


Abstract
The 48 human nuclear receptors (NRs) form a superfamily of transcription factors that regulate major physiological and pathological processes. Emerging evidence suggests that NR crosstalk can fundamentally change our understanding of NR biology, but detailed molecular mechanisms of crosstalk are lacking. Here, we report the molecular basis of crosstalk between the pregnane X receptor (PXR) and constitutive androstane receptor (CAR), where they form a novel heterodimer, resulting in their mutual inhibition. PXR and CAR regulate drug metabolism and energy metabolism. Although they have been broadly perceived as functionally redundant, a growing number of reports suggests a mutual inhibitory relation, but their precise mode of coordinated action remains unknown. Using methods including RNA sequencing, small-angle X-ray scattering and crosslinking mass spectrometry we demonstrate that the mutual inhibition altered gene expression globally and is attributed to the novel PXR–CAR heterodimerization via the same interface used by each receptor to heterodimerize with its functional partner, retinoid X receptor (RXR). These findings establish an unexpected functional relation between PXR, CAR and RXR, change the perceived functional relation between PXR and CAR, open new perspectives on elucidating their role and designing approaches to regulate them, and highlight the importance to comprehensively investigate nuclear receptor crosstalk.
INTRODUCTON
Nuclear receptors are members of a superfamily of transcription factors that regulate a myriad of physiological processes, including metabolism, inflammation, reproduction and cell growth (1,2). There are 48 human nuclear receptors, and their importance in pathophysiology is reflected by the fact that they are drug targets for over 15% of all approved drugs (3). Although most studies have been centered on the function of single nuclear receptors, the significance of considering the interplay among them and their signaling pathways has become very clear (4). It has been recognized that previously unknown crosstalk among nuclear receptors can challenge preconceived fundamental notions of target gene regulation with potential therapeutic implications, because manipulation of such crosstalk can be used as a strategy to alter gene expression pattern with beneficial clinical outcomes.
Although functional interplay among nuclear receptors has been observed, a fundamental but poorly explored frontier in understanding nuclear receptor function is that detailed molecular mechanisms of crosstalk are lacking (4). The proposed mechanisms include competition to overlapping DNA binding sites, competition for binding to common regulatory protein partners resulting in squelching, and direct interactions between different nuclear receptors (4–6). Many nuclear receptors are known to heterodimerize with the promiscuous nuclear receptor retinoid X receptor (RXR), which is required for their transcriptional activation (7). Aside from the obligatory association with RXR, physical interactions among nuclear receptors have largely remained a conceptualized notion based on indirect observations or partial experimental studies.
To investigate the molecular basis of nuclear receptor crosstalk, we chose the most important xenobiotic nuclear receptors human pregnane X receptor (hPXR) (NR1I2) and human constitutive androstane receptor (hCAR) (NR1I3) as a model. PXR and CAR are the major nuclear receptors that regulate the expression of genes encoding proteins involved in the metabolism and excretion of xenobiotics and endogenous metabolites, such as cytochrome P450 (CYP) drug-metabolizing enzymes, drug-conjugating enzymes, and drug transporters (8,9). Therefore, PXR and CAR have become important targets in controlling therapeutic and toxic drug effects. In addition, PXR and CAR have been recognized to regulate many cellular processes, such as maintaining energy homeostasis, cell proliferation, inflammation, and cancer development (10,11). They are considered functionally redundant because of their many overlapping roles in binding chemically diverse ligands and regulating the expression of common subsets of target genes (12). However, observations from cellular and animal studies indicate a possible mutual inhibitory relation between PXR and CAR, but the molecular basis is unknown (5,13,14). PXR and CAR represent nuclear receptors that heterodimerize with RXR, which are observed to be functionally interacting but without evidence for direct physical association. Mechanistic insights for PXR–CAR crosstalk will not only be critical to delineate the precise mode of coordination between PXR and CAR but will also shed light on uncharacterized crosstalk between other nuclear receptors.
We report here that hPXR and hCAR physically interact with each other, resulting in their mutual inhibition and alterations to their target gene expression profile. Using a combination of cell-based and biophysical methods, we mapped the heterodimerization interface between hPXR and hCAR to that used by RXR to interact with each receptor. We contemplate the possibility that similar heterodimerization interfaces can exist in other nuclear receptors that require RXR for transcriptional activation. Based on the numerous crystallographic studies, all receptors that heterodimerize with RXR share similar dimerization interfaces (15). Therefore, our work outlines the specifics for a potentially common crosstalk mechanism based on the formation of novel heterodimer complexes.
Our discovery of the inhibitory interactions between PXR and CAR through heterodimerization not only provides a novel molecular mechanism for variation in drug–drug interactions and a potential approach to their therapeutic regulation; it also sheds light on how these receptors coordinately regulate other physiologic processes, such as energy metabolism, and pathologic processes, such as cancer development. Furthermore, our work highlights the importance to comprehensively investigate the crosstalk among members of the nuclear receptor superfamily, through previously unrecognized physical association. Such crosstalk might constitute an unexplored space to define and target nuclear receptor physiology and pathophysiology.
MATERIALS AND METHODS
Materials
Lipofectamine 3000, penicillin–streptomycin, and puromycin stock solutions; Opti-MEM Reduced Serum Medium; phenol red–free Dulbecco's modified Eagle's medium; and DMSO were all purchased from Thermo Fisher Scientific (Atlanta, GA), and Fugene 6 was purchased from Promega (Madison, WI). Characterized fetal bovine serum (FBS) and charcoal/dextran treated FBS were purchased from HyClone (Logan, UT). Dual-Glo Luciferase Reporter Assay reagent was purchased from Promega (Madison, WI). The 96-well white tissue culture–treated plates and steadylite HTS reagent were purchased from PerkinElmer Life and Analytical Sciences (Boston, MA). Rifampicin (RIF) and CITCO were purchased from Tocris Bioscience (Minneapolis, MN). SPA70 was synthesized by WuXi App Tec (Wuhan, China). All tissue culture supplemental materials, such as tissue culture flasks and disposable pipettes, were purchased from Corning (Corning, NY).
Cell lines and cell culture
The human embryonic kidney cell line HEK-293 (ATCC CRL-1573) was obtained from American Culture Type Collection (ATCC) (Manassas, VA), and the cells were grown in culture in Dulbecco's modified Eagle's medium (DMEM) (Thermo Fisher Scientific) supplemented with 10% FBS and penicillin–streptomycin. Human hepatocellular carcinoma HepG2 cells (ATCC CRL-10741) were obtained from ATCC and were maintained in Eagle's minimum essential medium (EMEM) supplemented with 10% FBS and penicillin–streptomycin. Human hepatoma HepaRG PXR functional knockout (HepaRG PXR-KO) cells (cat. no. DCBMTOX1011), HepaRG 5F parental cells (cat. no. MTOX1010-1VL), HepaRG CAR-KO (cat. no. MTOX1012), and William's E medium (cat. no. W1878-500ML) were purchased from Sigma–Aldrich (St. Louis, MO). HepaRG Thaw, Plate and General-Purpose Medium Supplement (cat. no. HPRG770) and serum-free induction medium (cat. no. HPRG750) were purchased from GIBCO Life Technologies (Frederick, MD). Primary human hepatocytes were obtained from the Liver Tissue and Cell Distribution System at the University of Pittsburgh (donor #1 = case 19-008, donor #2 = case 19-010, donor #3 = case 20-011), and the cells were maintained in William's E medium supplemented with Primary Hepatocyte Maintenance Supplement (Thermo Fisher Scientific). All cells were grown in a humidified incubator at 37°C and 5% CO2. Cell lines were authenticated by short tandem repeat DNA profiling and were routinely verified to be mycoplasma free by using a MycoProbe Mycoplasma Detection Kit (R&D Systems, Minneapolis, MN).
Plasmids and transfection assays
GFP-hPXR and GFP-Vector control in pcDNA3.1 were purchased from Thermo Fisher Scientific. pcDNA3.1-MYC-RXRα (MYC-RXRα), pcDNA3.1-MYC-hCAR LBD (residues 100-348), and pcDNA3.1-MYC-hCAR (MYC-hCAR) were obtained from GenScript (Piscataway, NJ). GFP-hCAR1 (GFP-hCAR) has been described previously (16). pcDNA3-FLAG-hPXR (referred to here as FLAG-hPXR) wild-type and FLAG-PXR truncated proteins FLAG-hPXR LBD (comprising residues 100–434), FLAG-hPXR DBD (comprising residues 1–180), and FLAG-hPXR.2 (with residues 174–210 missing) have also been described previously (17). FLAG-hPXR with the T57D point mutation and FLAG-hPXR S350D were generated by site-directed mutagenesis as previously described (18,19). FLAG-hCAR1 in the pcDNA3.1 vector (FLAG-hCAR), the CYP2B6 promoter luciferase reporter (CYP2B6-luc 2.2 kb promoter), and the CYP3A4-luciferase reporter (CYP3A4-luc, in the pGL3 vector) have been described previously (20). FLAG-hCAR T38D in the pcDNA3.1 vector was prepared by Mutagenex Inc. (Suwanee, GA) (21). SF2-FLAG-hCAR was constructed by replacing hPXR in SF2-FLAG-hPXR with hCAR by using standard molecular biology methods (22). MYC-hCAR ΔP1 (with residues 310–325 of hCAR1 deleted) and MYC-hCAR C1 (with residues K311, E318, R320, E324 mutated to D311, K318, D320, K324, respectively) were generated by using the Q5 site directed mutagenesis kit (# E0554S, New England Biolabs, Ipswich, MA) and the following primers pairs 5′- TACGGGTACCAAATCCAG-3′ and 5′-ATACAGAAACCGATCCCG-3′, 5′-AAGCTCGATAGCATTAATAAGGCCTACGGGTACCAAATCCAG-3′ and 5′- AGCCAGCAGGCCTAGCAAATCCGCATACAGAAACCGATCCCG-3′3′, respectively. For mammalian two-hybrid assays, the Checkmate kit (Promega) with pG5-luc (a GAL4 promoter luciferase reporter construct) was used. The VP16 AD-hPXR (hPXR fused to Herpes Simplex Virus Protein 16 transcription activating domain, VP16 AD, in a pACT construct) and VP16 AD-hCAR plasmids were generated as described (19,20). VP16 AD–hPXR LBD containing residues 139–434 of the hPXR LBD was described previously (23). Full-length GAL4 DBD–hCAR (comprising residues 1–348 of hCAR fused to the GAL4 DNA binding domain, GAL4 DBD) and GAL4 DBD–hCAR LBD (comprising residues 103–348 fused to GAL4 DBD) were generated by PCR amplification, using the full-length pACT-hCAR1 (VP16 AD-hCAR) as a template and the following primers: 5′-CGGAATTCCCGGGGATCCCAATGGCCAGTAGGGAAGATGAGC-3′ and 5′-AGGGAAGCGGCCTTAGTTATTCAGCTGCAGATCTCCTGGAGC-3′ (to generate GAL4 DBD–hCAR1); and 5′-AGGGAAGCGGCCTTAGTTATTCAGCTGCAGATCTCCTGGAGC-3′ and 5′–CGGAATTCCCGGGGATCCCACCTGTGCAACTGAGTAAGGAGCAAG-3′ (to generate GAL4 DBD–hCAR1 LBD). Full-length GAL4 DBD–RXRα was generated as described (19). NEBuilder HiFi DNA Assembly Master Mix (New England Biolabs) was used to assemble the PCR products. HEK-293 cells were seeded for 24 h at 60% confluence before they were transfected with the appropriate plasmids (plasmid stocks were prepared at a concentration of 1 μg/μl), using Lipofectamine 3000 transfection reagents. Cells were harvested at 48 h post transfection.
Co-immunoprecipitation assays and Western blot analysis
HEK-293 cells were transfected with the respective plasmids for 24 h and were then incubated with or without the indicated amounts of compounds for an additional 24 h. HepaRG cells were seeded in collagen I coated 15 mm plates until confluent and differentiated with 1.5% DMSO for 2 weeks. Cells were harvested using Accutase cell detachment solution (Thermo Fisher Scientific), rinsed with cold phosphate-buffered saline (PBS), and lysed in Pierce IP lysis buffer (Thermo Fisher Scientific, cat. no. #87788) supplemented with 1× phosphatase (Sigma-Aldrich, cat. no. 4906837001) and 1× protease (Thermo Fisher Scientific, cat. no. 78438) inhibitors. Clarified cell lysates were quantified by BCA assays. Ten percent of each sample lysate was kept as input, and equal amounts of the sample lysates were used for co-immunoprecipitation (co-IP) pull downs. Immunoprecipitation was performed using a Dynabeads Protein G Immunoprecipitation Kit (Thermo Fisher Scientific, cat. no. 10007D). Briefly, 5 μl of mouse anti-GFP or anti-FLAG antibody and 50 μl of protein G magnetic beads were added to each sample. For immunoprecipitation of endogenous hCAR from HepaRG cells, 10 μl of mouse anti-hCAR antibody or 5 μl of immunoglobulin G (IgG) (Santa Cruz Biotech, cat. no. sc-2025) control was added to 50 μl of beads, respectively. The samples were incubated overnight at 4°C with gentle rotation. Bead–antibody–protein complexes were washed three times with IP wash buffer (50 mM Tris–HCl, pH 7.4, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 0.1% NP40, 1× protease inhibitors), then samples were eluted in 1× NuPage sample buffer with reducing agent and heating for 10 min at 70°C. Proteins were separated by gel electrophoresis on NuPage 4–12% bis–Tris gels (Thermo Fisher Scientific) in 1× NuPage MES running buffer (Thermo Fisher Scientific). The proteins were then transferred to a nitrocellulose membrane by using an iBlot gel transfer system (Thermo Fisher Scientific). Membranes were blocked with 5% skim milk for 1 h and probed with the indicated primary antibodies overnight at 4°C. Mouse anti-FLAG M2 (cat. no. F3165), rabbit anti-FLAG M2 (cat. no. F7425) monoclonal antibodies, mouse anti-β-actin (cat. no. A5441) and mouse anti-CYP2B6 (cat. no. SAB4100370) antibodies were obtained from Sigma–Aldrich (St. Louis, MO); mouse anti-hCAR (cat. no. PP-N4111-00) was obtained from R&D Systems (Minneapolis, MN); rabbit anti-hCAR (Ab186869), rabbit anti-CYP2C9 (Ab150364), rabbit monoclonal anti-GFP (cat. no. Ab290) and mouse monoclonal [9F9.F9] anti-GFP (cat. no. Ab1218) antibodies were obtained from Abcam (Cambridge, MA); and mouse anti-hPXR antibody (H-11) (cat. no. sc-48340) was purchased from Santa Cruz Biotech (Dallas, TX). Mouse monoclonal anti-CYP3A4 (K03) antibodies has been reported previously (23,24). Incubation with primary antibodies was followed by incubation for 1 h with secondary goat anti-mouse and goat anti-rabbit antibodies (IR-dye 800CW and IR-dye 680CW; LI-COR Biosciences, Lincoln, NE). Protein bands were visualized with an Odyssey Near-Infrared Fluorescence Imaging System (LI-COR Biosciences) and quantified using Image Studio version 3.1 software (LI-COR Biosciences). To quantitate the protein bands, the intensity of each band was normalized to that of the corresponding control band (β-actin, or the bait protein band in co-IP experiments) to generate the relative band intensity. The relative band intensity of the indicated control sample (e.g. DMSO vehicle or empty vector control) was set as 1 for comparisons with test samples.
