

Abstract
Dysregulated c-myc is a determinant of multiple myeloma (MM) progression. Translation of c-myc can be achieved by an mTOR-mediated, cap-dependent mechanism or a cap-independent mechanism where a sequence in the 5’UTR of mRNA, termed the IRES (internal ribosome entry site), recruits the 40S ribosomal subunit. This mechanism requires the RNA-binding factor hnRNP A1 (A1) and becomes critical when cap-dependent translation is inhibited during endoplasmic reticulum (ER) stress. We, thus, studied the role of A1 and the myc IRES in myeloma biology. A1 expression correlated with enhanced c-myc expression in patient samples. Expression of A1 in MM lines was mediated by c-myc itself, suggesting a positive feed-back circuit where myc induces A1 and A1 enhances myc translation. We then deleted the A1 gene in a myc-driven murine myeloma model. A1-deleted MM cells demonstrated down-regulated myc expression and were inhibited in their growth in vivo. Decreased myc expression was due to reduced translational efficiency and depressed IRES activity. We also studied the J007 inhibitor which prevents A1’s interaction with the myc IRES. J007 inhibited myc translation and IRES activity and diminished myc expression in murine and human MM lines as well as primary samples. J007 also inhibited tumor outgrowth in mice after subcutaneous or intravenous challenge and prevented osteolytic bone disease. When c-myc was ectopically re-expressed in A1-deleted MM cells, tumor growth was re-established. These results support the critical role of A1-dependent myc IRES-translation in myeloma.
Keywords: myeloma, c-myc, oncogene translation, mTOR, hnRNP A1
INTRODUCTION
Multiple myeloma (MM) cells can protect themselves from ER stress-induced death by restraining mTORC1 activity to limit unnecessary protein translation (1–5). Indeed, loss-of-function alleles of mTOR confer susceptibility to plasmacytoma development in mice (6). MTORC1 activates so-called “cap-dependent” translation by phosphorylating 4EBP-1 which liberates eIF-4E, allowing eIF-4E to interact with the cap structure at the 5’ end of transcripts.
Although inhibition of mTOR protects against ER stress, key oncoproteins must be translated in MM cells to maintain viability. One of these is c-myc, a factor in MM progression (7–9). During periods of mTOR inhibition, c-myc can be alternatively translated through a cap-independent mechanism, mediated by an internal ribosome entry site (IRES (10–12)). IRESes are sequences in the 5’UTR of transcripts that directly recruit the 40S ribosomal subunit for translation initiation in a process facilitated by RNA-binding proteins termed IRES trans-activating factors (ITAFs (11–14)). The myc IRES is located in exon 1 and relies on several ITAFs for optimal function (14,15). One of these is hnRNP A1 (16) which is a mandatory requirement for myc IRES activity. Since IRES-dependent translation does not require mTOR activity, there is no risk of enhanced global translation and ER stress.
Although prior work with cultured human lines (10), suggests a role for hnRNP A1 in MM, in vivo supporting data have been lacking. Thus, in the current study, we focused on a myc-driven murine model of MM. The results provide support for myc IRES activity and hnRNP A1 as key determinants of MM progression.
METHODS
Cell lines, plasmids, reagents-
The IL6Myc-1 is a continuously cultured MM line derived from an IL6Myc transgenic mouse (17), maintained in IMDM with 10% FBS. The pRF reporter was a gift from Dr. A. Willis. The myc IRES was cloned into pRF as described (16) to obtain pRmF. The p27 leader was subcloned upstream of the myc ORF as previously described (18) to generate the p27-myc ORF construct. Editing of the A1 gene utilized two separate guide RNAs for targeting the mouse A1 gene with the sequences: taccgtcatgtctaagtccg and tacctcggacttagacatga which were inserted into LentiCRISPRv2 (Addgene, Watertown, MA). Cells were transfected with the packaged lentiviruses. Single cell clones were screened for A1 gene deletion. The antibody used to identify A1 protein expression was purchased from Santa Cruz (antibody 4B10:sc-32301). Primary MM cells were purified from bone marrow aspirates as previously described (10).
Evaluation of translation-
Polysome analysis was performed as described (18, 19). Briefly, after separation of extracts on a 15–50% sucrose gradient, fractions were collected and UV absorbance at 254nm measured to differentiate monosome from polysome fractions. Associated myc RNA or actin RNA was quantified by qt-PCR.
A1 reporter expression assay-
As reported (20), the pSH CAT, containing the CAT reporter under the control of the A1 promoter was transfected into RPMI8226 cells with over-expression constructs of myc, MAF or IRF-4 or shRNA targeting these transcription factors. CAT assay read-outs were assayed after 24 hrs as described (20).
Myc IRES activity-
Dicistronic reporter constructs were transfected using Effectene Reagent (Qiagen) and normalized for transfection efficiency by co-transfection with pSVβGal. Transfection efficiencies were 5–15%. After 18 hrs, cells were harvested, followed by detection of Renilla luciferase, Firefly luciferase and beta-galactosidase activities (16). All luciferase activity is normalized to the luciferase values obtained for pRF in the absence of treatments, which is designated as a value of ‘1’.
In vivo tumor models-
All animal studies have been conducted in accordance with and with the approval of the Greater Los Angeles VA Healthcare Center and Medical College of Wisconsin. NOD/SCID mice were injected subcutaneously (SQ) with 1.5×106 IL6Myc-1 cells. For IV challenge, B-cell-deficient NOD/SCID mice were conditioned with irradiation (3 Gy) 4 hrs before injecting 1.5×106 luciferase-expressing cells IV. IV-challenged mice (6/group) were randomly divided into groups to receive either DMSO or inhibitor starting on day 7. After sacrifice, bones were imaged ex vivo using microCT (IVIS SpectrumCT). The scan was performed at 50 kV with 1mA and reconstructed to a voxel size of 50um. Three-dimensional parameters BV/TV (bone volume over total volume) were analyzed with the help of the BoneJ2 (v.7.0.7) plugin of imageJ (v.1.53c).
Max splice assay-
Total RNA was purified from MM cells by Qiagen RNAeasy kit and cDNA was generated with high capacity cDNA Reverse Transcription Kit (applied Biosystems). MAX exon 5 alternative splicing was assayed using primers designed to constitutive exons flanking alternative exons (as in (21)). The primers were: MAX (85F), 5’-tcagtcccatcactccaagg-3’; MAX (85R), 5’-gcacttgacctcgccttct-3’; Reverse primers were 32P-end labeled and PCR reactions were amplified for 22 cycles and resolved by denaturing PAGE and imaged.
IP-qt-PCR assay and qRT-PCR assays for RNA expression-
As described (22), cells were lysed and extracts cleared by centrifugation and immunoprecipitated with anti-eIF-4E or control IgG. Associated RNA was subjected to qRT-PCR assays which were performed using Taqman qPCR technology in the 7900HT system (Applied Biosystems; Myc Taqman primer reference Mm00487804-m1).
RNA sequencing-
Total RNA was extracted using RNeasy kit (Qiagen) following the manufacturer’s instructions. The cDNA library preparation and RNA sequencing were performed by the UCLA Center for Genomics & Bioinformatics, as previously described (23). Briefly, the cDNA library was prepared using KAPA Stranded mRNA-Seq Kit (Roche). Reads were obtained by Illumina HiSeq 3000 sequencer for Single End 50bp. Partek Flow Genomic Analysis software was used for bioinformatics analysis including reads alignment, gene feature annotation and differential gene expression analysis. Reads were aligned to the mouse (mm10) reference genome using STAR-2.7.8a aligner. Ensembl Transcripts released 102 gtf was used for gene feature annotation. The differential gene expressions were examined using DESeq2 algorithm. QIAGEN Ingenuity Pathway Analysis (IPA) software was used for pathway enrichment analysis of differentially expressed genes. The RNA seq data was deposited in NCBI’s Gene Expression Omnibus and are accessible through GSE185925 accession number.
Statistics-
Unless otherwise described, the student t-test was used to determine p values.
