

Abstract
Both opioids and nonsteroidal anti-inflammatory drugs (NSAIDS) produce deleterious side effects and fail to provide sustained relief in patients with chronic inflammatory pain. Peripheral neuroinflammation (PN) is critical for initiation and development of inflammatory pain. A better understanding of molecular mechanisms underlying PN would facilitate the discovery of new analgesic targets and the development of new therapeutics. Emerging evidence suggests that peripheral sensory neurons are not only responders to painful stimuli, but are also actively engaged in inflammation and immunity, whereas the intrinsic regulatory mechanism is poorly understood. Here we report the expression of proton-selective ion channel Hv1 in peripheral sensory neurons in rodents and humans, which was previously shown as selectively expressed in microglia in mammalian central nervous system. Neuronal Hv1 was up-regulated by PN or depolarizing stimulation, which in turn aggravates inflammation and nociception. Inhibiting neuronal Hv1 genetically or by a newly discovered selective inhibitor YHV98-4 reduced intracellular alkalization and ROS production in inflammatory pain, mitigated the imbalance in downstream SHP-1-pAKT signaling, and also diminished pro-inflammatory chemokine release to alleviate nociception and morphine-induced hyperalgesia and tolerance. Thus, our data reveal neuronal Hv1 as a novel target in analgesia strategy and managing opioids-related side effects.
Subject terms: Mechanisms of disease, Ion channel signalling
Introduction
Opioids such as morphine are the most potent analgesics discovered so far. However, chronic use of opioids produces detrimental side effects including addiction, tolerance and hyperalgesia. Presently, the most common default option to opioids is nonsteroidal anti-inflammatory drugs (NSAIDs), which also produce gastrointestinal and cardiovascular adverse effects.1 Recent clinical data suggest that neither opioids nor NSAIDs provide much sustained effective relief of pain in patients with chronic noncancer pain conditions, and consequently, their use for persistent inflammatory pain should be limited.2–4
Peripheral neuroinflammation (PN) is critical for the initiation and development of chronic inflammatory pain. A better understanding of the molecular mechanisms underlying PN should facilitate the discovery of new analgesic targets and the development of new therapeutics for chronic inflammatory pain. Although non-neuronal cells are considered as key players in chronic neuroinflammation, recent studies suggest that peripheral sensory neurons display cell autonomous regulation on nociception by inflammation,5,6 and inflammation is also involved in opioids-induced analgesic tolerance and hyperalgesia.7 The accumulation of NADPH oxidases (NOX)-dependent reactive oxygen species (ROS) contributes to neuroinflammation and pain hypersensitivity, and restoring the balance of ROS production represents potential therapeutic strategies for treating inflammatory pain.8 Nevertheless, the intrinsic regulator of NOX-dependent ROS in sensory neurons in pathological pain remains poorly understood. In the mammalian nervous system, hydrogen voltage gated channel 1 (Hv1), was shown to be selectively expressed and function in the CNS-resident microglia, and microglial Hv1 was required for NOX-dependent ROS generation and enhanced brain damage from ischemic stroke.9–14 In contrast, unequivocal evidence for the expression of Hv1 in mammalian neurons has been lacking for decades.
In this study, we report the striking expression of Hv1 in sensory neurons in dorsal root ganglion (DRG). In parallel, we also report a new family of Hv1 inhibitor targeting an intermediate conformational state of the channel which yielded notable selectivity over a wide range of ion channels containing the similar functional domain by using a structure-based approach. Using the new inhibitor along with genetic approaches, we provide evidence to show neuronal Hv1 mediates inflammatory pain and morphine-induced analgesic tolerance and hyperalgesia by regulating intracellular proton homeostasis and NOX-dependent ROS production.
Results
Expression of Hv1 and its up-regulation by inflammation in the peripheral nervous system
Although Hv1 was reported to be absent from central mammalian neurons,10,15,16 we detected the expression of Hv1 in peripheral DRG neurons using mRNA in situ hybridization (RNAscope technique) as well as immunohistochemistry (IHC). Hv1 expression was markedly up-regulated in mice or rats under chronic inflammation induced by the hind paw injection of Complete Freund’s adjuvant (CFA) (Fig. 1a, b; Supplementary information, Fig. S1a–d). The up-regulation was also observed in mice under chronic neuropathy induced by spared nerve injury (SNI) (Fig. 1c). Remarkably, an acute noxious painful stimulation lasting for 5 min induced by formalin injection was sufficient to induce a strong up-regulation of Hvcn1 (Fig. 1d). Hvcn1 was present in all NeuN-expressing (Rbfox3+) neurons (Fig. 1e), including different subtypes of nociceptors (Supplementary information, Fig. S1e–j). Consistent with our in situ data, previous studies using single-cell sequencing also identified Hvcn1 expression in different subtypes of DRG neurons.17–19 We confirmed the previous reports by identifying the presence of NOX1 and NOX4 in DRG neurons (Supplementary information, Fig. S1k, l), which were functionally coupled to Hv1.20 By contrast, we only detected relatively weaker Hvcn1 expression in a small subset of macrophages (less than 16%) in DRG in Cx3cr1GFP mice. Macrophage Hvcn1 expression contributes less than 8% of total Hvcn1 expression in DRG, which was not further up-regulated in the CFA mice (Fig. 1f). In the mice foot skin, Hv1 distribution was partially colocalized with the unmyelinated nerve endings marker PGP9.5, and was also up-regulated in the CFA mice (Supplementary information, Fig. S1m, n). Notably, in the Hvcn1−/− mice we generated (Supplementary information, Fig. S2), the Hv1 protein or mRNA expression was barely detectable in DRG or in foot skin under the naïve as well as CFA condition (Fig. 1g; Supplementary information, Fig. S1o, p). In the normal abdominal skin tissue and scar tissue from humans, we also detected Hv1 colocalized with PGP9.5 (Fig. 1h, i; Supplementary information, Fig. S1q, r). In the dorsal horn of the spinal cord in mice, we found Hvcn1 mRNA or protein expression level was low and sparse, which was restricted within a small subset of microglia and was slightly up-regulated in CFA condition (Supplementary information, Fig. S3).
Fig. 1. Expression of Hvcn1 in peripheral nervous system.

a Typical RNAscope images showing the expression of Hvcn1 in DRG in naïve mice and CFA model. b–d Quantifications of Hvcn1 expression in different conditions using RNAscope (CFA model, n = 31–54 sections from 10–15 mice, Mann–Whitney test; SNI model, n = 3–7 sections from three mice, two sample t-test; Formalin model, n = 7–11 sections from three mice, one-way ANOVA followed by Tukey’s multiple comparison test). e Typical RNAscope images showing Hvcn1 expression in Rbfox3+ neurons. f Images and quantifications showing Hvcn1 expression in GFP+ cells in CFA model in comparison to naïve mice (n = 8–14 sections from 6 mice, Mann–Whitney test). g Typical RNAscope images and quantifications showing the expression of Hvcn1 in DRG in Hvcn1−/− mice in comparison to WT mice under CFA condition. (n = 7–10 sections from three mice, two sample t-test). h, i RNAscope images for Hvcn1 and IHC images for PGP9.5 in the normal abdominal skin and scar tissue from humans. j Typical RNAscope images showing Hvcn1 expression in TNF-α treated DRG neurons. k–n Quantifications of Hvcn1 expression in DRG neurons in different conditions (n ≥ 6 mice for each condition, two sample t-test, Mann–Whitney test or Kruskal–Wallis test followed by Dunn’s multiple comparisons test). Data in b, c, d, f, g, and k–n are shown as means ± SEM. n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
We further found that inflammatory cytokines such as tumor necrosis factor-α (TNF-α, 10 ng/mL) or interleukin-1β (IL-1β, 50 ng/mL) was sufficient to induce up-regulation of Hv1 in acutely dissociated DRG neurons (Fig. 1j–l). This up-regulation was also observed by the application of Phorbol 12-myristate 13-acetate (PMA, 500 ng/mL), an agent commonly used to activate NOX resulting in ROS production,10 or by sustained action potential firings induced by high extracellular K+ (Fig. 1m, n). The increase of Hvcn1 expression induced by high K+ could be inhibited by a non-selective NOX inhibitor diphenyleneiodonium chloride (DPI, 10 μM) or by a selective NOX1/4 inhibitor GKT137831 (GKT, 10 μM) (Fig. 1n). Thus, the expression of neuronal Hv1 is highly sensitive to inflammatory signals or nociceptor hyperactivity which are hallmarks of peripheral inflammatory pain.
Discovery of a potent selective Hv1 channel inhibitor, YHV98-4
Although Hv1 inhibitors including Zn2+, Hanatoxin and 2GBI have been reported,21–24 they have poor selectivity, and none of them has yet been shown the efficacy in vivo. To better study Hv1 in sensory neurons, we set to discover small-molecule modulators for Hv1 using structure-based virtual screening. Ion channels are dynamic proteins and usually have several conformational states (Fig. 2a). Here we examined three conformations of human Hv1 (hHv1) for the virtual screening (Supplementary information, Fig. S4). The first conformation is the activated-state model of hHv1 (Supplementary information, Fig. S4a, b) based on the activated-state structure of the ciona intestinalis voltage sensor-containing phosphatase (ci-VSP) voltage sensing domain (VSD) (PDB code: 4G7V).25 The second conformation is the resting-state model of hHv1 (Supplementary information, Fig. S4a, c) based on a crystal structure of mouse Hv1 (mHv1cc) (PDB code: 3WKV).26 The third conformation was a recently reported solution NMR structure of hHv1 VSD in a micellar environment (PDB code: 5OQK),27 which was proposed to represent an intermediate conformation between the resting and activated states (Supplementary information, Fig. S4d).
Fig. 2. Structure-based discovery of Hv1 inhibitors.

a Strategy to design active compounds targeting Hv1 channel. b Pocket in the intermediate state of Hv1 used for virtual screening. Representative docking poses are shown as yellow sticks. c Chemical structure of YHV98-1. d Typical whole-cell current traces showing the effect of YHV98-1 on Hv1. e Dose-response curve for YHV98-1 on Hv1 (n ≥ 5). f View of the interactions between YHV98-1 and Hv1. Hydrogen bonds are shown as red dash. g Effects of YHV98-1 on Hv1 mutants (n ≥ 5, WT vs mutations, one-way ANOVA followed by Dunnett’s multiple comparison’s test). h MD snapshots showing the conformations of the water permeable pathway of Hv1. Water molecules are shown in blue. YHV98-1 is shown in yellow. i Chemical structures of YHV98-1 derivatives with substituents at the pyridine ring group. j Summary for the effects of 10 μM YHV98-1 and its derivatives on Hv1 channel (n ≥ 5, one-way ANOVA followed by Dunnett’s multiple comparison’s test). k Typical whole-cell current traces showing effects of YHV98-4 on Hv1. l Dose-response curve for YHV98-4 on Hv1 (n ≥ 5). m Comparison of the binding poses of YHV98-1 and YHV98-4. n Summary for the effects of YHV98-4 on other ion channels (n ≥ 5, Hv1 vs other channels, one-way ANOVA followed by Dunnett’s post hoc test). Data in e, g, j, l, and n are shown as means ± SEM, n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001.
We used Fpocket 2.0 server28 to evaluate whether the activated- and resting-state conformations as well as the intermediate conformation contain druggable pockets that could be used for virtual screening. A druggability score greater than 0.5 (the threshold) means that the pocket might be druggable.28 The activated conformation showed a pocket with a druggability score as high as 0.93 which may be the binding site of 2GBI, a reported inhibitor of Hv1.29 Notably, from the intracellular side of the intermediate conformation, we identified a cavity with a druggability score as high as 0.99 which was formed by the S0 segment and the intracellular ends of the four transmembrane segments (S1–S4) (Supplementary information, Fig. S4d). Because the corresponding pockets in the activated and resting conformations have much lower druggability scores (Supplementary information, Fig. S4b, c), we performed virtual screening targeting the pocket in the intermediate conformation with the Specs database (Fig. 2b). A few hits were tested for their effects on hHv1 overexpressed in human embryonic kidney (HEK)-293T cells by using the whole-cell patch-clamp recording, which led to the discovery of a Hv1 inhibitor, YHV98-1 (Fig. 2c). Under an acidic intracellular solution (pHi at 6.0), 10 μM YHV98-1 inhibited the outward currents to ~63% of control measured at 20 mV (Fig. 2d). YHV98-1 inhibition was dose-dependent, with a half-maximum inhibitory concentration (IC50) of 1.8 ± 0.4 μM (Fig. 2e). As control, in non-transfected HEK-293T cells, the outward currents elicited upon membrane depolarization were tiny (15.9 ± 1.6 pA at 20 mV, n = 6).
Virtual screening suggested a potential binding mode of YHV98-1 within the channel (Supplementary information, Fig. S5a). To further characterize the binding mode of the inhibitor to Hv1 with the consideration of protein flexibility, we performed a 200-ns molecular dynamics (MD) simulation for the YHV98-1-bound state of Hv1 on the best pose provided by an induced fit docking (Supplementary information, Fig. S5b, c). In the MD simulation, YHV98-1 bound configuration generally remained stationary (Fig. 2f; Supplementary information, Fig. S5d). Residues R89 and D174 interacted with YHV98-1 through water bridges. In addition to R89 and D174, several residues including S97, L108, I155, F161, and S219 were distributed around YHV98-1 and may contribute to ligand binding. We calculated the per-residue effective binding energy based on the MD simulation trajectory to evaluate potential contributions of the residues to the ligand-channel binding. The residues contributing more than –3 KJ/mol of binding energy include R89, S97, L108, I155, F161, D174, and S219 (Supplementary information, Fig. S5e). We mutated these residues and examined the inhibitory effects of YHV98-1 on proton current mediated by Hv1 mutants. A saturating concentration of YHV98-1 (30 μM) showed reduced inhibitory effects on R89A, S97A, L108A, I155A, F161A, and S219A, expect for the mutant D174A, which yielded a non-functional channel (Fig. 2g; Supplementary information, Fig. S6). These mutagenesis results are consistent with the computational predictions.
