

Abstract
Epstein-Barr virus (EBV; human herpesvirus 4; HHV-4) and Kaposi sarcoma-associated herpesvirus (KSHV; human herpesvirus 8; HHV-8) are human gammaherpesviruses that have oncogenic properties. EBV is a lymphocryptovirus, whereas HHV-8/KSHV is a rhadinovirus. As lymphotropic viruses, EBV and KSHV are associated with several lymphoproliferative diseases or plasmacytic/plasmablastic neoplasms. Interestingly, these viruses can also infect epithelial cells causing carcinomas and, in the case of KSHV, endothelial cells, causing sarcoma. EBV is associated with Burkitt lymphoma, classic Hodgkin lymphoma, nasopharyngeal carcinoma, plasmablastic lymphoma, lymphomatoid granulomatosis, leiomyosarcoma, and subsets of diffuse large B cell lymphoma, post-transplant lymphoproliferative disorder, and gastric carcinoma. KSHV is implicated in Kaposi sarcoma, primary effusion lymphoma, multicentric Castleman disease, and KSHV-positive diffuse large B cell lymphoma. Pathogenesis by these two herpesviruses is intrinsically linked to viral proteins expressed during the lytic and latent lifecycles. This comprehensive review intends to provide an overview of the EBV and KSHV viral cycles, viral proteins that contribute to oncogenesis, and the current understanding of the pathogenesis and clinicopathology of their related neoplastic entities.
Keywords: Kaposi sarcoma-associated virus/human herpesvirus 8, Epstein-Barr virus, Kaposi sarcoma, primary effusion lymphoma, multicentric Castleman disease, Burkitt lymphoma, nasopharyngeal carcinoma, post-transplant lymphoproliferative disease, LANA, K1, K15, EBNAs, LMP1, LMP2A
Introduction
The human gammaherpesviruses Epstein-Barr virus (EBV; human herpesvirus-4 or HHV-4) and Kaposi sarcoma-associated herpesvirus (KSHV; human herpesvirus-8 or HHV-8) are etiologic agents of a specific set of lymphoid and non-lymphoid neoplasms [1]. Like other herpesviruses, EBV and KSHV are double-stranded linear DNA viruses that exhibit a biphasic lifecycle (i.e., lytic and latent forms). Infections tend to persist in the host. Both viruses are known to cause human cancers when the infected hosts are immunosuppressed. EBV infects ~90% of the world’s adults and is considered an oncogenic viral agent for several distinct human neoplasms, including nasopharyngeal carcinoma (NPC) [2], Burkitt lymphoma (BL) [3], classic Hodgkin lymphoma (CHL) [4], EBV-positive diffuse large B cell lymphoma (EBV+ DLBCL) [5], DLBCL associated with chronic inflammation [6], plasmablastic lymphoma (PBL) [7], lymphomatoid granulomatosis [8], and subsets of post-transplant lymphoproliferative disorder (PTLD) [9], gastric carcinoma (GC) [10], and leiomyosarcoma [11]. Of note, EBV was the very first human tumor virus identified. KSHV is implicated in Kaposi sarcoma (KS) [12], primary effusion lymphoma (PEL) [13, 14], multicentric Castleman disease (MCD) [15], and KSHV-positive diffuse large B cell lymphoma [16]. The numerous neoplasms caused by EBV and KSHV are likely due to their large genomes that encode a myriad of viral genes, which in turn enable these viruses to express proteins that modify the cellular environment.
Viral life cycle and oncogenesis
The primary route of transmission for both EBV and KSHV is via bodily fluids including saliva [17–19]. The virus lifecycle can be delineated into two phases, latency and lytic replication (Figure 1). As the default viral lifecycle of EBV and KSHV, latency is key for these viruses to persist in the infected cells. During latency, the viral genomes assume a circular/ring structure known as an episome. The viruses are replicated during host cell division and then segregated into daughter cells [20]. Only few viral genes are expressed in the latent state and no virions are produced during the latent stage. In contrast, viral lytic replication occurs following reactivation from latency or following infection in some cell types depending on the virus. Infectious virions are produced as a result of lytic infection. During lytic replication, viral gene expression is highly orchestrated in three sequential phases, including the immediate early (IE), delayed early (DE), and late (L) phase. IE genes do not require protein synthesis to be expressed (indicating that host cell factors are capable of activating the promoters of IE genes) and most IE genes are viral transcription factors. DE gene expression is dependent upon protein expression but not DNA synthesis and DE genes e.g., DNA polymerase is involved in replication of the viral DNA. Finally, L genes are expressed after DNA synthesis. Late proteins generally encode viral capsid and envelope proteins, but also include tegument proteins that are able to function immediately upon infection.
Figure 1.
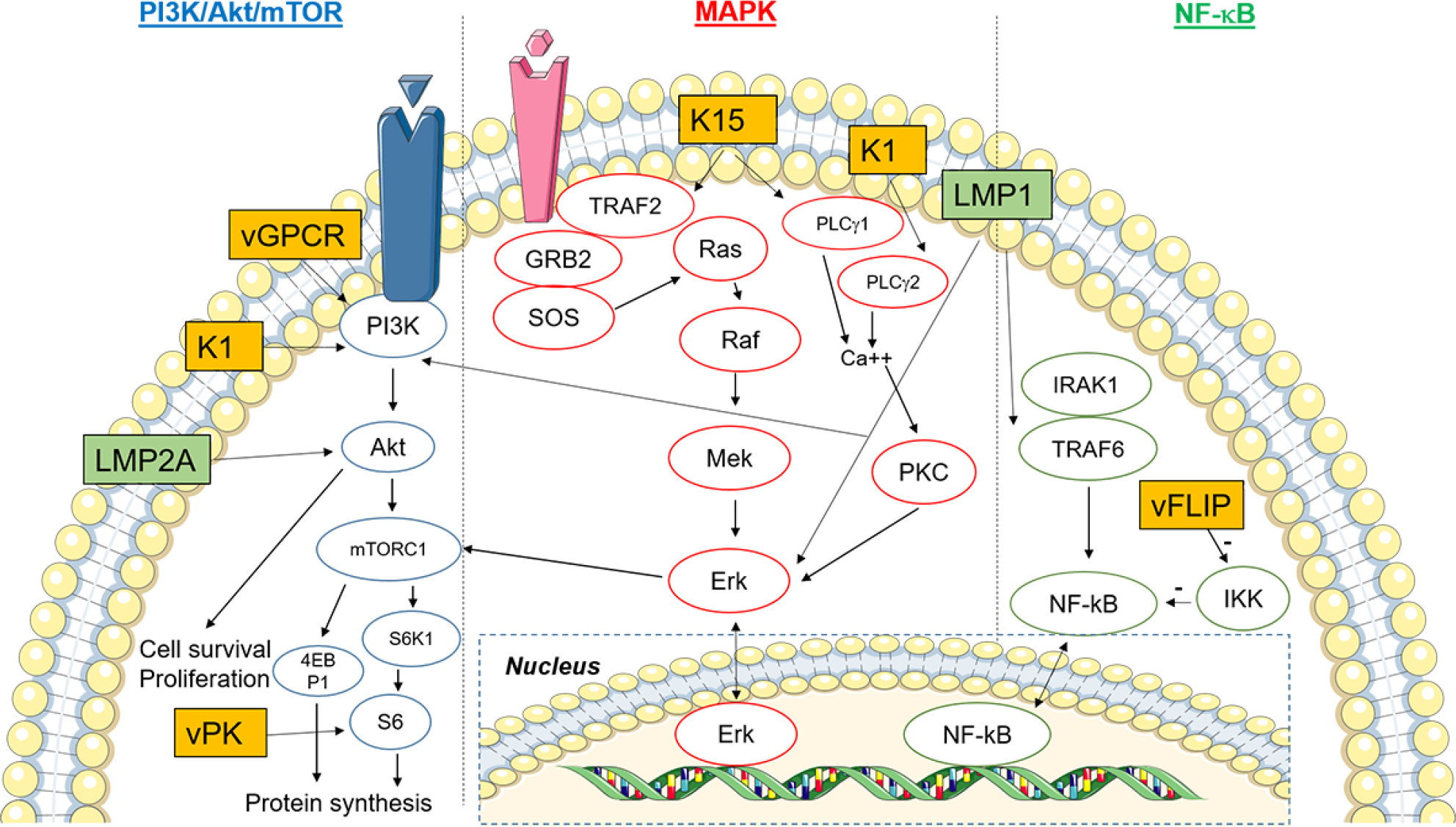
Key signaling pathways modulated by Epstein-Barr virus (EBV) and Kaposi sarcoma-associated virus (KSHV) proteins. The pathways shown include PI3K/Akt/mTOR, MAPK, and NF-ΚB. KSHV oncoproteins (K1, K15, vGPCR, vFLIP, vPK) are in orange boxes, whereas EBV oncoproteins (LMP1, LMP2A) are colored green. Please refer to the main text for detailed description of these pathways. Figure was created using artwork images from https://smart.servier.com/image-set-download/.
Typically, in the immunocompetent host, EBV and KSHV persist in infected B cells for many years without causing noticeable pathology. However, such long-term viral persistence via latency is postulated to contribute to human cancer when the host becomes immunocompromised. However, evidence suggests that certain lytic proteins are also involved through autocrine/paracrine effects that can enhance transformation [21–23].
Viral oncogenic signaling
EBV and KSHV have evolved to hijack multiple cellular signaling pathways involved in tumorigenesis and oncogenesis. Here we will focus on three major pathways modulated by viral proteins: i) phosphatidylinositol-4,5-bisphosphate 3 kinases (PI3K)/protein kinase B (Akt)/mammalian target of rapamycin (mTOR) pathway; ii) mitogen-activated protein kinase (MAPK) pathway; and iii) nuclear factor-κB (NF-κB) pathway (Figure 1). There is crosstalk amongst these pathways, adding to the complexity of the interplay between host cell factors and viral proteins [24–26]. We have decided to select the three pathways above because they are well-known in viral and non-viral oncogenesis and can be used to highlight the roles of EBV and KSHV oncoproteins. In addition to these three major pathways, there is a plethora of other cellular pathways perturbed by both viruses (e.g., Notch, Wnt/β-catenin, JAK/STAT, TGF-β, p53, Toll-like receptors, etc reviewed in [27–30], but they are beyond the scope of this review.
PI3K/Akt/mTOR pathway
There are four classes of PI3K (IA, IB, II, and III) [31]. Class IA and IB PI3Ks are heterodimeric proteins comprised of a regulatory subunit (p85α, p85β, p55α, p55γ and p50α) and a catalytic subunit (p110α, p110β, p110δ and p110γ) [32]. Classes IA and IB catalyze the conversion of phosphatidylinositol-4,5-bisphosphate (PIP2) into phosphatidylinositol-3,4,5-triphosphate (PIP3). This enables pleckstrin homology (PH)-domain containing proteins to localize to the plasma membrane [32]. As a phosphatase, phosphatase and tensin homology (PTEN) reverses the reaction, converting PIP3 back to PIP2 [33]. Loss of PTEN expression or function thus removes the brake on PI3K-mediated signaling. PIP3 recruits phosphoinositide-dependent kinase 1 (PDK1) to the plasma membrane, where PDK1 activates Akt by phosphorylation. By inhibiting tuberous sclerosis complex 2 (TSC2), Akt activates mTOR complex 1 (mTORC1), resulting in induction of protein synthesis [34]. In brief, mTORC1 phosphorylates p70 S6 kinase (S6KB1) and eukaryotic translation initiation factor 4E-binding protein 1 (4EBP1). The phosphorylation of p70 S6 kinase (S6KB1) leads to downstream phosphorylation of the S6 ribosome for protein translation. Phosphorylated 4EBP1 unleashes 4EBP1’s inhibitory effect on eukaryotic initiation factor 4E (eIF4E) and this unlocks the cap-dependent translational machinery [35]. Thus, the PI3K/Akt/mTOR signaling axis activates biogenesis and cellular proliferation. mTORC1 signaling has also been shown to regulate lipid metabolism and autophagy [36]. Other Akt downstream effects also include promoting cell cycle progression via transactivation of MYC and CCND1 genes and repressing apoptosis (by inhibiting pro-apoptotic proteins such as Fas ligands) [37, 38].
MAPK pathway
The MAPK pathway can be physiologically initiated by growth factors, cytokines, and various stress (e.g. heat, oxidation, and radiation) [39]. In this signaling cascade through a series of phosphorylation events, a stimulus activates MAPKK kinase (MAPKKK) [24], which in turn activates a MAPK kinase (MAPKK). The activated MAPKK subsequently phosphorylates and activates a downstream MAPK. The activated MAPK ultimately translocates into the nucleus and activates various transcriptions factors. Through this signaling cascade, the signal is dramatically amplified. The four canonical MAPK families in mammalian systems include extracellular-signal-regulated kinase (ERK) 1/2, ERK5, c-Jun N-terminal kinase (JNK)/stress-activated protein kinase (SAPK), and p38 kinase [40]. For example, in the “classical” MAPK pathway, extracellular ligand (growth factor or cytokine) binds to a receptor (e.g., G protein-coupled receptor (GPCR)), which phosphorylates and activates the MAPKKK Raf. Phosphorylated Raf then phosphorylates and activates the MAPKK MEK1/2, which in turn phosphorylates and activates the MAPK Erk1/2. Activated Erk1/2 then activates the substrate p90 S6 kinase (RSK), which subsequently activates various transcription factors including AP-1 [24]. As mentioned above, RSK also activates S6 and eIF4B that are a part of the mTORC1 signaling. The MAPK signaling events are critical for cell proliferation, cell cycle regulation, migration, and survival [40, 41].
NF-κB pathway
There are two major NF-κB pathways, namely, the canonical/classical and non-canonical/alternative pathways. The transcription factor (p65/p50 heterodimer) in the canonical pathway is inactivated and cytoplasmically sequestered by an inhibitor of NF-κB (composed of KKα/IKKβ/IKKγ (IKKγ is also known as NEMO)). The classical NF-κB pathway can be stimulated by tumor necrosis factor (TNF), IL-1, Toll-like receptor ligands, B cell receptor, or T cell receptor [42]. This is followed by activation of IκB kinase (IKK) complex, which then phosphorylates and degrades IκB via ubiquitination and proteasomal degradation. This subsequently enables the NF-κB heterodimer to translocate into the nucleus, whereby it activates several genes that are important for cell proliferation, angiogenesis, and inflammation [43].
The non-canonical NF-κB pathway can be induced by lymphotoxin, receptor activator of NF-κB ligand (RANKL), CD40 ligand, and B cell activating factor of the TNF family [42]. The NF-κB heterodimeric complex here is RelB/p52 instead of p65/p50. The non-canonical pathway is repressed by p100 in lieu of IκBα. Similar to IκBα in the canonical pathway, proteasomal degradation of p100 frees the NF-κB complex from cytoplasmic sequestration, permitting downstream activation of the pathway [44]. This alternative pathway is important for secondary lymphoid organ establishment and maintenance [43].
Individual KSHV/EBV viral proteins that modulate the above three major signaling pathways are described in subsequent sections of this review.
EBV lytic cycle
In order to enter epithelial cells, the virus binds to αvβ integrins and ephrin A2 receptor on epithelial cells [45, 46]. The viral glycoprotein gp350/gp220 allows EBV to bind CD21 on B cells, and fusion is triggered by a complex composed of gH/gL (gp110/gp85) and gB (gp25) [47, 48]. Next, the viral capsid enters the cytoplasm through endocytosis, followed by transportation along the microtubule to the nuclear membrane [49], where the viral genome is injected into the nucleus through the nuclear pore and viral replication ensues. Viral replication can also occur when EBV-infected cells are reactivated from latency (Figure 2).
Figure 2.
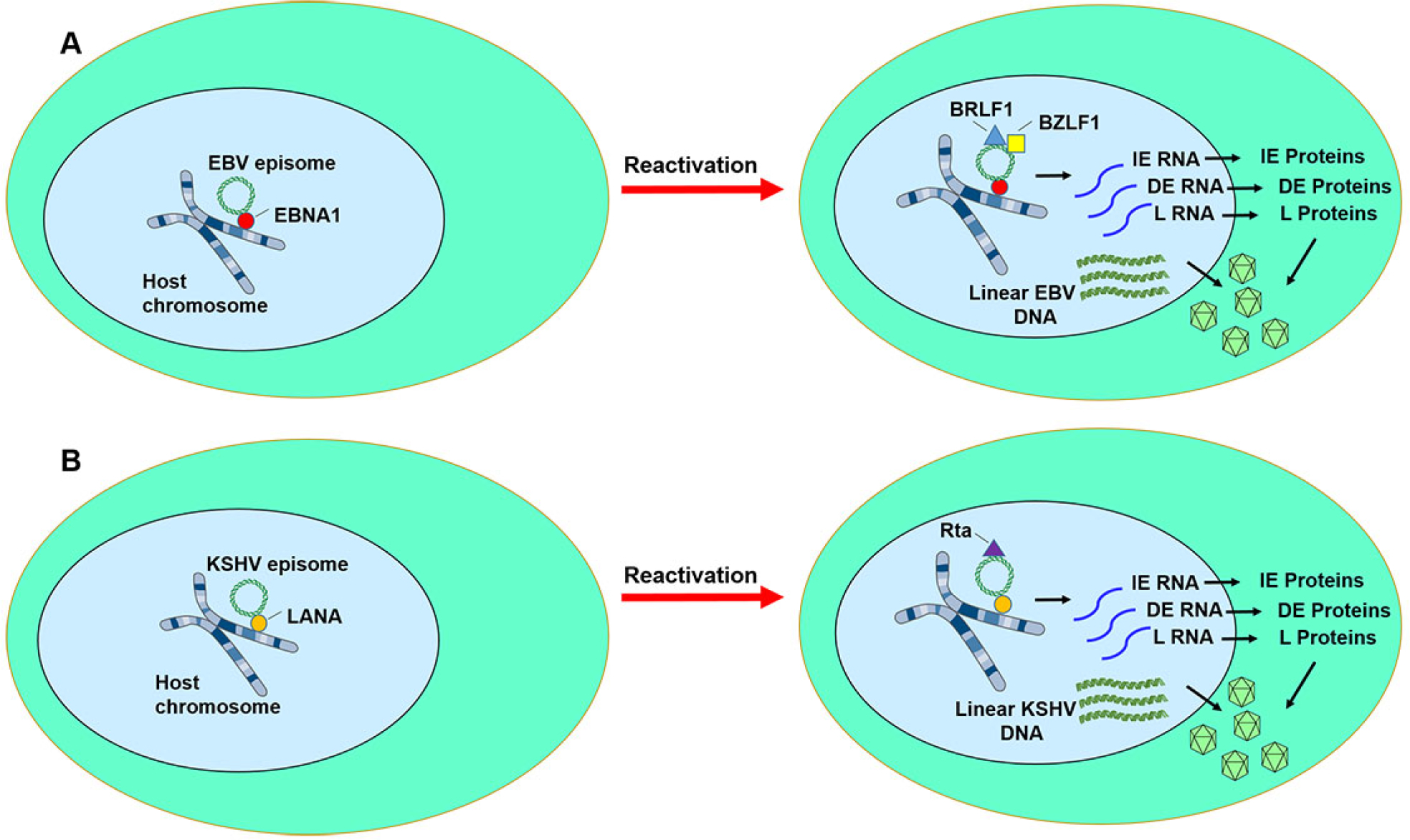
Epstein-Barr virus (EBV) and Kaposi sarcoma-associated virus (KSHV) latency and reactivation. A) During EBV latent infection, very limited numbers of viral gene such as Epstein-Barr nuclear antigen-1 (EBNA1) and latent membrane proteins (LMP1 and LMP2) are expressed. EBNA1 (red circle) tethers the EBV episome (green ring) to the host chromosome. When reactivation is triggered, immediate early genes (e.g., BZLF1 and BRLF1) initiate lytic viral transcription, resulting in the orderly expression of immediate early (IE) genes, delayed early (DE) genes, and late (L) genes. Linear viral DNA molecules are replicated. Viral particles (blue pyramid structures) are produced and then released. B) During KSHV latent infection, very limited number of viral genes such as LANA (orange circle) are expressed. Latency-associated nuclear antigen (LANA) tethers the KSHV episome (green ring) to the host chromosome. When reactivation is triggered, the immediate early gene, RTA/ORF50 initiates lytic viral transcription, resulting in the orderly expression of immediate early (IE) genes, delayed early (DE) genes, and late (L) genes. Viral particles (blue pyramid structures) are produced and then released. Figure was created using artwork images from https://smart.servier.com/image-set-download/.
The immediate early (IE) lytic genes BZLF1 (Zta or ZEBRA) and BRLF1 (Rta) initiate lytic EBV replication. EBV Zta has a preference for the CpG sequences on the methylated viral genome [50]. This results in upregulated expression of ~30 early lytic genes. Linear viral DNA is also replicated. EBV can be reactivated from latency by chemical or biological induction using reagents including 12-O-tetradecanoylphorbol-13-acetate (TPA), 5-aza-deoxycytidine (5-aza), calcium ionophore, sodium butyrate, histone deacetylase inhibitor, anti-immunoglobulin, hypoxia, or TGF-β [51].
Following infection of epithelial cells, EBV can subsequently cross the mucosa and spread into the blood stream to infect primary B cells and memory B cells.
EBV latent cycle (0, I-III)
Latency is established after a brief period of abortive lytic replication [52–55]. Viral latency is classified into 4 types (types 0, I, II, and III) based on the latent genes expressed (Figure 2, Table 1). During latency 0 (exemplified in healthy individuals), EBER-1, EBER-2, and miRNAs are expressed. EBER-1, EBER-2, BARTs, miRNAs, and EBNA1 are expressed during latency I (e.g., Burkitt lymphoma). Latency II (exemplified by nasopharyngeal carcinoma and classic Hodgkin lymphoma) additionally includes expression of LMP1, LMP2A, and LMP2B. In latency III (characterized by EBV-positive large B cell lymphoma and EBV-positive post-transplant lymphoproliferative disorder), EBNA2, EBNA3A, EBNA3B, EBNA3C, and EBNA-leading peptide (LP) are expressed in addition to the genes expressed in latency II. Latency is primarily observed in infected B lymphoblasts [56]. The critical roles of EBV lytic and latent cycles during oncogenesis is nicely described in a review by Münz [57].
Table 1.
EBV cancers and their respective latent states and gene products.
| Healthy individuals | GC or EN NK/T-cell lymphoma | BL | CHL | NPC | EBV+ LBCL or PTLD | |
|---|---|---|---|---|---|---|
| EBNA1 | + | + | + | + | + | |
| LMP1 | + | + | + | |||
| LMP2 | + | + | + | + | ||
| EBER | + | + | + | + | + | + |
| BART miRNA | + | + | + | + | ||
| EBNA2 | + | |||||
| EBNA3A/3B/3C | + | |||||
| Latency state | 0 | I | I | II | I/II | III |
Abbreviations: NPC: nasopharyngeal carcinoma; BL: Burkitt lymphoma; CHL: classic Hodgkin lymphoma; EBV: Epstein Barr virus; EN: extranodal; GC: gastric carcinoma; LBCL: Large B-cell lymphoma; NK: natural killer; NPC: nasopharyngeal carcinoma; PTLD: posttransplant lymphoproliferative disorder
EBV oncogenic viral proteins
Latent membrane protein 1 (LMP1)
LMP1 is frequently detected in nasopharyngeal carcinoma, classic Hodgkin lymphoma, Burkitt lymphoma, HIV+ lymphomas, EBV+ PTLD, and EBV+ gastric carcinoma [58–62]. LMP1 functions as a constitutively active tumor necrosis factor (TNF) receptor CD40. It has been shown to activate the MAPK, PI3K (Akt), c-Jun N-terminal kinase (JNK), epidermal growth factor receptor (EGFR), and NF-κB signaling pathways, which regulate cellular proliferation, cell cycle progression, motility, and anti-apoptosis via BCL2 expression [63] (Figure 1). The carboxyl-terminal activating regions 1 (CTAR1) and CTAR2, have been identified within the cytoplasmic carboxy terminal domain of LMP1 that activates NF-κB: CTAR1 recruits the TNF receptor–associated factors (TRAFs such as TRAF1/2 or TRAF3/5 heterodimers), which in turn activate NF-κB-inducing kinase (NIK) and inhibitor of κB kinase α (IKKα) leading to stimulation of the noncanonical NF-κB pathway, while also activating the canonical pathway [64–66]. CTAR1 also activates PI3K, which in turns activates Akt and glycogen synthase kinase 3β (GSK3β) [67]. CTAR2 recruits TRAF2 and TRAF6 using adapter molecules to activate the canonical NF-κB pathway [68].
LMP1 CTAR2 also activates c-Jun N-terminal kinase pathway by recruiting TRADD and TRAF2 [69, 70]. Furthermore, LMP1 downregulates the cyclin-dependent kinase inhibitors p27Kip1 and p16INK4a, as well as upregulates the inhibitor of differentiation 1 (Id1) and inhibitor of differentiation 3 (Id3), cyclin-dependent kinase 2 (CDK2), and retinoblastoma (Rb) [71–73].
LMP1 has transforming activity in epithelial cells, B cells, and fibroblasts in vitro [65, 74, 75]. Of note, activation of the PI3K-Akt pathway, but not NF-κB, is required for Rat-1 fibroblast transformation [76]. In NPC cells, LMP1 has been shown to inhibit necroptosis through targeting receptor-interacting protein kinase 1 (RIPK1) and RIPK3 ubiquitination [77]. In murine models, LMP1 expression induces B cell lymphoma [62].
Latent membrane protein 2A (LMP2A)
LMP2A maintains viral latency in infected B cells. LMP2A contains immunoreceptor tyrosine-based activation motifs (ITAM) that enable it to function like a B cell receptor (BCR) and activate PI3K/Akt pathway, via activation of the tyrosine kinases Src and Lyn and PY motifs [78–82] (Figure 1). It thus drives B-cell development and provides a survival signal for EBV-infected cells independent of BCR [82, 83]. Interestingly, a high level of LMP2A can block BCR expression and signaling by a dominant-negative effect [84, 85]. It effectively excludes BCR from lipid rafts and directs Lyn and Syk kinases away from the BCR and to the ubiquitin-proteasome degradation pathway [85–88]. LMP2A also enhances production of IL-10 by activating Bruton’s tyrosine kinase (BTK) and signal transducer and activator of transcription 3 (STAT3) [89]. LMP2A interacts with many cellular proteins as highlighted in a review on the LMP2A signalosome [90].
LMP2A expression is frequently detected in NPC, HL, and EBV+ gastric carcinoma [91–93]. In a gastric carcinoma line, LMP2A overexpression can prevent apoptosis induced by TGF-β1 [94]. This appears to be due to up-regulation of survivin expression by LMP2A [95]. LMP2A transgenic mice were characterized by a lack of surface immunoglobulin rearrangement resulting in BCR-negative B-cells, and yet these B-cells developed and survived via LMP2A ITAM signaling without the need for normal BCR signaling [82, 96]. B-cells from the LMP2A transgenic mice were sensitive to apoptosis with specific inhibitors of Ras, PI3K, and Akt, suggesting that LMP2A activates these cellular molecules to enhance B-cell survival [97].
Epstein-Barr virus nuclear antigen (EBNA)1
EBNA1 protein is crucial for latency and replication of the EBV genome. It tethers the circularized EBV episomes via the latent origin oriP to the host chromatin during cell division [98–103]. Binding to host chromosomes is mediated by EBNA1’s RG-rich residues, arginine-rich and glycine rich regions, its association with EBP2 and other proteins, and/or G-quadruplex RNA binding action [100, 104–108]. This enables episomal maintenance in latently infected B cells [109–111]. In addition, EBNA1 can also enhance cellular and viral gene expression by binding to various promoters [112, 113].
