

Abstract
Antimicrobial peptides (AMPs) are promising pharmaceutical candidates for the prevention and treatment of infections caused by multidrug-resistant ESKAPE pathogens, which are responsible for the majority of hospital-acquired infections. Clinical translation of AMPs has been limited, in part by apparent toxicity on systemic dosing, and by instability arising from susceptibility to proteolysis. Peptoids (sequence-specific oligo-N-substituted glycines) resist proteolytic digestion, and thus are of value as AMP mimics. Only a few natural AMPs such as LL-37 and polymyxin self-assemble in solution; whether antimicrobial peptoids mimic these properties has been unknown. Here we examine the antibacterial efficacy and dynamic self-assembly in aqueous media of eight peptoid mimics of cationic antimicrobial peptides designed to self-assemble, and two non-assembling controls. These amphipathic peptoids self-assembled in different ways, as determined by small angle X-ray scattering; some adopt helical bundles, while others form core-shell ellipsoidal or worm-like micelles. Interestingly, many of these peptoid assemblies show promising antibacterial, anti-biofilm and anti-abscess activity in vitro in both media and host-mimicking conditions, as well as in vivo. While self-assembly correlated overall with antibacterial efficacy, this correlation was imperfect. Certain self-assembled morphologies seem better suited for antibacterial activity. In particular, a peptoid exhibiting a high fraction of long, worm-like micelles showed reduced antibacterial, antibiofilm and anti-abscess activity against ESKAPE pathogens compared with peptoids that form ellipsoidal or bundled assemblies. This is the first report of self-assembling peptoid antibacterials with activity against in vivo biofilm infections relevant to clinical medicine.
Keywords: Peptoids, micelles, antibacterial, biofilm, abscess, infection
Graphical Abstract
Introduction
Natural antimicrobial peptides (AMPs), and their synthetic mimics 1 have emerged as promising therapeutics for treating infections caused by multi-drug resistant pathogens due to their broad-spectrum activity against both Gram-negative and Gram-positive bacteria 2. AMPs are diverse, short (generally < 40 amino acids), amphipathic, positively charged (+2 to +9 net charge) biomolecules found in all forms of life 3. In addition to their own potent antimicrobial activity, AMPs act synergistically in conjunction with traditional antibiotics. This property may reduce the induction of antimicrobial tolerance and resistance 4. We have developed peptidomimetics, known as peptoids, which are synthetic oligomers that mimic peptide structures 5. Peptoids are based on the same sequence of backbone atoms as natural peptides, but are less susceptible to proteolysis and enzymatic degradation because their functional side chains are appended to the backbone nitrogen (N)-atom rather than to the α-carbon atom 6. Peptoids are therefore sequence-specific N-substituted glycines. Peptoid AMP mimics studied to date have consisted of a relatively small number of different monomers, and typically were less than ~13 monomers in length 7. N-substitution of the peptoid backbone prevents it from serving as a hydrogen bond donor. Nonetheless, peptoids with certain sequences form stable secondary structures, such as helices 8-9.
It has been previously suggested that a relatively high number of peptoid residues is required to achieve sufficient levels of attractive side chain interactions for self-assembly, since backbone-backbone hydrogen bonding is restricted and flexibility is increased in peptoids relative to peptides 10. Covalent attachment of lipophilic tail residues can promote self-assembly by enhancing intermolecular hydrophobic interactions, inducing the formation of micellar macromolecular assemblies 11-12. Self-assembly of short, water-soluble, linear peptoids has also been demonstrated in the absence of chirality, hydrogen-bonding and charge group deionization 10. Therefore, in some instances, increased flexibility of the peptoid backbone can aid self-assembly due to accommodation of π−π stacking and hydrogen bonding between side chains 10. Further, we previously showed in Molchanova et al. how sequence length, degree of halogen substitution and halogen identity all impact on the self-assembly of peptoids, which in turn affects the antimicrobial activity 13. In the present work, we explored the relationship between self-assembly and biological activity against ESKAPE pathogens of a series of ten peptoids 14 (with and without lipid tails or halogen substitution), based on the previously described Peptoid 15 referred to herein as TM1, and another peptoid previously referred to as Peptoid 2 or 1-C134mer 15-17 (referred to herein as TM5).
The ESKAPE pathogens are six species of bacteria that are the primary cause of nosocomial (hospital-acquired) infections exhibiting virulence and multi-drug resistance 18. These species, including Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa and Enterobacter sp., have the propensity to form biofilms through a process of surface attachment, production of extracellular matrix, and maturation 18. Biofilm-forming bacteria often exist in densely populated communities (>107 CFU/mL) and are associated with ~65% and ~80% of all microbial and chronic infections, respectively 19. Current treatment regimens for biofilm infections are not standardized, and typically feature physical or mechanical removal of the biofilm (debridement) followed by antibiotic therapy using broad-spectrum antimicrobials 20. These treatments are often ineffective due to the ability of components of the biofilm to interfere with antibiotic activity 21 and the slower metabolic activity of organisms encapsulated deep within the biofilm architecture 22-23. Thus, there is an urgent need for new therapeutics with low induction of canonical resistance mechanisms in conjunction with potent antibiofilm activity 24-25.
We found that while the relationship between self-assembly and biofunctionality of the peptoids was complex, all active antimicrobial peptoids did form stable supramolecular assemblies. Several newly described supramolecular peptoid assemblies exhibited antimicrobial, anti-biofilm and anti-abscess activity against selected ESKAPE pathogens both in standard laboratory media and host-mimicking conditions in vitro and in vivo. This activity persisted even though small-angle X-ray scattering (SAXS) data showed that the peptoids adopted distinct macromolecular structures. Remarkably, certain peptoids retained their anti-biofilm activity in the context of polymicrobial infections comprised by clinical isolates of P. aeruginosa and S. aureus in host-mimicking conditions in vitro, while appearing to be safe and non-toxic to human cells even at concentrations as high as 256 μg/mL. Therefore, certain self-assembling peptoids described here offer good potential for development as anti-infective agents for the prevention and treatment of nosocomial infections caused by ESKAPE pathogens.
Results
Peptoids self-assembled into different defined nanostructures
Ten different peptoid AMP mimics, which were previously reported as antivirals active against Herpes Simplex Virus HSV-1 and SARS-CoV-2 14, were investigated herein as self-assembling antibacterial AMP mimics. LL-37 is an example of a self-assembling human antibacterial peptide that is also active against SARS-CoV-2 26-27. Of these ten peptoids, four were designed with terminal N-decyl or N-tridecyl alkyl tails, while all peptoids comprised α-chiral, aromatic Nspe and cationic lysine-like NLys residues (Figure 1). A detailed discussion of how the studied library of peptoids were designed was described previously 14. We investigated the self-assembly of these peptoids (except TM3) and of the natural human antimicrobial peptide LL-37 using small-angle X-ray scattering (SAXS) (Figure 2). Based on theoretical model analysis (Supplementary Information section 1.2 for details of models), the self-assembled structures of the peptoids in aqueous solution were determined (Figure 3). The peptoids without alkyl chains formed helical bundles either as dimers, trimers or/and tetramers (Figure 2A, 3A). The 12mer peptoid TM1, which prior studies have shown to be helical in secondary structure in association with anionic lipid micelles 5, 28, assembled mostly into dimers, but also demonstrated a smaller fraction of monomers and larger bundles (trimers/tetramers could not be distinguished in this mixture). TM6, an 11-mer version of TM1 lacking one C-terminal Nspe monomer, formed dimers and monomers with no larger bundles. This suggests that Nspe monomers are important for the intermolecular assembly of this class of peptoids, as seen in prior studies 29. TM7, a 6-mer version comprising one-half of TM1, was the only peptoid in the series that exhibited Gaussian chain morphology without higher order structure (with just a 0.003% fraction of larger aggregates (dimensions of 110Å x 350Å x >1000Å)), consistent with its shorter length. Helicity also might be important for self-assembly of this class of water-soluble peptoids due to their three-faced helical structures, and the known driving forces of assembly of amphipathic helices into bundles or coiled-coils to bury the hydrophobic side chains on two faces of the TM1 peptoid helix30. Helicity was previously found to be dependent on peptoid chain length 9.
