

Abstract
Alzheimer’s disease (AD) is the most prevalent dementia in the world. Its cause(s) are presently largely unknown. The most common explanation for AD, now, is the amyloid cascade hypothesis, which states that the cause of AD is senile plaque formation by the amyloid β peptide, and the formation of neurofibrillary tangles by hyperphosphorylated tau. A second, burgeoning theory by which to explain AD is based on the infection hypothesis. Much experimental and epidemiological data support the involvement of infections in the development of dementia. According to this mechanism, the infection either directly or via microbial virulence factors precedes the formation of amyloid β plaques. The amyloid β peptide, possessing antimicrobial properties, may be beneficial at an early stage of AD, but becomes detrimental with the progression of the disease, concomitantly with alterations to the innate immune system at both the peripheral and central levels. Infection results in neuroinflammation, leading to, and sustained by, systemic inflammation, causing eventual neurodegeneration, and the senescence of the immune cells. The sources of AD-involved microbes are various body microbiome communities from the gut, mouth, nose, and skin. The infection hypothesis of AD opens a vista to new therapeutic approaches, either by treating the infection itself or modulating the immune system, its senescence, or the body’s metabolism, either separately, in parallel, or in a multi-step way.
1. Introduction
Presently, Alzheimer’s disease (AD) is one of the most important public health concerns [1]. It remains the most common cause of dementia in the world [1–4]. Despite huge scientific efforts and financial outlay, we still do not know what is the cause of this disease, which is perhaps more appropriately defined as a syndrome [5–7]. More than 1000 clinical trials have failed, and all ongoing attempts to identify treatment do not seem to be promising [8–10]. The prevailing hypothesis to explain the pathomechanism(s) of AD puts the amyloid beta peptide (Aβ) at center stage and is defined as the beta amyloid cascade hypothesis [11–13]. All attempts to modulate by any means the concentration of Aβ in patients’ brains have resulted so far in a failure to improve the clinical status of patients suffering from any stage of AD. Thus, there is an urgent need to reconsider the causes of AD, which may and should lead us to the discovery of new innovative measures to prevent and treat AD [8–10]. A new hypothesis has emerged, which puts infection or microbial/microbiome challenge in the forefront of AD [5, 14].
AD is a chronic disease, and the pathophysiological processes leading ultimately to its overt symptoms start decades before the clinical manifestations may appear, triggered by age-related changes [15, 16], such as immune system modifications, inflammaging (increased levels of proinflammatory cytokines without overt signs of any inflammation), increase in gut leakage, and microbiome shift (dysbiosis), as well as the appearance of senescent cells in the gut and the brain; all factors that favor the development of AD [5, 7]. This makes it very difficult to cure, but in the meantime, this may convey hope as it can be prevented in the “incubation period” preceding the appearance of cognitive decline to avoid the full-blown disease, if appropriate predictive biomarkers can be discovered. It is, however, of interest that the development from the emergence of the first clinical symptoms [mild cognitive impairment (MCI (MCI))] to full-blown AD takes about 10–15 years. This time may also be used to slow down the progression or even cure it if the cause(s) of AD could be found or if biomarkers could allow it to be efficiently tracked.
2. What is the Prevailing Hypothesis and Why Is It Unsatisfactory?
Since the first description of AD by Alois Alzheimer, extracellular Aβ plaques and intracellular hyperphosphorylated tau deposition (called neurofibrillary tangles) unrelated or only indirectly related to the formation of Aβ have become the pathological hallmarks of AD [17, 18].
This gave rise to the amyloid hypothesis of AD which has been adopted by the majority of the AD scientific community. Everything in AD research, clinical trials and ultimately in memory clinics has been oriented and driven by the Aβ hypothesis [12, 19, 20].
However, the lack of useful and productive progress toward mechanistic understanding of AD calls for a revaluation of the Aβ cascade hypothesis. The amyloid hypothesis states that the production of Aβ from its amyloid precursor protein (APP) in neurons and astrocytes by β-secretase (BACE) together with a presenilin-containing complex called γ-secretase is the primary cause of AD [21–23]. Thus, the formation of Aβ is the starting point that initiates all the other observed pathological phenomena associated with AD and culminates in the deposition of amyloid plaques in the brain [13]. It also triggers the intracellular deposition of hyperphosphorylated tau. Both of these phenomena (formation of plaques and of neurofibrillary tangles) result in neurodegeneration (synapse degeneration and then neuronal cell death) and, more importantly, neuroinflammation [24–30]. It has subsequently been found that AD has many different genetic risk (susceptibility) factors, such as ApoE-ε4, TREM-2, and TOMM40 [31–37].
However, as appealing as this hypothesis may appear, many observations made over decades have spoken against it. One of the most important, yet constantly overlooked details is that these hallmarks exist in the brain of 20–30% of non-demented healthy elderly, while in contrast, an almost identical proportion of patients suffering from AD do not have these hallmarks [5, 38]. Evidence suggesting a role for events preceding and precipitating deposition of Aβ-containing plaques emerged almost a decade ago from the laboratory of Dr. Rudolf Tanzi, who had demonstrated the antimicrobial properties of Aβ and first described it as an antimicrobial peptides (AMP) [39]. These crucial observations were later confirmed by other laboratories, which found that Aβ acts as an AMP against many different micro-organisms [40, 41], which suggested the “Antimicrobial Protection Hypothesis” of AD. Moreover, several different microorganisms have been demonstrated in the brains of AD patients [42–53]. Nevertheless, the most important argument against the Aβ hypothesis of AD is, as already mentioned, the lack of success of almost all trials that directly targeted Aβ accumulation through vaccination or monoclonal antibodies or its production by the BACE inhibitors [54, 55]. An additional finding supports the antimicrobial role of Aβ generated in the brain, as a decrease in Aβ production during the clinical trials lead occasionally to the emergence of some type of infections in the brain [56]. Thus, based on these facts, the infection hypothesis of AD pathogenesis was developed, slowly conceptualized, and finally clearly published in a recent editorial [14]; see Fig. 1. Other possible hypotheses of mechanisms leading to AD have also been advanced [57–59].
Fig. 1.
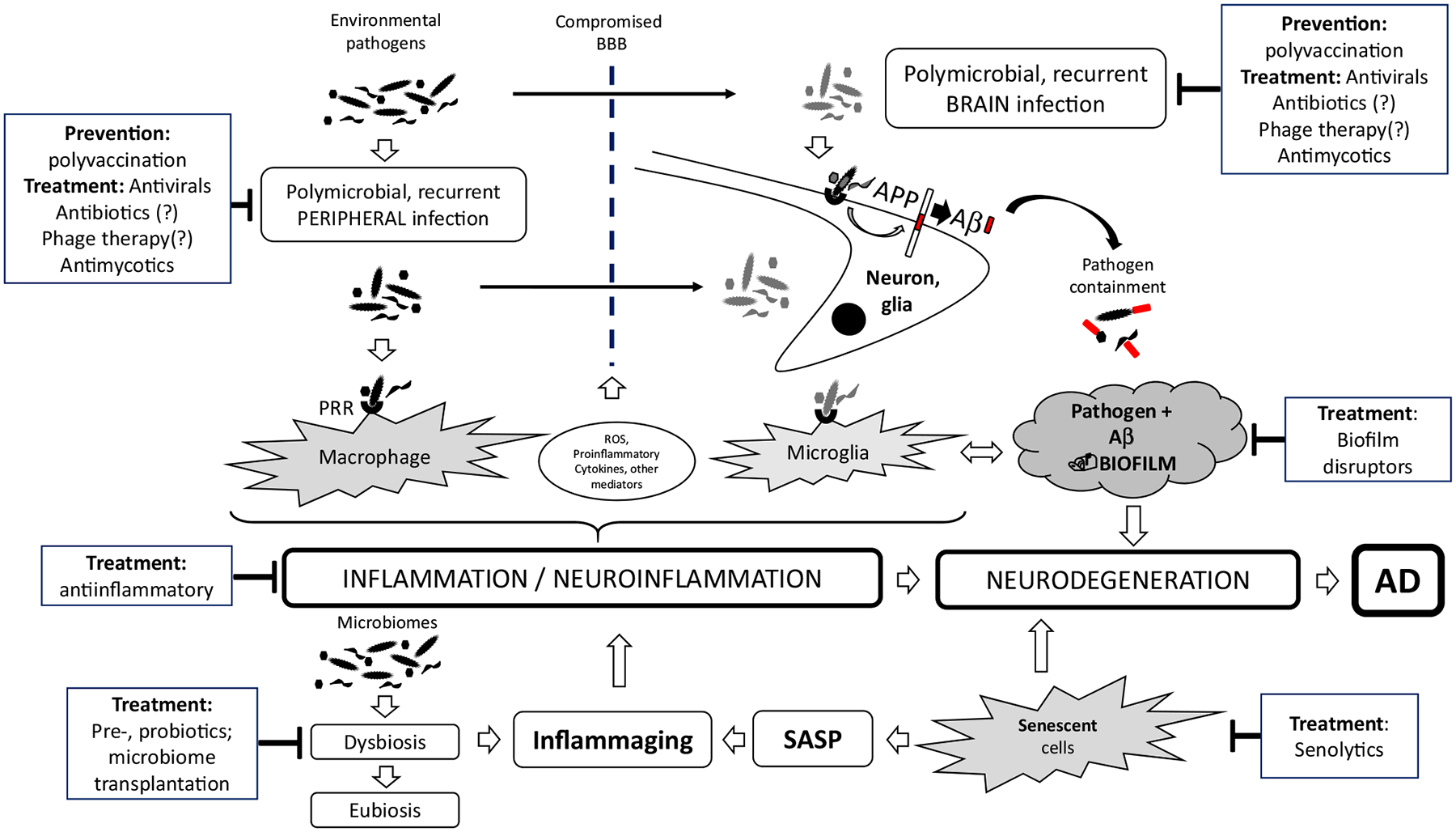
Possible intervention checkpoints according to the infection hypothesis. This figure depicts the various putative players in the development of AD, considering the infection hypothesis as well as the individual future targets for intervention. Aβ amyloid beta peptide, AD Alzheimer’s disease, APP amyloid precursor protein, BBB blood–brain barrier, PRR pattern recognition receptors, ROS reactive oxygen species, SASP senescence-associated secretory phenotype
3. Other Existing Theories
It should be mentioned that over the years a few researchers have promoted different ideas about AD etiology. Among them was the vascular hypothesis, which appeared in the 1990 s [59]. A study in nuns (The Nun Study) demonstrated that even if, pathologically, amyloid plaques could be detected in the brain, the clinical diagnosis of AD was established only when these lesions were accompanied by atherosclerotic lesions in the brain, regardless of the age of the nun [60]. Later, it was shown that ischemia and shear stress were also able to generate the production of Aβ [61, 62]. These ideas led to the integration of vascular problems and associated diseases (e.g., hypertension) as risk factors for AD [63, 64].
Another theory, the mitochondrial cascade hypothesis, authored by Swerdlow et al. [65], proposed that mitochondrial dysfunction resulting from aging, genetic predisposition, or environmental factors results in the production of reactive oxygen species (ROS) that damage brain cell functions, resulting in typical AD pathology. The mitochondrial cascade hypothesis, similarly to the Aβ hypothesis, cannot stand alone as a causative factor for neurodegeneration, but requires internal or external stresses acting on various brain cells, such as neurons, microglia, and astrocytes. All infections stimulate ROS production and may interact directly with mitochondria, perturbing mitochondrial DNA (mtDNA) and mitochondrial homeostasis (fission, fusion, and mitophagy), leading to mitochondrial dysfunction [58]. Thus, this hypothesis can be easily integrated into the infection hypothesis.
4. What Evidence Supports The Infection Hypothesis for AD?
There are numerous epidemiological and experimental discoveries that support that AD may be an infectious disease. Already, many years ago, epidemiological evidence has linked the treatment of rheumatoid arthritis (RA) to the prevention of AD. McGeer et al. [66] showed that RA patients who are receiving anti-inflammatory treatment develop AD much less often than others. This observation was confirmed by an updated meta-analysis of cohort studies [82]. Sixteen cohort studies, including 236,022 participants, published between 1995 and 2016, were included in this systematic review. Current evidence suggests that nonsteroidal anti-inflammatory drug (NSAID) exposure might be significantly associated with reduced risk of AD, but again, the need for prospective studies with individual NSAIDs is badly needed. Initially, this protection was suggested to be linked to the decrease in the neurotoxic effect of Aβ-induced neuroinflammation [67, 68]. However, more recently, RA was linked to the mouth bacterial pathogen Porphyromonas gingivalis [69–73]. Thus, the question may arise of whether the treatment of RA inflammation, which indirectly also decreased AD progression by reducing neuroinflammation, could somehow treat the common root, namely an infectious origin.
Epidemiologically, the first and strongest evidence was brought to the community by dentists [74–77]. They observed that people who suffer from periodontitis develop AD much more often than those who do not present this alteration in the mouth [78, 79]. Since these epidemiological observations, numerous experimental data have supported the link between periodontitis-induced systemic inflammation, oral dysbiosis, and altered immune response and AD [77, 80–89]. It should, however, be mentioned that some studies did not confirm these associations [90]. Increased AD incidence was linked to the presence of biofilms produced by the cornerstone bacteria P. gingivalis [80, 91]; however, recently, other bacteria were found to be involved, such as Treponema denticola and Tannerella forsythia [81]. The bacterial effect might be direct (bacteria entering the brain via the lingual nerve or the olfactory bulb) or indirect via their virulence products that stimulate the production of Aβ; the effect is the same: appearance in the brain of a structure resembling bacterial biofilm, called senile or Aβ plaques [75, 77, 78, 87, 92]. Indeed, it was recently postulated that amyloid plaques are biofilms [48]. This was recently supported by a study demonstrating the presence of one of the most important virulence factors of P. gingivalis, gingipain, in the brains of healthy and AD patients [52]. This latter group showed also [by quantitative polymerase chain reaction (qPCR)] the presence of P. gingivalis in the brains of healthy subjects as well as in the brains of patients suffering from AD [52]. Our unpublished data also demonstrated by qPCR the presence of P. gingivalis in AD brains (manuscript in preparation).
Yet another bit of information associating the development of AD with bacterial infections is the role of calreticulin (CRT) and galectin-3 in the brain. The decreased expression of CRT in the neurons of AD patients was first demonstrated almost 2 decades ago [93]. CRT is a multi-functional protein, which has since been associated with a chaperone function for APP; thus, the more CRT is present in the neuron, the more stable the APP becomes and less Aβ is produced resulting in its aggregates (plaques) [94]. On the other hand, CRT production is upregulated by Aβ oligomers, at least in vitro [95]. Serum levels of CRT are considered a negative biomarker of AD development and progression [96]. This may make sense, as CRT has been very recently shown to be secreted also by activated macrophages and microglia and to act as an opsonin facilitating the phagocytosis of bacteria invading the brain (albeit so far only in a rat model) [97]. Thus, we could imagine/propose a scenario where an infection leads to production and release of Aβ, which aggregates and upregulates the production and secretion of CRT, which in turn binds/opsonizes bacteria for microglia-executed phagocytosis; thus, more intracerebral infection could lead to decreased levels of CRT (as it would be used up by opsonization and phagocytosis), which would favor AD.
Very recently a study in Taiwan showed that those suffering from herpes simplex virus-1 (HSV-1) infection and treated with antiviral drugs had a reduced incidence of AD [98]. This retrospective cohort study from Taiwan showed the 10-year incidence of dementia in a group of 8362 subjects aged 50 years or over who were newly diagnosed with HSV-1 or HSV-2 infection was 2.56-fold greater than that in the control group (95% confidence interval 2.351–2.795; P < 0.001). More strikingly, anti-herpetic medication reduced the risk of developing dementia by approximately 91%. These results strongly support a potential causative link between HSV-1 infection and AD, mainly in genetically susceptible subjects [36]. This observation suggests that AD is linked somehow to viral infections [53, 99–101]. However, this still does not clearly demonstrate whether HSV-1 is the cause or the consequence of AD, but highly suggests that HSV-1 may be also involved in its pathogenesis. Interestingly, decades ago, Itzhaki showed experimentally that HSV-1 DNA is present in the brain plaques of persons suffering from AD [43]. This indicated that viral infection may play a role in the development or progression of AD and that the secretion of Aβ may be a reactive phenomenon to control infection. It may have some AMP effect or may be a general acute phase reaction to a strong stress as many other peptides in the organism during infection, such as LL-37, are affected [102, 103]. Very recently, the Lovheim group demonstrated an association between HSV-1 carriage and declining episodic memory function, only among ApoE-ε4 carriers, while the other alleles such as ε2 and ε3 did not show such an association in cross-sectional and longitudinal studies of a large population-based cohort [37]. Thus, the Lovheim group [36, 37] showed for the first time in a prospective epidemiological analysis that the host genetic background interacts with HSV-1 carriage to increase the risk for developing AD. The primary strengths of their studies include many cases with closely matched controls from the same population, combined with thorough clinical AD diagnosis. These studies further confirm the interaction between ApoE-ε4 heterozygosity (APOE-ε2/ε4 or ε3/ε4) and HSV-1 carriage, which increased the risk of AD by approximately fivefold, whereas the presence of only one factor did not. A calculated genetic risk score (GRS), based on nine additional risk genes (ABCA7, BIN1, CD33, CLU, CR1, EPHA1, MS4A4E, NECTIN2, and PICALM), also correlated with anti–HSV-1 immunoglobulin G (IgG) for increased risk of subsequent AD. The present findings suggest that the ApoE-ε4 allele and other AD genetic risk factors might potentiate the risk of developing HSV-1–associated AD. Another very recent study in a cohort in Bordeaux, France, further confirmed these relationships between ApoE4 and HSV-1 being a strong risk factor for AD development [104]. Together, these data could provide new insights into the possible mechanisms by which the genetic treat of ApoE-ε4 associated with HSV-1 carriage may be involved in the development of AD.
