

Abstract
Background
epi-cblC is a recently discovered inherited disorder of intracellular vitamin B12 metabolism associating hematological, neurological, and cardiometabolic outcomes. It is produced by an epimutation at the promoter common to CCDC163P and MMACHC, which results from an aberrant antisense transcription due to splicing mutations in the antisense PRDX1 gene neighboring MMACHC. We studied whether the aberrant transcription produced a second epimutation by encompassing the CpG island of the TESK2 gene neighboring CCDC163P.
Methods
We unraveled the methylome architecture of the CCDC163P–MMACHC CpG island (CpG:33) and the TESK2 CpG island (CpG:51) of 17 epi-cblC cases. We performed an integrative analysis of the DNA methylome profiling, transcriptome reconstruction of RNA-sequencing (RNA-seq), chromatin immunoprecipitation sequencing (ChIP-Seq) of histone H3, and transcription expression of MMACHC and TESK2.
Results
The PRDX1 splice mutations and activation of numerous cryptic splice sites produced antisense readthrough transcripts encompassing the bidirectional MMACHC/CCDC163P promoter and the TESK2 promoter, resulting in the silencing of both the MMACHC and TESK2 genes through the deposition of SETD2-dependent H3K36me3 marks and the generation of epimutations in the CpG islands of the two promoters.
Conclusions
The antisense readthrough transcription of the mutated PRDX1 produces an epigenetic silencing of MMACHC and TESK2. We propose using the term 'epi-digenism' to define this epigenetic disorder that affects two genes. Epi-cblC is an entity that differs from cblC. Indeed, the PRDX1 and TESK2 altered expressions are observed in epi-cblC but not in cblC, suggesting further evaluating the potential consequences on cancer risk and spermatogenesis.
Supplementary Information
The online version contains supplementary material available at 10.1186/s13148-022-01271-1.
Keywords: Epi-cblC; Secondary epimutation; Promoter hypermethylation; MMACHC; TESK2; Methylmalonic aciduria and homocystinuria, cblC type
Introduction
Epigenetic regulation of gene expression through DNA methylation is a source of genetic variation and can induce transcriptional haploinsufficiency [1]. Epigenetic diseases can be caused by aberrant DNA methylation marks, also called epimutations, which represent an underlying molecular mechanism of disease. In the setting of inherited metabolic disorders, we have previously reported a new type of inherited defect of vitamin B12 (cobalamin, cbl) metabolism with an epimutation in the promoter of the MMACHC gene, which we called epi-cblC [2]. Patients with epi-cblC have the same clinical presentation as cblC type, an autosomal recessive inherited disorder of intracellular vitamin B12 metabolism with combined methylmalonic aciduria and homocystinuria (OMIM phenotype ID: 277400) due to mutations in the MMACHC gene (metabolism of cobalamin-associated C; OMIM gene ID, 609831). In contrast, epi-cblC cases harbor an epimutation with an aberrant methylation of the CpG island (CpG:33) in the MMACHC promoter that silences the expression of the MMACHC gene. MMACHC belongs to a gene trio in which it is a sense gene flanked by CCDC163P and PRDX1 in the opposite orientation (trio with antisense (reverse, R1)/sense (forward, F2)/antisense (reverse, R3)) [2]. We found mutations in PRDX1 that produced an antisense transcript encompassing the MMACHC/CCDC163P bidirectional promoter, resulting in an H3K36me3 mark and the generation of the epimutation [2]. The splice acceptor variants induced readthrough transcripts extending beyond the normal poly(A) addition site, thus skipping the transcription termination signal of PRDX1 [2]. Most epi-cblC patients had a combination of alleles, with an epimutation in one allele and an MMACHC pathogenic genetic variant carried in trans [2]. One patient had a bi-allelic MMACHC epimutation due to the homozygous PRDX1:c.515-1G > T variant transmitted by both parents. In this homozygous case, we found that the bi-allelic epimutation produces the complete silencing of MMACHC in the patient’s fibroblasts [2]. No PRDX1 variant was detected in one of two epi-cblC cases reported in China, suggesting that the epimutation could also be triggered by a non-genetic mechanism [2]. Importantly, we demonstrated the presence of the MMACHC secondary epimutation in DNA from sperm, showing that the epimutation escaped erasure in spermatozoa [2]. This observation may be explained by the ubiquitously high expression of PRDX1 in germ cells [2].
The aberrant antisense transcript of PRDX1 encompasses the CCDC163P/MMACHC bidirectional promoter. TSK2 is an antisense gene neighboring CCDC163P, with the CpG island ‘CpG:51’ in its promoter region. However, whether the aberrant transcription encompasses the CpG island ‘CpG:51’ and produces a TESK2 epimutation was not considered. To address this hypothesis, we gathered data from the 17 epi-cblC patients described from Europe and North America. We unraveled the methylome architecture of the CCDC163P/MMACHC CpG island (CpG:33) and the TESK2 CpG island (CpG:51) by studying the epigenome-wide DNA methylation profile of the 17 epi-cblC cases [2, 3]. We performed an integrative analysis of the DNA methylome profiling, RNA-sequencing (RNA-seq) with de novo transcriptome reconstruction, reverse transcription-quantitative polymerase chain reaction (RT-qPCR), and chromatin immunoprecipitation sequencing (ChIP-Seq). We revealed epimutations in both the CCDC163P/MMACHC CpG island (CpG:33) and the TESK2 CpG island (CpG:51) that produced silencing in both genes.
Methods
Overview of the study design
First, we used an EWAS approach to look for epigenomic signatures associated with the epi-cblC phenotype. In a second step, we investigated whether the DNA methylation signatures revealed in the EWAS translate in terms of chromatin remodeling (ChIP-seq analysis), gene expression (RNA-seq and RT-qPCR analyses), and transcript structure (splicing analysis). We studied fibroblasts from skin biopsies since most reference centers use these cells for phenotyping inherited disorders of intracellular vitamin B12 metabolism.