Mammalian two-hybrid assay
For mammalian two-hybrid assays, the CheckMate system (Promega) was used. The pG5-luc construct that contains five GAL4 binding motifs was used to detect the interaction between hPXR, RXRα, or hCAR. GAL4 DBD and VP16 AD vector constructs were used as controls, and Renilla luciferase expressed from the GAL4 DBD plasmid was used as an internal transfection control. At 24 h after transfection of HEK-293 cells in six-well plates (1 μg of pG5-luc and 0.25 μg each of VP16 AD, GAL4 DBD with or without the fused hPXR, RXRα, or hCAR as indicated), the cells were transferred to 96-well plates and incubated for an additional 24 h in phenol red–free medium with 5% charcoal-treated FBS, with or without the indicated amount of compound (1 μM CITCO, 5 μM rifampicin, or 10 μM SPA70). A Dual-Glo Luciferase Reporter Assay kit was used to measure both firefly and Renilla luciferase activity. Comparisons were made to cells transfected with hCAR, hPXR, RXRα or the empty vector as indicated.
Luciferase reporter assays
HepG2 cells were transfected in six-well plates with 1 μg of CYP2B6-luc reporter plasmid along with 0.25 μg of FLAG-hCAR with or without 0.25 μg FLAG-hPXR, FLAG-hPXR DBD, or FLAG-hPXR LBD. As a transfection control, 0.1 μg of TK-Renilla luciferase plasmid was used. For all other experiments, 1 μg of CYP2B6-luc or CYP3A4-luc reporter plasmid was transfected along with the indicated amounts of hPXR, hCAR and/or RXRα plasmid combinations. In these experiments, appropriate amount of the pcDNA3.1 empty vector was added to ensure that the total amount of DNA used in each transfection was the same.
RNA isolation and quantitative real-time polymerase chain reaction analysis (RT-qPCR)
Differentiated HepaRG cells were seeded in 24-well plates at 0.4 × 106 cells/well and maintained in general-purpose medium for 3 days. Cells were then transfected with non-targeting siRNAs (siNT; D-001210–02-05) or siRNAs targeting PXR (siPXR; NR1I2 SMARTpool [M-003415–02,] or targeted NR1I2 [D-003415-02]) (from Dharmacon, Lafayette, CO) at a final concentration of 25 nM for 48 h. The cells were then treated with the respective chemicals for an additional 24 h in William's E medium supplemented with serum-free induction medium. Differentiated hPXR knockout (hPXR KO) cells and their parental control cells (the 5F clone) were seeded in 24-wells at 0.4 × 106 cells/well for 3 days. The cells were then treated with the indicated amounts of compound for 24 h, after which qPCR and western blot assays were performed. Primary human hepatocytes grown in six-well plates were treated with siRNAs for 48 h then treated with the respective chemicals as shown for an additional 24 h in William's E medium supplemented with serum-free induction medium. Total RNA was isolated from the cells by using Maxwell 16 LEV simplyRNA Tissue Kits (Promega). cDNA was generated from 2 μg of RNA by using the SuperScript™ VILO™ cDNA Synthesis Kit (Thermo Fisher Scientific). Diluted cDNA was used to perform quantitative RT-PCR, using the TaqMan gene expression assays specific for CYP2B6 (Hs04183483_g1), PXR (Hs01114267_m1), CYP3A4 (Hs00604506_m1) (Applied Biosystems, Thermo Fisher Scientific, Waltham, MA). 18S rRNA (Hs03928990_g1) and GAPDH (Hs02786624_g1) were used as reference genes, and qPCR was performed using the 7900HT Fast Real-Time PCR System (Applied Biosystems, Waltham, MA). Fold induction values were calculated according to the following equation: fold change = 2−ΔΔCt, where ΔCt represents the differences in cycle threshold numbers between the target gene and the reference gene and ΔΔCt represents the relative change in these differences between the control and treatment groups.
RNA-seq and data analysis
Total RNA was extracted from each cell line using the Maxwell 16 LEV automation system and simplyRNA purification kit (Promega, Cat. No. 1280) according to the manufacturer's protocol. The extracted RNA was quantified by spectrophotometry using a NanoDrop 8000 (Thermo Fisher Scientific, Waltham, MA) to ensure a reading of OD260/OD280 between 1.8 and 2.0. RNA quality was further checked by TapeStation 4200 high sensitivity RNA screen tape (Agilent, Santa Clara, CA) before library generation. Only high-quality samples with an RNA integrity number (RIN) value greater than or equal to 8 were used to construct the sequencing library. The RNA was fragmented using fragmentation reagent. In cDNA synthesis, the first-strand cDNA was generated using random hexamer-primed reverse transcription, then was followed by a second-strand cDNA synthesis. Libraries were prepared from total RNA with the TruSeq Stranded mRNA Library Preparation Kit (Illumina, San Diego, CA). Libraries were quantified using the Quant-iT PicoGreen dsDNA assay (Thermo Fisher Scientific). One hundred cycle paired-end sequencing was performed using Illumina NovaSeq 6000 to produce 100 bp paired-end reads.
To analyze the RNA-seq data, raw sequence files were merged across lanes according to sample and subjected to a first round of quality control using the FastQC tool [www.bioinformatics.babraham.ac.uk/projects/fastqc/]. Illumina universal adapters were trimmed from all samples using Trim Galore [https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/] and samples were then subjected to a second round of quality control. Reads were mapped to the Human Hg38 reference genome using Bowtie2 (25) in sensitive mode. Gene-level quantification was obtained using RSEM (26) to produce counts and normalized counts per million (CPM). Statistical testing to determine differential expression was performed in R using t-statistics modified to incorporate strength across genes to the variance estimation using the Limma (27) and Voom (28) packages alongside edgeR (29). Adjusted P-values represent the FDR (q-value) from Limma. Gene ontology (GO) term enrichment analysis was performed by using differentially expressed genes from the hPXR KO HepaRG cells as input. Genes up- or down-regulated were chosen based on log2(FC) values of ≥1 or ≤1, respectively, compared to the WT control HepaRG cells. The adjusted P-value was also set at <0.05 to keep only statistically significant genes as input. Each set of genes was tested for enrichment within the Enrichr database (30) using the R interface for Enrichr (github.com/wjawaid/enrichR). Specifically, the ‘GO Biological Process’ set of terms were used.
Chromatin immunoprecipitation (ChIP)-qPCR and data analysis
ChIP assays were performed according to a published protocol (31). Thirty million differentiated HepaRG control (5F) and hPXR KO cells grown in collagen I coated plates were crosslinked with 1% formaldehyde for 10 min at room temperature. The reaction was quenched for 5 min with 125 mM glycine and cells were washed 2× with cold PBS. Cells were scraped in PBS into Eppendorf tubes and were lysed in 50 mM HEPES [pH 7.9], 140 mM NaCl, 1 mM EDTA, 0.25% Triton X-100, 0.5% NP-40 and 10% glycerol for 10 min on ice. The lysate was centrifuged at 500 × g for 5 min to pellet nuclei. Nuclei were washed twice with 10 mM Tris (pH 8.0), 200 mM NaCl, 1 mM EDTA and 0.5 mM EGTA and twice with shearing buffer [10 mM Tris (pH 8.0), 1 mM EDTA, 0.1% SDS]. The pellet was resuspended for each wash followed by centrifugation at 500 × g for 5 min. The final pellets were resuspended in 1 ml shearing buffer and transferred to 1 ml milliTUBEs with AFA fibers (Covaris, Woburn, MA). Chromatin was sheared in a Covaris E220 Focused-ultrasonicator for 8 min using the manufacturer's recommended standard settings to yield an average fragment size of 300 bp. Sheared chromatin was centrifuged at 16 000 × g for 10 min to pellet insoluble material. 100 μl of clarified chromatin was put aside as input and 20 μl of anti-hCAR antibody or IgG (as negative control) with 100 μl of protein G beads were added per sample and incubated at 4°C overnight with gentle rotation. Beads were washed for 3 min each at 4°C with rotation with low salt wash [20 mM Tris (pH 8.0), 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS], high salt wash [20 mM Tris (pH 8.0), 500 mM NaCl, 2 mM EDTA, 1% Triton X-100, 0.1% SDS], lithium chloride wash [10 mM Tris (pH 8.0), 250 mM LiCl, 1 mM EDTA, 1% NP-40, 1% sodium deoxycholate], and twice with TE [10 mM Tris (pH 8.0), 1 mM EDTA]. Bound complexes were eluted twice with 125 μl of 100 mM sodium bicarbonate, 1% SDS for 15 min at 37°C. 1 U of proteinase K (New England Biolabs, cat. # P8107S) was added to the inputs and eluates with 40 mM Tris (pH 8.0), 200 mM NaCl, and 10 mM EDTA, and samples were incubated at 65°C for 2 h. DNA was purified with the QIAquick PCR Purification Kit (QIAGEN, Germantown, MD). After purification, DNA was subjected to amplification by denaturation at 95°C for 2 min, followed by 40 cycles of denaturation at 95°C for 15 s and annealing/extension at 60°C for 1 min using PowerUp SYBR Green Master Mix (Thermo Fisher Scientific) in an Applied Biosystems 7500 Fast Real-Time PCR System. Primers for CYP2B6 promoter (32), and negative control (33) were:
CYP2B6 promoter: 5′-TGTACTTTCCTGACCCTGAAGA-3′ and 5′-CTGCAATGAGCACCCAATCT-3′;
Negative control: 5′-ATGGTTGCCACTGGGGATCT-3′ and 5′-TGCCAAAGCCTAGGGGAAGA-3′. Results are expressed as percent input (% INPUT).
Immunofluorescence staining
HepG2 cells were grown in culture as previously described, and a set of plasmids were transfected into cells in 12-well culture plates. Briefly, HepG2 cells were plated at 150 000 cells/well. Next day, GFP-hCAR, FLAG-hPXR, FLAG-hPXR T57D, FLAG-hCAR T38D and/or GFP-hPXR were transfected individually or in combination, as indicated, using a TransIT-2020 Transfection Reagent Kit (MIR5400, Mirus Bio, Madison, WI) or Fugene 6 Transfection Reagent according to the protocol. Forty-eight hours later, the transfected cells were re-plated into 384-well plates at 5000 cells/well for immunofluorescence imaging of GFP or imaging after immunostaining with anti-FLAG antibodies. Twenty-four hours after being re-plated, the cells were fixed with 4% paraformaldehyde in 1× PBS at room temperature for 10 min, then washed with 1× PBS and permeabilized with 0.5% Triton X-100 in 1× PBS at room temperature for 10 min. The cells were further processed by blocking with 5% fetal bovine serum, 0.1% Triton X-100 in 1× PBS at room temperature for 30 min. After blocking, the cells were incubated with primary antibody (rabbit anti-FLAG, diluted 1:1000) at 4°C overnight. Next day, the cells were briefly rinsed with 1× PBS then incubated for 1 h with secondary antibodies (anti-rabbit Alexa594 or anti-rabbit Alexa647, both diluted 1:200) and Vybrant DyeCycle Violet Stain for nuclear staining. Images were then acquired with a CellVoyager CV8000 high-content analysis system (Yokogawa, Tokyo, Japan). The 60 × water immersion objective lens were used.
Simulated model of the heterodimer hPXR LBD – hCAR LBD
The RXRα LBD in the hCAR LBD–RXRα LBD structure (PDB code 1XVP) was replaced with the hPXR LBD structure (PDB code 5X0R) by alignment using The PyMOL Molecular Graphics System, Version 2.4.0 (Schrödinger, LLC, New York, NY). Analysis of the initial hCAR-hPXR interface helped us identify a segment of the hCAR LBD (AKLLGLLAELRSINEA, designated peptide ‘P’), which was used for docking of ‘Peptide ‘P’ to the hPXR LBD structure using FlexPepDock (34,35) in order to optimize the positions of the residues. The reported crystal structures of hPXR LBD and hCAR LBD display missing sections, which were filled with MODWEB (36,37). The docked ‘peptide P’ and hPXR LBD were replaced by the gap-filled structures through alignment. The resulting hPXR LBD-hCAR LBD complex was further refined by molecular dynamics simulations (MD simulations).
MD simulations were carried out in GROMACS (38) (2020.1 release) with the CHARMM36 force field (39) (March 2019 release). The proteins were enclosed in in a cubic box with at least 2.0 nm from the box edge filled with solvent. The system was neutralized by the addition of Na+ and Cl− ions. Energy minimization was performed to prevent steric clashes and improper geometry, which was followed by equilibration in the NVT (constant number of particles, volume, and temperature) and NPT (number of particles, pressure, and temperature) phases. The trajectories of the 50 ns MD simulation were analyzed using the GROMACS package and further visualized in The PyMOL Molecular Graphics System and Chimera X (40).
Expression and purification of hPXR, RXRα and hCAR for biophysical experiments
Bacterial expression constructs for hPXR LBD, RXRα LBD and hCAR LBD (all containing a His tag at the N-terminus with an SRC-1 fragment tethered at the C-terminus) were obtained from GenScript (Piscataway, NJ). A DNA fragment encoding MKKGHHHHHHG sequence followed by hPXR LBD (residues 140–434), RXRα LBD (residues 227–462) or hCAR LBD (residues 103–348), a pentapeptide linker (GGSGG), a peptide fragment of SRC-1 (residues 678–700), an ochre stop codon sequence (TAA) and an opal stop codon (TGA) were cloned into a pET-3a vector between NdeI and BamHI cloning sites. Expression and purification of the three nuclear receptors were performed as previously described (41) with minor modifications.
Dynamic light-scattering experiments
The interactions between hPXR LBD and hCAR LBD and that between hCAR LBD and RXRα LBD were monitored by dynamic light scattering (DLS), using a DynaPro Plate Reader II (Wyatt Technologies, Santa Barbara, CA). Additional details are indicated in the instrument manual (DYNAMICS User's Guide [M1400 Rev. K] (https://wyatt.com/files/literature/app-notes/dls-cuvette/ph-effects-stability). Assays were performed using purified hCAR-LBD, RXRα LBD and hPXR-LBD at a concentration of 12 μM in 25 mM HEPES, pH 7.9, containing 150 mM NaCl, 5% glycerol, and 5 mM DTT. Measurements were performed on Dynamics using these parameters: Isotemp mode set at 25°C; data collection in DLS mode; DLS acquisition time: 10 s; read intervals: 1; number of acquisitions: 10; and auto-attenuation mode enabled. Signals from the samples were measured using an Aurora 384-well plate (Wyatt Technologies) after the samples had been incubated at room temperature for 1 h. Peptide ‘P’ (AKLLGLLAELRSINEA, an hCAR peptide corresponding to the hCAR heterodimer interface region) and peptide ‘C’ (ADLLGLLAKLDSINKA, in which residues with negative charges in peptide P were replaced with residues with positive charges) were used at a concentration of 120 μM. Data were analyzed using Dynamics 7.8 software and plotted with OriginPro.