Almost at the same time, Miklossy and others have demonstrated the presence of other microbes, such as the spirochete Borrelia burgdorferi, in blood, cerebrospinal fluid, and brain tissue [105, 106]. They also hypothesized that this bacterium may produce a biofilm that would constitute the amyloid plaque, protecting bacteria from various stress in the brain [107]. Balin et al. have demonstrated the existence of Chlamydia pneumoniae in plaques [50]. They later observed that systemic infection with C. pneumoniae in turn increased the occurrence of AD [51]. All these data have converged to promote and justify the development of the infection hypothesis, stating that accumulation of Aβ is not the primary cause of AD, but is itself the consequence of infection. Aβ would then play its pathogenic role as stated by the amyloid hypothesis [5, 108].
Subsequently, the demonstration of Treponema in plaques reinforced the infection hypothesis. In the sexually transmitted infection syphilis, caused by Treponema pallidum, the tertiary stage is accompanied by a particular dementia status [109], which occurs in most cases several decades after the primary infection [109]. This is a very important similitude as this makes plausible the role of a bacterium of the genus Treponema in the pathogenesis of AD. Furthermore, another virus, HIV, has been associated with neurodegenerative disorders [HIV-associated neurodegenerative disorder (HAND)] [110, 111]. A neurodegenerative disorder related to HIV infection has been reported to cause a severe form of dementia [112]. Since the efficacious treatment of HIV by combination antiretroviral therapy (cART), patients live much longer with the virus, reaching old age, and their neurocognitive disorder has become much milder in its clinical manifestations [113–115]. In these patients, HAND resembles AD more and more, even including the production of Aβ in response to the virus [116, 117]. Interestingly, HIV suppresses production of Aβ at early stages of the infection as a protection against the AMP role of Aβ, which reinforces its AMP role [118].
The latest microorganisms abundantly found post-mortem in the brains of AD patients are pathogenic fungi [119]. The most important species were Candida albicans and the Malassezia sp. [120, 121]. We do not know how fungi may be involved in the development of AD, and this needs further investigations.
All this experimental evidence points toward the involvement of microbes in the pathogenesis of AD [14] (Table 1). These results also indicate that it would be very difficult to identify one microorganism as the unique cause. It was suggested that AD is a polymicrobial disease [120, 122]. Nevertheless, one bacterium may be more important than the others, namely P. gingivalis. Its cornerstone role in periodontitis, where it orchestrates the formation of biofilms, could be duplicated in AD. In support of this theory, a recent paper found the P. gingivalis virulence factors, gingipains, in the post-mortem brains of AD patients [52]. In summary, all of the experimental data gathered so far, suggest a potential causality between infections and AD [123].
Table 1.
The most frequently involved microorganisms in Alzheimer’s disease
| Viruses |
| HSV-1 |
| HIV |
| HHV-6 and HHV-7 |
| Bacteria |
| Borrelia burgdorferi |
| Treponema denticola |
| Chlamydia pneumoniae |
| Porphyromonas gingivalis |
| Fungi |
| Candida albicans |
| Malassezia furfur |
HHV human herpesvirus, HSV herpes simplex virus
Before further describing the putative pathomechanism that could explain how microorganisms may induce AD, we will describe the changes in the immune system that are a necessary corollary to allow infections to promote AD development, a process that may be the target for future treatments.
5. The Innate and Adaptive Immune System in AD
The immune system has the role to defend the organism against external and internal challenges [124, 125]. In many circumstances, the immune system may be activated for a longer period than necessary when a challenge is maintained for a long time or is reactivated from time to time [126]. This means that inflammation that plays a beneficial role in acute infection may become chronic and detrimental to the host organism and even generate disease [126].
In the case of AD, neuroinflammation is a fundamental part of its pathogenesis [13, 16, 24, 127–130]. According to the amyloid hypothesis, neuroinflammation is generated and maintained chronically by Aβ [13]. In the infection hypothesis, it is the result of the penetration of the microbes or their products into the brain and is meant to help in the elimination of the infection, at least at the beginning of the pathogen invasion into the brain [15]. However, as infection becomes chronic, neuroinflammation also becomes chronic and destructive [40, 41]. Neuroinflammation in AD is characterized by microglial and astrocyte activation, inflammasome activation via NLRP3, complement activation, and altered cytokine production shifted towards pro-inflammatory cytokines such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, and IL-6 [131]. All of these characteristic features of neuroinflammation may be found typically during infections as well [132].
Indeed, in AD, neuroinflammation is sustained mainly by the systemic and the local innate immune system. Systematically, the activated innate peripheral immune cells such as natural killer (NK) cells, neutrophils, and monocytes are, on the one hand, able to cross the blood–brain barrier (BBB) and create destruction in the brain directly or by their products, such as the pro-inflammatory cytokines or chemokines, which cross the BBB and act on resident brain immune innate cells, such as microglia and astrocytes, as demonstrated in humans and in animal models of sepsis [133–137]. Furthermore, Bu et al. have shown in an association study that the systemic infectious burden measured by antimicrobial antibodies increased the risk of AD [138]. This study points again towards the polymicrobial nature of AD. Thus, peripheral infections, inflammation, and stress were linked to microglial activation via the NFκB/NLRP3 pathway via pro-inflammatory cytokines [139–141]. Together, these data suggest that systemic immune activation has central effects and vice versa [77, 82, 142].
The brain has a powerful innate system composed of microglia (brain macrophages), astrocytes, and even neurons. They may destroy microorganisms or produce efficient antimicrobial peptides, the most important being the cathelicidin LL-37 [143–145]. Microglia, in response to stress, develop an inflammatory response (pathogen-associated molecular patterns or damage-associated molecular patterns) and secrete pro-inflammatory cytokines [146–148]. Importantly, microglia may also modulate astrocyte reactivity by IL-1α, TNF-α, and C1q, and such stimulated astrocytes may acquire a pro-inflammatory A1 phenotype [149, 150]. These “good” innate cells may be turned into “bad” cells by several microbial products, including lipopolysaccharides (LPS) and gingipains, resulting in their loss of ability to eliminate invaders and decrease the Aβ burden, and in the activation of their senescence as well as in increasing their attack against neurons [89, 131, 151, 152]. In summary, under microbial pressure, the brain’s innate immune system deviates from a defensive to a killing role, resulting in neuroinflammation, senescence and neuronal death. Again, one trigger suspected to play a pathogenic role in AD is microbes and their products such as LPS.
The demonstration by Soscia et al. that Aβ is an AMP gave a new impetus to the infection hypothesis [39]. They tested Aβ against bacteria and fungi and found it more powerful than even LL-37. More recently, we and others have demonstrated that, like LL-37, Aβ may also inactivate viruses, including HSV-1 [41], influenza [153], and retroviruses [118]. It was also shown that when infected by HSV-1, neurons were able to secrete substantial amounts of Aβ, which inhibited HSV-1 infection of other neurons [154]. This indicates that Aβ is not only a pathological peptide as supposed originally, but has a well-defined physiological role and is produced under very well-defined conditions. Moreover, Aβ was more powerful than interferon (IFN) type I. Recently, an interesting finding showed that Aβ may also have anticancer properties [155] as well as BBB repair properties [156]. The most important cells producing Aβ are neurons and astrocytes. This is not surprising as the latter together with microglia play a crucial role in the brain host defense either clearing waste or secreting defensins [157, 158].
The role of Aβ as an AMP has since been tested in many animal and experimental models. It was shown in a murine model of Salmonella enterica and Salmonella typhimurium infection that endogenous as well as exogenous Aβ could prevent infection in the brain [159, 160]. These authors hypothesized that the mechanism of action of Aβ was by formation of amyloid aggregates (plaques) using the microbial surface [161]. This led to the formulation of the Antimicrobial Protection Hypothesis [162]. However, they never linked biofilm formation to plaque formation, as had been hypothesized by Miklossy [48]. Together these data again strongly support the notion that Aβ is a newly recognized member of the large AMP family combating infections in humans.
All these findings provide an answer to why evolution would promote, and even select for, an enzymatic system (β- and γ-secretase) if the result had no pro-survival value and, as believed, was only detrimental (leading to AD). Now, based on the convincing observations described above, we can say that generation of Aβ has a clear pro-survival role.
In fact, both secretases have multiple targets with distinct, different physiological roles, which likely evolved in early vertebrates many million years before APP and Aβ [163–165]. Also, the APP and its cleavage products seem to be multifunctional. Yet, based on their studies, Moore et al. venture to say, “Although not as deeply conserved as BACE1, the Aβ peptide has been conserved for at least 430 million years. This indicates that Aβ, too, has essential functions that have thus far escaped discovery” [165], which indirectly supports our notion that antimicrobial effects of Aβ might be an (evolutionarily conserved) “bonus” feature.
The adaptive immune system also showed important changes in AD [166]. Naïve T cells decreased and CD8+ memory T cells increased. This situation is identical to what is observed during normal aging, but also in chronic infections, independent of age, such as cytomegalovirus (CMV) infections [167, 168]. This suggests that just like the innate immune system, the adaptive immune system is also chronically stimulated and its capacity to fight infections is not always efficient [169]. Thus, the immune system shows similar properties in AD patients to those found in many other chronic infectious diseases with, of course, specificities typical to its localization in the brain.
Furthermore, this constant stimulation of the immune system via what is called inflammaging results in the exhaustion of the immune cells, resulting in an increase of cellular senescence, which is also evident in microglial cells [170]. This cellular senescence, via the senescence-associated secretory phenotype (SASP), further supports and amplifies the notion of inflammaging [171–174]. SASP of microglia and astrocytes is sustained by the activation of two main intracellular inflammatory pathways that are intimately linked with NFκB and the inflammasome pathways [175–177]. The NOD receptor pathway via NLRP3 mediates the production of IL-1β, IL-18, and caspase-1, which increase in AD brains. Moreover, IL-1β has been shown to contribute to the permeability of the BBB, favoring the passage of microorganisms and their by-products [7, 178, 179]. These pathways may not only induce senescence but also what is called pyroptosis, which is an inflammation-triggered programmed cell death, especially in microglia [180].
6. What is the Pathomechanism that Microorganism(s) Use to Cause AD?
We suggest two overlapping pathways for microbes to induce AD.
The first involves direct migration of the microorganisms to the brain via either the lingual nerve or the olfactory bulb and crossing the permeabilized BBB, composed mainly of astrocytes, endothelial cells, and pericytes [181, 182]. For a long time, the brain was considered a privileged organ, as it was protected by a well sealed BBB; however, it has been shown that even in the early stages of AD, the BBB becomes more permeable [183]. This also may occur during the process of aging [184] as well as during systemic inflammatory responses elicited by microbial infections such as viruses or bacteria, with or without direct brain infection [185, 186]. Microbes have evolved to be able to make the BBB permeable, partly by subverting pericytes and/or endothelial cells by inducing either their apoptosis or by using the complement system receptor 3 (CR3) to their advantage to make their way to the brain [187–189]. Neurons would respond by producing Aβ, to try to destroy the invading microbes [39–41]. In the meantime, the microglia and astrocytes are also stimulated and produce other antimicrobial peptides, pro-inflammatory cytokines, free radicals, and proteases to destroy the microorganism [158, 190, 191]. Moreover, the complement system is activated, and this favors phagocytosis [192]. Finally, the adaptive immune system is also activated either to produce cytotoxic effector CD8 + T cells or antibodies via B cells [193]. Thus, in a normal situation, the invading microorganism may be totally eliminated or imprisoned in biofilm, seen as plaques, which protect the microbial community from destruction [48]. This process may occur for decades preceding clinical manifestations of AD, and many reactivation or reinfection cycles may lead to chronic neuroinflammation and plaque deposition resulting in massive neuronal death.
Another non-mutually exclusive pathway may be the passage not of the entire microorganism but only its virulence factors, such as LPS, gingipains, extracellular RNAs, or peptidyl arginine deiminase (PAD) enzymes [52, 86–89, 194, 195] or other. These substances may occur permanently in the organism and originate from any of the microbial communities/reservoirs of the organism, such as gut microbiome, mouth, or neurobiome [196–200]. These microbial products or metabolites may mediate their deleterious actions by being incorporated in extracellular vesicles (EVs) [201]. Indeed, many microorganisms, including P. gingivalis, are also able to release EVs containing gingipain and fimbriae, which modulates intestinal permeability as well as the function of the innate immune system, thus favoring an inflammatory status [88, 202, 203]. In this way these by-products will stimulate the immune system with the production of inflammatory mediators, which will chronically induce the same processes as the direct presence of the microorganism itself [89, 204].
As mentioned, these microbes or their virulence products may originate from various microbial reservoirs in the body including microorganisms that have reached the brain via the first pathomechanism. The most important microbial reservoir in humans is in the gut, which leads to the notion of the gut–brain axis. This means that there is a constant communication between the gut and the brain and vice versa throughout life [205–208]. Indeed, the direct presence of microbes and/or their by-products has been demonstrated in the brains of AD patients, but interestingly also in the brains of healthy aged subjects, hence the notion of a “neurobiome” [52]. Studies of the gut microbiome in aged people showed a tendency towards an increase in Gram-negative bacteria [209], which was also shown in MCI patients [210]. This becomes even more problematic when the immune system manifests some maladaptation with aging which permits the clinical development of AD through the translocation of microbes that are normally commensal (dysbiosis) [211–213] and are contained within the gut by the local immune system which has induced a tolerogenic state [214]. It has also been demonstrated that dysbiosis of the gut microbiome may promote various inflammatory disorders which have provoked microglial activation during the development of AD [211, 215–217]. Thus, this suggests that the gut microbiome or better, its dysbiosis, is involved in regulating microglial activation and neuroinflammation in AD.
Another important axis for the development of AD could be the mouth–brain axis, involving mainly P. gingivalis [77, 218]. P. gingivalis produces various virulence factors such as LPS, flagella, and toxic proteases called gingipains [219]. The LPS may activate astrocytes and transform them to the proinflammatory A1 phenotype by stimulating Toll like receptor (TLR)-4 [220]. Gingipains have been found in the brains of healthy subjects and AD patients, and have been proposed to be involved in the pathophysiology of AD [52]. In periodontitis, these virulence factors, mainly gingipains (lysine-gingipain and arginine-gingipain A/B), have been shown to play a role in host colonization, inactivation of the host immune response, and iron and nutrient acquisition [221, 222]. Gingipains may also activate various innate receptors such as TREM1, TREM2, TLR-4, CR1, and NLRP3 [223–226], which may result in the activation of the inflammasome [227]. This activation in turn facilitates plaque formation and may amplify the inflammatory reaction via release of ASC specks [228, 229]. Interestingly, the activation of this inflammasome results in pyroptosis which eliminates the cell infected by P. gingivalis and limits replication of this bacteria [230]. Furthermore, this phenomenon does not always require the presence of live P. gingivalis; released gingipains may penetrate cells and have similar effects [231, 232]. These processes involving the mentioned receptors, the inflammasome, and P. gingivalis or its gingipains will ultimately kill neurons and favor amyloid plaque deposition and IL-1β release. This will further help to permeabilize the BBB. Gingipains are also able to cleave IgG1 and IgG3, mainly by gingipain K, and in this way, the adaptive branch of the immune defense of the organism can be compromised [233, 234]. Another important virulence factor of P. gingivalis is peptidylarginine deiminase (PPAD), which catalyzes the citrullination of both bacterial and host proteins [235, 236]. PPAD helps P. gingivalis evade destruction by neutrophils by impairing phagocytosis and bacteria-induced NETosis [235]. Furthermore, when PPAD citrullinates cationic antimicrobial peptides such as LP9 and LL-37, it efficiently neutralizes them [237]. Gingipains can also deactivate them by proteolytic degradation [238, 239], which may be followed by PPAD citrullination of exposed arginine residues. All of these products from P. gingivalis help it to evade elimination by both the innate and adaptive immune systems. It is of note that the direct role of P. gingivalis and its products in the development and progression of AD, even if they have been found in the brain of AD patients, will require further studies.
Inflammaging is sustained by an imbalance between the innate and adaptive immune systems together with the senescence of the cells constituting the central nervous system (CNS), including neurons, microglia, and astrocytes. The concomitant processes of inflammaging, programmed cellular senescence, and dysbiosis further favor the leakage of the gut, resulting in the passage of bacteria (pathogenic and/or commensal) [238] and their products into the brain, including those which may contribute to AD, such as the curli [239].
One other recently described phenomenon which can lead to sustained neuroinflammation is the mechanism of “trained” innate immunity [240]. This process captures the constant inflammatory state seen in the innate immune system during aging, AD, and other chronic diseases [126]. Once monocytes have been activated, any new unrelated stimulation will result in higher response from these cells [241]. This property of the innate immune system is reminiscent of memory in the adaptive immune system and can lead to maintenance of a basic, constant activation in cells like microglia, which will likely contribute to constant neuronal destruction.
All these experimental results point to the fact that Aβ is deposited in the brain decades before the clinical manifestation of AD, suggesting that AD is related to chronic mutually sustaining inflammatory processes in the CNS and in the periphery as a result of a long-lasting antimicrobial response culminating in plaque deposition [15, 162, 242, 243].
Whatever the pathway that microorganisms employ to cause AD, better understanding of these processes could suggest new innovative strategies to prevent or intervene in the progression of AD.