RESULTS
A1 expression in MM-
We first mined a public data base (24–26) of RNA expression in patients with plasma cell dyscrasias. As previously described (7–9), c-myc expression increased as MM disease activity increased (mean fold increase vs normal plasma cells shown below fig 1A). A1 expression also increased significantly as disease activity increased (Fig 1A; p<0.0001 by ANOVA). In a post-hoc analysis (Tukey-Kramer test, suppl fig S1A), statistically significant differences were identified between MM diagnoses (smoldering, at diagnosis, or at relapse) versus normal or MGUS diagnoses. In contrast myc expression continues to increase during the course of tumor progression. This suggests that A1 dysregulation may be critical for early stages of myelomagenesis but not later stages of progression where continued myc dysregulation occurs. In a second data base (MMRF CoMMpass cohort), evaluating symptomatic patients, c-myc expression also correlated with A1 expression (Fig 1B). In addition, A1 expression correlated with decreased survival (suppl fig S1B). Fig 1C (upper panel) demonstrates that A1 protein expression is also present in primary MM cells which was comparable to the RPMI8226 MM cell line. A1 immunoprecipitated from these MM cells is bound to the myc IRES (Fig 1C, lower panel), suggesting that it can function as a myc ITAF in primary cells.
Figure 1: hnRNP A1 expression in myeloma.
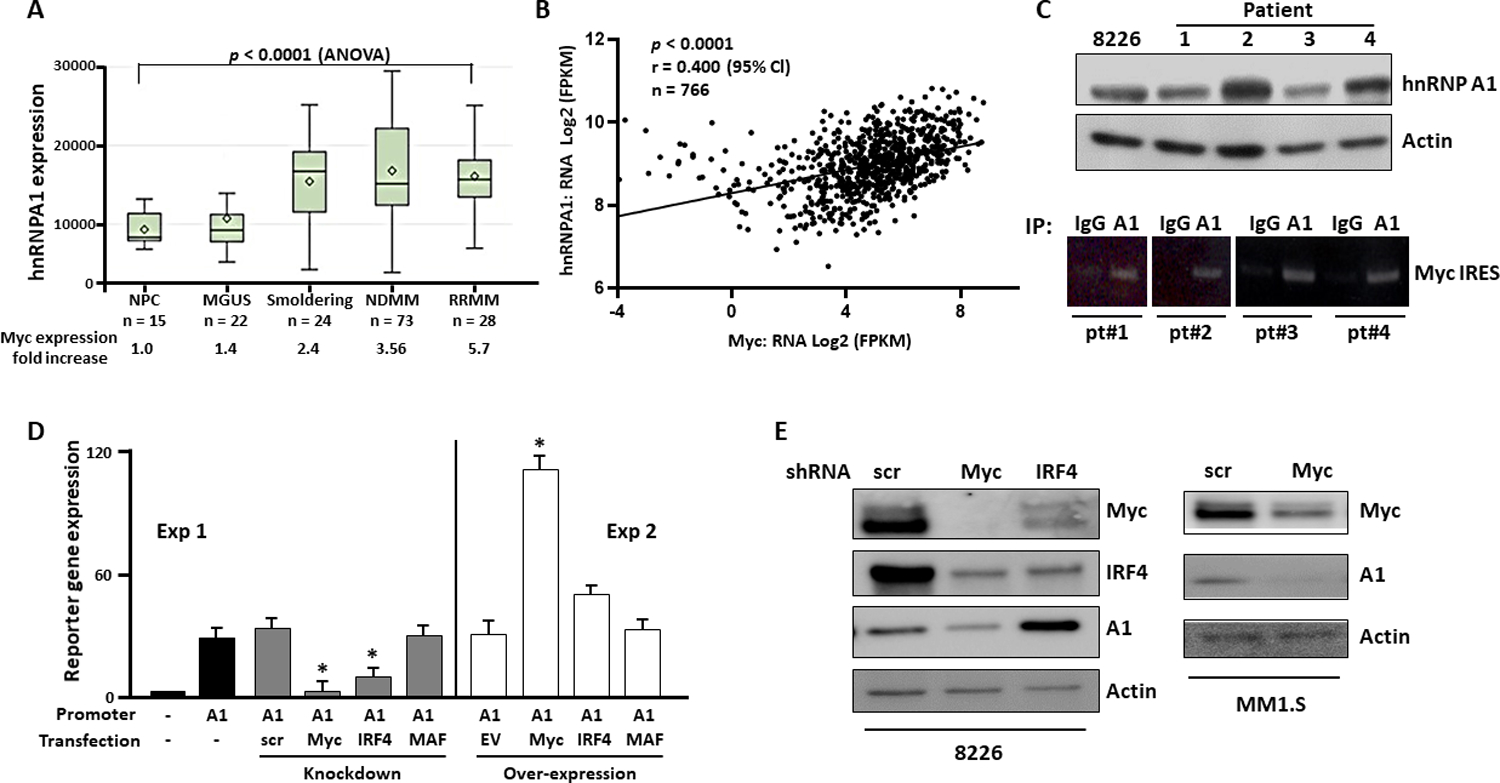
A) A1 expression in plasma cells from normal controls (NL) or patients taken from GSE6477 in GEO omnibus; In box and whisker plots, diamonds=means, cross-lines=medians, top and bottom of boxes are 75 and 25 percentiles and whiskers are ranges; B) Correlation of A1 expression with myc expression in 766 MM patients in the MMRF CoMMpass study (https://research.themmrf.org/); C) Upper panel- Immunoblot for A1 expression in plasma cells from MM patients and the RPMI8226 MM cell line; lower panel- patient cell extracts immunoprecipitated with αIG or αA1 and associated myc IRES identified by RT-PCR; D) A1 reporter assay in RPMI8226 cells following over-expression or knockdown (by shRNA) of myc, IRF4 or MAF oncogenes. Data are mean+/−SD, n=3; *= different from the controls (scrambled (scr) sequence or empty vector (EV)), p<0.05; E) Immunoblot following shRNA knockdown of scrambled sequence (scr), myc or IRF4.
To identify mechanisms of A1 RNA expression, we targeted the MM-associated transcription factors myc, IRF4 and MAF in A1 reporter assays (Fig 1D). At 24 hrs after shRNA was added to silence myc, IRF4 or MAF, there was minimal cytotoxicity (7+/−3% for myc, 5+/−2% for IRF4 and 6+/−1% for MAF knock downs (n=3)). In the RPMI8226 MM cell line, knockdown of myc or IRF4 resulted in decreased reporter expression (left panel). There was also a significant increase when myc was ectopically over-expressed (fig 1D, right panel). Although IRF4 over-expression also resulted in enhanced A1 reporter expression, this did not reach statistical significance. In contrast, there was no effect of MAF knockdown or over-expression. Western blot experiments (Fig 1E) supported the importance of myc as an A1 inducer. We successfully knocked down myc and IRF-4 in the RPMI8226 cell line (left panel). As stated above, both knockdowns resulted in minimal cytotoxicity at 24 or 48 hrs (<10%) although, by 72 hrs, there was excessive death (>40%) attesting to the previously reported (8, 27) myc and IRF4 addiction in MM. C-myc knockdown successfully inhibited A1 expression. Myc knockdown in MM1.S cells (fig 1E, right) also inhibited A1 expression further supporting the notion that c-myc drives A1 expression. The data with IRF4 is not as clear. In an auto-regulatory circuit, IRF4 induces myc and myc induces IRF4 (27) and our immunoblot assay (Fig 1E) is consistent with this autoregulatory loop. However, IRF4 knockdown, although clearly inhibiting A1 reporter expression (Fig 1D), in contrast, increased A1 protein expression (Fig 1E). It is possible that an IRF4 knockdown results in reduced A1 promoter activity but the loss of IRF4 may also derepress factors required for post-transcriptional processes resulting in overall accumulation of A1 at the protein level. The explanation for this dichotomy will require additional future experimentation. Nevertheless, although the role of IRF4 in A1 expression is still unclear, the data clearly indicate that c-myc significantly participates in driving A1 expression in MM cells.