EBNA1 is known to exert an indirect effect on the canonical NF-κB signaling pathway by inhibiting IKK phosphorylation and nuclear translocation of p65 [114]. In addition, EBNA1 can interact with USP7 that modulates p53 and Mdm2 by preventing their degradation [115, 116]. EBNA1 can also lead to the disruption of promyelocytic leukemia (PML) nuclear bodies, resulting in destabilization of p53, inhibition of apoptosis, and impaired DNA damage repair [117]. By forming a complex with Sp1/Sp1-like proteins bound to their cis-element at the survivin promoter, EBNA1 up-regulates survivin and prevents apoptosis in EBV-infected B lymphoma cells [118]. EBNA1 also perturbs STAT1 and transforming growth factor-β (TGF-β) signaling [119].
Although some studies suggest a role for EBNA1 in tumor formation in murine models [120, 121], recent studies by Kang et al. [122, 123] did not find similar results in nude mice. It is possible that EBNA1’s main roles are in maintenance of viral genomes and gene expression that are needed for tumorigenesis, but that it does not directly exert an oncogenic effect. Regardless, the consistent expression of EBNA1 in all EBV-associated cancers makes it an attractive potential therapeutic target [124]. There are numerous cellular proteins that interact with EBNA1 that we cannot extensively cover here, and interested readers are encouraged to explore them in [125].
In terms of disease relevance, EBNA1 expression was associated with significantly enhanced CD25 expression in the Hodgkin lymphoma cell line L428 that could translate into increased likelihood for lymphomagenesis in nonobese diabetic-SCID mice [126]. The level of EBNA1 expression correlated with Burkitt lymphoma cells survival by modulating apoptosis [127]. Furthermore, RNA interference/suppression of EBNA1 inhibited proliferation of EBV-positive Burkitt’s lymphoma cells [128]. In NPC, EBNA1 altered protein expression involved in oxidative stress responses and metastasis [129]. EBNA1 can also degrade PML proteins leading to PML nuclear body loss in NPC and EBV+ gastric carcinoma [117, 130]. EBNA1 inhibitors have also been developed [124, 131, 132].
EBNA2 and EBNA leading peptide (EBNA-LP)
EBNA2 is critical and necessary for viral transformation and immortalization of primary B-cells [133–135]. Although it does not directly bind DNA, EBNA2 targets DNA through binding to the C-promoter binding factor 1 (CBF1) that interacts with RBP-Jκ, which also modulates DNA binding by Notch [136–138]. Thus, EBNA2 can functionally mimic dysregulated Notch1 [139, 140].
EBNA2 functions to activate host cell gene expression, including MYC, CD21, CD23 [141–144]. MYC gene transactivation is thought to drive hyperphysiological B cell proliferation [145]. EBNA2 also transactivates viral latent genes via the promoters of LMP1, LMP2A, and LMP2B [146–153]. EBNA2 enhances transcription by recruiting transcription factors and cofactors through the activation domain as well as by association with hSNF5/INI1 and recruitment of the human SWI-SNF complex [154–156]. EBNA2 also upregulates transcription of TET2 and physically binds to TET2 protein to demethylate genes important for B cell transformation [157]. Furthermore, EBNA2 can transcriptionally suppress the expression of immunoglobulin M expression [158]. In Burkitt lymphoma lines with t(8;14) translocation, expression of c-Myc is activated under the immunoglobulin chain locus, suggesting that EBNA2 uses the same mechanism to control both IgM and c-Myc expression [158].
EBNA-LP is transcribed along with EBNA2 and has been shown to cooperate with EBNA2 to cause G0/G1 transition during immortalization and to transactivate cellular genes (e.g., RBP-Jκ, MYC, Pu.1, and EBF1) and viral genes [159–161]. However, EBNA-LP transgenic mice did not show increased tumor development but exhibited dilated cardiomyopathy [162].
EBNA3 proteins (EBNA3A, EBNA3B, EBNA3C)
All three EBNA3 proteins can antagonize the effect of EBNA2 on viral gene transactivation [163–165]. EBNA3A and EBNA3C are important for inhibition of plasmacytic differentiation and induction of B cell transformation, as they have been shown to repress p16INK4A and p14ARF in lymphoblast lines [166]. EBNA3A can induce histone modifications of the chemokine genes, CXCL9 and CXCL10 [167, 168]. As a viral oncoprotein, EBNA3C can cooperate with activated Ras to immortalize and transform primary rodent fibroblasts and disrupt cell cycle checkpoint [169, 170]. EBNA3C has been shown to block p53-mediated apoptosis via different mechanisms including direct binding and inhibition of p53 and induction of Aurora kinase B [171–174]. Unlike EBNA3A and EBNA3C, EBNA3B is thought to be dispensable for B cell transformation in vitro [175, 176]. EBNA3A and EBNA3C can recruit cellular proteins such as chromatin remodeling factors e.g. RBP-Jκ, histone deacetylases, and CtBP leading to deregulated gene expression [177–183]. Additional details of EBNA3 proteins are reviewed in [184–186].
EBV-encoded small RNAs (EBERs)
EBERs (EBER1 and EBER2) are confined to the nucleus and the most abundant viral transcripts during EBV latency (> 1 million copies per infected cell), which allow for very sensitive detection of EBV infection using in situ hybridization on tissue [187–191]. The EBERs modulate cellular proliferation, cell survival, and production of cytokines/autocrine factors [192]. They have been shown to interact with cellular La protein and the ribosomal protein, L22 [193, 194]. EBERs were shown to contribute to IL10-induced growth, malignant phenotypes, and resistance to apoptosis in BL [192, 195]. In addition, EBERs have been shown to induce the expression of insulin-like growth factor 1 (IGF-1) in EBV-positive gastric carcinoma cells, and nasopharyngeal carcinoma-derived cells, IL-9 in EBV-infected T cells, and IL-6 in lymphoblastoid lines [196–199]. EBERs released from EBV-infected patient sera can activate signaling from Toll-like receptor 3 in EBV-transformed lymphocytes and peripheral mononuclear cells, thus activating the type I interferon response [200, 201]. Additional roles of EBERs in EBV infection, oncogenesis, and modulation of immunity are reviewed in [191, 202, 203].
KSHV lytic lifecycle
KSHV is known to infect multiple cell types, including epithelial cells, endothelial cells, and various hematopoietic cells such as B cells, T cells, dendritic cells, and monocytes [204–206]. Multiple cellular receptors have been discovered for KSHV entry. They include the cysteine transporter xCT, ephrin receptor tyrosine kinase A2, DC-SIGN, and integrins [207–211]. KSHV enters the host cells primarily through clathrin- or actin-mediated endocytosis [212]. The envelope glycoproteins gB, gH, and gL of KSHV are necessary for fusion of the viral and cell membranes [213, 214]. KSHV binding to integrin also stimulates focal adhesion kinase (FAK) [215], which activates a host of downstream signaling cascades (including the PI3K/Akt and MAPK, and NF-κB pathways). These events are important for viral entry (e.g., PI3K), viral capsid transition along the microtubule by utilizing dynein motors to the nuclear membrane (e.g., PI3K), cell survival, and viral and host gene expression (e.g., MAPK and NF-κB) [216–220]. After the linear KSHV genomic DNA enters the nucleus through the nuclear core complex, it can be replicated via the lytic pathway or enter the latent state with the formation of viral episomal DNA.
Lytic replication is essential for the release and spread of KSHV to other cells (Figure 2, Table 2). The KSHV IE gene known as replication and transcription activator (RTA) is encoded by open reading frame (ORF)50. RTA alone is necessary and sufficient to initiate lytic replication [221, 222]. Alternatively, KSHV reactivation from the latent state can be triggered by caspase 3-dependent apoptotic stress, in the absence of RTA activity [223, 224]. KSHV can also chemically switch from latency to lytic replication by 12-O-tetradecanoylphorbol-13-acetate (TPA), valproic acid (VPA), and sodium butyrate (NaB) [225, 226]. Examples of oncogenic viral proteins in the lytic cycle include K1, K15, viral interleukin 6 (vIL-6), and viral G protein-coupled receptor (vGPCR).
Table 2.
| KS | PEL | MCD | |
|---|---|---|---|
| LANA (ORF73) | + | + | + |
| K8 | + | + | + |
| K10 | −/+ | + | + |
| K11 | + | + | |
| K15 | + | ||
| ORF59/PF8 | + | + | |
| ORF65 | + | ||
| K2 | + | + | |
| vIL6 | + | + | |
| K12/kaposin | + | + | + |
| vFLIP | + | + | + |
| vCyclin | + | + | + |
| K9/vIRF-1 | + | ||
| K10.5/LANA2 | + | + | |
| K10 | −/+ | + | + |
Katano, H., Sato, Y., Kurata, T., Mori, S. & Sata, T. (2000) Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease, Virology. 269, 335–44.
Chen, D., Gao, Y. & Nicholas, J. (2014) Human herpesvirus 8 interleukin-6 contributes to primary effusion lymphoma cell viability via suppression of proapoptotic cathepsin D, a cointeraction partner of vitamin K epoxide reductase complex subunit 1 variant 2, J Virol. 88, 1025–38.
Abere, B., Mamo, T. M., Hartmann, S., Samarina, N., Hage, E., Ruckert, J., Hotop, S. K., Busche, G. & Schulz, T. F. (2017) The Kaposi’s sarcoma-associated herpesvirus (KSHV) non-structural membrane protein K15 is required for viral lytic replication and may represent a therapeutic target, PLoS Pathog. 13, e1006639.
There is good evidence that viral latent proteins play oncogenic roles, by inducing cellular proliferation, inhibiting apoptosis, and evading immunity. However, KSHV lytic viral genes have also been shown to contribute to oncogenesis, as use of the nucleoside analogue ganciclovir could markedly lower the risk of KS in acquired immunodeficiency syndrome (AIDS) patients [227]. It is postulated that during lytic infection, inflammatory and angiogenic factors are induced, exerting an autocrine/paracrine effect on proliferation.
KSHV latent lifecycle (Figure 2, Table 2)
During KSHV latency, a handful of viral genes are expressed including latency-associated nuclear antigen (LANA), viral cyclin (vCyclin), and viral FADD-like interleukin-1-beta-converting enzyme-inhibitory protein (vFLIP), Kaposin, and microRNAs. In addition, viral interleukin-6 (vIL-6), K1, and K15 are expressed at a low level. LANA is needed to maintain latent replication, by spatially linking the viral episome with host chromatin [228, 229]. LANA can also serve as a transcriptional modulator of multiple viral and cellular genes [230]. LANA also binds to the promoter of RTA and other lytic genes to shut off their expression [230]. Of note, latently infected tumor cells make up the majority of KS and PEL tumors, while MCD displays more lytic gene expression.
KSHV oncogenic proteins
K1
The K1 transmembrane protein is expressed at low levels during latency but at much higher levels during the lytic cycle [231]. Like the B cell receptor and EBV LMP2A, the cytoplasmic tail of K1 contains a functional ITAM for signal transduction [232–235]. Unlikely BCR signaling which is transient, K1 constitutively activates the PI3K (via its p85 regulatory unit)/Akt/mTOR pro-survival pathway [236–238] in the absence of ligand and blocks Fas induction of apoptosis in B cells that is dependent upon heat shock proteins [239, 240] (Figure 1). Like vGPCR, K1 expression stimulates production and release of VEGF. This provides a positive feedback loop to activate PI3K/Akt/mTOR signaling [232–235, 241] via the ITAM signaling domain of K1 [232–235]. To activate MAPK signaling, K1 phosphorylates SH2-containing signaling molecules such as spleen tyrosine kinase (Syk), which signals through the MEK1/2-ERK1/2 pathway and PLCγ2 to mobilize calcium, leading to downstream nuclear localization of NFAT and NFAT binding with AP-1 [235]. More recently K1 has been reported to bind the γ subunit of 5’adenosine monophosphate-activated protein kinase (AMPKγ1) which was important for K1’s ability to enhance cell survival [237]. K1 oncogenicity was demonstrated by its ability to transform rodent fibroblasts [233] and immortalize primary human umbilical vein endothelial cells [241]. K1-expressing transgenic mice developed sarcomatoid tumor and/or lymphoma, thought to be mediated by activation of NF-κB and the B-cell transcription factor, Oct2 [242, 243].
K15
Similar to the K1 protein, K15 is a transmembrane viral protein. K15 activates the mitogen-activated protein kinase (MAPK) pathway, NF-κB pathway, and PLCγ1 through tumor necrosis factor receptor-associated factor 2 (TRAF2)’s interaction with its SH2-binding site [244, 245] (Figure 1). K15 shares signaling activities of both LMP1 and LMP2A [238, 244–246]. Like LMP2A, K15 can block BCR signaling [247]. It can bind to HAX-1 in vitro and in vivo to exert an antiapoptotic effect [248]. K15 has been shown to be important for viral lytic replication and is expressed in KS, PEL, and MCD [247–249].
Latency-associated nuclear antigen (LANA)
ORF73 encodes for LANA, which is crucial for maintaining the KSHV episome as well as promoting latent viral replication [250]. LANA binds to histones and this allows KSHV to tether the viral episomes to the host chromatin [14]. LANA has pleiotropic effects on various important cellular pathways, such as p53 [251], retinoblastoma-E2F [252], mitogen-activated protein kinase (MAPK) [252], c-Myc [253, 254], β-catenin/Wnt [255], and Notch [256] signaling. These various pathways have been implicated in oncogenesis. LANA-expressing transgenic mice developed B-cell hyperplasia reminiscent of MCD [257, 258]. Of note, only a subset of older LANA-expressing transgenic mice developed B cell lymphomas highlighting that although LANA can promote cell survival, it may need additional genetic alterations or other viral/cellular oncoproteins for lymphomagenesis [257].
LANA expression can be detected in KS, PEL, and MCD in vivo [259]. For clinical diagnostic purposes, immunohistochemistry for LANA protein is used to establish KSHV infection in its associated malignancies [250, 260, 261].
Viral cyclin (vCyclin)
vCyclin (ORF72) is a viral homologue of cellular cyclin D [262, 263]. Physiologically, cyclin D forms a complex with cyclin-dependent kinases (CDK)6 and CDK4 to phosphorylate Rb, resulting in the release of the E2F transcription factor. KSHV vCyclin interacts with cyclin-dependent kinase (CDK)6 and, to a lesser extent, other CDKs, to degrade CDK inhibitors, phosphorylate Rb and histone H1, and promote cell cycle progression [264–266]. Unlike cellular cyclin D1, vCyclin/CDK complexes are insensitive to CDK inhibitors and can degrade the CDK inhibitor p27Kip [264, 267]. vCyclin/CDK6 complex can phosphorylate the histone chaperone, nucleophosmin leading to genomic instability [268]. vCyclin is also necessary to overcome replicative senescence in primary human lymphatic endothelial cells [269].
In the absence of p53, lymphomagenesis was noted in vCyclin transgenic mice, implicating p53 loss is important to expand neoplastic clones with vCyclin-induced aneuploidy [270, 271]. In KS, pro-apoptotic vCyclin/CDK6 complex can phosphorylate and inactivate Bcl-2, suggesting that in addition to cell proliferation, modulation of apoptosis in infected host cells may also be important for the development of Kaposi’s sarcoma [266, 272].
Viral FADD-like interleukin-1-beta-converting enzyme-inhibitory protein (vFLIP)/K13
vFLIP/K13 (ORF71) blocks apoptosis induced by death receptors in KS lesions [273, 274] and PEL cells [275]. vFLIP structurally resembles the short form of cellular FLIP by not possessing the caspase-like domain of the long form of cellular FLIP. Mechanistically, vFLIP contains two death-effector domains to interact with the adaptor protein FADD, and this inhibits the recruitment and activation of the protease FLICE by the CD95 death receptor [273]. As a result, vFLIP-expressing cells can be protected from apoptosis induced by CD95 or other death domain-containing receptors [273]. vFLIP can promote cellular transformation through interaction of TRAF2 and TRAF4 that lead to activation of NF-κB that likely confers anti-apoptotic effect and pro-survival benefit [273, 275–277] (Figure 1). This also contributes to the spindle cell morphology and proinflammatory cytokine milieu as seen in KS [278]. In PEL cell lines, vFLIP can lead to constitutive activation of NF-κB and induction of cellular IL-6 expression by persistently phosphorylating and activating IKBα [279, 280]. In line with this, treatment of PEL cells in a murine system with an NF-κB inhibitor induced apoptosis, suppressed tumor growth, and prolonged survival [281]. Furthermore, by interacting with Atg3, vFLIP can prevent cell death from autophagy and this protective effect can be reversed using FLIP-derived short peptides that bind vFLIP and Atg3 [282]. Viral FLIP binds heat shock protein 90 (Hsp90), which is important for signaling and tumorigenicity [283]. Thus, targeting vFLIP could be a potential therapeutic option for KSHV-associated malignancies.
Transgenic mice expressing vFLIP exhibited B cell transdifferentiation and acquired expression of histiocytic/dendritic cell markers. These mice showed hematologic features typical of PEL and MCD [284]. vFLIP expression in endothelial cells of transgenic mice resulted in increased proinflammatory cytokine (e.g., IL6 and IL10) expression as well as aberrantly increased myeloid cells that might support a role in establishing the tumor microenvironment of various KSHV-associated lesions [285].
Viral G protein-coupled receptor (vGPCR)
vGPCR encoded by ORF74 is a chemokine receptor and cellular homologue of interleukin 8 (IL-8) receptors such as CXCR1 and CXCR2 [286–288]. Unlike its cellular homologue, vGPCR is constitutively active and does not respond to ligands [286–288]. As a viral oncoprotein expressed in the lytic cycle, vGPCR has been shown to transform endothelial cells and induce sarcomagenesis by hijacking the PI3K/Akt/mTOR pathway [289–293], MAPK pathway [294, 295], and NF-κB pathway [288, 296, 297] (Figure 1). Specifically, vGPCR constitutively activates p44/p42 MAPK signaling and PI3K/Akt signaling [295]. The signaling activity of vGPCR results in phosphorylation of regulatory tyrosine residues in Shp2, which is required for vGPCR-mediated activation of MEK in the MAPK pathway, NF-κB, and AP-1 [288]. Through MAPK signaling, vGPCR can also stimulate HIF-1α transcription [294]. In addition, vGPCR has also been shown to activate the canonical β-catenin/Wnt pathway [298]. KSHV vGPCR activates Rac1 and VEGF, both important for KS angiogenesis via autocrine/paracrine growth factor secretion [294, 299, 300]. Transgenic expression of vGPCR induced KS-like angioproliferative disease in a mouse model [301–303]. Conversely, siRNA knockdown of vGPCR in a KSHV bacterial artificial chromosome (BAC) cell culture system inhibited angiogenesis and KS-like tumorigenesis [304]. These studies indicate a critical role of vGPCR in the pathobiology of KS.
Viral protein kinase (vPK)
vPK is encoded by ORF36 and its expression can be induced by a hypoxic environment [305, 306]. It is a nuclear protein with serine-threonine kinase activity [305]. Functioning as a cyclin-dependent kinase (CDK), vPK has been shown to phosphorylate retinoblastoma and lamin A/C proteins [307]. In addition, it can phosphorylate mitogen-activated kinases 4 and 7 (MKK4 and MKK7), thereby activating c-Jun N-terminal kinase (JNK) pathway [308]. KSHV vPK was reported to structurally and functionally mimic the cellular protein S6 kinase (S6KB1) [309]. For example, it can phosphorylate the ribosomal S6 protein and eukaryotic initiation factor 4E (eIF4E) downstream of the PI3K/Akt/mTOR pathway, resulting in enhanced global protein synthesis, anchorage-independent cellular proliferation, as well as endothelial tubule formation [309] (Figure 1). Transgenic vPK mice were prone to developing lymphoproliferative disorder and lymphoma of B cell origin [310].
Clinical Diseases associated with EBV and KSHV
Clinicopathologic presentation of human neoplastic diseases associated with EBV
Burkitt lymphoma
Burkitt lymphoma (BL) is a highly aggressive B cell lymphoma that tends to involve extranodal sites and patients can present with tumor lysis [311]. While highly proliferative, with a doubling time of about a day, BL is frequently curable due to its high sensitivity to chemotherapy. Intensive chemotherapy confers long-term overall survival in the vast majority of cases. EBV can be variably detected in the three clinical/epidemiologic BL variants (more than 95% in endemic BL, 20–30% in sporadic BL, and 30% in immunodeficiency-associated BL). EBV exhibits a latency I program in BL, expressing EBNA1, LMP2, EBER, and BART miRNA. Endemic BL is most common in children (male-to-female ratio of 2:1) in equatorial Africa and Papua New Guinea [3, 312, 313]. In addition to EBV, it is associated with polymicrobial infections with malaria and arboviruses [314–316]). Endemic BL involves facial bones (e.g., mandible and orbit) in more than 50% of cases. Sporadic BL has a low incidence and accounts for 1–2% of tumors in western countries. It tends to involve the abdomen but may also destroy facial structures. Immunodeficiency-associated BL is most common in the setting of human immunodeficiency virus (HIV) infection and often presents in the lymph nodes and bone marrow. Its incidence has decreased since the introduction of highly active antiretroviral therapy (HAART) for HIV/AIDS[317].
Under the microscope, BL shows a diffuse proliferation of intermediate-sized lymphoid cells with round to mildly irregular nuclear contours, few nucleoli, deeply basophilic and often vacuolated and “squared off” cytoplasm. Scattered tingible-body macrophages are often noted in the background, imparting a “starry-sky” pattern (Figure 3, A–B). There tend to be many mitotic figures and apoptotic debris. The neoplastic cells express B cell antigens, such as CD20, PAX5, and CD79a. They demonstrate a germinal center phenotype (CD10 and BCL6 positive) and lack BCL2 [318, 319] (Figure 3, C–D). By definition, BL expresses c-Myc protein, which correlates with the rearrangement of the MYC oncogene at t(8;14), t(2;8), or t(8;22), and has a very high Ki-67 proliferative index by Ki-67 (virtually 100%) [319]. Next-generation sequencing frequently reveals mutations in TCF3 (that modulates germinal center gene expression and B cell receptor signaling via the PI3K pathway) or its negative regulator ID3 [320–322].
Figure 3.
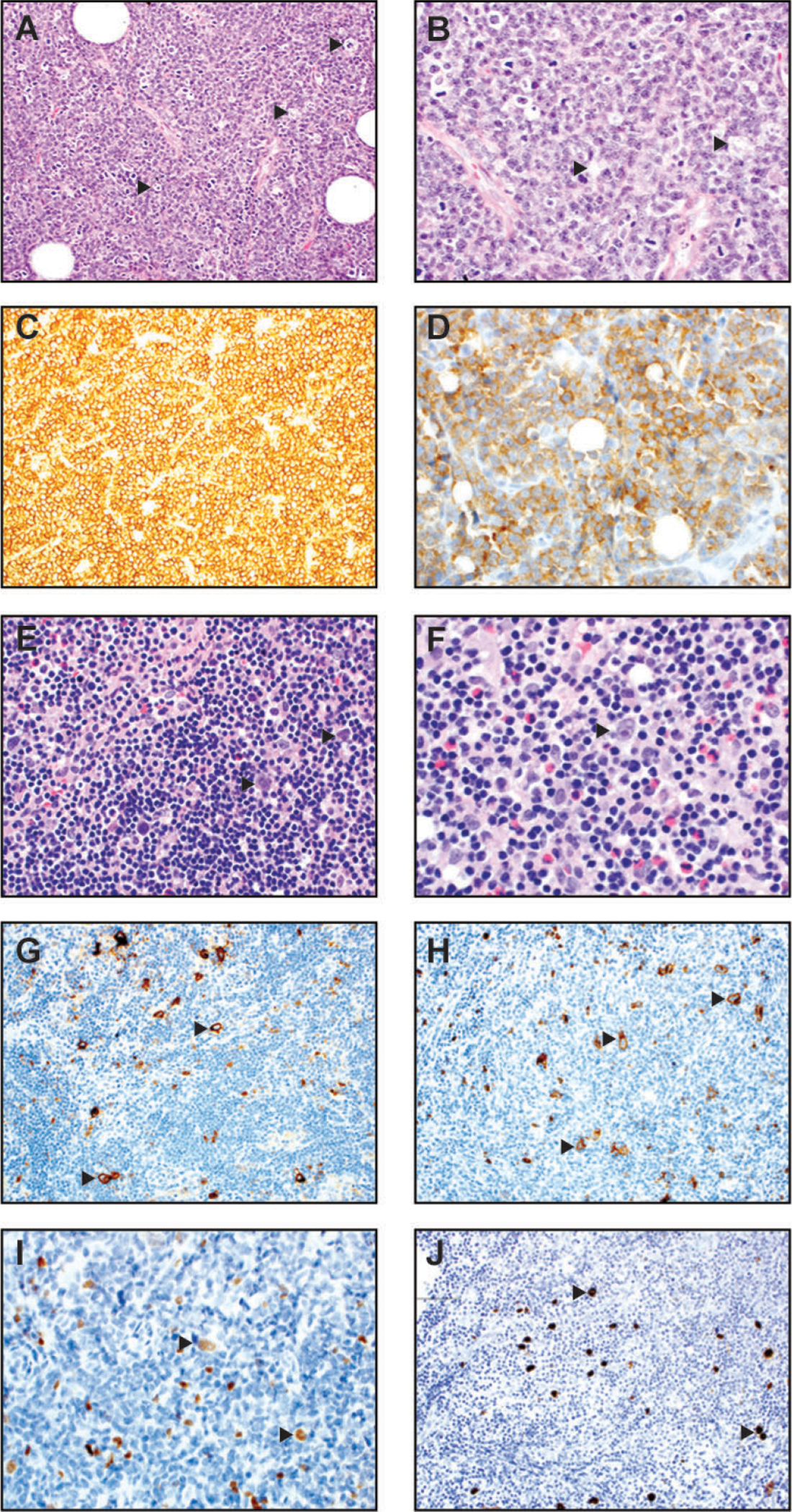
Burkitt lymphoma (A-D) and classic Hodgkin lymphoma, mixed cellular type (E-J). (A) At medium power of H&E (200x), a characteristic starry-sky pattern is present. The “stars” (arrowheads) are represented by tingible-body macrophages and the “sky” is composed of neoplastic lymphoid cells. (B) At high magnification of H&E slide (400x), the neoplastic cells are intermediate-sized, with slightly irregular nuclear contours, few nucleoli, and basophilic cytoplasm. Arrowheads mark tingible-body macrophages. Many mitotic figures and apoptotic debris are also present. (C) The neoplastic cells are strongly positive for the B cell marker CD20 (200x). (D) They are strongly positive for CD10 (400x) and BCL6 (not shown) consistent with a germinal center phenotype. (E) In this lymph node, the high-power magnification of H&E (400x) shows binucleate Reed-Sternberg (RS) cells (highlighted by arrowheads) and mononucleate Hodgkin (H) cells within a polymorphous background composed of small lymphocytes, histiocytes, eosinophils, and plasma cells. (F) A higher power magnification of H&E (600x) shows similar morphologic features, with an arrow pointing to a binucleate Reed-Sternberg H&E (600x). (G) Immunohistochemistry for CD30 (200x) highlights scattered RS and H cells (arrowheads). (H) Immunohistochemistry for CD15 (200x) highlights scattered RS and H cells (arrowheads). (I) The neoplastic RS and H cells (arrowheads) demonstrate variably weak positivity for PAX-5 immunostain. (J) The neoplastic RS and H cells (arrowheads) in this case are positive for EBV-encoded small RNA by in situ hybridization (400x).