Figure 1.
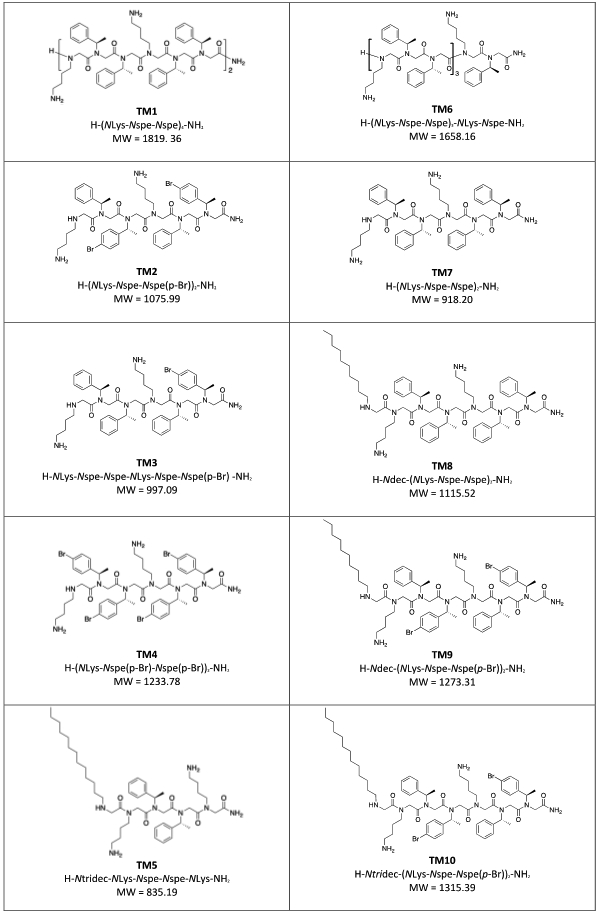
Chemical structures of the peptoids, TM1-10, included in this study, as previously presented in ref (13).
Figure 2.

Comparison of small angle X-ray scattering data of peptoids (4mM) and LL-37 plotted together with best fit (red line) using models described in the supplementary information. Peptoids could be distinguished according to their class, and are presented as groups of peptoids and peptide (A) or lipopeptoids (B).
Figure 3.

Morphology of peptoid/peptide aggregates based on best fit analysis of SAXS data. A) Monomers or helical bundles B) Core-shell ellipsoidal or worm-like micelles. The percentage of larger aggregates referrers to the presence of very small fraction of larger filaments seen from the sharp upturn at low Q in the scattering patterns.
*Structure above the critical micelle concentrations (CMC), which was undetectably low (in the order of 1 μg/ml).
The halogen-substituted peptoids TM2 and TM4 formed larger helical bundles (estimated to be tetramers based on theoretical modeling), likely through an effective “hydrophobic” interaction between the heavy bromine para-benzyl substituent atoms. TM2 (which includes two Nspe monomers with bromine substitutions) included a 0.005% fraction of larger aggregates (dimensions of 120Å x 280Å x >1000Å), seen as a sharp upturn at low Q (~0.009-0.03) (Figure 2A), while TM4 did not exhibit any of these larger aggregates. Interestingly, the SAXS pattern of TM4 was very similar to that of the human peptide LL-37 (included for comparison) (Figure 2A and 3A).
The scattered intensity obtained from the alkylated lipopeptoids exhibited a classical core-shell scattering pattern with clear oscillations, thus indicating the presence of micellar structures, which is expected due to the enhanced intermolecular hydrophobic interactions when introducing the alkyl tails 11-12. Interestingly, model analysis determined that TM5, a C13-terminated peptoid pentamer, formed ellipsoidal micellar assembles with Rcore = 13 Å, an eccentricity (ε) of 1.6 and a dR of 7 Å. Peptoid TM8, a C10-terminated heptamer, also formed ellipsoidal micellar assemblies but with a slightly smaller Rcore = 10 Å reflecting the shorter aliphatic tail, an ε = 1.9, and a slightly thicker shell as shown by a dR = 9 Å. TM8 exhibited an upturn at low Q indicating the presence of a small (~0.08%) fraction of bigger aggregates (dimensions of 150Å x 280Å x >1000Å). TM9 and TM10 are brominated versions of TM8 with either N-decyl or N-tridecyl amino-terminal tails, which assembled into mixtures of ellipsoids and longer worm-like micelles. While TM9 exhibited a relatively small (0.1%) fraction of worm-like micelles, this fraction was significantly higher for TM10 (0.4%).
Self-assembling peptoids inhibit the growth of ESKAPE pathogens
We previously showed that, at low micromolar concentrations (12.5 μM), TM1 inhibits growth of P. aeruginosa 17 and other clinically relevant Gram-negative and Gram-positive pathogens, including Escherichia coli, K. pneumoniae, E. faecalis and S. aureus 31. Here, we built on these studies using clinical isolates of the ESKAPE pathogens to assess the efficacy of this library of ten different peptoids, which contained eight novel compounds. TM1 inhibited growth of all ESKAPE pathogens at 1.56-12.5 μg/mL (Table 1). TM2 inhibited E. faecium and P. aeruginosa at the same concentrations as TM1 (1.56 and 12.5 μg/mL, respectively), but higher concentrations were needed to inhibit the growth of the other bacterial species. Similarly, TM4 inhibited E. faecium and P. aeruginosa at lower concentrations (0.78 and 6.25 μg/mL, respectively) than TM1, but equal or higher concentrations were needed to inhibit other species. TM5 inhibited growth of E. faecium, S. aureus and P. aeruginosa at concentrations equal to or less than those for TM1, whereas TM8 showed equal or improved activity toward E. faecium, A. baumannii and P. aeruginosa. In contrast, TM3, TM7, TM9 and TM10 exhibited equal or worse inhibitory activity toward all ESKAPE pathogens. This is intriguing since TM9 and TM10 formed worm-like micelles, which seems to be detrimental to the activity of these peptoids against ESKAPE pathogens.
Table 1.
Minimum inhibitory concentration (MIC; μg/ml) of peptoids against ESKAPE pathogens in Mueller-Hinton Broth (MHB) except for E. faecium #1-1, which was determined in TSB supplemented with 1% glucose.
| TM1 | TM2 | TM3 | TM4 | TM5 | TM7 | TM8 | TM9 | TM10 | |
|---|---|---|---|---|---|---|---|---|---|
| E. faecium #2-1 | 1.56 | 1.56 | 6.25 | 0.78 | 1.56 | 25 | 0.78 | 3.13 | 12.5 |
| S. aureus USA300 LAC | 1.56 | 6.25 | 50 | 6.25 | 1.56 | 100 | 3.13 | 1.56 | 12.5 |
| K. pneumoniae KPLN649 | 6.25 | 12.5 | 50 | 12.5 | 25 | >100 | 12.5 | 50 | >100 |
| A. baumannii AB5075 | 3.13 | 25 | >100 | 3.13 | 12.5 | >100 | 3.13 | 6.25 | 100 |
| P. aeruginosa LESB58 | 12.5 | 12.5 | 25 | 6.25 | 3.13 | 50 | 6.25 | 12.5 | 25 |
| E. cloacae 218R1 | 6.25 | 100 | >100 | 12.5 | 25 | >100 | 12.5 | 12.5 | 50 |
No apparent cytotoxicity of peptoids in vitro using primary human gingival cell cultures
Peptoids TM1, TM2, TM3, TM4, TM5, TM6, and TM9 (which we considered to be of interest as lead antibacterial compounds) exhibited no apparent cytotoxicity in vitro when applied to the apical surface of 3-dimensional tissue models of primary human gingival epithelial cells grown at the air-liquid interface, at concentrations as high as 256 μg/mL (Figure S4). These results are similar to those described for TM4, TM5, and TM9 with oral epithelial cells in a recently published paper 14.