DNA methylome study design and patients description
We performed a case–control EWAS to look for significant epigenomic signatures associated with an epi-cblC phenotype. The epi-cblC phenotype was defined by one of the three following conditions: i) composite MMACHC epimutation/MMACHC mutation, ii) heterozygous MMACHC epimutation, iii) homozygous MMACHC epimutation. We studied 17 patients with proven MMACHC epimutation, as previously described [2, 3]. All patients exhibited aberrant methylation on the CpG island ‘CpG:33’ on the CCDC163–MMACHC bidirectional promoter (Chr1: 45,965,587–45,966,049, GRCh37). Of the 17 patients with MMACHC epimutation, three were assessed at the Reference Center for Inborn Errors of Metabolism, University Hospital of Nancy, France (CHU-12122, a female patient with composite MMACHC epimutation/MMACHC mutation (composite epi-cblC disease) who died at 1-month of life from cardiometabolic decompensation and two relatives with heterozygous MMACHC epimutation [CHU-14061, the father, and CHU-14067, the grandfather]) [2] and three were assessed at the Department of Human Genetics, Montreal, Canada (WG-3838, a female patient with a composite epi-cblC disease who died at the age of two months from a sudden cardiac arrest with an acute renal failure, her father [CDH-867] who had a heterozygous MMACHC epimutation, and a 59-year-old male patient with a composite epi-cblC disease [WG-4152]) [2]. The remaining eleven patients were assessed at the Molecular and Cell Biology Laboratory of Neurometabolic Diseases and Paediatric Neurology Unit and Laboratories, Meyer Children’s Hospital, Florence, Italy. Of the eleven patients, eight had a composite epi-cblC disease and three corresponded to a trio composed of a proband exhibiting a homozygous MMACHC epimutation and the two parents with heterozygous MMACHC epimutation [3]. In total, of the 17 patients analyzed in the study, eleven had a composite MMACHC epimutation/MMACHC mutation, five had a heterozygous MMACHC epimutation, and one had a homozygous MMACHC epimutation. For the control group, we used in silico data generated using the Infinium Human Methylation 450 k BeadChip array from the MARTHA cohort, which included 350 unrelated European-ancestry ambulatory subjects recruited in Marseille (France) between January 1994 and October 2005 [4]. All adult patients and proband’s parents assessed at the Reference Center for Inborn Errors of Metabolism at the University Hospital of Nancy gave their informed written consent for performing the analyses, as previously reported [2]. The Columbia family provided informed consent, and the study was approved by the Columbia IRB [2]. Informed consent was obtained from all participants included in the Italian study, as previously reported (Ethics Committee of the Tuscany Region (No. CS_01/2021) [3].
DNA methylome analysis, quality controls, and statistical analyses
We carried out bisulfite conversion of 600 ng of DNA extracted from whole blood using the EZ DNA Methylation kit (Zymo Research, Proteigene, Saint-Marcel, France). The genome-wide profiling of DNA methylome was determined using the Infinium Human Methylation 450 k BeadChip array (Illumina, Paris, France) of the Infinium MethylationEPIC BeadChip array (Illumina, Paris, France), according to the manufacturer’s instructions. The arrays were scanned on an Illumina iScan® system, and raw methylation data were extracted using Illumina’s Genome Studio methylation module. For each CpG probe, the methylation level was described as a β value, ranging between 0 (fully unmethylated CpG probe) and 1 (fully methylated CpG probe). Background correction and normalization were implemented using the SWAN method (R Package Minfi) [5]. Probe annotation information, including sequence and chromosome location for Infinium Human Methylation 450 k and Infinium MethylationEPIC BeadChip arrays, was retrieved from the Manifest Files. We visually inspected the genome-wide distribution of the CpG probes according to their β value. We performed principal component analysis (PCA) to assess the clustering of methylation profiles according to the whole methylation landscape of the analyzed samples. The top ten principal components (eigenvectors, EV) were calculated with their respective eigenvalue. PCA plots were used to report on the three top eigenvalues. We compared the mean β values of each CpG probe between the two subgroups using the t test with Bonferroni correction. Output data included the mean β values in each subgroup, the difference of β values, the nominal P-value, and the Bonferroni corrected P-value. We used the smoothed P-value transformation by converting nominal P-values obtained from the t test to smoothed P-values using a window radius of 3, as previously reported [2, 3, 6]. All statistical analyses were performed using the SNP & Variation Suite (v8.8.1; Golden Helix, Inc., Bozeman, MT, USA) and MedCalc, version 19.5.3 (MedCalc Software, Ostend, Belgium).
Whole-genome chromatin Immunoprecipitation sequencing
ChIP-Seq was performed on patient’s fibroblasts and control fibroblast lines without inherited metabolic defects in the cobalamin pathway, as previously reported [2]. According to the manufacturer's instructions, the immunoprecipitation and reverse crosslinking were performed with the IP-Star SX-8G (Diagenode) using the Auto-Histone ChIP kit (Diagenode). ChIP-Seq libraries were prepared using the Kappa Library Preparation Kit Illumina Platforms (Kappa Biosystems) and Illumina TruSeq adapters (Illumina) according to the manufacturer’s instructions. Samples were sequenced using the HiSeq 2000 with a selected read length of 50 bp. Duplicated reads were removed using PICARD. Peaks were called using Model-based Analysis of ChIP-Seq MACS2 with input DNA as control and using the broad peak mod [7]. In order to generate Wiggle (WIG) and Tiled Data Format (TDF) files, HOMER (4.7) [8] and Integrative Genomics Viewer (2.3.67) [9] were used. Tags were normalized to 10 million reads to generate tracks for visualization.
RNA sequencing
As previously described, RNA depleted of rRNA transcripts was extracted from fibroblasts obtained by skin biopsies of CHU-12122 and WG-3838 epi-cblC cases, 3 cblC cases with the c.270_271insA mutation, 3 cblG cases (another inherited defect of vitamin B12 intracellular metabolism due to mutations in methionine synthase (MTR) gene) and 4 control fibroblasts using RiboMinus™ (Thermo Scientific, Villebon, France) [2]. cDNA libraries were prepared using the TruSeqTM RNA Sample Preparation Kit (Illumina, San Diego, CA, USA). RNA sequencing was performed using the Illumina Hi-scan sequencer. Reads were classified according to known 5′ and 3′ boundaries of annotated genes. RNA-sequencing FASTQ files for the two composite epi-cblC samples compiled 150-nucleotide-long paired-end reads. The control group originated from a previous study [10]. All samples were regrouped and analyzed using the same bioinformatics pipeline, with quality control, adapter removal, read alignment, and manipulation, as previously described [11]. The alignment step was performed on the human reference genome GRCh38, with two splice-aware tools, HISAT v2.0.4 [12] and STAR v2.5.1b [13], that accounted for splicing junctions and genetic polymorphisms. From this point on, as we observed and hypothesized that the RNA splicing mechanisms were abnormal in the genomic region spanning from TESK2 to PRDX1, we could not rely on reference-based transcriptome reconstruction methods, which would not efficiently report on the real isoform population and abundances in this region. We extracted the high-quality reads aligning inside the genomic interval chr1:45,443,800–45,543,000 (TESK2 to PRDX1) and performed de novo transcriptome assembly with Trinity v2.6.5 [14]. The Trinity pipeline was launched with default parameters for paired-end and unstranded reads, with the ‘–jacard_clip’ flag to account for high gene density, with possible overlapping at these genomic coordinates. Besides the two composite epi-cblC samples, 4 controls were suitable for the analysis and were used as controls against the composite epi-cblC cases. All samples presented with enough coverage in high-quality reads in the region of interest (> 10,000) to achieve transcript reconstruction, providing enough read depth for unbiased estimation of their expression levels. Reconstructed transcripts were then aligned with blastn (with parameters ‘-evalue 1e-10’ and ‘-max_target_seqs 3’) against a cDNA database obtained with BioMart (https://www.ensembl.org/biomart/) and comprising all known isoforms of the four genes TESK2, CCDC163, MMACHC, and PRDX1. Criteria for a successful transcript assignment to a known gene product of interest were as follows: i) biotype = protein-coding; ii) select the highest alignment length among candidates; iii) subject length = alignment length and is > 300 nt; iv) a number of mismatches allowed < 4; and v) a maximum number of gaps = 1. Finally, all transcripts were quantified using Kallisto v0.43 [15], which reported the normalized expression levels as estimated read counts and transcript-per-million (TPM) unit.