7. What are Possible Interventions Targeting the Prevention or Cure of AD?
The obvious treatments which come to mind are treatments by specific agents aimed at containing or direct elimination of the aforementioned microorganisms, such as antivirals, antibacterials, and antifungal products. In the case of viruses, the most relevant would appear to be the antiviral drugs that penetrate the BBB and which are very effective even in herpes viral encephalitis, such as valacyclovir [101, 244, 245]. Unfortunately, although HSV-1 has been implicated in AD, we do not know if other viruses are also involved, and even for HSV-1, we do not know exactly when and how it may cause AD (and, as mentioned above, rather assume that the cause is prolonged and polymicrobial). It may be experimentally challenging to determine when, how, and at what dose to use such agents [100]. Nevertheless, each time that we have an infectious burst such as herpes labialis or herpes genitalis or zoster, we should treat the patients most vigorously, regardless of age. If we consider data from the Taiwanese study mentioned above, each of these treatments should decrease the incidence of AD. Other viruses may also be involved, and so we will have to discover antiviral agents that cross the BBB to control them.
Are there any direct trials targeting any stages of AD with antiviral treatment? In fact, there is one ongoing, one which has been just finished, and some may be actively planned [100]. This is due to the uncertainty of the viral mechanisms causing AD. Another factor is knowing at what time to treat. Considering the long “incubation period” of AD, it would be logical to treat any viral infection at any time it manifests itself, which would be of great advantage in decreasing the deleterious effect not only on the immunobiography/inflammaging, but also the viral contribution to the chronicity of such an accumulation of burden involving several infections. To avoid unnecessary treatment of non-susceptible individuals, one of the best periods would be when memory problems are starting to appear in the subjective memory complaint (SMC) and MCI stages. In this way, we could assess whether this treatment would at least delay the progression towards AD. Logically, a pulse repeated intervention would be needed, but this again would have to be demonstrated. The advantage of valacyclovir and related drugs is that they have very few side effects even in elderly subjects. The epidemiological study from Taiwan seems to indicate that it could be a rewarding intervention. If successful in SMC or MCI, it could be envisaged at earlier times.
Devanand in his paper of 2018 [100] mentions a phase II, proof-of-concept, randomized, double-blind, placebo-controlled, 18-month treatment trial of 130 patients (65 valacyclovir, 65 placebo) with mild AD [Mini-Mental State Examination (MMSE) range 20–28] who tested positive for antibodies to HSV-1 or HSV-2. Valacyclovir dosage will be 2–4 g daily. The dose range was stated safe and is known to lead to CNS penetration with high cerebrospinal fluid (CSF) levels, which should increase the chance of efficacy. The hypothesis was that, in comparison with patients treated with placebo, patients treated with valacyclovir would show a smaller decline on the Alzheimer’s Disease Assessment Scale–Cognition 11-item scale (ADAS-Cog11) (cognitive measure; 0–78 weeks) and the Alzheimer’s Disease Cooperative Study–Activities of Daily Living scale (ADCS-ADL) (function measure; 0–78 weeks). The authors state that if the trial is successful, they will continue with a phase III trial. Indeed, there is a trial registered as ClinicalTrials.gov Identifier NCT03282916. This trial plans to use valacyclovir in MCI/AD patients to establish whether this treatment will restore or decrease cognitive functions. We should wait for the results of these trials to be published in forthcoming years, probably in 2022. Another study, the VALZ-Pilot study (NCT02997982), investigated the effects of valaciclovir treatment in individuals with AD or MCI of AD type. This study enrolled 36 persons for 4 weeks treatment then followed them for another 12 months. The study finished in March 2020, and no results are available yet. It will be interesting to have the results to plan larger phase III studies. A study (Apovir study) used Apovir (Apodemus AB, Solna), a combination of the experimental anti-enterovirus agent pleconaril, originally developed to treat the common cold, and the hepatitis C treatment ribavirin. This was reported at the Clinical Trials on Alzheimer’s Disease (CTAD) conference in Barcelona (2018) (https://www.ctad-alzheimer.com/files/files/CTAD%20ABSTRACT.pdf) as a phase 2a clinical trial including 69 people with mild AD given Apovir or placebo for 9 months. There was a very high dropout rate in the Apovir group because of side effects. However, the ADASCog improved slightly in patients taking Apovir versus placebo, by 3 points. This result was considered inconclusive, and the authors suggested that further studies were merited. These data were not yet published. Currently, there are no other ongoing clinical trials with antivirals for AD.
It is worthwhile to mention that several antibiotics have been tried to treat or at least to slow down the progression in prodromal as well as in mild to moderate AD [246]. The most used antibiotics in these clinical trials were doxycycline, minocycline, and rifampin. In a clinical trial, Loeb et al. (2004) [246] used oral doxycycline at 200 mg and rifampin 300 mg daily for 3 months in prodromal and mild to moderate AD. The end point was Standardized Alzheimer’s Disease Assessment Scale cognitive subscale (SADAScog) score at 6 months. This trial concluded that there were no major adverse events, and therapy with doxycycline and rifampin may have a therapeutic role in patients with mild to moderate AD; however, the mechanism could not be established, as it seemed unlikely to be due to the effect on C. pneumoniae. A few years later Molloy et al. [247] published the DARAD trial, which used doxycycline and rifampin for treatment of AD. This was a multicenter, blinded, randomized, 2 × 2 factorial controlled trial set at 14 geriatric outpatient clinics in Canada for 12 months. This study did not confirm the results of Loeb et al. [246]; instead, there was a significant deterioration in SADAScog over time with both rifampin and doxycycline in comparison with placebo. Another recent clinical trial, with minocycline, reported by Howard et al. [248], used an experimental design of 1:1:1 in a semifactorial design for patients to receive minocycline (400 mg/day or 200 mg/day) or placebo for 24 months. This clinical trial also found that minocycline did not delay the progress of cognitive or functional impairment in people with mild AD during a 2-year period and also found that 400 mg of minocycline was poorly tolerated in this population. These contradictory results can be explained by the fact that antibiotics target directly the infectious agents, which may not be present at the stage of the disease when they were used. The diferences in patient selection as well as the period of administration and various cognitive outcomes may also explain the apparent contradictions. Furthermore, as recently reported by Balducci and Forloni [249], doxycycline, which crosses the BBB, has had compelling preclinical results in mouse models of AD against Aβ oligomers and neuroinflammation. However, by targeting β-amyloid oligomers, as have many other trials, it may not be efficacious at the later stages of the disease. Another interesting question is the relationship between microbiota, AD, and dysbiosis. Recently, a review discussed this relationship [250], raising the possibility that broad-spectrum antibiotics can greatly affect the composition of the gut microbiota, reduce its biodiversity, and delay colonization for a long period after administration, which suggests that the action of antibiotics in AD could be wide and even opposite, depending on the type of antibiotic and on the specific role of the microbiome in AD pathogenesis. All these antibiotics also modulate the neuroinflammation; however, neuroinflammation may be somehow protective at some stages, by countering the microbial presence, rather than being only the cause for neurodegeneration [250]. More studies at different stages of AD are warranted to assess the exact role of antibiotics in the treatment of AD.
It is well known that P. gingivalis is almost impossible to eliminate by conventional antibiotics. Two other possibilities exist which would neutralize the virulence factors of these microorganisms. In animal studies, recently developed small molecule inhibitors COR286, COR271, and COR388 have been shown to protect animals from neurodegeneration, decrease the P. gingivalis load, and also decrease the burden of Aβ [52]. One small molecule is under clinical trial by Cortexyme to neutralize gingipains [52]. The second strategy involves vaccination of individuals with virulence factors [251–254]. Trials of vaccines to prevent or cure periodontitis are currently being considered [255]. We eagerly await the conclusion of these studies to see whether, by targeting the virulence factors, we can prevent or decrease the progression of AD. Of course, there are other virulence factors which could be targeted from any of the microorganisms. In the meantime, another possibility for treatment would be the use of peptoids (short peptidomimetics), which have been shown to be very effective antimicrobial substances in vitro and in mice [58, 256, 257]. Furthermore, in this line, the expression of natural antimicrobial peptides like LL-37 may be induced systemically [258, 259]. Recently, it was demonstrated to be effective against Staphylococcus aureus biofilms [260], and so it may also be useful against other biofilms, such as those created by P. gingivalis. The cytotoxic properties of LL-37 may limit its effective use [261, 262]. Nevertheless, new engineered peptides and peptidomimetics may be developed.
However, it should also be noted that considering the polymicrobial nature of AD, one antimicrobial agent might not be enough to treat this disease. A combined multi-target designed treatment should be envisaged.
There may be other possible treatments. The immune system may also be influenced by an anti-inflammatory treatment in a pulse form in later life or as soon as any chronic inflammatory disease manifests itself in the organism. The modulation by probiotics may also be beneficial to maintain the health of various microbiomes in the organism. Recently, a large epidemiological study showed that Bacteroides species were less represented in AD patients, suggesting that manipulation of the microbiota may be advantageous for AD [263]. Recently, a bioengineered curli was used as a restorative therapy for the intestinal barrier [264]. Curli patterned on bacterial models may promote tolerance against certain bacteria in the intestinal tract. They act by inhibiting instead of stimulating the TLRs (TLR-2 and TLR-4) [265].
Furthermore, immunotherapy as in the case of cancer may also be possible. Indeed, microbes have also been shown to pervert T cell co-receptors to decrease immune activation and evade detection [266]. In this context, it is worthwhile to mention that P. gingivalis is able to subvert programmed cell death protein 1 (PD-1), to further escape the host immune response [267].
Of course, other general supportive therapies which may reinvigorate the immune system, making the microbiome healthier through nutrition, exercise, or the administration of ketone bodies, may be envisaged. Modulation of dysbiosis by any means may alleviate the burden of neuroinflammation and microglial activation. In this line, a very recent study by Nagpal et al. [268] used modified a Mediterranean-ketogenic diet (MMKD) to modulate the gut microbiome in subjects with MCI. Their data suggested that in MCI patients, the gut microbiome has specific characteristics and MMKD can modulate the gut microbiome and metabolites in association with AD biomarkers such as Aβ in the CSF [270]. However, these authors did not perform any cognitive tests, so their observations remain to be validated at the clinical level. Cunnane’s group investigated a medium-chain triglyceride ketogenic diet and showed improvement in the executive functions of MCI patients; however, its effect on the microbiome was not studied [269]. In vitro studies showed that exposure of human macrophages to short-chain fatty acid butyrate may increase macrophage antimicrobial activity through histone deacetylase 3 (HDAC3) inhibition [270]. In small studies in China targeting gut dysbiosis, GV-971 (a mixture of acidic linear oligosaccharides) reversed cognitive impairment by decreasing neuroinflammation [271].
If we consider the role of senescent cells and their secretory phenotype (SASP) in the pathogenesis of AD related to infection, inflammation, and altered autophagy and mitophagy, one obvious treatment would be to eliminate these cells, as has already been suggested as an anti-aging treatment [272, 273]. Indeed, in this context, ciprofloxacin has been shown to modulate the accumulation of senescent DNA in SASP and, as such, played a senolytic role [274]. Further trials would be warranted to confirm this effect. Another molecule which may act as a senolytic is rapamycin, which targets the inhibition of mTOR [275, 276]. Furthermore, recent studies have demonstrated that mTOR inhibition resulted in the restoration of the intestinal barrier damaged by P. gingivalis [277, 278]. Interestingly, lithium has been shown also to modulate mTOR and GSK3β, which protect the intestinal barrier by decreasing EC senescence as well as the integrity of the BBB [279]. In this way, manipulation of mTOR may become a multi-effect treatment eliminating senescent cells, restoring integrity of the gut barrier, and restoring the altered gut microbiota which occurs with aging [280].
Another molecule, azithromycin, an anti–P. gingivalis macrolide antibiotic, also has mTOR-modulating properties and has senolytic effects, and may be useful in AD treatment [281, 282]. Concomitantly, other known antibiotics, such as minocycline and rifampicin, aside from inhibiting the NLRP3 pathway, may facilitate the removal of senescent cells [184, 283]. Thus, the use of antibiotics that double as senolytics links infection, inflammation, and cell senescence, which are accentuated by external and internal factors such as aging.
Thus, an obvious means to treat the infectious pathomechanism of AD would be the modulation of NLRP3 activation. This was shown in the case of fluoxetine, a selective serotonin reuptake inhibitor [284]. Indeed, a recent trial showed that fluoxetine has been able to decrease the progression from MCI to AD [285]. Along the same line of evidence, since defective mitochondria stimulate the NLRP3 pathway, the elimination of these defective mitochondria by increasing mitophagy may also be an effective therapy. Interestingly, some antibiotics, such as tetracycline, seemed to be able to increase mitophagy in AD [286]. Obviously, direct inflammasome inhibitory substances may also have a therapeutic role in AD. Among the most promising, as already mentioned, are short-chain fatty acids such as butyrate [287].
Another interesting therapeutic approach may stem from observations showing that glucagon-like peptide-1 (GLP-1) facilitates immune tolerance [288, 289] and may be upregulated by LPS stimulation. This generated the suggestion that GLP-1 may behave as an AMP [290]. Moreover, GLP-1 seemed to inhibit the development of A1 inflammatory astrocytes [291]. This has led to a new trial in AD using a well-known drug used in type 2 diabetes, liraglutide, which is a GLP-1 receptor agonist [292].
Yet another group of molecules which may be considered in AD therapy because they target the infection hypothesis at its origins are iron chelators [293]. Iron is essential for bacterial growth; thus, its chelation may enhance body defenses and diminish the microbial load. Moreover, recently, iron has also been shown to contribute to cell senescence [294] via stimulation of the mTOR pathway and inhibition of mitophagy [295]. Thus, iron chelators such as deferoxamine are mTOR inhibitors [296]. A natural in vivo iron chelator, lactoferrin, has been shown to bind LPS and thus to deactivate NLRP3 [297]. It has also been demonstrated to be an AMP with anti–P. gingivalis activity [298, 299]. So, lactoferrin may become a powerful treatment for AD [300–303].
New developments may include small molecules that target mitochondria such as MitoQ, Mdicvi-1, and SS31, which have proved to be efficient in preventing mitochondrial dysfunction and restoring mitochondrial homeostasis in cell cultures and in experimental animals; however, their use alone or in combination in humans awaits clinical trials [304]. In addition to iron chelators, mito-modulators have also been proposed to counteract the dysfunction of mitochondria in AD that has possibly been induced by microbial by-products such as gingipains. The overproduction of ROS associated with infection and microglia stimulation may be targeted by endogenous antioxidants such as reduced glutathione (GSH) [305] as well as by exogenous antioxidants, which are found in various nutrients as well as in diets such as the Mediterranean diet [306].
However, the most rewarding treatment would be prevention. In this way, we can imagine that vaccines against the microorganisms that are involved may be developed. An agent capable of destroying biofilms would also be a major breakthrough to treat the mouth microbiome and as such prevent AD.
Figure 1 and Table 2 summarizes potential interventions.
Table 2.
Potential interventions
| Targeting microoganisms directly |
| Antiviral agents |
| Antibacterial agents (antibiotics) |
| Antifungal agents |
| Immune-modulating treatment |
| Vaccination |
| Anti-inflammatory treatment |
| Checkpoint inhibitors |
| Cell biological treatment |
| Senolytics |
| Antimicrobial peptides |
| Iron chelators and mito-modulators |
| Supportive treatment |
| Probiotics/prebiotics |
| Ketone bodies |
| Nutritional support |
| Physical exercise |
8. Can We Learn from “Why” to Find “How” to Prevent or Treat “When”?
While various interventions are possible, we still have not identified the reason(s) why a pathogen would migrate to the brain. Understanding the events leading to pathogenic migration and colonization of the brain should help develop prophylactic strategies to reduce AD onset.
The direct relationship between amyloid plaques and presence of pathogens in the brain has not been firmly established despite strong circumstantial evidence. We do know that amyloid plaques are also present in individuals without AD. Similarly, individuals with atherosclerotic plaques are not all equally at risk of calcification and blood vessel disruption. This strongly suggests that pathogen migration to the brain may be independent of amyloid plaque formation per se.
Is it then possible to prevent this migration? Would that be enough to prevent the onset of AD? The contribution of ApoE isoforms in the susceptibility to AD can also be due to the fact ApoE4 facilitates entry to the brain [307]. Burgos et al. [308] have found that mice in a humanized mouse model expressing human ApoE4 have high levels of HSV-1 in the brain compared to ApoE3 humanized mice, while no difference was observed in viral load in other organs. A systematic study of other pathogens would be necessary to understand the array of pathogens that might gain increased access to the brain with the ApoE4 carrier.
Other mechanisms such as crossing the BBB have been put forward. Lachenmaier et al. [309] demonstrated that Toxoplasma gondii modulated gene expression of brain endothelial cells to promote its own migration through the BBB. They suggested that this enhanced migration could be via a “Trojan horse” mechanism with infected cells having a CD11b + CD11c ± phenotype. Another intriguing characteristic of T. gondii invasion of the brain was its propensity to develop low metabolic activity [310] upon entry into the CNS. This fine activity balance may also exist for a series of other pathogens located in the brain, but it may be disrupted during acute events.
Current concepts and data would imply that the brain of individuals without AD is free of pathogens. However, it appears that many or most individuals have microorganisms in their brain, as revealed by the recent report of a brain symbiotic ecosystem where restricted types of microorganisms can survive without inducing pathology (neurobiome) [52].
Could this be the result of an efficient immune cell–pathogen interaction specific for the brain environment? The logical consequence would be that a trigger is needed to disrupt this fine equilibrium just as in the gut where the well-arranged balance between microbes and the intestine is periodically disrupted and results in dysbiosis.
It is largely unknown which acute stress or repetitive acute stresses may be responsible for the activation of the metabolic switch leading to pathogen proliferation and subsequent sequelae. This will require intense research. A few possibilities exist: [1] brain inflammation associated with microvasculature defects; (2i) severe gut dysbiosis associated with leakage sensed in the brain; [3] acute infectious disease; and [4] major organ failure leading to transfer of biological reserves from the brain to the corresponding organ/system. Independent of the cause, understanding the brain symbiotic ecosystem (neurobiome/neurodysbiosis) and its regulation will enable better control of the events associated with AD onset.