Size-exclusion chromatography (SEC) experiments
Elution volumes for hPXR LBD, hCAR LBD, and hPXR LBD + hCAR LBD- were analyzed using a Superdex 200 Increase 10/300 GL column (Cytiva, Marlborough, MA, cat. no. 28990944) on an ÄKTA pure chromatography system (Cytiva). The column was equilibrated with 25 mM HEPES, pH 7.9, 150 mM NaCl, 5% glycerol, 5 mM DTT and was previously calibrated using the Gel Filtration High Molecular Weight Calibration Kit (Cytiva, cat. no. 28403842). Purified hCAR LBD and hPXR LBD were present at a concentration of 12 μM. The size-exclusion chromatograms were plotted with OriginPro.
Crosslinking mass spectrometry
Sample preparation
The experiment was performed with a previously reported protocol (42). hCAR LBD and hPXR LBD were mixed at 1:1 molar ratio and the binding reaction was carried out at room temperature (RT) for 1 h. Buffer was exchanged into a crosslinking buffer (20 mM HEPES, 150 mM NaCl, pH 7.8) by using Pur-A-Lyzer Mini Dialysis Kit (10–250 μl, MWCO 25 kDa). A total of 90 μg protein complex was crosslinked with DSSO (43) using a 1:320 protein-to-crosslinker molar ratio. The reaction was carried out at RT for 1 h, and then quenched with excess ammonium bicarbonate (ABC) for 30 min at RT. Denaturation buffer was added to reach 8 M urea concentration followed by reduction with dithiothreitol (DTT) and alkylation with iodoacetamide (IAA). Then the sample was diluted with 50 mM ABC to a final concentration of 2 M urea. Trypsin was added to a protease-to-substrate ratio of 1:10 (w/w), and the sample was incubated at 37°C for 3 h. AspN was added to the trypsin digested sample with a protease-to-substrate ratio of 1:10 (w/w) at 37°C overnight. Digestion reaction was stopped with 10% trifluoroacetic acid (TFA). A control sample with the same amount of DMSO added instead of DSSO was also included. All other experiment conditions for the control sample were the same as the cross-linked sample.
Microgram-scale basic pH LC fractionation
Fractionation of the digested samples was performed by basic pH RPLC using a microscale HPLC system (Agilent 1120) coupled with a three-way flow splitter (IDEX Health & Science). The samples were speed vacuumed to almost dry to avoid sample loss, and resuspended with mobile phase A (10 mM ammonium formate, pH 8.0), centrifuged at 21 000 × g for 5 min. With the flow splitter closed, the supernatant was loaded onto an XBridge C18 column (1 mm × 50 mm, 3.5 μm beads, Waters) at a flow rate of 50 μl/min by 95% of mobile phase A. After loading, the flow splitter was opened and the peptides were eluted with a 45 min gradient from 5–90% of mobile phase B (90% acetonitrile, 10 mM ammonium formate, pH 8.0). The HPLC pumps were operated at 800 μl/min and the column was eluted at 30 μl/min. Eight fractions were manually collected.
Acidic pH LC–MS/MS data acquisition and analysis
The LC–MS/MS analysis of pre-fractionated samples was performed using UltiMate 3000 RSLCnano system coupled with a Thermo Scientific Orbitrap Q Exactive HF. Mobile phase A consisted of 0.2% formic acid (FA), 3% DMSO in water and mobile phase B consisted of 0.2% FA, 3% DMSO in 67% acetonitrile. Peptides were loaded into a CoAnn C18 column (75 μm × 20 cm, 1.9 μm particles) at a flow rate of 250 nL/min with the following gradient: 12–22% mobile phase B over 30 min, 22–40% mobile phase B in 30 min, 40–60% mobile phase B in 7 min, 60–95% mobile phase B in 3 min, stay at 95% mobile phase B for 3 min, followed by 15 min equilibration (44).
MS data were acquired using a ‘high-high’ acquisition method (high resolution on both MS1 and MS2). MS1 scans were detected in the Orbitrap at 120K resolution in the m/z range 400–2000 and AGC target of 1 × 106 with a maximum injection time of 50 ms. Ions with charge states from 2+ to 8+ were selected for fragmentation by stepped high energy collision dissociation (HCD) at 27%, 30% 33% with AGC of 1 × 105, maximum injection time of 100 ms, and detected in the Orbitrap at 60K resolution.
Data analysis
Raw data were converted to mzXML format using MSconvert and searched with MeroX2.0 (45). Settings for crosslinking search were as follows: maximum total missed cleavage, 3; peptide length, 5–30 residues; static modification, carbamidomethylation of cysteines; dynamic modification, oxidation of methionine; cross linker, DSSO, assumed reaction site at lysine, serine, threonine, and tyrosine; modification of fragments, with 54.0105 Da and 85.9826 Da mass modification; MS1 precision, 10 ppm; MS2 precision, 10 ppm; RISEUP Mode with two maximum missing ions; Prescore, 10% intensity; FDR cut off, 5%; Score cut off, –1; include cRAP database; decoy database generation, shuffle sequence but keep protease sites.
Results from the MeroX search were manually validated using the following five critera. (i) MS1 mass accuracy; (ii) number of signature fragment ions within 5 ppm mass shift; (iii) number of b and y ions within 5 ppm mass shift; (iv) number of branched fragment ions within 5 ppm mass shift; (v) if shown in control run.
Small-angle X-ray scattering studies of the hPXR LBD-hCAR LBD heterodimer
Experiments involving small-angle X-ray scattering (SAXS) in line with SEC were performed at LIX-beamline (16-ID) of the National Synchrotron Light Source II (Upton, NY) (46). Data were collected at a wavelength of 0.819 Å in a detector configuration, yielding accessible scattering angle of 0.006 < q < 3.0 Å−1, where q is the momentum transfer, defined as q = 4 π sin(θ)/λ (λ is the wavelength and 2θ is the scattering angle). A 65 μl solution of 250 μM hPXR LBD and hCAR LBD was injected at 0.45 ml/min into a superdex 200 increase 5/150 sizing column (GE healthcare) equilibrated at RT with 25 mM HEPES, pH 7.9, containing 150 mM NaCl, 5 mM DTT and 5% glycerol. Eluent from the column flowed into a flow-cell, where X-ray data was collected at 2 s exposure. Scattering profiles were analyzed using the ATSAS Suite 3.0 (47). The program DAMMIF (47) was used to reconstruct an ab initio molecular envelope from the SAXS data with P23 symmetry bias enforced. An ab initio averaged/filtered beam model was calculated from the models of 20 individual simulations. The final averaged and filtered volume featured a central domain that could readily accommodate the hPXR LBD-hCAR LBD complex, which was confirmed by superimposition using supercomb from the ATSAS Suite.
Statistical analysis
Data are expressed as the mean ± SD of at least three independent experiments, and statistical significance was established if the P value was <0.05. Student's t-test was used to compare the means of two groups as specified, followed by the Holm–Šidák multiple comparison test. For all other data, analysis was performed, and statistical significance was established by using one-way ANOVA followed by Dunnett's multiple comparison test for all samples compared to the control or by using two-way ANOVA followed by Šidák's multiple comparison test. All qRT-PCR graphs were generated using GraphPad PRISM version 8.
RESULTS
Reduction of hPXR levels elevates hCAR transcriptional activity
To investigate how hPXR affected the function of hCAR, we first used HepaRG wild type (WT) cells, which endogenously express hPXR and hCAR (23). Knockdown of hPXR with siRNA targeting hPXR (siPXR) (Figure 1A) significantly elevated the levels of CYP2B6 (a major transcriptional target of hCAR) in the presence and absence of CITCO, a dual agonist of hPXR and hCAR (Figure 1B). When hPXR was knocked out (hPXR KO), CYP2B6 was more robustly elevated (Figure 1C), and the absence of hPXR protein in these cells was confirmed (Figure 1D). In addition, hPXR knockout did not increase the hCAR protein level (Supplementary Figure S1). These data clearly indicate that the presence of hPXR is inhibitory to hCAR transcriptional activity in HepaRG cells.
Figure 1.

Reduction of hPXR levels leads to an increase in hCAR-dependent CYP2B6 expression in human HepaRG cells. (A, B) HepaRG cells were transfected with non-targeting siRNA (siNT) or pooled siRNA targeting hPXR (siPXR) at 25 nM for 48 h then treated with DMSO or CITCO for an additional 24 h. RT-qPCR was performed for (A) hPXR or (B) CYP2B6 RNA. FC, fold change over siNT and DMSO treated cells. (C) CYP2B6 mRNA levels in parental wild-type (WT) cells compared to those in hPXR KO HepaRG cells treated with DMSO or 1 μM CITCO. FC, fold change over DMSO treated WT cells. (D) Western blot of CYP2B6 and hPXR proteins in WT and hPXR KO HepaRG cells. β-actin was used as the protein loading control. * P < 0.05; *** P < 0.0005.
We further used primary human hepatocytes to confirm the inhibitory effect of hPXR on hCAR. Previous studies have shown that hCAR and hPXR are sequestered in the cytoplasm of hepatocytes and translocate to the nucleus upon agonist binding, whereas in immortalized cells in culture, these receptors are localized primarily to the nucleus (48). Consistent with hCAR being sequestered in the cytoplasm of hepatocytes in the absence of an agonist, the basal levels of CYP2B6 were low (Figure 2A). Both CITCO (a dual agonist of hPXR and hCAR) and RIF (a specific agonist of hPXR) induced the expression of CYP2B6 (Figure 2A), indicating that both hPXR and hCAR regulate the expression of CYP2B6 in primary hepatocytes. Knockdown of hPXR with an individual siPXR (Figure 2A) or with pooled siPXRs (Supplementary Figure S2A) reduced the inducing effect of RIF on CYP2B6 but elevated the effect of CITCO on CYP2B6, thus demonstrating the inhibitory effect of hPXR on hCAR. As expected, CYP3A4, which is regulated mainly by hPXR, was induced by RIF but the induction was robustly reduced by siPXR (Figure 2B and see also Supplementary Figure S2B). The knockdown of hPXR was confirmed at both the mRNA level (Figure 2C and See also Supplementary Figure S2C) and the protein level (Figure 2D and see also Supplementary Figure S2D). Similar observations were made in primary hepatocytes from two different donors (Supplementary Figures S3 and S4), further confirming that in physiologically relevant human primary hepatocytes, hPXR is inhibitory to hCAR.
Figure 2.

hPXR knockdown in primary human hepatocytes enhances agonist-induced CAR-dependent CYP2B6 gene expression. Primary human hepatocytes (from donor #3) were treated with siNT or an individual siPXR at 25 nM for 48 h. This was followed by the indicated treatments (0.1% DMSO, 1 μM CITCO, or 5 μM RIF) for an additional 24 h. (A–C) The levels of CYP2B6, CYP3A4, and hPXR mRNA were measured by RT-qPCR. FC, fold change over siNT and DMSO treated cells. (D) Endogenous hPXR protein levels were analyzed by western blotting. β-actin was used as the loading control. ** P < 0.005; *** P < 0.0005; ns, not significant.
To demonstrate the genome-wide consequences of depletion of hPXR on gene expression, we performed RNA-seq analysis in HepaRG WT and hPXR KO cells. Knockout of hPXR altered gene expression globally (Figure 3A and see also Supplementary Figure S5). Among the highly upregulated genes following knockout of hPXR are genes involved in xenobiotic metabolism (Supplementary Table S1), such as UGT2B15, UGT2B10, UGT2B4, CYP2C9, CYP2B6 and CYP3A4 (Figure 3A). The observation that CYP3A4 level is elevated when hPXR is knocked out is consistent with previously published data using HepaRG cells (49) and a mouse model (14). We confirmed the elevated protein levels of CYP2B6 (Figure 1D), CYP2C9 and CYP3A4 (Figure 3B). CYP2B6 is a major transcriptional target of hCAR. ChIP-qPCR analysis showed that hPXR knockout in HepaRG cells increases hCAR recruitment to the endogenous CYP2B6 promoter (Supplementary Figure S6). Together, our data demonstrate that hPXR is inhibitory to hCAR transcriptional activity.
Figure 3.

Depletion of hPXR increases the expression of genes related to xenobiotic metabolism. (A) Volcano plot of RNA-seq data showing differential expression of hPXR KO versus WT control HepaRG cells. UGT2B15, UGT2B10, UGT2B4, CYP3A4, CYP2C9 and CYP2B6 are among the highly upregulated genes (red) following functional knockout of hPXR. Downregulated genes are shown in blue. Genes with adjusted P-values (adj. P-value) of ≥0.05 or having |log2(fold-change)| values ≤1 are shown in gray. Fold change, hPXR KO over wild type (WT) HepaRG cells. (B) Western blot of CYP3A4 and CYP2C9 proteins in WT and hPXR KO cells. β-Actin was used as the protein loading control.
hPXR interacts with hCAR, leading to mutual inhibition in cells
The observation that reducing hPXR protein levels led to elevated hCAR activity prompted us to hypothesize that hPXR and hCAR interacted physically, a concept that has never been proposed or tested before. To test our hypothesis, we co-expressed GFP-tagged hPXR (GFP-hPXR) or the GFP vector (as a negative control) and FLAG-tagged hCAR1 (FLAG-hCAR) or the FLAG vector (as a negative control) in HEK-293 cells then performed immunoprecipitation assays using either anti-GFP antibody or anti-FLAG antibody. As shown in Figure 4A, in the anti-GFP immunoprecipitation, FLAG-hCAR was co-immunoprecipitated with GFP-hPXR, but not with the GFP control (left panel). Reciprocally, in the anti-FLAG immunoprecipitation, GFP-hPXR was co-immunoprecipitated with FLAG-hCAR but not with the FLAG vector control (right panel). These data indicated that hPXR interacts with hCAR in the absence of a ligand.
Figure 4.