Classic Hodgkin lymphoma
Classic Hodgkin lymphoma (CHL) accounts for 90% of Hodgkin lymphomas. It exhibits a biphasic age distribution with the first peak at 15–35 years and second peak in older adult age. CHL tends to present with localized lymphadenopathy. Cervical lymph nodes are the most common affected (~75% of cases). Constitutional B-symptoms are noted in ~40% of patients.
In CHL, latency II is observed, with EBV expressing EBNA1, LMP1, LMP2, and EBER. The neoplastic Hodgkin/Reed-Sternberg (HRS) cells tend to be the minor population that is admixed in a background of inflammatory cells. HRS cells appear to have arisen from late germinal center or early postgerminal center B cells. These cells harbor crippling immunoglobulin genes rearrangements and would normally die, but they are rescued by NF-κB activation through LMP1 and LMP2 [83, 323, 324]. Furthermore, LMP1 induces Bmi-1 expression via NF-κB to provide a survival advantage for CHL [325].
Based on the inflammatory and fibrotic background, CHL has 4 different histologic subtypes: nodular sclerosis (NS), mixed cellularity (MC), lymphocyte-rich (LR), and lymphocyte-depleted (LD). EBV is detected more frequently in the mixed cellularity and lymphocyte-depleted subtypes than the nodular sclerosis and lymphocyte-rich subtypes [4]. EBV infection is implicated in blocking apoptosis in these cells. Hodgkin cells contain a single large nucleus with vesicular chromatin, a single large eosinophilic nucleolus, and abundant cytoplasm. Reed-Sternberg cells are binucleate/multinucleate with otherwise similar cytomorphology.
NS CHL is the most common CHL subtype in the United States and developed countries and accounts for 70% of cases. It is grossly and histologically characterized by thick fibrous bands surrounding nodules composed of inflammatory cells and neoplastic HRS cells. The inflammatory milieu frequently comprises eosinophils, neutrophils, lymphocytes, plasma cells, and histiocytes. EBV can be detected in neoplastic HRS cells in 10–25% of cases. Mixed cellularity (MC) subtype is the second most common subtype of CHL, accounting for 20–25% of cases. In patients with HIV, the MC subtype comprises the majority of all CHL subtypes, especially as CD4 counts decrease [326]. The neoplastic cells are often positive for EBV (75% of cases) (Figure 3, E–J). It is also characterized by a polymorphous background, but prominent fibrous bands of the NS subtype must be absent. Lymphocyte-depleted (LD) subtype is the rarest subtype of CHL and tends to present at older age and late stage [327]. It has an abundance of HRS cells. Similar to the MC subtype, EBV is frequently present in HRS cells (~75% of cases) [327]. In the LR subtype of CHL, EBV is detected more frequently than the NS subtype but less commonly than the MC and LD subtypes. Regardless of histologic subtype, the HRS cells express strong diffuse CD30 and dim PAX5 in virtually all cases and CD15 in most cases [328, 329] (Figure 3, E–J). However, HRS cells have a downregulated B cell program and lack most other B cell markers, such as CD79a, OCT2, and Bob.1 that is thought be related to the reprogramming effect of LMP1 [330]. CD20 expression may be absent or present with variable intensities [328, 329, 331].
EBV-positive diffuse large B-cell lymphoma
EBV-positive diffuse large B cell lymphoma (DLBCL), not otherwise specified (NOS) was previously termed “EBV-positive diffuse large B cell lymphoma (DLBCL) of the elderly” by the World Health Organization (WHO) Classification of Tumours of Haematopoietic and Lymphoid Tissues [332–335]. It is thought to arise due to immunosenescence. EBV-positive DLBCL, NOS can present in lymph nodes or extranodally, such as the gastrointestinal tract or lungs [336]). It is clinically aggressive and has a poor prognosis compared to EBV-negative DLBCL [336–338], although younger ages confer much better prognosis [333]. The prevalence of EBV in DLBCL varies with age from 6.7% for individuals younger than 50 years old [334] to 20–30% in patients more than 90 years old [339].
By definition, this diagnostic entity is a large B cell lymphoma with EBV expression (Figure 4A). EBV-positive DLBCL exemplifies latency III, where EBNA1, LMP1, LMP2, EBER, EBNA2, and EBNA3A/3B/3C are expressed. It may contain some HRS-like cells or show histopathologic resemblance to PTLD, despite the fact that clinically there should be no history of immunosuppression. By immunohistochemistry, the lymphoma cells express pan-B cell markers such as CD20, PAX5, and CD79a (Figure 4, B–D). They show non-germinal center phenotype (e.g., CD10 negative, BCL6 positive, and MUM1) [333, 340, 341]. They can be CD30-positive but CD15 is usually negative. In situ hybridization for EBV is positive in numerous lymphoma cells.
Figure 4.
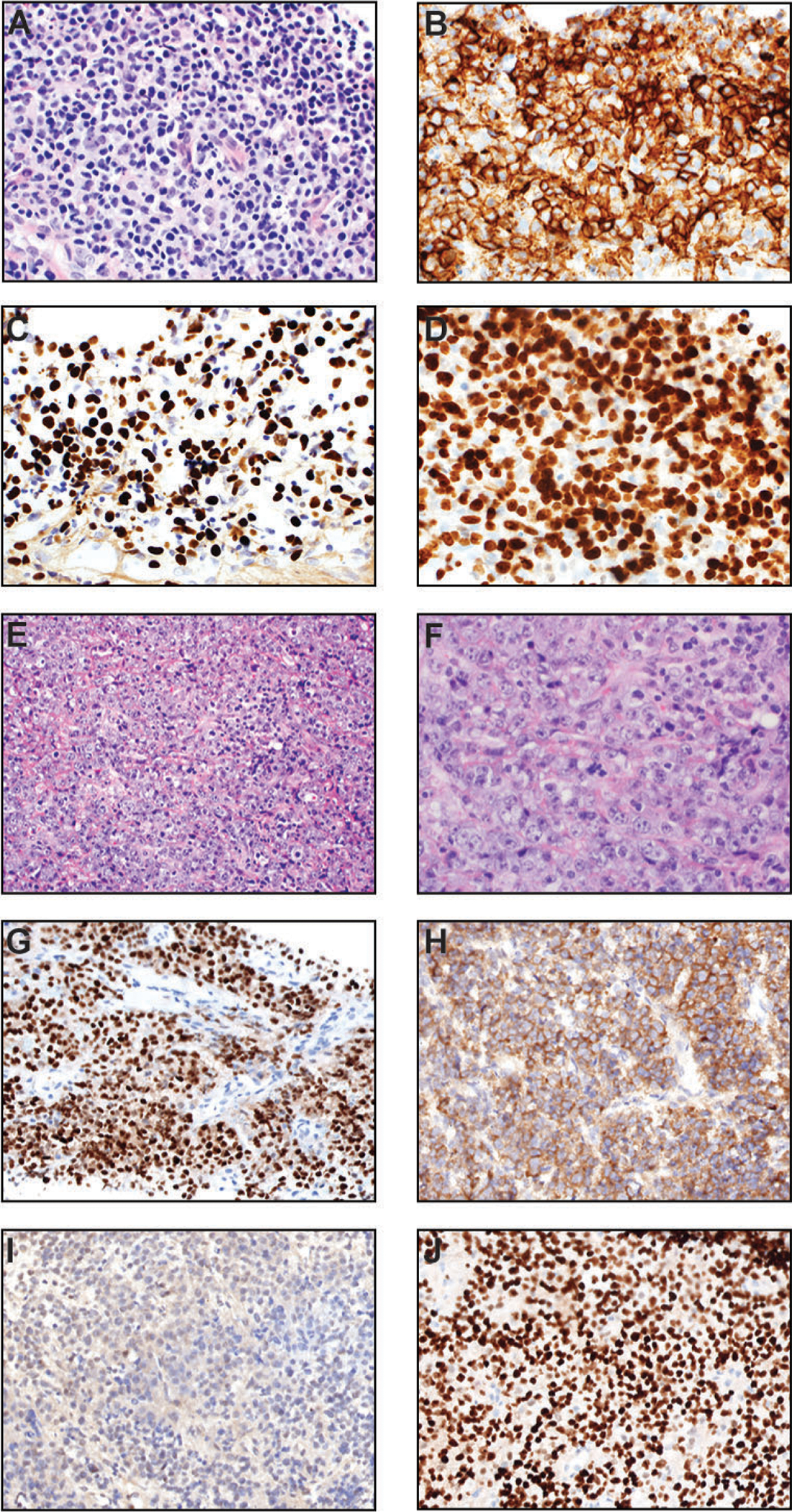
EBV-positive diffuse large B cell lymphoma, not otherwise specified (NOS) (A-D) and plasmablastic lymphoma (E-J). (A) H&E section (400x) of this case shows variable sizes, but there is a significant number of large cells. (B) CD20 immunostain (400x) highlights diffuse proliferation of neoplastic cells. (C) These lymphomatous cells are positive for EBV-encoded small RNA (400x). (D) Ki-67 immunostain (400x) exhibits a very high proliferative index. (E) At this medium power of H&E (200x), the plasmablastic lymphoma shows a diffuse pattern with scattered tingible-body macrophages. (F) At this higher power magnification of H&E (400x), the lymphomatous cells show pleomorphism with vesicular chromatin. Some nuclei contain single prominent nucleoli (immunoblastic morphology). Mitotic figures and karyorrhectic debris are frequently present. (G) In situ hybridization (ISH) for EBV-encoded small RNA (400x) shows positivity in essentially all lymphoma cells. (H) Kappa ISH (400x) in this case demonstrates that the tumor cells express the immunoglobulin kappa light chain. (I) Lambda ISH (400x) in this same case is negative in lymphoma cells, confirming kappa light chain restriction. (J) The PBL cells typically show diffuse and strong nuclear expression of MUM1/IRF-4 (400x). In this case, CD138 (syndecan-1) is also diffusely positive (not shown) to support plasmacytic differentiation.
Another type of DLBCL associated with EBV is termed DLBCL associated with chronic inflammation. The transformation and proliferation of EBV-positive cells are thought be stimulated by cytokine-mediated local immunodeficiency as a result of prolonged chronic suppuration/inflammation within a restricted anatomic space. The prototypic type of this aggressive lymphoma is pyothorax-associated lymphoma (PAL), a heavily male predominant disease that has been most frequently reported in Japan [6, 342]. Other body cavities can be involved by DLBCL associated with chronic inflammation. The morphological and immunophenotypic features of DLBCL associated with chronic inflammation are similar to those of EBV-positive DLBCL.
Plasmablastic lymphoma
Plasmablastic lymphoma (PBL) is a highly aggressive large B cell type lymphoma with plasmacytic differentiation. It arises in the setting of immunodeficiency. It most frequently occurs in HIV/AIDS individuals but is also present in other immunosuppressive states including autoimmune therapy, transplant therapy-related immune suppression, and immune senescence [7, 343, 344]. The prognosis is poor with a median survival of less than 1 year [345, 346]. EBV is quite frequently detected in the neoplastic cells. Clinically, PBL commonly presents as an extranodal mass, with the oral cavity/head and neck region being the most common site (44% of PBL), followed by the gastrointestinal tract mucosa (14% of PBL). [347]. Nodal involvement is much less common (7% of all PBL) but is the most common site of PBL presentation in the post-transplant setting [345, 348]. Of note, a small subset of cases has been reported in immunocompetent individuals [347]. Latency I is typically noted in PBL with EBV expressing EBNA1 and EBER; however, latency III can be seen in those patients with HIV infection or posttransplant PBL [345]. PBL is treated aggressively with CHOP (cyclophosphamide, doxorubicin, vincristine, prednisone) chemotherapy regimen and/or radiation [345]. In an AIDS-associated PBL cell line, IL-6 dependence on cell survival and proliferation was observed, with apoptosis induced by an mTOR inhibitor [349].
On microscopic examination, PBL has two morphologic variants: 1) “monomorphic” variant composed predominantly of immunoblasts with prominent single nucleoli and minimal to no plasma cell differentiation and 2) “plasmacytic” variant with marked plasmacytic differentiation. PBL is a very aggressive lymphoma, with high mitotic activity and a proliferative index measured by Ki-67 labeling is >90%, and karyorrhexis is often noted (Figure 4, E–F). Numerous tingible body macrophages with a starry-sky pattern are quite common. The large neoplastic cells have a plasma cell immunophenotype. They are positive for CD138, MUM1, CD38, BLIMP-1, and CD79a (weakly positive in ~50% of cases) [347, 350] (Figure 4, G–J). In 75% of the cases, in situ hybridization for EBER is positive, whereas LMP1 antigen is negative (EBV latency type 1), although rare cases do express LMP1 (EBV latency type 3) typically in the HIV-positive or post-transplant settings [345]. Unlike “HHV-8/KSHV positive large B-cell lymphoma”, KSHV is negative in all cases of PBL [347, 351]. PBL is typically negative or very weak for the B cell antigens CD20, CD22, and PAX5. These features help distinguish PBL from diffuse large B cell lymphoma, which should express several markers of B-cell lineage. Other markers that are frequently expressed in PBL cells are c-Myc (which is related to MYC gene rearrangement), CD30, and epithelial membrane antigen (EMA).
Nasopharyngeal carcinoma
Nasopharyngeal carcinoma (NPC) has a high prevalence in southeast Asia and the southern part of China [352]. It is most common in the 4th to 6th decade with a male predominance (male-to-female ratio of ~ 2–3:1). NPC tends to metastasize to locoregional lymph nodes [353]. The 5-year overall survival for stage IV is about 75%. It is a squamous cell carcinoma arising in the nasopharynx and has a very strong association with EBV infection (>95%) with an increasing incidence in the United States [2, 354]. Other risk factors include dietary intake of nitrosamine in fermented salt-preserved food, cigarette smoking, and exposure to radiation or chemicals such as formaldehyde in the occupational setting [355, 356]. Prognostic factors include older age, male gender, cranial nerve involvement, metastatic disease, high serum lactate dehydrogenase prior to therapy, certain HLA types, EGFR overexpression, and high neutrophil-to-lymphocyte ratio. NPC is clinically treated with radiation and/or chemotherapy [357]. In NPC, EBV exhibits a latency II program, expressing EBNA1, LMP1, LMP2, EBER, BART miRNA, but LMP1 protein expression can be absent in some cases (likely related to the BART miRNA interaction with the nonterminal region of LMP1) [358–360].
Interestingly, expression of LMP1 can promote epithelial–mesenchymal transition via its positive effect on Snail that suppresses E-cadherin expression and may predict metastasis of NPC [361]. As a potential immunotherapy for NPC, EBV-specific cytotoxic T cell (CTL) lines can be generated from NPC patients, which showed promising safety and antitumor activity with good clinical responses [362].
Microscopically, syncytial neoplastic cells in nests, sheet, or dispersed tumor cells with the morphology of keratinizing (WHO type I) or nonkeratinizing (WHO type II) squamous cell carcinoma are present (Figure 5A). The nonkeratinizing type is strongly associated with EBV. The neoplastic cells have large nuclei, vesicular chromatin, and prominent nucleoli. Mitotic figures are present. They are often accompanied by reactive lymphoplasmacytic infiltrate (Figure 5A). The neoplastic cells show strong reactivity to squamous markers, including cytokeratin (CK) 5/6, p63, and p40 [363] (Figure 5, B–D). Markers for epithelial differentiation/carcinoma, including AE1/3 and CAM5.2, are also positive.
Figure 5.
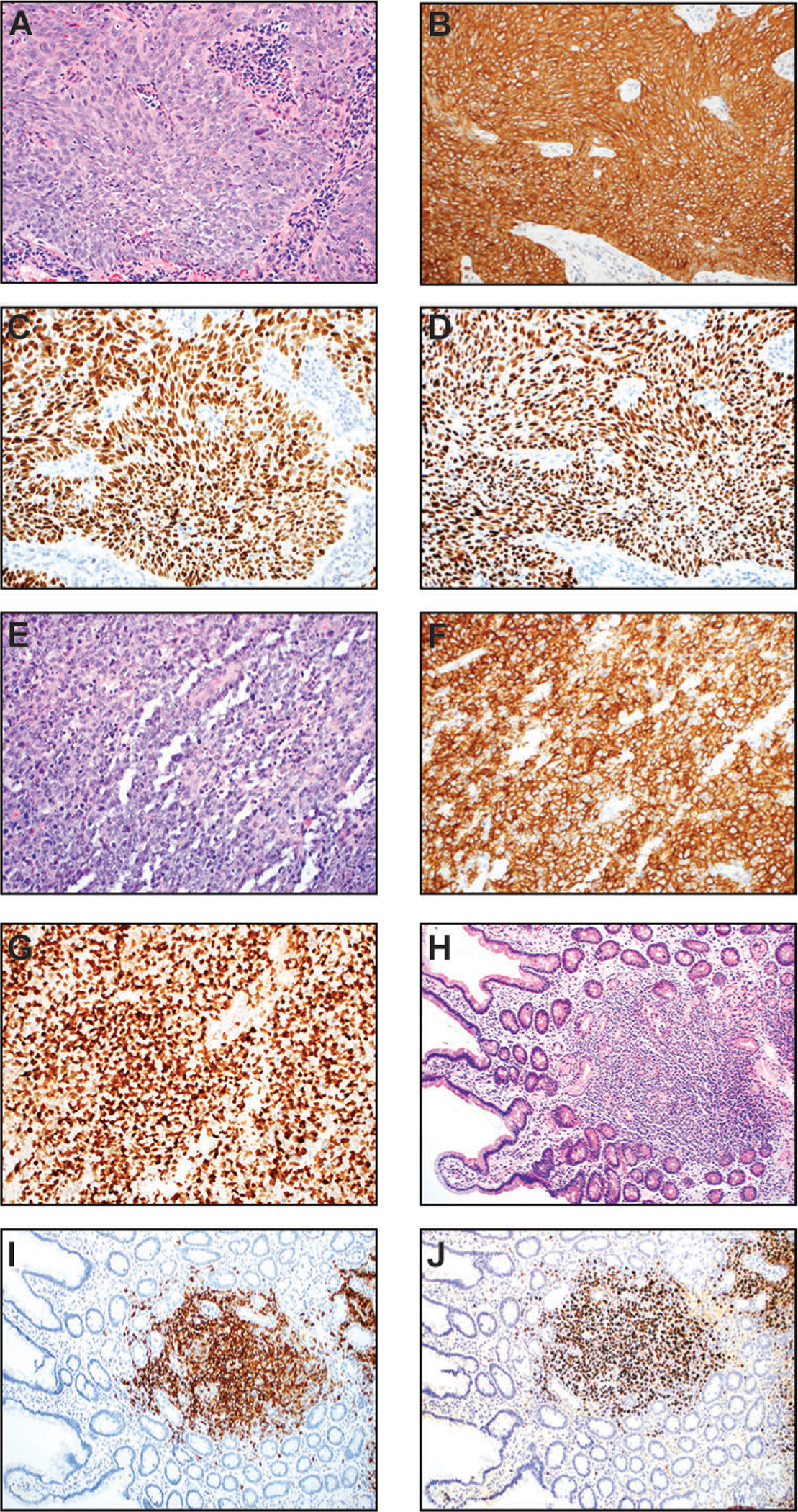
Nasopharyngeal carcinoma (NPC) (A-D) and posttransplant lymphoproliferative disorder (PTLD) (E-J). (A) In this medium power H&E image (200x), the NPC grows in syncytial nests and show a spindled cell morphology. The neoplastic cells are large with oval nuclei, vesicular chromatin, and prominent nucleoli. (B) CK5/6 immunostain (200x) is diffusely and strongly positive in tumor cells, supporting squamous differentiation. (C) EBV-encoded small RNA (200x) is virtually positive in all tumor nuclei. (D) The tumor nuclei are also diffusely and strongly positive for p63, another squamous cell marker (200x). (E-G): Monomorphic PTLD (diffuse large B-cell lymphoma type) in the central nervous system of a patient with prior renal transplant (15 years ago). (E) H&E (200x) shows a diffuse infiltrate of atypical large lymphoid cells with vesicular chromatin. Abundant apoptotic debris and mitotic figures are present. (F) CD20 immunostain (200x) confirms B-cell origin and large cell sizes. (G) Ki-67 (200x) immunohistochemistry highlights numerous neoplastic lymphoid cells with a very high proliferation index. (H-J) Duodenal polymorphic PTLD in a patient who had multiple stem cell transplants. (H) H&E section at a low power magnification (100x) shows mucosal involvement by a mixed hematolymphoid infiltrate. (I) CD20 (100x) demonstrates that the majority of cells are B-cells with variable sizes. (J) In situ hybridization for EBV-encoded small RNA (100x) highlights numerous EBV-positive lymphoid cells.
NPC is characterized by latent expression of EBNA1, LMP1, LMP2, EBER, and BART miRNA. In situ hybridization for EBER is very sensitive for highlighting the neoplastic cells (Figure 5C) but is negative in the background lymphocytes and plasma cells [364, 365].
Post-transplant lymphoproliferative disorder
EBV-positive post-transplant lymphoproliferative disorder (PTLD) exemplifies latency III, with expression of EBNA1, EBNA2, EBNA3A/3B/3C, LMP1, LMP2, and EBER. PTLD is a lymphoid proliferation that arises from immunosuppression in the setting of solid organs or hematopoietic stem cell (HSC) transplantation. It more commonly occurs within the first year after transplantation. Of note, solid organ transplantation (host-derived PTLD) has a higher risk for PTLD than HSC transplantation (donor-derived PTLD). The risk factors include negative EBV serostatus in the transplant recipients, type of allografts (e.g., incidence rate of ~20% in intestinal organ recipients versus 2% in renal organ recipients), pediatric population, and intensity/type of immunosuppressive agents [366]. Many cases (>90%) are serologically EBV-positive. Interestingly, EBV-negative cases tend to occur later, ~ 4–5 years post-transplant, and have a worse prognosis [367, 368]. Some EBV-negative PTLD cases were shown to be KSHV associated [369–371]. The clinical presentation and symptomatology are variable and include constitutional symptoms (such as fatigue, fever, and weight loss) and lymphadenopathy, likely related to involvement site (e.g., nodal vs. extranodal) and PTLD type.
Pathologically, PTLD is quite heterogeneous and includes 4 major categories per the current WHO classification. They include non-destructive PTLD and destructive PTLD (further subclassified into polymorphic, monomorphic, and classic Hodgkin lymphoma PTLD). Non-destructive PTLD has preserved tissue architecture and includes plasmacytic hyperplasia, infectious mononucleosis, and florid follicular hyperplasia PTLD. Non-destructive PTLD generally responds well to attenuation or cessation of immunosuppressives.
By definition, polymorphic PTLD does not fulfill the criteria for any lymphoma or plasma cell neoplasm. Instead, it displays a full range of B cell maturation, from immunoblasts to plasma cells (Figure 5, H–J). The atypical cells are predominantly small to intermediate-sized lymphoid cells, but occasional larger cells and HRS-like cells may be present. Monomorphic PTLD consists of transformed cells typically at one stage of maturation and is classified according to the WHO-defined lymphoma or plasma cell neoplasm it most recapitulates. The most common form is diffuse large B-cell lymphoma (DLBCL) (Figure 5, E–G), which is followed by NK/T cell lymphoma, plasma cell neoplasm, and classic Hodgkin lymphoma. Monomorphic DLBCL type can have plasmacytic differentiation and HRS-like cells that impart some polymorphic appearance; however, the predominant cells should be transformed large cells. For monomorphic B cell PTLD, the atypical lymphoid cells are positive for the B cell markers (such as CD20, PAX5, and CD79a) and show frequent expression of CD30 (Figure 5, E–G). EBER stain is very useful to detect EBV status in tissue for clinicopathologic assessment of PTLD (Figure 5J). In EBV-positive cases, PTLD tends to show non-germinal center phenotype. By contrast, EBV-negative cases usually have a germinal center phenotype. Almost all cases of destructive PTLD have clonal immunoglobulin gene rearrangement, which is usually not detected in non-destructive PTLD. Polymorphic PTLD tends to be driven largely by EBV, whereas monomorphic PTLD may have secondary driver genetic alterations [372–375]. Most polymorphic PTLD and a smaller subset of monomorphic PTLD may regress with reduction of immunosuppressive regimen. With more localized presentation, surgery and/or radiation therapy may be given to the patients. For more persistent/refractory cases, anti-CD20 therapy (rituximab) or R-CHOP (rituximab-CHOP) can be used. It is unclear why a subset of PTLD (20–40%) are EBV negative. It may be related to technical difficulties, other viral (e.g., KSHV) or antigenic induction of PTLD, or loss of EBV after transformation (“hit-and-run” hypothesis) [368, 370, 371, 376].
Extranodal natural killer (NK)/T cell lymphoma, nasal type
Like CHL, latency II is observed in extranodal NK/T cell lymphoma, nasal type. Expression of EBNA1, LMP1 (variable), LMP2, and EBER is often observed. Extranodal NK/T cell lymphoma, nasal type is the most common lymphoma in the sinonasal tract [377]. It is most frequently seen in Asia, but also common in Mexico and South American countries [377, 378]. There is a male predominance. The typical presentation is a low-stage locally destructive lesion in the nasal cavity involving maxillary sinuses and palate of older patients. EBV-encoded small RNA is detected in virtually all cases [378]. Interestingly, a low viral titer in serum or tissue is associated with a more favorable prognosis. Survival is approximately 25–50% at 5 years.
Pathologically, the neoplastic cells display variable sizes with vesicular chromatin and prominent nucleoli. Increased mitotic figures and angiocentricity or angioinvasion growth patterns are often present (Figure 6, A–C). Increased expression of cell adhesion molecules and chemokine receptors has been observed and correlated with LMP1 expression, highlighting its putative role in angiogenesis [379]. There is also a polymorphic inflammatory infiltrate and necrosis [378, 380]. In situ hybridization staining for EBV-encoded small RNA can help highlight the neoplastic NK/T cells (Figure 6D). The neoplastic cells are often diffusely positive for CD56 and the cytotoxic markers such as TIA-1, granzyme B, and perforin [378, 380] (Figure 6, E–H).
Figure 6.
