

Abstract
Background:
Eczema herpeticum (EH) is a rare complication of atopic dermatitis (AD) caused by disseminated herpes simplex virus (HSV) infection. The role of rare and/or deleterious genetic variants in disease etiology is largely unknown. This study aimed to identify genes that harbor damaging genetic variants associated with HSV infection in AD with a history of recurrent eczema herpeticum (ADEH+).
Methods:
Whole genome sequencing (WGS) was performed on 49 recurrent ADEH+ (≥3 EH episodes), 491 AD without a history of eczema herpeticum (ADEH−) and 237 non-atopic control (NA) subjects. Variants were annotated, and a gene-based approach (SKAT-O) was used to identify genes harboring damaging genetic variants associated with ADEH+. Genes identified through WGS were studied for effects on HSV responses and keratinocyte differentiation.
Results:
Eight genes were identified in the comparison of recurrent ADEH+to ADEH− and NA subjects: SIDT2, CLEC7A, GSTZ1, TPSG1, SP110, RBBP8NL, TRIM15, and FRMD3. Silencing SIDT2 and RBBP8NL in normal human primary keratinocytes (NHPKs) led to significantly increased HSV-1 replication. SIDT2-silenced NHPKs had decreased gene expression of IFNk and IL1b in response to HSV-1 infection. RBBP8NL-silenced NHPKs had decreased gene expression of IFNk, but increased IL1b. Additionally, silencing SIDT2 and RBBP8NL also inhibited gene expression of keratinocyte differentiation markers keratin 10 (KRT10) and loricrin (LOR).
Conclusion:
SIDT2 and RBBP8NL participate in keratinocyte's response to HSV-1 infection. SIDT2 and RBBP8NL also regulate expression of keratinocyte differentiation genes of KRT10 and LOR.
Keywords: atopic dermatitis, eczema herpeticum, genetics, herpes simplex virus, SIDT2, whole genome sequencing
Graphical Abstract
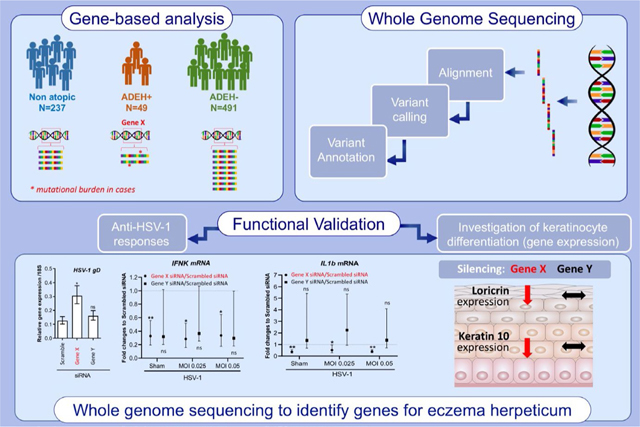
Whole genome sequencing was used to search genome-wide for deleterious genetic variants that are differentially enriched in ADEH+compared to ADEH-and non-atopic controls. A gene-based comparison between cases and controls was used prioritize genes enriched for such variants to be followed up for functional validation. Functional validation reveals two genes: SIDT2 and RBBP8NL are involved in keratinocyte differentiation and responses against HSV-1 infection.
1 |. INTRODUCTION
Atopic dermatitis (AD) is the most common inflammatory skin disease.1–6 A subset of AD patients have disseminated herpes simplex virus (HSV) infections, a rare complication known as eczema herpeticum (ADEH+).7 HSV-1 and HSV-2 are common human pathogens that are often present in latency in human sensory neurons and autonomous ganglions, typically causing mild and self-restricted infections.8 In ADEH+patients, however, HSV-1 infections lead to systemic infections including HSV viremia and meningitis that can be life-threatening.
Previous studies have demonstrated that genetic factors play a role in ADEH+pathogenesis. Genetic variants of thymic stromal lymphopoietin (TSLP), interferon regulatory factor 2 (IRF2), and filaggrin (FLG) have been implicated in ADEH+risk.9–11 TSLP is an epithelial-derived cytokine involved in the pathogenesis of allergic disorders12; IRF2 is a transcription factor that is profoundly involved in keratinocyte biology and interferon signaling regulation13; and FLG is a major skin barrier protein that is also associated with AD and the atopic march.14 FLG null mutations are the strongest genetic risk factors for ADEH+ (odds ratio, OR = 10.1).10 Relying on targeted deep sequencing of the IFNα receptor (IFNAR1), type II IFN (IFNg), IFNγ receptor (IFNgR1), and IL12 receptor (IL12RB1) genes, six rare IFNGR1 missense variants, including 3 damaging variants (Val14Met, Val61Ile, and Tyr397Cys), were found to confer a higher risk for ADEH+.15 Variants Val14Met and Tyr397Cys were found to lead to partial IFNGR1 deficiency.15 No associations were identified between IFNAR1/IFNg/IL12RB1 and ADEH+risk.15 Moreover, it has been observed that peripheral blood mononuclear cells from ADEH+subjects have reduced innate immune responses to vaccinia virus and HSV-1 stimulation compared to peripheral blood mononuclear cells from ADEH-subjects,16,17 supporting the potential role of genetic defects in the innate immune pathways of ADEH+patients.
To further define genetic risk factors for ADEH+, we have leveraged whole genome sequencing (WGS) on 777 subjects from the National Institute of Allergy and Infectious Diseases/Atopic Dermatitis Research Network (ADRN) to identify genes harboring deleterious genetic variants that are associated with recurrent ADEH+ (≥3 EH episodes) compared to ADEH-and non-atopic controls (NA). The genes prioritized by the WGS were validated for their biological function in keratinocyte host-defense responses against HSV-1 and keratinocyte differentiation. Additionally, we look specifically at deleterious gene variants in the FLG gene and the previously reported variants for ADEH+.
2 |. METHODS
2.1 |. Study subjects
Study subjects included unrelated non-Hispanic European American individuals from the ADRN registry. Patients were asked to self-identify their race and ethnicity from a list of United States Census categories. Additionally, principal component analysis (PCA) was performed to confirm the self-reported race and ethnicity. ADEH+subjects were defined as patients with AD who had at least three previous physician-documented EH episode.7 ADEH-subjects were defined as patients with AD (defined, at minimum, by evidence of pruritus and eczematous dermatitis with chronic or relapsing history; for patients who are < 4 years old, eczema must have been present for at least 6 months) and no history of EH. Non-atopic (NA) subjects were defined as healthy controls without personal history of a common allergic condition and without a first-degree family member with history of the common allergic conditions. All samples used for this study were obtained following written informed consent from participants. The University of Colorado, Johns Hopkins University, Northwestern University Feinberg School of Medicine, the Ann and Robert H. Lurie Children's hospital of Chicago, Oregon Health & Science University, Boston Children's Hospital, University of California at San Diego, University of Rochester Medical Center, University of Southern California, Icahn School of Medicine at Mount Sinai, and National Jewish Health Institutional Review Board approved the conduct of this study.
2.2 |. Measurements of AD severity
All study participants underwent a detailed history, physical examination, disease severity assessment, and blood draw. For AD patients only, disease severity was assessed by the Rajka-Langeland18 and the Eczema Area and Severity Index (EASI)19 scoring systems. For all study participants, blood samples were sent to Quest Diagnostics Laboratory for a complete blood count (CBC) with differential and to the Dermatology, Allergy and Clinical Immunology (DACI) Laboratory at The Johns Hopkins Asthma and Allergy Center for total serum IgE (tIgE) and multi-allergen and individual-specific IgE measurements. Using the UniCap 250 system (Pharmacia and Upjohn), the DACI Laboratory performed the following tests on serum samples from all ADEH+and ADEH-subjects: tIgE (kU/L) and allergen-specific Phadia ImmunoCAP®, including food (FX5E), mite-roach (HX2), animal dander (EX2), weed (WX1), grass (GX2), tree (TX3 and RTX10), and mold (MX2). The total eosinophil count (cells/mm3) was calculated from the CBC with differential.