Quantification of MMACHC, TESK2, and PRDX1 aberrant transcripts mRNA expression
Besides quantification from RNA-seq data, we also quantified the expression of MMACHC and TESK2 transcripts and PRDX1 aberrant transcript using reverse transcription-quantitative polymerase chain reaction (RT-qPCR). Fibroblasts from an epi-cblC case (CHU-12122) and control fibroblasts were cultured in DMEM medium supplemented with 10% v/v heat-inactivated fetal bovine serum, 1% v/v pyruvate, and 1% v/v penicillin–streptomycin. Cells were maintained at 37 °C and in 5% CO2. All the reagents were obtained from Sigma-Aldrich. RNA was extracted using NucleoSpin RNA Plus (Macherey–Nagel, Hœrdt, France). After the extraction, a DNase digestion step was carried out to remove DNA contamination using the DNA-free™ DNA Removal kit (Thermo Fisher Scientific France, Illkirch-Graffenstaden, France). RNA was reverse-transcribed using PrimeScript™ RT Master Mix (Takara Bio Europe, Saint-Germain-en-Laye, France); 2 µL of cDNA was used for qPCR with TB Green Premix Ex Taq (Tli RNase H Plus) (Takara Bio Europe, Saint-Germain-en-Laye, France) in a 20 µL reaction volume with the forward and the reverse primers specific for each of the studied genes (Additional file 3: Table S1) at a concentration of 0.2 µM. PCRs were carried out in 96-well plates using the CFX Connect Real-Time PCR Detection System (Bio-Rad, Marnes-la-Coquette, France) with the following temperature cycling: 95 °C for 30 s followed by 40 cycles consisting of 95 °C for 5 s followed by 60 °C for 30 s and finally a melt curve analysis from 65 °C to 95 °C to detect unspecific PCR products. All steps were done following the manufacturer’s instructions. Statistical analyses for RT-q-PCR were done using the CFX Maestro software. The forward and reverse primers for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) of MMACHC, TESK2 transcripts, and PRDX1 aberrant transcript are reported in Additional file 3: Table S1.
Results
Epigenome-wide association study for the epi-cblC phenotype
All the DNA methylome profiles passed the quality checks and exhibited a valid β value density distribution (Additional file 1: Figure S1). In PCA on genome-wide DNA methylome profiles, we found a clustered distribution according to patients’ gender (EV2) and the epi-cblC phenotype (EV1) (Fig. 1). Additionally, we performed a PCA analysis that only considered CpG probes on chromosome 1. The PCA plot reporting PC1 vs. PC2 from chromosome 1-based PCA analysis was similar to that obtained from the genome-wide PCA analysis (PC1 vs. PC3) (Additional file 1: Figure S2). The EWAS revealed a significant locus in chromosome 1 in association with the epi-cblC phenotype (Fig. 2A). The visualization of this top epigenomic signature confirmed the significant association at the CpG island ‘CpG:33’ on the CCDC163–MMACHC bidirectional promoter but also revealed a second significant association at the CpG island ‘CpG:51’ on the promoter region of the TESK2 gene (Figs. 2B and 3). The mean-beta values of the CpG probes located in the CpG islands CpG:33 and CpG:51 had a fully unmethylated profile among controls and a hemimethylated profile among cases (Table 1 and Figs. 2B and 3).
Fig. 1.

A 2-D plot using the two top eigenvectors (PC1, PC2) derived from the primary component analysis on the genome-wide methylome landscape of the studied patients and controls. B 2-D plot using the PC1 and PC3 eigenvectors derived from the primary component analysis on the genome-wide methylome landscape of the studied patients and controls
Fig. 2.

A Epi-Manhattan plot reporting the results of the epigenome-wide association study that compared 17 patients with MMACHC epimutation (epi-cblC disease, isolated MMACHC epimutation, biallelic MMACHC epimutation) with controls. The − log10 P-value reports the t-test that compared the mean β values between the two groups. The horizontal line indicates a P-value threshold of 1 × 10−100. The top significant hit in chromosome 1 corresponds to the CpG island (CpG:33) on the bidirectional promoter of CCDC163–MMACHC and the CpG island (CpG:51) on the promoter region of the TESK2 gene. B Epigrams reporting the methylation levels of the top CpG probes from the epigenome-wide association study results that compared 17 patients with MMACHC epimutation (epi-cblC disease, isolated MMACHC epimutation, biallelic MMACHC epimutation) (black bars) with controls (gray bars). The horizontal lines correspond to β value thresholds of 0.2, below which the CpG probe is considered to be fully unmethylated. Above 0.6, the CpG probe is considered fully methylated. A β value between 0.2 and 0.6 indicates a hemimethylated CpG probe. All the CpG probes located in CpG islands CpG:33 (CCDC163P–MMACHC bidirectional promoter), upstream the 5’ end of the TESK2 promoter, and the CpG:51 (TESK2 promoter) were fully unmethylated among controls and exhibited a hemimethylated profile among cases
Fig. 3.

A Zoomed view of the epi-Manhattan plot reporting the epigenome-wide association study results that compared 17 patients with MMACHC epimutation (epi-cblC disease, isolated MMACHC epimutation, biallelic MMACHC epimutation) with controls. The − log10 P-value reports the t-test that compared the mean β values between the two groups. The horizontal line indicates a P-value threshold of 1 × 10−100. The zoomed view of the top epigenomic signature in chromosome 1 confirmed the significant association at the CpG island ‘CpG:33’ on the CCDC163–MMACHC bidirectional promoter but also revealed a second significant association at the CpG island ‘CpG:51’ on the promoter region of the TESK2 gene. B Positions of the CpG islands CpG:33 and CpG:51 according to the CpG Islands UCSC annotation. C Mean β values among the 17 patients with MMACHC epimutation (epi-cblC disease, isolated MMACHC epimutation, biallelic MMACHC epimutation) (red dots) and controls (blue dots). The horizontal lines correspond to β value thresholds of 0.2, below which the CpG probe is considered to be fully unmethylated. Above 0.6, the CpG probe is considered fully methylated. A β value between 0.2 and 0.6 indicates a hemimethylated CpG probe. D Genomic annotation according to RefSeq Genes 105.20201022 v2, NCBI
Table 1.
