

Abstract
Antimicrobial resistance is one of the major problems in current practical medicine. The spread of genes coding for resistance determinants among bacteria challenges the use of approved antibiotics, narrowing the options for treatment. Resistance to carbapenems, last resort antibiotics, is a major concern. Metallo-β-lactamases (MBLs) hydrolyze carbapenems, penicillins and cephalosporins, becoming central to this problem. These enzymes diverge with respect to serine-β-lactamases, by exhibiting a different fold, active site, and catalytic features.
Dilucidating their catalytic mechanism has been a big challenge in the field that has limited the development of useful inhibitors. This review covers exhaustively the details of the active-site chemistries, the diversity of MBL alleles, the catalytic mechanism against different substrates, and how this information has helped developing inhibitors. We also discuss here different aspects critical to understand the success of MBLs in conferring resistance: the molecular determinants of their dissemination, their cell physiology, from the biogenesis to the processing involved in the transit to the periplasm, and the uptake of the Zn(II) ions upon metal starvation conditions, such as those encountered during an infection. In this regard, the chemical, biochemical and microbiological aspects provide an integrative view of the current knowledge of MBLs.
Graphical Abstract
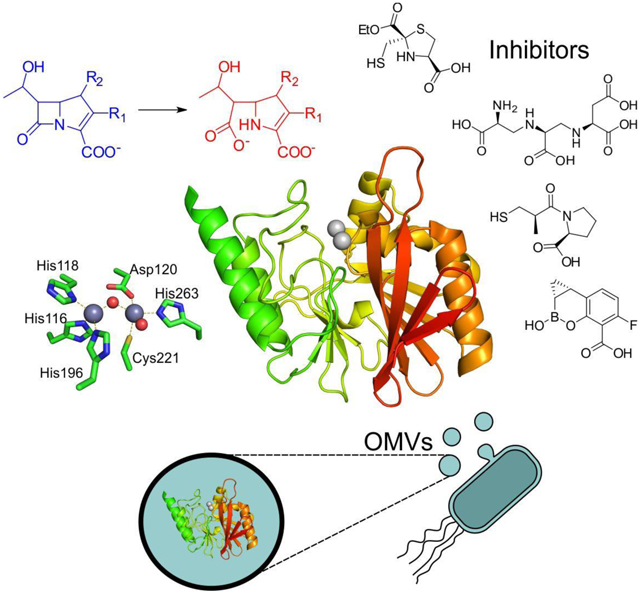
1. Introduction - Metallo-β-lactamases: from a biochemical curiosity to major players in antimicrobial resistance
The serendipitous discovery of benzylpenicillin by Alexander Fleming in 1928,1, 2 followed by the chemical characterization of this compound as a β-lactam by Howard Florey, Ernst Chain, Edward Abraham and coworkers,3, 4 stands as the dawn of the modern antibiotic era. Since then, different antibiotics were discovered or synthesized to combat the action of pathogenic and opportunistic bacteria.5
β-lactams antibiotics represent more than 60% of the injectable antibiotics used in the USA.5 The efficacy of these life-saving drugs is being continuously challenged by the development and dissemination of different resistance mechanisms in bacteria.6–8 Since β-lactams are natural products, many microorganisms have intrinsic resistance mechanisms to the bactericidal action of these compounds. But the overuse and misuse of antibiotics, either in livestock or by over and self-medication in many countries has exerted a huge evolutionary pressure that selected the most resistant bacteria. Consequently, antimicrobial resistance (AMR) has become one of the major issues in public health. A report commissioned by the UK government has estimated that, if the current trend of resistance spread continues, AMR may become the main cause of death in 2050.9 The current pandemic situation has further aggravated this problem, since antibiotics are usually prescribed to hospitalized patients affected by COVID-19.10
The main resistance mechanism to β-lactam drugs is the expression of β-lactamases, enzymes tailored to cleave the four-membered ring of these antibiotics. The study of β-lactamases has been pivotal for the understanding of antimicrobial resistance and for the development of new drugs. At the moment, the most threatening enzymes are the extended spectrum β-lactamases (ESBLs) and the carbapenemases. Carbapenems are the latest generation of β-lactam antibiotics and, as such, they are currently employed as last-resort drugs in Intensive Care Units. Thus, among β-lactamases, carbapenemases represent the main scourge in the clinics.
Metallo-β-lactamases (MBLs) represent one class of β-lactamases, accounting for 10% of the total of these enzymes. Despite this seemingly small number, MBLs are relevant due to their ability to hydrolyze carbapenems. MBLs are a common resistance mechanism in carbapenem-resistant enterobacteria (CRE) and frequently in carbapenem-resistant non-fermentative bacteria such as Pseudomonas aeruginosa and Acinetobacter baumannii. Many bacterial outbreaks are due to strains expressing different MBLs, that represent an important challenge in current chemotherapy.
This critical scenario was not foreseen at the time MBLs were initially reported.11–13 The first known MBL was an extracellular enzyme from Bacillus cereus (BcII), characterized as a zinc-dependent enzyme in 1966.13 Since this early discovery, MBLs were considered as a biochemical curiosity for almost three decades, until MBL-coding genes disseminated worldwide among pathogenic bacteria. From the scientific point of view, MBLs represent a fascinating system to study that keeps on posing challenges to researchers.
The study of MBLs has represented a separate field within the area of β-lactamases. Their understanding requires the knowledge coming from inorganic chemistry to understand the substrate recognition and catalytic mechanism, that differs from the rest of β-lactamases, posing additional challenges for inhibitor design. This approach is also essential to analyze the interplay between zinc homeostasis and trafficking in bacterial and host cells at the infection sites and the impact in MBL-mediated resistance. From the biochemical aspect, the ubiquity of the MBL fold in all kingdoms of life shows the versatility of this protein scaffold to acquire variate functionalities, representing at the same time a unique system to study protein evolution. Finally, in the last years the study of MBLs has led to new questions more related to the bacterial physiology, such as which is the impact of their localization within the bacterial cell, their secretion into vesicles, their biogenesis and degradation.
There are many excellent reviews in the literature that cover different aspects of the research efforts on MBLs,6, 14–24 which are targeted to biochemists, medicinal chemists, or clinical microbiologists. Here we provide a comprehensive review of the most recent progress in the study of MBLs covering all aspects. The current review describes the active-site chemistry and catalytic mechanism, the attempts to design MBL inhibitors, the diversity in the active-site chemistry and topologies found in natural MBL alleles and the impact of the use of antibiotics in their current evolution. We also present and discuss the molecular mechanisms involved in the expression, regulation, processing and degradation of MBLs within the cell, the genetics involved in the dissemination among different bacteria, the molecular mechanisms of zinc homeostasis at the host-pathogen interface, their secretion into outer membrane vesicles and a discussion of different phenotypic, chemical and genetic methods to identify MBLs in the clinics.
This review is mostly focused on the developments of the last decade, in which the study of MBLs has blossomed and expanded from the traditional microbiological reports and structural studies aimed to elucidate their catalytic mechanism, to novel strategies for inhibitor design, and the understanding of their physiology in the bacterial cell. We have included a historical account of the early discoveries of MBLs dating back from 1966. We aim to provide a hitchhiker’s guide for the MBL world that can be accessed by newcomers and aficionados with different scientific backgrounds. The review was written with an integrative perspective, but also considering that each section could be read separately, depending on the interest of each reader.
2. β-Lactam antibiotics and mechanisms of bacterial resistance
2.1. Classes of β-lactam antibiotics
β-Lactam antibiotics are by far the most utilized antibacterials in clinical settings, making up to 65% of all injectable antibiotic prescriptions in the US.5, 8, 25–29 This is due to their wide action spectrum, targeting most bacterial groups and being generally well tolerated, with low toxicity compared to other antibiotics. Because of these factors, the rising prevalence of resistance towards these life-saving drugs is a source of enormous concern.
The molecular targets of β-lactam antibiotics are the cell-wall transpeptidases, historically known as Penicillin-Binding Proteins (PBPs).6, 30 β-Lactams act by covalently binding and inhibiting the activity of essential enzymes involved in the last steps of peptidoglycan synthesis. All β-lactam antibiotics contain their namesake four-membered β-lactam ring, a 2-azetidinone group which is the key to their antibacterial activity (Figure 1). This ring is rather uncommon in other natural products.
Figure 1.
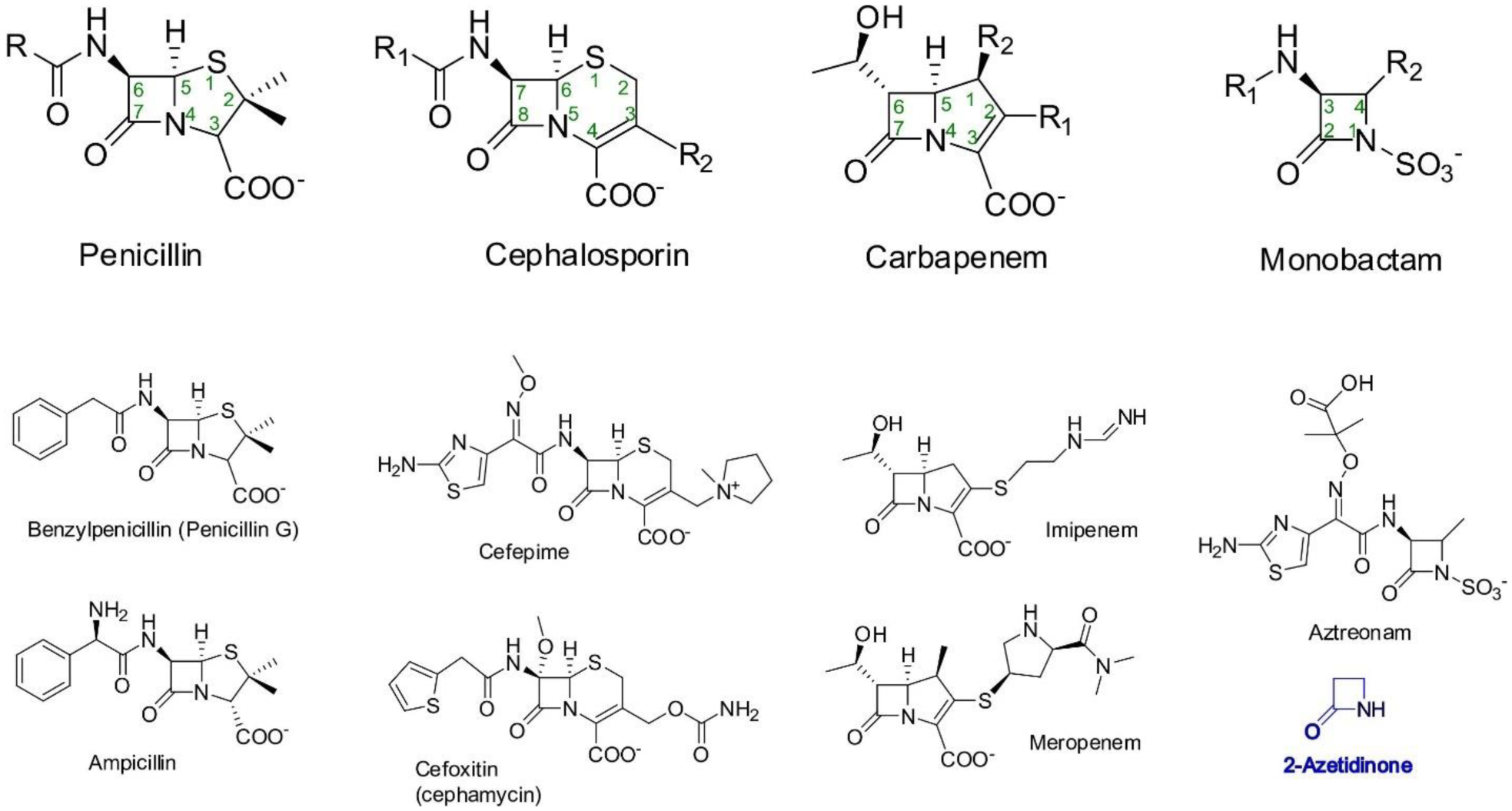
(Top) Structures of the four main classes of β-lactam antibiotics. (Bottom) Representative structures of antibiotics from each subclass, and of 2-azetidinone (shown in blue), the simplest compound containing a β-lactam ring.
This moiety possesses structural similarity to the terminal D-Ala-D-Ala dimer of peptidoglycan building blocks and allows the drugs to act as substrate mimics for the transpeptidase domains of PBPs, which catalyze the crosslinking of peptidoglycan strands.26 The active-site serine of these proteins performs a nucleophilic attack on the β-lactam carbonyl and forms an acyl-enzyme complex, and the slow hydrolysis of this intermediate leads to functionally irreversible inhibition of the affected transpeptidases.17 This in turn weakens the cell wall, leaving the cell susceptible to lysis via osmotic pressure. Additionally, β-lactam antibiotics also trigger the activation of lytic transglycosilase enzymes, leading to further destruction of the peptidoglycan sacculus integrity.31, 32 These effects make β-lactam antibiotics potent bactericides, although there may be further complexity yet to be explored regarding the ultimate mechanism by which they kill bacterial cells.33 It should also be noted that the specific effects of a given β-lactam antibiotic on bacterial physiology depend on the main PBP(s) that each one targets. For example, aztreonam leads to the formation of filamentous cells due to the inhibition of PBP3 that blocks cell division.34 Apart from their antibiotic function, recent studies have revealed a potential use of β-lactams as drugs targeting the central nervous system,35, 36 in particular regarding their ability to increase expression of the glutamate transporter (GLT1). These effects could be used in therapy for diseases such as Amyotrophic Lateral Sclerosis (ALS),36 and may be linked to the existence of eukaryotic homologs of metallo-β-lactamases acting as targets of these drugs in nervous system cells (see Section 4.9).
Beyond their common characteristics, β-lactam antibiotics present a large degree of structural heterogeneity, which determines their division into four major classes: penicillins, cephalosporins, carbapenems and monobactams (Figure 1).
Penicillins were the first β-lactam antibiotics to be discovered, with the identification of penicillin G (benzylpenicillin) by Alexander Fleming in 1928 as a product of the Penicillium chrysogenum (formerly P. notatum) fungus.37 They were also the first to see clinical use, boosted by tremendous efforts to optimize large scale production for the treatment of Allied soldiers in the later stages of World War II.5 These facts defined the beginning the modern antibiotic era. The structure of penicillins contains the β-lactam ring fused to a five-membered thiazolidine ring (Figure 1). In all penicillins, position 3 of this bicyclic structure contains a carboxyl group and a geminal dimethyl group is present in position 4. An amino group attached to the β-lactam ring at the C-6 position completes the 6-aminopenicillanic acid (6-APA) core. All current clinically used penicillins (with the exception of penicillin G and penicillin V) are chemically derived from 6-APA by the attachment of varying substituents to the 6-amino group,29, 38, 39 resulting in altered antibacterial action spectrum, pharmacological properties and stability towards β-lactamases. Few other chemical modifications to the 6-APA core have afforded useful drugs,39 among which can be noted temocillin, with the addition of a methoxy group to the C-6 position.
The first cephalosporin was discovered in a fungus belonging to the Acremonium genus (formerly Cephalosporium), isolated from a sewage outfall in Sardinia by Giuseppe Brotzu in the 1940s.40 The fungus produced cephalosporin C, which was isolated and characterized by Abraham.29, 40 This compound served as the starting point for the development of a large variety of semi-synthetic derivatives starting in the 1960s. More than 30 cephalosporins have been approved for clinical treatments, usually classified in five succesive generations,5, 38, 41 although not all of them are widely available and the use of a portion of them has since been discontinued. The chemical structure of cephalosporins contains the β-lactam cycle fused to a 6-membered dihydrothiazine ring. This cephem core structure has an attached carboxyl group at position 4, while position 3 is an attachment point for substituents (R2, Figure 1). Similarly to penicillins, cephalosporins contain an amide group linked to the β-lactam ring at C-7. Different substituents (R1, Figure 1) have been introduced linked to the carbonyl atom of this group. Cephamycins are generally considered within the cephalosporin class, possessing the same basic structure but with an additional methoxy group directly attached to the carbon atom at position 7. This modification improves stability against certain β-lactamases.5, 42
Carbapenems were identified as secondary metabolites of the Gram-positive bacterium Streptomyces cattleya, from which the isolation of thienamycin was first reported in 197943 (although its antimicrobial activity was described in 197628, 44). Thienamycin showed a potent, broad spectrum activity,45 even against bacteria resistant to cephalosporins and penicillins due to production of β-lactamases.43 However, it is spontaneously degraded in solution and this instability prevented its clinical use.44 The development of an N-formimidoyl derivative of this compound, termed imipenem, afforded stability and allowed its introduction as a clinical antibiotic in 1985.5, 46 Similarly to penicillins, the β-lactam group in carbapenems is fused to a five-membered ring, although their dihydropyrrole ring is unsaturated and lacks a sulfur atom. This second ring contains a carboxyl group at position 3, with the presence of variable substituents at position 2. Unlike penicillins and cephalosporins, imipenem has an α-oriented hydroxyethyl side chain next to the β-lactam. The orientation of this substituent has made imipenem and other carbapenems refractory to degradation by most β-lactamases.28, 47 Since imipenem can be degraded by human renal dehydropeptidase (DHP), it is administered in conjunction with the DHP inhibitor cilastatin to prolong its lifetime in vivo.5, 48 Other carbapenems developed later (such as meropenem, ertapenem, faropenem) contain a methyl group at position 1, which prevents degradation by DHP.47, 48
Finally, monobactams are monocyclic β-lactams which possess a sulfonate group attached to the β-lactam nitrogen (Figure 1). Currently, the only monobactam antibiotic in clinical use is aztreonam (originally named azthreonam),49 a synthetic analog of an antibiotic produced by the Gram-negative bacterium Chromobacterium violaceum (termed SQ 26,180).50, 51 Various soil bacteria produce monobactams and other monocyclic β-lactams,52, 53 including Pseudomonas, Gluconobacter, Agrobacterium and Nocardia, with other characterized natural and synthetic compounds including sulfazecin,54 tigemonam,55 carumonam56 and nocardicyn A.53 Rising antibiotic resistance has renewed interest in monobactams, with novel compounds in development (see below).
Other types of β-lactam compounds, such as clavams, do not bind to PBPs and thus have no antibiotic activity by themselves, but possess clinical applications as inhibitors of β-lactamases (see Section 6.1.1).
2.2. Spectrum of action of β-lactams
β-Lactam antibiotics are effective against a large variety of bacteria. Resistance mechanisms to these drugs range from the expression of intrinsic or acquired β-lactamases, low permeability to the drugs or low affinity of their PBP proteins for the antibiotics, resulting in reduced susceptibility (see below). The permeability barrier of the Gram-negative cell wall presented a restriction towards early penicillins, until the generation of new derivatives by chemical modification allowed for a widened action spectrum.57
Early penicillins such as penicillin G and penicillin V presented activity towards Gram-positive bacteria such as staphylococci and streptococci, although with great susceptibility to β-lactamases.5 Because of this, penicillins with lower susceptibility to staphylococcal lactamases were developed, such as methicillin, oxacillin and cloxacillin. These drugs, however, still had limited action towards most Gram-negative bacteria.39 The development of extended spectrum penicillins such as ampicillin and amoxicillin allowed the treatment of infections caused by many Gram-negative bacilli, including Enterobacteria.57 Furthermore, some of these penicillins (such as piperacillin, azlocillin and ticarcillin) display antipseudomonal activity, and are also broad spectrum antibiotics, being active against susceptible Enterobacteria and anaerobes.57, 58 The increasing prevalence of β-lactamase producers has reduced the use of penicillins as single therapy drugs, although they continue to be prescribed partnered with β-lactamase inhibitors with similar pharmacokinetic properties.5
Cephalosporins are generally active against staphylococci and streptococci among Gram-positive bacteria, although but not towards methicillin-resistant strains of the former.59, 60 Others such as Enterococcus faecalis and E. faecium, or Listeria monocytogenes, are generally not susceptible to cephalosporins.59 Drugs targeting methicillin-resistant Staphylococcus aureus (MRSA) have also been developed, including ceftaroline61 and ceftobiprole.62 These possess the ability to bind to the low affinity PBP2a that grants reduced susceptibility to β-lactams to MRSA isolates63, 64 (see Section 2.3.2). Many genera of Gram-negative bacteria are susceptible to cephalosporins, including Escherichia, Salmonella, Proteus and Neisseria.59 On the other hand, Pseudomonas are only susceptible to specific antipseudomonal cephalosporins,58, 60 such as cefepime, ceftazidime and the more recently introduced ceftolozane.65 The effectiveness of cephalosporins against Gram-negative anaerobes is limited, although cephamycins such as cefoxitin and cefotetan show greater activity towards these bacteria.60, 66 Although successive generations of cephalosporins have been developed to improve (among other characteristics) their stability towards degradation by bacterial enzymes, the effectiveness of many cephalosporins as single agents has been challenged by the dissemination of wide-spectrum β-lactamases.5 Similarly to penicillins, this can be in part remedied by their co-administration with β-lactamase inhibitors. Also of note is the novel siderophore cephalosporin cefiderocol67 (formerly S-649266) which has recently obtained clinical approval. The attached catechol moiety in cefiderocol chelates ferric ions and allows entry through the outer membrane receptors of iron-scavenging compounds,68 which are required for effective bacterial growth during infection. This “Trojan horse” strategy increases the periplasmic concentrations of the drug and restricts the possibility of permeability-altering mutations granting resistance. Furthermore, cefiderocol is stable towards hydrolysis by a wide range of lactamases,69 including metallo-β-lactamases (MBLs), and is proposed for treating carbapenem-resistant and multi-drug resistant Gram-negative bacteria.67, 70
Carbapenems are arguably the most versatile β-lactam agents. They possess a wide spectrum of action, with activity against Gram-positive and both aerobic and anaerobic Gram-negative bacteria.28, 47, 48 There are some variations of potency against these different groups of bacteria among carbapenems, with meropenem possessing slightly greater effectiveness in vitro than imipenem against Gram-negative bacteria, but with reduced activity towards Gram-positive bacteria.48 Additionally, both meropenem and doripenem have greater antibacterial activity than imipenem against P. aeruginosa strains which lack the OprD porin71 (see below). Meanwhile, ertapenem has limited activity towards non-fermentative Gram-negative organisms such as Acinetobacter spp. and P. aeruginosa.47 Most carbapenems are administered parenterally (e.g. intravenously or intramuscularly) due to poor low oral bioavailability.28, 72 However, the oral drug tebipenem was clinically approved for pediatric use in Japan in 2009, and is currently undergoing trials for an expanded use worldwide.73 These compounds are poorly hydrolyzed by many β-lactamases, allowing their use in cases of infection by microbes resistant to other antibiotics of the class.5, 74 Because of these characteristics, carbapenems are considered last-resort drugs and usually reserved to treat life-threatening infections by multirresistant bacteria.28 However, they are not impervious to degradation. The emergence of multiple types of transmissible carbapenemases (such as MBLs) in the last two decades threatens their clinical efficiency.75–77
Regarding monobactams, the only drug currently approved for clinical use is aztreonam, which is effective towards both Enterobacteria and P. aeruginosa, but with poor activity towards Gram-positive organisms and Gram-negative anaerobes such as Bacteroides.34, 49 The fact that monobactams are the only β-lactam antibiotics refractory to degradation by wide-spectrum MBLs generated considerable interest in them, following the increased prevalence of these enzymes. However, aztreonam can be inactivated by many serine-β-lactamases (SBLs) and, since many clinical isolates produce both types of β-lactamases, it cannot be used effectively to combat MBL-mediated resistance. This could change with the introduction of monobactams with greater stability to SBLs such as BOS-228 (formerly LYS228, currently in clinical trials),78, 79 or by combination of aztreonam with new SBL inhibitors80 (see Section 6.1.2).
2.3. Mechanisms of resistance.
2.3.1. Antibiotic degradation by β-lactamases.
Among the various strategies bacteria have developed to avoid the action of β-lactam antibiotics, the most common in Gram-negative organisms is the production of β-lactamases (EC 3.5.2.6), enzymes that degrade the offending compounds7 (Figure 2). Two main types of β-lactamases can be identified on the basis of their structures and catalytic mechanisms: serine-β-lactamases (SBLs) and metallo-β-lactamases (MBLs).6, 76 SBLs use a catalytic serine residue to perform a nucleophilic attack on the β-lactam carbonyl, resembling the action of serine-dependent proteases.17 Multiple classes of SBLs exists but they all share this basic catalytic scheme with some differences that will be outlined below. On the other hand, MBLs are zinc-binding metalloproteins in which the attacking nucleophile is a hydroxide ion, activated and positioned by the Zn(II) cations and assisting residues from the active site.21 These differences in the mechanism determine that MBLs are in general not affected by inhibitors targeting SBLs (see Section 5).
Figure 2.
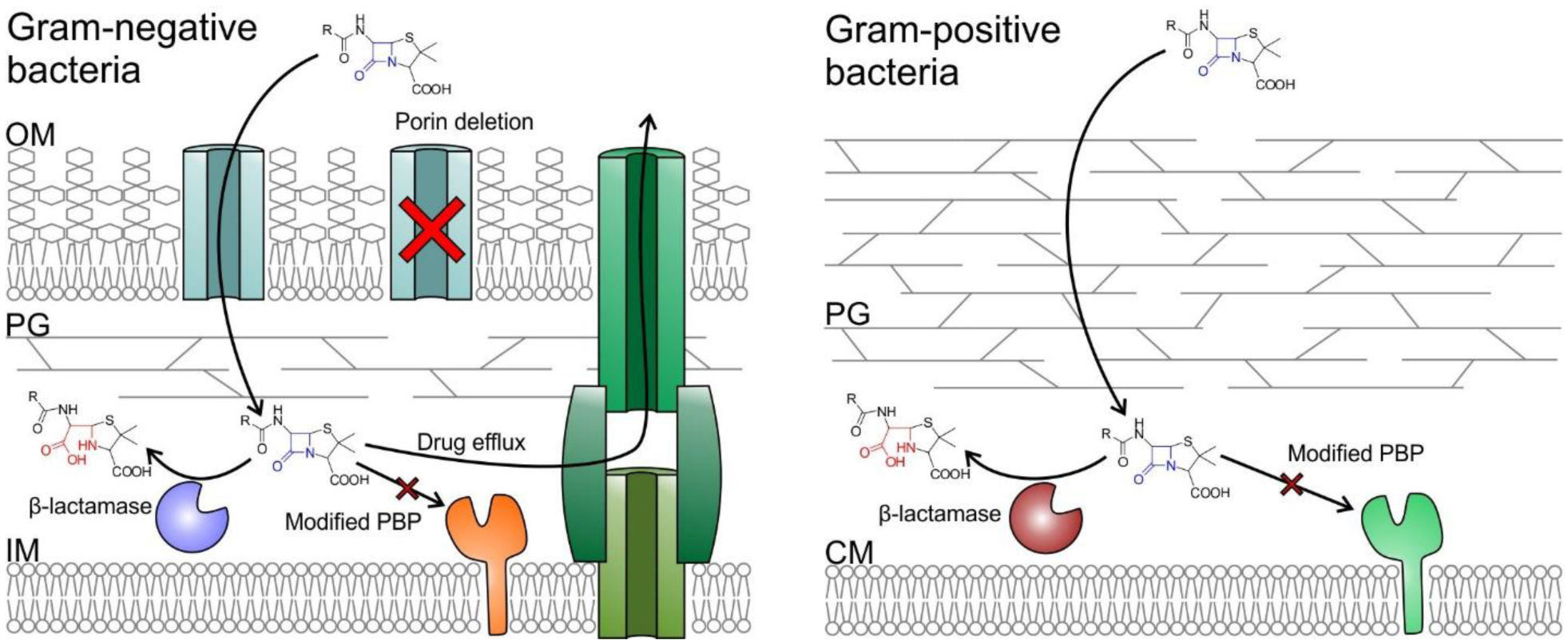
Representation of the main mechanisms of β-lactam resistance in Gram-negative (left) and Gram-positive (right) bacteria. OM (outer membrane), PG (peptidoglycan), IM (inner membrane), CM (cytoplasmic membrane). The β-amino acid moiety resulting from β-lactam cleavage is shown in red.
Many bacterial species carry within their chromosome genes encoding β-lactamases, and thus are intrinsically resistant to certain classes of β-lactam antibiotics.16, 81 Even in cases in which the native β-lactamase presents limited hydrolytic capability towards specific antibiotics, significant resistance can arise due to the selection of strains hyperproducing the enzyme.81–83 Additionally, β-lactamases are frequently observed in clinical settings either encoded in plasmids, or associated with other types of mobile genetic elements such as transposons and integrons7, 16 (see Section 9). This greatly increases the potential of the enzymes to spread to other hosts, and has resulted in the selection of bacterial strains impervious to one or more classes of these drugs that derive from strains that were typically susceptible to β-lactam antibiotics. Furthermore, other resistance determinants are usually present together with the β-lactamases, with their coding genes carried within cassettes in the mobile genetic element. This results in multi-drug resistant (MDR) and extensively drug resistant (XDR) bacteria,84, 85 that present a formidable challenge to established antimicrobial chemotherapy schemes and can force the use of drugs previously sidelined for their serious toxicity, such as colistin.
2.3.2. Altered PBPs
The production of β-lactamases is the main mechanism of resistance among Gram-negative bacteria, and was the first to be observed in clinical settings for Gram-positive organisms.86, 87 However, a reduction in β-lactam susceptibility among the latter group is most often mediated by an alternative strategy: the production of PBPs with reduced affinity for β-lactam antibiotics (Figure 2), modifying the drug target instead of directly neutralizing the chemical threat.88–90
The first identified example of this resistance mechanism arose after the introduction of methicillin, a penicillin stable to staphylococcal β-lactamases. Methicillin-resistant S. aureus (MRSA) strains were isolated91 in which the expression of an alternative PBP (PBP2a) allowed the organism to resist most β-lactam antibiotics.88, 90, 92 This is due to the fact that PBP2a reacts very slowly with these drugs, and the acylation rate of the enzyme can be up to 3 orders of magnitude slower compared to the native PBPs of S. aureus.89 The slow acylation of the enzyme by β-lactams is caused by the closed conformation of the active site blocking access by the antibiotic, as observed in the first crystal structure obtained for the protein.93 However, it was not clear how such a structure could still allow the enzyme to carry out its physiological role. This apparent paradox was dispelled when it was observed that the presence of peptidoglycan fragments increased the rate of acylation of PBP2a by β-lactams,94 pointing out the existence of an allosteric regulation mechanism that would enable access to the active site when PBP2a is actively catalyzing its native reaction while allowing it to otherwise remain occluded and protected from inactivation by these drugs.90 This was subsequently confirmed with the identification of the allosteric site at which peptidoglycan fragments bind, which is located at a distance of 60 Å from the active site in a domain contiguous to that possessing the transpeptidase activity.95 When this allosteric site is occupied, a conformational change is transmitted to the active site via the breaking and formation of a series of salt bridges, which results in the opening of the active-site cavity. Notably, the anti-MRSA cephalosporin ceftaroline can also bind to the allosteric domain of PBP2a, albeit in a different site to that observed for peptidoglycan, and can similarly trigger a conformational change allowing a second molecule of the antibiotic to bind within the active site.95 The protein, coded by the mecA gene, is not constitutively expressed but is instead regulated by the MecR1/MecI system.92 Upon exposure to β-lactams, the membrane sensor protein MecR1 activates the proteolytic activity of its cytoplasmic domain, cleaving the MecI repressor and allowing production of the alternative PBP.92
Various Enterococcus species intrinsically possess reduced β-lactam susceptibility due to the production of a PBP transpeptidase with low affinity for the drugs, termed PBP5fm.96, 97 The constitutive expression of the protein confers moderate resistance, with higher levels resulting from hyperproduction and/or the accumulation of mutations that further reduce its reactivity towards β-lactam antibiotics.98–100 Similarly, S. aureus isolates presenting mild resistance towards these antibiotics while lacking both β-lactamases and the mecA gene have been identified. Their resistance phenotype is attributed to the presence of mutations in their intrinsic PBPs that reduce their acylation rate.101 Streptococcus pneumoniae also attains resistance to β-lactam antibiotics through modifications of its native PBPs, either by point mutations or the generation of mosaic genes, possibly using fragments acquired by horizontal gene transfer from commensal streptococci.89, 102
Deletion of PBPs or accumulation of mutations that reduce their reactivity toward the antibiotics have also been observed to confer resistance to β-lactams in Gram-negative bacteria.89 Decreased susceptibility in P. aeruginosa clinical strains was associated with the lack of PBP4 and an altered production of PBP1a and PBP1b, in combination with overexpression of AmpC and efflux pumps and inactivation of the porin OprD.103 Additionally, point mutations in the essential PBP3 were observed in P. aeruginosa isolates obtained from cystic fibrosis patients as part of their adaptation to β-lactam therapies.104, 105 Amino acid substitutions in PBP3 have also been reported to decrease β-lactam susceptibility in clinical isolates of Haemophilus influenzae,106 and mutations in PBP1a contribute to resistance in Helicobacter pylori.107 Point mutations in PBPs and generation of mosaic genes (both frequently in PBP2) were reported in Neisseria gonorrhoeae and N. meningitidis as a mechanisms of PBP modification that reduce their susceptibility to β-lactams.108
2.3.3. Reduced permeability
The ability of β-lactams to reach their molecular targets within bacteria is absolutely essential for their efficacy. The cell wall of Gram-negative bacteria presents a daunting challenge in this aspect, as their outer membrane acts as a selective permeability barrier precluding access to many potential chemotherapeutic agents.109, 110 Certain hydrophobic drugs, such as macrolides and aminoglycosides, can traverse this obstacle by directly diffusing across the outer lipid bilayer.110 Meanwhile, hydrophilic drugs such as β-lactams and fluoroquinolones enter the periplasm through facilitated diffusion, using the pathways provided by certain Outer Membrane Proteins (OMPs), called porins.111, 112 These β-barrel proteins, also called porins, span the outer membrane and provide a hydrophilic channel allowing the passage of low molecular weight solutes.110, 113 The various porins produced by a given bacteria differ by the width and selectivity of their internal pore, and compounds may preferentially enter the cell through one particular channel. As a result, alteration in the number and composition of porins in the outer membrane, via porin deletion, mutation, or modified expression levels, can significantly impact on the antibiotic susceptibility of bacterial isolates109, 113 (Figure 2). This is often associated with other resistance mechanisms such as β-lactamase production or expression of drug efflux systems.
The main trimeric porins of E. coli are OmpC, OmpF and PhoE. These proteins act as general channels allowing the entrance to compounds smaller than 600 Da, although their pore characteristics differ, and the former two favor cationic substrates while the latter preferentially allows permeation of anionic species.112 These roles are covered by orthologues possessing a high degree of structural homology in other Enterobactales. Replacement of OmpF with the narrower-pore OmpC (or their corresponding homologs) as the major OM channel correlates with a dimished effectiveness of β-lactams in E. coli and other Enterobacterales such as K. pneumoniae, S. enterica and E. cloacae.112, 113 Loss of both porins renders the cell virtually impermeable to these drugs,113 albeit at the cost of possibly impairing bacterial fitness.114 In particular, mutations or truncation of OmpK35 or OmpK36 (OmpF and OmpC homologs, respectively) in K. pneumoniae has been implicated in the development of resistance to ceftazidime/avibactam.115, 116 The impact of general porin loss on fitness can be mitigated by increased production of specialized OMPs, as is the case for overproduction of maltoporin LamB in K. pneumoniae isolates lacking OmpK36.117
A less drastic mechanism is the alteration of the channel itself, with both steric factors and charge distribution impacting the permeability of a given compound through a specific porin.118 Mutations in K. pneumoniae OmpK36 located at the so-called constriction region, which affords the main permeability barrier along the pore, have been observed in various isolates with reduced β-lactam susceptibility.119–121
P. aeruginosa lacks general porins, and instead relies in an ensemble of more than 20 OMPs with greater substrate specificity.122 This renders its outer membrane less permeable towards most compounds, and grants it intrinsic resistance to a variety of antibiotics including many β-lactams.122, 123 The OprD (OccD1) porin is the main pathway used by carbapenems to enter P. aeruginosa.124 Mutation, reduced expression or truncation of the protein leads to reduced susceptibility to carbapenems.125–127 However, these mutant strains present lower resistance to to meropenem than to imipenem, suggesting the presence of an alternative entry route for the former.128
Acinetobacter species generally show high levels of intrinsic resistance towards most β-lactams and other antibiotics, possibly due to the restrictive nature of their OM pores,129, 130 but are generally still susceptible to carbapenems in absence of other resistance mechanisms.131 This would be caused by the presence of multiple specific channels allowing entry of these drugs into the periplasmic space, with the main pathway utilizing the CarO porin.131 The native function of these proteins is the uptake of basic amino acids, as was observed for OprD in P. aeruginosa. Loss of CarO and other putative entry points such as OprD/OccAB1 and Omp33/36, results in reduced susceptibility to carbapenems.131, 132
2.3.4. Drug efflux
Bacteria produce a variety of efflux pumps that allow extrusion of toxic substances outside the cell, together with roles in virulence and biofilm formation. Many of these systems possess a wide substrate range, and can contribute to antibiotic resistance by pumping the drugs away from their targets133–135 (Figure 2). Although Gram-negative bacteria express multiple families of efflux pumps with varying architectures and mechanisms, the main contributors to reduced antibiotic susceptibility are those belonging to the Resistance Nodulation Division (RND) family.136 These are tripartite systems spanning the inner membrane, periplasm and outer membrane, and allowing removal of compounds from both the bacterial cytoplasm and periplasm.
The archetypal RND system is the AcrAB-TolC efflux pump from E. coli, consisting on the AcrB transporter located on the inner membrane, the TolC outer membrane channel, and the periplasmic adaptor protein AcrA linking both together.137 The system is powered by the proton motive force across the inner membrane. RND systems show a high degree of conservation, and homologues of AcrB are present in numerous Gram-negative bacteria, such as P. aeruginosa (MexB), A. baumannii (AdeB) and N. gonorrhoeae (MtrD).134
Overexpression of efflux pumps has been implicated in decreased susceptibility to antibiotics, often linked to MDR phenotypes. As such, increased production of the AdeABC system in A. baumannii has been linked to increased resistance to β-lactams among other antibiotics,138, 139 and the AcrAB-TolC pump has also been implicated in reduced susceptibility to β-lactams in K. pneumoniae.140, 141 Coupled with the low permeability of its outer membrane, P. aeruginosa can produce multiple drug efflux systems such as MexAB-OprM, MexXY-OprA, MexCD-OprJ and MexEF-OprN, contributing to MDR phenotypes in this organism, including β-lactam resistance.127, 142
3. β-Lactamases: Classification and substrate profile
3.1. Ambler classification. Classes A, B, C, D
The Ambler classification system, also called the molecular classification system, divides β-lactamases into four classes on the basis of their primary sequence.143, 144 Ambler classes A, C and D are Serine-β-lactamases (SBLs), as all involve the action of an essential serine residue which is responsible of the nucleophilic attack on the β-lactam carbonyl to form an acyl-enzyme intermediate that is later deacylated by the action of a water molecule (Figure 3).145–148 These three classes of SBLs differ mechanistically in the identity and interactions of the general base of the reaction.6, 17, 149–151 Although their sequences are very divergent, all SBL classes have evolved from the transpeptidase domain of PBPs (the targets of β-lactam antibiotics) approximately three billions of years ago.152 This evolution resulted in a drastic substrate change: while DD peptidases cannot hydrolyze β-lactams, β-lactamases are unable to turnover D-Ala-D-Ala terminating peptides. In contrast to these transpeptidases, SBLs have evolved the ability to hydrolyze β-lactams by accelerating the deacylation step, destroying the covalent adduct with the antibiotic that would otherwise inactivate the protein (Figure 4). SBLs possess a highly conserved αβα fold153 that is shared with PBP transpeptidase and D,D-carboxypeptidase domains, β-lactam sensors such as BlaR1, and even some serine proteases.154–158
Figure 3.
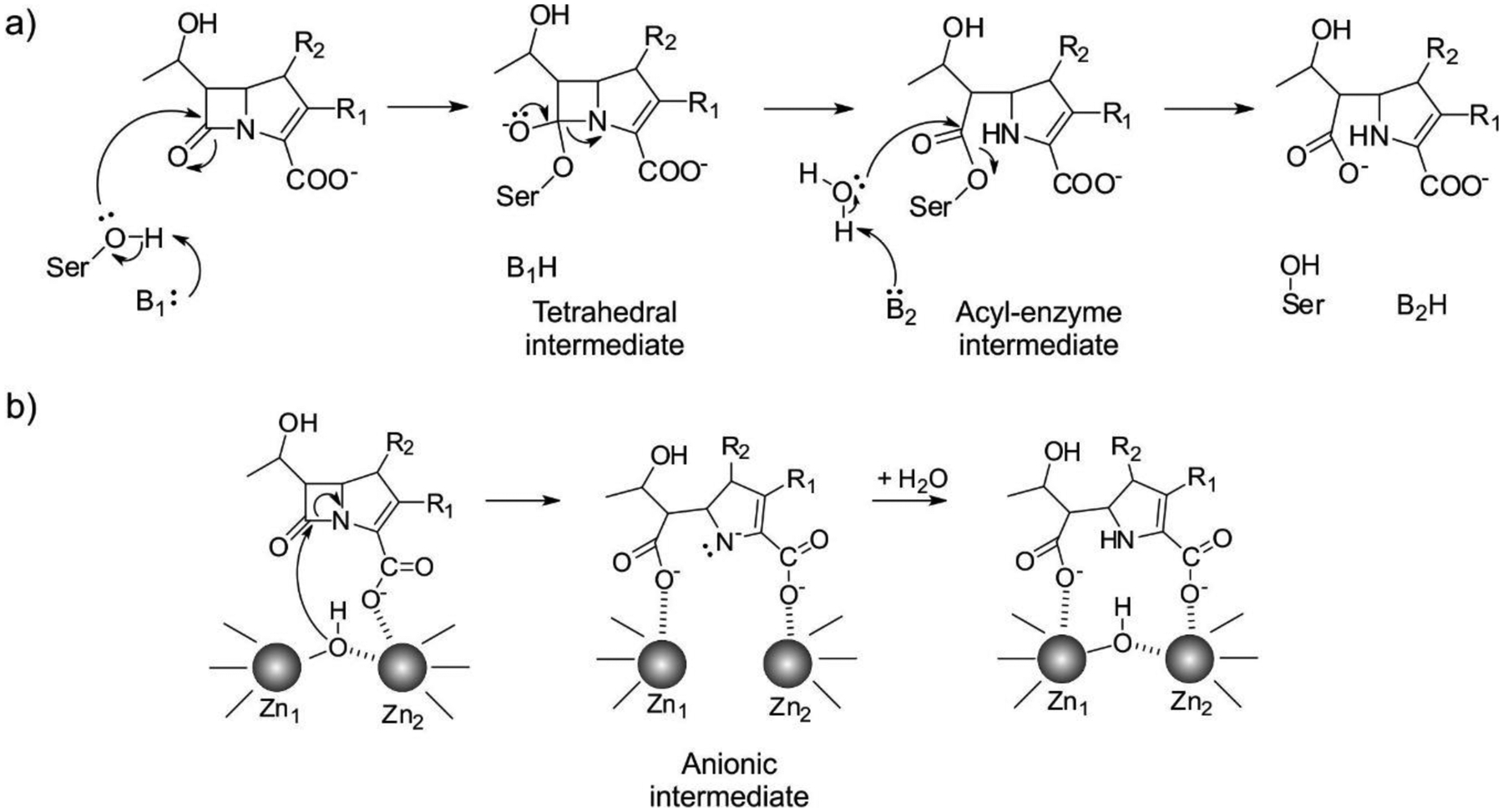
Minimalistic outlines of β-lactam hydrolysis mechanisms by SBLs (a) and MBLs (b). B1 is a general base activating the nucleophilic Ser residue in SBLs, and B2 is a general base involved in the deacylation step of SBL-mediated hydrolysis. The specific residues involved in the mechanism are discussed in the text.
Figure 4.
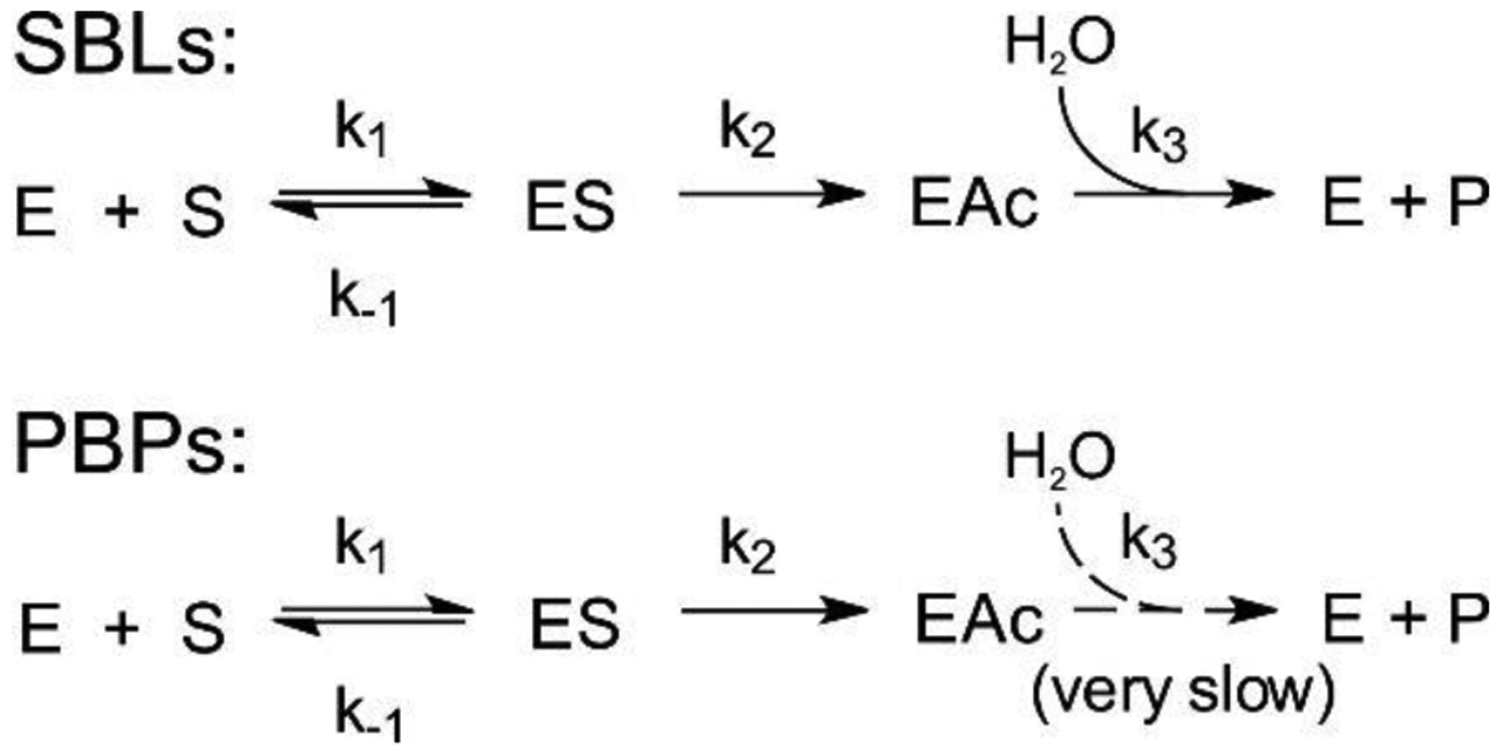
Reaction scheme of SBLs and PBPs with β-lactam antibiotics, showing the formation of the Michaelis complex, enzyme acylation and the final deacylation step that regenerates the enzyme.
Ambler class B enzymes are Metallo-β-Lactamases (MBLs), that can bind 1 or 2 zinc ions in their active sites, and use a hydroxide ion as a nucleophile for the ring opening of their substrates, without forming a covalently bound intermediate (Figure 3).19, 21, 23, 159 MBLs do not share any sequence nor structural homology with SBLs, and their evolutionary origins are unclear (see below).
Class A
Enzymes belonging to class A are, together with those from class D, the ones that display the largest diversity in their substrate spectra.6, 17, 149, 160 The first β-lactamases to acquire clinical relevance were class A proteins from staphylococci, such as PC-1/BlaZ from S. aureus, whose prevalence rapidly increased after the therapeutic introduction of penicillin. Such resistance was in fact observed as early as 1942.161 These are narrow-spectrum penicillinases, and although they are plasmid-encoded they have not spread from the host in which they were initially detected. Other functionally similar Gram-positive enzymes such as the chromosomal BlI from Bacillus cereus and other species from the genus were used in pioneering in vitro studies of lactamases.162 The initial transmissible plasmid-borne lactamases also belong to class A, arising and becoming widely disseminated in Gram-negative bacteria. These were the TEM-1163 and SHV-1 enzymes,164 the latter found to be derived from a chromosomal SBL from K. pneumoniae, with activity against aminopenicillins (such as ampicillin) and early cephalosporins. The introduction of extended-spectrum oxyiminocephalosporins and β-lactamase inhibitors led to the selection of variants of these enzymes with a widened substrate spectrum or resistance to inhibitors, giving rise to the Extended Spectrum β-Lactamases (ESBLs) and Inhibitor Resistant TEM enzymes (IRTs), respectively.160 A limited number of substitutions, even 2 or 3, are able to elicit these evolutionary advantages. More recently, other families of enzymes became the dominant class A plasmid-borne ESBLs, such as CTX-M lactamases,165, 166 widespread in E. coli, and GES, prevalent in Enterobacterales, P. aeruginosa and A. baumannii.167
A small number of class A lactamases have carbapenemase activity.75, 160, 168 The most clinically significant of these is the KPC family of plasmid-mediated enzymes, which has spread worldwide and are the dominant carbapenemases in the US. Although originating in K. pneumoniae, KPC enzymes have also been found in numerous Enterobacterales and P. aeruginosa.169 Their substrate spectrum is not limited to carbapenems, as they can hydrolyze penicillins, cephalosporins and aztreonam. Other carbapenemases from class A include certain GES enzymes,170 and the chromosomal proteins IMI/Nmc-A171 and SME172 from Enterobacter spp. and Serratia marcescens, respectively.
The expression of class A enzymes is regulated in various organisms. The Staphylococcus aureus β-lactamase BlaZ/PC1 is under the regulation of the BlaR/BlaI system, and is inducible by the presence of β-lactams.173 In Gram-negative organisms, the chromosomal β-lactamases from Proteus vulgaris,174 NmcA from Enterobacter cloacae175 and PenA from Burkholderia spp.176 are regulated by AmpR homologs (see Section 7.2). The mobilized IMI-2 enzyme is codified in a plasmid together with this regulation system.177
Owing to the diversity of class A lactamases, with distant members possessing 30% or lower sequence identity, a standard numbering scheme termed ABL was created to assign common residue numbers to structurally equivalent positions.178 This helps comparing functionally important positions among enzymes in spite of insertions or deletions altering the actual residue numbers in each enzyme. In this review we will use the consensus ABL numbering scheme to refer to the catalytic residues.
SBLs catalyze the hydrolysis of β-lactams via the nucleophilic attack of a serine side chain hydroxyl group on the substrate carbonyl group. The catalytic serine (Ser70 in class A SBLs) is located in a cleft in the interface of the α and α/β halves of the protein and must be deprotonated by a general base prior to the attack.17 Two residues have been suggested for this role: Glu166179, 180 and Lys73181, 182 (the latter present in a conserved SXXK motif together with Ser70). A vast amount of experimental and computational studies have shown that Lys73 is neutral,181, 183–186 thus confirming its role as general base in the activation of the catalytic Ser residue. After the nucleophilic attack, a negatively charged tetrahedral intermediate is formed, which is stabilized by a positively charged cavity termed the oxyanion hole.147 This species leads to the accumulation of an acyl-enzyme intermediate.145–147 After ring opening, the former β-lactam amide nitrogen is protonated by Ser130, and a water molecule activated by Glu166 hydrolyzes the acyl-enzyme complex, releasing the substrate and regenerating the active site. Glu166 is part of the Ω-loop, located at the entrance of the active site that plays a key role in defining substrate selectivity.160 This extended loop is shared with class C and D SBLs.187
Class B
The Ambler class B enzymes (or metallo-β-lactamases) are the main focus of this review article, and their features will be covered in the rest of this article in deeper detail.
Class C
The E. coli chromosomal class C enzyme was the very first β-lactamase to be reported, in 1940 (with the organism originally referred to as Bacillus coli).188 Decades later its coding gene was identified and the enzyme was named AmpC.81, 189 Additional class C enzymes are similarly encoded in the chromosomes of many Enterobacterales, Pseudomonas spp., Acinetobacter spp., and other Gram-negative bacteria.81Although these native lactamases are generically referred to as AmpC, specific names referring to the organism are preferred, such as the ADC (Acinetobacter-derived cephalosporinase), and PDC enzymes from Acinetobacter and Pseudomonas, respectively.16 Plasmid-borne class C enzymes have also been identified, including the FOX, CMY, MIR and DHA families.81, 190 There are chromosomally-encoded variants of these enzymes, representing possible origins for the mobilized forms. A standard residue numbering scheme (termed SANC)191 has recently been proposed for class C proteins.
The catalytic mechanism of class C enzymes is not fully understood. While possessing a similar S64-X-X-K67 catalytic motif, these SBLs lack the conserved Glu166 of class A enzymes. Thus, the identity of the general base for enzyme acylation and deacylation is not clear. A conserved Tyr150 may carry out this role,192 but experimental evidence contradicts its capability to exist in the tyrosinate form.193 Alternatively, Lys67 may act as a general base in acylation,194 as supported by high resolution crystal structures195 by Shoichet and hybrid QM/MM simulations.196 Class A and C β-lactamases have evolved from different peptidase ancestor enzymes,187 and this is reflected in their different mechanisms.
Production of the intrinsic enzymes is typically regulated, and their expression is induced in presence of β-lactam antibiotics, commonly via the AmpR system81 (see Section 7.2). There is however variability in AmpC regulation among different bacterial hosts, and E. coli lacks AmpR and cannot produce its chromosomic enzyme at clinically significant levels in the absence of promoter mutations,197 while P. aeruginosa possesses multiple homologs of the AmpD amidase.198 Either loss of AmpD or mutations on AmpR lead to β-lactamase overexpression or constitutive expression, increasing antibiotic resistance.199
Class C lactamases are mainly cephalosporinases, with hydrolysis rates for some of these substrates high enough for the process to be diffusion-limited.200 They also possess the ability to degrade benzylpenicillin and other penicillins, while the hydrolysis of cephamycins, oxyiminocephalosporins, carbapenems and aztreonam is very slow. However, most of these poor substrates are bound with sub-micromolar affinities,81 which can allow the enzymes to confer reduced susceptibility when overexpressed and in combination with porin deletion and/or drug efflux systems.
