

Abstract
Obesity is a chronic, relapsing condition characterized by excess body fat. Its prevalence has increased globally since the 1970s, and the number of obese and overweight people is now greater than those underweight. Obesity is a multifactorial condition, and as such, many components contribute to its development and pathogenesis. This is the first of three companion reviews that consider obesity. This review focuses on the genetics, viruses, insulin resistance, inflammation, gut microbiome, and circadian rhythms that promote obesity, along with hormones, growth factors, and organs and tissues that control its development. It shows that the regulation of energy balance (intake vs. expenditure) relies on the interplay of a variety of hormones from adipose tissue, gastrointestinal tract, pancreas, liver, and brain. It details how integrating central neurotransmitters and peripheral metabolic signals (e.g., leptin, insulin, ghrelin, peptide YY3-36) is essential for controlling energy homeostasis and feeding behavior. It describes the distinct types of adipocytes and how fat cell development is controlled by hormones and growth factors acting via a variety of receptors, including peroxisome proliferator-activated receptor-gamma, retinoid X, insulin, estrogen, androgen, glucocorticoid, thyroid hormone, liver X, constitutive androstane, pregnane X, farnesoid, and aryl hydrocarbon receptors. Finally, it demonstrates that obesity likely has origins in utero. Understanding these biochemical drivers of adiposity and metabolic dysfunction throughout the life cycle lends plausibility and credence to the “obesogen hypothesis” (i.e., the importance of environmental chemicals that disrupt these receptors to promote adiposity or alter metabolism), elucidated more fully in the two companion reviews.
Graphical Abstract
1. Introduction and secular trends
Obesity is a chronic, relapsing condition characterized by excess body fat [1, 2]. It is among the most critical global health issues and a growing pandemic affecting adults, children, and infants [3–5]. Obesity rates have tripled since the 1970s, and the prevalence of adult obesity in the U.S. increased from 30.5% in 2000 to 42.4% in 2018, a 40% increase in frequency in less than two decades [6] . Currently, there are more obese individuals globally than those who are underweight [7–9].
However, this increase in the prevalence of obesity is not restricted to humans. In 2011, Klimentidis and colleagues [10] reported average mid-life body weights of many animals, including domestic dogs and cats, non-human primates, and rodents, regardless of living conditions (both feral and living in research colonies), have also increased. These changes in animal weights across numerous species suggest that similar environmental changes have impacted both animals and humans to promote obesity. Although changes in human behavior undoubtedly play a role in the manifestation of obesity, specific hormonal and biochemical mechanisms outside of their immediate control likely contribute.
This is the first of three companion reviews focusing on obesity and obesogens. This first review delineates the organs and mechanisms responsible for regulating metabolism and their tight control by hormones and their respective receptors. Disruption of this regulation by hormones or other environmental stimuli can lead to obesity anytime during the life cycle, including prenatally. The companion review will elucidate the chemistry and pathophysiological action of obesogens — environmental chemicals designed for a specific purpose, such as pesticides, flame retardants or plasticizers, but which have side-effects that interfere with hormone action — which can lead to altered metabolism and ultimately to obesity. This second review will also establish the causal link between obesogens and obesity, explicitly providing evidence supporting the “obesogen hypothesis,” and discuss research gaps that must be explored. The third review will focus on direct and indirect assays to detect obesogens.
2. Obesity and disease
2.1. The relationship between obesity and non-communicable disease
Obesity is thought to be unhealthy primarily because it is associated with what are known collectively as non-communicable diseases (NCDs), including hypertension [11] , type 2 diabetes (T2D) [12, 13], dyslipidemia, [14], nonalcoholic fatty liver disease (NAFLD) [15], and cardiovascular disease (CVD) [16]. Currently, 54% of adults greater than 60 years of age manifest one or more of these diseases [17]. Obesity is also associated with increased prevalence of and mortality from at least thirteen types of cancers, including stomach, colon, liver, pancreas, breast, prostate, and renal cancer [18]. Obesity is also associated with increased rates of dementia [19]. Corresponding with the increasing prevalence of obesity, the prevalence and severity of all these diseases have increased over a 50-year time frame [20], resulting in increased mortality [21] and healthcare costs [22]. Perhaps even more concerning is that these diseases are now noted in 19-35% of youth and increasing prevalence [23–25].
2.2. Relationship between obesity and communicable diseases
Obesity is also associated with increased susceptibility to nosocomial infections, wound infections, and influenza pandemics [26, 27]. The health effects of obesity include increased risk of COVID-19-related deaths, intensive care admission, and hospitalization of 48, 74, and 113%, respectively [28].
2.3. Is obesity always unhealthy?
Obesity, however, is not always associated with increased morbidity or mortality. Despite being obese, 20% of obese individuals appear metabolically healthy [29–32]. However, it is uncertain if these individuals are healthy or have fewer metabolic abnormalities than those with metabolically unhealthy obesity [33, 34]. Indeed, prospective studies suggest that some obese individuals characterized as metabolically healthy are at increased risk of developing metabolic disease [35]. Conversely, up to 40% of normal-weight adults harbor metabolic perturbations similar to those in obesity, including T2D, NAFLD, and CVD [36, 37]. 87.8% of adults exhibit metabolic dysfunction, while 73.6% are overweight or obese [38]; thus, a sizable portion of normal-weight individuals also show metabolic dysfunction. Abdominal computed tomography (CT) evidence demonstrates that normal-weight individuals can manifest increased visceral fat; that is, they are “thin on the outside, fat on the inside” [39].
The dissociation of obesity and metabolic dysfunction becomes readily apparent by contrasting two pathophysiologic hormonal conditions. The “Little Women of Loja” are a founder-effect cohort in Ecuador of patients with growth hormone insensitivity due to a growth hormone receptor mutation. They become markedly obese in adulthood yet are protected from chronic metabolic diseases such as T2D and CVD. Despite their obesity, they manifest low serum insulin and triglyceride concentrations, reflecting improved insulin sensitivity and metabolic health [40]. Conversely, patients with either genetic or acquired lipodystrophy have a paucity of subcutaneous and visceral fat due to the dysfunctional expression of adipose tissue. They are leptin-deficient yet manifest extreme hyperglycemia, hyperinsulinemia, fatty liver disease, and other aspects of metabolic disease [41]. These two disease entities suggest that the linkage between body fat and metabolic function is not linear but significantly more complex than previously conceived.
2.4. Body mass index (BMI) is not the best measure of metabolic perturbation
The most common metric of adiposity, BMI [42], highlights the problem in differentiating between obesity and metabolic disease. For instance, while obesity prevalence (based on BMI) and diabetes prevalence correlate, they are not concordant. There are countries that are obese without being diabetic, such as Iceland, Mongolia, and Micronesia. At the same time, there are countries that are diabetic without being obese, such as India, Pakistan, and China [43]. This is further elaborated looking at years of life lost from diabetes vs. obesity [44].
Furthermore, different populations manifest variations in types of adipose tissue, skeletal muscle (e.g., body-builders, elite athletes), other lean-body mass components (e.g., bone), as well as significant racial/ethnic differences in body composition that ultimately portend different health risks in different populations [45]. Individuals can have a normal BMI but still be metabolically unhealthy, e.g., South-East Asians [46]. Thus BMI, while helpful because it is easy to apply and correlates with morbidity and mortality in large population studies [47], is an inadequate measure of metabolic perturbation in individuals and some populations. For any given BMI individuals may be insulin sensitive or insulin resistant [48].
In contrast, waist circumference may be a better measure of adiposity as it considers the metabolically deleterious ectopic fat mass that correlates with metabolic disorders and insulin resistance [46] [13]. Another non-invasive approach to differentiating fat mass from lean mass is air displacement plethysmography [49]. Beyond these and other techniques [39], it may be helpful to identify and use biomarkers specific for metabolically unhealthy obesity.
2.5. Three fat depots
A possible key to the challenges imposed by using BMI as a clinical “vital sign” may be the existence of three separate fat depots within the body with divergent effects on the development of metabolic disease [50]. These fat depots respond differently to the various differentiation signals and therefore may be differentially vulnerable to the multiple stressors that can alter these signals.
2.5.1. Subcutaneous adipose tissue (SAT)
The largest fat depot is subcutaneous adipose tissue (SAT). SAT drains into the systemic circulation; therefore, increased SAT-derived cytokines reaching the liver will be diluted compared with cytokines derived from visceral fat depots that drain into the portal circulation. Excess SAT will increase cytokine levels [51] and predispose to metabolic disease; however, a relatively large pannus (approximately 10 kg) is necessary before metabolic dysfunction is apparent. This can be shown in four ways. First, patients with lipedema (excess subcutaneous fat in the abdomen and thighs, due to a lymphatic disorder) have increased subcutaneous fat yet maintain metabolic health [52]. Second, the paucity of SAT impairs glucose metabolism and increases the risk of heart disease, hypertension, stroke, and diabetes in patients with lipodystrophy [53]. Third, studies of liposuction of SAT manifest no metabolic improvement [54]. Fourth, thiazolidinediones (TZD’s) improve insulin sensitivity and metabolic health at the expense of increasing SAT deposition and weight gain [55]. Thus, SAT tends to confer less metabolic morbidity and, in many cases, may even be protective against chronic metabolic disease [32].
2.5.2. Visceral adipose tissue (VAT)
In comparison, visceral adipose tissue (VAT), present mainly in the mesentery and omentum, is more metabolically active, more cellular, vascular, and innervated than subcutaneous fat, and it drains into the portal circulation [50, 56]. A mere two kg of excess VAT will increase portal vein cytokine levels, increasing hepatic insulin resistance leading to a higher risk of T2D [57]. One hypothesis to explain the insulin resistance caused by VAT is the “portal-visceral” paradigm [58] which states that increased adiposity leads to increased portal and systemic free fatty acid (FFA) flux. Recently an alternative theory has been proposed: “ectopic lipid deposition” [59], based on the fact that lipid content in the liver and/or muscle is a strong predictor of insulin resistance [60].
2.5.3. Ectopic (liver and muscle) fat
In contrast to lipid stored in WAT, which is, for the most part, adaptive, ectopic lipid in the liver and muscle is non-adaptive and associated with metabolic dysfunction. Liver fat is the most harmful to metabolic health [61]. It predicts whether people will develop T2D or CVD [62, 63]. It causes insulin resistance locally and directly; only 0.25 kg of liver fat accumulation is required to increase hepatic insulin resistance in children [64], which can even occur in normal-weight people [15]. Excess ectopic fat can lead to NAFLD, leading to cirrhosis or liver cancer. Children with fatty liver disease (FLD) also have excess pancreatic fat, impairing insulin secretion [65]. Dietary sugar and branched-chain amino acids (arising from de novo lipogenesis (DNL)) increase liver fat. Therefore, liver fat and VAT, primarily determine insulin resistance, and better predict metabolic dysfunction than SAT or total body fat.
These adipose tissue depots are sexually dimorphic; on average, men have more visceral fat, women have larger subcutaneous fat stores [66]. Given the relative impact of these fat depots on metabolic health, this sexual dimorphism may explain sex differences in metabolic disease risk until menopause, when the reduction in estrogen results in increasing LDL, triglycerides, visceral fat, and morbidity and mortality in females [67, 68].
3. Adipose tissue development and function
Adipose tissue grows by increased cell number and size during in utero life, childhood, and adolescence; after that, the number of adipocytes remains stable if weight remains stable. In humans, adipose tissue appears between the 14th and 24th week of gestation, and in mice, adipose tissue is apparent between postnatal days one and seven [69]. Adipose tissue depots are highly dynamic and plastic organs. Changes resulting from weight gain or loss involve coordinating multiple cell types, including immune cells, endothelial cells, fibroblasts, and neurons [69]. An increase in adipose cell number (hyperplasia) is protective against unhealthy obesity as it results in less inflammation. An increased adipocyte size (hypertrophy) is a marker of metabolic risk in obesity, with coordinated increases in inflammation [70]. A decrease in body weight will only change fat cell size, while weight gain can increase adipocyte size and number [71]. Adipocyte turnover rate is similar for all weight categories, with about 10% of adipocytes renewed annually, i.e., the average life of an adipocyte is about ten years [72].
Adipocytes derive from a common mesenchymal stem cell (MSC) precursor-like muscle and bone. Various transcription factors expressed during fetal life commit these precursors to alternative lineages. Myostatin and glucocorticoids (see below) are essential factors that inhibit muscle and promote adipocyte differentiation [73]. Furthermore, there are distinct types of adipocytes: white, brown, beige, and pink [74]. Specific cell surface markers distinguish between white, brown, and beige adipocytes [75], resulting in distinct functional roles. Pink adipocytes form during pregnancy, lactation and post-lactation from subcutaneous white adipocytes, which transform into milk-producing glands in breast tissue [76].
3.1. White adipose tissue (WAT)
White adipose tissue (WAT) is adapted explicitly for fat storage. White adipocytes have an expansive fat vacuole with a perilipin border and very few mitochondria, hence their white color. These cells are found in fat depots throughout the body; more white adipocytes provide greater fat storage and less likelihood of metabolic dysfunction, depending on their location. Liver and muscle cells can also store fat, but they do not have fat vacuoles under normal physiological conditions. WAT is an endocrine organ that secretes over 50 different hormones, growth factors, enzymes (e.g., dipeptidyl peptidase 4), and cytokines, including adiponectin, leptin, resistin, visfatin, apelin, bone morphogenic protein 2, endotropin, angiopoietins, adipokines, and noncoding RNAs sometimes found in extracellular vesicles [77]. These endocrine signals play a critical role in mediating the role of WAT in metabolic regulation. The characteristics of healthy WAT include smaller, more numerous adipocytes, adiponectin secretion, insulin sensitivity, low basal lipolysis, high vascularity, normal oxidative capacity of mitochondria, low macrophage infiltration, macrophages with an anti-inflammatory profile and a subcutaneous location [78, 79]. The characteristics of WAT in unhealthy obesity include increased secretion of leptin and resistin, decreased secretion of adiponectin, adipocyte hypertrophy, decreased insulin sensitivity, increased basal lipolysis, decreased vascular density, increased macrophage infiltration, pro-inflammatory macrophage profile, and predominantly visceral location [80].
3.2. Brown adipose tissue (BAT)
Brown adipose tissue (BAT) is composed of adipocytes designed for thermogenesis [81]. They are enriched in the interscapular, axillary, cervical, perirenal and periaortic areas. In humans, they are especially abundant in newborns, protecting against cold exposure. Brown adipocytes have little capacity for fat deposition, are highly vascularized, and are filled with mitochondria, hence their brown color. BAT dissipates chemical energy to produce heat either as a defense against cold [82] or as energy expenditure to compensate for food intake. They express high uncoupling protein-1 (UCP-1) levels, which dissociates energy utilization from ATP production to generate heat to maintain body temperature in cold environments [83]. They are the primary site for sympathetic (adrenergic)-mediated adaptive thermogenesis through stimulation of β3-adrenergic receptors. This leads to an increase in peroxisome proliferation activated receptor (PPARγ) coactivator-1α and thyroid hormone leading to mitochondrial biogenesis [84, 85]. In humans, BAT derives from a myogenic factor 5 (myf5) positive cell lineage, a muscle mesenchymal cell origin, the same as that of myocytes [86]. In animals, brown adipocytes derives from the mesodermal germ layer [87]. Thus far, efforts to transform WAT into BAT to improve insulin sensitivity have not been successful.
3.3. Beige adipose tissue
Beige adipocytes are distinguished from white adipocytes by their multicolor lipid droplets. They develop in subcutaneous adipose tissue from a distinct set of adipocyte precursor cells that are distinct from SAT [88] that emerge from a myf5 negative lineage or via the transdifferentiation of existing white adipocytes [89] and take up residence in SAT [90, 91]. In response to persistent sympathetic stimulation, e.g., prolonged cold exposure or exercise, white adipocytes can increase their mitochondria number and UCP-1 expression, thereby becoming beige adipocytes [92]. The appearance of beige adipocytes within WAT does appear to confer improvements in metabolic health [93].
4. Receptors involved in the control of energy metabolism
MSCs differentiate into mature white adipocytes (WAT) under the control of various cellular receptors, which act as transcription factors (Table 1) to control four physiological processes: proliferation, mitotic cloning, early differentiation, and terminal differentiation. Other liver-based receptors alter liver metabolism to direct energy disposition into adipose tissue for storage [94]. Lastly, systemic hormones that bind to their receptors in the liver, brain, and adipose tissue alter food intake versus energy expenditure or storage (Table 2). This section describes these receptors involved in controlling energy storage vs. expenditure and their role in obesity.
Table 1:
Transcription factors involved in obesity and obesogen action
| Adipose Tissue Transcription Factors | Abbreviation | Agonists promote: |
|---|---|---|
| Peroxisome proliferator-activated receptor gamma (heterodimer with RXR) | PPARγ | Increased adiposity, improved insulin sensitivity |
| Peroxisome proliferator-activated receptor alpha | PPARα | Decreased serum triglycerides, LDL cholesterol, increased HDL cholesterol, protection against diet-induced obesity |
| Peroxisome proliferator-activated receptor beta/delta | PPARβ/δ | Decreased LDL cholesterol, insulin, increased HDL cholesterol, protection against diet-induced obesity |
| Retinoid X receptor (heterodimer with PPARγ) | RXR | Improved insulin sensitivity, reduced hyperglycemia, hypertriglyceridemia, hyperinsulinemia, reduced weight gain and food intake |
| Liver Transcription Factors | ||
| Liver X receptor | LXR | Decreased visceral adiposity, increased subcutaneous adiposity, increased cholesterol, increased plasma and hepatic triglycerides |
| Pregnane X receptor | PXR | Enhanced hepatic lipid accumulation, fatty liver disease, increased hyperglycemia |
| Constitutive androstane receptor | CAR | Improved insulin sensitivity, glucose and lipid metabolism, protection against diet-induced obesity, decreased glyuose levels |
| Farnesoid X receptor | FXR | Increased adiposity, improved insulin resistance, increased glucose intolerance, reduced hepatic lipid accumulation |
| Aryl hydrocarbon receptor | AhR | Hepatic lipid accumulation, insulin resistance |
Table 2:
Systemic hormone receptors and their role in obesity and obesogen action
| Hormone receptor | Abbreviation | Agonists promote: |
|---|---|---|
| Insulin receptor | IR | Increased adiposity |
| Estrogen receptors | ER (α, β) | Increased adiposity |
| Androgen receptor | AR | Decreased adiposity, lipid accumulation |
| Glucocorticoid receptor | GR | Increased adiposity |
| Thyroid hormone receptors | TR (α, β) | α- increased adiposity, increased cholesterol, triglycerides β- reduction in cholesterol, triglycerides, and adiposity |
4.1. Adipose tissue transcription factors
4.1.1. Peroxisome Proliferator-Activated Receptors (PPARs)
Human adipose tissue differentiation is dependent on the actions of PPARγ [95], integral to adipose tissue differentiation and widely considered the master regulator of adipogenesis. Treatment of pre-adipocytes and/or MSCs with PPARγ agonists robustly promotes triglyceride accumulation [96, 97]. Adipogenesis is comprised of two phases: the determination phase and terminal differentiation. The determination phase of adipogenesis transitioning to the terminal differentiation phase is marked by changes in expression levels of the PPARγ isoforms PPARγ1 and PPARγ2 [98]. PPARγ operates as a physiological lipid sensor activated by the combined concentration of weakly activating fatty acids and eicosanoids. In its absence, adipose tissue is sparse and poorly regulated [99]. PPARγ has a large promiscuous ligand-binding pocket that creates a vulnerable access point for multiple obesogenic ligands to enter the adipocyte life cycle at various stages. To establish the necessity of this pathway to adipogenesis, Rosen et al. demonstrated that PPARγ-null cells do not generate adipocytes; cells lacking both copies of PPARγ could not be induced to differentiate, cells with one copy of PPARγ exhibited an intermediate degree of differentiation, and wild-type cells with both copies exhibited robust differentiation and efficacious expression of adipocyte-specific molecular markers [100].
The other PPAR isoforms, α and β/δ, have received less attention, though also have important roles in adipogenesis and obesity. Brun et al. reported that receptor isoform-specific activation failed to induce adipogenesis and triglyceride accumulation for PPARβ/δ, but did for PPARα, if with lower magnitude and potency than activation of PPARγ [101]. Activation of both α and β/δ isoforms in mice promotes anti-obesogenic effects, with improved lipids, triglycerides, and resistance to diet induced obesity [102–106]. In humans, treatment with PPARα agonists decreases serum triglycerides, LDL cholesterol, and increases HDL cholesterol [107, 108]. Human therapeutic testing in humans is limited, though treatment of monkeys with PPARβ/δ agonists lowered LDL cholesterol, triglycerides, insulin, and increased HDL cholesterol [109].
4.1.2. Retinoid X receptor (RXR)
RXR functions as a heterodimer with PPARγ, supporting a key role for RXR in adipogenesis [100]. Specific activation of RXR can promote both adipogenic differentiation and preadipocyte proliferation [110–112]. RXR heterodimers during the early stages of differentiation, transitioning to majority occupancy by PPARγ: RXR heterodimers in the later stages of differentiation [113]. Recent research described RXR activation as an essential signal for adipocyte cell lineage commitment in MSCs and promoting subsequent differentiation [114]. Adipocyte differentiation induced via RXR activation appears to create a functionally distinct adipocyte from those generated via PPARγ [115]. These RXR-induced white adipocytes demonstrated decreased glucose uptake and adiponectin expression. They failed to induce adipocyte browning pathways, suggesting a dysfunctional white adipose that promotes or exacerbates obesity and/or diabetes risk [115]. RXR global knock-out mouse models are embryonic lethal [116], though tissue-specific knockouts provide some context for the role of the receptor in metabolic health. Mice with ablated adipocyte-specific RXR receptors are resistant to diet and chemical-induced obesity have inhibited pre-adipocyte differentiation and inhibited lipolysis during fasting [117]. Exposure of rodents to RXR agonists can sensitize diabetic and obese mice to insulin [118] and reduce hyperglycemia, hypertriglyceridemia, hyperinsulinemia, weight gain, and food intake [118–120]. In humans, treatment with RXR agonists increases plasma triglycerides and decreases thyroid hormone [121–123].
4.2. Liver transcription factors
4.2.1. Liver X receptor (LXR)
The liver X receptor (LXR) forms a permissive heterodimer with RXR, and ligands can activate either receptor [124]. The α-isoform of the receptor is primarily expressed in the adipose, liver, intestine, and kidney, while the β-isoform is ubiquitously expressed [124]. LXRs mediate cholesterol transport, degradation, uptake, and fatty acid and triglyceride synthesis [124–126]. LXR agonism has been reported to increase triglyceride accumulation, differentiation of adipocytes, stimulation of adipogenic gene expression, and pre-adipocyte proliferation in a variety of cell and animal models [110, 127, 128], mediated at least in part via PPARγ [127, 128]. Knockdown experiments in rodents have described a primary role for LXRα in lipolysis [129] and LXRβ in cholesterol regulation [130]. LXR-null mice (both isoforms) have inhibited lipid metabolism, elevated thyroid hormone production, and are resistant to diet-induced obesity [126]; while LXRβ-knockout mice have reduced adiposity, are glucose-intolerant and accumulate lipids in pancreatic islets [130]. Adipocytes are reduced in size in LXR-deficient mice [128], with increased energy expenditure and reduced lipid accumulation in the brown adipose [131]. Mice treated with LXR agonists decreased visceral adiposity and increased SAT [132], reduced energy expenditure, and increased triglyceride accumulation in BAT [131]. LXR expression is increased in obese humans, and receptor polymorphisms are associated with increased metabolic health risks [133]. Despite some beneficial effects on adipose distribution, treatment with LXR agonists in humans resulted in increased cholesterol, plasma and hepatic triglycerides, and other negative metabolic markers [134].
4.2.2. Pregnane X receptor ((PXR) and constitutive androstane receptor (CAR)
The pregnane X receptor (PXR) and the constitutive androstane receptor (CAR) are closely related liver-enriched receptors that regulate xenobiotic-metabolizing enzymes, glucose and energy homeostasis, immune function, and lipid metabolism [135, 136]. PXR appears to act indirectly via PPARγ [136], and CAR may promote effects via PPARα [137]. PXR knockouts exhibit inhibition of diet-induced obesity, insulin resistance, and fatty liver disease in rodent models [136, 138]. PXR agonists induce hepatic triglyceride accumulation and promote fatty liver disease [139]. Treatment with CAR agonists enhances insulin sensitivity, improves glucose and lipid metabolism, and protects against diet-induced obesity [135, 140]. In humans, CAR activation via phenobarbital decreases glucose levels. It improves insulin sensitivity in diabetes and people with liver hyperplasia and cancer [135]. PXR activation via rifampicin, statins, and other pharmaceuticals appears to induce hyperglycemia and increase the risk of T2D [138].
4.2,3. Farnesoid X receptor (FXR)
The farnesoid X receptor (FXR) regulates bile acid synthesis, enterohepatic circulation, lipid metabolism, and other bile acid-associated receptors [141, 142]. It is expressed in mature adipocytes and differentiated 3T3-L1 cells [143]. FXR agonists increase adipocyte differentiation and enhance insulin-associated signaling, whereas FXR antagonists inhibit these processes [143, 144]. FXR antagonists have also been described as anti-adipogenic and pro-apoptotic [145]. FXR-knockout mice have decreased adiposity, reduced leptin, elevated free fatty acids, increased energy expenditure [144, 146–148]. These animals also have impaired insulin resistance and glucose tolerance [144, 149]. FXR agonist treatment in diet-induced obese mice exacerbated weight gain and glucose intolerance [150]. However, other researchers have reported beneficial metabolic effects of agonist treatment [142, 151]. In humans, FXR expression is downregulated in obese individuals [141, 152]. Treatment with FXR agonists reduces hepatic lipid accumulation, increases glucose uptake, reduces high-density lipoprotein (HDL), increases low-density lipoprotein (LDL) cholesterol, and reduces insulin resistance [141, 142, 151].