2.3 |. Whole genome sequencing (WGS)
2.3.1 |. DNA quality and whole genome sequencing
WGS was performed on DNA extracted from whole blood on Illumina TruSeq HiSeq 2000/HiSeqX instruments. All samples were sequenced as paired-end, non-indexed runs with variability in read length; 761 samples with 100 bp, 42 samples with 125 bp, and one sample with 150 bp read length. To address the variability in read length, best practices for WGS were followed with variants called across all samples using a unified variant caller, and all variants prioritized manually examined as described below. Sequencing runs were performed based on the HiSeq 2000/HiSeqX User Guide, using Illumina TruSeq SBS v3 Reagents. Illumina HiSeq Control Software (HCS) and real-time analysis (RTA) used on HiSeq 2000/HiSeqX sequencing runs for real-time image analysis and base calling.
2.3.2 |. Variant calling and generation of a multi-sample variant call file (VCF)
A total of 804 subjects were sent for WGS, including 250 NA, 499 ADEH−, and 55 ADEH+. Five samples were excluded from analysis for clinical and/or sequencing data quality reasons, including low-quality, duplicate, or contaminated sequencing data and remaining 799 were run through the QC pipeline (see below). There were 237 NA, 491 ADEH−, and 49 ADEH+individuals included in the final analysis (777 total: Table 1, detailed overview in Figure S1). Sequence variants were jointly called across all final 777 samples using the Platypus variant caller version 0.8.1for autosomes.20 The program output multi-sample variant call format (VCF) files that were reformatted to blocked GNU zip format (BGZF) for input to PLINK/Seq 0.10.21,22 Filtering steps were carried out using VCFtools and BCFtools23,24 to include variants passing site quality tests by Platypus were kept, total number of reads containing the variant >7, and not mapping to known segmental duplications. Only genotypes with Phred-scaled quality score ≥20 and number of reads covering the variant in each sample ≥7 were retained. Finally, while best practices were adopted for variant calling and variant filtering for quality control as described above, given the variability in read length two additional steps were taken to ensure that the association results were not driven by technical artifacts: (1) Using the first five principal components (PCs) derived as described below (Figure S2) pairwise PCs were examined to see whether there were any differences by the three sequencing batches, and none were observed (Figure S3). (2) A set of 15 samples (5 ADEH+, 5 ADEH−, and 5 NA) were further examined for each of the final variants prioritized in the analyses using the aligned sequence files in Integrative Genomics Viewer using the Human hg19 reference build.25 All Platypus variant calls were determined to be concordant and of high quality to the reads/coverage in the aligned sequence files.
TABLE 1.
Available subjects with whole-genome sequencing data within the Atopic Dermatitis Research Network included in the gene-based tests for eczema herpeticum
| Trait | Atopic Dermatitis without Eczema Herpeticum (ADEH−) Cases | Atopic Dermatitis with Recurrent Eczema Herpeticum (ADEH+) Cases | Non-atopic (NA) Controls |
p-value ADEH+ vs. ADEH− ADEH− vs. NA ADEH+ vs. NA |
|---|---|---|---|---|
| N | 491 | 49 | 237 | |
| Males (N; %) | 221 (45.0%) | 24 (49.0%) | 72 (30.4%) | 0.5950 |
| < 0.001 | ||||
| 0.0134 | ||||
| Agea (Years); mean (SD) | 28.0 (18.4) | 21.4 (15.6) | 37.4 (13.8) | 0.0162 |
| < 0.001 | ||||
| < 0.001 | ||||
| Total IgE(kU/L) mean (range) | 2188.4 (2.0–82020.0) | 3775.8 (14.4–2416.0) | 23.8 (2.2–306.0) | < 0.001 |
| < 0.001 | ||||
| < 0.001 | ||||
| Eosinophils mean (range) | 337.1 (4.0–2502.0) | 454.8 (22.0–1409.0) | 114.2 (0–421.0) | 0.0052 |
| < 0.001 | ||||
| < 0.001 | ||||
| Phadiatop (kUA/L) mean (range) | 103.9 (0.1–5632.0) | 215.8 (0.2–4181.0) | 0.3 (0.1–0.4) | 0.0041 |
| < 0.001 | ||||
| < 0.001 | ||||
| EASI mean (range) | 14.4 (0.3–66.8) | 15.4 (0–55.0) | N/A | 0.8803 |
| N/A | ||||
| N/A | ||||
| Rajka-Langeland score mean (SD) | 6.9 (1.5) | 6.9 (1.7) | N/A | 0.8812 |
| N/A | ||||
| N/A |
Abbreviation: SD, standard deviation.
Age at recruitment, not age at diagnosis.
2.4 |. Sample quality control and population structure analysis
Quality control (QC) checks were performed using genotype data available from the Illumina OMNI genotyping array also delivered by Illumina along with the WGS data. Samples excluded for QC reasons are detailed in Figure S1. There were 15,231 variants that went into PCA (after pruning for linkage disequilibrium, LD pruning). PLINK parameters used for LD pruning were as follows: a window size of 50, 5 SNP overlap, and R2 = 0.5 (or VIF = 2). SNPs with MAF <0.01 and HWE<1E-6 were filtered out from the analysis. PCA was performed using SmartPCA26 on the ADRN samples including 2094 unrelated samples from five Thousand Genomes Project (TGP)27 super-populations as reference. In total, 4 samples with PC1 or PC2 values greater than six standard deviations away from the median of ADRN were dropped. Subsequent to the QC step where samples were selected and SNPs genotypes were discarded, PCs were recalculated on ADRN samples only, that is, excluding the TGP reference populations. The first five PCs were used selected to be used covariates in analysis on the basis of the variation explained as summarized in the scree plot (Figure S2).
2.5 |. Gene variant annotation and gene-based tests for association
Variant annotation was performed using ANNOVAR.28 Missense variants had exonic functional annotations of either nonsynonymous, stop-gain, or stop-loss. A variant was defined as damaging for our analyses if it was labeled as deleterious or “D” by both SIFT and PolyPhen-2 HDIV algorithms. A variant is labeled deleterious in SIFT if the score is ≥0.05. In PolyPhen, a variant is labeled “probably damaging” if the score is ≤0.957, “possibly damaging” if 0.453 ≤ score ≤0.956. Variants with less than 5% TGP global frequency, or absent in the database, were labeled rare/novel. SKAT-O tests incorporated sample phenotype and the 5 PCs as covariates. Gene-based tests were performed using the SKAT-O R package (SNP-set (Sequence) Kernel Association Test, Optimal) with two variant sets per pairwise comparison29: Set 1 included all missense and damaging variants observed in the set of samples, and Set 2 included only missense and damaging variants that were rare/not presented in the external TGP data. Three gene-based tests were implemented based on phenotype definitions: ADEH+vs. ADEH, ADEH+vs. NA, and ADEH-vs. NA. Since no genes passed the Bonferroni multiple-comparison correction threshold (p < 0.05/n where n is the number of specific genes tests as described in results), not unexpected given the small sample size of the primary ADEH+case group, all genes with a p < 0.001 were moved forward to functional follow-up. As an additional means of gene selection, Gene Ontology consortium which uses the PANTHER classification system (http://geneontology.org/page/go-enrichment-analysis) was used to perform overrepresentation tests for gene set enrichment analysis based on the number of observed significant genes vs number of expected significant genes incorporating a Bonferroni correction for multiple testing. Genes with SKAT gene-based p ≤ 0.05 (794 genes) and p < 0.01 (104 genes) were tested. Unfortunately, there were no pathways that were enriched for these significant genes. Therefore, no additional information beyond a hard statistical threshold of p < 0.001 was used for prioritization of genes.
Single-variant tests were not performed genome-wide due to the serious lack of power; we had virtually no power for the ORs~1.5 which are typical of GWAS. Nonetheless, we performed single-variants test for three specific cases. First, given the prior documented role loss of function (LOF) FLG mutations play in AD and ADEH+, we tested specifically for FLG loss of function (LOF) variants for association with ADEH+using an additive linear association model with the same covariates. Second, we performed association tests for 21 of 27 previously identified GWAS SNPs,30 and third, we tested an additional 15 of 18 SNPs previously identified in genetics studies of EH.9,11,15
2.6 |. Keratinocyte culture and HSV-1 sources
Normal human primary keratinocytes (NHPK) were purchased from Thermo Fisher Scientific (Carlsbad, CA) and maintained in EpiLife medium containing 0.06 mM CaCl2 and S7 supplemental reagent in 5% CO2 and 37°C. HSV-1 (VR-733) was purchased from ATCC (Manassas, VA).