Class D
Class D enzymes are known as OXA’s, since they were initially identified by their capacity to hydrolyze oxacillin.201, 202 Although the first discovered enzymes were plasmid-borne narrow-spectrum penicillinases, this group is highly heterogeneous both from the functional and sequence homology point of view. Indeed, oxacillin hydrolysis is not a common feature of all class D enzymes, and the hundreds of currently known OXAs show a large degree of variability in their substrate spectrum. Some of them maintain a narrow spectrum while others possess ESBL characteristics, and some OXA enzymes are clinically relevant carbapenemases. As a result, several “subfamilies” of OXA lactamases are recognized, with particular substrate spectrum and host distribution.201–203 OXA-51 and related enzymes are ubiquitous in Acinetobacter baumannii, where they are chromosomally-encoded. Despite being poor carbapenemases, overexpression of these proteins can confer reduced susceptibility to these drugs. Other OXA families prevalent among Acinetobacter spp. are more efficient carbapenemases, being either native or acquired enzymes.203 These include the families of OXA-23, OXA-24/40, OXA-58 and OXA-143. Class D carbapenemases, mainly from the OXA-48 family, are also a significant cause of carbapenem resistance among Enterobacterales. OXA-48-like enzymes are especially widespread among K. pneumoniae.204 Many of these Carbapenem-hydrolyzing class D β-lactamase (CHDL) families present relatively modest hydrolysis rates for carbapenems, and tend to act in coordination with other resistance mechanisms such as efflux pumps to provide high levels of resistance. Several ESBLs from the OXA group are plasmid-borne enzymes derived from the narrow spectrum OXA-10 by a small number of point mutations, including OXA-11, OXA-13 and OXA-28.203 They provide resistance to P. aeruginosa towards the antipseudomonal cephalosporin ceftazidime and other extended spectrum cephalosporins. OXA-2-like ESBLs such as OXA-15 and OXA-32 have also been identified in this organism, while OXA-21 (from this group) was observed in A. baumannii. Class D lactamases have also been reported in Gram-positive organisms, as intrinsic enzymes in isolates from the Bacillus and Clostridioides genera. Similarly to what was previously mentioned for class A and C enzymes, there is a standard numbering scheme for class D β-lactamases, termed DBL.205
As elegantly demonstrated by the Mobashery group, the catalytic activity of class D enzymes is made possible by an unusual post-translational modification, in which the active-site lysine (equivalent to Lys73 and Lys67 in class A and class C enzymes, respectively) is N-carboxylated.206, 207 This process, facilitated by a highly hydrophobic site that lowers the lysine side chain pKa, allows it to act as a general base during catalysis. The carboxylation reaction is spontaneous and reversible, and thus an adequate pH (>= 7) and levels of bicarbonate in the reaction medium are required to obtain optimal enzyme activity. Intriguingly, Vakulenko et al. have reported that enzymes considered to be narrow spectrum, such as OXA-2 and OXA-10, possess similar carbapenem-hydrolysis parameters in vitro when compared to CHDLs such as OXA-23, OXA-24/40 and OXA-48.208 Furthermore, the former are able to confer clinically significant levels of carbapenem resistance when expressed in A. baumannii, but not in E. coli or P. aeruginosa. Thus, the established functional classifications within class D may need to be re-evaluated, given the large influence of the bacterial host on the observed phenotype, and the lack of adequate supplementation of bicarbonate in the reaction medium being a potential source of contradictions among different publications on the reported kinetic parameters of these enzymes.
3.2. Functional classification
An alternative system of classification of β-lactamases based on their activity profile and susceptibility to inhibitors was proposed by Bush, Jacoby and Medeiros.209, 210 This functional classification denotes three major groups by the numbers 1 to 3, with subdivisions within each group identified by one or more letters (e.g. 2a). The emphasis on the enzyme’s substrate profile and effective inhibitors is intended to aid physicians and clinical microbiologists, informing suitable therapeutic options upon molecular identification the resistance mechanism. In many cases, the groups are defined in terms of relative hydrolytic activity versus key substrates such as benzylpenicillin or early cephalosporins, due to the initial limitations in obtaining pure preparations of the proteins for determination of true catalytic parameters.
In spite of the different criteria used for each scheme, there is a significant alignment of the functional groupings and the Ambler classification. Group 1 corresponds to cephalosporinases that present a preference for cephalosporin substrates over benzylpenicillin, and is composed of all class C enzymes. Meanwhile, group 2 is composed by the Ambler class A and D β-lactamases, and is divided into several subclasses owing to the large heterogeneities in their substrate and inhibition profiles. Of these, class D lactamases exclusively compose subgroup 2d and its subdivisions, while the rest of the subgroups are made up of class A enzymes. Of particular clinical importance are enzymes from groups 2be, containing Extended Spectrum β-Lactamases (ESBLs) such as CTX-M and PER, and group 2f, which includes the class A carbapenemases such as KPC and GES. Group 3 contains the class B enzymes, i.e., metallo-β-lactamases. Two subgroups are defined: 3a contains the wide substrate spectrum MBLs, encompassing Ambler subclasses B1 and B3, while 3b is composed by MBLs which only efficiently degrade carbapenems, corresponding to Ambler subclass B2.
3.3. β-Lactamase databases
The widespread access and decreasing costs of DNA sequencing technologies have allowed β-lactamase detection at the molecular level, with direct identification of the coding gene, to accompany the traditional phenotypic and PCR-based assays used to assess the causes of antibiotic resistance. This has also allowed an increased ability to detect new β-lactamases and novel clinical variants of previously identified ones. Many of the main enzyme families now possess hundreds of known alleles. Therefore, it is crucial to possess adequate tools to access and interpret this growing corpus of biological sequence information. While most of these data are routinely deposited by authors in general databases such as NCBI’s GenBank, there is a need for specialized, curated repositories to guide the work of both clinical and research laboratories.
The main online database serving the β-lactamase community was maintained for many years by Karen Bush, George Jacoby and Timothy Palzkill at the Lahey Clinic website (originally hosted at http://www.lahey.org/Studies/, now at http://externalwebapps.lahey.org/studies/). In addition to tracking known β-lactamase sequences, the database managers assigned the corresponding numbers to newly reported alleles. Starting from 2015, this task has been taken up by NCBI personnel, with allele submission guidelines available at https://www.ncbi.nlm.nih.gov/pathogens/submit-beta-lactamase/. Agreement on a centralized authority to assess reports of new β-lactamases and assign variant numbers is of great importance to avoid the confusions caused by variants with duplicated names or missing sequence information. Additional databases are focused on specific classes and families of enzymes, including LACED211 (http://www.laced.uni-stuttgart.de/) which compiles information class A and B lactamases, and the Institute Pasteur database which contains mainly data from OKP, LEN and OXY lactamases from Klebsiella (https://bigsdb.pasteur.fr/klebsiella/klebsiella.html)
More recently, the Beta Lactamase Data Base (BLDB, http://www.bldb.eu/) was established by Bogdan Iorga, Thierry Nass and collaborators, covering all β-lactamase classes and providing a wider range of information compared to previously developed repositories.212 In addition to references to the corresponding nucleotide and amino acid sequence of each lactamase, the database provides data regarding their origin organism, publication of first description, sub-family (if any), available structures and existence of engineered mutants. Information on the kinetic parameters of antibiotic hydrolysis is also available for a subset of the proteins, as are pre-calculated sequence alignments and phylogenetic trees of each β-lactamase class and enzyme family.
An analysis of the information contained in BLBD reveals the staggering amount of β-lactamases that have been identified to date. As of this writing (January 2021), the site reports a total of 6789 enzymes, with 1589 of them belonging to molecular class A, 710 to class B, 3475 to class C and 1015 class D. However, this tally includes cases such as lactamases which are duplicated due to being assigned more than one name, reported enzymes with missing sequences or allele numbers which were assigned but with no known sequence. By our own analysis of the database entries, the number of unique protein sequences is slightly lower at 6330, of which 1283, 676, 3415 and 956 belong to classes A, B, C and D, respectively.
Lack of standardized naming conventions, such as to what should constitute a new family of β-lactamases, have led to disparities such as OXA-1 and OXA-2 sharing a similar (or even lower) sequence identity to that observed between different MBL families, such as NDM and VIM. The identification with different numbers for alleles separated by as little as a single amino acid substitution has also been a point of contention in comparison to practices in the study of other resistance mechanisms, although it may be justified by massive impact that a few substitutions may have in properties such as the substrate spectrum of a β-lactamase.
Apart from enzymes which are identified by unique names, a cursory search of the NCBI GenBank database using BLAST will reveal numerous homologous proteins for many of them, corresponding generally to sequences obtained from environmental bacteria that were either isolated or observed through metagenomic analyses. This reveals a vast reservoir of additional β-lactamases existing beyond the clinically relevant enzymes of the present, but with the potential to be mobilized from their currently innocuous context into pathogenic bacteria.
4. MBLs: B1, B2, B3 – folding, evolution, superfamily, active sites, phylogeny
4.1. General overview
MBLs are Zn(II)-dependent metalloproteins, which belong to class B in the Ambler molecular classification and group 3 of the functional classification system. MBLs are unique by their unsually broad substrate spectrum, being able to hydrolyze penicillins, cephalosporins and carbapenems and even β-lactam-based SBL inhibitors such as clavulanic acid and sulbactam, i.e., all classes of bicyclic β-lactams.14, 17, 19–21, 23 Only 3% of MBLs (a particular subclass) are exclusive carbapenemases. This makes MBL unique by the fact that all of them can efficiently hydrolyze carbapenems, thus representing a challenge for the clinical use of these antibiotics. Only monobactams are universally spared from hydrolysis by these versatile hydrolases. In contrast to SBLs, there are no clinically useful inhibitors for MBLs yet. As a result, the resistance conferred by these enzymes cannot be countered at this time. However, a varied set of promising compounds which abolish their activity have been identified, some of them currently under clinical trials (see Section 6).
According to the BLDB database,212 710 MBLs have been reported (January 2021), that are classified into three subclasses: B1 (509), B2 (22) and B3 (179), and there are in total 380 structures deposited in the PDB, encompassing native and mutant enzymes, as well as adducts with inhibitors and other ligands such as significant species of the catalytic mechanism.
MBLs consist of a single globular domain with a highly conserved structure despite the low sequence homology among different members of this protein family (Figure 7). Their characteristic αβ/βα tertiary structure is also shared with a set of proteins widely distributed among the three domains of the tree of life, able to catalyze various functions unrelated to antibiotic resistance. These proteins define the so-called MBL superfamily,20, 213–216 a diverse group of proteins, mostly metallohydrolases (see section 4.9). Since the first solved structure of this superfamily was BcII, the metallo-β-lactamase from B. cereus by Carfi, Dideberg and coworkers,217 it is known as the MBL fold. The large similarities in the linear succession of the secondary structure elements between the two αβ halves of these proteins suggest the possibility that they were the result of an ancestral gene duplication event, although no candidate for the initial gene has been identified and there is no sequence homology between the two halves.
Figure 7.
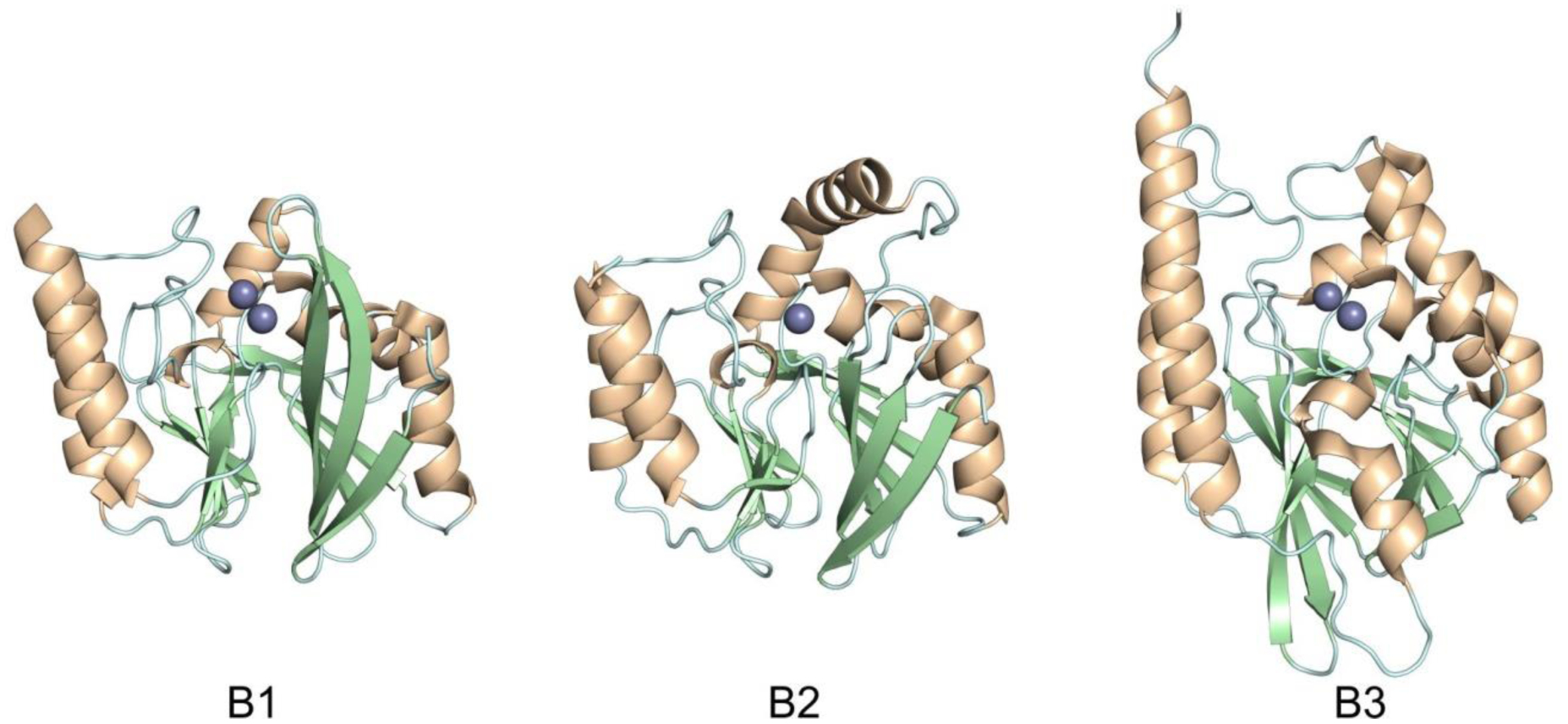
Structures showing the general fold of the B1, B2 and B3 subclasses of MBLs. Enzymes represented are B1: IMP-1 (PDB 1DD6), B2: CphA (PDB 1X8G) and B3: BJP-1 (3LVZ). Zn(II) ions are shown as grey spheres.
The low degree of primary structure conservation within MBLs (with sequence homologies as low as 10% between the most distant enzymes) makes it difficult to compare different proteins (Figure 9). This led to the need of a standard numbering scheme for these enzymes, called BBL numbering (Figure 6). This consensus numbering was obtained from a structural alignment of a set of representative MBLs.218, 219 It allows the identification of structurally conserved residues by assigning them a common residue number, regardless of their actual position in each MBL sequence, and is usually employed when discussing the structural features of these enzymes. There are still papers being published nowadays that do not follow the BBL numbering, generating confusion in the literature. This section, and all the current review, uses the most recent BBL numbering scheme219 to indicate residue positions within MBLs. It should be noted that there are certain discrepancies in the numbering between the first published report218 and the most recent description of the BBL scheme, and that some residues are still usually described in the literature using the numbers assigned in the former. To avoid confusion, the older numbering will also be indicated when necessary. Nevertheless, given that a large number of new MBLs have been discovered since the latest update of the BBL scheme in 2004, it imperious to revise this consensus numbering and foment its use in future publications.
Figure 9.
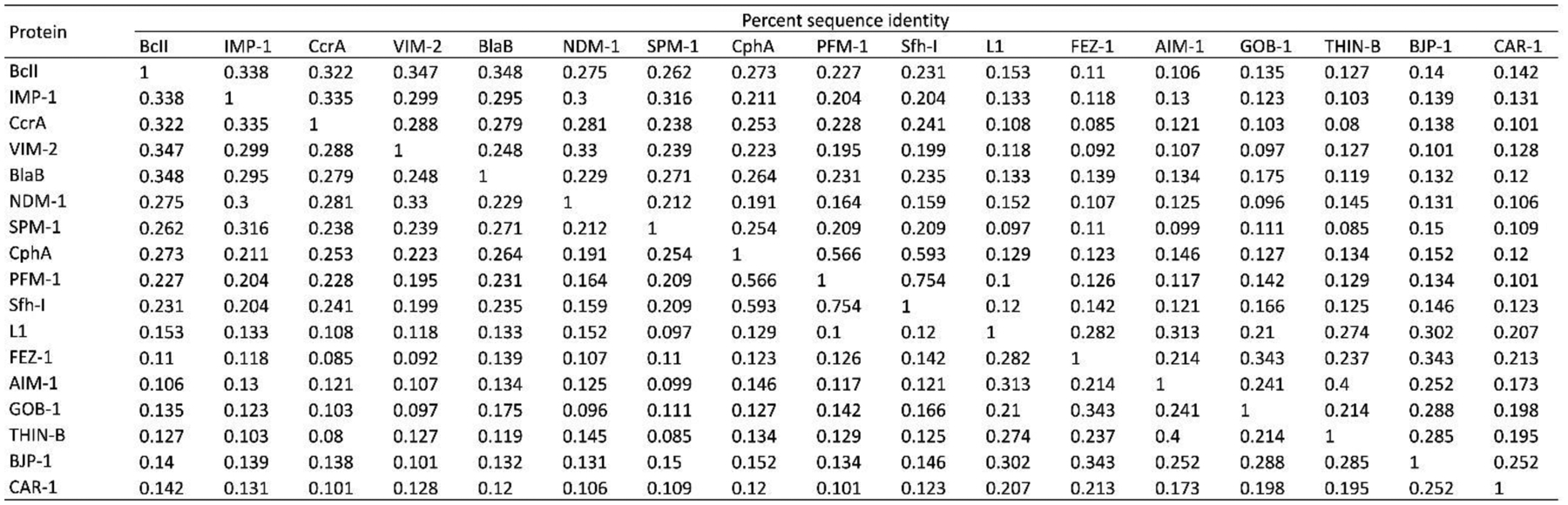
Percent sequence identity matrix for a representative set of B1, B2 and B3 MBLs. Sequences (without leader peptides) were aligned using PSI-Coffee, and the resulting alignment was manually edited in Jalview to match as closely as possible the latest published BBL alignment.219 The percent sequence identity among each pair of MBLs was calculated by dividing the number of matching residues by the average of the length of the two proteins.
Figure 6.
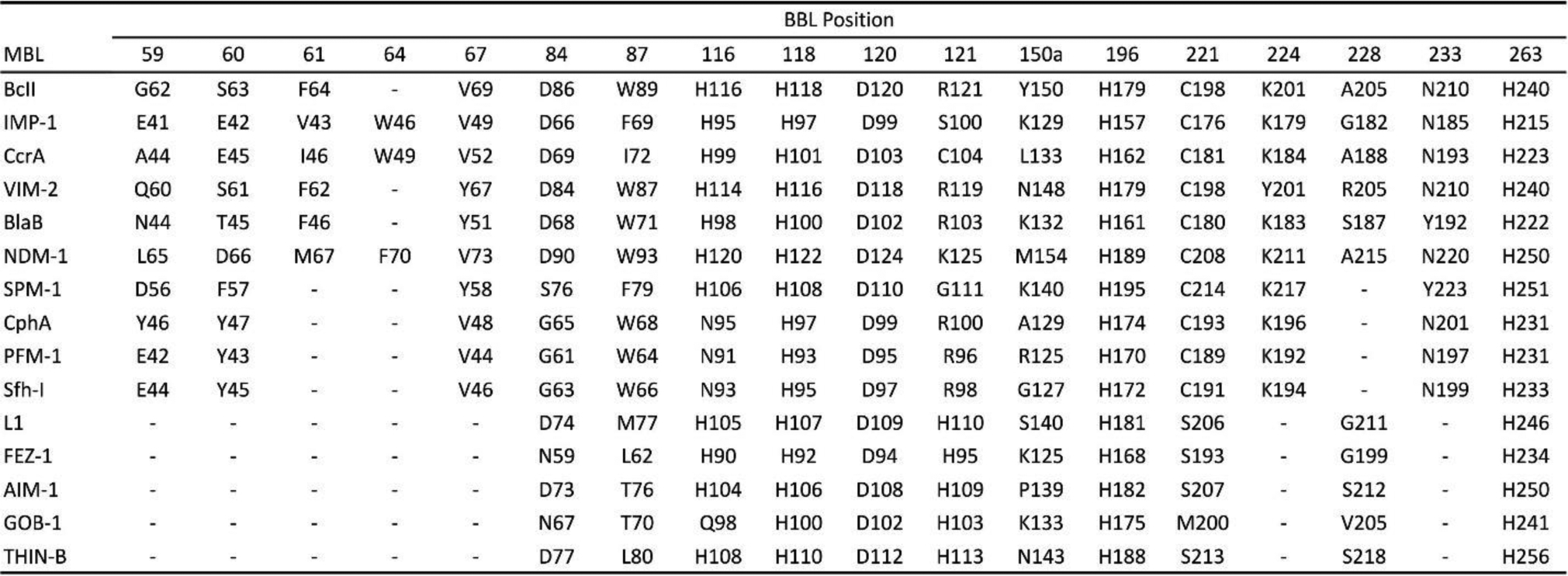
Correlation between consensus BBL numbering positions218, 219 and actual residue numbers in various MBLs.
The active site of MBLs is placed between two β-sheets that define the boundaries of the two halves of the protein (Figure 7). One or two zinc ions, responsible for substrate binding and catalysis, are coordinated in this position by a set of ligands located in different loops that define the active site. All MBLs contain a characteristic H/N116-X117-H118-X119-D120-H/X121 metal binding motif in the N-terminal half of the protein that provides three or four zinc ligands, and is conserved also in members of the MBL superfamily. Other two loops located in the C-terminal half provide two extra metal ligands. Differences among MBLs have led to the definition of three MBL subclasses: B1, B2 and B3 (Figure 7).220 This classification is primarily based on sequence homology, but each subclass is also characterized by a conserved set of metal ligands and occupancies of the two metal binding sites, as well as a defined substrate spectrum. A recent phylogenetic analysis by Kristiansson and co-workers proposed the further subdivision of subclass B1 and subclass B3 MBLs into a set of generally monophyletic groups, which will be discussed in Section 9.221
Evolutionarily, B3 enzymes form a separate clade from B1 and B2 MBLs, and are more closely related to members of the MBL superfamily devoid of lactamase activity than to B1 and B2 MBLs.222 Hall and Barlow have concluded that the β-lactamase activity has evolved independently within each subclass223 (see Section 4.8). Based on this divergence, Hall, Bush and Frère had a debate about whether the Ambler classification of MBLs should be reconsidered or not.224, 225 Here we will present general features distinguishing B1, B2 and B3 enzymes, and particular details will be discussed in more detail in Section 4.7.
Zn(II) ions are essential for the ability of MBLs to confer resistance, being responsible of substrate binding and catalysis. B1 MBLs possess two metal binding sites, known as the Zn1 and Zn2 sites. The Zn1 ion is coordinated to residues His116, His118 and His196 (also known as 3H site), while Zn2 is bound to Asp120, Cys221, His263 (DCH site) and a water molecule (Figure 8). A bridging hydroxide completes the coordination sphere of both sites. As a result, the Zn1 site adopts a tetrahedral coordination geometry, while the Zn2 is trigonal bipiramidal. Some crystal structures have revealed the presence of additional water ligands in either site, but this represents the canonical binuclear site of B1 MBLs. The Zn-Zn distance ranges from 3.5 to 3.9 Å in the resting state.226–229 This ligand set is highly conserved within B1 enzymes (with only one exception, the enzyme SPS-1230), and mutation of the coordinating residues generally leads to variants in which in general both the catalytic parameters and the zinc binding features are impaired.231–237 The assignment of a hydroxide character to the bridging ligand is derived from the short Zn1-OH bond length (1.9 Å)226 and the lack of an acid pKa influencing the pH dependence of substrate hydrolysis238 (see discussion in Section 5). Instead, the Zn2-OH distance is 2.2 – 2.5 Å, revealing a stronger interaction of this ligand with the Zn1 site (Figure 8). The essentiality and functional roles of the two metal binding sites have been matter of intense debate in the literature. The consensus picture is that Zn1 positions the nucleophilic hydroxide while Zn2 acts in substrate binding by coordinating the C3/C4 carboxylate present in the non-β-lactam ring of the antibiotic, and stabilizing an anionic intermediate.239–242 Zn1 has been suggested to polarize the β-lactam carbonyl, but this role has also been attributed to active-site residues such as Asn233.226, 227, 243, 244 These aspects will be discussed in detail in Section 5. The active site in B1 MBLs is a shallow groove flanked by two loops, generally termed L3 and L10 (corresponding to BBL positions ca. 59–67 and 223–241, respectively). As a result, B1 enzymes can accommodate and hydrolyze penicillins, cephalosporins and carbapenems. Subclass B1 encompasses most clinically relevant MBLs, including the widely disseminated plasmid-borne enzymes of the NDM, VIM and IMP families. The enzyme SPS-1 from Sediminispirochaeta smaragdinae is a notable exception within B1 enzymes, since His116 is replaced by a glycine residue and the resulting vacant coordination position of the Zn1 site is occupied by a water molecule, demonstrating a degree of plasticity in the metal ligand site (Figure 8). SPS-1 can hydrolyze carbapenems and certain cephalosporins but not penicillins.230
Figure 8.
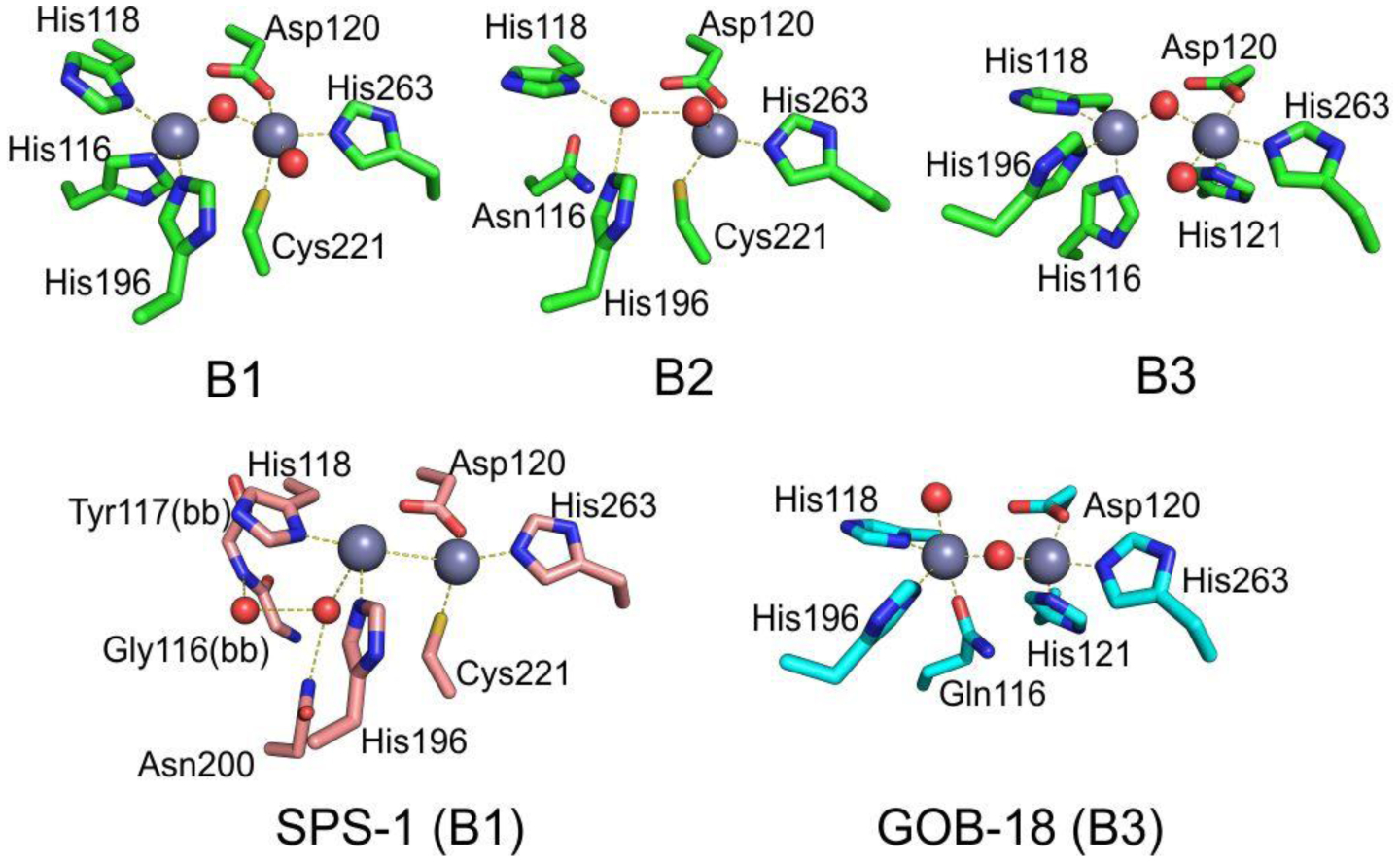
Typical active-site metal coordination geometry for B1, B2, and B3 MBLs (Top). The non-standard Zn(II) coordination spheres for the enzymes SPS-1 (B1) and GOB-18 (B3) are displayed (Bottom). Zn(II) ions and water molecules / hydroxide ions are displayed as grey and red spheres, respectively. For SPS-1, Gly116(bb) and Tyr117(bb) indicate that only the backbone atoms for these residues are represented, while only side chains are represented for all other residues. In images showing binuclear enzymes, Zn1 is displayed at the left, and Zn2 is displayed at the right. The PDB codes for the structures used are: 5N5G (B1 – VIM-1), 3SD9 (B2 – Sfh-I), 3LVZ (B3 – BJP-1), 6CQS (SPS-1), 5K0W (GOB-18).
Proteins belonging to the B2 subclass are evolutionarily related to B1 enzymes, but possess key differences in metal coordination and the active-site topology. Crystal structures are available for only two B2 enzymes: CphA245 and Sfh-I,246 the latter being the only one corresponding to the resting state form of a native B2 MBL. These MBLs are monometallic, with only the Zn2 position (at the DCH site) occupied in the catalytically active form (Figure 8). This is due to a reduced metal affinity in the Zn1 site caused by the substitution of His116 by an asparagine residue. Metal binding to the Zn1 site inhibits the hydrolytic capabilities of these enzymes, confirming that they are active as the mononuclear species.247, 248 The metal site in the resting state form is tetrahedral, with an apical water ligand completing the coordination geometry at 2.24 Ǻ from the Zn(II) ion, suggesting that this ligand might not be the attacking nucleophile (see below). B2 MBLs are exclusive carbapenemases, while hydrolysis of other beta-lactam substrates is undetectable or irrelevant to elicit resistance when expressed in bacteria. This substrate selectivity has been attributed to a narrower active site compared to B1 and B3 MBLs, despite second sphere residues also play a role in shaping the substrate profile of B2 enzymes (see Section 4.7.2). Instead of being flanked by two loops, the entrance of the active site is defined by an elongated and kinked α3 helix that allows that restricts substrate access.245, 249 Overall, the active site is deeper and narrower than in B1 and B3 enzymes.245, 246
MBLs from subclass B3 are broad spectrum and have two metal binding sites, like B1 MBLs. The structure of L1 from S. maltophilia was the first B3 structure available, and still represents the model for B3 enzymes.250 While Zn1 ligands are similar to those in B1 enzymes (3H site), there is no Cys ligand at the Zn2 site (in contrast to B1 and B2 MBLs). Cys221 is substituted by a residue which does not participate in metal binding, and which varies among different B3 enzymes. Instead, residue His121 (present in the metal binding domain and next to the conserved D120) coordinates this metal ion, leading to a modified ligand geometry for the Zn2 site (DHH site), which is rotated ca. 80° with respect to the one in B1 sites250 (Figure 8). There are some exceptions to this ligand configuration, as GOB enzymes possess a glutamine in position 116, and have been shown to be active both in bimetallic forms and as mono-zinc enzymes, with only the Zn2 site occupied, similarly to B2 MBLs.251–254 The active site in B3 enzymes is also flanked by two loops that are more extended than those from B1 lactamases (Figure 7). The B3 enzyme L1 is a tetramer,255, 256 an exception since all other MBLs are monomeric proteins. In the case of L1, mutations that prevent formation of the tetrameric form lead to impaired catalytic activity.257
4.2. Zn(II) binding: Does it take two to tango?
Abraham and Newton reported in 1956 an unusual hydrolytic activity of Bacillus cereus 569/H cultures against cephalosporin C.11 In 1962 they informed that the penicillinase activity fell rapidly to 15 % of its original value after incubation at 60 °C, while 90 % of the cephalosporinase activity was retained, concluding that “culture fluids of strains of B. cereus contain a cephalosporinase which may be a β-lactamase”.12 Protein preparations showed a loss of cephalosporinase activity, while retaining part of the penicillinase activity. Based on this, Sabath and Abraham concluded in 1966 that this new cephalosporinase required a cofactor that was lost during the purification.13 Upon identifying the inactivation of this activity by addition of EDTA, they showed that this enzyme was a Zn(II)-dependent cephalosporinase. The characterization of this enzyme as a broad spectrum lactamase led to the name β-lactamase II,262 as the organism also produced two additional lactamases: β-lactamase I and β-lactamase III (both SBLs). This enzyme, then renamed as BcII, is an extracellular protein, as is β-lactamase I. Another highly similar MBL was reported in strain 5/B/6 of this organism.263 Since this host is not a clinical pathogen, BcII was regarded as a biochemical curiosity without clinical impact for almost three decades. These studies were fundational to the field and were exploited when MBLs became a clinical threat. The carbapenemase activity of MBLs was recognized much later, since thienamycin (the first known carbapenem) was discovered in 1976.44
In 1970, Kuwabara purified and obtained obtained the first crystals of BcII.264 Pioneering biochemical and spectroscopic studies allowed Davies and Abraham in 1974 to identify the presence of two metal binding sites, and a Cys as metal ligand in a must-read paper for the field.265 Metal substitution experiments were crucial to obtain the first glympses into the enzyme active site. They also observed that metal binding to the apo (metal-depleted) enzyme elicited conformational changes. Later, a series of NMR experiments revealed that 3 His residues were involved in one metal binding site, and a fourth His could bind the zinc at the second site.266–269 These studies paved the way for a series of elegant pioneering mechanistic studies by Stephen G. Waley and coworkers in Oxford in the mid-1980s that set the bases for the elucidation of the chemical mechanism of MBLs that was finally defined along the next 30 years.270, 271 There were many controversies on the mechanism that will be discussed in Section 5.
A big step ahead in the study of MBLs was the simultaneous report of the amino acid272 and gene273 sequences of BcII, that confirmed the lack of evolutionary link with SBLs. Expression of BcII (an extracellular protein) in E. coli resulted in an efficient processing of the signal peptide and secretion as a single product retained into the periplasm.273 Thus, despite its origin, BcII has been frequently expressed in Gram negative bacteria as a control for MBL-mediated resistance, and even for directed evolution studies in E.coli.274 Gene cloning enabled the first mutagenesis studies of BcII, that identified His118275 and Asp120276 as critical for function. This evidence led to the initial suggestion of Asp120 as a possible general base for catalysis.
The first crystal structure of BcII was obtained in 1987 for the Cd(II) derivative at 3.5 Å.277 This resolution did not allow assessing in detail the metal coordination environment, but it suggested the possible binding of a second metal-ion equivalent and the involvement of Asp120 as a metal ligand. A new era in the study of MBLs started in 1995 when Carfi, Dideberg and coworkers reported the structure of BcII at 2.5 Å with a single Zn(II) in the active site,217 bound to three His residues and a solvent molecule with a tetrahedral geometry, i.e., resembling the active site of carbonic anhydrase. The subsequent crystal structure of the Bacteroides fragilis enzyme CcrA/CfiA reported in 1996 by Concha, Herzberg and coworkers showed for the first time the binuclear Zn(II) site of B1 MBLs.227, 228 In 1998, two crystal structures of BcII soaked in a buffer with higher Zn(II) concentration by the Sutton and Dideberg groups226, 278 and a spectroscopic study from the Vila lab279 confirmed that BcII could host a binuclear site identical to the one reported for CcrA. The same year, Spencer and coworkers informed the structure of the tetrameric B3 enzyme L1 from S. maltophilia.250 The first structure of a B2 lactamase (CphA from Aeromonas hydrophila) with a single Zn(II) ion was reported in 2005 by Garau, Dideberg and coworkers,245 although not in the free form. Indeed, the first structure of the mono-Zn(II) site of a B2 enzyme in the unbound form was available in 2011 for Sfh-I from S. fonticola, which was solved by the Spencer lab.246
The discussion of the metal-ion stoichiometry required for catalysis in B1 and B3 enzymes has been a central issue in the field of MBL structure-function analysis (see also Section 5). In the case of B1 enzymes, most crystal structures of the mono-Zn(II) variants show the metal ion localized at the 3H site, and Cys221 is oxidized to different degrees: sulfinic acid in BcII,280 sulfenic acid in SPM-1281 and sulfonic acid in VIM-2.282 Different authors280, 281 have concluded that Cys oxidation is the consequence of dissociation of Zn2. This suggests that Zn2 is more labile to dissociation than Zn1, but this is contradictory with the affinity constants (see below). The absence of Zn2 and Cys oxidation disrupts the active-site hydrogen bond network and elicits changes in the position of the putative Zn2 ligands. The structure of mono-Zn(II) NDM-1,283 shows a Zn1 ion with large B factors bound to His116 and His118, but beyond binding distance to the third His ligand of the 3H site (3.2 Å), and lacking a bound water. This raises the question whether this site can effectively bind and hydrolyze substrates. The structure of mono-Zn(II) L1,260 instead shows that the absence of the Zn2 ion does not elicit any distortion in the metal site. However, even the structure of apo-L1 shows an ordered active site, with the metal ligands adopting almost the same position than in the bi-Zn(II) enzyme,260, 284 which is not the case for apo-BcII,278 showing a disorded active site. In the case of B1 enzymes it seems unlikely that these structures can represent an active species, based on the disorder elicited by the absence of the Zn2 that removes some structural features essential for substrate binding (Section 4.4) and catalysis (Section 5).
In solution, there is a completely different scenario, since there is contradictory evidence regarding the metal binding affinities and the cooperativity of binding of the two metal-ion equivalents. As a result, different laboratories have conflicting evidence regarding the possibility of having in solution mono-Zn(II) species with the metal ion localized in only one site. Addition of substoichiometric Zn(II), Cd(II) or Co(II) to BcII do not lead to sequential binding of both sites, and different spectroscopies have revealed a mixture of population of mononuclear and binuclear sites, some of them in fast equilibrium.285–290
The Crowder group has reported the sequential binding of Zn(II) to several MBLs monitored by EXAFS spectroscopy, linked to the report of activity for the mono-Zn(II) variants of Bla2,291 L1292 and NDM-1293 with the metal ion localized at the 3H site. These reports are consistent with the crystal structures of mono-Zn(II) L1 and NDM-1. However, the zinc binding constants for the first and second equivalent to L1 are very similar and would make it not possible to isolate the monometallated variant (2.6 and 5.7 nM).290, 294 Metal binding experiments suggest a different view, since various studies have indicated the presence of positive cooperativity for Zn(II) binding in enzymes such as BcII,285–287, 295–297 CcrA298 and IMP-1,299 which would lead to formation of binuclear species even under substoichiometric metal concentrations. Wommer and coworkers also reported that substrate binding results in a significant increase in the Zn(II) binding affinity in several MBLs,294 a finding that is consistent with slower koff values when external ligands are bound to the enzymes.
In spite of contradictory in vitro evidence and the presence of activity in mono-metallic forms of various enzymes, there is a general consensus supporting that the physiologically relevant forms of the B1 and B3 enzymes are bimetallic.300 Inhibitor design to B1 and B3 enzymes has been focused to inhibit the binuclear species. In the case of BcII, it was shown that impairing the binding affinity of the Zn2 site resulted in low resistance levels to antibiotics unless there is excess Zn(II) in the external milieu,300 indicating that the active species in the periplasm is the binuclear form.
4.3. MBLs have unique metal sites
MBL sites present some unique coordination chemistry features. The Zn1 site in B1 and B3 enzymes resembles the one of carbonic anhydrase, with a tetrahedral His3Wat ligand set.301 However, there are some differences. First, the pKa of the Zn(II)-bound water is much lower in MBLs than in carbonic anhydrase.302, 303 Second, two His ligands are in a His-X-His sequence in both enzymes, but these His are bound to the zinc through their Nε2 in carbonic anhydrase, while in MBLs, the coordinating N atoms are Nε2 (H116) and Nδ1 (H118). This is unusual, since Nδ1 ligation entails more steric requirements than Nε2 ligation, and Nδ1 coordination is generally favored for His ligands distant in the protein sequence.304
The binuclear site is also unusual in featuring Zn1-Zn2 distances (3.5 to 3.9 Å) longer than in most binuclear zinc hydrolases and by the lack of a protein ligand bridging the two metal ions. Metallohydrolases usually feature one or two carboxylate residues binding the two metal ions in a μ-fashion.305, 306 In principle, this configuration would enable a larger flexibility to the MBL sites, a fact that is compatible with their broad spectrum substrates, that requires the accommodation of ligands with different geometries. This hypotheses has been substantiated by the observation of even longer Zn-Zn distances during turnover (up to 4.6 Å).307
The presence of a cysteine residue in a catalytic zinc site (in B1 and B2 MBLs) is rare308, 309 and especially so in oxidizing environments such as the periplasm and extracellular medium under aerobic growth. Analysis of the BcII C221D mutant, in which this residue was replaced by the conserved aspartate present in superfamily enzymes, demonstrated that the presence of cysteine in this position significantly boosts the affinity towards Zn(II).300 While the mutant enzyme showed similar catalytic efficiency in vitro under Zn(II)-replete conditions, its capability to confer resistance without metal-ion supplementation was highly impaired. Similarly, IMP-1 mutant C221D possesses reduced Zn(II) binding affinity, but its activity was restored by addition of excess Zn(II) to the assay media.233, 310 Thus, the unusual presence of this residue as a metal ligand is an adaptation that helps MBLs constitute their active species under conditions of low Zn(II) availability, such as those that would be experienced by bacterial pathogens in infection sites (see Section 10).311, 312
In general, substitutions in any of the metal ligands leads to both reduced Zn(II) binding and impaired catalytic efficiency, as has been observed for replacement of the Cys or His residues.231–237, 313, 314 However, the most drastic effects in activity have been observed upon mutagenesis studies of Asp120, present in all MBL subclasses.231, 276, 315–318
4.4. Substrate binding to MBLs
Trapping substrate binding to enzymes is usually challenging. In the case of MBLs, countless undocumented efforts have been devoted to trap an intact substrate molecule in the active site of these enzymes to obtain a structural description of the Michaelis complex (ES) of a β-lactam bound to an MBL (a model of an intact cephalosporin substrate bound to NDM-1 is displayed in Figure 10-a). Different strategies have been attempted using poorly active metal derivatives, mutants of essential residues (mostly, Asp120) combined to substrates hydrolyzed with low kcat and low KM values. There is a report of faropenem bound to Cd(II)-NDM-1,319 but a closer analysis320 led to the conclusion that the refined electron density could not be described by an intact substrate molecule. The closest experimental structural picture to an ES complex is the adduct of a cyclobutanone synthesized by Dmitrienko complexed to SPM-1 reported by the Spencer and Schofield groups.243 The structure shows the hydrated form of the cyclobutanone in the active site, with a tetrahedral C6 with two oxygen atoms (Figure 10-b), mimicking the transition state generated upon the nucleophilic attack. These oxygen atoms are not bound to Zn1, suggesting that this species was formed in solution and not within the active site. The carboxylate interacts with Zn2 and Lys224. This is the most common pattern of substrate binding, but others will be analyzed when discussing the features of each protein family (Section 4.7).
Figure 10.
a) Structural model of non-hydrolyzed cephalexin bound to NDM-1. Structural model kindly provided by Dr. Nisanth N. Nair.321 b) Structure of cyclobutanone inhibitor bound to SPM-1243 (PDB 5NDB). Zn(II) ions are shown as grey spheres and water molecules / hydroxide ions as red spheres, while bound compounds are shown as colored sticks and protein residues as white sticks (Zn(II) ligands are displayed as thinner sticks). Interactions between the compound and the protein residues and Zn(II) ions are indicated with yellow dashed lines, while coordination interactions of Zn(II) ions by protein residues are shown as green dashed lines.
Substrate binding has been monitored by stopped-flow tryptophan fluorescence under pseudo-first order conditions, revealing conformational changes in the active site coupled to substrate binding.322 This strategy has been employed to study substrate binding to L1,322 BcII239 and ImiS,323 enabling measurement of the binding constant KS, and individual kinetic constants. However, substrate binding in the literature is mostly analyzed by comparing the Michaelis constant, KM, that includes other parameters. In the case of penicillin and cephalosporin binding to L1 and BcII, the main complex that accumulates retains the intact amide bond (except for the particular case of nitrocefin, discussed below). This approach also allowed assessing the movement of a loop flanking the active site in L1 is kinetically linked to the formation of a reaction intermediate.324 Imipenem binding to BcII experiences a conformational rearrangement during turnover, with the accumulation of two intermediates (Figure 11).239 The time course of carbapenem binding to the B2 enzyme ImiS also showed two phases, revealing that this particular behavior of carbapenems in the active site during turnover is not exclusive of B1 enzymes.323 This can be related to the finding of two intermediate species in the hydrolysis of carbapanems by MBLs.241
Figure 11.
Binding of imipenem (solid lines) or cefotaxime (dashed lines) to apo-BcII (gray lines) and to Zn(II)-BcII (black lines) followed by Trp fluorescence. The absence of changes in the fluorescence of apo-BcII upon substrate addition reveals the lack of binding to this variant. Reproduced with permission from reference239 Rasia et al. Copyright 2004 American Society for Biochemistry and Molecular Biology.
Fluorescence experiments also demonstrated that apo-BcII (demetallated) is not able to bind β-lactam substrates (Figure 11). The X-ray structures of apo-MBLs show that the active site becomes disordered when the metal ions are removed, but the enzyme is folded and stable. Therefore, substrate binding in B1 MBLs is driven by electrostatic interactions with the metal ions. A crystal structure of apo-L1 virtually identical to that of the bi-Zn(II) species was not able to bind any ligand in cristallo, extending these results to B3 lactamases.284 This is unusual, since the metal ions in zinc enzymes are mainly responsible of the chemistry while substrate recognition depends on the presence of specific patches in the active site: for example, apo-carboxypeptidase A is able to bind peptide substrates through hydrophobic interactions.325 These results are in line with the lack of an optimized substrate recognition patch and the shape of the active site of broad spectrum B1 and B3 MBLs.
Substrate binding is steered by the electrostatic interaction of the carboxylate group of the β-lactam with the metal site. Docking experiments,227, 326, 327 as well as the different crystal structures of products and intermediates229, 328, 329 reveal that binding of the invariant carboxylate group (at C3 in penicillins and carbapenems and C4 in cephalosporins) to Zn2 orients the β-lactam ring close to the nucleophilic hydroxide (Figure 10). Other conserved active-site residues also contribute to substrate binding. Lys224, present in most B1 and B2 MBLs, establishes H-bond interactions with the carboxylate group of the substrate, and a similar role is fulfilled by Arg228 in many VIM family enzymes, which lack this residue (see Section 4.7.1.2). In the B3 enzyme L1, there are no positively charged residues in this position, and the carboxylate interacts with S221 and S225 (223 in the older BBL numbering).250, 330 Meanwhile, Asn233 (B1 and B2) or Tyr229 (B3) contact the β-lactam carbonyl, at least in some cases, and may assist in its polarization prior to the nucleophilic attack. Both of these positions are situated in the active-site loop L10, which constitutes one of the walls of the active site. These minimalistic binding features enable the active site of broad spectrum MBLs to bind and hydrolyze efficiently a wide variety of bicyclic compounds, some of them decorated with bulky substituents. Despite the metal ions are essential for substrate binding, the active-site residues present in positions 224, 228, 221 and 225 play a key role in adequately steering the carboxylate group. Indeed, as elegantly shown by Meyer and coworkers,331 different β-lactam substrates bind a bi-Zn(II) model complex unproductively, with the carboxylate making a μ-η1:η1 bridging interaction with the two Zn(II) ions (Figure 12). This result highlights the relevance of the active-site architecture for the adequate positioning of the substrate.
Figure 12.
Crystal structure of the adduct of a bi-Zn(II) model complex synthesized by Meyer and coworkers with sulbactam. The oxygen atoms of the carboxylate from sulbactam (O1 and O2) bind the binuclear metal center. Reproduced with permission from reference331 Meyer et al. Copyright 2012 American Chemical Society.
Loop L3 in B1 enzymes and an analogous loop in B3 MBLs conform the other wall flanking the active site, and these loops are also crucial for substrate binding (Figure 14). The L3 loop extends between β-strands 3 and 4, and spans residues ca. 59 to 67 in BBL numbering, while its functional equivalent in B3 enzymes (loop α3-β7) spans BBL residues ca. 148 to 164. The loop acts as a flexible flap, allowing it to close over substrates and inhibitors upon their binding to the Zn(II) ions. Motions and conformational changes of this segment have been experimentally correlated with enzyme catalysis, and various positions along it establish hydrophobic contacts with the substrate. A conserved hydrophobic amino acid (mostly aromatic) is usually found at the tip of the loop in position 64, being Trp for CcrA and IMP-1, Phe for NDM-1, although it is Ala and amenable to substitution without great impact in antibiotic hydrolysis in VIM-2.332 Residues 61 and 67 at the base of the loop, together with the nearby residue at position 87 (generally Trp or Phe), form a hydrophobic patch that interacts with substrate R1 substituents. Mutagenesis experiments have shown that substitution or deletion of amino acids in this loop can impact catalytic parameters or influence the substrate preference of the enzyme.232, 333–336 Furthermore, engineering the L3 loop of VIM-2 or IMP-1 in the scaffold of NDM-1 resulted in changes in the accumulation of a reaction intermediate in the catalytic mechanism evidenced by pre-steady-state kinetics,337 as was previously shown for mutations in this region within BcII and IMP-1.333 More detailed experiments are discussed in Section 4.7.
Figure 14.
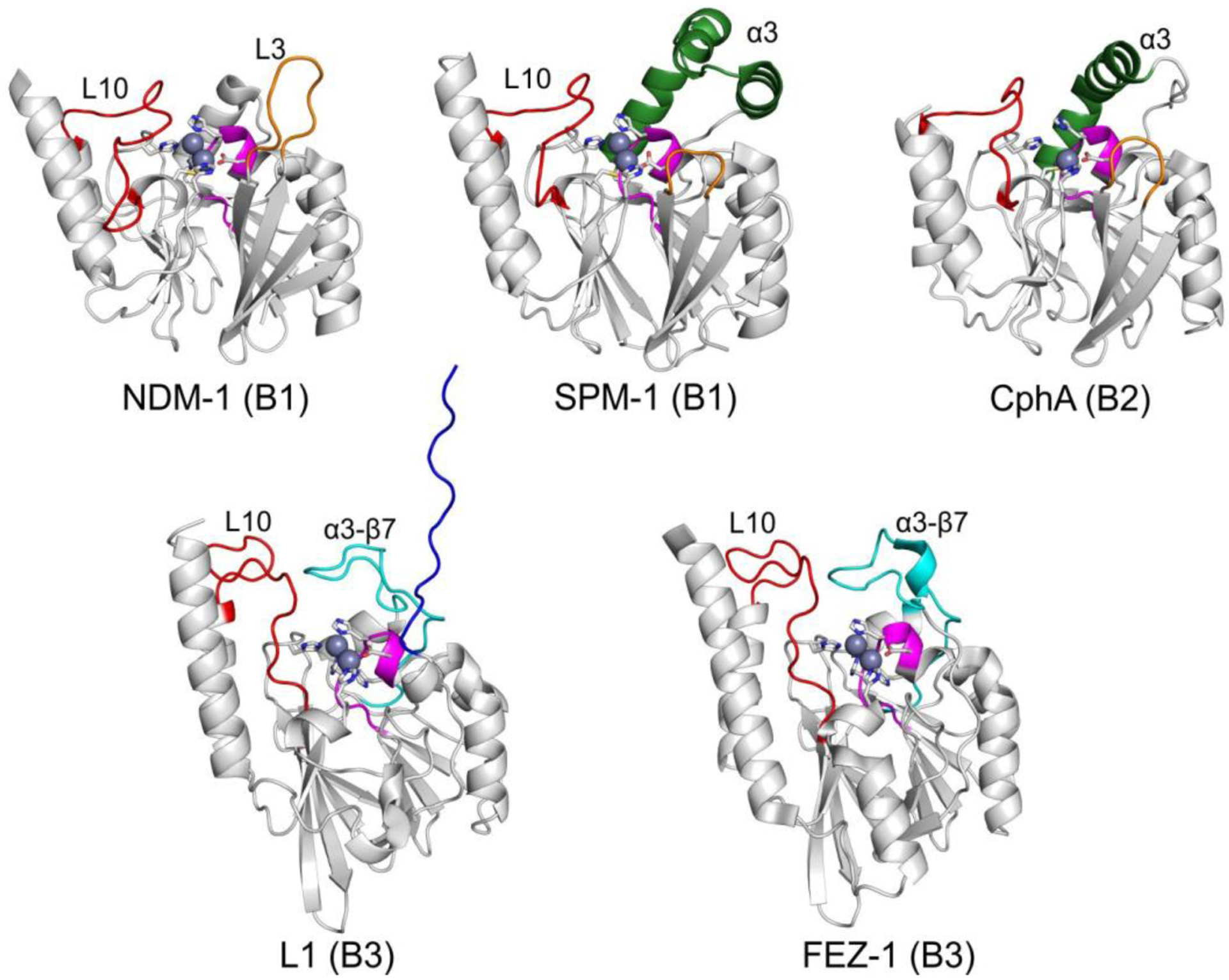
Structures of MBLs from each subclass highlighting key structural features. Zn(II) ions are shown as grey spheres and the metal ligands as white sticks. The L3 loop (and its shorter counterpart in B2 enzymes) is shown in orange, while the α3-β7 loop in B3 enzymes, the L7 loop and the L10 loop are shown in cyan, magenta and red, respectively. The extended α3 helix present in SPM and B2 enzymes is displayed in dark green, and the extended N-terminus in the L1 enzyme is highlighted in blue. PDB codes for the structures: NDM-1 – 4EXY, SPM-1 – 4BP0, CphA – 1X8G, L1 – 1SML, FEZ-1 – 1K07.
