serve as rationally designed inhibitors of biomolecular complexes, but the process of miniaturization also leads to loss of important binding contacts and affinity. One potential strategy for creating high affinity protein mimics is to engage protein binding pockets which may not be accessible to natural side chains. We have developed a combined computational and experimental approach to develop protein mimics with noncanonical side chains. The computational method AlphaSpace provides a topographical map of the protein surface and identifies underutilized targetable pockets which may be better occupied by nonnatural side-chain groups.5,6,38 The experimental approach builds on the computational method and allows screening of side-chain fragments at specific pockets. Here we described the results of this combined approach to develop peptidomimetics for KIX—a coactivator which has been difficult to target using conventional design and screening strategies.
Fragment screening is a well-established method for identifying hits, but a perpetual challenge for any fragment screening method is to coax the library to interrogate a specific pocket. Protein tethering offers an avenue for pocket specific screening of fragments.19,20 Building on this strategy, we tested a peptide tethering approach in which the peptide is expected to localize to a specific site on the cognate receptor as guided by native binding contacts—a chosen pocket can then be screened to optimize binding. While this study focused on cysteine-reactive chemistry, we expect that peptide tethering will be expanded beyond the scope of cysteine residues to accommodate different residues and chemistries.
A combination of the computational topographic mapping and peptide tethering afforded a peptidomimetic that binds the KIX domain of coactivators p300/CBP with >2000-fold improved affinity as compared to the wild-type sequence, resulting in a submicromolar inhibitor of KIX/MLL complex formation. Importantly, each fragment provides an additive boost in affinity. We and others have targeted several domains on this coactivator including the TAZ1 domain,31,39 histone acetyltransferase domain,40 and bromodomain,41 and we anticipate that the identified peptide will add to the list of ligands available to modulate p300/CBP linked gene expression.42
Supplementary Material
ACKNOWLEDGMENTS
We thank the National Institutes of Health (R35GM130333) for financial support of this work.
Footnotes
Supporting Information
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/jacs.1c09666.
Synthesis and characterization of peptides and constrained helices, fragment screening procedure, protein expression, purification and binding assays, and HSQC NMR analysis (PDF)
The authors declare no competing financial interest.
Contributor Information
Ashley E. Modell, Department of Chemistry, New York University, New York, New York 10003, United States
Frank Marrone, III, Department of Chemistry, New York University, New York, New York 10003, United States.
Nihar R. Panigrahi, Department of Chemistry, New York University, New York, New York 10003, United States
Yingkai Zhang, Department of Chemistry, New York University, New York, New York 10003, United States.
Paramjit S. Arora, Department of Chemistry, New York University, New York, New York 10003, United States.
REFERENCES
- (1).Pelay-Gimeno M; Glas A; Koch O; Grossmann TN Structure-Based Design of Inhibitors of Protein–Protein Interactions: Mimicking Peptide Binding Epitopes. Angew. Chem., Int. Ed 2015, 54, 8896–8927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Modell AE; Blosser SL; Arora PS Systematic Targeting of Protein-Protein Interactions. Trends Pharmacol. Sci 2016, 37, 702–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Braisted AC; Wells JA Minimizing a binding domain from protein A. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 5688–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Berdan VY; Klauser PC; Wang L Covalent peptides and proteins for therapeutics. Bioorg. Med. Chem 2021, 29, 115896. [DOI] [PubMed] [Google Scholar]
- (5).Torner JM; Yang Y; Rooklin D; Zhang Y; Arora PS Identification of Secondary Binding Sites on Protein Surfaces for Rational Elaboration of Synthetic Protein Mimics. ACS Chem. Biol 2021, 16, 1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Rooklin D; Modell AE; Li H; Berdan V; Arora PS; Zhang Y Targeting Unoccupied Surfaces on Protein–Protein Interfaces. J. Am. Chem. Soc 2017, 139, 15560–15563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Thakur JK; Yadav A; Yadav G Molecular recognition by the KIX domain and its role in gene regulation. Nucleic Acids Res. 2014, 42, 2112–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).De Guzman RN; Goto NK; Dyson HJ; Wright PE Structural basis for cooperative transcription factor binding to the CBP coactivator. J. Mol. Biol 2006, 355, 1005–13. [DOI] [PubMed] [Google Scholar]