We also docked YHV98-1 into the activated and resting states of Hv1. The glide docking scores indicate that YHV98-1 does not interact well with these two states of Hv1 (Supplementary information, Fig. S7a). Moreover, MD simulations revealed that the binding pose between YHV98-1 and the activated-state or the resting-state was not stable. YHV98-1 escaped from the initial binding position to the simulated water environment in the 50-ns MD simulations (Supplementary information, Fig. S7b). In contrast, the binding of YHV98-1 in the intermediate state was stable during the 200-ns MD simulation (Fig. 2f; Supplementary information, Fig. S5d).
Previous studies suggested that proton conduction in Hv1 may occur under the environment of water wires in the pathway.30–32 In addition, Gianti et al. indicated that molecules may inhibit the channel by interrupting the water wires.33 We next examined the influences of YHV98-1 on water permeable pathway of the intermediate state Hv1 in the MD simulation. For comparison, we carried out an additional MD simulation on the apo (ligand-free) Hv1. Similar to previous studies,30–32,34 the MD simulation on the apo Hv1 supported that the inner cavity of Hv1 is a water permeable pathway (Fig. 2h, left). Compared to the apo Hv1, the permeability of the water molecules in the YHV98-1-bound Hv1 channel was reduced (Fig. 2h, right). However, YHV98-1 did not block the water permeable pathway completely. This might explain the experimental observation that YHV98-1 with a saturating concentration could only partially inhibit Hv1 current (Fig. 2e).
Pharmacokinetic stability predicted by XenoSite server35 suggested that the pyridine ring was a potential site that may affect the stability of the molecule in vivo (Supplementary information, Fig. S8). The pyridine ring of YHV98-1 is located in a relatively hydrophobic cavity (Fig. 2f) and its modification to more hydrophobic groups may potentially increase affinity. To test this idea, we synthesized three analogs of YHV98-1 by modifying the pyridine ring (Fig. 2i). The most potent analog YHV98-4 has a chlorobenzene group instead of the pyridine ring group (Fig. 2i–k). YHV98-4 inhibited Hv1 channel with improved affinity compared to YHV98-1 and the IC50 for YHV98-4 inhibition of Hv1 is 0.7 ± 0.6 μM (Fig. 2l). The binding poses of YHV98-1 and YHV98-4 were similar (Fig. 2m). Importantly, single dose pharmacokinetic analysis suggested that YHV98-4 achieved a better exposure in the plasma (4465 ng/mL vs 3746 ng/mL) and longer half-life (1.75 h vs 0.82 h) than YHV98-1 (Supplementary information, Table S1), making it a better compound for in vivo application.
Hv1 channel and the other VSDs-containing voltage gated cation channels share a similar structural arrangement. Therefore, we examined the selectivity of YHV98-4 against several other channels containing VSDs, including hERG, KCNQ2, BK, and Nav1.7 channel. Electrophysiological assays revealed that 20 μM YHV98-4 has no inhibitory effects on these channels (Fig. 2n; Supplementary information, Fig. S9). Furthermore, 20 μM YHV98-4 did not inhibit transient receptor potential V1 (TRPV1) or A1 (TRPA1), which also contains VSD-like domain (Fig. 2n; Supplementary information, Fig. S9). In the subsequent studies, we used YHV98-4 as the major pharmacological tool to probe the biological functions of Hv1.
Hv1 mediates voltage-gated H+ currents in sensory neurons
We sought to measure the outward proton currents mediated by Hv1 channels in DRG neurons. Voltage-gated outward currents present in DRG neurons from naïve mice, CFA mice, or Hvcn1−/− mice with an acidic pHi (pH 5.5) in the recording pipette. Previous study identified confounding Cl− channel-mediated inward anion current when studying proton currents in human cardiac fibroblast.36 Indeed, we also found part of the current was mediated by volume-regulated anion channel (VRAC), as they could be partially (~30%) inhibited by DCPIB (20 μM) or Tamoxifen (10 μM), two VRAC inhibitors (Supplementary information, Fig. S10).37–39 To better assess the role of Hv1 in DRG neurons, we recorded the voltage-gated outward currents in the presence of DCPIB (defined thereafter as “control” currents). For neurons from naïve or CFA mice, the control current was markedly inhibited by 100 μM Zn2+, a well-established inhibitor for Hv1 (Fig. 3a, b).40 We quantified the Zn2+-sensitive current (IControl−) and the inhibitory effect of Zn2+ in different conditions: First, the Zn2+-sensitive current in DRG neurons of the CFA mice was significantly up-regulated compared to that of the naïve mice (Fig. 3c). Second, at pHi 7.3, the Zn2+-sensitive current was still present but significantly reduced. Last, in the Hvcn1−/− mice, the Zn2+-sensitive current and the Zn2+ inhibition was tiny even at pHi 5.5 (Fig. 3c). In a similar experimental setting, for naïve or CFA mice, the control current could be markedly inhibited by 20 μM YHV98-4 (Fig. 3d, e). It should be noted that YHV98-4 had no effect on the VRAC-mediated currents (Supplementary information, Fig. S9g). The YHV98-4-sensitive current (IControl−I+YHV98-4) was significantly up-regulated in DRG neurons from CFA mice. At pHi 7.3, the YHV98-4-sensitive current was still present but significantly reduced in CFA mice (Fig. 3f). In the Hvcn1−/− mice, the YHV98-4-sensitive current and the inhibition was tiny even at pHi 5.5 (Fig. 3f). The YHV98-4-sensitive current was also significantly up-regulated in DRG neurons from SNI mice (Supplementary information, Fig. S11). We further determined the reversal potential of the YHV98-4-sensitive current by tail current recordings with different pHi gradients in DRG neurons (Fig. 3g).41 The reversal potentials at different pHi determined experimentally were close to the prediction by the Nernst Equation for a proton-selective membrane with a deviation due to proton depletion (Fig. 3g). The proton depletion was also previously observed and discussed.10,41 We noticed very fast activation kinetics of both YHV98-4 sensitive and Zn2+-sensitive current (Fig. 3c, f), which was similar to the proton current in snail neurons,42 contrasting to the slow activation kinetics of Hv1 current in mammalian microglia.10 This fast kinetics of the proton current might be ideal for adapting the rapid electric activities in neurons, and may be determined by some neuron-specific regulators. Having determined the specificity of YHV98-4 on Hv1, we assessed if acutely blocking Hv1 has any effect on neuronal excitability. We found that YHV98-4 had no effect on the resting membrane potential nor on the action potential firing properties of DRG neurons (Supplementary information, Fig. S12). Overall, the data above suggest: (1) Hv1-mediated rapid outward H+ current was functionally present in DRG neurons from naïve mice, which was up-regulated in inflammation. (2) YHV98-4 effectively and selectively inhibited Hv1-mediated currents in DRG neurons.
Fig. 3. Hv1-mediated proton currents in DRG neurons.

a Left: typical whole-cell current traces showing the effect of Zn2+ on Hv1 current in DRG neurons. Right: current–voltage (I-V) relationships for DRG neurons with or without Zn2+ (n = 7 cells from 6 mice, two-way repeated measures ANOVA). b I-V relationships for DRG neurons with or without Zn2+ under CFA condition (n = 8 cells from 6 mice, two-way repeated measures ANOVA). c Left: typical whole-cell current traces of Zn2+ sensitive current under different condition. Middle: I-V curves of the experiment represented in left. (n = 7–8 cells from at least 6 mice, two-way repeated measures ANOVA followed by Dunnett’s multiple comparisons test). Right: Zn2+ inhibition rate under different condition. (n = 7–8 cells from at least 6 mice, one-way ANOVA followed by Dunnett’s multiple comparisons test). d Left: typical whole-cell current traces showing the effect of YHV98-4 on Hv1 current in DRG neurons. Right: I-V relationships for DRG neurons with or without YHV98-4 (n = 10 cells from eight mice, two-way repeated measures ANOVA). e I-V relationships for DRG neurons with or without YHV98-4 under CFA condition (n = 8 cells from six mice, two-way repeated measures ANOVA). f Left: typical whole-cell current traces of YHV98-4 sensitive current under different condition. Middle: I-V curves of the experiment represented in left. (n = 6–10 cells from at least 6 mice, two-way repeated measures ANOVA followed by Dunnett’s multiple comparisons test). Right: YHV98-4 inhibition rate under different condition. (n = 6–10 cells from at least 6 mice, one-way ANOVA followed by Dunnett’s multiple comparisons test). g Left: Typical Hv1 tail current traces. The YHV98-4 sensitive current was recorded at various potentials from −80 mV to +60 mV following a depolarizing pre-pulse to +60 mV for 1.5 s. Middle: Reversal potentials were determined by tail current recordings. (n = 6–8 cells from six mice). Right: Reversal potential at different pHo/pHi gradients in DRG neurons. Data were fitted by a continuous line (black line) with a slope of −31.49 mV/∆pH. The Nernst potential for a proton-selective membrane has a slope of −58 mV/∆pH (red dashed line). h Top: schematic of construction of AAVs. Bottom, left: I-V curves of Zn2+ sensitive current in DRG neuron from Hv1 knockdown mice (AAV-Hv1 KD) or control group (AAV-Control). (n = 9–10 cells from at least 7 mice, two-way repeated measures ANOVA). Bottom, right: Zn2+ inhibition rate at 20 mV under different condition in Hv1 knockdown mice or control group. (n = 9–10 cells from at least 7 mice, two sample t-test). All data are shown as means ± SEM. **P < 0.01, ***P < 0.001.
We used DRG injection of AAV constructs as a strategy for acute knockdown of Hvcn1 specifically in DRG neurons (Fig. 3h). The AAV injection infected a part but not all the neurons in DRG (Supplementary information, Fig. S13a–d). As a result, the Hvcn1 knockdown mice by AAV2/9-hSyn-Hv1-miRNA (AAV-Hv1 KD) showed heterogeneous but overall strong reduction in mRNA (~75%) or protein expression (~59%) of Hv1 in sensory neurons, compared to the control mice (AAV2/9-hSyn-NC-miRNA, AAV-Control) (Supplementary information, Fig. S13a–d). No viral expression was identified in macrophages, in which the Hv1 expression was barely detectable in both groups (Supplementary information, Fig. S13e). We recorded the Zn2+-sensitive current in DRG neurons under CFA condition guided by the mCherry fluorescence in both groups which represented neurons with confirmed AAV infection and the data suggest that in mCherry-positive neurons from the AAV-Hv1 KD but not the AAV-Control mice, the Zn2+-sensitive current was virtually absent (Fig. 3h).
Inhibiting Hv1 reduces intracellular alkalization and ROS production
Next, we examined if YHV98-4 regulates intracellular H+ homeostasis in DRG neurons by using 5-fluorescein isothiocyanate (FITC) as a fluorescent probe for intracellular pH measurement. Consistent with the previous report that intracellular alkalization of DRG neuron was associated with pain,43 we found that: (1) the resting pHi of DRG neurons in CFA mice was more alkalized than that under naïve condition; (2) the Hvcn1−/− mice or AAV-Hv1 KD DRGs had lower pHi under naïve condition than the control mice, and they were also less alkalized than the control mice under CFA condition (Fig. 4a; Supplementary information, Fig. S14a–b). DRG neurons were acid loaded using the rebound acidification technique10,43 by perfusing cells with 20 mM NH4Cl, and the pHi recovery was measured. After intracellular acidification upon NH4Cl washout, the pHi recovery was barely detectable in WT DRG neurons with or without YHV98-4 treatment or in those from Hvcn1−/− mice in an extracellular solution containing high Na+ (Fig. 4b). To exclude the involvement of Na+-dependent mechanism (such as Na+/H+ exchanger), a Na+ free solution (NMDG) was applied to further assess action potential-induced pHi change. Under this condition, pHi rapidly recovered when cells were depolarized by a high K+ solution, which was significantly reduced in the presence of YHV98-4 and in neurons from Hvcn1−/− mice (Fig. 4b, c; Supplementary information, Fig. S14c). This reduced recovery was also observed in DRG neurons from AAV-Hv1 KD mice (Fig. 4c). PMA is used to specifically activate NOX and induce NOX-dependent ROS production. We found that PMA induced a slow intracellular alkalization in DRG neurons, which was inhibited by YHV98-4 or Hvcn1 KD (Fig. 4d, e). Intraplantar (i.pl.) administration of PMA (50 pmol/paw) in mice produced a significant mechanical allodynia (Supplementary information, Fig. S14d), which further supported the idea that intracellular alkalization in DRG was associated with pain. Thus, Hv1 is not only critical for H+ homeostatic change following neuroinflammation and pain but also involved in pH rapid recovery following neuronal activation.
Fig. 4. Hv1-mediated intracellular proton homeostasis and ROS production.