Certain peptoids exhibit antibiofilm activity against ESKAPE pathogens
We further investigated the anti-biofilm activity of TM peptoids against all ESKAPE pathogens (Figure 4). At 1.56 μg/mL, TM1 exhibited slight but significant inhibition of biofilm formation by S. aureus, K. pneumoniae, A. baumannii, P. aeruginosa and E. cloacae relative to a PBS control. At 6.25 μg/mL, TM1 reduced the biomass of pre-formed biofilms for S. aureus, K. pneumoniae and P. aeruginosa and reduced metabolic activity of all ESKAPE pathogens in the biofilm growth state.
Figure 4.

Biofilm inhibition and eradication of ESKAPE pathogens at the lowest concentration of peptoid tested. In MBIC assays, 1.56 μg/ml of peptoid was used. In MBEC assays, 6.25 μg/ml of peptoid was used to treat A. baumannii but 12.5 μg/ml was used to treat all other species relative to PBS (%). Biofilm inhibition was measured by crystal violet (CV) staining, and eradication was measured by CV staining and tetrazolium chloride (TTC) reduction. Results from three independent experiments (n = 3) are displayed as mean using a grey-scale gradient where green indicates less biofilm (<75%) and red indicates more biofilm (> 120%).
TM2 inhibited biofilm formation by E. faecium and E. cloacae only, but slightly reduced the biomass of pre-formed biofilms for K. pneumoniae and P. aeruginosa and attenuated biofilm metabolic activity for all species except P. aeruginosa. TM3 and TM4 only exhibited significant biofilm inhibition against E. cloacae and showed minor reduction of pre-formed biofilm biomass for S. aureus and/or P. aeruginosa. However, these peptoids reduced biofilm metabolic activity for E. faecium, S. aureus and K. pneumoniae. TM5 inhibited biofilm formation by K. pneumoniae and P. aeruginosa but reduced biofilm biomass of S. aureus and K. pneumoniae and reduced biofilm metabolic activity for the same pathogens as well as for E. faecium and P. aeruginosa.
TM8, TM9 and TM10 inhibited biofilm formation by all pathogens except E. faecium and A. baumannii, although the effect for TM9 against S. aureus biofilm formation was not significant. The biomass of S. aureus and K. pneumoniae pre-formed biofilms was impacted across these peptoids and metabolic activity was reduced for all pathogens except P. aeruginosa as well as, in the case of TM10, E. faecium, A. baumannii and E. cloacae.
Peptoids exhibited superior anti-biofilm activity against S. aureus in both mono- and polymicrobial biofilms
The host environment is an important factor to consider for the assessment of novel therapeutic treatments. It has become increasingly recognized that the host environment affects drug activity 32-33. Therefore, we selected P. aeruginosa and S. aureus to represent Gram-negative and Gram-positive organisms, respectively, to further assess peptoid activity under host-mimicking conditions. Tissue culture medium supplemented with serum and glucose (DMEM-FBS-G) was used to assess antimicrobial and anti-biofilm activity of the peptoids. Overall peptoid antimicrobial activity was enhanced against S. aureus under these conditions (Table S1). In order to assess the anti-biofilm activity of the peptoids, we performed eradication experiments of monomicrobial P. aeruginosa, S. aureus and polymicrobial P. aeruginosa-S. aureus preformed biofilms in DMEM-FBS-G. While none of the peptoids reduced the biomass of P. aeruginosa biofilms significantly under these conditions (Figure 5A), all of the peptoids showed >50% biomass reduction against S. aureus at all concentrations tested (Figure 5B). Intriguingly, all of the peptoids achieved a reduction of polymicrobial biofilm mass by ~50%, except for TM9 (Figure 5C). We further determined 31.25 μg/mL as a potential, effective anti-biofilm concentration across all investigated peptoids against both mono- and polymicrobial biofilms (Figure 6). At this concentration, TM1, TM5, TM6 and TM8 reduced 17-25% of P. aeruginosa biomass when compared to the PBS-treated control biofilms (Figure 6A). Intriguingly, TM8 reduced P. aeruginosa significantly by 1,000-fold and although not significant, TM1 (p = 0.09) and TM6 (p = 0.22) also visually reduced P. aeruginosa bacterial numbers by ~1,000-fold and, 100-fold, respectively (Figure 6B). Against S. aureus, all of the peptoids visually reduced biomass by at least 70% except TM6, which had a 28% reduction. TM8 was the only peptoid that significantly reduced S. aureus biomass (by 81%) (Figure 6C). S. aureus cells were significantly reduced by at least 10,000-fold in wells treated with TM1, TM2 and TM6 (i.e., CFU recovery below limit of detection) (Figure 6D).
Figure 5.

Effect of peptoids under host-mimicking conditions on mono- and polymicrobial biofilm eradication. (A) P. aeruginosa LESB58 (5 × 105 CFU/ml) and (B) S. aureus USA300 LAC (2.5 × 107 CFU/ml) mono- and (C) polymicrobial biofilms were grown for 20-24 h in DMEM-FBS-G prior to treatment with peptoids. Biofilms were stained with crystal violet (0.1%) after an additional 24 h. Values were normalised to the biofilm growth control, which is indicated by the dotted line. Data from three independent experiments (n = 3) are presented as the mean ± SEM.
Figure 6.

Effect of peptoids on mono- and polymicrobial biofilms. Peptoids (31.25 μg/ml) were used to treat biofilms comprising (A,B) P. aeruginosa LESB58 (5 × 105 CFU/ml), (C,D) S. aureus USA300 LAC (2.5 × 107 CFU/ml) or (E,F) both species. Biofilms were grown for 20-24 h in DMEM-FBS-G prior to treatment and re-incubated for another 20-24 h. (A,C,E) Biofilm was quantified by CV staining (%) and (B,D,F) and bacterial recovery from biofilms (CFU/ml) determined on selective agar plates. The dotted line indicates the limit of detection (LOD) at 102 CFU. Data from three independent experiments each (n = 3) are shown as (A,C) mean ± SEM or geometric mean ± geometric SD. * P < 0.05, ** P < 0.01 according to Kruskal-Wallis test with Dunn’s correction.
Overall, a >50% reduction of biomass was observed for all peptoids against P. aeruginosa-S. aureus polymicrobial biofilms (Figure 6E). TM1 and TM2 reduced biomass significantly (p=0.003) by 73% and (p=0.04) by 66%, respectively. Biofilms treated with TM4 showed higher biomass staining, but significantly (p=0.04) reduced S. aureus within the biofilm below the limit of detection of 102 CFU/mL. TM1 and TM6 also reduced S. aureus within the biofilm significantly (p=0.03 and p=0.002, respectively) below the limit of detection, however, the majority of peptoids reduced viable S. aureus within the biofilm by ~10,000 fold, thus indicating that the biofilms were P. aeruginosa dominant. By comparison, the majority of the peptoids reduced P. aeruginosa by ~10-fold within the polymicrobial biofilm. TM1 and TM6 were the only peptoids that significantly reduced P. aeruginosa, by 100-fold (p=0.02) and 1,000-fold (p=0.001), respectively (Figure 6F).
Peptoids reduced S. aureus abscess size and bacterial load in vivo
We formed skin abscesses in mice using P. aeruginosa and S. aureus as representatives of the Gram-negative and Gram-positive ESKAPE pathogens (Figure 7). One hour post infection, peptoids were administered at their maximum tolerated dose (2.5 mg/kg for TM1, TM2, and TM4, and 1.25 mg/kg for TM5, and TM8; Table S3) and compared to a PBS control. TM1 reduced P. aeruginosa abscess size by a factor of ~2 (from 79.6 mm2 to 36.8 mm2) and significantly reduced bacterial load ~5-fold (from 4.9 x 108 CFU/mL to 8.2 x 107 CFU/mL). TM4 also reduced abscess sizes ~2-fold to 35.5 mm2 but did not significantly impact on bacterial load (3.2 x 108 CFU/mL). Other peptoids did not significantly impact on abscess size or bacterial load in vivo when compared to the PBS control. Intriguingly, more peptoids maintained their activity toward S. aureus infection in vivo. TM1 and TM2 showed the greatest reduction of abscess size (>90%, from 47.2 mm2 to 4.6 and 2.7 mm2, respectively), respectively, and also the greatest reduction in bacterial load (by >10,000-fold, from 2.3 x 108 CFU/mL to 4.9 x 104 for TM1, and by >100,000-fold to 8.5 x 102 CFU/mL for TM2). TM4 and TM5 also reduced abscess size by ~80% to 5.6 and 10.2 mm2, respectively, and reduced bacterial load by ~1,000-fold. TM8 insignificantly reduced S. aureus abscess size as well as bacterial burden.