4.2.4. Aryl hydrocarbon receptor (AhR)
The aryl hydrocarbon receptor (AhR) is a member of the basic-helix-loop-helix (bHLH)–PER-ARNT-SIM (PAS) superfamily of transcriptional factors [153]. Like other members of the PAS superfamily, the AhR transduces signals in response to the environment, including changes in oxygen levels, altered redox potential, and changes in circadian rhythm, by binding various small-molecule ligands [153]. AhR signaling in metabolism deregulation is exemplified in diet- and obesogen-induced obesity. AhR inhibition prevented and reversed obesity, making it an attractive target for the control of obesity. Indeed, MSCs contain AhR receptors and their activation by kynurenine (Kyn) or benzo[a] pyrene (BaP) (AhR agonists) inhibit terminal adipogenic differentiation [154]. AhR levels and activity are most significant in the liver’s hepatocytes [155, 156]. The majority of fat metabolism and cholesterol biosynthesis occurs in the liver [157], much of which is dependent on AhR signaling [158, 159]. Thus, the contention that the liver is the site where AhR signaling operates in the manifestation of obesity is not surprising and is supported by recent studies showing that the conditional/inducible knockout of the AhR gene in hepatocytes caused obesity resistance in female mice fed a HFD [160].
4.3. Systemic hormone receptors
4.3.1. Insulin receptor (IR)
The insulin receptor (IR) and signaling cascade are essential to fat cell differentiation. For instance, lack of fetal pancreatic insulin secretion during the third trimester results in Transient Diabetes Mellitus of the Newborn, characterized by a lack of adipose tissue in all fat depots [161]. Similarly, leprechaunism, a genetic defect of the IR, leads to a virtual absence of neonatal adipose tissue and extreme insulin resistance [162]. Insulin can stimulate both the insulin receptor and the insulin growth factor-1 receptor (IGF-1R) to induce adipogenesis; however, knockout of IGF-1R only lost 25% of its WAT, while knockout of the insulin receptor resulted in a 95% loss of WAT, with a concomitant 50% increase in BAT [163]. These effects are enacted through insulin’s mediation of Forkhead Box O (FOXO) transcription factors in various fat-specific depots [164].
4.3.2. Estrogen receptor (ER)
Estrogen acts via both membrane and nuclear (ERα and ERβ) receptors [165, 166] Signaling through the nuclear estrogen receptor (ER) is an important factor in SAT development, particularly at times when CNS leptin signaling for reproductive competency becomes necessary (e.g., puberty, pregnancy) [167]. Indeed, estrogen deficiency during gestation, as seen in aromatase deficiency, results in reduced SAT and the appearance of liver fat [155, 156], both of which can be abrogated by estrogen replacement [157] suggesting that these two fat depots are malleable postnatally. Furthermore, estrogen is positively associated with obesity in children across the sexes [168], starting at puberty. In humans, decreased estrogen levels at menopause are associated with increased abdominal obesity. Estrogen replacement therapy may reduce this weight gain [169]. Estrogens act on adipocytes to inhibit lipogenesis and help modulate energy expenditure and food consumption [170]. Studies have described a role for estrogens in promoting preadipocyte proliferation and regulating adipocyte number in various depots [171, 172], presumably mediated by IGF-1R and PPARγ [171]. Some additional insight can be gained through examination of estrogen receptor knock-out (ERKO) mice, which supports differential roles for estrogen receptors alpha and beta. αERKO mice exhibit increased adipose tissue, fat pad weights, adipocyte size, adipocyte numbers, and dysregulated lipoprotein profiles relative to wild type control animals [173, 174], and over time develop both impaired glucose tolerance and insulin resistance [173]. This is mirrored by studies demonstrating that ovariectomized female mice also exhibit increased adiposity, primarily in the abdominal depot [175], and aromatase deficient (ArKO) mice have significantly elevated adipose and fat pad weights as well as hyperplasia and hypertrophy of adipocytes, reduced physical activity levels, reduced glucose oxidation, and decreased lean body mass [176–178]. There are also data describing inhibitory effects of estrogens on adipocyte differentiation [179], possibly mediated through the G-protein-coupled ER (GPER) rather than the nuclear ER [180].
4.3.3. Androgen receptor (AR)
Androgens also play a crucial role in adipogenesis and adipose tissue development. Androgens are generally considered anti-obesogenic, with decreased androgens associated with increased adiposity [181, 182]. Dihydrotestosterone inhibits triglyceride accumulation and adipogenic gene expression in human MSCs and preadipocytes from various adipose depots [183]; co-treatment with anti-androgens inhibited these effects. Neither had any apparent impact on preadipocyte proliferation [183]. Treatment with flutamide, a non-steroidal anti-androgen, has been demonstrated to promote triglyceride accumulation in 3T3-L1 and OP9 preadipocytes [110], again without impacting preadipocyte proliferation. Other research has suggested that some of these effects may occur through inhibition of adipocyte lineage commitment [184, 185]. In agreement with the data indicating that androgens are anti-obesogenic, androgen receptor knock-out (ARKO) mice have increased adiponectin secretion, increased adiposity and body weights, increased triglycerides, and decreased insulin sensitivity [186–190]. Androgen replacement, as expected, protects against these metabolic effects, including obesity [188]. Androgen treatment inhibits adipogenesis in adipose tissue samples collected from both sexes [191] and reduces human fat mass [192]. Anti-androgens such as flutamide can improve lipid profiles in women [193, 194]. In men, they are generally associated with a favorable metabolic phenotype and reduced adiposity [195]. There are sex-specific effects of androgens [196], as demonstrated by elevated androgens and excess adiposity in women with polycystic ovary syndrome (characterized by excess androgens) and increased obesity, hypertension, dyslipidemia, insulin resistance, and metabolic dysfunction in men with hypogonadism, e.g., low testosterone [192, 196].
4.3.4. Glucocorticoid receptor (GR)
The glucocorticoid receptor (GR), and its related signaling cascade, are primary drivers of visceral fat differentiation [197] and metabolic dysfunction [198]. VAT is differentially responsive to glucocorticoids compared to SAT [199]). Cushing’s syndrome, a disease characterized by high glucocorticoid levels, exhibits excessive visceral fat [200]. Furthermore, VAT possesses high levels of 11β-hydroxysteroid dehydrogenase type 1, which converts inactive cortisone to active cortisol, increasing visceral fat differentiation and growth [201]. Extensive evidence supports the role of GR in lipid metabolism, adipose formation, and maintenance [202]. GR agonist treatments (typically dexamethasone) promote triglyceride (TG) accumulation and pre-adipocyte proliferation in diverse mesenchymal and pre-adipocyte models [110, 203], while GR antagonists inhibit differentiation [204]. Some studies have reported mediation of effects of GR on adipocyte differentiation in part through PPARγ [205]; however, other studies have not confirmed these results [206]. Silencing GR inhibits cortisol’s adipogenic effects and decreases leptin and adiponectin [207], suggesting specificity to GR. GR activation may be necessary for an intermediate commitment state before differentiation of preadipocytes [208], though it could also be due to varied receptor expression in this model [110]. Although tissue-specific knockouts have been developed, GR knock-out (GRKO) mice are perinatally lethal due to respiratory failure [209]. Local GR knock-out promoted a favorable metabolic profile via 11(-hydroxysteroid dehydrogenase overexpression. It reduced fat accumulation and food intake, improved insulin sensitivity and glucose tolerance, and increased energy expenditure [210].
4.3.5. Thyroid hormone receptor (TR)
The thyroid hormone receptor (TR) is essential for metabolic health regulation [211], mainly via maintaining lipid and carbohydrate metabolism, blood pressure, and muscle mass [124, 212]. Receptor isoforms (α, β1, β2) play distinct roles in metabolic health. TRα primarily regulates thermogenesis, while TRβ primarily regulates cholesterol metabolism and lipogenesis [212]. TRβ also governs several genes and enzymes essential for pre-adipocyte proliferation and adipocyte differentiation, either directly or via PPARγ [212]. TR agonist treatments are pro-adipogenic [110, 213, 214]. TRα-null mice are leaner than wild-type counterparts, with reduced adiposity, higher energy expenditure, increased oxidative capacity of fat, and resistance to diet-induced obesity [215]. Mice with a point mutation in TRα showed increased visceral adiposity, serum leptin, fasting insulin, fasting glucose, and an inhibited cold-induced adaptive thermogenesis [216]. TRβ-null mice demonstrated an increased energy expenditure [217], with no reported effects on adiposity. Conversely, synthetic TRβ agonists cause a reduction in cholesterol, triglycerides, and adiposity [218, 219], suggesting a potential therapeutic mechanism. In humans, low thyroxine (T4) and triiodothyronine (T3) along with high thyroid-stimulating hormone (TSH) levels result in hypothyroidism that is characterized by weight gain, that can be treated successfully with supplementation [220–222]. High T3 and T4 and low TSH characterizing hyperthyroidism are associated with reduced weight, fat-free mass, and adiposity [223, 224].
These cellular systems, which play a critical role in the differentiation and growth of fat cells and weight control, can be targets of various exogenous obesogens, as delineated in the companion review. They can promote energy deposition and either primary (via liver dysfunction) or secondary (via obesity) chronic metabolic disease when activated or inhibited.
5. Neuroendocrinology of obesity
5.1. Homeostatic mechanisms
Regulation of energy balance, and hence weight status, relies on integrating peripheral hunger and satiety signals by the central nervous system (CNS) controlling feeding behavior and activity [225, 226]. Feeding behavior is often dichotomized as homeostatic or hedonic [227]. Homeostatic feeding is defined as that which is required to meet physiological/survival needs and is based on increasing the motivation to eat when energy stores are depleted. In contrast, hedonic feeding is driven by sensory perception or pleasure. The hedonic drive may override homeostatic regulation even in the face of abundant energy stores via increased desire to consume highly palatable foods [227, 228].
Key CNS regions involved in the homeostatic pathway include the neuroendocrine hypothalamus and the nucleus of the solitary tract (NST) in the lower brainstem (Figure 1). There are two distinct populations of neurons within the arcuate nucleus (ARC) of the hypothalamus. One population expresses the anorexigenic neuropeptide precursor pro-opiomelanocortin (POMC), while the other population expresses the orexigenic neuropeptides neuropeptide Y (NPY) and agouti-related protein (AgRP). These neurons, in turn, project to other hypothalamic nuclei, including the paraventricular nucleus (PVN), which integrate emotional and stress responses as well as exert physiological control over metabolism via the release of thyrotropin-releasing hormone (TRH) and corticotropin-releasing hormone (CRH) [229]. Serotonergic neurons from the dorsal raphe nucleus (DRN), which receive afferent input from the spinal column and many parts of the brainstem, project to the ARC, NTS and paraventricular nucleus (PVN). Depleting CNS serotonin results in hyperphagia and obesity, while elevated CNS serotonin resulted in anorexia and decreased energy intake [230, 231].
Figure 1. Endocrine control of Metabolism.
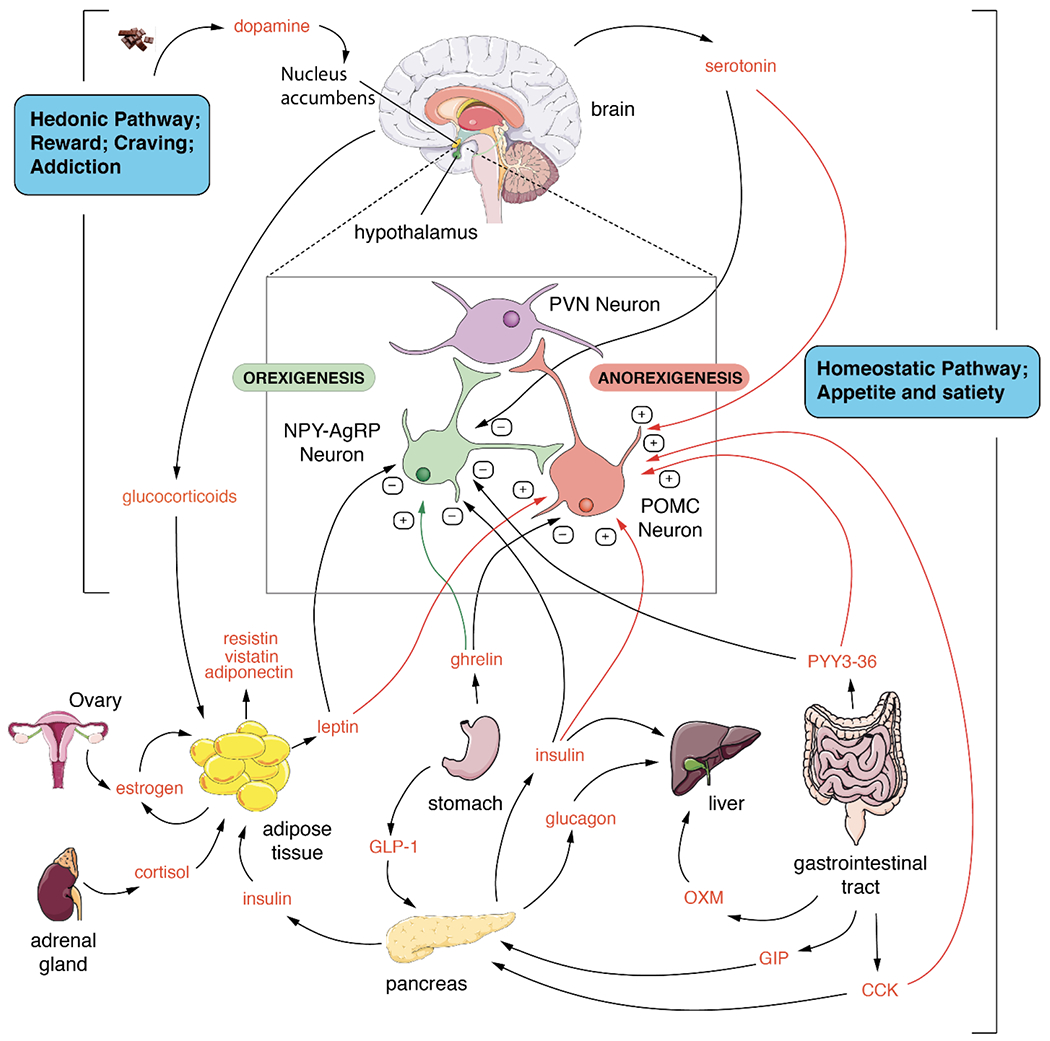
The main tissues involved in controlling metabolism are shown along with the main endocrine and paracrine modulators of metabolic function. Many positive and negative control points in the tissues and the brain are integrated to control metabolism and body weight. The homeostatic pathway is the largest and most complex and likely regulates the set point for metabolism via control of the appetite and satiety centers in the brain. The hedonic center controls emotional eating and food addiction which can override the homeostatic system.
The integration of central neurotransmitters and peripheral metabolic signals to control energy homeostasis are well known [232] and they exert central effects that alter food-seeking behavior and, ultimately obesity. Key peripheral hormones involved in energy homeostasis include but are not limited to leptin, ghrelin, insulin, peptide YY (PYY3-36) and cholecystokinin (CCK) [231, 233]. Leptin is synthesized by adipose tissue and is secreted in proportion to fat mass; insulin is secreted by pancreatic β-cells in response to elevated blood glucose levels, and PYY3-36 is produced by enteroendocrine L-cells of the gut and released into the circulation postprandially. Hence increased leptin, insulin and PYY3-36 levels represent the fed state and/or a surfeit of stored energy. These three hormones bind receptors in POMC neurons, stimulating POMC release and bind receptors in NPY neurons that suppress NPY/AgRP signaling with a resultant decrease in orexigenic tone. The gonadal hormone estradiol also targets POMC neurons in the ARC, indirectly repressing the synthesis of NPY and AgRP and thereby reducing caloric intake [234]. Oxyntomodulin (OXM) and glucagon-like peptide-1 (GLP-1) are also produced by enteroendocrine L-cells of the gut and are released postprandially. GLP-1 and OXM inhibit appetite: OXM may also influence energy balance via increased activity-related energy expenditure. CCK released post-prandially from the small intestine reduces food intake via binding CCK1 receptors on the afferent vagal nerve, resulting in POMC in the NST [235].
Ghrelin, dubbed “the hunger hormone,” is a 28 amino acid peptide hormone produced by the stomach [236]. Acylation of ghrelin with an octanoyl group at serine 3 is necessary for this hormone to cross the blood-brain barrier. Ghrelin production rises in response to fasting and falls post-prandially [236]. In contrast to leptin, insulin and PYY3-36, ghrelin stimulates the production of NPY/AgRP neuronal activity leading to inhibition of POMC neuronal signaling, resulting in increased food intake.
5.2. Hedonic mechanisms
The neurological substrates and circuits involved in binge eating and food addiction have been previously reviewed [237]. It has been proposed that binge eating and addiction interplay between the dopaminergic reward centers (nucleus accumbens (NA), amygdala, ventral tegmental area (VTA)) and the prefrontal impulsivity-control networks (prefrontal cortex, the anterior cingulate cortex, and the insular cortex). Binge eating disorders are thought to be due to a transition from reward- or goal-directed drive to consume food to an impulsive drive to consume food that involves changes in how the ventral and dorsal striatum communicate. This change may be due to the expression of two different receptors for the neurotransmitter dopamine in the striatum. Eventually, this change decreases reward sensitivity (less dopamine release) and is associated with decreased inhibition of reward from the impulsivity network (prefrontal cortex, etc.).
Highly palatable foods increase the release of dopamine in the NA [238]. The mesolimbic dopamine pathway, which demonstrates increased dopamine signaling from nuclei in the VTA onto neurons in the NA in response to common drugs of abuse, is thought to drive hedonic feeding [239]. Cellular and molecular changes like those associated with prolonged limbic system activation by drugs of abuse are also seen in rodents chronically exposed to highly palatable foods [240, 241]. Compared to lean women, obese women have increased activation of several limbic areas when presented with pictures of highly palatable foods [228]. Hence alterations in reward pathways may promote obesity when exposed to highly palatable foods, and/or chronic exposure to highly palatable foods may result in alterations in reward pathways that activate the hedonic feeding drive. Both the homeostatic and hedonic pathways control obesity. The hormones and neurotransmitters that control them are subject to modifications that can lead to obesity via exposures to obesogens.
5.3. Importance of the leptin setpoint
In most humans, bodyweight remains relatively stable over long periods. It is autoregulated, without conscious control, similar to that of body temperature and blood pressure by leptin signal transduction, which functions as a “servomechanism” for bodyweight [242]. An acute change of 5-10 pounds of weight gain or loss quickly returns to normal when factors perturbing weight are removed. This ability to maintain weight results from a metabolic body weight setpoint [243], a physiological value around which the normal weight range fluctuates. The actual location of the body’s weight setpoint is not known. It is thought to have a genetic basis. Still, it may also be modified by epigenetic changes due to outside stressors such as environmental chemicals or changes in nutrition, especially during development. The concept of the leptin setpoint concept and its regulation [244] argues that “homeostatic obesity” may occur due to an environmental stressor promoting defective leptin signaling (e.g., leptin resistance) at the ARC, resulting in “brain starvation”; and thus altering the threshold for leptin signaling resulting in both reduced energy expenditure and increased food intake [245]. Similarly, “hedonic obesity” can result from defective leptin signaling at the VTA, resulting in failure to extinguish the food reward signal, resulting in continued consumption [246]. In each case, obesity results from overriding the setpoint due to interference with the leptin signal, both at the ARC and VTA [244].
Although still debated, the likely culprit that promotes leptin resistance at both nuclei is hyperinsulinemia [247] and resultant CNS insulin resistance (Figure 2). Neurons in both CNS nuclei co-localize insulin and leptin receptors on their cell surface [248]. POMC neurons, exposed to a high insulin concentration in vitro, silence their firing in response to leptin administration, resulting in leptin resistance [249]. Hyperinsulinemia blocks leptin receptor signaling in the hypothalamus through three separate mechanisms; at insulin receptor substrate-2 (IRS-2) [250], at protein tyrosine phosphatase-1B (PTP-1B) [251], and finally at phosphoinositol-3-kinase (PI3-kinase) [252]. Thus, insulin resistance and hyperinsulinemia can lead to central alterations of CNS leptin signaling, resulting in “brain starvation,” which stimulates appetite and persistent weight gain [253].
Figure 2. The interplay between leptin and insulin signaling pathways.
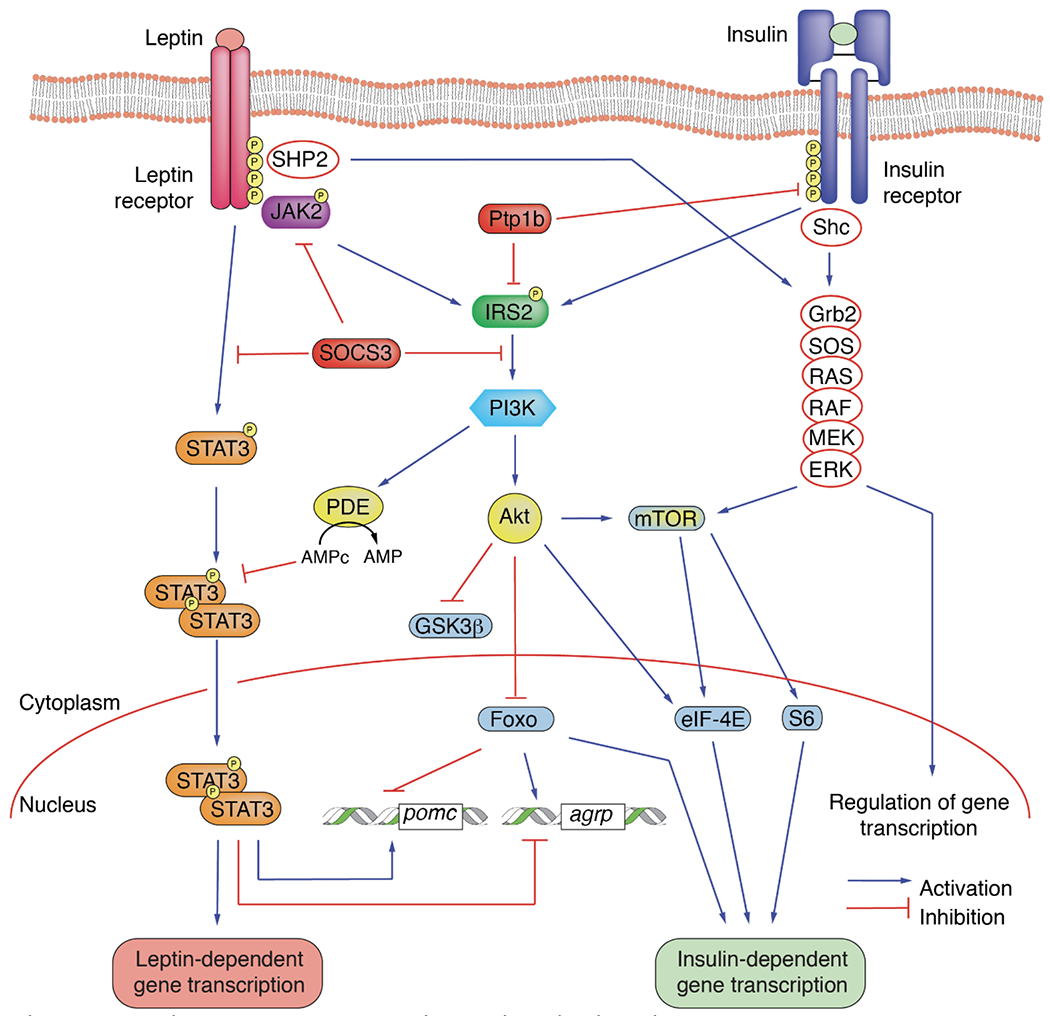
The insulin receptor and the leptin receptor recruit the low-abundance-message insulin receptor substrate-2. Lack of available insulin receptor substrate 2 for the leptin receptor due to hyperinsulinemia could result in defective leptin signal transduction. Alternatively, insulin induction of suppressor of cytokine signaling-3 could inactivate the leptin receptor through alterations in tyrosine phosphorylation.
Abbreviations, Agrp: Agouti-Related Neuropeptide; Akt: AKR Thymoma, protein kinase B; eIF-4E: Eukaryotic translation Initiation Factor 4E; ERK: Extracellular signal-regulated Kinase; Foxo: Forkhead box; Grb2: Growth factor receptor-Bound protein 2; GSK3β: Glycogen Synthase Kinase 3-beta; IRS2: Insulin Receptor Substrate 2; JAK2: Janus Kinase 2; MEK: MAPK/ERK Kinase; mTOR: mammalian Target Of Rapamycin; PDE Phosphodiesterase; PI3K: PhosphatidylInositol 3-Kinase; pomc: Proopiomelanocortin; Ptp1b: Protein-Tyrosine Phosphatase 1B; RAF: Rapidly Accelerated Fibrosarcoma kinase; RAS: Rat Sarcoma virus protein; S6: S6 ribosomal protein; Shc: Src homology 2 domain-containing; SHP2: Src Homology region 2-containing Protein tyrosine phosphatase 2; SOCS3: Suppressor Of Cytokine Signaling 3; STAT3: Signal Transducer and Activator of Transcription 3
6. Heterogeneous nature of control of weight and adiposity
As noted above, energy metabolism, and thus weight as a secondary metric, is tightly controlled by an integrated system of organs and hormones dependent on genetics and the environment. The complex changes in metabolism that result in obesity account for the difficulty in understanding, controlling, and treating obesity: no two patients have the same genetic backgrounds and environmental stimuli. It also illustrates that the pathogenesis of obesity involves more than just overeating and lack of exercise. Below we will discuss each of these factors and their role in obesity.