A1 gene deletion prevents c-myc translation-
To investigate a role for A1 in vivo, we used a myc-driven model (17, 28) where plasmacytomas arise in mice doubly transgenic for IL-6 and myc within the B cell lineage at 4–5 months of age. RNA expression profiling in splenocytes, harvested prior to development of tumors, demonstrated a >4 x fold increase in A1 expression relative to litter mates singly transgenic for only the IL-6 gene (Fig 2A). An advantage of this model was the ability to harvest an individual plasma cell tumor, whose tumor cells remained viable during continuous culture, allowing genetic manipulation. This transgenic line, designated IL6Myc-1, expresses high levels of CD138, produces an IgA-kappa monoclonal protein, and is clonogenic in vitro and in vivo (17). Following subcutaneous challenge, it grows as a solid tumor. Following IV challenge, mice develop hind limb paralysis and osteolytic bone lesions (17).
Figure 2: Myc expression in A1-deleted cells.
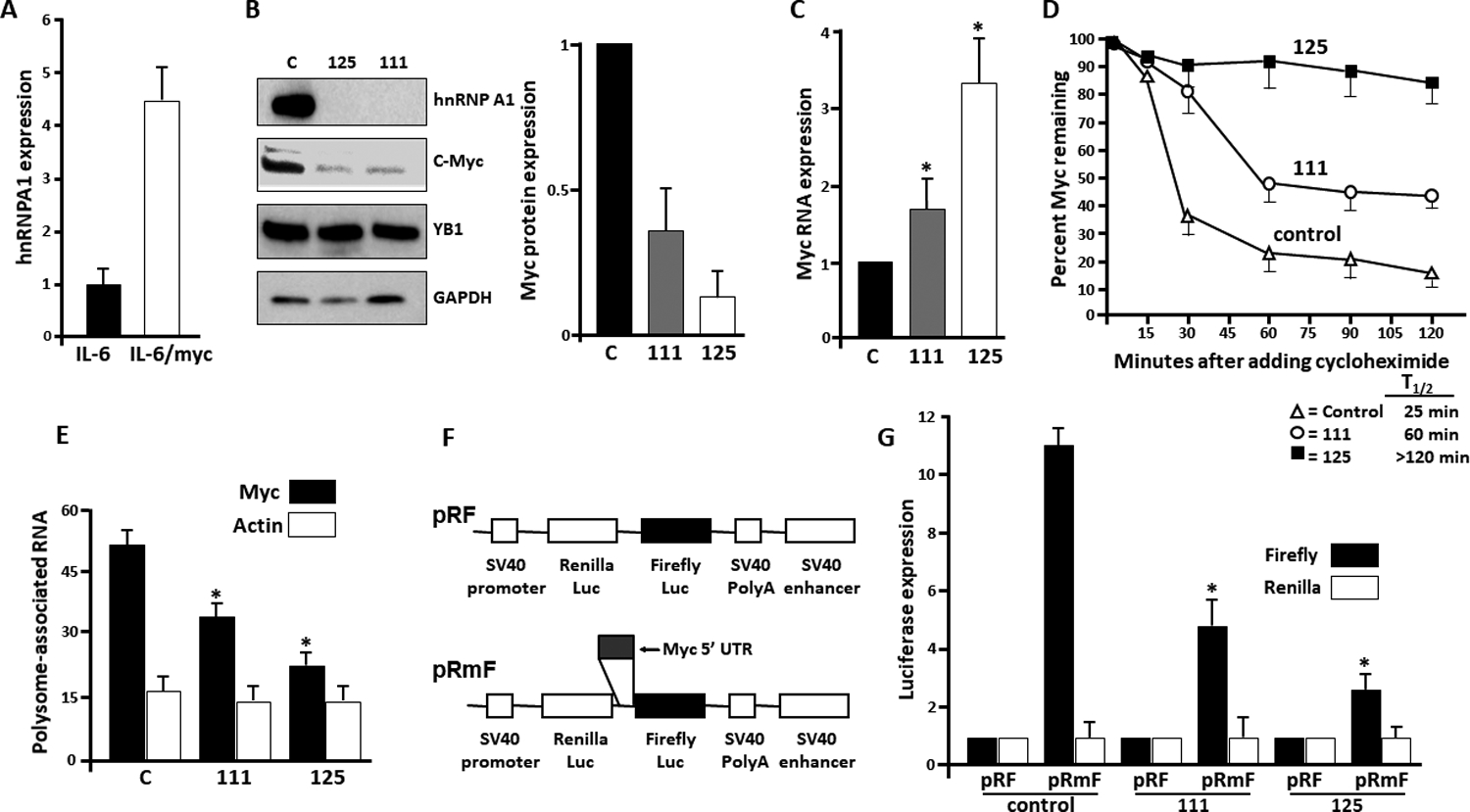
A) Relative A1 expression (by qRT-PCR) in splenocytes from mice doubly transgenic for IL-6+myc genes (IL-6/myc) or singly transgenic for only IL-6 gene. Data are mean+/−SD, n=10; B) left panel- Immunoblot in the IL6/Myc-1 myeloma line (‘C’ for control) vs the same line with a deleted A1 gene (125 and 111 lines); right panel- relative myc expression in the three lines (c-myc/GAPDH ratios by densitometry, mean+/−SD, n=4); C) Relative myc RNA expression (vs GAPDH) in the 3 lines. Data are mean+/−SD, n=4;*=p<0.05 versus control D) Pulse-chase experiment for myc protein stability in the 3\ lines with extrapolated half life shown below graph; E) polysome profiling assay for translation showing % of c-myc or actin RNA associated with polysomes, mean+/−SD, n=3; *=significant (p<0.05) decreased vs control (C) cells. F) Schematic of pRF and pRmF vectors; G) Relative reporter expression of firefly or Renilla luciferase in 3 cell lines transfected with pRF or pRmF vectors. Data are mean+/−SD, n=3; *=significantly decreased (p<0.05) versus pRmF in control cells.
We performed CRISPR-Cas9 deletion of the A1 gene in IL6Myc-1 cells by targeting two independent sequences in exon 1 (termed sequence 11 or 12). A non-targeting gRNA was used as a control. Single cell-derived clones were then generated. All clones from targeted sequences demonstrated deletion of the A1 gene and loss of A1 expression (supplemental figure S2A). Additionally, all clones had diminished expression of c-myc (suppl fig S2A). We selected 1 clone from each of the two targeted sequences to expand and study, designated #125 and #111. Both exhibited loss of A1 expression and decreased c-myc expression when compared to control cells from the non-targeted clone (fig 2B, left and right panels). The inhibitory effect on c-myc expression was specific as A1 deletion had no effect on YB1 expression. YB-1 was used as a specificity control as it is also a known myc-IRES ITAF (29).
We first tested if decreases in steady state myc RNA or myc protein half-life could explain diminished myc protein levels in A1-deleted cells. However, instead of a decrease, there were significant increases (Figs 2C & 2D). These alterations may be compensatory mechanisms. To assess translational efficiency, we performed a polysome analysis. This assay is based upon the observation that well translated transcripts are associated with polysomes and poorly translated mRNAs are monosomal. Thus, polysomes were separated from monosomes and associated RNAs were quantified by qt-PCR. Fig 2E demonstrates a striking decrease in myc RNA associated with polysomes in the 111 and 125 A1-deleted cells, indicating inhibited myc translation. In contrast, translation of actin was not suppressed.
To assess effects on cap-dependent versus IRES-dependent translation, we utilized the dicistronic reporter assay where cells were transfected with reporter constructs shown in fig 2F. The myc 5’UTR, containing its IRES, was subcloned into the intra-cistronic space between the Renilla and Firefly (FF) luciferase ORFs in the pRF vector to yield the pRmF vector. The pRmF reporter’s FF luciferase translation is driven by the myc 5’UTR and is a reflection of IRES-dependent translation whereas Renilla expression is due to cap-dependent translation. Results in fig 2G confirm the presence of the 5’UTR in pRmF reporter increases FF expression without effect on Renilla in control IL6/Myc-1 cells. It is also apparent that relative FF expression is decreased in A1-deleted 111 and 125 cell lines versus control cells. Thus, loss of A1 inhibits IRES-dependent translation of myc.