Figure 7.

In vivo activity of maximum tolerated concentration of peptoids against clinical isolates of P. aeruginosa (A,B) and S. aureus (C,D). Mice were subcutaneously inoculated with ~2.5-5 x 107 CFU P. aeruginosa LESB58 or ~3-5.5 x 107 CFU S. aureus USA300 LAC and treated with peptoid or PBS one h later. After three days, mice were euthanized, abscesses measured (A,C), and then collected for bacterial enumeration (B,D). Results displayed as median with whiskers to min and max (A,C) or geometric mean ± geometric SD (B,D). * P < 0.05, ** P < 0.01 different from PBS according to Kruskal-Wallis test. n = 10. Limit of detection (LOD) displayed as dotted line at 102 CFU.
Discussion
Antimicrobial resistance is rapidly accelerating, thus creating an urgent need for new antibacterial drugs. Here, we explored a family of peptoids comprising two well studied compounds TM1 and TM5, and eight peptoids (TM2-TM4, TM6-TM10) which are variants and molecular hybrids of TM1 and TM5. We used these peptoids to treat ESKAPE pathogens, and investigated their activity against Gram-negative P. aeruginosa and Gram-positive S. aureus under host-mimicking conditions and in a very-challenging high-density cutaneous mouse abscess infection model. These peptoids varied in chain length (6mer-12mer), net positive charge and hydrophobicity by inclusion of varying numbers of Nspe monomers, covalently bound alkyl chains and halogen substitutions.
The majority of these peptoids exhibited antimicrobial activity against all ESKAPE pathogens, except for the 6-mer TM7, which was found to have no antimicrobial or anti-biofilm activity (Table 1, Figure 4). This correlated with TM7 being the only peptoid not found to self-assemble to some degree in solution, which was likely due to its short length. All of the other peptoids investigated by SAXS revealed strong intermolecular interactions promoting self-assembly into larger bundled or micellar structures(Fig 1). The critical micellar concentrations (CMCs) as estimated by modeling of SAXS data generally seem to be below the MIC range (in the order of 1 μg/ml) , which supports our hypothesis that sufficient self-assembly contributed to both antimicrobial and anti-biofilm activity of peptoids. Self-assembly into defined multimers have rarely been observed in natural AMPs despite of their amphiphilic properties.34 However, one exception is the human cathelicidin LL-37 27, 35, which formed larger tetrameric helical bundles according to the current study (Figures 2, 3). In a previous study TM1, TM6 and LL-37 were all found to cross bacterial membranes, bind to DNA, and also rapidly aggregate bacterial ribosomes in vitro and in vivo, and these phenomena have therefore been suggested as key mechanisms of killing for both cationic, amphipathic peptoids and peptides 36. We hypothesize that these newly discovered supramolecular peptoid assemblies disassociate when they come in contact with anionic bacterial membranes, explaining the rapid bacterial membrane permeabilization that has been observed for these peptoids (unpublished data, manuscript in preparation).
Other peptoids with intriguing activity are TM3 and TM10; both exhibited antimicrobial activity against P. aeruginosa that was comparable to TM1 (the antimicrobial activity of TM3 and TM10 was at least 4-fold decreased against other ESKAPE pathogens). TM10 is a lipopeptoid with similar structure to TM5, TM8 and TM9 (Figure 1, Figure 3), however TM10 forms a higher proportion of worm-like micelles (TM5 and TM8 formed only ellipsoidal micelles, while TM9 formed a mixture of 90 % ellipsoidal and 10 % worm-like micelles, and TM10 60 % ellipsoidal and 40 % worm-like micelles). We hypothesize that worm-like physical morphology, if too stable, can inhibit antibacterial and antiviral activity, which may therefore account for TM10’s reduced activity when compared to its analogs TM5, TM8, and TM9, which exhibited antimicrobial activity within 2-4-fold of TM1. Furthermore, TM10 also recently demonstrated lower anti-viral activity when compared to TM9 14. Given the structural similarity of TM9 and TM10, which differ only in the lengths of their alkyl chains (C10 for TM9, C13 for TM10), these results suggest that both hydrophobicity and micellar aggregation number contribute to the biological function of peptoids. In line with this, the activity of the YGAAKKAAKAAKKAAKAA (AKK) peptides conjugated to fatty acids of varying lengths was lost when the minimal active concentration exceeded the CMC 37. While the conjugation of the AKK peptide with fatty acid tails increased their affinity for anionic lipid membranes, the self-assembled structure (obtained at concentrations above the CMC) apparently could inhibit the efficient binding of the peptide to bacterial cell membranes 37, and we hypothesize also intracellular mechanisms of action, although those were not mentioned in that report. Thus, for alkylated AMPs, optimal activity may require a proper balance between hydrophobicity and self-assembly, which is different for various biological activities (e.g. TM10 showed no antibiofilm activity against P. aeruginosa). However, as this hypothesis is based only on results obtained with TM10 together with limited literature references, further studies are warranted that focus more specifically on how morphology and stability of self-assembled structures affects bioactivity, especially as regards the antibacterial activity of the ellipsoidal micellar vs. more extended wormlike structures.
This study suggests that the chemical structure of antimicrobial peptoids is not the only factor that needs to be considered when assessing biological activity: we must also consider their propensity to self-assemble in a physiological aqueous environment. Moreover, various features of the host environment including pH, nutrient availability and the presence of albumin also can impact on peptoid activity 38. The host environment is known to affect bacterial virulence 39 and antibiotic efficacy 33 and can also affect interspecies interactions. Under physiologically relevant conditions, peptoid antimicrobial activity was enhanced against S. aureus when compared to results in nutrient-rich laboratory medium (Table S1). Furthermore, the peptoids exhibited potent anti-biofilm activity against S. aureus in mono- and polymicrobial biofilms but were less active vs. P. aeruginosa (Figure 5, Figure 6).
The higher antimicrobial and anti-biofilm activity of these cationic peptoids toward S. aureus compared to P. aeruginosa could be attributed at least partially to acidification observed in the S. aureus-inoculated wells (Figure S5). It is known that many AMPs, including several that have successfully completed clinical trials 40, have pH-dependent activity. This may be caused by protonation of amino acid residues, which can influence their interaction with bacterial membranes 41 and other targets and thereby promote peptide synergy with other membrane targeting antibiotics and host defense molecules 42-43. In an analogous manner, the net positive charge of the presently described cationic peptoids may have increased under the low pH (acidic) conditions observed 44-45, thus increasing their affinity for the slightly anionic (lipo)teichoic acids in the cell wall of S. aureus.
TM1 and TM6 retained significant activity toward P. aeruginosa and S. aureus polymicrobial biofilms, whereas other peptoids studied herein were less effective towards at least one of the species compared to their respective activity in monomicrobial infection (Figure 6). TM1 and TM6 are self-assembling peptoids that form a mixture of monomers and mainly dimeric helical bundles. However, they lack halogen substitution and an alkyl tail, both of which are known to increase hydrophobicity and, consequently, the tendency for supramolecular assembly 11, 13, 46. In contrast, peptoids with halogen substituents, such as TM2 (half-substituted with bromination of every other phenyl ring) and TM4 (fully substituted), did not exhibit any anti-biofilm activity against P. aeruginosa in monomicrobial or polymicrobial biofilms (Figure 6). Previously, it was observed that a library of peptoids similar in structure to the peptoids studied here (and also based on TM1-analogs) were effective against polymicrobial biofilms comprising Candida albicans, E. coli and S. aureus 47. However, as here, C. albicans was less susceptible to the peptoids in polymicrobial biofilms than monomicrobial biofilms. These data highlight the complexity of treating infections based on polymicrobial biofilms, and how the interactions between species may impact the effectiveness of treatment strategies using AMPs and peptoids.