Despite this ability to bind and hydrolyze many substrates, it is surprising to note that the simpler, smaller monobactams are hydrolyzed by MBLs with very low turnover numbers and thus, MBLs are unable to confer resistance to monobactams in bacteria. The best reported performance of an MBL towards aztreonam is that of NDM-1 (kcat ≈ 0.014 s−1, KM of 9 mM).338 While all bicyclic β-lactams possess a carboxylate group in the C3/C4 position which binds to the Zn2 site, monobactams have a sulfonate group (Figure 1). Aztreonam binding to BcII339 and NDM-1338 is weak and, according to NMR experiments, it binds Zn2 through the sulfonate group in a non-productive fashion, since the β-lactam ring is located far from the nucleophile (Figure 13).339 Computational studies suggest an alternative non-productive binding of aztreonam to NDM-1 in which the sulfonate moiety replaces the bridging hydroxide between the two Zn(II) ions.340 A minimalistic 2-azetidinone with a carboxylate group was also refractory to hydrolysis by BcII. We conclude that the bicyclic scaffolds of penicillins, cephalosporins and carbapenem are required for productive binding to all MBLs. It has been speculated that the non-null activity of NDM-1 toward aztreonam could evolve towards more efficient variants,338 as observed for TEM-1. We speculate that in the case of broad spectrum B1 and B3, a productive binding of a mononuclear β-lactam may require an active-site restriction that would result in an activity tradeoff compromising the catalytic efficiency toward bicyclic substrates. In this sense, the combined use of aztreonam with other β-lactams may be successful for the treatment of infectious caused by MBL producers.
Figure 13.
Structural model of aztreonam binding to the active site of BcII. Structural model based on NMR data from Poeylaut-Palena et al.339.
Finally, Schofield, Brem and coworkers also reported the hydrolytic activity of several MBLs against the DBO inhibitor avibactam.338 However, as the authors themselves indicate, the reported catalytic parameters are imprecise due to the low turnover numbers. No data about how this drug binds MBLs are available. The clinical impact of these findings, particularly in future evolutionary events, remains to be addressed.
4.5. Active-site Flexibility
Substrate binding to MBL active sites occurs primarily through coordination of conserved elements of β-lactam antibiotics to the Zn(II) ions. This alteration of the metal coordination sphere is reflected in an increased Zn-Zn distance in binuclear MBLs during catalysis and when bound to products or inhibitors, as observed in crystal structures and by spectroscopic techniques such as EXAFS (see Section 5). This rearrangement is enabled by the lack of a residue bridging both zinc ions and is coupled to the flexibility of the loops that provide the metal ligands. Residues belonging to the L7 loop (Figure 14), containing the main metal binding motif H116XH118XD120, were observed to present alternate conformations in a series of BcII structures with varying metal content and oxidation state of Cys221.341 B1 MBLs possess two near-universally conserved Gly residues preceding the Zn2 ligand Cys221, originating the subclass-characteristic motif G219G220C221 (the only known exception so far being the recently discovered environmental MBL ANA-1, with Ala219). These two contiguous glycine residues endow Cys221 with a considerable flexibility, and allow B1 enzymes to accommodate disparate substrates for hydrolysis. In this regard, Cys221 was observed to present dual conformations in structures of BcII, possibly reflecting a displacement of the ligand when the Zn2 site is vacant given its partial metal occupancy.341 Meanwhile, B2 MBLs possess a corresponding G219N220C221 motif, which would restrict the conformational flexibility of Cys221 and may partially account for the carbapenemase-only nature of these β-lactamases. Indeed, engineering a B1 motif in B2 enzyme CphA via the N220G mutation (in combination with the N116H substitution) resulted in an expanded substrate spectrum, supporting this hypothesis.342
The flexibility of active-site loops helps in accommodating the diverse chemical structures present in β-lactam substrate side chains, contributing to their broad substrate spectrum. The L3 loop in B1 MBLs (Figure 14) has been characterized in numerous studies to undergo conformational changes upon binding of substrates and inhibitors, as it would act as a flap closing over the active site and contacting the substrate substituents. The flexibility of this region is revealed by the either high B factors or lack of electron density observed for it in crystal structures. NMR studies have characterized flexibility in the pico- to nanosecond timescale in the loop that are quenched upon binding of ligands to the active site,343, 344 in agreement with this often disordered segment becoming visible in ligand-bound crystal structures. Solution NMR structures of BcII have indicated an altered conformation of the loop with respect to that observed in crystals, and also a shift in the region upon binding of R-thiomandelic acid to the active site, displaying a hinge-like motion to contact this inhibitor.345 This displacement was observed together with conformational changes in the loop containing the main metal binding motif and helix α2, and displacement of the Zn(II) ions with respect to the free protein. Results obtained by rapid freeze quench (RFQ) followed by double electron electron resonance (DEER) in a doubly spin labelled NDM-1 enzyme can be accounted for by a closing motion of this loop over the active site during catalysis, in the millisecond timescale.346 The link between loop mobility and substrate hydrolysis is also illustrated by a BcII mutant with expanded substrate spectrum, which was shown by NMR relaxation experiments to possess increased dynamics of the active-site environment in the catalytically relevant micro- to millisecond timescale with respect to the wild type protein.347 The altered sites map mainly to active-site loops (L3, L7, L10), and are caused by second sphere mutations G262S (next to ligand H263) and N70S (base of L3).
SPM-1, although belonging to subclass B1, possesses a much shorter L3 loop and an extended α3 helix, features reminiscent of B2 MBLs. This segment can adopt two different conformations (“open” and “closed”) as observed in structures obtained under different crystallization conditions, either extending away from or bordering the active site (Figure 15).281, 348 The mobility of this segment of the protein was studied in solution by 19F NMR, revealing that the α-helix is sampling two distinct conformations in slow exchange in solution.348 However, when the 19F label was introduced in a different, contiguous position in α3, observations indicated either a single conformation or rapid exchange.349 When the 19F probe was introduced in the L3 loop, a single resonance was observed. Binding of β-lactams caused a shift in resonances for the 19F label located in loop L3 but not in α3, implying a larger role in substrate binding for the former.
Figure 15.
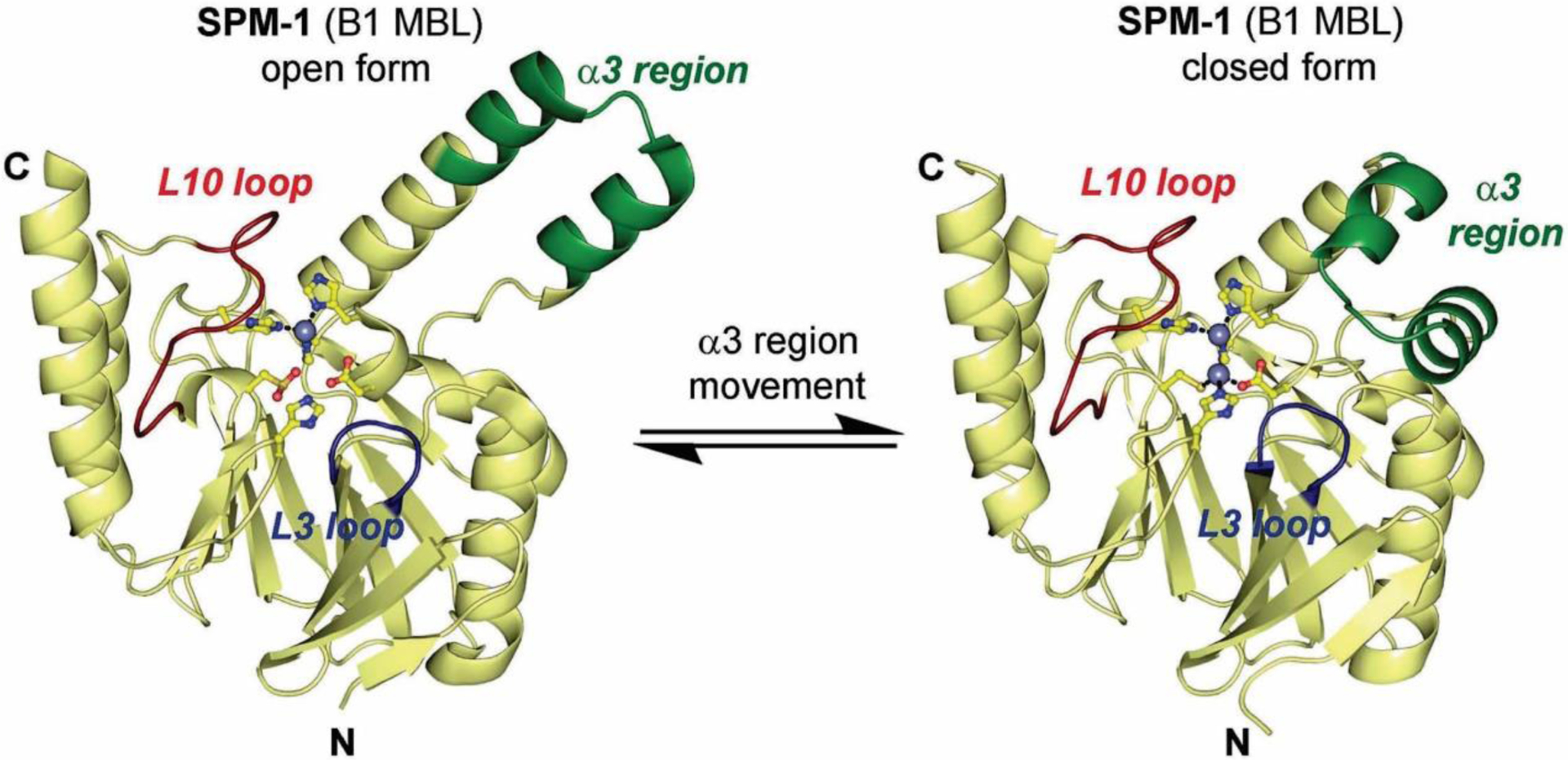
Equilibrium between the ‘open’ (PDB 2FHX) and ‘closed’ (PDB 4BP0) conformations of SPM-1. Mobile regions are highlighted in blue (L3 loop), red (L10 loop) and green (α3 region). Reproduced with permission from reference348 Brem et al. Published by The Royal Society of Chemistry.
An exception to these observations for L3 loop motion in B1 enzymes was encountered in a RFQ-DEER characterization of spin-labeled CcrA.350 This study revealed a movement of ca. 9 Å of this loop away from the active site upon binding of the chromogenic cephalosporin chromacef. This study was not further deepened later, but it could suggest the existence of distinct behaviors for the loop in different MBLs or depending on the substrate.
The extended α3 helix in B2 MBLs (Figure 14) has been proposed to carry out a similar functionality to loop L3 in B1 enzymes. An analysis combining RFQ with electron paramagnetic resonance (EPR) of Zn(II)- and Co(II)-bound ImiS, containing a spin label within the aforementioned helix, revealed motions of this segment during catalysis.351
B3 MBLs also have a protruding loop bordering their active site (α3-β7 loop, Figure 14) that is posited to participate in substrate binding and catalysis. Stopped-flow studies with a mutant of the L1 enzyme containing a Trp residue in this loop displayed variations in fluorescence that were in synchrony with substrate binding and the formation of a reaction intermediate, and support the existence of loop motion during the catalytic cycle.324
All in all, these observations suggest that the motions of extended loops and helical segments flanking the MBL active site play a key role in the hydrolytic capabilities of these enzymes. Additionally, they illustrate the importance of complementing the high-resolution static images provided by X-ray crystallography with the dynamic information afforded by spectroscopic techiniques in deciphering the function of MBLs and other proteins.
4.6. Second-shell residues
A conserved network of H-bonds has been identified in the active-site floor of B1 enzymes, which connects metal ligands (His116, Asp120, Cys221) with each other, via residues known as second-shell residues (including positions 69,70, 84, 115, 121 and 262) (Figure 16).281 As in other metalloenzymes, these second-shell residues act to both fine-tune the orientations and polarize the residues directly coordinating the Zn(II) ions.352, 353 The highly conserved Asp84 (also present in superfamily members) takes part in this network, and is found to assume a conformation outside of the Ramachandran-allowed zones in crystal structures, probably indicating an important role in maintaining the protein folding. Asp84 is also conserved in some B3 enzymes, such as L1, AIM and THIN-B. This residue interacts with the positively charged Arg/Lys121 present in most B1 MBLs (although it is replaced by serine in IMP and cysteine in CcrA enzymes, respectively) (Figure 16). Gly/Ser262 is also an important second sphere residue, generally interacting with Arg/Lys121, and capable of influencing metal binding and substrate selectivity in B1 lactamases. A G262S substitution in a BcII mutant was shown to increase the catalytic efficiency of the enzyme towards a poorly hydrolyzed substrate, expanding the catalytic spectrum in exchange for lowered Zn(II) affinity, although this could be offset by another second-shell substitution (N70S).274, 354, 355 Similarly, the opposite mutation (S262G) has been associated with a narrowing of the substrate spectrum but with increased hydrolysis of newer carbapenems in the transition of IMP-1 to IMP-6, possibly as a result of evolutionary adaptation towards the use of these antibiotics.356–358 A series of experiments analyzing the specific role of second-shell ligands, either generated by mutagenesis or present in natural allelic variants, will be discussed for each MBL family in Section 4.7.
Figure 16.
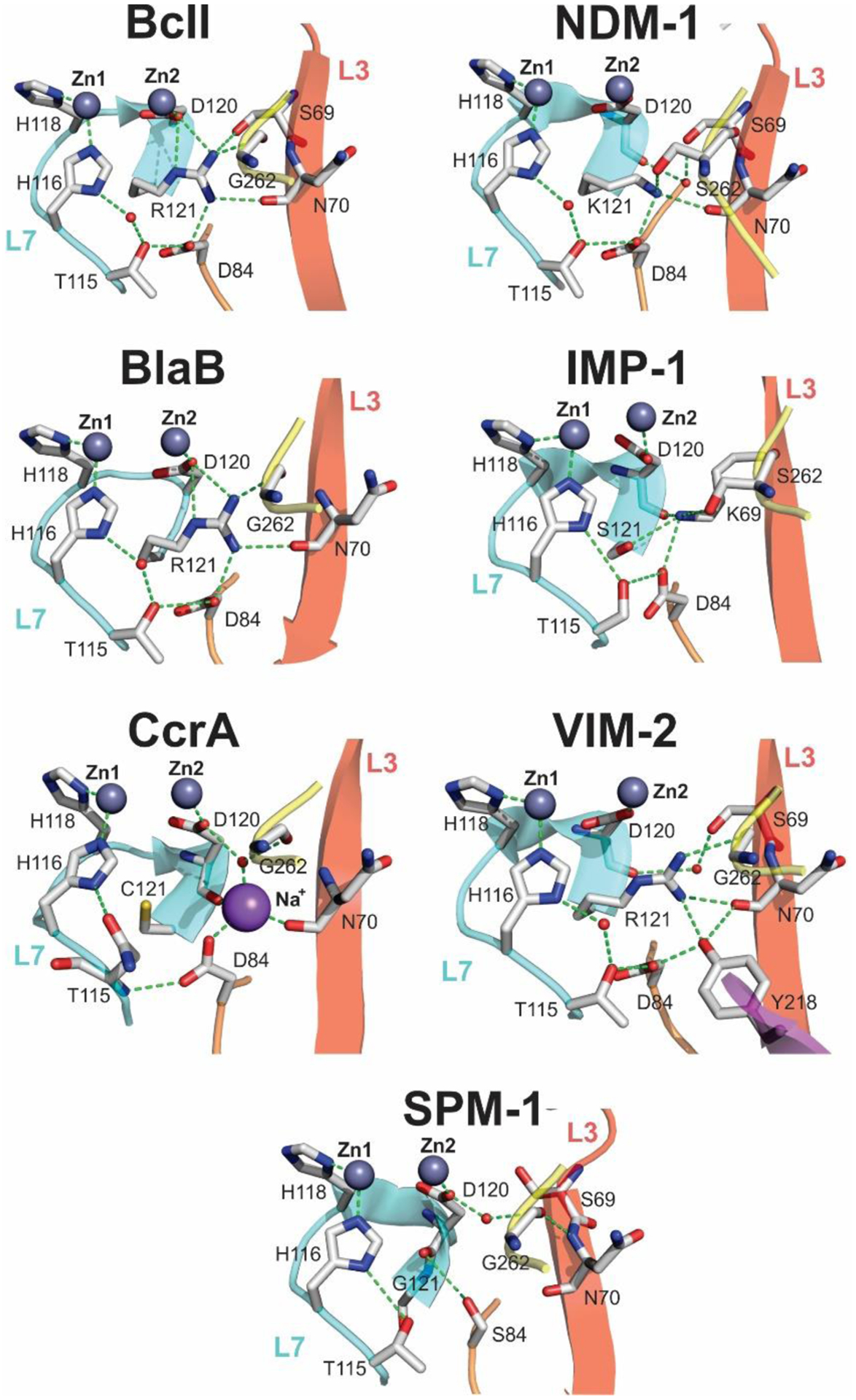
Active-site bonding networks in subclass B1 MBLs281 : BcII (PDB 1BC2), NDM-1 (PDB 6TWT), BlaB (PDB 1M2X), IMP-1 (PDB 1DDK), CcrA (PDB 1ZNB), VIM-2 (PDB 1KO3) and SPM-1 (PDB 4BP0). Hydrogen bonding and ionic interactions are shown as dashed green lines. The different protein regions connected through the conserved interaction network are depicted as cartoon representations of different colors. Zinc ions are rendered as grey spheres, sodium ions as magenta spheres and water molecules as red spheres
4.7. Representative MBLs. Main enzymes families of each subclass
This section intends to cover the features of the most important MBLs, analyzing the natural allelic variants observed in the clinics, and the impact of these mutations in the catalytic efficiency, stability and resistance profile. Also the unique features of each protein family and the level of divergence is discussed.
The comparison of activity data reported in the literature of different MBLs (either from different families or within allelic variants) is highly challenging due to different reasons. First, experiments to measure kinetic data are not always performed under similar conditions (buffer, temperature, pH, ionic strength, added Zn(II)). Measurement of the metal content of the enzyme is of critical importance, to assess the amount of active enzyme and to check for potential mismetallation. Second, kcat values refer to the amount of folded and active enzyme, a value that in the case of MBLs depends on the amount of properly metallated enzyme. Some authors correct the measured kcat value based on the Zn/MBL ratio. In this review, we will compare activity data performed in conditions as close as possible. Changes in the kinetic parameters of at least one order of magnitude will be considered as relevant.
The comparative analysis of resistance data (MIC values) is even more difficult, since different expression vectors are used with widely different promoter strengths or plasmid copy numbers, with or without the addition of molecules inducing expression. Also, the bacterial strains used not only differ from the clinical ones, resulting in different genetic bakcgrounds, but it is very likely that most groups utilize E.coli strains to measure MIC values even for proteins for which E. coli is not the natural host. Finally, it is advisable to quantitate the periplasmic levels of the enzymes in the bacterial periplasm in order to analyze the impact of mutations in expression, activity and stability in vivo. This concern also applies to the comparison of any other microbiological data.
In the following discussions of the most relevant MBLs, substitutions present in natural alleles will be indicated in parentheses with respect to a certain reference variant of the enzyme. For example, VIM-4 (VIM-1 S228R) indicates that VIM-4 differs from VIM-1 by the S228R substitution. Meanwhile, lab-generated mutants will be referenced as the name of the base enzyme followed by the corresponding mutations (e.g. VIM-13 R228S indicates a mutant in which Arg228 of VIM-13 was replaced by Ser and no other residues were modified).
4.7.1. Subclass B1
The B1 subclass contains the first MBL to be discovered, the chromosomally-encoded BcII enzyme from Bacillus cereus 569/H/9, which was first regarded as an outlier. Its finding was linked to the initial idea that MBLs were not clinically relevant. This notion was dispelled upon the finding of two Bacteroides fragilis isolates resistant to cephamycins and imipenem in 1986,359 β-lactam compounds used for the treatment of this anaerobic pathogen. This resistance (unusual at that time) was due to the chromosomal expression of an MBL, originally named CfiA, and later CcrA (for carbapenem and cephamycin resistance),360 that presented high homology with BcII in the purported active-site residues. At that time, the imipenemase activity of a B1 MBL from Myroides odoratus (formerly Flavobacterium odoratum),361 characterized as TUS-1 two decades later,362 and the B3 enzyme L1 had also been reported.363
The whole scenario changed upon the report of the first plasmid-borne MBL, the IMP-1 (Imipenemase) enzyme, identified in 1991.364 The rise of genes coding for these resistance determinants associated with plasmids and other mobile genetic elements allowed for a dramatically increased spread potential amongst different pathogens, and paved the way for MBLs to become a threat to the clinical efficacy of β-lactam antibiotics. Subclass B1 contains most of the clinically relevant and plasmid-encoded MBLs. Among them, the most frequently isolated MBLs worldwide belong to the NDM, VIM and IMP families of plasmid-associated lactamases.16, 75 Other acquired MBLs (either plasmid-borne or located within other mobile genetic elements) within this subclass are SPM-1,365 of great local importance in Brazil, DIM-1,366 FIM-1,367 HMB-1,368 TMB-1,369 KHM-1,370 and the GIM371 and SIM enzyme families.372
In addition to the already mentioned BcII, CcrA/CfiA and TUS-1, this subclass contains various other native chromosomal enzymes such as Bla2 (Bacillus anthracis),373 BlaB (Elizabethkingia meningoseptica),374 IND (Chryseobacterium indologenes),375 JOHN-1 (Flavobacterium johnsoniae),376 MUS-1 (Myroides odoratimimus),377 EBR-1 (Empedobacter brevis)378 and CGB-1 (Chryseobacterium gleum).362
4.7.1.1. IMP family
IMP-1, the first transferable MBL, was reported the literature in 1991 after being identified in a clinical isolate of P. aeruginosa obtained in Japan in 1988.16, 364 The gene was located in a conjugative plasmid, and the enzyme was afterwards also detected in isolates of S. marcescens and other Enterobacterales.379 The discovery of IMP-1, soon followed by other plasmid-borne class B lactamases, determined the transition of MBLs from an enzymological curiosity to a clinical threat. With 80 natural variants detected so far, IMP enzymes have not experienced a wide geographical dissemination as other carbapenemases such as VIM, NDM or KPC. However, they represent a persistent cause of carbapenem resistance in Japan and Southeast Asia, and have also began spreading in the Middle East and Australia.16 Overall, IMP variants present 77%–99.6% of sequence identity, giving rise to a diffuse dendogram (Figure 17) that suggests a polyphyletic origin of IMPs,380 contrasting with the VIM group. IMP alleles differ by up to 54 amino acids, as in the IMP-53/IMP-46 pair. Amino acid substitutions are located primarily in the peptide leader sequences, second coordination sphere and positions distant from the active site, and active-site loops to a lesser degree (Figure 18).
Figure 17.
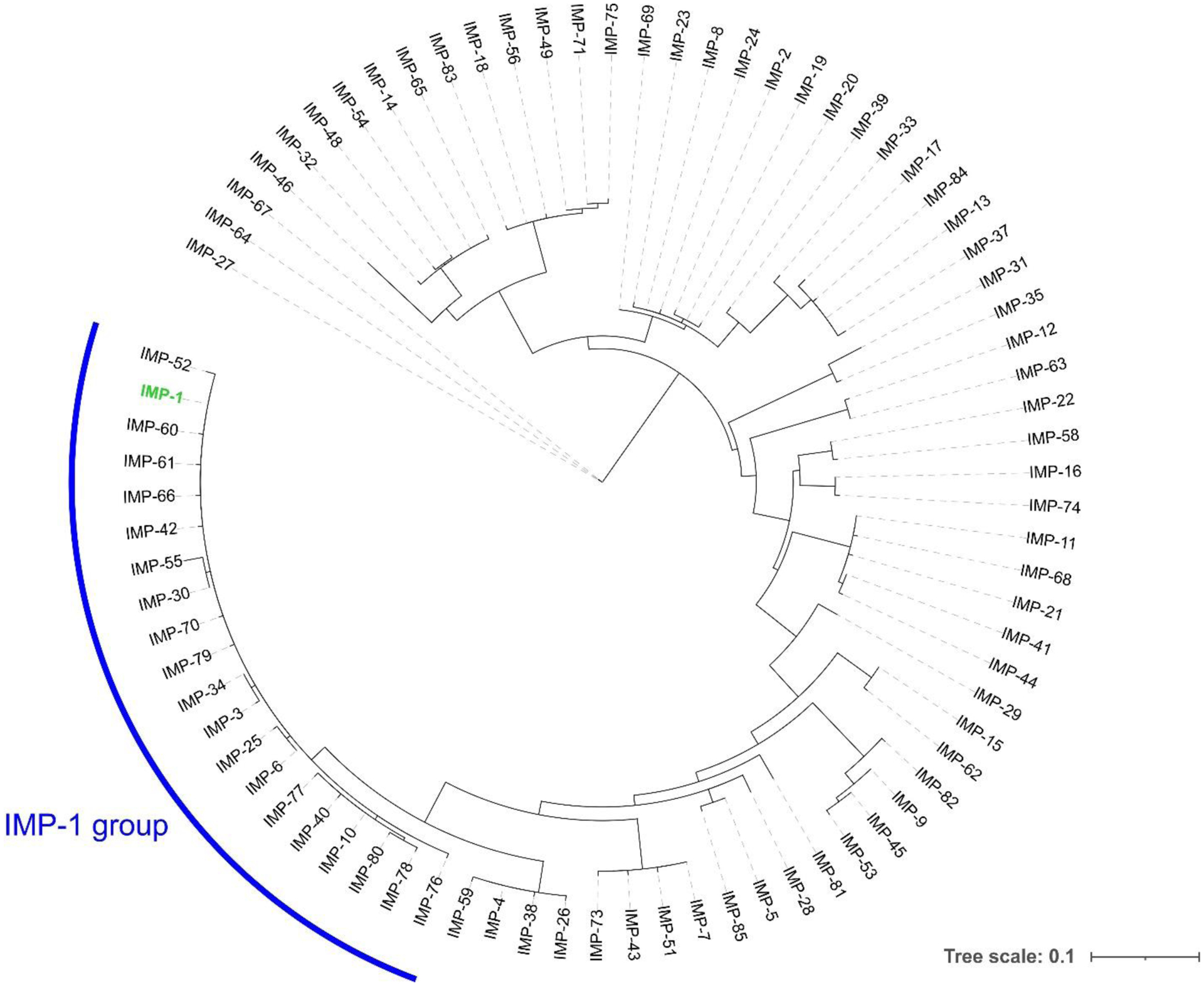
Phylogenetic tree of the IMP enzyme family. The location of IMP-1 is shown in green. Protein sequences were aligned using Clustal Omega, phylogenetic trees were constructed using PhyML (via the phylogeny.fr web server) and tree representations were generated with iTOL.
Figure 18.
Structure of IMP-1 (PDB 4C1G) highlighting the positions presenting sequence variation within the IMP family. The positions are colored (from black to red) in the cartoon representation according to an increasing absolute frequency of mutation within all IMP enzymes with respect to IMP-1. The side chains of positions with a frequency of substitution in the top 4 categories are shown as sticks.
In terms of substrate-profile and protein structure, IMP enzymes generally behave as typical broad-spectrum MBLs, with some exceptions (see below). IMP-1 exhibits average kcat/KM values of ~105 – 106 M−1s−1 for penicillins, ~106–107 M−1s−1 for cephalosporins and ~106 M−1s−1 for carbapenems; the slightly lower preference for penicillins stems from higher KM values compared to the other substrates. Apart from monobactams, IMP enzymes are unable to hydrolyze temocillin (kcat/KM values ≤ 102 M−1s−1), and generally confer low resistance levels (if any) against piperacillin even though recombinant enzymes exhibit catalytic efficiencies of ~104 – 106 M−1s−1 for this antibiotic.233, 381–389
Studies conducted with the IMP family, mainly IMP-1, provided key insights into the mode of binding of β-lactam antibiotics to the active sites of MBLs. Residues from the L3 and L10 loops as well as from the second-shell sphere were shown to be critical for the recognition and hydrolysis of substrates (Figure 19). Among early studies, Palzkill and coworkers performed pioneering codon randomization experiments on IMP-1 that shed light into the role of several residues near the active-site cleft.235, 335 The selection criterion was based on the ability of the randomized mutants to confer resistance towards ampicillin, cefotaxime, imipenem and cephaloridine, showing that the requirements for some residues are highly substrate-dependent. Residues directly involved in metal binding were unable to tolerate amino acid substitutions. Site-directed mutagenesis of His116, His118, Cys221 or His263 provoked a marked decrease in catalytic efficiencies due to a drop in enzyme metal binding affinities. Under Zn(II) supplementation, catalytic efficiencies were partially or completely restored.233 Saturation mutagenesis at Cys221 revealed strict limitations upon substituting this position due in large part to a loss in IMP-1 stability, suggesting that residue Cys221 contributes to enzyme stability by binding to Zn2.310 IMP-1 C221D and C221G mutants were the only mutants capable of conferring resistance in E. coli. The discovery of glycine as a functional residue in vivo was surprising, since it cannot serve as a zinc ligand. However, purified C221G mutant bound 1 equivalent of Zn(II), and catalytic efficiencies as well as MIC values were strongly dependent on the amount of exogenous Zn(II), indicating that binding of Zn2 requires excess Zn(II). The absence of C221G in natural variants of IMP-1 thus reflects how conditions of Zn(II) limitation encountered during bacterial infections excludes substitutions impairing metal-binding affinities (see Section 10). In the case of Asp120, D120A and D120E substitutions provoked a remarkable reduction in enzyme activity without affecting the overall structure nor the metal content of the enzymes.390 The fact that the activity of these IMP-1 mutants was not recovered in the presence of an excess of Zn(II) ion indicated that Asp120 is important for the hydrolysis of β-lactams. According to pre-steady state kinetic studies, the rate-determining step for the IMP-1-catalyzed reaction of nitrocefin proceeded without significant accumulation of an anionic intermediate, unlike other MBLs.299, 390 This suggested that Asp120 is not the proton donor to the anionic intermediate. Instead, based on pH-dependence activity experiments and the crystallographic structure of D120E mutant, it was proposed that the critical role of Asp120 is to orientate the bridging H2O/OH- for nucleophilic attack without affecting its pKa, while also aiding in positioning the Zn(II) ions and His263390 (see Section 5 for a more detailed explanation on the catalytic mechanism of MBLs).
Figure 19.
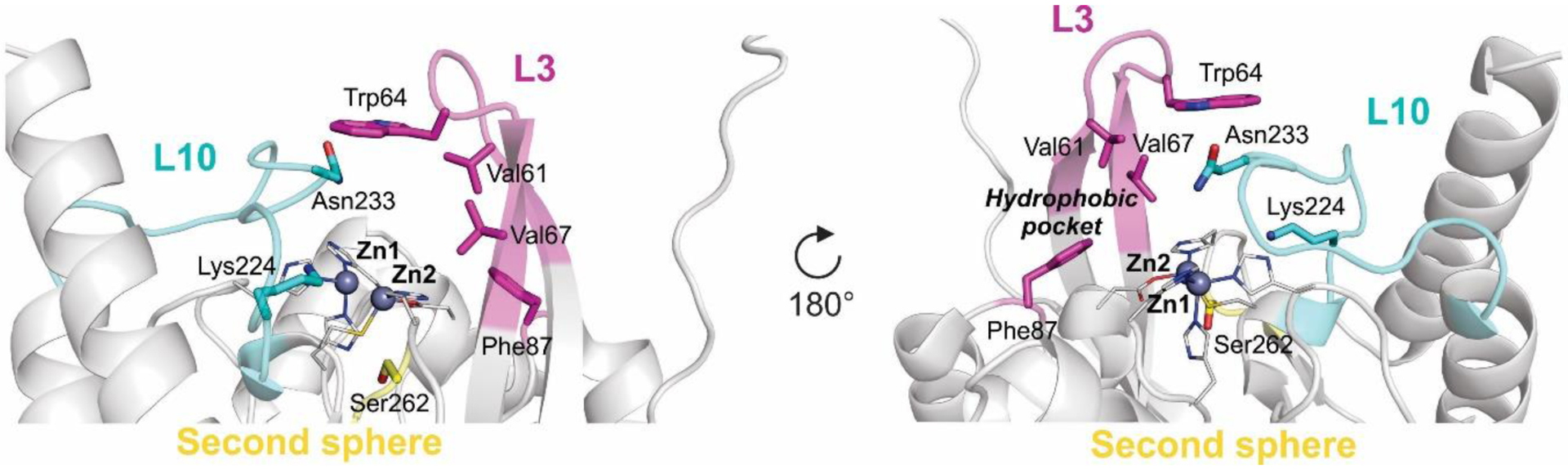
Front and back view of IMP-1 showing the active-site loops (L10 and L3) and second-shell sphere regions involved in substrate recognition and turnover. Side chains of most relevant residues are shown as sticks, metal ligands as lines and Zn(II) ions as spheres (PDB 5EV6). L3 loop and Phe87 are shown in magenta, L10 in cyan.
4.7.1.1.1. Substrate binding and turnover in IMP enzymes
Mutagenesis studies and computational simulations, as well as crystal structures of IMP-1 in complex with a mercaptocarboxylate inhibitor (similar in structure to benzylpenicillin) and IMP-13 associated to different hydrolyzed carbapenems, identified several residues involved in substrate binding. These are principally located in the second-shell sphere, L3 and L10 loops.
4.7.1.1.1.1. Second-shell residues
One position that gained considerable attention during the 2000s is second-shell residue 262. With key contributions from Oelschlaeger and coworkers, residue 262 was shown to fine-tune the substrate profile of IMP enzymes without a direct interaction with substrates nor affecting metal binding. Along the IMP family, position 262 is occupied by a Ser (80%, including IMP-1) or Gly (19%, including IMP-3 and IMP-6) residue, with the exception of IMP-80 that harbors an Ala. The effect of ubiquitous substitutions, such as S262G or V67F (see below), inside the IMP family are more easily tracked when analyzing IMP variants by a network approach instead of classical phylogenetic trees.391
In general, S262G substitution provokes a reduction in catalytic efficiencies towards penicillins (benzylpenicillin and ampicillin), cephalosporins with a pyridinium moiety at the C3 position (cephaloridine and ceftazidime) and carbapenems with positively charged group at the C2 position (imipenem).356–358, 392 These changes were reflected in MIC values.357, 358, 392 Instead, for cephalosporins with an acetoxy group at C3 (cephalotin, cefotaxime and cefoxitin) and carbapenems with a neutral group at C2 (meropenem, panipenem and doripenem), the S262G substitution either does not affect or slightly improve the catalytic efficiencies (carbapenems). For this group of substrates, the S262G substitution was shown to markedly increase MIC values of meropenem in IMP-6, and even further in IMP-25 (IMP-1 S262G G235S), suggesting that this mutation (requiring a single-nucleotide change) is an adaptation towards newer carbapenems, in detriment of penicillinase activity.357, 392 In this case, variations in meropenem MICs correlate with changes in kcat and not kcat/KM, which are similar, suggesting that MBLs would be saturated at the MIC conditions given that KM values are very low in all cases.357 In addition, pre-steady state kinetic studies showed that S262G and G235S substitutions in IMP-25 affect the energetic barriers and kinetic constants of β-lactam hydrolysis, stabilizing the anionic intermediate for meropenem393 (see Section 5). According to these studies, two possible scenarios have been proposed. On one side, IMP-6 might be an ancestor of IMP-1, and in this context, the role of mutation G262S would have been to broaden the substrate profile. On the other side, if IMP-1 is an ancestor of IMP-6, the effect of mutation S262G would have been to improve the activity towards newer carbapenems at the expense of the penicillinase activity. These hypotheses heavily rely on assumptions of the evolutionary timeline of these enzymes that cannot be verified.391 Finally, position 262 seems to act independently of the presence of other substitutions, as similar effects were observed when comparing IMP-7 vs IMP-51 (IMP-7 S262G)394 and IMP-11 vs. IMP-68 (IMP-11 S262G),389 i.e., a drop in MIC values of imipenem, ceftazidime and cephaloridine, and an increase in MIC values of newer carbapenems. In the same vein, Crowder, Bonomo and coworkers have recently demonstrated that the IMP-1-group (comprising 20 variants, see Figure 17) is evolving in response to the structural differences introduced by the carbapenems meropenem and ertapenem.395 Alleles harboring the S262G and/or V67F (see next) substitutions exhibited higher MICs toward newer carbapems, in detriment of penicillins and imipenem, being IMP-78 (IMP-1 S262G/V67F) the variant exhibiting the most prominent effects. These studies evidence a lack of significant epistatic effects between residue 262 and other positions in the protein presenting substitutions. In the case of BcII, strong epistatic interactions between positions 262 and 70 were observed.355 However, in the IMP family this position (His70) is strictly conserved, suggesting that it is already optimized.
Based on computational simulations, the remote effect of residue 262 was described by a “domino effect” involving the substituent at C3 or C2, His263 and position 262, that influences the stability of reaction intermediates.396 Codon randomization studies indeed indicated that the sequence requirements at Ser262 are very stringent among mutants selected for resistance to type II substrates, while different amino acid types were observed among mutants selected for cefotaxime resistance.335 In the same vein, site-directed mutagenesis analysis at position 262 and second-shell residues proximal to it showed that the hydrogen bond network below the active site indeed plays a crucial role in determining substrate specificities.356, 397, 398 Molecular dynamics simulations suggested that apart from the domino effect, the hydrogen bond network below the active site affects the conformation of L3 loop involved in substrate binding.399
Less investigated second-shell residues include position 119, within the metal-binding motif, which is mostly Ser as in IMP-1, Gly in 11 alleles and Asp in IMP-46. The structures of IMP-13 in complex with hydrolyzed carbapenems showed that Ser119 is able to form hydrogen-bond interactions with the substrate substituent at C6 or C7, or C7/C8 carboxylate upon C-N cleavage.400 Substitutions in Ser119, either by Ser or Asp, are always accompanied by the S115T mutation, suggesting synergy between both positions. Zhang et. al. identified epistatic effects between these two second-shell positions: S115T suppresses the thermal stability and expression defect introduced by S119G.401
4.7.1.1.1.2. L10 loop
Initial randomization studies identified the L10 loop of IMP-1 (residues 222–238) as a region involved in binding and/or turnover of carbapenems and penicillins, to a minor degree.235, 335 Residue Lys224, present at the N-terminal edge of the L10 loop, is highly conserved in B1 and B2 MBLs except for VIMs (see Section 4.7.1.2). This residue is invariant across the entire IMP family. As observed for other MBLs, in the IMP family Lys224 interacts, together with Zn2, with the C3/C4 carboxylate present in penicillins, cephalosporins and carbapenems, with displacement of the apical water coordinated to Zn2 in the native structures.382, 400, 402 This stands as one of the major interactions involved in substrate binding and positioning into the active site. Accordingly, substitution of Lys224 by Arg, Ala or Glu in IMP-1 impaired the catalytic efficiency of the enzyme towards benzylpenicillin, cephalosporins and imipenem, due to marked increases in KM values, especially in the case of K224E.403 These mutants rendered enzymes with 1.2 equivalents of Zn(II) ions per molecule, indicating that the identity of the residue in position 224 also affects metal binding. Codon randomization studies revealed that although Lys224 is critical for the hydrolysis of ampicillin, imipenem and cefotaxime, Gln224 was observed at equal frequency among mutants selected for cephaloridine resistance.335 This is because K224Q exhibits a large increase in kcat for cephalosporins that partly compensates for the reduced binding, so that the catalytic efficiency is reduced only slightly compared to that of the wild type enzyme. In this way, it has been proposed that position 224 also affects catalysis.
Asn233 located in the L10 loop is highly conserved among B1 and B2 MBLs. Different roles, or not role at all, have been proposed for this residue, including substrate binding through interaction with carbonyl oxygen of substrate β-lactam ring or participation in the catalytic mechanism.23 This reside is widely conserved across the IMP family, except in alleles 14, 31, 32, 35, 46, 48, 54, 59 and 65, which harbor a N233Y substitution, and IMP-55 with Lys233. Initial mutagenesis studies on IMP-1 suggested that Asn233 is essential only for imipenem hydrolysis.235, 310, 403 IMP-1 mutants such as N233A, N233Y, N233D or N233S in general exhibited catalytic efficiencies and/or MICs that were similar or even higher than those of wild type IMP-1 for penicillins and cephalosporins. For imipenem, instead, substitution of Asn233 generated marked increases in KM with a ≥ ten-fold decrease in catalytic efficiencies. Later studies performed over a collection of IMP-1 mutants encoding each of the 19 possible amino acid substitutions at position 233, indicated that although the sequence requirements at this position is more stringent for resistance to ampicillin or imipenem compared to cephalosporins, most substituted enzymes were able to provide a high level of resistance towards a broad range of β-lactams.404 As before, variations in in catalytic efficiencies were mainly governed by KM, suggesting that position 233 is involved in penicillin/carbapenem binding and/or initial steps of the catalytic mechanism by interaction with reaction intermediates. In this sense, docking studies guided by the structure of IMP-1 in complex with mercaptocarboxylate inhibitor suggested that the side chain of Asn233 interact with the carbonyl oxygen of benzylpenicillin.382 Comparison between the crystal structures of IMP-13 alone and bound to hydrolyzed carbapenems indicated that Asn233 moves closer to the active site in order to hydrogen-bond the C7/C8 carboxylate generated upon C-N cleavage, suggesting that Asn233 is able to stabilize reaction intermediates.400 Furthermore, Asn233 was shown to interact with Trp64 from L3 loop, which is involved in strong π-sulfur interactions with the sulfur atom present in the linker region of all carbapenems, thereby contributing to the position of the core scaffold of these substrates. In line with mutagenesis studies, molecular dynamics simulations showed that these interactions are absent for cephalotin.402 Finally, given that imipenem hydrolysis imposes the most stringent sequence requirements on IMP-1 position 233, it has been proposed that naturally occurring molecules, such as thienamycin, that inspired the design of synthetic carbapenems, may have driven the conservation of asparagine at position 233.404
Other residues from L10 loop that shape the substrate profile of IMP-1 include Pro225, Gly232 and Asp236. Codon randomization studies revealed that Asp236, whose side-chain forms a hydrogen bond with the zinc ligand His118, is essential for conferring resistance against ampicillin or imipenem but not for cephalosporins.335, 382, 400, 405, 406 Pro225 is required for resistance against imipenem, cephaloridine and ampicillin but not for cefotaxime, while Gly232 is essential for imipenem and cephaloridine but not for ampicillin or cefotaxime.335 Asp236, Pro225 and Gly232 are invariant across the entire IMP family.
The identity of position 235 (Gly in 67 alleles, Asp in 12 alleles and Ser in IMP-25) was shown to modulate the resistance towards meropenem.357 Apart from an increase in meropenem kcat and MICs in IMP-25 (IMP-6 G235S) compared to IMP-6, G235S substitution moderately affected MICs and kcat/KM of cephalosporins, and increased MIC values of ampicillin although catalytic efficiencies were slightly reduced. It has been proposed that Ser235 exerts its effect by hydrogen-bonding with Asn233.
4.7.1.1.1.3. L3 loop
The L3 loop, encompassing residues 60–67, is mobile and adopts different conformations across the IMP family. Its conformation is highly sensitive to substrate/inhibitor binding, giving insight into its role in substrate binding. Several residues from this loop, or proximal to it, were investigated in their role in substrate binding and turnover.
The crystal structure of IMP-1 bound to mercaptocarboxylate inhibitor provided the first clues regarding the role of L3 loop in substrate binding.382 In this case, inhibitor binding triggers conformational change of L3 that closes over the active site and interacts with the inhibitor through Trp64, placed at the tip of the loop (Figure 20). This observation led to the general proposal suggesting that L3 loop closes over the active site upon substrate binding. In addition, a hydrophobic pocket delineated by residues Val61, Val67 and Phe87 was identified at the base of the loop. This pocket was hypothesized to accommodate the phenyl (in substituent at C6) and axial methyl groups of benzylpenicillin (Figure 21-a).333, 382 Recent crystal structures of IMP-13 in complex with hydrolyzed carbapenems confirmed this hypothesis, since L3 closes over the active site upon substrate binding, forming a tunnel-like structure of hydrophobic nature (Figure 21-b).400
Figure 20.
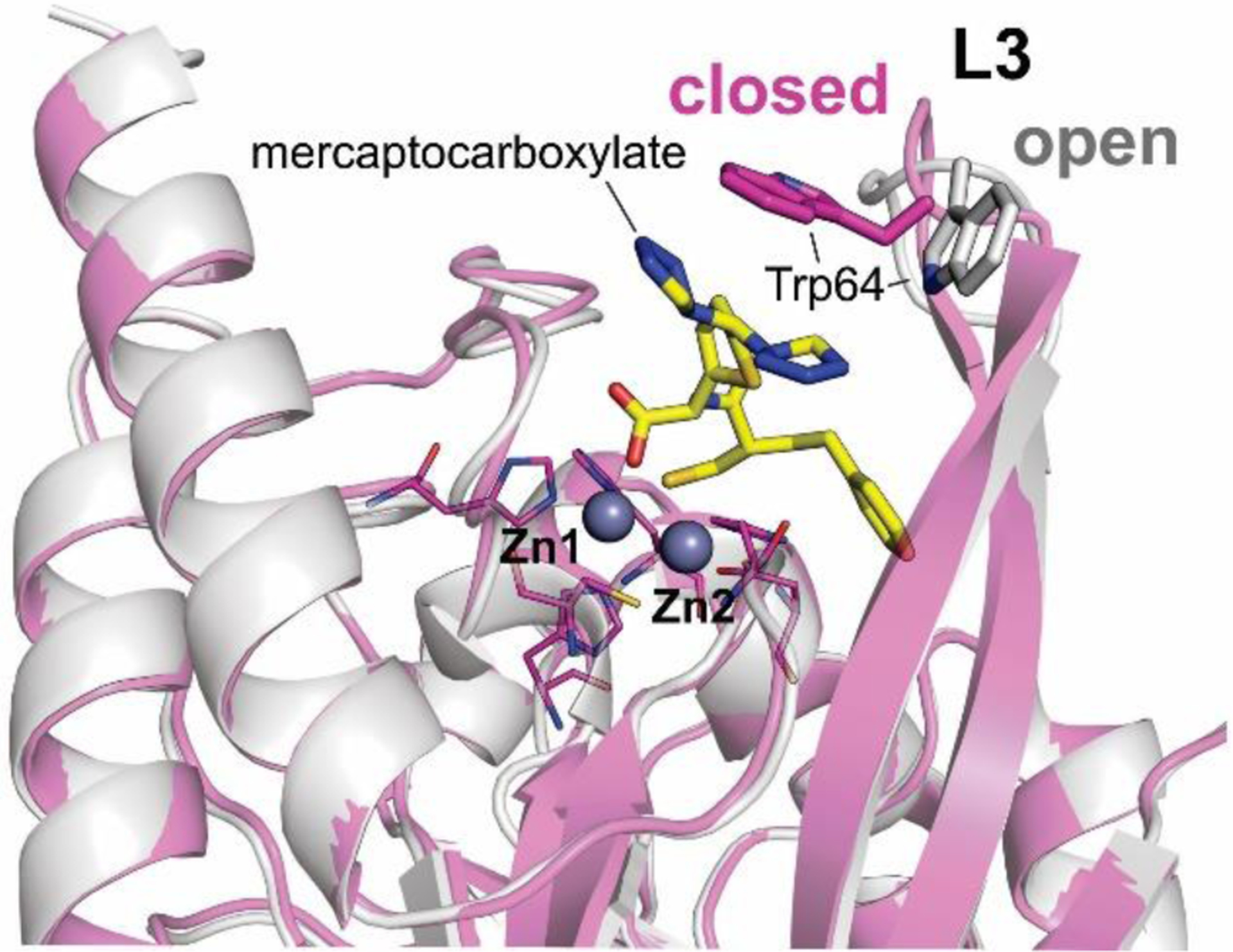
Structures of unbound IMP-1 (PDB 1DDK, open) and IMP-1 in complex with a mercaptocarboxylate inhibitor (PDB 1DD6, closed). The change in the position of Trp64 side chain is highlighted.
Figure 21.
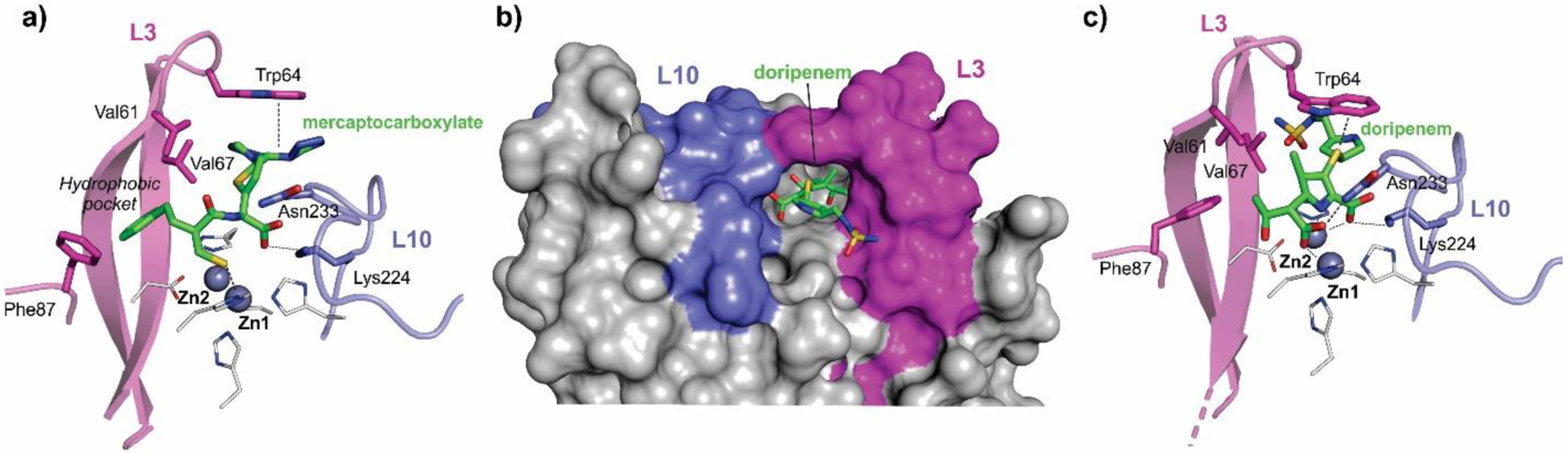
Binding of substrates/inhibitors to IMP enzymes. a) Cartoon representation of of IMP-1 active site in complex with mercaptocarboxylate inhibitor (PDB 1DD6). b) Surface of IMP-13 in complex with hydrolyzed doripenem (PDB 6S0H). c) Cartoon representation of IMP-13 active site with hydrolyzed meropenem (PDB 6S0H). Residues involved in interactions (dashed lines) with inhibitor/substrate are shown as sticks. L3 loop and Phe87 are shown in magenta, L10 in cyan.
In IMP-1, either deletion of the entire L3 loop (including Val61, Trp64 and Val67) or replacement of Trp64 by Ala, albeit to a minor extent, resulted in marked increments in values of KM for most substrates, as well as a decreased affinity for inhibitors.333 Regarding the catalytic efficiencies, L3 affected principally penicillins and cephalosporins, with minor effects over imipenem. In the case of nitrocefin and cefoxitin, in which KM is similar to KS, it was shown that Trp64 is involved in substrate binding. Accordingly, IMP-13 structures and NMR studies revealed that Trp64 located at the tip of the loop bridges the gap between the loop backbone and the active-site residues, forming a closed tunnel that is accompanied by a loss in L3 flexibility.400 The sulfur atom present in the linker region of substituents at C2 of all carbapenems created strong π-sulfur interactions with the aromatic ring of Trp64, thereby contributing to the position of the core scaffold of all carbapenem substrates (Figure 21-c). In the case of imipenem, multiple conformations of substituent at C2 were observed, indicating that the orientation for the π-sulfur interaction is not always optimal. In addition, Trp64 showed interactions with the pyrroline methyl group present in all carbapenems other than imipenem. The lack of this interaction could lead to a reduction in binding interactions to the loop and may contribute to the observed higher KM for imipenem (ca. 50 μM compared to low μM or high nM for meropenem and ertapenem, respectively) and a greater flexibility of L3, as concluded from MD calculations, which would aid in product release.384, 400 This explains the higher kcat values observed for imipenem compared to other carbapenems. The interactions observed in IMP-13 may extend to other alleles. Indeed, the catalytic efficiencies of IMP-13 towards carbapenems are similar to those of IMP-1.384 The substrate profile of IMP-13, however, exhibits some particularities, the most prominent being the kcat/KM of ticarcillin, which is 3 orders of magnitude higher than that of IMP-1. This variation may be the result of one or several substitutions, as mature IMP-13 and IMP-1 differ by 31 residues.