2.7 |. siRNA knockdown and NHPK treatment
Targeted siRNA duplexes of the 8 genes prioritized from the WGS analysis, and non-targeting scrambled siRNA duplexes were purchased from Dharmacon (Louisville, CO). The targeting sequences for each gene are listed in Table S1. NHPK cells were seeded in 24-well plates at 1×105 per well. The following day, cells were transfected with siRNA duplexes (final concentration of 10 nM) using Lipofectamine 2000 according to the manufacturer's instructions (Invitrogen, Carlsbad, CA). 48 h later, cells were then stimulated with HSV-1 (MOI of 0, 0.025, or 0.05) for an additional 24 hours. To investigate the impact of candidate genes on keratinocyte differentiation, after transfection of siRNA duplexes, NHPK cells were then cultured in EpiLife medium containing 1.3 mM CaCl2 for 5 days prior to harvest for assays. All silencing experiments were examined for silencing efficiency using real-time qRT-PCR (primers used listed in Table S2).
2.8 |. Total RNA extraction and qRT-PCR
Total RNA was extracted using the RNeasy Mini Kit according to the manufacturer's guidelines (Qiagen, MD). RNA was then reverse-transcribed into cDNA using superScript® III reverse transcriptase from Invitrogen (Portland, OR) and analyzed by real-time qRT-PCR using an ABI Prism 7000 sequence detector (Applied Biosystems, Foster City, CA). Primers and probes for human 18s (Hs99999901_s1), IFNk (Hs00737883_m1), IL1b (Hs01555410_m1), IFNb1 (Hs01077958_s1), keratin 10 (KRT10) (Hs00166289), FLG (Hs00856927_g1), loricrin (LOR) (Hs01894962-s1), and involucrin (IVL) (Hs00846307_s1) were purchased from Applied Biosystems (Foster City, CA). Primer sequences of HSV-1 gD gene were as follows: forward 5'- CGGCCGTGTGACACTATCG-3'; reverse 5'-CTCGTAAAATGGCCCCTCC-3'. The probe sequence was 5'-CCATACCGACCACA CCGACGAACC-3', with the probe labeled at its 5'-end with 6-carboxyfluorescein. Primers and probes were synthesized at Integrated DNA Technologies (Coralville, IA). Expression quantities of all target genes in test samples were normalized to the corresponding 18s levels.
2.9 |. Viral plaque assay
Vero cells were maintained in Minimum Essential Medium (MEM) with 5% of FBS. Cells were plated into 24-well dishes at 2 × 105 to form monolayers. The following day, HSV-1–infected NHPK cells with culture supernatants were disrupted completely by three cycles of freeze and thaw to release the infectious particles. The infectious solutions were then added to Vero cell monolayers with serial dilutions. After 2 hours of incubation, infectious media were removed, and the cells were covered with 2% methylcellulose made in MEM containing 2% FBS. The cells were then cultured in the incubator with 5% CO2. After 3 days, viral plaque formation was visualized by 1% crystal violet staining.
2.10 |. Western blot protein detection
Scrambled- and specific gene-siRNA–silenced differentiated NHPK Cells were lysed in 2× Laemmli sample buffer (Bio-Rad), and proteins were run on Western blots. Antibodies against β-actin (clone W16197A) and KRT10 (clone DE-K10) were purchased from BioLegend (San Diego, CA); antibody against IVL (MA5–11803) was purchased from Thermo Fisher Scientific (Carlsbad, CA); and antibody against LOR was a kind gift from Dr. Dennis Roop (University of Colorado).31 Antibodies against IL1β and IFNκ were purchased from R&D biosystems (Minneapolis, MN).
2.11 |. Statistical analysis
Relative gene expression values were log-transformed for analysis. Comparisons in log-transformed relative gene expression between multiple target siRNA and scrambled siRNA were made using Dunnett's test to account for multiple comparisons with scrambled siRNA set as the control. Log-transformed gene expression values were fit using linear mixed models, where data from multiple independent experiments were fit in the same model (when applicable) and accounted for by including a random intercept for experiment; models were stratified by siRNA and HSV-1 (MOI). For plaque-forming units, similar models were fit, including a covariate for HSV-1 (MOI) and stratified by siRNA. For each model, t tests were used to compare log-transformed relative gene expression values between target siRNA and scrambled siRNA within each HSV-1 (MOI). For all models, the geometric mean (95% CI) of relative gene expression in the presence of target siRNA was calculated and presented. Statistical significance was set at the α = 0.05 significance level. All analyses were conducted using SAS software version 9.4.
3 |. RESULTS
3.1 |. WGS analyses identified ADEH+-associated damaging and rare genetic variants
In total, 237 NA, 491 ADEH−, and 49 recurrent ADEH+individuals were included in the gene-based analysis as described in Table 1, with 33,769 total damaging variants observed across the final set of 777 subjects. These variants mapped to a total of 11,526 coding genes. When limited to those rare or novel (<5%) variants compared to the TGP, 32,551 total deleterious variants were observed, mapping to a total of 11,391 coding genes. The specific results pertaining to each of the six sets of analyses performed (based on phenotype: ADEH+vs ADEH−, ADEH+vs NA, and ADEH-vs NA and public catalog frequency of variants: All damaging vs. rare/novel damaging) are presented in Figure S4 and Table S3. No genes passed the Bonferroni threshold (p-value < 0.05/11,526 and p-value < 0.05/11,391) for the two sets of gene-based tests performed, which was not unexpected given the small sample size of the primary recurrent ADEH+case group. As a result, genes with a p < 0.001 were moved forward to functional follow-up. Table 2 is the overview of the 8 genes for which we observed a gene-based p < 0.001 in the comparison of ADEH+vs ADEH-or ADEH+vs. NA, but not in the ADEH–vs NA group. Results were robust to the addition of sex as a covariate. They are RBBP8NL (RBBP8N-terminal like), FRMD3 (FERM domain–containing 3), TPSG1 (tryptase gamma 1), GSTZ1 (glutathione S-transferase zeta 1), SIDT2 (SID1 transmembrane family member 2), CLEC7A (C-type lectin domain containing 7A), SP110 (SP110 nuclear body protein), and TRIM15 (tripartite motif–containing 15). The detailed information of the missense/damaging mutations in the 8 genes, including allele frequency in the three groups and amino acids change at each variant enriched in recurrent ADEH+subjects, is shown in Table 3.
TABLE 2.
The top 8 genes with genetic variants significantly enriched in ADEH+compared to ADEH−and NA (p < 0.001) revealed by the SKAT-O gene-based tests
| ADEH+ vs ADEH− |
ADEH+ vs NA |
ADEH− vs NA |
|||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All missense/damaging |
Rare missense/damaging |
All missense/damaging |
Rare missense/damaging |
All missense/damaging |
Rare missense/damaging |
||||||||
| Gene | Chr | # SNVs | p-value | # SNVs | p-value | # SNVs | p-value | # SNVs | p-value | # SNVs | p-value | # SNVs | p-value |
| RBBP8NL | 20 | 4 | 0.0014 | 3 | 0.0014 | 3 | 0.0001 | 2 | 0.0001 | 5 | 0.3000 | 4 | 0.3000 |
| TRIM15 | 6 | 2 | 0.0003 | 2 | 0.0003 | 3 | 0.108 | 3 | 0.108 | 3 | 0.0435 | 3 | 0.0435 |
| CLEC7A | 12 | 3 | 0.158 | 2 | 0.537 | 2 | 0.0003 | 1 | 0.032 | 3 | 0.0305 | 2 | 0.1380 |
| SIDT2 | 11 | 4 | 0.0006 | 4 | 0.0006 | 3 | 0.0003 | 3 | 0.0003 | 5 | 0.7040 | 5 | 0.7040 |
| FRMD3 | 9 | 2 | 0.0034 | 2 | 0.0034 | 1 | 0.0005 | 1 | 0.0005 | 2 | 0.2400 | 2 | 0.2400 |
| TPSG1 | 16 | 3 | 0.129 | 2 | 0.101 | 3 | 0.0025 | 2 | 0.0007 | 3 | 0.0122 | 2 | 0.0044 |
| GSTZ1 | 14 | 3 | 0.0684 | 1 | 0.453 | 2 | 0.0007 | NA | NA | 3 | 0.1790 | 1 | 0.7410 |
| SP110 | 2 | 3 | 0.0009 | 3 | 0.0009 | 4 | 0.0043 | 4 | 0.0043 | 2 | 0.7020 | 2 | 0.7020 |
TABLE 3.