hPXR interacts with hCAR, resulting in their mutual inhibition. (A) hPXR co-immunoprecipitated with hCAR. HEK-293 cells were co-transfected with FLAG-hCAR (or FLAG vector) and GFP-hPXR (or GFP vector) as shown. Co-IP assays were carried out using either anti-GFP (left panel) or anti-FLAG (right panel) antibodies at 48 h post-transfection. This was followed by immunoblot analysis with anti-FLAG or anti-GFP. (B) hPXR interacted with hCAR in a mammalian two-hybrid assay performed in HEK-293 cells using full-length hPXR (VP16 AD–hPXR) and hCAR (GAL4 DBD–hCAR). The interaction between hPXR and hCAR was measured as the relative activity of the pG5-luc luciferase reporter (the relative luciferase activity was obtained by normalizing firefly luciferase to Renilla luciferase and compared hCAR and hPXR co-expression to the expression of hCAR alone) at 48 h post-transfection. FC, fold change over cells transfected with GAL4 DBD and VP16 AD vectors (negative controls). (C) Activation of the CYP2B6 promoter was reduced by hPXR in a dose-responsive manner. HepG2 cells were co-transfected with the CYP2B6 luciferase reporter (CYP2B6-luc) together with the indicated amounts of hCAR and hPXR plasmids. CYP2B6 promoter reporter activity was measured at 48 h post-transfection. CYP2B6 promoter activity from hCAR and hPXR co-transfection was compared to that in cells transfected with hCAR alone. FC, fold change over cells transfected with hCAR alone. (D) Co-expression of hPXR and hCAR reduced CYP3A4 promoter activity. HepG2 cells were co-transfected with the CYP3A4 luciferase reporter plasmid and the indicated amounts of hCAR and hPXR. CYP3A4 promoter activity was measured at 48 h post-transfection. FC, fold change over cells transfected with hCAR alone. Statistical comparisons were made to hCAR alone (B, C) or hPXR alone (D). *** P < 0.0005.
We further examined the interaction between hPXR (fused to the transcription activating domain of VP16, VP16 AD) and hCAR (fused to the GAL4 DNA binding domain, GAL4 DBD) by performing mammalian two-hybrid assays in HEK-293 cells. GAL4 luciferase reporter was used as a readout for the interaction between hPXR and hCAR. As shown in Figure 4B, GAL4 DBD–hCAR increased the luciferase reporter activity, which was further enhanced by co-expression of VP16 AD–hPXR, indicating that the VP16 transcription activating domain is brought into proximity with the GAL4 DBD through the hPXR–hCAR interaction and providing additional evidence that hPXR interacts with hCAR in the absence of a ligand. As a control, Supplementary Figure S7 showed that both hPXR and hCAR interact with RXRα. The interaction between hPXR and hCAR was not enhanced but instead, moderately reduced by treatment of their ligands in both co-immunoprecipitation (co-IP) and mammalian two-hybrid assays (See Supplementary Figures S8A and S8B, respectively).
To further confirm that the hPXR–hCAR interaction led to the inhibition of transactivation by hCAR of its primary target gene, CYP2B6, we co-expressed various amounts of hPXR with hCAR and a CYP2B6 luciferase reporter (under the control of the CYP2B6 promoter) in HepG2 cells. hCAR alone is constitutively active, and this constitutive activity was diminished by hPXR in a dose-dependent manner (Figure 4C).
To determine whether the hPXR–hCAR interaction was mutually inhibitory, we co-expressed hPXR, hCAR, or both, along with a CYP3A4 luciferase reporter (under the control of the CYP3A4 promoter), in HepG2 cells. As expected, both hPXR and hCAR individually transactivated the CYP3A4 promoter (Figure 4D). Interestingly, co-expression of hPXR and hCAR decreased, instead of increasing, the hPXR activity (Figure 4D), indicating that there was mutual inhibition of the two receptors. In addition, overexpression of hCAR in HepaRG cells enhanced the basal levels of CYP2B6 and CYP3A4. However, the ability of the hPXR-specific agonist RIF to induce both CYP2B6 and CYP3A4 was compromised by overexpression of hCAR (See Supplementary Figures S9A and S9B), which is consistent with a previous report (50) and is indicative of the inhibitory effect of hCAR on hPXR. In contrast to the effect of hCAR overexpression, hCAR knockout in HepaRG cells (hCAR KO) increased the inducing effect of RIF on both CYP2B6 and CYP3A4 (Supplementary Figures S9C and S9D). Consistent with previous reports, hCAR knockout abolished the effect of CITCO on inducing CYP2B6 (51) but increased its effect on inducing CYP3A4 (23,51). Together, these data suggest that hPXR interacts physically with hCAR, resulting in mutual inhibition of the respective transcription activities of these receptors.
hPXR interaction with hCAR is independent of DNA binding
The discovery of the inhibitory physical interaction between hPXR and hCAR prompted us to investigate whether such interaction depended on the DNA binding ability of the receptor. PXR and CAR share common structural features with other members of the nuclear receptor superfamily, including a modular architecture consisting of the N-terminus DNA-binding domain (DBD) and the C-terminus ligand binding domain (LBD). A flexible hinge connects the two domains. Two zinc-fingers within the DBD are essential for DNA engagement by the nuclear receptors. hPXR and hCAR target distinct and overlapping DNA motifs. The CYP2B6 promoter contains a phenobarbital-responsive element module (PBREM) consisting of three direct repeats (DR4) to which hCAR and hPXR bind (9,52). The CYP3A4 promoter contains PXR response elements (PXREs), which contain a direct repeat (DR3) and an everted repeat (ER6) that are recognized by hCAR and hPXR, respectively (9). Competition for these binding motifs has been proposed as a mechanism for the observed hPXR–hCAR functional interference (52,53). If the inhibitory interaction of hPXR with hCAR was dependent on DNA binding, then an hPXR mutant that failed to bind DNA would prevent such inhibitory interaction. Our previous study revealed that mutation of hPXR threonine 57 to aspartate (T57D) within a zinc finger in the DBD led to a total loss of hPXR DNA binding and function (18). In an immunoprecipitation assay, both wild type (WT) hPXR and hPXR T57D interacted with hCAR (Figure 5A), surprisingly suggesting that hPXR interacts with hCAR independently of its DNA binding.
Figure 5.
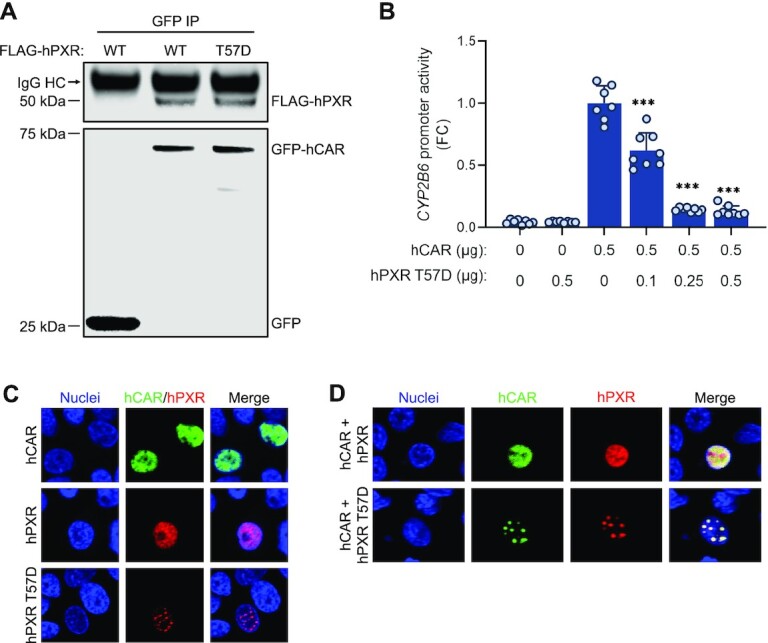
Receptor DNA binding is not required for hPXR–hCAR interaction. (A) hCAR interacts with the DNA binding–defective hPXR T57D mutant. HEK-293 cells were co-transfected with GFP-hCAR and FLAG-hPXR or FLAG-hPXR T57D. The GFP vector was used as a control. Co-IP assays were performed at 48 h post-transfection, followed by immunoblotting with anti-GFP and anti-FLAG. (B) The FLAG-hPXR T57D mutant (hPXR T57D) inhibits hCAR. HepG2 cells were transfected with the CYP2B6-luc reporter plasmid together with the indicated amounts of hCAR and hPXR T57D. CYP2B6 promoter activity was measured at 48 h post-transfection. Empty vector plasmid was used as a control. FC, fold change over cells transfected with hCAR alone. The effect of hPXR T57D on hCAR was determined by comparing its activity to that in cells transfected with hCAR alone. *** P < 0.0005. (C, D) hPXR co-localizes with hCAR. HepG2 cells were transfected with (C) individual GFP-hCAR (hCAR), FLAG-hPXR (hPXR), or the FLAG-hPXR T57D mutant (hPXR T57D) or (D) co-transfected with GFP-hCAR/FLAG-hPXR (hCAR + hPXR) or GFP-hCAR/FLAG-hPXR T57D mutant (hCAR + hPXR T57D). This was followed by immunostaining using antibodies against FLAG (for hPXR) to determine the protein localization at 48 h post-transfection. Nuclei were stained with Vybrant DyeCycle Violet Stain.
We further determined whether the interaction between hPXR T57D and hCAR led to inhibition of hCAR. Similar to WT hPXR (Figure 4C), hPXR T57D inhibited the ability of hCAR to transactivate the CYP2B6 promoter in a dose-dependent manner (Figure 5B). Mutating threonine 38 to aspartate in hCAR (T38D) has been reported to promote the formation of an inactive hCAR homodimer and greatly reduced its DNA-binding and transcription activity (48). WT hCAR decreased transactivation of the CYP3A4 promoter by hPXR (Figure 4D). hCAR T38D also inhibited the ability of hPXR to transactivate the CYP3A4 promoter in a dose-dependent manner but did so to a lesser degree than did the WT hCAR (see Supplementary Figure S10A). To provide additional evidence that hPXR interacted with hCAR independently of DNA binding, we transfected HepG2 cells with GFP-hCAR (hCAR) with or without FLAG-hPXR (hPXR) or FLAG-hPXR T57D then performed immunofluorescence imaging of GFP and immunostaining with anti-FLAG antibodies. As seen in Figure 5C, when individually expressed, hPXR and hCAR were localized and distributed uniformly within the nuclei whereas hPXR T57D displayed a unique punctate pattern within the nuclei (Figure 5C), consistent with our previous report (18). Strikingly, when co-expressed with hPXR T57D, hCAR also displayed the distinctive punctate pattern within the nuclei (Figure 5D), from which we infer that hPXR T57D physically interacts with and alters the cellular distribution of hCAR. When expressed alone, hCAR T38D was localized heterogeneously but mainly in the cytosol and displayed a punctate pattern (See Supplementary Figure S10B). When co-expressed with hPXR, hCAR T38D sequestered some hPXR in the cytosol and changed its distribution from uniform to punctate (see Supplementary Figure S10C). In addition, we showed that endogenous hPXR physically interacts with endogenous hCAR in HepaRG cells (Supplementary Figure S11A). Together, these results suggest that hPXR physically interacts with hCAR and that the inhibitory interaction between hPXR and hCAR is independent of DNA binding by the receptors.
hPXR and hCAR interact through their LBDs
The observation that hPXR interaction with hCAR was independent of DNA binding led us to examine which hPXR domains were responsible for the interaction with hCAR. Figure 6A illustrates the hPXR plasmids used in co-IP experiments with hCAR: full-length FLAG-hPXR (hPXR FL) or truncated FLAG-hPXR plasmids (containing the hPXR LBD, the hPXR DBD, or the hPXR.2 isoform missing 37 amino acids within the LBD [hPXR.2]). Co-IP assays showed that the hPXR LBD, but not the hPXR DBD, interacted with hCAR (Figure 6B). hPXR.2 also interacted with hCAR (Figure 6C). We further confirmed the physical interaction of the hPXR LBD and hCAR LBD by using mammalian two-hybrid assays, with the hCAR LBD fused to the GAL4 DBD (GAL4 DBD–hCAR LBD) and the hPXR LBD fused to the VP16 activating domain (VP16 AD–hPXR LBD). GAL4 DBD–hCAR LBD alone increased the GAL4 promoter reporter activity, which was further enhanced when VP16 AD–hPXR LBD was co-expressed (Figure 6D), indicating that hPXR and hCAR interact through their LBDs. The physical interaction between hPXR LBD and hCAR LBD was confirmed in a co-immunoprecipitation assay in HEK-293 cells co-transfected with MYC-hCAR LBD and FLAG-hPXR LBD (Supplementary Figure S11B). Next, we examined the effects of different hPXR domains on the transactivation activity of hCAR. We co-expressed a CYP2B6 luciferase reporter and hCAR with the full-length hPXR (hPXR FL), the hPXR LBD or the hPXR DBD. Our results showed that both hPXR FL and the hPXR LBD, but not the hPXR DBD, suppressed hCAR transactivation activity (Figure 6E). Similarly, both hCAR and hCAR LBD suppressed hPXR transactivation of the CYP3A4 luciferase reporter (Supplementary Figure S12). Taken together, our data indicate that the hPXR LBD interacts with the hCAR LBD to mutually inhibit their transactivation activities.
Figure 6.

The hPXR ligand-binding domain is sufficient for interaction with hCAR. (A) Schematics showing hPXR in full-length (hPXR FL) or truncated (hPXR LBD, hPXR DBD, hPXR.2) forms. hPXR.2 lacks 37 residues (174–210) within the LBD. (B) The hPXR LBD interacts with hCAR. HEK-293 cells were co-transfected with MYC-hCAR, along with FLAG vector control (Ctrl), full-length hPXR (FL), FLAG-hPXR LBD (LBD), or FLAG-hPXR DBD (DBD). Co-IP assays using anti-FLAG antibodies were performed at 48 h post-transfection, followed by immunoblot analysis with anti-FLAG and anti-MYC. (C) hPXR.2 interacts with hCAR. HEK-293 cells were co-transfected with GFP-hCAR (or GFP vector) and hPXR.2 isoform. A co-IP assay was performed at 48 h post transfection, followed by immunoblot analysis with anti-GFP and anti-hPXR. (D) The hPXR LBD interacts with the hCAR LBD in a mammalian two-hybrid assay. HEK-293 cells were transfected with VP16 AD–hPXR LBD, GAL4 DBD–hCAR LBD, or vector controls (VP16 AD and GAL4 DBD) as indicated. At 48 h post-transfection, pG5 luciferase activity resulting from hPXR LBD and hCAR LBD co-expression was measured (the relative luciferase activity was obtained by normalizing firefly luciferase to Renilla luciferase) and compared to that with hCAR LBD expression alone. FC, fold change over cells transfected with vector controls (VP16 AD and GAL4 DBD). (E) The hPXR LBD inhibits hCAR activity. HepG2 cells were transfected with the CYP2B6-luc reporter in combination with hCAR (0.25 μg) with or without 0.25 μg of full-length hPXR (hPXR FL), hPXR LBD, or hPXR DBD as shown. At 48 h post-transfection, the CYP2B6 promoter activity resulting from hPXR and hCAR co-expression was measured and compared to that with hCAR alone. *** P < 0.0005; ns, not significant. Immunoglobulin heavy chain (IgG HC, 55 kDa) and light chain (IgG LC, 25 kDa) were indicated with an arrow in (B). FC, fold change over cells transfected with vector controls (VP16 AD and GAL4 DBD).
hPXR heterodimerizes with hCAR
Structural model of the hPXR–hCAR heterodimer complex
Our findings indicate that there are direct interactions between hPXR and hCAR through their respective LBDs, resulting in the potential formation of a novel heterodimer complex. We envisaged that the hPXR LBD and hCAR LBD could engage each other to form a heterocomplex similar to the well-established hPXR LBD–RXRα and hCAR–RXRα pairs, particularly because the LBDs of hPXR, hCAR and RXRα are structurally very similar; the root mean square deviation (RMSD) between the structures of the hPXR LBD and the RXRα LBD (PDB code: 4J5W) is 1.5 Å, and that between the hCAR LBD and the RXRα LBD (PDB code: 1XVP) is 1.7 Å (54,55). An initial structural replacement of the RXRα LBD with the hPXR LBD (PDB code: 5X0R) in the hCAR LBD–RXRα LBD structure (PDB code: 1XVP) enabled us to identify a segment of the hCAR LBD (310-AKLLGLLAELRSINEA-325, designated peptide ‘P’) as part of the dimerization interface, which forms part of helix 10 in the hCAR structure. Peptide ‘P’ was docked to the hPXR LBD structure, and after alignment of the hCAR LBD structure to the resulting docked peptide ‘P,’ the molecular interactions of the hPXR LBD–hCAR LBD complex were further refined by molecular dynamics simulations (Figure 7A). The model shown in Figure 7A was the basis for the design of peptide ‘P,’ which was intended to disrupt the heterodimerization by competing with the hCAR LBD in binding to the hPXR LBD in subsequent experiments. Four residues within peptide ‘P’ were mutated to residues with opposing charges (ADLLGLLAKLDSINKA, named peptide ‘C’) that the model shown in Figure 7A indicated were important for interactions with the hPXR LBD, thus rendering peptide ‘C’ incapable of disturbing the heterodimerization.