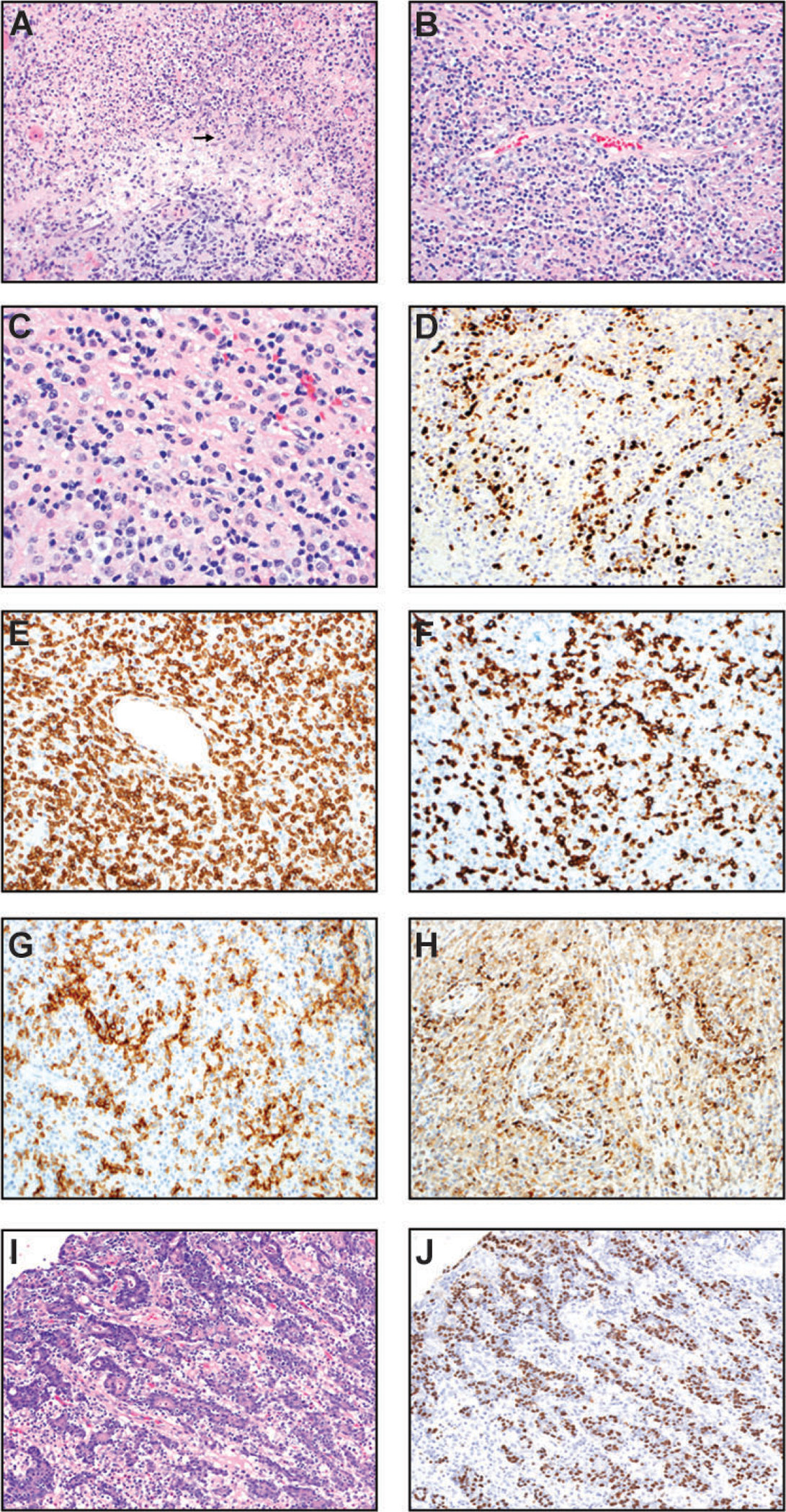
Extranodal natural killer (NK)/T cell lymphoma, nasal type (A-H) and gastric carcinoma associated with EBV (I-J). (A & B) H&E sections (200x) show angiodestruction by invading lymphoma cells with associated necrosis and karyorrhectic debris (arrow in Panel A). (C) In this high-power H&E image (400x), most lymphoma cells are small to intermediate-sized with irregular nuclear contours/folds. Many cells possess moderate amounts of clear/vacuolated cytoplasm. (D) In situ hybridization for EBV-encoded RNA (200x) is positive in the nuclei of most lymphoma cells. (E) CD3 (200x) demonstrates that the majority of the lymphoid infiltrate is T cells. (F) CD8 immunostain is positive in the neoplastic T cells with angiocentric pattern (200x). (G) CD56 immunostain highlights the angiocentric lymphoma cells (200x). (H) TIA-1 immunostain highlights the angiocentric lymphoma cells. (I-J) Gastric carcinoma associated with EBV: Gastric carcinoma with lymphoid stroma. (I) H&E (200x) slide shows epithelioid tumor cells in fused glands and cords. There are intraepithelial lymphocytes as well as mature lymphocytes scattered in the background. (J) In situ hybridization for EBV-encoded RNA (200x) is positive in the nuclei of the neoplastic epithelial cells but not the background lymphocytes.
Gastric carcinoma associated with EBV
Gastric carcinoma with lymphoid stroma is also known as medullary carcinoma or lymphoepithelioma-like carcinoma. This is a rare subtype of gastric adenocarcinoma that is associated with EBV and accounts for 10% of all gastric carcinoma [381, 382]. It shows a male predominance and a predilection for arising in the proximal stomach [383]. The mean patient age is 60 years. EBV positivity in gastric carcinoma is associated with a more favorable prognosis compared to EBV-negative cases [384]. Gastric carcinoma associated with EBV is characterized by type II latency and typically expresses EBNA1, LMP1, LMP2A, and EBER.
The tumor can be grossly ulcerated or saucer-like with thickened wall. The neoplastic epithelial cells often proliferate in sheets and are associated with dense lymphoid infiltrate and intraepithelial lymphocytes (Figure 6I). The inflammatory background is composed of lymphocytes, histiocytes, neutrophils, and plasma cells and likely related to the immunogenic effects of EBV infection. The neoplastic cells contain abundant amounts of cytoplasm, enlarged nuclei, vesicular chromatin, and often prominent nucleoli. Interestingly, approximately one third of gastric carcinoma with lymphoid stroma cases are positive for EBV-encoded small RNA [385] (Figure 6J).
Clinicopathologic presentation of human neoplastic diseases associated with KSHV
Kaposi sarcoma
Kaposi sarcoma is an endothelial neoplasm. This is different from primary effusion lymphoma and multicentric Castleman disease, which are lymphoproliferative disorders of B cell origin. All the neoplastic endothelial cells are infected by KSHV [386, 387]. The classic presentation of Kaposi sarcoma (KS) is that of an elderly Mediterranean man (“classic KS”). AIDS-associated KS is the most common and aggressive form currently, although the advent of antiretroviral therapy for AIDS has reduced its incidence [388]. Other forms include endemic KS (e.g., children or African descents) and post-transplant/iatrogenic KS. Like PTLD, reduction of immunosuppressives may be efficacious for iatrogenic KS. KS lesions can involve skin, mucous membranes, and visceral organs. They often present as violaceous patches, plaques, or nodules. Interestingly, the mTOR inhibitor rapamycin showed dramatic KS remission in transplant recipients, highlighting the importance of activating the PI3K/Akt/mTOR pathway by viral oncoproteins in KS tumorigenesis [389, 390].
Histologically, KS is characterized by proliferation of spindle-shaped endothelial cells forming slit-like vascular spaces (Figure 7A). Extravasated red blood cells with hemosiderin and associated inflammatory cells can often be identified. Immunohistochemistry for LANA protein highlights KSHV-positive cells with classic speckled/punctate nuclear staining pattern. The endothelial origin can also be confirmed by immunohistochemistry for vascular markers such as ERG (Figure 7, B–D).
Figure 7.
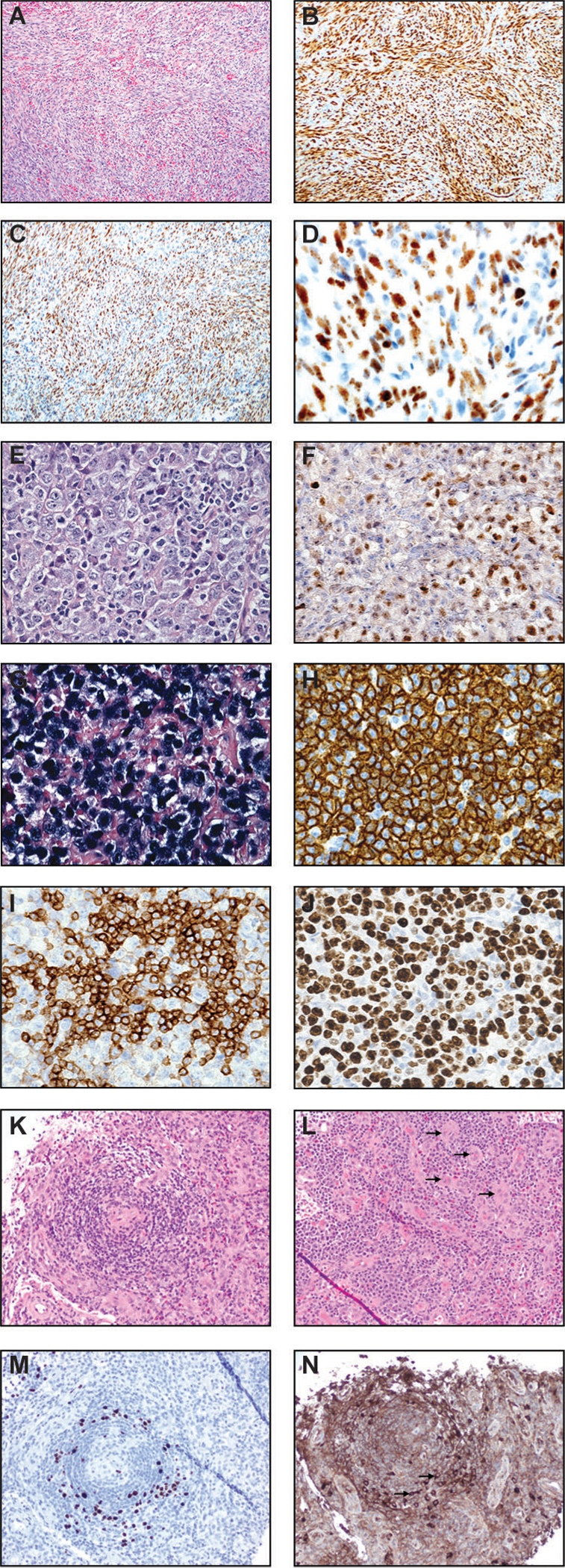
Kaposi sarcoma (A-D), primary effusion lymphoma (PEL) (E-J), and multicentric Castleman disease (K-N). (A) H&E image (100x) of a lymph node involved by Kaposi sarcoma shows intersecting fascicles of spindled cells. There are intervening slit-like/sieve-like spaces with entrapped red blood cells. (B) ERG immunostain (100x) is positive in essentially all spindled cells. Other vascular markers such as CD31 and CD34 are also positive in KS (not shown). (C) KSHV LANA immunostain (100x) is positive in virtually all nuclei of Kaposi sarcoma cells. (D) KSHV LANA immunostain at a high power (400x) demonstrates the classic punctate/speckled staining of KS nuclei. Primary effusion lymphoma (PEL) (E-J). (E) H&E image at high power field (600x) displays large and pleomorphic PEL cells with anaplastic nuclei, variably prominent nucleoli, and abundant cytoplasm. There is plasmablastic morphology. Mitotic figures and mummified cells are easily identified. (F) KSHV LANA stain (600x) shows positive staining in PEL nuclei, a key to the diagnosis. (G) In situ hybridization stain for EBV-encoded small RNA (600x) highlights neoplastic cells in this PEL case. (H) CD30 immunostain (600x) shows diffuse and strong membranous and cytoplasmic staining of PEL cells. (I) MUM1/IRF-4 stain (600x) demonstrates plasmacytic differentiation. (J) Ki-67 immunostain (600x) demonstrates a high proliferation index. Multicentric Castleman disease (K-N). (K) H&E image (200x) shows a regressed germinal center concentrically involved (“onion skinning” pattern) by numerous lymphoid cells and plasma cells/plasmablasts. (L) H&E image (200x) demonstrates numerous plasma cells in clusters and increased vessels (arrows). (M) KSHV LANA stain (200x) highlights plasmablasts. (N) Lambda immunostain (200x) highlights monotypic plasmablasts.
Primary effusion lymphoma
Primary effusion lymphoma (PEL), also known as body cavity-based lymphoma, is a highly aggressive and fatal disease that is mainly seen in severely immunocompromised AIDS individuals [387, 391, 392]. All cases are high stage and patients have a median survival of less than 6 months [393]. The lymphoma is typically seen in body cavities (e.g., pleural, pericardial, and peritoneal effusion fluids) but it can also present as an extranodal mass (so-called extracavitary or solid variant). [391, 394, 395]. Additionally, these patients may have concurrent KS or multicentric Castleman disease [391, 396, 397]. PEL cells are of post-germinal center origin. By definition, the neoplastic cells are KSHV-positive. Indeed, PEL cells harbor KSHV genome at a high copy number (50–150 per cell) [394]. Many KSHV latent products such as LANA, vCyclin, vFLIP, Kaposin, vIRF3 can be detected in PEL cells. Coinfection with EBV is commonly detected, particularly in the HIV setting, therefore, EBV is likely a cofactor, but is not causative [398, 399]. PEL cells are highly addicted to the PI3K/Akt/mTOR and MAPK pathways, and single or dual inhibitors of these kinases were tested in vitro which showed promising results [26, 400, 401]. The proteasome inhibitor bortezomib targets the NF-κB pathway and was found to be efficacious by inducing apoptosis in PEL cells [402–404]. More recently, inhibitors of fatty acid synthase and sphingosine kinase 2 also induced apoptosis in PEL cells, suggesting the potential therapeutic relevance of targeting dysregulated pathways of metabolism [405, 406].
Histologically, PEL cells are large sized with pleomorphic nuclei, prominent nucleoli, and abundant amounts of basophilic/amphophilic cytoplasm with or without vacuoles (Figure 7, E–F). They may resemble plasmablasts, immunoblasts, or occasional Hodgkin/Reed-Sternberg cells (binucleated/multinucleated forms). Stains for KSHV (LANA and viral IL-6) and EBV (e.g., in situ hybridization for EBV-encoded RNA) are useful, with detectable EBV co-infection in about half of the cases [398, 399]. They are often negative for B cell markers (CD20, PAX-5), but can be detected by CD45 and plasmacytic markers (CD138, CD38, MUM1, EMA) [395, 407] (Figure 7, G–J). Increased c-Myc expression is also a known phenomenon and has been shown to be driven by KSHV LANA [253, 254].
Multicentric Castleman disease
Multicentric Castleman disease (MCD) is also called multicentric angiofollicular hyperplasia. Clinically, the patient often shows systemic symptoms, including fever, night sweats, weight loss, generalized lymphadenopathy, hepatomegaly, splenomegaly, and autoimmune phenomena. Interestingly, serum interleukin-6 (IL-6) can be detected, at least in part, due to the effect of viral IL-6 and LANA [408]. The plasma cell variant of MCD has a very strong association with KSHV infection, especially in the AIDS setting [15, 387]. Tocilizumab and siltuximab, both monoclonal antibodies against human IL-6 receptor, have been investigated to treat MCD and found to produce clinical responses without severe toxicities or complications [409–412]. Unlike KS and PEL, KSHV lytic proteins are more commonly expressed in MCD [413]. A small pilot study investigated the utility of targeting the two KSHV lytic genes ORF36 and ORF21, using zidovudine and valganciclovir in KSHV+ MCD, showed promising clinical responses and survival [414]. This highlights the potential of targeting lytic KSHV infection in KSHV-associated MCD.
Morphologically, the lymph node shows extensive vascular proliferation in the germinal centers. Plasma cells and plasmablasts are increased in the mantle zones and interfollicular areas (Figure 7, K–N). Atypical plasmacytoid cells known as plasmablasts in the mantles are often present and they are often monotypic for IgM heavy chain and lambda light chain expression (Figure 7, K–N).
HHV-8/KSHV-positive diffuse large B-cell lymphoma, NOS
Large B-cell lymphomas can be positive for KSHV LANA antigen and found in patients with a systemic KSHV infection [16, 351, 407, 415, 416]. They are termed “HHV-8/KSHV-positive diffuse large B-cell lymphoma, NOS” [16]. They most commonly arise from MCD, and the patients often present with splenomegaly as well as other KSHV-associated lesions such as KS and PEL [351, 396, 397, 415]. However, they can occasionally be seen without evidence of MCD [407, 415]. The neoplastic cells efface the architecture of the involved lymph node, spleen, or other sites such as marrow and peripheral blood [16, 351, 416]. They morphologically resemble plasmablasts (or immunoblasts); they are positive for LANA, cytoplasmic IgM, and lambda light chain but negative for CD45 and CD20 [16, 351, 407, 416]. However, immunoblastic morphology has also been reported. These patients often have other KSHV-associated lesions such as KS and PEL [396, 397].
Finally, KSHV and EBV have been concurrently associated with germinotropic lymphoproliferative disorder [415, 417], which is not associated with HIV/AIDS and is not considered a malignant neoplasm. It is thus not further discussed in this review.
Concluding remarks
Both KSHV and EBV are oncogenic gammaherpesviruses that are implicated in several human neoplastic diseases. Oncogenesis by either virus is related to the viral oncoproteins of the lytic and latent states that are both crucial for the viruses to replicate, propagate, and persist in in a lifelong manner. These viral proteins function by perturbing physiologic signaling pathways, thereby enhancing pro-survival, anti-apoptotic, and immunoevasive properties of EBV and KSHV. As highlighted in this review, animal models have generated very informative data for understanding the oncogenic properties of these viral proteins in vivo. The neoplastic diseases seen in mouse models appear to mirror the histopathology observed in the human counterparts. Although current mainstay therapies do not target the lifecycles and viral proteins of EBV and KSHV, rational design of drugs and vaccines that prevent viral entry into specific cell types, lytic and latent replication, and the oncogenic and immune evasive function of various EBV and KSHV proteins may enable more effective prevention and treatment of the human cancer associated with these viruses.
Acknowledgements
We apologize that we had to omit many important publications due to space restrictions. The authors are supported by NIH grants CA096500, DE028211, CA163217, CA228172, CA254564, and CA019014.
Abbreviations:
- 4EBP1
4E-binding protein 1
- 5-aza
5-aza-deoxycytidine
- AIDS
acquired immunodeficiency syndrome
- Akt
protein kinase B
- BL
Burkitt lymphoma
- BTK
Bruton’s tyrosine kinase
- CDK
cyclin-dependent kinase
- CHL
classic Hodgkin lymphoma
- CHOP
cyclophosphamide, doxorubicin, vincristine, prednisone
- CK
cytokeratin
- DE
delayed early
- DLBCL
diffuse large B cell lymphoma
- EBNA
Epstein-Barr virus nuclear antigen
- EBERs
EBV-encoded small RNAs
- EBV
Epstein-Barr virus
- EGFR
epidermal growth factor receptor
- eIF4E
eukaryotic initiation factor 4E
- ERK
extracellular-signal-regulated kinase
- GC
gastric carcinoma
- GPCR
G protein-coupled receptor
- HAART
highly active antiretroviral therapy
- HHV-4
human herpesvirus 4
- HHV-8
human herpesvirus 8
- HIV
human immunodeficiency virus
- HRS
Hodgkin/Reed-Sternberg
- HSC
hematopoietic stem cell
- IE
immediate early
- Ig
immunoglobulin
- IL
interleukin
- ITAM
immunoreceptor tyrosine-based activation motifs
- JNK
c-Jun N-terminal kinase
- KSHV
Kaposi sarcoma-associated herpesvirus
- L
late
- LANA
latency-associated nuclear antigen
- LD
lymphocyte-depleted
- LMP
Latent membrane protein
- LR
lymphocyte-rich
- MAPK
mitogen-activated protein kinase
- MC
mixed cellularity
- MCD
multicentric Castleman disease
- MKK
mitogen-activated kinase
- mTOR
mammalian target of rapamycin
- NaB
sodium butyrate
- NF-κB
nuclear factor-κB
- NK
natural killer
- NPC
nasopharyngeal carcinoma
- NS
nodular sclerosis
- ORF
open reading frame
- PAL
pyothorax-associated lymphoma
- PDK1
phosphoinositide-dependent kinase 1
- PBL
plasmablastic lymphoma
- PEL
primary effusion lymphoma
- PH
pleckstrin homology
- PI3K
phosphatidylinositol-4,5-bisphosphate 3 kinase
- PIP2
phosphatidylinositol-4,5-bisphosphate
- PIP3
phosphatidylinositol-3,4,5-triphosphate
- PTEN
phosphatase and tensin homology
- PTLD
post-transplant lymphoproliferative disorder
- RANKL
receptor activator of NF-κB ligand
- Rb
retinoblastoma
- R-CHOP
rituximab, cyclophosphamide, doxorubicin, vincristine, prednisone
- RSK
p90 S6 kinase
- RTA
replication and transcription activator
- S6KB1
p70 S6 kinase
- SAPK
stress-activated protein kinase
- STAT3
signal transducer and activator of transcription 3
- TGF-β
transforming growth factor-β
- TNF
tumor necrosis factor
- TPA
12-O-tetradecanoylphorbol-13-acetate
- TRAF
tumor necrosis factor receptor-associated factor
- TSC2
tuberous sclerosis complex 2
- vCyclin
viral cyclin
- vFLIP
viral FADD-like interleukin-1-beta-converting enzyme-inhibitory protein
- vGPCR
viral G protein-coupled receptor
- vIL-6
viral interleukin 6
- VPA
valproic acid
- WHO
World Health Organization
Footnotes
Competing financial interests
K.W.W., L.W., J.R.M., and B.D. have no competing financial interests.