a–e pHi at rest (a n = 151–1245 cells for each group from at least 4 mice, Two-way ANOVA followed by Tukey’s multiple comparisons test) or in response to different stimulations of DRG neurons from WT, Hvcn1−/− or Hvcn1 knockdown mice (b–e n = 19–56 cells for each group from at least 4 mice, Kruskal–Wallis test followed by Dunn’s multiple comparisons test). f Images and quantifications for DHE staining in DRG section from naïve, or vehicle or YHV98-4 (10 mg/kg, i.p.) treated mice. (n = 16–18 DRGs from at least eight mice, one-way ANOVA followed by Tukey’s multiple comparison test). g Quantifications for DHE staining in DRG section from naïve, or WT control or Hvcn1−/− mice after CFA injection (n = 29–34 sections from 4 mice, one-way ANOVA followed by Tukey’s multiple comparison test). h Quantifications for DCFH-DA staining in DRG section from naïve, or AAV-Control or AAV-Hv1 KD mice after CFA injection (n = 9–10 DRGs from 3 mice, One-way ANOVA followed by Tukey’s multiple comparisons test). i–l Images for DCFH-DA staining and quantifications showing the ROS production in acutely dissociated DRG neurons in different conditions (n = 204–3652 cells from 8–12 mice, Kruskal–Wallis followed by Dunn’s multiple comparisons test). Data in a, c, e–l are shown as means ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Hv1-mediated ROS production has been well documented in neutrophils.10,44,45 As expected, the Hv1 inhibitors, YHV98-4 and Zn2+, both significantly inhibited PMA induced ROS production in neutrophils (Supplementary information, Fig. S14e). Next, we examined the role of neuronal Hv1 function in ROS production. We detected significant increase in ROS production detected by dihydroethidium (DHE) staining in the DRG of CFA mice compared to naïve mice (Fig. 4f), which could be reversed by YHV98-4 treatment [10 mg/kg, intraperitoneal injection (i.p.)]. Consistently, DRGs from Hvcn1−/− mice or AAV-Hv1 KD mice showed significant reduced ROS production than those from control mice (Fig. 4g, h; Supplementary information, Fig. S14f, g). In acutely dissociated DRG neurons, YHV98-4 significantly reduced lipopolysaccharide (LPS)-induced ROS production (Fig. 4i), or PMA induced NOX-mediated ROS production reflected by 2’,7’-dichlorodihydrofluorescein diacetate (DCFH-DA) staining (Fig. 4j; Supplementary information, Fig. S14h). Interestingly, overt neuronal activation induced by high K+ or by activation of TRP channels by 100 μM allyl isothiocyanates (AITC, for TRPA1) plus 1 μM capsaicin (for TRPV1) increased ROS production which was also inhibited by YHV98-4 (Fig. 4k, l; Supplementary information, Fig. S14i, j). Increased ROS production was observed in SNI mice, which could be also attenuated by in vivo administration of YHV98-4 (Supplementary information, Fig. S14k). Thus, our data using pharmacological inhibition and genetic intervention together support that inhibiting neuronal Hv1 reduces ROS production in neuroinflammation or neuronal hyperactivation.
Inhibiting Hv1 alleviates pain and inflammation
Inhibiting neuronal Hv1 reduces ROS production. Because high ROS level leads to pain sensitization and reducing ROS levels can alleviate pathological pain,8,46,47 next, we tested whether inhibiting Hv1 may treat inflammation and pain. Indeed, in the chronic inflammatory pain developed in the CFA mice, YHV98-4 displayed dose-dependent effects on alleviating mechanical allodynia in the von Frey test with a fast onset (30 min) after i.p. injection, with a maximal effect at a dose of 10 mg/kg (data for male mice in Fig. 5a, and data for female mice in Supplementary information, Fig. S15a). Hereafter, 10 mg/kg i.p. injection was used for the following animal studies. YHV98-4 also reduced heat hyperalgesia in the Hargreaves test (data for male mice in Fig. 5b, and data for female mice in Supplementary information, Fig. S15b) and paw flinches, a behavior which was often observed in rodents in pain conditions (Fig. 5c).48 Moreover, 10 mg/kg YHV98-4 had higher maximal possible effect (MPE) in alleviating mechanical allodynia and paw flinches than a medium dose of diclofenac sodium [30 mg/kg, oral (p.o.)], one of the first-line NSAIDS for the treatment of pain and inflammation (Fig. 5a, c).49 In SNI mice, YHV98-4 was effective in alleviating mechanical allodynia and heat hyperalgesia (Supplementary information, Fig. S15c, d). YHV98-4 had no effect on rotarod test or grip strength test (Supplementary information, Fig. S15e, f), suggesting that YHV98-4 had no effect on the locomotion activities in mice.
Fig. 5. Analgesic and anti-inflammatory effects of YHV98-4 in chronic pain.

a–c Comparison of the effects of YHV98-4 (i.p.) and diclofenac sodium (p.o.) on mechanical allodynia (a) heat hyperalgesia (b) and paw flinches (c) in CFA mice. Paw flinches on day 5 after CFA injection (n = 8–12, Two Way ANOVA followed by Dunnett’s multiple comparisons test or Mann–Whitney test for a Two Way ANOVA followed by Dunnett’s multiple comparisons test or two sample t-test for b Two Way ANOVA followed by Dunnett’s multiple comparisons test, Mann–Whitney test or two sample t-test for c). AUC, area under the curve. d–f Basal mechanical allodynia (d) thermal hyperalgesia (e) and paw flinches (f) under CFA in WT or Hvcn1−/− mice (n = 16–27 for d, e and n = 6 for f two way repeated measures ANOVA followed by Dunnett’s multiple comparisons test or two sample t-test). g–i Basal mechanical allodynia (g) thermal hyperalgesia (h) and paw flinches (i) under CFA in AAV-Control or AAV-Hv1 KD mice (n = 9–10, two way repeated measures ANOVA followed by Dunnett’s multiple comparisons test or two sample t-test). j Quantifications for paw edema (n = 6–16, one-way ANOVA followed by Tukey’s multiple comparison test). k, l quantifications for CD4+ cells (k) or F4/80+ cells (l) in situ expressions from foot skin sections in different conditions (n = 6–7 sections from 6–7 mice, one-way ANOVA followed by Tukey’s multiple comparison test). Data are shown as means ± SEM. n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Was YHV98-4 truly targeting neuronal Hv1 to be analgesic? We performed additional target validation experiments. The Hvcn1−/− mice exhibited normal nociceptive sensitivity in naïve condition but reduced mechanical allodynia, heat hyperalgesia and paw flinches compared to WT control mice under CFA condition (Fig. 5d–f). Similar observations were found in the Hvcn1−/− mice in SNI condition (Supplementary information, Fig. S15g, h). As expected, YHV98-4 was completely inactive in the Hvcn1−/− mice in all the pain-related tests (Supplementary information, Fig. S15i–m). To further confirm that YHV98-4 acts as an analgesic by targeting peripheral neuronal Hv1 but not central microglia Hv1, we measured the pain sensitivity in AAV-Hv1 KD mice and AAV-Control mice. The virus injection itself did not modify the pain sensitivity in these mice (Supplementary information, Fig. S16a, b). The AAV-Hv1 KD mice exhibited normal nociceptive sensitivity but reduced mechanical allodynia, heat hyperalgesia and paw flinches compared to the AAV-Control mice under CFA condition (Fig. 5g–i), and YHV98-4 was completely inactive in the AAV-Hv1 KD mice (Supplementary information, Fig. S16c, d). These results suggest that Hv1 in sensory neurons was required for the analgesic effects of YHV98-4. To further test the hypothesis that ROS accumulation is mechanistically involved in the role of Hv1 in pain, we used phenyl-N-t-butyl nitrone (PBN), a ROS scavenger which was previously shown to reduce pain hypersensitivity by reducing ROS production.50–53 In AAV-Control mice, PBN (50 mg/kg, i.p.)54 was effective in alleviating mechanical and thermal allodynia under CFA condition. In contrast, PBN displayed little effects in AAV-Hv1 KD mice, in which the ROS production was largely reduced (Supplementary information, Fig. S16e, f). These data suggested that ROS accumulation is required for the production of the analgesic effect by inhibiting Hv1.
Last, CFA-induced inflammation was evaluated by measuring the skin histopathology. Similar to diclofenac sodium, YHV98-4 significantly alleviated paw edema by reducing epidermal and dermal thickening (Fig. 5j; Supplementary information, Fig. S17a). We also performed staining for CD4+ T-cell and F4/80+ macrophage. Our results showed that CD4+ T-cells and F4/80+ macrophages were increased in CFA-induced inflammatory skin. Both YHV98-4 (10 mg/kg, i.p.) and diclofenac sodium significantly alleviated the immune cells infiltration reflected by reduced numbers of CD4+ T-cell and F4/80+ macrophage per square millimeter (Fig. 5k, l; Supplementary information, Fig. S17b, c).
Inhibiting Hv1 restores impaired ROS-SHP-1-PI3K/pAKT-CXCL1 pathway and attenuates morphine-induced hyperalgesia and tolerance
We explored the signaling pathway underlying the Hv1-depentent anti-inflammatory and analgesic effect mediated by YHV98-4. Src homology 2 domain-containing tyrosine phosphatase 1 (SHP-1) is a key protein tyrosine phosphatase. The down-regulation of SHP-1 is involved in the generation of pain,55,56 whereas the underlying mechanism remains incompletely understood. The activity of SHP-1 is highly sensitive to ROS.45 We observed a significant decrease in total SHP-1 as well as phosphorylated SHP-1 (pSHP-1) in DRG sections in situ under CFA condition, which could be reversed by YHV98-4 (Fig. 6a, b). Consistently, we also observed an increased SHP-1 and pSHP-1 in Hvcn1−/− DRG (Supplementary information, Fig. S18). Moreover, YHV98-4-induced analgesia was abolished by the SHP-1 inhibitor sodium stibogluconate (SSG) (Fig. 6c, d). SSG itself aggravated mechanical allodynia in the Hvcn1−/− mice under CFA condition but showed no effect in WT control mice (Fig. 6e, f), suggesting inhibiting SHP-1 could enhance pain hypersensitivity under CFA condition but only in the absence of Hv1. Taken together, our data suggest that SHP-1 is a downstream signaling molecule following Hv1-mediated ROS production in DRG neurons and contributes to YHV98-4-evoked analgesia under neuroinflammation. The reduced SHP-1 activity may disinhibit its downstream PI3K/pAKT pathway,45 which was also involved in pathological pain.57,58 To support this, we found that inhibiting SHP-1 using SSG was sufficient to increase the pAKT expression in DRG neurons (Fig. 6g). In CFA mice, we observed an increase of pAKT expression in situ in DRG, which was also reversed by YHV98-4 (Fig. 6h). It should be noted that ion channels including TRP and ASICs are sensitive to both ROS and extracellular proton. Their relationship to Hv1 in DRG remains to be explored individually and systematically.
Fig. 6. YHV98-4 restores impaired ROS-mediated inflammatory pathway.

a, b IHC images and quantifications for SHP-1 (a) and pSHP-1 (b) in situ expressions from DRG sections in different conditions (n = 10–18 sections from 9–10 mice, one-way ANOVA followed by Tukey’s multiple comparison test). c, d von Frey test (c) and Hargreaves test (d) in CFA mice treated with YHV98-4 (10 mg/kg, i.p.) with or without SSG (10 mg/kg, i.p.) (n = 8 mice, Two way repeated measures ANOVA followed by Sidak’s multiple comparisons test). e, f The effect of SSG (10 mg/kg, i.p.) on mechanical allodynia (e) and thermal hyperalgesia (f) in Hvcn1−/− mice or WT mice under CFA condition (n = 8, Two way repeated measures ANOVA followed by Sidak’s multiple comparisons test). g Typical IHC images and quantifications showing pAKT expression in SSG stimulated (10 μg/mL, 6 h) DRG neurons with or without YHV98-4 (n = 8 fields of view from 4 mice, Kruskal–Wallis test followed by Dunn’s multiple comparisons test). h Typical IHC images and quantifications showing pAKT expression in DRG neurons in situ from CFA mice or naïve mice (n = 15–17 sections from 12 mice, one-way ANOVA followed by Tukey’s multiple comparison test). i, j Quantification for release of cytokines and chemokines in different conditions (n = 8, Kruskal–Wallis test followed by Dunn’s multiple comparisons test or one-way ANOVA followed by Tukey’s multiple comparisons test). k Quantification for release of CXCL1 in different conditions (n = 6, one-way ANOVA followed by Tukey’s multiple comparisons test). l Schematic diagram of Hv1-mediated inflammatory signaling pathway. Data in a–k are shown as means ± SEM. n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Multiplex analysis for the protein extraction from mouse skin revealed significant increases in a variety of pro-inflammatory cytokines and chemokines in CFA mice in comparison to the naïve mice. Among those CFA-induced factors, the upregulation of CXCL1, IL-1β, IL-9, MIP-2, and MIP-1β could be significantly reversed by YHV98-4 (Fig. 6i; Supplementary information, Fig. S19). To further examine the anti-inflammatory effects mediated by neuronal Hv1, we also measured the cytokine and chemokine release in AAV-Hv1 KD and AAV-Control mice under CFA condition. Our results revealed significant decreases in CXCL1, Eotaxin, IL-6 and INF-γ in AAV-Hv1 KD mice (Fig. 6j; Supplementary information, Fig. S20). Thus, the two independent assays together highlighted the reduced level of CXCL1, a chemokine previously identified in pain,59,60 following neuronal Hv1 inhibition. Furthermore, wortmannin (WM), a PI3K inhibitor, was sufficient to reverse the increased CXCL1 release in DRG under CFA condition as a positive control (Fig. 6k). Taken together, our data suggest that inhibiting neuronal Hv1 restored the imbalanced SHP-1-PI3K/pAKT-CXCL1 pathway in inflammation (Fig. 6l).