6.1. Genetics of obesity
Twin studies indicate that 40-70% of obesity may be due to genetics [254]. This figure is misleading because twins have genetic, epigenetic, and environmental similarities. The most common form of obesity is polygenic obesity. Genome-wide association studies (GWAS) of obesity have identified over 300 gene variants associated with obesity, including MCR4, BDNF, and PC1. These genes only account for about 3-5% of individual variation in the genetic risk for obesity [42]. However, GWAS evaluation of metabolic syndrome suggests that only 15% can be explained by genetics alone; the other 85% is explained by the environment [255].
While genes certainly play a role in obesity pathogenesis, genetics alone cannot explain the increase in obesity rates over the last 40 years, as genetics have not changed significantly over that short time frame. Thus, the focus must turn to environmental factors as the cause of the current obesity pandemic.
6.2. Environment and obesity
Environmental factors that promote obesity include a host of alterations, including nutrition, overeating and binge eating disorders/food addiction, time of eating, physical inactivity, drugs, viruses, and the gut microbiome. Various obesogens exert their actions within one or more of these pathways, and their actions will be discussed in the review following this one. We recognize that there are other environmental factors involved in obesity, such as the built environment, social disparities, and economic policies; however, they will not be covered here.
6.3. Nutrition and obesity
The traditional explanation for obesity has been that it results from an imbalance between calorie intake and energy expenditure. Therefore, excess caloric intake accumulates fat whenever calorie intake is greater than energy expenditure. However, this explanation is overly simplistic. Increased caloric intake correlates with weight gain but does not explain the mechanism for the increased calorie intake. In addition, obesity has multifactorial origins and, nutrition is more complicated than just counting calories. Indeed, all calories are not equivalent. An attractive alternative hypothesis is that hyperinsulinemia is the primary factor driving energy storage and weight gain [252, 256]. According to this model, increasing fat deposition resulting from an exaggerated insulin response to specific foods (e.g., refined carbohydrate and sugar), which foments positive energy balance, leads to obesity [256]. Indeed, the typical Western diet promotes obesity by increasing calorically dense (high fat and high sugar) diets, which promote insulin secretion, insulin resistance, and resultant weight gain. Furthermore, hyperinsulinemia interferes with leptin signal transduction to increase food intake [252].
6.3.1. Calories.
While it is true that all calories possess the same capacity for heat generation, equivalent to 4,184 joules of energy, this is not equivalent to how much energy is needed to store it. The efficiency of capturing calories and transforming them into chemical energy in the human body is highly uneven. Understanding these various phenomena show that, in fact, “a calorie is not a calorie,” and there is an actual difference between eating a handful of almonds and a donut, even if their calorie count is identical [257].
6.3.2. Fiber.
Of the 160 calories in a handful of almonds, the body only absorbs 130. The other 30 are prevented from absorption because the fiber in the almonds prevents early absorption in the duodenum allowing the bacteria in the jejunum and ileum to utilize the remaining 30 for their purposes. Thus, even though 160 calories were ingested, not all of them reach the systemic circulation because the microbiome metabolizes it.
There are two types of fiber — soluble (e.g., pectin, inulin) and insoluble (e.g., cellulose, chitin). Together they form a gel on the inside of the duodenum, reducing intestinal absorption by 25-30%. Thus, the two fibers reduce the absorption of mono- and disaccharides and slow the catabolism of starches. Reduced absorption means reduced transport to the liver, preventing the liver from turning excess energy into fat and, thus, preventing hepatic insulin resistance [258]. A sizeable portion of what is eaten stays in the intestine for bacteria to metabolize it. When the gut bacteria are starved of nutrients, they will hydrolyze the protective mucin layer that overlays intestinal epithelial cells, denuding the intestine resulting in “leaky gut” and inflammation while increasing the risk of autoimmune disease and food allergy [259]. Fiber also helps food transit the intestine faster, thereby generating the “satiety signal” (the gut hormone peptide YY3-36, which is transmitted to the brain) sooner, reducing consumption of second portions.
Gut bacteria metabolize soluble fiber into short-chain fatty acids such as butyrate. These feed the colon’s microbiome and are absorbed into the bloodstream, where they are anti-inflammatory and suppress pancreatic insulin secretion, lowering insulin levels [260].
6.3.3. Protein.
Suppose a branched-chain amino acid is is metabolized for energy metabolism. In that case, the amino group is removed in the liver by branched-chain amino-acyl transferase (BCAAT) to convert it into an organic acid (e.g., aspartate to oxaloacetate). Instead of phosphorylating carbohydrates , which costs only one ATP, it costs two ATPs to metabolize amino acids. The amount of energy required for digestion and absorption of food is known as the thermic effect of food (TEF). Fats generate about 2–3% of TEF, carbohydrates about 6–8%, and protein about 25–30% of TEF [261]. In other words, it takes more energy to burn a protein than carbohydrate.
6.3.4. Fat.
All dietary fats would liberate nine calories per gram if burned in a bomb calorimeter. But omega-3 fatty acids are required for cell membrane maintenance and therefore are not burned. Conversely, mitochondria cannot burn trans-fats; humans do not have the enzyme to cleave the trans-double bond. Over the past 50 years, dietary advice has been predicated on the idea that dietary fat is intimately involved in the pathogenesis of obesity and metabolic syndrome. The implication was that the removal of dietary fat should improve health. One restriction study and two substitution studies assessed the effects of removing saturated fat from the diet. The restriction study was the Women’s Health Initiative which studied women who reduced their consumption of saturated fat from 30% to 10% of calories and showed no effect on either weight loss or CVD [262, 263]. In the Sydney Diet Heart Study, men who had experienced a heart attack had the saturated fat removed from their diet and replaced with linoleic acid, an omega-6 fatty acid, which can be pro-inflammatory. All subjects experienced a decline in their low-density lipoprotein (LDL) levels, yet their risk for heart attack increased by 62%, and their mortality risk increased by 70% [264]. Finally, the Minnesota Coronary Study followed 9,000 patients over five years at state mental hospitals and nursing homes, where meals were controlled by removing saturated fat and substituting linoleic acid. The study found similar findings as in Sydney, LDL declined, but heart attacks and deaths increased [265]. These data show that the focus on “a calorie is a calorie” in obesity and obesity-associated comorbidities are incorrect. The nature of the calories consumed is likely much more important than the actual number of calories.
6.3.5. Sugar.
Added sugar (sucrose) consists of equal amounts of glucose and fructose. Both provide the same number of calories, but they are metabolized differently in the liver and the brain. All body tissues can metabolize glucose. Only 20% of a glucose load goes to the liver, and insulin turns it into glycogen (liver starch). All fructose is metabolized in the liver and is unaffected by insulin. An excess of fructose overwhelms the capacity of hepatic mitochondria to utilize it, and the excess is converted into liver fat, driving insulin resistance. Fructose does not shut off the hunger hormone ghrelin, so the brain does not respond appropriately to satiety signals. Fructose is also addictive. Indeed, data show that the adverse effect of sugar on obesity is driven by fructose and not glucose. Fructose, especially when consumed during development and early childhood, causes weight gain [266].
6.3.6. Other carbohydrates.
There are three inherent issues about carbohydrates that play a role in whether they are causative or protective against obesity and metabolic syndrome.
6.3.6.1. Sugar vs. starch.
Sugars are mono- and disaccharides (1 or 2 linked molecules, glucose and fructose), while starches are complex carbohydrates composed of many linked molecules (glucose only). Sugars have one or no bonds to break. They are digested and absorbed quickly in the duodenum, especially when outside a food matrix (e.g., soda, fruit juice), and rapidly move to the liver. Starch has more bonds to break and is digested and absorbed more slowly. Fructose is only found in dietary sugar, not in dietary starch.
6.3.6.2. Type of starch.
There are two basic types of starch: amylose (e.g., beans, lentils, legumes — linked together by alpha-1,4-glucosidyl bonds, which are digested and absorbed slowly) and amylopectin (e.g., wheat, pasta, rice, potatoes — linked together by both alpha-1,4-glucosidyl and alpha-1,6-glucosidyl bonds, which are digested and absorbed rapidly). Amylose is healthier because it is a string of glucose with two ends; therefore, only two enzymes at a time can digest it, resulting in slow digestion and absorption. The longer it stays in the intestine, the greater the chance for gut bacteria to utilize it. Amylopectin is more like a tree of glucoses, with many branch points. Various enzymes can hydrolyze it simultaneously, releasing glucose more rapidly, which is more likely to be absorbed early, flood the liver and pancreas, and generate a more significant insulin response.
6.3.6.3. Glycemic index vs. glycemic load.
A primary goal of improving metabolic health is to lower insulin levels. One way to do so is to eat foods that produce a reduced postprandial glucose excursion (an indirect proxy for insulin), such as with amylose. Higher glucose spikes upon eating are associated with more inflammation [267] and higher mortality [268]. Tables of specific foods and their inherent GI are readily available [269]. Some propose that a low-GI diet will keep blood glucose down and help weight loss [270]. However, the real reason that GI works is that it reduces insulin spikes. Indeed, the glycemic load of carbohydrates consumed is a more important predictor of weight gain than the actual number of calories [271].
6.4. Exercise and obesity
Management of obesity is generally related to interventions in terms of lifestyle, mainly through changes in diet directed at caloric restriction and adherence to exercise programs. Indeed, such interventions, especially when recommended together, are well known to lead to beneficial health effects, e.g., improved mitochondrial function, decreased muscle and liver fat, decreased insulin resistance, reduced inflammation, decreased level of serum triglycerides, weight loss, and reduction in waist circumference [272]. Accordingly, other metabolic diseases such as NAFLD or T2D also benefit from such recommendations [273–275].
Mechanistically, physical activity can counteract insulin resistance [276, 277], notably by interfering with the metabolism of bioactive lipids such as ceramide diacylglycerols [278]. Other possible mechanisms to improve insulin sensitivity might relate to a positive impact of exercise on mitochondria-associated ER membranes (MAMs) that play critical roles in calcium homeostasis, lipid transport, and metabolism [279]. Exercise can also mitigate immunemetabolic disturbances [280–282] due to the production of soluble factors (myokines, hepatokines, osteokines) as well as cytokines and adipokines [280, 283]. It is noteworthy that myokines can crosstalk with metabolic organs (adipose tissue, liver, pancreas), favoring thermogenic activity, glucose production, and insulin secretion [283, 284]. Similarly, extracellular vesicles produced by skeletal muscle might be involved in crosstalk, possibly due to their role as transporters of myokines [285] and for micro-RNAs (miRNAs) [286]. Physical exercise can modulate miRNA expression [287] [288]. Such epigenetic effects of exercise might explain why maternal exercise can have a beneficial action on the metabolic health of offspring [289, 290]. Lastly, an emerging role for lactate, known to be produced by glycolysis upon physical activity, has recently been reported as an appetite-regulatory molecule, thereby controlling energy intake [290, 291].
6.5. Viruses and obesity
Surprisingly, infection with seven different viruses and a scrapie agent has been associated with obesity in animals and humans [292]. Lyons first described the development of massive obesity in a high percentage of Swiss albino mice that survived infection with the canine distemper virus [293]. Subsequent mechanistic studies suggest that obesity resulted from damage to and altered neurochemistry in the hypothalamus of affected mice [292]. Chickens infected with Rous-associated virus type 7 developed obesity accompanied by stunting, dyslipidemia characterized by high serum triglyceride levels, enlarged fatty livers, anemia, and immunosuppression. Of particular interest, obesity develops despite no significant increase in caloric intake. Although this phenotype is associated with depressed thyroid hormone levels [294], damage to the CNS may also develop this obesity phenotype [292]. SMAM-1 is an avian adenovirus that causes increased accumulation of fat and fatty liver along with a paradoxical decrease in serum lipids in infected chickens. Neither canine distemper virus nor Rous-associated virus seven nor SMAM-1 virus is thought to infect humans.
Three additional viruses that are known to infect humans can also cause obesity in animals: human Adenovirus 5 (Ad-5) in mice, Adenovirus 36 (Ad-36) in chickens, mice, rats, and marmosets, and Adenovirus 37 (Ad-37) in chickens [292]. A role for Ad-36 (but not Ad-5 or Ad-37) in promoting human obesity was first suggested by the observation that the prevalence of serum neutralizing antibodies for Ad-36 was significantly higher in obese individuals than non-obese individuals (30% vs. 11%, respectively). Furthermore, the average BMI of seropositive individuals was substantially higher than that of seronegative individuals. Twin pairs discordant for Ad-36 seropositivity had significantly higher BMIs and body fat than seronegative twins. Both serum cholesterol levels and serum triglyceride levels were paradoxically lower in seropositive subjects [295]. Most, but not all, subsequent studies corroborate the relationship between Ad-36 and obesity. A meta-analysis of 11 case-controlled studies found that compared with non-obese controls, Ad-36 infection increased obesity risk by a pooled OR of 1.60. A subgroup analysis revealed exceptionally high risk in children (OR = 1.95) [296].
Mammals infected with Ad-36 have increased body weights and reduced serum levels of cholesterol and triglycerides, and infected rats also show an increase in insulin sensitivity and glucose uptake. Upregulation of C/EBPβ, C/EBPα, PPARγ and glycerol-3-phosphate dehydrogenase expression in infected animals stimulates preadipocyte differentiation. In vitro studies show that Ad-36 accelerates differentiation and proliferation of 3T3-L1 preadipocytes and primary human adipose-derived stem/stromal cells from human volunteers, with enhanced accumulation of lipid. Consistent with increased glucose uptake in rats, human skeletal muscle infected with Ad-36 demonstrates higher gene expression and glucose transporter GLUT1/GLUT4 receptor abundance independent of insulin [297]. Ad-36 infection also suppressed the expression of leptin mRNA and leptin release in 3T3-LI cells and rat primary adipocytes. Adipose tissue of infected rats showed two to five-fold lower levels of leptin mRNA and several-fold higher levels of acetyl Co-A carboxylase-1 and fatty acid synthase expression compared to weight and adiposity matched controls [298]. Finally, glucose, insulin, insulin resistance (HOMA-IR), and leptin levels were all found to be significantly lower in obese human subjects seropositive for Ad-36 compared to obese seronegative subjects [299].
Hence Ad-36 infection is associated with obesity in humans via virally induced hyperplasia and hypertrophy of SAT paired with an alteration of key peripherally and centrally acting adipokines. It is unlikely that this association is simply due to increased susceptibility to adenovirus infection associated with obesity because the rates of seropositivity for Ad-2 and Ad-37 do not differ between weight classes. However, reverse causality cannot yet be ruled out. The obesity phenotype associated with Ad-36 infection appears to be that of “healthy obesity” with paradoxically lower lipids, glucose, insulin, and insulin resistance. This is reminiscent of changes induced by thiazolidinediones, a class of anti-diabetic drugs that act as PPARγ agonists and paradoxically promote weight gain) and improve insulin sensitivity via selective adipogenesis and fatty acid uptake SAT rather than VAT. Further work is needed to characterize the Ad-36 associated obesity phenotype, including risks, responses to interventions, and interaction with behaviors and obesogens.
7. Pathophysiologic mechanisms that promote obesity and/or NCDs
7.1. Insulin resistance and obesity
Insulin resistance is defined as the decreased tissue response to insulin-mediated cellular actions. It has been proposed that insulin resistance results from reduced glucose transport, particularly impairment of GLUT4, which may be due to increased reactive oxygen species (ROS) from fatty acid oxidation via an unknown mechanism [300]. As stated earlier, while obesity and insulin resistance overlap, they are not the same, as some patients are obese without being insulin resistant, and some are insulin resistant without being obese [301]. This is because not all tissues are equally insulin resistant; only liver, muscle and brain insulin resistance lead to hyperinsulinemia and obesity [302], while adipose tissue insulin resistance prevents further fat accumulation.
7.1.1. Hepatic insulin resistance.
The liver is the primary target of insulin action. Following a glucose load, insulin travels directly to the liver via the portal vein, where it binds to the insulin receptor recruiting and activating the PI3K pathway, eliciting two key actions at the level of gene transcription. First, insulin stimulates the phosphorylation of the forkhead protein FoxO1, which prevents it from entering the nucleus [303, 304]. This diminishes the expression of two genes required for gluconeogenesis (phosphoenolpyruvate carboxykinase and glucose 6-phosphatase), thereby shutting down glucose synthesis leading to diminished hepatic glucose output. Second, insulin induces sterol regulatory element-binding protein (SREBP)-1c, which increases transcription of genes required for fatty acid and triglyceride biosynthesis (ATP-citrate lyase, acetyl-coenzyme A carboxylase, and fatty acid synthase, which constitute the process of de novo lipogenesis (DNL)). Excess fat synthesized by DNL that is not exported out of the liver as VLDL is instead stored as triglyceride (TG) droplets within the hepatocyte [305], contributing to fatty liver disease, increasing the enzyme c-jun N-terminal kinase-1 (JNK-1), altering the phosphorylation status of insulin receptor substrate-1 (IRS-1) [306] and inducing hepatic insulin resistance [307].
7.1.2. Brain insulin resistance.
There are several postulated mechanisms for the development of CNS insulin resistance. As stated earlier, hyperinsulinemia can drive CNS insulin resistance by down-regulating the insulin receptor at the level of the ARC, resulting in defective leptin signal transduction resulting in “brain starvation” and in a lack of attenuation of reward in the NA, both leading to increased food intake and weight gain [252]. Another proposal for the mechanism of CNS insulin resistance is the production of ceramide due to fatty liver disease, which can induce brain oxidative stress and inflammation leading to CNS insulin resistance [308]. Lastly, as seen in systemic insulin resistance, increased peripheral free fatty acids may be taken up by microglia in the ARC to induce hypothalamic neuroinflammation and CNS insulin resistance [309].
7.1.3. Adipose tissue insulin resistance.
If any given adipocyte (subcutaneous or visceral) remains insulin sensitive, insulin-induced lipoprotein lipase will drive fatty acids from circulating VLDL and chylomicrons to promote fat storage in vesicles insulin-induced GLUT4 will increase glucose uptake to make glycerol, and triacylglycerol deposition can continue [310–312]. Increases in glucose uptake also increase branched-chain hydroxy-fatty acid esters, which protect GLUT4 levels and reduce inflammation to increase insulin sensitivity in adipose tissue [70]. However, continued expansion of adipose tissue causes cells to become hypoxic and die and secrete inflammatory mediators that promote insulin resistance by releasing TNF-alpha and other cytokines [313] [314]. Adipocyte insulin resistance disinhibits hormone-sensitive lipase (HSL), leading to free fatty acid release into the systemic circulation, which leads to hepatic inflammation and hepatic insulin resistance [315, 316]. Cell death also reduces adiponectin production, promoting hepatic insulin resistance [317].
7.2. Inflammation and obesity
A state of low-grade inflammation and immune dysfunction may account for some of the links between obesity and chronic metabolic diseases [46]. Obese humans and rodents show increased production of pro-inflammatory cytokines and adipokines such as monocyte chemotactic protein 1 (MCP1), interleukin-6 (IL-6), tumor necrosis factor-alpha (TNF-α), IL-1β, and leptin, along with infiltration of macrophages with pro-inflammatory M1-like phenotype in adipose tissue [318], [319]. Resistin secretion from macrophages occurs during inflammation and enhances the production of pro-inflammatory cytokines such as TNF-α, IL-6 and IL-12 [320]. Resistin also plays a role in the upregulation of the expression of chemokines and adhesion molecules.
Both adaptive and innate immunity are also disturbed in obesity. There are reports of a decrease in naive CD4+ T cells and regulatory T cells (T regs), an increase in CD4+ Th17 and Th22 pro-inflammatory subsets, CD8 cytotoxic T cells, and natural killer (NK) cells [27]. Inflammation also makes obese patients more prone to develop infectious and non-infectious diseases. In addition to immune mechanisms, endothelial and adipose dysfunction coupled with viral receptor expression and insulin resistance can account for these harmful effects [28].
The immune system plays both a homeostatic and pathologic role in diseases of metabolic organs. The balance between pro-and anti-inflammatory processes is critical to achieving metabolic homeostasis. Immune system imbalance is observed in metabolic diseases; however, many pathways connecting obesogens, inflammation, and metabolic disease(s) remain unknown[321]. There is a close relationship between nutrient excess and activation of innate immunity, which is seen in the pancreas, liver, skeletal muscle, adipose tissue, gut, and blood vessels, i.e., the organs playing critical roles in the maintenance of energy homeostasis [317]. Adipose tissue inflammation, linked with the processes governing fibrosis and insufficient angiogenesis within adipose tissue, contributes to adipocyte expansion and systemic inflammation [322, 323].
Resident M2-like macrophages in adipose tissue promote the health of normal adipose tissue and influence energy metabolism and mitochondrial function in adipocytes [324]. Adipose tissue inflammation is associated with a perturbation of the homeostatic role of adipose tissue-resident macrophages [325]. Obesity leads to an altered immune state in adipose tissue where enlarged adipocytes produce chemokines and chemotactic adipokines such as MCP1 and leukotriene B4 (LTB4) that attract monocytes [326]. Adipocytes also can secrete other chemokines that attract eosinophils, invariant natural killer T cells (iNKTs) and T-cells to adipose tissue [327, 328]. Upon recruitment of pro-inflammatory cells, cytokines such as TNF-α, IFN-γ, and IL-17 promote M1 polarization of adipose tissue macrophages [328, 329]. T-helper cells are polarized to Th1 cells via M1 macrophages and adipocytes secreted leptin [327]. Through the production of TNF-α and IFN-γ by Th1 cells, the vicious cycle of macrophage activation is invoked, subsequently remodeling the adipose tissue [328–330]. This shift towards pro-inflammation in adipose tissue was also observed in obese mice [328, 329]. The crosstalk between adipocytes and immune cells progressively creates a pro-inflammatory environment in adipose tissue. Additionally, pro-inflammatory cytokines cause insulin resistance through direct serine phosphorylation of the insulin receptor substrate-1 or 2 [326]. Together these studies point to an association of enhanced pro-inflammatory and reduced anti-inflammatory state with obesity and metabolic dysfunction.
7.3. Gut microbiome and obesity
Research in the last decade has unraveled a significant role of the host gut microbiome in defining the development of obesity and its promotion of metabolic consequences in organ systems such as liver, lung, kidney, reproductive organs, and the heart [331]. Diseases such as NAFLD, chronic hypertension, polycystic ovarian syndrome, CVD, and chronic kidney disease (CKD) have all been associated with obesity [332, 333]. Importantly, in all the complications associated with an obese phenotype, the host gut microbiome has been found to play a profound role in disease progression leading to poor outcomes [334].
The gut microbiome is a large body of bacteria, viruses, and fungi that reside in a complete symbiotic environment in the small intestine, the large intestine and the colon [335], [336]. Host microbiomes play a crucial role in physiological homeostasis, forming a critical part of the gut, brain, metabolic, and immune health. These resident bacteria and viruses are intertwined with the host’s metabolism and often contribute to small molecule intermediates that help the host maintain normal metabolic processes, e.g., short-chain fatty acids [337]. Conversely, defined as a change in the relative abundance of the millions of bacteria, viruses, and fungi needed for the symbiotic environment, a modified microbiome is associated with obesity [338, 339]. The development of genetic sequencing methods and adoption of more sophisticated bioinformatics tools have led to significant progress to identify abundance among phyla, order, family, genus and species of the host microbiome, empowering studies to examine the linkage of dysregulated microbiota to metabolic dysfunction.
Some of the first studies of an altered microbiome accurately identified an increase in Firmicutes abundance over the more common Bacteroidetes in obesity [340, 341]. Later, this altered microbiome signature was also found in FLD, confirming that diet and accumulation of free fatty acids play a role in the altered diversity and abundance of the host bacteriome [342]. Several studies have identified the role of specific phyla such as Porphyromonadaceae, Enterococcaceae and Lactobacillaceae in obesity [343]. More importantly, obesity is associated with low-level inflammation primarily from elevated levels of TNF-α, IL1β and MCP-1[344]. An altered microbial diversity and increases in the abundance of Firmicutes or Porphyromonadaceae species may be responsible for a “leaky gut” and increased levels of these cytokines in the circulation [345]. Obesity is often associated with a low inflammatory trigger and fat accumulation that skew the healthy microbiome towards an increased abundance of the gut bacteria known to decrease gut and immune health. Transplant studies showed proof of concept; for example, cohousing mice harboring microbiota from an obese twin with mice containing microbiota from a lean twin prevented increased body mass and metabolic phenotypes in the obese cage mates [346].
Disruptions of the gut microbiome may lead to altered production of important bacteria (and host)-derived molecules, including short-chain fatty acids (fermentation products), tryptophan metabolites including indole-3-carboxylic acid, 10-oxo-12-ocadecenoic acid (from lactate), bile acids ( mediators of thermogenesis) with a known impact on obesity and metabolism [347–350]. The altered bacterial composition may also disrupt intestinal barrier functions and affect distant organs, including adipose tissue, pancreas, brain, and liver, thus promoting metabolic inflammation and obesity [351]. Gut microbiota dysbiosis can promote white adipose tissue inflammation and expansion [352]. Alterations in the gut microbiome strongly impact NAFLD development [353]. The gut is also a critical organ in developing metabolic syndrome [354].
7.4. Circadian rhythms and obesity
In mammals, the circadian clock is responsible for regulating 24-hour rhythms of behavior and physiology. A central clock controls this timing system in the suprachiasmatic nucleus (SCN) of the hypothalamus and peripheral clocks in peripheral tissues (liver, adipose tissue, skeletal muscle, etc.), playing a pivotal role in many biological processes. The circadian clock regulates several metabolic processes, including lipid and glucose metabolism and mitochondrial oxidative phosphorylation [355–359]. Interestingly, gut microbiota may also regulate their host circadian rhythms via the metabolites they produce, such as short-chain fatty acids [360]. In this context, any change in circadian rhythms can impact metabolic health, resulting in pathologies such as obesity, T2D, metabolic syndrome, or NAFLD [361–364].