Genetic variants enriched in recurrent ADEH+ subjects showing the frequency of heterozygous (Ref/Alt) and homozygous (Alt/Alt) carriers within each ADRN phenotype group and the reference and alternate alleles (Ref/Alt) frequency in the TGP super-populations as reference
| Variant |
Frequency of carriers (Ref/Alt +Alt/Alt)a |
TGP Frequency |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Gene symbol | dbSNP ID | ADEH+ (n = 49) | ADEH− (n = 491) | NA (n = 237) | Amino acid change | AFR (Ref/Alt) | AMR (Ref/Alt) | EAS (Ref/Alt) | EUR (Ref/Alt) | SAS (Ref/Alt) |
| SIDT2 | rs142171036 | 8% | 0.4% | 0% | p. Arg710Cys | C: 100%/T: 0% | C: 100%/T: 0% | C: 100%/T: 0% | C: 99.9%/T: 0.1% | C: 100%/T: 0% |
| CLEC7A | rs16910526 | 28% +2% | 14.2% +1.2% | 10% +0.4% | Stop-gained | A: 98.9%/C: 1.1% | A: 96.3%/C:3.7% | A: 99.6%/C: 0.4% | A: 92.9%/C: 7.1% | A: 90.9%/C: 9.1% |
| rs140318683 | 2% | 0.8% | 0% | p. Leu183Phe | G: 100% A: 0% | G: 100%/A: 0% | G: 100%/A: 0% | G: 99.5%/A: 0.5% | G: 100%/A: 0% | |
| GSTZ1 | rs7972 | 26% +4% | 14.6% +0.8% | 11.2% +0.8% | p. Gly42Arg | G: 99.6%/A:.04% | G: 97.3%/A: 2.7% | G: 100%/A: 0% | G: 88.9%/A: 11.1% | G: 98.3%/A: 1.7% |
| TPSG1 | rs61587627 | 6% | 4.4% | 1.2% | p.Thr100Lys | G: 98.1%/T: 1.9% | G: 97.3%/T: 2.7% | G: 90.3%/T: 9.7% | G: 98.9%/T: 1.1% | G: 98.7%/T:1.3% |
| rs187607214 | 4% | 0.6% | 0% | p. Cys51 Tyr | C: 99.9%/T: 0.1% | C: 100%/T: 0% | C: 100%/T: 0% | C: 99.8%/T: 0.2% | C: 100%/T: 0% | |
| SP110 | rs372023963 | 2% | 0% | 0% | p. Thr628Met | NA | NA | NA | NA | NA |
| rs768938186 | 2% | 0% | 0% | p. Arg539Ter isoform b & d | NA | NA | NA | NA | NA | |
| rs149485401 | 6% | 1.2% | 0.8% | p. Gly483Arg isoform a & c | C: 100%/T: 0% | C: 99.1%/T: 0.9% | C: 100%/T: 0% | C: 98.1%/T: 1.9% | C: 100%/T: 0% | |
| RBBP8NL | rs200738153 | 14% | 3.2% | 1.6% | p. Leu267Pro | A: 99.9%/G: 0.1% | A: 99.7%/G: 0.3% | A: 100%/G: 0% | A: 99.2%/G: 0.8% | A: 99.8%/G: 0.2% |
| TRIM15 | rs34823152 | 26% | 7% +0.2% | 11.6% +0.4% | p. Leu235Val | C: 99.9%/G: 0.1% | C: 96.8%/G:3.2% | C: 99.8%/G: 0.2% | C: 95.1%/G: 4.9% | C: 98.9%/G: 1.1% |
| FRMD3 | rs4877747 | 16% | 4.4% | 2.8% | p. Asp485 Tyr | C: 99.9%/A: 0.1% | C: 97.0%/A: 3.0% | C: 93.1%/A: 6.9% | C: 97.6%/A: 2.4% | C: 96.0%/A: 4.0% |
Abbreviation: NA, variant not found in TGP.
If only one number is shown, it indicates only heterozygous carriers in each group; if two numbers are shown, then the first number is the frequency of heterozygous carriers, and the second number is the frequency of the homozygous carriers. Allele frequencies were obtained for Phase 3 release of TGP for five super-populations: AFR (African), AMR (Admixed American), EAS (East Asian), EUR (European), and SAS (South Asian). The frequencies were obtained from the Human (GRCh37.p13) genome on Ensembl.
3.2 |. Silencing gene expression of SIDT2 and RBBP8NL led to enhanced HSV-1 replication in NHPK
Since the epidermal keratinocyte is one of the major cell types involved in ADEH+, it was critical to investigate the role of the prioritized 8 genes in the response of keratinocytes to HSV-1 infection. To determine gene involvement in NHPK antiviral immune responses, we first investigated the gene expression of the 8 genes in keratinocytes in response to HSV-1 stimulation. Six genes SIDT2, SP110, RBBP8NL, GSTZ1, CLEC7A, and FRMD3 were expressed in keratinocytes, and SIDT2, SP110, RBBP8NL, and GSTZ1 were inhibited by HSV-1. However, TPSG1 and TRIM15 were hardly detected by PCR in NHPK at both sham and HSV-1–treated conditions (Figure S4). We silenced the expression of the 6 genes in NHPK and then infected the cells with HSV-1. The expression of the six genes was successfully inhibited by siRNA treatment (Figure S6). As shown in Figure 1A,B, HSV-1 gDmRNA was increased in SIDT2-, RBBP8NL-, and SP110-silenced NHPK compared to scrambled siRNA-treated NHPK after infection with HSV-1 at MOI of 0.025 or 0.05. Silencing of CLEC7A and FRMD3 led to decreased HSV-1 replication compared to scrambled siRNA-treated cells at MOI of 0.025 (Figure 1A), but could not replicate at MOI of 0.05 (Figure 1B). We performed multiple independent experiments, combined data across independent experiments, and validated that silencing SIDT2, RBBP8NL, and SP110 led to increased expression of HSV-1 gD gene (Figure 1C). We further performed viral plaque assays to evaluate infectious particles produced by NHPK silenced for RBBP8NL, SIDT2, and SP110. As shown in Figure 1D,E, SIDT2- and RBBP8NL-silenced NHPK produced increased numbers of viral particles at the majority of tested HSV-1 MOIs, while SP110-silenced NHPK did not. Since the production of infectious particles is a gold standard for evaluation of viral load, we conclude that SIDT2 and RBBP8NL are important for control of HSV-1 infection in NHPK.
FIGURE 1.