9. Searching for New Directions in Drug Discovery
The National Alzheimer’s Project Act by world leaders mandates a plan, which articulates the ultimate goal of preventing or effectively treating AD by the year 2025 [311]. To propose a possible pathway, it is important to put into perspective past failures, discuss novel opportunities, and understand the feasibility of delivering a drug by 2025. Several decades of research on competing hypotheses for explaining the cause of AD [e.g., cholinergic [312], amyloid [313], tau [314], glucose synthase kinase 3 [315], inflammation [316]] led to the development of drugs that reached clinical trials but failed. Despite billions of euros spent worldwide on drug development and clinical trials based largely on animal modeling, these have repeatedly failed to translate into effective interventions [317]. Under these hard-to-accept empirical observations, it is imperative to consider alternative hypotheses (e.g., the infection hypothesis), but also to consider drug development and research strategies that shy away from transgenic animal models that do not accurately reproduce human AD.
Indeed, recent technological leaps in stem cell research have led to the ground-breaking development of lab-grown human mini-brains, which reproduce the hallmarks of AD [318, 319]. This alternative model allows for testing of various in vivo–based hypotheses and extract correct and complex information. Combining these advances to the infection hypothesis of AD, as well as the Antimicrobial Protection Hypothesis of AD [160], provides clear targets and the framework for novel AD drug designs. Indeed, since Aβ is a powerful antimicrobial peptide that targets and neutralizes AD pathogens, it is reasonable to consider the development of a cocktail of novel and more powerful antimicrobial peptides based on the Aβ template. To achieve these ultimate goals, we envisage a multi-stage, closed-loop framework between in silico drug screening and drug testing in mini-brains as follows. First, data mining in existing databases (e.g., cAMP) and antimicrobial activity prediction via rational design [320] should generate analogs with improved activity. Second, state-of-the-art molecular simulations should be employed to determine the mechanism of action of Aβ against AD pathogens. Third, by combining information gained from step 1 and 2, and with further determination of physical–chemical descriptors of the generated analogs and Aβ, these can be used to train and screen potential antimicrobial peptides candidates via advanced machine-learning drug discovery software.
This final stage should involve testing against user-desired properties (e.g., half maximal inhibitory concentration (IC50)), as well as multiomics analysis. In this way, antimicrobial peptides sequences can be ranked in terms of the desired property, and those of poorest quality are rejected, allowing a new population to be selected.
Note that biofilm experiments in neural tissue based on multiomics data from patients and deceased frozen brains can be recreated in mini-brains and tested. Moreover, modern high-throughput technologies enable rapid and efficient simultaneous acquisition of multiomics data in the course of a single experiment [321]. This is significant since it departs from traditional experimental studies, which are usually carried out to isolate the effects of a single mechanism and not to investigate the interactions of many mechanisms. This leads to a set of results that seem conflicting; thus, it is difficult to interpret or understand the interactions of the underlying mechanisms leading to the pathogenesis of the disease.
The observables of such a modeling approach could in principle be integrated with a drug discovery process and therefore lead to a systematic and holistic screening of antimicrobial peptides with high therapeutic efficacy against AD pathogens. Therefore, novel biological models and experimental approaches, as well as multiomics acquisition devices provide unique opportunities to study and accelerate drug development in the context of novel hypotheses of AD by coupling it to advanced data analysis and state-of-the-art in silico drug screening. Moreover, this proposed pathway has the potential to reduce the overall cost of drug development.
10. Conclusion: Perspectives
It seems clear that it will be difficult to find one pathogen to explain the whole spectrum of AD in the spirit of the infection hypothesis. From the available data, it seems that we should think instead about a causative polymicrobial community which affects the immune/inflammatory reactions in the brain and in the periphery, and which interacts with various factors such as genetics, environment, and age. Thus, more properly, AD may be considered a complex syndrome involving dysregulation of the brain’s immune system. Obviously, future treatments (and/or prevention) of AD will not be one simple molecule but a multimodal complex treatment. This will combine most probably antimicrobial, senolytic, and anti-inflammatory agents with pro-mitophagy treatments. In this way, prevention and even treatment of AD will most probably become feasible. Many clinical investigations and trials will be necessary before we can arrive at this stage.
Key Points.
Experimental and epidemiological data increasingly support the involvement of infections in the development of Alzheimer’s disease (AD), with the sources of AD-involved microbes being various body microbiome communities from the gut, mouth, nose, and skin.
Infection results in neuroinflammation, leading to and sustained by systemic inflammation, causing neurodegeneration, and the senescence of the immune cells preceding the clinical manifestations.
The infection hypothesis and the Antimicrobial Protection Hypothesis of AD open the way to new therapeutic approaches.
Acknowledgements
The works presented in the article were supported by grants from the Canadian Institutes of Health Research (CIHR) (No. 106634 and No. PJT-162366) to AK and TF, the Société des médecins de l’Université de Sherbrooke and the Research Center on Aging of the CIUSSS-CHUS, Sherbrooke to TF and EF, the Centre de Recherches Cliniques de l’Université de Sherbrooke to EF, and the FRQS Audace to EF, TF, PBB, and J-PB; and by the Polish Ministry of Science and Higher Education statutory grant 02-0058/07/262 to JMW, by the Agency for Science Technology and Research (A*STAR) to AL, by Ikerbasque (The Basque Foundation for Science), partially supported by the Basque Government under the grant “Artificial Intelligence in BCAM number EXP. 2019/00432,” by the Basque Government through the BERC 2018-2021 program, and by the Ministry of Science, Innovation and Universities: BCAM Severo Ochoa accreditation SEV-2017-0718, and through project RTI2018-093860B-C21 funded by (AEI/FEDER, UE), with the acronym “MathNEURO,” to SR.
Funding
No outside funding was received in support of this publication, and the authors did not receive funding for writing this paper.
Footnotes
Conflict of interest The authors, Tamàs Fülöp, Usma Munawara, Anis Larbi, Mathieu Desroche, Serafim Rodrigues, Michele Catanzaro, Andrea Guidolin, Abdelouahed Khalil, François Bernier, Annelise E. Barron, Katsuiku Hirokawa, Pascale B. Beauregard, David Dumoulin, Jean-Philippe Bellenger, Jacek M. Witkowski, and Eric Frost, declare that they have no conflict of interest related to this article.
References
- 1.Reiss AB, Arain HA, Stecker MM, Siegart NM, Kasselman LJ. Amyloid toxicity in Alzheimer’s disease. Rev Neurosci. 2018;29(6):613–27. [DOI] [PubMed] [Google Scholar]
- 2.Mayeux R, Stern Y. Epidemiology of Alzheimer disease. Cold Spring Harb Perspect Med. 2012;2(8):a006239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hu H, Tan CC, Tan L, Yu JT. A mitocentric view of Alzheimer’s disease. Mol Neurobiol. 2017;54(8):6046–60. [DOI] [PubMed] [Google Scholar]
- 4.Reddy PH, Manczak M, Yin X, Grady MC, Mitchell A, Tonk S, Kuruva CS, Bhatti JS, Kandimalla R, Vijayan M, Kumar S, Wang R, Pradeepkiran JA, Ogunmokun G, Thamarai K, Quesada K, Boles A, Reddy AP. Protective effects of indian spice curcumin against amyloid-β in Alzheimer’s disease. J Alzheimers Dis. 2018;61(3):843–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fulop T, Witkowski JM, Bourgade K, Khalil A, Zerif E, Larbi A, Hirokawa K, Pawelec G, Bocti C, Lacombe G, Dupuis G, Frost EH. Can an infection hypothesis explain the beta amyloid hypothesis of Alzheimer’s disease? Front Aging Neurosci. 2018;24(10):224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fulop T, Lacombe G, Cunnane S, Le Page A, Dupuis G, Frost EH, Bourgade-Navarro K, Goldeck D, Larbi A, Pawelec G. Elusive Alzheimer’s disease: can immune signatures help our understanding of this challenging disease? Part 1: clinical and historical background. Discov Med. 2013;15(80):23–32. [PubMed] [Google Scholar]
- 7.Osorio C, Kanukuntla T, Diaz E, Jafri N, Cummings M, Sfera A. The post-amyloid era in Alzheimer’s disease: trust your gut feeling. Front Aging Neurosci. 2019;26(11):143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cummings JL, Morstorf T, Zhong K. Alzheimer’s disease drug-development pipeline: few candidates, frequent failures. Alzheimers Res Ther. 2014;6:37–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cummings J, Ritter A, Zhong K. Clinical trials for disease-modifying therapies in Alzheimer’s disease: a primer, lessons learned, and a blueprint for the future. J Alzheimers Dis. 2018;64(s1):S3–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Long JM, Holtzman DM. Alzheimer disease: an update on pathobiology and treatment strategies. Cell. 2019;179(2):312–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hardy JA, Higgins GA. Alzheimer’s disease: the amyloid cascade hypothesis. Science. 1992;256(5054):184–5. [DOI] [PubMed] [Google Scholar]
- 12.Hardy J, Allsop D. Amyloid deposition as the central event in the aetiology of Alzheimer’s disease. Trends Pharmacol Sci. 1991;12:383–8. [DOI] [PubMed] [Google Scholar]
- 13.McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126:479–97. [DOI] [PubMed] [Google Scholar]
- 14.Itzhaki RF, Lathe R, Balin BJ, Ball MJ, Bearer EL, Braak H, Bullido MJ, Carter C, Clerici M, Cosby SL, Del Tredici K, Field H, Fulop T, Grassi C, Griffin WS, Haas J, Hudson AP, Kamer AR, Kell DB, Licastro F, Letenneur L, Lövheim H, Mancuso R, Miklossy J, Otth C, Palamara AT, Perry G, Preston C, Pretorius E, Strandberg T, Tabet N, Taylor-Robinson SD, Whittum-Hudson JA. Microbes and Alzheimer’s disease. J Alzheimers Dis. 2016;51(4):979–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Le Page A, Dupuis G, Frost EH, Larbi A, Pawelec G, Witkowski JM, et al. Role of the peripheral innate immune system in the development of Alzheimer’s disease. Exp Gerontol. 2018;107:59–66. [DOI] [PubMed] [Google Scholar]
- 16.McManus RM, Heneka MT. Role of neuroinflammation in neurodegeneration: new insights. Alzheimers Res Ther. 2017;9:14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hanger DP, Lau DH, Phillips EC, Bondulich MK, Guo T, Woodward BW, et al. Intracellular and extracellular roles for tau in neurodegenerative disease. J. Alzheimers Dis 2014;40(Suppl. 1):S37–45. [DOI] [PubMed] [Google Scholar]
- 18.Sun X, Chen WD, Wang YD. β-Amyloid: the key peptide in the pathogenesis of Alzheimer’s disease. Front. Pharmacol 2015;6:221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Roher AE, Maarouf CL, Kokjohn TA. Familial presenilin mutations and sporadic Alzheimer’s disease pathology: is the assumption of biochemical equivalence justified? J Alzheimers Dis. 2016;50(3):645–58. [DOI] [PubMed] [Google Scholar]
- 20.Karran E, De Strooper B. The amyloid cascade hypothesis: are we poised for success or failure? J Neurochem. 2016;139(Suppl. 2):237–52. [DOI] [PubMed] [Google Scholar]
- 21.Siegel G, Gerber H, Koch P, Bruestle O, Fraering PC, Rajendran L. The Alzheimer’s disease γ-secretase generates higher 42:40 ratios for β-amyloid than for p3 peptides. Cell Rep. 2017;19(10):1967–76. [DOI] [PubMed] [Google Scholar]
- 22.Chow VW, Mattson MP, Wong PC, Gleichmann M. An overview of APP processing enzymes and products. NeuroMol Med. 2010;12:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Galante D, Corsaro A, Florio T, Vella S, Pagano A, Sbrana F, Vassalli M, Perico A, D’Arrigo C. Differential toxicity, conformation and morphology of typical initial aggregation states of Aβ1–42 and Aβpy3-42 beta-amyloids. Int J Biochem Cell Biol. 2012;44(11):2085–93. [DOI] [PubMed] [Google Scholar]
- 24.Bolós M, Perea JR, Avila J. Alzheimer’s disease as an inflammatory disease. Biomol Concepts. 2017;8:37–43. [DOI] [PubMed] [Google Scholar]
- 25.McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR Jr, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH. The diagnosis of dementia due to Alzheimer’s disease: recommendations from the National Institute on Aging-Alzheimer’s Association workgroups on diagnostic guidelines for Alzheimer’s disease. Alzheimers Dement. 2011;7(3):263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Serrano-Pozo A, Mielke ML, Gómez-Isla T, Betensky RA, Growdon JH, Frosch MP, Hyman BT. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am J Pathol. 2011;179(3):1373–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holtzman DM, Carrillo MC, Hendrix JA, Bain LJ, Catafau AM, Gault LM, Goedert M, Mandelkow E, Mandelkow EM, Miller DS, Ostrowitzki S, Polydoro M, Smith S, Wittmann M, Hutton M. Tau: from research to clinical development. Alzheimers Dement. 2016;12(10):1033–9. [DOI] [PubMed] [Google Scholar]
- 28.Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. 2017;12(1):50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rogers J, Cooper NR, Webster S, Schultz J, McGeer PL, Styren SD, Civin WH, Brachova L, Bradt B, Ward P. Complement activation by beta-amyloid in Alzheimer disease. Proc Natl Acad Sci USA. 1992;89(21):10016–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126(4):479–97. [DOI] [PubMed] [Google Scholar]
- 31.Kanatsu K, Tomita T. Molecular mechanisms of the genetic risk factors in pathogenesis of Alzheimer disease. Front Biosci (Landmark Ed). 2017;1(22):180–92. [DOI] [PubMed] [Google Scholar]
- 32.Takatori S, Wang W, Iguchi A, Tomita T. Genetic risk factors for Alzheimer disease: emerging roles of microglia in disease pathomechanisms. Adv Exp Med Biol. 2019;1118:83–116. [DOI] [PubMed] [Google Scholar]
- 33.Tao Q, Ang TFA, DeCarli C, Auerbach SH, Devine S, Stein TD, Zhang X, Massaro J, Au R, Qiu WQ. Association of chronic low-grade inflammation with risk of Alzheimer disease in ApoE4 carriers. JAMA Netw Open. 2018;1(6):e183597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yu L, Lutz MW, Wilson RS, Burns DK, Roses AD, Saunders AM, Yang J, Gaiteri C, De Jager PL, Barnes LL, Bennett DA. APOE ε4-TOMM40 ‘523 haplotypes and the risk of Alzheimer’s disease in older Caucasian and African Americans. PLoS One. 2017;12(7):e0180356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tasaki S, Gaiteri C, Mostafavi S, De Jager PL, Bennett DA. The molecular and neuropathological consequences of genetic risk for Alzheimer’s dementia. Front Neurosci. 2018;8(12):699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lövheim H, Norman T, Weidung B, Olsson J, Josefsson M, Adolfsson R, Nyberg L, Elgh F. Herpes simplex virus, APOEɛ4, and cognitive decline in old age: results from the Betula Cohort Study. J Alzheimers Dis. 2019;67(1):211–20. [DOI] [PubMed] [Google Scholar]
- 37.Lopatko Lindman K, Weidung B, Olssonc J, Josefssone M, Kokf E, Johanssong A, Eriksson S, Hallmansh G, Elgh F, Lovheim H. A genetic signature including apolipoprotein Eε4 potentiates the risk of herpes simplex–associated Alzheimer’s disease. Alzheimer’s Dement Transl Res Clin Interv. 2019;5:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Barroeta-Espar I, Weinstock LD, Perez-Nievas BG, Meltzer AC, Chong STM, Amaral AC, Murray ME, Moulder KL, Morris JC, Cairns NJ, Parisi JE, Lowe VJ, Petersen RC, Kofler J, Ikonomovic MD, López O, Klunk WE, Mayeux RP, Frosch MP, Wood LB, Gomez-Isla T. Distinct cytokine profiles in human brains resilient to Alzheimer’s pathology. Neurobiol Dis. 2019;121:327–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Soscia SJ, Kirby JE, Washicosky KJ, Tucker SM, Ingelsson M, Hyman B, Burton MA, Goldstein LE, Duong S, Tanzi RE, Moir RD. The Alzheimer’s disease-associated amyloid beta-protein is an antimicrobial peptide. PLoS One. 2010;5(3):e9505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bourgade K, Garneau H, Giroux G, Le Page AY, Bocti C, Dupuis G, Frost EH, Fülöp T Jr. β-Amyloid peptides display protective activity against the human Alzheimer’s disease-associated herpes simplex virus-1. Biogerontology. 2015;16(1):85–98. [DOI] [PubMed] [Google Scholar]