ROS and CXCL1 are both involved in opioid-induced hyperalgesia and tolerance,8,61 two of the limitations for opioids use in chronic pain. We assessed if YHV98-4 could attenuate these adverse effects induced by morphine. The mouse group continuously treated with morphine administration (10 mg/kg, i.p.) for 6 days (termed MH group mice) showed markedly increased mechanical allodynia, which did not appear in the mouse group co-administrated of morphine and YHV98-4 for 6 days (termed MY group mice) (Fig. 7a, b). Subsequent single dose of morphine administration (1, 5, or 10 mg/kg) in the MH group mice showed little analgesic effect, suggesting the occurrence of morphine tolerance; whereas in the MY group morphine still showed analgesic effects which were not significantly different from those in the naïve mice (Fig. 7c). Strikingly, a single dose of YHV98-4 still exhibited analgesic effect in the MH group mice (Fig. 7d). As expected, we also detected significant higher ROS production in the DRG of MH group mice than in the MY group mice (Fig. 7e). Thus, repeated administration of YHV98-4 largely attenuated morphine-induced hyperalgesia and tolerance. Consistently, morphine-induced hyperalgesia and tolerance and high level of ROS production were present in AAV-Control mice but absent in AAV-Hv1 KD mice (Fig. 7f–j). Thus, our data using pharmacological inhibition and genetic intervention together support that neuronal Hv1 channel mediates opioid-induced hyperalgesia and tolerance associated with ROS production.
Fig. 7. YHV98-4 or Hvcn1 knockdown attenuates morphine-induced side effects.

a Schematic illustrating the experimental protocol for measuring morphine-induced side effects. b Mechanical allodynia measured in MH mice and MY mice (n = 8, one-way repeated measures ANOVA within MY group mice, two sample t-test or Mann–Whitney test for MY vs MH). c Quantification of the MPE of morphine in different conditions (n = 8, Kruskal–Wallis test followed by Dunn’s multiple comparisons test). d YHV98-4-induced analgesia in MH mice (n = 8, Mann–Whitney test). e DHE staining and quantifications showing the ROS production in DRG section in mice (n = 3–6 mice, Kruskal–Wallis test followed by Dunn’s multiple comparisons test). f Schematic illustrating the experimental protocol for measurements of morphine-induced side effects in Hvcn1 knockdown mice. g, h The time course of the mechanical allodynia and MPE following systemic administration of morphine (n = 10, one-way repeated measures ANOVA within AAV-Hv1 KD or AAV-Control group mice, two way repeated measure ANOVA for AAV-Hv1 KD vs AAV-Control group mice). i Quantification of the MPE of morphine in different conditions (n = 10, Mann–Whitney test). j DCFH-DA staining and quantifications showing the ROS production in DRG section from AAV-Hv1 KD and AAV-Control mice (n = 2–3 DRGs per mice, from 4–5 mice, Kruskal–Wallis test followed by Dunn’s multiple comparisons test). Data are shown as means ± SEM. n.s., not significant, *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001.
Discussion
Most of the previous work on Hv1 channel has focused on immune cells which are mostly microglia in the nervous system, and these studies in general have limitations in Hv1-based therapeutics due to the lack of suitable selective inhibitors for in vivo application. Here we report a few advances: (1) we discovered the first line of inhibitors for in vivo application by targeting an intermediate conformational state of the channel which yielded improved selectivity; (2) we modify the conventional view that Hv1 was restricted to microglia in the nervous system by showing its unexpected function in DRG neurons. We used the Hvcn1−/− mice and neuron-specific Hv1 knockdown along with the newly discovered inhibitor YHV98-4 in vitro and in vivo in most experimental paradigms in this study to support our conclusions: Hv1 serves as a key cell autonomous mechanism in inflammatory pain and up-scales neuroinflammation in a positive-feedback fashion. The previous study showed that the deletion of µ opioid receptor in sensory neurons abrogates morphine tolerance without disrupting analgesia.62 Our study further supports the idea that targeting PN represents a promising strategy for minimizing opioids side effects. The inhibitory effects on morphine-induced hyperalgesia and tolerance by YHV98-4 suggest Hv1 as a novel target for the development of adjuvant therapy to retain the beneficial effects of morphine for chronic pain, including cancer pain control.
The discovery of selective modulator of voltage-gated ion channels has been challenging due to the scarcity of druggable sites maintaining selectivity. The VSD is emerging as a new drug design site.63 Indeed, small-molecule modulators can regulate Kv, Nav and Hv channels by binding to the extracellular side of the water-filled crevice of the VSD.29,64–66 Now we move one more step forward by showing that the intracellular part of the water-filled crevice is also a druggable site. In addition, the current work supports the idea that intermediate conformational states of ion channels can be targeted for designing modulators with improved selectivity.67 Further comprehensive screenings are needed to characterize the selectivity of YHV98s against a wider range of ion channels and receptors. The inhibitor reported here would enable in vivo dissection of the role of Hv1 in many diseases including respiratory diseases, immune diseases, cancer and neurological diseases.
The current study has a few limitations which may require further investigation. Firstly, both treatment with YHV98-4 and knockdown of Hv1 reduced the release of several pro-inflammatory cytokines, suggesting an anti-inflammatory effect of inhibiting Hv1 in DRG neurons. On the other hand, although our ex vivo electrophysiological experiments suggest YHV98-4 did not have an “acute” effect on action potential properties of DRG neurons, additional approaches such as in vivo electrophysiology or in vivo Ca2+ imaging are needed to further clarify the role of Hv1 in neuronal excitability as a potential analgesic mechanism. Secondly, although the Hv1 currents (isolated by Zn2+ or YHV98-4) were detectable when the membrane potential of DRG neurons were above 20 mV, the operational time window for Hv1 is still limited as the neurons only reach such a depolarized state when action potential occurs and the membrane potential falls back instantaneously. The temporal contribution, constraints and limitations of Hv1 currents in the function of DRG neurons require further study. Thirdly, the possible side effects of inhibiting Hv1 remains to be assessed systematically. Fourthly, the transcriptional mechanism underlying the up-regulation of Hv1 expression under inflammation needs to be further explored. Fifthly, our study revealed SHP-1-PI3K/pAKT-CXCL1 as a disrupted downstream signaling pathway of Hv-1-mediated ROS production in pathological pain; nevertheless, ROS targets multiple signaling pathways, whether their activities are also impacted by Hv1 remains to be identified in the future study using high-throughput proteomic or transcriptomic methods. Lastly, the role of Hv1 in microglia or in neurons in the central nervous system in nociception and pain-associated mood disorders remains to be elaborated, the fulfillment of which would be valuable in assessing the role of Hv1 in chronic pain.
Materials and methods
Ethics statement on mouse and human skin studies
All experiments with animals were approved by the Animal Research Committee of East China Normal University (PROTOCOL No. m20190205) and the Animal Research Committee of West China Hospital of Sichuan University (PROTOCOL No. 2018175 A). For tissue collection, animals were given a lethal dose of pentobarbital intraperitoneally or euthanized by carbon dioxide.
The study protocol for human skin sample collection was reviewed and approved by Institutional Review Board and Biomedical Ethics Committee of West China Hospital of Sichuan University. The skin samples were collected under the guidance of the West China Biobank. The biobanking procedures were certified by the China Human Genetic Resources Management Office (2016, No. 406) as a part of West China Biobanks.
Animals
BALB/c, C57BL/6 mice and Sprague-Dawley rats were used in most animal studies. Male and female mice aged 8–10 weeks were used for behavioral tests unless stated otherwise. Animals were housed in a conventional facility at 23 ± 2 °C on a 12 h light-dark cycle with food and water ad libitum.
Human skin preservation
The normal abdominal skin tissues and scar tissues from human donors were immediately immersed in optimum cutting temperature medium (O.C.T., SAKURA, USA) and 15 μm frozen skin sections were prepared and stored at −20 °C for further analysis.
Hvcn1 Knockout mice generation
Adult, age-matched male Hvcn1 knockout (Hvcn1−/−) and WT control littermates were used for all related tests. Hvcn1−/− mice were generated using CRISPR-Cas9 technique on a C57BL/6 N background from Cyagen Inc (China). CRISPR-Cas9 genome editing system with two single-guide RNAs (sgRNA1 (matching forward strand of gene): TTGAAACACTTTACGGTGGTTGG; sgRNA2 (matching reverse strand of gene): CAAGCACGAGGAGGGCGTCCAGG) flanking Exon 4 and 5 was used to generate a 4254-bp deletion mutation.
Mouse DRG neuron culture
Mice were euthanized. The DRGs were collected in a 35-mm tissue culture dish and digested in 2.5 mg/mL collagenase (GibcoTM) for 30 min at 37 °C, followed by 1% trypsin (GibcoTM) for another 30 min. Digested ganglia were gently washed with neurobasal medium and mechanically dissociated by passage through pipet. Neurons were seeded on laminin-coated (Sigma-Aldrich) coverslip in 35-mm tissue culture dish (Corning) and maintained at 37 °C in an atmosphere of 5% CO2 in neurobasal medium (GibcoTM) containing 2% B27 (GibcoTM) supplement, 1% glutamine (Gibco), 0.5% penicillin and streptomycin.
Isolation of mouse neutrophil from bone marrow
Mice were anesthetized by injection of pentobarbital (i.p.) and sacrificed. Femur and tibia from both hind legs were removed and cleared off all soft tissues avoiding cutting the bone. The distal tip of each extremity was cut off and were flushed with HBSS medium (without calcium, magnesium, and phenol red) (Gibco) on ice using a 29 G needle. Then, the bone marrow was centrifuged at 300× g for 6 min. The supernatant was discarded and the pellet was resuspended in 3 mL HBSS medium. The cells were overlaid on a three-layer Percoll (GE Healthcare) density gradient of 72%, 64%, and 52%, respectively, diluted in HBSS (100% Percoll = nine parts Percoll and one part 10× HBSS) and centrifuged at 1200× g for 30 min without brake at room temperature. Neutrophils were harvested from 64%/72% interface after removing the cell layers from the upper phase carefully. After washed with HBSS, neutrophils were kept in RPMI 1640 (Gibco) with 10% FBS used within 6 h.
Homology modeling for the activated- and resting-state structural models of hHv1
The crystal structures of Ci-VSP (PDB code: 4G7V)25 and murine-Hv1 (mHv1) (PDB code:3WKV)26 were used as templates for building the activated- and resting-state models of hHv1, respectively. The hHv1, mHv1 and Ci-VSP sequences were aligned using Clustal Omega server68 and the aligned sequences were subsequently analyzed by ESPript 3.0 to visualize conserved regions69 (Supplementary information, Fig. S4a). The alignment result is identical to the result reported by Yasushi Okamura et al.70 Structural models of hHv1 were then generated by using the MODELER software.71 Models with the lowest root mean square deviation to the templates were selected for further analysis.
Virtual screening
We used Glide software (Schrödinger, LLC) to screen the Specs database (www.specs.net). The pan-assay interference compounds (PAINS) were removed before virtual screening. The three-dimensional (3D) conformations of the remaining about 220,000 compounds were generated by the ligPrep module of Maestro (Schrödinger, LLC) with the OPLS_2005 force field. Possible ionization states of each compound were generated in the pH range of 7.0 ± 2.0 using Ionizer. Possible tautomer forms were also generated for each ligand. Compounds were screened using the high-throughput virtual screening (HTVS) module followed by the standard precision (SP) docking module in Glide.72 The Glide G-score was used to rank the results list. Finally, 12 hits were manually selected for electrophysiological assay.
MD simulation
To obtain more accurate starting conformation for MD simulation, Inducted Fit docking protocol in Maestro (Schrödinger, LLC) was used to optimized the interactions of YHV98-1 with the hHv1 channel. The binding model of YHV98-1 to hHv1 provided by virtual screening was used as the initial input for the Induced Fit docking. The Extended Sampling Protocol was used, which could generate up to 20 poses per ligand by automated docking. In each case, the centroid of the grid box was fixed on YHV98-1 in ligand-channel complex and allowed residue refinement within 12 Å of the ligand poses. All obtained docking complexes were inspected visually to select one that displayed the closest compliance with the common binding mode.
The structural model of the complex of YHV98-1 and Hv1 obtained from Induced Fit docking was inserted in a 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) lipid bilayer to establish the YHV98-1-bound simulation system. The NMR structure of hHv1 without ligand was inserted in a POPC bilayer to build the apo Hv1 simulation system. The docking conformations between YHV98-1 and the activated- or resting-state of Hv1 channel with the lowest glide Gscores were put into POPC bilayer to build another two simulation systems. MD simulations were performed using GROMACS 5.1.473 with force field CHARMM36.74,75 The topology files of YHV98-1 were generated using the CGenFF tool76 and converted to GROMACS format with the cgenff_charmm2gmx script. The molecular mechanics Poisson-Boltzmann surface area (MMPBSA)77 approach was used to estimate the binding free energy of ligand-channel interaction.
Chemicals
YHV98-1 and its derivatives YHV98-2, YHV98-3 and YHV98-4 were purchased from Specs and their purity was > 95%. These compounds are used in the initial electrophysiological screening. Then, YHV98-4 was synthesized in the lab for the following studies in this paper. The synthesis routes and characterization of YHV98-4 are outlined in the following section. Morphine hydrochloride injection (10 mg/mL) was obtained from Northeast Pharmaceutical Group, Shenyang Pharmaceutical Co., Ltd., SFDA Approval No. H21022436 (Liaoning, China).
For electrophysiology, stock solutions of YHV98-1 and its derivatives (20 mM) were prepared in dimethyl sulfoxide (DMSO) and diluted in the extracellular solution before use. For animal studies, YHV98-4 was dissolved in 10% DMSO + 10% tween-80 + 80% (20% SBE-β-CD). Sodium stibogluconate (Sigma-Aldrich) was dissolved in saline. Wortmannin (Sigma-Aldrich) was prepared in DMSO (5 mM) and diluted in saline before use. Phenyl-N-t-butyl nitrone was diluted in saline before use. Meanwhile, the solvents were used as vehicle controls.