Silencing SIDT2 and RBBP8NL led to enhanced HSV-1 replication in NHPK. NHPK were transfected with scrambled and candidate gene siRNA duplexes for 48 hours. The cells were then infected with HSV-1 with indicated MOI for 24 hours. HSV-1 gD expression was evaluated with qRT-PCR in samples treated with different MOI of HSV-1 at 0.025 (A) and 0.05 (B). (C) HSV-1gD expression was evaluated by qRT-PCR in NHPK treated with scrambled SIDT2, RBBP8NL, and SP110 siRNA. Results of three (SP110) or four (SIDT2, RBBP8NL) independent experiments were combined. For panels A, B and C, data are presented as the geometric mean (95% CI) of the fold change between target siRNA and scrambled siRNA. In panels A and B, comparisons of log-transformed relative gene expression were made using Dunnett's test with Scrambled siRNA as the control. In panel C, comparisons of log-transformed relative gene expression were made using t tests. Significant differences had *p-value < 0.05 and **p < 0.01; ns = not significant. (D) Representative pictures of HSV-1 viral plaque assays. (E) The quantitative results of viral plaque-forming units from five independent experiments
3.3 |. Silencing SIDT2 and RBBP8NL altered antiviral responses in NHPK
To elucidate the mechanism by which HSV-1 replication is enhanced in SIDT2- and RBBP8NL-silenced NHPK, we examined cytokine production upon HSV-1 stimulation. Type I interferon (IFN) cytokines are often produced and released by host cells in response to viral stimulation.31 These cytokines can induce an “antiviral state” of host cells through autocrine and paracrine mechanisms. Besides the innate immune function, type I IFNs can also activate the adaptive immune memory against re-infection by the same pathogen.32 In addition to IFNs, inflammatory cytokines are also induced in host cells by invading viruses to activate inflammatory responses. We therefore investigated gene expression of type I IFN IFNk,IFNb1, and inflammatory cytokine IL1b. We determined that both SIDT2 and RBBP8NL were efficiently inhibited by siRNA treatment in NHPK (Figure S7A and C). As shown in Figure 2A,B, expression of IFNk and IL1b mRNA was reduced in SIDT2-silenced NHPK under both sham and HSV-1–stimulated conditions compared to scrambled siRNA-treated controls. However, gene expression of IFNb1 had no significant difference in SIDT2-silenced NHPK compared to scrambled siRNA-treated NHPK (Figure S7B). Interestingly, silencing RBBP8NL led to reduced IFNk gene expression, but enhanced IL1b gene expression in both sham- and HSV-1–treated NHPK (MOI of 0.05) compared to the scrambled siRNA-treated controls (Figure 2A,B). Same as SIDT2 silencing, IFNb1 gene expression had no difference between RBBP8NL-silenced and scrambled siRNA-treated NHPK (Figure S7D). We performed Western blotting assays to determine the protein expression of IL1β and IFNκ in cell lysates, and confirmed that IL1β and IFNκ proteins were decreased in SIDT2 and RBBP8NL-RBBP8NL-silenced NHPK at both sham and HSV-1 (HSV-1 0.05) treated conditions (Figure 2C).
FIGURE 2.

Silencing SIDT2 and RBBP8NL altered immune responses to HSV-1 stimulation in NHPK. NHPK were transfected with SIDT2 siRNA, RBBP8NL siRNA, and scrambled siRNA duplexes for 48 hours. The cells were then infected with HSV-1 with indicated MOI for 24 hours. IFNk (A) and IL1b (B) were evaluated by real-time qRT-PCR. For (A), data from five independent experiments are shown. For (B), data from five (RBBP8NL) and four (SIDT2) independent experiments are shown. Data are presented as the geometric mean (95% CI) of relative gene expression. Comparisons of log-transformed relative gene expression values between specific gene siRNA and scrambled siRNA under varying MOI were made using t tests. Statistical significance is represented as ns = not significant, *p < 0.05 and **p < 0.01. (C) IL1β and IFNκ protein expression in cell lysates determined by Western blot assays
3.4 |. Silencing SIDT2 and RBBP8NL altered NHPK differentiation
Since dysregulation of keratinocyte differentiation is a feature of AD development,33 we investigated whether SIDT2 and RBBP8NL regulated expression of genes involved in keratinocyte differentiation, including KRT10,IVL,FLG, and LOR.34 As shown in Figure 3A, silencing SIDT2 led to decreased expression of KRT10 mRNA compared to the scrambled siRNA treatment in differentiated NHPK. Silencing RBBP8NL led to decreased expression of FLG, LOR, and KRT10 mRNA compared to the scrambled siRNA treatment in differentiated NHPK. We further performed Western blot assays using NHPK from multiple donors, and validated the alteration of LOR, IVL, and KRT10 protein expression in the two siRNA-silenced differentiated NHPK (Figure 3B). Although LOR mRNA was not reduced in SIDT2-silenced differentiated NHPK, LOR protein was reduced (Figure 3B). These results suggest that LOR had post-transcriptional regulation in SIDT2-silenced NHPK. LOR protein and KRT10 protein were reduced in RBBP8NL-silenced differentiated NHPK compared to scrambled siRNA-treated controls (Figure 3B).
FIGURE 3.

Silencing SIDT2 and RBBP8NL altered NHPK differentiation. NHPK cells were transfected with SIDT2 siRNA, RBBP8NL siRNA, and scrambled siRNA duplexes for 24 hours, and then differentiated in EpiLife with 1.3 mM CaCl2 for 5 days. (A) Expression of FLG, LOR, IVL, and KRT10 in differentiated NHPK were evaluated by real-time qRT-PCR. Data of three independent experiments are shown. Data are presented as the geometric mean (95% CI) of relative gene expression. Comparisons of log-transformed relative gene expression values between target siRNA and scrambled siRNA were made using t tests. Significant differences had *p-value < 0.05 and ns = not significant. (B) Western blot assay to detect protein expression in differentiated NHPK; cell lysates were prepared from NHPK of three different donors. *Note: Hard edges/pixilation in panel 3B have been editorially validated in source images
3.5 |. Genetic determinants of ADEH+in previously identified AD and ADEH genes
Five previously documented LOF FLG mutations were each tested individually for association with ADEH+ (Table 4).10 The strongest association was observed for R501X with an increasing risk–there is a near doubling of risk when ADEH+is compared to NA controls (OR = 8.44, p = 2.5 × 10−5) in contrast to when ADEH-is compared to NA controls (OR = 4.66, p = 0.0001). However, we note that the 95% CIs for the two estimates are overlapping, and this if further reflected a non-significant results for the comparison of ADEH+to ADEH− (OR = 1.65, p = NS). While less significant, similar doubling in effects sizes was noted for R2447X, while the 2282del4 LOF mutation had similar effects for AD irrespective of EH status. No associations were significant for S3247X, and3702delG was not observed in this ADRN dataset.
TABLE 4.
Association between five previously documented FLG variants and risk of ADEH+ and ADEH− showing the carrier count within each group, the odds ratio (OR), 95% confidence interval (CI) and p-value for each comparison, and the allele frequencies in the TGP super-populations as reference
| Carrier Count |
ADEH− vs NA |
ADEH+ vs ADEH− |
ADEH+ vs NA |
TGP Frequency (Ref/Alt) |
||||
|---|---|---|---|---|---|---|---|---|
| FLG Variant | ADEH+ | ADEH− | NA | OR [95% CI] p-value |
OR [95% CI] p-value |
OR [95% CI] p-value] |
AFR AMR EAS EUR SAS |
|
| S3247X (WT = CC; MUT = AA) (rs150597413) |
GG | 48 | 481 | 236 | 4.87 [0.7–213.6] 0.116 |
1.00 [0.02–7.3] 1 |
4.88 [0.06–386.2] 0.3136 |
G/T |
| 100%/0% | ||||||||
| 100%/0% | ||||||||
| 100%/0% | ||||||||
| GT | 1 | 10 | 1 | 99.8%/0.2% | ||||
| TT | 0 | 0 | 0 | 100%/0% | ||||
| R2447X (WT = CC; MUT = TT) (rs138726443) |
GG | 46 | 474 | 237 | 17.20 [undefineda] 0.0026 |
1.79 [0.3–6.6] 0.4165 |
34.78 [undefineda] 0.0049 |
G/A |
| 100%/0% | ||||||||
| 99.7%/0.3% | ||||||||
| 100%/0% | ||||||||
| GA | 3 | 17 | 0 | 99.7%/0.3% | ||||
| AA | 0 | 0 | 0 | 100%/0% | ||||
| R501X (WT = CC; MUT = TT) (rs61816761) |
GG | 38 | 428 | 230 | 4.66 [2.1–10.2] 0.0001 |
1.65 [0.8–3.2] 0.1377 |
8.44 [3.1–22.7] 2.4957×10−5 |
G/A |
| 99.8/0.2% | ||||||||
| 99.4/ 0.6% | ||||||||
| 100/0% | ||||||||
| GA | 11 | 58 | 7 | 99.0/1.0% | ||||
| AA | 0 | 5 | 0 | 99.9/0.1% | ||||
| 2282del4 (MUT = delCAGT) |
WT | 40 | 419 | 225 | 3.14 [1.7–5.8] 0.0003 |
1.21 [0.6–2.4] 0.5917 |
3.98 [1.6–10.2] 0.0038 |
NA |
| HET | 7 | 65 | 12 | |||||
| MUT | 1 | 7 | 0 | |||||
| 3702delG (MUT = delG) |
No Carriers for this deletion in the dataset. | NA | ||||||
Abbreviation: NA, variant not found in TGP.