The addition of a terminal alkyl tail to peptoids was shown here to enable the supramolecular assembly of core-shell micellar structures, obviously with a significantly higher aggregation number than for the helical bundles formed by TM1 and TM6. Peptoids TM5, TM8 and TM9 all formed ellipsoidal micellar assemblies, with aggregation numbers of approximately 98, 103 and 117 peptoids on average, respectively. These high aggregation numbers are remarkable and suggest that there are intermolecular interactions beyond hydrophobic forces, for example potential hydrogen bonding between NLys groups and π−π stacking between Nspe groups. Judging from the aggregation number alone, the physical stability of these ellipsoids would be substantial. This may be beneficial for the effective drugability of these peptoids, since these supramolecular peptoid assemblies seem to act as a sort of vehicle-free self-controlled delivery system 48. This approach is advantageous in that it allows for the elimination of any need for physical encapsulation or for the covalent conjugation of pharmaceutical excipients 49. In the subcutaneous abscess infection model presented in this study, mice were injected with peptoids dissolved in PBS without the inclusion of excipients or of delivery vehicles, thus demonstrating this benefit.
Our murine abscess model of infection provides insights into the activity of peptoids in human skin infections. The high densities of bacteria in this model make treatment very challenging, since antibiotics do not work well against high density infections. Peptoids exhibited significant reduction of bacterial burden under physiologically relevant conditions, when tested against S. aureus and little reduction of P. aeruginosa reflective of our in vitro anti-biofilm data (Figure 5, Figure 6). The pH of the murine skin abscess remained slightly acidic throughout the course of infection (Figure S6). Although P. aeruginosa did not influence the pH of in vitro media when compared to the sterile control (Figure S6), the antibacterial activity of TM1 was reduced (2-fold) by adjusting the pH to 5.4-5.9 (Table S2). This might at least partially explain why TM1 reduced the P. aeruginosa bacterial load only modestly (~10-fold) in vivo (Figure 7). However, peptoids TM1 and TM4 did substantially reduce the sizes of abscesses formed by P. aeruginosa when compared to untreated controls, indicating that the anti-abscess activity of peptoids is distinct from their direct antimicrobial activity. This could involve an inhibition of inflammation, as several naturally occurring AMPs have demonstrated previously 50, or the inhibition of other host-mediated responses to high-density infections not yet explored. None of TM1, TM2 or TM4 caused toxic side-effects at a concentration of 2.5 mg/kg, whereas TM5 and TM8 did. Thus, TM5 and TM8 were administered intra-abscess at a lower concentration of 1.25 mg/kg. Since activity of peptoids could be concentration-dependent, comparison of peptoids used at different concentrations was limited.
This constitutes the first report of discrete, limited supramolecular peptoid assemblies with antibacterial activity, and further demonstrates that supramolecular assembly has complex effects on bioactivity. This new understanding of how the self-assembly of antimicrobial peptoids can affect both their in vitro and in vivo activity against ESKAPE pathogens can assist us with future molecular design projects. It is notable that many of the best antibacterial peptoids studied here, also exhibit antiviral properties, which is also true of the human host defense peptide LL-37 itself. Finally, our results reveal that several of the novel peptoids reported here simultaneously possess antimicrobial, anti-biofilm and anti-abscess activity in standard laboratory media as well as in host- mimicking conditions. Since the selected peptoids studied here retained their activity in diverse, physiologically relevant conditions, they should be considered for further therapeutic development, particularly for treating high-density skin wound infections. We also confirm that these peptoids exhibit no apparent cytotoxicity in vitro with primary human cells at concentrations of up to 256 μg/mL, making them exciting drug candidates as a novel class of anti-infectives.
Materials and Methods
Source of Peptoids and stock solutions
The peptoids TM1-10 (see Figure 1) were synthesized manually as previously described 14 (Supplementary Information subsection 1.1 for details). Peptoids were dissolved in phosphate buffered saline (PBS), pH=7.5 (Gibco) and prepared at 2.5 mg/mL stock solutions prior to storage at −80°C.
Small Angle X-ray Scattering (SAXS)
Unless noted otherwise, SAXS experiments were performed at ALS beamline 12.3.1 LBNL Berkeley, California, USA 51, with a detector distance of 2 meter and X-ray wavelength of λ=1.27 Å, covering a Q range of 0.009 Å−1 to 0.4 Å−11. The data set was calibrated to an absolute intensity scale using water as a primary standard. All experiments were performed at 20°C and data were processed as previously described 52.
TM7 and TM10 were measured using a Bruker NANOSTAR equipped with a microfocus X-ray source (IμS Cu, Incoatec, Germany) and a VÅNTEC-2000 detector. Raw scattering data was calibrated to absolute intensity scale using water as a primary standard and radially averaged in order to obtain the 1D scattered intensity profile as a function of the scattering vector, with a wavelength of 1.54 Å. The modelling fit analysis of the scattering data is explained in detail in subsection 1.2 of the Supplemental Information.
Bacterial strains and growth conditions
Bacterial strains used in this study were E. faecium #2-1 (BEI resources, NR31909), S. aureus USA300 LAC 53 K. pneumoniae KPLN649 54, A. baumannii AB5075 55, P. aeruginosa LESB58 56 and E. cloacae 218R1 57. All organisms were streaked onto Lysogeny Broth (LB) or double Yeast Tryptone (dYT) agar plates from frozen stocks and grown at 37°C. When needed, overnight liquid subcultures were grown from a single colony with shaking (250 rpm) for no more than 16 h. Bacterial growth was monitored using a spectrophotometer (Eppendorf, Missisauga, ON).
Cytotoxicity assay
Normal human gingival epithelial cells, obtained from MatTek (EpiGingival) were grown at an air-liquid interface at 37°C. Serial dilutions of peptoid were prepared from stocks, resulting in a final concentration of 64-256 μM. Peptoid (100 μl) was applied to the apical surface of cultures for 3 h. Cell viability was quantified using the CyQUANT MTT Cell Viability Assay (ThermoFisher), following the manufacturer’s instructions. OD540 was scanned and survival relative to untreated cultures (%) was calculated. The experiment was performed in triplicate. Ethanol was used as a positive control.
Biofilm growth conditions
A. baumannii, P. aeruginosa and E. cloacae biofilms were grown in MHB for 18-24 h. S. aureus biofilms were grown in MHB incubated shaking (200 rpm) for 24 h. K. pneumoniae and E. faecium biofilms were grown in TSB supplemented with 0.1% and 1% glucose respectively for 48 h. For host-mimicking conditions, P. aeruginosa and S. aureus biofilms were grown in Gibco high glucose Dulbecco’s minimal Eagle’s medium (DMEM) supplemented with 5% fetal bovine serum (FBS) (USA origin) and 1% glucose (G) (Sigma-Aldrich), referred to as DMEM-FBS-G. Biofilms were formed as previously described 58 with minor modifications (see subsection 1.3 in Supplemental Information).
Antimicrobial activity of peptoids
Bacterial susceptibility to peptoids was determined across ESKAPE pathogens using the broth microdilution assay 59 in microtiter plates (Falcon, #351172). Details are included in subsection 1.4 in Supplemental Information.
Antimicrobial activity of peptoids under host-mimicking conditions
MIC of peptoids were determined using an adapted microdilution broth method 59 in polypropylene 96-well plates (Greiner Bio-One, #655201) using MHB (Oxoid) and DMEM-FBS-G. Details are included in subsection 1.5 in Supplemental Information. All tests were performed in triplicate following the Clinical and Laboratory Standards Institute guidelines 59-60.