Trp64 is strictly conserved across the entire IMP family. However, substitution of Trp64 by residues such as Phe, Ser or Asn result in functional mutants.333 In the case of cephalosporins, in which IMP-1 exhibits low KM values, either deletion of L3 loop or W64A substitution increased the values of kcat towards these antibiotics, suggesting that L3 loop favors binding of cephalosporins but in an suboptimal orientation for hydrolysis.333
Codon randomization of IMP-1 showed that positions 61, 67 and 87 from the hydrophobic pocket tolerate a variety of mutations depending on the antibacterial used for selection.333 However, there was a bias towards hydrophobic amino acids among the functional mutants, with the most stringent sequence requirements observed for ampicillin hydrolysis (wild-type residues Val67 and Phe87 were present in 90% of the mutants, and there was a strict requirement for hydrophobic residues at position 61). In the case of residue 67, the most common residue along the IMP family is Val (present in 53 alleles, including IMP-1) followed by Phe (17 alleles), Ile (in 5 alleles) and Ala (3 alleles). The effect of these substitutions was studied in IMP-10 (IMP-1 V67F), IMP-1 V67I and IMP-1 V67A.336, 407 All variants exhibited significantly higher catalytic efficiencies than IMP-1 toward cephalosporins and carbapenems with neutral R groups (cephalothin, cefotaxime, meropenem and doripenem). IMP-1 and the two mutants IMP-1 V67A and IMP-1 V67I showed comparable catalytic efficiencies toward β-lactams with positively charged R2 groups (ceftazidime and imipenem), while those of IMP-10 were 2–6 times higher. In the case of penicillins, the situation was opposite: IMP-1 was the most efficient enzyme, closely followed by IMP-1-V67I, while IMP-1-V67A and IMP-10 were significantly less efficient.336, 408 In this way, the most common substitution V67F may reflect an adaptation towards carbapenems, especially the newer ones (meropenem and doripenem), at the expense of penicillins. Indeed, this was the case in MIC values. The rationale for this observation may be that antibiotics with positively charged R2 groups interact less favorably with hydrophobic residues. With Phe67, instead, there would be favorable pi-cation interactions. A similar effect was observed when comparing IMP-44 (IMP-11 V67F) with IMP-11, both alleles exhibiting several substitutions with respect to IMP-1 (91% sequence identity).409 MICs of doripenem and meropenem were each 4-fold higher for E. coli expressing IMP-43 than E. coli expressing IMP-7. In the case of penicillins, although the V67F substitution generated a 6–7 fold drop in catalytic efficiencies, variations in MICs were moderate or null, indicating that the action of this substitution may be partially affected by the presence of other substitutions. Similarly, in the case of carbapenems, catalytic efficiencies did not correlate with MICs, except for meropenem. In variant IMP-26, exhibiting 95.5% sequence identity with IMP-1, the presence of V67F substitution was associated with a preference of the enzyme for meropenem (MIC and catalytic efficiency) and doripenem (MIC) compared to imipenem, even though there is no allelic variant of IMP-26 differing exclusively in position 67 for comparison.410
Residue 87 is occupied by Phe in 64 IMP alleles followed by Ile (9 alleles), Ser (2 alleles), Val (2 alleles) and Thr (IMP-48). Mutational analysis in IMP-18 (harboring Ile87) revealed that this position affects the hydrolysis of penicillins and several cephalosporins.406 In agreement with codon randomization studies performed in IMP-1, the mutant IMP-18 I87F showed kcat/KM values for ampicillin, benzylpenicillin and ceftazidime 5-fold, 7-fold and 4-fold, respectively, higher than those of wild-type IMP-18.386, 406 Similarly, E. coli cells expressing the single mutant IMP-1 F87V exhibited lower MIC values for amoxicillin and ceftazidime, and similar or slightly lower MIC values for carbapenems, compared to IMP-1.407 The catalytic efficiencies followed a similar trend; IMP-1 F87V exhibited >ten-fold drop in kcat/KM values of benzylpenicillin, ceftazidime and doripenem while not affecting those of imipenem, meropenem and panipenem. Both studies showed that values of KM are lower for most substrates when Phe87 is present, suggesting the presence of favorable hydrophobic interactions between the aromatic ring of Phe and the substituent at C6 or C7 of the substrates. As discussed before, the identity of the residues comprising the hydrophobic pocket is very stringent for penicillins.335 However, Gly87 was the most frequent residue selected in functional mutants conferring resistance against cefotaxime. It is possible that removal of bulky side chains at position 87 leaves more space for the bulky R1 substituent of cefotaxime.
Substitutions at both positions 67 and 87 have been identified in five IMP alleles. The combination V67F/F87S, evaluated in the pair IMP-44 (IMP-11 V67F/F87S) vs. IMP-11, was shown to increase the catalytic efficiencies and MICs of all carbapenems while being detrimental for penicillins.409 Similarly, IMP-40 (IMP-1 V67F/F87S) was the variant presenting the lowest ability to confer resistance against penicillins of the IMP-1-group, together with IMP-78 (IMP-1 V67F/S262G), while exhibiting increased resistance towards all carbapenems compared to IMP-1, especially for ertapenem.395 On the other hand, E. coli expressing IMP-70 (IMP-1 V67F/F87V) exhibited a 2-fold increment in MIC values of meropenem and doripenem, a drop in MIC values of penicillins and cephalosporins, and similar resistance levels against impenem and panipenem when compared to cells expressing IMP-1.407 Comparison between the catalytic efficiencies of IMP-70 (IMP-1 V67F/F87V), IMP-10 (IMP-1 V67F), IMP-1 F87V and IMP-1 suggests that the effects of positions 67 and 87 are additive instead of epistatic, as would be expected considering the spatial closeness of these residues in the enzyme structure. While IMP-70 and IMP-10 showed slightly higher hydrolytic activities than IMP-1 against meropenem, IMP-70 exhibited similar enzyme activities against doripenem, imipenem and panipenem. In this way, the F67V substitution, but not F87V, would govern the increment in meropenem hydrolysis observed in IMP-70. It is possible that residue 87 plays some role in enzyme stability as observed for VIM-2.332
In the case of position 61, Val is strictly conserved in the entire IMP family except for alleles IMP-31 (Ile61) and IMP-35 (Asp61). The roles of Val61 and Val67 in substrate binding were envisaged in the crystal structures of IMP-13 in complex with hydrolyzed carbapenems.400 Compared to the resting enzyme, the largest active-site-facing changes seen in the L3 loop occur between residues Val61 and Val67, with these two residues moving towards the carbapenems to form hydrophobic interactions, especially in the case of newer carbapenems. The residues located in the middle of the L3 loop, Val61 and Trp64, show more significant changes, moving approximately 9–10 Å in order to cover the substrate during catalysis, thereby forming an a tunnel-like structure of hydrophobic nature above the active site.
Other residues were shown to affect L3 loop conformation (Figure 22). Comparison of the structures of IMP-1 and IMP-2 or IMP-13 indicated that the amino acid substitution at position 68 on one of the β-strands forming the base of L3 loop (Pro in IMP-1 versus Ser in IMP-2 or Thr in IMP-13) may be a staple factor affecting the flexibility of the loop.382, 400, 405 While in IMP-1 L3 adopts an open and disordered conformation, in IMP-2 it forms a closed conformation in which the side chain of Trp64 is oriented so as to cover the active site.405 Catalytic efficiencies of IMP-2 are overall similar to those by IMP-1, except for ampicillin, in which kcat/KM value is 23-fold lower.411 These differences, however, might be consequence of several substitutions (IMP-2 differs from IMP-1 by 36 residues). In the case of IMP-13 (harboring Thr68), structures were obtained with L3 in a open (PDB 6R79) or closed (PDB 6R78) conformation.400 In the open conformation, although the tip of the loop is further out of the active site, the β-strand of the loop at this position is closer to the active site compared to IMP-1. It is noteworthy that the position of the tip of L3 loop in IMP-13 may be distorted due to interactions with another molecule of the cell unit in the crystal structure. Even in the case of IMP-1, different crystallization conditions or buffer molecules bound to the active site seems to affect the orientation of Trp64.382, 412 Another allelic variant harboring Thr68 is IMP-18.386 The crystal structure of this enzyme revealed that the conformation of the L3 loop slides sideways into a different direction (over susbtituents at C6 or C7 of the substrates) with respect to the open or closed conformations observed in IMP-1.406 However, as with IMP-13, the L3 loop is engaged in interactions with the active site of a different molecule in the cell unit. IMP-18 has the particularity of exhibiting low catalytic efficiencies towards carbapenems other than imipenem (~103 and ~104 M−1s−1 for meropenem and ertapenem respectively),386 although this may be the outcome of multiple susbtitutions (IMP-18 differs from IMP-1 by 48 residues). Notwithstanding, mutagenesis analysis of IMP-18 indicated that residue 68 has a key effect on the catalytic efficiency of many substrates, especially meropenem; the T68P substitution resulted in a > ten-fold increase in kcat/KM value for this substrate.406 IMP-18 also harbors a N62K substitution compared to IMP-1. However, this position exerted only moderate effects on enzyme activity. All in all, It is likely that the Pro32 to Thr/Ser mutation leads to a more flexible loop, as a result of the more constrained dihedral angles of proline.
Figure 22.
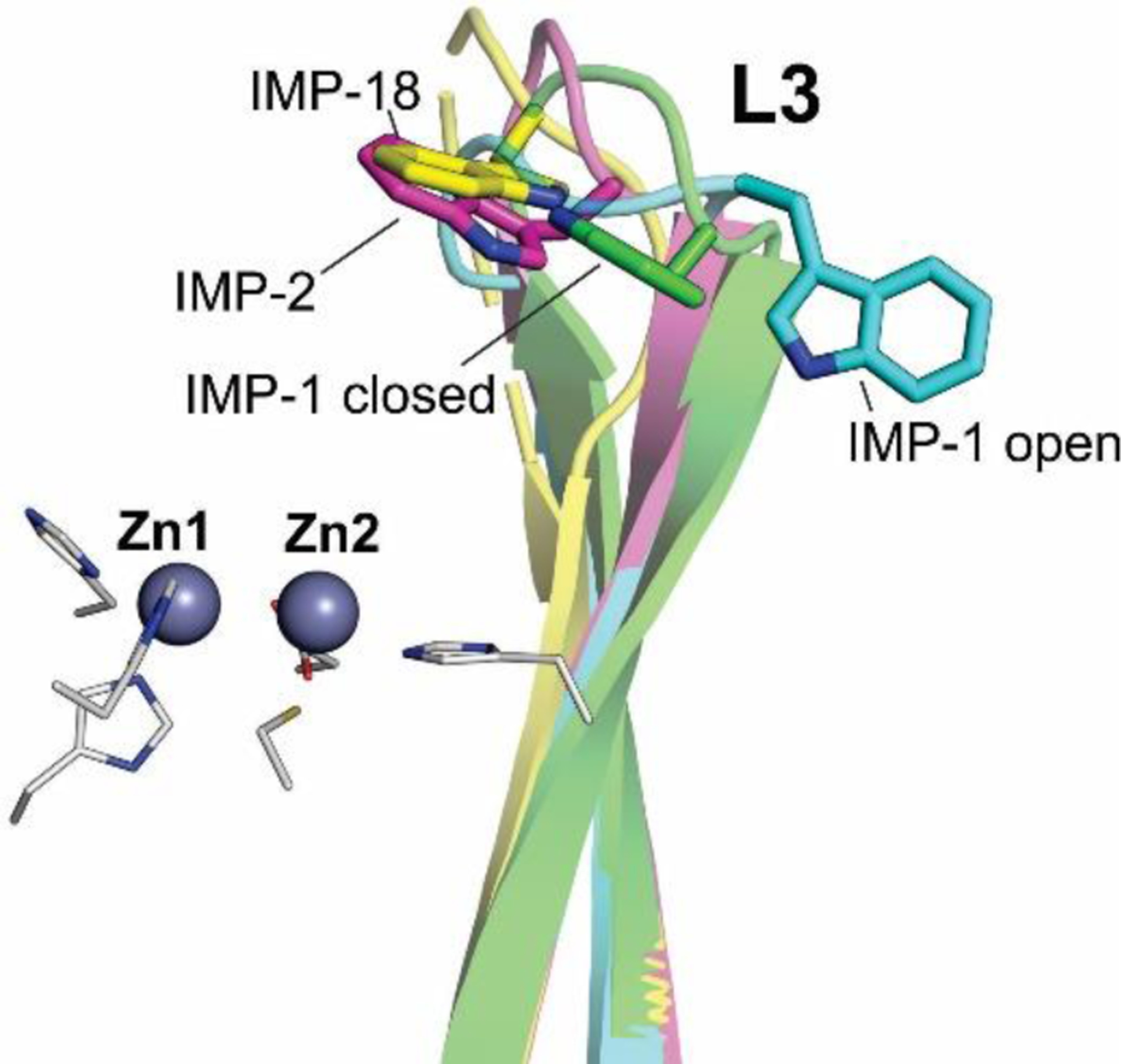
Conformation of the L3 loop from IMP-1 (PDB 1DDK, open), IMP-1 in complex with mercaptocarboxylate (PDB 1DD6, closed), IMP-2 (PDB 4UBQ) and IMP-18 (PDB 5B3R).
Oelschlaeger and co-workers evaluated the role of position 59 located in one of the β-strands forming the L3 loop.413 Residue 59 is Glu in the entire IMP family except for IMP-30 and IMP-55 alleles, in which a Lys is present. IMP-30, which deviates from IMP-1 by the single mutation E59K, presented similar kinetics parameters to IMP-1, with the exception of ceftazidime, for which catalytic efficiency and E. coli MIC were six-fold and two-fold higher, respectively, compared to IMP-1. This allele possesses the highest catalytic efficiency towards ceftazidime of all described IMP variants, and also compared to VIM variants,414 SPM-1415 and NDM-1.416 In silico simulations inferred that in IMP-30 Lys59 stabilizes an enzyme-ceftazidime complex during the catalytic cycle through an interaction with the negatively charged R1 group of ceftazidime, while in IMP-1 the R1 carboxylate occasionally forms a salt bridge with Lys150a.413
Finally, residues Gly65 and Gly63 were shown to be essential for resistance against all substrates, except ampicillin for Gly63.335 Indeed, substitutions at these positions are rarely found across the IMP family, and these include G65S, G65N or G63R. Given that Gly65 and Gly63 contribute to the formation of the β-hairpin turn at the tip of L3 loop, it is possible that these residues are involved in structure stability and/or function of the enzyme.335
4.7.1.1.2. Other characterized IMP alleles
Several biochemical characterizations and resistance level profiles are available for many other clinical IMP alleles. In these cases, however, variations in substrate profiles are difficult to assign to particular amino acid substitutions given that these variants either exhibit multiple substitutions or there are no closely related alleles existing or characterized for comparison. These include IMP-2,411 IMP-13384 and IMP-18386 variants already discussed, and IMP-12,383 IMP-16,385 IMP-27,417 IMP-28,418 IMP-29,387 IMP-31,388 IMP-33419 and IMP-55.420
IMP-16 is atypical in that is unable to hydrolyze cefoxitin.385 In addition, penicillins are poorer substrates than carbapenems and cephalosporins. This variant differs from IMP-74, yet uncharacterized, by a V67F substitution, and from IMP-1 by 30 residues in the mature protein.
IMP-27 lacks hydrolytic activity against piperacillin and exhibits low (~104 M−1s−1) catalytic efficiency for ceftazidime, which explains the susceptibility towards these antibiotics of the P. mirabilis isolate harboring blaIMP-27.417 Furthermore, as explained in Section 9, blaIMP-27 is the only IMP gene identified in a class 2 integron.
IMP-28 showed overall lower MICs and catalytic efficiencies compared to IMP-1, except for cefotaxime.418 The latter was characterized by a systematic drop in kcat values. However, there is no information regarding the metal content of the enzyme (affecting kcat) or the reaction media used in the assays.
IMP-29 exhibits catalytic efficiencies that are slightly higher (2–7-fold for penicillins) or lower (1–2 orders of magnitude for cephalosporins, 1 order of magnitude for imipenem, and <1 order of magnitude for meropenem and ertapenem) compared to IMP-1.387 Mature IMP-29 differs by 24 residues from IMP-1. It is noteworthy that IMP-29 was able to confer resistance to E. coli against ceftazidime (MIC of 16 mg/ml) even though the catalytic efficiency of this antibiotic was ~103 M−1s−1.
IMP-31 showed overall lower catalytic efficiencies for all substrates compared to IMP-1, attributed to lower kcat values (up to 50-fold) except for cefotaxime.388 Accordingly, blaIMP-31-expressing E. coli showed generally lower MICs of all tested β-lactam antibiotics. As with IMP-28, however, no information regarding the metal content of the enzyme was reported. blaIMP-31 and blaIMP-35, its closest relative (97% sequence identity), were identified in class I integrons displaying a similar array of gene cassettes. In fact, these variants were found in isolates from adjacent geographical regions in Western Europe.
Finally, IMP-33 displays five amino acid substitutions compared to its closest relative IMP-13: A38S, A39S, E109K, I223V, and M302L.419 In spite of these substitutions, MIC values of E. coli expressing IMP-33 were similar to those of IMP-13, with ≤ 2-fold variations.
4.7.1.1.3. Concluding remarks and future perspectives
The studies on the IMP family were seminal to expand the knowledge of the role of active-site loops and second sphere residues in the activity profile of these enzymes. Substitutions in several of these positions, alone or combined, are quite frequent in the IMP family and generally result in an improvement of the enzyme activity towards newer carbapenems in detriment of penicillins (and imipenem in many cases). There are still several residues that exhibit a high frequency of substitution in clinical alleles and are located at distant positions from the active site. Although many of them are expected to affect the protein fold, further studies are required to assess the role of these distant mutations. Recent studies on the IMP-1-subgroup revealed that carbapenem hydrolysis (instead of zinc limitation) is the major force driving the evolution of this cluster.395 This agrees with the fact that IMP-1 displays moderate in vivo tolerance to Zn(II) deprivation,421 and higher metal-binding affinities compared to VIM-2 and NDM-1,395 suggesting that this enzyme is partially tailored to resist conditions of nutritional immunity. However, given the great heterogeneity of the IMP family, further studies are required to test this hypothesis in the remaining IMP groups.
4.7.1.2. VIM family
VIM-1 was identified in 1999 in a clinical strain of P. aeruginosa from Verona, Italy by Rossolini, Amicosante and coworkers.422 Soon after, VIM-2 was discovered in an isolate of the same bacterium, originally detected in France in 1996 by Nordmann, Poirel and Naas.423 To date 71 clinical alleles of the enzyme have been reported (although sequences are available for only 69 of them, as VIM-21 and VIM-22 have been assigned but no sequence can be found for either), forming a monophyletic group divided into five clusters (Figure 23). The two larger ones are centered on VIM-1/VIM-4 (with VIM-4 possessing a S228R substitution with respect to VIM-1424 and VIM-2, respectively. A large number of VIM alleles differ from these core enzymes by one or two point mutations, while VIM-1 and VIM-2 present 25 substitutions with respect to each other. Meanwhile, two smaller groups exist around VIM-5 and VIM-13,425 both associated to the VIM-1/VIM-4 group, with the VIM-13 cluster being more divergent. Alleles among all these 4 clusters share ≥ 89% sequence identity. Finally, there is an additional more divergent enzyme, VIM-7,426 with ca. 75 % sequence identity to other VIM family members and with two recently identified related alleles (VIM-61 and VIM-69).
Figure 23.
Phylogenetic tree of the VIM enzyme family. The 5 groups present within the family are highlighted in colors, and the locations of the reference enzymes within each group are shown in green. Protein sequences were aligned using Clustal Omega, phylogenetic trees were constructed using PhyML (via the phylogeny.fr web server) and tree representations were generated with iTOL.
The most frequently encountered allele is VIM-2 followed by VIM-1, with a marked host preference between them, as VIM-1 is prevalently found in Enterobacterales while VIM-2 is almost exclusively present in P. aeruginosa isolates.427–429 This differential host selectivity has been attributed to the efficiency of the signal peptide processing upon export to the periplasm in different bacterial hosts430 (see Section 9).
Early studies on VIM-1 and VIM-2 by Docquier and Rossolini identified these enzymes as broad spectrum MBLs with no clear preference among penicillins, cephalosporins or carbapenems, with most substrates presenting catalytic efficiencies on the order of 105 to 106 M−1 s−1.431 These enzymes presented some particularities in their hydrolysis kinetics which are generally conserved among VIM enzymes, including a combination of very tight affinity towards carbapenems (KM values often below 10 μM) and smallturnover numbers (around 10 s−1) compared to other MBLs that usually present considerably larger values for both parameters. Additionally, cephalosporins with bulky C3 substituents, especially ceftazidime and cefepime, are usually poor substrates of VIM enzymes, with catalytic efficiencies mostly on the order of 104 M−1 s−1. Active-site mutations, however, can improve hydrolysis of these substrates (see below). Finally, although temocillin, a penicillin containing a C6-methoxy group, is also a poor substrate of both VIM-1 and VIM-2 with kcat/KM values around 2 × 104 M−1 s−1, it is hydrolyzed at a much higher efficiency than that observed for IMP-1 (< 1 × 102 M−1 s−1).431, 432
VIM enzymes show variability in the identity of residues 224 and 228, both located in loop L10. In particular, VIM lactamases lack residue Lys224, responsible of interacting with the C3/C4 carboxylate group from bicyclic β-lactams in most B1 and B2 enzymes. This residue is replaced by a tyrosine in VIM-2, and the crystallographic structure of this enzyme suggests that Arg228 (instead of Tyr224) could play a role equivalent to Lys224.282 However, this residue in position 228 is not conserved across VIM MBLs, since 68% possess arginine at this position, while 7% and 23% present leucine and serine residues, respectively, and one allele (VIM-70) has a glycine residue. Regarding position 224, the proportion of VIM variants that possess a histidine, tyrosine and leucine residue is 39%, 46% and 13%, respectively, while VIM-52 is the only VIM allele identified so far that contains arginine. The crystal structure of VIM-1 (possessing His224 and Ser228) revealed that a water molecule forming a hydrogen bond with the backbone carbonyl from Cys221 could fulfill this role in this variant.259 Indeed, a water molecule was noted to be present in this position in numerous available crystal structures of VIM enzymes (Figure 25), and its conserved interactions with the substrate C3/C4 carboxyl group (Figure 26) were proposed to play a role in buffering the interplay of the different residues present in positions 224 and 228 within this MBL family, as a structural module providing an anchoring position for the carboxylate group from the substrate.259
Figure 25.
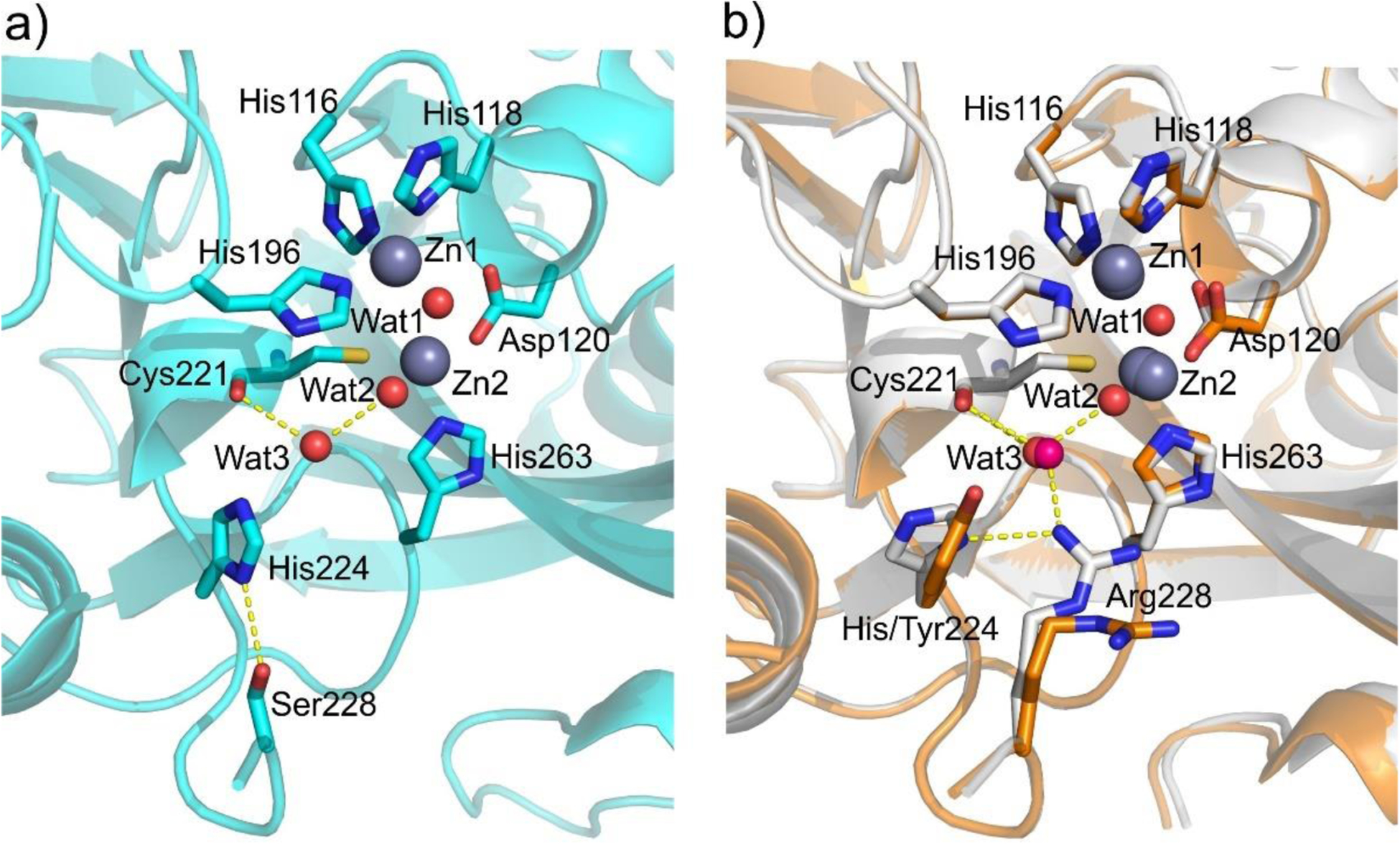
Comparison of VIM active sites.259 a) Active site of VIM-1, showing positions of His224 and Ser228 and location of Cys221-bound water Wat3. Hydrogen bonding interactions are shown as dashed lines (PDB 5N5G). b) Active site superpositions of VIM-2 (PDB 4BZ3, orange) and VIM-4 (PDB 2WHG, white) showing variations at positions 224 and 228.
Figure 26.
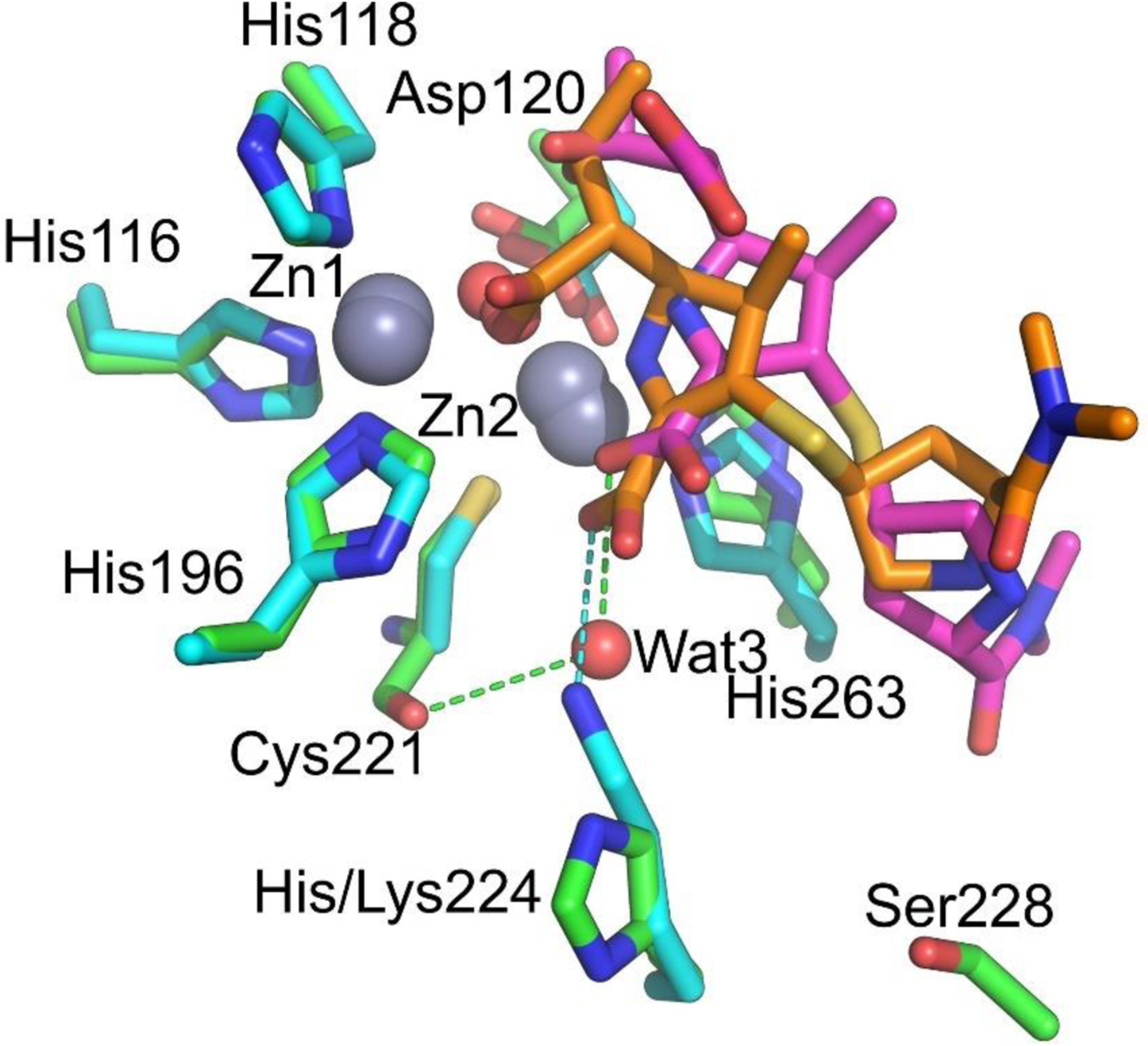
Binding of hydrolyzed meropenem to VIM-1 (PDB 5N5I, green) and NDM-1 (PDB 5N0H, gray).259 Bound hydrolyzed meropenem is shown in pink and orange for VIM-1 and NDM-1, respectively. Interactions of substrate C3/C4 carboxylate with active site residues or water molecules are shown as dashed lines.
The tolerance of VIM-2 to mutations was explored by Chen, Tokuriki and coworkers by deep mutational scanning.237 Interestingly, this work does not only identified essential residues for activity, but also for stability and peptide signal processing, despite the work was performed in E. coli, a non-frequent host for VIM-2.
The substrate spectrum and kinetics of VIM enzymes is largely controlled by the identity of residues at a small number of key positions, mostly in the L10 loop, regardless of their position in the phylogenetic tree. In particular, the largest variation among alleles is generally observed for the hydrolysis of cephalosporins with bulky C2/C3 substituents, especially ceftazidime and (to a lower degree) cefepime (see below). Their hydrolysis is often improved in VIM variants in which Arg228 is replaced by serine or leucine residues, leading to a larger active-site cavity enabling the accommodation of large substituents. On the other hand, this can impair hydrolysis of other substrates that benefit from the interaction of Arg228 with their C3/C4 carboxylate. Position 224 was also observed to be of great relevance in influencing substrate preference (see below). While Tyr224 in VIM-2 was proposed to contribute to tight substrate binding, it is replaced by histidine or leucine in other alleles.
Although the substitution of Arg228 is considered to increase ceftazidime hydrolysis ostensibly due to changes in the active site that allow it to better accommodate the substituents of this substrate, it should be noted that VIM-2 displays a KM value for ceftazidime 10-fold lower than VIM-1 (containing Ser228). This is compensated by a lower kcat, which leads to similar kcat/KM for both enzymes.431 VIM-1 also has a kcat/KM value for penicillin G two orders of magnitude lower than that of VIM-2 and VIM-4 (VIM-1 S228R), and similarly large differences are also observed for ampicillin among VIM-1 and VIM-2.431, 433 Thus, the interaction with Arg228 may aid in better binding of these substrates. In general, the presence of Arg228 led to a similar substrate profile in VIM-4 to that of VIM-2, as the enzyme also presented higher catalytic efficiencies for cephalosporins and carbapenems than VIM-1, and similar to those of VIM-2.433
The interplay of positions 224 and 228 on substrate spectrum can be further illustrated by VIM-13, which possesses 19 substitutions with respect to VIM-1, among them S228R and H224L.434 VIM-13 confers considerably lower resistance towards ceftazidime and cefepime than VIM-1, by 5 and 4 binary dilutions, respectively. Bou and coworkers constructed VIM-13 single and double mutants introducing the residues present at positions 224 and 228 in VIM-1.435 VIM-13 R228S was able to confer increased resistance towards ceftazidime, matching that of VIM-1, but it did not markedly increase cefepime MICs, while the L224H substitution did not have a significant impact on the resistance against either antibiotic. However, the combination of both mutations was synergistic, and VIM-13 R228S L224H presented nearly identical resistance levels to VIM-1 despite the remaining 17 mutations with respect to this enzyme.435 Similarly, VIM-19 (VIM-1 N215K S228R) was found to confer considerably lower resistance towards ceftazidime and cefepime with respect to VIM-1 but showed higher MICs towards carbapenems.436 This correlates with a 10-fold reduction in kcat/KM for ceftazidime and cefepime for VIM-19 with respect to VIM-1, together a 6-fold increase in the catalytic efficiency towards carbapenems, and 8- to 100-fold increases in this parameter for cefotaxime, benzylpenicillin and piperacillin. The R228L substitution differentiates VIM-24 from VIM-2, resulting in an increase of ceftazidime and cefepime MICs in VIM-24, while the resistance levels for other antibiotics were generally unaltered, as shown by Mojica, Bonomo and coworkers.437 This was accompanied by increases in kcat/KM in VIM-24 vs. VIM-2 by 6-fold and 3-fold for ceftazidime and cefepime, respectively, together with similar reductions in catalytic efficiency in substrates with kcat/KM > 106 M−1 s−1. Surprisingly, codon randomization at this position identified additional mutations, such as R228Q and R228M, that not only led to even higher resistance to ceftazidime and cefepime but also higher MICs for all other β-lactams, although currently no such substitutions have been observed in natural VIM alleles.437 In contrast, VIM-23 (VIM-2 R228S) was reported to confer lower resistance than VIM-2 for ceftazidime, cefepime, cefotaxime and ertapenem by 2 binary dilutions, although smaller reductions were also observed for other β-lactams,438 and subsequent studies have observed MICs for ceftazidime to be increased in this allele.425, 439 Meanwhile, VIM-60 (VIM-2 R228L H254R; note that position 254 was 252 in the older BBL numbering) led to greater resistance than VIM-2 for most antibiotics when both enzymes were expressed in the same background, while in contrast kcat/KM values for both enzymes were generally within 2-fold of each other.440 The largest increases in kcat/KM for VIM-60 vs. VIM-2 were 3- to 7- fold increments in catalytic efficiency for the 4-generation cephalosporins (cefepime, cefozopran and cefpirome) for which ≥ 4-fold higher MICs had been observed in cells producing VIM-60.
Loop L3 and residue Trp87 (located at the base of this loop) have been proposed to be involved in substrate binding in various MBLs. However, saturation mutagenesis experiments in VIM-2 by Docquier et al. showed that positions 61, 65 (64 in the older BBL numbering scheme) and 67 in this loop can tolerate substitutions without impacting the capability of the enzyme to confer resistance.332 In particular, substitution A65W led to minor changes in kinetic parameters with no impact in the resistance profile while the reciprocal mutation in IMP-1 was shown to greatly increase KM for most substrates. In contrast, Trp87 was not amenable to substitution in VIM-2, with mutations greatly reducing the resistance conferred due to an impaired protein stability with a limited impact on catalysis.332 These results show that the contribution of loop L3 to enzyme function and substrate recognition cannot be generalized for all B1 MBLs.
The highly divergent VIM-7 is better at hydrolyzing penicillins than either VIM-1 or VIM-2 and also possesses higher kcat/KM for carbapenems than VIM-1, with comparatively lower catalytic efficiencies towards cephalosporins, as shown by Samuelsen, Spencer and coworkers. Meanwhile, cefepime and ceftazidime are very poor substrates (kcat/KM ~1 × 104 M−1 s−1) of VIM-7.414 Among a large number of mutations with respect to VIM-2 (68 substitutions and a deletion of 1 residue close to the N-terminus of the signal peptide), VIM-7 presents the Y218F and Y224H substitutions within the L10 loop and a cluster of mutations within loop L3. Mutants reverting said changes in L10 were analyzed, and the F218Y substitution was found to increase the catalytic efficiencies for most substrates, with kcat/KM values at least 1 order of magnitude higher for cephalosporins and penicillins with respect to VIM-7 mainly due to larger hydrolysis rates.441 This can be accounted for by the presence of additional H-bonds established by the Tyr218 side chain in the mutant with Asn70 at the base of the L3 loop, the conserved Asp84 and Arg121 (also observed for this residue in VIM-2 and VIM-4), as shown by the structure solved by Samuelsen, Leiros, Spencer and others.442 The H224Y substitution was also found to increase the catalytic efficiency of VIM-7, but mainly towards imipenem and the cephalosporins cefepime, ceftazidime and cefoxitin.441 The Tyr224 side chain was observed in the crystal structure of the mutant to establish H-bonds with the Ala231 backbone oxygen and with the side chain of Zn(II) ligand His196 via a water molecule. This mutant presented an altered conformation of the Arg228 present in VIM-7, resulting in a more open active site which could better accommodate the ceftazidime C3 side chain.441
Meanwhile, the presence of the same substitution found at position 218 in VIM-7 was observed within the context of VIM-15 (VIM-2 Y218F) to lead to considerably higher catalytic efficiencies than VIM-2, with 10- to 40-fold higher kcat/KM values for most substrates. However, differences in resistance of strains expressing both enzymes were minor, with the largest change being a 4-fold higher cefotaxime MIC for cells producing VIM-15.443 Regarding substitutions at position 224, VIM-26 (VIM-1 H224L) was found to generally confer lower resistance towards β-lactams than VIM-1, coupled with lower catalytic activity toward cephalosporins but higher kcat/KM values vs penicillins.444 The crystal structure of the enzyme revealed that the combination of Leu224 and Ser228 led to a larger and electrically neutral cavity to bind C2/C3 substituents of β-lactam substrates. The related variant VIM-39 (VIM-1 T33A H224L) was reported to present significantly improved catalytic efficiencies with respect to VIM-1, with ≥ 10-fold increases in kcat/KM for most substrates, and smaller increments with respect to VIM-26. However, while VIM-39 conferred higher resistance levels than VIM-26, MICs for cells producing the former enzyme were very close to those expressing VIM-1, and thus generally did not correlate with the differences observed in catalytic performance.445 A substitution at position 224 is also present in VIM-31 (VIM-2 Y224H H254R), which was found to confer slightly lower resistance levels than VIM-2, including a 4-fold reduction in ceftazidime MICs.446 A general reduction in catalytic efficiencies was also observed for this variant vs. VIM-2, due to a combination of lower kcat and higher KM values. A 7-fold increase in KM was observed for ceftazidime, indicating a role for Tyr224 in binding of this substrate, as had previously been suggested based on the 10-fold higher KM for ceftazidime in VIM-1 (containing His224) with respect to VIM-2. The crystal structure of this variant revealed narrower active-site conformation with altered positions for the L3 and L10 loops with respect to VIM-2, and similar to that of VIM-4 (also containing His224).447 Arg228 was observed to interact with the side chain of His224 in VIM-31, which may restrict the flexibility of this residue and could hamper its interaction with the substrate.
Variant VIM-6 (VIM-2 Q59R N165S) displayed >10-fold increases in catalytic efficiency towards penicillins with respect to VIM-2, along with other 4- to 7-fold increases for various cephalosporins and imipenem, with many kcat/KM values on the order of 107 M−1 s−1.448 This enzyme was subsequently observed to confer substantially higher levels of resistance towards ceftazidime compared to VIM-2.425, 439 In contrast, variant VIM-11 (VIM-2 N165S) presented similar kinetic parameters to VIM-2, with 2- to 4-fold increases in kcat/KM for cephalosporins with bulky C3 substituents (cefepime, ceftazidime, cefpirome) that were not correlated in the resistance conferred by this variant, although there were indications of lower levels of VIM-11 in periplasmic extracts that could reflect lower expression of the enzyme than VIM-2.449
These observations were complemented and expanded by a characterization of VIM variants by Galán and coworkers.425 A phylogenetic analysis suggested that VIM-1 has arisen from VIM-4 (via the R228S substitution) and identified positions under selective pressure both in the VIM-1/VIM-4 and VIM-2 clusters. The strongest evidence of selective pressure was found for residues 57, 59, 224 and 228, followed by positions 165, 215, and 254, and a lower degree of evidence for position 218. Characterization of resistance conferred by simple and double combinations of natural mutations at these positions within VIM-2 showed that simple mutants tended to increase ceftazidime MICs, particularly via the R228S and R228L substitutions, and this was generally accompanied a by a decrease in resistance to ertapenem and piperacillin/tazobactam. Meanwhile, double mutants that included the R228S substitution almost universally led to a global decrease in resistance, and in contrast those containing the R228L mutation led to further increases in ceftazidime resistance while alleviating the antagonism for hydrolysis of other antibiotics. A similar pattern was observed within the VIM-1/VIM-4 group, with the R228S and N165S mutations greatly increasing MICs towards ceftazidime (and cefepime), while double mutants including these substitutions additionally improved resistance towards cefotaxime and to carbapenems to a lesser degree. All in all, this led to the proposal that ceftazidime would be the main driving force for diversification and selection of mutations within VIM alleles, also supported by in vitro evolution experiments. Interestingly, double mutants leading to large increases in carbapenem MICs were detected within both VIM clusters, but of these only VIM-27 (VIM-4 A57S R228S) was observed to occur in nature, in spite of the individual mutations being present in clinical alleles, which would suggest a lower impact of carbapenems in selective pressure within this MBL family. On the other hand, Papagiannitsis and coworkers reported that VIM-27 conferred lower imipenem resistance than VIM-1, from which it differs only by the A57S substitution.450
It must be noted however that such differences in catalytic efficiencies and substrate preferences have not been observed in all studies, and that there may be other factors under selection within VIM alleles. A comparison of the kinetic parameters of VIM-1, VIM-2, VIM-5 (VIM-1 A130K, H224L, E225A, S228R, K291T) and VIM-38 (VIM-5 A316V) by Schofield and coworkers revealed minor (within 2-fold) differences in kcat/KM for ceftazidime and other β-lactams.451 Instead, this study revealed considerable different inhibition profiles and thermostabilities of these enzymes, with Tm values ranging from 60°C to 83°C. These changes could reflect different active-site structures and an altered propensity for in vivo proteolysis and/or aggregation, but this has not been tested. A study of variants within the VIM-1/VIM-4 and VIM-2 clusters by Crowder et al. did find appreciable differences in ceftazidime resistance among alleles, and also identified significant variations regarding the impact of limiting Zn(II) conditions.439 Various alleles showed a greatly increased capability to confer resistance upon metal starvation (see Section 10), particularly VIM-20 (VIM-2 H254R), VIM-28 (VIM-4 H224L), VIM-37 (VIM-4 A57S) and VIM-59 (VIM-1 H254R). The H254R substitution (H229R in VIM-2 residue numbers) in VIM-20 did not appreciably alter its kinetic parameters nor the resistance it conferred under standard test conditions versus VIM-2, but the increased resistance under metal depletion was found instead to be correlated with the formation of salt bridge between this arginine residue with E188 in the crystal structure of the enzyme, which led to greater thermostability of the purified protein and in vivo stability under upon Zn(II) limitation. A similar salt bridge could potentially form in VIM-59 and explain its better tolerance to Zn(II) starvation.439 Thus, as also observed for allelic variants of NDM,452 the ability of VIM enzymes to endure and confer resistance under metal starvation seems to be a characteristic under selective pressure.
VIM-12 is a chimeric variant with a sequence corresponding to VIM-1 but with about 45 residues of its C-terminus replaced by those of VIM-2, and could have originated from a recombination event of these two alleles.453 According to the described proportions of each variant contained in the chimera, VIM-12 is phylogenetically closer to VIM-1 (8 substitutions) than to VIM-2 (17 substitutions). Although the active-site positions of the enzyme are those of VIM-1, including L10 residues (His224, Ser228), it was reported to possess a very restricted substrate spectrum: only penicillin G was hydrolyzed efficiently, while imipenem was very poorly degraded, and no hydrolysis of meropenem or ceftazidime was observed. The structural bases of these observations are unknown. Another unusual VIM variant, differing from VIM-2 by 8 substitutions and a C-terminal extension, was reported as VIM-48,454 but its sequence does not correspond to allele VIM-48 as identified in BLDB and it does not currently appear in this database. These mutations lead to 2 to 4-fold increases in resistance to some β-lactams, but do not result in significant changes in the enzyme kinetics.
In summary, positions within the L10 loop, particularly 224 and 228 are key determinants of the catalytic profile of VIM enzymes. Evidence points to selective pressure to grant higher resistance towards ceftazidime having driven the diversification of VIM alleles, but further studies will be required to ascertain the intriguing roles posited in recent studies for optimization in stability and tolerance to Zn(II) deficient conditions in the evolution of these enzymes.
4.7.1.3. SPM-1
First reported in Brazil in 2002 by Gales, Walsh and coworkers,455 SPM-1 (São Paulo MBL) is noteworthy among acquired lactamases in that it has been isolated almost exclusively in P. aeruginosa, and is associated to one specific clone (ST277).456, 457 Furthermore, it has not had significant geographic expansion beyond this country despite representing an important clinical carbapenemase there.16, 457–459 There have been only isolated reports of the enzyme in the rest of the world, and only in one instance it has been observed to be produced by a different organism, namely one reported isolate of Acinetobacter baumannii.460. Although SPM-1 was originally described to be codified in a plasmid,455 subsequent studies observed the blaSPM-1 gene to be located within mobile genetic elements in the chromosome of the corresponding P. aeruginosa isolates.461, 462
SPM-1 is a broad spectrum MBL, with some preference towards cephalosporins.415 The structure of the enzyme is unusual, in that it lacks the conserved Asp84 and has a much shorter L3 loop, possessing instead a 24-residue insertion in helix α3 that leads to an extended helical segment bordering the active site in a manner similar to B2 enzymes (Figure 14). Deletion of this extension of α3 impairs the catalytic activity of SPM-1 between three- and 32-fold, i.e., less than deletion of the L3 loop in other B1 enzymes.281 Different crystallization conditions trapped a closed conformation of the α3 region that is more reminiscent of the conformation present in B2 enzymes348 (Figure 15). The open conformation featured an active site with an oxidized Cys221 and a vacant Zn2 site, while the enzyme with the closed conformation possessed a canonical binuclear B1 center. MS and NMR data further supported the mobile nature of these structural elements in solution.348, 349 The fact that SPM-1 contains features from both B1 and B2 subclasses allowed the design of a chimeric protein in which the insertion sequence present in the exclusive B2 carbapenemases replaced the 24-residue insertion in SPM-1.463 This resulted in a selective cephalosporinase, confirming that this section of the active site can tune the substrate spectrum of MBLs.
SPM-1 also posseses a particular H-bond network below the active site, since it is disrupted by the presence of residues S84 and G121 at the second coordination sphere, instead of the typical D84/R121 pair present in most B1 enzymes281 (Figure 16). This combination of residues has been shown to optimize the Zn(II) binding abilities and the resistance profile of SPM-1 against anti-pseudomonal cephalosporins, while sacrificing the catalytic efficiency against substrates towards which the pathogen is naturally resistant.464 This is a unique example of host adaptability in MBLs by means of second-shell residues.464
Two recently described MBLs from environmental bacteria, PAN-1465 and SPS-1,230 possess high sequence identity to SPM-1, including the distinct insertion leading to the extended α3 helix. The crystal structure of SPS-1 reveals the close similarity to SPM-1.230 This suggests the existence of a larger set of MBLs with similar structural characteristics to SPM-1.
4.7.1.4. NDM family
The New Delhi Metallo-beta-lactamase (NDM-1) was first reported in literature in 2009 by Walsh, Toleman and coworkers.416 It was identified in isolates of Klebsiella pneumoniae and Escherichia coli from a Swedish patient, who was transferred home after multiple hospitalizations in India due to bacterial infections. Retrospective studies have detected the presence of the coding gene in Enterobacterales isolated in the region in 2006 and 2007, indicating a period of silent dissemination before its discovery.466
Among clinical MBLs, NDM-1 showed upon its discovery the greatest similarity to VIM-2, (32% sequence identity). More recently, the B1 MBL FIM-1 shows higher homology (40% identity).367 Several MBLs from environmental sources, such as MYX-1 from Myxococcus xanthus, ANA-1 from Anaeromyxobacter sp.,467 and especially ElBla2 from Erythrobacter litoralis,468 also display high homology to NDM-1, with 32%, 33% and 55% sequence identity, respectively.
NDM-1 has been detected in a wide variety of bacterial hosts, but most clinical isolates producing the enzyme correspond to Enterobacterales (especially E. coli and K. pneumoniae), P. aeruginosa and A. baumannii.469–471 The coding gene has been found mainly within plasmids belonging to different incompatibility groups, and its global dispersion is not associated to any particular clone, plasmid or genetic structure.472 The immediate genetic environment of the blaNDM-1 gene usually contains an ISAba125 sequence in the 5’ direction, either complete or truncated, and the bleomycin resistance gene bleMBL in the 3’ direction.472, 473
A feature that distinguishes NDM-1 from all other MBLs of clinical importance is the presence of a bacterial lipidation signal (termed lipobox) within the signal peptide of the protein. This sequence was identified on the basis of a bioinformatic analysis of the NDM-1 signal peptide,474 and the membrane localization of the enzyme was experimentally confirmed in two independent studies.421, 474 This sequence is recognized by a widely conserved protein lipidation system, and directs the protein to the inner bilayer of the outer membrane in Gram negative bacteria instead of resulting in a soluble periplasmic protein as other MBLs (see Section 8.1). A recent report has challenged these findings, suggesting that NDM-1 is a soluble periplasmic enzyme with a suboptimal signal peptide inefficiently processed by signal peptidase I.475 This study, however, did not include cell fractioning experiments to analyze the localization of the enzyme or the ratios of soluble and membrane-bound fractions. The lipidated form of NDM-1 has not been characterized biochemically, but processing of the enzyme in E. coli was found to be impaired by globomycin,421 a specific lipoprotein biogenesis inhibitor (see Section 8.1), confirming the post-translational modification. A recent study has characterized the conformation of NDM-1 in contact with the membrane surface, and also found residual binding to membranes of a soluble mutant of the enzyme, as a result of a membrane-interaction surface in the protein.476
The presence of ISAba125 upstream of NDM-1 was analyzed in detail by Toleman, Spencer and Walsh, who found that the intervening 100 bp region between this insertion sequence and the MBL gene was conserved among a set of NDM-1 genetic environment sequences, and that this segment together with the first 19 bases of blaNDM-1 aligned perfectly with the N-terminus and upstream sequence of an aphA6 gene from Acinetobacter baumanii.477 These findings suggested that NDM-1 is a chimera, with its first 6 amino acids being derived from the aminoglycoside resistance protein AphA6 by an in-frame fusion of the aphA6 gene with that of a shorter precursor MBL (the sequence of this protein would correspond to that starting from Met7 in the current NDM-1). Furthermore, various lines of evidence point to this event having occurred in Acinetobacter, among them the strong association of this bacterium and ISAba125 sequences. This putative precursor protein also possesses a lipidation signal, as determined by the bioinformatic tool LipoP.477
So far, 29 natural alleles of this enzyme have been reported. NDM variants share a higher degree of sequence identity than that observed for other MBL enzyme families, as they differ in most cases by only 1 to 3 point mutations (Table 2, Figure 27). The most divergent members are NDM-10 (with 5 amino acid substitutions) and NDM-18 (which contains a duplication of a 5-residue stretch close to its N-terminus). The lipidation signal in the leader peptide is conserved in all alleles identified so far, but at the moment the membrane localization has been experimentally confirmed only for alleles NDM-1 to NDM-16.452
Table 2.
Mutations present in NDM allelic variants, indicated both in the numbering corresponding to the primary sequence of the protein, and also using BBL numbering.
| Allelic variant | Mutations with respect to NDM-1 | |
|---|---|---|
| Primary sequence numbering | BBL numbering | |
| NDM-1 | - | - |
| NDM-2 | P28A | P22A |
| NDM-3 | D95N | D89N |
| NDM-4 | M154L | M150aL |
| NDM-5 | V88L, M154L | V82L, M150aL |
| NDM-6 | A233V | A248V |
| NDM-7 | D130N, M154L | D126N, M150aL |
| NDM-8 | D130G, M154L | D126G, M150aL |
| NDM-9 | E152K | E149K |
| NDM-10 | R32S, G36D, G69S, A74T, G200R | R26S, G30D, G63S, A68T, G207R |
| NDM-11 | M154V | M150aV |
| NDM-12 | M154L, G222D | M150aL, G235D |
| NDM-13 | D95N, M154L | D89N, M150aL |
| NDM-14 | D130G | D126G |
| NDM-15 | M154L, A233V | M150aL, A248V |
| NDM-16 | R264H | R305H |
| NDM-17 | V88L, M154L, E170K | V82L, M150aL, E177K |
| NDM-18 | Duplication of G42-F46 (GDQRF) | Duplication of G36-F40 (GDQRF) |
| NDM-19 | D130N, M154L, A233V | D126N, M150aL, A248V |
| NDM-20 | V88L, M154L, R270H | V82L, M150aL, R311H |
| NDM-21 | G69S, V88L, M154L | G63S, V82L, M150aL |
| NDM-22 | M248L | M261L |
| NDM-23 | I101L | I95L |
| NDM-24 | V88L | V82L |
| NDM-25 | A55S | A49S |
| NDM-26 | V88L, M154L, G222S | V82L, M150aL, G235S |
| NDM-27 | D95N, A233V | D89N, A248V |
| NDM-28 | A266V | A307V |
| NDM-29 | D130N | D126N |
Figure 27.
Phylogenetic tree of the NDM enzyme family. The location of NDM-1 is shown in green. Protein sequences were aligned using Clustal Omega, phylogenetic trees were constructed using PhyML (via the phylogeny.fr web server) and tree representations were generated with iTOL.