- 41.Bourgade K, Le Page A, Bocti C, Witkowski JM, Dupuis G, Frost EH, Fülöp T Jr. Protective effect of amyloid-β peptides against herpes simplex virus-1 infection in a neuronal cell culture model. J Alzheimers Dis. 2016;50(4):1227–41. [DOI] [PubMed] [Google Scholar]
- 42.Wozniak MA, Itzhaki RF, Shipley SJ, Dobson CB. Herpes simplex virus infection causes cellular beta-amyloid accumulation and secretase upregulation. Neurosci Lett. 2007;429(2–3):95–100. [DOI] [PubMed] [Google Scholar]
- 43.Itzhaki RF. Herpes and Alzheimer’s disease: subversion in the central nervous system and how it might be halted. J Alzheimers Dis. 2016;54(4):1273–81. [DOI] [PubMed] [Google Scholar]
- 44.Lövheim H, Olsson J, Weidung B, Johansson A, Eriksson S, Hall-mans G, Elgh F. Interaction between cytomegalovirus and herpes simplex virus type 1 associated with the risk of Alzheimer’s disease development. J Alzheimers Dis. 2018;61(3):939–45. [DOI] [PubMed] [Google Scholar]
- 45.Lövheim H, Gilthorpe J, Adolfsson R, Nilsson LG, Elgh F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 2015;11(6):593–9. [DOI] [PubMed] [Google Scholar]
- 46.Carter CJ. Genetic, transcriptome, proteomic and epidemiological evidence for blood-brain barrier disruption and polymicrobial brain invasion as determinant factors in Alzheimer’s disease. J. Alzheimers Dis. Rep 2017;1:125–57. 10.3233/ADR-170017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Miklossy J. Alzheimer’s disease—a neurospirochetosis. Analysis of the evidence following Koch’s and Hill’s criteria. J Neuroinflamm. 2011;8:90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Miklossy J. Bacterial amyloid and DNA are important constituents of senile plaques: further evidence of the spiro-chetal and biofilm nature of senile plaques. J Alzheimers Dis. 2016;53(4):1459–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Miklossy J, McGeer PL. Common mechanisms involved in Alzheimer’s disease and type 2 diabetes: a key role of chronic bacterial infection and inflammation. Aging (Albany NY). 2016;8(4):575–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Balin BJ, Gérard HC, Arking EJ, Appelt DM, Branigan PJ, Abrams JT, Whittum-Hudson JA, Hudson AP. Identification and localization of Chlamydia pneumoniae in the Alzheimer’s brain. Med Microbiol Immunol. 1998;187(1):23–42. [DOI] [PubMed] [Google Scholar]
- 51.Balin BJ, Little CS, Hammond CJ, Appelt DM, Whittum-Hudson JA, Gérard HC, Hudson AP. Chlamydophila pneumoniae and the etiology of late-onset Alzheimer’s disease. J Alzheimers Dis. 2008;13(4):371–80. [DOI] [PubMed] [Google Scholar]
- 52.Dominy SS, Lynch C, Ermini F, Benedyk M, Marczyk A, Konradi A, Nguyen M, Haditsch U, Raha D, Griffin C, Holsinger LJ, Arastu-Kapur S, Kaba S, Lee A, Ryder MI, Potempa B, Mydel P, Hellvard A, Adamowicz K, Hasturk H, Walker GD, Reynolds EC, Faull RLM, Curtis MA, Dragunow M, Potempa J. Porphyromonas gingivalis in Alzheimer’s disease brains: evidence for disease causation and treatment with small-molecule inhibitors. Sci Adv. 2019;5(1):eau3333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Itzhaki RF. Herpes simplex virus type 1 and Alzheimer’s disease: possible mechanisms and signposts. FASEB J. 2017;31(8):3216–26. [DOI] [PubMed] [Google Scholar]
- 54.Sacks CA, Avorn J, Kesselheim AS. The failure of solanezumab—how the FDA saved taxpayers billions. N Engl J Med. 2017;376:1706–8. [DOI] [PubMed] [Google Scholar]
- 55.Mehta D, et al. Why do trials for Alzheimer’s disease drugs keep failing? A discontinued drug perspective for 2010–2015. Expert Opin Investig Drugs. 2017;26:735–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ferrer I, Boada Rovira M, Sánchez Guerra ML, Rey MJ, Costa-Jussá F. Neuropathology and pathogenesis of encephalitis following amyloid-beta immunization in Alzheimer’s disease. Brain Pathol. 2004;14(1):11–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lanzillotta C, Di Domenico F, Perluigi M, Butterfield DA. Targeting mitochondria in Alzheimer disease: rationale and perspectives. CNS Drugs. 2019;33(10):957–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Oliver DMA, Reddy PH. Small molecules as therapeutic drugs for Alzheimer’s disease. Mol Cell Neurosci. 2019;96:47–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.de la Torre J The vascular hypothesis of Alzheimer’s Disease: a key to preclinical prediction of dementia using neuroimaging. J Alzheimers Dis. 2018;63(1):35–52. [DOI] [PubMed] [Google Scholar]
- 60.Snowdon DA, Greiner LH, Mortimer JA, Riley KP, Greiner PA, Markesbery WR. Brain infarction and the clinical expression of Alzheimer disease. The Nun Study. JAMA. 1997;277(10):813–7. [PubMed] [Google Scholar]
- 61.Li L, Zhang X, Yang D, Luo G, Chen S, Le W. Hypoxia increases Abeta generation by altering beta- and gamma-cleavage of APP. Neurobiol Aging. 2009;30(7):1091–8. [DOI] [PubMed] [Google Scholar]
- 62.Trumbore CN. Shear-induced amyloid formation of IDPs in the brain. Prog Mol Biol Transl Sci. 2019;166:225–309. [DOI] [PubMed] [Google Scholar]
- 63.Xu W, Tan L, Wang HF, Jiang T, Tan MS, Tan L, Zhao QF, Li JQ, Wang J, Yu JT. Meta-analysis of modifiable risk factors for Alzheimer’s disease. J Neurol Neurosurg Psychiatry. 2015;86(12):1299–306. [DOI] [PubMed] [Google Scholar]
- 64.de Heus RAA, Olde Rikkert MGM, Tully PJ, Lawlor BA, Claassen JAHR, NILVAD Study Group. Blood pressure variability and progression of clinical Alzheimer disease. Hypertension. 2019;74(5):1172–80. [DOI] [PubMed] [Google Scholar]
- 65.Swerdlow RH, Burns JM, Khan SM. The Alzheimer’s disease mitochondrial cascade hypothesis: progress and perspectives. Biochim Biophys Acta. 2014;1842(8):1219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McGeer PL, Schulzer M, McGeer EG. Arthritis and anti-inflammatory agents as possible protective factors for Alzheimer’s disease: a review of 17 epidemiologic studies. Neurology. 1996;47(2):425–32. [DOI] [PubMed] [Google Scholar]
- 67.McGeer PL, McGeer E, Rogers J, Sibley J. Anti-inflammatory drugs and Alzheimer disease. Lancet. 1990;335(8696):1037. [DOI] [PubMed] [Google Scholar]
- 68.Zhang C, Wang Y, Wang D, Zhang J, Zhang F. NSAID exposure and risk of Alzheimer’s disease: an updated meta-analysis from cohort studies. Front Aging Neurosci. 2018;28(10):83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.de Smit M, Westra J, Vissink A, van der Meer DB, Brouwer E, van Winkelhoff AJ. Periodontitis in established rheumatoid arthritis patients: a cross-sectional clinical, microbiological and serological study. Arthritis Res Ther. 2012;14(5):R222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Lundberg K, Wegner N, Yucel-Lindberg T, Venables PJ. Periodontitis in RA-the citrullinated enolase connection. Nat Rev Rheumatol. 2010;6(12):727–30. [DOI] [PubMed] [Google Scholar]
- 71.Wegner N, Wait R, Sroka A, Eick S, Nguyen KA, Lundberg K, Kinloch A, Culshaw S, Potempa J, Venables PJ. Peptidylarginine deiminase from Porphyromonas gingivalis citrullinates human fibrinogen and α-enolase: implications for autoimmunity in rheumatoid arthritis. Arthritis Rheum. 2010;62(9):2662–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Mangat P, Wegner N, Venables PJ, Potempa J. Bacterial and human peptidylarginine deiminases: targets for inhibiting the autoimmune response in rheumatoid arthritis? Arthritis Res Ther. 2010;12(3):209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Quirke AM, Lugli EB, Wegner N, Hamilton BC, Charles P, Chowdhury M, Ytterberg AJ, Zubarev RA, Potempa J, Culshaw S, Guo Y, Fisher BA, Thiele G, Mikuls TR, Venables PJ. Heightened immune response to autocitrullinated Porphyromonas gingivalis peptidylarginine deiminase: a potential mechanism for breaching immunologic tolerance in rheumatoid arthritis. Ann Rheum Dis. 2014;73(1):263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Noble JM, Scarmeas N, Celenti RS, Elkind MS, Wright CB, Schupf N, Papapanou PN. Serum IgG antibody levels to periodontal microbiota are associated with incident Alzheimer disease. PLoS One. 2014;9:e114959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hashioka S, Inoue K, Miyaoka T, Hayashida M, Wake R, Oh-Nishi A, Inagaki M. The possible causal link of periodontitis to neuropsychiatric disorders: more than psychosocial mechanisms. Int J Mol Sci. 2019;20(15). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Stein PS, Desrosiers M, Donegan SJ, Yepes JF, Kryscio RJ. Tooth loss, dementia and neuropathology in the Nun study. J Am Dent Assoc. 2007;138(10):1314–22. [DOI] [PubMed] [Google Scholar]
- 77.Konkel JE, O’Boyle C, Krishnan S. Distal consequences of oral inflammation. Front Immunol. 2019;25(10):1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Singhrao SK, Olsen I. Assessing the role of Porphyromonas gingivalis in periodontitis to determine a causative relationship with Alzheimer’s disease. J Oral Microbiol. 2019;11:1563405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Poole S, Singhrao SK, Kesavalu L, Curtis MA, Crean S. Determining the presence of periodontopathic virulence factors in short-term postmortem Alzheimer’s disease brain tissue. J. Alzheimers Dis 2013;36:665–77. [DOI] [PubMed] [Google Scholar]
- 80.Sadrameli M, Bathini P, Alberi L. Linking mechanisms of periodontitis to Alzheimer’s disease. Curr Opin Neurol. 2020;33(2):230–8. [DOI] [PubMed] [Google Scholar]
- 81.Leblhuber F, Huemer J, Steiner K, Gostner JM, Fuchs D. Knock-on effect of periodontitis to the pathogenesis of Alzheimer’s disease? Wien Klin Wochenschr. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dubar M, Delatre V, Moutier C, Sy K, Agossa K. Awareness and practices of general practitioners towards the oral-systemic disease relationship: a regionwide survey in France. J Eval Clin Pract. 2019. 10.1111/jep.13343. [DOI] [PubMed] [Google Scholar]
- 83.Shaik MM, Ahmad S, Gan SH, Abuzenadah AM, Ahmad E, Tabrez S, Ahmed F, Kamal MA. How do periodontal infections affect the onset and progression of Alzheimer’s disease? CNS Neurol Disord Drug Targets. 2014;13(3):460–6. [DOI] [PubMed] [Google Scholar]
- 84.Sochocka M, Sobczyński M, Sender-Janeczek A, Zwolińska K, Błachowicz O, Tomczyk T, Ziętek M, Leszek J. Association between periodontal health status and cognitive abilities. The role of cytokine profile and systemic inflammation. Curr Alzheimer Res. 2017;14(9):978–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sudhakara P, Gupta A, Bhardwaj A, Wilson A. Oral dysbiotic communities and their implications in systemic diseases. Dent J (Basel). 2018;6(2):10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hayashi K, Hasegawa Y, Takemoto Y, Cao C, Takeya H, Komohara Y, Mukasa A, Kim-Mitsuyama S. Continuous intracerebroventricular injection of Porphyromonas gingivalis lipopolysaccharide induces systemic organ dysfunction in a mouse model of Alzheimer’s disease. Exp Gerontol. 2019;120:1–5. [DOI] [PubMed] [Google Scholar]
- 87.Nie R, Wu Z, Ni J, Zeng F, Yu W, Zhang Y, Kadowaki T, Kashiwazaki H, Teeling JL, Zhou Y. Porphyromonas gingivalis infection induces amyloid-β accumulation in monocytes/macrophages. J Alzheimers Dis. 2019;72(2):479–94. [DOI] [PubMed] [Google Scholar]
- 88.Han EC, Choi SY, Lee Y, Park JW, Hong SH, Lee HJ. Extracellular RNAs in periodontopathogenic outer membrane vesicles promote TNF-α production in human macrophages and cross the blood-brain barrier in mice. FASEB J. 2019;33(12):13412–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Liu Y, Wu Z, Nakanishi Y, Ni J, Hayashi Y, Takayama F, Zhou Y, Kadowaki T, Nakanishi H. Infection of microglia with Porphyromonas gingivalis promotes cell migration and an inflammatory response through the gingipain-mediated activation of protease-activated receptor-2 in mice. Sci Rep. 2017;7(1):11759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gil Montoya JA, Barrios R, Sanchez-Lara I, Ramos P, Carnero C, Fornieles F, Montes J, Santana S, Luna JD, Gonzalez-Moles MA. Systemic inflammatory impact of periodontitis on cognitive impairment. Gerodontology. 2020;37(1):11–8. [DOI] [PubMed] [Google Scholar]
- 91.Sakanaka A, Takeuchi H, Kuboniwa M, Amano A. Dual lifestyle of Porphyromonas gingivalis in biofilm and gingival cells. Microb Pathog. 2016;94:42–7. [DOI] [PubMed] [Google Scholar]
- 92.Patrick KL, Bell SL, Weindel CG, Watson RO. Exploring the “multiple-hit hypothesis” of neurodegenerative disease: bacterial infection comes up to bat. Front Cell Infect Microbiol. 2019;28(9):138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Taguchi J, Fujii A, Fujino Y, Tsujioka Y, Takahashi M, Tsuboi Y, Wada I, Yamada T. Different expression of calreticulin and immunoglobulin binding protein in Alzheimer’s disease brain. Acta Neuropathol. 2000;100(2):153–60. [DOI] [PubMed] [Google Scholar]
- 94.Johnson RJ, Xiao G, Shanmugaratnam J, Fine RE. Calreticulin functions as a molecular chaperone for the beta-amyloid precursor protein. Neurobiol Aging. 2001;22(3):387–95. [DOI] [PubMed] [Google Scholar]
- 95.Joerchel S, Raap M, Bigl M, Eschrich K, Schliebs R. Oligomeric beta-amyloid(1–42) induces the expression of Alzheimer disease-relevant proteins in cholinergic SN56.B5.G4 cells as revealed by proteomic analysis. Int J Dev Neurosci. 2008;26(3–4):301–8. [DOI] [PubMed] [Google Scholar]
- 96.Lin Q, Cao Y, Gao J. Serum calreticulin is a negative biomarker in patients with Alzheimer’s disease. Int J Mol Sci. 2014;15(12):21740–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Cockram TOJ, Puigdellívol M, Brown GC. Calreticulin and galectin-3 opsonise bacteria for phagocytosis by microglia. Front. Immunol, 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Tzeng NS, Chung CH, Lin FH, et al. Anti-herpetic medications and reduced risk of dementia in patients with herpes simplex virus infections—a nationwide, population-based cohort study in Taiwan. Neurotherapeutics. 2018;15(2):417–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Watson AM, Prasad KM, Klei L, Wood JA, Yolken RH, Gur RC, Bradford LD, Calkins ME, Richard J, Edwards N, Savage RM, Allen TB, Kwentus J, McEvoy JP, Santos AB, Wiener HW, Go RC, Perry RT, Nasrallah HA, Gur RE, Devlin B, Nimgaonkar VL. Persistent infection with neurotropic herpes viruses and cognitive impairment. Psychol Med. 2013;43(5):1023–31. [DOI] [PubMed] [Google Scholar]
- 100.Devanand DP. Viral hypothesis and antiviral treatment in Alzheimer’s disease. Curr Neurol Neurosci Rep. 2018;18(9):55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Qin QS, Li Y. Herpes viral infections and antimicrobial protection for Alzheimer’s disease: implications for prevention and treatment. J Med Virol. 2019;91:1368–77. [DOI] [PubMed] [Google Scholar]
- 102.Fabisiak A, Murawska N, Fichna J. LL-37: Cathelicidin-related antimicrobial peptide with pleiotropic activity. Pharmacol Rep. 2016;68(4):802–8. [DOI] [PubMed] [Google Scholar]
- 103.De Lorenzi E, Chiari M, Colombo R, Cretich M, Sola L, Vanna R, Gagni P, Bisceglia F, Morasso C, Lin JS, Lee M, McGeer PL, Barron AE. Evidence that the human innate immune peptide LL-37 may be a binding partner of amyloid-β and inhibitor of fibril assembly. J Alzheimers Dis. 2017;59(4):1213–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Linard M, Letenneur L, Garrigue I, Doize A, Dartigues JF, Helmer C. Interaction between APOE4 and herpes simplex virus type 1 in Alzheimer’s disease. Alzheimers Dement. 2020;16(1):200–8. [DOI] [PubMed] [Google Scholar]
- 105.MacDonald AB, Miranda JM. Concurrent neocortical borreliosis and Alzheimer’s disease. Hum Pathol. 1987;18(7):759–61. [DOI] [PubMed] [Google Scholar]
- 106.Miklossy J, Khalili K, Gern L, Ericson RL, Darekar P, Bolle L, Hurlimann J, Paster BJ. Borrelia burgdorferi persists in the brain in chronic lyme neuroborreliosis and may be associated with Alzheimer disease. J Alzheimers Dis. 2004;6(6):639–49. [DOI] [PubMed] [Google Scholar]
- 107.Feng J, Zhang S, Shi W, Zubcevik N, Miklossy J, Zhang Y. Selective essential oils from spice or culinary herbs have high activity against stationary phase and biofilm Borrelia burgdorferi. Front Med (Lausanne). 2017;11(4):169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Fülöp T, Itzhaki RF, Balin BJ, Miklossy J, Barron AE. Role of microbes in the development of Alzheimer’s disease: state of the art—an international symposium presented at the 2017 IAGG Congress in San Francisco. Front Genet. 2018;10(9):362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tuddenham S, Ghanem KG. Neurosyphilis: knowledge gaps and controversies. Sex Transm Dis. 2018;45(3):147–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Antinori A, Arendt G, Becker JT, Brew BJ, Byrd DA, Cherner M, Clifford DB, Cinque P, Epstein LG, Goodkin K, Gisslen M, Grant I, Heaton RK, Joseph J, Marder K, Marra CM, McArthur JC, Nunn M, Price RW, Pulliam L, Robertson KR, Sacktor N, Valcour V, Wojna VE. Updated research nosology for HIV-associated neurocognitive disorders. Neurology. 2007;69:1789–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Robertson KR, Smurzynski M, Parsons TD, Wu K, Bosch RJ, Wu J, McArthur JC, Collier AC, Evans SR, Ellis RJ. The prevalence and incidence of neurocognitive impairment in the HAART era. AIDS. 2007;21:1915–21. [DOI] [PubMed] [Google Scholar]