References
- 1.Bouvard V, Baan R, Straif K, Grosse Y, Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C, Galichet L, Cogliano V & Group, W. H. O. I. A. f. R. o. C. M. W. (2009) A review of human carcinogens--Part B: biological agents, Lancet Oncol. 10, 321–2. [DOI] [PubMed] [Google Scholar]
- 2.Pathmanathan R, Prasad U, Sadler R, Flynn K & Raab-Traub N (1995) Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma, N Engl J Med. 333, 693–8. [DOI] [PubMed] [Google Scholar]
- 3.Epstein MA, Achong BG & Barr YM (1964) Virus Particles in Cultured Lymphoblasts from Burkitt’s Lymphoma, Lancet. 1, 702–3. [DOI] [PubMed] [Google Scholar]
- 4.Carbone A & Gloghini A (2018) Epstein Barr Virus-Associated Hodgkin Lymphoma, Cancers (Basel). 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adam P, Bonzheim I, Fend F & Quintanilla-Martinez L (2011) Epstein-Barr virus-positive diffuse large B-cell lymphomas of the elderly, Adv Anat Pathol. 18, 349–55. [DOI] [PubMed] [Google Scholar]
- 6.Fukayama M, Ibuka T, Hayashi Y, Ooba T, Koike M & Mizutani S (1993) Epstein-Barr virus in pyothorax-associated pleural lymphoma, Am J Pathol. 143, 1044–9. [PMC free article] [PubMed] [Google Scholar]
- 7.Delecluse HJ, Anagnostopoulos I, Dallenbach F, Hummel M, Marafioti T, Schneider U, Huhn D, Schmidt-Westhausen A, Reichart PA, Gross U & Stein H (1997) Plasmablastic lymphomas of the oral cavity: a new entity associated with the human immunodeficiency virus infection, Blood. 89, 1413–20. [PubMed] [Google Scholar]
- 8.Wilson WH, Kingma DW, Raffeld M, Wittes RE & Jaffe ES (1996) Association of lymphomatoid granulomatosis with Epstein-Barr viral infection of B lymphocytes and response to interferon-alpha 2b, Blood. 87, 4531–7. [PubMed] [Google Scholar]
- 9.Singavi AK, Harrington AM & Fenske TS (2015) Post-transplant lymphoproliferative disorders, Cancer Treat Res. 165, 305–27. [DOI] [PubMed] [Google Scholar]
- 10.Oda K, Tamaru J, Takenouchi T, Mikata A, Nunomura M, Saitoh N, Sarashina H & Nakajima N (1993) Association of Epstein-Barr virus with gastric carcinoma with lymphoid stroma, Am J Pathol. 143, 1063–71. [PMC free article] [PubMed] [Google Scholar]
- 11.McClain KL, Leach CT, Jenson HB, Joshi VV, Pollock BH, Parmley RT, DiCarlo FJ, Chadwick EG & Murphy SB (1995) Association of Epstein-Barr virus with leiomyosarcomas in young people with AIDS, N Engl J Med. 332, 12–8. [DOI] [PubMed] [Google Scholar]
- 12.Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM & Moore PS (1994) Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma, Science. 266, 1865–9. [DOI] [PubMed] [Google Scholar]
- 13.Cesarman E, Chang Y, Moore PS, Said JW & Knowles DM (1995) Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in AIDS-related body-cavity-based lymphomas, N Engl J Med. 332, 1186–91. [DOI] [PubMed] [Google Scholar]
- 14.Cotter MA 2nd & Robertson ES (1999) The latency-associated nuclear antigen tethers the Kaposi’s sarcoma-associated herpesvirus genome to host chromosomes in body cavity-based lymphoma cells, Virology. 264, 254–64. [DOI] [PubMed] [Google Scholar]
- 15.Soulier J, Grollet L, Oksenhendler E, Cacoub P, Cazals-Hatem D, Babinet P, d’Agay MF, Clauvel JP, Raphael M, Degos L & et al. (1995) Kaposi’s sarcoma-associated herpesvirus-like DNA sequences in multicentric Castleman’s disease, Blood. 86, 1276–80. [PubMed] [Google Scholar]
- 16.Oksenhendler E, Boulanger E, Galicier L, Du MQ, Dupin N, Diss TC, Hamoudi R, Daniel MT, Agbalika F, Boshoff C, Clauvel JP, Isaacson PG & Meignin V (2002) High incidence of Kaposi sarcoma-associated herpesvirus-related non-Hodgkin lymphoma in patients with HIV infection and multicentric Castleman disease, Blood. 99, 2331–6. [DOI] [PubMed] [Google Scholar]
- 17.Cohen JI (2000) Epstein-Barr virus infection, N Engl J Med. 343, 481–92. [DOI] [PubMed] [Google Scholar]
- 18.Ablashi DV, Chatlynne LG, Whitman JE Jr. & Cesarman E (2002) Spectrum of Kaposi’s sarcoma-associated herpesvirus, or human herpesvirus 8, diseases, Clin Microbiol Rev. 15, 439–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Koelle DM, Huang ML, Chandran B, Vieira J, Piepkorn M & Corey L (1997) Frequent detection of Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) DNA in saliva of human immunodeficiency virus-infected men: clinical and immunologic correlates, J Infect Dis. 176, 94–102. [DOI] [PubMed] [Google Scholar]
- 20.Speck SH & Ganem D (2010) Viral latency and its regulation: lessons from the gamma-herpesviruses, Cell Host Microbe. 8, 100–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Manners O, Murphy JC, Coleman A, Hughes DJ & Whitehouse A (2018) Contribution of the KSHV and EBV lytic cycles to tumourigenesis, Curr Opin Virol. 32, 60–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Miller G, El-Guindy A, Countryman J, Ye J & Gradoville L (2007) Lytic cycle switches of oncogenic human gammaherpesviruses, Adv Cancer Res. 97, 81–109. [DOI] [PubMed] [Google Scholar]
- 23.Ma SD, Hegde S, Young KH, Sullivan R, Rajesh D, Zhou Y, Jankowska-Gan E, Burlingham WJ, Sun X, Gulley ML, Tang W, Gumperz JE & Kenney SC (2011) A new model of Epstein-Barr virus infection reveals an important role for early lytic viral protein expression in the development of lymphomas, J Virol. 85, 165–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mendoza MC, Er EE & Blenis J (2011) The Ras-ERK and PI3K-mTOR pathways: cross-talk and compensation, Trends Biochem Sci. 36, 320–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hussain AR, Ahmed SO, Ahmed M, Khan OS, Al Abdulmohsen S, Platanias LC, Al-Kuraya KS & Uddin S (2012) Cross-talk between NFkB and the PI3-kinase/AKT pathway can be targeted in primary effusion lymphoma (PEL) cell lines for efficient apoptosis, PLoS One. 7, e39945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Anders P, Bhende PM, Foote M, Dittmer DP, Park SI & Damania B (2015) Dual inhibition of phosphatidylinositol 3-kinase/mammalian target of rapamycin and mitogen activated protein kinase pathways in non-Hodgkin lymphoma, Leuk Lymphoma. 56, 263–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jha HC, Banerjee S & Robertson ES (2016) The Role of Gammaherpesviruses in Cancer Pathogenesis, Pathogens. 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luo Y, Liu Y, Wang C & Gan R (2021) Signaling pathways of EBV-induced oncogenesis, Cancer Cell Int. 21, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Charostad J, Nakhaie M, Dehghani A & Faghihloo E (2020) The interplay between EBV and KSHV viral products and NF-kappaB pathway in oncogenesis, Infect Agent Cancer. 15, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hayward SD, Liu J & Fujimuro M (2006) Notch and Wnt signaling: mimicry and manipulation by gamma herpesviruses, Sci STKE. 2006, re4. [DOI] [PubMed] [Google Scholar]
- 31.Bhatt AP & Damania B (2012) AKTivation of PI3K/AKT/mTOR signaling pathway by KSHV, Front Immunol. 3, 401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Engelman JA, Luo J & Cantley LC (2006) The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism, Nat Rev Genet. 7, 606–19. [DOI] [PubMed] [Google Scholar]
- 33.Cantley LC & Neel BG (1999) New insights into tumor suppression: PTEN suppresses tumor formation by restraining the phosphoinositide 3-kinase/AKT pathway, Proc Natl Acad Sci U S A. 96, 4240–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hsieh AC, Truitt ML & Ruggero D (2011) Oncogenic AKTivation of translation as a therapeutic target, Br J Cancer. 105, 329–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gingras AC, Gygi SP, Raught B, Polakiewicz RD, Abraham RT, Hoekstra MF, Aebersold R & Sonenberg N (1999) Regulation of 4E-BP1 phosphorylation: a novel two-step mechanism, Genes Dev. 13, 1422–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Saxton RA & Sabatini DM (2017) mTOR Signaling in Growth, Metabolism, and Disease, Cell. 168, 960–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gera JF, Mellinghoff IK, Shi Y, Rettig MB, Tran C, Hsu JH, Sawyers CL & Lichtenstein AK (2004) AKT activity determines sensitivity to mammalian target of rapamycin (mTOR) inhibitors by regulating cyclin D1 and c-myc expression, J Biol Chem. 279, 2737–46. [DOI] [PubMed] [Google Scholar]
- 38.Manning BD & Cantley LC (2007) AKT/PKB signaling: navigating downstream, Cell. 129, 1261–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang W & Liu HT (2002) MAPK signal pathways in the regulation of cell proliferation in mammalian cells, Cell Res. 12, 9–18. [DOI] [PubMed] [Google Scholar]
- 40.Kim EK & Choi EJ (2015) Compromised MAPK signaling in human diseases: an update, Arch Toxicol. 89, 867–82. [DOI] [PubMed] [Google Scholar]
- 41.Amaral T, Sinnberg T, Meier F, Krepler C, Levesque M, Niessner H & Garbe C (2017) The mitogen-activated protein kinase pathway in melanoma part I - Activation and primary resistance mechanisms to BRAF inhibition, Eur J Cancer. 73, 85–92. [DOI] [PubMed] [Google Scholar]
- 42.Taniguchi K & Karin M (2018) NF-kappaB, inflammation, immunity and cancer: coming of age, Nat Rev Immunol. 18, 309–324. [DOI] [PubMed] [Google Scholar]
- 43.Zhang Q, Lenardo MJ & Baltimore D (2017) 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology, Cell. 168, 37–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sun SC (2011) Non-canonical NF-kappaB signaling pathway, Cell Res. 21, 71–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen J, Sathiyamoorthy K, Zhang X, Schaller S, Perez White BE, Jardetzky TS & Longnecker R (2018) Ephrin receptor A2 is a functional entry receptor for Epstein-Barr virus, Nat Microbiol. 3, 172–180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zhang H, Li Y, Wang HB, Zhang A, Chen ML, Fang ZX, Dong XD, Li SB, Du Y, Xiong D, He JY, Li MZ, Liu YM, Zhou AJ, Zhong Q, Zeng YX, Kieff E, Zhang Z, Gewurz BE, Zhao B & Zeng MS (2018) Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry, Nat Microbiol. 3, 1–8. [DOI] [PubMed] [Google Scholar]
- 47.Haan KM, Kwok WW, Longnecker R & Speck P (2000) Epstein-Barr virus entry utilizing HLA-DP or HLA-DQ as a coreceptor, J Virol. 74, 2451–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chesnokova LS, Nishimura SL & Hutt-Fletcher LM (2009) Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8, Proc Natl Acad Sci U S A. 106, 20464–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hutt-Fletcher LM (2007) Epstein-Barr virus entry, J Virol. 81, 7825–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wille CK, Nawandar DM, Panfil AR, Ko MM, Hagemeier SR & Kenney SC (2013) Viral genome methylation differentially affects the ability of BZLF1 versus BRLF1 to activate Epstein-Barr virus lytic gene expression and viral replication, J Virol. 87, 935–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Murata T (2014) Regulation of Epstein-Barr virus reactivation from latency, Microbiol Immunol. 58, 307–17. [DOI] [PubMed] [Google Scholar]
- 52.Inagaki T, Sato Y, Ito J, Takaki M, Okuno Y, Yaguchi M, Masud H, Watanabe T, Sato K, Iwami S, Murata T & Kimura H (2020) Direct Evidence of Abortive Lytic Infection-Mediated Establishment of Epstein-Barr Virus Latency During B-Cell Infection, Front Microbiol. 11, 575255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wen W, Iwakiri D, Yamamoto K, Maruo S, Kanda T & Takada K (2007) Epstein-Barr virus BZLF1 gene, a switch from latency to lytic infection, is expressed as an immediate-early gene after primary infection of B lymphocytes, J Virol. 81, 1037–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jochum S, Moosmann A, Lang S, Hammerschmidt W & Zeidler R (2012) The EBV immunoevasins vIL-10 and BNLF2a protect newly infected B cells from immune recognition and elimination, PLoS Pathog. 8, e1002704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Wang C, Li D, Zhang L, Jiang S, Liang J, Narita Y, Hou I, Zhong Q, Zheng Z, Xiao H, Gewurz BE, Teng M & Zhao B (2019) RNA Sequencing Analyses of Gene Expression during Epstein-Barr Virus Infection of Primary B Lymphocytes, J Virol. 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yuan J, Cahir-McFarland E, Zhao B & Kieff E (2006) Virus and cell RNAs expressed during Epstein-Barr virus replication, J Virol. 80, 2548–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Munz C (2019) Latency and lytic replication in Epstein-Barr virus-associated oncogenesis, Nat Rev Microbiol. 17, 691–700. [DOI] [PubMed] [Google Scholar]
- 58.Fahraeus R, Fu HL, Ernberg I, Finke J, Rowe M, Klein G, Falk K, Nilsson E, Yadav M, Busson P & et al. (1988) Expression of Epstein-Barr virus-encoded proteins in nasopharyngeal carcinoma, Int J Cancer. 42, 329–38. [DOI] [PubMed] [Google Scholar]
- 59.Young L, Alfieri C, Hennessy K, Evans H, O’Hara C, Anderson KC, Ritz J, Shapiro RS, Rickinson A, Kieff E & et al. (1989) Expression of Epstein-Barr virus transformation-associated genes in tissues of patients with EBV lymphoproliferative disease, N Engl J Med. 321, 1080–5. [DOI] [PubMed] [Google Scholar]
- 60.Ok CY, Li L & Young KH (2015) EBV-driven B-cell lymphoproliferative disorders: from biology, classification and differential diagnosis to clinical management, Exp Mol Med. 47, e132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Chen YP, Zhang WN, Chen L, Tang LL, Mao YP, Li WF, Liu X, Zhou GQ, Sun Y, Kang TB, Zeng MS, Liu N & Ma J (2015) Effect of latent membrane protein 1 expression on overall survival in Epstein-Barr virus-associated cancers: a literature-based meta-analysis, Oncotarget. 6, 29311–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Shair KH, Bendt KM, Edwards RH, Nielsen JN, Moore DT & Raab-Traub N (2012) Epstein-Barr virus-encoded latent membrane protein 1 (LMP1) and LMP2A function cooperatively to promote carcinoma development in a mouse carcinogenesis model, J Virol. 86, 5352–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Shair KH, Schnegg CI & Raab-Traub N (2008) EBV latent membrane protein 1 effects on plakoglobin, cell growth, and migration, Cancer Res. 68, 6997–7005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Izumi KM, Cahir McFarland ED, Ting AT, Riley EA, Seed B & Kieff ED (1999) The Epstein-Barr virus oncoprotein latent membrane protein 1 engages the tumor necrosis factor receptor-associated proteins TRADD and receptor-interacting protein (RIP) but does not induce apoptosis or require RIP for NF-kappaB activation, Mol Cell Biol. 19, 5759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Izumi KM & Kieff ED (1997) The Epstein-Barr virus oncogene product latent membrane protein 1 engages the tumor necrosis factor receptor-associated death domain protein to mediate B lymphocyte growth transformation and activate NF-kappaB, Proc Natl Acad Sci U S A. 94, 12592–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Mosialos G, Birkenbach M, Yalamanchili R, VanArsdale T, Ware C & Kieff E (1995) The Epstein-Barr virus transforming protein LMP1 engages signaling proteins for the tumor necrosis factor receptor family, Cell. 80, 389–99. [DOI] [PubMed] [Google Scholar]
- 67.Mainou BA, Everly DN Jr. & Raab-Traub N (2007) Unique signaling properties of CTAR1 in LMP1-mediated transformation, J Virol. 81, 9680–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Song YJ & Kang MS (2010) Roles of TRAF2 and TRAF3 in Epstein-Barr virus latent membrane protein 1-induced alternative NF-kappaB activation, Virus Genes. 41, 174–80. [DOI] [PubMed] [Google Scholar]
- 69.Eliopoulos AG, Blake SM, Floettmann JE, Rowe M & Young LS (1999) Epstein-Barr virus-encoded latent membrane protein 1 activates the JNK pathway through its extreme C terminus via a mechanism involving TRADD and TRAF2, J Virol. 73, 1023–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Eliopoulos AG & Young LS (1998) Activation of the cJun N-terminal kinase (JNK) pathway by the Epstein-Barr virus-encoded latent membrane protein 1 (LMP1), Oncogene. 16, 1731–42. [DOI] [PubMed] [Google Scholar]
- 71.Everly DN Jr., Mainou BA & Raab-Traub N (2004) Induction of Id1 and Id3 by latent membrane protein 1 of Epstein-Barr virus and regulation of p27/Kip and cyclin-dependent kinase 2 in rodent fibroblast transformation, J Virol. 78, 13470–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Ohtani N, Brennan P, Gaubatz S, Sanij E, Hertzog P, Wolvetang E, Ghysdael J, Rowe M & Hara E (2003) Epstein-Barr virus LMP1 blocks p16INK4a-RB pathway by promoting nuclear export of E2F4/5, J Cell Biol. 162, 173–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Li HM, Zhuang ZH, Wang Q, Pang JC, Wang XH, Wong HL, Feng HC, Jin DY, Ling MT, Wong YC, Eliopoulos AG, Young LS, Huang DP & Tsao SW (2004) Epstein-Barr virus latent membrane protein 1 (LMP1) upregulates Id1 expression in nasopharyngeal epithelial cells, Oncogene. 23, 4488–94. [DOI] [PubMed] [Google Scholar]
- 74.Nicholson LJ, Hopwood P, Johannessen I, Salisbury JR, Codd J, Thorley-Lawson D & Crawford DH (1997) Epstein-Barr virus latent membrane protein does not inhibit differentiation and induces tumorigenicity of human epithelial cells, Oncogene. 15, 275–83. [DOI] [PubMed] [Google Scholar]
- 75.Kaye KM, Izumi KM, Mosialos G & Kieff E (1995) The Epstein-Barr virus LMP1 cytoplasmic carboxy terminus is essential for B-lymphocyte transformation; fibroblast cocultivation complements a critical function within the terminal 155 residues, J Virol. 69, 675–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mainou BA, Everly DN Jr. & Raab-Traub N (2005) Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K, Oncogene. 24, 6917–24. [DOI] [PubMed] [Google Scholar]
- 77.Liu X, Li Y, Peng S, Yu X, Li W, Shi F, Luo X, Tang M, Tan Z, Bode AM & Cao Y (2018) Epstein-Barr virus encoded latent membrane protein 1 suppresses necroptosis through targeting RIPK1/3 ubiquitination, Cell Death Dis. 9, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Scholle F, Bendt KM & Raab-Traub N (2000) Epstein-Barr virus LMP2A transforms epithelial cells, inhibits cell differentiation, and activates Akt, J Virol. 74, 10681–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mancao C & Hammerschmidt W (2007) Epstein-Barr virus latent membrane protein 2A is a B-cell receptor mimic and essential for B-cell survival, Blood. 110, 3715–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Fukuda M & Longnecker R (2007) Epstein-Barr virus latent membrane protein 2A mediates transformation through constitutive activation of the Ras/PI3-K/Akt Pathway, J Virol. 81, 9299–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Rovedo M & Longnecker R (2008) Epstein-Barr virus latent membrane protein 2A preferentially signals through the Src family kinase Lyn, J Virol. 82, 8520–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Merchant M, Caldwell RG & Longnecker R (2000) The LMP2A ITAM is essential for providing B cells with development and survival signals in vivo, J Virol. 74, 9115–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caldwell RG, Wilson JB, Anderson SJ & Longnecker R (1998) Epstein-Barr virus LMP2A drives B cell development and survival in the absence of normal B cell receptor signals, Immunity. 9, 405–11. [DOI] [PubMed] [Google Scholar]
- 84.Fruehling S, Lee SK, Herrold R, Frech B, Laux G, Kremmer E, Grasser FA & Longnecker R (1996) Identification of latent membrane protein 2A (LMP2A) domains essential for the LMP2A dominant-negative effect on B-lymphocyte surface immunoglobulin signal transduction, J Virol. 70, 6216–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Dykstra ML, Longnecker R & Pierce SK (2001) Epstein-Barr virus coopts lipid rafts to block the signaling and antigen transport functions of the BCR, Immunity. 14, 57–67. [DOI] [PubMed] [Google Scholar]
- 86.Fruehling S & Longnecker R (1997) The immunoreceptor tyrosine-based activation motif of Epstein-Barr virus LMP2A is essential for blocking BCR-mediated signal transduction, Virology. 235, 241–51. [DOI] [PubMed] [Google Scholar]
- 87.Casola S, Otipoby KL, Alimzhanov M, Humme S, Uyttersprot N, Kutok JL, Carroll MC & Rajewsky K (2004) B cell receptor signal strength determines B cell fate, Nat Immunol. 5, 317–27. [DOI] [PubMed] [Google Scholar]
- 88.Ikeda M, Ikeda A, Longan LC & Longnecker R (2000) The Epstein-Barr virus latent membrane protein 2A PY motif recruits WW domain-containing ubiquitin-protein ligases, Virology. 268, 178–91. [DOI] [PubMed] [Google Scholar]
- 89.Incrocci R, Barse L, Stone A, Vagvala S, Montesano M, Subramaniam V & Swanson-Mungerson M (2017) Epstein-Barr Virus Latent Membrane Protein 2A (LMP2A) enhances IL-10 production through the activation of Bruton’s tyrosine kinase and STAT3, Virology. 500, 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Portis T, Cooper L, Dennis P & Longnecker R (2002) The LMP2A signalosome--a therapeutic target for Epstein-Barr virus latency and associated disease, Front Biosci. 7, d414–26. [DOI] [PubMed] [Google Scholar]
- 91.Heussinger N, Buttner M, Ott G, Brachtel E, Pilch BZ, Kremmer E & Niedobitek G (2004) Expression of the Epstein-Barr virus (EBV)-encoded latent membrane protein 2A (LMP2A) in EBV-associated nasopharyngeal carcinoma, J Pathol. 203, 696–9. [DOI] [PubMed] [Google Scholar]
- 92.Brooks L, Yao QY, Rickinson AB & Young LS (1992) Epstein-Barr virus latent gene transcription in nasopharyngeal carcinoma cells: coexpression of EBNA1, LMP1, and LMP2 transcripts, J Virol. 66, 2689–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Murray PG, Young LS, Rowe M & Crocker J (1992) Immunohistochemical demonstration of the Epstein-Barr virus-encoded latent membrane protein in paraffin sections of Hodgkin’s disease, J Pathol. 166, 1–5. [DOI] [PubMed] [Google Scholar]
- 94.Fukuda M, Ikuta K, Yanagihara K, Tajima M, Kuratsune H, Kurata T & Sairenji T (2001) Effect of transforming growth factor-beta1 on the cell growth and Epstein-Barr virus reactivation in EBV-infected epithelial cell lines, Virology. 288, 109–18. [DOI] [PubMed] [Google Scholar]
- 95.Hino R, Uozaki H, Inoue Y, Shintani Y, Ushiku T, Sakatani T, Takada K & Fukayama M (2008) Survival advantage of EBV-associated gastric carcinoma: survivin up-regulation by viral latent membrane protein 2A, Cancer Res. 68, 1427–35. [DOI] [PubMed] [Google Scholar]
- 96.Caldwell RG, Brown RC & Longnecker R (2000) Epstein-Barr virus LMP2A-induced B-cell survival in two unique classes of EmuLMP2A transgenic mice, J Virol. 74, 1101–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Portis T & Longnecker R (2004) Epstein-Barr virus (EBV) LMP2A mediates B-lymphocyte survival through constitutive activation of the Ras/PI3K/Akt pathway, Oncogene. 23, 8619–28. [DOI] [PubMed] [Google Scholar]
- 98.Yates JL & Guan N (1991) Epstein-Barr virus-derived plasmids replicate only once per cell cycle and are not amplified after entry into cells, J Virol. 65, 483–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Adams A (1987) Replication of latent Epstein-Barr virus genomes in Raji cells, J Virol. 61, 1743–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wu H, Kapoor P & Frappier L (2002) Separation of the DNA replication, segregation, and transcriptional activation functions of Epstein-Barr nuclear antigen 1, J Virol. 76, 2480–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Deutsch MJ, Ott E, Papior P & Schepers A (2010) The latent origin of replication of Epstein-Barr virus directs viral genomes to active regions of the nucleus, J Virol. 84, 2533–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hodin TL, Najrana T & Yates JL (2013) Efficient replication of Epstein-Barr virus-derived plasmids requires tethering by EBNA1 to host chromosomes, J Virol. 87, 13020–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Grogan EA, Summers WP, Dowling S, Shedd D, Gradoville L & Miller G (1983) Two Epstein-Barr viral nuclear neoantigens distinguished by gene transfer, serology, and chromosome binding, Proc Natl Acad Sci U S A. 80, 7650–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hung SC, Kang MS & Kieff E (2001) Maintenance of Epstein-Barr virus (EBV) oriP-based episomes requires EBV-encoded nuclear antigen-1 chromosome-binding domains, which can be replaced by high-mobility group-I or histone H1, Proc Natl Acad Sci U S A. 98, 1865–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Nayyar VK, Shire K & Frappier L (2009) Mitotic chromosome interactions of Epstein-Barr nuclear antigen 1 (EBNA1) and human EBNA1-binding protein 2 (EBP2), J Cell Sci. 122, 4341–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Kapoor P & Frappier L (2003) EBNA1 partitions Epstein-Barr virus plasmids in yeast cells by attaching to human EBNA1-binding protein 2 on mitotic chromosomes, J Virol. 77, 6946–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Shire K, Ceccarelli DF, Avolio-Hunter TM & Frappier L (1999) EBP2, a human protein that interacts with sequences of the Epstein-Barr virus nuclear antigen 1 important for plasmid maintenance, J Virol. 73, 2587–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Norseen J, Johnson FB & Lieberman PM (2009) Role for G-quadruplex RNA binding by Epstein-Barr virus nuclear antigen 1 in DNA replication and metaphase chromosome attachment, J Virol. 83, 10336–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Chittenden T, Lupton S & Levine AJ (1989) Functional limits of oriP, the Epstein-Barr virus plasmid origin of replication, J Virol. 63, 3016–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Lupton S & Levine AJ (1985) Mapping genetic elements of Epstein-Barr virus that facilitate extrachromosomal persistence of Epstein-Barr virus-derived plasmids in human cells, Mol Cell Biol. 5, 2533–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Chen MR, Zong J & Hayward SD (1994) Delineation of a 16 amino acid sequence that forms a core DNA recognition motif in the Epstein-Barr virus EBNA-1 protein, Virology. 205, 486–95. [DOI] [PubMed] [Google Scholar]
- 112.Lu F, Wikramasinghe P, Norseen J, Tsai K, Wang P, Showe L, Davuluri RV & Lieberman PM (2010) Genome-wide analysis of host-chromosome binding sites for Epstein-Barr Virus Nuclear Antigen 1 (EBNA1), Virol J. 7, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Canaan A, Haviv I, Urban AE, Schulz VP, Hartman S, Zhang Z, Palejev D, Deisseroth AB, Lacy J, Snyder M, Gerstein M & Weissman SM (2009) EBNA1 regulates cellular gene expression by binding cellular promoters, Proc Natl Acad Sci U S A. 106, 22421–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Valentine R, Dawson CW, Hu C, Shah KM, Owen TJ, Date KL, Maia SP, Shao J, Arrand JR, Young LS & O’Neil JD (2010) Epstein-Barr virus-encoded EBNA1 inhibits the canonical NF-kappaB pathway in carcinoma cells by inhibiting IKK phosphorylation, Mol Cancer. 9, 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Li M, Chen D, Shiloh A, Luo J, Nikolaev AY, Qin J & Gu W (2002) Deubiquitination of p53 by HAUSP is an important pathway for p53 stabilization, Nature. 416, 648–53. [DOI] [PubMed] [Google Scholar]