The substitutions observed in NDM variants occur outside the active site, and do not correspond to residues involved in substrate recognition or from the second coordination sphere (Figure 28), in contrast to the most common mutations reported in the IMP or VIM families. As a result, NDM variants do not show clear differences in substrate preferences.452, 478, 479 However, various alleles have been informed to confer higher resistance levels than NDM-1, in particular towards carbapenems (see below). Remarkably, resistance levels for strains expressing NDM variants were observed by different groups to be higher when the enzyme was expressed using its native promoter (derived from ISAba125) rather than a promoter from the expression plasmid, most noticeably against carbapenems.478, 480–483 The causes of this phenomenon are not known.
Figure 28.
Structure of NDM-1 (PDB 4EXY) highlighting the positions presenting sequence variation within the NDM enzyme family. The positions are colored in the cartoon representation according to increasing absolute frequency of mutation within NDM enzymes belonging to each group. The side chains of those positions with a frequency of substitution in the top 4 categories are shown as sticks.
The most frequently mutated position within NDM variants is, by far, residue 150a (residue 154 in the enzyme sequence), with the M150aL substitution being present in 12 alleles and an additional allele containing the M150aV substitution (Figure 29, Table 2). The greatest reported resistance corresponds to variants containing the V82L, M150aL or D126G/N substitutions.
Figure 29.
Network diagram of the NDM family, showing residue substitutions generating the different allelic variants. The color coding indicates the number of mutations with respect to NDM-1, as indicated in the figure.
Both NDM-5 (NDM-1 V82L M150aL) and the related variant NDM-17 (NDM-1 V82L M150aL E177K) were reported to confer 2- to 8-fold higher resistance towards carbapenems than NDM-1.480, 483 E. coli cells producing NDM-21 (NDM-1 G63S V82L M150aL) presented similar carbapenems MICs compared to those expressing NDM-5, and thus would also confer higher resistance to these β-lactams than NDM-1, but no direct comparison to this latter enzyme is available.484 NDM-20 (NDM-1 V82L, M150aL, R311H) conferred 4-fold higher resistance than NDM-1 towards ertapenem and ceftazidime, while the resistance against imipenem and most other substrates was unaltered.485 NDM-24 (NDM-1 V82L) presented a 4-fold increase in ertapenem MIC with respect to NDM-1 despite this conservative replacement, with minor or no changes for other substrates.486
Variant NDM-7 (NDM-1 D126N M150aL) presented increased MICs by ≥ 2-fold only for various carbapenems vs. NDM-1,481 while another report found identical MICs for carbapenems for cells expressing NDM-7 vs. those producing NDM-4 (NDM-1 M150aL).487 Increases in MICs for certain cephalosporins were also later reported for strains expressing NDM-5 and NDM-7 vs. NDM-1,488 and both NDM-7 and the related triple mutant NDM-19 (NDM-1 D126N M150aL A248V) were reported to confer resistance levels that were 2–4 binary dilutions higher than NDM-1 for cefepime and various carbapenems.489 While NDM-19 was observed to confer similar resistance levels than NDM-7 under standard conditions, MICs for cells expressing the former enzyme were higher in Zn(II)-depleted media. Meanwhile, NDM-4 (NDM-1 M150aL) was found to increase MICs for imipenem and ertapenem by 2-fold with respect to NDM-1,490 and an about 4-fold increment in meropenem and imipenem MICs was also observed for NDM-14 (NDM-1 D126G) while values for other β-lactams were outside the measured range.482
On the other hand, only minor differences in MICs vs. were reported for cells producing NDM-1 vs. those expressing variants NDM-3 (NDM-1 D89N),491 NDM-8 (NDM-1 D126G M150aL),492 NDM-12 (NDM-1 M150aL G235D)493 and NDM-13 (NDM-1 D89N M150aL).494 The more divergent variant NDM-10 (NDM-1 R26S G30D G63S A68T G207R) was observed to be present in a K. pneumoniae isolate presenting high levels of resistance towards β-lactams, although this bacterium possessed various SBLs in addition to the enzyme and no comparison was performed with NDM-1 in an isogenic background.495 Curiously, subsequent studies have indicated that this variant confers significantly lower resistance than NDM-1.452, 479 These findings cannot account for the selection of this variant in a clinical environment.
Regarding the in vitro enzyme kinetics, NDM-1 displays a wide substrate spectrum, like most B1 MBLs, with catalytic efficiencies on the order of 105–106 M−1 s−1 for penicillins, cephalosporins and carbapenems.416, 478 Studies comparing β-lactam hydrolysis by NDM-1 with the subsequent alleles revealed very similar kinetic parameters.478 Only slight differences for catalytic efficiencies vs. NDM-1 (≤ 2-fold variations in kcat/KM for all or most substrates) were observed for NDM-3,491 NDM-4,490 NDM-8,492 NDM-9 (NDM-1 E149K),496 NDM-13 (NDM-1 D89N M150aL)494 and NDM-14 (NDM-1 D126G).482 Although NDM-12 (NDM-1 M150aL G235D) presented slightly lower kcat/KM values for most β-lactams compared to NDM-1, it displayed ≥10-fold reductions in this parameter for cefoxitin, cefradine and moxalactam, accompanied by a a 4-fold reduction in MICs for cefoxitin.493 However, it should be noted that changes in resistance were not always correlated with determined kinetic parameters, since kcat/KM values for carbapenems were found to be very similar between NDM-14 and NDM-1,482 and the same situation was reported for NDM-4 and NDM-1490 in spite both enzymes were reported to confer greater resistance to these β-lactams. A similar phenomenon was observed for NDM-24.486 Conversely, a report comparing the first 8 NDM alleles found increases in resistance towards carbapenems for NDM-4, NDM-5, NDM-6 and NDM-7, despite the kinetic parameters across the variants were very similar for all tested β-lactams.482
In contrast to the previously described observations regarding MIC levels, two studies comparing alleles NDM-1 to NDM-16452 and NDM-1 to NDM-17479 found no major differences in resistance conferred by these enzymes, except for the particular case of NDM-10 (that will not be discussed further). Kinetic parameters for variants NDM-1 through NDM-17 were also reported to be very similar, generally with only 2- to 3-fold variations in kcat and KM and with NDM-1 displaying the largest kcat/KM values for all substrates tested: ampicillin, chromacef and meropenem.479 However, significant differences in resistance among alleles were observed in MIC assays in a Zn(II)-depleted extracellular milieu, elicited by the addition of metal chelators (either EDTA or dipicolinic acid – DPA) (Figure 30). Most variants were able to confer higher resistance than NDM-1 under such conditions.452, 479 Indeed, strains expressing various alleles were able to outcompete those producing NDM-1 when grown together in presence of β-lactams and metal chelators.452 Furthermore, the difference in resistance under low Zn(II) was found to be larger for double and triple mutants than those containing a single substitution. The accumulation of mutations within NDM alleles progressively improves their tolerance to conditions mimicking the environment encountered by the enzyme in infection sites, as a result of metal chelation by the vertebrate innate immune system (see Section 10.2). In particular, the most frequent mutation among NDM alleles, M150aL, was observed to increase the Zn(II) affinity of the enzyme,452, 497 while no such effect was observed for substitution M150aV (present only in NDM-11). The presence of a Leu residue in this position was reported to act as a buttress for His118, which may explain its effects over Zn(II) binding despite not being located directly in the active-site cavity.497 NDM variants were also reported to be active in mono-Zn(II) forms, and the M150aL substitution to increase catalytic activity of these mononuclear enzymes.479 Meanwhile, substitution A248V was observed to increase the in vivo stability of the enzyme towards proteolysis.452 Increases in thermostability have also been found for most NDM alleles in comparison with NDM-1.478, 479
Figure 30.
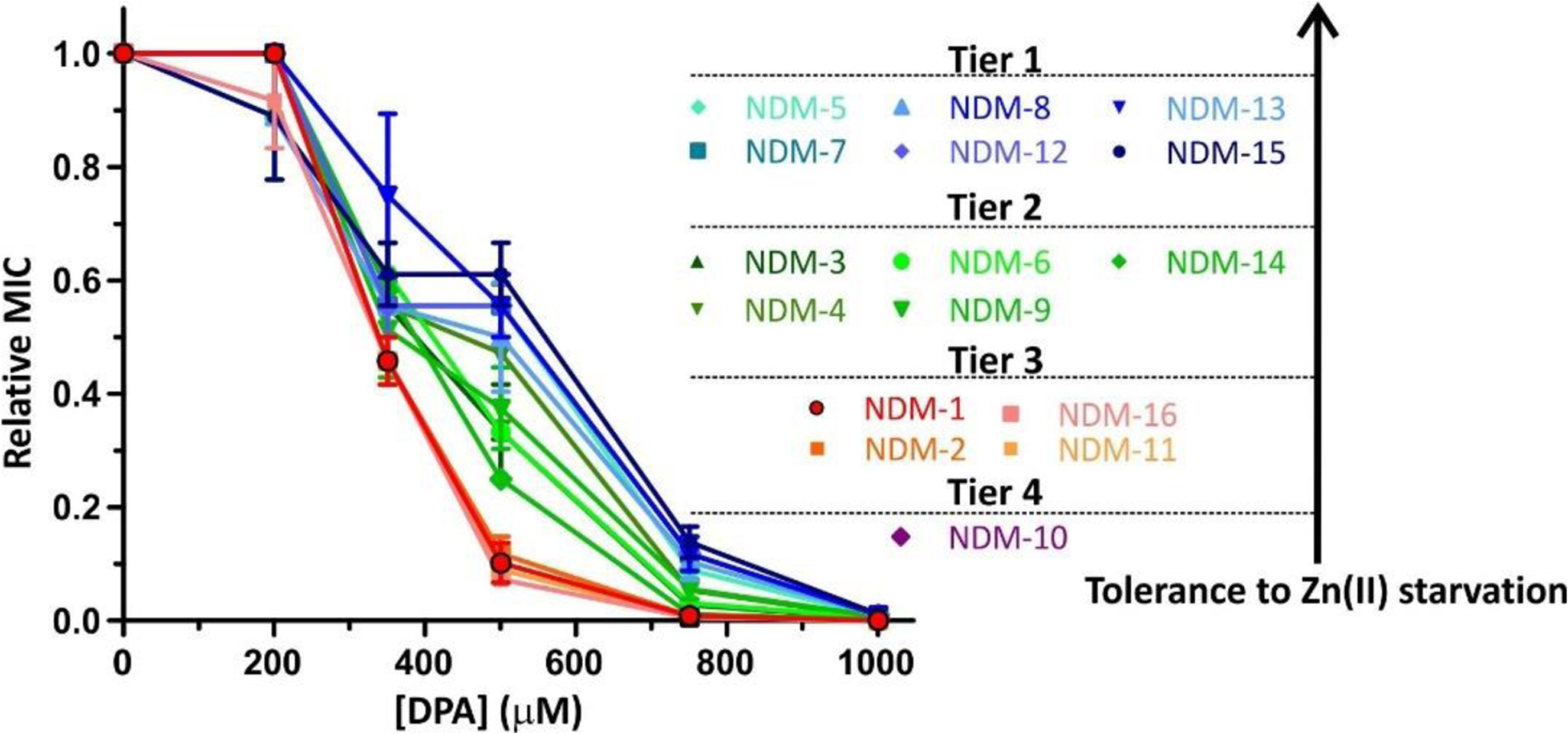
Allelic variants of NDM display an increased tolerance to Zn(II) starvation with respect to that of NDM-1.452 MIC values of cefotaxime for E. coli cells expressing different NDM variants in growth medium supplemented with increasing concentrations of dipicolinic acid (DPA), shown relative to the MIC in 0 μM DPA.
In vitro mutagenesis studies have provided additional information regarding the structural determinants for enzyme function in NDM-1. A saturation mutagenesis study of NDM-1 targeting 31 positions around the active site followed by selection using ampicillin, cefotaxime or imipenem was carried out by Palzkill and coworkers, in order to evaluate important positions for hydrolysis of each β-lactam class. A group of residues were found to be critical for hydrolysis of all substrates, as the WT amino acid was always predominantly selected independently of the antibiotic.236 These include all Zn(II) ligands and conserved residues Asp84 and Asp199, in agreement with previous studies.498, 499 Another set of residues were found to be context-dependent, in which the selected residue varied according to the antibiotic used: Trp87, Lys121, Gly195, Thr197, Gly220, Lys224, Gly232, Asn233, Gly262.236 Mutations on these positions mostly affected imipenem resistance. A previous report had also found substrate dependent impact on catalysis and resistance of certain substitutions at positions 87, 224 and 233, along with residues in the L3 loop.500 Based the previously mentioned results, the triple mutant NDM-1 K224R G232A N233Q was constructed, which displayed a nearly 600-fold and 250-fold reductions in imipenem and meropenem kcat/KM values, respectively. Meanwhile the activity for ampicillin was maintained and a 50-fold reduction was observed for cefotaxime. The crystal structure of this mutant showed an altered L10 loop, with the Gln233 side chain oriented out of the active site and replaced instead with that of Leu234, while Ala232 protruding into the active site could cause steric clashes on the C2/C3 substituents of the substrate.236 In concordance with previous observations for IMP-1 and AIM-1, the hydrolysis of carbapenems in wide-spectrum MBLs appears to have more stringent active-site structure requirements than that of other β-lactams.
The role of loop L3 within NDM-1 was explored in several works. Replacement of the native loop for those present in VIM-2 and IMP-1 resulted in different conformations, revealing that the sequence of the loop rather than the protein scaffold determine the spatial arrangement of the loop (Figure 31). It was also shown that the more closed conformations of the loop elicited a larger accumulation of the reaction intermediate.337 Removal of different residues from the loop or substitution of hydrophobic side chains by polar or charged residues lead to higher KM values although effects were not uniform for all substrates tested.501 In the case of imipenem, KM increases in these artificial loop variants were partially compensated by increments in kcat, but this did not occur for ampicillin, while KM values with the chromogenic cephalosporin CENTA were actually lower in some L3 deletion mutants. Additionally, enzyme inhibition for both WT NDM-1 and most mutants was observed at high concentrations of CENTA. In contrast, deletion of residues 149–150a (i.e. comprising the positions of mutations M150aL/V and E149K observed in natural alleles) led to decreases in both kcat and KM. All in all, modifications at both these positions still resulted in active enzymes, and the reductions observed in kcat/KM were <10-fold. This suggests that while the L3 loop seems to contribute to substrate binding in NDM-1, it is not essential for enzyme functionality. Meanwhile, substitution of W87, located next to the base of the loop, resulted in lower capability to confer resistance and stability in mutant NDM-1 W87A.502
Figure 31.
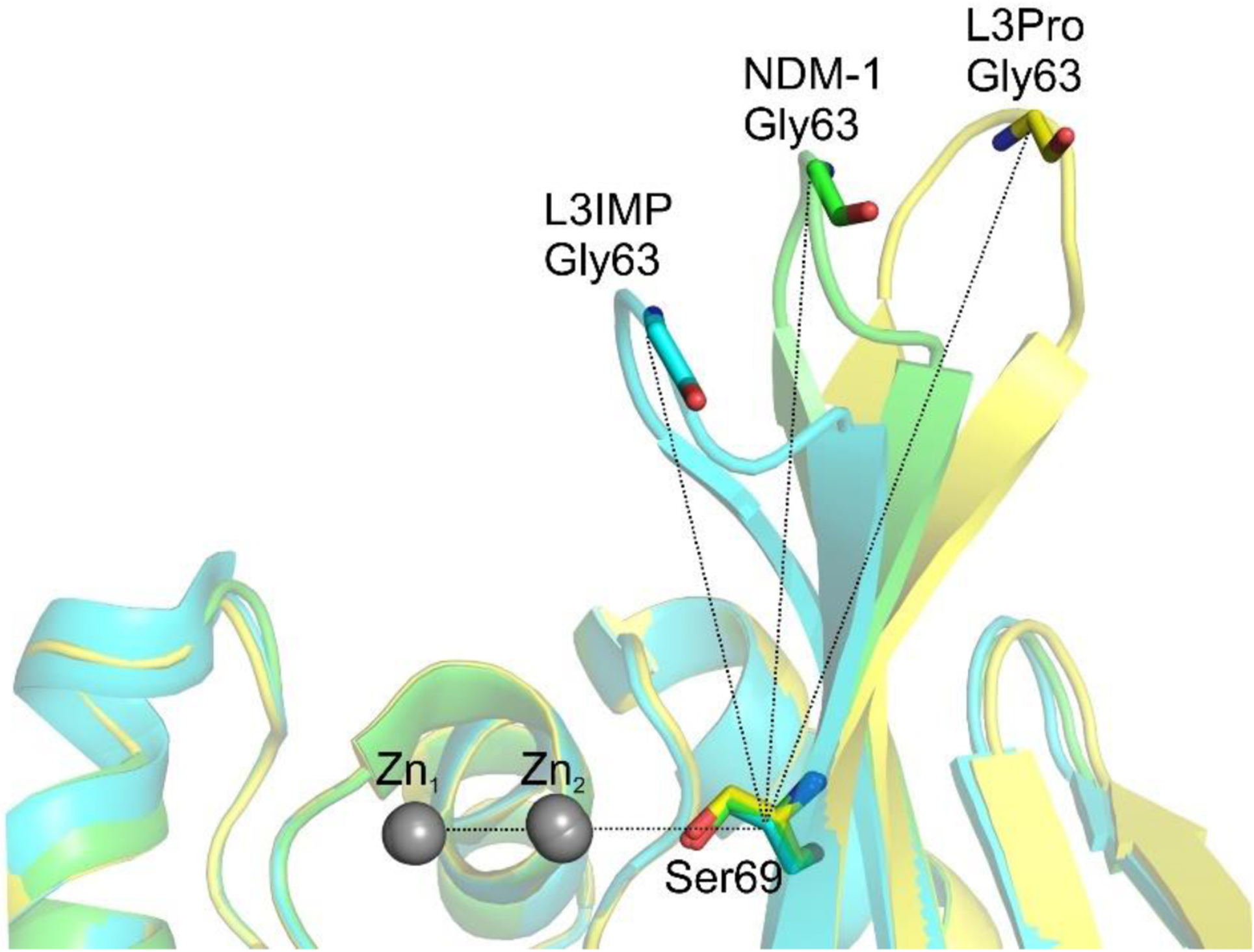
Angle determined by the loop L3 and the plane of the active site of NDM-1 (PDB 3SPU, green), and L3IMP (PDB 6C6I, cyan) and L3Pro (PDB 6CAC, yellow) mutants.337 Angles between Zn1, Cα of Ser69 and Cα of Gly63 are as follows: L3IMP, 68°; NDM-1, 88°; and L3Pro, 110°. L3IMP corresponds to NDM-1 in which the L3 loop (residues 57 to 68) was replaced by the one from IMP-1; L3Pro is a mutant of NDM-1 with a Pro inserted after Ala68.
Residue Gln119, located between the Zn(II) ligands His118 and Asp120, is highly tolerant to mutation in NDM-1. A set of six mutants with diverse substitutions (Q119C/F/E/Y/V/K) displayed either similar or higher kcat/KM compared to WT NDM-1 for many substrates.503 In particular, 9- to 70-fold increases in kcat/KM were observed for various substrates (benzylpenicillin, carbenicillin, meropenem, cefotaxime, ceftazidime, cefepime) in mutant NDM-1 Q119K with respect to the WT while the hydrolysis of others was less affected. Increases in kcat/KM ≥ 10-fold were observed for several β-lactams due to substitutions Q119E, Q119V and Q119Y, with the latter displaying a 90-fold higher kcat/KM for ceftazidime than WT NDM-1. In contrast the Q119D substitution had been previously reported to severely reduce catalytic activity of NDM-1, possibly due to impaired positioning of the adjacent Zn(II) ligands.500, 504
While other B1 MBLs present a Trp at position 244, NDM-1 possesses a Tyr residue at this site (Tyr229 in NDM primary sequence numbering), the same as observed for B2 and B3 MBLs. The Tyr side chain was observed to establish an H-bond with the backbone of Leu222 in NDM-1, adjacent to Zn2 ligand Cys221. Although reintroducing the Trp observed in other B1 enzymes led to up to 7-fold greater kcat/KM values in mutant NDM-1 Y244W with respect to WT NDM-1, this variant conferred lower β-lactam resistance levels.505 Meanwhile, replacement of Leu222 (Leu209 in NDM primary sequence numbering) via the L222F mutation caused to nearly 100-fold reductions in kcat values vs. NDM-1 for all substrates tested, leading 70- to 100-fold lower kcat/KM for cephalosporins and carbapenems, although Zn(II) binding was not impaired. The impact on kcat/KM for cephalosporins was less drastic, with only 3 to 5-fold lower values than NDM-1, due to compensation by smaller KM values.506 However, the combination of both mutations in NDM-1 L222F Y244W restored catalytic activity with respect to NDM-1 L222F, with similar kcat/KM values to WT NDM-1.507
In summary, multiple lines of evidence point to mutations found within NDM allelic variants as having adaptive roles, instead of being merely the result of genetic drift. Further studies are necessary, however, to fully characterize the impact of these mutations, in particular regarding the capability of NDM variants to confer resistance vs. NDM-1. Future analyses characterizing a large number of NDM alleles are required to resolve the differences among different studies with respect to the kinetic parameters and the resistance capabilities of these enzymes.
4.7.2. Subclass B2
The B2 subclass is a small group within MBLs initially reported by Rossolini and coworkers, but with a marked distinction from all other class B β-lactamases since they are strict carbapenemases508–511 which are active with only one metal ion bound, in the Zn2 (DCH) site,245, 246 with a high binding affinity.510 Indeed, the affinity of CphA towards Zn(II) is in the low pM range, being the strongest reported affinity among all MBLs.510 Meanwhile, the low affinity binding of a second Zn(II) ion leads to non-competitive enzyme inhibition.247, 248 Most of the known B2 proteins belong to the CphA family of enzymes from aquatic bacterium Aeromonas hydrophyla512 and other species from the genus.16 CphA has been characterized in detailed by Amicosante, Galleni and coworkers.234, 247, 313, 513, 514 ImiS, a related B2 enzyme from Aeromonas veronii used as a model in numerous in vitro studies by the Crowder lab,323, 351, 515, 516 possesses a 96% identity to CphA and is classified as a CphA allele in the BLDB database (CphA11). Other members are SfhI from Serratia fonticola,517 the recently described PFM family,518 from Pseudomonas spp., and YEM-1 from Yersinia mollaretti.519 Notably, this last enzyme has been recently reported to possess an even more restricted substrate spectrum: it efficiently hydrolyzes imipenem but binds various other carbapenems with very high KM.519 The low affinity towards these substrates was shown to be partially alleviated by the introduction of the Y67V substitution, targeted to this residue of the active-site wall.519
B2 MBLs also possess high homology among them (>55% sequence identity, see Figure 9), in contrast to B1 and B3 subclasses. All B2 enzymes are chromosomally encoded. This fact, together with the fact that their carriers are environmental bacteria and not primary human pathogens, determines that B2 β-lactamases lack clinical relevance at the moment. However, an increase in clinical isolates of carbapenem resistant Aeromonas spp. producing CphA has been reported.16
The restricted substrate spectrum of B2 MBLs has been attributed at least in part from the fact that the active-site cavity is more occluded compared to the more versatile B1 and B3 MBLs245, 249 (Figure 32). This is due to the presence of an extended α3 helix, thus reducing the range of substrate side chains that can be accommodated. This α3 helix is followed by an unusual proline-rich loop that was mutated to test its role in the substrate profile. However, individual and additive mutations of some of these Pro residues to Ala resulted in a destabilization of the protein, without broadening the substrate specificity.514 Instead, the combination of mutations N116H N220G was shown to greatly expand the catalytic efficiency of CphA against various cephalosporins and penicillins, albeit at the cost of reduced imipenem hydrolysis.342 These mutants did not alter the active-site access, suggesting that second sphere residues also determine the restricted substrate profile in B2 MBLs. The effect was not observed for the N220G single mutant, and only to a minor extent in the case of the N116H mutant. Both substitutions mimic features of B1 enzymes: the first restores the missing ligand in the Zn1 site, while the second replaces the G219N220C221 motif typical of subclass B2 by the G219G220C221 sequence present in subclass B1 and related to active-site dynamics.341 The N220G mutation was shown to increase flexibility of the polypeptide backbone around the Cys221 ligand, leading to the presence of dual conformation for the Zn2 site in the corresponding crystal structure, one of them corresponding to the WT position of the metal ion and another 1.5 Å apart.245 As a result of combining the two substitutions, the enzyme binds the second Zn(II) with increased affinity and is activated rather than inhibited by it. All in all, this suggests that the differences in substrate spectrum between B1 and B2 enzymes are also determined by subtler structural variations apart from the narrower active-site architecture.
Figure 32.
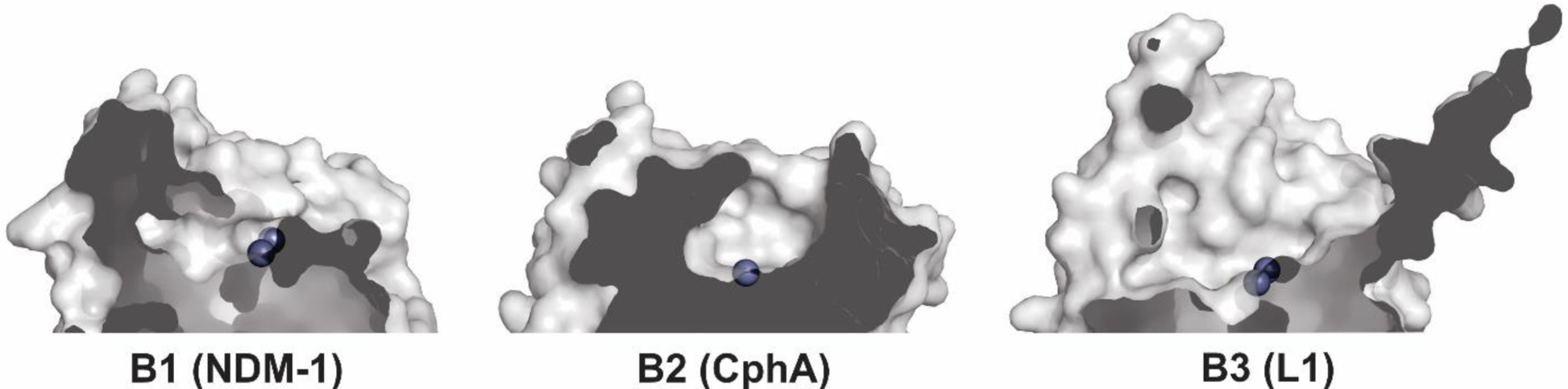
Active-site topology in B1, B2 and B3 enzymes. Structures of NDM-1 (B1) (PDB 3S0Z), CphA (B2) (PDB 1X8G) and L1 (B3) (PDB 1SML) are shown as surfaces. Zn(II) ions are depicted as grey spheres.
4.7.3. Subclass B3
The B3 subclass of MBLs is much more divergent than the two other groups, with a lower degree of sequence identity among its members compared to other subclasses. It mostly consists of chromosomic MBLs, which include the L1 (Stenotrophomonas maltophilia),520 GOB (Elizabethkingia meningoseptica)521 and BJP (Bradyrhizobium spp.)522 enzyme families, FEZ-1 (Fluoribacter gormanii),523 the closely related CAU-1 and Mlb1b enzymes (Caulobacter crescentus, also known as Caulobacter vibroides),524, 525 THIN-B (Janthinobacterium lividum),526 POM (Pseudomonas otitidis),527 CAR-1 (Pectobacterium atrosepticum),528 SPR-1 (Serratia proteomaculans),529 PEDO-1 and 2 (Pedobacter spp.),530 and the Erythrobacter spp. enzymes EAM-1, ECM-1, EFM-1, ELM-1 and EVM-1.531 Additional B3 MBLs with confirmed activity have also been identified from metagenomic screenings, with yet unknown bacterial hosts. These include the enzymes Rm3 from soil exposed to industrial effluents,532, 533 MEMA-1 from agricultural soil534 and various LRA enzymes from Alaskan soil.535
Similar to B1 MBLs, subclass B3 enzymes generally possess a wide substrate spectrum and can degrade penicillins, cephalosporins and carbapenems. There are, however, some exceptions. CAR-1 was reported to degrade cephalosporins and penicillins, but with almost no hydrolytic activity towards carbapenems.528 LRA-8 degraded penicillins and carbapenems but not the cephalosporins cefuroxime and cephalothin, although it did hydrolyze the chromogenic cephalosporin nitrocefin.536 SPR-1 was found to efficiently degrade only penicillins, with very poor hydrolysis of cephalosporins and carbapenems.529 Mbl1b, despite showing a wide substrate spectrum, displayed substrate inhibition kinetics with various cephalosporins and penicillins, while it hidrolyzed imipenem, meropenem and nitrocefin with simple Michaelis-Menten kinetics.524 This inhibition behavior was not observed for the highly homologous enzyme CAU-1, despite it did display low affinities for most β-lactams assayed, with KM values generally above 300 μM. This intringuing features have not been rationalized on structural bases.
4.7.3.1. L1 family
The S. maltophilia chromosomic enzyme L1 was among the first MBLs to be discovered, only second to BcII. L1 was identified in 1982 by Mitsuhashi and coworkers in clinical strains of the organism (then termed Pseudomonas maltophilia), together with the class A cephalosporinase L2.363 The enzyme was inhibited by EDTA and reactivated by various divalent metal ions, suggesting the discovery of a novel MBL. This first report also noted the inducible expression of both enzymes (see Section 7.2), its unique tetrameric nature, and highlighted its capability to hydrolyze imipenem, when this pioneering carbapenem antibiotic was still not available for general clinical use. A subsequent study by Waley et al. confirmed that L1 was an MBL, and found that none of the Cys residues in the protein was required for activity nor bound to Zn(II), thus suggesting a different metal coordination compared to BcII.537
Stenotrophomonas maltophilia is an emergent opportunistic human pathogen causing difficult-to-treat infections, which primarily affects inmunocompromised patients and those suffering from cystic fibrosis.538 In addition to the resistance towards carbapenems and other β-lactams conferred by the tandem of L1 and L2, it possesses intrinsic aminoglycoside and quinolone resistance enzymes together with multiple drug efflux pumps, which make it a multi-drug resistant organism.539 This illustrates the fact that clinically relevant MBLs do exist outside the B1 subclass, although attention is usually focused on this group due to its widespread enzymes associated to mobile genetic elements.
The determination of the full amino acid sequence of L1 in 1994 revealed a large divergence with respect to the available sequences of BcII and CphA,520 which suggested the need for further subdivisions within the Ambler class B enzymes. This was further supported by the novel metal coordination geometry and other unique features observed upon resolution of its crystal structure in 1998 by Spencer et al.250 Zn2 was found to be ligated to Asp120, His121 and His263 due to the replacement of Cys221 by a Ser, which only indirectly interacts with the metal ion through a hydrogen bond interaction with the apical water ligand. This leads to a rotation of 76° of the plane of the equatorial ligands with respect to that of B1 and B2 MBLs, while Zn1 maintains the canonical MBL ligands His116, His118 and His196. A set of second-shell residues aid in orienting the Zn(II) ligands, via hydrogen bonds (either directly or mediated by a water molecule) or aromatic interactions. These include Trp38, Asp67, Asp84, Asp153, Phe156, Asp220, Ser221 and Ser225 (223 in the older BBL numbering).250 While Asp84 is not conserved in B3 enzymes as is in B1 MBLs, this residue was also found to be outside the allowed Ramachandran regions in L1 and to participate in stabilizing interactions of the protein structure.250
The active site of the enzyme is delimited by two extended loops subtended between α3-β7 and between β12-α5. While the latter is equivalent in position to the L10 loop in B1 and B2 enzymes, the former stems from a different segment of the primary structure to loop L3, but it is also endowed with significant mobility and participates in substrate binding and catalysis324, 540 (see Section 4.5). Ser221 and Ser225 from loop L10 (loop β12-α5) assist substrate binding by interacting with the C3/C4 carboxylate similarly to Lys224 in B1 enzymes, while residues from the α3-β7 loop (Phe156, Ile162) and extended N-terminus (Tyr33, 32 in the older BBL numbering) form a hydrophobic patch to accommodate the C6/C7 substituent.330 However, these interactions are not critical, as replacement of Ser225 (and other β12-α5 loop positions) or mutations in the mentioned α3-β7 loop residues only had a relatively mild effect on catalytic parameters.540
An intramolecular disulfide bridge exists in L1 between Cys256 and Cys296 (290 in the older BBL numbering) that connects the α5-β12 loop to the C-terminal alpha helix,250 which was later also observed to be present in FEZ-1, SMB-1 and AIM-1 (this enzyme features two additional disulfides: Cys33-Cys67 and Cys208-Cys213).541–543 BJP-1 also forms an internal disulfide bridge but at a different position, involving residues Cys200 and Cys220,261 while GOB lacks a disulfide bridge as it only contains 1 Cys residue, buried within its protein structure.254 LRA-12 also lacks a disulfide bridge despite possessing 2 Cys residues.544 This feature has not been observed in MBLs from the other two subclasses. The structure of L1 also revealed an extended N-terminal loop involved in inter-subunit contacts during oligomerization, which also participates in substrate binding.250, 330
Tetramerization of L1 depends mainly on hydrophobic interactions among the different chains.250 Met175 plays a key role in this association. This residue from chain A fits into a hydrophobic cavity in subunit B formed by Leu150d, Pro213, Tyr237 and Pro238 (these are Leu154, Pro213, Tyr236 and Pro237 in the older BBL numbering, respectively) and viceversa (the same pattern of contacts also exists for chains C and D). This interaction is essential for the formation of the quaternary structure of L1, as the introduction of the M175D mutation resulted in a monomeric enzyme with greatly impaired substrate binding and hydrolysis.257 Additional contacts between the A and B (and C and D) subunits are made by residues in the C-terminus of helix α3 with their counterparts in the other chain. Finally, the aforementioned extended N-terminus participates in hydrophobic and electrostatic interactions between chains A and C (and B and D), with two leucine residues (Leu27 and Leu30 – Leu26 and Leu29 in the older BBL numbering, respectively) from this segment binding to hydrophobic cavities on the surface of the other chain. Deletion of the N-terminal extension still resulted in a tetrameric enzyme, although it affected Zn(II) content, substrate binding and hydrolysis.257 However, the WT enzyme presents negative cooperativity for nitrocefin hydrolysis, which was abolished by mutations in this protein stretch, confirming that the N-terminus is indeed involved in intersubunit interactions.257
There currently are 118 reported alleles of L1 (named L1–1 to L1–118).545, 546 The marked sequence variations in L1 enzymes among S. maltophilia isolates were highlighted by Walsh et al.,546 which together with a previous report547 identified the existence of a total of 5 aleles of L1, named L1-a through L1-e (now named L1–1 to L1–5). Substantial variations in catalytic parameters were observed for L1-e with respect to L1-a, with the former presenting a reduction of around 2 orders of magnitude in kcat/KM for both imipenem and nitrocefin. While the coding genes for both L1 and L2 were reported to be present in a large plasmid-like elements,546 the sequenced genome of the organism later revealed that both genes are located within the chromosome.548 Mojica, Bonomo and coworkers reported a comprehensive analysis of L1 and L2 sequences among a large collection of S. maltophilia clinical isolates from the USA identifying a large number of new alleles of both proteins, with variants of the L1 enzyme presenting 99% to 78% sequence identity.545 Frequent substitutions were observed in the second sphere position Asp68 and within the α3-β7 and β12-α5 loops that surround the active site, which may affect substrate preferences and catalytic parameters. Meanwhile, key positions involved in tetramerization were unaltered, confirming that this oligomeric state is critical for enzyme functionality. The requirement for tetramerization, unique for L1, as well as this recently reported variability of mutants, claims for a detailed study.
4.7.3.2. Other notable B3 enzymes
The B3 subclass includes other clinically important enzymes. The opportunistic pathogen Elizabethkingia meningoseptica (formerly Flavobacterium meningosepticum and Chryseobacterium meningosepticum) is unique since it possesses two different chromosomal MBLs. These are the subclass B3 GOB enzyme and the B1 enzyme BlaB, in addition to the class A SBL CME.521 Multiple alleles of the enzymes have been reported, and variants have also been identified in other Elizabethkingia species as well.549 Although infections by E. meningoseptica and other bacteria from the genus are rare, they are emergent pathogens that can cause life-threatening infections.550 BlaB has been shown to be the main determinant of carbapenem resistance of this bacterium due to its higher levels of expression with respect to GOB.551
GOB enzymes present a different metal ligand set compared to other B3 enzymes, with a H116Q substitution, resembling the H116N mutation of B2 MBLs. However, this does not impair metal binding, since di-Zn(II) GOB-1 and GOB-18 are fully active enzymes.253, 254 The coordination position normally occupied by the His116 side chain was shown to be taken up by the Gln116 side chain amide nitrogen in the GOB-18 crystal structure254 (see Figure 8).Expression of GOB enzymes as periplasmic proteins in E. coli, results in di-Zn(II) enzymes. Instead, when GOB is expressed and purified from the cytoplasm an inactive Fe(III)-bound form is isolated, but it can be reconstituted giving rise to an active mono-Zn(II) species.251 Notably, the PEDO-1 and PEDO-2 enzymes and other recently reported B3 MBLs from metagenomic studies such as LRA12, LRA17 and LRA19 also possess a Gln116 residue.
Although most B3 MBLs are chromosomally encoded, some new members of the subclass are contained in mobile genetic elements, with evidences of horizontal gene transfer. LMB-1, first identified in a clinical isolate of Enterobacter cloacae from Austria, was reported to be plasmid-borne and has been recently been found in a Citrobacter freundii isolate in Argentina.552, 553 AIM-1, although reported to be located in the chromosome of P. aeruginosa isolates, was found associated to a mobile genetic element.554 and was recently described in a K. pneumoniae strain.555 Codon randomization in different active-site positions of AIM-1 revealed that the conserved Ser221 is relevant for resistance against several antibiotics, while other active positions (excluding metal ligands) show some tolerance to mutations and possible routes for activity enhancement. In particular, mutation F119M was shown to increase resistance to cefoxitin.556 SMB-1 was similarly observed to be contained in the chromosome within a gene module allowing for potential horizontal gene transfer.557 This emergence of mobilized enzymes belonging to subclass B3 is a serious concern, alerting of a possible spread of these MBLs to pathogenic and opportunistic bacteria.
In addition to the distinct coordination sphere observed for GOB enzymes, an additional variant to the B3 Zn(II) ligands was observed for the enzyme SPR-1 from Serratia proteomaculans.529 Although the protein presented 33% sequence identity with L1, three substitutions in the Zn binding positions (H118R, H121Q and H263K) lead to a very unusual putative coordination sphere. The crystal structure of SPR-1 has not been obtained yet, precluding confirmation of the identified Zn ligands and their mode of binding to these metal ions. Both Arg and Lys residues are uncommon metal ligands, and Asn262 as was proposed by the authors as a potential alternative ligand to Lys263. The purified protein presented only one Zn(II), but assays suggested that an additional metal ion is incorporated upon substrate binding.
The diversity of B3 MBLs and variability in metal coordination has been recently highlighted by a study analyzing the presence of homologous proteins in a collection of bacterial genomes.558 Such enzymes were observed in 1.2 % of all genomes examined, with a total of 1449 homologues. Most of them possessed the canonical HHH Zn1 and DHH Zn2 sites, but 162 have a GOB-like QHH/DHH ligand set, and 47 present the HRH/DQK motif observed in SPR-1. A crystal structure obtained for the CSR-1 enzyme, belonging to the latter group, displayed no Zn(II) ions in its active site, suggesting a markedly reduced metal affinity caused by this set of ligands. Additionally, a group of 90 putative B3 MBLs presented a novel coordination sphere composition, with the residues EHH and DHH in their Zn1 and Zn2 sites, respectively.
The diversity of B3 enzymes and their closer relatedness to members of the superfamily, make this subclass highly appealing for further research. The interest in B3 lactamases is further spurred by the finding of a large number L1 variants in S. maltophilia, an emerging opportunistic pathogen affecting cystic fibrosis patients, and the finding of B3 MBLs coded in plasmids.
4.8. MBL Phylogeny and evolutionary origin
The division of MBLs into three separate subclasses reflects the characteristic active-site structures and substrate spectrum of each group but it does not accurately recapitulate their phylogeny. As mentioned before, all MBLs share a common αβ/βα fold with an heterogeneous set of proteins from all three domains of life, constituting the MBL superfamily (see Section 4.9). The origin of the superfamily would therefore correspond to an ancestral protein existing before the divergence of the Eukarya, Archaea and Eubacteria domains.
B1 and B2 enzymes, despite active site and susbtrate profile differences, constitute a phylogenetic group distinct from the more divergent B3 subclass222 (Figure 33). While they are grouped into a single class, the divergence among B1+B2 and B3 enzymes is similar to that among A, C and D SBLs. Analysis of phylogenetic relations with non-β-lactamase members of the MBL superfamily suggests that β-lactamase activity arose separately in the B1+B2 and B3 enzymes.223 Thus, these two groups of MBLs are descendants of different ancestral proteins devoid of β-lactamase activity. The origin of the B1+B2 group was gauged at around 1000 million years ago, while B3 MBLs would be more ancient, with an estimated origin at 2200 million years ago.223 A later study aiming to reconstruct the most recent common ancestor (MRCA) of MBLs generally supports the hypothesis of a dual acquisition of β-lactamase activity within the superfamily.559 Therefore, at the moment it is considered unlikely all MBL subclasses are the result of divergent evolution from a common precursor with β-lactamase activity.
Figure 33.
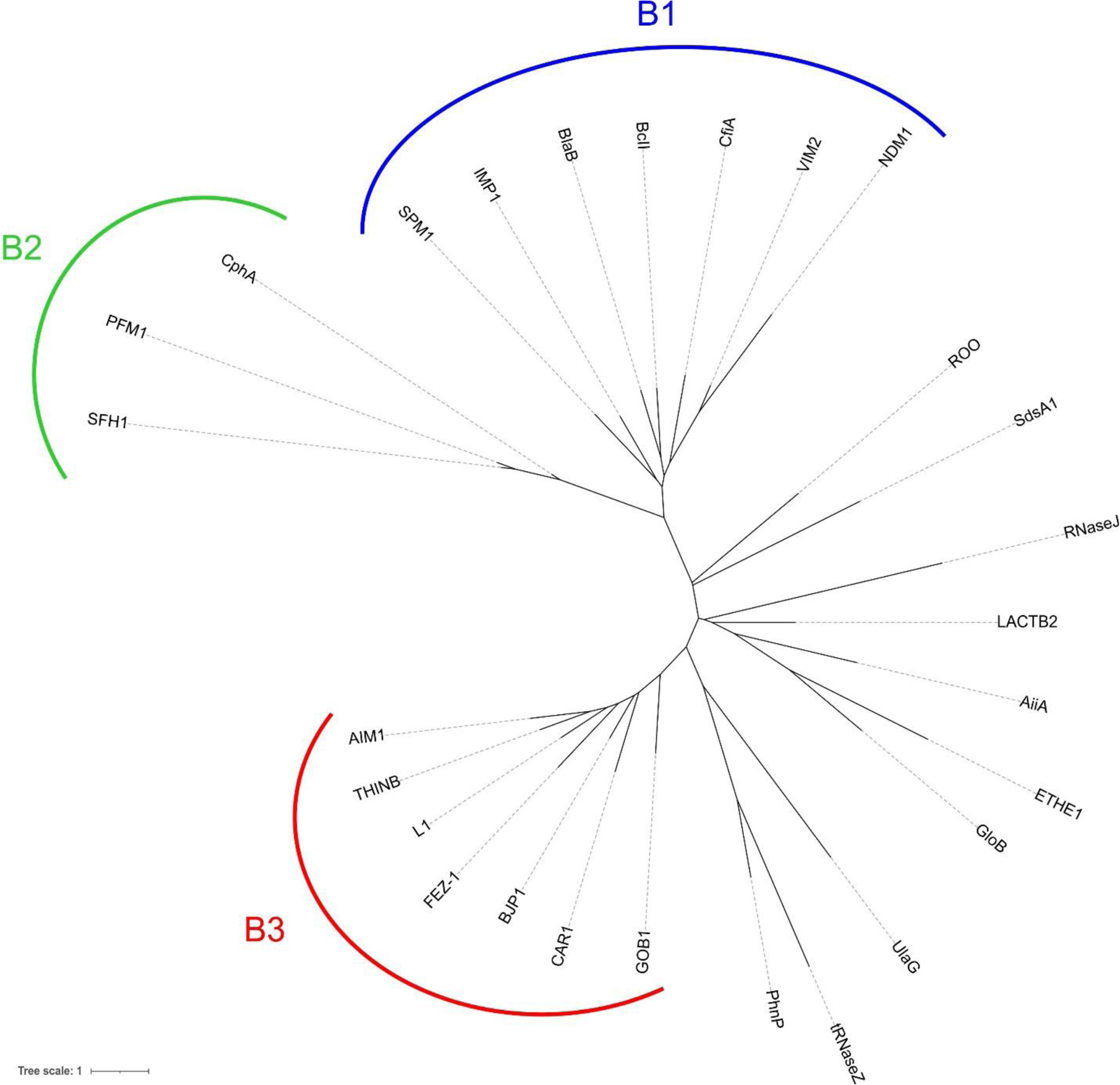
Phylogenetic tree showing the clustering of the different subclasses of MBLs with respect to other members of the MBL superfamily. Protein sequences were aligned with PSI-Coffee, alignments were trimmed using JalView to remove highly gapped and poor-quality regions, and phylogenetic trees were calculated using PhyML (via the phylogeny.fr web server). Tree representations were constructed using iTOL.
4.9. MBL superfamily
The crystal structure of BcII led to the definition of the MBL fold.217 In the subsequent years, a large number of proteins of varied functions were identified with structural homology and conserved sequence motifs shared with MBLs, which were annotated as members of the MBL superfamily. This is an ancestral group of proteins widely distributed among the three domains of life, carrying out a large variety of roles,20, 213–216 and possesses as today more than 100,000 identified members on the Pfam database (clan Metallo-HOrase, CL0381). Among proteins assigned to the superfamily in Pfam, 78.8% belong to Bacteria, 16.3% to Eukarya, and 4.9% to Archaea.
Proteins belonging to the MBL superfamily display a wide repertoire of roles in their host organisms.20, 213, 214 Most of these proteins are metallohydrolases (like MBLs) acting on diverse substrates, able to target from small substrates to large nucleic acid polymers. Meanwhile, other enzymes belonging to the superfamily act as oxidoreductases.
Despite the high sequence divergence, all proteins in the superfamily share the αβ/βα folding with a conserved localization for the metal binding site. Many of these proteins possess additional domains associated to their specific function.213, 216 In spite of their sequence heterogeneity, superfamily proteins share several structurally conserved features.213, 214 The characteristic metal binding stretch H116-X-H-X-D-H121, identical to that present in B3 MBLs is present in most superfamily members, together with the additional metal ligands H196 and H263. A characteristic that distinguishes most binuclear sites in the superfamily from antibiotic-degrading MBLs is the presence of the conserved aspartate residue D221 acting as a bridging ligand between the two metal sites. This is a major structural difference with respect to MBLs, which contain in this position the metal ligand Cys221 (subclasses B1 and B2) or a non-coordinating residue (subclass B3). Finally, the nearly ubiquitous D84 (BBL numbering is used to refer to the positions), which similarly to MBLs is found in non-Ramanchandran allowed conformations in superfamily protein crystal structures suggesting its importance in maintaining the domain fold.
The identity and number of metal ions bound to the active site also varies, with most proteins having binuclear sites. Many superfamily proteins bind Zn(II) ions, including most hydrolases such as the alkylsulfatase SdsA1,560 the MPH methylparathion hydrolase,561 the NAPE-hydrolyzing phospholipase D,562 isothiocyanate hydrolases such as SaxA,563 and lactonases such as the quorum-quenching N-acyl homoserine lactone hydrolases AiiA from Bacillus thuringiensis564 and AiiB from Agrobacterium tumefaciens.565 Many MBL superfamily enzymes acting in nucleic acids processing are also Zn(II)-dependent enzymes. These include the tRNAse Z proteins (acting in the cleavage of 3’ sequences of tRNA precursors),566, 567 RNase J (prokaryotic enzymes acting on mRNA processing and degradation),568 enzymes involved in DNA interstrand crosslink repair or V(D)J recombination (such SNM1A, SNM1B and SNM1C),216, 569 and the cleavage and polyadenylation specificity factor (CPSF) protein CPSF73 (acting in 3’ processing of polyadenylated mRNA precursors).570 The mRNA processing protein CPSF100 contains an MBL superfamily domain and can form heterodimers with CPSF73, but it lacks metal binding capacity due to loss of part of its ligand residues.216
Other superfamily members possess a binuclear iron site, including the phosphorylcholine esterase Pce571 and the redox enzymes from the flavodiiron proteins (FDPs) group such as rubredoxin:oxygen oxidoreductase (ROO) from Desulfovibrio gigas572 and E. coli nitric oxide reductase FlRd.573 Meanwhile, persulfide dioxygenases (PDO) such as human ETHE1 bind a single iron ion and catalyze the detoxification of hydrogen sulfide, converting the persulfide formed by non-enzymatic reaction of H2S and glutathione to sulfite using molecular oxygen as an electron acceptor.574 Homologs of this enzyme have been reported in plants and bacteria.575 PqqB is a superfamily protein participating in the biosynthesis pathway for the redox cofactor PQQ (pyrroloquinoline quinone) in bacteria. Although it was initially believed to act as a transporter for PQQ,576 PqqB was recently shown to possess Fe-dependent hydroxylase activity and proposed to carry out a hitherto uncharacterized step in the production of this cofactor.577 Another Fe-dependent protein is the diiron β-hydroxilase CmlA, which catalyzes a key step in the synthesis of chloramphenicol.578 Manganese-dependent enzymes also exist within the superfamily, such as the mononuclear lactonase UlaG, which participates in L-ascorbate metabolism in Enterobacteria,579 and the phosphodiesterase PhnP, which binds two Mn(II) ions in its active site and acts in organophosphonate catabolism in E. coli.580, 581
Some proteins in this superfamily display an outstanding plasticity regarding their metal-ion requirements. Glyoxalase II is widely distributed across both bacteria and eukaryotes, and acts in tandem with glyoxalase I and glutathione to detoxify 2-oxo-aldehydes such as the glycolysis and lipid metabolism sideproduct methylglyoxal.582 Glyoxalase II from various sources has been purified with varying levels of Zn(II), Mn(II) and Fe(II)/Fe(III),583, 584 and the Salmonella Typhimurium enzyme (GloB) has been shown to have similar levels of activity with all three ions bound to its binuclear active site.585 In contrast, E. coli tRNase Z ZipD/ElaC was reported to lack endonuclease activity when its Zn(II) ions were replaced by Fe(II).586
Many proteins in the superfamily are still uncharacterized or their functions are not yet fully understood. Out of the 18 MBL superfamily proteins identified in the human genome, the functions of 5 are still unknown (HAGHL, MBLAC1, MBLAC2, PNKD, CMAHP), and at least one of these proteins (PNKD) is involved in human genetic disease.216 The previously uncharacterized LACTB2 has been recently shown to act as a mitocondrial endoribonuclease.587 Recent studies have identified MBLAC1 as a likely ortholog of Caenorhabditis elegans protein Swip-10, which is involved in regulation of glutamate-dependent neuron excitability.35 Interestingly, although its native substrate is unknown, MBLAC is expressed in brain tissue and was shown to bind the cephalosporin ceftriaxone.35 Thus, it could constitute the molecular target responsible for the reported effects on nervous system function by this antibiotic, which acts to increase expression of the glutamate transporter GLU-1, and could be applicable to the treatment of various neurodegenerative diseases. Proteins playing key roles in natural bacterial competence such as ComEC in Bacillus subtilis, and its homologs such as ComA in N. gonorrhoeae and Rec2 in Haemophilus influenzae, contain a MBL fold domain close to their C-terminus.588 The proteins also contain an N-terminal domain of unknown function (DUF4131) and a central competence domain, containing transmembrane helixes that would constitute a pore for DNA entry. Although the physiological activity of the MBL domain has not been elucidated, it has recently been proposed to act as a nuclease, possibly transforming double stranded DNA into single stranded forms during its incorporation into the cell.588 Type II polyketide synthases (PKS), multienzyme complexes participating in biosynthesis of various antibiotics and secondary metabolites in Streptomyces and other organisms, possess a modular architecture with different activities contained in separate polypeptides, of which proteins annotated as dehydratase/cyclases were identified as belonging to the MBL superfamily.213, 589 A more recent analysis has shown that one of these enzymes, ActIV, participates in the synthesis of the antibiotic actinorhodin acting as a bifunctional thioesterase/cyclase.590 It catalyzes a ring formation via aldol condensation and the subsequent release of the product from the polyketide synthase complex through hydrolysis of a thioester bond, the latter similar to the reaction carried out by glyoxalase II. To our knowledge, however, a detailed biochemical and structural characterization of these enzymes and their metal requirements has not been carried out. Similarly, the Aspergillus terreus enzyme atrochrysone carboxyl ACP thioesterase (ACTE) was found to belong to the superfamily. This enzyme catalyzes the release of products from a type I polyketide synthase, and was shown to be metal-dependent from its inhibition by EDTA.591 The homologue AdaB from fungus Aspergillus niger, evidenced to be a binuclear Mn(II) enzyme, was observed to catalyze a Claisen-like condensation for product release instead of a hydrolysis.592
While characterized MBLs are almost universally monomeric, with the exception of the tetrameric B3 enzyme L1, various members of the superfamily require the formation of dimers or oligomers to carry out their functions. Pseudomonas aeruginosa alkylsulfatase SdsA1 allows the bacterium to degrade the detergent SDS and resist high concentrations of the compound.560 Apart from the N-terminal MBL domain, the enzyme contains an association domain that allows it to form tightly interlocking dimers with greater stability towards the detergent, and an additional C-terminal domain possessing a hydrophobic grove that assists the binding of long aliphatic chains.560 Other examples of dimer-forming MBL superfamily proteins include ROO,572 tRNAase Z,566, 586 PhnP581 and ETHE1,574 while E. coli nitric oxide reductase FlRd is tetrameric,573 and UlaG forms hexamers (trimers of UlaG dimers).579
There is some overlap between the catalytic abilities of different groups of enzymes within the MBL superfamily. Indeed, multiple enzymes have been shown to possess the ability to catalyze reactions from other groups within the superfamily. Although the catalytic efficiency for these promiscuous activities tend to be low (kcat/KM ≤ 102 M−1 s−1), this implies that the catalytic profile within the superfamily varies as a continuum within sequence space, and enzymes could have transitioned smoothly from one function to another during evolution.215 This “functional connectivity” among MBL superfamily proteins also provides information regarding the potential evolutionary linkage of different member families, especially in cases where sequence divergence has occurred to the extent that sequence homology is nearly undetectable.215, 593 However, the presence of these promiscuous activities should be interpreted cautiously, as they can occur serendipitously in enzymes not showing close phylogenetic ties.