- (9).Goto NK; Zor T; Martinez-Yamout M; Dyson HJ; Wright PE Cooperativity in Transcription Factor Binding to the Coactivator CREB-binding Protein (CBP): the mixed lineage leukemia protein (mll) activation domain binds to an allosteric site on the kix domain. J. Biol. Chem 2002, 277, 43168–43174. [DOI] [PubMed] [Google Scholar]
- (10).Denis CM; Chitayat S; Plevin MJ; Wang F; Thompson P; Liu S; Spencer HL; Ikura M; LeBrun DP; Smith SP Structural basis of CBP/p300 recruitment in leukemia induction by E2A-PBX1. Blood 2012, 120, 3968–77. [DOI] [PubMed] [Google Scholar]
- (11).Ramaswamy K; Forbes L; Minuesa G; Gindin T; Brown F; Kharas MG; Krivtsov AV; Armstrong SA; Still E; de Stanchina E; Knoechel B; Koche R; Kentsis A Peptidomimetic blockade of MYB in acute myeloid leukemia. Nat. Commun 2018, 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Best JL; Amezcua CA; Mayr B; Flechner L; Murawsky CM; Emerson B; Zor T; Gardner KH; Montminy M Identification of small-molecule antagonists that inhibit an activator: coactivator interaction. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 17622–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Majmudar CY; Hojfeldt JW; Arevang CJ; Pomerantz WC; Gagnon JK; Schultz PJ; Cesa LC; Doss CH; Rowe SP; Vasquez V; Tamayo-Castillo G; Cierpicki T; Brooks CL III; Sherman DH; Mapp AK Sekikaic acid and lobaric acid target a dynamic interface of the coactivator CBP/p300. Angew. Chem., Int. Ed. Engl 2012, 51, 11258–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Rutledge SE; Volkman HM; Schepartz A Molecular recognition of protein surfaces: high affinity ligands for the CBP KIX domain. J. Am. Chem. Soc 2003, 125, 14336–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Lodge JM; Majmudar CY; Clayton J; Mapp AK Covalent Chemical Cochaperones of the p300/CBP GACKIX Domain. Chembiochem 2018, 19, 1907–1912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Lodge JM; Rettenmaier TJ; Wells JA; Pomerantz WC; Mapp AK FP Tethering: a screening technique to rapidly identify compounds that disrupt protein-protein interactions. Med. Chem. Commun 2014, 5, 370–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Joy ST; Henley MJ; De Salle SN; Beyersdorf MS; Vock IW; Huldin AJL; Mapp AK A Dual-Site Inhibitor of CBP/p300 KIX is a Selective and Effective Modulator of Myb. J. Am. Chem. Soc 2021, 143, 15056–15062. [DOI] [PubMed] [Google Scholar]
- (18).Rooklin D; Wang C; Katigbak J; Arora PS; Zhang Y AlphaSpace: Fragment-Centric Topographical Mapping To Target Protein-Protein Interaction Interfaces. J. Chem. Inf Model 2015, 55, 1585–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Erlanson DA; Braisted AC; Raphael DR; Randal M; Stroud RM; Gordon EM; Wells JA Site-directed ligand discovery. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 9367–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Erlanson DA; Wells JA; Braisted AC Tethering: Fragment-Based Drug Discovery. Annu. Rev. Biophys. Biomol. Struct 2004, 33, 199–223. [DOI] [PubMed] [Google Scholar]
- (21).Bancet A; Raingeval C; Lomberget T; Le Borgne M; Guichou J-F; Krimm I Fragment Linking Strategies for Structure-Based Drug Design. J. Med. Chem 2020, 63, 11420–11435. [DOI] [PubMed] [Google Scholar]
- (22).Sadowsky JD; Burlingame MA; Wolan DW; McClendon CL; Jacobson MP; Wells JA Turning a protein kinase on or off from a single allosteric site via disulfide trapping. Proc. Natl. Acad. Sci. U. S. A 2011, 108, 6056–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Mabonga L; Kappo AP Protein-protein interaction modulators: advances, successes and remaining challenges. Biophys Rev. 2019, 11, 559–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sijbesma E; Somsen BA; Miley GP; Leijten-van de Gevel IA; Brunsveld L; Arkin MR; Ottmann C Fluorescence Anisotropy-Based Tethering for Discovery of Protein–Protein Interaction Stabilizers. ACS Chem. Biol 2020, 15, 3143–3148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Ostrem JM; Peters U; Sos ML; Wells JA; Shokat KM K-Ras(G12C) inhibitors allosterically control GTP affinity and effector interactions. Nature 2013, 503, 548–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Nazaré M; Matter H; Will DW; Wagner M; Urmann M; Czech J; Schreuder H; Bauer A; Ritter K; Wehner V Fragment Deconstruction of Small, Potent Factor Xa Inhibitors: Exploring the Superadditivity Energetics of Fragment Linking in Protein–Ligand Complexes. Angew. Chem., Int. Ed 2012, 51, 905–911. [DOI] [PubMed] [Google Scholar]