Figure 7.
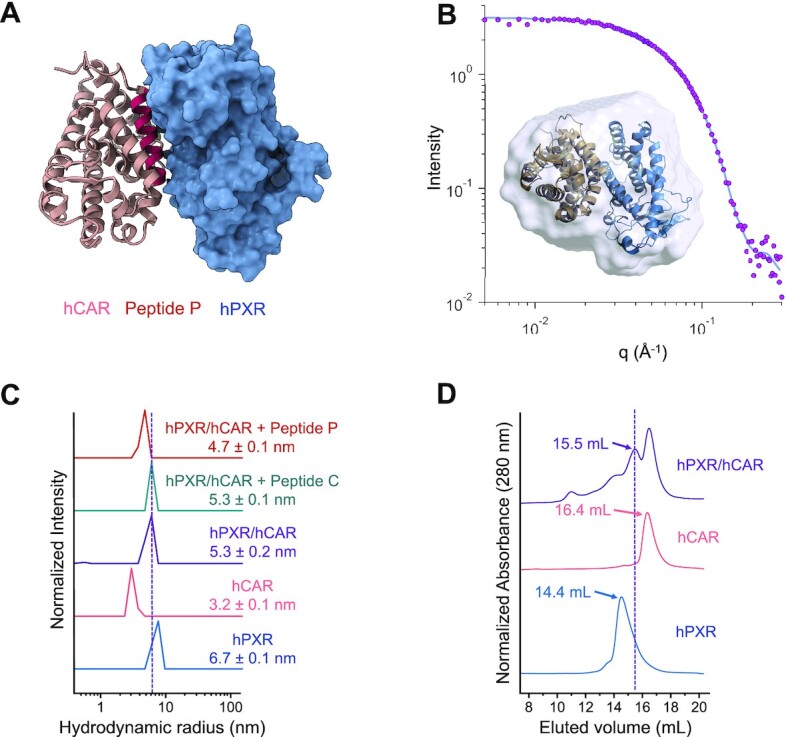
hPXR heterodimerizes with hCAR. (A) A simulated model of the hPXR LBD-hCAR LBD complex. The region corresponding to the AKLLGLLAELRSINEA peptide (peptide ‘P’) is shown in red. hPXR LBD is depicted as surface representation (blue) and hCAR LBD is illustrated as cartoon (light brown). (B) SAXS experiments determined the shape of the hPXR (blue)-hCAR (light brown) complex by defining a molecular envelope (light blue surface representation) where the simulated heterodimer model can be readily accommodated (inset). The envelop was created by ab initio averaged/filtered simulated SAXS curves (solid blue line), which were fitted to the SAXS data (pink spheres). (C) Dynamic light scattering (DLS) analysis illustrated as regularization histograms of hPXR LBD, hCAR LBD, and the appearance of a new species suggesting the formation of the hPXR LBD-hCAR LBD complex. The heterodimer was partially disrupted by peptide ‘P’, but not by the mutated peptide ‘C’ (ADLLGLLAKLDSINKA). (D) Size-exclusion chromatography (SEC) experiments corroborate the formation of the hPXR LBD-hCAR LBD complex, eluting at 15.5 ml. The elution volume of the heterodimer is different from those of the monomeric hCAR LBD (16.4 ml) and the homodimeric form of hPXR LBD (14.4 ml).
Small-angle X-ray scattering studies corroborate the hPXR LBD-hCAR LBD heterodimer model
To experimentally reinforce the structural model for the hPXR LBD-hCAR LBD complex obtained from MD simulations, we used small-angle x-ray scattering (SAXS), a technique that provides information about the shape and size of a macromolecule in solution (56). SAXS can outline the shape of the hPXR LBD-hCAR LBD complex by generating a molecular envelope in which the MD simulated structural model can be readily accommodated (Figure 7B, inset). The envelope was constructed using ab initio averaged/filtered simulated SAXS curves (Figure 7B, solid blue line), which are in good agreement with the SAXS data (Figure 7B, pink spheres). Based on the linear fit of the Guinier region of the SAXS data, the hPXR LBD-hCAR LBD complex is present in solution as a monodisperse and homogenous population with a calculated radius of gyration (Rg) of 26.67 Å (Supplementary Figure S13A). Analysis of the Kratky plot indicates that the heterodimer is compact and globular (Supplementary Figure S13B), and the maximal interatomic distance (Dmax) within the complex is 100 Å based on the pair-wise distance distribution function (Supplementary Figure S13C).
Dynamic light scattering studies of hPXR–hCAR complex formation
Dynamic light scattering (DLS) experiments were performed to further evaluate the interactions between hPXR and hCAR to form the novel heterodimer complex elucidated by MD simulations and SAXS studies. DLS is a non-destructive qualitative technique used to determinate the size distribution of particles in solution (57). The calculated hydrodynamic radius, based on the quantitation of scattered light from the molecules in solution, is representative of the size of the protein (58). Another parameter that is obtained from DLS experiments is the polydispersity index (PdI), which is a measurement of the heterogeneity of each species. A PdI value lower than 20% is indicative of a homogeneous (monodisperse) species, while that higher than 20% is considered a heterogeneous species (i.e. more than one oligomeric species). The hydrodynamic radius of hPXR LBD was determined to be 6.7 nm, and the calculated PdI value of 12.5% indicates a homogeneous species (Figure 7C). On the other hand, hCAR LBD displayed a hydrodynamic radius of 3.2 nm, which is also present as a homogeneous species based on the PdI value of 17%. Although phosphorylated hCAR may form an inactive homodimer in cells (16), it is widely accepted that hCAR LBD is a monomer in solution, and the notable difference in the hydrodynamic radius between hPXR LBD and hCAR LBD leads to the indication that hPXR LBD is detected as a homodimer as previously reported (59). Crystal structures of hPXR LBD show that the β1′ strand of each monomer engages in the homodimerization by interlocking the corresponding W-223 and Y-225. A solution containing both receptors gives rise to a new population with a hydrodynamic radius of 5.3 nm, suggesting the formation of a new oligomeric species, with a hydrodynamic radius greater than that of the monomeric hCAR LBD and smaller than that of the homodimeric hPXR LBD. The high PdI value of 23.5% indicates multiple species within the distribution which cannot be resolved into separate distributions because there is no five-fold difference in size among the species. The presence of peptide ‘P’ decreased the hydrodynamic radius to 4.7 nm and increased the PdI value to 27.8%, suggesting that peptide ‘P’ partially disrupted the hPXR LBD-hCAR LBD complex by competing with hCAR LBD in binding to the hPXR LBD at the heterodimerization interface (Figure 7A). The large PdI value indicates a polydisperse and heterogeneous sample with multiple species represented within the histogram, which cannot be separated into individual distributions because the species do not differ from each other by 5-fold in size. In contrast, the control peptide ‘C’ had no effect on the size of the hydrodynamic radius of the hPXR LBD-hCAR LBD complex, because mutations of key interacting residues prevent it from interacting with hPXR LBD, and therefore, it is unable to disrupt the heterodimer. hCAR is known to heterodimerize with RXRα. As expected, DLS studies with the well-established hCAR LBD-RXRα LBD complex revealed similar profiles as those for hPXR LBD-hCAR LBD, confirming the resolving power of DLS to distinguish between monomer and heterodimer LBDs (Supplementary Figure S14).
Size-exclusion chromatographic analysis confirms direct interactions of the hPXR LBD and hCAR LBD
Size-exclusion chromatography (SEC) separates proteins on the basis of their molecular weight driven by the geometry-dependent (shape) partition of the macromolecules between a continuous liquid phase and the porous interior of a gel or cross-linked bead (60). Larger proteins elute out of the column and are detected at an earlier volume (mL) than smaller proteins. Based on the eluted volume, the size of each protein can be determined provided that the SEC column has been previously calibrated with protein standards of known molecular weight. This technique was applied to corroborate the formation of the hPXR LBD-hCAR LBD heterodimer (Figure 7D). hPXR LBD showed a single peak eluting at 14.4 ml, while hCAR LBD eluted at 16.4 ml also as a single peak. The earlier elution of hPXR LBD compared to that of hCAR LBD corroborate that hPXR LBD is a homodimer and hCAR LBD is a monomer, confirmed by the calculated molecular weight based on the calibration curve generated with the protein standards (See Supplementary Figure S15). The SEC profile of the hPXR LBD-hCAR LBD complex showed a peak at 16.4 ml, with the same elution volume as in the hCAR LBD sample. A second major peak at 15.5 ml corresponds to a new species that is not seen in the hPXR LBD or hCAR LBD preparations, suggesting the detection of the hPXR LBD-hCAR LBD complex. The elution volume of the heterodimer (15.5 ml) is in between those of hCAR LBD (16.4 ml) and hPXR LBD (14.4 ml), following the same trend observed in DLS experiments: the homodimeric hPXR LBD is larger in size than the hPXR LBD-hCAR LBD complex, and in turn, the latter has a larger molecular weight than the monomeric hCAR LBD.
Even though SEC and DLS provided evidence of the formation of a new species in the presence of hPXR LBD and hCAR LBD, it is noteworthy to point out that these two techniques are complementary to each other instead of being redundantly overlapping. DLS measures a closed system, where the equilibrium between hPXR LBD, hCAR LBD and hPXR LBD-hCAR LBD complex is not disturbed. In contrast, in a separation method such as SEC, where there is a continuous flow of the liquid phase through the column, there can be perturbations of the equilibrium between association and dissociation of the heterodimer (61), and ionic and hydrophobic interactions between the protein and the column material can potentially distort the elution of proteins (62). Hence, some differences can arise between these two techniques. For instance, a clear distinction can be observed in SEC between the signal resulting from hCAR LBD and that from the heterodimer in samples containing both hPXR LBD and hCAR LBD, which cannot be appreciated in DLS. In addition, there is contrasting differences in the resolving power of species with certain size differences between the two methods.
RXRα decreases hCAR–hPXR interaction
RXRα is a heterodimeric partner of both hPXR and hCAR (63), and our model shows that hPXR interacts with hCAR through the same protein-protein interacting interface used for heterodimerization with RXR. Therefore, we investigated whether RXRα disrupted hPXR–hCAR interaction and released the inhibition on hCAR in a cellular environment. When GFP-hPXR was co-expressed with either MYC-tagged RXRα (MYC-RXRα) or FLAG-hCAR, GFP-hPXR co-immunoprecipitated with MYC-RXRα or FLAG-hCAR confirming the interactions between hPXR and RXRα and between hPXR and hCAR (Figure 8A). In the presence of increasing amounts of MYC-RXRα (0, 0.5, 1 and 2 μg), the amount of FLAG-hCAR that co-immunoprecipitated with GFP-hPXR decreased as the amount of MYC-RXRα increased, indicating that the formation of heterodimers (hPXR–hCAR, hPXR–RXRα and hCAR–RXRα) is affected by the relative expression level of each receptor and suggesting that there is a competition for heterodimeric partners among the three receptors (Figure 8A). When the segment of the hCAR corresponding to the sequence of peptide P (AKLLGLLAELRSINEA) was mutated to mimic the sequence of peptide C (ADLLGLLAKLDSINKA) (to generate MYC-hCAR C1) or was deleted (to generate MYC-hCAR ΔP1), both MYC-hCAR C1 and MYC-hCAR ΔP1 displayed reduction in interaction with GFP-hPXR compared to wild type hCAR (Supplementary Figure S16A). As expected, both MYC-hCAR C1 and MYC-hCAR ΔP1 decreased their activity in activating the CYP2B6 promoter (Supplementary Figure S16B). These studies confirmed that the peptide P sequence (AKLLGLLAELRSINEA) is part of hPXR–hCAR heterodimerization interface, and supporting the hPXR–hCAR heterodimer model proposed in Figure 7A.
Figure 8.
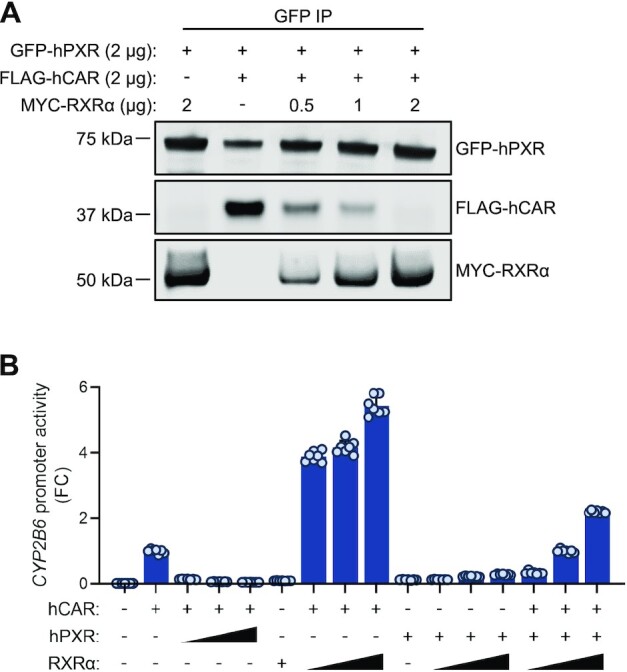
RXRα disrupts hPXR–hCAR interaction and releases the inhibitory effect of hPXR on hCAR. (A) Expression of RXRα decreases the hPXR–hCAR interaction in a dose-responsive manner. HEK-293 cells were co-transfected with GFP-hPXR (2 μg) and FLAG-hCAR (2 μg) with or without MYC-RXRα in increasing amounts as indicated. Co-IP assays were carried out at 48 h post-transfection. (+) represents 2 μg of hPXR, hCAR, or RXRα, whereas (−) represents 2 μg of pcDNA3.1 vector control. (B) Expression of RXRα releases the inhibitory effect of hPXR on hCAR in a dose-responsive manner. HepG2 cells were co-transfected with the CYP2B6-luc reporter in combination with hCAR, hPXR or RXRα in the amounts indicated. Triangles represent increasing receptor plasmid doses (0.05, 0.1 and 0.25 μg), (+) represents 0.25 μg of receptor plasmid and (−) represents variable amounts of pcDNA3.1 vector control, calculated to make the total amount of transfected DNA equal. CYP2B6 promoter transactivation was measured at 48 h post-transfection. FC, fold change over cells transfected with hCAR alone.