Multiple groups within the superfamily where shown to possess moonlightning β-lactamase activity.215 MBLs have been reported as well to display promiscuous phosphodiesterase, phosphotriesterase and phosphonatase activities,215 while B1 enzymes MIM-1 and MIM-2 were shown to possess significant lactonase activity and may participate in quorum quenching.594 The capability of MBLs to adopt activities carried out by other superfamily proteins is illustrated by a directed evolution study in which enzymes such as NDM-1 and VIM-2 were subjected to multiple rounds of mutagenesis to optimize their phosphonate monoester hydrolase (PMH) activity, achieving over a 104 fold improvement in kcat/KM values with a limited number of mutations.595 Another example of catalytic flexibility is PNGM-1, a MBL-fold protein discovered in a metagenomic screening of deep sea sediments. While it is described as a B3 MBL, conferring resistance to β-lactam antibiotics and possessesing 15–18% sequence identity with that lactamase subclass, its catalytic efficiency towards these substrates is limited (kcat/KM = 102–103 M−1 s−1 for penicillins, cephalosporins and carbapenems).596 Additionally, phylogenetic analysis shows the protein to be more closely related to tRNase Z enzymes, and both its active site structure and in vitro RNase degradation assays display characteristics compatible with this protein class.597, 598 As such, although its physiological function has not been characterized, the fact that PNGM-1 displays dual β-lactamase and tRNase Z activities may provide clues regarding the potential origin of B3 MBLs. Similarly, MBL-fold proteins from humans have been shown to possess residual β-lactamase activity.599
The distinction between actual MBLs and other superfamily members is blurred by the recent report of the protein VarG, from Vibrio cholerae.600 This enzyme was found to confer resistance to various β-lactam antibiotics, and its expression to be inducible by β-lactams (see Section 7.2), but given its unusual characteristics it does not belong to any of the three known MBL subclasses. Its active site is predicted to contain 3 His residues for Zn1, but 2 Asp and 1 His for Zn2, a coordination geometry not observed for any other previously characterized MBL but which could correspond to MBL superfamily members. However, a crystal structure for this enzyme is required to confirm these features. VarG was purified as a dimeric protein, and it showed positive cooperativity for β-lactam substrates during turnover. It hydrolyzes carbapenems with relatively high efficiency (kcat/KM around 105 M−1 s−1), showing relatively poor catalytic efficiencies towards cephalosporins and penicillins (kcat/KM of about 102 to 103 M−1 s−1).600 Further characterization is necessary to ascertain whether VarG represents an entirely new subclass of bona fide MBLs or possibly a MBL superfamily protein recently repurposed to act as a resistance determinant.
The study of members of the MBL superfamily is of interest to track possible evolutionary origins, particularly of B3 enzymes. However, the finding of residual lactamase activity on these enzymes cannot be taken as a confirmation of their identity as MBLs.
5. Catalytic mechanism of MBLs
5.1. Challenges and approaches to study the chemistry of β-lactam hydrolysis
β-Lactam hydrolysis by β-lactamases is a simple, hydrolytic reaction that cleaves the 2-azetidinone ring giving rise to a β-amino acid that is no longer active against the target peptidase domains from PBPs. Despite this simplicity, identifying the subtleties of the catalytic mechanism of MBL-mediated hydrolysis was subject of a long controversy along more than three decades.19, 21, 601–603 Early attempts to sketch a mechanism for MBLs were inspired in the previous knowledge of antibiotic hydrolysis by SBLs.227, 604 There are, however, substantial differences. First, the active-site features of MBLs give rise to a different substrate recognition. Second, metal-mediated hydrolysis is chemically different and this results in a different mechanism.
β-Lactam cleavage involves three chemical steps (Figure 3): (1) the attack of an activated nucleophile to the carbonyl group, (2) C-N bond cleavage and (3) protonation of the nitrogen atom. In the case of SBLs, the activated hydroxyl group from a Ser residue is the nucleophile, giving rise to a tetrahedral intermediate (due to the sp3 character of the carbon atom after the nucleophilic attack). This species involves a covalent bond between the former carbonyl carbon of the β-lactam and the oxygen of the Ser residue (Figure 3). This negatively charged tetrahedral intermediate is stabilized by interactions with the oxyanion hole. Next, fission of the C-N bond leads to a covalent acyl-enzyme species (Figure 3). The last reaction step is the deacylation of this covalent adduct aided by nitrogen protonation. Many mechanistic-based SBL inhibitors are suicide substrates in which the deacylation step is extremely slow.
Currently, it is widely accepted that there are two main steps in the MBL-catalyzed reaction: the nucleophilic attack on the carbonyl and the protonation of the N atom (Figure 3). Nevertheless, within this general scheme, there have been several controversial issues involved in the debate of the mechanism of MBL-mediated catalysis: (1) whether some of these events are synchronous or occur in discrete steps with accumulation of reaction intermediates, (2) the identity of these intermediates, (3) the identities of the nucleophile and the proton donor, (4) the rate-determining step of the reaction and, last but not least, (5) the role and essentiality of each Zn(II) site in this mechanism. The diversity in active-site topology, zinc ligands and metal stoichiometry of MBLs from different subclasses (and even within subclasses, as is the case for SPS-1 and GOB enzymes) has made it difficult to sketch a common mechanism. However, a recent consensus mechanism has been achieved as the result of the combination of biochemical, spectroscopic, computational and structural studies. There are still some minor undefined issues.
Classical biochemical and biophysical approaches, such as steady-state296, 297, 604–606 and pre-steady state kinetics240–242, 270, 271, 287, 322, 607 in different MBLs and site-directed mutants have contributed to outline the currently accepted mechanism. X-ray crystallography of MBLs in complex with hydrolyzed products (and some reaction intermediates) has also provided structural views of the binding mode of the different catalytic species within the active site of several MBLs.229, 245, 328–330 The presence of one or two Zn(II) ions has also been exploited by the use of different spectroscopies that allow interrogating the metal site during catalytic turnover. Zn(II) is a d10 transition metal ion, and as such does not possess unpaired electrons nor any d-d electronic transitions in the visible range, being silent to most spectroscopies. However, Zn(II) centers in proteins can be studied by X-ray Absorption Spectroscopy (XAS), mostly by EXAFS (Extended X-Ray Absorption Fine Structure).285, 291, 293, 515 EXAFS provides information about the identity of the coordinating atoms to the metal center and ligand-metal distances and Zn-Zn distances (in binuclear sites). These techniques, when coupled with Rapid Freeze-Quench (RFQ) devices, enable the trapping and characterization of possible intermediates in the millisecond time scale. An alternative approach is the replacement of the native metal ion by other divalent ion surrogates that can reproduce their binding features and are amenable to be studied by different spectroscopic techniques. This is the case for Co(II), a d7 metal ion, that has been shown to be the best spectroscopic probe for zinc sites in proteins, in its high-spin (S=3/2) form.279, 608–610 Co(II)-substituted MBLs have been extensively characterized by different spectroscopic techniques. The utility of UV-visible spectroscopy is boosted by the presence of ligand to metal charge-transfer in the presence of Cys ligand and d-d absorption bands that are sensitive to the geometry and coordination number of the Co(II) ion, and provide sensitive probes to analyze the catalytic cycle.279 Electronic spectroscopy in real time under pre-steady state conditions coupled to stopped-flow equipments has been extensively used for mechanistic studies (Figure 35, Figure 41, Figure 45). Magnetic techniques are slower, and as a result, EPR and ENDOR in Co(II)-MBLs have been exploited in combination with RFQ devices.240, 241, 287, 299, 351, 611 Cd(II) is also a possible metal substituent, allowing use of113Cd NMR and Perturbed Angular Correlation (PAC) spectroscopies to study the active-site structure.252, 288, 612 Metal substitution by other metals, such as Cu(II), Mn(II), Cu(II) and Ni(II) have been attempted, but the catalytic performance of these derivatives is much lower than those observed with Co(II) or Cd(II).605, 613 Indeed, it has been suggested that the inhibitory activity of MBLs by some Cu(II) complexes could be due to metal replacement. Fe(II) substitution has rendered active derivatives of BcII and VIM-2, but it requires manipulation in gloveboxes since the metal is prone to be oxidized to Fe(III), giving rise to an inactive variant.614
Figure 35.
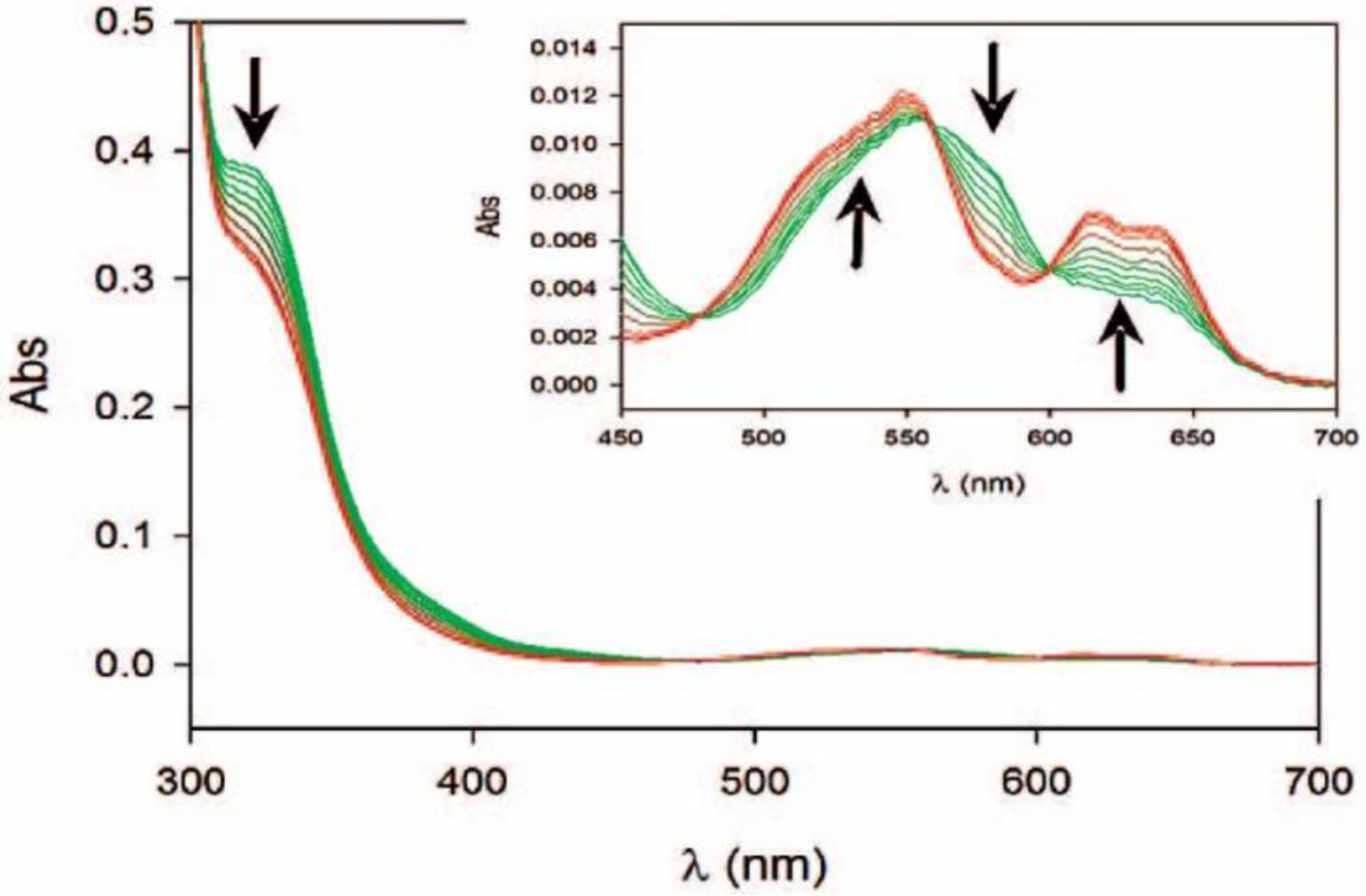
Photodiode array stopped-flow spectra of di-Co(II) BcII showing changes in the ligand-to-metal charge transfer (LMCT, 343 nm) and ligand field bands (d-d, 500–650 nm) during the hydrolysis of benzylpenicillin. The inset shows an amplification of the ligand field bands (in the 450–700 nm region of the spectra). The arrows indicate whether there is an increase or a decrease in the intensity of the corresponding absorption bands in the transition from an ES complex to the resting state enzyme. Adapted with permission from reference287 Llarrull et al. Copyright 2008 American Chemical Society.
Figure 41.
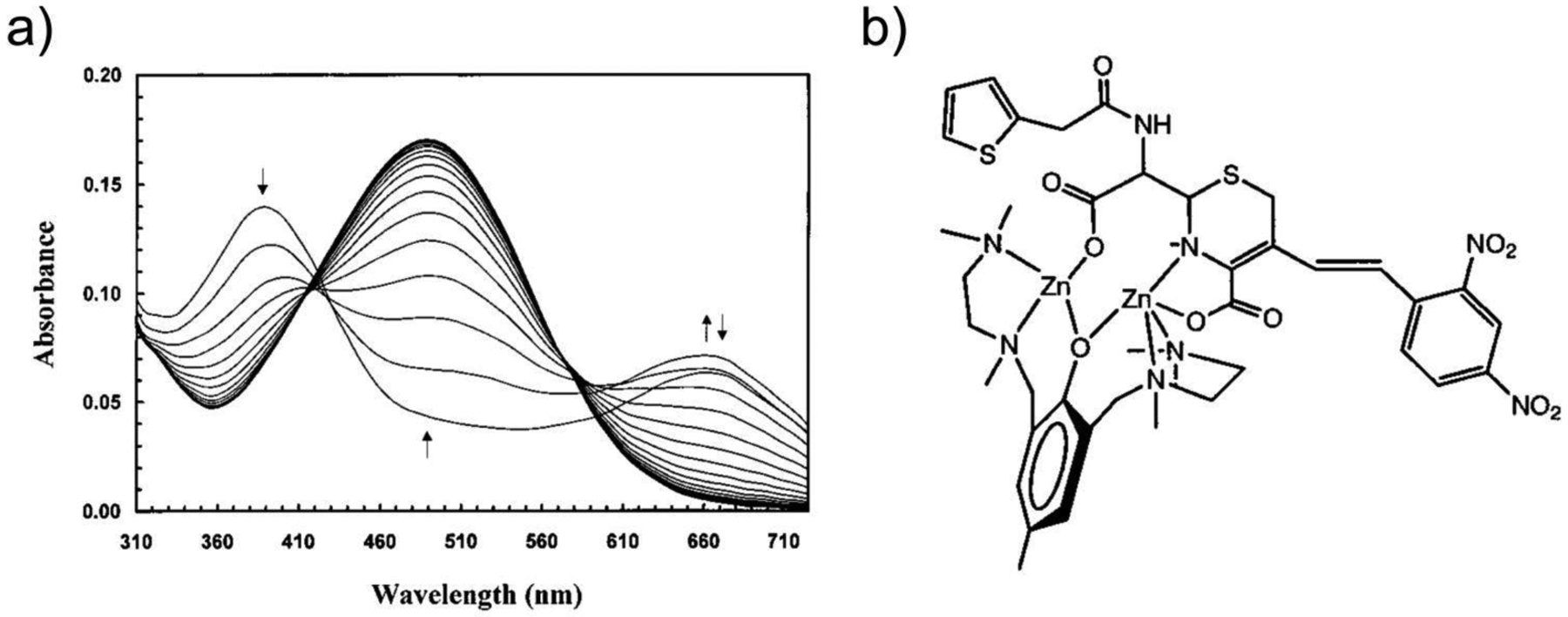
a) Spectral changes during nitrocefin hydrolysis by CcrA, monitored by stopped-flow absorbance meassurements. The first spectrum was acquired at 1.28 ms after mixing, and each subsequent spectrum 2.56 ms after its predecessor. Reproduced with permission from reference607 Wang and Benkovic. Copyright 1998 American Chemical Society. b) Structure postulated for the anionic reaction intermediate of nitrocefin hydrolyzed by a binuclear Zn(II) complex. Adapted with permission from reference654 Kaminskaia, Lippard et al. Copyright 2001 American Chemical Society.
Figure 45.
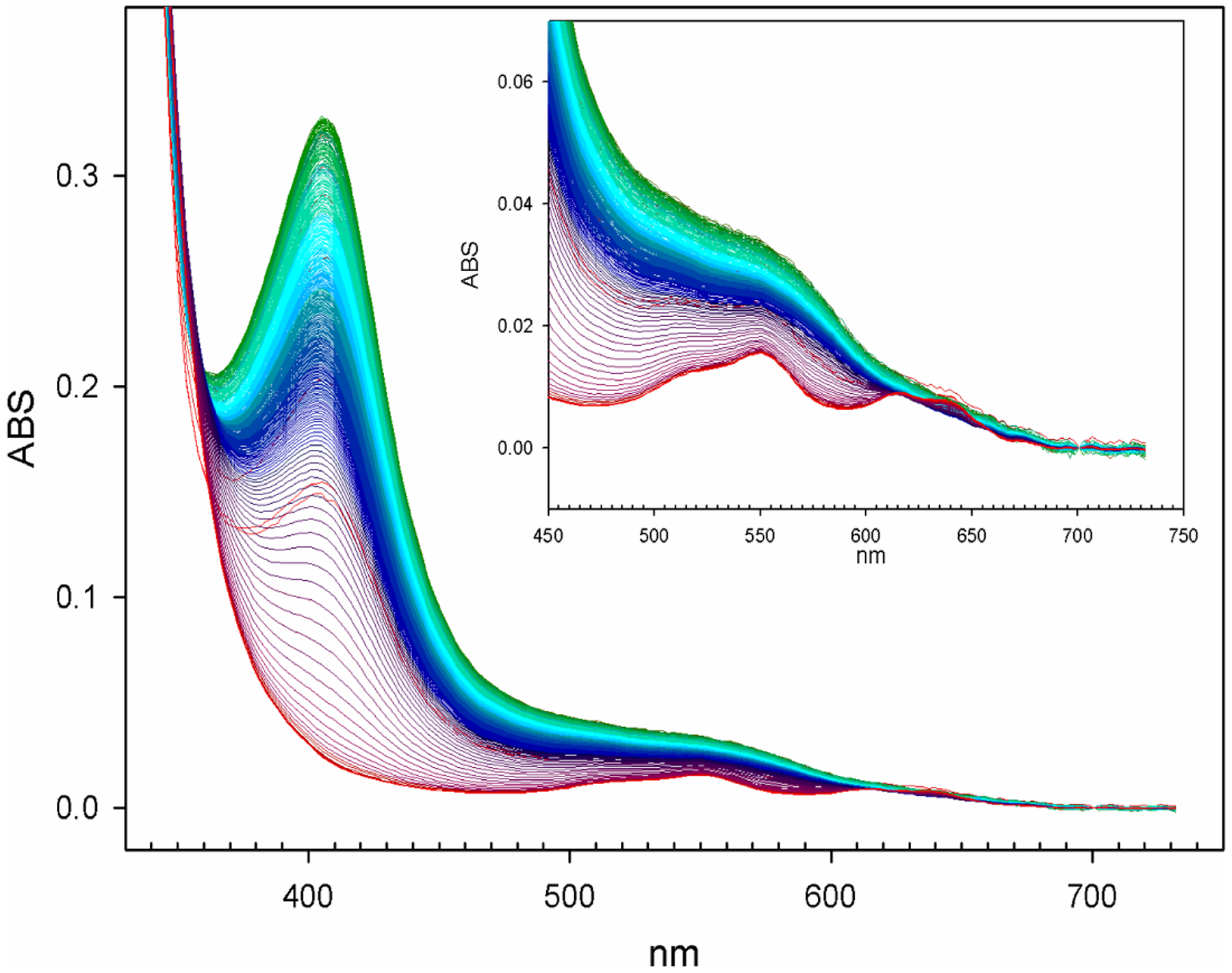
Spectral changes during hydrolysis of imipenem by bi-Co(II) BcII, monitored by stopped-flow absorbance meassurements. The absorption at 408 nm is due to the anionic intermediate, obscuring the Cys-Co(II) LMCT band. The inset shows changes in the ligand field band during turnover, that reveal modifications in geometry of the Co(II) center. Adapted with permission from reference240 Tioni, Llarrull, Vila and coworkers. Copyright 2008 American Chemical Society.
Another approach to identify mechanistic intermediates is the study of the time evolution of spectroscopic features of the β-lactam substrates during the reaction. The β-lactam chromophore absorbs at 235–300 nm, depending on the substrate. Therefore, β-lactam hydrolysis can be followed kinetically by monitoring the fall in absorbance at the maximum wavelength corresponding to each antibiotic. Even though the changes in the extinction coefficients can be low for some substrates, this absorption feature can be safely exploited for steady-state kinetics study. However, working under single turnover conditions leads to overlap of the β-lactam absorption with protein chromophores in this region making it difficult to follow the hydrolysis rates. For these reasons, the development of chromogenic β-lactam compounds with absorption features in the visible range that change upon hydrolysis (Figure 40), and with large extinction coefficients, has been essential for mechanistic studies as well as for multi-plate screening methods that use shorter path lengths, as reviewed by Fast and Sutton.615 Among β-lactams, cephalosporins provide an adequate scaffold, since leaving groups at C-3 can be dissociated upon β-lactam hydrolysis, eliciting larger spectral changes (see below).616
Figure 40.
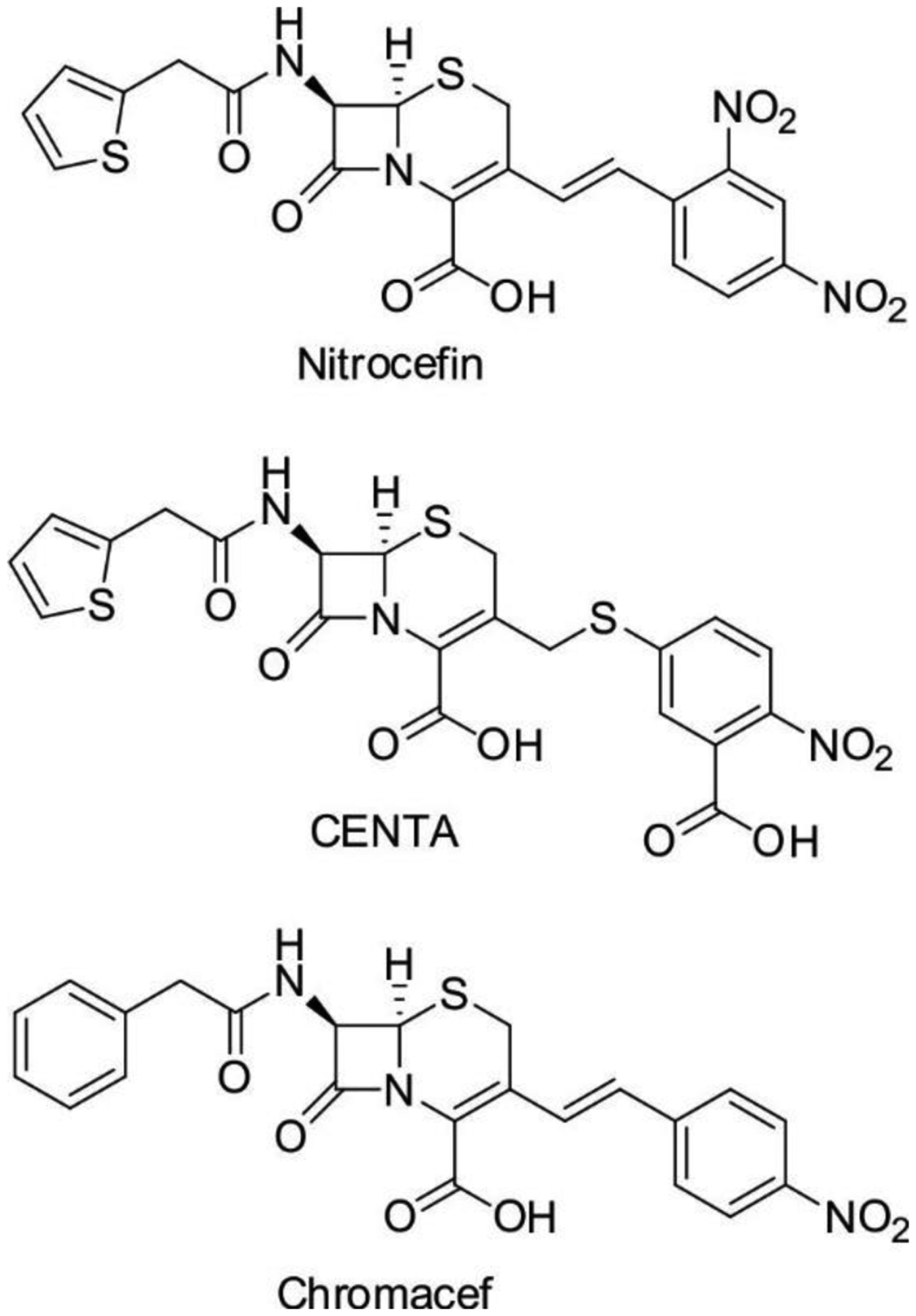
Chromogenic cephalosporin substrates.
Computational simulations of the reaction mechanism have been of great help in validating different hypotheses arising from experimental data. In this regard, quantum mechanics-molecular mechanics (QM-MM) hybrid calculations describing the metal site with a density functional theory (DFT) approach, and the protein scaffold with a classical description, proved to be valuable.244, 249, 321, 326, 327, 617–628 Numerous computational studies have been performed to analyze the interaction of MBLs with substrates, intermediates and products. Despite Zn(II) is a closed-shell transition metal ion, these calculations have faced the challenge of describing the intrinsic flexibility of the metal site, that in MBLs is larger compared to other binuclear hydrolases due to the requirement of hosting a large number of substrates.
5.2. Role of Zn(II) in metallohydrolases
Zn(II) is a trace element essential for life, second to iron in the list of most abundant transition metal ions in living organisms.303, 629 Zn(II) is a strong Lewis acid but, in contrast to iron, is devoid of redox activity. The full-shell d10 configuration results in a ligand field stabilization energy (LFSE) equal to zero. Thus, Zn(II) has no preferences for a defined geometry or coordination number, and it is found in geometries ranging from 4- to 6- coordination in proteins. Zn(II) is an intermediate hard-soft metal ion in terms of the Pearson classification, thus being able to coordinate equally well with N- and O- (hard) and S- (soft) ligands. Zn(II) is characterized by a fast ligand exchange rates (kex H2O = 2×107 s−1) that enables rapid binding and detachment of protein ligand groups and exogenous ligands.630 As a result, compared to other divalent 3d ions, Zn(II) can provide Lewis acid chemistry, nucleophile activation and rapid ligand dissociation (this is particularly important for metal-bound water molecules) that elicit an efficient turnover in hydrolysis reactions. In this regard, different roles have been postulated for the Zn(II) ions in mono-Zn1, mono-Zn2 and binuclear variants of MBLs: (1) as a Lewis acid polarizing the C=O bond resulting in a more electron-deficient carbon atom,226, 604 (2) favoring the formation of a transient negative charge in a tetrahedral intermediate,604, 631 (3) lowering the pKa of the Zn(II)-bound water molecule which facilitates nucleophilic attack to the β-lactam carbonyl,604 (4) providing an anchoring point for the negatively charged carboxylate group at C3 or C4 in β-lactams, thus steering substrate binding,239 (5) stabilizing the formation of an anionic intermediate240, 241, 393, 607 and (6) orienting/polarizing a water molecule to act as a proton donor.241 There is compelling evidence supporting some of these proposed roles, while other postulates are not supported by experimental results. These aspects, together with the distinct activities of the differently metallated forms in MBLs will be discussed in this section.
The relative activity of the mononuclear and the binuclear forms, as well as their physiological relevance, has been matter of an intense debate, particularly in B1 MBLs (See Section 4.2).285, 287–293 Crucial to disentangle this dilemma is the possibility of isolating the Zn1-only or Zn2-only variants of the different enzymes, with contradictory results from different laboratories. These experiments are further complicated by the report of quite diverse binding affinity constants for the zinc sites, since these values strongly depend on the method employed. Moreover, the affinities of MBLs towards Zn(II) are in the low nM range, which is much lower than the affinities of other zinc enzymes (for example, for carbonic anhydrase and thermolysin, the affinity is pM632, 633), and for carboxypeptidase is sub-nM.634 In addition, the Cys ligand of the Zn2 site in B1 enzymes is prone to oxidation, which is sometimes irreversible and prevents metal binding at the Zn2 site.280–282 This has been repeatedly reported in crystal structures of mono-Zn1 variants, that are the result of Cys oxidation, in many cases resulting from synchrotron radiation, but also to the different susceptibility of oxidation in solution from the Cys221 ligand. In particular, VIM-2 and SPM-1 have been proved difficult to characterize in the binuclear form due to this phenomenon.281, 282 Despite these controversies, there is a growing consensus that the binuclear form of B1 MBLs is the active species in the bacterial periplasm.300 This aspect will be discussed later in this review in the context of the cell biology of MBLs and zinc homeostasis in bacteria (Section 10).
5.3. Identity of the nucleophile and impact of pH in the activity of MBLs
Two possible candidates were initially suggested as attacking nucleophiles for the mono-Zn(II) enzymes: the Zn(II)-bound water molecule, and a deprotonated Asp120.242, 604 Assuming a nucleophilic Asp120 would result in the formation of an intermediate anhydride species. However, addition of methanol, hydroxylamine or methoxylamine to the reaction of BcII and CcrA did not result in trapping of any anhydride intermediate. These experiments by the groups of Page and Benkovic supported that a Zn(II)-bound hydroxide could be the attacking nucleophile.242, 604
Nucleophile activation by metal ions does not require a general base to deprotonate the water molecule. The pKa of a Zn(II)-bound water is tuned by the immediate environment, ranging from 9.5 in Zn(H2O)6++ to 6.8 in human carbonic anhydrase II (whose active site resembles the Zn1 site of B1 and B2 lactamases), or to 5.8 in carboxypeptidase A. In this way, the study of the pH dependence of the catalytic performance of zinc enzymes has helped in revealing the pKa of the zinc-bound water molecule. Initial experiments from Page, Frère and Galleni on BcII revealed that the enzyme is inactivated at low pH with a slope of 2, suggesting the involvement of two protons in this event.604 The pKa of this inactivation was shown to depend on the hydrolyzed substrate, the Zn(II) concentration on the reaction buffer231, 635 and the identity of the metal ion (when Zn(II) was replaced by Co(II), Mn(II) or Cu(II)).605 The different chemical properties of each metal ion (in particular, the Lewis acidity and the ionic ratio) are known to tune the ability of polarizing the water molecule and finally change its pKa. This pKa changes significantly upon replacement of the native zinc by Co(II) (6.3), Cd(II) (8.7) or Mn(II) (8.5) in BcII.605 This trend supports that the role of Zn1 is to provide the nucleophilic hydroxide, rather than to act as a Lewis acid to polarize the carbonyl group and stabilize the early tetrahedral high energy intermediate.
The acidic limb of the pH dependence of kcat shifted to lower pH values upon addition of extra Zn(II). This reversible inactivation was correlated to the finding of Zn(II) dissociation at acidic pH by Rasia and Vila,635 later confirmed by crystal structures from the Sutton lab indicating the protonation of Asp120 at low pH in BcII,280 that detaches from Zn2. The Zn1-Wat distance increases from 1.9 Å to 2.3 Å at low pH, confirming the protonation of the zinc-bound water molecule, and therefore accounting for the anomalous slope observed in the pH-dependent experiments. This water ligand detaches from the metal site at pH 4.5. IMP-1390 showed no titratable groups between pH 4.6–9.0, and the inactivation at lower pH values was also correlated to Zn(II) dissociation.636 Overall, the acidic pKa limb in B1, B2 and B3 lactamases has been detected at low pH or even undetected238, 317, 319, 323 confirming that the pKa of the attacking nucleophile is < 5 in all cases, and that enzyme inactivation at low pH is mostly due to Zn(II) dissociation from the active site.
Some authors have argued that the presence of a second Zn(II) ion could help in further lowering the water pKa.601 However, the simultaneous binding to two Zn(II) ions renders a less powerful nucleophile. In binuclear hydrolases, a terminal hydroxide is expected to be a more efficient nucleophile than a bridging hydroxide.637 This notion has been supported by model compound studies,638 and to the observation that the Zn1-O bond is shorter than the Zn2-O bond in all binuclear MBLs.226 This favors the hypothesis (supported by QM calculations)327 suggesting that substrate binding takes place also by coordination of the carboxylate group to Zn2, with a simultaneous detachment of the bridging hydroxide from this metal site, releasing a potent nucleophile.241
The impact of the different substrates in the pKa is less clear. Since the hydrolyzed products are dicarboxylates, they are expected to act as moderate chelators or inhibitors, and this phenomenon could be reinforced at higher pH values. Thus, the different chelating power of the hydrolyzed products could explain the distinct pKa values for each substrate.
5.4. Identity of the proton donor
Solvent Kinetic Isotope Effects (SKIE) have been of help in identifying the rate-determining step of the reaction. All mechanistic studies in B1, B2, and B3 enzymes displayed normal SKIE (Dkcat / Hkcat > 1), revealing that the rate-determining step involves a proton transfer.315, 604, 639, 640 A situation in which the nucleophilic attack of a metal-bound water is rate-limiting should give rise to an inverse isotope effect (Dkcat / Hkcat < 1), confirming that the protonation, the last step of the reaction is rate-limiting for all MBLs).641, 642 Proton inventory studies from the Crowder lab also suggested one proton in flight involved in this step.315 Last but not least, since the Zn(II)-bound hydroxide is consumed in the first step of the reaction, a new water molecule is require to bind Zn1 during the mechanism to regenerate the nucleophile. This rearrangement may be linked to the final protonation step. The possible proton donors that have been suggested are: Asp120, the newly formed carboxylate after hydrolysis, a Zn(II)-bound water molecule, and a bulk water molecule.
Mutation of Asp120 is deleterious for the activity of all MBLs.231, 276, 315–318, 390 This led to the proposal of this residue as the proton donor in the last step of the reaction. However, Asp120 mutants also showed a normal SKIE, suggesting that the rate-limiting step is the proton transfer.318 Since kcat is not significantly altered in the isosteric D120N mutant in BcII,318 IMP-1390 and L1,317 this suggests that Asp120 is not the proton donor in the last step of the reaction. Instead, structural analysis of different Asp mutants support the conclusion that the main role of Asp120 is to adequately position Zn2 for substrate binding and catalysis.316, 318, 390 The most accepted hypothesis for a proton donor is a water molecule, as discussed below for the different substrate families.
5.5. Mechanism of hydrolysis of the different families of β-lactam compounds
β-Lactamases are similar to peptidases in that they catalyze the hydrolysis of an amide bond, and they pursue this chemistry by means of activated serine residues (such as trypsin, chymotrypsin or thrombin) or a Zn(II)-bound water molecule (such as the carboxypeptidases, aminopeptidases, matrix metalloproteases and ADAM proteins). However, β-lactam hydrolysis presents different challenges compared to the hydrolysis of peptides and linear amides and this results in different catalytic mechanisms. Peptide bonds are planar, while bicyclic β-lactams are butterfly-shaped molecules with the nitrogen adopting a pyramidal configuration. This results in the lone pair of the N atom oriented to the α side of the antibiotic molecule. The hydrolysis of planar peptides is governed by stereoelectronic effects, so that the nucleophilic attack takes place antiperiplanar to the direction of the N lone pair.643 The V shape of β-lactam substrates overrides the stereoelectronic effect in such a way that both the nucleophilic attack and nitrogen protonation occur from the less hindered α side. Thus, the catalytic machinery of β-lactamases should provide an adequate orientation of the nucleophile and proton donor fulfilling these geometric requirements.
In addition to these common features, the families of bicyclic antibiotics can be distinguished by the different chemical nature of the ring fused to the 2-azetidinone moiety: a saturated five-membered ring (penicillins), an unsaturated six-membered ring (cephalosporins) and an unsaturated five-membered ring (carbapenems) (Figure 1). These differences, as well as the different substituents decorating these rings, determine the possibility of stabilizing possible anionic intermediates during the catalytic cycle. Another important difference to consider is the different chirality of C6 in carbapenems (S) compared to penicilins and cephalosporins (R) (Figure 1). This gives rise to unique features in the mechanism of hydrolysis of each family of substrates.
5.5.1. Mechanism of penicillin hydrolysis
Penicillin absorption occurs at 230 nm and, therefore, is much difficult to be followed under single turnover conditions due to interference from protein absorption. Thus, Co(II)-substitution has been useful to study the mechanism of penicillin hydrolysis by MBLs. Co(II)-MBLs are slightly less active than the native Zn(II) variants, being useful to obtain a bona fide description of the mechanism, providing more chances to trap transient mechanistic species.605 The absorption spectra of Co(II)-substituted MBLs inform on the geometry of the metal binding sites (Figure 36).279, 292 Co(II)-substituted B1 enzymes have a a characteristic pattern of ligand field bands between 500–650 nm from both sites, that in the resting state is dominated by the intense features of the tetrahedral Co1 site. In the UV range, an intense Cys→Co(II) ligand-to-metal charge-transfer band (LMCT) at 340 nm reports changes at the Co2 site. These particular signatures enable following independently changes in the Co1 and the Co2 site upon ligand binding (i.e., inhibitor binding or during turnover). This is not possible in Co(II)-substituted B3 enzymes, lacking a Cys residue. Co(II)Co(II)-L1,292, 644 Co(II)Co(II) AIM-1611 and Co(II)-GOB252 only present features in the visible range due to the ligand field bands that, in the case of binuclear enzymes, present overlapped features of the two binding sites. EPR286, 351, 645, 646 and EXAFS285 of the Co(II) derivatives at cryogenic temperatures have also been employed to retrieve information of the active sites and during catalysis. Seminal studies in the 1980s by Stephen Waley on Co(II)-substituted BcII unveiled the first details of the mechanism one decade before the availability of a crystal structure (indeed, without an accurate description of the metal stoichiometry and assuming a mononuclear enzyme as the active species).270, 271 This work provided the first experimental evidences of changes in the coordination geometry of the metal ions during turnover.
Figure 36.
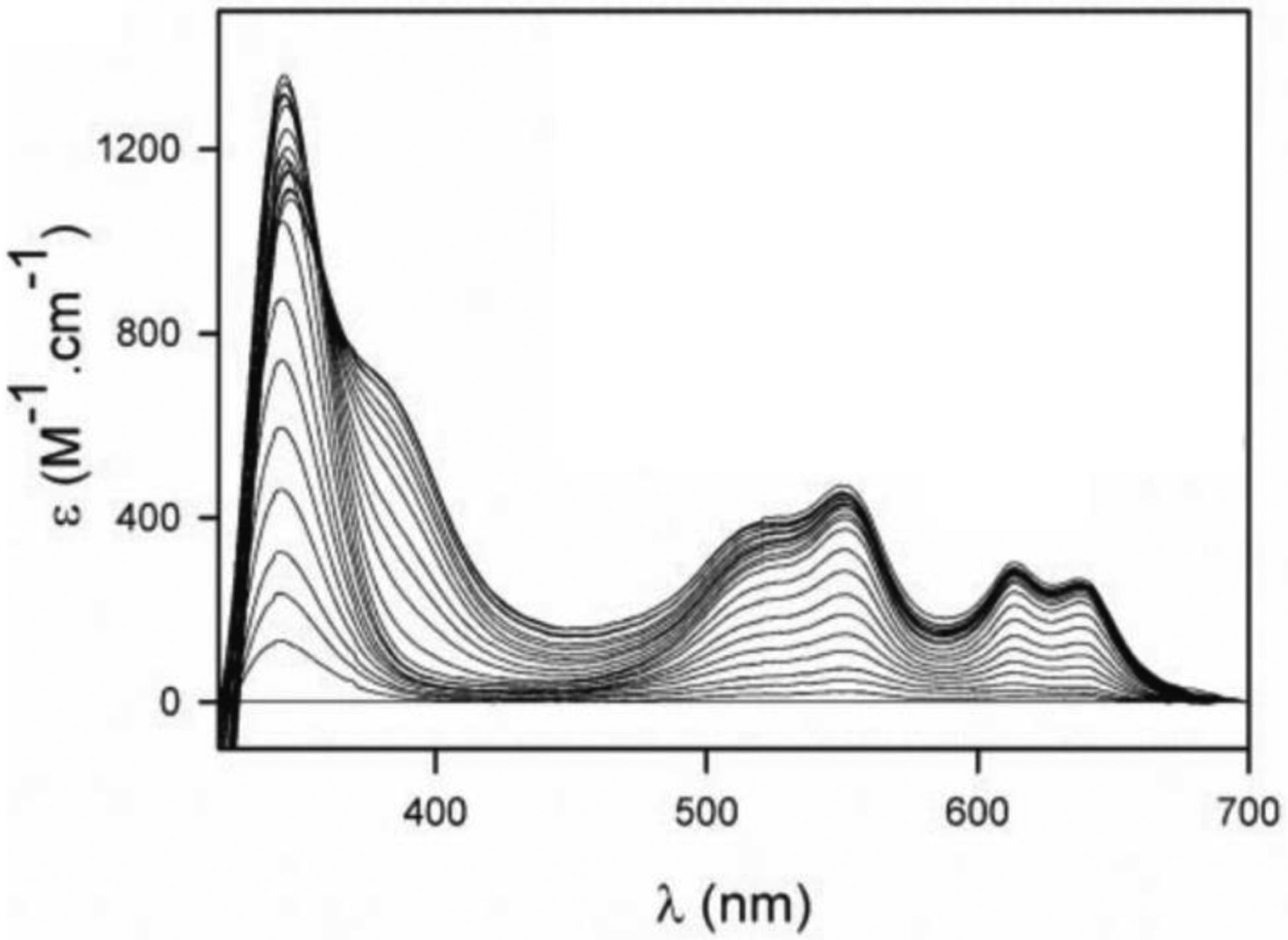
Titration of apo-BcII with Co(II). The simultaneous growth of the LMCT band and the ligand field bands shows that (under these conditions) both metal sites are simultaneously loaded. Adapted with permission from reference286 Llarrull et al. Copyright 2007 American Society for Biochemistry and Molecular Biology.
Hydrolysis of penicillin G (benzylpenicillin) by Co(II) BcII takes place by a branched mechanism.270, 271 Steady-state kinetics following the absorption features of the substrate showed a biphasic behavior, that Page and coworkers attributed to the dissociation of one of the Co(II) ions during catalysis, giving rise to a less active (or inactive) mono-Co(II) species.296 A pre-steady state study of penicillin hydrolysis by BcII loaded with different amounts of Co(II) by Llarrull, Vila and coworkers287 confirmed the branched pathway reported by Waley, that was accounted for by the assumption of two active species: the di-Co(II) form, and a mono-Co(II) BcII with the metal ion localized in the Co2 (DCH) site. This model is based on the evidence that mono-Co(II) BcII is a mixture of two mono-Co(II) forms in equilibrium, where the metal ion is switching between the Co1 (3H) and the DCH site.287 Substrate binding shifts this equilibrium to the DCH site, providing the anchoring point to the C3 carboxylate group. In the mono-Co(II)-DCH species, the suggested nucleophile is a water molecule activated by hydrogen bonding interactions with His118 and His196, similar to the mechanism of mononuclear B2 enzymes (see below). The spectroscopic data also support that in the enzyme-substrate (ES) complexes the two metal ions interact with the substrate, changing their coordination features. No reaction intermediates could be trapped in penicillin hydrolysis by BcII.
Figure 37 displays the minimal mechanism proposed for penicillin hydrolysis. This mechanism considers a terminal (not bridging) hydroxide as the attacking nucleophile, simultaneous with the activation of the carbonyl moiety by Zn1 and/or Asn233 in the first step of the reaction. The proton donor is supposed to be a water molecule. We postulate that Wat2, the water molecule bound to Zn1 in the resting state enzyme, moves to the bridging position synchronous to the nucleophilic attack and becomes the proton donor, regenerating the nucleophile at EP. This is the most plausible hypothesis since it does not require the assumption of a second water molecule that needs to be located at the same face of the antibiotic. However, this hypothesis cannot be definitively discarded and it also depicted in Figure 37.
Figure 37.
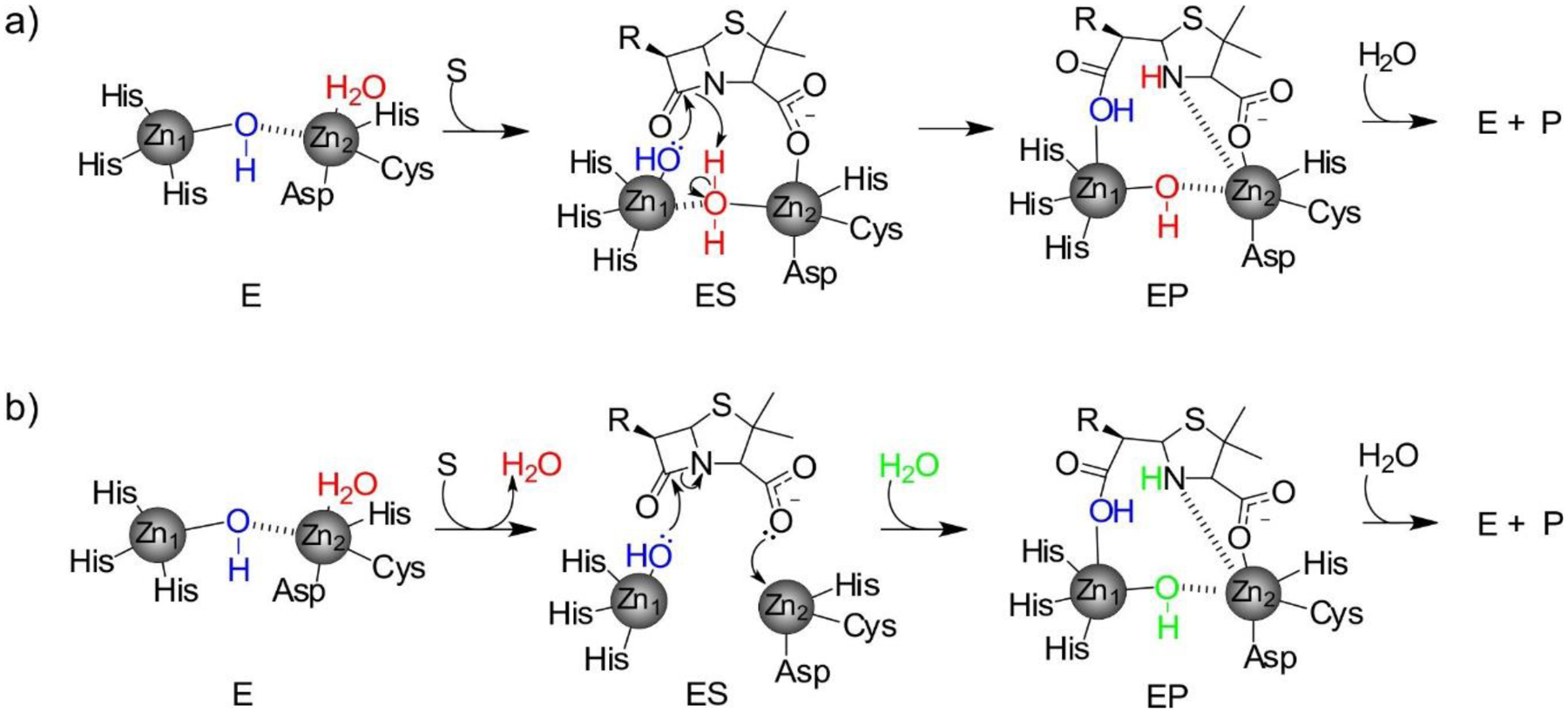
Mechanism of penicillin hydrolysis by bi-Zn(II) MBLs, with two proposed pathways for the protonation step. a) The nucleophilic hydroxide (blue) detaches from Zn2 upon substrate binding in ES. The water ligand originally bound to Zn2 (red) becomes a bridging ligand, becoming acidic and acting as the proton donor and regenerating the nucleophilic hydroxide. Based on the work from Llarrull, Vila and coworkers287. b) The Zn2-bound apical water (red) is detached from the metal site upon substrate binding. An additional water molecule from the bulk solvent (green) is incorporated into the active site in the subsequent step, acting as the proton donor. Based on the work from Nair and coworkers.244
X-ray crystallography of adducts with penicillins have provided pictures of the enzyme-product (EP) complexes with NDM-1. Indeed, the first structural snapshot of a complexed B1 enzyme was the structure of NDM-1 with hydrolyzed ampicillin, reported in 2011 by Zhang and Hao.647 Soon after, King, Strynadka and coworkers229 informed the structures of EP adducts of NDM-1 with methicillin, benzylpenicillin and oxacillin (Figure 38). These structures reveal the same binding pattern: (1) the C-N distance is 2.9 Å, confirming that the cleavage of the β-lactam ring has occurred, (2) the newly generated C-7 carboxylate binds Zn1 with one oxygen atom, while the other oxygen interacts with Asn233, (3) the N and the C3 carboxylate group coordinate to Zn2, (4) the C3 carboxylate forms a salt bridge with Lys224, which adopts the same orientation in all complexes, (5) a similar long Zn1-Zn2 distance compared to the unbound enzyme is found (ca. 4.6 Å, the longest intermetal distance reported for MBLs) and (6) there is a bridging water/hydroxide located closer to Zn1 (2.0 Å) than to Zn2 (3.0 Å).258 These features are preserved regardless the substrate, pH and crystallization conditions. These structures are consistent with the spectroscopic data describing the mechanism of binuclear B1 enzymes. The N atom replaces a water molecule in the apical position of the coordination sphere of Zn2, that remains pentacoordinated. The puckering of the thiazolidine ring suggests an sp3 hybridization for the N atom, that is expected to be already protonated. The bridging water molecule in the EP adduct is also favorably oriented and activated by Zn(II) binding to provide a proton to the N atom. This water molecule is likely the apical Wat molecule bound to Zn2 in the resting state enzyme.
Figure 38.
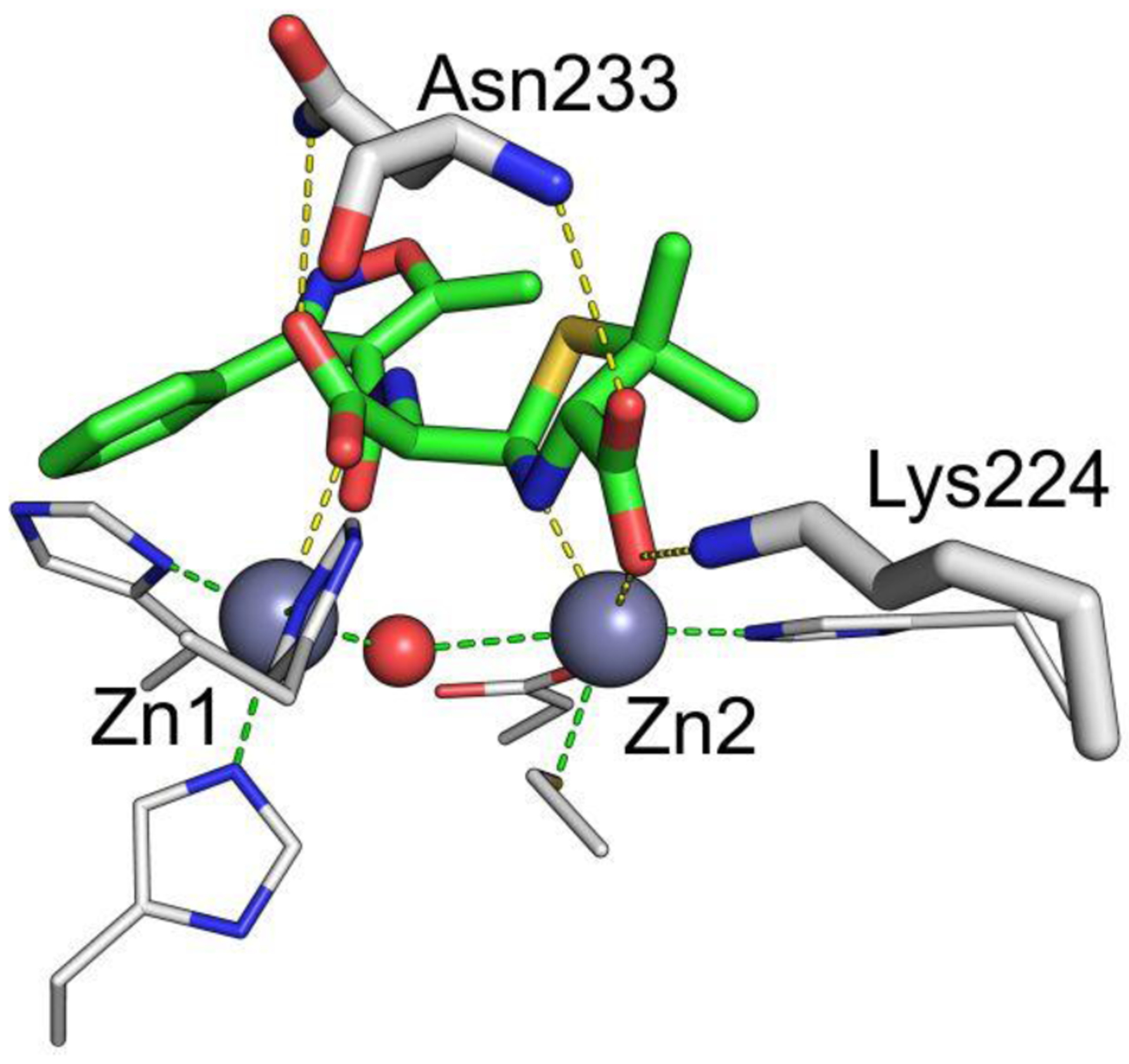
Structure of NDM-1 bound to hydrolyzed oxacillin (PDB 4EYB). Zn(II) ions and water molecules / hydroxide ions are shown as grey and red spheres, respectively, while the ligand is shown as green sticks and protein residues as white sticks. Interactions between the Zn(II) ions and their coordination residues are shown as green dashed lines, and interactions of the ligand with the protein are shown as yellow dashed lines.