Top significant CpG probe in the epigenome-wide association study on the epi-cblC phenotype
| CpG probe | Chr | Position* | CpG island | Locus | t-test, P-value | t-test, Bonf. P-value | β-value (Cases) | β-value (Controls) | Δβ-value |
|---|---|---|---|---|---|---|---|---|---|
| cg12630522 | 1 | 45956424 | CpG:51 | TESK2 (promoter) | 3.86E-223 | 1.74E-217 | 0.55 | 0.01 | 0.54 |
| cg19250177 | 1 | 45956646 | CpG:51 | TESK2 (promoter) | 7.56E-204 | 3.42E-198 | 0.46 | 0.01 | 0.45 |
| cg25827112 | 1 | 45956773 | CpG:51 | TESK2 (promoter) | 4.96E-192 | 2.25E-186 | 0.46 | 0.06 | 0.41 |
| cg27633763 | 1 | 45956828 | CpG:51 | TESK2 (promoter) | 1.22E-94 | 5.53E-89 | 0.49 | 0.03 | 0.47 |
| cg19551082 | 1 | 45956882 | CpG:51 | TESK2 (promoter) | 3.62E-148 | 1.64E-142 | 0.32 | 0.06 | 0.27 |
| cg00103132 | 1 | 45956932 | Upstream 5’ end CpG:51 | Upstream 5’ end TESK2 | 4.42E-212 | 2.00E-206 | 0.42 | 0.04 | 0.38 |
| cg00609097 | 1 | 45956974 | Upstream 5’ end CpG:51 | Upstream 5’ end TESK2 | 6.61E-195 | 2.99E-189 | 0.48 | 0.05 | 0.43 |
| cg24296786 | 1 | 45957014 | Upstream 5’ end CpG:51 | Upstream 5’ end TESK2 | 2.75E-199 | 1.24E-193 | 0.53 | 0.05 | 0.48 |
| cg04132845 | 1 | 45957038 | Upstream 5’ end CpG:51 | Upstream 5’ end TESK2 | 1.96E-218 | 8.85E-213 | 0.46 | 0.06 | 0.41 |
| cg15605315 | 1 | 45957053 | Upstream 5’ end CpG:51 | Upstream 5’ end TESK2 | 3.07E-196 | 1.39E-190 | 0.50 | 0.03 | 0.46 |
| cg05765466 | 1 | 45957060 | Upstream 5’ end CpG:51 | Upstream 5’ end TESK2 | 8.34E-203 | 3.77E-197 | 0.46 | 0.08 | 0.38 |
| cg13848568 | 1 | 45965625 | CpG:33 | CCDC163P–MMACHC (bidirectional promoter) | 1.87E-138 | 8.44E-133 | 0.39 | 0.04 | 0.35 |
| cg00081251 | 1 | 45965679 | CpG:33 | CCDC163P–MMACHC (bidirectional promoter) | 2.89E-182 | 1.31E-176 | 0.52 | 0.07 | 0.45 |
| cg09323228 | 1 | 45965727 | CpG:33 | CCDC163P–MMACHC (bidirectional promoter) | 1.18E-171 | 5.33E-166 | 0.49 | 0.05 | 0.44 |
| cg27393325 | 1 | 45965846 | CpG:33 | CCDC163P–MMACHC (bidirectional promoter) | 2.16E-195 | 9.79E-190 | 0.52 | 0.02 | 0.50 |
| cg22536808 | 1 | 45965870 | CpG:33 | CCDC163P–MMACHC (bidirectional promoter) | 7.73E-167 | 3.50E-161 | 0.42 | 0.00 | 0.42 |
| cg14836864 | 1 | 45965990 | CpG:33 | CCDC163P–MMACHC (bidirectional promoter) | 2.33E-167 | 1.05E-161 | 0.43 | 0.03 | 0.40 |
| cg03108114 | 1 | 45966048 | CpG:33 | CCDC163P–MMACHC (bidirectional promoter) | 1.51E-208 | 6.82E-203 | 0.54 | 0.02 | 0.51 |
Chr chromosome; Bonf. Bonferroni; Δβ-value difference between case and control β values
*Genomic positions are reported according to GRCh37
Genome-wide chromatin landscape profiling in fibroblasts of patients with epi-cblC
We assessed ChIP-Seq data generated on case and control fibroblasts previously described in reference 2 to further characterize the epigenetic changes at the MMACHC and TESK2 loci. We observed a significant accumulation of trimethylated lysine 36 on histone H3 (H3K36me3) and no mark of H3K4me3 in the CCDC163–MMACHC bidirectional promoter region (CpG:33) and in the TESK2 promoter region (CpG:51) among patients with an epi-cblC phenotype in comparison with controls (Fig. 4). Overall, the region spanning from mid-PRDX1 to TESK2 harbored a continuous enrichment of H3K36me3 marks in epi-cblC patients as compared to controls (Fig. 4).
Fig. 4.

Results of ChIP-Seq analyses at the genomic region encompassing the CCDC163–MMACHC bidirectional promoter region (CpG:33) and in the TESK2 promoter region (CpG:51). Genomic panels show normalized coverage for histone H3 trimethylated lysine 36 (H3K36me3) and H3K4me3 marks in patients with an epi-cblC phenotype and controls. The dashed rectangles indicate the CCDC163–MMACHC bidirectional promoter region (CpG:33) and the TESK2 promoter region (CpG:51). The same scale has been set in all panels. ChIP-Seq showed a significant accumulation of the H3K36me3 and a mirrored profile of H3K4me3 in the CCDC163–MMACHC bidirectional promoter region (CpG:33) and in the TESK2 promoter region (CpG:51) among patients with an epi-cblC phenotype in comparison with controls. In contrast, the histone H3K36me3 and H3K4me3 marks are similar in the PRDX1 promoter region. The RNAseq showed a high expression of PRDX1 exons and low or no expression of MMACHC exons in case WG-3838 epi-cblC and cblC fibroblasts
The epimutations in CCDC163–MMACHC bidirectional promoter and TESK2 promoter result from PRDX1 antisense transcription
The RNA-seq analyses showed aberrant splicing and wrong splice junction usage in the genomic region encompassing TESK2, CCDC163, MMACHC, and PRDX1 (Fig. 5). The RNA-seq analyses showed the absence of MMCHC sense transcripts in case WG-3838, a result which may be consistent with a splicing defect produced by the c.80A > G variant in MMACHC, which may then lead to total degradation of the mRNA (Fig. 5). In contrast, in case CHU-12122, we detected transcripts with the c.270_271insA mutation (heterozygous c.270_271insA, p.Arg91LysfsTer14) (Fig. 5). The RNA-Seq and RT-qPCR analyses of case fibroblasts showed a PRDX1 readthrough transcription through the CCDC163–MMACHC bidirectional promoter region (CpG:33) but also through the TESK2 promoter region (CpG:51) (Figs. 5 and 6). In epi-cblC fibroblasts, after de novo transcriptome reconstruction in the region spanning from PRDX1 to TESK2, we observed a dramatic increase in the PRDX1 aberrant transcripts as compared with control fibroblasts (Fig. 5). We further assessed the expression of MMACHC, TESK2, and PRDX1 aberrant transcripts in fibroblasts of patients with composite epi-cblC. In comparison with controls, fibroblasts from the CHU-12122 patient with epi-cblC showed a 22-fold increase of PRDX1 aberrant transcript and a dramatic decrease of both MMACHC (12-fold decrease) and TESK2 (fourfold decrease) mRNA, in RT-qPCR analyses normalized to GAPDH and TBP (Fig. 6B). The canonical transcriptions of TESK2 and MMACHC, but not PRDX1, were also decreased in the RNA-seq analysis of CHU-12122 and WG-3838 fibroblasts (Fig. 6C). In contrast, the canonical transcription of MMACHC and TESK2 was similar to that of PRDX1 in RNA-seq and RT-qPCR analysis of control cells (Fig. 6C).