- 112.Nookala AR, Kumar A. Molecular mechanisms involved in HIV-1 Tat-mediated induction of IL-6 and IL-8 in astrocytes. J Neuroinflammation. 2014;11:214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Cole MA, Margolick JB, Cox C, Li X, Selnes OA, Martin EM, Becker JT, Aronow HA, Cohen B, Sacktor N, Miller EN. Longitudinally preserved psychomotor performance in longterm asymptomatic HIV-infected individuals. Neurology. 2007;69:2213–20. [DOI] [PubMed] [Google Scholar]
- 114.Milanini B, Valcour V. Differentiating HIV-associated neurocognitive disorders from Alzheimer’s disease: an emerging issue in geriatric neuroHIV. Curr HIV/AIDS Rep. 2017;14:123–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ellis RJ, Badiee J, Vaida F, Letendre S, Heaton RK, Clifford D, Collier AC, Gelman B, McArthur J, Morgello S, McCutchan JA, Grant I. CD4 nadir is a predictor of HIV neurocognitive impairment in the era of combination antiretroviral therapy. AIDS. 2011;25:1747–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Clifford DB, Fagan AM, Holtzman DM, Morris JC, Teshome M, Shah AR, Kauwe JS. CSF biomarkers of Alzheimer disease in HIV-associated neurologic disease. Neurology. 2009;73:1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Everall I, Vaida F, Khanlou N, Lazzaretto D, Achim C, Letendre S, Moore D, Ellis R, Cherner M, Gelman B, Morgello S, Singer E, Grant I, Masliah E, National Neuro ATC. Clinico neuropatho-logic correlates of human immunodeficiency virus in the era of antiretroviral therapy. J Neurovirol. 2009;15:360–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Fulop T, Witkowski JM, Larbi A, Khalil A, Herbein G, Frost EH. Does HIV infection contribute to increased beta-amyloid synthesis and plaque formation leading to neurodegeneration and Alzheimer’s disease? J Neurovirol. 2019;25(5):634–47. [DOI] [PubMed] [Google Scholar]
- 119.Alonso R, Pisa D, Fernández-Fernández AM, Rábano A, Carrasco L. Fungal infection in neural tissue of patients with amyotrophic lateral sclerosis. Neurobiol Dis. 2017;108:249–60. [DOI] [PubMed] [Google Scholar]
- 120.Carter CJ. Genetic, transcriptome, proteomic, and epidemiological evidence for blood-brain barrier disruption and polymicrobial brain invasion as determinant factors in Alzheimer’s disease. J Alzheimers Dis Rep. 2017;1(1):125–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Alonso R, Pisa D, Aguado B, Carrasco L. Identification of fungal species in brain tissue from Alzheimer’s Disease by next-generation sequencing. J Alzheimers Dis. 2017;58(1):55–67. [DOI] [PubMed] [Google Scholar]
- 122.Pisa D, Alonso R, Fernández-Fernández AM, Rábano A, Carrasco L. Polymicrobial infections in brain tissue from Alzheimer’s disease patients. Sci Rep. 2017;7(1):5559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hill AB. The environment and disease: association or causation? Bull World Health Organ. 1965;58:295–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Monti D, Ostan R, Borelli V, Castellani G, Franceschi C. Inflammaging and human longevity in the omics era. Mech Ageing Dev. 2017;165(Pt B):129–38. [DOI] [PubMed] [Google Scholar]
- 125.Müller L, Di Benedetto S, Pawelec G. The immune system and its dysregulation with aging. Subcell Biochem. 2019;91:21–43. [DOI] [PubMed] [Google Scholar]
- 126.Fülöp T, Larbi A, Witkowski JM. Human inflammaging. Gerontology. 2019;65(5):495–504. [DOI] [PubMed] [Google Scholar]
- 127.Su F, Bai F, Zhou H, Zhang Z. Microglial toll-like receptors and Alzheimer’s disease. Brain Behav Immun. 2016;52:187–98. [DOI] [PubMed] [Google Scholar]
- 128.Venegas C, Heneka MT. Danger-associated molecular patterns in Alzheimer’s disease. J Leukoc Biol. 2017;101:87–98. [DOI] [PubMed] [Google Scholar]
- 129.Yang SH. Cellular and molecular mediators of neuroinflammation in Alzheimer disease. Int Neurourol J. 2019;23(Suppl 2):S54–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Heneka MT, Carson MJ, El Khoury J, Landreth GE, Brosseron F, Feinstein DL, Jacobs AH, Wyss-Coray T, Vitorica J, Ransohoff RM, Herrup K, Frautschy SA, Finsen B, Brown GC, Verkhratsky A, Yamanaka K, Koistinaho J, Latz E, Halle A, Petzold GC, Town T, Morgan D, Shinohara ML, Perry VH, Holmes C, Bazan NG, Brooks DJ, Hunot S, Joseph B, Deigendesch N, Garaschuk O, Boddeke E, Dinarello CA, Breitner JC, Cole GM, Golenbock DT, Kummer MP. Neuroinflammation in Alzheimer’s disease. Lancet Neurol. 2015;14(4):388–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Mosher KI, Wyss-Coray T. Microglial dysfunction in brain aging and Alzheimer’s disease. Biochem Pharmacol. 2014;88(4):594–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mawanda F, Wallace R. Can infections cause Alzheimer’s disease? Epidemiol Rev. 2013;35:161–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Giridharan VV, Masud F, Petronilho F, Dal-Pizzol F, Barichello T. Infection-induced systemic inflammation is a potential driver of Alzheimer’s disease progression. Front Aging Neurosci. 2019;28(11):122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gasparotto J, Ribeiro CT, da Rosa-Silva HT, Bortolin RC, Rabelo TK, Peixoto DO, Moreira JCF, Gelain DP. Systemic inflammation changes the site of RAGE expression from endothelial cells to neurons in different brain areas. Mol Neurobiol. 2019;56(5):3079–89. [DOI] [PubMed] [Google Scholar]
- 135.Gasparotto J, Girardi CS, Somensi N, Ribeiro CT, Moreira JCF, Michels M, Sonai B, Rocha M, Steckert AV, Barichello T, Quevedo J, Dal-Pizzol F, Gelain DP. Receptor for advanced glycation end products mediates sepsis-triggered amyloid-β accumulation, Tau phosphorylation, and cognitive impairment. J Biol Chem. 2018;293(1):226–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Wang LM, Wu Q, Kirk RA, Horn KP, Ebada Salem AH, Hoffman JM, Yap JT, Sonnen JA, Towner RA, Bozza FA, Rodrigues RS, Morton KA. Lipopolysaccharide endotoxemia induces amyloid-β and p-tau formation in the rat brain. Am J Nucl Med Mol Imaging. 2018;8(2):86–99. [PMC free article] [PubMed] [Google Scholar]
- 137.Ehler J, Barrett LK, Taylor V, Groves M, Scaravilli F, Wittstock M, Kolbaske S, Grossmann A, Henschel J, Gloger M, Sharshar T, Chretien F, Gray F, Nöldge-Schomburg G, Singer M, Sauer M, Petzold A. Translational evidence for two distinct patterns of neuroaxonal injury in sepsis: a longitudinal, prospective translational study. Crit Care. 2017;21(1):262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bu XL, Yao XQ, Jiao SS, Zeng F, Liu YH, Xiang Y, Liang CR, Wang QH, Wang X, Cao HY, Yi X, Deng B, Liu CH, Xu J, Zhang LL, Gao CY, Xu ZQ, Zhang M, Wang L, Tan XL, Xu X, Zhou HD, Wang YJ. A study on the association between infectious burden and Alzheimer’s disease. Eur J Neurol. 2015;22(12):1519–25. [DOI] [PubMed] [Google Scholar]
- 139.Couturier J, Stancu IC, Schakman O, Pierrot N, Huaux F, Kienlen-Campard P, Dewachter I, Octave JN. Activation of phagocytic activity in astrocytes by reduced expression of the inflammasome component ASC and its implication in a mouse model of Alzheimer disease. J Neuroinflammation. 2016;27(13):20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D, Remus A, Tzeng TC, Gelpi E, Halle A, Korte M, Latz E, Golenbock DT. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature. 2013;493(7434):674–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Rea IM, Gibson DS, McGilligan V, McNerlan SE, Alexander HD, Ross OA. Age and age-related diseases: role of inflammation triggers and cytokines. Front Immunol. 2018;9(9):586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Abbayya K, Puthanakar NY, Naduwinmani S, Chidambar YS. Association between periodontitis and Alzheimer’s disease. N Am J Med Sci. 2015;7(6):241–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bergman P, Termén S, Johansson L, Nyström L, Arenas E, Jonsson AB, Hökfelt T, Gudmundsson GH, Agerberth B. The antimicrobial peptide rCRAMP is present in the central nervous system of the rat. J Neurochem. 2005;93(5):1132–40. [DOI] [PubMed] [Google Scholar]
- 144.Lee M, Shi X, Barron AE, McGeer E, McGeer PL. Human antimicrobial peptide LL-37 induces glial-mediated neuroinflammation. Biochem Pharmacol. 2015;94(2):130–41. [DOI] [PubMed] [Google Scholar]
- 145.Brandenburg LO, Varoga D, Nicolaeva N, Leib SL, Wilms H, Podschun R, Wruck CJ, Schröder JM, Pufe T, Lucius R. Role of glial cells in the functional expression of LL-37/rat cathelin-related antimicrobial peptide in meningitis. J Neuropathol Exp Neurol. 2008;67(11):1041–54. [DOI] [PubMed] [Google Scholar]
- 146.Jung YJ, Chung WS. Phagocytic roles of glial cells in healthy and diseased brains. Biomol Ther (Seoul). 2018;26(4):350–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Ashraf GM, Tarasov VV, Makhmutova A, Chubarev VN, Avila-Rodriguez M, Bachurin SO, Aliev G. The possibility of an infectious etiology of Alzheimer disease. Mol Neurobiol. 2019;56(6):4479–91. [DOI] [PubMed] [Google Scholar]
- 148.Linnartz B, Wang Y, Neumann H. Microglial immunoreceptor tyrosine-based activation and inhibition motif signaling in neuroinflammation. Int J Alzheimers Dis. 2010;22:2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Clarke LE, Liddelow SA, Chakraborty C, Münch AE, Heiman M, Barres BA. Normal aging induces A1-like astrocyte reactivity. Proc Natl Acad Sci USA. 2018;115(8):E1896–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Vilalta A, Brown GC. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018;285(19):3566–75. [DOI] [PubMed] [Google Scholar]
- 151.von Bernhardi R, Eugenín-von Bernhardi L, Eugenín J. Microglial cell dysregulation in brain aging and neurodegeneration. Front Aging Neurosci. 2015;20(7):124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Lana D, Ugolini F, Nosi D, Wenk GL, Giovannini MG. Alterations in the interplay between neurons, astrocytes and microglia in the rat dentate gyrus in experimental models of neurodegeneration. Front Aging Neurosci. 2017;11(9):296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.White MR, Kandel R, Tripathi S, Condon D, Qi L, Taubenberger J, Hartshorn KL. Alzheimer’s associated β-amyloid protein inhibits influenza A virus and modulates viral interactions with phagocytes. PLoS One. 2014;9(7):e101364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Bourgade K, Dupuis G, Frost EH, Fülöp T. Anti-viral properties of amyloid-β peptides. J. Alzheimers Dis 2016;54:859–78. [DOI] [PubMed] [Google Scholar]
- 155.Maqbool M, Hoda N. GSK3 Inhibitors in the therapeutic development of diabetes, cancer and neurodegeneration: past, present and future. Curr Pharm Des. 2017;23(29):4332–50. [DOI] [PubMed] [Google Scholar]
- 156.Brothers HM, Gosztyla ML, Robinson SR. The physiological roles of amyloid-β peptide hint at new ways to treat Alzheimer’s disease. Front Aging Neurosci. 2018;25(10):118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Morizawa YM, Hirayama Y, Ohno N, Shibata S, Shigetomi E, Sui Y, Nabekura J, Sato K, Okajima F, Takebayashi H, Okano H, Koizumi S. Reactive astrocytes function as phagocytes after brain ischemia via ABCA1-mediated pathway. Nat Commun. 2017;8(1):28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Ransohoff RM, Brown MA. Innate immunity in the central nervous system. J Clin Invest. 2012;122(4):1164–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Kumar DK, Eimer WA, Tanzi RE, Moir RD. Alzheimer’s disease: the potential therapeutic role of the natural antibiotic amyloid-β peptide. Neurodegener Dis Manag. 2016;6(5):345–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kumar DK, Choi SH, Washicosky KJ, Eimer WA, Tucker S, Ghofrani J, Lefkowitz A, McColl G, Goldstein LE, Tanzi RE, Moir RD. Amyloid-β peptide protects against microbial infection in mouse and worm models of Alzheimer’s disease. Sci Transl Med. 2016;8(340):340ra72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Golde TE. Alzheimer disease: host immune defence, amyloid-β peptide and Alzheimer disease. Nat Rev Neurol. 2016;12(8):433–4. [DOI] [PubMed] [Google Scholar]
- 162.Moir RD, Lathe R, Tanzi RE. The antimicrobial protection hypothesis of Alzheimer’s disease. Alzheimers Dement. 2018;14(12):1602–14. [DOI] [PubMed] [Google Scholar]
- 163.Venugopal C, Demos CM, Rao KS, Pappolla MA, Sambamurti K. Beta-secretase: structure, function, and evolution. CNS Neurol Disord Drug Targets. 2008;7(3):278–94 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Smolarkiewicz M, Skrzypczak T, Wojtaszek P. The very many faces of presenilins and the γ-secretase complex. Proto-plasma. 2013;250(5):997–1011. 10.1007/s00709-013-0494-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Moore DB, Gillentine MA, Botezatu NM, Wilson KA, Benson AE, Langeland JA. Asynchronous evolutionary origins of Aβ and BACE1. Mol Biol Evol. 2014;31(3):696–702. 10.1093/molbev/mst262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Larbi A, Pawelec G, Witkowski JM, Schipper HM, Derhovanessian E, Goldeck D, Fulop T. Dramatic shifts in circulating CD4 but not CD8 T cell subsets in mild Alzheimer’s disease. J Alzheimers Dis. 2009;17(1):91–103. [DOI] [PubMed] [Google Scholar]
- 167.Derhovanessian E, Larbi A, Pawelec G. Biomarkers of human immunosenescence: impact of cytomegalovirus infection. Curr Opin Immunol. 2009;21(4):440–5. [DOI] [PubMed] [Google Scholar]
- 168.Pawelec G, Derhovanessian E, Larbi A, Strindhall J, Wikby A. Cytomegalovirus and human immunosenescence. Rev Med Virol. 2009;19(1):47–56. [DOI] [PubMed] [Google Scholar]
- 169.Shen-Orr SS, Furman D, Kidd BA, Hadad F, Lovelace P, Huang YW, Rosenberg-Hasson Y, Mackey S, Grisar FA, Pickman Y, Maecker HT, Chien YH, Dekker CL, Wu JC, Butte AJ, Davis MM. Defective signaling in the JAK-STAT pathway tracks with chronic inflammation and cardiovascular risk in aging humans. Cell Syst. 2016;3(4):374–384.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Kritsilis M, Rizou VS, Koutsoudaki PN, Evangelou K, Gorgoulis VG, Papadopoulos D. Ageing, cellular senescence and neurodegenerative disease. Int J Mol Sci. 2018;19(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Tsai CY, Shen CY, Liao HT, Li KJ, Lee HT, Lu CS, Wu CH, Kuo YM, Hsieh SC, Yu CL. Molecular and cellular bases of immunosenescence, inflammation, and cardiovascular complications mimicking “inflammaging” in patients with systemic lupus erythematosus. Int J Mol Sci. 2019;20(16). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Chen Y, Liu S, Leng SX. Chronic low-grade inflammatory phenotype (CLIP) and senescent immune dysregulation. Clin Ther. 2019;41(3):400–9. [DOI] [PubMed] [Google Scholar]
- 173.Olivieri F, Prattichizzo F, Grillari J, Balistreri CR. Cellular senescence and inflammaging in age-related diseases. Mediators Inflamm. 2018;17(2018):9076485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Sanada F, Taniyama Y, Muratsu J, Otsu R, Shimizu H, Rakugi H, Morishita R. Source of chronic inflammation in aging. Front Cardiovasc Med. 2018;22(5):12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Yamazaki Y, Baker DJ, Tachibana M, Liu CC, van Deursen JM, Brott TG, Bu G, Kanekiyo T. Vascular cell senescence contributes to blood-brain barrier breakdown. Stroke. 2016;47(4):1068–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Burton DGA, Stolzing A. Cellular senescence: immunosurveillance and future immunotherapy. Ageing Res Rev. 2018;43:17–25. [DOI] [PubMed] [Google Scholar]
- 177.Bossù P, Ciaramella A, Salani F, Vanni D, Palladino I, Caltagirone C, Scapigliati G. Interleukin-18, from neuroinflammation to Alzheimer’s disease. Curr Pharm Des. 2010;16(38):4213–24. [DOI] [PubMed] [Google Scholar]