To determine whether RXRα could release the suppressive effects of hPXR on hCAR activity, we expressed hCAR alone or with hPXR and/or RXRα, together with a CYP2B6 luciferase reporter, in HepG2 cells. hCAR is constitutively active, and the constitutive activity was repressed by hPXR but enhanced by RXRα (Figure 8B). Importantly, the repressive effect of hPXR on hCAR was released by RXRα in a dose-dependent manner (Figure 8B). Co-expression of hPXR and RXRα only minimally activated the CYP2B6 promoter, suggesting that when RXRα, hCAR and hPXR were co-expressed, the CYP2B6 promoter was activated mainly by hCAR–RXRα. The transactivating activity of hCAR–RXRα is higher in the absence of hPXR than in its presence, further confirming the inhibitory effect of hPXR. Similarly, the activity of hPXR (measured by using a CYP3A4 luciferase reporter) was repressed by hCAR but moderately enhanced by RXRα, and the transactivating activity of hCAR–RXRα on CYP3A4 promoter is decreased in the presence of hPXR (Supplementary Figure S17), confirming the mutual inhibition of hPXR and hCAR, and suggesting that when the three receptors are present, their ultimate transactivating outcome depends on the levels of each receptor which affects the dynamics of the formation of hPXR-RXRα, hCAR-RXRα and hPXR-hCAR heterodimers. We previously showed that an S350D mutation in the LBD of hPXR compromised its heterodimerization with RXRα, leading to greatly reduced transactivating function (19). Interestingly, the S350D mutant inhibited hCAR, but to a lesser extent than did WT hPXR (Supplementary Figure S18A). The S350D mutant also interacted with hCAR, but the interaction was weaker than that seen with WT hPXR (See Supplementary Figure S18B), which is consistent with the reduced inhibitory effect of the S350D mutant and suggests that residue S350 of hPXR plays a role in its heterodimerization with hCAR in a manner similar to that previously observed in hPXR–RXRα heterodimerization. Taken together, our data support the hypothesis that hPXR heterodimerizes with hCAR by using the same interface that is used by RXRα to heterodimerize with each receptor. Together with the discovery that hPXR-hCAR are mutually inhibitory, the data offers explanation for the observations of the mutual functional interference between hPXR and hCAR and define their functional relationship, which is inhibitory rather than redundant.
Crosslinking mass spectrometry studies validate the model of the hPXR LBD-hCAR LBD complex
Chemical crosslinking coupled with mass spectrometry (XL-MS) was applied to probe the interactions between hPXR and hCAR in solution, which was used to corroborate the protein-protein interacting interface of the hPXR-hCAR model. XL-MS has become a powerful technique to investigate the topology and formation of protein complexes, yielding complementary information to guide in the refinement of structural models (64,65). The crosslinking reagent DSSO is a small compound with two reactive chemical groups that primarily form covalent bonds with lysine residues (65). With the two chemical moieties separated by a spacer arm of approximately 10 Å (43), DSSO can bridge a distance of ∼24–30 Å between α carbons (Cα) (43,45,65,66). XL-MS analysis of hPXR LBD-hCAR LBD samples treated with DSSO followed by enzymatic digestion resulted in seven validated interlinked peptides, which are the types of crosslinked peptides formed between sections of hPXR LBD and hCAR LBD that provide protein-protein interaction information (See Supplementary Table S2). Reliability in the XL-MS results is manually confirmed by the values of the MeroX scores and mass accuracy.
The intermolecular peptide with the highest MeroX score corresponds to DQISLLK277(hPXR)GAAF interlinked to K251(hCAR)LQLQ (peptide 1), with a distance spanning 26 Å in the MD simulated model (Figure 9 and See also Supplementary Table S3). Thus, the presence of the interlinked peptide 1 supports the proposed heterodimer interface for the hPXR LBD-hCAR LBD complex because the crosslink between K277 (hPXR) and K251 (hCAR) extends within the upper limit of 26–30 Å for distances between Cα of lysine residues. The interlinked peptides 2 and 5 also fall within the cutoff distance, supporting the heterocomplex model. On the other hand, peptides 3, 4, 6 and 7 have longer distances than what is typically observed in DSSO-based crosslinked peptides. However, the identification of interlinked peptides with longer distances than 30 Å is not uncommon, which may be attributed in part to highly flexible and dynamic regions within proteins (64). hPXR and hCAR are very malleable proteins with fluid segments, and they can be subject to structural rearrangements as a mechanism for ligand entry to the ligand binding pocket, which is encased deep within the LBD (10). B factor (temperature factor) values extracted from hPXR and hCAR crystal structures can provide indications of flexible residues and regions within the crystallized protein, where higher values reflect increased mobility (67). The hPXR residues K198, K234 and K204 implicated in the respective crosslinking peptides 3, 4 and 6 show larger b factor values than the hPXR lysine residues of the remaining peptides (See Supplementary Table S3). They are situated in loops or disordered sections of hPXR, where regions with larger b factor values are illustrated in red and increased thickness of the cartoon representation (see Supplementary Figure S19). In addition, compared to the apo PXR structure, the crystal structure of the rifampicin bound PXR lacks density in several regions, including those comprising these residues (Supplementary Figure S20). The hPXR residue K332 in the crosslinked peptide 7 is located at the edge of helix α7 and adjacent to a loop connecting helices α7 and α8; loops are known to be flexible in solution and could therefore change the distance between the two helices in solution, potentially having a downstream effect in the distance of the interlinked peptide 7. In addition, helix α7 of many nuclear receptors, which in hPXR contains K332, is reported to be part of a highly plastic region that is important in ligand binding events (68).
Figure 9.

Crosslinking mass spectrometry experiments validate the model of the hPXR LBD-hCAR LBD complex. hPXR LBD (blue) and hCAR LBD (red) are depicted in cartoon representation, with K251 from hCAR and K277 from hPXR shown as spheres. The distance of 26 Å (dashed line) between the two lysine residues is within the upper limit for a DSSO-based interlinked peptide, whose sequence is indicated in the insert with the reacted lysine residues colored in red.
DISCUSSION
Given the importance of nuclear receptors in major physiological processes, they have become major drug targets for the development of therapeutics. Most of the research has been conducted regarding their function and regulation of their target genes as individual nuclear receptor. However, the functional correlation and interplay among nuclear receptors have emerged as a significantly unexplored area, which challenges our existing understanding of the biology of nuclear receptors. Detailed mechanisms that explain the observed crosstalk remain lacking, particularly for those nuclear receptors that affect each other's activity through direct physical interactions. In the present study, we determined the molecular basis for the crosstalk between PXR and CAR, where we describe their heterodimerization as the reason for their mutual inhibition. We mapped the heterodimerization interface between hPXR and hCAR to that used by RXR to interact with each receptor. We speculate that similar heterodimerization interfaces to that of the hPXR-hCAR can exist in other nuclear receptors, resulting in alterations to their target gene expression profile, hence, delineating a potentially common crosstalk mechanism. Based on crystallographic studies, all receptors that heterodimerize with RXR contain the φKφφ motifs (where φ is a hydrophobic residue) and share similar dimerization interfaces (15). Therefore, it is quite possible that nuclear receptors that heterodimerize with RXR may form novel heterodimer complexes through the same interface shared with RXR, such as in the case of the hPXR-hCAR complex.
One of the major breakthroughs in understanding how drug metabolism is regulated at the molecular level came from the pioneering work of multiple laboratories that first established PXR (69–71) and CAR (72–75) as master xenobiotic receptors that promiscuously bind compounds of diverse structure and transcriptionally regulate genes encoding proteins involved in drug metabolism, such as CYPs. The CYP gene products, such as CYP3A and CYP2B, are essential for the metabolism of endobiotics such as steroid hormones and xenobiotics such as prescription drugs. It is well established that CYP3A is regulated mainly by PXR, whereas CYP2B is regulated mainly by CAR (9,76), although PXR and CAR are reported to regulate CYP2B and CYP3A reciprocally (12,52). Therefore, CYP3A and CYP2B are regulated by PXR and CAR in both an overlapping and a preferential manner. The fact that PXR and CAR share ligands and target genes led to the perception that PXR and CAR were functionally redundant (12,77–79). However, such a perception conflicts with the observations that the protein levels of one receptor inversely correlate with the activity of the other receptor (5,13,23,50,80).These findings opened the possibility of the existence of crosstalk between PXR and CAR, but left uncertainty if they regulate their target genes in an independent or coordinated manner. Our study has established that hPXR and hCAR form a heterodimer and are mutually inhibitory rather than functionally redundant. This explains the inverse correlation between the activity of one receptor and the protein levels of the other.
The LBD of PXR is responsible for providing the major contacts for protein-protein interactions with RXR and coregulatory proteins. hPXR forms a homodimer, where W-223 and Y-225 within the LBD are critical residues in the hPXR homodimer interface (59). Based on crystal structures and functional studies with W-223-A/Y-225-A double mutants, PXR homodimerization does not seem to affect hPXR heterodimerization with RXRα (59). Similarly, phosphorylated hCAR forms an inactive homodimer in cells (16) and it is suggested that the hCAR–hCAR homodimer interface resides at a location opposite the hCAR–RXRα heterodimer interface (15,55,81). Therefore, the heterodimerization of PXR with CAR may not be affected by their homodimerization status. In fact, crystallographic studies indicate that several nuclear receptors have a homodimerization interface that is different from the heterodimerization interface with RXR (Supplementary Figure S21). The observation that RXRα releases the inhibitory effect of hPXR on hCAR in a dose-dependent manner suggests that there is a dynamic interaction between hPXR, hCAR, and RXRα as hetero- and homodimeric partners and a more complex mechanism of target gene regulation than is currently appreciated. In addition to the known hPXR–hPXR, hPXR–RXRα, hCAR–hCAR and hCAR–RXRα dimers, we have advanced the current model by establishing that hCAR heterodimerizes with hPXR, which explains some unexpected, published data and will facilitate the correct elucidation of hPXR–hCAR regulation. Our current findings support a scenario in which hPXR–hCAR heterodimerization is facilitated by the same dimer interface used by hPXR to heterodimerize with RXRα.
Previous in vivo studies in animals showed evidence that PXR inhibits CAR. Clinical hypothyroidism affects several metabolic processes, including drug metabolism. In mice with hypothyroidism (induced by a low-iodine diet), the induction of Cyp2b10 (a target of mouse CAR [mCAR]) was unexpectedly elevated in PXR knockout (KO) mice but abolished in CAR KO and PXR/CAR double-KO mice, suggesting that Cyp2b10 induction depends on CAR but is suppressed by PXR (13). Similarly, PXR KO, but not CAR KO or PXR/CAR double KO, increased bilirubin clearance in mice as a result of the increased expression of selective bilirubin transporters such as multidrug resistance–associated protein 2 (MRP2) (5). In cultured cells, mCAR constitutively activates MRP2 expression but co-expression of mouse PXR (mPXR) decreases it, suggesting that ligand-free mPXR inhibits CAR-induced expression of MRP2 (5). In another study, basal levels of Cyp3a11 were increased when mPXR was knocked out in mice, revealing an unexpected role for mPXR in repressing Cyp3a11 basal expression. Activation of mCAR further enhanced the Cyp3a11 levels in mPXR KO mice, demonstrating that mCAR regulates Cyp3a11 in vivo independently of mPXR. Whether mPXR repressed basal Cyp3a11 expression by inhibiting mCAR was not further investigated (14). Although these observations suggest that PXR inhibits CAR, the mechanism of inhibition was unclear.
Consistent with our data on hPXR and hCAR, it was reported that ligand-free mPXR could suppress both constitutive and ligand-induced MRP2 expression (5). Ligand-bound mPXR did not reduce the constitutive activity of mCAR in inducing MRP2, but ligand-bound mPXR alone also robustly induced MRP2. We also observed that ligand of hPXR reduced the inhibitory effect of hPXR on hCAR. The mechanism by which ligand affects PXR–CAR interaction warrants further investigation. Saini et al. also proposed competition for the common coactivator SRC-1 in a target gene–specific manner as the underlying mechanism for the crosstalk between mCAR and mPXR, but they did not investigate the interaction between mCAR and mPXR (5). Our findings provide mechanistic insights into the hPXR–hCAR crosstalk by providing evidence that hPXR interacts with hCAR. Although our data do not exclude competition for common coactivators as a contributing factor in hPXR–hCAR mutual inhibition, it is highly unlikely that the availability of common coactivators is a major limiting factor for hPXR/hCAR activity as these coactivators are shared among many nuclear receptors.
The use of a cell model in which only hPXR or hCAR is expressed precludes observation of the coordinated action of hPXR and hCAR. The human hepatocellular carcinoma cell line HepG2 and primary hepatocytes are commonly used to study hPXR function. However, hepatocytes are not readily available and often display considerable donor-to-donor variation. Although HepG2 is the most widely used cell line in drug metabolism studies (82), its ‘hepatocyte-like’ features (i.e., endogenous expression of hPXR and hCAR) have diminished, resulting in the need to express hPXR or hCAR ectopically when investigating one of these receptors. Recently, the HepaRG cell line (consisting of terminally differentiated hepatic cells derived from a human hepatic progenitor cell line that retains many characteristics of hepatocytes, such as the expression of hPXR and hCAR) has been used as an alternative to hepatocytes and has facilitated observations of the inhibitory relationship between hPXR and hCAR (23,51). The value of using a relevant cell model such as HepaRG resulted in the recent findings that CITCO and phenobarbital, previously thought to be specific hCAR activators, also function as hPXR agonists, suggesting the need to re-examine our current understanding of ligand-induced, receptor-specific regulation of target genes (23,51). In HepaRG cells, hCAR KO elevated basal levels of the hPXR target genes CYP3A7, CYP3A5 (80), and CYP3A4 (23). Moreover, stable overexpression of hCAR in HepaRG cells resulted in increased basal CYP3A4 (probably because of the constitutive activity of hCAR in regulating the basal level of CYP3A4) but reduced CYP3A4 induction by the specific hPXR agonist RIF (50), suggesting that hCAR suppresses the RIF-induced activity of hPXR. Therefore, although hPXR and hCAR appear to have overlapping functions as individual receptors when overexpressed in cell lines (most cell lines express undetectable endogenous levels of hPXR and/or hCAR), they appear to be mutually inhibitory when co-expressed in more physiologically relevant liver cell models, but the mechanism of this mutual inhibition was unknown.