A1-deleted MM cells are inhibited in tumor growth-
Although containing diminished c-myc levels, A1-deleted 111 and 125 cells were capable of in vitro expansion, doubling in viable cell numbers by 72 hrs (Fig 3A). While in vitro growth was comparable to control cultures at 24 and 48 hrs, there was a modest decrease at 72 hrs which was maintained at 96 hrs but not more impressive (30–35% decrease in viable tumor cell numbers in 111 and 125 cultures). Mice were next challenged SQ with control IL6Myc-1 cells (designated wild type (WT) in figure 3B) or A1-deleted tumor cells. Individual mice received WT control inocula in one flank (9 inocula) or 111 cell challenges (n=9) in the opposite flank (Group 1) or WT control inocula in one flank (n-9) and 125 cell challenges in the opposite flank (n=9) (Group 2). As shown in figs 3B and 3C, outgrowth of 111 and 125 tumors were inhibited compared to WT control tumors. This was especially true for 125 tumors which were extremely small (Figure 3D). Immunoblot of A1-deleted tumors demonstrated corresponding decreases in c-myc protein (Figure 3E) and absence of hnRNP A1 (latter examples shown in suppl fig S2B).
Figure 3: Depressed growth of A1-deleted tumors-.
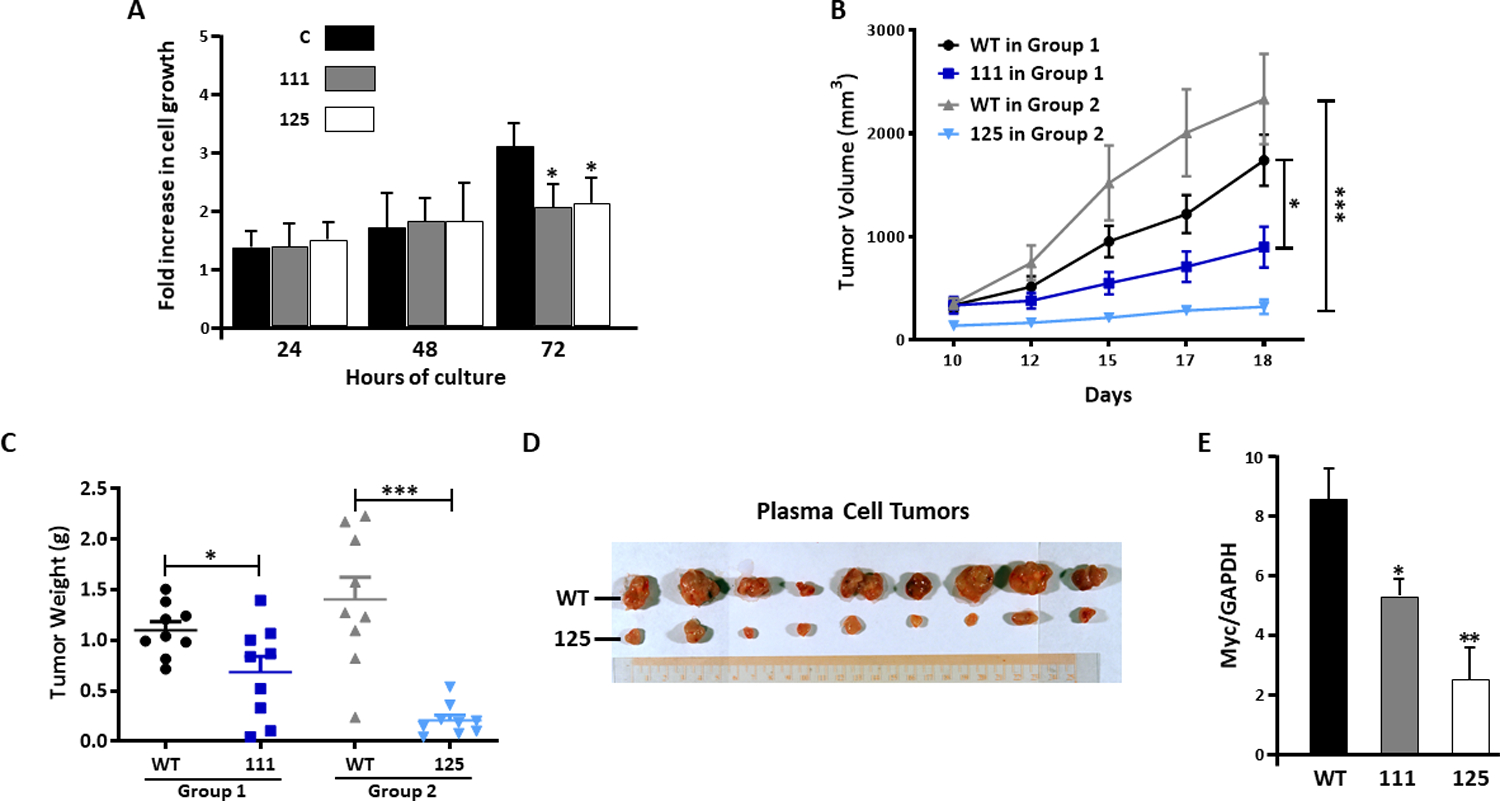
A) A1-containing control (C) or A1-deleted 111 or 125 cells were seeded at 105/ml and growth assessed 24–72 hours later. Data are fold increase in viable cell numbers, mean+/−SD, n=3. *=decrease vs control (C) by T-test, p<0.05; B) Growth of wild type (WT) vs 111 or 125 cell lines following subcutaneous challenge. Data represent mean+/−SD, 9 mice/group (6 female, 3 male); C) Weights of tumors from mice of experiment shown in ‘B’. Data are mean+/−SEM; D) Individual tumors dissected from mice 18 days after challenge; E) Immunoblot detection of c-myc (vs GAPDH) expression in lysates of tumors obtained from experiment shown in ‘B’ and ‘C’. *=significantly decreased, p<0.05; **= p<0.01.
Effects of a myc IRES inhibitor-
We previously identified an inhibitor, C11, which prohibited myc IRES activity (10). C11 was biochemically modified to yield a more efficacious inhibitor, J007 (Fig 4A) (detailed synthesis in reference #22). IL6/Myc-1 cells exposed to J007 demonstrated a concentration-dependent decrease in myc expression (fig 4B). J007 was also effective in the RPMI8226 line and primary MM specimens (fig 4B). Inhibited myc expression in IL6/Myc-1 cells was not associated with decreases in myc RNA abundance (fig 4C) nor protein stability (suppl fig S3). Polysome profiling (fig 4D) demonstrates an inhibitory effect of J007 (50nM) on translational efficiency of myc. In the dicistronic reporter assay, J007 significantly decreased FF expression while having no effect on Renilla expression (fig 4E). These results indicated that J007 could inhibit IRES-dependent translation in MM cells. However, the ability of the inhibitor, used alone, to curtail overall protein expression was surprising since cap-dependent translation of myc should proceed unfettered. Thus, we performed additional assays to rule out IRES-independent adverse effects on c-myc. As A1 is also a splicing factor (21), we tested if a J007-A1 interaction regulates Max splicing. Max is spliced via A1 to generate Delta Max, a truncated version of the Max protein which includes exon 5 (21). As shown in fig 4F, J007 exposure did not alter Max splicing in wild type cells. As a positive control, the A1 deletion in 125 cells markedly reduced exon 5-containing Delta Max transcript levels. Furthermore, we tested effects of J007 on cap-dependent translation of myc by immunoprecipitating eIF-4E and assessing the relative amounts of associated c-myc mRNA. As shown in fig 4G, J007 did not inhibit c-myc RNA association with eIF-4E ruling out an effect on the mTOR/4EBP-1/eIF-4E cascade as an explanation for J007’s inhibition of myc expression.