Synthesis of YHV98-4 (N-(4-chlorophenyl)-2-(3-oxo-3,4-dihydro-2H-benzo[b][1,4]thiazin-2-yl)acetamide)
Scheme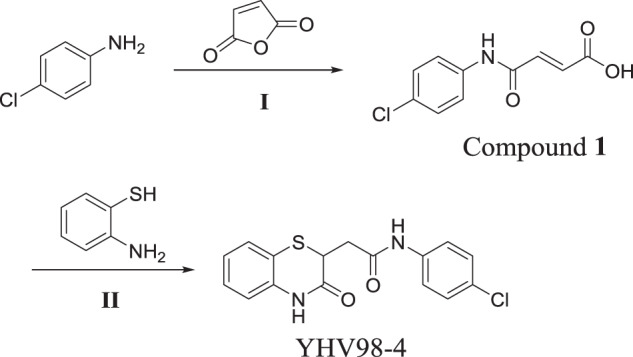
Furan-2,5-dione (4.61 g, 47.01 mmol) in tetrahydrofuran (50 mL) was added to a solution of 4-chloroaniline (5.0 g, 39.19 mmol) in tetrahydrofuran (50 mL) at room temperature. The mixture was stirred at room temperature overnight. TLC showed the reaction was completed. The thick precipitate was filtered off, washed with tetrahydrofuran and dried to obtain the desired product (E)-4-((4-chlorophenyl)amino)-4-oxobut-2-enoic acid (Compound 1) (8.22 g, yield 93%) as a light yellow solid1.H NMR (400 MHz, DMSO-d6): δ 13.00 (brs, 1H), 10.46 (s, 1H), 7.65 (d, J = 8.8 Hz, 2H), 7.38 (d, J = 8.8 Hz, 2H), 6.46 (d, J = 12.0 Hz, 1H), 6.31 (d, J = 12.0 Hz, 1H).
2-aminobenzenethiol (1.83 g, 14.62 mmol) was added to a solution of Compound 1 (3.0 g, 13.30 mmol) in DMF (30 mL) at room temperature and the mixture was heated to 150 °C for 3 h. TLC showed the reaction was completed. The mixture was cooled to room temperature and poured into water, the resulting solid was filtrated to obtain the crude product. The crude product was added tert-Butyl methyl ether/ tetrahydrofuran (4:1), stirred for 30 minutes at room temperature, and filtrated to obtain the desired product 1-(1-naphthalen)-biguanide (YHV98-4) (2.25 g, yield 51%) as a yellow solid, of which the purity was determined as 99.2% by HPLC. LC-MS: m/z = 333.1[M + H]+1.H NMR (400 MHz, DMSO-d6) δ10.72 (s, 1H), 10.21 (s, 1H), 7.66-7.58 (m, 2H), 7.37-7.33 (m, 3H), 7.24-7.20 (m, 1H), 7.04-6.99 (m, 2H), 3.95 (dd, J = 8.8, 6.0 Hz, 1H), 2.99 (dd, J = 15.6, 6.0 Hz, 1H), 2.60 (dd, J = 15.6, 8.8 Hz, 1H).
Electrophysiological measurements on recombinant ion channels
Electrophysiological recordings of WT Hv1 and its mutants were performed in transiently transfected HEK-293T cells. The cDNAs of the channels were transfected into HEK-293T cells. The cells were then cultured in Dulbecco’s modified Eagle’s medium supplemented with 10% bovine calf serum. Electrophysiological recordings were carried out 4–16 h after reseeding, which corresponds to 18–30 h after transfection. The transfected cells were identified by the fluorescent signal from the green fluorescent protein. Currents were recorded in the whole-cell patch-clamp configuration with EPC10 (HEKA) at 23–25 °C. The pipettes for whole-cell recordings were pulled from borosilicate glass capillaries (World Precision Instruments) and had a resistance of 3–7 MΩ. 60%–80% of the voltage error due to the series resistance was compensated by a circuit in the amplifier. The current signals were filtered at 2 kHz and digitized at 10 kHz. The external solution contained (in mM) 75 N-methyl-D-glucamine (NMDG), 1 CaCl2, 1 MgCl2, and 180 HEPES, pH 7.2. The pipette solution contained (in mM) 65 NMDG, 3 MgCl2, 1 EGTA and 110 MES, pH 6.0. Current–voltage relationships were evoked from a holding potential of −60 mV to test pulses from −80 to +80 mV for 2 s in 10-mV intervals. Data were analyzed with Clampfit 10.4 (Molecular Devices).
Electrophysiology tests of hKCNQ2 were performed with transfected CHO-K1 cells. Electrophysiological tests of hERG, BK and Nav1.7 were performed with stable cell lines. The CHO-hERG stable cell line was generated in-house and was based on a standard CHO-K1 cell line. The CHO-human BK and HEK-293T-Nav1.7 stable cell lines were generated by Ion Channel Explore. Electrophysiology tests of hTRPV1 and hTRPA1 were performed with transfected HEK-293T cells. Electrophysiology tests of VRAC was performed with HEK-293T cell. Whole-cell recordings of these ion channels were performed with patch-clamp amplifiers (EPC10, HEKA or Axon 700B, Molecular Devices) at 23–25 °C. The current signals were filtered at 2 kHz and digitized at 10 kHz. The pipettes for whole-cell recordings were pulled from borosilicate glass capillaries (World Precision Instruments) and had a resistance of 3–7 MΩ. For recordings of KCNQ2, pipette solution contained (in mM) 150 KCl, 3 MgCl2, 5 EGTA, 10 HEPES (pH 7.3 with KOH). Extracellular solution contained (in mM) 145 NaCl, 5 KCl, 1 CaCl2, 3 MgCl2, 10 HEPES (pH 7.4 with NaOH). For recordings of hERG channel, the standard pipette solution contained (in mM) 140 KCl, 2 MgCl2, 10 EGTA, 1 CaCl2, and 10 HEPES (pH 7.3, adjusted with KOH), and the external solution contained (in mM) 150 NaCl, 5 KCl, 0.5 CaCl2, 1.2 MgCl2, and 10 HEPES (pH 7.3, adjusted with NaOH). For recordings of BK channel, the standard pipette solution contained (in mM) 140 KCl, 1 MgCl2·6H2O, 10 HEPES, 10 EGTA, 9.75 CaCl2·2H2O (pH 7.2, adjusted with KOH), and the external solution contained (in mM) 140 NaCl, 3.5 KCl, 1 MgCl2·6H2O, 2 CaCl2·2H2O, 1.25 NaH2PO4, 10 HEPES, 10 D-Glucose (pH 7.4, adjusted with NaOH). For recordings of Nav1.7 channel, the internal solution contained (in mM) 50 CsCl, 10 NaCl, 10 HEPES, 60 CsF, 20 EGTA (pH 7.2, adjusted with CsOH), and the external solution contained (in mM) 140 NaCl, 3.5 KCl, 1 MgCl2·6H2O, 2 CaCl2·2H2O, 10 D-Glucose, 10 HEPES, 1.25 NaH2PO4·2H2O (pH 7.4, adjusted with NaOH). For recordings of the TRPV1 and TRPA1 currents, the internal solution contained (in mM) 140 CsCl, 2 MgCl2·6H2O, 5 CaCl2·2H2O, 10 HEPES, 1 Mg-ATP, 10 EGTA (pH 7.2, adjusted with CsOH), and the external solution contained (in mM) 140 NaCl, 5 KCl, 2 MgCl2·6H2O, 2 CaCl2·2H2O, 10 HEPES, 5 D-Glucose (pH 7.4, adjusted with NaOH). For recordings of the VRAC current, the internal solution contained (in mM) 110 CsCl, 5.17 CaCl2, 10 EGTA and 10 Tris (pH 7.2, adjusted with CsOH), and the external solution contained (in mM) 140 NaCl,10 HEPES, 2 CaCl2, 1 MgCl2, and 4 CsCl (pH 7.4, adjusted with NaOH). The hypotonic solutions were made by adding water (1/3 v:2/3 v) to the isotonic solutions. For recordings of KCNQ2, currents were evoked by a 2 s depolarization step from −90 mV to +60 mV in 10 mV increments at a holding potential of −80 mV, followed by a hyperpolarization step to −120 mV to record the tail current. For recordings of hERG, the outward current of hERG channels was elicited by a 2.5 s depolarization step to +30 mV from a holding potential of −80 mV, followed by a 4 s repolarization step to −50 mV to measure the tail current. For recordings of BK, currents were evoked by a 2 s depolarization step to +60 mV from a holding potential of −80 mV. For recordings of Nav1.7, currents were recorded at a holding potential of −120 mV for 30 ms and depolarized to 0 mV for 20 ms, then hyperpolarized to −50 mV. For recordings of TRPA1 and TRPV1, currents were recorded using a ramp protocol from −100 mV to +100 mV over a period of 300 ms at a holding potential of 0 mV. For recordings of VRAC, current–voltage relationships obtained from the symmetric voltage ramps applied every 5 s (±100 mV, 300 ms duration).
Acutely dissociated DRG neuron electrophysiology
The pipettes for whole-cell recordings were pulled from borosilicate glass capillaries (World Precision Instruments). The resulting pipettes had resistances ranging from 5 to 10 MΩ as measured in the bath. The bath solution (extracellular solution) contained (in mM): 140 NaCl, 3 KCl, 1.3 MgCl2, 10 HEPES, 2.4 CaCl2, 10 D-glucose (pH 7.3, adjusted with NaOH). The vehicle solution (extracellular solution for dissolve DCPIB, ZnCl2 or YHV98-4) contained (in mM): 100 CsMeSO3, 10 D-glucose, 100 HEPES, 25 sucrose (pH 7.3, adjusted with CsOH). The pipette solution (intracellular solution) contained (in mM): 130 CsMeSO3, 1 EGTA, and either 50 MES (pH 5.5), 50 Bis-Tris (pH 6.5) or 50 HEPES (pH 7.3), pH adjusted with CsOH. To record the voltage-gated currents, the voltage steps from −20 mV to +80 mV were applied for 1.5 s. The tail current was determined by YHV98-4 sensitive current. The tail current was recorded at various potentials from −80 mV to +60 mV after activation of the outward current by a voltage step to +60 mV for 1.5 s, and averaged in the 5–15 ms interval after the start of the test pulse were used for analyzing. Stimulation and data acquisition were controlled by the PATCHMASTER software package (HEKA) or Digidata 1440 A (Molecular Devices), and data were analyzed with the pCLAMP program (Version 10).
Measurement of AP and RMP
The DRG neurons for recording action potential (AP) and resting membrane potential (RMP) were harvested from CFA mice. The extracellular solution contained (in mM): 140 NaCl, 3 KCl, 1.3 MgCl2, 10 HEPES, 2.4 CaCl2, 10 D-glucose (pH 7.3, adjusted with NaOH). The pipette solution (intracellular solution) contained (in mM): 135 KCl, 10 NaCl, 5 EGTA, 50 Bis-Tris, and 4 Mg-ATP, (pH 6.5, adjusted with KOH). Rheobase was determined by measuring the minimum amplitude of a 500-ms square wave depolarizing current injection (from 0 pA to 150 pA with an increment of 5 pA) that was required to generate APs. The response to supra-threshold stimuli was assessed by stimulating neurons with depolarizing current injections equal to 2.5× rheobase. DRG neuron excitability was assessed by counting total number of APs discharged in response to a 500-ms depolarizing current injections equal to 2.5× rheobase. The RMP was determined under current clamp at zero current for at least 30 s.
Pharmacokinetics study
Pharmacokinetics of YHV98-1 and YHV98-4 was analyzed in male C57BL/6 mice (n = 5). Plasma and brain concentrations were determined using LC-MS/MS after a single i.p. injection dose (10 mg/kg) of a compound as a clear solution (YHV98-1 in 5% DMSO + 95% water with 40% HP-β-CD; YHV98-4 in 10% DMSO + 10% tween80 + 80% (20% SBE-β-CD) at a concentration of 1.0 mg/mL. Blood samples were collected into heparinized test tube at each time point (0.083 h, 0.25 h, 0.5 h, 1.0 h, 2.0 h, 4.0 h, and 6.0 h) and centrifuged at 8000× g for 10 min to generate plasma samples. Brains were collected after myocardial perfusion with normal saline and homogenated with normal saline (1:2, W/V) to generate brain samples. LC-MS/MS methods to quantify YHV98-1 or YHV98-4 in plasma samples and brain were developed. Plasma and brain were extracted with acetonitrile (ACN), mixed and centrifuged. The supernatants were transferred to autosampler vials with glass inserts for analysis. The samples were analyzed with an Agilent 1290 Infinity II HPLC system coupled to a 6470 LC/TQ mass spectrometer (Agilent, USA), which was equipped with an Applied Biosystems electrospray ionization (ESI) source and operated with Analysis of Mass Hunter Workstation Data Acquisition (Agilent, USA). Chromatographic separation was fitted with an Agilent Poroshell 120 EC-C18 2.1 × 50 mm, 1.9 μm column. The mobile phase consisted of A: 0.1% formic acid in water, and B: 0.1% formic acid in ACN. Standard curves were prepared by spiking compounds into control plasma and brain were used to determine drug concentrations. Area under the curve (AUC), Tmax, Cmax, T1/2, CL (clearance) and Mean Retention Time (MRT) were calculated by non-compartmental analysis using WinNonlin (Phoenix Version 6.1, Pharsight Corporation, Mountain View, CA) with mean concentration at each time point.