Minimal biofilm inhibition (MBIC) and eradication (MBEC) assays of ESKAPE pathogens
Biofilm formation conditions across species are included in subsection 1.6 of the Supplemental Information. Data from three biological replicates (n = 3) are presented as the percentage of biofilm, relative to the vehicle control (PBS) at the lowest concentration of tested peptoid (6.25-12.5 μg/mL). Biofilm inhibition and eradication was measured by crystal violet (CV) staining and/or 2,3,5-triphenyl tetrazolium chloride (TTC) reduction as previously described 61.
Biofilm eradication experiments under host-mimicking conditions
The eradication methodology was adapted from Haney et al. 62 with minor modifications (see subsection 1.7 in Supplemental Information). For eradication experiments with 31.25 μg/mL peptoids, biofilms were scraped with a sterile cotton swab, submerged in 1 mL of MHB, vortexed and further used for serial dilutions. Dilutions were plated onto LB agar for bacterial enumeration. For polymicrobial cultures selective agar plates were used: Pseudomonas cetrimide agar (Oxoid) to select for P. aeruginosa and 7.5% NaCl plates to select for S. aureus. Experiments were repeated three times with five technical replicates each.
Ethics statement
Animal experiments were performed in accordance with the Canadian Council on Animal Care (CCAC) guidelines and were approved by the University of British Columbia Animal Care Committee (protocol A19-0064) and the University of Otago Animal Welfare Office (protocol 19-125). Details are included in subsection 1.8 of the Supplemental Info.
Subcutaneous abscess infection
Peptoid susceptibility of P. aeruginosa and S. aureus was assessed in vivo using a subcutaneous abscess model, as previously described 63. Modifications are described in subsection 1.8 of the Supplemental Info.
For in vivo pH measurements, mice were subcutaneously injected with 50 μl of P. aeruginosa, S. aureus or a mixture (1:1) of P. aeruginosa and S. aureus (corresponding to 1.25-2.5 × 107 CFU). One h post-infection, mice were punctured with an 18G needle and the InLab Nano Combination Electrode was inserted to measure the voltage (mV). This process was repeated daily and mice were euthanized on day three.
Supplementary Material
Acknowledgment:
JEN, HJ and RL gratefully acknowledge NordForsk (Project no. 82004) for financial support. JEN also acknowledge financial support from UiO:Life Science and the Norwegian PhD School of Pharmacy. NM and HJ were also funded by the Danish Council for Independent Research, Technology and Production (Project no. 4005-00029). AEB, JF, JSL and JEN acknowledge funding from the U.S. Public Health Services (an NIH Pioneer Award to Annelise Barron, NIH/NIA grant # 1DP1 OD029517-01). MAA holds a UBC Killam Doctoral Scholarship, Four-Year Fellowship and CIHR Vanier Graduate Scholarship. We also gratefully acknowledge funding to REWH from the Canadian Institutes for Health Research grant FDN-154287 and Michael Smith Foundation for Health Research grant 17774. REWH holds a Canada Research Chair in Health and Genomics and a UBC Killam Professorship. DP acknowledges funding from the University of Otago Research Grant, the Otago Medical Research Foundation Grant (AG388) and Maxwell Biosciences. We also thank Dr. Allan Gamble at the University of Otago for his help with the pH microelectrode experiments. We also thank Erika Figgins at the University of Louisville for support with the in-vitro cytotoxicity experiments. Parts of this work was conducted at the Advanced Light Source (ALS), a national user facility operated by Lawrence Berkeley National Laboratory on behalf of the Department of Energy, Office of Basic Energy Sciences, through the Integrated Diffraction Analysis Technologies (IDAT) program, supported by DOE Office of Biological and Environmental Research. Additional support comes from the National Institute of Health project ALS-ENABLE (P30 GM124169) and a High-End Instrumentation Grant S10OD018483. We thank Dr. Gregory Hura and Kathryn Burnett at ALS for support during the SAXS experiment. Work at the Molecular Foundry was supported by the Office of Science, Office of Basic Energy Sciences, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231. We gratefully acknowledge Dr. Michael Connolly and Dr. Behzad Rad at the Molecular Foundry for assistance with peptoid synthesis and sample preparation equipment. We acknowledge use of the Norwegian national infrastructure for X-ray diffraction and scattering (RECX).
Abbreviations
- A. Baumannii
Acinetobacter baumannii
- AMP
antimicrobial peptide
- C. Albicans
Candida albicans
- CFU
colony forming unit
- CMC
critical micellar concentration
- CV
crystal violet
- DMEM
Dulbecco’s minimal Eagle’s medium
- DNA
deoxyribonucleic acid
- dYT
double Yeast Tryptone
- E. Cloacae
Enterobacter cloacae
- E. Faecium
Enterococcus faecium
- ESKAPE
Enterococcus faecium, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii,; Pseudomonas aeruginosa, and Enterobacter sp.
- FBS
fetal bovine serum
- K. pneumoniae
Klebsiella pneumoniae
- LB
Lysogeny Broth
- MHB
Mueller Hinton Broth
- NLys
N-(4-aminobutyl)glycine
- Nspe
(S)-N-(1-phenylethyl)amine
- P. aeruginosa
Pseudomonas aeruginosa
- PBS
phosphate buffered saline
- S. Aureus
Staphylococcus aureus
- SAXS
small angle X-ray scattering
- TTC
2,3,5-triphenyl tetrazolium chloride
Footnotes
Supporting Information.
Synthesize and purification of peptoids; theoretical modelling of SAXS data; experimental procedure for cytotoxicity assays and more detailed information on Biofilm and MIC assays; SAXS data at different peptoid concentrations; cytotoxicity data; data on antimicrobial activity under host mimicking conditions; in-vivo toxicity data; and data on pH of the localized abscess infection as a function of time.