- (27).Shuker SB; Hajduk PJ; Meadows RP; Fesik SW Discovering High-Affinity Ligands for Proteins: SAR by NMR. Science 1996, 274, 1531–1534. [DOI] [PubMed] [Google Scholar]
- (28).Henchey LK; Porter JR; Ghosh I; Arora PS High specificity in protein recognition by hydrogen-bond-surrogate alpha-helices: selective inhibition of the p53/MDM2 complex. Chembiochem 2010, 11, 2104–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Chapman RN; Dimartino G; Arora PS A highly stable short alpha-helix constrained by a main-chain hydrogen-bond surrogate. J. Am. Chem. Soc 2004, 126, 12252–3. [DOI] [PubMed] [Google Scholar]
- (30).Patgiri A; Yadav KK; Arora PS; Bar-Sagi D An orthosteric inhibitor of the Ras-Sos interaction. Nat. Chem. Biol 2011, 7, 585–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Kushal S; Lao BB; Henchey LK; Dubey R; Mesallati H; Traaseth NJ; Olenyuk BZ; Arora PS Protein domain mimetics as in vivo modulators of hypoxia-inducible factor signaling. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 15602–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Watkins AM; Bonneau R; Arora PS Side-Chain Conformational Preferences Govern Protein–Protein Interactions. J. Am. Chem. Soc 2016, 138, 10386–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bullock BN; Jochim AL; Arora PS Assessing Helical Protein Interfaces for Inhibitor Design. J. Am. Chem. Soc 2011, 133, 14220–14223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Jochim AL; Arora PS Systematic Analysis of Helical Protein Interfaces Reveals Targets for Synthetic Inhibitors. ACS Chem. Biol 2010, 5, 919–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Toto A; Giri R; Brunori M; Gianni S The mechanism of binding of the KIX domain to the mixed lineage leukemia protein and its allosteric role in the recognition of c-Myb. Protein Sci. 2014, 23, 962–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Palazzesi F; Barducci A; Tollinger M; Parrinello M The allosteric communication pathways in KIX domain of CBP. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 14237–14242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Arai M; Dyson HJ; Wright PE Leu628 of the KIX domain of CBP is a key residue for the interaction with the MLL transactivation domain. FEBS Lett. 2010, 584, 4500–4504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Sadek J; Wuo MG; Rooklin D; Hauenstein A; Hong SH; Gautam A; Wu H; Zhang Y; Cesarman E; Arora PS Modulation of virus-induced NF-κB signaling by NEMO coiled coil mimics. Nat. Commun 2020, 11, 1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Lao BB; Grishagin I; Mesallati H; Brewer TF; Olenyuk BZ; Arora PS In vivo modulation of hypoxia-inducible signaling by topographical helix mimetics. Proc. Natl. Acad. Sci. U. S. A 2014, 111, 7531–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Lasko LM; Jakob CG; Edalji RP; Qiu W; Montgomery D; Digiammarino EL; Hansen TM; Risi RM; Frey R; Manaves V; Shaw B; Algire M; Hessler P; Lam LT; Uziel T; Faivre E; Ferguson D; Buchanan FG; Martin RL; Torrent M; Chiang GG; Karukurichi K; Langston JW; Weinert BT; Choudhary C; de Vries P; Kluge AF; Patane MA; Van Drie JH; Wang C; McElligott D; Kesicki E; Marmorstein R; Sun C; Cole PA; Rosenberg SH; Michaelides MR; Lai A; Bromberg KD Discovery of a selective catalytic p300/CBP inhibitor that targets lineage-specific tumours. Nature 2017, 550, 128–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Romero FA; Murray J; Lai KW; Tsui V; Albrecht BK; An L; Beresini MH; de Leon Boenig G; Bronner SM; Chan EW; Chen KX; Chen Z; Choo EF; Clagg K; Clark K; Crawford TD; Cyr P; de Almeida Nagata D; Gascoigne KE; Grogan JL; Hatzivassiliou G; Huang W; Hunsaker TL; Kaufman S; Koenig SG; Li R; Li Y; Liang X; Liao J; Liu W; Ly J; Maher J; Masui C; Merchant M; Ran Y; Taylor AM; Wai J; Wang F; Wei X; Yu D; Zhu BY; Zhu X; Magnuson S GNE-781, A Highly Advanced Potent and Selective Bromodomain Inhibitor of Cyclic Adenosine Monophosphate Response Element Binding Protein, Binding Protein (CBP). J. Med. Chem 2017, 60, 9162–9183. [DOI] [PubMed] [Google Scholar]
- (42).Breen ME; Mapp AK Modulating the masters: chemical tools to dissect CBP and p300 function. Curr. Opin. Chem. Biol 2018, 45, 195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.