Figure 4: Effect of J007-.
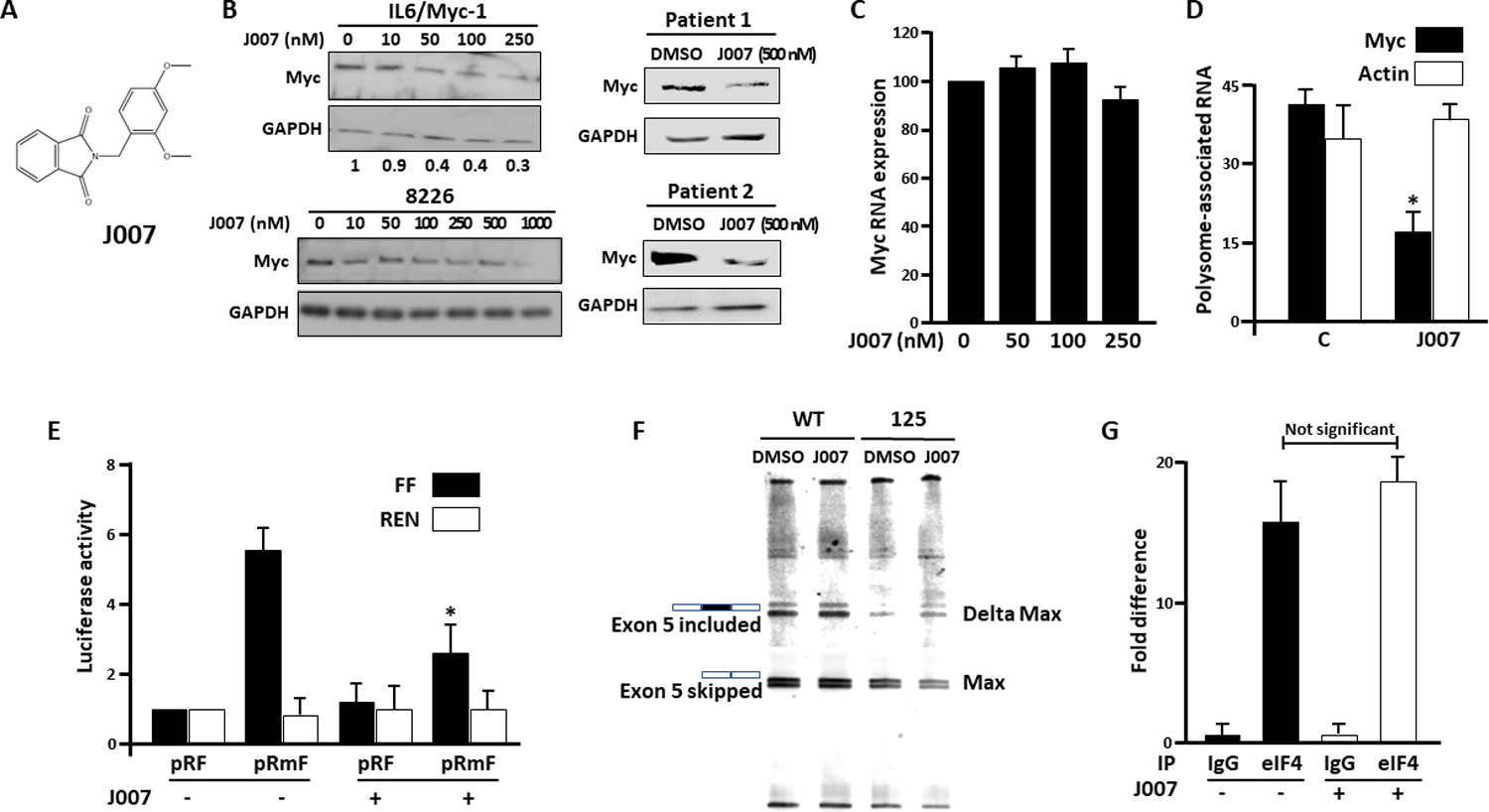
A) Structure of J007; B) Immunoblot of murine IL6/Myc-1, RPMI8226 or patient MM cells after exposure to J007 for 24 hours. IL6/Myc-1, RPMI8226 and primary cell viabilities were maintained >85% after exposure; Below IL6/Myc-1 blot are relative myc/GAPDH expression levels from 3 experiments (means); C) Relative myc RNA expression (percent of control (J007=0)) in cells exposed to J007 for 24 hours (mean+/−SD, n=3); D) Translational efficiency in cells treated +/− J007 (100nM), mean+/−SD, n=3; *=significantly (p<0.05) decreased versus control. E) Dicistronic reporter assay in cells treated +/− J007 (at 100nM) for 24 hours. Data are mean+/−SD, n=3. *= different from untreated cells, p<0.05. F) Wild type (WT) or A1-deleted 125 cells were treated +/− J007 at 100 nM and splicing analysis for Max exon 5 was performed; G) Wild type cells treated +/− J007 (100 nM for 24 hrs) were lysed and immunoprecipitated (IP) using either eIF-4E or control IgG antibody. Bound c-myc RNA was detected via qt-PCR. Data represent bound myc RNA (mean+/−SD, n=3) versus background amount in IgG control immunoprecipitates (arbitrarily designated ‘1’).
To test J007 in vivo, mice were challenged with SQ inocula of control IL6Myc-1 tumor cells in one flank and 111 tumor cells in the opposite flank. Half were treated with J007 and half with vehicle. The 111 MM cells are moderately inhibited in tumor growth (fig 3B & 3C) but should not be further inhibited by J007 as the drug’s target (A1) is absent. As shown in figure 5A, J007 prevented tumor outgrowth from control IL6Myc-1 inocula but had no effect on growth of 111 tumors, documenting the A1 specificity of the anti-tumor response. J007-treated mice demonstrated no toxicity or weight loss (supplemental fig S4A). Immunoblot analyses confirmed inhibited myc expression in wild type control tumors but no effect in 111 tumors (Figure 5B). In a separate experiment, treatment of tumor-challenged mice with J007 resulted in prolonged survival (Figure 5C).
Figure 5: Effect of J007 in vivo.