Fig. 5.

Overview of the transcription anomalies with aberrant splicing and wrong splice junction usage in the genomic region encompassing TESK2, CCDC163, MMACHC, and PRDX1. For every three samples, two tracks are represented: i) a sashimi plot with splice junction usage (thickness of the line is proportional to read depth supporting the splice junctions; in red: forward strand processing; in blue: reverse strand processing), and ii) an exhaustive read coverage in squished mode, with horizontal blue-grey lines indicating inserts between paired-end reads; In this view, well-defined horizontal gaps represent exons when introns are successfully spliced. Alignment was made with HISAT2 on the reference genome hg38
Fig. 6.
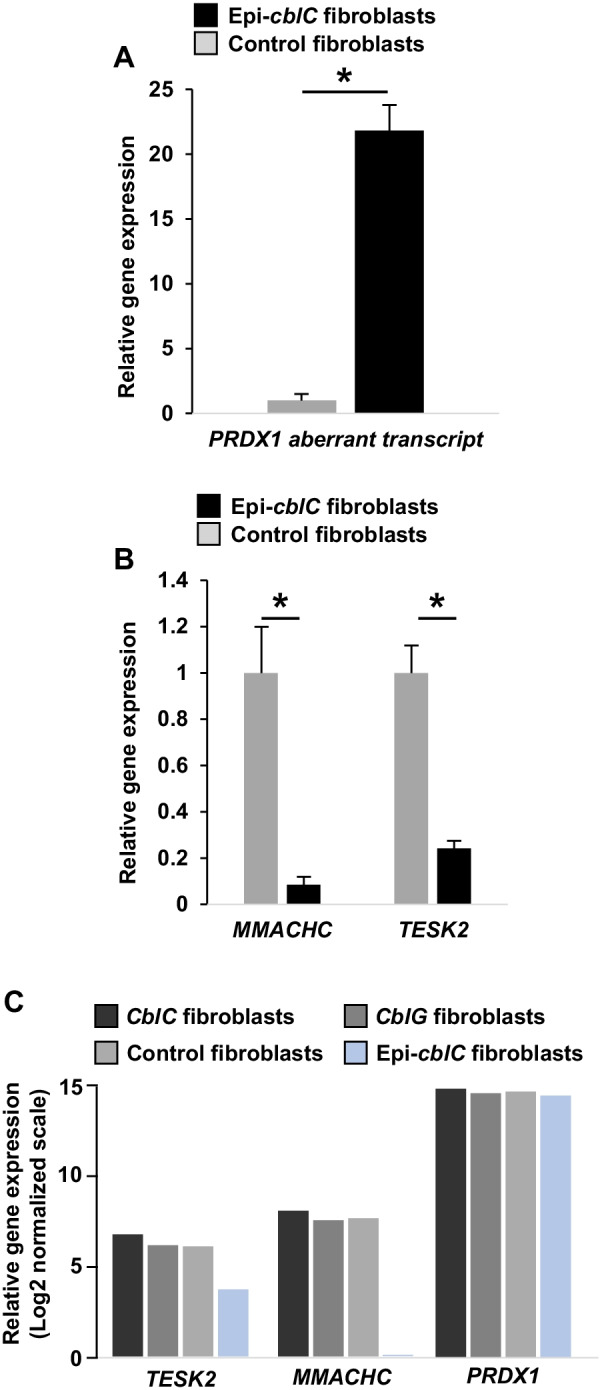
A Relative expression levels of PRDX1 aberrant transcript in CHU-12122 epi-cblC fibroblasts to control fibroblasts normalized to GAPDH and TBP; B relative expression levels of MMACHC and TESK2 mRNAs in epi-cblC fibroblasts relative to control fibroblasts normalized to GAPDH and TBP (*P < 0.001, n = 5 analysed cell flasks). C Expression levels of canonical transcripts for TESK2, MMACHC and PRDX1 genes in RNA-seq analyses of fibroblasts from epi-cblC (n = 2), cblC (n = 3) and cblG (n = 3) cases and control fibroblasts (n = 4). Expressions in log scale
Discussion
We found that the two PRDX1 mutations reported in 17 epi-cblC cases produced antisense readthrough transcripts encompassing the CCDC163P/MMACHC bidirectional promoter and the TESK2 promoter, resulting in H3K36me3 marks, generation of epimutations in the two CpG islands and silencing of MMACHC and TESK2 genes. These results showed that epi-cblC is a clinical entity that must be distinguished from cblC with regard to the potential consequences that may be produced by TESK2 silencing. Epi-cblC is an example of an altered expression of two genes through a common epigenetic mechanism, which led us to introduce the term 'epi-digenism.'