- 116.Holowaty MN, Zeghouf M, Wu H, Tellam J, Athanasopoulos V, Greenblatt J & Frappier L (2003) Protein profiling with Epstein-Barr nuclear antigen-1 reveals an interaction with the herpesvirus-associated ubiquitin-specific protease HAUSP/USP7, J Biol Chem. 278, 29987–94. [DOI] [PubMed] [Google Scholar]
- 117.Sivachandran N, Sarkari F & Frappier L (2008) Epstein-Barr nuclear antigen 1 contributes to nasopharyngeal carcinoma through disruption of PML nuclear bodies, PLoS Pathog. 4, e1000170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lu J, Murakami M, Verma SC, Cai Q, Haldar S, Kaul R, Wasik MA, Middeldorp J & Robertson ES (2011) Epstein-Barr Virus nuclear antigen 1 (EBNA1) confers resistance to apoptosis in EBV-positive B-lymphoma cells through up-regulation of survivin, Virology. 410, 64–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Wood VH, O’Neil JD, Wei W, Stewart SE, Dawson CW & Young LS (2007) Epstein-Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFbeta signaling pathways, Oncogene. 26, 4135–47. [DOI] [PubMed] [Google Scholar]
- 120.AlQarni S, Al-Sheikh Y, Campbell D, Drotar M, Hannigan A, Boyle S, Herzyk P, Kossenkov A, Armfield K, Jamieson L, Bailo M, Lieberman PM, Tsimbouri P & Wilson JB (2018) Lymphomas driven by Epstein-Barr virus nuclear antigen-1 (EBNA1) are dependant upon Mdm2, Oncogene. 37, 3998–4012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wilson JB, Bell JL & Levine AJ (1996) Expression of Epstein-Barr virus nuclear antigen-1 induces B cell neoplasia in transgenic mice, EMBO J. 15, 3117–26. [PMC free article] [PubMed] [Google Scholar]
- 122.Kang MS, Lu H, Yasui T, Sharpe A, Warren H, Cahir-McFarland E, Bronson R, Hung SC & Kieff E (2005) Epstein-Barr virus nuclear antigen 1 does not induce lymphoma in transgenic FVB mice, Proc Natl Acad Sci U S A. 102, 820–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kang MS, Soni V, Bronson R & Kieff E (2008) Epstein-Barr virus nuclear antigen 1 does not cause lymphoma in C57BL/6J mice, J Virol. 82, 4180–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Messick TE, Smith GR, Soldan SS, McDonnell ME, Deakyne JS, Malecka KA, Tolvinski L, van den Heuvel APJ, Gu BW, Cassel JA, Tran DH, Wassermann BR, Zhang Y, Velvadapu V, Zartler ER, Busson P, Reitz AB & Lieberman PM (2019) Structure-based design of small-molecule inhibitors of EBNA1 DNA binding blocks Epstein-Barr virus latent infection and tumor growth, Sci Transl Med. 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wilson JB, Manet E, Gruffat H, Busson P, Blondel M & Fahraeus R (2018) EBNA1: Oncogenic Activity, Immune Evasion and Biochemical Functions Provide Targets for Novel Therapeutic Strategies against Epstein-Barr Virus- Associated Cancers, Cancers (Basel). 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kube D, Vockerodt M, Weber O, Hell K, Wolf J, Haier B, Grasser FA, Muller-Lantzsch N, Kieff E, Diehl V & Tesch H (1999) Expression of epstein-barr virus nuclear antigen 1 is associated with enhanced expression of CD25 in the Hodgkin cell line L428, J Virol. 73, 1630–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kennedy G, Komano J & Sugden B (2003) Epstein-Barr virus provides a survival factor to Burkitt’s lymphomas, Proc Natl Acad Sci U S A. 100, 14269–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Hong M, Murai Y, Kutsuna T, Takahashi H, Nomoto K, Cheng CM, Ishizawa S, Zhao QL, Ogawa R, Harmon BV, Tsuneyama K & Takano Y (2006) Suppression of Epstein-Barr nuclear antigen 1 (EBNA1) by RNA interference inhibits proliferation of EBV-positive Burkitt’s lymphoma cells, J Cancer Res Clin Oncol. 132, 1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Cao JY, Mansouri S & Frappier L (2012) Changes in the nasopharyngeal carcinoma nuclear proteome induced by the EBNA1 protein of Epstein-Barr virus reveal potential roles for EBNA1 in metastasis and oxidative stress responses, J Virol. 86, 382–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sivachandran N, Dawson CW, Young LS, Liu FF, Middeldorp J & Frappier L (2012) Contributions of the Epstein-Barr virus EBNA1 protein to gastric carcinoma, J Virol. 86, 60–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li N, Thompson S, Schultz DC, Zhu W, Jiang H, Luo C & Lieberman PM (2010) Discovery of selective inhibitors against EBNA1 via high throughput in silico virtual screening, PLoS One. 5, e10126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Jiang L, Xie C, Lung HL, Lo KW, Law GL, Mak NK & Wong KL (2018) EBNA1-targeted inhibitors: Novel approaches for the treatment of Epstein-Barr virus-associated cancers, Theranostics. 8, 5307–5319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Cohen JI, Wang F, Mannick J & Kieff E (1989) Epstein-Barr virus nuclear protein 2 is a key determinant of lymphocyte transformation, Proc Natl Acad Sci U S A. 86, 9558–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Hammerschmidt W & Sugden B (1989) Genetic analysis of immortalizing functions of Epstein-Barr virus in human B lymphocytes, Nature. 340, 393–7. [DOI] [PubMed] [Google Scholar]
- 135.Kempkes B, Spitkovsky D, Jansen-Durr P, Ellwart JW, Kremmer E, Delecluse HJ, Rottenberger C, Bornkamm GW & Hammerschmidt W (1995) B-cell proliferation and induction of early G1-regulating proteins by Epstein-Barr virus mutants conditional for EBNA2, EMBO J. 14, 88–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Henkel T, Ling PD, Hayward SD & Peterson MG (1994) Mediation of Epstein-Barr virus EBNA2 transactivation by recombination signal-binding protein J kappa, Science. 265, 92–5. [DOI] [PubMed] [Google Scholar]
- 137.Grossman SR, Johannsen E, Tong X, Yalamanchili R & Kieff E (1994) The Epstein-Barr virus nuclear antigen 2 transactivator is directed to response elements by the J kappa recombination signal binding protein, Proc Natl Acad Sci U S A. 91, 7568–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tong X, Drapkin R, Reinberg D & Kieff E (1995) The 62- and 80-kDa subunits of transcription factor IIH mediate the interaction with Epstein-Barr virus nuclear protein 2, Proc Natl Acad Sci U S A. 92, 3259–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Strobl LJ, Hofelmayr H, Marschall G, Brielmeier M, Bornkamm GW & Zimber-Strobl U (2000) Activated Notch1 modulates gene expression in B cells similarly to Epstein-Barr viral nuclear antigen 2, J Virol. 74, 1727–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Sakai T, Taniguchi Y, Tamura K, Minoguchi S, Fukuhara T, Strobl LJ, Zimber-Strobl U, Bornkamm GW & Honjo T (1998) Functional replacement of the intracellular region of the Notch1 receptor by Epstein-Barr virus nuclear antigen 2, J Virol. 72, 6034–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kaiser C, Laux G, Eick D, Jochner N, Bornkamm GW & Kempkes B (1999) The proto-oncogene c-myc is a direct target gene of Epstein-Barr virus nuclear antigen 2, J Virol. 73, 4481–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Wang F, Gregory C, Sample C, Rowe M, Liebowitz D, Murray R, Rickinson A & Kieff E (1990) Epstein-Barr virus latent membrane protein (LMP1) and nuclear proteins 2 and 3C are effectors of phenotypic changes in B lymphocytes: EBNA-2 and LMP1 cooperatively induce CD23, J Virol. 64, 2309–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Wang F, Gregory CD, Rowe M, Rickinson AB, Wang D, Birkenbach M, Kikutani H, Kishimoto T & Kieff E (1987) Epstein-Barr virus nuclear antigen 2 specifically induces expression of the B-cell activation antigen CD23, Proc Natl Acad Sci U S A. 84, 3452–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Wang F, Kikutani H, Tsang SF, Kishimoto T & Kieff E (1991) Epstein-Barr virus nuclear protein 2 transactivates a cis-acting CD23 DNA element, J Virol. 65, 4101–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Zhao B, Zou J, Wang H, Johannsen E, Peng CW, Quackenbush J, Mar JC, Morton CC, Freedman ML, Blacklow SC, Aster JC, Bernstein BE & Kieff E (2011) Epstein-Barr virus exploits intrinsic B-lymphocyte transcription programs to achieve immortal cell growth, Proc Natl Acad Sci U S A. 108, 14902–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Abbot SD, Rowe M, Cadwallader K, Ricksten A, Gordon J, Wang F, Rymo L & Rickinson AB (1990) Epstein-Barr virus nuclear antigen 2 induces expression of the virus-encoded latent membrane protein, J Virol. 64, 2126–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Nitsche F, Bell A & Rickinson A (1997) Epstein-Barr virus leader protein enhances EBNA-2-mediated transactivation of latent membrane protein 1 expression: a role for the W1W2 repeat domain, J Virol. 71, 6619–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Harada S & Kieff E (1997) Epstein-Barr virus nuclear protein LP stimulates EBNA-2 acidic domain-mediated transcriptional activation, J Virol. 71, 6611–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Tsang SF, Wang F, Izumi KM & Kieff E (1991) Delineation of the cis-acting element mediating EBNA-2 transactivation of latent infection membrane protein expression, J Virol. 65, 6765–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Wang F, Tsang SF, Kurilla MG, Cohen JI & Kieff E (1990) Epstein-Barr virus nuclear antigen 2 transactivates latent membrane protein LMP1, J Virol. 64, 3407–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Woisetschlaeger M, Strominger JL & Speck SH (1989) Mutually exclusive use of viral promoters in Epstein-Barr virus latently infected lymphocytes, Proc Natl Acad Sci U S A. 86, 6498–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Woisetschlaeger M, Yandava CN, Furmanski LA, Strominger JL & Speck SH (1990) Promoter switching in Epstein-Barr virus during the initial stages of infection of B lymphocytes, Proc Natl Acad Sci U S A. 87, 1725–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Woisetschlaeger M, Jin XW, Yandava CN, Furmanski LA, Strominger JL & Speck SH (1991) Role for the Epstein-Barr virus nuclear antigen 2 in viral promoter switching during initial stages of infection, Proc Natl Acad Sci U S A. 88, 3942–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Wu DY, Krumm A & Schubach WH (2000) Promoter-specific targeting of human SWI-SNF complex by Epstein-Barr virus nuclear protein 2, J Virol. 74, 8893–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Johannsen E, Koh E, Mosialos G, Tong X, Kieff E & Grossman SR (1995) Epstein-Barr virus nuclear protein 2 transactivation of the latent membrane protein 1 promoter is mediated by J kappa and PU.1, J Virol. 69, 253–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Sjoblom A, Jansson A, Yang W, Lain S, Nilsson T & Rymo L (1995) PU box-binding transcription factors and a POU domain protein cooperate in the Epstein-Barr virus (EBV) nuclear antigen 2-induced transactivation of the EBV latent membrane protein 1 promoter, J Gen Virol. 76 (Pt 11), 2679–92. [DOI] [PubMed] [Google Scholar]
- 157.Lu F, Wiedmer A, Martin KA, Wickramasinghe P, Kossenkov AV & Lieberman PM (2017) Coordinate Regulation of TET2 and EBNA2 Controls the DNA Methylation State of Latent Epstein-Barr Virus, J Virol. 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Jochner N, Eick D, Zimber-Strobl U, Pawlita M, Bornkamm GW & Kempkes B (1996) Epstein-Barr virus nuclear antigen 2 is a transcriptional suppressor of the immunoglobulin mu gene: implications for the expression of the translocated c-myc gene in Burkitt’s lymphoma cells, EMBO J. 15, 375–82. [PMC free article] [PubMed] [Google Scholar]
- 159.Peng CW, Xue Y, Zhao B, Johannsen E, Kieff E & Harada S (2004) Direct interactions between Epstein-Barr virus leader protein LP and the EBNA2 acidic domain underlie coordinate transcriptional regulation, Proc Natl Acad Sci U S A. 101, 1033–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Peng CW, Zhao B & Kieff E (2004) Four EBNA2 domains are important for EBNALP coactivation, J Virol. 78, 11439–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Sinclair AJ, Palmero I, Peters G & Farrell PJ (1994) EBNA-2 and EBNA-LP cooperate to cause G0 to G1 transition during immortalization of resting human B lymphocytes by Epstein-Barr virus, EMBO J. 13, 3321–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Huen DS, Fox A, Kumar P & Searle PF (1993) Dilated heart failure in transgenic mice expressing the Epstein-Barr virus nuclear antigen-leader protein, J Gen Virol. 74 (Pt 7), 1381–91. [DOI] [PubMed] [Google Scholar]
- 163.Le Roux A, Kerdiles B, Walls D, Dedieu JF & Perricaudet M (1994) The Epstein-Barr virus determined nuclear antigens EBNA-3A, −3B, and −3C repress EBNA-2-mediated transactivation of the viral terminal protein 1 gene promoter, Virology. 205, 596–602. [DOI] [PubMed] [Google Scholar]
- 164.Radkov SA, Bain M, Farrell PJ, West M, Rowe M & Allday MJ (1997) Epstein-Barr virus EBNA3C represses Cp, the major promoter for EBNA expression, but has no effect on the promoter of the cell gene CD21, J Virol. 71, 8552–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Zhao B & Sample CE (2000) Epstein-barr virus nuclear antigen 3C activates the latent membrane protein 1 promoter in the presence of Epstein-Barr virus nuclear antigen 2 through sequences encompassing an spi-1/Spi-B binding site, J Virol. 74, 5151–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Maruo S, Zhao B, Johannsen E, Kieff E, Zou J & Takada K (2011) Epstein-Barr virus nuclear antigens 3C and 3A maintain lymphoblastoid cell growth by repressing p16INK4A and p14ARF expression, Proc Natl Acad Sci U S A. 108, 1919–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Harth-Hertle ML, Scholz BA, Erhard F, Glaser LV, Dolken L, Zimmer R & Kempkes B (2013) Inactivation of intergenic enhancers by EBNA3A initiates and maintains polycomb signatures across a chromatin domain encoding CXCL10 and CXCL9, PLoS Pathog. 9, e1003638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Skalska L, White RE, Franz M, Ruhmann M & Allday MJ (2010) Epigenetic repression of p16(INK4A) by latent Epstein-Barr virus requires the interaction of EBNA3A and EBNA3C with CtBP, PLoS Pathog. 6, e1000951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Parker GA, Crook T, Bain M, Sara EA, Farrell PJ & Allday MJ (1996) Epstein-Barr virus nuclear antigen (EBNA)3C is an immortalizing oncoprotein with similar properties to adenovirus E1A and papillomavirus E7, Oncogene. 13, 2541–9. [PubMed] [Google Scholar]
- 170.Parker GA, Touitou R & Allday MJ (2000) Epstein-Barr virus EBNA3C can disrupt multiple cell cycle checkpoints and induce nuclear division divorced from cytokinesis, Oncogene. 19, 700–9. [DOI] [PubMed] [Google Scholar]
- 171.Cai Q, Guo Y, Xiao B, Banerjee S, Saha A, Lu J, Glisovic T & Robertson ES (2011) Epstein-Barr virus nuclear antigen 3C stabilizes Gemin3 to block p53-mediated apoptosis, PLoS Pathog. 7, e1002418. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 172.Saha A, Murakami M, Kumar P, Bajaj B, Sims K & Robertson ES (2009) Epstein-Barr virus nuclear antigen 3C augments Mdm2-mediated p53 ubiquitination and degradation by deubiquitinating Mdm2, J Virol. 83, 4652–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Yi F, Saha A, Murakami M, Kumar P, Knight JS, Cai Q, Choudhuri T & Robertson ES (2009) Epstein-Barr virus nuclear antigen 3C targets p53 and modulates its transcriptional and apoptotic activities, Virology. 388, 236–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Jha HC, Yang K, El-Naccache DW, Sun Z & Robertson ES (2015) EBNA3C regulates p53 through induction of Aurora kinase B, Oncotarget. 6, 5788–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Chen A, Divisconte M, Jiang X, Quink C & Wang F (2005) Epstein-Barr virus with the latent infection nuclear antigen 3B completely deleted is still competent for B-cell growth transformation in vitro, J Virol. 79, 4506–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Tomkinson B, Robertson E & Kieff E (1993) Epstein-Barr virus nuclear proteins EBNA-3A and EBNA-3C are essential for B-lymphocyte growth transformation, J Virol. 67, 2014–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Cooper A, Johannsen E, Maruo S, Cahir-McFarland E, Illanes D, Davidson D & Kieff E (2003) EBNA3A association with RBP-Jkappa down-regulates c-myc and Epstein-Barr virus-transformed lymphoblast growth, J Virol. 77, 999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Johannsen E, Miller CL, Grossman SR & Kieff E (1996) EBNA-2 and EBNA-3C extensively and mutually exclusively associate with RBPJkappa in Epstein-Barr virus-transformed B lymphocytes, J Virol. 70, 4179–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Robertson ES, Grossman S, Johannsen E, Miller C, Lin J, Tomkinson B & Kieff E (1995) Epstein-Barr virus nuclear protein 3C modulates transcription through interaction with the sequence-specific DNA-binding protein J kappa, J Virol. 69, 3108–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Robertson ES, Lin J & Kieff E (1996) The amino-terminal domains of Epstein-Barr virus nuclear proteins 3A, 3B, and 3C interact with RBPJ(kappa), J Virol. 70, 3068–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Radkov SA, Touitou R, Brehm A, Rowe M, West M, Kouzarides T & Allday MJ (1999) Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription, J Virol. 73, 5688–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Knight JS, Lan K, Subramanian C & Robertson ES (2003) Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines, J Virol. 77, 4261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Hickabottom M, Parker GA, Freemont P, Crook T & Allday MJ (2002) Two nonconsensus sites in the Epstein-Barr virus oncoprotein EBNA3A cooperate to bind the co-repressor carboxyl-terminal-binding protein (CtBP), J Biol Chem. 277, 47197–204. [DOI] [PubMed] [Google Scholar]
- 184.Allday MJ, Bazot Q & White RE (2015) The EBNA3 Family: Two Oncoproteins and a Tumour Suppressor that Are Central to the Biology of EBV in B Cells, Curr Top Microbiol Immunol. 391, 61–117. [DOI] [PubMed] [Google Scholar]
- 185.Bhattacharjee S, Ghosh Roy S, Bose P & Saha A (2016) Role of EBNA-3 Family Proteins in EBV Associated B-cell Lymphomagenesis, Front Microbiol. 7, 457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Subramanian C, Knight JS & Robertson ES (2002) The Epstein Barr nuclear antigen EBNA3C regulates transcription, cell transformation and cell migration, Front Biosci. 7, d704–16. [DOI] [PubMed] [Google Scholar]
- 187.Lerner MR, Andrews NC, Miller G & Steitz JA (1981) Two small RNAs encoded by Epstein-Barr virus and complexed with protein are precipitated by antibodies from patients with systemic lupus erythematosus, Proc Natl Acad Sci U S A. 78, 805–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Rymo L (1979) Identification of transcribed regions of Epstein-Barr virus DNA in Burkitt lymphoma-derived cells, J Virol. 32, 8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Gulley ML & Raab-Traub N (1993) Detection of Epstein-Barr virus in human tissues by molecular genetic techniques, Arch Pathol Lab Med. 117, 1115–20. [PubMed] [Google Scholar]
- 190.Chang KL, Chen YY, Shibata D & Weiss LM (1992) Description of an in situ hybridization methodology for detection of Epstein-Barr virus RNA in paraffin-embedded tissues, with a survey of normal and neoplastic tissues, Diagn Mol Pathol. 1, 246–55. [PubMed] [Google Scholar]
- 191.Skalsky RL & Cullen BR (2015) EBV Noncoding RNAs, Curr Top Microbiol Immunol. 391, 181–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Komano J, Maruo S, Kurozumi K, Oda T & Takada K (1999) Oncogenic role of Epstein-Barr virus-encoded RNAs in Burkitt’s lymphoma cell line Akata, J Virol. 73, 9827–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Fok V, Friend K & Steitz JA (2006) Epstein-Barr virus noncoding RNAs are confined to the nucleus, whereas their partner, the human La protein, undergoes nucleocytoplasmic shuttling, J Cell Biol. 173, 319–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Fok V, Mitton-Fry RM, Grech A & Steitz JA (2006) Multiple domains of EBER 1, an Epstein-Barr virus noncoding RNA, recruit human ribosomal protein L22, RNA. 12, 872–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Kitagawa N, Goto M, Kurozumi K, Maruo S, Fukayama M, Naoe T, Yasukawa M, Hino K, Suzuki T, Todo S & Takada K (2000) Epstein-Barr virus-encoded poly(A)(−) RNA supports Burkitt’s lymphoma growth through interleukin-10 induction, EMBO J. 19, 6742–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Iwakiri D, Sheen TS, Chen JY, Huang DP & Takada K (2005) Epstein-Barr virus-encoded small RNA induces insulin-like growth factor 1 and supports growth of nasopharyngeal carcinoma-derived cell lines, Oncogene. 24, 1767–73. [DOI] [PubMed] [Google Scholar]
- 197.Iwakiri D, Eizuru Y, Tokunaga M & Takada K (2003) Autocrine growth of Epstein-Barr virus-positive gastric carcinoma cells mediated by an Epstein-Barr virus-encoded small RNA, Cancer Res. 63, 7062–7. [PubMed] [Google Scholar]
- 198.Yang L, Aozasa K, Oshimi K & Takada K (2004) Epstein-Barr virus (EBV)-encoded RNA promotes growth of EBV-infected T cells through interleukin-9 induction, Cancer Res. 64, 5332–7. [DOI] [PubMed] [Google Scholar]
- 199.Wu Y, Maruo S, Yajima M, Kanda T & Takada K (2007) Epstein-Barr virus (EBV)-encoded RNA 2 (EBER2) but not EBER1 plays a critical role in EBV-induced B-cell growth transformation, J Virol. 81, 11236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Iwakiri D, Zhou L, Samanta M, Matsumoto M, Ebihara T, Seya T, Imai S, Fujieda M, Kawa K & Takada K (2009) Epstein-Barr virus (EBV)-encoded small RNA is released from EBV-infected cells and activates signaling from Toll-like receptor 3, J Exp Med. 206, 2091–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Nanbo A, Inoue K, Adachi-Takasawa K & Takada K (2002) Epstein-Barr virus RNA confers resistance to interferon-alpha-induced apoptosis in Burkitt’s lymphoma, EMBO J. 21, 954–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Samanta M & Takada K (2010) Modulation of innate immunity system by Epstein-Barr virus-encoded non-coding RNA and oncogenesis, Cancer Sci. 101, 29–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Iwakiri D & Takada K (2010) Role of EBERs in the pathogenesis of EBV infection, Adv Cancer Res. 107, 119–36. [DOI] [PubMed] [Google Scholar]
- 204.Parsons CH, Adang LA, Overdevest J, O’Connor CM, Taylor JR Jr., Camerini D & Kedes DH (2006) KSHV targets multiple leukocyte lineages during long-term productive infection in NOD/SCID mice, J Clin Invest. 116, 1963–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Gregory SM, Wang L, West JA, Dittmer DP & Damania B (2012) Latent Kaposi’s sarcoma-associated herpesvirus infection of monocytes downregulates expression of adaptive immune response costimulatory receptors and proinflammatory cytokines, J Virol. 86, 3916–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Myoung J & Ganem D (2011) Infection of primary human tonsillar lymphoid cells by KSHV reveals frequent but abortive infection of T cells, Virology. 413, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Kaleeba JA & Berger EA (2006) Kaposi’s sarcoma-associated herpesvirus fusion-entry receptor: cystine transporter xCT, Science. 311, 1921–4. [DOI] [PubMed] [Google Scholar]
- 208.Hahn AS, Kaufmann JK, Wies E, Naschberger E, Panteleev-Ivlev J, Schmidt K, Holzer A, Schmidt M, Chen J, Konig S, Ensser A, Myoung J, Brockmeyer NH, Sturzl M, Fleckenstein B & Neipel F (2012) The ephrin receptor tyrosine kinase A2 is a cellular receptor for Kaposi’s sarcoma-associated herpesvirus, Nat Med. 18, 961–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Chakraborty S, Veettil MV, Bottero V & Chandran B (2012) Kaposi’s sarcoma-associated herpesvirus interacts with EphrinA2 receptor to amplify signaling essential for productive infection, Proc Natl Acad Sci U S A. 109, E1163–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Rappocciolo G, Jenkins FJ, Hensler HR, Piazza P, Jais M, Borowski L, Watkins SC & Rinaldo CR Jr. (2006) DC-SIGN is a receptor for human herpesvirus 8 on dendritic cells and macrophages, J Immunol. 176, 1741–9. [DOI] [PubMed] [Google Scholar]
- 211.Kumar B & Chandran B (2016) KSHV Entry and Trafficking in Target Cells-Hijacking of Cell Signal Pathways, Actin and Membrane Dynamics, Viruses. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Akula SM, Naranatt PP, Walia NS, Wang FZ, Fegley B & Chandran B (2003) Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) infection of human fibroblast cells occurs through endocytosis, J Virol. 77, 7978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Akula SM, Pramod NP, Wang FZ & Chandran B (2001) Human herpesvirus 8 envelope-associated glycoprotein B interacts with heparan sulfate-like moieties, Virology. 284, 235–49. [DOI] [PubMed] [Google Scholar]
- 214.Naranatt PP, Akula SM & Chandran B (2002) Characterization of gamma2-human herpesvirus-8 glycoproteins gH and gL, Arch Virol. 147, 1349–70. [DOI] [PubMed] [Google Scholar]
- 215.Krishnan HH, Sharma-Walia N, Streblow DN, Naranatt PP & Chandran B (2006) Focal adhesion kinase is critical for entry of Kaposi’s sarcoma-associated herpesvirus into target cells, J Virol. 80, 1167–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Sharma-Walia N, Naranatt PP, Krishnan HH, Zeng L & Chandran B (2004) Kaposi’s sarcoma-associated herpesvirus/human herpesvirus 8 envelope glycoprotein gB induces the integrin-dependent focal adhesion kinase-Src-phosphatidylinositol 3-kinase-rho GTPase signal pathways and cytoskeletal rearrangements, J Virol. 78, 4207–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Naranatt PP, Akula SM, Zien CA, Krishnan HH & Chandran B (2003) Kaposi’s sarcoma-associated herpesvirus induces the phosphatidylinositol 3-kinase-PKC-zeta-MEK-ERK signaling pathway in target cells early during infection: implications for infectivity, J Virol. 77, 1524–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Naranatt PP, Krishnan HH, Smith MS & Chandran B (2005) Kaposi’s sarcoma-associated herpesvirus modulates microtubule dynamics via RhoA-GTP-diaphanous 2 signaling and utilizes the dynein motors to deliver its DNA to the nucleus, J Virol. 79, 1191–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Sharma-Walia N, Krishnan HH, Naranatt PP, Zeng L, Smith MS & Chandran B (2005) ERK1/2 and MEK1/2 induced by Kaposi’s sarcoma-associated herpesvirus (human herpesvirus 8) early during infection of target cells are essential for expression of viral genes and for establishment of infection, J Virol. 79, 10308–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Naranatt PP, Krishnan HH, Svojanovsky SR, Bloomer C, Mathur S & Chandran B (2004) Host gene induction and transcriptional reprogramming in Kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8)-infected endothelial, fibroblast, and B cells: insights into modulation events early during infection, Cancer Res. 64, 72–84. [DOI] [PubMed] [Google Scholar]