As will be discussed in detail in the companion review, all of these tissues, organs and mechanisms that control metabolism and weight gain are possible sites of obesogen action.
8. Fetal origins of obesity
Excessive weight gain can start at any time across the lifespan. Still, the prenatal and neonatal environment has long been considered a critical period for the origins of obesity and cardio-metabolic disease in humans, as described in the Developmental Origins of Health and Disease (DOHaD) hypothesis [365]. For example, undernutrition during pregnancy leads to a low birth weight linked to an increased incidence of obesity, CVD and T2D later in life [366]. Substantial evidence from animal and human studies indicates that the conditions of the in utero environment can contribute to the risk of obesity and metabolic disease in childhood and adulthood [367, 368] by increasing the sensitivity or susceptibility to gain weight. The DOHaD paradigm applies to nutrition during development and stress and environmental chemical exposures, including obesogens. Developmental exposure to obesogens also leads to obesity later in life and even across generations (discussed in detail in the companion review).
8.1. Birth status and future obesity
Birth weight appears to have a U-shaped association with later cardio-metabolic risk. Both low and high birth weights have been associated with a greater risk of obesity and metabolic disease [369–372]. Restricted fetal growth may be followed by rapid or aberrant growth in infancy, and early-life “catch-up growth” has been linked to a greater risk of obesity and greater abdominal fat mass in childhood [373, 374]. Consistent with this pattern, specific prenatal environmental exposures have been associated with lower infant birth weight (236,237) but greater adiposity later in childhood or adulthood [375–378]. Conversely, excessive prenatal somatic growth, independent of maternal type 2 diabetes, leads to obesity and metabolic syndrome in the offspring [379].
8.2. Small for gestational age (SGA), developmental programming, and future obesity
Documentation of the relationship of SGA with adult obesity and cardiovascular disease started with studies of the Dutch famine during World War II and its aftermath [380]. Several studies of newborns born SGA demonstrate that they are hyperinsulinemic and insulin resistant at birth, exhibit rapid catch-up growth in the early post-natal period, and develop obesity in childhood, with persistent insulin resistance and late development of the metabolic syndrome. An analysis of Indian newborns born in India vs. the U.K. [381] demonstrates that despite weighing 700 gm less at birth, their glucose and insulin levels are markedly elevated. They show increased adiposity and become insulin resistant and obese during early childhood [382].
Animal models of gestational caloric restriction recapitulate the human condition and lead to SGA and insulin resistance at the birth of the offspring [383, 384]. Several investigators have postulated that prenatal metabolic perturbation alters the normal ontogeny and organization of the hypothalamus and, ultimately, post-natal regulation of energy balance, mediated through changes in leptin action in utero. Leptin-deficient mice (ob/ob) exhibit maldevelopment of hypothalamic architecture, with aberrant projections of neurons from the arcuate nucleus (the site of leptin receptors) to the paraventricular nucleus (the site of the melanocortin-4 receptors) [385]. However, a single leptin injection at birth can restore normal hypothalamic development and neurotransmission [386]. This effect of neonatal leptin is also seen in a rat model of maternal undernutrition. Offspring of pregnant rats placed on food restriction during gestation are SGA, insulin resistant, and leptin-deficient at birth. However, with adequate nutrition after the neonatal period, these animals become obese as adults, particularly when placed on high-fat chow after weaning [387], suggesting that the neonatal leptin overrode a developmental programming signal for future obesity.
8.3. Large for gestational age (LGA), epigenetics, and future obesity
Overnutrition during pregnancy, as experienced with maternal obesity, gestational diabetes, and excess weight gain, is also associated with greater adiposity and risk of obesity in the offspring [388–391]. Babies born LGA are hyperinsulinemic at birth [392], demonstrating a doubling of the prevalence of insulin resistance and metabolic syndrome [393]. In contrast, obese women who underwent bariatric surgery reduced the incidence of LGA offspring and their future risk for obesity [394]. Offspring of mothers with obesity and hyperglycemia during pregnancy demonstrate epigenetic changes at birth [395], which may link the early life environment to later obesity phenotype [396–398]. Overnutrition during this sensitive period may produce lasting changes in offspring body composition, either via direct influences on organ and adipose development or via epigenetic regulation of gene expression [397, 399, 400].
8.4. Prematurity, prenatal stress, glucocorticoids, epigenetics, and future obesity
Although there are no studies documenting hyperinsulinemia at birth in premature infants due to blood drawing issues, follow-up of these babies into early childhood also demonstrates increased weight gain and insulin resistance with compensatory insulin secretion that is inappropriately high for their degree of weight gain [401]. Thus, some aspect of prematurity leads to alteration in developmental programming and obesity and insulin resistance in later life.
Prenatal stress may alter the newborn through glucocorticoid effects or autonomic CRH effects [401]. For instance, glucocorticoid exposure during pregnancy results in lower birth weight [402, 403] and predisposition to obesity in later life. Similarly, offspring born of prenatal stress demonstrate altered glucose and insulin homeostasis upon reaching adulthood [404] and are more vulnerable to a high fat diet in later life [405].
Prenatal stress, likely working through CRH manufactured in the placenta, or possibly through maternal cortisol, which overwhelms placental 11bHSD2 (which normally protects the fetus), may alter fetal metabolism in several ways. Prenatal stress may also cause epigenetic changes in the methylation status of the CpG island in the NR3C1 gene, coding for the glucocorticoid receptor, which predicts altered neonatal stress reactivity [406].
Thus, the high glucocorticoid levels associated with chronic stress may act as a feedforward system, recruiting a major chronic stress response network [407] that alters the normal output of autonomic, neuroendocrine and behavioral systems, biasing the usual responses toward increased HPA axis activity as well as accentuating fear-like behavior. Increased activity of this system increases ingestion of high energy-density foods [408] with subsequent increased VAT stores and the metabolic syndrome.
9. Conclusions
This review focused on the tissues/organs, hormones, pathways, and mechanisms that play key roles in metabolism to induce adipose tissue, resulting in obesity. Obesity is a multifunctional, multi-tissue, multi-hormone, multi-receptor, and multi-mechanism disease. When obesity results from increased VAT or liver fat with large fat cells, inflammation, and insulin and leptin resistance, it is also associated with several metabolic disorders, including T2D, NAFLD, CVD and some cancers. On the other hand, when the obesity results from increased SAT with small adipocytes, limited inflammation, and normal insulin and leptin activity, there is, in some cases, a lack of corresponding metabolic perturbation, at least initially. The multifunctional nature of obesity results from the many highly coordinated interacting factors that play a role in obesity, including genetics and environmental factors. The environment includes nutrition, exercise, viruses, microbiome, circadian rhythms (this review), and environmental chemicals (discussed in the companion review). Environmental factors act on the complex interacting organ systems that control metabolism, including adipose tissue, the gastrointestinal tract, muscle, pancreas, liver, and various parts of the brain, that integrate the control of feeding behavior, including homeostatic hedonic eating. Control of adipose tissue development, as well as the number and size of adipocytes, depends on the activity and interaction of a variety of transcription factors, including the two master regulators of adipogenesis: PPARγ; and RXR, which can activate adipogenesis either alone or as a heterodimer with PPARγ. The hormones insulin, estrogen, androgen, glucocorticoid, and thyroid hormone also play important roles in metabolism and adipogenesis by binding to their receptors. Other liver transcription factors modulate specific metabolic signals, which can lead to disease when dysfunctional. LXR, although not activated by specific hormones, regulates adipocyte differentiation, cholesterol transport, and triglyceride accumulation. PXR and CAR appear to act in conjunction with PPARγ and regulate glucose and energy homeostasis and immune and lipid metabolism. FXR regulates bile acid synthesis and lipid metabolism, and AhR can result in hepatic insulin resistance. Activation of these receptors can result in hyperinsulinemia, leptin resistance, and weight gain.
The tissues and organs controlling metabolism, and therefore weight gain, communicate via a network of hormones and neurotransmitters. For example, leptin, resistin, and adiponectin are elicited from adipocytes; ghrelin and GIP from the stomach; CCK, GLP-1, OXM, and PYY3-36 from the gastrointestinal tract; insulin and glucagon from the pancreas; NPY-AgRP and POMC from hypothalamic neurons; dopamine from the VTA and NA; as well as estrogen, androgen, thyroid hormone, and cortisol from their respective endocrine glands.
While obesity can occur throughout the lifespan, it can have its origins during fetal development and infancy and is thus regulated by changes in epigenetic regulation or developmental programming of gene expression. These perturbations are particularly susceptible to environmental influences.
This review of metabolism highlights the organs and mechanisms responsible for regulating adiposity. It also sets the hypothesis that environmental chemicals capable of disrupting these mechanisms, termed obesogens, can lead to obesity. Numerous compounds, some nutritional and some endocrine-disrupting chemicals (EDCs), can affect the hormonal control of adipose tissue differentiation, development, growth, and/or maintenance. These changes subsequently result in differential effects on fat depots that can ultimately affect metabolic dysfunction. Thus, we proffer that the change in prevalence and severity of obesity is, at least in part, due to the occurrence and accumulation of various environmental alterations — in the form of either poor nutrition or obesogenic EDCs — in an otherwise genetically susceptible population, the most susceptible of which is the fetus. The second companion article will review the plausibility, mechanism, and evidence for these obesogens — in vitro, animals, and humans.
Essential Points.
There is an expanding global obesity and non-communicable disease pandemic.
Obesity is a multifactorial, multi-organ, multi-hormone, and multi-mechanism disease.
Genetic and environmental influences control adiposity and weight gain.
Understanding the tissues/organs, hormones, and mechanisms involved in obesity set the stage for understanding the evidence for the obesogen hypothesis.
Funding
Christopher Kassotis, NIH, R00ES30405
Dominique Lagadic-Gossman, European Union Horizon 2020 Research and Innovation Program, Oberon #825712
Vesna Munic Kos, Swedish Research Council for Sustainable Development (FORMAS) #2019-00375
Troy Roepke, NIH, R01MH12 3544, P30ES005022, USDA/NIFA NJ6195
Jan Vondracek, Czech Science Foundation #21-005335, Institute of Biophysics of the Czech Academy of Science, RVO-68081707
Robert Barouki, European Union Horizon 2020 Research and Innovation Program, Oberon #825712
Amita Bansal, Diabetes Australia #S5610040
Mathew Cave, NIH, R35ES028373, R01ES032189, T32ES011564, P42ES023716, P30ES030283, R21ES031510
Saurabh Chatterjee, NIH, P20GM103641, P01ES028942, P01AT003961, DoD-IIRFA W81XWH1810374
Mahua Choudhury, Morris L Lichtenstein Jr Medical Research Foundation.
David Collier, NIH, P30ES025128
Abbreviations
- Ad
Adenovirus
- AgRP
agouti-related protein
- AhR
aryl hydrocarbon receptor
- Akt
AKR Thymoma, protein kinase B
- AR
androgen receptor
- ARC
arcuate nucleus
- ARKO
Androgen receptor knock-out
- ATP
adenosine triphosphate
- BAT
brown adipose tissue
- BaP
benzo[a]pyrene
- BCAAT
branched-chain amino-acyl transferase
- bHLH
basic-helix-loop-helix
- BMI
body mass index
- CAR
constitutive androstane receptor
- CCK
cholecystokinin
- CKD
chronic kidney disease
- CNS
central nervous system
- CRH
corticotropin-releasing hormone
- CT
computed tomography
- CVD
cardiovascular disease
- DNL
de novo lipogenesis
- DOHaD
Developmental Origins of Health and Disease
- DRN
dorsal raphe nucleus
- EDC
endocrine disrupting chemical
- eIF-4E
Eukaryotic translation Initiation Factor 4E
- ER
estrogen receptor
- ERK
extracellular signal-regulated Kinase
- ERKO
estrogen receptor knock-out
- FFA
free fatty acid
- FLD
fatty liver disease
- FOXO
Forkhead box O
- FXR
farnesoid X receptor
- GI
glycemic index
- GIP
gastric inhibitory polypeptide
- GLP
glucagon-like peptide
- GLUT
glucose transporter
- GPER
G-protein-coupled estrogen receptor
- GR
glucocorticoid receptor
- Grb2
Growth factor receptor-Bound protein 2
- GRKO
glucocorticoid receptor knock-out
- GSK3b
Glycogen Synthase Kinase 3-beta
- GWAS
genome-wide association studies
- HDL
high-density lipoprotein
- HFD
high-fat diet
- HOMA-IR
homeostatic model assessment of insulin resistance
- IFN-γ
interferon gamma
- IGF
insulin growth factor
- IGF1R
insulin growth factor-1 receptor
- IL
interleukin
- iNKT
invariant natural killer T cells
- IR
insulin receptor
- IRS
insulin receptor substrate
- JAK2
Janus Kinase 2
- JNK-1
c-jun N-terminal kinase-1
- Kyn
kynurenine
- LDL
low-density lipoprotein
- LTB4
leukotriene B4
- LXR
liver X receptor
- MAM
mitochondria-associated endoplasmic reticulum membranes
- MCP1
monocyte chemotactic protein-1
- MEK
MAPK/ERK Kinase
- miRNA
micro-RNA
- MRI
magnetic resonance imaging
- MSC
mesenchymal stem cell
- mTOR
mammalian Target Of Rapamycin
- myf5
myogenic factor 5
- NA
nucleus accumbens
- NAFLD
non-alcoholic fatty liver disease
- NK
natural killer cells
- NPY
neuropeptide Y
- NST
nucleus of the solitary tract
- OXM
oxyntomodulin
- PAS
PER-ARNT-SIM
- PDE
phosphodiesterase
- PI3K
phosphoinositol-3-kinase
- POMC
pro-opiomelanocortin
- PPAR
peroxisome proliferation activated receptor
- PTP-1B
protein tyrosine phosphatase-1B
- PVN
paraventricular nucleus
- PXR
pregnane X receptor
- PYY3-36
peptide YY3-36
- RAF
Rapidly Accelerated Fibrosarcoma kinase
- RAS
Rat Sarcoma virus protein
- ROS
reactive oxygen species
- RXR
retinoid X receptor
- S6
S6 ribosomal protein
- SAT
subcutaneous adipose tissue
- SCD
stearoyl-CoA desaturase
- SCN
suprachiasmatic nucleus
- Shc
Src homology 2 domain-containing protein
- SHP2
Src Homology region 2-containing Protein tyrosine phosphatase 2
- SOCS3
Suppressor Of Cytokine Signaling 3
- SREBP
sterol regulatory element-binding protein
- STAT3
Signal Transducer and Activator of Transcription 3
- T2D
type 2 diabetes
- T3
triiodothyronine
- T4
thyroxine
- TCDD
2,3,7,8-tetrachlorodibenzo-p-dioxin
- TEF
thermic effect of food
- TG
triglyceride
- TNF-α
tumor necrosis factor-alpha
- TR
thyroid hormone receptor
- TRH
thyrotropin releasing hormone
- TSH
thyroid-stimulating hormone
- TZDs
thiazolidinediones
- UCP-1
uncoupling protein-1
- VAT
visceral adipose tissue
- VLDL
very-low-density lipoproteins
- VTA
ventral tegmental area
- WAT
white adipose tissue
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Summary: None of the authors have anything to declare regarding writing this review.
References
- [1].Jastreboff AM, Kotz CM, Kahan S, Kelly AS, Heymsfield SB, Obesity as a Disease: The Obesity Society 2018 Position Statement, Obesity (Silver Spring) 27(1) (2019) 7–9. [DOI] [PubMed] [Google Scholar]
- [2].Mechanick JI, Garber AJ, Handelsman Y, Garvey WT, American Association of Clinical Endocrinologists’ position statement on obesity and obesity medicine, Endocr Pract 18(5) (2012) 642–8. [DOI] [PubMed] [Google Scholar]
- [3].Jaacks LM, Vandevijvere S, Pan A, McGowan CJ, Wallace C, Imamura F, Mozaffarian D, Swinburn B, Ezzati M, The obesity transition: stages of the global epidemic, The lancet. Diabetes & endocrinology 7(3) (2019) 231–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Blüher M, Obesity: global epidemiology and pathogenesis, Nat Rev Endocrinol 15(5) (2019) 288–298. [DOI] [PubMed] [Google Scholar]
- [5].Morales Camacho WJ, Molina Díaz JM, Plata Ortiz S, Plata Ortiz JE, Morales Camacho MA, Calderón BP, Childhood obesity: Aetiology, comorbidities, and treatment, Diabetes Metab Res Rev 35(8) (2019) e3203. [DOI] [PubMed] [Google Scholar]
- [6].Hales CM, Carroll MD, Fryar CD, Ogden CL, Prevalence of Obesity Among Adults and Youth: United States, 2015-2016, NCHS data brief (288) (2017) 1–8. [PubMed] [Google Scholar]
- [7].Williams EP, Mesidor M, Winters K, Dubbert PM, Wyatt SB, Overweight and Obesity: Prevalence, Consequences, and Causes of a Growing Public Health Problem, Current obesity reports 4(3) (2015) 363–370. [DOI] [PubMed] [Google Scholar]
- [8].Ng M, Fleming T, Robinson M, Thomson B, Graetz N, Margono C, Mullany EC, Biryukov S, Abbafati C, Abera SF, Abraham JP, Abu-Rmeileh NM, Achoki T, AlBuhairan FS, Alemu ZA, Alfonso R, Ali MK, Ali R, Guzman NA, Ammar W, Anwari P, Banerjee A, Barquera S, Basu S, Bennett DA, Bhutta Z, Blore J, Cabral N, Nonato IC, Chang JC, Chowdhury R, Courville KJ, Criqui MH, Cundiff DK, Dabhadkar KC, Dandona L, Davis A, Dayama A, Dharmaratne SD, Ding EL, Durrani AM, Esteghamati A, Farzadfar F, Fay DF, Feigin VL, Flaxman A, Forouzanfar MH, Goto A, Green MA, Gupta R, Hafezi-Nejad N, Hankey GJ, Harewood HC, Havmoeller R, Hay S, Hernandez L, Husseini A, Idrisov BT, Ikeda N, Islami F, Jahangir E, Jassal SK, Jee SH, Jeffreys M, Jonas JB, Kabagambe EK, Khalifa SE, Kengne AP, Khader YS, Khang YH, Kim D, Kimokoti RW, Kinge JM, Kokubo Y, Kosen S, Kwan G, Lai T, Leinsalu M, Li Y, Liang X, Liu S, Logroscino G, Lotufo PA, Lu Y, Ma J, Mainoo NK, Mensah GA, Merriman TR, Mokdad AH, Moschandreas J, Naghavi M, Naheed A, Nand D, Narayan KM, Nelson EL, Neuhouser ML, Nisar MI, Ohkubo T, Oti SO, Pedroza A, Prabhakaran D, Roy N, Sampson U, Seo H, Sepanlou SG, Shibuya K, Shiri R, Shiue I, Singh GM, Singh JA, Skirbekk V, Stapelberg NJ, Sturua L, Sykes BL, Tobias M, Tran BX, Trasande L, Toyoshima H, van de Vijver S, Vasankari TJ, Veerman JL, Velasquez-Melendez G, Vlassov VV, Vollset SE, Vos T, Wang C, Wang X, Weiderpass E, Werdecker A, Wright JL, Yang YC, Yatsuya H, Yoon J, Yoon SJ, Zhao Y, Zhou M, Zhu S, Lopez AD, Murray CJ, Gakidou E, Global, regional, and national prevalence of overweight and obesity in children and adults during 1980-2013: a systematic analysis for the Global Burden of Disease Study 2013, Lancet 384(9945) (2014) 766–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Ogden CL, Carroll MD, Fryar CD, Flegal KM, Prevalence of Obesity Among Adults and Youth: United States, 2011-2014, NCHS data brief (219) (2015) 1–8. [PubMed] [Google Scholar]
- [10].Klimentidis YC, Beasley TM, Lin HY, Murati G, Glass GE, Guyton M, Newton W, Jorgensen M, Heymsfield SB, Kemnitz J, Fairbanks L, Allison DB, Canaries in the coal mine: a cross-species analysis of the plurality of obesity epidemics, Proc Biol Sci 278(1712) (2011) 1626–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Landsberg L, Aronne LJ, Beilin LJ, Burke V, Igel LI, Lloyd-Jones D, Sowers J, Obesity-related hypertension: pathogenesis, cardiovascular risk, and treatment: a position paper of The Obesity Society and the American Society of Hypertension, J Clin Hypertens (Greenwich) 15(1) (2013) 14–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Andolfi C, Fisichella PM, Epidemiology of Obesity and Associated Comorbidities, J Laparoendosc Adv Surg Tech A 28(8) (2018) 919–924. [DOI] [PubMed] [Google Scholar]
- [13].Stefan N, Causes, consequences, and treatment of metabolically unhealthy fat distribution, The lancet. Diabetes & endocrinology 8(7) (2020) 616–627. [DOI] [PubMed] [Google Scholar]
- [14].Chan JC, Cheung JC, Stehouwer CD, Emeis JJ, Tong PC, Ko GT, Yudkin JS, The central roles of obesity-associated dyslipidaemia, endothelial activation and cytokines in the Metabolic Syndrome--an analysis by structural equation modelling, International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 26(7) (2002) 994–1008. [DOI] [PubMed] [Google Scholar]
- [15].Loomis AK, Kabadi S, Preiss D, Hyde C, Bonato V, St Louis M, Desai J, Gill JM, Welsh P, Waterworth D, Sattar N, Body Mass Index and Risk of Nonalcoholic Fatty Liver Disease: Two Electronic Health Record Prospective Studies, The Journal of clinical endocrinology and metabolism 101(3) (2016) 945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Mathieu P, Lemieux I, Després JP, Obesity, inflammation, and cardiovascular risk, Clin Pharmacol Ther 87(4) (2010) 407–16. [DOI] [PubMed] [Google Scholar]
- [17].Virani SS, Alonso A, Aparicio HJ, Benjamin EJ, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Cheng S, Delling FN, Elkind MSV, Evenson KR, Ferguson JF, Gupta DK, Khan SS, Kissela BM, Knutson KL, Lee CD, Lewis TT, Liu J, Loop MS, Lutsey PL, Ma J, Mackey J, Martin SS, Matchar DB, Mussolino ME, Navaneethan SD, Perak AM, Roth GA, Samad Z, Satou GM, Schroeder EB, Shah SH, Shay CM, Stokes A, VanWagner LB, Wang NY, Tsao CW, Heart Disease and Stroke Statistics-2021 Update: A Report From the American Heart Association, Circulation 143(8) (2021) e254–e743. [DOI] [PubMed] [Google Scholar]
- [18].Koroukian SM, Dong W, Berger NA, Changes in Age Distribution of Obesity-Associated Cancers, JAMA network open 2(8) (2019) e199261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Stephan BCM, Birdi R, Tang EYH, Cosco TD, Donini LM, Licher S, Ikram MA, Siervo M, Robinson L, Secular Trends in Dementia Prevalence and Incidence Worldwide: A Systematic Review, J Alzheimers Dis 66(2) (2018) 653–680. [DOI] [PubMed] [Google Scholar]
- [20].Benziger CP, Roth GA, Moran AE, The Global Burden of Disease Study and the Preventable Burden of NCD, Glob Heart 11(4) (2016) 393–397. [DOI] [PubMed] [Google Scholar]
- [21].Global, regional, and national age-sex specific mortality for 264 causes of death, 1980-2016: a systematic analysis for the Global Burden of Disease Study 2016, Lancet 390(10100) (2017) 1151–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Bhattacharya J, Bundorf MK, The incidence of the healthcare costs of obesity, Journal of health economics 28(3) (2009) 649–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Xanthakos SA, Lavine JE, Yates KP, Schwimmer JB, Molleston JP, Rosenthal P, Murray KF, Vos MB, Jain AK, Scheimann AO, Miloh T, Fishbein M, Behling CA, Brunt EM, Sanyal AJ, Tonascia J, Progression of Fatty Liver Disease in Children Receiving Standard of Care Lifestyle Advice, Gastroenterology 159(5) (2020) 1731–1751.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bjornstad P, Drews KL, Caprio S, Gubitosi-Klug R, Nathan DM, Tesfaldet B, Tryggestad J, White NH, Zeitler P, Long-Term Complications in Youth-Onset Type 2 Diabetes, The New England journal of medicine 385(5) (2021) 416–426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Lawrence JM, Divers J, Isom S, Saydah S, Imperatore G, Pihoker C, Marcovina SM, Mayer-Davis EJ, Hamman RF, Dolan L, Dabelea D, Pettitt DJ, Liese AD, Trends in Prevalence of Type 1 and Type 2 Diabetes in Children and Adolescents in the US, 2001-2017, Jama 326(8) (2021) 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Dobner J, Kaser S, Body mass index and the risk of infection - from underweight to obesity, Clin Microbiol Infect 24(1) (2018) 24–28. [DOI] [PubMed] [Google Scholar]