- 178.Wang Y, Jin S, Sonobe Y, Cheng Y, Horiuchi H, Parajuli B, Kawanokuchi J, Mizuno T, Takeuchi H, Suzumura A. Interleukin-1β induces blood-brain barrier disruption by downregulating Sonic hedgehog in astrocytes. PLoS One. 2014;9(10):e110024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Bester J, Soma P, Kell DB, Pretorius E. Viscoelastic and ultra-structural characteristics of whole blood and plasma in Alzheimer-type dementia, and the possible role of bacterial lipopolysaccharides (LPS). Oncotarget. 2015;6(34):35284–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Man SM, Karki R, Kanneganti TD. Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev. 2017;277(1):61–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Sweeney MD, Zhao Z, Montagne A, Nelson AR, Zlokovic BV. Blood-brain barrier: from physiology to disease and back. Physiol Rev. 2019;99(1):21–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Villabona-Rueda A, Erice C, Pardo CA, Stins MF. The evolving concept of the blood brain barrier (BBB): from a single static barrier to a heterogeneous and dynamic relay center. Front Cell Neurosci. 2019;20(13):405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Nation DA, Sweeney MD, Montagne A, Sagare AP, D’Orazio LM, Pachicano M, Sepehrband F, Nelson AR, Buennagel DP, Harrington MG, Benzinger TLS, Fagan AM, Ringman JM, Schneider LS, Morris JC, Chui HC, Law M, Toga AW, Zlokovic BV. Blood-brain barrier breakdown is an early biomarker of human cognitive dysfunction. Nat Med. 2019;25(2):270–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, Toga AW, Jacobs RE, Liu CY, Amezcua L, Harrington MG, Chui HC, Law M, Zlokovic BV. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Nwafor DC, Brichacek AL, Mohammad AS, Griffith J, Lucke-Wold BP, Benkovic SA, Geldenhuys WJ, Lockman PR, Brown CM. Targeting the blood-brain barrier to prevent sepsis-associated cognitive impairment. J Cent Nerv Syst Dis. 2019;11:1179573519840652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Cain MD, Salimi H, Gong Y, Yang L, Hamilton SL, Heffernan JR, Hou J, Miller MJ, Klein RS. Virus entry and replication in the brain precedes blood-brain barrier disruption during intranasal alphavirus infection. J Neuroimmunol. 2017;308:118–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Al-Obaidi MMJ, Desa MNM. Mechanisms of blood brain barrier disruption by different types of bacteria, and bacterial-host interactions facilitate the bacterial pathogen invading the brain. Cell Mol Neurobiol. 2018;38(7):1349–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Miner JJ, Diamond MS. Mechanisms of restriction of viral neuroinvasion at the blood-brain barrier. Curr Opin Immunol. 2016;38:18–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Shi Q, Chowdhury S, Ma R, Le KX, Hong S, Caldarone BJ, Stevens B, Lemere CA. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 Sci Transl Med. 2017;9(392). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Williams WM, Castellani RJ, Weinberg A, Perry G, Smith MA. Do β-defensins and other antimicrobial peptides play a role in neuroimmune function and neurodegeneration? ScientificWorld-Journal. 2012;2012:905785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Frost GR, Li YM. The role of astrocytes in amyloid production and Alzheimer’s disease. Open Biol. 2017; 7(12). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Orsini F, De Blasio D, Zangari R, Zanier ER, De Simoni MG. Versatility of the complement system in neuroinflammation, neurodegeneration and brain homeostasis. Front Cell Neurosci. 2014;7(8):380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Miller KD, Matullo CM, Milora KA, Williams RM, O’Regan KJ, Rall GF. Immune-mediated control of a dormant neurotropic RNA virus infection. J Virol. 2019;93(18). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhao Y, Jaber V, Lukiw WJ. Secretory products of the human GI tract microbiome and their potential impact on Alzheimer’s disease (AD): detection of lipopolysaccharide (LPS) in AD hippocampus. Front Cell Infect Microbiol. 2017;11(7):318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Louw C, Gordon A, Johnston N, Mollatt C, Bradley G, Whiteley CG. Arginine deiminases: therapeutic tools in the etiology and pathogenesis of Alzheimer’s disease. J Enzyme Inhib Med Chem. 2007;22(1):121–6. [DOI] [PubMed] [Google Scholar]
- 196.Odamaki T, Kato K, Sugahara H, Hashikura N, Takahashi S, Xiao JZ, Abe F, Osawa R. Age-related changes in gut microbiota composition from newborn to centenarian: a cross-sectional study. BMC Microbiol. 2016;25(16):90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Riccio P, Rossano R. Undigested food and gut microbiota may cooperate in the pathogenesis of neuroinflammatory diseases: a matter of barriers and a proposal on the origin of organ specificity. Nutrients. 2019;11(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Sochocka M, Donskow-Łysoniewska K, Diniz BS, Kurpas D, Brzozowska E, Leszek J. The gut microbiome alterations and inflammation-driven pathogenesis of Alzheimer’s disease-a critical review. Mol Neurobiol. 2019;56(3):1841–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Maurer K, Rahming S, Prvulovic D. Dental health in advanced age and Alzheimer’s disease: a possible link with bacterial toxins entering the brain? Psychiatry Res Neuroimaging. 2018;30(282):132–3. [DOI] [PubMed] [Google Scholar]
- 200.Sato S, Kiyono H, Fujihashi K. Mucosal immunosenescence in the gastrointestinal tract: a mini-review. Gerontology. 2015;61(4):336–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Rodrigues M, Fan J, Lyon C, Wan M, Hu Y. Role of extracellular vesicles in viral and bacterial infections: pathogenesis, diagnostics, and therapeutics. Theranostics. 2018;8(10):2709–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Mantri CK, Chen CH, Dong X, Goodwin JS, Pratap S, Paromov V, Xie H. Fimbriae-mediated outer membrane vesicle production and invasion of Porphyromonas gingivalis. Microbiologyopen. 2015;4(1):53–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Singhrao SK, Olsen I. Are Porphyromonas gingivalis outer membrane vesicles microbullets for sporadic Alzheimer’s disease manifestation? J Alzheimers Dis Rep. 2018;2(1):219–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Ilievski V, Zuchowska PK, Green SJ, Toth PT, Ragozzino ME, Le K, Aljewari HW, O’Brien-Simpson NM, Reynolds EC, Watanabe K. Chronic oral application of a periodontal pathogen results in brain inflammation, neurodegeneration and amyloid beta production in wild type mice. PLoS One. 2018;13(10):e0204941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Junges VM, Closs VE, Nogueira GM, Gottlieb MGV. Crosstalk between gut microbiota and central nervous system: a focus on Alzheimer’s disease. Curr Alzheimer Res. 2018;15(13):1179–90. [DOI] [PubMed] [Google Scholar]
- 206.Abdel-Haq R, Schlachetzki JCM, Glass CK, Mazmanian SK. Microbiome-microglia connections via the gut-brain axis. J Exp Med. 2019;216:41–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Bell JS, Spencer JI, Yates RL, Yee SA, Jacobs BM, DeLuca GC. Invited review: From nose to gut—the role of the microbiome in neurological disease. Neuropathol Appl Neurobiol. 2019;45(3):195–215. [DOI] [PubMed] [Google Scholar]
- 208.Heiss CN, Olofsson LE. The role of the gut microbiota in development, function and disorders of the central nervous system and the enteric nervous system. J Neuroendocrinol. 2019;31(5):e12684. [DOI] [PubMed] [Google Scholar]
- 209.Salazar N, Valdés-Varela L, González S, Gueimonde M, de Los Reyes-Gavilán CG. Nutrition and the gut microbiome in the elderly. Gut Microbes. 2017;8(2):82–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Li B, He Y, Ma J, Huang P, Du J, Cao L, Wang Y, Xiao Q, Tang H, Chen S. Mild cognitive impairment has similar alterations as Alzheimer’s disease in gut microbiota. Alzheimers Dement. 2019;15(10):1357–66. [DOI] [PubMed] [Google Scholar]
- 211.Zhuang ZQ, Shen LL, Li WW, Fu X, Zeng F, Gui L, Lü Y, Cai M, Zhu C, Tan YL, Zheng P, Li HY, Zhu J, Zhou HD, Bu XL, Wang YJ. Gut microbiota is altered in patients with Alzheimer’s disease. J Alzheimers Dis. 2018;63(4):1337–46. [DOI] [PubMed] [Google Scholar]
- 212.Lin L, Zheng LJ, Zhang LJ. Neuroinflammation, gut microbiome, and Alzheimer’s disease. Mol Neurobiol. 2018;55(11):8243–50. [DOI] [PubMed] [Google Scholar]
- 213.Giau VV, Wu SY, Jamerlan A, An SSA, Kim SY, Hulme J. Gut microbiota and their neuroinflammatory implications in Alzheimer’s disease. Nutrients. 2018;10(11). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Hwang JS, Im CR, Im SH, Immune disorders and its correlation with gut microbiome. Immune Netw. 12: 129–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Cerovic M, Forloni G, Balducci C. Neuroinflammation and the gut microbiota: possible alternative therapeutic targets to counteract Alzheimer’s disease? Front Aging Neurosci. 2019;18(11):284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Cattaneo A, Cattane N, Galluzzi S, Provasi S, Lopizzo N, Festari C, Ferrari C, Guerra UP, Paghera B, Muscio C, Bianchetti A, Volta GD, Turla M, Cotelli MS, Gennuso M, Prelle A, Zanetti O, Lussignoli G, Mirabile D, Bellandi D, Gentile S, Belotti G, Villani D, Harach T, Bolmont T, Padovani A, Boccardi M, Frisoni GB, INDIA-FBP Group. Association of brain amyloidosis with pro-inflammatory gut bacterial taxa and peripheral inflammation markers in cognitively impaired elderly. Neurobiol Aging. 2017;49:60–8. [DOI] [PubMed] [Google Scholar]
- 217.Vogt NM, Kerby RL, Dill-McFarland KA, Harding SJ, Merluzzi AP, Johnson SC, Carlsson CM, Asthana S, Zetterberg H, Blennow K, Bendlin BB, Rey FE. Gut microbiome alterations in Alzheimer’s disease. Sci Rep. 2017;7(1):13537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Pritchard AB, Crean S, Olsen I, Singhrao SK. Periodontitis, microbiomes and their role in Alzheimer’s disease. Front Aging Neurosci. 2017;24(9):336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Xie H. Biogenesis and function of Porphyromonas gingivalis outer membrane vesicles. Future Microbiol. 2015;10(9):1517–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Henneberger C, Steinhäuser C. Astrocytic TLR4 at the crossroads of inflammation and seizure susceptibility. J Cell Biol. 2016;215(5):607–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Gui MJ, Dashper SG, Slakeski N, Chen Y-Y, Reynolds EC. Spheres of influence: Porphyromonas gingivalis outer membrane vesicles. Mol. Oral Microbiol 2016;31:365–78. [DOI] [PubMed] [Google Scholar]
- 222.Grenier D, Roy S, Chandad F, Plamondon P, Yoshioka M, Nakayama K, Mayrand D. Effect of inactivation of the Arg- and/or Lysgingipain gene on selected virulence and physiological properties of Porphyromonas gingivalis. Infect Immun. 2003;71:4742–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Jay TR, von Sauken VE, Landreth GE. TREM2 in neurodegenerative diseases. Mol Nuerodegener. 2017;12:56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 224.Minoretti P, Gazzaruso C, Vito CD, Emanuele E, Bianchi MP, Coen E, Reino M, Geroldi D. Effect of the functional toll-like receptor 4 Asp299Gly polymorphism on susceptibility to late-onset Alzheimer’s disease. Neurosci Lett. 2006;391:147–9. [DOI] [PubMed] [Google Scholar]
- 225.Brouwers N, Van Cauwenberghe C, Engelborghs S, Lambert J-C, Bettens K, Le Bastard N, Pasquier F, Montoya AG, Peeters K, Mattheijssens M, Vandenberghe R, Deyn PP, Cruts M, Amouyel P, Sleegers K, Van Broeckhoven C. Alzheimer risk associated with a copy number variation in the complement receptor 1 increasing C3b/C4b binding sites. Mol. Psychiatry 2012;17:223–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Tan M-S, Yu J-T, Jiang T, Zhu X-C, Wang H-F, Zhang W, Wang Y-L, Jiang W, Tan L. NLRP3 polymorphisms are associated with late-onset Alzheimer’s disease in Han Chinese. J Neuroimmunol. 2013;265:91–5. [DOI] [PubMed] [Google Scholar]
- 227.Olsen L, Yilmaz O. Modulation in inflammasome activity by Porphyromonas gingivalis in periodontitis and associated systemic diseases. J Oral Microbiol. 2016;8:30385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Venegas C, Kumar S, Franklin BS, Dierkes T, Brinkschulte T, Tejera D, Vieira-Saecker A, Schwartz S, Santarelli F, Kummer MP, Griep A, Gelpi E, Beilharz D, Riedel D, Golenbock DT, Geyer M, Walter J, Latz E, Heneka H. Microglia-derived ASC specks cross-seed amyloid-β in Alzheimer’s disease. Nature. 2017;552:355–61. [DOI] [PubMed] [Google Scholar]
- 229.Franklin BS, Latz E, Schmidt FI. The intra- and extracellular functions of ASC specks. Immunol Rev. 2018;281(1):74–87. [DOI] [PubMed] [Google Scholar]
- 230.Mariathasan S, Weiss DS, Dixit VM, Monack DM. Innate immunity against Francisella tularensis is dependent on the ASC/caspase-1 axis. J Exp Med. 2005;202:1043–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Cecil JD, O’Brien-Simpson NM, Lenzo JC, Holden JA, Singleton W, Perez-Gonzalez A, Mansell A, Reynolds EC. Outer membrane vesicles prime and activate macrophage inflammasomes and cytokine secretion in vitro and in vivo. Front Immunol. 2017;8:1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Fleetwood AJ, Lee MKS, Singleton W, Achuthan A, Lee M-C, O’Brien-Simpson NM, Cook AD, Murphy AJ, Dashper SG, Reynolds EC, Hamilton JA. Metabolic remodeling, inflammasome activation, and pyroptosis in macrophages stimulated by Porphyromonas gingivalis and its outer membrane vesicles. Front Cell Infect Microbiol. 2017;7:351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Vincents B, Guentsch A, Kostolowska DA, von Pawel-Rammingen U, Eick S, Jan Potempa J, Abrahamson M. Cleavage of IgG1 and IgG3 by gingipain K from Porphyromonas gingivalis may compromise host defense in progressive periodontitis. FASEB J. 2011;25:3741–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Arndt Guentsch, Christiane Hirsch, Wolfgang Pfister, Bjarne Vincents, Magnusm Abrahamson, Aneta Sroka, Jan Potempa, Sigrun Eick. Cleavage of IgG1 in GCF is associated with presence of Porphyromonas gingivalis. J Periodontal Res. 2013;48(4):458–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Stobernack T, du Teil Espina M, Mulder LM, Palma Medina LM, Piebenga DR, Gabarrini G, Zhao X, Janssen KMJ, Hulzebos J, Brouwer E, Sura T, Becher D, van Winkelhoff AJ, Götz F, Otto A, Westra J, van Dijl JM. A secreted bacterial peptidylarginine deiminase can neutralize human innate immune defenses. mBio. 2018;9:e01704–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Gabarrini G, Palma Medina LM, Stobernack T, Prins RC, du Teil Espina M, Kuipers J, Chlebowicz MA, Rossen JWA, van Winkelhoff AJ, van Dijl JM. There’s no place like OM: vesicular sorting and secretion of the peptidylarginine deiminase of Porphyromonas gingivalis. Virulence. 2018;9(1):456–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Al-Adwani S, Wallin C, Balhuizen MD, Veldhuizen EJA, Coorens M, Landreh M, Végvári Á, Smith ME, Qvarfordt I, Lindén A, Gräslund A, Agerberth B, Bergman P. Studies on citrullinated LL-37: detection in human airways, antibacterial effects and biophysical properties. Sci Rep. 2020;10(1):2376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Seo DO, Holtzman DM. Gut microbiota: from the forgotten organ to a potential key player in the pathology of Alzheimer disease. J Gerontol A Biol Sci Med Sci. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Friedland RP, Chapman MR. The role of microbial amyloid in neurodegeneration. PLoS Pathog. 2017;13:e1006654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Netea MG, van der Meer JW. Trained immunity: an ancient way of remembering. Cell Host Microb. 2017;21:297–300. [DOI] [PubMed] [Google Scholar]
- 241.Baëhl S, Garneau H, Lorrain D, Viens I, Svotelis A, Lord JM, Cabana F, Larbi A, Dupuis G, Fülöp T. Alterations in monocyte phenotypes and functions after a hip fracture in elderly individuals: a 6-month longitudinal study. Gerontology. 2016;62(5):477–90. [DOI] [PubMed] [Google Scholar]
- 242.Penke B, Bogar F, Fulop L. Beta-amyloid and the pathomechanisms of Alzheimer’s disease: a comprehensive view. Molecules (Basel, Switzerland). 2017;22:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Kagan BL, Jang H, Capone R, Teran Arce F, Ramachandran S, Lal R, et al. Antimicrobial properties of amyloid peptides. Mol Pharm. 2011;9(4):708–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Wu JJ, Brentjens MH, Torres G, Yeung-Yue K, Lee P, Tyring SK. Valacyclovir in the treatment of herpes simplex, herpes zoster, and other viral infections. J Cutan Med Surg. 2003;7(5):372–81. [DOI] [PubMed] [Google Scholar]
- 245.Itzhaki RF. Herpes simplex virus type 1 and Alzheimer’s disease: increasing evidence for a major role of the virus. Front Aging Neurosci. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Loeb MB, Molloy DW, Smieja M, Standish T, Goldsmith CH, Mahony J, Smith S, Borrie M, Decoteau E, Davidson W, McDougall A, Gnarpe J, O’DONNell M, Chernesky M. A randomized, controlled trial of doxycycline and rifampin for patients with Alzheimer’s disease. J Am Geriatr Soc. 2004;52(3):381–7. [DOI] [PubMed] [Google Scholar]
- 247.Molloy DW, Standish TI, Zhou Q, Guyatt G, DARAD Study Group. A multicenter, blinded, randomized, factorial controlled trial of doxycycline and rifampin for treatment of Alzheimer’s disease: the DARAD trial. Int J Geriatr Psychiatry. 2013;28(5):463–70. [DOI] [PubMed] [Google Scholar]