- 221.Sun R, Lin SF, Gradoville L, Yuan Y, Zhu F & Miller G (1998) A viral gene that activates lytic cycle expression of Kaposi’s sarcoma-associated herpesvirus, Proc Natl Acad Sci U S A. 95, 10866–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Lukac DM, Renne R, Kirshner JR & Ganem D (1998) Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein, Virology. 252, 304–12. [DOI] [PubMed] [Google Scholar]
- 223.Prasad A, Remick J & Zeichner SL (2013) Activation of human herpesvirus replication by apoptosis, J Virol. 87, 10641–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Prasad A, Lu M, Lukac DM & Zeichner SL (2012) An alternative Kaposi’s sarcoma-associated herpesvirus replication program triggered by host cell apoptosis, J Virol. 86, 4404–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Shin HJ, DeCotiis J, Giron M, Palmeri D & Lukac DM (2016) Correction for Shin et al., Histone Deacetylase Classes I and II Regulate Kaposi’s Sarcoma-Associated Herpesvirus Reactivation, J Virol. 90, 5845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Darst RP, Haecker I, Pardo CE, Renne R & Kladde MP (2013) Epigenetic diversity of Kaposi’s sarcoma-associated herpesvirus, Nucleic Acids Res. 41, 2993–3009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Martin DF, Kuppermann BD, Wolitz RA, Palestine AG, Li H & Robinson CA (1999) Oral ganciclovir for patients with cytomegalovirus retinitis treated with a ganciclovir implant. Roche Ganciclovir Study Group, N Engl J Med. 340, 1063–70. [DOI] [PubMed] [Google Scholar]
- 228.Domsic JF, Chen HS, Lu F, Marmorstein R & Lieberman PM (2013) Molecular basis for oligomeric-DNA binding and episome maintenance by KSHV LANA, PLoS Pathog. 9, e1003672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Hellert J, Weidner-Glunde M, Krausze J, Richter U, Adler H, Fedorov R, Pietrek M, Ruckert J, Ritter C, Schulz TF & Luhrs T (2013) A structural basis for BRD2/4-mediated host chromatin interaction and oligomer assembly of Kaposi sarcoma-associated herpesvirus and murine gammaherpesvirus LANA proteins, PLoS Pathog. 9, e1003640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Hu J, Yang Y, Turner PC, Jain V, McIntyre LM & Renne R (2014) LANA binds to multiple active viral and cellular promoters and associates with the H3K4methyltransferase hSET1 complex, PLoS Pathog. 10, e1004240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Chandriani S & Ganem D (2010) Array-based transcript profiling and limiting-dilution reverse transcription-PCR analysis identify additional latent genes in Kaposi’s sarcoma-associated herpesvirus, J Virol. 84, 5565–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Lee H, Guo J, Li M, Choi JK, DeMaria M, Rosenzweig M & Jung JU (1998) Identification of an immunoreceptor tyrosine-based activation motif of K1 transforming protein of Kaposi’s sarcoma-associated herpesvirus, Mol Cell Biol. 18, 5219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lee H, Veazey R, Williams K, Li M, Guo J, Neipel F, Fleckenstein B, Lackner A, Desrosiers RC & Jung JU (1998) Deregulation of cell growth by the K1 gene of Kaposi’s sarcoma-associated herpesvirus, Nat Med. 4, 435–40. [DOI] [PubMed] [Google Scholar]
- 234.Lagunoff M, Majeti R, Weiss A & Ganem D (1999) Deregulated signal transduction by the K1 gene product of Kaposi’s sarcoma-associated herpesvirus, Proc Natl Acad Sci U S A. 96, 5704–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Lee BS, Lee SH, Feng P, Chang H, Cho NH & Jung JU (2005) Characterization of the Kaposi’s sarcoma-associated herpesvirus K1 signalosome, J Virol. 79, 12173–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Tomlinson CC & Damania B (2004) The K1 protein of Kaposi’s sarcoma-associated herpesvirus activates the Akt signaling pathway, J Virol. 78, 1918–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Anders PM, Zhang Z, Bhende PM, Giffin L & Damania B (2016) The KSHV K1 Protein Modulates AMPK Function to Enhance Cell Survival, PLoS Pathog. 12, e1005985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Steinbruck L, Gustems M, Medele S, Schulz TF, Lutter D & Hammerschmidt W (2015) K1 and K15 of Kaposi’s Sarcoma-Associated Herpesvirus Are Partial Functional Homologues of Latent Membrane Protein 2A of Epstein-Barr Virus, J Virol. 89, 7248–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Wang S, Wang S, Maeng H, Young DP, Prakash O, Fayad LE, Younes A & Samaniego F (2007) K1 protein of human herpesvirus 8 suppresses lymphoma cell Fas-mediated apoptosis, Blood. 109, 2174–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Wen KW & Damania B (2010) Hsp90 and Hsp40/Erdj3 are required for the expression and anti-apoptotic function of KSHV K1, Oncogene. 29, 3532–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Wang L, Dittmer DP, Tomlinson CC, Fakhari FD & Damania B (2006) Immortalization of primary endothelial cells by the K1 protein of Kaposi’s sarcoma-associated herpesvirus, Cancer Res. 66, 3658–66. [DOI] [PubMed] [Google Scholar]
- 242.Prakash O, Tang ZY, Peng X, Coleman R, Gill J, Farr G & Samaniego F (2002) Tumorigenesis and aberrant signaling in transgenic mice expressing the human herpesvirus-8 K1 gene, J Natl Cancer Inst. 94, 926–35. [DOI] [PubMed] [Google Scholar]
- 243.Berkova Z, Wang S, Sehgal L, Patel KP, Prakash O & Samaniego F (2015) Lymphoid hyperplasia and lymphoma in KSHV K1 transgenic mice, Histol Histopathol. 30, 559–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Brinkmann MM, Glenn M, Rainbow L, Kieser A, Henke-Gendo C & Schulz TF (2003) Activation of mitogen-activated protein kinase and NF-kappaB pathways by a Kaposi’s sarcoma-associated herpesvirus K15 membrane protein, J Virol. 77, 9346–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Gramolelli S, Weidner-Glunde M, Abere B, Viejo-Borbolla A, Bala K, Ruckert J, Kremmer E & Schulz TF (2015) Inhibiting the Recruitment of PLCgamma1 to Kaposi’s Sarcoma Herpesvirus K15 Protein Reduces the Invasiveness and Angiogenesis of Infected Endothelial Cells, PLoS Pathog. 11, e1005105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Abere B, Samarina N, Gramolelli S, Ruckert J, Gerold G, Pich A & Schulz TF (2018) Kaposi’s Sarcoma-Associated Herpesvirus Nonstructural Membrane Protein pK15 Recruits the Class II Phosphatidylinositol 3-Kinase PI3K-C2alpha To Activate Productive Viral Replication, J Virol. 92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Choi JK, Lee BS, Shim SN, Li M & Jung JU (2000) Identification of the novel K15 gene at the rightmost end of the Kaposi’s sarcoma-associated herpesvirus genome, J Virol. 74, 436–46. [PMC free article] [PubMed] [Google Scholar]
- 248.Sharp TV, Wang HW, Koumi A, Hollyman D, Endo Y, Ye H, Du MQ & Boshoff C (2002) K15 protein of Kaposi’s sarcoma-associated herpesvirus is latently expressed and binds to HAX-1, a protein with antiapoptotic function, J Virol. 76, 802–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Abere B, Mamo TM, Hartmann S, Samarina N, Hage E, Ruckert J, Hotop SK, Busche G & Schulz TF (2017) The Kaposi’s sarcoma-associated herpesvirus (KSHV) non-structural membrane protein K15 is required for viral lytic replication and may represent a therapeutic target, PLoS Pathog. 13, e1006639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Ballestas ME, Chatis PA & Kaye KM (1999) Efficient persistence of extrachromosomal KSHV DNA mediated by latency-associated nuclear antigen, Science. 284, 641–4. [DOI] [PubMed] [Google Scholar]
- 251.Friborg J Jr., Kong W, Hottiger MO & Nabel GJ (1999) p53 inhibition by the LANA protein of KSHV protects against cell death, Nature. 402, 889–94. [DOI] [PubMed] [Google Scholar]
- 252.Radkov SA, Kellam P & Boshoff C (2000) The latent nuclear antigen of Kaposi sarcoma-associated herpesvirus targets the retinoblastoma-E2F pathway and with the oncogene Hras transforms primary rat cells, Nat Med. 6, 1121–7. [DOI] [PubMed] [Google Scholar]
- 253.Bubman D, Guasparri I & Cesarman E (2007) Deregulation of c-Myc in primary effusion lymphoma by Kaposi’s sarcoma herpesvirus latency-associated nuclear antigen, Oncogene. 26, 4979–86. [DOI] [PubMed] [Google Scholar]
- 254.Liu J, Martin HJ, Liao G & Hayward SD (2007) The Kaposi’s sarcoma-associated herpesvirus LANA protein stabilizes and activates c-Myc, J Virol. 81, 10451–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Fujimuro M, Wu FY, ApRhys C, Kajumbula H, Young DB, Hayward GS & Hayward SD (2003) A novel viral mechanism for dysregulation of beta-catenin in Kaposi’s sarcoma-associated herpesvirus latency, Nat Med. 9, 300–6. [DOI] [PubMed] [Google Scholar]
- 256.Lan K, Verma SC, Murakami M, Bajaj B, Kaul R & Robertson ES (2007) Kaposi’s sarcoma herpesvirus-encoded latency-associated nuclear antigen stabilizes intracellular activated Notch by targeting the Sel10 protein, Proc Natl Acad Sci U S A. 104, 16287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Fakhari FD, Jeong JH, Kanan Y & Dittmer DP (2006) The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma, J Clin Invest. 116, 735–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Sin SH, Fakhari FD & Dittmer DP (2010) The viral latency-associated nuclear antigen augments the B-cell response to antigen in vivo, J Virol. 84, 10653–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Dupin N, Fisher C, Kellam P, Ariad S, Tulliez M, Franck N, van Marck E, Salmon D, Gorin I, Escande JP, Weiss RA, Alitalo K & Boshoff C (1999) Distribution of human herpesvirus-8 latently infected cells in Kaposi’s sarcoma, multicentric Castleman’s disease, and primary effusion lymphoma, Proc Natl Acad Sci U S A. 96, 4546–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Kedes DH, Lagunoff M, Renne R & Ganem D (1997) Identification of the gene encoding the major latency-associated nuclear antigen of the Kaposi’s sarcoma-associated herpesvirus, J Clin Invest. 100, 2606–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Mariggio G, Koch S, Zhang G, Weidner-Glunde M, Ruckert J, Kati S, Santag S & Schulz TF (2017) Kaposi Sarcoma Herpesvirus (KSHV) Latency-Associated Nuclear Antigen (LANA) recruits components of the MRN (Mre11-Rad50-NBS1) repair complex to modulate an innate immune signaling pathway and viral latency, PLoS Pathog. 13, e1006335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Cesarman E, Nador RG, Bai F, Bohenzky RA, Russo JJ, Moore PS, Chang Y & Knowles DM (1996) Kaposi’s sarcoma-associated herpesvirus contains G protein-coupled receptor and cyclin D homologs which are expressed in Kaposi’s sarcoma and malignant lymphoma, J Virol. 70, 8218–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Li M, Lee H, Yoon DW, Albrecht JC, Fleckenstein B, Neipel F & Jung JU (1997) Kaposi’s sarcoma-associated herpesvirus encodes a functional cyclin, J Virol. 71, 1984–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Swanton C, Mann DJ, Fleckenstein B, Neipel F, Peters G & Jones N (1997) Herpes viral cyclin/Cdk6 complexes evade inhibition by CDK inhibitor proteins, Nature. 390, 184–7. [DOI] [PubMed] [Google Scholar]
- 265.Godden-Kent D, Talbot SJ, Boshoff C, Chang Y, Moore P, Weiss RA & Mittnacht S (1997) The cyclin encoded by Kaposi’s sarcoma-associated herpesvirus stimulates cdk6 to phosphorylate the retinoblastoma protein and histone H1, J Virol. 71, 4193–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Ojala PM, Tiainen M, Salven P, Veikkola T, Castanos-Velez E, Sarid R, Biberfeld P & Makela TP (1999) Kaposi’s sarcoma-associated herpesvirus-encoded v-cyclin triggers apoptosis in cells with high levels of cyclin-dependent kinase 6, Cancer Res. 59, 4984–9. [PubMed] [Google Scholar]
- 267.Ellis M, Chew YP, Fallis L, Freddersdorf S, Boshoff C, Weiss RA, Lu X & Mittnacht S (1999) Degradation of p27(Kip) cdk inhibitor triggered by Kaposi’s sarcoma virus cyclin-cdk6 complex, EMBO J. 18, 644–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sarek G, Jarviluoma A, Moore HM, Tojkander S, Vartia S, Biberfeld P, Laiho M & Ojala PM (2010) Nucleophosmin phosphorylation by v-cyclin-CDK6 controls KSHV latency, PLoS Pathog. 6, e1000818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.DiMaio TA, Vogt DT & Lagunoff M (2020) KSHV requires vCyclin to overcome replicative senescence in primary human lymphatic endothelial cells, PLoS Pathog. 16, e1008634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Verschuren EW, Klefstrom J, Evan GI & Jones N (2002) The oncogenic potential of Kaposi’s sarcoma-associated herpesvirus cyclin is exposed by p53 loss in vitro and in vivo, Cancer Cell. 2, 229–41. [DOI] [PubMed] [Google Scholar]
- 271.Verschuren EW, Hodgson JG, Gray JW, Kogan S, Jones N & Evan GI (2004) The role of p53 in suppression of KSHV cyclin-induced lymphomagenesis, Cancer Res. 64, 581–9. [DOI] [PubMed] [Google Scholar]
- 272.Ojala PM, Yamamoto K, Castanos-Velez E, Biberfeld P, Korsmeyer SJ & Makela TP (2000) The apoptotic v-cyclin-CDK6 complex phosphorylates and inactivates Bcl-2, Nat Cell Biol. 2, 819–25. [DOI] [PubMed] [Google Scholar]
- 273.Thome M, Schneider P, Hofmann K, Fickenscher H, Meinl E, Neipel F, Mattmann C, Burns K, Bodmer JL, Schroter M, Scaffidi C, Krammer PH, Peter ME & Tschopp J (1997) Viral FLICE-inhibitory proteins (FLIPs) prevent apoptosis induced by death receptors, Nature. 386, 517–21. [DOI] [PubMed] [Google Scholar]
- 274.Sturzl M, Hohenadl C, Zietz C, Castanos-Velez E, Wunderlich A, Ascherl G, Biberfeld P, Monini P, Browning PJ & Ensoli B (1999) Expression of K13/v-FLIP gene of human herpesvirus 8 and apoptosis in Kaposi’s sarcoma spindle cells, J Natl Cancer Inst. 91, 1725–33. [DOI] [PubMed] [Google Scholar]
- 275.Guasparri I, Keller SA & Cesarman E (2004) KSHV vFLIP is essential for the survival of infected lymphoma cells, J Exp Med. 199, 993–1003. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 276.Chaudhary PM, Jasmin A, Eby MT & Hood L (1999) Modulation of the NF-kappa B pathway by virally encoded death effector domains-containing proteins, Oncogene. 18, 5738–46. [DOI] [PubMed] [Google Scholar]
- 277.Guasparri I, Wu H & Cesarman E (2006) The KSHV oncoprotein vFLIP contains a TRAF-interacting motif and requires TRAF2 and TRAF3 for signalling, EMBO Rep. 7, 114–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Grossmann C, Podgrabinska S, Skobe M & Ganem D (2006) Activation of NF-kappaB by the latent vFLIP gene of Kaposi’s sarcoma-associated herpesvirus is required for the spindle shape of virus-infected endothelial cells and contributes to their proinflammatory phenotype, J Virol. 80, 7179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.An J, Sun Y, Sun R & Rettig MB (2003) Kaposi’s sarcoma-associated herpesvirus encoded vFLIP induces cellular IL-6 expression: the role of the NF-kappaB and JNK/AP1 pathways, Oncogene. 22, 3371–85. [DOI] [PubMed] [Google Scholar]
- 280.Liu L, Eby MT, Rathore N, Sinha SK, Kumar A & Chaudhary PM (2002) The human herpes virus 8-encoded viral FLICE inhibitory protein physically associates with and persistently activates the Ikappa B kinase complex, J Biol Chem. 277, 13745–51. [DOI] [PubMed] [Google Scholar]
- 281.Keller SA, Hernandez-Hopkins D, Vider J, Ponomarev V, Hyjek E, Schattner EJ & Cesarman E (2006) NF-kappaB is essential for the progression of KSHV- and EBV-infected lymphomas in vivo, Blood. 107, 3295–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Lee JS, Li Q, Lee JY, Lee SH, Jeong JH, Lee HR, Chang H, Zhou FC, Gao SJ, Liang C & Jung JU (2009) FLIP-mediated autophagy regulation in cell death control, Nat Cell Biol. 11, 1355–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 283.Nayar U, Lu P, Goldstein RL, Vider J, Ballon G, Rodina A, Taldone T, Erdjument-Bromage H, Chomet M, Blasberg R, Melnick A, Cerchietti L, Chiosis G, Wang YL & Cesarman E (2013) Targeting the Hsp90-associated viral oncoproteome in gammaherpesvirus-associated malignancies, Blood. 122, 2837–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Ballon G, Chen K, Perez R, Tam W & Cesarman E (2011) Kaposi sarcoma herpesvirus (KSHV) vFLIP oncoprotein induces B cell transdifferentiation and tumorigenesis in mice, J Clin Invest. 121, 1141–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Ballon G, Akar G & Cesarman E (2015) Systemic expression of Kaposi sarcoma herpesvirus (KSHV) Vflip in endothelial cells leads to a profound proinflammatory phenotype and myeloid lineage remodeling in vivo, PLoS Pathog. 11, e1004581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Arvanitakis L, Geras-Raaka E, Varma A, Gershengorn MC & Cesarman E (1997) Human herpesvirus KSHV encodes a constitutively active G-protein-coupled receptor linked to cell proliferation, Nature. 385, 347–50. [DOI] [PubMed] [Google Scholar]
- 287.Bais C, Santomasso B, Coso O, Arvanitakis L, Raaka EG, Gutkind JS, Asch AS, Cesarman E, Gershengorn MC & Mesri EA (1998) G-protein-coupled receptor of Kaposi’s sarcoma-associated herpesvirus is a viral oncogene and angiogenesis activator, Nature. 391, 86–9. [DOI] [PubMed] [Google Scholar]
- 288.Bakken T, He M & Cannon ML (2010) The phosphatase Shp2 is required for signaling by the Kaposi’s sarcoma-associated herpesvirus viral GPCR in primary endothelial cells, Virology. 397, 379–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Martin D, Galisteo R, Molinolo AA, Wetzker R, Hirsch E & Gutkind JS (2011) PI3Kgamma mediates kaposi’s sarcoma-associated herpesvirus vGPCR-induced sarcomagenesis, Cancer Cell. 19, 805–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Sodhi A, Montaner S, Patel V, Gomez-Roman JJ, Li Y, Sausville EA, Sawai ET & Gutkind JS (2004) Akt plays a central role in sarcomagenesis induced by Kaposi’s sarcoma herpesvirus-encoded G protein-coupled receptor, Proc Natl Acad Sci U S A. 101, 4821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Montaner S, Sodhi A, Pece S, Mesri EA & Gutkind JS (2001) The Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor promotes endothelial cell survival through the activation of Akt/protein kinase B, Cancer Res. 61, 2641–8. [PubMed] [Google Scholar]
- 292.Sodhi A, Chaisuparat R, Hu J, Ramsdell AK, Manning BD, Sausville EA, Sawai ET, Molinolo A, Gutkind JS & Montaner S (2006) The TSC2/mTOR pathway drives endothelial cell transformation induced by the Kaposi’s sarcoma-associated herpesvirus G protein-coupled receptor, Cancer Cell. 10, 133–43. [DOI] [PubMed] [Google Scholar]
- 293.Jham BC, Ma T, Hu J, Chaisuparat R, Friedman ER, Pandolfi PP, Schneider A, Sodhi A & Montaner S (2011) Amplification of the angiogenic signal through the activation of the TSC/mTOR/HIF axis by the KSHV vGPCR in Kaposi’s sarcoma, PLoS One. 6, e19103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Sodhi A, Montaner S, Patel V, Zohar M, Bais C, Mesri EA & Gutkind JS (2000) The Kaposi’s sarcoma-associated herpes virus G protein-coupled receptor up-regulates vascular endothelial growth factor expression and secretion through mitogen-activated protein kinase and p38 pathways acting on hypoxia-inducible factor 1alpha, Cancer Res. 60, 4873–80. [PubMed] [Google Scholar]
- 295.Smit MJ, Verzijl D, Casarosa P, Navis M, Timmerman H & Leurs R (2002) Kaposi’s sarcoma-associated herpesvirus-encoded G protein-coupled receptor ORF74 constitutively activates p44/p42 MAPK and Akt via G(i) and phospholipase C-dependent signaling pathways, J Virol. 76, 1744–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Martin D, Galisteo R, Ji Y, Montaner S & Gutkind JS (2008) An NF-kappaB gene expression signature contributes to Kaposi’s sarcoma virus vGPCR-induced direct and paracrine neoplasia, Oncogene. 27, 1844–52. [DOI] [PubMed] [Google Scholar]
- 297.Pati S, Cavrois M, Guo HG, Foulke JS Jr., Kim J, Feldman RA & Reitz M (2001) Activation of NF-kappaB by the human herpesvirus 8 chemokine receptor ORF74: evidence for a paracrine model of Kaposi’s sarcoma pathogenesis, J Virol. 75, 8660–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Angelova M, Ferris M, Swan KF, McFerrin HE, Pridjian G, Morris CA & Sullivan DE (2014) Kaposi’s sarcoma-associated herpesvirus G-protein coupled receptor activates the canonical Wnt/beta-catenin signaling pathway, Virol J. 11, 218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Montaner S, Sodhi A, Servitja JM, Ramsdell AK, Barac A, Sawai ET & Gutkind JS (2004) The small GTPase Rac1 links the Kaposi sarcoma-associated herpesvirus vGPCR to cytokine secretion and paracrine neoplasia, Blood. 104, 2903–11. [DOI] [PubMed] [Google Scholar]
- 300.Bais C, Van Geelen A, Eroles P, Mutlu A, Chiozzini C, Dias S, Silverstein RL, Rafii S & Mesri EA (2003) Kaposi’s sarcoma associated herpesvirus G protein-coupled receptor immortalizes human endothelial cells by activation of the VEGF receptor-2/ KDR, Cancer Cell. 3, 131–43. [DOI] [PubMed] [Google Scholar]
- 301.Yang TY, Chen SC, Leach MW, Manfra D, Homey B, Wiekowski M, Sullivan L, Jenh CH, Narula SK, Chensue SW & Lira SA (2000) Transgenic expression of the chemokine receptor encoded by human herpesvirus 8 induces an angioproliferative disease resembling Kaposi’s sarcoma, J Exp Med. 191, 445–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Guo HG, Sadowska M, Reid W, Tschachler E, Hayward G & Reitz M (2003) Kaposi’s sarcoma-like tumors in a human herpesvirus 8 ORF74 transgenic mouse, J Virol. 77, 2631–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Jensen KK, Manfra DJ, Grisotto MG, Martin AP, Vassileva G, Kelley K, Schwartz TW & Lira SA (2005) The human herpes virus 8-encoded chemokine receptor is required for angioproliferation in a murine model of Kaposi’s sarcoma, J Immunol. 174, 3686–94. [DOI] [PubMed] [Google Scholar]
- 304.Mutlu AD, Cavallin LE, Vincent L, Chiozzini C, Eroles P, Duran EM, Asgari Z, Hooper AT, La Perle KM, Hilsher C, Gao SJ, Dittmer DP, Rafii S & Mesri EA (2007) In vivo-restricted and reversible malignancy induced by human herpesvirus-8 KSHV: a cell and animal model of virally induced Kaposi’s sarcoma, Cancer Cell. 11, 245–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Park J, Lee D, Seo T, Chung J & Choe J (2000) Kaposi’s sarcoma-associated herpesvirus (human herpesvirus-8) open reading frame 36 protein is a serine protein kinase, J Gen Virol. 81, 1067–71. [DOI] [PubMed] [Google Scholar]
- 306.Haque M, Wang V, Davis DA, Zheng ZM & Yarchoan R (2006) Genetic organization and hypoxic activation of the Kaposi’s sarcoma-associated herpesvirus ORF34–37 gene cluster, J Virol. 80, 7037–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Kuny CV, Chinchilla K, Culbertson MR & Kalejta RF (2010) Cyclin-dependent kinase-like function is shared by the beta- and gamma- subset of the conserved herpesvirus protein kinases, PLoS Pathog. 6, e1001092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Hamza MS, Reyes RA, Izumiya Y, Wisdom R, Kung HJ & Luciw PA (2004) ORF36 protein kinase of Kaposi’s sarcoma herpesvirus activates the c-Jun N-terminal kinase signaling pathway, J Biol Chem. 279, 38325–30. [DOI] [PubMed] [Google Scholar]
- 309.Bhatt AP, Wong JP, Weinberg MS, Host KM, Giffin LC, Buijnink J, van Dijk E, Izumiya Y, Kung HJ, Temple BR & Damania B (2016) A viral kinase mimics S6 kinase to enhance cell proliferation, Proc Natl Acad Sci U S A. 113, 7876–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Anders PM, Montgomery ND, Montgomery SA, Bhatt AP, Dittmer DP & Damania B (2018) Human herpesvirus-encoded kinase induces B cell lymphomas in vivo, J Clin Invest. 128, 2519–2534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Blum KA, Lozanski G & Byrd JC (2004) Adult Burkitt leukemia and lymphoma, Blood. 104, 3009–20. [DOI] [PubMed] [Google Scholar]
- 312.Burkitt D (1962) A children’s cancer dependent on climatic factors, Nature. 194, 232–4. [DOI] [PubMed] [Google Scholar]
- 313.Booth K, Burkitt DP, Bassett DJ, Cooke RA & Biddulph J (1967) Burkitt lymphoma in Papua, New Guinea, Br J Cancer. 21, 657–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.van den Bosch CA (2004) Is endemic Burkitt’s lymphoma an alliance between three infections and a tumour promoter?, Lancet Oncol. 5, 738–46. [DOI] [PubMed] [Google Scholar]
- 315.Johnston WT, Mutalima N, Sun D, Emmanuel B, Bhatia K, Aka P, Wu X, Borgstein E, Liomba GN, Kamiza S, Mkandawire N, Batumba M, Carpenter LM, Jaffe H, Molyneux EM, Goedert JJ, Soppet D, Newton R & Mbulaiteye SM (2014) Relationship between Plasmodium falciparum malaria prevalence, genetic diversity and endemic Burkitt lymphoma in Malawi, Sci Rep. 4, 3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.van den Bosch C (2012) A Role for RNA Viruses in the Pathogenesis of Burkitt’s Lymphoma: The Need for Reappraisal, Adv Hematol. 2012, 494758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Lim ST, Karim R, Nathwani BN, Tulpule A, Espina B & Levine AM (2005) AIDS-related Burkitt’s lymphoma versus diffuse large-cell lymphoma in the pre-highly active antiretroviral therapy (HAART) and HAART eras: significant differences in survival with standard chemotherapy, J Clin Oncol. 23, 4430–8. [DOI] [PubMed] [Google Scholar]
- 318.Dogan A, Bagdi E, Munson P & Isaacson PG (2000) CD10 and BCL-6 expression in paraffin sections of normal lymphoid tissue and B-cell lymphomas, Am J Surg Pathol. 24, 846–52. [DOI] [PubMed] [Google Scholar]
- 319.Lai R, Arber DA, Chang KL, Wilson CS & Weiss LM (1998) Frequency of bcl-2 expression in non-Hodgkin’s lymphoma: a study of 778 cases with comparison of marginal zone lymphoma and monocytoid B-cell hyperplasia, Mod Pathol. 11, 864–9. [PubMed] [Google Scholar]
- 320.Sander S, Calado DP, Srinivasan L, Kochert K, Zhang B, Rosolowski M, Rodig SJ, Holzmann K, Stilgenbauer S, Siebert R, Bullinger L & Rajewsky K (2012) Synergy between PI3K signaling and MYC in Burkitt lymphomagenesis, Cancer Cell. 22, 167–79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Love C, Sun Z, Jima D, Li G, Zhang J, Miles R, Richards KL, Dunphy CH, Choi WW, Srivastava G, Lugar PL, Rizzieri DA, Lagoo AS, Bernal-Mizrachi L, Mann KP, Flowers CR, Naresh KN, Evens AM, Chadburn A, Gordon LI, Czader MB, Gill JI, Hsi ED, Greenough A, Moffitt AB, McKinney M, Banerjee A, Grubor V, Levy S, Dunson DB & Dave SS (2012) The genetic landscape of mutations in Burkitt lymphoma, Nat Genet. 44, 1321–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Stadtman ER (1988) Biochemical markers of aging, Exp Gerontol. 23, 327–47. [DOI] [PubMed] [Google Scholar]