The previous reports of the clinical and metabolic presentations of the 17 epi-cblC cases showed similar manifestations, compared to the cblC type of inherited disorders of cobalamin metabolism, with combined methylmalonic aciduria and homocystinuria, hypotonia, failure to thrive, megaloblastic anemia, pulmonary hypertension, and fatal neonatal cardio-metabolic decompensation [2, 3, 16–18]. Of note, the clinical phenotype of the epi-cblC homozygote appeared to be more severe than homozygous cases with MMACHC c.271dupA, which is the most common mutation reported in European patients with cblC [3, 18]. In addition, some manifestations are frequent in epi-cblC, including skeletal deformity, metabolic acidosis with or without hyperammonemia, and recurrent severe infections [3]. The presence of the PRDX1 mutation and the potential clinical consequences of TESK2 silencing in epi-cblC cases suggest that it should be considered as a distinct entity. PRDX1 encodes Peroxiredoxin 1, a versatile protein involved in cell defense against cellular oxidative stress, influencing cell growth, differentiation, and apoptosis [19]. PRDX1 enhances the natural killer cell cytotoxicity [20] and inhibits the function of oncoproteins such as c-Abl [21] and c-Myc [19]. The role of PRDX1 in suppressing tumors has been confirmed in Prdx1-knockout mouse models [22, 23]. We suggest periodically monitoring subjects bearing the c.515-1G > T variant to test the hypothesis of a potential increased risk of cancer. This potential risk could be related to the decreased expression of PRDX1 evidenced in RNA-seq analyses (Fig. 5). The PRDX1:c.515-1G > T variant leads to the skipping of the last exon, which encodes one of the two cysteine residues which are essential for the catalytic activity [2, 3]. We also observed a clear reduction of the expression of TESK2, which could impact the disease course of epi-cblC cases. TESK2 encodes a putative 555-amino acid protein with a kinase domain that exhibits a dual specific protein kinase activity on both serine/threonine and tyrosine residues [24]. The low expression of TESK2 is associated with poor survival of patients with lung adenocarcinoma and premetastatic stage in human lung-to-brain metastasis [25]. TESK2 is mainly expressed in the testis and prostate and influences cofilin phosphorylation and actin scaffold in testicular Sertoli cells [24, 26]. It also influences myogenic differentiation through actin remodeling [27]. CCDC163P was also silenced and was predicted to be the coding gene of a transmembrane protein, which has a main transcriptional expression in the thyroid, brain, and testis [28]. However, the protein has not been characterized and it is not associated with pathological outcomes [28].
The RNA-seq data analysis showed that the readthrough transcription of PRDX1 was prolongated through the MMACHC/CCDC163P bidirectional promoter and the TESK2 promoter in fibroblasts of epi-cblC patients (Fig. 5). The H3K36me3 mark was the predominant common characteristic of histone marks at both MMACHC and TESK2 promoters (Fig. 4). This mark is deposited only by the histone-lysine N-methyltransferase SETD2 at K36 of histone H3, where it binds the active form of RNA polymerase II [29]. Subsequently, the H3K36me3 mark allows the recruitment of DNMT3B1 through the binding of the heterochromatin protein 1 (HP1). This explains why the de novo methylation of CpG islands is preferentially targeted to genomic regions with elevated H3K36me3 levels through the recruitment of DNMT3B1 in mouse stem cells [30]. Based on these data and our data on RNA-seq and ChIP-seq of patient fibroblasts, we propose a common mechanism that produces the writing of the two epimutations through the successive recruitment of SETD2, HP1, and DNMT2B in epi-cblC disease. The RNA-seq analyses showed the activation of numerous cryptic splice sites that participated in the readthrough transcription of PRDX1 and its prolongation through the MMACHC, CCDC163P, and TESK2 genes. The activation of these cryptic splice sites could involve SETD2. Indeed the SETD2 methyltransferase not only mediates the co-transcriptional methylation in histone H3 but it also has other functions that include the regulation of pre-mRNA splicing [31].
Digenic inheritance (DI) concerns pathologies with multigenic etiology implicating more than one gene [32]. Examples of unequivocal digenism in known diseases have been reviewed previously [32]. One of the categories of digenism is the inheritance of a single primary mutation that establishes the diagnosis and a second DNA variant, which modifies the phenotype [32]. In the case of epi-cblC, the epimutation in the CCDC163P/MMACHC promoter produces the main phenotype of the disease while the epimutation in the promoter of TESK2 produces its decreased expression with potential clinical consequences [25]. Therefore, we propose introducing the term ‘epi-digenism’ and considering epi-cblC as an example of epidigenism since the silencing of MMACHC and TESK2 are produced by a shared epigenetic mechanism on the two promoters.
Recently, a comprehensive genome-wide analysis of human epivariations analyzed in silico data generated from 23,116 individuals with the Illumina 450 k methylation array and reported 4,452 unique autosomal epivariations, including 384 in disease-causing OMIM genes [33]. The epivariation in the CCDC163P/MMACHC promoter was the most frequent among the 384 hypermethylated in the studied individuals [17]. Of note, one of the subjects had epivariations in TESK2 and CCDC163P/MMACHC promoters [17]. This suggests that both epivariations could be triggered by environmental factors, as reviewed recently for CCDC163P/MMACHC promoter [17]. In addition, several EWAS from the EWAS catalog (http://ewascatalog.org/?cpg=cg0036120) showed the association of hypermethylated CpG in CCDC163P/MMACHC and TESK2 promoters with sex, age, child abuse, rheumatoid arthritis, and five-year Increase in epigenomic biological age of subjects with chronic HIV Infection [34–37].
In conclusion, our results show that epi-cblC should be distinguished from cblC, with potential consequences on spermatogenesis and cancer risk produced by TESK2 silencing. The coverage of readthrough transcripts through a large region that encompasses MMACHC, CCDC163P and TESK2 involves the activation of numerous splice cryptic sites. Epi-cblC is an example of altered expression of two neighboring genes through a shared epigenetic pathomechanism. We propose introducing the term 'epi-digenism' to characterize this type of disorder.
Supplementary Information
Additional file 1. Supplemental Figure S1. Genome-wide density distribution of CpG probes in the 17 DNA methylome profiles of the 17 patients with MMACHC epimutation (epi-cblC disease, isolated MMACHC epimutation, biallelic MMACHC epimutation).
Additional file 2. Supplemental Figure S2. (A) 2-D plot using the two top eigenvectors (PC1, PC3) derived from the primary component analysis on the genome-wide methylome landscape of the studied patients and controls. (B) 2-D plot using the PC1 and PC2 eigenvectors derived from the primary component analysis on the methylome landscape of chromosome 1 of the studied patients and controls.
Additional file 3. Supplemental Table S1. Forward and reverse primers for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) of MMACHC, TESK2 transcripts and PRDX1 aberrant transcript.
Acknowledgements
We would like to thank the mother, father, and relatives of cases for their valuable contribution and help in the collection of data and samples used to study the entire families, and the scientists of the McGill University and Genome Quebec and the Genomic Platform of the FR3209 CNRS-Inserm, University of Lorraine, for expert advice and performing high throughput sequencing.
Author contributions
AO contributed to methodology; software; validation; formal analysis; investigation; resources; writing of the draft; visualization; CCh, CCa, YS, SH, PR, DSF, LF, AG, SG , SM, MRB, GP, AT, CM, FM, FF, MD, SM, GM, MAD, RG, PEM, DT, MP, AB, DW, TP, AM, FH, BAR, WKC, FF, JM, DSR, and AM contributed to investigation; resources; writing—review and editing; JLG contributed to conceptualization; methodology; validation; formal analysis; investigation; resources; writing of the draft—review and editing; visualization; supervision; project administration; funding acquisition. All authors read and approved the final manuscript.