The physical interaction and mutual inhibition between hPXR and hCAR predict that the combined activity of hPXR and hCAR depends on the expression level of each receptor, which would explain the individual variability in drug responses, including adverse responses, seen in human populations (83,84). The observation that the LBD of the receptor is sufficient for the inhibitory interaction indicates that isoforms of hPXR and hCAR (as long as they contain the LBD) (85,86) contribute to regulating the activity of hPXR–hCAR. Accumulating evidence implicates PXR and CAR in energy homeostasis, cell proliferation, inflammation, tissue injury and repair, immune responses, and cancer development (10,11,87–89). Now that we know hPXR and hCAR to be mutually inhibitory, the individual and combined roles of hPXR and hCAR in these cellular processes need to be re-examined. As the protein levels of hPXR and hCAR determine the combined activity of the receptors and the interaction between hPXR and hCAR does not require ligand binding, chemical tools that selectively degrade one receptor, or that disrupt the hPXR–hCAR interaction, will be valuable in dissecting the roles of hPXR and hCAR.
In addition to RXR, co-activators, and co-repressors, post-translational modifications may affect hPXR–hCAR interaction. For example, a phosphomimetic hPXR mutant, S350D, compromises hPXR interaction with RXR (19), and we have shown here that the S350D mutation decreases hPXR interaction with hCAR, possibly via the same mechanism that compromises the hPXR–RXR interaction. In another study, phosphorylation at Thr38 of hCAR promoted the formation of an inactive hCAR homodimer (16). Consistent with this, we have shown here that a T38D phosphomimetic mutant of hCAR reduces the inhibitory effect of hCAR on hPXR, probably because of the increased hCAR homodimerization.
To date, no PXR response elements in gene promoters that mediate the inhibitory effect of hPXR have been reported. Our findings that hPXR can repress hCAR-mediated CYP2B6 gene expression through protein–protein interaction with hCAR might explain the recent finding that PXR downregulates twice as many genes as it induces in primary human hepatocytes (90). It should be pointed out that, in that study, RIF and CITCO were intended to be specific agonists of hPXR and hCAR, respectively. However, we now know that CITCO is a dual agonist of both hPXR and hCAR (23). Therefore, in future receptor-specific gene expression studies, both ligand specificity and protein–protein interactions should be considered. Furthermore, we cannot rule out the possibility that both hPXR and hCAR heterodimerize with many other nuclear receptors. The finding that hPXR heterodimerizes with hCAR opens new perspectives on the regulation of hPXR and hCAR in the context of systematic protein–protein interactions that involve other nuclear receptors and the polypharmacologic properties of their ligands.
To summarize, we first confirmed the inhibitory effect of hPXR on hCAR in cell models that co-express hPXR and hCAR, such as human primary hepatocytes, then we revealed the previously unknown physical interaction between hPXR and hCAR. Through a combination of cellular and biophysical assays, we mapped the heterodimerization interface between hPXR and hCAR to that used by RXR to interact with each receptor. Many spliced variants of both hPXR and hCAR have been identified and shown to contribute to individual variability in drug responses (85,86). Furthermore, hPXR and hCAR are known to play broader roles beyond drug metabolism (10,11,88). Therefore, our discovery of the inhibitory interactions between hPXR and hCAR not only provides a novel molecular mechanism for variation in drug–drug interactions and a potential approach to their therapeutic regulation; it also sheds light on how these receptors coordinately regulate other physiologic processes, such as energy metabolism, and pathologic processes, such as cancer development. Furthermore, our work highlights the importance to comprehensively investigate the crosstalk among members of the nuclear receptor superfamily, through previously unrecognized physical association. Such crosstalk might constitute an unexplored space to define and target nuclear receptor physiology and pathophysiology.
DATA AVAILABILITY
Raw and processed RNA-seq data can be obtained in the GEO database under accession code GSE149001.
Supplementary Material
ACKNOWLEDGEMENTS
The authors thank ALSAC for support; Keith A. Laycock, PhD, ELS (St. Jude Department of Scientific Editing) for editing the manuscript; the Hartwell Center at St. Jude Children's Research Hospital for synthesizing the hCAR peptides and performing RNA-seq; Meritxell B Cutrona for technical assistance; Alireza Abdolvahabi, Sandra Kietlinska, and members of the Chen laboratory for valuable discussions. Human primary hepatocytes were obtained through the Liver Tissue and Cell Distribution System (Pittsburgh, PA), which is funded by NIH contract # HHSN275201700005C. This research used the Life Science X-ray Scattering (LIX) beamline at the National Synchrotron Light Source-II, a DOE, Office of Science User Facility operated for the DOE, Office of Science by Brookhaven National Laboratory under Contract DE-SC0012704. The LIX beamline is part of the Life Science Biomedical Technology Research resource, primarily supported by the National Institutes of Health, National Institute of General Medical Sciences under Grant P41 GM111244 and by the DOE Office of Biological and Environmental Research under Grant KP1605010, with additional support from NIH Grant S10 OD012331. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Contributor Information
Monicah N Bwayi, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Efren Garcia-Maldonado, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Sergio C Chai, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Boer Xie, Center for Proteomics and Metabolomics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Shirish Chodankar, National Synchrotron Light Source II, Brookhaven National Laboratory, Upton, NY 11973, USA.
Andrew D Huber, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Jing Wu, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Kavya Annu, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
William C Wright, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Hyeong-Min Lee, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Jayaraman Seetharaman, Department of Structural Biology, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Jingheng Wang, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Cameron D Buchman, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Junmin Peng, Center for Proteomics and Metabolomics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA; Department of Structural Biology, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA; Department of Developmental Neurobiology, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
Taosheng Chen, Department of Chemical Biology and Therapeutics, St. Jude Children's Research Hospital, Memphis, TN 38105-3678, USA.
SUPPLEMENTARY DATA
Supplementary Data are available at NAR Online.
FUNDING
National Institute of General Medical Sciences of the National Institutes of Health [R35GM118041 to T.C.]. Funding for open access charge: National Institutes of Health.
Conflict of interest statement. None declared.
REFERENCES
- 1. Evans R.M. The nuclear receptor superfamily: a rosetta stone for physiology. Mol. Endocrinol. 2005; 19:1429–1438. [DOI] [PubMed] [Google Scholar]
- 2. Laudet V. Evolution of the nuclear receptor superfamily: early diversification from an ancestral orphan receptor. J. Mol. Endocrinol. 1997; 19:207–226. [DOI] [PubMed] [Google Scholar]
- 3. Santos R., Ursu O., Gaulton A., Bento A.P., Donadi R.S., Bologa C.G., Karlsson A., Al-Lazikani B., Hersey A., Oprea T.I.et al.. A comprehensive map of molecular drug targets. Nat. Rev. Drug Discov. 2017; 16:19–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. De Bosscher K., Desmet S.J., Clarisse D., Estebanez-Perpina E., Brunsveld L.. Nuclear receptor crosstalk - defining the mechanisms for therapeutic innovation. Nat. Rev. Endocrinol. 2020; 16:363–377. [DOI] [PubMed] [Google Scholar]
- 5. Saini S.P., Mu Y., Gong H., Toma D., Uppal H., Ren S., Li S., Poloyac S.M., Xie W.. Dual role of orphan nuclear receptor pregnane x receptor in bilirubin detoxification in mice. Hepatology. 2005; 41:497–505. [DOI] [PubMed] [Google Scholar]
- 6. Singhal H., Greene M.E., Tarulli G., Zarnke A.L., Bourgo R.J., Laine M., Chang Y.F., Ma S., Dembo A.G., Raj G.V.et al.. Genomic agonism and phenotypic antagonism between estrogen and progesterone receptors in breast cancer. Sci. Adv. 2016; 2:e1501924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Evans R.M., Mangelsdorf D.J.. Nuclear receptors, RXR, and the big bang. Cell. 2014; 157:255–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Maglich J.M., Parks D.J., Moore L.B., Collins J.L., Goodwin B., Billin A.N., Stoltz C.A., Kliewer S.A., Lambert M.H., Willson T.M.et al.. Identification of a novel human constitutive androstane receptor (CAR) agonist and its use in the identification of CAR target genes. J. Biol. Chem. 2003; 278:17277–17283. [DOI] [PubMed] [Google Scholar]
- 9. Pascussi J.M., Gerbal-Chaloin S., Drocourt L., Maurel P., Vilarem M.J.. The expression of CYP2B6, CYP2C9 and CYP3A4 genes: a tangle of networks of nuclear and steroid receptors. Biochim. Biophys. Acta. 2003; 1619:243–253. [DOI] [PubMed] [Google Scholar]
- 10. Chai S.C., Wright W.C., Chen T.. Strategies for developing pregnane x receptor antagonists: implications from metabolism to cancer. Med. Res. Rev. 2020; 40:1061–1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Mackowiak B., Hodge J., Stern S., Wang H.. The roles of xenobiotic receptors: beyond chemical disposition. Drug Metab. Dispos. 2018; 46:1361–1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Xie W., Barwick J.L., Simon C.M., Pierce A.M., Safe S., Blumberg B., Guzelian P.S., Evans R.M.. Reciprocal activation of xenobiotic response genes by nuclear receptors SXR/PXR and CAR. Genes Dev. 2000; 14:3014–3023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Park Y.J., Lee E.K., Lee Y.K., Park D.J., Jang H.C., Moore D.D.. Opposing regulation of cytochrome P450 expression by CAR and PXR in hypothyroid mice. Toxicol. Appl. Pharmacol. 2012; 263:131–137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Staudinger J.L., Goodwin B., Jones S.A., Hawkins-Brown D., MacKenzie K.I., LaTour A., Liu Y., Klaassen C.D., Brown K.K., Reinhard J.et al.. The nuclear receptor PXR is a lithocholic acid sensor that protects against liver toxicity. Proc. Natl. Acad. Sci. U.S.A. 2001; 98:3369–3374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Suino K., Peng L., Reynolds R., Li Y., Cha J.Y., Repa J.J., Kliewer S.A., Xu H.E.. The nuclear xenobiotic receptor CAR: structural determinants of constitutive activation and heterodimerization. Mol. Cell. 2004; 16:893–905. [DOI] [PubMed] [Google Scholar]
- 16. Shizu R., Osabe M., Perera L., Moore R., Sueyoshi T., Negishi M.. Phosphorylated nuclear receptor CAR forms a homodimer to repress its constitutive activity for ligand activation. Mol. Cell. Biol. 2017; 37:e00649-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Elias A., Wu J., Chen T.. Tumor suppressor protein p53 negatively regulates human pregnane x receptor activity. Mol. Pharmacol. 2013; 83:1229–1236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Pondugula S.R., Dong H., Chen T.. Phosphorylation and protein-protein interactions in PXR-mediated CYP3A repression. Expert Opin. Drug Metab. Toxicol. 2009; 5:861–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wang Y.M., Chai S.C., Lin W., Chai X., Elias A., Wu J., Ong S.S., Pondugula S.R., Beard J.A., Schuetz E.G.et al.. Serine 350 of human pregnane x receptor is crucial for its heterodimerization with retinoid x receptor alpha and transactivation of target genes in vitro and in vivo. Biochem. Pharmacol. 2015; 96:357–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Cherian M.T., Lin W., Wu J., Chen T.. CINPA1 is an inhibitor of constitutive androstane receptor that does not activate pregnane x receptor. Mol. Pharmacol. 2015; 87:878–889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Cherian M.T., Chai S.C., Wright W.C., Singh A., Alexandra Casal M., Zheng J., Wu J., Lee R.E., Griffin P.R., Chen T. CINPA1 binds directly to constitutive androstane receptor and inhibits its activity. Biochem. Pharmacol. 2018; 152:211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ong S.S., Goktug A.N., Elias A., Wu J., Saunders D., Chen T.. Stability of the human pregnane x receptor is regulated by E3 ligase UBR5 and serine/threonine kinase DYRK2. Biochem. J. 2014; 459:193–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lin W., Bwayi M., Wu J., Li Y., Chai S.C., Huber A.D., Chen T.. CITCO directly binds to and activates human pregnane x receptor. Mol. Pharmacol. 2020; 97:180–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Schuetz E.G., Schinkel A.H., Relling M.V., Schuetz J.D.. P-glycoprotein: a major determinant of rifampicin-inducible expression of cytochrome P4503A in mice and humans. Proc. Natl. Acad. Sci. U.S.A. 1996; 93:4001–4005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Langmead B., Salzberg S.L.. Fast gapped-read alignment with bowtie 2. Nat. Methods. 2012; 9:357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Li B., Dewey C.N.. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinf. 2011; 12:323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Ritchie M.E., Phipson B., Wu D., Hu Y., Law C.W., Shi W., Smyth G.K.. limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res. 2015; 43:e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Law C.W., Chen Y., Shi W., Smyth G.K.. voom: precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol. 2014; 15:R29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Robinson M.D., McCarthy D.J., Smyth G.K.. edgeR: a bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics. 2010; 26:139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A.et al.. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016; 44:W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Huber A.D., Wright W.C., Lin W., Majumder K., Low J.A., Wu J., Buchman C.D., Pintel D.J., Chen T.. Mutation of a single amino acid of pregnane x receptor switches an antagonist to agonist by altering AF-2 helix positioning. Cell. Mol. Life Sci. 2021; 78:317–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hariparsad N., Chu X., Yabut J., Labhart P., Hartley D.P., Dai X., Evers R.. Identification of pregnane-X receptor target genes and coactivator and corepressor binding to promoter elements in human hepatocytes. Nucleic Acids Res. 2009; 37:1160–1173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Surapureddi S., Rana R., Reddy J.K., Goldstein J.A.. Nuclear receptor coactivator 6 mediates the synergistic activation of human cytochrome P-450 2C9 by the constitutive androstane receptor and hepatic nuclear factor-4alpha. Mol. Pharmacol. 2008; 74:913–923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. London N., Raveh B., Cohen E., Fathi G., Schueler-Furman O.. Rosetta flexpepdock web server–high resolution modeling of peptide-protein interactions. Nucleic Acids Res. 2011; 39:W249–W253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Raveh B., London N., Schueler-Furman O.. Sub-angstrom modeling of complexes between flexible peptides and globular proteins. Proteins. 2010; 78:2029–2040. [DOI] [PubMed] [Google Scholar]