- [27].Korakas E, Ikonomidis I, Kousathana F, Balampanis K, Kountouri A, Raptis A, Palaiodimou L, Kokkinos A, Lambadiari V, Obesity and COVID-19: immune and metabolic derangement as a possible link to adverse clinical outcomes, American journal of physiology. Endocrinology and metabolism 319(1) (2020) E105–E109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Popkin BM, Du S, Green WD, Beck MA, Algaith T, Herbst CH, Alsukait RF, Alluhidan M, Alazemi N, Shekar M, Individuals with obesity and COVID-19: A global perspective on the epidemiology and biological relationships, Obes Rev 21(11) (2020) e13128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Chan JM, Rimm EB, Colditz GA, Stampfer MJ, Willett WC, Obesity, fat distribution, and weight gain as risk factors for clinical diabetes in men, Diabetes Care 17 (1994) 961–969. [DOI] [PubMed] [Google Scholar]
- [30].McLaughlin T, Abbasi F, Cheal K, Chu J, Lamendola C, Reaven GM, Use of metabolic markers to identify overweight individuals who are insulin resistant, Ann. Int. Med 139 (2003) 802–809. [DOI] [PubMed] [Google Scholar]
- [31].Chen DL, Liess C, Poljak A, Xu A, Zhang J, Thoma C, Trenell M, Milner B, Jenkins AB, Chisholm DJ, Samocha-Bonet D, Greenfield JR, Phenotypic characterization of insulin-resistant and insulin-sensitive obesity., J. Clin. Endocrinol. Metab 100(11) (2015) 4082–4091. [DOI] [PubMed] [Google Scholar]
- [32].Samocha-Bonet D, Dixit VD, Kahn CR, Leibel RL, Lin X, Nieuwdorp M, Pietiläinen KH, Rabasa-Lhoret R, Roden M, Scherer PE, et al. , Metabolically healthy and unhealthy obese--the 2013 Stock Conference report, Obes. Rev 15 (2014) 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Smith GI, Mittendorfer B, Klein S, Metabolically healthy obesity: facts and fantasies, J Clin Invest 129(10) (2019) 3978–3989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Blüher M, Metabolically Healthy Obesity, Endocr Rev 41(3) (2020) 405–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Zhou Z, Macpherson J, Gray SR, Gill JMR, Welsh P, Celis-Morales C, Sattar N, Pell JP, Ho FK, Are people with metabolically healthy obesity really healthy? A prospective cohort study of 381,363 UK Biobank participants, Diabetologia 64(9) (2021) 1963–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Abbasi F, Chu JW, Lamendola C, McLaughlin T, Hayden J, Reaven GM, Reaven PD, Discrimination between obesity and insulin resistance in the relationship with adiponectin, Diabetes 53(3) (2004) 585–590. [DOI] [PubMed] [Google Scholar]
- [37].Voulgari C, Tentolouris N, Dilaveris P, Tousoulis D, Katsilambros N, Stefanadis C, Increased heart failure risk in normal-weight people with metabolic syndrome compared with metabolically healthy obese individuals, J. Am. Coll. Cardiol 58(13) (2011) 1343–1350. [DOI] [PubMed] [Google Scholar]
- [38].Araújo J, Cai J, Stevens J, Prevalence of Optimal Metabolic Health in American Adults: National Health and Nutrition Examination Survey 2009-2016, Metab. Syndr. Relat. Disord 17(1) (2019) 46–52. [DOI] [PubMed] [Google Scholar]
- [39].Thomas EL, Fitzpatrick JA, Malik SJ, Taylor-Robinson SD, Bell JD, Whole body fat: content and distribution, Prog. Nucl. Magn. Reson. Spectrosc 73 (2013) 56–80. [DOI] [PubMed] [Google Scholar]
- [40].Rosenbloom AL, Guevara Aguirre J, Rosenfeld RG, Fielder PJ, The little women of Loja--growth hormone-receptor deficiency in an inbred population of southern Ecuador, N Engl J Med. 323(20) (1990) 1367–1374. [DOI] [PubMed] [Google Scholar]
- [41].Chehab FF, Obesity and lipodystrophy--where do the circles intersect?, Endocrinology 149(3) (2008) 925–934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Schwartz MW, Seeley RJ, Zeltser LM, Drewnowski A, Ravussin E, Redman LM, Leibel RL, Obesity Pathogenesis: An Endocrine Society Scientific Statement, Endocrine reviews 38(4) (2017) 267–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Basu S, Yoffe P, Hills N, Lustig RH, The relationship of sugar to population-level diabetes prevalence: an econometric analysis of repeated cross-sectional data, PLoS One 8(2) (2013) e57873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Sepúlveda J, Murray C, The state of global health in 2014, Science 345(6202) (2014) 1275–1278. [DOI] [PubMed] [Google Scholar]
- [45].Park YW, Zhu S, Palaniappan L, Heshka S, Carnethon MR, Heymsfield SB, The metabolic syndrome: prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988-1994, Archives of internal medicine 163(4) (2003) 427–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Gallagher EJ, LeRoith D, Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality, Physiological reviews 95(3) (2015) 727–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Calle EE, Thun MJ, Petrelli JM, Rodriguez C, Heath CW Jr., Body-mass index and mortality in a prospective cohort of U.S. adults, The New England journal of medicine 341(15) (1999) 1097–105. [DOI] [PubMed] [Google Scholar]
- [48].Wildman RP, Muntner P, Reynolds K, McGinn AP, Rajpathak S, Wylie-Rosett J, Sowers MR, The obese without cardiometabolic risk factor clustering and the normal weight with cardiometabolic risk factor clustering: prevalence and correlates of 2 phenotypes among the US population (NHANES 1999-2004), Archives of internal medicine 168(15) (2008) 1617–24. [DOI] [PubMed] [Google Scholar]
- [49].Dempster P, Aitkens S, A new air displacement method for the determination of human body composition, Med Sci Sports Exerc 27(12) (1995) 1692–7. [PubMed] [Google Scholar]
- [50].Ibrahim MM, Subcutaneous and visceral adipose tissue: structural and functional differences, Obes Rev 11(1) (2010) 11–8. [DOI] [PubMed] [Google Scholar]
- [51].Ter Horst R, van den Munckhof ICL, Schraa K, Aguirre-Gamboa R, Jaeger M, Smeekens SP, Brand T, Lemmers H, Dijkstra H, Galesloot TE, de Graaf J, Xavier RJ, Li Y, Joosten LAB, Rutten JHW, Netea MG, Riksen NP, Sex-Specific Regulation of Inflammation and Metabolic Syndrome in Obesity, Arterioscler Thromb Vase Biol 40(7) (2020) 1787–1800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Torre YS, Wadeea R, Rosas V, Herbst KL, Lipedema: friend and foe, Hormone molecular biology and clinical investigation 33(1) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Porter SA, Massaro JM, Hoffmann U, Vasan RS, O’Donnel CJ, Fox CS, Abdominal subcutaneous adipose tissue: a protective fat depot?, Diabetes care 32(6) (2009) 1068–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Mohammed BS, Cohen S, Reeds D, Young VL, Klein S, Long-term effects of large-volume liposuction on metabolic risk factors for coronary heart disease, Obesity (Silver Spring) 16(12) (2008) 2648–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Bastien M, Poirier P, Brassard P, Arsenault BJ, Bertrand OF, Després JP, Costerousse O, Piché ME, Effect of PPARγ agonist on aerobic exercise capacity in relation to body fat distribution in men with type 2 diabetes mellitus and coronary artery disease: a 1-yr randomized study, American journal of physiology. Endocrinology and metabolism 317(1) (2019) E65–e73. [DOI] [PubMed] [Google Scholar]
- [56].Kabir M, Catalano KJ, Ananthnarayan S, Kim SP, Van Citters GW, Dea MK, Bergman RN, Molecular evidence supporting the portal theory: a causative link between visceral adiposity and hepatic insulin resistance., Am. J. Physiol. Endocrinol. Metab 288(2) (2004) E454–E461. [DOI] [PubMed] [Google Scholar]
- [57].Gruzdeva OV, Borodkina AD, Akbasheva OE, Dileva YA, Antonova LV, Matveeva VG, Uchasova EG, Ivanov SV, Belik EV, Fanaskova EV, Karetnikova VN, Kokov AN, Barbarash OL, Influence of visceral obesity on the secretion of adipokines with epicardial adipocytes in patients with coronary heart disease, Ter Arkh 90(10) (2018) 71–78. [DOI] [PubMed] [Google Scholar]
- [58].Björntorp P, How should obesity be defined?, J Intern Med 227(3) (1990) 147–9. [DOI] [PubMed] [Google Scholar]
- [59].Ravussin E, Smith SR, Increased fat intake, impaired fat oxidation, and failure of fat cell proliferation result in ectopic fat storage, insulin resistance, and type 2 diabetes mellitus, Ann N Y Acad Sci 967 (2002) 363–78. [DOI] [PubMed] [Google Scholar]
- [60].D’Adamo E, Cali AM, Weiss R, Santoro N, Pierpont B, Northrup V, Caprio S, Central role of fatty liver in the pathogenesis of insulin resistance in obese adolescents, Diabetes Care 33(8) (2010) 1817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Britton KA, Fox CS, Ectopic fat depots and cardiovascular disease, Circulation 124(24) (2011) e837–41. [DOI] [PubMed] [Google Scholar]
- [62].Lustig RH, Mulligan K, Noworolski SM, Gugliucci A, Erkin-Cakmak A, Wen MJ, Tai VW, Schwarz JM, Isocaloric fructose restriction and metabolic improvement in children with obesity and metabolic syndrome, Obesity (Silver Spring) 24 (2016) 453–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Schwarz JM, Noworolski SM, Erkin-Cakmak A, N.J. K, Wen MJ, Tai VW, Jones GM, Palii SP, Velasco-Alin M, Pan K, Patterson BW, Gugliucci A, Lustig RH, Mulligan K, Impact of dietary fructose restriction on liver fat, de novo lipogenesis, and insulin kinetics in children with obesity, Gastroenterology 153 (2017) 743–752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Lee EH, Kim JY, Yang HR, Association between ectopic pancreatic and hepatic fat and metabolic risk factors in children with non-alcoholic fatty liver disease, Pediatric obesity 16(10) (2021) e12793. [DOI] [PubMed] [Google Scholar]
- [65].Isserow JA, Siegelman ES, Mammone J, Focal fatty infiltration of the pancreas: MR characterization with chemical shift imaging, Am. J. Roentgenol 173(5) (1999) 1263–1265. [DOI] [PubMed] [Google Scholar]
- [66].Blaak E, Gender differences in fat metabolism, Current opinion in clinical nutrition and metabolic care 4(6) (2001) 499–502. [DOI] [PubMed] [Google Scholar]
- [67].Kannel WB, Hjortland MC, McNamara PM, Gordon T, Menopause and risk of cardiovascular disease: the Framingham study, Ann Intern Med 85(4) (1976) 447–52. [DOI] [PubMed] [Google Scholar]
- [68].Derby CA, Crawford SL, Pasternak RC, Sowers M, Sternfeld B, Matthews KA, Lipid changes during the menopause transition in relation to age and weight: the Study of Women’s Health Across the Nation, Am J Epidemiol 169(11) (2009) 1352–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Qian S, Tang Y, Tang Q-Q, Adipose tissue plasticity and the pleiotropic roles of BMP signaling, The Journal of biological chemistry 296 (2021) 100678–100678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Smith U, Kahn BB, Adipose tissue regulates insulin sensitivity: role of adipogenesis, de novo lipogenesis and novel lipids, Journal of Internal Medicine 280(5) (2016) 465–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Arner P, Fat Tissue Growth and Development in Humans, Nestle Nutrition Institute workshop series 89 (2018) 37–45. [DOI] [PubMed] [Google Scholar]
- [72].Spalding KL, Arner E, Westermark PO, Bernard S, Buchholz BA, Bergmann O, Blomqvist L, Hoffstedt J, Naslund E, Britton T, Concha H, Hassan M, Ryden M, Frisen J, Arner P, Dynamics of fat cell turnover in humans, Nature 453(7196) (2008) 783–7. [DOI] [PubMed] [Google Scholar]
- [73].Feldman BJ, Streeper RS, Farese RV Jr., Yamamoto KR, Myostatin modulates adipogenesis to generate adipocytes with favorable metabolic effects, Proceedings of the National Academy of Sciences of the United States of America 103(42) (2006) 15675–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Lynes MD, Tseng Y-H, Deciphering adipose tissue heterogeneity, Annals of the New York Academy of Sciences 1411(1) (2018) 5–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Ussar S, Lee KY, Dankel SN, Boucher J, Haering MF, Kleinridders A, Thomou T, Xue R, Macotela Y, Cypess AM, Tseng YH, Mellgren G, Kahn CR, ASC-1, PAT2, and P2RX5 are cell surface markers for white, beige, and brown adipocytes, Sci Transl Med 6(247) (2014) 247ra103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cinti S, Pink Adipocytes, Trends in endocrinology and metabolism: TEM 29(9) (2018) 651–666. [DOI] [PubMed] [Google Scholar]
- [77].Funcke J-B, Scherer PE, Beyond adiponectin and leptin: adipose tissue-derived mediators of interorgan communication, Journal of lipid research 60(10) (2019) 1648–1684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Heinonen S, Jokinen R, Rissanen A, Pietiläinen KH, White adipose tissue mitochondrial metabolism in health and in obesity, Obesity Reviews 21(2) (2020) e12958. [DOI] [PubMed] [Google Scholar]
- [79].Vishvanath L, Gupta RK, Contribution of adipogenesis to healthy adipose tissue expansion in obesity, J Clin Invest 129(10) (2019) 4022–4031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Goossens GH, The Metabolic Phenotype in Obesity: Fat Mass, Body Fat Distribution, and Adipose Tissue Function, Obesity facts 10(3) (2017) 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Carpentier AC, Blondin DP, Virtanen KA, Richard D, Haman F, Turcotte ÉE, Brown Adipose Tissue Energy Metabolism in Humans, Frontiers in endocrinology 9 (2018) 447–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Cypess AM, Chen Y-C, Sze C, Wang K, English J, Chan O, Holman AR, Tal I, Palmer MR, Kolodny GM, Kahn CR, Cold but not sympathomimetics activates human brown adipose tissue in vivo, Proceedings of the National Academy of Sciences of the United States of America 109(25) (2012) 10001–10005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Sidossis L, Kajimura S, Brown and beige fat in humans: thermogenic adipocytes that control energy and glucose homeostasis, The Journal of clinical investigation 125(2) (2015) 478–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, Floering LM, Spiegelman BM, Collins S, p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene, Molecular and cellular biology 24(7) (2004) 3057–3067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Guilherme A, Yenilmez B, Bedard AH, Henriques F, Liu D, Lee A, Goldstein L, Kelly M, Nicoloro SM, Chen M, Weinstein L, Collins S, Czech MP, Control of Adipocyte Thermogenesis and Lipogenesis through β3-Adrenergic and Thyroid Hormone Signal Integration, Cell reports 31(5) (2020) 107598–107598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Seale P, Bjork B, Yang W, Kajimura S, Chin S, Kuang S, Scimè A, Devarakonda S, Conroe HM, Erdjument-Bromage H, Tempst P, Rudnicki MA, Beier DR, Spiegelman BM, PRDM16 controls a brown fat/skeletal muscle switch, Nature 454(7207) (2008) 961–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Carobbio S, Guenantin A-C, Bahri M, Rodriguez-Fdez S, Honig F, Kamzolas I, Samuelson I, Long K, Awad S, Lukovic D, Erceg S, Bassett A, Mendjan S, Vallier L, Rosen BS, Chiarugi D, Vidal-Puig A, Unraveling the Developmental Roadmap toward Human Brown Adipose Tissue, Stem Cell Reports 16(3) (2021) 641–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Oguri Y, Shinoda K, Kim H, Alba DL, Bolus WR, Wang Q, Brown Z, Pradhan RN, Tajima K, Yoneshiro T, Ikeda K, Chen Y, Cheang RT, Tsujino K, Kim CR, Greiner VJ, Datta R, Yang CD, Atabai K, McManus MT, Koliwad SK, Spiegelman BM, Kajimura S, CD81 Controls Beige Fat Progenitor Cell Growth and Energy Balance via FAK Signaling, Cell 182(3) (2020) 563–577.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Chen Y, Ikeda K, Yoneshiro T, Scaramozza A, Tajima K, Wang Q, Kim K, Shinoda K, Sponton CH, Brown Z, Brack A, Kajimura S, Thermal stress induces glycolytic beige fat formation via a myogenic state, Nature 565(7738) (2019) 180–185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Barbatelli G, Murano I, Madsen L, Hao Q, Jimenez M, Kristiansen K, Giacobino JP, De Matteis R, Cinti S, The emergence of cold-induced brown adipocytes in mouse white fat depots is determined predominantly by white to brown adipocyte transdifferentiation, American journal of physiology. Endocrinology and metabolism 298(6) (2010) E1244–53. [DOI] [PubMed] [Google Scholar]
- [91].Pilkington A-C, Paz HA, Wankhade UD, Beige Adipose Tissue Identification and Marker Specificity-Overview, Frontiers in endocrinology 12 (2021) 599134–599134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Stine RR, Shapira SN, Lim H-W, Ishibashi J, Harms M, Won K-J, Seale P, EBF2 promotes the recruitment of beige adipocytes in white adipose tissue, Molecular metabolism 5(1) (2015) 57–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Heeren J, Münzberg H, Novel aspects of brown adipose tissue biology, Endocrinology and metabolism clinics of North America 42(1) (2013) 89–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Moseti D, Regassa A, Kim W-K, Molecular Regulation of Adipogenesis and Potential Anti-Adipogenic Bioactive Molecules, International journal of molecular sciences 17(1) (2016) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Lehrke M, Pascual G, Glass CK, Lazar MA, Gaining weight: the Keystone Symposium on PPAR and LXR, Genes & development 19(15) (2005) 1737–1742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Chawla A, Schwarz EJ, Dimaculangan DD, Lazar MA, Peroxisome proliferator-activated receptor (PPAR) gamma: adipose-predominant expression and induction early in adipocyte differentiation, Endocrinology 135(2) (1994) 798–800. [DOI] [PubMed] [Google Scholar]
- [97].Lehmann JM, Moore LB, Smith-Oliver TA, Wilkison WO, Willson TM, Kliewer SA, An antidiabetic thiazolidinedione is a high affinity ligand for peroxisome proliferator-activated receptor y (PPARy), The Journal of biological chemistry 270(22) (1995) 12953–12956. [DOI] [PubMed] [Google Scholar]
- [98].Farmer SR, Transcriptional control of adipocyte formation, Cell Metab 4(4) (2006) 263–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Vigouroux C, Fajas L, Khallouf E, Meier M, Gyapay G, Lascols O, Auwerx J, Weissenbach J, Capeau J, Magré J, Human peroxisome proliferator-activated receptor-gamma2: genetic mapping, identification of a variant in the coding sequence, and exclusion as the gene responsible for lipoatrophic diabetes, Diabetes 47(3) (1998) 490–2. [DOI] [PubMed] [Google Scholar]
- [100].Rosen ED, Sarraf P, Troy AE, Bradwin G, Moore K, Milstone DS, Spiegelman BM, Mortensen RM, PPAR gamma is required for the differentiation of adipose tissue in vivo and in vitro, Mol Cell 4(4) (1999) 611–7. [DOI] [PubMed] [Google Scholar]
- [101].Brun RP, Tontonoz P, Forman BM, Ellis R, Chen J, Evans RM, Spiegelman BM, Differential activation of adipogenesis by multiple PPAR isoforms, Genes Dev 10(8) (1996) 974–84. [DOI] [PubMed] [Google Scholar]
- [102].Wang YX, Lee CH, Tiep S, Yu RT, Ham J, Kang H, Evans RM, Peroxisome-proliferator-activated receptor delta activates fat metabolism to prevent obesity, Cell 113(2) (2003) 159–70. [DOI] [PubMed] [Google Scholar]
- [103].Peters JM, Lee SS, Li W, Ward JM, Gavrilova O, Everett C, Reitman ML, Hudson LD, Gonzalez FJ, Growth, adipose, brain, and skin alterations resulting from targeted disruption of the mouse peroxisome proliferator-activated receptor beta(delta), Molecular and cellular biology 20(14) (2000) 5119–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Guerre-Millo M, Gervois P, Raspe E, Madsen L, Poulain P, Derudas B, Herbert JM, Winegar DA, Willson TM, Fruchart JC, Berge RK, Staels B, Peroxisome proliferator-activated receptor alpha activators improve insulin sensitivity and reduce adiposity, The Journal of biological chemistry 275(22) (2000) 16638–42. [DOI] [PubMed] [Google Scholar]
- [105].Jeong S, Yoon M, Fenofibrate inhibits adipocyte hypertrophy and insulin resistance by activating adipose PPARalpha in high fat diet-induced obese mice, Exp Mol Med 41(6) (2009) 397–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Tsuchida A, Yamauchi T, Takekawa S, Hada Y, Ito Y, Maki T, Kadowaki T, Peroxisome proliferator-activated receptor (PPAR)alpha activation increases adiponectin receptors and reduces obesity-related inflammation in adipose tissue: comparison of activation of PPARalpha, PPARgamma, and their combination, Diabetes 54(12) (2005) 3358–70. [DOI] [PubMed] [Google Scholar]
- [107].Das M, Irvin MR, Sha J, Aslibekyan S, Hidalgo B, Perry RT, Zhi D, Tiwari HK, Absher D, Ordovas JM, Arnett DK, Lipid changes due to fenofibrate treatment are not associated with changes in DNA methylation patterns in the GOLDN study, Frontiers in genetics 6 (2015) 304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Forcheron F, Cachefo A, Thevenon S, Pinteur C, Beylot M, Mechanisms of the triglyceride- and cholesterol-lowering effect of fenofibrate in hyperlipidemic type 2 diabetic patients, Diabetes 51(12) (2002) 3486–91. [DOI] [PubMed] [Google Scholar]
- [109].Oliver WR Jr., Shenk JL, Snaith MR, Russell CS, Plunket KD, Bodkin NL, Lewis MC, Winegar DA, Sznaidman ML, Lambert MH, Xu HE, Sternbach DD, Kliewer SA, Hansen BC, Willson TM, A selective peroxisome proliferator-activated receptor delta agonist promotes reverse cholesterol transport, Proceedings of the National Academy of Sciences of the United States of America 98(9) (2001) 5306–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [110].Kassotis CD, Masse L, Kim S, Schlezinger JJ, Webster TF, Stapleton HM, Characterization of adipogenic chemicals in three different cell culture systems: implications for reproducibility based on cell source and handling, Scientific reports 7 (2017) 42104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [111].Tontonoz P, Singer S, Forman BM, Sarraf P, Fletcher JA, Fletcher CD, Brun RP, Mueller E, Altiok S, Oppenheim H, Evans RM, Spiegelman BM, Terminal differentiation of human liposarcoma cells induced by ligands for peroxisome proliferator-activated receptor gamma and the retinoid X receptor, Proceedings of the National Academy of Sciences of the United States of America 94(1) (1997) 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Canan Koch SS, Dardashti LJ, Cesario RM, Croston GE, Boehm MF, Heyman RA, Nadzan AM, Synthesis of retinoid X receptor-specific ligands that are potent inducers of adipogenesis in 3T3-L1 cells, J Med Chem 42(4) (1999) 742–50. [DOI] [PubMed] [Google Scholar]
- [113].Nielsen R, Pedersen TA, Hagenbeek D, Moulos P, Siersbaek R, Megens E, Denissov S, Borgesen M, Francoijs KJ, Mandrup S, Stunnenberg HG, Genome-wide profiling of PPARgamma:RXR and RNA polymerase II occupancy reveals temporal activation of distinct metabolic pathways and changes in RXR dimer composition during adipogenesis, Genes Dev 22(21) (2008) 2953–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Shoucri BM, Martinez ES, Abreo TJ, Hung VT, Moosova Z, Shioda T, Blumberg B, Retinoid X Receptor Activation Alters the Chromatin Landscape To Commit Mesenchymal Stem Cells to the Adipose Lineage, Endocrinology 158(10) (2017) 3109–3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Shoucri BM, Hung VT, Chamorro-García R, Shioda T, Blumberg B, Retinoid X Receptor Activation During Adipogenesis of Female Mesenchymal Stem Cells Programs a Dysfunctional Adipocyte, Endocrinology 159(8) (2018) 2863–2883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Sucov HM, Dyson E, Gumeringer CL, Price J, Chien KR, Evans RM, RXR alpha mutant mice establish a genetic basis for vitamin A signaling in heart morphogenesis, Genes Dev 8(9) (1994) 1007–18. [DOI] [PubMed] [Google Scholar]
- [117].Imai T, Jiang M, Chambon P, Metzger D, Impaired adipogenesis and lipolysis in the mouse upon selective ablation of the retinoid X receptor alpha mediated by a tamoxifen-inducible chimeric Cre recombinase (Cre-ERT2) in adipocytes, Proceedings of the National Academy of Sciences of the United States of America 98(1) (2001) 224–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Mukherjee R, Davies PJ, Crombie DL, Bischoff ED, Cesario RM, Jow L, Hamann LG, Boehm MF, Mondon CE, Nadzan AM, Paterniti JR Jr., Heyman RA, Sensitization of diabetic and obese mice to insulin by retinoid X receptor agonists, Nature 386(6623) (1997) 407–10. [DOI] [PubMed] [Google Scholar]