References
- 1.Namivandi-Zangeneh R; Wong EH; Boyer C, Synthetic Antimicrobial Polymers in Combination Therapy: Tackling Antibiotic Resistance. ACS Infectious Diseases 2021, 7 (2), 215–253. [DOI] [PubMed] [Google Scholar]
- 2.Sinha R; Shukla P, Antimicrobial peptides: recent insights on biotechnological interventions and future perspectives. Protein Pept Lett 2019, 26 (2), 79–87. DOI: 10.2174/0929866525666181026160852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jenssen H; Hamill P; Hancock REW, Peptide antimicrobial agents. Clin Microbiol Rev 2006, 19 (3), 491–511. DOI: 10.1128/cmr.00056-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pletzer D; Mansour SC; Hancock REW, Synergy between conventional antibiotics and anti-biofilm peptides in a murine, sub-cutaneous abscess model caused by recalcitrant ESKAPE pathogens. PLoS Pathog 2018, 14 (6), e1007084. DOI: 10.1371/journal.ppat.1007084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Chongsiriwatana NP; Patch JA; Czyzewski AM; Dohm MT; Ivankin A; Gidalevitz D; Zuckermann RN; Barron AE, Peptoids that mimic the structure, function, and mechanism of helical antimicrobial peptides. Proc Natl Acad Sci U S A 2008, 105 (8), 2794. DOI: 10.1073/pnas.0708254105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Park M; Jardetzky TS; Barron AE, NMEGylation: a novel modification to enhance the bioavailability of therapeutic peptides. Biopolymers 2011, 96 (5), 688–693. DOI: 10.1002/bip.21607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mojsoska B; Zuckermann RN; Jenssen H, Structure-activity relationship study of novel peptoids that mimic the structure of antimicrobial peptides. Antimicrob Agents Ch 2015, 59 (7), 4112–4120. DOI: 10.1128/Aac.00237-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Brandt W; Herberg T; Wessjohann L, Systematic conformational investigations of peptoids and peptoid–peptide chimeras. Biopolymers 2011, 96 (5), 651–668. DOI: 10.1002/bip.21620. [DOI] [PubMed] [Google Scholar]
- 9.Sanborn TJ; Wu CW; Zuckermann RN; Barron AE, Extreme stability of helices formed by water-soluble poly-N-substituted glycines (polypeptoids) with α-chiral side chains. Biopolymers: Original Research on Biomolecules 2002, 63 (1), 12–20. [DOI] [PubMed] [Google Scholar]
- 10.Castelletto V; Seitsonen J; Tewari KM; Hasan A; Edkins RM; Ruokolainen J; Pandey LM; Hamley IW; Lau KHA, Self-assembly of minimal peptoid sequences. ACS macro letters 2020, 9 (4), 494–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Lau KHA; Castelletto V; Kendall T; Sefcik J; Hamley IW; Reza M; Ruokolainen J, Self-assembly of ultra-small micelles from amphiphilic lipopeptoids. Chemical Communications 2017, 53 (13), 2178–2181. [DOI] [PubMed] [Google Scholar]
- 12.Sternhagen GL; Gupta S; Zhang Y; John V; Schneider GJ; Zhang D, Solution self-assemblies of sequence-defined ionic peptoid block copolymers. Journal of the American Chemical Society 2018, 140 (11), 4100–4109. [DOI] [PubMed] [Google Scholar]
- 13.Molchanova N; Nielsen JE; Sørensen KB; Prabhala BK; Hansen PR; Lund R; Barron AE; Jenssen H, Halogenation as a tool to tune antimicrobial activity of peptoids. Scientific reports 2020, 10 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Diamond G; Molchanova N; Herlan C; Fortkort JA; Lin JS; Figgins E; Bopp N; Ryan LK; Chung D; Adcock RS; Sherman M; Barron AE, Potent antiviral activity against HSV-1 and SARS-CoV-2 by antimicrobial peptoids. Pharmaceuticals (Basel) 2021, 14 (4). DOI: 10.3390/ph14040304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Uchida M; McDermott G; Wetzler M; Le Gros MA; Myllys M; Knoechel C; Barron AE; Larabell CA, Soft X-ray tomography of phenotypic switching and the cellular response to antifungal peptoids in Candida albicans. Proceedings of the National Academy of Sciences 2009, 106 (46), 19375–19380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chongsiriwatana NP; Miller TM; Wetzler M; Vakulenko S; Karlsson AJ; Palecek SP; Mobashery S; Barron AE, Short alkylated peptoid mimics of antimicrobial lipopeptides. Antimicrobial agents and chemotherapy 2011, 55 (1), 417–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kapoor R; Wadman MW; Dohm MT; Czyzewski AM; Spormann AM; Barron AE, Antimicrobial peptoids are effective against Pseudomonas aeruginosa biofilms. Antimicrob Agents Ch 2011, 55 (6), 3054–7. DOI: 10.1128/AAC.01516-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mulani MS; Kamble EE; Kumkar SN; Tawre MS; Pardesi KR, Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: a review. Frontiers in microbiology 2019, 10, 539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Jamal M; Ahmad W; Andleeb S; Jalil F; Imran M; Nawaz MA; Hussain T; Ali M; Rafiq M; Kamil MA, Bacterial biofilm and associated infections. J Chin Med Assoc 2018, 81 (1), 7–11. DOI: 10.1016/j.jcma.2017.07.012. [DOI] [PubMed] [Google Scholar]
- 20.Hughes G; Webber MA, Novel approaches to the treatment of bacterial biofilm infections. Br J Pharmacol 2017, 174 (14), 2237–2246. DOI: 10.1111/bph.13706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sabnis A; Ledger EVK; Pader V; Edwards AM, Antibiotic interceptors: creating safe spaces for bacteria. PLoS Pathog 2018, 14 (4), e1006924–e1006924. DOI: 10.1371/journal.ppat.1006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Rabin N; Zheng Y; Opoku-Temeng C; Du Y; Bonsu E; Sintim HO, Biofilm formation mechanisms and targets for developing antibiofilm agents. Future Med Chem 2015, 7 (4), 493–512. DOI: 10.4155/fmc.15.6. [DOI] [PubMed] [Google Scholar]
- 23.Stokes JM; Lopatkin AJ; Lobritz MA; Collins JJ, Bacterial metabolism and antibiotic efficacy. Cell Metab 2019, 30 (2), 251–259. DOI: 10.1016/j.cmet.2019.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Centers for Disease Control and Prevention 2019. AR threats report. https://www.cdc.gov/drugresistance/biggest-threats.html (accessed 26 Aug).
- 25.Orazi G; O’Toole GA, “It takes a village”: mechanisms underlying antimicrobial recalcitrance of polymicrobial biofilms. J Bacteriol 2019, 202 (1), e00530–19. DOI: 10.1128/JB.00530-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang C; Wang S; Li D; Chen P; Han S; Zhao G; Chen Y; Zhao J; Xiong J; Qiu J, Human Cathelicidin Inhibits SARS-CoV-2 Infection: Killing Two Birds with One Stone. ACS infectious diseases 2021. [DOI] [PubMed] [Google Scholar]
- 27.Engelberg Y; Landau M, The Human LL-37 (17-29) antimicrobial peptide reveals a functional supramolecular structure. Nature communications 2020, 11 (1), 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Patch JA; Barron AE, Helical peptoid mimics of magainin-2 amide. Journal of the American Chemical Society 2003, 125 (40), 12092–12093. [DOI] [PubMed] [Google Scholar]
- 29.Huang K; Wu CW; Sanborn TJ; Patch JA; Kirshenbaum K; Zuckermann RN; Barron AE; Radhakrishnan I, A threaded loop conformation adopted by a family of peptoid nonamers. Journal of the American Chemical Society 2006, 128 (5), 1733–1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Woolfson DN, The design of coiled-coil structures and assemblies. Advances in protein chemistry 2005, 70, 79–112. [DOI] [PubMed] [Google Scholar]
- 31.Czyzewski AM; Jenssen H; Fjell CD; Waldbrook M; Chongsiriwatana NP; Yuen E; Hancock RE; Barron AE, In vivo, in vitro, and in silico characterization of peptoids as antimicrobial agents. PLoS One 2016, 11 (2), e0135961. DOI: 10.1371/journal.pone.0135961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Belanger CR; Huei-Yi Lee A; Pletzer D; Dhillon BK; Falsafi R; Hancock REW, Identification of novel targets of azithromycin activity against Pseudomonas aeruginosa grown in physiologically relevant media. Proc Natl Acad Sci 2020, 202007626. DOI: 10.1073/pnas.2007626117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ersoy SC; Heithoff DM; Barnes L; Tripp GK; House JK; Marth JD; Smith JW; Mahan MJ, Correcting a fundamental flaw in the paradigm for antimicrobial susceptibility testing. EBioMedicine 2017, 20, 173–181. DOI: 10.1016/j.ebiom.2017.05.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tian X; Sun F; Zhou XR; Luo SZ; Chen L, Role of peptide self-assembly in antimicrobial peptides. Journal of Peptide Science 2015, 21 (7), 530–539. [DOI] [PubMed] [Google Scholar]