5.5.2. Mechanism of cephalosporin hydrolysis
The effectiveness and reactivity of cephalosporins is highly dependent on the identity of the C3 substituent.643, 648 The chemistry of cephalosporins is unique among β-lactams since, after C-N bond cleavage, they generally undergo tautomerization of the C3-C4 double bond to the C4-N5 position in the dihydrothiazine ring, that can end up in different reactions,649 such as (1) elimination of the C3 substituent resulting in an exocyclic C3-C3’ double bond, (2) protonation at C3 or (3) protonation at the C3 substituent (Figure 39). The absorption of the β-lactam chromophore in cephalosporin is ca. 250–260 nm, but the cephalosporin core can be decorated with different chromophoric groups at the C3 position that can be exploited for mechanistic studies.615 The most widely used chromogenic cephalosporin is nitrocefin (Figure 40), with a dinitrostyryl moiety that gives rise to its spectral features.650 Other popular chromophoric cephalosporins are CENTA651, 652 and chromacef653 (Figure 40). The synthesis of fluorogenic cephalosporins has been employed for in cell studies or high-throughput screening assays of lactamase inhibitors. In particular, nitrocefin is yellow (absorbing at 390 nm), and its hydrolysis product is red (λmax = 490 nm).
Figure 39.
possible reactions occurring after hydrolysis of the β-lactam ring in cephalosporins.
In 1998, Benkovic and coworkers reported the accumulation of a reaction intermediate during the hydrolysis of nitrocefin hydrolysis by di-Zn(II) CcrA.607 This intermediate was identified based on its strong absorption at 665 nm, while intact nitrocefin and hydrolyzed nitrocefin present absorption maxima at 390 and 490 nm, respectively (Figure 41-a). This red-shifted spectral feature was attributed to an enzyme-bound anionic intermediate in which the N atom is negatively charged after C-N bond cleavage.242, 607 The spectrum of this intermediate was reproduced by treating hydrolyzed nitrocefin with a strong base, strongly supporting this proposal. A similar reaction intermediate in which nitrocefin hydrolysis was catalyzed by a synthetic binuclear Zn(II) model complex by the Lippard group allowed its characterization by infrared and 13C NMR spectroscopy, confirming the proposed structure (Figure 41-b).638, 654 QM-MM calculations also supported the stabilization of the anionic nitrogen by coordination to Zn2.625 The stability and feasibility of this intermediate have been matter of debate, since the electron-withdrawing π-conjugated dinitrostyryl moiety in nitrocefin can be expected to stabilize a negative charge, but this may not be the case for clinically relevant cephalosporins, with other substituents. The pKa values of this nitrogen in other systems is not expected to be low enough to stabilize a negative charge, suggesting that this intermediate could be an artifact resulting from the use of nitrocefin. However, in the absence of an available proton, Zn2 could act as a superacid, stabilizing the negative charge developed in the nitrogen atom in the anionic intermediate. DFT calculations on the mechanism of cefotaxime and cephalexin hydrolysis by dinuclear enzymes support the stabilization of this intermediate as a general feature.321, 620 The lack of experimental evidence supporting accumulation of this intermediate in other cephalosporins can be attributed either to a reduced stability compared to the nitrocefin intermediate, or to a possible overlap of the absorption band of this intermediate with the absorption in the UV range of the protein chromophores.
The rate-limiting step in this reaction is the protonation of this intermediate, a proposal consistent with a solvent kinetic isotope effect of 2.9.242 A water molecule is the most likely proton donor, either an incoming water molecule from the bulk solvent that binds the metal center, or the water molecule bound to Zn2 in the resting state enzyme that replaces the vacant position left by the attacking hydroxide and finally ends bound to Zn1. Again, Zn(II) coordination is expected to lower the pKa of this water molecule, favoring N protonation, and regenerating the nucleophilic hydroxide in EP. Similar intermediates have been informed for the hydrolysis of nitrocefin or chromacef by the binuclear forms of NDM-1, VIM-2 and L1.640, 655, 656This mechanism provided the first direct evidence of a catalytic role for the Zn2 site.
Different spectroscopies were used to interrogate the metal site during nitrocefin turnover. Co(II) substitution allowed identification of geometry changes in nitrocefin hydrolysis by L1.645 RFQ-EXAFS on L1 also showed that the Zn-Zn distance increases from 3.4 in the resting state to 3.7 Å in the intermediate.307 The latter distance is closer to that corresponding to the enzyme-product adduct (3.6 Å). This work by Crowder and Tierney provided a direct evidence of a scissoring motion of the metal site during turnover. This is also in line with the loss of coupling between the two Co(II) ions in di-Co(II)-NDM-1 observed by EPR in this intermediate.293
Different studies further supported the role of the Zn2 site in the formation of the anionic intermediate. Mono-Zn(II) GOB presents the Zn(II) at the DHH (Zn2 site) and shows accumulation of this intermediate in the absence of a Zn(II) ion at the Zn1 site.252 In contrast, a mono-Zn(II) form of L1, with the metal ion localized at the 3H site, showed a poor activity against nitrocefin and was not stable to accumulate the anionic intermediate.292 This intermediate also accumulates to different extents in the Zn(II)Zn(II)-, Zn(II)Co(II)-, Co(II)Co(II)-, and Co(II)Cd(II) derivatives of NDM-1, with the finding that the product release rates correlate to the the Lewis acidity of the metal at the DCH site (Zn(II) > Co(II) > Cd(II)).
The accumulation of the nitrocefin intermediate is different among MBLs: there is barely no intermediate formed with BcII657 or IMP-1.333 Minor perturbations, such as as second sphere mutations that change the hydrogen bond network or loop changes can lead to large changes in the accumulation of this and other intermediates.354 Insertion of the B3 loop from IMP-1 into BcII resulted in accumulation of the intermediate.333 The intermediate in IMP-25, an allelic variant with second sphere (S262G) and loop L10 (G235S) mutations leads to a more populated intermediate than in IMP-1.393 In the case of B3 enzymes, a study of mutations in loop α3-β7 (ca. residues 148–164) showed that loop motions correlated with the rate of formation of the nitrocefin intermediate.540 Overall, these results demonstrate that several factors determine the degree of accumulation of this intermediate, such as the position and the charge at the Zn2 site, and the interactions and conformation of loops L3 and L10 flanking the active site. In any case, the study of this intermediate was crucial for the dissection of MBL-mediated catalysis of cephalosporins.
No crystal structures have been reported of MBLs complexed with nitrocefin. However, several structures of MBLs with bound forms of cephalosporins are available. Indeed, the first description of a product bound to a binuclear MBL was the crystal structure of the hydrolyzed oxacephem moxalactam bound to the B3 enzyme L1 reported in 2005 by Spencer and coworkers.330 The structure reveals that moxalactam has experienced the elimination of the 1-methyl-5-thiotetrazole substituent at C3, giving rise to an 3′-exo-methylene group. This structure shows several similarities with the EP adducts of penicillins, i.e., product binding to the metal site occurs through a direct interaction of the two carboxylate moieties (the C4-carboxylate with Zn2 and the C8 carboxylate with Zn1), and the β-lactam nitrogen (at 2.4 Ǻ from Zn2). The C4-carboxylate also forms hydrogen bonding interactions with Ser221 and Ser225 (Ser223 in the older BBL numbering), despite these residues have been shown not to be essential for catalysis. A bridging water is preserved between the two Zn(II) ions. As a result, both metal ions increase their coordination number to penta- (Zn1) and hexa-coordinated (Zn2). The Zn-Zn distance is 3.68 Å, in agreement with the EXAFS data of the EP complex of L1 with nitrocefin,307 i.e., larger than in the resting state, but much shorter than the distance observed in the adducts with hydrolyzed penicillins. A series of structures of L1 bound to hydrolyzed moxalactam reported by the Joachimiak group in which the native metal ion has been replaced by Cu(II), Ni(II) or Cd(II) revealed similar binding patterns.284 These structures confirm the presence of a bridging water molecule in the EP.
Feng, Wang and Lui reported a series of complexes of NDM-1 with hydrolyzed cephalexin and a cephalosporoate intermediate of cefuroxime, with similar binding modes.329 The binding mode is similar to that observed for hydrolyzed penicillins, also preserving the hydrogen bonding interaction of the C8-carboxylate with Asn233, lacking in the structure of moxalactam with L1. In the case of the cephalexin complex, the electron density is consistent with a protonated sp3 C-3 from the α face, in agreement with NMR data of the hydrolyzed product. In this case, the most likely proton donor would be a Zn2-bound water molecule. The Zn-Zn distance is as long as in the penicillin adducts (4.5 Å), also including a bridging water closer to Zn1. Trp93 also shows an favorable S-π interaction with the dihydrothiazine ring, resembling that observed for the penicillin adducts.
The structure of NDM-1 with cefuroxime (Figure 43) presents a different scenario.329 The planarity among C6, N5, C4 and the C4 carboxylate C16 indicates that the double bond has already tautomerized from C3-C4 to C4-N5. Even if the C3 atom is sp3, the presence of the C3 carbamoyl substituent suggests that this structure represents an intermediate species. The C3 atom adopts a different chirality as that of the cephalexin product, suggesting that protonation is likely to occur from the β-face through a bulk water molecule. The bridging water molecule is preserved, and the Zn-Zn distance is 3.8 Å. This picture reveals similarities with the binding mode of penicillins, and trapping of this intermediate in a substrate with a good leaving group reveals that protonation (at the C atom in this case) is rate-limiting, as reported also for nitrocefin and the other chromophoric cephalosporins.
Figure 43.
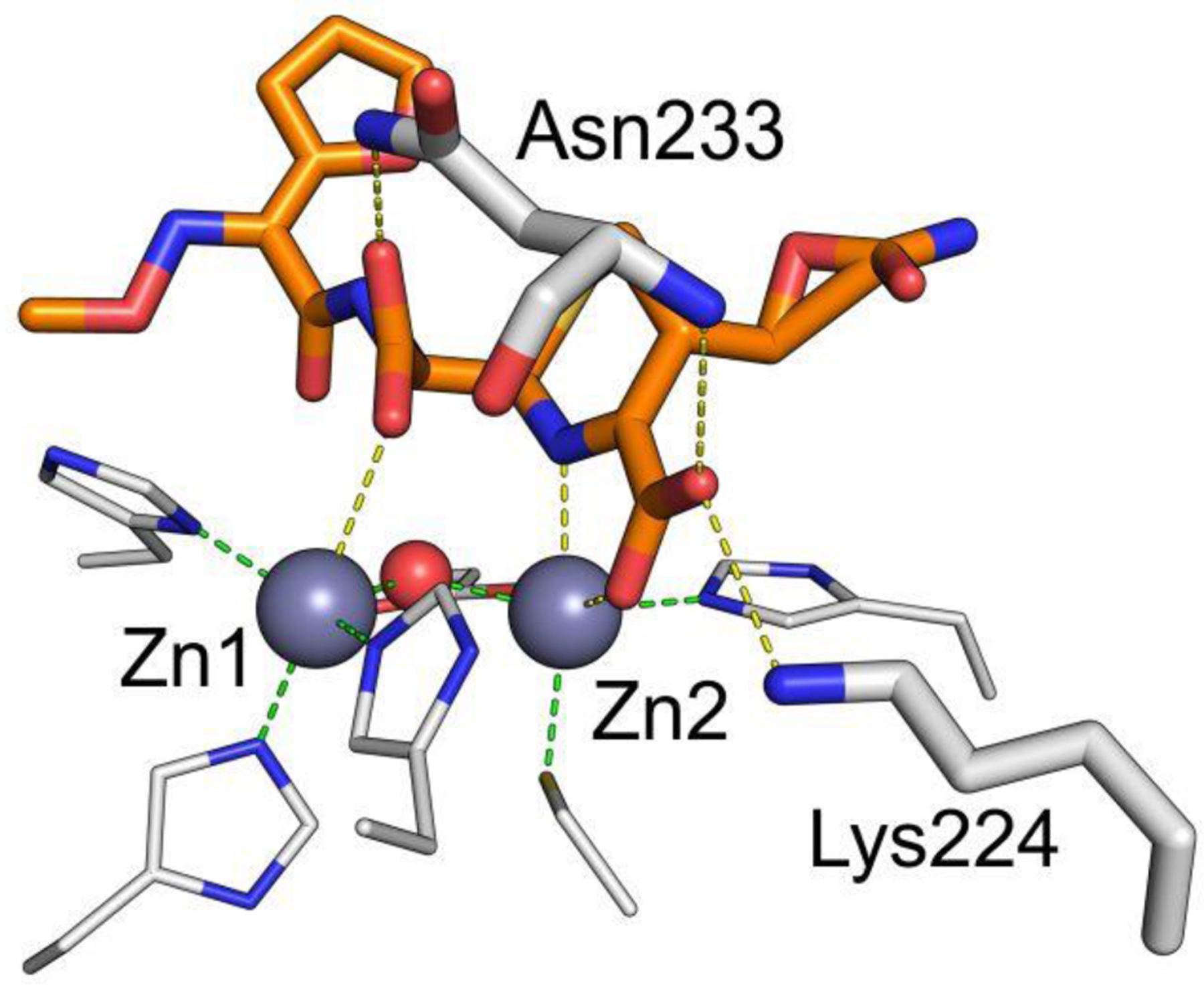
Structure of NDM-1 bound to hydrolyzed cefuroxime329 (PDB 4RL0). Zn(II) ions and water molecules / hydroxide ions are shown as grey and red spheres, respectively, while the ligand is shown as orange sticks and protein residues as white sticks. Interactions between the Zn(II) ions and their coordination residues are shown as green dashed lines, and interactions of the ligand with the protein are shown as yellow dashed lines.
5.5.3. Mechanism of carbapenem hydrolysis
Carbapenems are the only common substrates to all MBLs, thus enabling the mechanistic study of B1, B2 and B3 enzymes. Knowles and coworkers showed in the early 1980s658, 659 that carbapenem hydrolysis is followed by tautomerization of the double bond in the pyrroline ring from C2-C3 (the enamine Δ2 tautomer) to C3-N4 (the imine Δ1 tautomer), equivalent to the rearrangement taking place in some cephalosporins (Figure 44). If this tautomerization occurs within the active site and the two tautomers are present, N or C2 protonation can occur. C2 protonation can take place from the α or the β face of the ring, thus involving different proton donors. If the carbapenem is protonated at the N in the active site, when the Δ2 tautomer is released, it equilibrates in solution to generate the more stable Δ1 tautomer with a 1:1 ratio of the α and β diastereomers, which correspond to R and S stereochemistries at the C2, respectively.660 Thus, a stereoselective protonation at C2 or a diasteromeric ratio differing from 1:1 suggests that carbon protonation has taken place (at least partially) within the active site.
Figure 44.
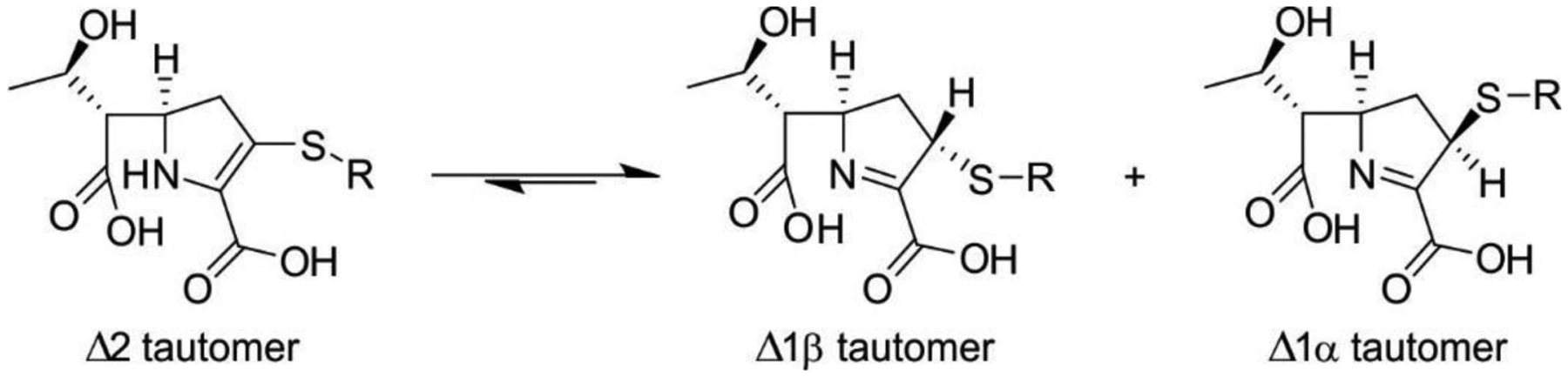
possible tautomers in hydrolyzed carbapenems.
The absorption of the β-lactam ring in carbapenems is red-shifted compared to penicillins and cephalosporins (λmax = 300 nm for imipenem) and can be better followed, despite partially overlapping with the aromatic protein chromophores. The detection of reaction intermediates in carbapenem hydrolysis has been challenging due to the limited spectroscopic properties of these compound. However, pre-steady state studies on imipenem hydrolysis by di-Zn(II) BcII revealed formation of a reaction intermediate at 390 nm, also present in the mechanism of di-Co(II) BcII, but shifted at 407 nm, making it more amenable to characterization (Figure 45).240 The different absorption maxima upon metal replacement confirms the direct involvement of the metal ion in this intermediate. RFQ-Resonance Raman experiments enabled the assignment of this band to the deprotonated form of a ring-opened pyrroline derivative that resembles the anionic intermediates identified in cephalosporin hydrolysis.240 This intermediate was also observed in meropenem hydrolysis by the B1 enzymes SPM-1 and IMP-25.348, 393
Further pre-steady state studies of imipenem hydrolysis by B1, B2 and B3 enzymes showed that carbapenem hydrolysis by enzymes from the three subclasses can be described by a common, branched mechanism.241 This mechanism involves two productive reaction intermediates: EI1 (the previously reported one, at 390 nm) and EI2, absorbing at 340 nm. Similar intermediates at 375 and 336 nm were reported for meropenem hydrolysis, confirming a general mechanism for carbapenem hydrolysis. This general scheme is valid for binuclear enzymes as well as for mono-Zn(II) enzymes with the metal at the Zn2 site, such as Sfh-I and GOB-18. The mechanism of B1 enzymes will be discussed first, on a comparative bases with the mechanism of penicillin and cephalosporin hydrolysis, and the particular features of the mechanism of mono-Zn(II) MBLs will be examined later.
Hydrolysis of imipenem by bi-Zn(II)-NDM-1 under single turnover showed accumulation of the same reaction intermediates EI1 and EI2, while formation and decay of the Michaelis complex (ES) occurred in the dead time of the equipment (2 ms).241 Co(II) substitution also disclosed changes in the coordination geometry of the metal sites during turnover. RFQ- EXAFS enabled following changes in the Zn-Zn distance at different steps of the reaction. This distance increased from 3.42 Ǻ in the resting state to 3.82 Å in EI1, and then relaxed to 3.51 Å in the EP complex.
Both intermediates correspond to anionic species and, based on carbapenem chemistry, they may differ in favoring protonation at N or at C2 and maybe requiring two different proton donors. The α:β diastereomer ratio for the imipenem hydrolysis products by different MBLs revealed an excess of the β diastereomer, in contrast to the acid-induced hydrolysis. This ratio can be accounted for only by assuming that, in addition to protonation at the N atom, a diastereoselective protonation at C-2 takes place within the enzyme active site, whose product is the Δ1β diastereomer. It was proposed then that the two productive intermediates could correspond to anionic species with charge localization at the N or C2 atoms.
Several structures of NDM-1 bound to hydrolyzed carbapenems229, 328 reveal a similar interaction with Zn2 through the C3-carboxylate and the N atom, and involvement of Lys224, resembling the EP complexes with penicillins and cephalosporins (Figure 47). However, all these structures lack a bridging water/hydroxide while showing a C7-carboxylate bridging the two Zn(II) ions. The Zn1-Zn2 distance is 4.0 Å, i.e., lengthened with respect to the unbound form, but shorter than in other complexes. These structures suggest that the Zn2 site is able to stabilize several negatively charged ligands in addition to the DCH ligand set. The L3 loop is more closed in these structures compared to penicillin- or cephalosporin-bound structures, possibly due to the smaller substituents at C6 in carbapenems. Feng and coworkers reported a series of structures of NDM-1 with hydrolyzed imipenem and carbapenems, that the authors attributed to the EI1, EI2 and EP complexes. In agreement with the previous EP adducts, these structures lacked a bridging water.328 Joachimiak reported the structures of L1 complexed to hydrolyzed imipenem and meropenem. The binding pattern of meropenem (PDB 6UAH) resembles those reported for the adducts with NDM-1, while in the imipenem complex (PDB 6UAF) the C7 carboxylate is not binding Zn1, and a bridging water molecule is present.
Figure 47.
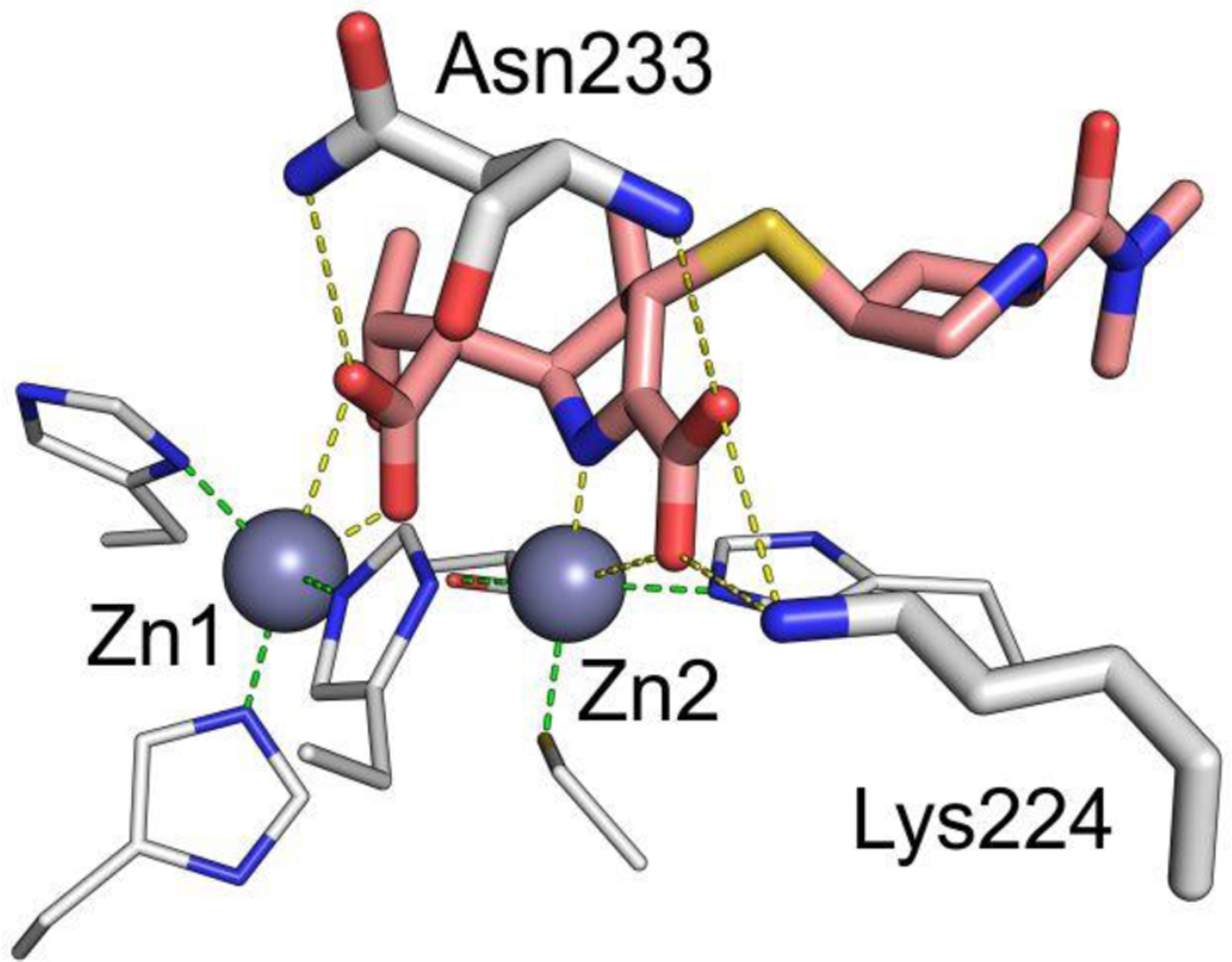
Structure of NDM-1 bound to meropenem328 (PDB 5YPN). Zn(II) ions are shown as grey spheres, while the ligand is shown as orange sticks and protein residues as white sticks. Interactions between the Zn(II) ions and their coordination residues are shown as green dashed lines, and interactions of the ligand with the protein are shown as yellow dashed lines.
The EP or EI structures of carbapenems with NDM-1 lack a bridging water/hydroxide, that would result after protonation of the N atom, while C2 protonation has been proposed to occur by water molecules located at the β face of the carbapenem, close to the hydrophilic wall defined by loop L10. This can be due to the dissociation of the water ligand at the Zn2 site upon carbapenem binding, precluding the possibility of N protonation. It is also likely that the EP species with the bound water could be unstable, leading to dissociation of the hydrolyzed product that may not be trapped in the crystal. The latter explanation is supported by QM-MM calculations. The lack of a bridging water molecule was attributed by Feng and coworkers to the unique S configuration of C6 in carbapenems (differing from penicillins and cephalosporins).328 After hydrolysis, both oxygens from the newly formed C6 carboxylate bind Zn1 in a bidentate fashion, precluding the presence of bridging water molecule.
Wachino and coworkers reported the EP complexes of B3 lactamase SMB-1 with hydrolyzed imipenem, meropenem and doripenem revealing a tetrahedral Δ1 tautomer protonated from the β face.661 In line with these results, in 2017 Feng, Wang and Liu confirmed the existence of pyrroline tautomerization inside the enzyme active site.328 Different structures were consistent with binding of Δ2 (EI1) in one imipenem- and three meropenem-bound structures and others with a bound Δ1 (EI2 or EP) form in five imipenem- and two meropenem structures bound to NDM-1. All trapped molecules in Δ1 (EI2 or EP) show S chirality at C2, implying protonation from the β face). No α diasteromers could be detected by this group. Feng and coworkers proposed a linear reaction mechanism based on the finding of a single reaction product, but this conclusion does not account for the kinetic studies.
Lohans, Schofield and coworkers have questioned the conclusions about the stereochemistry of the C2-protonation,662 since the different tautomeric forms equilibrate rapidly in solution, and the stereochemistry of the final products may not reflect the protonation within the enzyme active site. However, the consistent lack of a water molecule in the α face of carbapenems in the different adducts would support protonation by the β face leading to a steroselective protonation at the C2 atom.241
There are few theoretical mechanistic studies of carbapenem hydrolysis. Nair and coworkers reported a QM-MM study calculation on the reaction coordinate of the hydrolysis of meropenem by NDM-1 that support formation of a stable anionic intermediate species.244 DFT calculations reveal that the negative charge on this intermediate is delocalized among the N4 and C3 atoms of the pyrroline ring and the sulfur atom at the C4-side chain.241
Imipenem hydrolysis by two mono-Zn(II) MBLs (the B3 GOB-18 and the B2 Sfh-I) revealed different accumulations of the intermediate species.241 In contrast to binuclear lactamases, both cases revealed a significant accumulation of the Michaelis complex (ES). This has been attributed to a slower reactivity of the nucleophile in mono-Zn2 enzymes, which would not be metal-activated. Instead, in binuclear MBLs, metal activation of the nucleophile accelerates the first step of the reaction, resulting in a very short-lived ES complex. In the case of GOB, substrate binding was accompanied by an increase of the coordination number at the ES, while in Sfh-I the metal ion remained 4-coordinated, suggesting that binding of the C3-carboxylate to the zinc in the latter case takes place by replacing the bound water. Similar conclusions were obtained for Co(II)-GOB-18 and Co(II)-ImiS.241
The structure of the B2 enzyme CphA in complex with hydrolyzed biapenem showed a form of biapenem resulting from a molecular rearrangement after the nucleophilic attack.245 The N4 atom and the C3 carboxylate bind the Zn(II) ion, as observed for binuclear enzymes. The C3 carboxylate also interacts with Lys224 and Asn233, thus presenting similar binding features as B1 enzymes despite the absence of the Zn1 ion. The structure of resting state mono-Zn(II) Sfh-I reveals a water molecule making hydrogen bond interactions with His118 (at 2.0 Ǻ) and His196 (at 2.5 Ǻ).
The crystal structure of di-Zn(II) CphA shows that the inhibitory metal ion binds His118 and His196 (metal ligands from the canonical 3H site in B1 enzymes), a water molecule and a sulfate ion. Asn116 does not interact with the metal ion at this site.248 Binding of the second Zn(II) does not affect the overall structure nor the active-site entrance. Thus, inhibition is not due to any steric effect elicited by zinc binding. The position of the Zn2 is not altered either. Binding of the second Zn equivalent prevents His118 and His196 to activate the attacking nucleophile, while not being able to provide a geometry adequate for metal-based nucleophile activation as in binuclear B1 MBLs. This proposal is supported by the low activity of the His118Ala and His196Ala mutants.313
In summary, the main mechanistic difference of B2 enzymes with the dinuclear B1 and B3 MBLs is the fact that the metal ion is not involved in the activation of the water nucleophile. However, the two-fold role of Zn(II) in B2 enzymes in binding the substrate and stabilizing the anionic intermediate resembles the role proposed for the Zn2 site in B1 and B3 enzymes, despite the different active-site topologies and metal site content.
5.6. Reflexions on the catalytic mechanism of MBLs
All bicyclic β-lactams present a common binding pattern in mono and binuclear MBLs, in which the Zn2 ion steers the productive binding by interacting with the carboxylate group and positioning the β-lactam ring to the attacking nucleophile. Mechanistic differences among the different types of substrates (accumulation of anionic intermediates, N or C protonation) are due to the specific nature of each substrate in particular, either the particular stereochemistry at C6 in carbapenems, or the different substituents in cephalosporins.
The mechanism proposed for mono-Zn(II) enzymes with the metal ion at the Zn2 site confirms the non-essentiality of a metal-activated nucleophile. Indeed, engineering the position of the Zn2 site in such a way that is not able to stabilize the anionic intermediate despite the activated nucleophile is present, results in drop of three to four orders of magnitude in the catalytic efficiency of the enzymes. Hao and coworkers have argued that the Zn-Zn distance, instead of the position of the Zn2 ion, is crucial for the enzyme activity. In other words, MBLs can prescind from the Zn1 site, but not from the Zn2 site. This conclusion is in conflict with in vitro reports of the activity of mono-Zn1 enzymes, but at the moment there are not known MBLs in which the Zn2 site has been abolished resulting in an active enzyme.
The unique coordination chemistry of MBLs makes them suitable to perform a precise and at the same type promiscuous chemistry. The lack of a bridging protein ligand endows the metal site with a high flexibility during catalysis that is accompanied by the fact that all metal ligands are located in loops. This metal site dynamics results in unprecedented lengthening of the Zn-Zn distances that cannot be afforded by traditional binuclear hydrolases with carboxylate bridging ligands, that are generally shaped to target only one substrate. The herein discussed mechanism reflect the malleability of this protein scaffold that still has many uncovered and controversial issues to unveil.
6. MBL Inhibitors
The main concern regarding the dissemination of genes coding for MBLs among Gram-negative bacteria is the inability of the available SBL inhibitors to block the action of these metalloenzymes.15, 17–19, 22, 80, 663–668 Despite significant efforts since the mid 1990’s, several thousands of MBL inhibitors have been reported in the literature, but no one has made it to the clinics yet. There are recent progresses that are promising. Cefiderocol is a siderophore-modified cephalosporin active against Gram-negative bacteria recently approved by the FDA, that has been reported to be refractory to the action of MBLs.669–671 However, the molecular details by which this antibiotic escape the hydrolytic action of MBLs are still unknown, and the adaptation of current MBLs to this substrate cannot be discarded in the near future. On the other hand, two boronate compounds that can target MBLs and SBLs, taniborbactam (previously known as VNRX-5133) and QPX7728 are currently in phase III clinical trials. Here we will discuss the challenges faced in the development of MBL inhibitors, and the different strategies, that depict a tour-de-force in medicinal chemistry, and as such includes many important lessons to be learned.
6.1. Beta-lactamase (SBL) inhibitors
6.1.1. Traditional β-lactam based inhibitors
The emergence of the first plasmid-mediated β-lactamases in the 1970s spurred the development of compounds capable of counteracting these enzymes and restoring the efficacy of β-lactam antibiotics. The first clinically employed β-lactamase inhibitor was clavulanic acid 2 (Figure 50-a), a natural product from Streptomyces clavuligerus.672, 673 This compound belonged to a novel class of β-lactam drugs, the clavams, with structural similarities to penicillins, although with an oxygen atom substituting the sulfur in the second ring. Clavulanic acid possesses negligible antimicrobial activity by itself, but is a suicide inhibitor of many class A SBLs. The nucleophilic attack of the active Ser residue on clavulanic acid is followed by a very slow deacylation step, leading to an irreversible inactivation of the enzymes.674 Oxidation of the sulfur atom in the penicillin core to yield penicillanic acid sulfones lead to the discovery of two further inhibitors, sulbactam 3 and tazobactam 4 (Figure 50-a).675, 676 They possess a similar spectrum of activity to that of clavulanic acid, and also lack significant activity against bacteria by themselves. In spite of their clinical value, these drugs lack activity towards class B enzymes, and also certain SBLs from class A, C and D.7, 210
Figure 50.
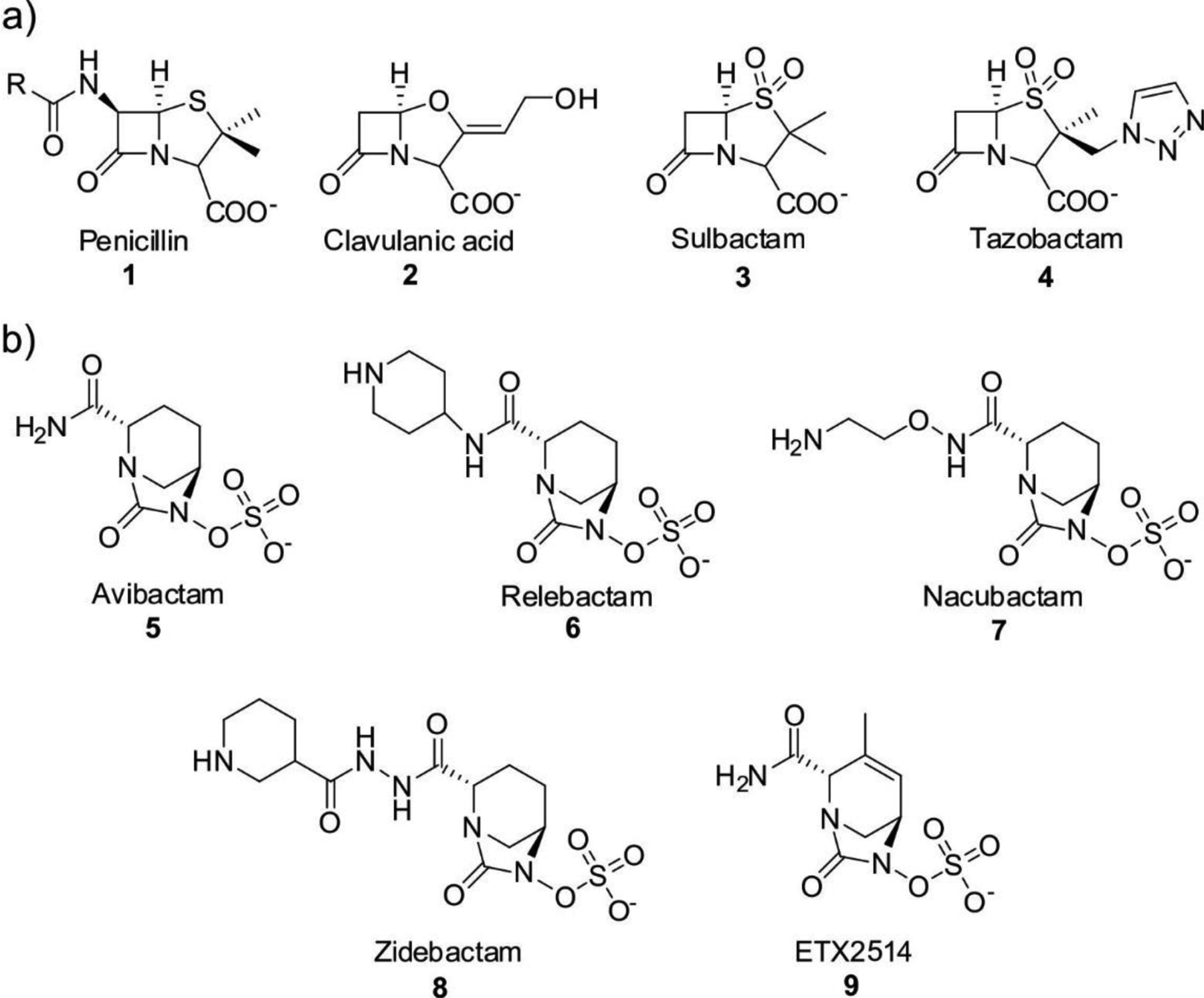
a) Structure of the penicillin core, and β-lactam based SBL inhibitors. b) Diazabyciclooctanone (DBO) β-lactamase inhibitors.
6.1.2. Diazabicyclooctanones
The significant gap in inhibitor coverage was finally being narrowed by the development of new classes of compounds.80, 677 The first family of newer inhibitors to be introduced for clinical applications was that of the diazabicyclooctanones (DBOs, Figure 50-b), among which avibactam 5 was approved by the FDA in 2015 for use in combination with ceftazidime as an efficient combinaction against SBLs.678, 679 A potent inhibitor of class A and C β-lactamases, avibactam also shows activity towards some class D enzymes while being, however, almost inactive towards MBLs. The action of MBLs against avibactam has already been discussed in this review (Section 4.4). Its mechanism of action is distinct from previous SBL inhibitors, with covalent but reversible binding to its target β-lactamases.678 The combination of avibactam with aztreonam is currently in phase III trials,79 thus offering a tandem that is potentially effective against both SBL and MBL producers.
Additional inhibitors of the DBO class are in the pipeline: relebactam 6 (Figure 50-b) was recently approved in combination with imipenem/cilastatin,680, 681 and others such as nacubactam 7 and zidebactam 8 are under clinical trials.79, 682, 683 Although incapable of inhibiting MBLs, nacubactam possesses intrinsic antibacterial through PBP inhibition, and its combination with meropenem showed effectiveness toward multidrug resistant Enterobacterales that included MBL producers.684 Finally, ETX2514 9 is a DBO compound in clinical trials that is engineered to improve inhibition towards class D β-lactamases, while maintaining potency towards A and C class enzymes.685 This is especially important for the treatment of infections by Acinetobacter spp., which produce a variety of OXA (class D) enzymes and frequently present multi-drug resistant phenotypes.
6.1.3. Boronates
Boronic acids represent the other novel class of SBL inhibitors.80, 668, 677 These compounds act as mimics of the tetrahedral transition state of β-lactam hydrolysis by SBLs, and reversibly bind the enzymes through covalent binding to their catalytic serine residue.668 Vaborbactam is a potent inhibitor of class A lactamases, clinically approved in combination with meropenem.686, 687 It is also active towards some class C enzymes, but cannot inhibit those belonging to class D or B. More novel members of the class have managed to expand the spectrum of inhibition of boronic acids. Taniborbactam and QPX7728 are bicyclic boronates that target MBLs and SBLs, and will be discussed in detail below.
6.2. Challenges for designing MBL inhibitors
There are many challenges that have impeded so far the availability of a clinical MBL inhibitor. Fortunately, one or two boronic acid derivatives are likely to be approved soon. Despite this great progress, there are several aspects that still deserve consideration and discussion in the arena of MBL inhibition.
The first obstacle is related to the different mechanism and structure of SBLs and MBLs. Inhibition of SBLs strongly relies in the presence of a covalent intermediate, that is lacking in MBL-mediated hydrolysis. This precludes the use of suicide substrates such as clavulanic acid and related compounds. Second, SBLs have a narrow and deep catalytic site, with a buried Ser residue, while the active site in most MBLs is located in a shallow groove. Third, the diversity in active-site topologies among B1, B2 and B3 lactamases, involving also the metal-ion stoichiometry, has made it difficult to design a common inhibitor targeting all MBLs. Fourth, the close link of MBLs to the MBL superfamily, including metalloproteins with similar folds and active site present in mammalian cells, raises the issue of possible off-target reactivity of MBL inhibitors. More recently, there has been progress in the use of zinc chelators or strategies for zinc replacement for other metal ions that give rise to inactive derivatives have been studied, but the concern of specificity is even higher when resorting to these approaches. There are excellent recent reviews about MBL inhibition reporting on specific NDM-1 inhibitors,18 inhibitors inspired in the mechanism,15, 19 compared strategies for inhibition,22, 615, 665, 666, 688 analyses of specific types of MBL inhibitors668 and patent reviews,663 to which the reader is referred.
Most studies reporting MBL inhibitors are limited to the report of the in vitro inhibitory effect quantitated in terms of IC50 or KI values, measured under a wide variety of conditions and using different substrates. Moreover, in many cases chromophoric cephalosporins such as nitrocefin are used as reported substrates, an approach that is useful for library screening. However, in many cases, inhibition of MBLs versus a clinical substrate is not reported. Since all MBLs are carbapenemases, and this hydrolytic capability raises the most important clinical concern, it is desirable that the most promising inhibitors are tested in vitro against MBLs using carbapenems as substrates. Based on these premises, in this review we will not present a comprehensive account of these values but will, instead, analyze the different strategies and ranges of inhibitory potencies achieved in some relevant cases, discussing the rationale of the design, the structural outcome (if available) and the limitations of some approaches.
Most of the original articles present different synthetic or screening strategies to find MBL inhibitors and test them, but most of them lack the characterization of the impact of these inhibitors in bacteria expressing MBLs and, most important, on clinical strains, that are more difficult to treat that laboratory strains. Finally, cytotoxicity, PK/PD studies or pre-clinical assays in animal models are very scarce in this field. Therefore, most of the comparative analysis in this section will refer to the chemical strategy and the relative success in achieving an in vitro inhibitor.
The different inhibitors can be classified based on the chemistry: targeting the Zn(II) ions (as most of the approaches consider) or targeting other structural aspects of the MBLs (such as allosteric inhibitors, or covalent inhibition aimed to modify some active-site residues). The mechanistic-based approach have relied mostly in the assumption of a high energy tetrahedral adduct similar to that present in SBLs, that has inspired phosphonate and boronate-based inhibitors, the latter being by far more successful, and mimicking the common anionic intermediate formed after C-N bond cleavage.
A recent review by Linciano, Donda and coworkers includes a thorough analysis of the different inhibitors assayed against NDM-1.18 The dendrogram shown in Figure 51 shows the distribution of NDM-1 inhibitors based on chemical structures and colored by inhibitory potencies. Since the work focused on a single MBL, the inhibition profiles differ for other MBLs, but this dendrogram depicts in a faithful manner the distribution of inhibitors tested for all MBLs.
Figure 51.
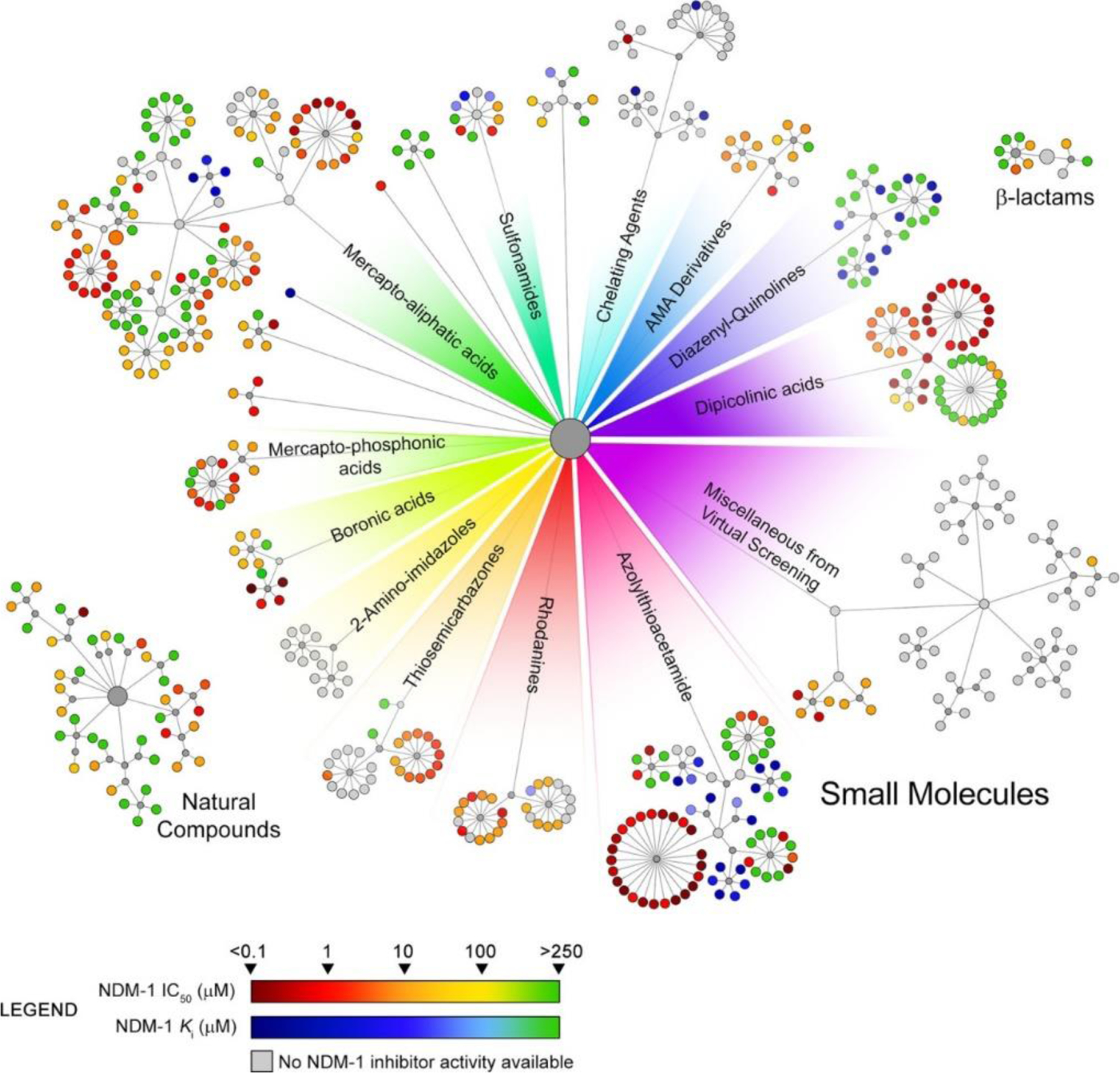
Dendrogram of NDM-1 inhibitors. Inhibitors are classified into three main categories (small molecules, natural compounds, and β-lactams) and clustered based on their chemical similarity. Each inhibitor is represented by a colored dot at the end of each branch. The color coding indicates the potency of the inhibitor against NDM-1, as indicated in the color bar at the bottom. Gray colors correspond to compounds with no inhibitory action. Reproduced with permission from reference18 Linciano et al. Copyright 2019 American Chemical Society.
6.3. MBL inhibitors inspired on β-lactam scaffolds
MBLs bind and hydrolyze bicyclic β-lactam compounds. Based on this, a number of efforts have been devoted to exploit the β-lactam scaffold for designing MBL inhibitors.
Buynak and coworkers have employed this approach aiming to a simultaneous inactivation of SBLs and MBLs. The introduction of a hydroxamate group at the C7 position of cephalosporins such as in compound 10 (Figure 52-a) resulted in a low-μM inhibitory potency against B1 enzymes.689 They also modified the penicillin scaffold by introducing a thiol moiety targeting MBLs,690 with the ability of acylate irreversibly SBLs. Compound 11 (Figure 52-a) showed an IC50 in the low-μM range against BcII, L1, TEM-1 and P99.
Figure 52.
a) MBL inhibitors inspired on β-lactam scaffolds. b) Crystal complex of L-15:NDM-1697 (PDB 4U4L) c) Crystal complex of D-15:IMP-1412 (PDB 5EV8). Zn(II) ions are shown as grey spheres, protein residues are shown as white sticks, while the bound ligands are shown as colored sticks and water molecules are shown as red spheres. Protein-ligand interactions are shown as dashed lines.
Pratt, Page and Frère synthesized thioxocephalosporins, in which the β-lactam reactivity is impaired by the presence of a thioamide group).691, 692 The 8-thioxocephalosporin 12a (Figure 52-a) was poorly hydrolyzed, and the hydrolysis product 12b was able to inhibit BcII (KI = 96 μM).691, 692
A library of carbapenems derivatives resulted in the identification of several moderate MBL inhibitors. J-110,441 13 (Figure 52-a), with a benzothiophene substituent at C2, was able to inhibit B1 and B3 enzymes, and showed synergy with imipenem against a S. marcescens clinical strain expressing IMP-1.693, 694 Other β-lactam-based compounds that lead to irreversible inhibition by generating active thiol species will be discussed later in Section 6.7.
The Franz lab has synthesized a modified cephalosporin (PcephPT 14a, Figure 52-a) that, upon hydrolysis, releases a chelator prodrug PT 14b (pyrithione)695 with activity against NDM-1.696 The action of this compound, however, is not through removing the metal ions but as a metal ligand.
Bisthiazolidines (BTZs) are more recent generation of bicyclic inhibitors designed as substrate mimics inspired on the structure of penicillins, but lacking the amide group and with a thiol as metal binding group, preserving the carboxylate (cf. L-15, D-15, L-16 and D-16 in Figure 52-a) BTZs are pan-MBL inhibitors, i.e., able to inhibit B1, B2 and B3 enzymes.412, 437, 697 Binding to the active site is steered by the thiol, that bridges the two Zn(II) ions in binuclear B1 and B3 MBLs. The carboxylate interacts with the conserved Asn223 and Lys224 residues (Figure 52-b,c) in B1 enzymes. Instead, in the case of the B2 Sfh-I, metal binding occurs through the carboxylate. The stereochemistry of the chiral centers only affected the inhibitory power in B2 enzymes. BTZs can restore efficacy of carbapenems against MBL-producing clinical strains.
6.4. Metal ligands as MBL Inhibitors
6.4.1. Thiol-based inhibitors
Zn(II) is a thiophilic metal ion, and thus thiol-containing molecules have been largely exploited as MBL inhibitors. Most of the first MBL inhibitors were thiol-based, such as cysteinyl dipeptides,698 thiomandelic acid,699 captopril700 and a mercaptocarboxylate compound partially mimicking a hydrolyzed penicillin.382
The pH dependence of the inhibition profile of cysteinyl dipeptides suggested that the thiol group displaced the bridging water/hydroxide in BcII.698 The strategy of using cysteinyl dipeptides was later revisited by Crowder and cowokers who screened a library of 90 homo-cysteinyl peptides, and were able to optimize binding up to nM affinity against L1.701
The binding mode of thiol groups was later demonstrated by the crystal structure of IMP-1 inhibited by the mercaptocarboxylate 17 (Figure 53-a) reported by Herzberg, Concha and coworkers.382 The compound is efficient against IMP-1 and L1 in the nM range. This molecule reproduces some of the chemical moieties present in benzylpenicillin: a linear amide, the carboxylate and an aromatic group. The thiol moiety indeed bridges the two Zn(II) ions, and makes electrostratic interaction with Asn233, Lys224 and hydrophobic contacts with L3 (Figure 53-c) mimicking some substrate-binding features. A similar strategy was recently reported targeting NDM-1.702 Despite the product mimicking features, the thiolate provide the driving force for binding, as described for the susbtrate-mimicking bisthiazolidines.
Figure 53.
a) Thiol-based inhibitors. b) Model of meropenem bound to NDM-1 (base PDB 3SPU). c – e) Crystal complexes of 17:IMP-1382 (c, PDB 1DD6), D-22:IMP-1706 (d, PDB 4C1G), and 24:NDM-1707 (e, PDB 6ZYP). Zn(II) ions are shown as grey spheres, protein residues are shown as white sticks (green sticks for residues involved in hydrophobic interactions with the ligand), while the bound ligands are shown as colored sticks and water molecules are shown as red spheres. Protein-ligand interactions are shown as dashed lines.
Thiomandelic acid 18 (Figure 53-a) was reported in 2001 as a potent sub-μM inhibitor of different B1 and B3 enzymes, being the first broad spectrum MBL inhibitor reported.612, 699 Instead, inhibition of the B2 lactamase CphA was weaker. The NMR structure of BcII complexed to R-thiomandelic acid345 solved in 2013 revealed that binding was due to the thiol moiety, and did not suggest any relevant role of the carboxylate in interactions with active-site residues. The impact of thiomandelic acid against clinically relevant strains expressing MBLs has been recently assessed.703
Thiol groups were combined in different synthetic inhibitors with other functional groups. Bebrone, Galleni and Frère reported a series of mercaptophosphonates as efficient broad-spectrum inhibitors of MBLs.704 The crystal structures of some adducts revealed that either the phosphonate or the thiol groups could bind the Zn(II) site. A boronic acid with a thiol group was synthesized aiming to build up an inhibitor targeting both MBLs and SBLs,705 with some impact in B1 enzymes in the low-μM range.