AAV virus generation and DRG microinjection
The neuron-specific Hv1 knockdown was using microRNA-based silencing technique, and the BLOCK-iT Pol II miR RNAi Expression Vector Kits (Invitrogen) were used. Six pre-miRNA sequences for mouse Hvcn1 gene knockdown (Hvcn1, NCBI gene ID: 74096) (AAV-Hv1 KD) and negative control sequence (AAV-Control) were designed (Invitrogen’s RNAi Designer), synthesized, and cloned into pAAV-CMV_bGI·mCherry·miRNAi vector (Taitool Bioscience). The KD efficiency was then evaluated by co-transfecting EGFP-tagged Hv1 with the Hv1 miRNA vectors in human embryonic kidney (HEK-293) cells, and the KD efficiency was indicated by the reduction of the fluorescence signal expressed by the EGFP-Hv1 vector. The most effective sequence was chosen as follows: AAV-Hv1 KD, 5’-GAGGATGAGCAGGTTCTTGAA-3’, and AAV-Control, 5’-AAATGTACTGCGCGTGGAGAC-3’. The selected oligos were then cloned into the linearized pAAV·hSyn·mCherry·miRNAi vector (Taitool Bioscience) using T4 DNA ligase. The titer of AAV2/9·hSyn·mCherry·Hv1-miRNAi and AAV2/9·hSyn·mCherry·NC-miRNAi were determined by qPCR.
DRG microinjection was adapted from recent studies.66,78 Mice were anesthetized by i.p. injection of pentobarbital. The skin on the lower lumbar back region was shaved by an electric clipper for aseptic surgery. A 1–2 cm midline incision was made and the muscles were separated by blunt dissection to expose the lateral aspect of L4 and L5 lumbar vertebra, and their transverse process. Partial lamina was removed to expose the DRGs. 1 μL virus solution (titer ≥ 0.7 × 1013) for each DRG was injected into one site in the L4 and L5 DRGs with a glass micropipette connected to a Hamilton syringe at a rate of 0.5 µL/min (The L3, L4 and L5 DRGs were injected for opioid-induced tolerance and hyperalgesia experiment or pHi measurement). The pipette was left in place for 5 min after injection. After pipette was removed, the muscle and skin were then closed using 4.0 monofilament nylon sutures. No signs of paresis or other abnormalities were shown in the injected mice.
Chronic inflammatory pain model
A volume of 20 μL CFA (Sigma-Aldrich) was injected subcutaneously into the left hind paw of the mouse to induce chronic inflammatory pain. After injection the syringe was maintained for at least 30 s to avoid overflow. Experiments were performed at least 24 h after CFA injection.
Formalin-induced inflammatory model
A volume of 20 μL formalin (2.5% formalin, diluted in saline) was injected subcutaneously into the left hind paw of mouse to induce acute inflammatory. After injection the syringe was maintained for at least 30 s to avoid overflow.
SNI model
SNI surgery was performed based on published protocols from previous study.79 Briefly, after disinfection with povidone iodide and 75% ethanol, a mice skin incision was made at the mid-thigh quantity to expose the sciatic nerve and its three branches by separating the muscle layers. The tibial and common peroneal nerves were tightly ligated with 5.0 silk threads, and a 1–2 mm section was removed between the proximal and distal parts of the nerves. Experiments were performed 7–21 days after SNI.
PMA-induced nociception
The experiments were carried out as previously described80 with some modifications. Briefly, a volume of 20 μL of PMA solution (50 pmol/paw) diluted in saline was injected intraplantarly under the plantar surface of the hind paw. And a volume of 20 μL 0.1 % DMSO as vehicle. The von Frey test was applied from 15 to 105 min following PMA injection.
von Frey test
The mice were placed in an elevated transparent cage (20 × 20 × 14 cm) with a wire mesh floor (0.5 × 0.5 cm). The mechanical paw withdrawal threshold was assessed using von Frey filaments with an ascending order (mice: 0.008, 0.02, 0.04, 0.07, 0.16, 0.4, 0.6, 1.0, 1.4, and 2.0 g, starting with 0.008 g and ending with 2.0 g, each filament was applied for 5 s with a 30 s interval). The tip of filament was perpendicularly targeted to the hind paw of the mice. The sufficient stimulation was held for 5 s. Rapid paw withdrawal or flinching was considered as a positive response and the bending force for which at least 60% of the application elicited positive response was recorded as the mechanical paw withdrawal threshold. Data are also expressed as the MPE (%) for von Frey, Hargreaves and paw flinches test, calculated as MPE (%) = 100 × [(post drug response − baseline response)/(pretreatment response − baseline response)]. The MPE (%) data from each animal were also converted to AUC.
Hargreaves test
The hind paw sensitivity to thermal noxious stimulus was assessed using a radiant heat source (model 37370; Ugo Basile, Varese, Italy), stimulus intensity was set to produce an approximate latency of 10 s at baseline, and a cut-off value was set at 20 s to avoid unexpected damage. Mice were allowed to acclimate in Plexiglas chambers with a glass floor for 3 days, and the duration to paw withdrawal was measured per mouse within a 5 min inter-stimulation period. Three trials were averaged to yield the withdrawal latency.
Paw flinches test
After 3 days acclimation, the CFA mice were placed in an elevated transparent cage (20 × 20 × 12 cm) with a wire mesh floor (0.5 × 0.5 cm). A 10 min duration was videoed by an action camera (SONY, HDR-AS50) for each mouse, and the number of left hind paw flinching was calculated by another experimenter unaware of the mouse treatments.
Rotarod test
Accelerating rotarods (YLS-31A, Jinan Yiyan Technology Development Co., Ltd., China) were used to measure the motor coordination of mice. The rotarod measures the ability of the mouse to maintain balance on a motor-driven, rotating rod. During 3 days of habituation, the mice were treated for two separated rounds of training with at least 10 min interval at 20 rpm for 5 min. For data acquisition, rotation was started at 4 rpm and accelerated over a 5 min period to 40 rpm. Animal falls were detected by a pressure sensitive lever and the time taken for the mice to fall from the rod was recorded.
Grip strength test
A Grip Strength Meter (47200, Ugo Basile) was used to measure forelimb grip strength according to previously described methods.79,81 Mice were accustomed to the placement of their forelimbs on the apparatus trapeze for 3 days before experiments were conducted. For data acquisition, a mouse was suspended gently by the tail and then was lowered to the extend so that it could grasp the force transducer (trapeze) of the grip strength meter. As the mouse grasped the bar, the peak pull force in grams was recorded. In each test, those trials in which only one forepaw, or the hindlimbs were used or during which the mouse turned or left the bar without resistance were excluded. Five consecutive measurements were performed at one-min intervals for each time point and the averaged result was analyzed.
Opioid-induced tolerance and hyperalgesia in mice
Opioid-induced tolerance and hyperalgesia model was performed as described previously.82 The WT mice were given single injection of morphine daily (10 mg/kg, i.p.) + YHV98-4 (10 mg/kg, i.p.) or morphine (10 mg/kg, i.p.) + YHV98-4 Vehicle (i.p.) for 6 consecutive days. The AAV-Control and AAV-Hv1 KD mice were given single daily injection of morphine (10 mg/kg, i.p.) for 6 consecutive days. The von Frey test was performed at 30 min before (baseline) and 15 min after (post drug) the morphine administration. The MPE (%) value was calculated to compare the antinociceptive effects and tolerance development. MPE (%) = 100 × [(post drug threshold − baseline threshold)/(cutoff threshold − baseline threshold)] (the cutoff threshold of 2 g was set to avoid injury).
Measurements of pHi using FITC
Cultured neurons grown on coverslips were loaded with 0.5 μM FITC for 1 h at 37 °C. The coverslips were perfused with HCO3−-free HEPES-buffered solution contained (in mM) 140 NaCl, 5 KCl, 1 MgCl2, 2 CaCl2, 5 D-glucose, 20 HEPES (pH 7.3). Intracellular pH measurements with FITC were made by determining the pH-dependent ratio of emission intensity (detected at 510–550 nm) when the dye was excited at 488 nm (pH-sensitive) versus the emission intensity when excited at 402 nm (pH-insensitive). Cells were acid loaded with 20 mM NH4+-containing solution (5 min) or 500 ng/mL PMA (5 min). For Na+-free solution, NaCl was replaced with NMDG-Cl. For Na+-free, high K+ solution, NaCl was replaced by KCl. For establishing standard pH curves between pH 6.0–8.0, 10 μM nigericin was added to solutions contained (in mM) 150 KCl, 1 MgCl2, 0.2 EGTA, 5 D-glucose, 20 HEPES.
Measurements of ROS
For in vitro study, the DRG neurons were incubated with 10 μM DCFH-DA (Sigma) probe, a fluorogenic dye used to measure ROS within cells, for 30 min at 37 °C and subsequently washed three times with HBSS. Then the neurons were respectively exposed to PMA (500 ng/mL) for 1 h, KCl (50 mM) for 5 min or AITC (100 μM) + capsaicin (1 μM) for 5 min. For stimulation with LPS, the neurons were incubated with LPS (3 mg/mL) or LPS + YHV98-4 (20 μM) for 24 h before DCFH-DA was applied. Celigo® Image Cytometer (Nexcelom, USA) or two-photon confocal scanning microscope (Nikon A1R MP, Japan) was used to take the florescent images of neurons at room temperature. All experiments were from three independent preparations.
For in vivo study, detection of superoxide production was performed by DHE (Sigma) staining,83,84 or DCFH-DA staining85,86 for DRGs treated with AAV virus injection. The CFA mice or morphine induced tolerance mice were euthanized. L4 and L5 DRG were dissociated rapidly and snap frozen with liquid nitrogen, cryostat sectioned (10 μm), incubated with 10 μM DHE or 10 μM DCFH-DA for 30 min at 37 °C in dark humid chamber and then washed three times with phosphatase buffer saline (PBS). The nucleus was visualized with DAPI. Ethidium fluorescence (excitation/emission at 550/610 nm) was examined by fluorescence microscopy (Olympus, BX53, Japan) or two-photon confocal scanning microscope (Nikon A1R MP, Japan), and evaluated by using ImageJ software.
Measurement of ROS in neutrophils
Neutrophils were seeded on coverslip in 35-mm tissue culture dish and assayed in the HBSS medium. For detecting the production of superoxide in neutrophils, neutrophils were incubated with 10 μM DHE at 37 °C for 30 min in dark and then washed with HBSS. Vehicle (0.1% DMOS in HBSS), 20 μM YHV98-4, and 100 μM ZnCl2 were added in the dish 20 min before recording. PMA (500 ng/mL) was added at t = 0 min. Fluorescent images were taken at t = 0 min and 30 min using a Two-photon confocal scanning microscope and analyzed using ImageJ software.
DRG neuron dissociation and fixation
After experiment animals were euthanized, the DRG were dissociated and fixed in 4% buffered paraformaldehyde (PFA) solution for 12 h and transferred to the 30% sucrose solution for another 24 h at 4 °C. After fixation, DRG were immediately immersed in optimum cutting temperature medium (O.C.T., SAKURA, USA). 15 μm frozen sections were prepared and stored at −20 °C for further analysis.
RNAscope in situ hybridization
Sequences of target probes, preamplifier, amplifier and label probes are proprietary and commercially available (Advanced Cell Diagnostics). In situ hybridization was performed on frozen DRG and frozen skin sections (15 μm) using RNAscope Multiplex Fluorescent Reagent Kit v2 (ACDbio, #323100) and RNAscope 4-Plex Ancillary Kit for Multiplex Fluorescent Kit v2 (ACDbio, #323120). The hybridization assay as described by vendor’s protocol. In situ probes are from ACDbio include: Trpa1 (#400211), Trpv1 (#313331), Trpm8 (#420451), Rbfox3 (#313311), Th (#317621), Ntrk2 (#423611), P2rx3 (#521611), hvcn1 (#521351), Hs-hvcn1 (#1037061). The specificity of the florescent signals was validated by RNAscope 3-plex Positive Control Probe (#320881) and RNAscope 3-plex Negative Control Probe (#320871). Fluorescent images were taken using a Two-photon confocal scanning microscope (Nikon A1R MP, Japan) and were analyzed using ImageJ software.
For DRG neuron RNAscope staining, the DRG neurons were respectively treated with TNF-α (10 ng/mL) for 4 h, IL-1β (50 ng/mL) for 6 h, PMA (500 ng/mL) for 0.5 h or high K+ (50 mM) for 5 min. For treat with DPI or GKT137831, the DPI (10 μM) or GKT137831 (10 μM) were applied 10 min before high K+ stimulation, then co-incubated with high K+ (50 mM) for 5 min.
IHC
The frozen skin, DRG sections and coverslip were treated with the same immunofluorescence protocol. All sections or coverslips were washed with PBS solution, and permeabilized with 0.5% Triton-X100 (Sigma-Aldrich, Germany) in PBS for 30 min. Preincubation was performed with 10% goat serum for 1 h at 37 °C. The following primary antibodies were used: rabbit anti-Hv1 (1:200; Abcam 117520), mouse anti-NeuN (1:500; Millipore MAB377), mouse anti-PGP9.5 (1:600; Abcam 8189), rabbit anti-SHP-1 (1:100; Abcam 131537), rabbit anti-phosphorylated SHP-1 (1:50; Abcam 51171), mouse AKT-phospho-S473 antibody (1:200; Proteintech 66444-1-Ig), mouse anti-Iba1(1:200; WAKO), rabbit anti NOX1 (1:200; Invitrogen PA5-79751) and rabbit anti NOX4 (1:200; Invitrogen PA5-86036). The following Alexa-conjugated secondary antibodies were used: goat anti-mouse 488 (1:1000, Abcam 150113), goat anti-rabbit 488 (1:1000, Abcam 150077), goat anti-rabbit 647 (1:500, Abcam 150079). After nucleus labeling with DAPI, the sections were mounted using anti-fade solution. For quantification of fluorescent intensity, sections from the two groups were stained and imaged with exactly the same protocol.
Measurement of hind paw edema
YHV98-4 (10 mg/kg, i.p.) was administrated twice a day for 2–3 days for paw edema and skin pathology study. The Plethysmometer (YLS-7C, Jinan Yiyan Technology Development Co., Ltd., China) was used to measure paw volume by calculating fluid volume displacement in a measurement chamber. Each hind paw was measured three times and an average was calculated.