- 36. Eswar N., John B., Mirkovic N., Fiser A., Ilyin V.A., Pieper U., Stuart A.C., Marti-Renom M.A., Madhusudhan M.S., Yerkovich B.et al.. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003; 31:3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pieper U., Eswar N., Braberg H., Madhusudhan M.S., Davis F.P., Stuart A.C., Mirkovic N., Rossi A., Marti-Renom M.A., Fiser A.et al.. MODBASE, a database of annotated comparative protein structure models, and associated resources. Nucleic Acids Res. 2004; 32:D217–D222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pronk S., Pall S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M.R., Smith J.C., Kasson P.M., van der Spoel D.et al.. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013; 29:845–854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Soteras Gutierrez I., Lin F.Y., Vanommeslaeghe K., Lemkul J.A., Armacost K.A., Brooks C.L. 3rd, MacKerell A.D. Jr. Parametrization of halogen bonds in the CHARMM general force field: improved treatment of ligand-protein interactions. Bioorg. Med. Chem. 2016; 24:4812–4825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Goddard T.D., Huang C.C., Meng E.C., Pettersen E.F., Couch G.S., Morris J.H., Ferrin T.E.. UCSF chimerax: meeting modern challenges in visualization and analysis. Protein Sci. 2018; 27:14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Wang W., Prosise W.W., Chen J., Taremi S.S., Le H.V., Madison V., Cui X., Thomas A., Cheng K.C., Lesburg C.A.. Construction and characterization of a fully active PXR/SRC-1 tethered protein with increased stability. Protein Eng. Des. Sel. 2008; 21:425–433. [DOI] [PubMed] [Google Scholar]
- 42. Myasnikov A., Zhu H., Hixson P., Xie B., Yu K., Pitre A., Peng J., Sun J.. Structural analysis of the full-length human LRRK2. Cell. 2021; 184:3519–3527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Kao A., Chiu C.L., Vellucci D., Yang Y., Patel V.R., Guan S., Randall A., Baldi P., Rychnovsky S.D., Huang L.. Development of a novel cross-linking strategy for fast and accurate identification of cross-linked peptides of protein complexes. Mol. Cell. Proteomics. 2011; 10:M110.002212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Wang H., Yang Y., Li Y., Bai B., Wang X., Tan H., Liu T., Beach T.G., Peng J., Wu Z.. Systematic optimization of long gradient chromatography mass spectrometry for deep analysis of brain proteome. J. Proteome Res. 2015; 14:829–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Iacobucci C., Gotze M., Ihling C.H., Piotrowski C., Arlt C., Schafer M., Hage C., Schmidt R., Sinz A.. A cross-linking/mass spectrometry workflow based on MS-cleavable cross-linkers and the MeroX software for studying protein structures and protein-protein interactions. Nat. Protoc. 2018; 13:2864–2889. [DOI] [PubMed] [Google Scholar]
- 46. Yang L., Antonelli S., Chodankar S., Byrnes J., Lazo E., Qian K.. Solution scattering at the life science X-ray scattering (LiX) beamline. J. Synchrotron Radiat. 2020; 27:804–812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Franke D., Petoukhov M.V., Konarev P.V., Panjkovich A., Tuukkanen A., Mertens H.D.T., Kikhney A.G., Hajizadeh N.R., Franklin J.M., Jeffries C.M.et al.. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J. Appl. Crystallogr. 2017; 50:1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Mutoh S., Osabe M., Inoue K., Moore R., Pedersen L., Perera L., Rebolloso Y., Sueyoshi T., Negishi M.. Dephosphorylation of threonine 38 is required for nuclear translocation and activation of human xenobiotic receptor CAR (NR1I3). J. Biol. Chem. 2009; 284:34785–34792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Preiss L.C., Liu R., Hewitt P., Thompson D., Georgi K., Badolo L., Lauschke V.M., Petersson C.. Deconvolution of cytochrome P450 induction mechanisms in HepaRG nuclear hormone receptor knockout cells. Drug Metab. Dispos. 2021; 49:668–678. [DOI] [PubMed] [Google Scholar]
- 50. van der Mark V.A., Rudi de Waart D., Shevchenko V., Elferink R.P.J.O., Chamuleau R.A.F.M., Hoekstra R.. Stable overexpression of the constitutive androstane receptor reduces the requirement for culture with dimethyl sulfoxide for high drug metabolism in HepaRG cells. Drug Metab. Dispos. 2017; 45:56–67. [DOI] [PubMed] [Google Scholar]
- 51. Li L., Welch M.A., Li Z., Mackowiak B., Heyward S., Swaan P.W., Wang H.. Mechanistic insights of phenobarbital-mediated activation of human but not mouse pregnane x receptor. Mol. Pharmacol. 2019; 96:345–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Faucette S.R., Sueyoshi T., Smith C.M., Negishi M., Lecluyse E.L., Wang H.. Differential regulation of hepatic CYP2B6 and CYP3A4 genes by constitutive androstane receptor but not pregnane x receptor. J. Pharmacol. Exp. Ther. 2006; 317:1200–1209. [DOI] [PubMed] [Google Scholar]
- 53. Faucette S.R., Zhang T.C., Moore R., Sueyoshi T., Omiecinski C.J., LeCluyse E.L., Negishi M., Wang H.. Relative activation of human pregnane x receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J. Pharmacol. Exp. Ther. 2007; 320:72–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Wallace B.D., Betts L., Talmage G., Pollet R.M., Holman N.S., Redinbo M.R.. Structural and functional analysis of the human nuclear xenobiotic receptor PXR in complex with RXRα. J. Mol. Biol. 2013; 425:2561–2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Xu R.X., Lambert M.H., Wisely B.B., Warren E.N., Weinert E.E., Waitt G.M., Williams J.D., Collins J.L., Moore L.B., Willson T.M.et al.. A structural basis for constitutive activity in the human CAR/RXRalpha heterodimer. Mol. Cell. 2004; 16:919–928. [DOI] [PubMed] [Google Scholar]
- 56. Putnam C.D., Hammel M., Hura G.L., Tainer J.A.. X-ray solution scattering (SAXS) combined with crystallography and computation: defining accurate macromolecular structures, conformations and assemblies in solution. Q. Rev. Biophys. 2007; 40:191–285. [DOI] [PubMed] [Google Scholar]
- 57. den Engelsman J., Garidel P., Smulders R., Koll H., Smith B., Bassarab S., Seidl A., Hainzl O., Jiskoot W.. Strategies for the assessment of protein aggregates in pharmaceutical biotech product development. Pharm. Res. 2011; 28:920–933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Al-Ghobashy M.A., Mostafa M.M., Abed H.S., Fathalla F.A., Salem M.Y.. Correlation between dynamic light scattering and size exclusion high performance liquid chromatography for monitoring the effect of pH on stability of biopharmaceuticals. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2017; 1060:1–9. [DOI] [PubMed] [Google Scholar]
- 59. Noble S.M., Carnahan V.E., Moore L.B., Luntz T., Wang H., Ittoop O.R., Stimmel J.B., Davis-Searles P.R., Watkins R.E., Wisely G.B.et al.. Human PXR forms a tryptophan zipper-mediated homodimer. Biochemistry. 2006; 45:8579–8589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Brooks D.E., Haynes C.A., Hritcu D., Steels B.M., Muller W.. Size exclusion chromatography does not require pores. Proc. Natl. Acad. Sci. U.S.A. 2000; 97:7064–7067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Philo J.S. Is any measurement method optimal for all aggregate sizes and types. AAPS J. 2006; 8:E564–E571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goyon A., Fekete S., Beck A., Veuthey J.L., Guillarme D. Unraveling the mysteries of modern size exclusion chromatography - the way to achieve confident characterization of therapeutic proteins. J. Chromatogr. B Analyt. Technol. Biomed. Life Sci. 2018; 1092:368–378. [DOI] [PubMed] [Google Scholar]
- 63. Dash A.K., Yende A.S., Jaiswal B., Tyagi R.K.. Heterodimerization of retinoid x receptor with xenobiotic receptor partners occurs in the cytoplasmic compartment: mechanistic insights of events in living cells. Exp. Cell. Res. 2017; 360:337–346. [DOI] [PubMed] [Google Scholar]
- 64. Hevler J.F., Lukassen M.V., Cabrera-Orefice A., Arnold S., Pronker M.F., Franc V., Heck A.J.R.. Selective cross-linking of coinciding protein assemblies by in-gel cross-linking mass spectrometry. EMBO J. 2021; 40:e106174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Khan K., Baleanu-Gogonea C., Willard B., Gogonea V., Fox P.L.. 3-Dimensional architecture of the human multi-tRNA synthetase complex. Nucleic Acids Res. 2020; 48:8740–8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Merkley E.D., Rysavy S., Kahraman A., Hafen R.P., Daggett V., Adkins J.N.. Distance restraints from crosslinking mass spectrometry: mining a molecular dynamics simulation database to evaluate lysine-lysine distances. Protein Sci. 2014; 23:747–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Sun Z., Liu Q., Qu G., Feng Y., Reetz M.T.. Utility of B-Factors in protein science: interpreting rigidity, flexibility, and internal motion and engineering thermostability. Chem. Rev. 2019; 119:1626–1665. [DOI] [PubMed] [Google Scholar]
- 68. Edman K., Hosseini A., Bjursell M.K., Aagaard A., Wissler L., Gunnarsson A., Kaminski T., Kohler C., Backstrom S., Jensen T.J.et al.. Ligand binding mechanism in steroid receptors: from conserved plasticity to differential evolutionary constraints. Structure. 2015; 23:2280–2290. [DOI] [PubMed] [Google Scholar]
- 69. Bertilsson G., Heidrich J., Svensson K., Asman M., Jendeberg L., Sydow-Backman M., Ohlsson R., Postlind H., Blomquist P., Berkenstam A.. Identification of a human nuclear receptor defines a new signaling pathway for CYP3A induction. Proc. Natl. Acad. Sci. U.S.A. 1998; 95:12208–12213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Kliewer S.A., Moore J.T., Wade L., Staudinger J.L., Watson M.A., Jones S.A., McKee D.D., Oliver B.B., Willson T.M., Zetterstrom R.H.et al.. An orphan nuclear receptor activated by pregnanes defines a novel steroid signaling pathway. Cell. 1998; 92:73–82. [DOI] [PubMed] [Google Scholar]
- 71. Lehmann J.M., McKee D.D., Watson M.A., Willson T.M., Moore J.T., Kliewer S.A.. The human orphan nuclear receptor PXR is activated by compounds that regulate CYP3A4 gene expression and cause drug interactions. J. Clin. Invest. 1998; 102:1016–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Baes M., Gulick T., Choi H.S., Martinoli M.G., Simha D., Moore D.D.. A new orphan member of the nuclear hormone receptor superfamily that interacts with a subset of retinoic acid response elements. Mol. Cell. Biol. 1994; 14:1544–1552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Forman B.M., Tzameli I., Choi H.S., Chen J., Simha D., Seol W., Evans R.M., Moore D.D.. Androstane metabolites bind to and deactivate the nuclear receptor CAR-beta. Nature. 1998; 395:612–615. [DOI] [PubMed] [Google Scholar]
- 74. Honkakoski P., Zelko I., Sueyoshi T., Negishi M.. The nuclear orphan receptor CAR-retinoid x receptor heterodimer activates the phenobarbital-responsive enhancer module of the CYP2B gene. Mol. Cell. Biol. 1998; 18:5652–5658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Kawamoto T., Sueyoshi T., Zelko I., Moore R., Washburn K., Negishi M.. Phenobarbital-responsive nuclear translocation of the receptor CAR in induction of the CYP2B gene. Mol. Cell. Biol. 1999; 19:6318–6322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Moore J.T., Moore L.B., Maglich J.M., Kliewer S.A.. Functional and structural comparison of PXR and CAR. Biochim. Biophys. Acta. 2003; 1619:235–238. [DOI] [PubMed] [Google Scholar]
- 77. Goodwin B., Redinbo M.R., Kliewer S.A.. Regulation of cyp3a gene transcription by the pregnane x receptor. Annu. Rev. Pharmacol. Toxicol. 2002; 42:1–23. [DOI] [PubMed] [Google Scholar]
- 78. Smirlis D., Muangmoonchai R., Edwards M., Phillips I.R., Shephard E.A.. Orphan receptor promiscuity in the induction of cytochromes p450 by xenobiotics. J. Biol. Chem. 2001; 276:12822–12826. [DOI] [PubMed] [Google Scholar]
- 79. Xie W., Yeuh M.F., Radominska-Pandya A., Saini S.P., Negishi Y., Bottroff B.S., Cabrera G.Y., Tukey R.H., Evans R.M.. Control of steroid, heme, and carcinogen metabolism by nuclear pregnane x receptor and constitutive androstane receptor. Proc. Natl. Acad. Sci. U.S.A. 2003; 100:4150–4155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Li D., Mackowiak B., Brayman T.G., Mitchell M., Zhang L., Huang S.M., Wang H.. Genome-wide analysis of human constitutive androstane receptor (CAR) transcriptome in wild-type and CAR-knockout HepaRG cells. Biochem. Pharmacol. 2015; 98:190–202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Shan L., Vincent J., Brunzelle J.S., Dussault I., Lin M., Ianculescu I., Sherman M.A., Forman B.M., Fernandez E.J.. Structure of the murine constitutive androstane receptor complexed to androstenol: a molecular basis for inverse agonism. Mol. Cell. 2004; 16:907–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Donato M.T., Tolosa L., Gomez-Lechon M.J.. Culture and functional characterization of human hepatoma hepg2 cells. Methods Mol. Biol. 2015; 1250:77–93. [DOI] [PubMed] [Google Scholar]
- 83. Mani S., Dou W., Redinbo M.R.. PXR antagonists and implication in drug metabolism. Drug Metab. Rev. 2013; 45:60–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Staudinger J.L. Clinical applications of small molecule inhibitors of pregnane x receptor. Mol. Cell. Endocrinol. 2019; 485:61–71. [DOI] [PubMed] [Google Scholar]
- 85. Brewer C.T., Chen T.. PXR variants: the impact on drug metabolism and therapeutic responses. Acta Pharm Sin B. 2016; 6:441–449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lamba J., Lamba V., Schuetz E.. Genetic variants of PXR (NR1I2) and CAR (NR1I3) and their implications in drug metabolism and pharmacogenetics. Curr. Drug Metab. 2005; 6:369–383. [DOI] [PubMed] [Google Scholar]
- 87. Hakkola J., Rysa J., Hukkanen J.. Regulation of hepatic energy metabolism by the nuclear receptor PXR. Biochim. Biophys. Acta. 2016; 1859:1072–1082. [DOI] [PubMed] [Google Scholar]
- 88. Oladimeji P.O., Chen T.. PXR: more than just a master xenobiotic receptor. Mol. Pharmacol. 2018; 93:119–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Xing Y., Yan J., Niu Y.. PXR: a center of transcriptional regulation in cancer. Acta Pharm Sin B. 2020; 10:197–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Kandel B.A., Thomas M., Winter S., Damm G., Seehofer D., Burk O., Schwab M., Zanger U.M.. Genomewide comparison of the inducible transcriptomes of nuclear receptors CAR, PXR and PPARalpha in primary human hepatocytes. Biochim. Biophys. Acta. 2016; 1859:1218–1227. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw and processed RNA-seq data can be obtained in the GEO database under accession code GSE149001.