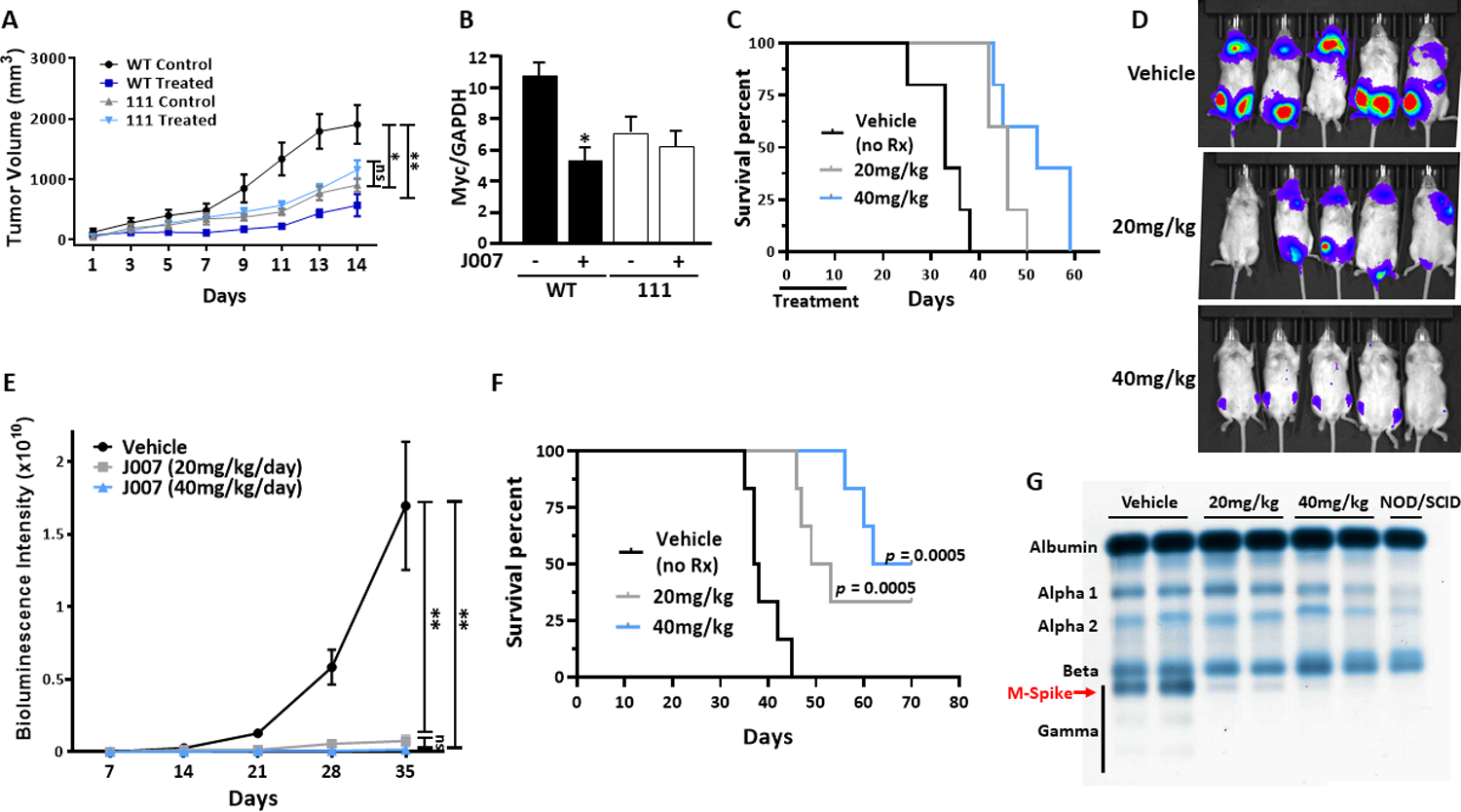
A) Mice (8/group; 6 female, 2 male) challenged SQ w/ WT control cells or A1-deleted 111 cells and treated +/− daily IP injections of J007 at 20 mg/kg; J007 significantly (p<0.01) inhibited growth of WT cells but had no effect on 111 growth; B) Immunoblot of myc expression in tumors obtained from mice shown in figure 5A; *=significantly inhibited (p<0.01) vs non-treatment; C) Survival of mice (5 mice/group; 3 female, 2 male) challenged with SQ tumors and treated +/− J007 with daily injections for 15 days; J007 significantly prolonged survival at both doses (p=0.0018); D) Bioluminescence of mice challenged with IV tumor cells, treated with vehicle or J007 and imaged day 28; Treatment was started day +7 after inoculation and continued until day +28; E) ROI signal of experiment shown in fig ‘D’; F) Survival of mice from experiment shown in ‘D’ (6 mice/group, 3 female, 3 male); G) Electrophoresis of serum from B cell-deficient NOD/SCID mice challenged IV with MM cells and treated as in fig 5F. Each lane comprises pooled sera from 3 mice in each group. “NOD/SCID” lane from non-tumor-bearing mice (gamma globulin fraction is ‘empty’ as mice are B cell deficient).
We also tested if J007 was efficacious in mice challenged IV with luciferase-expressing IL6Myc-1 where tumor grows within the skeleton. Vehicle-treated control mice demonstrated progressive tumor growth which was suppressed by both 20 and 40mg/kg of J007 (Figs 5D & 5E). In addition, while having no effect on weights (suppl fig S4B), both doses prolonged survival of IV-challenged mice (Fig 5F) and markedly diminished serum M-protein (Fig 5G). MicroCT demonstrated an inhibition of bone loss which accompanied tumor growth in vehicle-control mice (suppl fig S5).
Effects of combining J007 with pp242-
In a previous study (22), IRES inhibition had additive anti-tumor effects when administered with the mTOR inhibitor pp242. This was not surprising as both IRES-dependent myc translation would be prohibited as well as mTOR/cap-dependent translation (by pp242). We, thus, tested if J007 could interact with pp242 for heightened responses. Although mTORC1 was inhibited by pp242 in vitro (decreased phosphorylation of p70/S6/4EBP-1), there was no inhibition of c-myc expression (suppl fig S6A). Since mTOR inhibitors upregulate IRES activity (30, 31), the lack of pp242’s effect on myc expression further supports the primacy of cap-independent myc translation in the MM model. However, the addition of pp242 to a low concentration of J007 resulted in greater inhibition of c-myc expression as compared to J007 used alone (Supplemental figure S6B). To assess an interaction in vivo, tumor-challenged mice were treated with DMSO, pp242 (50 mg/kg/day), J007 (20 mg/kg/day) or the combination of drugs. As shown in supplemental fig S6C, both pp242 or J007, used alone, induced a significant albeit modest slowing of tumor growth. The combination of drugs resulted in enhanced anti-tumor effect.
Re-expression of c-myc in A1-deleted cells-
To confirm that diminished tumor growth in A1-deleted MM cells was due to c-myc inhibition, we attempted to ectopically re-express myc. However, as previous data suggested a dependence on A1-dependent IRES activity for myc translation, we were concerned that transfected myc would not be expressed in A1-deleted cells. Prior studies have identified sequences in the p27 mRNA leader that can direct cap-independent translation (32, 33) and the p27 IRES does not require A1 as an ITAF (18). Accordingly, we generated a construct in which we replaced the native 5’UTR of c-myc with the p27 leader containing its IRES. The transcription of c-myc-containing genes was driven by a CMV promoter. The A1-deleted 125 MM line was transfected with an empty vector, the myc ORF construct (no 5’UTR) or the p27IRES-myc ORF (p27 IRES upstream of the myc ORF). After selection, c-myc expression was compared to control WT cells. As shown in fig 6A, the inhibited myc protein expression in EV-transfected 125 cells is again identified (versus control WT cells). Down-regulated myc expression in 125 cells is not affected by transfection with CMV-myc ORF but the construct with upstream p27 IRES sequences allowed re-expression of myc protein. Fig 6B demonstrates the myc protein re-expression data from 3 independent experiments and shows that myc RNAs were faithfully expressed. These cell lines were then used to challenge mice. As shown in fig 6C & 6D, tumor growth was rescued in A1-deleted 125 cells only when transfected with the p27IRES-myc ORF construct. Immunoblot assay (fig 6E) confirms re-expression of myc correlates with rescued tumor outgrowth. These data confirm that the decreased myc expression in A1-deleted cells is a key determinant of inhibited tumor growth and, furthermore, underscore the importance of the 5’UTR in A1’s regulation of myc expression.
Figure 6: Re-expression of c-myc in A1-deleted cells.