CI could not be estimated due to 0 genotype counts in the control category. Allele frequencies were obtained for Phase 3 release of TGP for five super-populations: AFR (African), AMR (Admixed American), EAS (East Asian), EUR (European), and SAS (South Asian). The frequencies were obtained from the Human (GRCh37.p13) genome on Ensembl.
We also tested 35 variants that were found in our dataset out of 45 variants known to be associated with ADEH+and AD GWAS variants from Paternoster et al9,11,15,30 One intergenic variant rs61813875 mapping between genes CRCT1 and late cornified envelope gene LCE3E was found to be significant in ADEH+compared to NA controls (OR = 6.62, p = 0.0002) using a Bonferroni correction for multiple testing (Table S4A, B).
4 |. DISCUSSION
In this study, we used a WGS approach to analyze genome sequences of 49 patients with recurrent ADEH+ (≥3 episodes of EH) (an extreme trait) in comparison with 491 ADEH-subjects and 237 NA health subjects. Given the limitations in sample size for this rare and extreme phenotype, we relied on a gene-based approach iterating over several different options of inclusion of variants based on their allele frequency and classification of deleterious function. This approach resulted in the prioritization of 8 novel genes harboring rare/novel deleterious variants that may determine risk for recurrent ADEH+. While replication is the first sought next step to genetic association studies, this extreme phenotype and WGS-based approach presents challenges for this traditional approach–there are no available independent datasets with sequencing for ADHE+. Therefore, we sought to validate all the identified genes and driver variants using mechanistic approaches for functional validation. Additionally, we recognize several limitations with our approach that focuses on coding variants that were limited to those having a non-synonymous/stop-gain/stop-loss damaging annotation. In this approach, we focused on as our initial priority, variants called with high accuracy from the WGS data, that are the highest order of deleteriousness and straightforward to functional validate (ie, missense variants) bearing in mind the sample size considerations with 48 ADEH+cases. Further work is needed for more exhausting approaches that expand to non-coding regions, including structural variation, and potentially synonymous variants with in silico predictions of deleteriousness.
The 8 genes prioritized by the WGS approach in this study are all novel candidates in recurrent ADEH+disease. The biological functions of 4 genes in this list, RBBP8NL, FRMD3, TPSG1, and GSTZ1, are currently unknown. The other four genes are involved in various host-defense immune responses. TRIM15 functions in RIG-1–mediated antiviral immune responses.35,36 CLEC7A, also named dectin-1, is a glucan receptor involved in a wide range of inflammatory responses triggered by fungi, parasites, and allergens37–39; and SIDT2has been reported to be a double-stranded RNA transporter in antiviral immune responses.40 SP110has a host-defense function against mycoplasma tuberculosis in macrophages.41–43 Genetic mutations of SP110 cause a very rare genetic disease hepatic veno-occlusive disease with immunodeficiency.44,45 Based on a literature review, to date, none of these genes have been studied in the context of HSV infections in skin, nor have the genetic variants enriched in our current study of recurrent ADEH+subjects been investigated elsewhere. Our study is the first to demonstrate that silencing the RBBP8NL and SIDT2 genes lead to enhanced HSV-1 replication in NHPK. This suggests the two genes may have a role in the skin barrier to control HSV-1 infection. The fact that silencing the two genes in NHPK resulted in different immune, and inflammatory responses, that is, reduced IFNk and IL1b for SIDT2, reduced IFNk but increased IL1b for RBBP8NL (Figure 2), strongly suggests that the two genes use different mechanisms to control HSV-1 infection in keratinocytes.
Our data demonstrated that SIDT2 and RBBP8NL play a role in gene expression of KRT10, LOR, and FLG, the hallmarks for keratinocytes of different differentiation stages.33 Reduced expression of FLG,KRT10, and LOR inRBBP8NL-silenced NHPK implicated that deficient function of RBBP8NL may lead to abnormal keratinocyte differentiation. Data have shown that reduced gene expression of FLG or tight junction protein (such as CLAUDIN-1) led to enhanced HSV-1 replication in keratinocytes.10,46 The mechanism(s) by which silencing RBBP8NL led to increased HSV-1 replication in NHPK may be partially caused by impaired keratinocyte differentiation. Further investigation is warranted in the future to prove this speculation. We found that silencing SIDT2 led to decreased mRNA and protein expression of KRT10, while LOR protein was reduced in SIDT2-silenced NHPK cells without reduction of its mRNA levels. These data suggested that SIDT2 has a profound function in keratinocyte differentiation.
There are prior reports of SIDT2 in anti–HSV-1 immune responses. Nguyen et al demonstrated that SIDT2 knock-out mice were more susceptible to HSV-1–induced death and had significantly reduced levels of type I IFN IFNb, RANTES, and IL-6 in serum.40 This group further demonstrated that SIDT2 protein is located in the membrane of late endosomes and transports endocytosed double-stranded RNA (dsRNA) from the endosome to the cytoplasm for RIG-1 and MDA5 recognition and triggering of antiviral responses.40 Several other studies describe SIDT2 as a lysosome membrane protein that transports cytoplasmic RNA and DNA to lysosomes for degradation.47–50 It is still not clear how these two mechanisms co-exist in the same cellular system. In NHPK, we found that silencing SIDT2 led to reduced IFNk mRNA expression following stimulation with HSV-1 compared to cells treated with scrambled siRNA, while IFNb1 gene expression had no change. The same was observed after RBBP8NL silencing. Our data suggested that the endogenous expression of IFNk may be more important than IFNb1 in keratinocytes to control HSV-1 infection. Five missense mutations of SIDT2 were identified in this study (Table S3). Based on the allele frequencies of each missense mutation in different groups, only rs142171036 is significantly enriched in recurrent ADEH+subjects (Table 3). This is a rare genetic variant which appears in the 1000 Genomes Project database at a frequency of 0.2% (2 in 1000). In recurrent ADEH+subjects, the frequency of rs142171036 is 8%, 0.4% of ADEH-patients carried one mutated allele, whereas none of the NA controls carried a mutated allele. SIDT2 rs142171036 causes an arginine switch to cysteine at amino acid 710 (SIDT2-R710C) in the seventh transmembrane domain. The side chain of arginine is significantly different from the side chain of cysteine. RBBP8NL had 4 missense mutations identified in this cohort, but only genetic variant rs200738153 is significantly enriched in recurrent ADEH+subjects: 14% of recurrent ADEH+subjects carry one mutated allele, while only 3.2% of ADEH−patients and 1.6% of NA controls carry this allele, respectively. RBBP8NL rs200738153 causes a leucine switch to a proline at amino acid 267 (Table 3).
Further studies will be needed to investigate the biological significance of these specific genetic variants beyond the gene-based functional experiments performed here. The demographic information of the patients who have the alleles of the interested variants is provided in Table S5. Interestingly, patients with the SIDT2 gene variant had increased AD severity, eosinophilia, and higher IgE levels compared to patients with wild-type SIDT2 gene. No demographic differences were noted regarding the other gene variants identified. None of the patients had co-existence of both deleterious alleles. Nevertheless, we double silenced SIDT2 and RBBP8NL in NHPK cells and found that double silencing of the two genes did not have synergistic effects on antiviral responses and keratinocyte differentiation genes compared to single-gene silencing (data not shown). While our study is underpowered to perform genome-wide single variant approaches, a deeper dive into FLG LOF mutations confirm the importance of these LOF mutations in eczema herpeticum, with a notable near-doubling in effect size for ADEH+vs. ADEH−. This is consistent with previous observations that filaggrin deficiency is associated with skin barrier dysfunction, increased intracellular viral replication, and disseminated viral skin infection in animal models.51,52 In summary, the whole genome sequencing approach was successful in identifying potential novel genes harboring variants that increase the risk of recurrent ADEH+. Functional validation of the candidate genes further narrowed the number of targets and provided rationale to investigate the biological significance of these targets (SIDT2 and RBBP8NL) for their roles in conferring the risk of EH.