- [119].Sadasivuni MK, Reddy BM, Singh J, Anup MO, Sunil V, Lakshmi MN, Yogeshwari S, Chacko SK, Pooja TL, Dandu A, Harish C, Gopala AS, Pratibha S, Naveenkumar BS, Pallavi PM, Verma MK, Moolemath Y, Somesh BP, Venkataranganna MV, Jagannath MR, CNX-013-B2, a unique pan tissue acting rexinoid, modulates several nuclear receptors and controls multiple risk factors of the metabolic syndrome without risk of hypertriglyceridemia, hepatomegaly and body weight gain in animal models, Diabetol Metab Syndr 6(1) (2014) 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Emilsson V, O’Dowd J, Wang S, Liu YL, Sennitt M, Heyman R, Cawthorne MA, The effects of rexinoids and rosiglitazone on body weight and uncoupling protein isoform expression in the Zucker fa/fa rat, Metabolism 49(12) (2000) 1610–5. [DOI] [PubMed] [Google Scholar]
- [121].Farol LT, Hymes KB, Bexarotene: a clinical review, Expert Rev Anticancer Ther 4(2) (2004) 180–8. [DOI] [PubMed] [Google Scholar]
- [122].de Vries-van der Weij J, de Haan W, Hu L, Kuif M, Oei HL, van der Hoorn JW, Havekes LM, Princen HM, Romijn JA, Smit JW, Rensen PC, Bexarotene induces dyslipidemia by increased very low-density lipoprotein production and cholesteryl ester transfer protein-mediated reduction of high-density lipoprotein, Endocrinology 150(5) (2009) 2368–75. [DOI] [PubMed] [Google Scholar]
- [123].Pinaire JA, Reifel-Miller A, Therapeutic potential of retinoid x receptor modulators for the treatment of the metabolic syndrome, PPAR Res 2007 (2007) 94156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Shulman AI, Mangelsdorf DJ, Retinoid x receptor heterodimers in the metabolic syndrome, The New England journal of medicine 353(6) (2005) 604–15. [DOI] [PubMed] [Google Scholar]
- [125].Ulven SM, Dalen KT, Gustafsson JA, Nebb HI, LXR is crucial in lipid metabolism, Prostaglandins Leukot Essent Fatty Acids 73(1) (2005) 59–63. [DOI] [PubMed] [Google Scholar]
- [126].Kalaany NY, Gauthier KC, Zavacki AM, Mammen PP, Kitazume T, Peterson JA, Horton JD, Garry DJ, Bianco AC, Mangelsdorf DJ, LXRs regulate the balance between fat storage and oxidation, Cell Metab 1(4) (2005) 231–44. [DOI] [PubMed] [Google Scholar]
- [127].Seo JB, Moon HM, Kim WS, Lee YS, Jeong HW, Yoo EJ, Ham J, Kang H, Park MG, Steffensen KR, Stulnig TM, Gustafsson JA, Park SD, Kim JB, Activated liver X receptors stimulate adipocyte differentiation through induction of peroxisome proliferator-activated receptor gamma expression, Mol Cell Biol 24(8) (2004) 3430–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Juvet LK, Andresen SM, Schuster GU, Dalen KT, Tobin KA, Hollung K, Haugen F, Jacinto S, Ulven SM, Bamberg K, Gustafsson JA, Nebb HI, On the role of liver X receptors in lipid accumulation in adipocytes, Mol Endocrinol 17(2) (2003) 172–82. [DOI] [PubMed] [Google Scholar]
- [129].Stenson BM, Ryden M, Venteclef N, Dahlman I, Pettersson AM, Mairal A, Astrom G, Blomqvist L, Wang V, Jocken JW, Clement K, Langin D, Arner P, Laurencikiene J, Liver X receptor (LXR) regulates human adipocyte lipolysis, J Biol Chem 286(1) (2011) 370–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Gerin I, Dolinsky VW, Shackman JG, Kennedy RT, Chiang SH, Burant CF, Steffensen KR, Gustafsson JA, MacDougald OA, LXRbeta is required for adipocyte growth, glucose homeostasis, and beta cell function, J Biol Chem 280(24) (2005) 23024–31. [DOI] [PubMed] [Google Scholar]
- [131].Korach-Andre M, Archer A, Barros RP, Parini P, Gustafsson JA, Both liver-X receptor (LXR) isoforms control energy expenditure by regulating brown adipose tissue activity, Proceedings of the National Academy of Sciences of the United States of America 108(1) (2011) 403–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Archer A, Stolarczyk E, Doria ML, Helguero L, Domingues R, Howard JK, Mode A, Korach-Andre M, Gustafsson JA, LXR activation by GW3965 alters fat tissue distribution and adipose tissue inflammation in ob/ob female mice, J Lipid Res 54(5) (2013) 1300–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Dahlman I, Nilsson M, Jiao H, Hoffstedt J, Lindgren CM, Humphreys K, Kere J, Gustafsson JA, Arner P, Dahlman-Wright K, Liver X receptor gene polymorphisms and adipose tissue expression levels in obesity, Pharmacogenet Genomics 16(12) (2006) 881–9. [DOI] [PubMed] [Google Scholar]
- [134].Kirchgessner TG, Sleph P, Ostrowski J, Lupisella J, Ryan CS, Liu X, Fernando G, Grimm D, Shipkova P, Zhang R, Garcia R, Zhu J, He A, Malone H, Martin R, Behnia K, Wang Z, Barrett YC, Garmise RJ, Yuan L, Zhang J, Gandhi MD, Wastall P, Li T, Du S, Salvador L, Mohan R, Cantor GH, Kick E, Lee J, Frost RJ, Beneficial and Adverse Effects of an LXR Agonist on Human Lipid and Lipoprotein Metabolism and Circulating Neutrophils, Cell Metab 24(2) (2016) 223–33. [DOI] [PubMed] [Google Scholar]
- [135].Gao J, Xie W, Targeting xenobiotic receptors PXR and CAR for metabolic diseases, Trends Pharmacol Sci 33(10) (2012) 552–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Moreau A, Vilarem MJ, Maurel P, Pascussi JM, Xenoreceptors CAR and PXR activation and consequences on lipid metabolism, glucose homeostasis, and inflammatory response, Mol Pharm 5(1) (2008) 35–41. [DOI] [PubMed] [Google Scholar]
- [137].Zhou J, Febbraio M, Wada T, Zhai Y, Kuruba R, He J, Lee JH, Khadem S, Ren S, Li S, Silverstein RL, Xie W, Hepatic fatty acid transporter Cd36 is a common target of LXR, PXR, and PPARgamma in promoting steatosis, Gastroenterology 134(2) (2008) 556–67. [DOI] [PubMed] [Google Scholar]
- [138].He J, Gao J, Xu M, Ren S, Stefanovic-Racic M, O’Doherty RM, Xie W, PXR ablation alleviates diet-induced and genetic obesity and insulin resistance in mice, Diabetes 62(6) (2013) 1876–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Wada T, Gao J, Xie W, PXR and CAR in energy metabolism, Trends in endocrinology and metabolism: TEM 20(6) (2009) 273–9. [DOI] [PubMed] [Google Scholar]
- [140].Gao J, He J, Zhai Y, Wada T, Xie W, The constitutive androstane receptor is an anti-obesity nuclear receptor that improves insulin sensitivity, J Biol Chem 284(38) (2009) 25984–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Jiao Y, Lu Y, Li XY, Farnesoid X receptor: a master regulator of hepatic triglyceride and glucose homeostasis, Acta Pharmacol Sin 36(1) (2015) 44–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Prawitt J, Caron S, Staels B, How to modulate FXR activity to treat the Metabolic Syndrome, Drug Discov Today: Disease Mechanisms 6(1-4) (2009) e55–e64. [Google Scholar]
- [143].Rizzo G, Disante M, Mencarelli A, Renga B, Gioiello A, Pellicciari R, Fiorucci S, The farnesoid X receptor promotes adipocyte differentiation and regulates adipose cell function in vivo, Mol Pharmacol 70(4) (2006) 1164–73. [DOI] [PubMed] [Google Scholar]
- [144].Cariou B, van Harmelen K, Duran-Sandoval D, van Dijk TH, Grefhorst A, Abdelkarim M, Caron S, Torpier G, Fruchart JC, Gonzalez FJ, Kuipers F, Staels B, The farnesoid X receptor modulates adiposity and peripheral insulin sensitivity in mice, J Biol Chem 281(16) (2006) 11039–49. [DOI] [PubMed] [Google Scholar]
- [145].Yang JY, Della-Fera MA, Baile CA, Guggulsterone inhibits adipocyte differentiation and induces apoptosis in 3T3-L1 cells, Obesity (Silver Spring) 16(1) (2008) 16–22. [DOI] [PubMed] [Google Scholar]
- [146].Abdelkarim M, Caron S, Duhem C, Prawitt J, Dumont J, Lucas A, Bouchaert E, Briand O, Brozek J, Kuipers F, Fievet C, Cariou B, Staels B, The farnesoid X receptor regulates adipocyte differentiation and function by promoting peroxisome proliferator-activated receptor-gamma and interfering with the Wnt/beta-catenin pathways, J Biol Chem 285(47) (2010) 36759–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [147].Prawitt J, Abdelkarim M, Stroeve JH, Popescu I, Duez H, Velagapudi VR, Dumont J, Bouchaert E, van Dijk TH, Lucas A, Dorchies E, Daoudi M, Lestavel S, Gonzalez FJ, Oresic M, Cariou B, Kuipers F, Caron S, Staels B, Farnesoid X receptor deficiency improves glucose homeostasis in mouse models of obesity, Diabetes 60(7) (2011) 1861–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [148].Zhang Y, Ge X, Heemstra LA, Chen WD, Xu J, Smith JL, Ma H, Kasim N, Edwards PA, Novak CM, Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice, Mol Endocrinol 26(2) (2012) 272–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Maneschi E, Vignozzi L, Morelli A, Mello T, Filippi S, Cellai I, Comeglio P, Sarchielli E, Calcagno A, Mazzanti B, Vettor R, Vannelli GB, Adorini L, Maggi M, FXR activation normalizes insulin sensitivity in visceral preadipocytes of a rabbit model of MetS, The Journal of endocrinology 218(2) (2013) 215–31. [DOI] [PubMed] [Google Scholar]
- [150].Watanabe M, Horai Y, Houten SM, Morimoto K, Sugizaki T, Arita E, Mataki C, Sato H, Tanigawara Y, Schoonjans K, Itoh H, Auwerx J, Lowering bile acid pool size with a synthetic farnesoid X receptor (FXR) agonist induces obesity and diabetes through reduced energy expenditure, J Biol Chem 286(30) (2011) 26913–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [151].Rader DJ, Liver X Receptor and Farnesoid X Receptor as Therapeutic Targets, Am J Cardiology 100(11A) (2007) 15N–19N. [DOI] [PubMed] [Google Scholar]
- [152].Lu Y, Ma Z, Zhang Z, Xiong X, Wang X, Zhang H, Shi G, Xia X, Ning G, Li X, Yin Yang 1 promotes hepatic steatosis through repression of farnesoid X receptor in obese mice, Gut 63(1) (2014) 170–8. [DOI] [PubMed] [Google Scholar]
- [153].McIntosh BE, Hogenesch JB, Bradfield CA, Mammalian Per-Arnt-Sim proteins in environmental adaptation, Annual review of physiology 72 (2010) 625–45. [DOI] [PubMed] [Google Scholar]
- [154].de Almeida DC, Evangelista LSM, Câmara NOS, Role of aryl hydrocarbon receptor in mesenchymal stromal cell activation: A minireview, World J Stem Cells 9(9) (2017) 152–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Henderson Colin J., McLaughlin Lesley A., Osuna-Cabello M, Taylor M, Gilbert I, McLaren Aileen W., Wolf CR, Application of a novel regulatable Cre recombinase system to define the role of liver and gut metabolism in drug oral bioavailability, Biochemical Journal 465(3) (2015) 479–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [156].Shimizu Y, Nakatsuru Y, Ichinose M, Takahashi Y, Kume H, Mimura J, Fujii-Kuriyama Y, Ishikawa T, Benzo[a]pyrene carcinogenicity is lost in mice lacking the aryl hydrocarbon receptor, Proceedings of the National Academy of Sciences 97(2) (2000) 779–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Nguyen P, Leray V, Diez M, Serisier S, Le Bloc’h J, Siliart B, Dumon H, Liver lipid metabolism, Journal of animal physiology and animal nutrition 92(3) (2008) 272–83. [DOI] [PubMed] [Google Scholar]
- [158].Tanos R, Patel RD, Murray IA, Smith PB, Patterson AD, Perdew GH, Aryl hydrocarbon receptor regulates the cholesterol biosynthetic pathway in a dioxin response element-independent manner, Hepatology (Baltimore, Md.) 55(6) (2012) 1994–2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [159].Tanos R, Murray IA, Smith PB, Patterson A, Perdew GH, Role of the Ah receptor in homeostatic control of fatty acid synthesis in the liver, Toxicological sciences : an official journal of the Society of Toxicology 129(2) (2012) 372–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Girer NG, Carter D, Bhattarai N, Mustafa M, Denner L, Porter C, Elferink CJ, Inducible Loss of the Aryl Hydrocarbon Receptor Activates Perigonadal White Fat Respiration and Brown Fat Thermogenesis via Fibroblast Growth Factor 21, International journal of molecular sciences 20(4) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Beltrand J, Busiah K, Vaivre-Douret L, Fauret AL, Berdugo M, Cavé H, Polak M, Neonatal Diabetes Mellitus, Front Pediatr 8 (2020) 540718–540718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [162].Plamper M, Gohlke B, Schreiner F, Woelfle J, Mecasermin in Insulin Receptor-Related Severe Insulin Resistance Syndromes: Case Report and Review of the Literature, International journal of molecular sciences 19(5) (2018) 1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [163].Boucher J, Softic S, El Ouaamari A, Krumpoch MT, Kleinridders A, Kulkarni RN, O’Neill BT, Kahn CR, Differential Roles of Insulin and IGF-1 Receptors in Adipose Tissue Development and Function, Diabetes 65(8) (2016) 2201–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [164].Homan EP, Brandão BB, Softic S, El Ouaamari A, O’Neill BT, Kulkarni RN, Kim JK, Kahn CR, Differential roles of FOXO transcription factors on insulin action in brown and white adipose tissue, J Clin Invest 131(19) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Nadal A, Ropero AB, Laribi O, Maillet M, Fuentes E, Soria B, Nongenomic actions of estrogens and xenoestrogens by binding at a plasma membrane receptor unrelated to estrogen receptor alpha and estrogen receptor beta, Proceedings of the National Academy of Sciences of the United States of America 97(21) (2000) 11603–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [166].Fuentes N, Silveyra P, Estrogen receptor signaling mechanisms, Adv Protein Chem Struct Biol 116 (2019) 135–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Jasik CB, Lustig RH, Adolescent obesity and puberty: the “perfect storm”, Ann N Y Acad Sci 1135 (2008) 265–79. [DOI] [PubMed] [Google Scholar]
- [168].Crocker MK, Stern EA, Sedaka NM, Shomaker LB, Brady SM, Ali AH, Shawker TH, Hubbard VS, Yanovski JA, Sexual dimorphisms in the associations of BMI and body fat with indices of pubertal development in girls and boys, The Journal of clinical endocrinology and metabolism 99(8) (2014) E1519–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Davis SR, Castelo-Branco C, Chedraui P, Lumsden MA, Nappi RE, Shah D, Villaseca P, Understanding weight gain at menopause, Climacteric 15 (2012) 419–429. [DOI] [PubMed] [Google Scholar]
- [170].Cooke PS, Naaz A, Role of estrogens in adipocyte development and function, Exp Biol Med (Maywood) 229(11) (2004) 1127–35. [DOI] [PubMed] [Google Scholar]
- [171].Dieudonne MN, Pecquery R, Leneveu MC, Giudicelli Y, Opposite effects of androgens and estrogens on adipogenesis in rat preadipocytes: evidence for sex and site-related specificities and possible involvement of insulin-like growth factor 1 receptor and peroxisome proliferator-activated receptor gamma2, Endocrinology 141(2) (2000) 649–56. [DOI] [PubMed] [Google Scholar]
- [172].Roncari DA, Van RL, Promotion of human adipocyte precursor replication by 17beta-estradiol in culture, J Clin Invest 62(3) (1978) 503–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [173].Heine PA, Taylor JA, Iwamoto GA, Lubahn DB, Cooke PS, Increased adipose tissue in male and female estrogen receptor-alpha knockout mice, Proceedings of the National Academy of Sciences of the United States of America 97(23) (2000) 12729–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Ohlsson C, Hellberg N, Parini P, Vidal O, Bohlooly YM, Rudling M, Lindberg MK, Warner M, Angelin B, Gustafsson JA, Obesity and disturbed lipoprotein profile in estrogen receptor-alpha-deficient male mice, Biochem Biophys Res Commun 278(3) (2000) 640–5. [DOI] [PubMed] [Google Scholar]
- [175].Stubbins RE, Holcomb VB, Hong J, Nunez NP, Estrogen modulates abdominal adiposity and protects female mice from obesity and impaired glucose tolerance, Eur J Nutr 51(7) (2012) 861–70. [DOI] [PubMed] [Google Scholar]
- [176].Murata Y, Robertson KM, Jones ME, Simpson ER, Effect of estrogen deficiency in the male: the ArKO mouse model, Mol Cell Endocrinol 193(1-2) (2002) 7–12. [DOI] [PubMed] [Google Scholar]
- [177].Jones ME, Thorburn AW, Britt KL, Hewitt KN, Misso ML, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER, Aromatase-deficient (ArKO) mice accumulate excess adipose tissue, J Steroid Biochem Mol Biol 79(1-5) (2001) 3–9. [DOI] [PubMed] [Google Scholar]
- [178].Jones ME, Thorburn AW, Britt KL, Hewitt KN, Wreford NG, Proietto J, Oz OK, Leury BJ, Robertson KM, Yao S, Simpson ER, Aromatase-deficient (ArKO) mice have a phenotype of increased adiposity, Proceedings of the National Academy of Sciences of the United States of America 97(23) (2000) 12735–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Hong L, Colpan A, Peptan IA, Modulations of 17-beta estradiol on osteogenic and adipogenic differentiations of human mesenchymal stem cells, Tissue Eng 12(10) (2006) 2747–53. [DOI] [PubMed] [Google Scholar]
- [180].Zhu P, Yuen JM, Sham KW, Cheng CH, GPER mediates the inhibitory actions of estrogen on adipogenesis in 3T3-L1 cells through perturbation of mitotic clonal expansion, General and comparative endocrinology 193 (2013) 19–26. [DOI] [PubMed] [Google Scholar]
- [181].Blouin K, Boivin A, Tchernof A, Androgens and body fat distribution, J Steroid Biochem Mol Biol 108(3-5) (2008) 272–80. [DOI] [PubMed] [Google Scholar]
- [182].O’Reilly MW, House PJ, Tomlinson JW, Understanding androgen action in adipose tissue, J Steroid Biochem Mol Biol 143 (2014) 277–84. [DOI] [PubMed] [Google Scholar]
- [183].Gupta V, Bhasin S, Guo W, Singh R, Miki R, Chauhan P, Choong K, Tchkonia T, Lebrasseur NK, Flanagan JN, Hamilton JA, Viereck JC, Narula NS, Kirkland JL, Jasuja R, Effects of dihydrotestosterone on differentiation and proliferation of human mesenchymal stem cells and preadipocytes, Mol Cell Endocrinol 296(1-2) (2008) 32–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [184].Chazenbalk G, Singh P, Irge D, Shah A, Abbott DH, Dumesic DA, Androgens inhibit adipogenesis during human adipose stem cell commitment to preadipocyte formation, Steroids 78(9) (2013) 920–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [185].Singh R, Artaza JN, Taylor WE, Gonzalez-Cadavid NF, Bhasin S, Androgens stimulate myogenic differentiation and inhibit adipogenesis in C3H 10T1/2 pluripotent cells through an androgen receptor-mediated pathway, Endocrinology 144(11) (2003) 5081–8. [DOI] [PubMed] [Google Scholar]
- [186].Sato T, Matsumoto T, Yamada T, Watanabe T, Kawano H, Kato S, Late onset of obesity in male androgen receptor-deficient (AR KO) mice, Biochem Biophys Res Commun 300(1) (2003) 167–71. [DOI] [PubMed] [Google Scholar]
- [187].Yanase T, Fan W, Kyoya K, Min L, Takayanagi R, Kato S, Nawata H, Androgens and metabolic syndrome: lessons from androgen receptor knock out (ARKO) mice, J Steroid Biochem Mol Biol 109(3-5) (2008) 254–7. [DOI] [PubMed] [Google Scholar]
- [188].Fagman JB, Wilhelmson AS, Motta BM, Pirazzi C, Alexanderson C, De Gendt K, Verhoeven G, Holmang A, Anesten F, Jansson JO, Levin M, Boren J, Ohlsson C, Krettek A, Romeo S, Tivesten A, The androgen receptor confers protection against diet-induced atherosclerosis, obesity, and dyslipidemia in female mice, FASEB journal : official publication of the Federation of American Societies for Experimental Biology 29(4) (2015) 1540–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Yeh S, Tsai MY, Xu Q, Mu XM, Lardy H, Huang KE, Lin H, Yeh SD, Altuwaijri S, Zhou X, Xing L, Boyce BF, Hung MC, Zhang S, Gan L, Chang C, Generation and characterization of androgen receptor knockout (ARKO) mice: an in vivo model for the study of androgen functions in selective tissues, Proceedings of the National Academy of Sciences of the United States of America 99(21) (2002) 13498–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Fan W, Yanase T, Nomura M, Okabe T, Goto K, Sato T, Kawano H, Kato S, Nawata H, Androgen receptor null male mice develop late-onset obesity caused by decreased energy expenditure and lipolytic activity but show normal insulin sensitivity with high adiponectin secretion, Diabetes 54(4) (2005) 1000–8. [DOI] [PubMed] [Google Scholar]
- [191].Blouin K, Nadeau M, Perreault M, Veilleux A, Drolet R, Marceau P, Mailloux J, Luu-The V, Tchernof A, Effects of androgens on adipocyte differentiation and adipose tissue explant metabolism in men and women, Clinical endocrinology 72(2) (2010) 176–88. [DOI] [PubMed] [Google Scholar]
- [192].Mammi C, Calanchini M, Antelmi A, Cinti F, Rosano GM, Lenzi A, Caprio M, Fabbri A, Androgens and adipose tissue in males: a complex and reciprocal interplay, Int J Endocrinol 2012 (2012) 789653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Diamanti-Kandarakis E, Mitrakou A, Raptis S, Tolis G, Duleba AJ, The effect of a pure antiandrogen receptor blocker, flutamide, on the lipid profile in the polycystic ovary syndrome, The Journal of clinical endocrinology and metabolism 83(8) (1998) 2699–705. [DOI] [PubMed] [Google Scholar]
- [194].Ibanez L, Ong K, Ferrer A, Amin R, Dunger D, de Zegher F, Low-dose flutamide-metformin therapy reverses insulin resistance and reduces fat mass in nonobese adolescents with ovarian hyperandrogenism, The Journal of clinical endocrinology and metabolism 88(6) (2003) 2600–6. [DOI] [PubMed] [Google Scholar]
- [195].Ibanez L, De Zegher F, Flutamide-metformin therapy to reduce fat mass in hyperinsulinemic ovarian hyperandrogenism: effects in adolescents and in women on third-generation oral contraception, The Journal of clinical endocrinology and metabolism 88(10) (2003) 4720–4. [DOI] [PubMed] [Google Scholar]
- [196].Pasquali R, Obesity and androgens: facts and perspectives, Fertil Steril 85(5) (2006) 1319–40. [DOI] [PubMed] [Google Scholar]
- [197].Feldman BJ, Glucocorticoids influence on mesenchymal stem cells and implications for metabolic disease, Pediatr Res 65(2) (2009) 249–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Walker BR, Soderberg S, Lindahl B, Olsson T, Independent effects of obesity and cortisol in predicting cardiovascular risk factors in men and women, J Intern Med 247(2) (2000) 198–204. [DOI] [PubMed] [Google Scholar]
- [199].Pickering RT, Lee MJ, Karastergiou K, Gower A, Fried SK, Depot Dependent Effects of Dexamethasone on Gene Expression in Human Omental and Abdominal Subcutaneous Adipose Tissues from Obese Women, PLoS One 11(12) (2016) e0167337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Geer EB, Shen W, Strohmayer E, Post KD, Freda PU, Body composition and cardiovascular risk markers after remission of Cushing’s disease: a prospective study using whole-body MRI, The Journal of clinical endocrinology and metabolism 97(5) (2012) 1702–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [201].Masuzaki H, Paterson J, Shinyama H, Morton NM, Mullins JJ, Seckl JR, Flier JS, A transgenic model of visceral obesity and the metabolic syndrome, Science 294(5549) (2001) 2166–70. [DOI] [PubMed] [Google Scholar]
- [202].John K, Marino JS, Sanchez ER, Hinds TD Jr., The glucocorticoid receptor: cause of or cure for obesity?, American journal of physiology. Endocrinology and metabolism 310(4) (2016) E249–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Contador D, Ezquer F, Espinosa M, Arango-Rodriguez M, Puebla C, Sobrevia L, Conget P, Dexamethasone and rosiglitazone are sufficient and necessary for producing functional adipocytes from mesenchymal stem cells, Exp Biol Med (Maywood) 240(9) (2015) 1235–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Asada M, Rauch A, Shimizu H, Maruyama H, Miyaki S, Shibamori M, Kawasome H, Ishiyama H, Tuckermann J, Asahara H, DNA binding-dependent glucocorticoid receptor activity promotes adipogenesis via Kruppel-like factor 15 gene expression, Lab Invest 91(2) (2011) 203–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [205].Vidal-Puig AJ, Considine RV, Jimenez-Linan M, Werman A, Pories WJ, Caro JF, Flier JS, Peroxisome proliferator-activated receptor gene expression in human tissues. Effects of obesity, weight loss, and regulation by insulin and glucocorticoids, J Clin Invest 99(10) (1997) 2416–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Sargis RM, Johnson DN, Choudhury RA, Brady MJ, Environmental Endocrine Disruptors Promote Adipogenesis in the 3T3-L1 Cell Line through Glucocorticoid Receptor Activation, Obesity (Silver Spring, Md.) 18(7) (2010) 1283–1288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [207].Lee MJ, Fried SK, The glucocorticoid receptor, not the mineralocorticoid receptor, plays the dominant role in adipogenesis and adipokine production in human adipocytes, International journal of obesity (2005) 38(9) (2014) 1228–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [208].Pantoja C, Huff JT, Yamamoto KR, Glucocorticoid signaling defines a novel commitment state during adipogenesis in vitro, Mol Biol Cell 19(10) (2008) 4032–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [209].Whirledge S, DeFranco DB, Glucocorticoid Signaling in Health and Disease: Insights From Tissue-Specific GR Knockout Mice, Endocrinology 159(1) (2018) 46–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [210].Kershaw EE, Morton NM, Dhillon H, Ramage L, Seckl JR, Flier JS, Adipocyte-specific glucocorticoid inactivation protects against diet-induced obesity, Diabetes 54(4) (2005) 1023–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [211].Iwen KA, Schroder E, Brabant G, Thyroid hormones and the metabolic syndrome, European thyroid journal 2(2) (2013) 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [212].Obregon MJ, Thyroid hormone and adipocyte differentiation, Thyroid 18(2) (2008) 185–95. [DOI] [PubMed] [Google Scholar]