- 35.Sancho-Vaello E; François P; Bonetti E-J; Lilie H; Finger S; Gil-Ortiz F; Gil-Carton D; Zeth K, Structural remodeling and oligomerization of human cathelicidin on membranes suggest fibril-like structures as active species. Scientific reports 2017, 7 (1), 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chongsiriwatana NP; Lin JS; Kapoor R; Wetzler M; Rea JAC; Didwania MK; Contag CH; Barron AE, Intracellular biomass flocculation as a key mechanism of rapid bacterial killing by cationic, amphipathic antimicrobial peptides and peptoids. Sci Rep 2017, 7 (1), 16718. DOI: 10.1038/s41598-017-16180-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chu-Kung AF; Nguyen R; Bozzelli KN; Tirrell M, Chain length dependence of antimicrobial peptide–fatty acid conjugate activity. Journal of colloid and interface science 2010, 345 (2), 160–167. [DOI] [PubMed] [Google Scholar]
- 38.Sabath LD, Six factors that increase the activity of antibiotics in vivo. Infection 1978, 6 (1), S67–S71. DOI: 10.1007/BF01646069. [DOI] [Google Scholar]
- 39.Pulkkinen K; Pekkala N; Ashrafi R; Hämäläinen DM; Nkembeng AN; Lipponen A; Hiltunen T; Valkonen JK; Taskinen J, Effect of resource availability on evolution of virulence and competition in an environmentally transmitted pathogen. FEMS Microbiol Ecol 2018, 94 (5). DOI: 10.1093/femsec/fiy060. [DOI] [PubMed] [Google Scholar]
- 40.Malik E; Dennison SR; Harris F; Phoenix DA, pH dependent antimicrobial peptides and proteins, their mechanisms of action and potential as therapeutic agents. Pharmaceuticals 2016, 9 (4). DOI: 10.3390/ph9040067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jimenez CJ; Tan J; Dowell KM; Gadbois GE; Read CA; Burgess N; Cervantes JE; Chan S; Jandaur A; Karanik T; Lee JJ; Ley MC; McGeehan M; McMonigal A; Palazzo KL; Parker SA; Payman A; Soria M; Verheyden L; Vo VT; Yin J; Calkins AL; Fuller AA; Stokes GY, Peptoids advance multidisciplinary research and undergraduate education in parallel: Sequence effects on conformation and lipid interactions. Biopolymers 2019, 110 (4), e23256. DOI: 10.1002/bip.23256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sawyer JG; Martin NL; Hancock RE, Interaction of macrophage cationic proteins with the outer membrane of Pseudomonas aeruginosa. Infect Immun 1988, 56 (3), 693–8. DOI: 10.1128/iai.56.3.693-698.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lehrer RI; Lichtenstein AK; Ganz T, Defensins: antimicrobial and cytotoxic peptides of mammalian cells. Annu Rev Immunol 1993, 11, 105–28. DOI: 10.1146/annurev.iy.11.040193.000541. [DOI] [PubMed] [Google Scholar]
- 44.Walkenhorst WF; Klein JW; Vo P; Wimley WC, pH dependence of microbe sterilization by cationic antimicrobial peptides. Antimicrob Agents Ch 2013, 57 (7), 3312–3320. DOI: 10.1128/AAC.00063-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Gan BH; Gaynord J; Rowe SM; Deingruber T; Spring DR, The multifaceted nature of antimicrobial peptides: current synthetic chemistry approaches and future directions. Chemical Society Reviews 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gong H; Sani M-A; Hu X; Fa K; Hart JW; Liao M; Hollowell P; Carter J; Clifton LA; Campana M, How do self-assembling antimicrobial lipopeptides kill bacteria? ACS applied materials & interfaces 2020, 12 (50), 55675–55687. [DOI] [PubMed] [Google Scholar]
- 47.Luo Y; Bolt HL; Eggimann GA; McAuley DF; McMullan R; Curran T; Zhou M; Jahoda PC; Cobb SL; Lundy FT, Peptoid efficacy against polymicrobial biofilms determined by using propidium monoazide-modified quantitative PCR. Chembiochem 2017, 18 (1), 111–118. DOI: 10.1002/cbic.201600381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yang M; Xu D; Jiang L; Zhang L; Dustin D; Lund R; Liu L; Dong H, Filamentous supramolecular peptide–drug conjugates as highly efficient drug delivery vehicles. Chemical communications 2014, 50 (37), 4827–4830. [DOI] [PubMed] [Google Scholar]
- 49.Tyrrell ZL; Shen Y; Radosz M, Fabrication of micellar nanoparticles for drug delivery through the self-assembly of block copolymers. Progress in Polymer Science 2010, 35 (9), 1128–1143. [Google Scholar]
- 50.Alford MA; Baquir B; Santana FL; Haney EF; Hancock REW, Cathelicidin host defense peptides and inflammatory signaling: striking a balance. Front Microbiol 2020, 11 (1902). DOI: 10.3389/fmicb.2020.01902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Classen S; Hura GL; Holton JM; Rambo RP; Rodic I; McGuire PJ; Dyer K; Hammel M; Meigs G; Frankel KA, Implementation and performance of SIBYLS: a dual endstation small-angle X-ray scattering and macromolecular crystallography beamline at the Advanced Light Source. Journal of applied crystallography 2013, 46 (1), 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hura GL; Menon AL; Hammel M; Rambo RP; Poole Ii FL; Tsutakawa SE; Jenney FE Jr; Classen S; Frankel KA; Hopkins RC, Robust, high-throughput solution structural analyses by small angle X-ray scattering (SAXS). Nature methods 2009, 6 (8), 606–612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Centers for Disease C, P., Outbreaks of community-associated methicillin-resistant Staphylococcus aureus skin infections--Los Angeles County, California, 2002-2003. MMWR Morb Mortal Wkly Rep 2003, 52 (5), 88. [PubMed] [Google Scholar]
- 54.Behroozian S; Svensson SL; Davies J; Blaser MJ, Kisameet clay exhibits potent antibacterial activity against the ESKAPE pathogens. mBio 2016, 7 (1), e01842–15. DOI: doi: 10.1128/mBio.01842-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jacobs AC; Thompson MG; Black CC; Kessler JL; Clark LP; McQueary CN; Gancz HY; Corey BW; Moon JK; Si Y; Owen MT; Hallock JD; Kwak YI; Summers A; Li CZ; Rasko DA; Penwell WF; Honnold CL; Wise MC; Waterman PE; Lesho EP; Stewart RL; Actis LA; Palys TJ; Craft DW; Zurawski DV, AB5075, a highly virulent isolate of Acinetobacter baumannii, as a model strain for the evaluation of pathogenesis and antimicrobial treatments. mBio 2014, 5 (3), e01076–14. DOI: 10.1128/mBio.01076-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cheng K; Smyth RL; Govan JR; Doherty C; Winstanley C; Denning N; Heaf DP; van Saene H; Hart CA, Spread of beta-lactam-resistant Pseudomonas aeruginosa in a cystic fibrosis clinic. Lancet 1996, 348 (9028), 639–42. DOI: 10.1016/s0140-6736(96)05169-0. [DOI] [PubMed] [Google Scholar]
- 57.Marchou B; Bellido F; Charnas R; Lucain C; Pechère JC, Contribution of beta-lactamase hydrolysis and outer membrane permeability to ceftriaxone resistance in Enterobacter cloacae. Antimicrob Agents Ch 1987, 31 (10), 1589–95. DOI: 10.1128/aac.31.10.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Coffey BM; Anderson GG, Biofilm formation in the 96-well microtiter plate. In Pseudomonas methods and protocols, Springer: 2014; pp 631–641. [DOI] [PubMed] [Google Scholar]
- 59.Wiegand I; Hilpert K; Hancock REW, Agar and broth dilution methods to determine the minimal inhibitory concentration (MIC) of antimicrobial substances. Nat. Protoc 2008, 3 (2), 163–175. DOI: 10.1038/nprot.2007.521. [DOI] [PubMed] [Google Scholar]
- 60.Clinical and Laboratory Standards Institute, Performance standards for dilution antimicrobial susceptibility tests for bacteria that grow aerobically. 11th ed.; Clinical and Laboratory Standards Institute: Wayne, PA, 2018. [Google Scholar]
- 61.Haney EF; Trimble MJ; Hancock REW, Microtiter plate assays to assess antibiofilm activity against bacteria. Nature Protocols 2021, 16 (5), 2615–2632. DOI: 10.1038/s41596-021-00515-3. [DOI] [PubMed] [Google Scholar]
- 62.Haney EF; Trimble MJ; Cheng JT; Vallé Q; Hancock REW, Critical assessment of methods to quantify biofilm growth and evaluate antibiofilm activity of host defence peptides. Biomolecules 2018, 8 (2), 29. DOI: 10.3390/biom8020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Pletzer D; Mansour SC; Wuerth K; Rahanjam N; Hancock REW, New mouse model for chronic infections by Gram-negative bacteria enabling the study of anti-infective efficacy and host-microbe interactions. mBio 2017, 8 (1), e00140–17. DOI: 10.1128/mBio.00140-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.