Assessment of skin pathology
Mice were euthanized with carbon dioxide. Then, the hind paws were removed immediately, fixed in 10% buffered formalin for at least for 48 h and prepared for routine paraffin histology. The paraffin-embedded 5-μm-thick sections of the hind paw pads were stained by hematoxylin and eosin. Epidermal and Dermal thickness was measured on photomicrographs and three measurements were averaged for each sample. Skin specimens were assessed and scored to provide a semiquantitative measurement of dermal inflammation (0, none; 1, little; 2, mild; 3, moderate; 4, severe).87
Multiplex immunofluorescence staining (Multiplex IF)
Multiplex Immunofluorescence Staining was carried out as previously described,88 with some modifications. 4 mm sections of formalin fixed paraffin-embedded (FFPE) foot skin tissues were mounted on Superfrost Plus Microscope Slides (Fisher Scientific) and prepared for Multiplex IF. The slides were deparaffinized in xylene and rehydrated in graded ethanol. Antigen retrieval was performed in citrate buffer (pH 6.0) or EDTA Antigen Retrieval Solution (pH 9.0). Staining was performed manually using the primary antibodies against the following: T cell lymphocytes marker CD4 (1:10, Invitrogen MA5-12259), Macrophages marker F4/80 (1:200, Cell signaling technology 30325). Antibody detection was performed with Opal Polymer HRP Ms + Rb IHC detection reagent (PelkinElmer, Boston, MA), and the detection for each marker was completed before application of the next antibody. The Opal Polymer HRP Ms + Rb detection reagent was used for the primary antibody detection and Opal 7-Color Manual IHC kit, with 2 reactive fluorophores Opal 520, 690 and DAPI nuclear counterstain, these reagents were added according to the manufacturer’s instructions. Uniplex IF and Negative controls were stained with the same protocols.
CD4+ and F4/80+ cells in epidermis and dermis skin were measured by quantifying positively stained cells in two random ROI (0.2 mm2 each) in the foot skin. Six or seven specimens from each group were quantified. The average number of cells positive for each marker in the two ROI was shown in density per mm2.
Multiplex cytokine & chemokine analysis and ELISA
For multiplex cytokine & chemokines analysis, YHV98-4 (10 mg/kg, i.p.) was given twice a day for 2 days after CFA injection in WT mouse, AAV-Hv1 KD or AAV-Control mice. 15 days after DRG injection of AAV virus, the CFA was injected in AAV-Hv1 KD or AAV-Control mice. Mice were sacrificed 48 h after CFA injection, and the protein of foot skin was extracted using ProcartaPlex Cell Lysis Buffer (ThermoFisher Scientific). Protein concentrations were determined using the BCA Protein Assay Kit (Beyotime). The skin multiplex cytokine/chemokine measurement was done using Mouse Cytokine & Chemokine 26-plex (ThermoFisher Scientific).
For Elisa experiment, a volume of 5 μL Wortmanin (0.06 μg/μL) or vehicle (1% DMSO in saline) were given intrathecally 24 h after CFA injection and 4 h before sacrifice. Mice were sacrificed 48 h after CFA injection. DRG were removed and collected in cold NP40 (Beyotime) on ice containing 1 mM PMSF (Beyotime), then homogenized and separated by centrifugation (at 4 °C, 12,000× g for 10 min) to obtain protein. The level of CXCL1 was assessed by a mouse CXCL1 ELISA kit (Proteintech) according with the manufacturer’s instructions.
Statistical analysis
Statistical analyses were carried out using Origin 9.0 software (Origin Lab Corporation), GraphPad Prism 8.0.1 (GraphPad Software). Data were analyzed as described in the figure legends. Differences in measured variables were assessed by using a one-tailed or two-tailed Student’s t-test or nonparametric test for single comparisons, and one-way analysis of variance (ANOVA) or nonparametric test followed by indicated post hoc corrections for multiple comparison testing. The normality of the data distribution was determined using the Shapiro–Wilk test, Kolmogorov–Smirnov test and D’Agostino & Pearson test before appropriate statistical methods were chosen. No statistical methods were used to predetermine sample sizes.
Supplementary information
Acknowledgements
We thank Prof. Jun-Li Cao (Xuzhou Medical University, China), Prof. Xia Zhang (University of Ottawa, Canada) for helpful discussion on the manuscript, J. Zhang, Z. Yang, and L. Bai (Histology and Imaging platform, Core Facility of West China Hospital) for assistance with acquiring some ISH images; Li Li, Fei Chen and Chunjuan Bao (Institute of Clinical Pathology, West China Hospital) for processing histological staining. Yuandong Liu and Prof. Yang Tian (East China Normal University) for assistance in pH imaging. We thank the support of ECNU Multifunctional Platform for Innovation (001 and 011). This work was equally funded by the National Natural Science Foundation of China (32071003 to R.J.) and the Ministry of Science and Technology of China (2018ZX09711002 to Q.Z.), partially funded by the National Natural Science Foundation of China (81873808 to Y.Z, 31600832 to R.J., 31800699 to Q.Z.), the Fundamental Research Funds for the Central Universities (to H.Y. and 2018SCUH0086 to R.J.), the “XingFuZhiHua” funding of ECNU (44300-19311-542500/006 to H.Y.), the Department of Science and Technology of Sichuan Province (2020ZYD006 to R.J.) and the 1-3-5 Project for Disciplines of Excellence of West China Hospital of Sichuan University (ZYJC21034 to R.J.).
Author contributions
Q.Z. performed drug design and computations. Y.R. performed electrophysiology in DRG, DRG microinjection, behavioral tests, cytokine and chemokine measurement, ISH, IHC, and ROS imaging. Y.M performed mouse genetics on Hvcn1−/− mice, pharmacokinetics study, behavioral tests, skin histopathological study, and ROS imaging. P.G. performed electrophysiology in HEK293 cells. P.L. performed ISH, IHC, pH and ROS imaging. Y.Luo performed ISH, IHC, ROS imaging, ELISA and behavioral tests. J.M. performed IHC and pH imaging. Z.C. and J.F. performed electrophysiology in HEK293 cells. Y.Zhang performed IHC imaging. Y.Li performed ROS imaging. L.Y. and D.L. performed some behavioral tests. J.S. performed drug design. W.H. and X.X. acquired and prepared human skin tissues. Y.G. performed behavioral tests, and ROS imaging. L.M. synthetized YHV98-4. Y.Zuo and J.L. oversaw some behavioral tests. H.Y. and R.J. initiated, supervised the project, analyzed the experiments, and wrote the manuscript with input from all coauthors.
Competing interests
H.Y., R.J., and Q.Z. are inventors on a patent application (202011283354.3) submitted by East China Normal University, West China Hospital of Sichuan University and Shaoxing ZeroIn Biomedicines Co. Ltd. that cover the potential usage of YHV98-1, YHV98-4 and their derivatives.
Footnotes
These authors contributed equally: Qiansen Zhang, Yimin Ren, Yiqing Mo, Peipei Guo, Ping Liao, Yuncheng Luo.
Contributor Information
Huaiyu Yang, Email: hyyang@bio.ecnu.edu.cn.
Ruotian Jiang, Email: ruotianjiang@scu.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41422-022-00616-y.
References
- 1.Grosser T, Woolf CJ, FitzGerald GA. Time for nonaddictive relief of pain. Science. 2017;355:1026–1027. doi: 10.1126/science.aan0088. [DOI] [PubMed] [Google Scholar]
- 2.Woolf CJ. Capturing novel non-opioid pain targets. Biol. Psychiatry. 2020;87:74–81. doi: 10.1016/j.biopsych.2019.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gregori D, et al. Association of pharmacological treatments with long-term pain control in patients with knee osteoarthritis a systematic review and meta-analysis. J. Am. Med. Assoc. 2018;320:2564–2579. doi: 10.1001/jama.2018.19319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Busse JW, et al. Opioids for chronic noncancer pain a systematic review and meta-analysis. J. Am. Med. Assoc. 2018;320:2448–2460. doi: 10.1001/jama.2018.18472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ji RR, Chamessian A, Zhang YQ. Pain regulation by non-neuronal cells and inflammation. Science. 2016;354:572–577. doi: 10.1126/science.aaf8924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Donnelly CR, et al. STING controls nociception via type I interferon signalling in sensory neurons. Nature. 2021;591:275–280. doi: 10.1038/s41586-020-03151-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Puig S, Gutstein HB. Opioids: keeping the good, eliminating the bad. Nat. Med. 2017;23:272–273. doi: 10.1038/nm.4277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Grace PM, et al. Nitroxidative signaling mechanisms in pathological pain. Trends Neurosci. 2016;39:862–879. doi: 10.1016/j.tins.2016.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wu LJ. Voltage-gated proton channel Hv1 in microglia. Neuroscientist. 2014;20:599–609. doi: 10.1177/1073858413519864. [DOI] [PubMed] [Google Scholar]
- 10.Wu LJ, et al. The voltage-gated proton channel Hv1 enhances brain damage from ischemic stroke. Nat. Neurosci. 2012;15:565–573. doi: 10.1038/nn.3059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Li XF, et al. Microglial Hv1 exacerbates secondary damage after spinal cord injury in mice. Biochem. Biophys. Res. Commun. 2020;525:208–215. doi: 10.1016/j.bbrc.2020.02.012. [DOI] [PubMed] [Google Scholar]
- 12.Li XF, et al. Deficiency of the microglial Hv1 proton channel attenuates neuronal pyroptosis and inhibits inflammatory reaction after spinal cord injury. J. Neuroinflammation. 2020;17:263. doi: 10.1186/s12974-020-01942-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Murugan M, et al. The voltage-gated proton channel Hv1 contributes to neuronal injury and motor deficits in a mouse model of spinal cord injury. Mol. Brain. 2020;13:143. doi: 10.1186/s13041-020-00682-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li Y, et al. The voltage-gated proton channel Hv1 plays a detrimental role in contusion spinal cord injury via extracellular acidosis-mediated neuroinflammation. Brain Behav. Immun. 2021;91:267–283. doi: 10.1016/j.bbi.2020.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.De Simoni A, Allen NJ, Attwell D. Charge compensation for NADPH oxidase activity in microglia in rat brain slices does not involve a proton current. Eur. J. Neurosci. 2008;28:1146–1156. doi: 10.1111/j.1460-9568.2008.06417.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Schilling T, Eder C. Ion channel expression in resting and activated microglia of hippocampal slices from juvenile mice. Brain Res. 2007;1186:21–28. doi: 10.1016/j.brainres.2007.10.027. [DOI] [PubMed] [Google Scholar]
- 17.Li CL, et al. Somatosensory neuron types identified by high-coverage single-cell RNA-sequencing and functional heterogeneity. Cell Res. 2016;26:83–102. doi: 10.1038/cr.2015.149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Usoskin D, et al. Unbiased classification of sensory neuron types by large-scale single-cell RNA sequencing. Nat. Neurosci. 2015;18:145–153. doi: 10.1038/nn.3881. [DOI] [PubMed] [Google Scholar]
- 19.Wang KK, et al. Single-cell transcriptomic analysis of somatosensory neurons uncovers temporal development of neuropathic pain. Cell Res. 2021;31:904–918. doi: 10.1038/s41422-021-00479-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Seredenina T, Demaurex N, Krause KH. Voltage-gated proton channels as novel drug targets: from NADPH oxidase regulation to sperm biology. Antioxid. Redox Signal. 2015;23:490–513. doi: 10.1089/ars.2013.5806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cherny VV, DeCoursey TE. pH-dependent inhibition of voltage-gated H(+) currents in rat alveolar epithelial cells by Zn(2+) and other divalent cations. J. Gen. Physiol. 1999;114:819–838. doi: 10.1085/jgp.114.6.819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Alabi AA, Bahamonde MI, Jung HJ, Kim JI, Swartz KJ. Portability of paddle motif function and pharmacology in voltage sensors. Nature. 2007;450:370–375. doi: 10.1038/nature06266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hong L, Pathak MM, Kim IH, Ta D, Tombola F. Voltage-sensing domain of voltage-gated proton channel Hv1 shares mechanism of block with pore domains. Neuron. 2013;77:274–287. doi: 10.1016/j.neuron.2012.11.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhao R, et al. Role of human Hv1 channels in sperm capacitation and white blood cell respiratory burst established by a designed peptide inhibitor. Proc. Natl. Acad. Sci. USA. 2018;115:E11847–E11856. doi: 10.1073/pnas.1816189115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Li Q, et al. Structural mechanism of voltage-dependent gating in an isolated voltage-sensing domain. Nat. Struct. Mol. Biol. 2014;21:244–252. doi: 10.1038/nsmb.2768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Takeshita K, et al. X-ray crystal structure of voltage-gated proton channel. Nat. Struct. Mol. Biol. 2014;21:352–357. doi: 10.1038/nsmb.2783. [DOI] [PubMed] [Google Scholar]
- 27.Bayrhuber M, et al. Nuclear magnetic resonance solution structure and functional behavior of the human proton channel. Biochemistry. 2019;58:4017–4027. doi: 10.1021/acs.biochem.9b00471. [DOI] [PubMed] [Google Scholar]