- 323.Kanzler H, Kuppers R, Hansmann ML & Rajewsky K (1996) Hodgkin and Reed-Sternberg cells in Hodgkin’s disease represent the outgrowth of a dominant tumor clone derived from (crippled) germinal center B cells, J Exp Med. 184, 1495–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Cahir-McFarland ED, Carter K, Rosenwald A, Giltnane JM, Henrickson SE, Staudt LM & Kieff E (2004) Role of NF-kappa B in cell survival and transcription of latent membrane protein 1-expressing or Epstein-Barr virus latency III-infected cells, J Virol. 78, 4108–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Dutton A, Woodman CB, Chukwuma MB, Last JI, Wei W, Vockerodt M, Baumforth KR, Flavell JR, Rowe M, Taylor AM, Young LS & Murray PG (2007) Bmi-1 is induced by the Epstein-Barr virus oncogene LMP1 and regulates the expression of viral target genes in Hodgkin lymphoma cells, Blood. 109, 2597–603. [DOI] [PubMed] [Google Scholar]
- 326.Biggar RJ, Jaffe ES, Goedert JJ, Chaturvedi A, Pfeiffer R & Engels EA (2006) Hodgkin lymphoma and immunodeficiency in persons with HIV/AIDS, Blood. 108, 3786–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Klimm B, Franklin J, Stein H, Eichenauer DA, Haverkamp H, Diehl V, Fuchs M, Borchmann P & Engert A (2011) Lymphocyte-depleted classical Hodgkin’s lymphoma: a comprehensive analysis from the German Hodgkin study group, J Clin Oncol. 29, 3914–20. [DOI] [PubMed] [Google Scholar]
- 328.Zukerberg LR, Collins AB, Ferry JA & Harris NL (1991) Coexpression of CD15 and CD20 by Reed-Sternberg cells in Hodgkin’s disease, Am J Pathol. 139, 475–83. [PMC free article] [PubMed] [Google Scholar]
- 329.Schmid C, Pan L, Diss T & Isaacson PG (1991) Expression of B-cell antigens by Hodgkin’s and Reed-Sternberg cells, Am J Pathol. 139, 701–7. [PMC free article] [PubMed] [Google Scholar]
- 330.Vockerodt M, Morgan SL, Kuo M, Wei W, Chukwuma MB, Arrand JR, Kube D, Gordon J, Young LS, Woodman CB & Murray PG (2008) The Epstein-Barr virus oncoprotein, latent membrane protein-1, reprograms germinal centre B cells towards a Hodgkin’s Reed-Sternberg-like phenotype, J Pathol. 216, 83–92. [DOI] [PubMed] [Google Scholar]
- 331.Foss HD, Reusch R, Demel G, Lenz G, Anagnostopoulos I, Hummel M & Stein H (1999) Frequent expression of the B-cell-specific activator protein in Reed-Sternberg cells of classical Hodgkin’s disease provides further evidence for its B-cell origin, Blood. 94, 3108–13. [PubMed] [Google Scholar]
- 332.Beltran BE, Morales D, Quinones P, Medeiros LJ, Miranda RN & Castillo JJ (2011) EBV-positive diffuse large b-cell lymphoma in young immunocompetent individuals, Clin Lymphoma Myeloma Leuk. 11, 512–6. [DOI] [PubMed] [Google Scholar]
- 333.Nicolae A, Pittaluga S, Abdullah S, Steinberg SM, Pham TA, Davies-Hill T, Xi L, Raffeld M & Jaffe ES (2015) EBV-positive large B-cell lymphomas in young patients: a nodal lymphoma with evidence for a tolerogenic immune environment, Blood. 126, 863–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Hong JY, Yoon DH, Suh C, Huh J, Do IG, Sohn I, Jo J, Jung SH, Hong ME, Yoon H, Ko YH, Kim SJ & Kim WS (2015) EBV-positive diffuse large B-cell lymphoma in young adults: is this a distinct disease entity?, Ann Oncol. 26, 548–55. [DOI] [PubMed] [Google Scholar]
- 335.Uccini S, Al-Jadiry MF, Scarpino S, Ferraro D, Alsaadawi AR, Al-Darraji AF, Moleti ML, Testi AM, Al-Hadad SA & Ruco L (2015) Epstein-Barr virus-positive diffuse large B-cell lymphoma in children: a disease reminiscent of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly, Hum Pathol. 46, 716–24. [DOI] [PubMed] [Google Scholar]
- 336.Dojcinov SD, Venkataraman G, Pittaluga S, Wlodarska I, Schrager JA, Raffeld M, Hills RK & Jaffe ES (2011) Age-related EBV-associated lymphoproliferative disorders in the Western population: a spectrum of reactive lymphoid hyperplasia and lymphoma, Blood. 117, 4726–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Castillo JJ, Beltran BE, Miranda RN, Young KH, Chavez JC & Sotomayor EM (2016) EBV-positive diffuse large B-cell lymphoma of the elderly: 2016 update on diagnosis, risk-stratification, and management, Am J Hematol. 91, 529–37. [DOI] [PubMed] [Google Scholar]
- 338.Sato A, Nakamura N, Kojima M, Ohmachi K, Carreras J, Kikuti YY, Numata H, Ohgiya D, Tazume K, Amaki J, Moriuchi M, Miyamoto M, Aoyama Y, Kawai H, Ichiki A, Hara R, Kawada H, Ogawa Y & Ando K (2014) Clinical outcome of Epstein-Barr virus-positive diffuse large B-cell lymphoma of the elderly in the rituximab era, Cancer Sci. 105, 1170–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Shimoyama Y, Yamamoto K, Asano N, Oyama T, Kinoshita T & Nakamura S (2008) Age-related Epstein-Barr virus-associated B-cell lymphoproliferative disorders: special references to lymphomas surrounding this newly recognized clinicopathologic disease, Cancer Sci. 99, 1085–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 340.Oyama T, Ichimura K, Suzuki R, Suzumiya J, Ohshima K, Yatabe Y, Yokoi T, Kojima M, Kamiya Y, Taji H, Kagami Y, Ogura M, Saito H, Morishima Y & Nakamura S (2003) Senile EBV+ B-cell lymphoproliferative disorders: a clinicopathologic study of 22 patients, Am J Surg Pathol. 27, 16–26. [DOI] [PubMed] [Google Scholar]
- 341.Oyama T, Yamamoto K, Asano N, Oshiro A, Suzuki R, Kagami Y, Morishima Y, Takeuchi K, Izumo T, Mori S, Ohshima K, Suzumiya J, Nakamura N, Abe M, Ichimura K, Sato Y, Yoshino T, Naoe T, Shimoyama Y, Kamiya Y, Kinoshita T & Nakamura S (2007) Age-related EBV-associated B-cell lymphoproliferative disorders constitute a distinct clinicopathologic group: a study of 96 patients, Clin Cancer Res. 13, 5124–32. [DOI] [PubMed] [Google Scholar]
- 342.Nakatsuka S, Yao M, Hoshida Y, Yamamoto S, Iuchi K & Aozasa K (2002) Pyothorax-associated lymphoma: a review of 106 cases, J Clin Oncol. 20, 4255–60. [DOI] [PubMed] [Google Scholar]
- 343.Carbone A, Gloghini A, Canzonieri V, Tirelli U & Gaidano G (1997) AIDS-related extranodal non-Hodgkin’s lymphomas with plasma cell differentiation, Blood. 90, 1337–8. [PubMed] [Google Scholar]
- 344.Lin L, Zhang X, Dong M, Li L, Wang X, Zhang L, Fu X, Sun Z, Wu J, Li Z, Chang Y, Wang Y, Zhou Z, Zhang M & Chen Q (2017) Human immunodeficiency virus-negative plasmablastic lymphoma: A case report and literature review, Medicine (Baltimore). 96, e6171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Castillo JJ, Bibas M & Miranda RN (2015) The biology and treatment of plasmablastic lymphoma, Blood. 125, 2323–30. [DOI] [PubMed] [Google Scholar]
- 346.Morscio J, Dierickx D, Nijs J, Verhoef G, Bittoun E, Vanoeteren X, Wlodarska I, Sagaert X & Tousseyn T (2014) Clinicopathologic comparison of plasmablastic lymphoma in HIV-positive, immunocompetent, and posttransplant patients: single-center series of 25 cases and meta-analysis of 277 reported cases, Am J Surg Pathol. 38, 875–86. [DOI] [PubMed] [Google Scholar]
- 347.Colomo L, Loong F, Rives S, Pittaluga S, Martinez A, Lopez-Guillermo A, Ojanguren J, Romagosa V, Jaffe ES & Campo E (2004) Diffuse large B-cell lymphomas with plasmablastic differentiation represent a heterogeneous group of disease entities, Am J Surg Pathol. 28, 736–47. [DOI] [PubMed] [Google Scholar]
- 348.Borenstein J, Pezzella F & Gatter KC (2007) Plasmablastic lymphomas may occur as post-transplant lymphoproliferative disorders, Histopathology. 51, 774–7. [DOI] [PubMed] [Google Scholar]
- 349.Mine S, Hishima T, Suganuma A, Fukumoto H, Sato Y, Kataoka M, Sekizuka T, Kuroda M, Suzuki T, Hasegawa H, Fukayama M & Katano H (2017) Interleukin-6-dependent growth in a newly established plasmablastic lymphoma cell line and its therapeutic targets, Sci Rep. 7, 10188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Vega F, Chang CC, Medeiros LJ, Udden MM, Cho-Vega JH, Lau CC, Finch CJ, Vilchez RA, McGregor D & Jorgensen JL (2005) Plasmablastic lymphomas and plasmablastic plasma cell myelomas have nearly identical immunophenotypic profiles, Mod Pathol. 18, 806–15. [DOI] [PubMed] [Google Scholar]
- 351.Dupin N, Diss TL, Kellam P, Tulliez M, Du MQ, Sicard D, Weiss RA, Isaacson PG & Boshoff C (2000) HHV-8 is associated with a plasmablastic variant of Castleman disease that is linked to HHV-8-positive plasmablastic lymphoma, Blood. 95, 1406–12. [PubMed] [Google Scholar]
- 352.Henle W, Henle G, Ho HC, Burtin P, Cachin Y, Clifford P, de Schryver A, de-The G, Diehl V & Klein G (1970) Antibodies to Epstein-Barr virus in nasopharyngeal carcinoma, other head and neck neoplasms, and control groups, J Natl Cancer Inst. 44, 225–31. [PubMed] [Google Scholar]
- 353.Vokes EE, Liebowitz DN & Weichselbaum RR (1997) Nasopharyngeal carcinoma, Lancet. 350, 1087–91. [DOI] [PubMed] [Google Scholar]
- 354.Argirion I, Zarins KR, Ruterbusch JJ, Vatanasapt P, Sriplung H, Seymour EK & Rozek LS (2020) Increasing incidence of Epstein-Barr virus-related nasopharyngeal carcinoma in the United States, Cancer. 126, 121–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Yu MC, Mo CC, Chong WX, Yeh FS & Henderson BE (1988) Preserved foods and nasopharyngeal carcinoma: a case-control study in Guangxi, China, Cancer Res. 48, 1954–9. [PubMed] [Google Scholar]
- 356.Yu MC, Ho JH, Lai SH & Henderson BE (1986) Cantonese-style salted fish as a cause of nasopharyngeal carcinoma: report of a case-control study in Hong Kong, Cancer Res. 46, 956–61. [PubMed] [Google Scholar]
- 357.Chua MLK, Wee JTS, Hui EP & Chan ATC (2016) Nasopharyngeal carcinoma, Lancet. 387, 1012–1024. [DOI] [PubMed] [Google Scholar]
- 358.Cosmopoulos K, Pegtel M, Hawkins J, Moffett H, Novina C, Middeldorp J & Thorley-Lawson DA (2009) Comprehensive profiling of Epstein-Barr virus microRNAs in nasopharyngeal carcinoma, J Virol. 83, 2357–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Bell AI, Groves K, Kelly GL, Croom-Carter D, Hui E, Chan ATC & Rickinson AB (2006) Analysis of Epstein-Barr virus latent gene expression in endemic Burkitt’s lymphoma and nasopharyngeal carcinoma tumour cells by using quantitative real-time PCR assays, J Gen Virol. 87, 2885–2890. [DOI] [PubMed] [Google Scholar]
- 360.Lo AK, To KF, Lo KW, Lung RW, Hui JW, Liao G & Hayward SD (2007) Modulation of LMP1 protein expression by EBV-encoded microRNAs, Proc Natl Acad Sci U S A. 104, 16164–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Horikawa T, Yoshizaki T, Kondo S, Furukawa M, Kaizaki Y & Pagano JS (2011) Epstein-Barr Virus latent membrane protein 1 induces Snail and epithelial-mesenchymal transition in metastatic nasopharyngeal carcinoma, Br J Cancer. 104, 1160–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Straathof KC, Bollard CM, Popat U, Huls MH, Lopez T, Morriss MC, Gresik MV, Gee AP, Russell HV, Brenner MK, Rooney CM & Heslop HE (2005) Treatment of nasopharyngeal carcinoma with Epstein-Barr virus--specific T lymphocytes, Blood. 105, 1898–904. [DOI] [PubMed] [Google Scholar]
- 363.Franchi A, Moroni M, Massi D, Paglierani M & Santucci M (2002) Sinonasal undifferentiated carcinoma, nasopharyngeal-type undifferentiated carcinoma, and keratinizing and nonkeratinizing squamous cell carcinoma express different cytokeratin patterns, Am J Surg Pathol. 26, 1597–604. [DOI] [PubMed] [Google Scholar]
- 364.Plaza G, Manzanal AI, Fogue L, Santon A, Martinez-Montero JC & Bellas C (2002) Association of Epstein-Barr virus and nasopharyngeal carcinoma in Caucasian patients, Ann Otol Rhinol Laryngol. 111, 210–6. [DOI] [PubMed] [Google Scholar]
- 365.Hsiao JR, Jin YT & Tsai ST (1998) EBER1 in situ hybridization as an adjuvant for diagnosis of recurrent nasopharyngeal carcinoma, Anticancer Res. 18, 4585–9. [PubMed] [Google Scholar]
- 366.Dierickx D, Tousseyn T, Sagaert X, Fieuws S, Wlodarska I, Morscio J, Brepoels L, Kuypers D, Vanhaecke J, Nevens F, Verleden G, Van Damme-Lombaerts R, Renard M, Pirenne J, De Wolf-Peeters C & Verhoef G (2013) Single-center analysis of biopsy-confirmed posttransplant lymphoproliferative disorder: incidence, clinicopathological characteristics and prognostic factors, Leuk Lymphoma. 54, 2433–40. [DOI] [PubMed] [Google Scholar]
- 367.Ghobrial IM, Habermann TM, Macon WR, Ristow KM, Larson TS, Walker RC, Ansell SM, Gores GJ, Stegall MD & McGregor CG (2005) Differences between early and late posttransplant lymphoproliferative disorders in solid organ transplant patients: are they two different diseases?, Transplantation. 79, 244–7. [DOI] [PubMed] [Google Scholar]
- 368.Nelson BP, Nalesnik MA, Bahler DW, Locker J, Fung JJ & Swerdlow SH (2000) Epstein-Barr virus-negative post-transplant lymphoproliferative disorders: a distinct entity?, Am J Surg Pathol. 24, 375–85. [DOI] [PubMed] [Google Scholar]
- 369.Kapelushnik J, Ariad S, Benharroch D, Landau D, Moser A, Delsol G & Brousset P (2001) Post renal transplantation human herpesvirus 8-associated lymphoproliferative disorder and Kaposi’s sarcoma, Br J Haematol. 113, 425–8. [DOI] [PubMed] [Google Scholar]
- 370.Matsushima AY, Strauchen JA, Lee G, Scigliano E, Hale EE, Weisse MT, Burstein D, Kamel O, Moore PS & Chang Y (1999) Posttransplantation plasmacytic proliferations related to Kaposi’s sarcoma-associated herpesvirus, Am J Surg Pathol. 23, 1393–400. [DOI] [PubMed] [Google Scholar]
- 371.Dotti G, Fiocchi R, Motta T, Facchinetti B, Chiodini B, Borleri GM, Gavazzeni G, Barbui T & Rambaldi A (1999) Primary effusion lymphoma after heart transplantation: a new entity associated with human herpesvirus-8, Leukemia. 13, 664–70. [DOI] [PubMed] [Google Scholar]
- 372.Djokic M, Le Beau MM, Swinnen LJ, Smith SM, Rubin CM, Anastasi J & Carlson KM (2006) Post-transplant lymphoproliferative disorder subtypes correlate with different recurring chromosomal abnormalities, Genes Chromosomes Cancer. 45, 313–8. [DOI] [PubMed] [Google Scholar]
- 373.Poirel HA, Bernheim A, Schneider A, Meddeb M, Choquet S, Leblond V, Charlotte F, Davi F, Canioni D, Macintyre E, Mamzer-Bruneel MF, Hirsch I, Hermine O, Martin A, Cornillet-Lefebvre P, Patey M, Toupance O, Kemeny JL, Deteix P & Raphael M (2005) Characteristic pattern of chromosomal imbalances in posttransplantation lymphoproliferative disorders: correlation with histopathological subcategories and EBV status, Transplantation. 80, 176–84. [DOI] [PubMed] [Google Scholar]
- 374.Cesarman E, Chadburn A, Liu YF, Migliazza A, Dalla-Favera R & Knowles DM (1998) BCL-6 gene mutations in posttransplantation lymphoproliferative disorders predict response to therapy and clinical outcome, Blood. 92, 2294–302. [PubMed] [Google Scholar]
- 375.Vakiani E, Basso K, Klein U, Mansukhani MM, Narayan G, Smith PM, Murty VV, Dalla-Favera R, Pasqualucci L & Bhagat G (2008) Genetic and phenotypic analysis of B-cell post-transplant lymphoproliferative disorders provides insights into disease biology, Hematol Oncol. 26, 199–211. [DOI] [PubMed] [Google Scholar]
- 376.Srinivas SK, Sample JT & Sixbey JW (1998) Spontaneous loss of viral episomes accompanying Epstein-Barr virus reactivation in a Burkitt’s lymphoma cell line, J Infect Dis. 177, 1705–9. [DOI] [PubMed] [Google Scholar]
- 377.Swerdlow SH, Jaffe ES, Brousset P, Chan JK, de Leval L, Gaulard P, Harris NL, Pileri S, Weiss LM & International Lymphoma Study G (2014) Cytotoxic T-cell and NK-cell lymphomas: current questions and controversies, Am J Surg Pathol. 38, e60–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Chan JK (2017) Virus-associated neoplasms of the nasopharynx and sinonasal tract: diagnostic problems, Mod Pathol. 30, S68–S83. [DOI] [PubMed] [Google Scholar]
- 379.Liu A, Nakatsuka S, Yang WI, Kojya S & Aozasa K (2005) Expression of cell adhesion molecules and chemokine receptors: angioinvasiveness in nasal NK/T-cell lymphoma, Oncol Rep. 13, 613–20. [PubMed] [Google Scholar]
- 380.Li S, Feng X, Li T, Zhang S, Zuo Z, Lin P, Konoplev S, Bueso-Ramos CE, Vega F, Medeiros LJ & Yin CC (2013) Extranodal NK/T-cell lymphoma, nasal type: a report of 73 cases at MD Anderson Cancer Center, Am J Surg Pathol. 37, 14–23. [DOI] [PubMed] [Google Scholar]
- 381.Watanabe H, Enjoji M & Imai T (1976) Gastric carcinoma with lymphoid stroma. Its morphologic characteristics and prognostic correlations, Cancer. 38, 232–43. [DOI] [PubMed] [Google Scholar]
- 382.Hissong E, Ramrattan G, Zhang P, Zhou XK, Young G, Klimstra DS, Shia J, Fernandes H & Yantiss RK (2018) Gastric Carcinomas With Lymphoid Stroma: An Evaluation of the Histopathologic and Molecular Features, Am J Surg Pathol. 42, 453–462. [DOI] [PubMed] [Google Scholar]
- 383.Murphy G, Pfeiffer R, Camargo MC & Rabkin CS (2009) Meta-analysis shows that prevalence of Epstein-Barr virus-positive gastric cancer differs based on sex and anatomic location, Gastroenterology. 137, 824–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Camargo MC, Kim WH, Chiaravalli AM, Kim KM, Corvalan AH, Matsuo K, Yu J, Sung JJ, Herrera-Goepfert R, Meneses-Gonzalez F, Kijima Y, Natsugoe S, Liao LM, Lissowska J, Kim S, Hu N, Gonzalez CA, Yatabe Y, Koriyama C, Hewitt SM, Akiba S, Gulley ML, Taylor PR & Rabkin CS (2014) Improved survival of gastric cancer with tumour Epstein-Barr virus positivity: an international pooled analysis, Gut. 63, 236–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Falzarano SM, Mourmouras V, Mastrogiulio MG, La Magra C & Vindigni C (2009) Undifferentiated gastric carcinoma with lymphoid stroma (lymphoepithelioma-like carcinoma/medullary carcinoma), Pathologica. 101, 15–7. [PubMed] [Google Scholar]
- 386.Dittmer DP (2011) Restricted Kaposi’s sarcoma (KS) herpesvirus transcription in KS lesions from patients on successful antiretroviral therapy, mBio. 2, e00138–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Cesarman E & Knowles DM (1997) Kaposi’s sarcoma-associated herpesvirus: a lymphotropic human herpesvirus associated with Kaposi’s sarcoma, primary effusion lymphoma, and multicentric Castleman’s disease, Semin Diagn Pathol. 14, 54–66. [PubMed] [Google Scholar]
- 388.Biggar RJ, Chaturvedi AK, Goedert JJ, Engels EA & Study, H. A. C. M. (2007) AIDS-related cancer and severity of immunosuppression in persons with AIDS, J Natl Cancer Inst. 99, 962–72. [DOI] [PubMed] [Google Scholar]
- 389.Stallone G, Schena A, Infante B, Di Paolo S, Loverre A, Maggio G, Ranieri E, Gesualdo L, Schena FP & Grandaliano G (2005) Sirolimus for Kaposi’s sarcoma in renal-transplant recipients, N Engl J Med. 352, 1317–23. [DOI] [PubMed] [Google Scholar]
- 390.Campistol JM, Gutierrez-Dalmau A & Torregrosa JV (2004) Conversion to sirolimus: a successful treatment for posttransplantation Kaposi’s sarcoma, Transplantation. 77, 760–2. [DOI] [PubMed] [Google Scholar]
- 391.Nador RG, Cesarman E, Chadburn A, Dawson DB, Ansari MQ, Sald J & Knowles DM (1996) Primary effusion lymphoma: a distinct clinicopathologic entity associated with the Kaposi’s sarcoma-associated herpes virus, Blood. 88, 645–56. [PubMed] [Google Scholar]
- 392.Komanduri KV, Luce JA, McGrath MS, Herndier BG & Ng VL (1996) The natural history and molecular heterogeneity of HIV-associated primary malignant lymphomatous effusions, J Acquir Immune Defic Syndr Hum Retrovirol. 13, 215–26. [DOI] [PubMed] [Google Scholar]
- 393.Boulanger E, Gerard L, Gabarre J, Molina JM, Rapp C, Abino JF, Cadranel J, Chevret S & Oksenhendler E (2005) Prognostic factors and outcome of human herpesvirus 8-associated primary effusion lymphoma in patients with AIDS, J Clin Oncol. 23, 4372–80. [DOI] [PubMed] [Google Scholar]
- 394.Arvanitakis L, Mesri EA, Nador RG, Said JW, Asch AS, Knowles DM & Cesarman E (1996) Establishment and characterization of a primary effusion (body cavity-based) lymphoma cell line (BC-3) harboring kaposi’s sarcoma-associated herpesvirus (KSHV/HHV-8) in the absence of Epstein-Barr virus, Blood. 88, 2648–54. [PubMed] [Google Scholar]
- 395.Chadburn A, Hyjek E, Mathew S, Cesarman E, Said J & Knowles DM (2004) KSHV-positive solid lymphomas represent an extra-cavitary variant of primary effusion lymphoma, Am J Surg Pathol. 28, 1401–16. [DOI] [PubMed] [Google Scholar]
- 396.Teruya-Feldstein J, Zauber P, Setsuda JE, Berman EL, Sorbara L, Raffeld M, Tosato G & Jaffe ES (1998) Expression of human herpesvirus-8 oncogene and cytokine homologues in an HIV-seronegative patient with multicentric Castleman’s disease and primary effusion lymphoma, Lab Invest. 78, 1637–42. [PubMed] [Google Scholar]
- 397.Ascoli V, Signoretti S, Onetti-Muda A, Pescarmona E, Della-Rocca C, Nardi F, Mastroianni CM, Gastaldi R, Pistilli A, Gaidano G, Carbone A & Lo-Coco F (2001) Primary effusion lymphoma in HIV-infected patients with multicentric Castleman’s disease, J Pathol. 193, 200–9. [DOI] [PubMed] [Google Scholar]
- 398.Song JY & Jaffe ES (2013) HHV-8-positive but EBV-negative primary effusion lymphoma, Blood. 122, 3712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Cobo F, Hernandez S, Hernandez L, Pinyol M, Bosch F, Esteve J, Lopez-Guillermo A, Palacin A, Raffeld M, Montserrat E, Jaffe ES & Campo E (1999) Expression of potentially oncogenic HHV-8 genes in an EBV-negative primary effusion lymphoma occurring in an HIV-seronegative patient, J Pathol. 189, 288–93. [DOI] [PubMed] [Google Scholar]
- 400.Sin SH, Roy D, Wang L, Staudt MR, Fakhari FD, Patel DD, Henry D, Harrington WJ Jr., Damania BA & Dittmer DP (2007) Rapamycin is efficacious against primary effusion lymphoma (PEL) cell lines in vivo by inhibiting autocrine signaling, Blood. 109, 2165–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Bhatt AP, Bhende PM, Sin SH, Roy D, Dittmer DP & Damania B (2010) Dual inhibition of PI3K and mTOR inhibits autocrine and paracrine proliferative loops in PI3K/Akt/mTOR-addicted lymphomas, Blood. 115, 4455–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Keller SA, Schattner EJ & Cesarman E (2000) Inhibition of NF-kappaB induces apoptosis of KSHV-infected primary effusion lymphoma cells, Blood. 96, 2537–42. [PubMed] [Google Scholar]
- 403.Saji C, Higashi C, Niinaka Y, Yamada K, Noguchi K & Fujimuro M (2011) Proteasome inhibitors induce apoptosis and reduce viral replication in primary effusion lymphoma cells, Biochem Biophys Res Commun. 415, 573–8. [DOI] [PubMed] [Google Scholar]
- 404.Sarosiek KA, Cavallin LE, Bhatt S, Toomey NL, Natkunam Y, Blasini W, Gentles AJ, Ramos JC, Mesri EA & Lossos IS (2010) Efficacy of bortezomib in a direct xenograft model of primary effusion lymphoma, Proc Natl Acad Sci U S A. 107, 13069–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Bhatt AP, Jacobs SR, Freemerman AJ, Makowski L, Rathmell JC, Dittmer DP & Damania B (2012) Dysregulation of fatty acid synthesis and glycolysis in non-Hodgkin lymphoma, Proc Natl Acad Sci U S A. 109, 11818–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 406.Dai L, Trillo-Tinoco J, Bai A, Chen Y, Bielawski J, Del Valle L, Smith CD, Ochoa AC, Qin Z & Parsons C (2015) Ceramides promote apoptosis for virus-infected lymphoma cells through induction of ceramide synthases and viral lytic gene expression, Oncotarget. 6, 24246–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Engels EA, Pittaluga S, Whitby D, Rabkin C, Aoki Y, Jaffe ES & Goedert JJ (2003) Immunoblastic lymphoma in persons with AIDS-associated Kaposi’s sarcoma: a role for Kaposi’s sarcoma-associated herpesvirus, Mod Pathol. 16, 424–9. [DOI] [PubMed] [Google Scholar]
- 408.An J, Lichtenstein AK, Brent G & Rettig MB (2002) The Kaposi sarcoma-associated herpesvirus (KSHV) induces cellular interleukin 6 expression: role of the KSHV latency-associated nuclear antigen and the AP1 response element, Blood. 99, 649–54. [DOI] [PubMed] [Google Scholar]
- 409.Nishimoto N, Kanakura Y, Aozasa K, Johkoh T, Nakamura M, Nakano S, Nakano N, Ikeda Y, Sasaki T, Nishioka K, Hara M, Taguchi H, Kimura Y, Kato Y, Asaoku H, Kumagai S, Kodama F, Nakahara H, Hagihara K, Yoshizaki K & Kishimoto T (2005) Humanized anti-interleukin-6 receptor antibody treatment of multicentric Castleman disease, Blood. 106, 2627–32. [DOI] [PubMed] [Google Scholar]
- 410.Song SN, Tomosugi N, Kawabata H, Ishikawa T, Nishikawa T & Yoshizaki K (2010) Down-regulation of hepcidin resulting from long-term treatment with an anti-IL-6 receptor antibody (tocilizumab) improves anemia of inflammation in multicentric Castleman disease, Blood. 116, 3627–34. [DOI] [PubMed] [Google Scholar]
- 411.Galeotti C, Boucheron A, Guillaume S & Kone-Paut I (2012) Sustained remission of multicentric Castleman disease in children treated with tocilizumab, an anti-interleukin-6 receptor antibody, Mol Cancer Ther. 11, 1623–6. [DOI] [PubMed] [Google Scholar]
- 412.van Rhee F, Casper C, Voorhees PM, Fayad LE, van de Velde H, Vermeulen J, Qin X, Qi M, Tromp B & Kurzrock R (2015) A phase 2, open-label, multicenter study of the long-term safety of siltuximab (an anti-interleukin-6 monoclonal antibody) in patients with multicentric Castleman disease, Oncotarget. 6, 30408–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Katano H, Sato Y, Kurata T, Mori S & Sata T (2000) Expression and localization of human herpesvirus 8-encoded proteins in primary effusion lymphoma, Kaposi’s sarcoma, and multicentric Castleman’s disease, Virology. 269, 335–44. [DOI] [PubMed] [Google Scholar]
- 414.Uldrick TS, Polizzotto MN, Aleman K, O’Mahony D, Wyvill KM, Wang V, Marshall V, Pittaluga S, Steinberg SM, Tosato G, Whitby D, Little RF & Yarchoan R (2011) High-dose zidovudine plus valganciclovir for Kaposi sarcoma herpesvirus-associated multicentric Castleman disease: a pilot study of virus-activated cytotoxic therapy, Blood. 117, 6977–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Courville EL, Sohani AR, Hasserjian RP, Zukerberg LR, Harris NL & Ferry JA (2014) Diverse clinicopathologic features in human herpesvirus 8-associated lymphomas lead to diagnostic problems, Am J Clin Pathol. 142, 816–29. [DOI] [PubMed] [Google Scholar]
- 416.Oksenhendler E, Boutboul D, Beldjord K, Meignin V, de Labarthe A, Fieschi C, Dossier A, Agbalika F, Parravicini C, Tosato G, Gerard L & Galicier L (2013) Human herpesvirus 8+ polyclonal IgMlambda B-cell lymphocytosis mimicking plasmablastic leukemia/lymphoma in HIV-infected patients, Eur J Haematol. 91, 497–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Du MQ, Diss TC, Liu H, Ye H, Hamoudi RA, Cabecadas J, Dong HY, Harris NL, Chan JK, Rees JW, Dogan A & Isaacson PG (2002) KSHV- and EBV-associated germinotropic lymphoproliferative disorder, Blood. 100, 3415–8. [DOI] [PubMed] [Google Scholar]