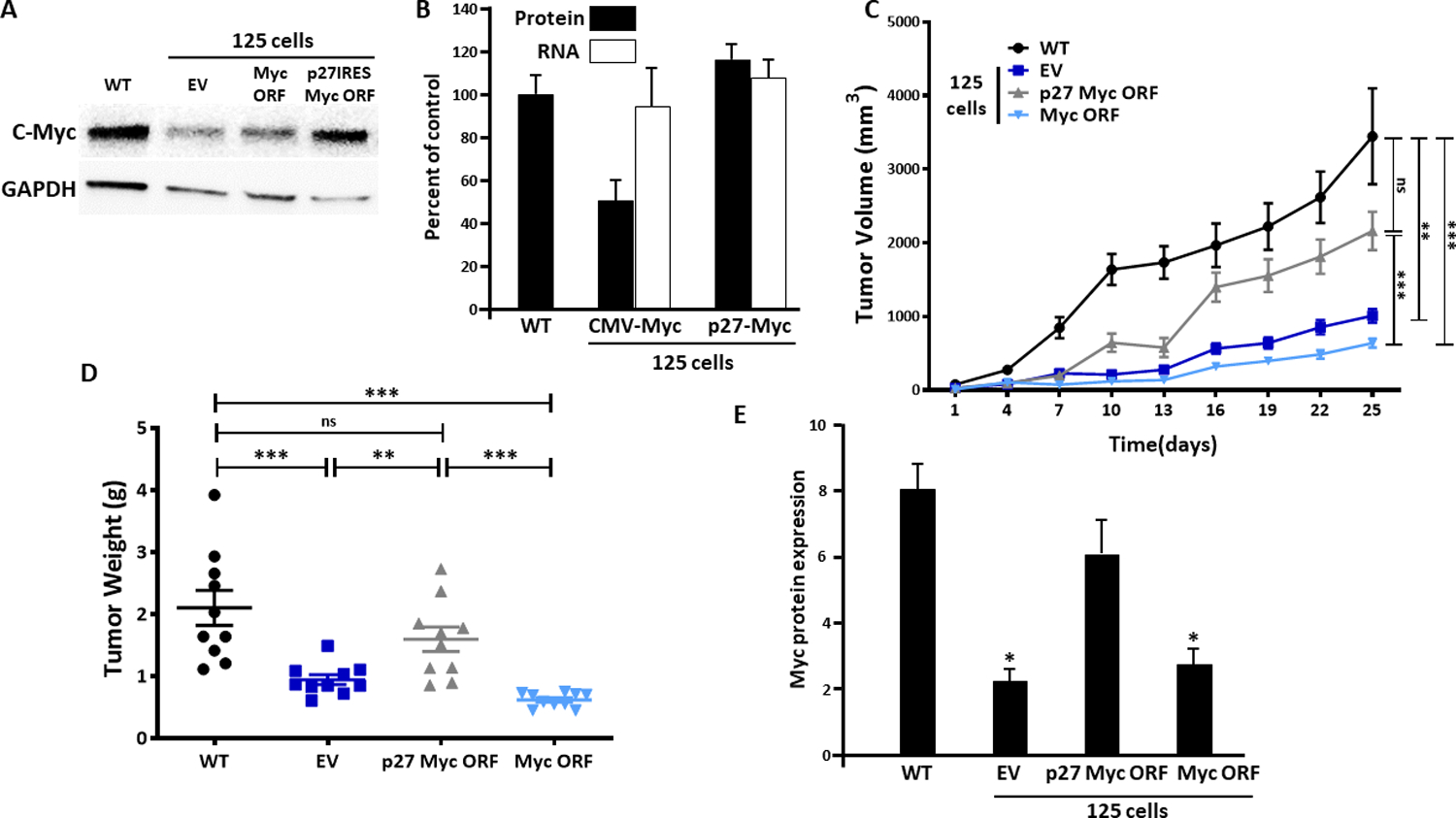
A) A1-deleted 125 cell line transfected with empty vector (EV), myc expression vector (myc ORF) or myc expression vector with inserted p27 IRES. Immunoblot demonstrates re-expression of c-myc in latter transfected cells; B) myc RNA (white bars) versus protein (black bars) expression in transfected lines, mean+/−SD, n=3; C) Tumor growth after challenge with the 4 cell lines (10 mice/group, 6 female, 4 male); D) Tumor weights from experiment shown in ‘C’; E) myc protein expression (vs GAPDH) in lysates of tumors from experiment shown in ‘C’, mean+/−SD; *=significantly lower than WT tumors.
Although our data indicated that down-regulation of myc translation mediates J007’s anti-MM effect, it was possible that other alterations could play a role. We, thus, performed an unbiased evaluation of altered gene expression in J007-treated MM cells. Genome wide RNAseq data was obtained from WT cells treated +/− J007 (1 uM for 72 hrs). The viability of both cell preparations was >97% at the time of RNA harvesting. We subsequently identified 257 genes that were upregulated greater than 2x fold and 230 genes that were downregulated at least 50% by J007. Gene set enrichment from the RNAseq assay is shown in supplemental figure 7A. As anticipated, the most significantly altered gene set was that of ‘myc targets’ (enrichment plot in suppl fig 7B. Of the myc target activation gene set, several significantly down-regulated genes were of particular interest for the myeloma model, such as heat shock protein 1, exportin 1, PCNA and several proteasomal subunits. However, it is interesting that the ‘angiogenesis’, ‘mTORC1 signaling’, ‘protein secretion’, ‘G2M checkpoint’, ‘unfolded protein response’ and ‘fatty acid metabolism’ gene sets were also decreased by J007 treatment. Some of these additional alterations may contribute to the anti-MM effect of J007.
DISCUSSION
These results support roles for A1 and myc IRES in MM. They also indict myc as an inducer of A1 expression in MM. A1 RNA expression was considerably higher in IL6/Myc transgenic B cells versus single IL6 transgenic mice and over-expression/knockdown experiments in human MM lines also support a role for myc. These results point to a positive feedback circuit where myc stimulates A1 RNA expression and A1 enhances myc translation.
It was remarkable that IRES targeting, by itself, was so effective because mTOR/cap-dependent translation of myc should have proceeded without restriction. In fact, short term exposure to the mTOR inhibitor, pp242, had no effect on myc expression (suppl fig 6). These results suggest that overall myc translation and expression in MM is more dependent on IRES activity than on mTOR activity. It is certainly possible that A1 may also enhance cap-dependent translation in addition to its role as a myc ITAF (34). Nevertheless, the rescue of myc expression by transfection which required a functional IRES upstream of the myc ORF, underscores the importance of A1-dependent IRES activity. In addition, although the mTORC1 pathway was a gene set significantly impacted by J007 in RNAseq analysis (suppl fig S7), J007 had no effect on binding of myc RNA to eIF-4E, indicating mTORC1/cap-dependent translation of myc was not constrained by J007.
A particular ER stress in myc-driven MM may explain heightened sensitivity to IRES disruption as depicted in the model in supplemental figure S8. ER stress inhibits mTORC1 (10) and promotes a shift to IRES-dependent translation via the unfolded protein response (UPR). This renders MM cells completely dependent on IRES activity for myc translation. In similar fashion, A1 mediates activation of IRES-dependent SREBP-1a translation in response to ER stress (35). Previous work with additional myc ITAFs (29, 36, 37) also supports the critical role of the myc IRES in MM cells.
An unbiased transcriptome analysis suggested additional J007-induced alterations that could participate in an anti-MM response. Some of these (for example, angiogenesis) may simply be sequelae of inhibited myc target activation. However, direct effects on G2M cell cycle transit, the unfolded protein response and fatty acid metabolism may be contributary and deserve to be tested.
Targeting of the myc oncogene has been a long-sought goal for many years (38, 39). The molecular structure of the myc gene and its RNA in MM may render it sensitive to IRES targeting. Although lymphoma cells with 8;14 translocations contain myc breakpoints which theoretically disrupt or decapitate the IRES, only 15% of MM specimens contain a myc translocation (40) and even MM cell lines with myc translocations contain an intact exon 1 (41). Thus, a strategy of targeting myc with a drug like J007 in MM is promising.