Funding
INSERM UMR_S 1256, Nutrition, Genetics, and Environmental Risk Exposure (NGERE). The methylome analyses were funded by FHU ARRIMAGE and the French Agence Nationale de la Recherche, PIA project 'Lorraine Université d’Excellence' (ANR-15-IDEX-04-LUE). Institutional grants were received from the Region Lorraine, i-SITE Lorraine University of Excellence (LUE) and FEDER (fonds européen de développement regional). Canadian Institutes for Health Research. W.K.C received support from the Simons Foundation and the JPB Foundation.
Availability of data and materials
Data are available for use in collaborative studies to researchers upon reasonable request (jean-louis.gueant@univ-lorraine.fr). Data will be provided following the review and approval of a research proposal (including a statistical analysis plan) and the completion of a data-sharing agreement. Responses to the request for the raw data will be judged by the IRB of INSERM UMR_S 1256.
Declarations
Ethics approval and consent to participate
All adult patients and proband’s parents assessed at the Reference Center for Inborn Errors of Metabolism at the University Hospital of Nancy gave their informed written consent for performing the analyses, as previously reported [2]. Informed consent was obtained from all participants included in the Italian study, as previously reported (Ethics Committee of the Tuscany Region (No. CS_01/2021) [3].
Consent for publication
Not applicable.
Competing interests
The authors declare no competing financial interests.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Zhou H, Brockington M, Jungbluth H, Monk D, Stanier P, Sewry CA, Moore GE, Muntoni F. Epigenetic allele silencing unveils recessive RYR1 mutations in core myopathies. Am J Hum Genet. 2006;79(5):859–868. doi: 10.1086/508500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gueant JL, Chery C, Oussalah A, Nadaf J, Coelho D, Josse T, Flayac J, Robert A, Koscinski I, Gastin I, et al. APRDX1 mutant allele causes a MMACHC secondary epimutation in cblC patients. Nat Commun. 2018;9(1):67. doi: 10.1038/s41467-017-02306-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cavicchi C, Oussalah A, Falliano S, Ferri L, Gozzini A, Gasperini S, Motta S, Rigoldi M, Parenti G, Tummolo A, et al. PRDX1 gene-related epi-cblC disease is a common type of inborn error of cobalamin metabolism with mono- or bi-allelic MMACHC epimutations. Clin Epigenet. 2021;13(1):137. doi: 10.1186/s13148-021-01117-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dick KJ, Nelson CP, Tsaprouni L, Sandling JK, Aissi D, Wahl S, Meduri E, Morange PE, Gagnon F, Grallert H, et al. DNA methylation and body-mass index: a genome-wide analysis. Lancet. 2014;383(9933):1990–1998. doi: 10.1016/S0140-6736(13)62674-4. [DOI] [PubMed] [Google Scholar]
- 5.Aryee MJ, Jaffe AE, Corrada-Bravo H, Ladd-Acosta C, Feinberg AP, Hansen KD, Irizarry RA. Minfi: a flexible and comprehensive Bioconductor package for the analysis of Infinium DNA methylation microarrays. Bioinformatics. 2014;30(10):1363–1369. doi: 10.1093/bioinformatics/btu049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gallet P, Oussalah A, Pouget C, Dittmar G, Chery C, Gauchotte G, Jankowski R, Gueant JL, Houlgatte R. Integrative genomics analysis of nasal intestinal-type adenocarcinomas demonstrates the major role of CACNA1C and paves the way for a simple diagnostic tool in male woodworkers. Clin Epigenet. 2021;13(1):179. doi: 10.1186/s13148-021-01122-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9(9):R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38(4):576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Robinson JT, Thorvaldsdottir H, Winckler W, Guttman M, Lander ES, Getz G, Mesirov JP. Integrative genomics viewer. Nat Biotechnol. 2011;29(1):24–26. doi: 10.1038/nbt.1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Quintana AM, Yu HC, Brebner A, Pupavac M, Geiger EA, Watson A, Castro VL, Cheung W, Chen SH, Watkins D, et al. Mutations in THAP11 cause an inborn error of cobalamin metabolism and developmental abnormalities. Hum Mol Genet. 2017;26(15):2838–2849. doi: 10.1093/hmg/ddx157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zgheib R, Battaglia-Hsu SF, Hergalant S, Quere M, Alberto JM, Chery C, Rouyer P, Gauchotte G, Gueant JL, Namour F. Folate can promote the methionine-dependent reprogramming of glioblastoma cells towards pluripotency. Cell Death Dis. 2019;10(8):596. doi: 10.1038/s41419-019-1836-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kim D, Paggi JM, Park C, Bennett C, Salzberg SL. Graph-based genome alignment and genotyping with HISAT2 and HISAT-genotype. Nat Biotechnol. 2019;37(8):907–915. doi: 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29(1):15–21. doi: 10.1093/bioinformatics/bts635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haas BJ, Papanicolaou A, Yassour M, Grabherr M, Blood PD, Bowden J, Couger MB, Eccles D, Li B, Lieber M, et al. De novo transcript sequence reconstruction from RNA-seq using the Trinity platform for reference generation and analysis. Nat Protoc. 2013;8(8):1494–1512. doi: 10.1038/nprot.2013.084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bray NL, Pimentel H, Melsted P, Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat Biotechnol. 2016;34(5):525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 16.Zhang X, Chen Q, Song Y, Guo P, Wang Y, Luo S, Zhang Y, Zhou C, Li D, Chen Y, et al. Epimutation of MMACHC compound to a genetic mutation in cblC cases. Mol Genet Genomic Med. 2021;9(6):e1625. doi: 10.1002/mgg3.1625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gueant JL, Siblini Y, Chery C, Schmitt G, Gueant-Rodriguez RM, Coelho D, Watkins D, Rosenblatt DS, Oussalah A. Epimutation in inherited metabolic disorders: the influence of aberrant transcription in adjacent genes. Hum Genet 2022. [DOI] [PubMed]
- 18.Huemer M, Diodato D, Schwahn B, Schiff M, Bandeira A, Benoist JF, Burlina A, Cerone R, Couce ML, Garcia-Cazorla A, et al. Guidelines for diagnosis and management of the cobalamin-related remethylation disorders cblC, cblD, cblE, cblF, cblG, cblJ and MTHFR deficiency. J Inherit Metab Dis. 2017;40(1):21–48. doi: 10.1007/s10545-016-9991-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ledgerwood EC, Marshall JW, Weijman JF. The role of peroxiredoxin 1 in redox sensing and transducing. Arch Biochem Biophys. 2017;617:60–67. doi: 10.1016/j.abb.2016.10.009. [DOI] [PubMed] [Google Scholar]