- [213].Darimont C, Gaillard D, Ailhaud G, Negrel R, Terminal differentiation of mouse preadipocyte cells: adipogenic and antimitogenic role of triiodothyronine, Mol Cell Endocrinol 98(1) (1993) 67–73. [DOI] [PubMed] [Google Scholar]
- [214].Jiang W, Miyamoto T, Kakizawa T, Sakuma T, Nishio S, Takeda T, Suzuki S, Hashizume K, Expression of thyroid hormone receptor alpha in 3T3-L1 adipocytes; triiodothyronine increases the expression of lipogenic enzyme and triglyceride accumulation, The Journal of endocrinology 182(2) (2004) 295–302. [DOI] [PubMed] [Google Scholar]
- [215].Pelletier P, Gauthier K, Sideleva O, Samarut J, Silva JE, Mice lacking the thyroid hormone receptor-alpha gene spend more energy in thermogenesis, burn more fat, and are less sensitive to high-fat diet-induced obesity, Endocrinology 149(12) (2008) 6471–86. [DOI] [PubMed] [Google Scholar]
- [216].Liu YY, Schultz JJ, Brent GA, A thyroid hormone receptor alpha gene mutation (P398H) is associated with visceral adiposity and impaired catecholamine-stimulated lipolysis in mice, J Biol Chem 278(40) (2003) 38913–20. [DOI] [PubMed] [Google Scholar]
- [217].Weiss RE, Murata Y, Cua K, Hayashi Y, Seo H, Refetoff S, Thyroid hormone action on liver, heart, and energy expenditure in thyroid hormone receptor beta-deficient mice, Endocrinology 139(12) (1998) 4945–52. [DOI] [PubMed] [Google Scholar]
- [218].Lu C, Cheng SY, Thyroid hormone receptors regulate adipogenesis and carcinogenesis via crosstalk signaling with peroxisome proliferator-activated receptors, J Mol Endocrinol 44(3) (2010) 143–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [219].Grover GJ, Mellstrom K, Malm J, Therapeutic potential for thyroid hormone receptor-beta selective agonists for treating obesity, hyperlipidemia and diabetes, Curr Vasc Pharmacol 5(2) (2007) 141–54. [DOI] [PubMed] [Google Scholar]
- [220].Bryzgalova G, Effendic S, Khan A, Rehnmark S, Barbounis P, Boulet J, Dong G, Singh R, Shapses S, Malm J, Webb P, Baxter JD, Grover GJ, Anti-obesity, anti-diabetic, and lipid lowering effects of the thyroid receptor beta subtype selective agonist KB-141, J Steroid Biochem Mol Biol 111(3-5) (2008) 262–7. [DOI] [PubMed] [Google Scholar]
- [221].Dale J, Daykin J, Holder R, Sheppard MC, Franklyn JA, Weight gain following treatment of hyperthyroidism, Clinical endocrinology 55(2) (2001) 233–9. [DOI] [PubMed] [Google Scholar]
- [222].Lonn L, Stenlof K, Ottosson M, Lindroos AK, Nystrom E, Sjostrom L, Body weight and body composition changes after treatment of hyperthyroidism, The Journal of clinical endocrinology and metabolism 83(12) (1998) 4269–73. [DOI] [PubMed] [Google Scholar]
- [223].Kolyvanos Naumann U, Furer J, Kaser L, Vetter W, [Hypothyroidism. Main symptoms: fatigue, weight gain, depression, myalgia, edema], Praxis (Bern 1994) 96(38) (2007) 1411–7. [DOI] [PubMed] [Google Scholar]
- [224].Bratusch-Marrain P, Schmid P, Waldhausl W, Schlick W, Specific weight loss in hyperthyroidism, Horm Metab Res 10(5) (1978) 412–5. [DOI] [PubMed] [Google Scholar]
- [225].Valassi E, Scacchi M, Cavagnini F, Neuroendocrine control of food intake, Nutr Metab Cardiovasc Dis 18(2) (2008) 158–68. [DOI] [PubMed] [Google Scholar]
- [226].Sohn JW, Network of Hypothalamic Neurons that Control Appetite, BMB reports (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Rossi MA, Stuber GD, Overlapping Brain Circuits for Homeostatic and Hedonic Feeding, Cell Metab 27(1) (2018) 42–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Lutter M, Nestler EJ, Homeostatic and hedonic signals interact in the regulation of food intake, The Journal of nutrition 139(3) (2009) 629–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [229].Matafome P, Seiça R, The Role of Brain in Energy Balance, Adv Neurobiol 19 (2017) 33–48. [DOI] [PubMed] [Google Scholar]
- [230].Heisler LK, Lam DD, An appetite for life: brain regulation of hunger and satiety, Current opinion in pharmacology 37 (2017) 100–106. [DOI] [PubMed] [Google Scholar]
- [231].Koliaki C, Liatis S, Dalamaga M, Kokkinos A, The Implication of Gut Hormones in the Regulation of Energy Homeostasis and Their Role in the Pathophysiology of Obesity, Current obesity reports 9(3) (2020) 255–271. [DOI] [PubMed] [Google Scholar]
- [232].Morton GJ, Meek TH, Schwartz MW, Neurobiology of food intake in health and disease, Nature reviews. Neuroscience 15(6) (2014) 367–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [233].Kleinridders A, Ferris HA, Cai W, Kahn CR, Insulin action in brain regulates systemic metabolism and brain function, Diabetes 63(7) (2014) 2232–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [234].Stincic TL, Rønnekleiv OK, Kelly MJ, Diverse actions of estradiol on anorexigenic and orexigenic hypothalamic arcuate neurons, Horm Behav 104 (2018) 146–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Zanchi D, Depoorter A, Egloff L, Haller S, Mählmann L, Lang UE, Drewe J, Beglinger C, Schmidt A, Borgwardt S, The impact of gut hormones on the neural circuit of appetite and satiety: A systematic review, Neurosci Biobehav Rev 80 (2017) 457–475. [DOI] [PubMed] [Google Scholar]
- [236].Makris MC, Alexandrou A, Papatsoutsos EG, Malietzis G, Tsilimigras DI, Guerron AD, Moris D, Ghrelin and Obesity: Identifying Gaps and Dispelling Myths. A Reappraisal, In Vivo 31(6) (2017) 1047–1050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [237].Kessler RM, Hutson PH, Herman BK, Potenza MN, Neuroscience and Biobehavioral Reviews The neurobiological basis of binge-eating disorder, Neuroscience and Biobehavioral Reviews 63 (2016) 223–238. [DOI] [PubMed] [Google Scholar]
- [238].Baik JH, Dopamine signaling in reward-related behaviors, Front Neural Circuits 7 (2013) 152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Baik JH, Dopaminergic Control of the Feeding Circuit, Endocrinol Metab (Seoul) 36(2) (2021) 229–239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [240].Wiss DA, Criscitelli K, Gold M, Avena N, Preclinical evidence for the addiction potential of highly palatable foods: Current developments related to maternal influence, Appetite 115 (2017) 19–27. [DOI] [PubMed] [Google Scholar]
- [241].Charbogne P, Gardon O, Martín-García E, Keyworth HL, Matsui A, Mechling AE, Bienert T, Nasseef MT, Robé A, Moquin L, Darcq E, Ben Hamida S, Robledo P, Matifas A, Befort K, Gavériaux-Ruff C, Harsan LA, von Elverfeldt D, Hennig J, Gratton A, Kitchen I, Bailey A, Alvarez VA, Maldonado R, Kieffer BL, Mu Opioid Receptors in Gamma-Aminobutyric Acidergic Forebrain Neurons Moderate Motivation for Heroin and Palatable Food, Biol Psychiatry 81(9) (2017) 778–788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [242].Rosenbaum M, Murphy EM, Heymsfield SB, Matthews DE, Leibel RL, Low dose leptin administration reverses effects of sustained weight-reduction on energy expenditure and circulating concentrations of thyroid hormones, The Journal of clinical endocrinology and metabolism 87(5) (2002) 2391–4. [DOI] [PubMed] [Google Scholar]
- [243].Yu YH, Vasselli JR, Zhang Y, Mechanick JI, Korner J, Peterli R, Metabolic vs. hedonic obesity: a conceptual distinction and its clinical implications, Obes Rev 16(3) (2015) 234–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [244].Yu YH, Making sense of metabolic obesity and hedonic obesity, J Diabetes 9(7) (2017) 656–666. [DOI] [PubMed] [Google Scholar]
- [245].Lustig RH, Sen S, Soberman JE, Velasquez-Mieyer PA, Obesity, leptin resistance, and the effects of insulin reduction, International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 28(10) (2004) 1344–8. [DOI] [PubMed] [Google Scholar]
- [246].Farooqi IS, Bullmore E, Keogh J, Gillard J, O’Rahilly S, Fletcher PC, Leptin regulates striatal regions and human eating behavior, Science 317(5843) (2007) 1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [247].Münzberg H, Myers MG Jr., Molecular and anatomical determinants of central leptin resistance, Nat Neurosci 8(5) (2005) 566–70. [DOI] [PubMed] [Google Scholar]
- [248].Figlewicz DP, Evans SB, Murphy J, Hoen M, Baskin DG, Expression of receptors for insulin and leptin in the ventral tegmental area/substantia nigra (VTA/SN) of the rat, Brain research 964(1) (2003) 107–15. [DOI] [PubMed] [Google Scholar]
- [249].Hill JW, Williams KW, Ye C, Luo J, Balthasar N, Coppari R, Cowley MA, Cantley LC, Lowell BB, Elmquist JK, Acute effects of leptin require PI3K signaling in hypothalamic proopiomelanocortin neurons in mice, J Clin Invest 118(5) (2008) 1796–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [250].Lin X, Taguchi A, Park S, Kushner JA, Li F, Li Y, White MF, Dysregulation of insulin receptor substrate 2 in beta cells and brain causes obesity and diabetes, J Clin Invest 114(7) (2004) 908–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Zabolotny JM, Bence-Hanulec KK, Stricker-Krongrad A, Haj F, Wang Y, Minokoshi Y, Kim YB, Elmquist JK, Tartaglia LA, Kahn BB, Neel BG, PTP1B regulates leptin signal transduction in vivo, Dev Cell 2(4) (2002) 489–95. [DOI] [PubMed] [Google Scholar]
- [252].Lustig RH, Childhood obesity: behavioral aberration or biochemical drive? Reinterpreting the First Law of Thermodynamics, Nat Clin Pract Endocrinol Metab 2(8) (2006) 447–58. [DOI] [PubMed] [Google Scholar]
- [253].Mietus-Snyder ML, Lustig RH, Childhood obesity: adrift in the “limbic triangle”, Annu Rev Med 59 (2008) 147–62. [DOI] [PubMed] [Google Scholar]
- [254].Aguilera CM, Olza J, Gil A, Genetic susceptibility to obesity and metabolic syndrome in childhood, Nutricion hospitalaria 28 Suppl 5 (2013) 44–55. [DOI] [PubMed] [Google Scholar]
- [255].Lusis AJ, Attie AD, Reue K, Metabolic syndrome: from epidemiology to systems biology, Nat Rev Genet 9(11) (2008) 819–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [256].Ludwig DS, Aronne LJ, Astrup A, de Cabo R, Cantley LC, Friedman MI, Heymsfield SB, Johnson JD, King JC, Krauss RM, Lieberman DE, Taubes G, Volek JS, Westman EC, Willett WC, Yancy WS, Ebbeling CB, The carbohydrate-insulin model: a physiological perspective on the obesity pandemic, Am J Clin Nutr (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [257].Stanhope KL, Goran MI, Bosy-Westphal A, et al. , Pathways and mechanisms linking dietary components to cardiometabolic disease: thinking beyond calories, Obes. Rev 19(9) (2018) 1205–1295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [258].Weickert MO, Pfeiffer AFH, Metabolic effects of dietary fiber consumption and prevention of diabetes, J. Nutr 138 (2008) 439–442. [DOI] [PubMed] [Google Scholar]
- [259].Desai MS, Seekatz AM, Koropatkin NM, et al. , A Dietary Fiber-Deprived Gut Microbiota Degrades the Colonic Mucus Barrier and Enhances Pathogen Susceptibility, Cell 167(5) (2016) 1339–1353.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [260].Ferrarese R, Ceresola ER, Preti A, Canducci F, Probiotics, prebiotics and synbiotics for weight loss and metabolic syndrome in the microbiome era, Eur. Rev. Med. Pharmacol. Sci 22(21) (2018) 7588–7605. [DOI] [PubMed] [Google Scholar]
- [261].Johnston CS, Day CS, Swan PD, Postprandial thermogenesis is increased 100% on a high-protein, low-fat diet versus a high-carbohydrate, low-fat diet in healthy, young women, J. Am. Coll. Nutr 21 (2002) 55–61. [DOI] [PubMed] [Google Scholar]
- [262].Howard BV, Manson JE, Stefanick ML, Beresford SA, Frank G, Jones B, Rodabough RJ, Snetselaar L, Thomson C, Tinker L, Vitolins M, Prentice R, Low-fat dietary pattern and weight change over 7 years: the Women’s Health Initiative Dietary Modification Trial, JAMA 295(1) (2006) 39–49. [DOI] [PubMed] [Google Scholar]
- [263].Howard BV, Van Horn L, Hsia J, Manson JE, Stefanick ML, Wassertheil-Smoller S, Kuller LH, LaCroix AZ, Langer RD, Lasser NL, Lewis CE, Limacher MC, Margolis KL, Mysiw WJ, Ockene JK, Parker LM, Perri MG, Phillips L, Prentice RL, Robbins J, Rossouw JE, Sarto GE, Schatz IJ, Snetselaar LG, Stevens VJ, Tinker LF, Trevisan M, Vitolins MZ, Anderson GL, Assaf AR, Bassford T, Beresford SA, Black HR, Brunner RL, Brzyski RG, Caan B, Chlebowski RT, Gass M, Granek I, Greenland P, Hays J, Heber D, Heiss G, Hendrix SL, Hubbell FA, Johnson KC, Kotchen JM, Low-fat dietary pattern and risk of cardiovascular disease: the Women’s Health Initiative Randomized Controlled Dietary Modification Trial., JAMA 295(6) (2006) 655–666. [DOI] [PubMed] [Google Scholar]
- [264].Ramsden CE, Zamora D, Majchrzak-Hong S, et al. , Re-evaluation of the traditional diet-heart hypothesis: analysis of recovered data from Minnesota Coronary Experiment (1968-73), BMJ (Clinical research ed.) 353 (2016) i1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [265].Frantz ID, Dawson EA, Ashman PL, et al. , Test of effect of lipid lowering by diet on cardiovascular risk. The Minnesota Coronary Survey. , Arteriosclerosis. 9 (1989) 129–135. [DOI] [PubMed] [Google Scholar]
- [266].Goran MI, Dumke K, Bouret SG, Kayser B, Walker RW, Blumberg B, The obesogenic effect of high fructose exposure during early development, Nature reviews. Endocrinology 9(8) (2013) 494–500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [267].Temelkova-Kurktschiev T, Siegert G, Bergmann S, et al. , Subclinical inflammation is strongly related to insulin resistance but not to impaired insulin secretion in a high risk population for diabetes, Metabolism 51(6) (2002) 743–749. [DOI] [PubMed] [Google Scholar]
- [268].de Vegt F, Dekker JM, Ruhé HG, et al. , Hyperglycaemia is associated with all-cause and cardiovascular mortality in the Hoorn population: the Hoorn Study, Diabetologia 42(8) (1999) 926–931. [DOI] [PubMed] [Google Scholar]
- [269].Foster-Powell K, Brand-Miller J, International tables of glycemic index, Am. J. Clin. Nutr 62(4) (1995) 871S–890S. [DOI] [PubMed] [Google Scholar]
- [270].Slabber M, Barnard HC, Kuyl JM, Dannhauser A, Schall R, Effects of a low-insulin-response, energy-restricted diet on weight loss and plasma insulin concentrations in hyperinsulinemic obese females, Am. J. Clin. Nutr 60(1) (1994) 48–53. [DOI] [PubMed] [Google Scholar]
- [271].Ludwig DS, Hu FB, Tappy L, Brand-Miller J, Dietary carbohydrates: role of quality and quantity in chronic disease, BMJ (Clinical research ed.) 361 (2018) k2340–k2340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [272].Lee HS, Lee J, Effects of Combined Exercise and Low Carbohydrate Ketogenic Diet Interventions on Waist Circumference and Triglycerides in Overweight and Obese Individuals: A Systematic Review and Meta-Analysis, Int J Environ Res Public Health 18(2) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [273].Magkos F, Hjorth MF, Astrup A, Diet and exercise in the prevention and treatment of type 2 diabetes mellitus, Nat Rev Endocrinol 16(10) (2020) 545–555. [DOI] [PubMed] [Google Scholar]
- [274].Li H, Dun Y, Zhang W, You B, Liu Y, Fu S, Qiu L, Cheng J, Ripley-Gonzalez JW, Liu S, Exercise improves lipid droplet metabolism disorder through activation of AMPK-mediated lipophagy in NAFLD, Life sciences 273 (2021) 119314. [DOI] [PubMed] [Google Scholar]
- [275].Thorp A, Stine JG, Exercise as Medicine: The Impact of Exercise Training on Nonalcoholic Fatty Liver Disease, Curr Hepatol Rep 19(4) (2020) 402–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [276].Kim JY, Jeon JY, Role of exercise on insulin sensitivity and beta-cell function: is exercise sufficient for the prevention of youth-onset type 2 diabetes?, Annals of pediatric endocrinology & metabolism 25(4) (2020) 208–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [277].Barber TM, Kyrou I, Randeva HS, Weickert MO, Mechanisms of Insulin Resistance at the Crossroad of Obesity with Associated Metabolic Abnormalities and Cognitive Dysfunction, International journal of molecular sciences 22(2) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [278].Imierska M, Kurianiuk A, Błachnio-Zabielska A, The Influence of Physical Activity on the Bioactive Lipids Metabolism in Obesity-Induced Muscle Insulin Resistance, Biomolecules 10(12) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [279].Sun Y, Ding S, ER-Mitochondria Contacts and Insulin Resistance Modulation through Exercise Intervention, International journal of molecular sciences 21(24) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [280].Gonzalez-Gil AM, Elizondo-Montemayor L, The Role of Exercise in the Interplay between Myokines, Hepatokines, Osteokines, Adipokines, and Modulation of Inflammation for Energy Substrate Redistribution and Fat Mass Loss: A Review, Nutrients 12(6) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [281].Rosa-Neto JC, Silveira LS, Endurance Exercise Mitigates Immunometabolic Adipose Tissue Disturbances in Cancer and Obesity, International journal of molecular sciences 21(24) (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [282].Soltani N, Marandi SM, Kazemi M, Esmaeil N, The Exercise Training Modulatory Effects on the Obesity-Induced Immunometabolic Dysfunctions, Diabetes, metabolic syndrome and obesity : targets and therapy 13 (2020) 785–810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [283].Laurens C, Bergouignan A, Moro C, Exercise-Released Myokines in the Control of Energy Metabolism, Frontiers in physiology 11 (2020) 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [284].Fan Z, Xu M, Exercise and Organ Cross Talk, Adv Exp Med Biol 1228 (2020) 63–76. [DOI] [PubMed] [Google Scholar]
- [285].Trovato E, Di Felice V, Barone R, Extracellular Vesicles: Delivery Vehicles of Myokines, Frontiers in physiology 10 (2019) 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [286].Vechetti IJ, Valentino T, Mobley CB, McCarthy JJ, The role of extracellular vesicles in skeletal muscle and systematic adaptation to exercise, The Journal of physiology 599(3) (2021) 845–861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [287].Improta Caria AC, Nonaka CKV, Pereira CS, Soares MBP, Macambira SG, Souza BSF, Exercise Training-Induced Changes in MicroRNAs: Beneficial Regulatory Effects in Hypertension, Type 2 Diabetes, and Obesity, International journal of molecular sciences 19(11) (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [288].Ehtesham N, Shahrbanian S, Valadiathar M, Mowla SJ, Modulations of obesity-related microRNAs after exercise intervention: a systematic review and bioinformatics analysis, Mol Biol Rep (2021). [DOI] [PubMed] [Google Scholar]
- [289].Harris JE, Baer LA, Stanford KI, Maternal Exercise Improves the Metabolic Health of Adult Offspring, Trends in endocrinology and metabolism: TEM 29(3) (2018) 164–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [290].Zheng J, Zhou LY, Xiao XH, Maternal exercise and its beneficial effects on glucose metabolism in offspring, Chin Med J (Engl) 133(7) (2020) 863–867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [291].McCarthy SF, Islam H, Hazell TJ, The emerging role of lactate as a mediator of exercise-induced appetite suppression, American journal of physiology. Endocrinology and metabolism 319(4) (2020) E814–E819. [DOI] [PubMed] [Google Scholar]
- [292].Atkinson RL, Viruses as an etiology of obesity, Mayo Clin Proc 82(10) (2007) 1192–8. [DOI] [PubMed] [Google Scholar]
- [293].Lyons MJ, Faust IM, Hemmes RB, Buskirk DR, Hirsch J, Zabriskie JB, A virally induced obesity syndrome in mice, Science 216(4541) (1982) 82–5. [DOI] [PubMed] [Google Scholar]
- [294].Carter JK, Ow CL, Smith RE, Rous-associated virus type 7 induces a syndrome in chickens characterized by stunting and obesity, Infect Immun 39(1) (1983) 410–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [295].Atkinson RL, Dhurandhar NV, Allison DB, Bowen RL, Israel BA, Albu JB, Augustus AS, Human adenovirus-36 is associated with increased body weight and paradoxical reduction of serum lipids, International journal of obesity (2005) 29(3) (2005) 281–6. [DOI] [PubMed] [Google Scholar]
- [296].Shang Q, Wang H, Song Y, Wei L, Lavebratt C, Zhang F, Gu H, Serological data analyses show that adenovirus 36 infection is associated with obesity: a meta-analysis involving 5739 subjects, Obesity (Silver Spring) 22(3) (2014) 895–900. [DOI] [PubMed] [Google Scholar]
- [297].Ponterio E, Gnessi L, Adenovirus 36 and Obesity: An Overview, Viruses 7(7) (2015) 3719–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [298].Vangipuram SD, Yu M, Tian J, Stanhope KL, Pasarica M, Havel PJ, Heydari AR, Dhurandhar NV, Adipogenic human adenovirus-36 reduces leptin expression and secretion and increases glucose uptake by fat cells, International journal of obesity (2005) 31(1) (2007) 87–96. [DOI] [PubMed] [Google Scholar]
- [299].Sapunar J, Fonseca L, Molina V, Ortiz E, Barra MI, Reimer C, Charles M, Schneider C, Ortiz M, Brito R, Manríquez V, Pavez M, Cerda A, Adenovirus 36 seropositivity is related to obesity risk, glycemic control, and leptin levels in Chilean subjects, International journal of obesity (2005) 44(1) (2020) 159–166. [DOI] [PubMed] [Google Scholar]
- [300].Fazakerley DJ, Krycer JR, Kearney AL, Hocking SL, James DE, Muscle and adipose tissue insulin resistance: malady without mechanism?, J Lipid Res 60(10) (2019) 1720–1732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [301].Samocha-Bonet D, Dixit VD, Kahn CR, Leibel RL, Lin X, Nieuwdorp M, Pietiläinen KH, Rabasa-Lhoret R, Roden M, Scherer PE, Klein S, Ravussin E, Metabolically healthy and unhealthy obese--the 2013 Stock Conference report, Obes Rev 15(9) (2014) 697–708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [302].Kitamura T, Kahn CR, Accili D, Insulin receptor knockout mice, Annual review of physiology 65 (2003) 313–32. [DOI] [PubMed] [Google Scholar]
- [303].Matsumoto M, Han S, Kitamura T, Accili D, Dual role of transcription factor FoxO1 in controlling hepatic insulin sensitivity and lipid metabolism, J Clin Invest 116(9) (2006) 2464–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [304].Naïmi M, Gautier N, Chaussade C, Valverde AM, Accili D, Van Obberghen E, Nuclear forkhead box O1 controls and integrates key signaling pathways in hepatocytes, Endocrinology 148(5) (2007) 2424–34. [DOI] [PubMed] [Google Scholar]
- [305].Lewis GF, Uffelman KD, Szeto LW, Steiner G, Effects of acute hyperinsulinemia on VLDL triglyceride and VLDL apoB production in normal weight and obese individuals, Diabetes 42(6) (1993) 833–42. [DOI] [PubMed] [Google Scholar]
- [306].Fu S, Watkins SM, Hotamisligil GS, The role of endoplasmic reticulum in hepatic lipid homeostasis and stress signaling, Cell Metab 15(5) (2012) 623–34. [DOI] [PubMed] [Google Scholar]