Supplementary Material
ACKNOWLEDGMENTS
This work was funded by NIH/NIAID Atopic Dermatitis Research Network grants 1U19AI117673-01 and 1UM1AI151958. The authors acknowledge the nurses of Clinical Translational Research Center at National Jewish Health for their hard work in recruiting human subjects for this study. Clinical Translational Research Center at National Jewish Health is supported in part by the Colorado Clinical and Translational Science Award/Colorado Clinical & Translational Sciences Institute grant UL1 RR025780 from National Center for Research Resources/NIH and UL1 TR000154 from NIH/National Center for Advancing Translational Sciences. The authors also thank Dr. Matthew Strand for his help on statistical analyses of the data. The authors have no financial conflict of interest related to this manuscript. Dr. Bin has nothing to disclose. Dr. Malley has nothing to disclose. Patricia Taylor, NP-C, has nothing to disclose. Ms. Boorgula, Mr. Chavan, Dr. Daya, Ms. Mathias, Mr. Shankar, Mr. Rafaels, Dr. Vergara, Mr. Potee, Ms Campbell, Dr. Hanifin, Dr. Simpson, Dr. Gallo, Dr. Hatta, Dr. Ong, Dr. Richers, Dr. Baraghoshi, Dr. Ruczinski, Dr. Barnes, Dr. Leung, and Dr. Mathias have nothing to disclose. Dr. Schneider reports other from Regeneron, personal fees from AbbVie, grants from Pfizer, outside the submitted work. Dr. Paller has been an investigator (without personal compensation) for AbbVie, AnaptysBio, Eli Lilly, Incyte, Leo, Janssen, Novartis, and Regeneron, and a consultant with honorarium for AbbVie, Asana, Boehringer Ingelheim, Dermavant, Dermira, Eli Lilly, Forte, Galderma, Incyte, Inmed, Leo, Lifemax, Novartis, Pfizer, RAPT, Regeneron, and Sanofi Genzyme, outside the submitted work. Dr. De Benedetto reports other from Kiniksa, grants from Pfizer, personal fees from Regeneron Sanofi, outside the submitted work. Dr. Beck reports grants and personal fees from AbbVie, personal fees from Allakos, personal fees from AstraZeneca, personal fees from Benevolent AIBio, personal fees from Incyte, personal fees from LEO Pharma, grants and personal fees from Lilly, personal fees from NAOS Bioderma, personal fees from Novartis, grants and personal fees from Pfizer, personal fees from Principia Biopharma, personal fees from Rapt Therapeutics, grants and personal fees from Regeneron, grants and personal fees from Sanofi/Genzyme, personal fees from UCB, personal fees from Vimalan, personal fees from Sanofi-Aventis, stock in Medtronics, other from 3M, other from Moderna, and other from Gilead, outside the submitted work. Dr. Guttman-Yassky is an employee of Mount Sinai and has received research funds (grants paid to the institution) from AbbVie, Almirall, Amgen, AnaptysBio, Asana Biosciences, Boehringer Ingelheim, Celgene, Dermavant, DS Biopharma, Eli Lilly, Galderma, Ichnos Sciences, Innovaderm, Janssen, Kiniska, Kyowa Kirin, Leo Pharma, Novan, Pfizer, Ralexar, Regeneron Pharmaceuticals, Inc., Sienna Biopharma, UCB and Union Therapeutics, and is a consultant for AbbVie, Aditum Bio, Almirall, Amgen, Asana Biosciences, AstraZeneca, Boehringer Ingelheim, Cara Therapeutics, Celgene, Concert, DBV, Dermira, DS Biopharma, Eli Lilly, EMD Serono, Escalier, Galderma, Ichnos Sciences, Incyte Kyowa Kirin, Leo Pharma, Mitsubishi Tanabe, Pandion Therapeutics, Pfizer, RAPT Therapeutics, Regeneron Pharmaceuticals, Inc., Sanofi, Sienna Biopharma, Target PharmaSolutions, and Union Therapeutics, outside of this work.
Funding information
National Institute of Allergy and Infectious Diseases, Grant/Award Number: 1U19AI117673-01 and 1UM1AI151958; National Center for Advancing Translational Sciences, Grant/Award Number: UL1 TR000154; National Center for Research Resources, Grant/Award Number: UL1 RR025780
Abbreviations:
- AD
atopic dermatitis
- ADEH−
atopic dermatitis without a history of eczema herpeticum
- ADEH+
atopic dermatitis with a history of eczema herpeticum
- ADRN
Atopic Disease Research Network
- CBC
complete blood count
- CI
confidence interval
- dsRNA
double-stranded RNA
- FLG
filaggrin
- GWAS
genome-wide association study
- HSV-1
Herpes simplex virus-1
- IFN
interferon
- Indel
insertion-deletion polymorphism
- IVL
involucrin
- KRT10
keratin 10
- LD
linkage disequilibrium
- LOF
loss of function
- LOR
loricrin
- MAF
minor allele frequency
- MDA5
melanoma differentiation-associated gene 5
- MOI
multiplicity of infection
- NA
non-atopic control
- NHPK
normal human primary keratinocyte
- OR
odds ratio
- PC
principal component
- PCA
principal component analysis
- PRR
pathogen recognition receptor
- QC
quality control
- RIG-1
retinoic acid–inducible gene I
- SNP
single nucleotide polymorphism
- SNV
single nucleotide variant
- TGP
Thousand Genomes Project
- VCF
variant call format
- WGS
whole genome sequencing
- AD
atopic dermatitis
- ADEH+
atopic dermatitis with a history of eczema herpeticum
- ADEH−
atopic dermatitis without a history of eczema herpeticum
- HSV-1
herpes simplex virus-1
- IFN
interferon
- RBBP8NL
RBBP8 N-terminal-like
- SIDT2
SID1 transmembrane family member 2
- siRNA
small interfering RNA
Footnotes
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section.
REFERENCES
- 1.Czarnowicki T, He H, Krueger JG, Guttman-Yassky E. Atopic dermatitis endotypes and implications for targeted therapeutics. J Allergy Clin Immunol. 2019;143:1–11. [DOI] [PubMed] [Google Scholar]
- 2.Leung DYM, Calatroni A, Zaramela LS, et al. The nonlesional skin surface distinguishes atopic dermatitis with food allergy as a unique endotype. Sci Transl Med. 2019;11(480):eaav2685. 10.1126/scitranslmed.aav2685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Simpson EL, Villarreal M, Jepson B, et al. Patients with atopic dermatitis colonized with staphylococcus aureus have a distinct phenotype and endotype. J Invest Dermatol. 2018;138:2224–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Brunner PM, Leung DYM, Guttman-Yassky E. Immunologic, microbial, and epithelial interactions in atopic dermatitis. Ann Allergy Asthma Immunol. 2018;120:34–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Noda S, Suarez-Farinas M, Ungar B, et al. The Asian atopic dermatitis phenotype combines features of atopic dermatitis and psoriasis with increased TH17 polarization. J Allergy Clin Immunol. 2015;136:1254–1264. [DOI] [PubMed] [Google Scholar]
- 6.Brunner PM, Guttman-Yassky E, Leung DY. The immunology of atopic dermatitis and its reversibility with broad-spectrum and targeted therapies. J Allergy Clin Immunol. 2017;139:S65–S76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Beck LA, Boguniewicz M, Hata T, et al. Phenotype of atopic dermatitis subjects with a history of eczema herpeticum. J Allergy Clin Immunol. 2009;124:260–269, 9 e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mark KE, Wald A, Magaret AS, et al. Rapidly cleared episodes of herpes simplex virus reactivation in immunocompetent adults. J Infect Dis. 2008;198:1141–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gao PS, Leung DY, Rafaels NM, et al. Genetic variants in interferon regulatory factor 2 (IRF2) are associated with atopic dermatitis and eczema herpeticum. J Invest Dermatol. 2012;132:650–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gao PS, Rafaels NM, Hand T, et al. Filaggrin mutations that confer risk of atopic dermatitis confer greater risk for eczema herpeticum. J Allergy Clin Immunol. 2009;124:507–513, e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gao PS, Rafaels NM, Mu D, et al. Genetic variants in thymic stromal lymphopoietin are associated with atopic dermatitis and eczema herpeticum. J Allergy Clin Immunol. 2010;125:1403–1407 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ziegler SF. Thymic stromal lymphopoietin and allergic disease. J Allergy Clin Immunol. 2012;130:845–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mercado N, Schutzius G, Kolter C, et al. IRF2 is a master regulator of human keratinocyte stem cell fate. Nat Commun. 2019;10:4676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Paller AS, Spergel JM, Mina-Osorio P, Irvine AD. The atopic march and atopic multimorbidity: Many trajectories, many pathways. J Allergy Clin Immunol. 2019;143:46–55. [DOI] [PubMed] [Google Scholar]