- 248.Howard R, Zubko O, Bradley R, Harper E, Pank L, O’Brien J, Fox C, Tabet N, Livingston G, Bentham P, McShane R, Burns A, Ritchie C, Reeves S, Lovestone S, Ballard C, Noble W, Nilforooshan R, Wilcock G, Gray R, Minocycline in Alzheimer Disease Efficacy (MADE) Trialist Group. Minocycline at 2 different dosages vs placebo for patients with mild Alzheimer disease: a randomized clinical trial. JAMA Neurol. 2019;77(2):164–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Balducci C, Forloni G. Doxycycline for Alzheimer’s disease: fighting β-amyloid oligomers and neuroinflammation. Front Pharmacol. 2019;3(10):738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.Angelucci F, Cechova K, Amlerova J, Hort J. Antibiotics, gut microbiota, and Alzheimer’s disease. J Neuroinflamm. 2019;16(1):108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.O’Brien-Simpson NM, Holden JA, Lenzo JC, Tan Y, Brammar GC, Walsh KA, Singleton W, Orth RKH, Slakeski N, Cross KJ, Darby IB, Becher D, Rowe T, Morelli AB, Hammet A, Nash A, Brown A, Ma B, Vingadassalom D, McCluskey J, Kleanthous H, Reynolds EC. A therapeutic Porphyromonas gingivalis gingipain vaccine induces neutralising IgG1 antibodies that protect against experimental periodontitis. NPJ Vacc. 2016;1(1):16022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Puth S, Hong SH, Park MJ, Lee HH, Lee YS, Jeong K, Kang IC, Koh JT, Moon B, Park SC, Rhee JH, Lee SE. Mucosal immunization with a flagellin-adjuvanted Hgp44 vaccine enhances protective immune responses in a murine Porphyromonas gingivalis infection model. Hum Vaccin Immunother. 2017;13(12):2794–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Wilensky A, Potempa J, Houri-Haddad Y, Shapira L. Vaccination with recombinant RgpA peptide protects against Porphyromonas gingivalis-induced bone loss. J Periodontal Res. 2017;52(2):285–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Reynolds EC, O’Brien-Simpson N, Rowe T, Nash A, McCluskey J, Vingadassalom D, Kleanthous H. Prospects for treatment of Porphyromonas gingivalis-mediated disease—immune-based therapy. J Oral Microbiol. 2015;18(7):29125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Choi JI, Seymour GJ. Vaccines against periodontitis: a forward-looking review. J Periodontal Implant Sci. 2010;40(4):153–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Kadowaki T, Baba N, Abe R, Takki M, Hashimoto T, Tsukuba S, Okazaki Y, Suda T, Akao K, Yamamoto Y. Suppression of pathogenicity of Porphyromonas gingivalis by newly developed gingipain inhibitors. Mol Pharmacol. 2004;66:1599–606. [DOI] [PubMed] [Google Scholar]
- 257.Czyzewski AM, Jenssen H, Fjell CD, Waldbrook M, Chongsiriwatana NP, Yuen E, Hancock RE, Barron AE. In vivo, in vitro, and in silico characterization of peptoids as antimicrobial agents. PLoS One. 2016;11(2):e0135961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Overhage J, Campisano A, Bains M, Torfs ECW, Rehm BHA, Hancock REW. Human host defense peptide LL-37 prevents bacterial biofilm formation. Infect Immun. 2008;9:4176–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Singh PK, Parseck MR, Greenberg EP, Welsh MJ. A component of innate immunity prevents bacterial biofilm development. Nature. 2002;417:552–5. [DOI] [PubMed] [Google Scholar]
- 260.Kang J, Dietz MJ, Li B. Antimicrobial peptide LL-37 is bactericidal against Staphylococcus aureus biofilms. PLoS One. 2019;14(6):e0216676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Svenson D, Wilk L, Morgelin M, Herwald H, Nilsson B-O. LL-37 induced host cell cytotoxicity depends on cellular expression of the globular C1q receptor (CD33). Biochem J. 2016;473:87–98. [DOI] [PubMed] [Google Scholar]
- 262.Johansson J, Gudmudsson GH, Rottenberg ME, Agerberth B. Conformation dependent antibacterial activity of the naturally occurring human peptide LL-37. J Biol Chem. 1998;273:3716–24. [DOI] [PubMed] [Google Scholar]
- 263.Saji N, Niida S, Murotani K, Hisada T, Tsuduki T, Sugimoto T. Analysis of the relationship between the gut microbiome and dementia: a cross-sectional study conducted in Japan. Sci Rep. 2019;9:1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Duraj-Thatte AM, Pravschotinunt P, Nash TR, Ward FR, Joshi N. Modulating bacterial and gut mucosal interactions with engineered biofilm matrix protein. Sci Rep. 2018;8:3475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.d’Hennezel E, Abubucker S, Murphy LO, Cullen TW. Total polysaccharide from the human gut microbiome silences toll like receptor signaling. mSystems. 2017;2:00046–e00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Attanasio J, Wherry EJ. Costimulatory and coinhibitory receptor pathways in infectious disease. Immunity. 2016;44:1052–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Groeger S, Jarzina F, Mamat U, Meyle J. Induction of B7-H1 receptor by bacterial cells fractions of Porphyromonas gingivalis on human oral epithelial cells: B7-H1 induction by Porphyromonas gingivalis fractions. Immunobiology. 2017;222:137–47. [DOI] [PubMed] [Google Scholar]
- 268.Nagpal R, Neth BJ, Wang S, Craft S, Yadav H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine. 2019;47:529–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Fortier M, Castellano CA, Croteau E, Langlois F, Bocti C, St-Pierre V, Vandenberghe C, Bernier M, Roy M, Descoteaux M, Whittingstall K, Lepage M, Turcotte ÉE, Fulop T, Cunnane SC. A ketogenic drink improves brain energy and some measures of cognition in mild cognitive impairment. Alzheimers Dement. 2019;15(5):625–34. [DOI] [PubMed] [Google Scholar]
- 270.Schulthess J, Pandey S, Capitani M, Rue-Albrecht KC, Arnold I, Franchini F, Chomka A, Ilott NE, Johnston DGW, Pires E, McCullagh J, Sansom SN, Arancibia-Carcamo VC, Uhlig HH, Powrie F. The short chain fatty acid butyrate imprints an antimicrobial program in macrophages. Immunity. 2019;50:432–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Wang X, Sun G, Feng T, Zhang J, Huang X, Wang T, Xie Z, Chu X, Yang J, Wang H, Chang S, Gong Y, Ruan L, Zhang G, Yan S, Lian W, Du C, Yang D, Zhang Q, Lin F, Liu J, Zhang H, Ge C, Xiao S, Ding J, Geng M. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019;29(10):787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Baar MP, Brandt RMC, Putavet DA, Klein JDD, Derks KWJ, Bourgeaois BRM. Targeted apoptosis of senescent cells restores tissue homeostasis in response to chemotoxicity and aging. Cell. 2017; pp 132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Kirkland JL, Tchokonia T, Zhu Y, Niedernhofer LJ, Robbins PD. The clinical potential of senolytic drugs. J Am Geriatr Soc. 2017;65:2297–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Németh A, Orgovan N, Sodar BW, Osteikoetxea X, Paloczi K, Szabo-Taylor KE. Antibiotic-induced release of small extracellular vesicles (exosomes) with surface-associated DNA. Sci Rep. 2017;7:8202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Wang R, Yu Z, Sunchu B, Shoaf J, Dang I, Zhao S. Rapamycin inhibits the secretory phenotype of senescent cells by Nfr2-independent mechanism. Aging Cell. 2017;16:565–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Wang S, Xie X, Lei T, Zhang K, Lai B, Zhang Z. Statins attenuate activation of the NLRP23 inflammasome by oxidized LDL or TNFalpha in vascular endothelial cells through a PXR dependent mechanism. Mol Pharmacol. 2017;92:256264. [DOI] [PubMed] [Google Scholar]
- 277.Xu G, Li Z, Ding L, Tang H, Guo S, Liang H. Intestinal mTOR regulates GLP-1 production in mouse L-cells. Diabetologia. 2015;58:1887–97. [DOI] [PubMed] [Google Scholar]
- 278.Ji Y, Luo X, Yang Y, Dai Z, Wu G, Wu Z. Endoplasmic reticulum stress-induced apoptosis in intestinal epithelial cells: a feedback regulation by mechanistic target of rapamycin complex 1 (mTORC1). J Anim Sci Biotechnol. 2018;9:38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 279.Martin SA, Souder DC, Miller KN, Clark JP, Sugar AK, Eliceiri KW. GSK3beta regulates brain energy metabolism. Cell Rep. 2018;23:1922–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 280.Salem I, Ramser A, Isham N, Ghannoum M. The gut microbiome as a major regulator of the gut skin axis. Front Microbiol. 2018;9:1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 281.Ozsvari B, Nuttal JR, Sotgia F, Lisanti MP. Azythromycin and roxithromycin define a new family of senolytic drugs that target senescent human fibroblasts. Aging. 2018;10:3294–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Weng D, Wu Q, Chen XQ, Du YK, Chen T, Li H. Azythromycin treats diffuse panbronchiolitis by targeting T cells via inhibition of mTOR pathway. Biomed Pharmacother. 2019;110:440–8. [DOI] [PubMed] [Google Scholar]
- 283.Lee GJ, Lim JJ, Hyun S. Minocycline treatment increases resistance to oxidative stress and extends lifespan in Drosophila via FOXO. Oncotarget. 2017;8:87879–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Du RH, Tan J, Sun XY, Lu M, Ding JH, Hu G. Fluoxetine inhibits NLRP3 inflammasome activation: implication in depression. Int J Neuropsychopharmacol. 2016;19:pyw037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Bartels C, Wagner M, Wolfsgruber S, Ehrenreich H, Schneider A. Impact of SSRI therapy on risk of conversion from mild cognitive impairment to Alzheimer’s dementia in individuals with previous depression. Am J Psychiatry. 2018;175:232–41. [DOI] [PubMed] [Google Scholar]
- 286.Xing Y, Liqi Z, Jian L, Quinghua Y, Qian Y. Doxycycline induces mitophagy and suppresses production of interferon-beta IPEC-J2 cells. Front Cel Infect Microbiol. 2017;7:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Feng Y, Wang Y, Wang P, Huang Y, Wang F. Short chain fatty acids manifest stimulative and protective effects on intestinal barrier functions through the inhibition of the NLRP3 inflammasome and autophagy. Cell Physiol Biochem. 2018;49:190–205. [DOI] [PubMed] [Google Scholar]
- 288.Bendlin BB. Antidiabetic therapies and Alzheimer disease. Dialogues Clin Neurosci. 2019;21(1):83–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 289.Yusta B, Baggio LL, Koehler J, Holland D, Cao X, Pinnell LJ. GLP-1R agonists modulate enteric immune responses through the intestinal intraepithelial lymphocyte (IEL) GLP1R. Diabetes. 2015;64:2537–49. [DOI] [PubMed] [Google Scholar]
- 290.Lebrun LJ, Lenaertz K, Kiers D, de Barros JPP, Le Guern N, Plesnik J. Enteroendocrine L cells sense LPS after gut barrier injury to enhance GLP-1 secretion. Cell Rep. 2017;21:1160–8. [DOI] [PubMed] [Google Scholar]
- 291.Yun SP, Kam TL, Panicker N, Kim S, Oh Y, Parl JS. Block of A1 astrocyte conversion by microglia is neuroprotective in models of Parkinson’s disease. Nat Med. 2018;24:931–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Kim DS, Choi HI, Wang Y, Luo Y, Hoffer BJ, Greig NH. A new treatment strategy for Parkinson’s disease through the gut brain axis the glucagon like peptide-1 receptor pathway. Cell Transp. 2017;26:1560–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Wang T, Xu SF, Fan YG, Li LB, Guo C. Iron pathophysiology in Alzheimer’s diseases. Adv Exp Med Biol. 2019;1173:67–104. [DOI] [PubMed] [Google Scholar]
- 294.Killilea DW, Atamna H, Liao C, Ames BN. Iron accumulation during cellular senescence in human fibroblasts in vitro. Anti-oxyd Redox Sign. 2003;5:507–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Bayeva M, Kechaduri A, Puig S, Chang HC, Patial S, Blackshear J. mTOR regulates cellular iron homeostasis through tristetrapolin. Cell Metab. 2012;16:645–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Inoue H, Hanawa N, Katsumata-Tsuboi R, Katsumata SI, Takahashi N, Uehara M. Down-regulation of senescence marker protein 30 by iron-specific chelator deferoxamine drives cell senescence. Biosci Biotechnol Biochem. 2018;82:900–3. [DOI] [PubMed] [Google Scholar]
- 297.Sfera A, Bullock K, Price A, Inderias L, Osorio C. Ferrosenescence of iron age of neurodegeneration? Mech Age Dev. 2018;174:63–75. [DOI] [PubMed] [Google Scholar]
- 298.Drago-Serrano ME, de la Graza-Amaya M, Luna JS, Campos-Rodrigues R. Lactoferin-lipopolysaccharide (LPS) binding as key to antibacterial and antiendotoxin effects. Int Immunopharmacol. 2012;12:1–9. [DOI] [PubMed] [Google Scholar]
- 299.Kruzel ML, Zimecki M, Acor JK. Lactoferrin in a context of inflammation-induced pathology. Front Immunol. 2017;8:1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.van Splunter M, Perdijk O, Fick-Brinkhof H, Feitsma AL, Floris-Vollenbroek EG, Meijer B, Brugman S, Savelkoul HFJ, van Hoffen E, van Neerven RJJ. Bovine lactoferrin enhances TLR7-mediated responses in plasmacytoid dendritic cells in elderly women: results from a nutritional intervention study with bovine lactoferrin, GOS and vitamin D. Front Immunol. 2018;20(9):2677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Bramanti TE, Holt SC. Roles of porphyrins and host iron transport proteins in regulation of growth of Porphyromonas gingivalis W50. J Bacteriol. 1991;173(22):7330–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Grenier D, Chen H, Ben Lagha A, Fournier-Larente J, Morin MP. Dual action of myricetin on Porphyromonas gingivalis and the inflammatory response of host cells: a promising therapeutic molecule for periodontal diseases. PLoS One. 2015;10(6):e0131758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Moon JH, Herr Y, Kim SW, Lee JY. In vitro activity of deferoxamine against Porphyromonas gingivalis. FEMS Microbiol Lett. 2011;323(1):61–7309. [DOI] [PubMed] [Google Scholar]
- 304.Reddy PH, Manczak M, Yin X, Reddy AP. Synergistic protective effects of mitochondrial division inhibitor 1 and mitochondria-targeted small peptide SS31 in Alzheimer’s disease. J Alzheimers Dis. 2018;62(4):1549–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Reddy PH. Mitochondrial medicine for aging and neurodegenerative diseases. NeuroMol Med. 2008;10(4):291–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Reddy PH. Mitochondrial oxidative damage in aging and Alzheimer’s disease: implications for mitochondrially targeted antioxidant therapeutics. J. Biomed. Biotechnol 2006;2006(3):31372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Burgos JS, Ramirez C, Sastre I, Valdivieso F. Effect of apolipoprotein E on the cerebral load of latent herpes simplex virus type 1 DNA. J Virol. 2006;80(11):5383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Burgos JS, Ramirez C, Sastre I, Bullido MJ, Valdivieso F. ApoE4 is more efficient than E3 in brain access by herpes simplex virus type 1. Neuroreport. 2003;14(14):1825–7. [DOI] [PubMed] [Google Scholar]
- 309.Lachenmaier SM, Deli MA, Meissner M, Liesenfeld O. Intracellular transport of Toxoplasma gondii through the blood-brain barrier. J Neuroimmunol. 2011;232(1–2):119–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 310.Kamerkar S, Davis PH. Toxoplasma on the brain: understanding host-pathogen interactions in chronic CNS infection. J Parasitol Res. 2012;2012:589295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.WHO. First WHO ministerial conference on global action against dementia: meeting report. Geneva: World Health Organization; 2015. [Google Scholar]
- 312.Francis PT, et al. The cholinergic hypothesis of Alzheimer’s disease: a review of progress. J Neurol Neurosurg Psychiatry. 1999;66(2):137–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.John Hardy, Selkoe Dennis J. The amyloid hypothesis of Alzheimer’s disease: progress and problems on the road to therapeutics. Science. 2002;297(5580):353. [DOI] [PubMed] [Google Scholar]
- 314.Allal Boutajangout, Thomas Wisniewski. Tau-based therapeutic approaches for Alzheimer’s disease—a mini-review. Gerontology. 2014;60(5):381–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Claudie Hooper, Richard Killick, Simon Lovestone. The gsk3 hypothesis of Alzheimer’s disease. J Neurochem. 2008;104(6):1433–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Spangenberg EE, Green KN. Inflammation in Alzheimer’s disease: lessons learned from microglia-depletion models. Brain Behav Immun. 2017;61:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 317.Golde Todd E, DeKosky Steven T, Galasko Douglas. Alzheimer’s disease: the right drug, the right time. Science. 2018;362(6420):1250. [DOI] [PubMed] [Google Scholar]
- 318.Choi SH, et al. A three-dimensional human neural cell culture model of Alzheimer’s disease. Nature. 2014;515:274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Marton Rebecca M. et al. Differentiation and maturation of oligodendrocytes in human three-dimensional neural cultures. Nat Neurosci. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Christopher Loose, et al. A linguistic model for the rational design of antimicrobial peptides. Nature. 2006;443:867–9. [DOI] [PubMed] [Google Scholar]
- 321.Hiroyuki Ohashi, et al. Next-generation technologies for multiomics approaches including interactome sequencing. Biomed Res Int. 2015;104209:104209. [DOI] [PMC free article] [PubMed] [Google Scholar]