- [307].Birkenfeld AL, Shulman GI, Nonalcoholic fatty liver disease, hepatic insulin resistance, and type 2 diabetes, Hepatology 59(2) (2014) 713–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [308].Sripetchwandee J, Chattipakorn N, Chattipakorn SC, Links Between Obesity-Induced Brain Insulin Resistance, Brain Mitochondrial Dysfunction, and Dementia, Front Endocrinol (Lausanne) 9 (2018) 496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [309].Valdearcos M, Douglass JD, Robblee MM, Dorfman MD, Stifler DR, Bennett ML, Gerritse I, Fasnacht R, Barres BA, Thaler JP, Koliwad SK, Microglial Inflammatory Signaling Orchestrates the Hypothalamic Immune Response to Dietary Excess and Mediates Obesity Susceptibility, Cell Metab 26(1) (2017) 185–197.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [310].Lewis GF, Carpentier A, Adeli K, Giacca A, Disordered fat storage and mobilization in the pathogenesis of insulin resistance and type 2 diabetes, Endocr Rev 23(2) (2002) 201–29. [DOI] [PubMed] [Google Scholar]
- [311].Lobo S, Bernlohr DA, Fatty acid transport in adipocytes and the development of insulin resistance, Novartis Found Symp 286 (2007) 113–21; discussion 121-6, 162-3, 196-203. [DOI] [PubMed] [Google Scholar]
- [312].Thompson BR, Lobo S, Bernlohr DA, Fatty acid flux in adipocytes: the in's and out's of fat cell lipid trafficking, Mol Cell Endocrinol 318(1-2) (2010) 24–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [313].Zhao P, Wong KI, Sun X, Reilly SM, Uhm M, Liao Z, Skorobogatko Y, Saltiel AR, TBK1 at the Crossroads of Inflammation and Energy Homeostasis in Adipose Tissue, Cell 172(4) (2018) 731–743.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [314].Bremer AA, Devaraj S, Afify A, Jialal I, Adipose tissue dysregulation in patients with metabolic syndrome, The Journal of clinical endocrinology and metabolism 96(11) (2011) E1782–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [315].Shoelson SE, Lee J, Goldfine AB, Inflammation and insulin resistance, J Clin Invest 116(7) (2006) 1793–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [316].Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, Sole J, Nichols A, Ross JS, Tartaglia LA, Chen H, Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance, J Clin Invest 112(12) (2003) 1821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [317].Saltiel AR, Olefsky JM, Inflammatory mechanisms linking obesity and metabolic disease, J Clin Invest 127(1) (2017) 1–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [318].Cildir G, Akincilar SC, Tergaonkar V, Chronic adipose tissue inflammation: all immune cells on the stage, Trends Mol Med 19(8) (2013) 487–500. [DOI] [PubMed] [Google Scholar]
- [319].Kolb R, Sutterwala FS, Zhang W, Obesity and cancer: inflammation bridges the two, Current opinion in pharmacology 29 (2016) 77–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [320].Steppan CM, Brown EJ, Wright CM, Bhat S, Banerjee RR, Dai CY, Enders GH, Silberg DG, Wen X, Wu GD, Lazar MA, A family of tissue-specific resistin-like molecules, Proceedings of the National Academy of Sciences of the United States of America 98(2) (2001) 502–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [321].Bansal A, Henao-Mejia J, Simmons RA, Immune System: An Emerging Player in Mediating Effects of Endocrine Disruptors on Metabolic Health, Endocrinology 159(1) (2018) 32–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [322].Crewe C, An YA, Scherer PE, The ominous triad of adipose tissue dysfunction: inflammation, fibrosis, and impaired angiogenesis, J Clin Invest 127(1) (2017) 74–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [323].Reilly SM, Saltiel AR, Adapting to obesity with adipose tissue inflammation, Nat Rev Endocrinol 13(11) (2017) 633–643. [DOI] [PubMed] [Google Scholar]
- [324].Russo L, Lumeng CN, Properties and functions of adipose tissue macrophages in obesity, Immunology 155(4) (2018) 407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [325].Li Y, Yun K, Mu R, A review on the biology and properties of adipose tissue macrophages involved in adipose tissue physiological and pathophysiological processes, Lipids Health Dis 19(1) (2020) 164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [326].Osborn O, Olefsky JM, The cellular and signaling networks linking the immune system and metabolism in disease, Nat Med. 18(3) (2012) 363–74. doi: 10.1038/nm.2627. [DOI] [PubMed] [Google Scholar]
- [327].Ouchi N, Parker JL, Lugus JJ, Walsh K, Adipokines in inflammation and metabolic disease, Nat Rev Immunol. 11(2) (2011) 85–97. doi: 10.1038/nri2921. Epub 2011 Jan 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [328].Sell H, Habich C, Eckel J, Adaptive immunity in obesity and insulin resistance, Nat Rev Endocrinol. 8(12) (2012) 709–16. doi: 10.1038/nrendo.2012.114. Epub 2012 Jul 31. [DOI] [PubMed] [Google Scholar]
- [329].Brestoff JR, Artis D, Immune regulation of metabolic homeostasis in health and disease, Cell. 161(1) (2015) 146–60. doi: 10.1016/j.cell.2015.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [330].Itoh M, Suganami T, Hachiya R, Ogawa Y, Adipose tissue remodeling as homeostatic inflammation, Int J Inflam 2011 (2011) 720926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [331].Hill JO, Understanding and addressing the epidemic of obesity: an energy balance perspective, Endocr Rev 27(7) (2006) 750–61. [DOI] [PubMed] [Google Scholar]
- [332].Sarwar R, Pierce N, Koppe S, Obesity and nonalcoholic fatty liver disease: current perspectives, Diabetes, metabolic syndrome and obesity : targets and therapy 11 (2018) 533–542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [333].Hruby A, Hu FB, The Epidemiology of Obesity: A Big Picture, PharmacoEconomics 33(7) (2015) 673–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [334].Durack J, Lynch SV, The gut microbiome: Relationships with disease and opportunities for therapy, J Exp Med 216(1) (2019) 20–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [335].Shreiner AB, Kao JY, Young VB, The gut microbiome in health and in disease, Curr Opin Gastroenterol 31(1) (2015) 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [336].Cani PD, Human gut microbiome: hopes, threats and promises, Gut 67(9) (2018) 1716–1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [337].Li M, van Esch B, Wagenaar GTM, Garssen J, Folkerts G, Henricks PAJ, Pro- and anti-inflammatory effects of short chain fatty acids on immune and endothelial cells, Eur J Pharmacol 831 (2018) 52–59. [DOI] [PubMed] [Google Scholar]
- [338].Castaner O, Goday A, Park YM, Lee SH, Magkos F, Shiow STE, Schroder H, The Gut Microbiome Profile in Obesity: A Systematic Review, Int J Endocrinol 2018 (2018) 4095789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [339].Abenavoli L, Scarpellini E, Colica C, Boccuto L, Salehi B, Sharifi-Rad J, Aiello V, Romano B, De Lorenzo A, Izzo AA, Capasso R, Gut Microbiota and Obesity: A Role for Probiotics, Nutrients 11(11) (2019) 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [340].Qin J, Li R, Raes J, Arumugam M, Burgdorf KS, Manichanh C, Nielsen T, Pons N, Levenez F, Yamada T, Mende DR, Li J, Xu J, Li S, Li D, Cao J, Wang B, Liang H, Zheng H, Xie Y, Tap J, Lepage P, Bertalan M, Batto JM, Hansen T, Le Paslier D, Linneberg A, Nielsen HB, Pelletier E, Renault P, Sicheritz-Ponten T, Turner K, Zhu H, Yu C, Li S, Jian M, Zhou Y, Li Y, Zhang X, Li S, Qin N, Yang H, Wang J, Brunak S, Dore J, Guarner F, Kristiansen K, Pedersen O, Parkhill J, Weissenbach J, Meta HITC, Bork P, Ehrlich SD, Wang J, A human gut microbial gene catalogue established by metagenomic sequencing, Nature 464(7285) (2010) 59–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [341].Ley RE, Turnbaugh PJ, Klein S, Gordon JI, Microbial ecology: human gut microbes associated with obesity, Nature 444(7122) (2006) 1022–3. [DOI] [PubMed] [Google Scholar]
- [342].Wang R, Tang R, Li B, Ma X, Schnabl B, Tilg H, Gut microbiome, liver immunology, and liver diseases, Cell Mol Immunol 18(1) (2021) 4–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [343].Aguilar T, Nava GM, Olvera-Ramirez AM, Ronquillo D, Camacho M, Zavala GA, Caamano MC, Acevedo-Whitehouse K, Rosado JL, Garcia OP, Gut Bacterial Families Are Associated with Body Composition and Metabolic Risk Markers in School-Aged Children in Rural Mexico, Childhood obesity (Print) 16(5) (2020) 358–366. [DOI] [PubMed] [Google Scholar]
- [344].Sekikawa A, Kadowaki T, Curb JD, Evans RW, Maegawa H, Abbott RD, Sutton-Tyrrell K, Okamura T, Shin C, Edmundowicz D, Kadota A, Choo J, El-Saed A, Ueshima H, Kuller LH, group EJS, Circulating levels of 8 cytokines and marine n-3 fatty acids and indices of obesity in Japanese, white, and Japanese American middle-aged men, J Interferon Cytokine Res 30(7) (2010) 541–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [345].Henao-Mejia J, Elinav E, Jin C, Hao L, Mehal WZ, Strowig T, Thaiss CA, Kau AL, Eisenbarth SC, Jurczak MJ, Camporez JP, Shulman GI, Gordon JI, Hoffman HM, Flavell RA, Inflammasome-mediated dysbiosis regulates progression of NAFLD and obesity, Nature 482(7384) (2012) 179–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [346].Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI, Gut microbiota from twins discordant for obesity modulate metabolism in mice, Science 341(6150) (2013) 1241214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [347].Agus A, Planchais J, Sokol H, Gut Microbiota Regulation of Tryptophan Metabolism in Health and Disease, Cell host & microbe 23(6) (2018) 716–724. [DOI] [PubMed] [Google Scholar]
- [348].Laurans L, Venteclef N, Haddad Y, Chajadine M, Alzaid F, Metghalchi S, Sovran B, Denis RGP, Dairou J, Cardellini M, Moreno-Navarrete JM, Straub M, Jegou S, McQuitty C, Viel T, Esposito B, Tavitian B, Callebert J, Luquet SH, Federici M, Fernandez-Real JM, Burcelin R, Launay JM, Tedgui A, Mallat Z, Sokol H, Taleb S, Genetic deficiency of indoleamine 2,3-dioxygenase promotes gut microbiota-mediated metabolic health, Nat Med 24(8) (2018) 1113–1120. [DOI] [PubMed] [Google Scholar]
- [349].Sanna S, van Zuydam NR, Mahajan A, Kurilshikov A, Vich Vila A, Vosa U, Mujagic Z, Masclee AAM, Jonkers D, Oosting M, Joosten LAB, Netea MG, Franke L, Zhernakova A, Fu J, Wijmenga C, McCarthy MI, Causal relationships among the gut microbiome, short-chain fatty acids and metabolic diseases, Nat Genet 51(4) (2019) 600–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [350].Xiao H, Kang S, The Role of the Gut Microbiome in Energy Balance With a Focus on the Gut-Adipose Tissue Axis, Front Genet 11 (2020) 297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [351].Tilg H, Zmora N, Adolph TE, Elinav E, The intestinal microbiota fuelling metabolic inflammation, Nature reviews. Immunology 20(1) (2020) 40–54. [DOI] [PubMed] [Google Scholar]
- [352].Virtue AT, McCright SJ, Wright JM, Jimenez MT, Mowel WK, Kotzin JJ, Joannas L, Basavappa MG, Spencer SP, Clark ML, Eisennagel SH, Williams A, Levy M, Manne S, Henrickson SE, Wherry EJ, Thaiss CA, Elinav E, Henao-Mejia J, The gut microbiota regulates white adipose tissue inflammation and obesity via a family of microRNAs, Sci Transl Med 11(496) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [353].Boursier J, Mueller O, Barret M, Machado M, Fizanne L, Araujo-Perez F, Guy CD, Seed PC, Rawls JF, David LA, Hunault G, Oberti F, Cales P, Diehl AM, The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota, Hepatology 63(3) (2016) 764–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [354].Fandriks L, Roles of the gut in the metabolic syndrome: an overview, J Intern Med 281(4) (2017) 319–336. [DOI] [PubMed] [Google Scholar]
- [355].Kohsaka A, Bass J, A sense of time: how molecular clocks organize metabolism, Trends in endocrinology and metabolism: TEM 18(1) (2007) 4–11. [DOI] [PubMed] [Google Scholar]
- [356].Gooley JJ, Circadian regulation of lipid metabolism, The Proceedings of the Nutrition Society 75(4) (2016) 440–450. [DOI] [PubMed] [Google Scholar]
- [357].Mayeuf-Louchart A, Zecchin M, Staels B, Duez H, Circadian control of metabolism and pathological consequences of clock perturbations, Biochimie 143 (2017) 42–50. [DOI] [PubMed] [Google Scholar]
- [358].Serin Y, Acar Tek N, Effect of Circadian Rhythm on Metabolic Processes and the Regulation of Energy Balance, Annals of nutrition & metabolism 74(4) (2019) 322–330. [DOI] [PubMed] [Google Scholar]
- [359].Guan D, Lazar MA, Interconnections between circadian clocks and metabolism, J Clin Invest 131(15) (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [360].Parkar SG, Kalsbeek A, Cheeseman JF, Potential Role for the Gut Microbiota in Modulating Host Circadian Rhythms and Metabolic Health, Microorganisms 7(2) (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [361].Mukherji A, Bailey SM, Staels B, Baumert TF, The circadian clock and liver function in health and disease, J Hepatol 71(1) (2019) 200–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [362].Pan X, Mota S, Zhang B, Circadian Clock Regulation on Lipid Metabolism and Metabolic Diseases, Adv Exp Med Biol 1276 (2020) 53–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [363].Saran AR, Dave S, Zarrinpar A, Circadian Rhythms in the Pathogenesis and Treatment of Fatty Liver Disease, Gastroenterology 158(7) (2020) 1948–1966.e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [364].Andriessen C, Schrauwen P, Hoeks J, The importance of 24-h metabolism in obesity-related metabolic disorders: opportunities for timed interventions, International journal of obesity (2005) 45(3) (2021) 479–490. [DOI] [PubMed] [Google Scholar]
- [365].Barker DJ, The fetal and infant origins of adult disease, BMJ (Clinical research ed.) 301(6761) (1990) 1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [366].Barker DJ, The origins of the developmental origins theory, J Intern Med 261(5) (2007) 412–7. [DOI] [PubMed] [Google Scholar]
- [367].Oken E, Gillman MW, Fetal origins of obesity, Obesity research 11(4) (2003) 496–506. [DOI] [PubMed] [Google Scholar]
- [368].Taylor PD, Poston L, Developmental programming of obesity in mammals, Exp Physiol 92(2) (2007) 287–98. [DOI] [PubMed] [Google Scholar]
- [369].Hales CN, Barker DJ, Clark PM, Cox LJ, Fall C, Osmond C, Winter PD, Fetal and infant growth and impaired glucose tolerance at age 64, BMJ (Clinical research ed.) 303(6809) (1991) 1019–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [370].Curhan GC, Willett WC, Rimm EB, Spiegelman D, Ascherio AL, Stampfer MJ, Birth weight and adult hypertension, diabetes mellitus, and obesity in US men, Circulation 94(12) (1996) 3246–50. [DOI] [PubMed] [Google Scholar]
- [371].Curhan GC, Chertow GM, Willett WC, Spiegelman D, Colditz GA, Manson JE, Speizer FE, Stampfer MJ, Birth weight and adult hypertension and obesity in women, Circulation 94(6) (1996) 1310–5. [DOI] [PubMed] [Google Scholar]
- [372].Rogers I, Group E-BS, The influence of birthweight and intrauterine environment on adiposity and fat distribution in later life, International journal of obesity and related metabolic disorders : journal of the International Association for the Study of Obesity 27(7) (2003) 755–77. [DOI] [PubMed] [Google Scholar]
- [373].Ong KK, Ahmed ML, Emmett PM, Preece MA, Dunger DB, Association between postnatal catch-up growth and obesity in childhood: prospective cohort study, BMJ (Clinical research ed.) 320 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [374].Ibáñez L, Ong K, Dunger DB, de Zegher F, Early development of adiposity and insulin resistance after catch-up weight gain in small-for-gestational-age children, The Journal of clinical endocrinology and metabolism 91(6) (2006) 2153–8. [DOI] [PubMed] [Google Scholar]
- [375].Halldorsson TI, Rytter D, Haug LS, Bech BH, Danielsen I, Becher G, Henriksen TB, Olsen SF, Prenatal Exposure to Perfluorooctanoate and Risk of Overweight at 20 Years of Age: A Prospective Cohort Study, Environ Health Perspect (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [376].Braun JM, Chen A, Romano ME, Calafat AM, Webster GM, Yolton K, Lanphear BP, Prenatal perfluoroalkyl substance exposure and child adiposity at 8 years of age: The HOME study, Obesity (Silver Spring) 24(1) (2016) 231–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [377].Vrijheid M, Fossati S, Maitre L, Marquez S, Roumeliotaki T, Agier L, Andrusaityte S, Cadiou S, Casas M, de Castro M, Dedele A, Donaire-Gonzalez D, Grazuleviciene R, Haug LS, McEachan R, Meltzer HM, Papadopouplou E, Robinson O, Sakhi AK, Siroux V, Sunyer J, Schwarze PE, Tamayo-Uria I, Urquiza J, Vafeiadi M, Valentin A, Warembourg C, Wright J, Nieuwenhuijsen MJ, Thomsen C, Basagana X, Slama R, Chatzi L, Early-Life Environmental Exposures and Childhood Obesity: An Exposome-Wide Approach, Environ Health Perspect 128(6) (2020) 67009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [378].Oken E, Levitan EB, Gillman MW, Maternal smoking during pregnancy and child overweight: systematic review and meta-analysis, International journal of obesity (2005) 32(2) (2008) 201–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [379].Boney CM, Verma A, Tucker R, Vohr BR, Metabolic syndrome in childhood: association with birth weight, maternal obesity, and gestational diabetes mellitus, Pediatrics 115(3) (2005) e290–6. [DOI] [PubMed] [Google Scholar]
- [380].Roseboom TJ, van der Meulen JH, Ravelli AC, Osmond C, Barker DJ, Bleker OP, Effects of prenatal exposure to the Dutch famine on adult disease in later life: an overview, Mol Cell Endocrinol 185(1-2) (2001) 93–8. [DOI] [PubMed] [Google Scholar]
- [381].Yajnik CS, Lubree HG, Rege SS, Naik SS, Deshpande JA, Deshpande SS, Joglekar CV, Yudkin JS, Adiposity and hyperinsulinemia in Indians are present at birth, The Journal of clinical endocrinology and metabolism 87(12) (2002) 5575–80. [DOI] [PubMed] [Google Scholar]
- [382].Arends NJ, Boonstra VH, Duivenvoorden HJ, Hofman PL, Cutfield WS, Hokken-Koelega AC, Reduced insulin sensitivity and the presence of cardiovascular risk factors in short prepubertal children born small for gestational age (SGA), Clinical endocrinology 62(1) (2005) 44–50. [DOI] [PubMed] [Google Scholar]
- [383].Petry CJ, Ozanne SE, Wang CL, Hales CN, Effects of early protein restriction and adult obesity on rat pancreatic hormone content and glucose tolerance, Horm Metab Res 32(6) (2000) 233–9. [DOI] [PubMed] [Google Scholar]
- [384].Simmons RA, Templeton LJ, Gertz SJ, Intrauterine growth retardation leads to the development of type 2 diabetes in the rat, Diabetes 50(10) (2001) 2279–86. [DOI] [PubMed] [Google Scholar]
- [385].Bouret SG, Simerly RB, Minireview: Leptin and development of hypothalamic feeding circuits, Endocrinology 145 (2004) 2621–2626. [DOI] [PubMed] [Google Scholar]
- [386].Pinto S, Roseberry AG, Liu H, Diano S, Shanabrough M, Cai X, Friedman JM, Horvath TL, Rapid rewiring of arcuate nucleus feeding circuits by leptin, Science 304(5667) (2004) 110–5. [DOI] [PubMed] [Google Scholar]
- [387].Vickers MH, Gluckman PD, Coveny AH, Hofman PL, Cutfield WS, Gertler A, Breier BH, Harris M, Neonatal leptin treatment reverses developmental programming, Endocrinology 146(10) (2005) 4211–6. [DOI] [PubMed] [Google Scholar]
- [388].Starling AP, Brinton JT, Glueck DH, Shapiro AL, Harrod CS, Lynch AM, Siega-Riz AM, Dabelea D, Associations of maternal BMI and gestational weight gain with neonatal adiposity in the Healthy Start study, Am J Clin Nutr 101(2) (2015) 302–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [389].Catalano PM, Drago NM, Amini SB, Maternal carbohydrate metabolism and its relationship to fetal growth and body composition, Am J Obstet Gynecol 172(5) (1995) 1464–70. [DOI] [PubMed] [Google Scholar]
- [390].Whitaker RC, Dietz WH, Role of the prenatal environment in the development of obesity, The Journal of pediatrics 132(5) (1998) 768–76. [DOI] [PubMed] [Google Scholar]
- [391].Lawlor DA, Smith GD, O’Callaghan M, Alati R, Mamun AA, Williams GM, Najman JM, Epidemiologic evidence for the fetal overnutrition hypothesis: findings from the mater-university study of pregnancy and its outcomes, Am J Epidemiol 165(4) (2007) 418–24. [DOI] [PubMed] [Google Scholar]
- [392].Silverman BL, Landsberg L, Metzger BE, Fetal hyperinsulinism in offspring of diabetic mothers. Association with the subsequent development of childhood obesity, Ann N Y Acad Sci 699 (1993) 36–45. [DOI] [PubMed] [Google Scholar]
- [393].Silverman BL, Rizzo TA, Cho NH, Metzger BE, Long-term effects of the intrauterine environment. The Northwestern University Diabetes in Pregnancy Center, Diabetes Care 21 Suppl 2 (1998) B142–9. [PubMed] [Google Scholar]
- [394].Kral JG, Biron S, Simard S, Hould FS, Lebel S, Marceau S, Marceau P, Large maternal weight loss from obesity surgery prevents transmission of obesity to children who were followed for 2 to 18 years, Pediatrics 118(6) (2006) e1644–9. [DOI] [PubMed] [Google Scholar]
- [395].Pinney S, Simmons R, Metabolic Programming, Epigenetics, and Gestational Diabetes Mellitus, Current Diabetes Reports 12(1) (2012) 67–74. [DOI] [PubMed] [Google Scholar]
- [396].Boyle KE, Patinkin ZW, Shapiro ALB, Bader C, Vanderlinden L, Kechris K, Janssen RC, Ford RJ, Smith BK, Steinberg GR, Davidson EJ, Yang IV, Dabelea D, Friedman JE, Maternal obesity alters fatty acid oxidation, AMPK activity, and associated DNA methylation in mesenchymal stem cells from human infants, Molecular metabolism 6(11) (2017) 1503–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [397].Heerwagen MJ, Miller MR, Barbour LA, Friedman JE, Maternal obesity and fetal metabolic programming: a fertile epigenetic soil, Am J Physiol Regul Integr Comp Physiol 299(3) (2010) R711–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [398].Allard C, Desgagne V, Patenaude J, Lacroix M, Guillemette L, Battista MC, Doyon M, Menard J, Ardilouze JL, Perron P, Bouchard L, Hivert MF, Mendelian randomization supports causality between maternal hyperglycemia and epigenetic regulation of leptin gene in newborns, Epigenetics 10(4) (2015) 342–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [399].Heindel JJ, vom Saal FS, Role of nutrition and environmental endocrine disrupting chemicals during the perinatal period on the aetiology of obesity, Mol Cell Endocrinol 304(1-2) (2009) 90–6. [DOI] [PubMed] [Google Scholar]
- [400].Barouki R, Melén E, Herceg Z, Beckers J, Chen J, Karagas M, Puga A, Xia Y, Chadwick L, Yan W, Audouze K, Slama R, Heindel J, Grandjean P, Kawamoto T, Nohara K, Epigenetics as a mechanism linking developmental exposures to long-term toxicity, Environment International 114 (2018) 77–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [401].Hofman PL, Regan F, Jackson WE, Jefferies C, Knight DB, Robinson EM, Cutfield WS, Premature birth and later insulin resistance, The New England journal of medicine 351(21) (2004) 2179–86. [DOI] [PubMed] [Google Scholar]
- [402].French NP, Hagan R, Evans SF, Godfrey M, Newnham JP, Repeated antenatal corticosteroids: size at birth and subsequent development, Am J Obstet Gynecol 180(1 Pt 1) (1999) 114–21. [DOI] [PubMed] [Google Scholar]
- [403].Bloom SL, Sheffield JS, Mclntire DD, Leveno KJ, Antenatal dexamethasone and decreased birth weight, Obstetrics and gynecology 97(4) (2001) 485–90. [DOI] [PubMed] [Google Scholar]
- [404].Entringer S, Wüst S, Kumsta R, Layes IM, Nelson EL, Hellhammer DH, Wadhwa PD, Prenatal psychosocial stress exposure is associated with insulin resistance in young adults, Am J Obstet Gynecol 199(5) (2008) 498.e1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [405].Tamashiro KL, Terrillion CE, Hyun J, Koenig JI, Moran TH, Prenatal stress or high-fat diet increases susceptibility to diet-induced obesity in rat offspring, Diabetes 58(5) (2009) 1116–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [406].Oberlander TF, Weinberg J, Papsdorf M, Grunau R, Misri S, Devlin AM, Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses, Epigenetics 3(2) (2008) 97–106. [DOI] [PubMed] [Google Scholar]
- [407].Dallman MF, Pecoraro NC, la Fleur SE, Chronic stress and comfort foods: self-medication and abdominal obesity, Brain Behav Immun 19(4) (2005) 275–80. [DOI] [PubMed] [Google Scholar]
- [408].Dallman MF, Pecoraro N, Akana SF, La Fleur SE, Gomez F, Houshyar H, Bell ME, Bhatnagar S, Laugero KD, Manalo S, Chronic stress and obesity: a new view of “comfort food”, Proceedings of the National Academy of Sciences of the United States of America 100(20) (2003) 11696–701. [DOI] [PMC free article] [PubMed] [Google Scholar]