- 20.Shau H, Gupta RK, Golub SH. Identification of a natural killer enhancing factor (NKEF) from human erythroid cells. Cell Immunol. 1993;147(1):1–11. doi: 10.1006/cimm.1993.1043. [DOI] [PubMed] [Google Scholar]
- 21.Wen ST, Van Etten RA. The PAG gene product, a stress-induced protein with antioxidant properties, is an Abl SH3-binding protein and a physiological inhibitor of c-Abl tyrosine kinase activity. Genes Dev. 1997;11(19):2456–2467. doi: 10.1101/gad.11.19.2456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Neumann CA, Krause DS, Carman CV, Das S, Dubey DP, Abraham JL, Bronson RT, Fujiwara Y, Orkin SH, Van Etten RA. Essential role for the peroxiredoxin Prdx1 in erythrocyte antioxidant defence and tumour suppression. Nature. 2003;424(6948):561–565. doi: 10.1038/nature01819. [DOI] [PubMed] [Google Scholar]
- 23.Egler RA, Fernandes E, Rothermund K, Sereika S, de Souza-Pinto N, Jaruga P, Dizdaroglu M, Prochownik EV. Regulation of reactive oxygen species, DNA damage, and c-Myc function by peroxiredoxin 1. Oncogene. 2005;24(54):8038–8050. doi: 10.1038/sj.onc.1208821. [DOI] [PubMed] [Google Scholar]
- 24.Rosok O, Pedeutour F, Ree AH, Aasheim HC. Identification and characterization of TESK2, a novel member of the LIMK/TESK family of protein kinases, predominantly expressed in testis. Genomics. 1999;61(1):44–54. doi: 10.1006/geno.1999.5922. [DOI] [PubMed] [Google Scholar]
- 25.Singh M, Venugopal C, Tokar T, McFarlane N, Subapanditha MK, Qazi M, Bakhshinyan D, Vora P, Murty NK, Jurisica I, et al. Therapeutic targeting of the premetastatic stage in human lung-to-brain metastasis. Cancer Res. 2018;78(17):5124–5134. doi: 10.1158/0008-5472.CAN-18-1022. [DOI] [PubMed] [Google Scholar]
- 26.Toshima J, Toshima JY, Takeuchi K, Mori R, Mizuno K. Cofilin phosphorylation and actin reorganization activities of testicular protein kinase 2 and its predominant expression in testicular Sertoli cells. J Biol Chem. 2001;276(33):31449–31458. doi: 10.1074/jbc.M102988200. [DOI] [PubMed] [Google Scholar]
- 27.Kawauchi K, Tan WW, Araki K, Abu Bakar FB, Kim M, Fujita H, Hirata H, Sawada Y. p130Cas-dependent actin remodelling regulates myogenic differentiation. Biochem J. 2012;445(3):323–332. doi: 10.1042/BJ20112169. [DOI] [PubMed] [Google Scholar]
- 28.Fagerberg L, Hallstrom BM, Oksvold P, Kampf C, Djureinovic D, Odeberg J, Habuka M, Tahmasebpoor S, Danielsson A, Edlund K, et al. Analysis of the human tissue-specific expression by genome-wide integration of transcriptomics and antibody-based proteomics. Mol Cell Proteom. 2014;13(2):397–406. doi: 10.1074/mcp.M113.035600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Krogan NJ, Kim M, Tong A, Golshani A, Cagney G, Canadien V, Richards DP, Beattie BK, Emili A, Boone C, et al. Methylation of histone H3 by Set2 in Saccharomyces cerevisiae is linked to transcriptional elongation by RNA polymerase II. Mol Cell Biol. 2003;23(12):4207–4218. doi: 10.1128/MCB.23.12.4207-4218.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Baubec T, Colombo DF, Wirbelauer C, Schmidt J, Burger L, Krebs AR, Akalin A, Schubeler D. Genomic profiling of DNA methyltransferases reveals a role for DNMT3B in genic methylation. Nature. 2015;520(7546):243–247. doi: 10.1038/nature14176. [DOI] [PubMed] [Google Scholar]
- 31.McDaniel SL, Strahl BD. Shaping the cellular landscape with Set2/SETD2 methylation. Cell Mol Life Sci. 2017;74(18):3317–3334. doi: 10.1007/s00018-017-2517-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deltas C. Digenic inheritance and genetic modifiers. Clin Genet. 2018;93(3):429–438. doi: 10.1111/cge.13150. [DOI] [PubMed] [Google Scholar]
- 33.Garg P, Jadhav B, Rodriguez OL, Patel N, Martin-Trujillo A, Jain M, Metsu S, Olsen H, Paten B, Ritz B, et al. A survey of rare epigenetic variation in 23,116 human genomes identifies disease-relevant epivariations and CGG expansions. Am J Hum Genet. 2020;107(4):654–669. doi: 10.1016/j.ajhg.2020.08.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mulder RH, Neumann A, Cecil CAM, Walton E, Houtepen LC, Simpkin AJ, Rijlaarsdam J, Heijmans BT, Gaunt TR, Felix JF, et al. Epigenome-wide change and variation in DNA methylation in childhood: trajectories from birth to late adolescence. Hum Mol Genet. 2021;30(1):119–134. doi: 10.1093/hmg/ddaa280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang BZ, Zhang H, Ge W, Weder N, Douglas-Palumberi H, Perepletchikova F, Gelernter J, Kaufman J. Child abuse and epigenetic mechanisms of disease risk. Am J Prev Med. 2013;44(2):101–107. doi: 10.1016/j.amepre.2012.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu Y, Aryee MJ, Padyukov L, Fallin MD, Hesselberg E, Runarsson A, Reinius L, Acevedo N, Taub M, Ronninger M, et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat Biotechnol. 2013;31(2):142–147. doi: 10.1038/nbt.2487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gross AM, Jaeger PA, Kreisberg JF, Licon K, Jepsen KL, Khosroheidari M, Morsey BM, Swindells S, Shen H, Ng CT, et al. Methylome-wide analysis of chronic HIV infection reveals five-year increase in biological age and epigenetic targeting of HLA. Mol Cell. 2016;62(2):157–168. doi: 10.1016/j.molcel.2016.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Supplemental Figure S1. Genome-wide density distribution of CpG probes in the 17 DNA methylome profiles of the 17 patients with MMACHC epimutation (epi-cblC disease, isolated MMACHC epimutation, biallelic MMACHC epimutation).
Additional file 2. Supplemental Figure S2. (A) 2-D plot using the two top eigenvectors (PC1, PC3) derived from the primary component analysis on the genome-wide methylome landscape of the studied patients and controls. (B) 2-D plot using the PC1 and PC2 eigenvectors derived from the primary component analysis on the methylome landscape of chromosome 1 of the studied patients and controls.
Additional file 3. Supplemental Table S1. Forward and reverse primers for reverse transcription-quantitative polymerase chain reaction (RT-qPCR) of MMACHC, TESK2 transcripts and PRDX1 aberrant transcript.
Data Availability Statement
Data are available for use in collaborative studies to researchers upon reasonable request (jean-louis.gueant@univ-lorraine.fr). Data will be provided following the review and approval of a research proposal (including a statistical analysis plan) and the completion of a data-sharing agreement. Responses to the request for the raw data will be judged by the IRB of INSERM UMR_S 1256.