- 15.Gao L, Bin L, Rafaels NM, et al. Targeted deep sequencing identifies rare loss-of-function variants in IFNGR1 for risk of atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol. 2015;136:1591–1600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Leung DY, Gao PS, Grigoryev DN, et al. Human atopic dermatitis complicated by eczema herpeticum is associated with abnormalities in IFN-gamma response. J Allergy Clin Immunol. 2011;127:965–973 e1–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bin L, Edwards MG, Heiser R, et al. Identification of novel gene signatures in patients with atopic dermatitis complicated by eczema herpeticum. J Allergy Clin Immunol. 2014;134:848–855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rajka G, Langeland T. Grading of the severity of atopic dermatitis. Acta Derm Venereol Suppl. 1989;144:13–14. [DOI] [PubMed] [Google Scholar]
- 19.Hanifin JM, Thurston M, Omoto M, Cherill R, Tofte SJ, Graeber M. The eczema area and severity index (EASI): assessment of reliability in atopic dermatitis. EASI Evaluator Group. Exp Dermatol. 2001;10:11–18. [DOI] [PubMed] [Google Scholar]
- 20.Rimmer A, Phan H, Mathieson I, et al. Integrating mapping-, assembly-and haplotype-based approaches for calling variants in clinical sequencing applications. Nat Genet. 2014;46:912–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chang CC, Chow CC, Tellier LC, Vattikuti S, Purcell SM, Lee JJ. Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience. 2015;4:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Purcell S, Neale B, Todd-Brown K, et al. PLINK: a tool set for whole-genome association and population-based linkage analyses. Am J Hum Genet. 2007;81:559–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Danecek P, Auton A, Abecasis G, et al. The variant call format and VCFtools. Bioinformatics. 2011;27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Danecek P, McCarthy SA. BCFtools/csq: haplotype-aware variant consequences. Bioinformatics. 2017;33:2037–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Thorvaldsdottir H, Robinson JT, Mesirov JP. Integrative Genomics Viewer (IGV): high-performance genomics data visualization and exploration. Brief Bioinform. 2013;14:178–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AL, Patterson NJ, Plenge RM, Weinblatt ME, Shadick NA, Reich D. Principal components analysis corrects for stratification in genome-wide association studies. Nat Genet. 2006;38:904–909. [DOI] [PubMed] [Google Scholar]
- 27.Genomes Project C, Auton A, Brooks LD, et al. A global reference for human genetic variation. Nature. 2015; 526:68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang K, Li M, Hakonarson H. ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res. 2010;38:e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lee S, Emond MJ, Bamshad MJ, et al. Optimal unified approach for rare-variant association testing with application to small-sample case-control whole-exome sequencing studies. Am J Hum Genet. 2012;91:224–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Paternoster L, Standl M, Waage J, et al. Multi-ancestry genome-wide association study of 21,000 cases and 95,000 controls identifies new risk loci for atopic dermatitis. Nat Genet. 2015;47:1449–1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mehrel T, Hohl D, Rothnagel JA, et al. Identification of a major keratinocyte cell envelope protein, loricrin. Cell. 1990;61:1103–1112. [DOI] [PubMed] [Google Scholar]
- 32.Fensterl V, Sen GC. Interferons and viral infections. BioFactors. 2009;35:14–20. [DOI] [PubMed] [Google Scholar]
- 33.Bin L, Leung DY. Genetic and epigenetic studies of atopic dermatitis. Allergy Asthma Clin Immunol. 2016;12:52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Candi E, Schmidt R, Melino G. The cornified envelope: a model of cell death in the skin. Nat Rev Mol Cell Biol. 2005;6:328–340. [DOI] [PubMed] [Google Scholar]
- 35.Uchil PD, Hinz A, Siegel S, et al. TRIM protein-mediated regulation of inflammatory and innate immune signaling and its association with antiretroviral activity. J Virol. 2013;87:257–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Uchil PD, Quinlan BD, Chan WT, Luna JM, Mothes W. TRIM E3 ligases interfere with early and late stages of the retroviral life cycle. PLoS Pathog 2008;4:e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito T, Hirose K, Norimoto A, et al. Dectin-1 plays an important role in house dust mite-induced allergic airway inflammation through the activation of CD11b+ dendritic cells. J Immunol. 2017;198:61–70. [DOI] [PubMed] [Google Scholar]
- 38.Saijo S, Fujikado N, Furuta T, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol. 2007;8:39–46. [DOI] [PubMed] [Google Scholar]
- 39.Zimara N, Chanyalew M, Aseffa A, et al. Dectin-1 positive dendritic cells expand after infection with leishmania major parasites and represent promising targets for vaccine development. Front Immunol. 2018;9:263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Nguyen TA, Smith BRC, Tate MD, et al. SIDT2 transports extracellular dsrna into the cytoplasm for innate immune recognition. Immunity. 2017;47:498–509 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Cai L, Deng SL, Liang L, et al. Identification of genetic associations of SP110/MYBBP1A/RELA with pulmonary tuberculosis in the Chinese Han population. Hum Genet. 2013;132:265–273. [DOI] [PubMed] [Google Scholar]
- 42.Leu JS, Chen ML, Chang SY, et al. SP110b controls host immunity and susceptibility to tuberculosis. Am J Respir Crit Care Med. 2017;195:369–382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fox GJ, Sy DN, Nhung NV, et al. Polymorphisms of SP110 are associated with both pulmonary and extra-pulmonary tuberculosis among the Vietnamese. PLoS One. 2014;9:e99496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cliffe ST, Bloch DB, Suryani S, et al. Clinical, molecular, and cellular immunologic findings in patients with SP110-associated veno-occlusive disease with immunodeficiency syndrome. J Allergy Clin Immunol. 2012;130:735–42 e6. [DOI] [PubMed] [Google Scholar]
- 45.Roscioli T, Cliffe ST, Bloch DB, et al. Mutations in the gene encoding the PML nuclear body protein Sp110 are associated with immunodeficiency and hepatic veno-occlusive disease. Nat Genet. 2006;38:620–622. [DOI] [PubMed] [Google Scholar]
- 46.De Benedetto A, Slifka MK, Rafaels NM, et al. Reductions in claudin-1 may enhance susceptibility to herpes simplex virus 1 infections in atopic dermatitis. J Allergy Clin Immunol. 2011;128:242–246 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Aizawa S, Contu VR, Fujiwara Y, et al. Lysosomal membrane protein SIDT2 mediates the direct uptake of DNA by lysosomes. Autophagy. 2017;13:218–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Aizawa S, Fujiwara Y, Contu VR, et al. Lysosomal putative RNA transporter SIDT2 mediates direct uptake of RNA by lysosomes. Autophagy. 2016;12:565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Contu VR, Hase K, Kozuka-Hata H, et al. Lysosomal targeting of SIDT2 via multiple YxxPhi motifs is required for SIDT2 function in the process of RNautophagy. J Cell Sci. 2017;130:2843–2853. [DOI] [PubMed] [Google Scholar]
- 50.Takahashi M, Contu VR, Kabuta C, et al. SIDT2 mediates gymnosis, the uptake of naked single-stranded oligonucleotides into living cells. RNA Biol. 2017;14:1534–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Oyoshi MK, Murphy GF, Geha RS. Filaggrin-deficient mice exhibit TH17-dominated skin inflammation and permissiveness to epicutaneous sensitization with protein antigen. J Allergy Clin Immunol. 2009;124:485–493, 93 e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kim BE, Bin L, Ye YM, Ramamoorthy P, Leung DYM. IL-25 enhances HSV-1 replication by inhibiting filaggrin expression, and acts synergistically with Th2 cytokines to enhance HSV-1 replication. J Invest Dermatol. 2013;133:2678–2685. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.