Supplementary Material
Acknowledgments
Supported by NIH grants RO1CA111448, RO1CA214246, RO1CA217820, RO1CA151354, the VA, the MMRF and The William G Schuett, Jr. MM Research Endowment
Footnotes
Conflicts: Authors disclose no conflicts
REFERENCES
- 1).Carrasco DR, Sukhdeo K, Protopopova M, et al. The factor XBP-1 drives myeloma pathogenesis. Cancer Cell 2007; 11:349–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2).Obeng EA, Carlson LM, Gutman DM, et al. Proteasome inhibitors induce a terminal UPR in myeloma cells. Blood 2006; 107:4907–4916 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3).Suzuki A, Iida S, Kato-Uranishi M, et al. ARK5 is regulated by MAF and mediates IGF-1-induced invasion in myeloma. Oncogene 2005; 24:6936–6944 [DOI] [PubMed] [Google Scholar]
- 4).Peterson TR, Laplante M, Thoreen CC, et al. DEPTOR is an mTOR inhibitor overexpressed in multiple myeloma cells and required for survival. Cell 2009; 137:873–886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5).Liu L, Ulbrich J, Muller J, et al. Deregulated MYC induces dependence upon AMPK-related kinase 5. Nature 2012; 483:608–612 [DOI] [PubMed] [Google Scholar]
- 6).Bliskovsky V, Ramsay ES, Scott J, et al. FRAP is a candidate gene for plasmacytoma resistance locus Pctr2 and can act as a tumor suppressor. Proc Natl Acad Sci 2003; 100:14982–14987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7).Bergsagel PL & Kuehl WM. Chromosome translocations in myeloma. Oncogene 2001; 20:5611–5622 [DOI] [PubMed] [Google Scholar]
- 8).Kuehl WM & Bergsagel PL. Myc addiction: A potential target in MM. Blood 2012; 120:2351–2352 [DOI] [PubMed] [Google Scholar]
- 9).Holien T, Vatsveen TK, Hella H, Waage A, Sundan A. Addiction to myc in myeloma. Blood 2012; 120:2450–2453 [DOI] [PubMed] [Google Scholar]
- 10).Shi Y, Yang Y, Hoang B, et al. Therapeutic potential of targeting IRES-dependent myc translation in myeloma cells during ER stress. Oncogene 2016; 35:1015–1024 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 11).Baird SD, Turcotte M, Korneluk RG, Holcik M. Searching for IRES. RNA 2006; 10:1755–1785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12).Hellen CU & Sarnow P. Internal ribosome entry sites in eukaryotic mRNA molecules.Genes & Dev 2001; 15:1593–1612 [DOI] [PubMed] [Google Scholar]
- 13).Gera J & Lichtenstein A (2013) IRES-dependent, cap-independent translation in myeloma. Chapter 4 in “Genetic & Molecular Epidemiology of multiple Myeloma”, Springer, ed. Lentzsch Suzanne [Google Scholar]
- 14).Stoneley M & Willis AE. Cellular IRESes: structures, trans-acting factors and regulation of gene expression. Oncogene 2004; 23:3200–3207 [DOI] [PubMed] [Google Scholar]
- 15).Subkhankulova T, Mitchell SA, Willis AE. IRES-mediated initiation of c-myc protein synthesis following genotoxic stress. Biochem J. 2001; 359:183–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16).Jo OD, Martin J, Bernath A, et al. hnRNP A1 regulates cyclin D1 and c-myc IRES function through AKT signaling. Journal of Biol Chem 2008; 283:23274–23287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17).Sun F, Cheng Y, Walsh SA, et al. Osteolytic disease in IL-6 and myc-dependent mouse model of human myeloma. Haematologica 2019; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18).Gera JF, Mellinghoff IK, Shi Y, Rettig M, Tran C, Hsu J-h, et al. AKT activity determines sensitivity to mTOR inhibitors by regulating cyclin D1 and c-myc expression J Biol Chem 2004; 279:2737–27461 [DOI] [PubMed] [Google Scholar]
- 19).Shi Y, Frost P, Hoang B, et al. MNK kinases facilitate c-myc IRES activity in rapamycin-treated myeloma cells. Oncogene 2013; 32:190–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20).Biamonti G, Bassi MT, Cartegni L, et al. hnRNP protein A1 gene expression. J. Mol. Biol 1993; 230:77–89 [DOI] [PubMed] [Google Scholar]
- 21).Babic I, Anderson ES, Tanaka K, et al. EGFR mutation-induced alternative splicing of Max contributes to growth of tumors in brain cancer. Cell Metab 2013; 17:1000–1008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22).Holmes B, Lee J, Landon K, et al. mTOR inhibition synergizes with reduced IRES-mediated translation of cyclin D1 and c-myc mRNAs to treat glioblastoma. Journal of Biolog Chem 2016; 291:14146–14159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23).Fisher AL, Sangkhae V, Balusikova K, et al. Iron-dependent apoptosis causes embryotoxicity in inflamed and obese pregnancy. Nature Communic 2021; 12:4026–4042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24).Chng WJ, Kumar S, Vanwier S, et al. Molecular dissection of hyperdiploid myeloma by gene expression profiling. Cancer Res 2007; 67:2982–2989 [DOI] [PubMed] [Google Scholar]
- 25).Anguiano A, Tuchman SA, Acharya C, et al. Gene expression profiles of tumor biology provide a novel approach to prognosis and guide the selection of therapeutic targets in myeloma. J Clinical Onc 2009; 27:4197–4203 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26).Chng WJ, Huang GF, Chung TH et al. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed myeloma. Leukemia 2011; 25:1026–1035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27).Shaffer AL, Tolga Emre NC, Lamy L, et al. IRF4 addiction in myeloma. Nature 2008; 454:226–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28).Rutsch S, Neppalli VT, Shin D-M, et al. IL-6 and myc collaborate in plasma cell tumor formation in mice. Blood 2010; 115:1746–1754 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29).Bommert KS, Effenberger M, Leich E, et al. The feed-forward loop between YB-1 and myc is essential for multiple myeloma cell survival. Leukemia 2013; 27:441–450 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30).Cloninger C, Bernath A, Bashir T, et al. Inhibition of SAPK2/p38 enhances sensitivity to mTORC1 inhibition by blocking IRES-mediated translation initiation in glioblastoma. Mol Cancer Ther 2011; 10:2244–2256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31).Thoreen CC, Chantranupong L, Keys HR et al. A unifying model for mTORC1-mediated regulation of mRNA translation. Nature 2012; 485:109–113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32).Kullmann M, Gopfert U, Siewe B, Hengst L. ELAV/Hu proteins inhibit p27 translation via an IRES element in the p27 5’UTR. Genes & Development 2002; 16:3087–3099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33).Miskimins WK, Wang G, Hawkinson M, Miskimins R. Control of cyclin-dependent kinase inhibitor p27 expression by cap-independent translation. Mol Cell Biol 2001; 21:4960–4967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34).Svitkin YV, Ovchinnikov LP, Dreyfuss G, Sonenberg N. General RNA binding proteins render translation cap dependent. The EMBO Journal 1996; 15:7147–7155 [PMC free article] [PubMed] [Google Scholar]
- 35).Damiano F, Rochira A, Tocci R, et al. hnRNP A1 mediates the activation of the IRES-dependent SREBP-1a mRNA translation in response to ER stress. Biochem J 2013; 449:543–553 [DOI] [PubMed] [Google Scholar]
- 36).Cobbold LC, Wilson LA, Sawicka K, et al. Upregulated c-myc expression in multiple myeloma by internal ribosome entry results from increased interactions with and expression of PTB-1 and YB-1. Oncogene 2010; 29:2884–2891 [DOI] [PubMed] [Google Scholar]
- 37).Chatterjee M, Rancso C, Stuhmer T, et al. The Y box protein YB-1 is associated with pregressive disease and mediates survival and drug resistance in multiple myeloma. Blood 2008; 111:3714–3722 [DOI] [PubMed] [Google Scholar]
- 38).Chaidos A, Caputo V, Gouvedenou K et al. Potent antimyeloma activity of the novel bromodomain inhibitors I-BET151 and I-BET762. Blood 2014; 123:697–705 [DOI] [PubMed] [Google Scholar]
- 39).Delmore JE, Issa GC, Lemieux ME, et al. BET bromodomain inhibition as a therapeutic strategy to target c-myc. Cell 2011; 146:904–917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40).Avet-Loiseau H, Gerson F, Magrangeas F, et al. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001; 98:3082–3086 [DOI] [PubMed] [Google Scholar]
- 41).Shou Y, Martello ML, Gabrea A, et al. Diverse karyotypic abnormalities of the c-myc locus associated with c-myc dysregulation and tumor progression in multiple myeloma. Proc Natl Acad Sci 2000; 97:228–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.