- 28.Le Guilloux V, Schmidtke P, Tuffery P. Fpocket: an open source platform for ligand pocket detection. BMC Bioinformatics. 2009;10:168. doi: 10.1186/1471-2105-10-168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hong L, Kim IH, Tombola F. Molecular determinants of Hv1 proton channel inhibition by guanidine derivatives. Proc. Natl. Acad. Sci. USA. 2014;111:9971–9976. doi: 10.1073/pnas.1324012111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Boonamnaj P, Sompornpisut P. Effect of ionization state on voltage-sensor structure in resting state of the Hv1 channel. J. Phys. Chem. B. 2019;123:2864–2873. doi: 10.1021/acs.jpcb.9b00634. [DOI] [PubMed] [Google Scholar]
- 31.Wood ML, et al. Water wires in atomistic models of the Hv1 proton channel. Biochim. Biophys. Acta. 2012;1818:286–293. doi: 10.1016/j.bbamem.2011.07.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ramsey IS, et al. An aqueous H+ permeation pathway in the voltage-gated proton channel Hv1. Nat. Struct. Mol. Biol. 2010;17:869–875. doi: 10.1038/nsmb.1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gianti E, Delemotte L, Klein ML, Carnevale V. On the role of water density fluctuations in the inhibition of a proton channel. Proc. Natl. Acad. Sci. USA. 2016;113:E8359–E8368. doi: 10.1073/pnas.1609964114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Geragotelis AD, et al. Voltage-dependent structural models of the human Hv1 proton channel from long-timescale molecular dynamics simulations. Proc. Natl. Acad. Sci. USA. 2020;117:13490–13498. doi: 10.1073/pnas.1920943117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zaretzki J, Matlock M, Swamidass SJ. XenoSite: accurately predicting CYP-mediated sites of metabolism with neural networks. J. Chem. Inf. Model. 2013;53:3373–3383. doi: 10.1021/ci400518g. [DOI] [PubMed] [Google Scholar]
- 36.El Chemaly A, et al. A voltage-activated proton current in human cardiac fibroblasts. Biochem. Biophys. Res. Commun. 2006;340:512–516. doi: 10.1016/j.bbrc.2005.12.038. [DOI] [PubMed] [Google Scholar]
- 37.Decher T, et al. DCPIB is a novel selective blocker of I-Cl,I-swell and prevents swelling-induced shortening of guinea-pig atrial action potential duration. Br. J. Pharmacol. 2001;134:1467–1479. doi: 10.1038/sj.bjp.0704413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Nilius B, Sehrer J, Droogmans G. Permeation properties and modulation of volume‐activated Cl−‐currents in human endothelial cells. Br. J. Pharmacol. 1994;112:1049–1056. doi: 10.1111/j.1476-5381.1994.tb13189.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shen MR, et al. Differential expression of volume-regulated anion channels during cell cycle progression of human cervical cancer cells. J. Physiol. 2000;529:385–394. doi: 10.1111/j.1469-7793.2000.00385.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ramsey IS, Moran MM, Chong JHA, Clapham DE. A voltage-gated proton-selective channel lacking the pore domain. Nature. 2006;440:1213–1216. doi: 10.1038/nature04700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Decoursey TE. Voltage-gated proton channels and other proton transfer pathways. Physiol. Rev. 2003;83:475–579. doi: 10.1152/physrev.00028.2002. [DOI] [PubMed] [Google Scholar]
- 42.Byerly L, Meech R, Moody W., Jr. Rapidly activating hydrogen ion currents in perfused neurones of the snail, Lymnaea stagnalis. J. Physiol. 1984;351:199–216. doi: 10.1113/jphysiol.1984.sp015241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fujita F, et al. Intracellular alkalization causes pain sensation through activation of TRPA1 in mice. J. Clin. Invest. 2008;118:4049–4057. doi: 10.1172/JCI35957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Morgan D, et al. Voltage-gated proton channels maintain pH in human neutrophils during phagocytosis. Proc. Natl. Acad. Sci. USA. 2009;106:18022–18027. doi: 10.1073/pnas.0905565106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Capasso M, et al. HVCN1 modulates BCR signal strength via regulation of BCR-dependent generation of reactive oxygen species. Nat. Immunol. 2010;11:265–272. doi: 10.1038/ni.1843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Sisignano M, Baron R, Scholich K, Geisslinger G. Mechanism-based treatment for chemotherapy-induced peripheral neuropathic pain. Nat. Rev. Neurol. 2014;10:694–707. doi: 10.1038/nrneurol.2014.211. [DOI] [PubMed] [Google Scholar]
- 47.Kim HK, et al. Reactive oxygen species (ROS) play an important role in a rat model of neuropathic pain. Pain. 2004;111:116–124. doi: 10.1016/j.pain.2004.06.008. [DOI] [PubMed] [Google Scholar]
- 48.Tappe-Theodor A, Kuner R. Studying ongoing and spontaneous pain in rodents - challenges and opportunities. Eur. J. Neurosci. 2014;39:1881–1890. doi: 10.1111/ejn.12643. [DOI] [PubMed] [Google Scholar]
- 49.Todd PA, Sorkin EM. Diclofenac sodium. A reappraisal of its pharmacodynamic and pharmacokinetic properties, and therapeutic efficacy. Drugs. 1988;35:244–285. doi: 10.2165/00003495-198835030-00004. [DOI] [PubMed] [Google Scholar]
- 50.Zhou YQ, et al. Reactive oxygen species scavengers ameliorate mechanical allodynia in a rat model of cancer-induced bone pain. Redox Biol. 2018;14:391–397. doi: 10.1016/j.redox.2017.10.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gwak YS, Hassler SE, Hulsebosch CE. Reactive oxygen species contribute to neuropathic pain and locomotor dysfunction via activation of CamKII in remote segments following spinal cord contusion injury in rats. Pain. 2013;154:1699–1708. doi: 10.1016/j.pain.2013.05.018. [DOI] [PubMed] [Google Scholar]
- 52.Lu JM, Gong NA, Wang YC, Wang YX. D-Amino acid oxidase-mediated increase in spinal hydrogen peroxide is mainly responsible for formalin-induced tonic pain. Br. J. Pharmacol. 2012;165:1941–1955. doi: 10.1111/j.1476-5381.2011.01680.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yoshizawa K, et al. Antinociceptive activity of the novel RAGE inhibitor, papaverine, in a mouse model of chronic inflammatory pain. Synapse. 2021;75:e22188. doi: 10.1002/syn.22188. [DOI] [PubMed] [Google Scholar]
- 54.Schwartz ES, Lee I, Chung K, Chung JM. Oxidative stress in the spinal cord is an important contributor in capsaicin-induced mechanical secondary hyperalgesia in mice. Pain. 2008;138:514–524. doi: 10.1016/j.pain.2008.01.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chen G, et al. PD-L1 inhibits acute and chronic pain by suppressing nociceptive neuron activity via PD-1. Nat. Neurosci. 2017;20:917–926. doi: 10.1038/nn.4571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Xiao X, et al. Shp-1 dephosphorylates TRPV1 in dorsal root ganglion neurons and alleviates CFA-induced inflammatory pain in rats. Pain. 2015;156:597–608. doi: 10.1097/01.j.pain.0000460351.30707.c4. [DOI] [PubMed] [Google Scholar]
- 57.Martin LJ, et al. Epiregulin and EGFR interactions are involved in pain processing. J. Clin. Invest. 2017;127:3359–3372. doi: 10.1172/JCI87406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen SP, et al. PI3K/Akt pathway: a potential therapeutic target for chronic pain. Curr. Pharm. Des. 2017;23:1860–1868. doi: 10.2174/1381612823666170210150147. [DOI] [PubMed] [Google Scholar]
- 59.Zhang ZJ, Jiang BC, Gao YJ. Chemokines in neuron-glial cell interaction and pathogenesis of neuropathic pain. Cell. Mol. Life Sci. 2017;74:3275–3291. doi: 10.1007/s00018-017-2513-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Zhang ZJ, Cao DL, Zhang X, Ji RR, Gao YJ. Chemokine contribution to neuropathic pain: Respective induction of CXCL1 and CXCR2 in spinal cord astrocytes and neurons. Pain. 2013;154:2185–2197. doi: 10.1016/j.pain.2013.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lin CP, et al. Role of spinal CXCL1 (GRO alpha) in opioid tolerance a human-to-rodent translational study. Anesthesiology. 2015;122:666–676. doi: 10.1097/ALN.0000000000000523. [DOI] [PubMed] [Google Scholar]
- 62.Corder G, et al. Loss of μ opioid receptor signaling in nociceptors, but not microglia, abrogates morphine tolerance without disrupting analgesia. Nat. Med. 2017;23:164–173. doi: 10.1038/nm.4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kornilov P, Peretz A, Attali B. Channel gating pore: a new therapeutic target. Cell Res. 2013;23:1067–1068. doi: 10.1038/cr.2013.89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peretz A, et al. Targeting the voltage sensor of Kv7.2 voltage-gated K+ channels with a new gating-modifier. Proc. Natl. Acad. Sci. USA. 2010;107:15637–15642. doi: 10.1073/pnas.0911294107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Ottosson NE, et al. A drug pocket at the lipid bilayer-potassium channel interface. Sci. Adv. 2017;3:e1701099. doi: 10.1126/sciadv.1701099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marvaldi L, et al. Importin alpha 3 regulates chronic pain pathways in peripheral sensory neurons. Science. 2020;369:842–846. doi: 10.1126/science.aaz5875. [DOI] [PubMed] [Google Scholar]
- 67.Ma YQ, et al. Discovery of an inhibitor for the TREK-1 channel targeting an intermediate transition state of channel gating. J. Med. Chem. 2020;63:10972–10983. doi: 10.1021/acs.jmedchem.0c00842. [DOI] [PubMed] [Google Scholar]
- 68.Sievers F, et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011;7:539. doi: 10.1038/msb.2011.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Robert X, Gouet P. Deciphering key features in protein structures with the new ENDscript server. Nucleic Acids Res. 2014;42:W320–W324. doi: 10.1093/nar/gku316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Okamura Y, Fujiwara Y, Sakata S. Gating mechanisms of voltage-gated proton channels. Annu. Rev. Biochem. 2015;84:685–709. doi: 10.1146/annurev-biochem-060614-034307. [DOI] [PubMed] [Google Scholar]
- 71.Eswar N, et al. Comparative protein structure modeling using Modeller. Curr. Protoc. Bioinformatics. 2006;15:5.6. 1–5.6. 30. doi: 10.1002/0471250953.bi0506s15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Friesner RA, et al. Glide: A new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004;47:1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- 73.Pronk S, et al. GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics. 2013;29:845–854. doi: 10.1093/bioinformatics/btt055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Pastor RW, MacKerell AD. Development of the CHARMM force field for lipids. J. Phys. Chem. Lett. 2011;2:1526–1532. doi: 10.1021/jz200167q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Huang J, MacKerell AD. CHARMM36 all-atom additive protein force field: Validation based on comparison to NMR data. J. Comput. Chem. 2013;34:2135–2145. doi: 10.1002/jcc.23354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Vanommeslaeghe K, et al. CHARMM general force field: a force field for drug-like molecules compatible with the CHARMM all-atom additive biological force fields. J. Comput. Chem. 2010;31:671–690. doi: 10.1002/jcc.21367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumari R, Kumar R, Lynn A, Consort OSDD. g_mmpbsa-A GROMACS Tool for High-Throughput MM-PBSA Calculations. J. Chem. Inf. Model. 2014;54:1951–1962. doi: 10.1021/ci500020m. [DOI] [PubMed] [Google Scholar]
- 78.Zhao XL, et al. A long noncoding RNA contributes to neuropathic pain by silencing Kcna2 in primary afferent neurons. Nat. Neurosci. 2013;16:1024–1031. doi: 10.1038/nn.3438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liao P, et al. Selective activation of TWIK-related acid-sensitive K+ 3 subunit-containing channels is analgesic in rodent models. Sci. Transl. Med. 2019;11:eaaw8434. doi: 10.1126/scitranslmed.aaw8434. [DOI] [PubMed] [Google Scholar]
- 80.Ferreira J, Triches KM, Medeiros R, Calixto JB. Mechanisms involved in the nociception produced by peripheral protein kinase c activation in mice. Pain. 2005;117:171–181. doi: 10.1016/j.pain.2005.06.001. [DOI] [PubMed] [Google Scholar]
- 81.Takeshita H, et al. Modified forelimb grip strength test detects aging-associated physiological decline in skeletal muscle function in male mice. Sci. Rep. 2017;7:42323. doi: 10.1038/srep42323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wang ZL, et al. Anti-PD-1 treatment impairs opioid antinociception in rodents and nonhuman primates. Sci. Transl. Med. 2020;12:eaaw6471. doi: 10.1126/scitranslmed.aaw6471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Geis C, Geuss E, Sommer C, Schmidt HHHW, Kleinschnitz C. NOX4 is an early initiator of neuropathic pain. Exp. Neurol. 2017;288:94–103. doi: 10.1016/j.expneurol.2016.11.008. [DOI] [PubMed] [Google Scholar]
- 84.Ibi M, et al. Involvement of NOX1/NADPH oxidase in morphine-induced analgesia and tolerance. J. Neurosci. 2011;31:18094–18103. doi: 10.1523/JNEUROSCI.4136-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kim UJ, Won R, Lee KH. Neuroprotective effects of okadaic acid following oxidative injury in organotypic hippocampal slice culture. Brain Res. 2015;1618:241–248. doi: 10.1016/j.brainres.2015.05.039. [DOI] [PubMed] [Google Scholar]
- 86.Kim HA, Lee KH, Lee BH. Neuroprotective effect of melatonin against kainic acid-induced oxidative injury in hippocampal slice culture of rats. Int. J. Mol. Sci. 2014;15:5940–5951. doi: 10.3390/ijms15045940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Yamamoto T, Takagawa S, Katayama I, Mizushima Y, Nishioka K. Effect of superoxide dismutase on bleomycin-induced dermal sclerosis: Implications for the treatment of systemic sclerosis. J. Invest. Dermatol. 1999;113:843–847. doi: 10.1046/j.1523-1747.1999.00758.x. [DOI] [PubMed] [Google Scholar]
- 88.Riquelme E, et al. Tumor microbiome diversity and composition influence pancreatic cancer outcomes. Cell. 2019;178:795–806. doi: 10.1016/j.cell.2019.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.