Dmitrienko undertook a systematic kinetic study of thiol-based inhibitors of IMP-1,708 concluding that these compounds inhibit MBLs by replacing the active nucleophile and not by metal removal, and that the presence of two thiol groups in the same molecule does nor result in a more potent inhibition. Finally, strong binding thiol inhibitors show a biphasic behavior, that has been interpreted as resulting from an initial binding to Zn2 then followed by a rearrangement to give rise to the bridging μ-sulfide mode. A similar biphasic mode was also reported for the binding of bisthiazolidines to VIM-2 and VIM-24, suggesting that this could be a general mechanism of binding for thiol inhibitors.437
The use of thiol compounds as MBL inhibitors has been questioned based on their tendence to be oxidized to the dimeric form or to different oxygen-containing variants.703 However, there is a significant number of thiol-containing drugs approved. Among them, captopril has been the best characterized thiol-based MBL inhibitor (see below). The Proschak group screened eleven of these drugs or their active metabolites against several B1 MBLs.709 Interestingly, the selected drugs showed a wide range of inhibitory potencies. Captopril 22, thiorphan 19, dimercaprol 20 and tiopronin 21 (Figure 53-a), showed inhibition at the low μM range and reduced the MIC of clinical isolates expressing different MBLs. Introduction of a piperazine scaffold led to improved inhibition.710
L-captopril 22a is an inhibitor of the angiotensin converting enzyme (ACE) regularly prescribed for the treatment of hypertension.711 In addition to the thiol group, its structure resembles some features present in hydrolyzed penicillins. L- and D-captopril, as well as a series of captopril analogues have been explored as MBL inhibitors in the last two decades.229, 260, 543, 700, 706, 712–716 The stereochemistry had different impact in the inhibition depending on the enzymes: both L- and D-captopril inhibit BcII, IMP-1 and VIM-2 with high efficiency, but the D-stereoisomer is much more efficient than the L- variant for NDM-1 and CphA, while both are poor inhibitors of SPM-1. Several crystal structures have revealed the binding modes of the different variants to distinct MBLs.229, 260, 543, 706, 713 The thiol group bridges both Zn(II) ions in all cases, while the carboxylate interacts with different active-site residues (Figure 53-d). In general, the most potent inhibition is obtained upon a larger number of hydrogen bonding interactions of the carboxylate group,706 in general with residue Lys224 (B1 enzymes), Arg228 (VIM enzymes). Schenk explored captopril analogues witha six-membered pyrrolidine scaffold.715
The recently reported 2-mercaptomethyl-thiazolidines (MMTZs, 23 and 24) achieved inhibition of B1,707 B2 and B3 enzymes (Hinchliffe et al., submitted). Despite their close resemblance to captopril and their variants, MMTZs achieved better potencies which did not depend on their stereochemistry (except for B2 enzymes) since they are more deeply buried within the active site of MBLs and, instead of making electrostatic interactions through the carboxylate group, the thioether of the thiazolidine ring forms conserved S-π interactions in all adducts (Figure 53-e). In summary, the hydrophobic interactions override the effect of the electrostatic ones leading to better inhibitors.
6.4.2. Carboxylate-based inhibitors
Screening of the Merck chemical collection by Toney and coworkers resulted in the finding of succinic acid derivatives as MBL inhibitors.717 Among them, a variant with aromatic groups 25 (Figure 54-a) inhibited IMP-1 in the nM range. The crystal structure of the adduct revealed binding one of the carboxylates to Zn2, Lys224 and Asn233, while the other carboxylate moiety bridged the two Zn(II) ions replacing the attacking nucleophile (Figure 54-b). Another screening found other succinic derivatives that were able to revert meropenem resistance in IMP-1 expressing E. coli strains.718
Figure 54.
a) Structure of carboxylate-based inhibitor 25. b) Crystal complex of 25:IMP-1717 (PDB 1JJT). Zn(II) ions are shown as grey spheres, protein residues are shown as white sticks, while the bound ligand is shown as colored sticks. Protein-ligand interactions are shown as dashed lines.
Different phtalic acid derivatives were also reported as MBL inhibitors in the μM range,719–721 as well as maleic acid derivatives displaying μM inhibition toward some B1 enzymes and active against MBL-producing P. aeruginosa strains.722–724
Crowder reported a series of N-heterocyclic dicarboxylic acids as MBL inhibitor, some of them with low-μM KI against the three subclasses with potency against E. coli laboratory strains expressing different MBLs.725 More recently, iminodiacetic acid was employed by Cohen and Crowder as a scaffold for obtaining more potent NDM-1 inhibitors. The best compound inhibited NDM-1 by binding, instead of stripping the metal ions with low μM affinity.726 A series of benzimidazole and benzoxazole-based compounds reported by the Franz group resulted in sub μM inhibitory potency through binding rather than chelation.727 It is therefore important to evaluate the effect of polycarboxylate molecules towards MBLs to assess whether they act as metal ligands or metal chelators.
6.4.3. Phosphonates
The tetrahedral geometry of phosphonate groups has been largely exploited as transition state mimics for different enzymes, including SBLs.728, 729 This approach has not been equally successful when applied to MBLs. For instance, none of the mercaptophosphonates described in Section 6.4.1 behaved as a transition state mimic.704 Crowder and Oelschlaeger synthesized a β-phospholactam active against some B1 and B3 enzymes.730 However, this compound is hydrolyzed to a phosphonate in solution, casting some doubt on the chemical identity of the inhibitor. The carboxylate group in mercaptocarboxylic acids was replaced by bioisosteric groups like phosphonate esters, phosphonic acids and NH-tetrazoles in a study by Leiros, Samuelsen and coworkers. The most efficient inhibitors were those containing the thiol group and a phosphonate ester or acid. Some of these substitutions improved the inhibitory power, but by means of hydrophobic interactions.731
A series of phosphonate-based pyridine-carboxylates synthesized by Dmitrienko exhibited inhibition in the low-μM range against VIM-2, NDM-1, IMP-1 and L1 (26, Figure 55-a). Compound 26 was active against different MBL producers, even towards the hard-to-deal Stenotrophomonas maltophilia. Spencer solved the structure of the adduct of 26 with IMP-1, that reveals that the phosphonate, instead of replacing the bridging hydroxide, forms a hydrogen bond interaction with it, while carboxylate interacts with Zn2 and Lys224 (Figure 55-b). Instead, L1 inhibition takes place by binding of the phosphonate to Zn1 that elicits dissociation of Zn2.732
Figure 55.
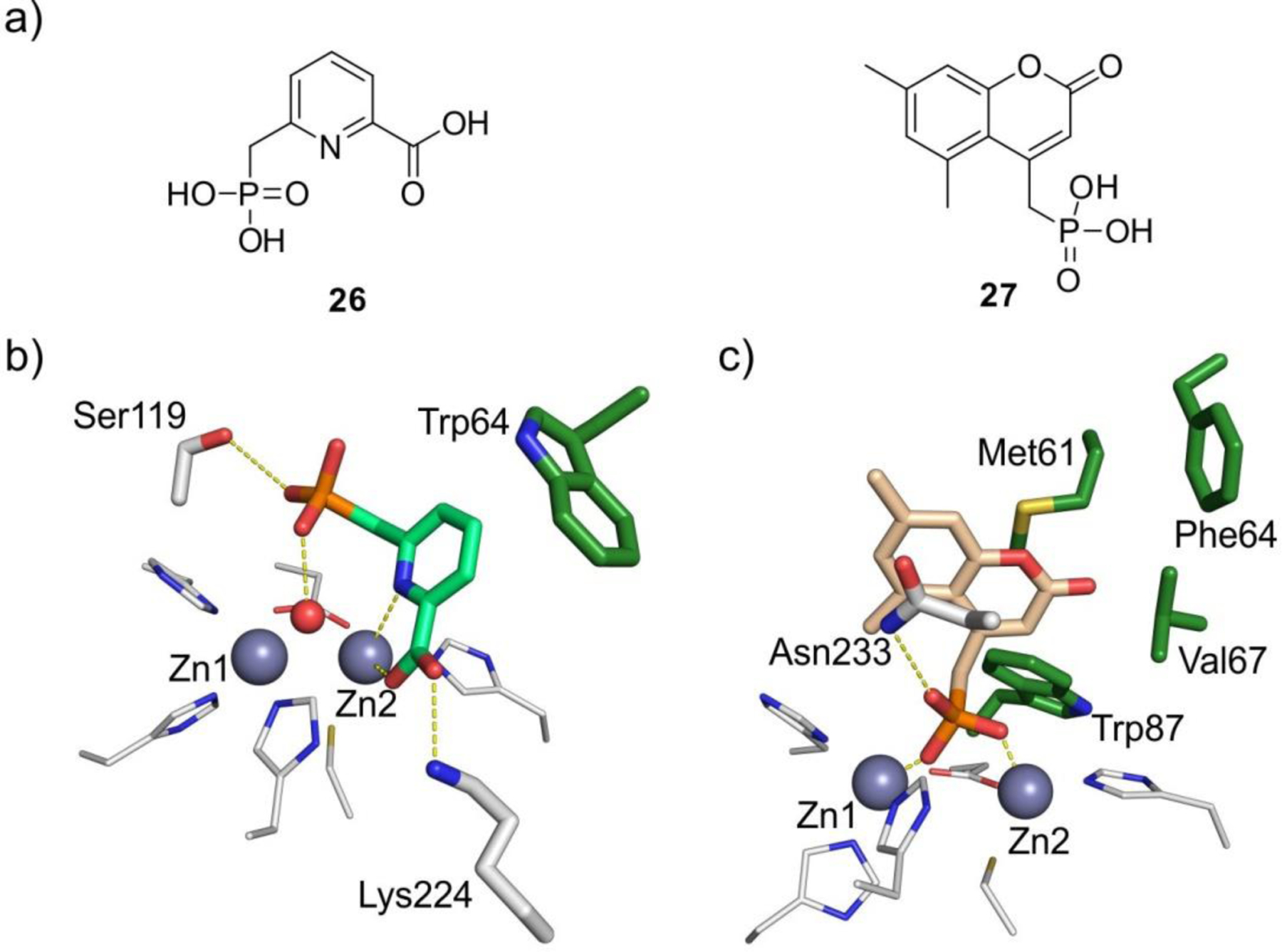
a) Phosphonate-based inhibitors. b) Crystal complex of 26:IMP-1732 (PDB 5HH4). c) Crystal complex of 27:NDM-1733 (PDB 6D1D). Zn(II) ions are shown as grey spheres, protein residues are shown as white sticks (green sticks for residues involved in hydrophobic interactions with the ligand), while the bound ligands are shown as colored sticks. Protein-ligand interactions are shown as dashed lines.
Chen and coworkers designed heteroaryl phosphonates targeting both SBLs and MBLs (27, Figure 55-a),733 which bound to the metal sites by replacing the bridging hydroxide, being the first series of phosphonates able to pursue this chemistry resulting in high inhibitory efficiency, aided by the interaction of the aromatic moieties with conserved aromatic groups in the active-site loops (Figure 55-c). Thus, hydrophobic interactions are key to the positioning of metal binding groups.
6.4.4. Boronates
Boronates have been proposed as transition state analogues mimicking the tetrahedral species formed in SBLs and MBLs soon after the nucleophilic attack. Boronate-based compounds have been explored as enzyme inhibitors for more than four decades.734 As recently reviewed by the Schofield group, this strategy is based in the ability of boron to mimic the sp2 configuration of the carbonyl group of the β-lactam in the substrates, and the sp3 geometry of the tetrahedral species in the transition state (or the high energy intermediate species) before C-N bond cleavage.668
The boron atom is a Lewis acid with a vacant p orbital that allows it to react with different electron-rich nucleophilic species.735 The B hybridization can be tuned according to the pKa, resulting in a trigonal planar sp2 boronic acid or ester, or a tetrahedral sp3 borate species. In case of borates, the negative charge is mostly localized on the boron atom (while in the transition states, the oxygen atoms bear the negative charge). The B-O and C-O bonds (and the B-C and C-C bonds) possess similar lengths, favoring their action as mimics.668
In 2017, vaborbactam 28 (Figure 56-a) became the first clinical boron-based SBL inhibitor approved by FDA. This boronate is prescribed paired to meropenem under the name of Vabomere to treat different SBL-producing bacteria.80 Vaborbactam is a cyclic boronate that inhibits class A and class C SBLs after experiencing the nucleophilic attack of the active Ser, and forming a reversible adduct. Vaborbactam is inactive against class D enzymes and MBLs.
Figure 56.
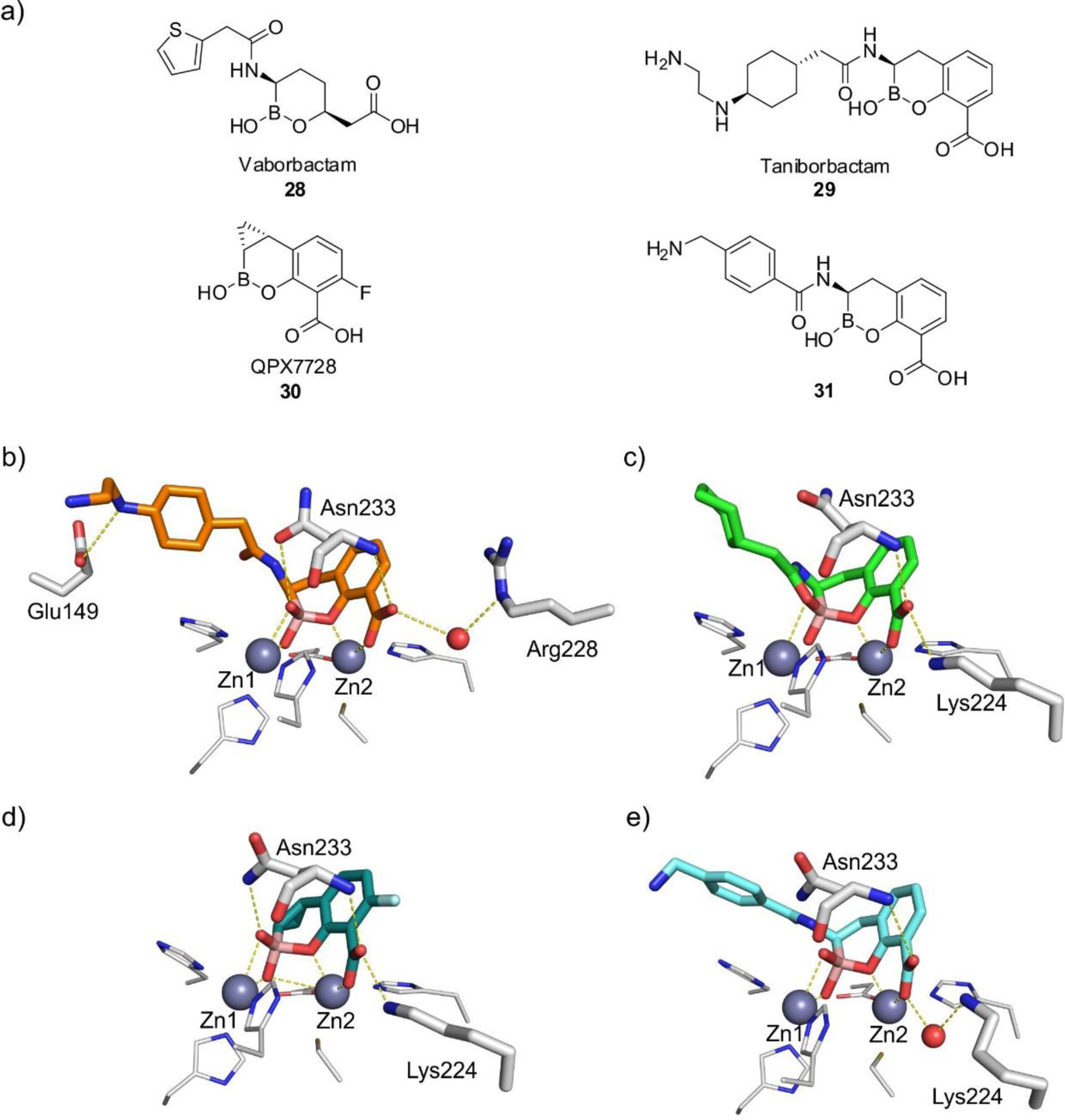
a) Boronate-based inhibitors. b-e) Crystal complexes of 29:VIM-2736 (b, PDB 6SP7), 29:NDM-1737 (c, PDB 6RMF), 30:NDM-1738 (d, PDB 6V1M) and 31:BcII631 (e, PDB 5FQB). Zn(II) ions are shown as grey spheres, protein residues are shown as white sticks, while the bound ligands are shown as colored sticks and water molecules are shown as red spheres. Protein-ligand interactions are shown as dashed lines.
The recent discovery of bicyclic boronates as successful MBL inhibitors (also targeting SBLs) revolutionized the field, making the difference with respect to monocylic boronates such as vaborbactam. Two molecules are of particular interest: taniborbactam 29 (previously VNRX-5133),736, 739 developed by VenatorX Pharmaceuticals (Figure 56-a) and QPX7728 30 (Figure 56-a),738, 740 designed by Qpex Biopharma. Taniborbactam is now in Phase 3 clinical trials combined with the fourth generation cephalosporin cefepime to treat complicated urinary tract infections, while QPX7728 has initiated Phase 1 clinical trials in intravenous and orally-administered forms.
Taniborbactam and QPX7728 are active against MBLs and SBLs. In contrast to vaborbactam, the fusion of the boronate ring to an aromatic group containing a carboxylic acid seems to fulfill the needs to bind the MBL site mimicking binding of the carboxylate moiety of the substrates to Zn2, and orienting the boron atom towards the attacking brigding nucleophile. This scaffold is also present in bicylic boronates synthesized by the Schofield group, and validated by crystal structures of the adducts with different MBLs. However, despite their success that may bring relief by allowing the treatment of several infections, the field of boronate-based inhibition is still new, and there are many open questions to be addressed.
The design of taniborbactam considered the inclusion of a primary amine that improved the permeability towards Gram-negative bacteria, consistent some general rules for antibiotic design.741 Taniborbactam inhibits NDM-1 and VIM-2 in the nM to low-μM range, being poorly active against B2 enzymes and inactive towards L1.736, 737 The impact on IMP-1 is controversional, with different evidence coming from distinct laboratories.736, 737, 739 The binding features to VIM-2 reveal a tetrahedral boron atom in which one of its oxygen atoms binds Zn1, i.e., acting as a transition state analogue (Figure 56-b). The carboxylate group reproduces the binding features found in enzyme-product adducts, interacting with Zn2 and Arg228 (Figure 56-b). The crystal structure of taniborbactam bound to NDM-1 revealed two bound species: one reproducing the binding features present in VIM-2, and other in which the oxygen atom from the amide side chain has reacted with the boron atom giving rise to a tricyclic compound (Figure 56-c).737 It is unclear whether this species is formed in solution or within the active site. Taniborbactam restored the antibiotic susceptibility of several MBL producers, with no toxicity against eukaryotic cells.
QPX7728 30 (Figure 56-a) is a bicyclic boronate developed by Qpex Biopharma which is presented as an ultra-broad spectrum lactamase inhibitor, since it is able to target class A ESBLs, class C P99, various OXA enzymes, NDM-1 and VIM-1. The binding features resemble those reported for taniborbactam (Figure 56-d), but the unprecedent inclusion of a cyclopropyl group favors hydrophobic interactions in the active site that potentiate its activity.738, 740, 742–745
The Schofield group reported a series of bicyclic boronates. Among them, compound 31 (Figure 6) displayed IC50 values against B1 enzymes from the nM to low-μM range. The impact on B2 and B3 enzymes was from moderate to null. This boronate also showed inhibitory activity against class A and D β-lactamases and one PBP. The adducts with BcII and VIM-2 revealed a similar binding mode than taniborbactam and QPX7728 (Figure 56-e).631, 746, 747
Overall, the use of bicyclic boronates to target all classes of β-lactamases appears highly promising. However, it is not still understood why these compounds are potent inhibitors, while monocyclic boronates are not. More work on this area is required, since mimicking the tetrahedral carbon of a high energy species seems to provide a common inhibitory strategy targeting both MBLs and SBLs.
6.5. Screening of chemical and natural product libraries
Many screenings were attempted in the early days on natural extracts and in chemical libraries of different origin. Different virtual screenings have been reviewed elsewhere.22, 748–755 Notably, deep learning approaches have not been applied yet to the discovery of MBL inhibitors.22
An early screening of Streptomyces extracts identified two phenazines which inhibit several MBLs, apparently by metal chelation with low μM efficiencies.756 Screening of extracts from Chaetomium funicola led to the identification of tricylic natural products with phenolic and quinone moieties which inhibited IMP-1, CcrA and BcII.757 A separate work identified the flavonoids galangin and quercetin as L1 inhibitors.758 The most successful screening of natural products libraries has been the one that led to the identification of the strong chelator Aspergillomarasmine A, which is discussed in Section 6.9.2.759
High throughput screening (HTS) efforts on chemical libraries have also provided important clues for the identification of key pharmacophore groups to inhibit MBLs. Indeed, the already discussed succinic acid inhibitors717 and some biphenyl tetrazoles760, 761 resulted from screening of the Merck chemical library. Schofield, Frère and colleagues employed MS to screen a dynamic combinatorial library of thiols and disulfides762 that allowed the identification of some sub-μM MBL inhibitors.763
Several NMR-based screening methods have been specifically developed for MBLs. The Schofield group has employed selective 19F labeling at the mobile loop of NDM-1 to monitor conformational changes and ligand binding.349, 764 A conventional NMR screening by using CPMG spectra enabled the discovery of VIM-2 inhibitors that do not bind the metal ions but instead interact with conserved residues at the active site, such as Arg228 and Asn233.765
An effort from Astra Zeneca combined target-cased whole-cell Screening with NMR spectroscopy to identify compounds inhibiting a specific enzymatic reaction in bacterial cells by following directly carbapenem hydrolysis by 1H NMR and the impact of adding different inhibitors.766 This method also provides in cell IC50 values, which can be compared with MIC values and in vitro IC50 values and understand possible differences between the inhibitory profile in bacteria and in the test tube.
Rhodanines are inhibitors of SBLs768 and PBPs.769 A HTS of the NIH libraries identified rhodanine ML302 32a (Figure 57a) as an inhibitor of IMP-1 and VIM-2,770 which was later characterized by Schofield and coworkers as a low-μM inhibitor of B1 MBLs.767, 771 Different rhodanines were reported by this group and by Oelschlaeger, He and coworkers, with high inhibitory potencies.772 Brem et al. suggested that their activity is due to the generation of a thienolate 32b through MBL-mediated hydrolysis, which is the inhibitory molecule by means of a thiol group that bridges both metal ions (Figure 57-a,b).767 The intact rhodanine molecule could be also found bound to the active site. Oelschlaeger and coworkers, instead, proposed that the intact rhodanine is the active species, since some rhodanine analogues are potent L1 inhibitors without experiencing hydrolysis. The different binding modes and activities of rhodanines require deeper research efforts.
Figure 57.
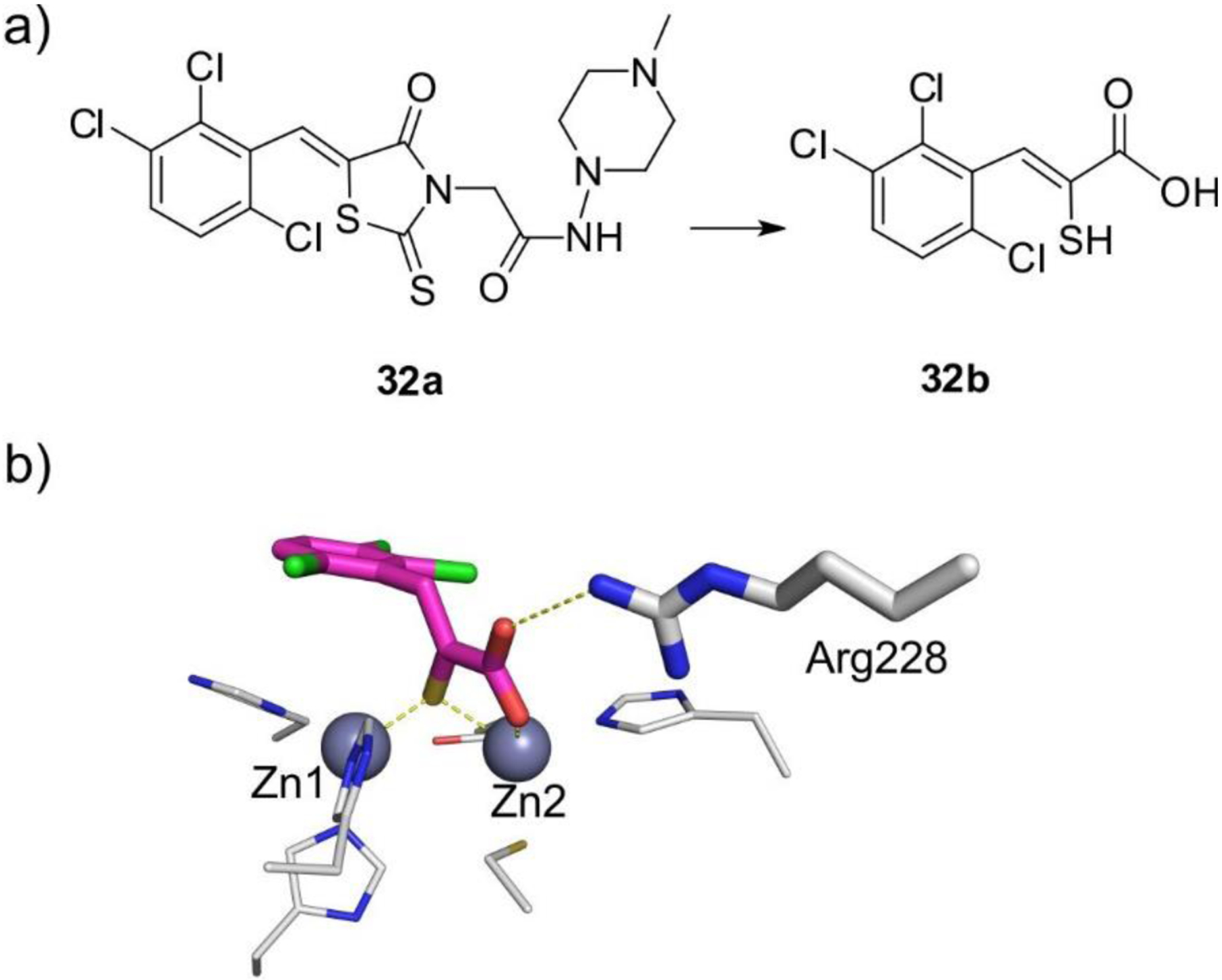
a) Rhodanine inhibitor ML302 32a, and its product generated upon MBL-mediated hydrolysis 32b. b) Crystal complex of 32b:VIM-2767 (PDB 4PVO). Zn(II) ions are shown as grey spheres, protein residues are shown as white sticks, while the bound ligands are shown as colored sticks. Protein-ligand interactions are shown as dashed lines.
6.6. Fragment-based inhibitor design
Fragment-based screening has arisen as a promising strategy to detect novel inhibitors, being successful in several cases where HTS have failed.773 This approach has been used to look for MBL inhibitors, as well.
The Leiros group has undertaken a fragment-based screening approach using surface plasmon resonance to identify fragments with MBL-binding ability.774, 775 This resulted in the identification of 2-(4-fluorobenzoyl)benzoic acid, with μM inhibitory efficiency. The crystal structure of the adduct with VIM-2 demonstrated binding of the carboxylate to the Zn(II) ions. Another hit from this screening was 3-formylchromone that covalently inactivates NDM-1 via the proposed formation of a Schiff’s base adduct with Lys224, discussed later.776 Fragment screening was also monitored combining a fluorescence-based assay with saturation transfer difference (STD) NMR spectroscopy.777 This resulted in a series of compounds with moderate inhibition activity towards VIM-2 and IMP-1.
In silico generated fragments were screened towards MBLs in which different fragments can be linked by using the thiol moiety that binds to the binuclear center.778 This strategy resulted in compounds with μM affinity, and offers and appealing strategy combining virtual screening and the conventional knowledge of thiol reactivity against MBLs.
Vella, Schenk and coworkers screened a 500-compound Maybridge library using CENTA as a reporter substrate, and identified a series of zinc-binding fragments with μM and sub-affinities towards IMP-1.779 There is clearly room to further explore this strategy for MBL inhibitors.
6.7. Allosteric MBL inhibitors
Allosteric enzyme inhibition is an alternative approach that is gaining increasing acceptance and success.780–782 This strategy has not reached its maturity in the case of MBL inhibitors, but there are several, isolated attempts, that are worth mentioning. Most of them lack in identifying a canonical allosteric site regulating MBL activity.
By using phage display technology and screening against VIM-4, Galleni et al. identified nanobody as inhibitor of this MBL. Alanine scanning of this nanobody allowed the identification of hydrophobic residues as responsible of binding to two protein located in loop L6 loop and at the end of the α2 helix (distant to the active site). Unfortunately, these results were not further explored.783
The SELEX (Systematic Evolution of Ligands by EXponential Enrichment) approached was used by Shaw and coworkers to identify a 10-mer single stranded DNA as a nM inhibitor of BcII.784 NMR experiments allowed the identification of a binding site located in protein structure at the opposite face of the active site, confirmed by mutagenesis experiments.785
The Dmitrienko group explored a library of synthetic peptides, identifying those containing Arg residues as inhibitors of VIM-2, acting mostly by inducing enzyme aggregation upon binding. The inhibitory effect was strongly substrate-dependent, suggesting the formation of a ternary complex before aggregation. Instead, the inhibitory effect on IMP-1 was moderate, confirming the relevance of the protein surface in these interactions.786
A self-assembled DNA nanoribbon was able to inhibit NDM-1, ImiS and L1 with a nM binding affinity, but unable to act against VIM-2. NDM-1 binds this nanoribbon through its minor groove.787
On a different note, a screening of nanomaterials against VIM-2 resulted in the identification of graphene oxide and carbon nanotubes as inhibitors, which act by means of hydrophobic interactions with the enzyme.788
Despite these limited and independent attempts to target MBL inhibition by means of allosteric inhibitors, there are several lessons to be learned from them. First, the diversity of MBLs which spans different families and a large allelic diversity among families, makes it difficult to find a conserved allosteric site to target. Second, interaction with hydrophobic patches seem to be more conserved than those with charged regions regarding interaction with inhibitors. Third, none of this efforts have provided a clear identification of an allosteric site regulating MBL activity that can be targeted in a rational fashion.
6.8. Irreversible metal-independent MBL inhibitors
6.8.1. Covalent binding to the Cys ligand
A series of sulfur-and selenium-containing reactive molecules have been identified as inhibitors of B1 and B2 MBLs based on their ability to react with the thiol moiety of Cys221, forming a covalent disulfide bridge or Se-S bond. Despite this strategy is efficient in delivering a covalent MBL inhibitor, it cannot be employed to target B3 enzymes. The strategy of disrupting a sulfur-based zinc binding site exploiting disulfide formation has been exploited in several metalloproteins.789, 790 From the thermodynamic point of view, Zn(II) -bound thiolates are more reactive toward electrophiles than free thiols. However, there is a kinetic limit that depends on the event of Zn(II) dissociation from the active site.
A group of mercaptoacetic acids were assayed as inhibitors of BcII, CcrA and CphA with moderate impact.791 The Lippard group reported a mechanistic study of the inhibition of CcrA with different thiolate molecules.792 Despite these compounds were not tested in bacteria, this work provided the chemical bases for the design of inhibitors exploting this strategy.
Hydrolysis of different sulfur-containing β-lactam antibiotics result in the generation of sulfur-reactive species that in situ react with the sulfur thiolate giving rise to a covalent adduct. Galleni, Frère and coworkers reported the inactivation of the B2 enzyme CphA with cefoxitin and moxalactam 33 (Figure 58-a).513 Damblon and Page studied in detail the mechanism of inhibition of BcII by cephalexin, that generates a dihydrothiazine moiety binding the Zn site an inhibiting the enzyme.793 These results could be exploited for designing prodrugs or suicide inhibitors as inhibitors releasing the active metabolite within the enzyme active site.
Figure 58.
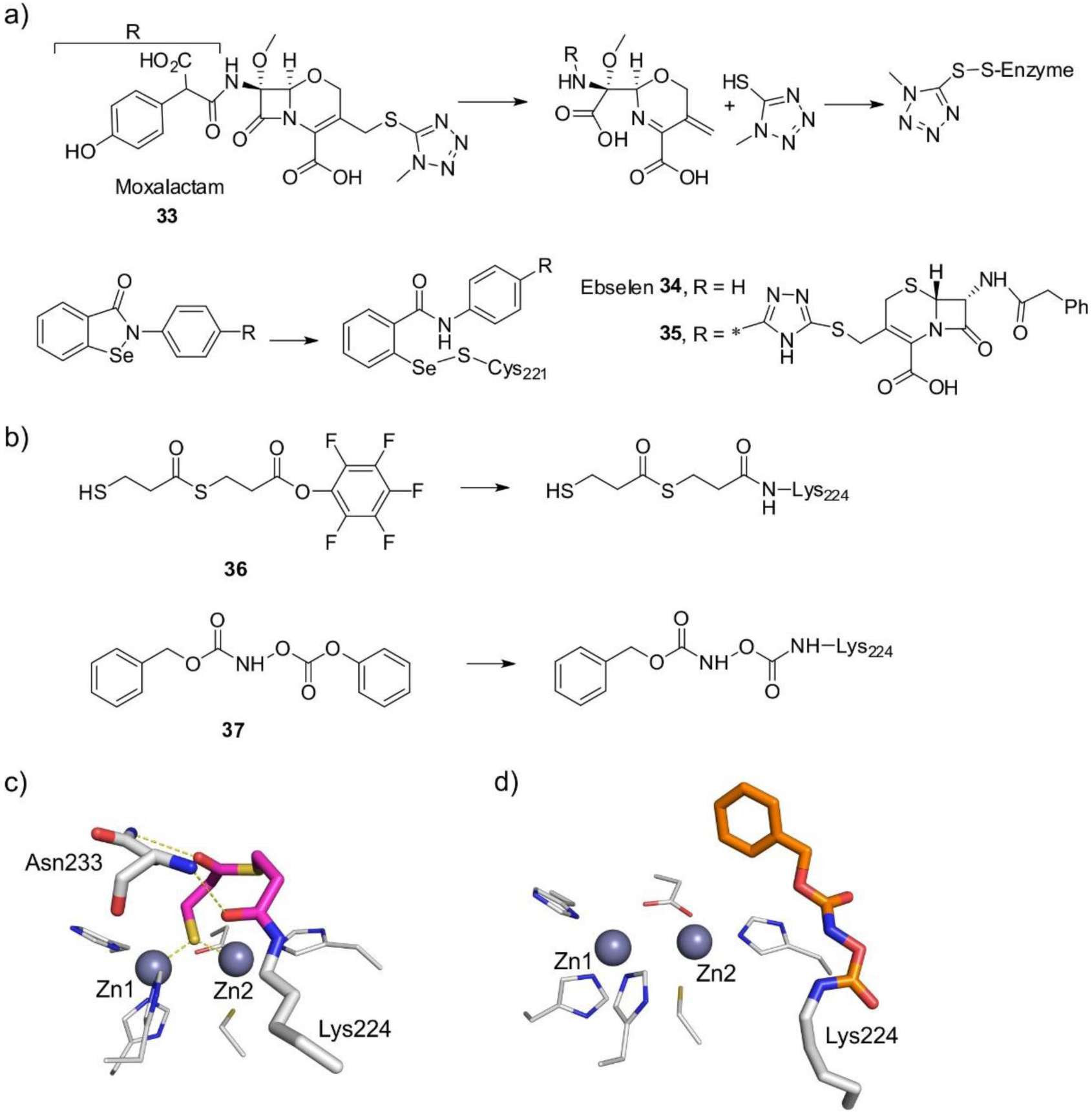
a) Inhibitors that covalently bind to the Cys ligand. b) Inhibitors that covalently bind to Lys224. c) Crystal complex of 36:IMP-1800 (PDB 1VGN) d) Crystal complex of 37:NDM-1801 (PDB 6OVZ). Zn(II) ions are shown as grey spheres, protein residues are shown as white sticks, while the bound ligands are shown as colored sticks. Protein-ligand interactions are shown as dashed lines.
A screening of disulfide compounds as potential NDM-1 inhibitors identified disulfiram, a drug currently used for the treatment of chronical alcoholism, as an irreversible inhibitor of NDM-1, IMP-1 and ImiS by covalently binding Cys221.794 This compound proved inactive against the B3 enzyme L1.
Ebselen 34 (Figure 58-a) is a synthetic organoselenium compound with anti-inflammatory activity that acts a covalent inhibitor of NDM-1 by forming a Se-S bond with the thiol of Cys221, as reported by Chen and coworkers.795 Ebselen is currently under clinical trials targeting different diseases based on its ability to inhibit glutathione-S-transferase. An ebselen scaffold linked to rhodamine B as a fluorescent label was reported by Chen, Liu and coworkers, that enables detection of binding in living cells.796 Its potency against B1 and B2 enzymes is in the low-μM range, but it cannot target B3 enzymes which lack the Cys ligand. Ebsulfur was inspired in ebselen, in which the Se atom is replaced by a sulfur, and shows a similar reactivity.797 A library of ebselen derivatives was prepared, that comprised attachment of a cephalosporin moiety (35, Figure 58-a).798, 799 These compounds could also inhibit L1, thus reaching inhibitory potency against MBLs from the three subclasses. In general, ebselen derivatives were also effective in restoring the efficacy of β-lactam antibiotics in different NDM-1 producers.
6.8.2. Covalent binding to Lys224
Since Lys224 is highly conserved among B1, different approached targeting covalent modification of this residue have been attempted. This strategy was pioneered by Kurosaki and colleagues who synthesized a series of activated esters of 3-mercaptopropionic acid 36 (Figure 58-b) that act as irreversible inhibitors of IMP-1.800 The thiol group binds the Zn(II) site, and Lys224 attacks the activated ester, forming a covalent amide bond, as confirmed by the crystal structure of the inhibited adduct (Figure 58-c)
Fast, Pratt and coworkers employed the same approach to inhibit NDM-1 by using an O-aryloxycarbonyl hydroxamate 37 (Figure 58-b) to target Lys224 (originally conceived as inhibitors of class C lactamases). The covalent adduct inhibits NDM-1 with a KI of 140 μM due to carbamylation of the Lys residue without affecting the metal site (Figure 58-d).801
Based on a fragment-based screening strategy, the Leiros group identified an NDM-1 inhibitor, 3-formylchromone, with an aldehyde group that reacts with Lys224 forming a Schiff base resulting in a KI of 580 nM.776 In contrast to other inhibitors forming a covalent bond, binding is reversible based on the Schiff chemistry involved.
An ebselen derivative was designed by Chen, Liu and coworkers that combines covalent binding to Cys221 and Lys224.796 This compound is based on an ebselen scaffold that provides Se-S reactivity linked to an activated ester that can also react with the amine group of Lys224. This molecule effectively inhibited NDM-1, IMP-1 and the B2 enzyme ImiS wth IC50 values in the low μM range. Despite these compounds cannot target B3 enzymes or MBLs from the VIM family, the dual covalent strategy is highly appealing and provides inspiration for future design.
6.9. Metal chelators as MBL inhibitors
6.9.1. Classical chelating agents
Zn(II) chelation is an efficient strategy for inactivating MBLs in the test tube and a straightforward method to differentiate whether the activity is due to an SBL or an MBL in several clinical microbiology assays (see Section 11). However, the use chelation therapy is not advised due to the side effects eliciting indiscriminate metal depletion from essential metal ions in the host. As such, it is only prescribed to treat heavy metal intoxication. Notwithstanding, many metal chelators have been assayed as MBL inhibitors.
EDTA 38 (ethylenediaminetetraacetic acid) (Figure 59) restores the efficacy of β-lactams in MBL producers and presents antimicrobial effectiveness by itself,802 but is toxic against eukaryotic cells by eliciting massive metal-ion chelation.803, 804 Ca(II)-EDTA (approved to treat heavy metal poisoning) showed synergy with β-lactams against P. aeruginosa and E. coli isolates producing different MBLs.805, 806
Figure 59.
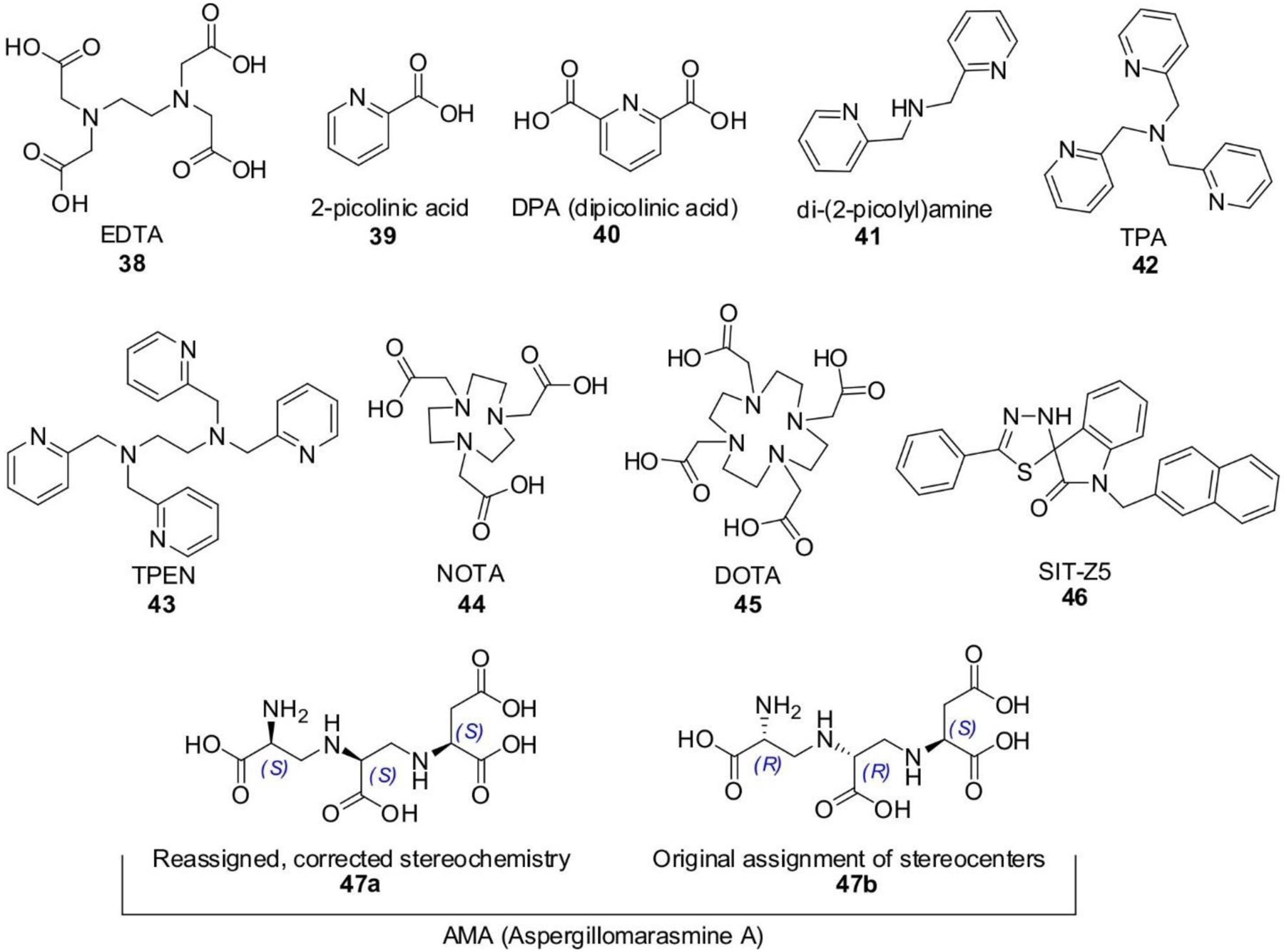
Metal chelator MBL inhibitors.
Piridine dicarboxylates represent an important group of chelators assayed against MBLs. 2-picolinic 39 and dipicolinic acid 40 (DPA, or pyridine-2,6-dicarboxylic acid) (Figure 59) have been extensively explored. DPA has been used as alternative to EDTA to identify MBLs.381, 432, 807 A thorough analysis of the inhibitory effect of different picolinic acid in MBLs by Galleni and coworkers was able to trap pyridine-2,4-dicarboxylic acid bound to the Zn(II) ion in CphA in a bidentate fashion.808 DPA was also identified as an MBL inhibitor in a fragment-based drug discovery approach.809–811 A series of DPA derivatives were synthesized with inhibitory potencies ranging from μM to nM. The inclusion of additional metal binding groups did not necessarily result in better inhibition. Some derivatives acted as metal chelators, while others were able to form a stable MBL-Zn(II)-inhibitor ternary complex.
A group of amine-based compounds: di-(2-picolyl)amine 41, tris(2-pyridylmethyl)amine 42 (TPA) and N, N, N′, N′ -Tetrakis(2-pyridylmethyl)ethylenediamine 43 (TPEN) (Figure 59) have been able to restore the sensitivity to meropenem on MBL producers.804, 812 Different cyclam derivatives such as NOTA 44 (2,2′,2”-(1,4,7-triazacyclononane-1,4,7-triyl)triacetic acid) and DOTA 45 (1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid) (Figure 59) were active against different carbapenem-resistant bacteria expressing NDM, VIM or IMP enzymes.813
SIT-Z5 46 (Figure 59) is a spiro-indoline-thiadiazole compound that shows specific Zn(II) chelation and disrupts metal-ion homeostasis in E. coli. SIT-Z5 was able to restore the efficacy of meropenem against a clinical NDM-1 expressing K. pneumoniae. Instead, the compound was unable to inhibit IMP-1.814
6.9.2. Aspergillomarasmine A and derivatives
Aspergillomarasmine A 47a (AMA) (Figure 59) is a potent metal chelator isolated from Aspergillus versicolor. Despite this compound was originally reported in 1965,815 its use as MBL inhibitor started after a screening of NDM-1 inhibitors towards DMSO-dissolved natural product extracts derived from environmental microorganisms by King, Wright and coworkers.759 AMA is a potent inhibitor of NDM-1 and VIM-2 (KI’s of 11 and 7 nM, respectively), but showing less potency against IMP-7 (KI > 500 μM) and inactive against SPM-1, AIM and IMP-1. AMA had already been reported as an inhibitor of the Zn(II)-dependent angiotensin-converting enzyme.816
AMA is able to restore the efficacy of meropenem on clinical strains of non fermenters and Enterobacterales expressing NDM or VIM. The effect of AMA depends on the MBL and the antibiotic partner, possibly due to the different Zn(II) binding affinities of each enzyme.817 Last but not least, AMA does not show toxicity against mice nor to eukaryotic cells and in combination with meropenem was effective on mice infected with K. pneumoniae NDM-1 producers.759
Two total synthesis of AMA enabled the reassignment of the absolute configuration of some of the stereocenters (cf. 47a vs 47b) (Figure 59).818, 819 A series of related stereoisomers were synthesized, but none of them could improve the performance of AMA, despite most of them showed IC50 values in the same range.820, 821 The carboxylate groups are essential for inhibition, but the structure of AMA is highly tolerant to stereochemical changes. Crowder and coworkers showed that AMA inhibits MBLs by a metal sequestration mechanism, and discarded the formation of a tertiary complex in which AMA binds the enzymes to remove the metal ions.822
6.10. Metal replacement strategies
Metal-based drugs (also known as metallodrugs) have been employed in medicine for more than three centuries to treat different diseases.823, 824 Bismuth and gold compounds are representative of this family of compounds, and have been recently recognized as a new strategy to inhibit MBLs. The chemotherapeutic effect of bismuth drugs occurs by binding of Bi(III) to target metalloenzymes, particularly with Cys and His residues in their active sites. Sun and coworkers have recently shown that several bismuth compounds, particularly colloidal bismuth subcitrate 48 (CBS, commercially known as De-Nol) (Figure 60-a) was a potent inhibitor of IMP-4, VIM-2 and NDM-1, with IC50 values in the low-μM range.825 CBS is an approved drug to treat H. pylori infections leading to ulcers. CBS was also able to restore the activity of meropenem activity against some clinical NDM-1 producers in a mouse infection model. The action of CBS is based on the ability of the Bi(III) ion to displace both Zn(II) ions from the metal site of MBLs. A crystal structure of Bi(III)-substituted NDM-1 (Figure 60-b) revealed binding of one Bi(III) ion to Cys221, Asp120, His116 and His196 and a water molecule (i.e., ligands from both Zn1 and Zn2 sites).825 This binding is irreversible, since excess Zn(II) cannot revert this effect on the Bi(III)-inhibited enzyme. The prolonged use of bismuth drugs does not elicit resistance, and in vitro experiments exposing different generation of E. coli cells expressing NDM-1 did not show resistance.
Figure 60.
a) Structure of inhibitors leading to metal replacement in MBLs. b) Metal coordination sphere of NDM-1 bound to Bi(III)825 (PDB 5XP9). c) Metal coordination sphere of NDM-1 bound to Au(I) ions826 (PDB 6LHE). Bi(III) and Au(I) ions are shown as purple and gold spheres, respectively, while protein residues are shown as sticks and water molecules are shown as red spheres. Ligand-metal interactions are shown as dashed lines.
The Sun group also reported the use of auranofin 49 (AUR) (Figure 60-a), a gold-based anti-rheumatic drug, as inhibitor of NDM-1.826 AUR was also shown to target also the Zn(II) - dependent enzyme MCR-1, an enzyme responsible for the resistance to colistin. Similar to BCS, the action of AUR is due to the displacement of the Zn(II) cofactors from the active sites in both enzymes. In contrast to Bi(III), two Au(I) ions occupy the Zn1 and Zn2 site, with a Au-Au distance of 3.8 Å (Figure 60-c).826 Successful experiments on mice as infection models also support the potency of this drug in vivo. It is notable that AUR is able to target at the same time two Zn(II)-dependent enzymes that are completely unrelated in terms of structure, function and active site, simultaneously tackling two distinct resistance mechanisms.
Silver nanoparticles inhibit NDM-5, inducing protein dimerization.827 This has been attributed to interaction of Ag(I) with the metal site, but this hypothesis has not been explored. Ruthenium complexes are MBL inhibitors and restore the efficiency of meropenem towards MBL-producing bacteria. The action of Ru(III) takes place apparently also by metal replacement, involving ligand Cys221.828
Finally, cisplatin and a series of Pd(II) complexes were able to inhibit NDM-1, VIM-2, IMP-1 and ImiS with μM affinity, with no impact on L1. Some of these compounds were able to restore the efficacy of meropenem against MBL producers. Inhibition was irreversible, and occurred by replacement of one equivalent of Zn(II) by Pt(II) or Pd(II), binding to Cys221. This was assessed by absorption spectroscopy and ICP, but there are no crystal structures that account for the fine details of the mechanism of action of these metallodrugs.829
In summary, metallodrugs appear as a novel strategy, employing well-known compounds with low or no toxicity effects that can be employed to treat MBL-based infections targeting the metal site without resorting to a non-selective chelating strategy.
6.11. Perspectives
There have been several thousands of MBL inhibitors reported in the literature. To the moment, none of them has made it into the clinic, despite the recent progress with bicyclic boronates gives some hope. During more than 30 years of inhibitor research, inhibitors inspired on metal binding groups, β-lactam scaffolds, chelators, and metallodrugs have been explored. This has resulted in a deep understanding of the chemistry of MBLs, complementing structural and mechanistic studies. The transition state and intermediate species common to all MBLs provide an appealing strategy to overcome the difficulties imposed by the active-site variability among different MBLs subclasses, since there are common mechanistic features.
Thiol-based compounds provide the best Zn(II) binding groups, but the sensitivity of thiols to oxidation is a challenge to be faced. The development of prodrugs in which the thiol group is protected and can be released within the cell deserves to be further explored.
Zn(II) chelation and displacement are gaining increasing interest, despite the general skepticism against the indiscriminate action of these approaches. The chelator AMA and some metallodrugs open new avenues to be explored. This approach, however, should not overlook escape mutations on MBLs than can offer resistance to these inhibitors by improving the Zn(II) affinity, an issue of great interest to the field.
Taniborbactam will possibly be the first approved MBL inhibitor. There are, however, many issues to address in the chemistry of boronates and MBLs. Understanding the lack of activity of linear and monocyclic boronates against MBLs may be useful to design better boronate-based inhibitors.
7. Expression, regulation and processing of MBLs
7.1. Biogenesis and export mechanisms
MBLs are produced in the bacterial cytoplasm and must be exported across the cytoplasmic membrane to be able to protect the peptidoglycan synthesis machinery. Therefore, MBLs are synthesized as precursors containing a signal peptide directing the protein to a suitable export system.251, 830 All MBLs identified so far possess a Sec-type signal peptide, which directs their translocation by the SecA-SecYEG system. This is a complex in which the SecYEG proteins form the translocon pore while SecA acts as a cytoplasmic ATPase driving the export, and transports proteins in their unfolded forms.831, 832 In contrast, the Tat system allows translocation of fully folded polypeptides.833 While certain SBLs are secreted via the Tat pathway,830, 834–837 at the moment there are no known MBLs undergoing this processing. As a result, folding and metal-ion acquisition by MBLs takes place in the periplasmic space in Gram-negative bacteria, following their translocation as unstructured polypeptides. Since most bacteria lack specific mechanisms regulating Zn(II) homeostasis in the periplasm, the availability of this ion for MBLs strictly depends on its extracellular levels. This fact has marked consequences on MBL-mediated resistance under metal starvation conditions (see Section 10).
This mechanism of biogenesis has been reported for L1, and characterized in greater detail for the B3 enzyme GOB-18.830, 838 L1 was found to be exported by the Sec system in E. coli, while its companion SBL L2 was secreted through the Tat system, consistent with predictions based on their signal peptides.830 In the case of GOB-18, expression in E. coli strains conditionally deficient for SecA or SecY resulted in lower periplasmic levels of the enzyme and reduced β-lactam resistance. Instead, Tat-deficient cells showed similar levels of GOB-18 compared to the parental strain, discarding a possible role of the Tat pathway in export of this protein.838 The formation of the active species of MBLs also depends on the interaction of their precursor polypeptide with specific cytoplasmic chaperones such as DnaK or, to a lesser degree, the Trigger factor protein, that prevent its degradation by proteases and collaborate in its uptake by the translocation system. Instead, the Sec-dedicated SecB chaperone played no role. Thus, the DnaK system has been proposed as an antibiotic target to disrupt MBL function.838 Deletion of the low molecular weight PBP DacD/PBP6b was shown to impact periplasmic levels and resistance conferred by GOB-18 in E. coli and Salmonella enterica, possibly due to increased proteolytic degradation of the enzyme in the periplasm.839 This suggests that this protein, ubiquitous in Enterobacterales, could assist in the biogenesis of folded and metal-bound MBLs in the periplasm, although the underlying mechanism is still unknown. These studies only represent the starting point of the study of MBL processing, which will require a thorough analysis of MBLs from different subclasses in different bacteria.
Folding and metal acquisition by MBLs occurs in the periplasmic space. It is likely that this mechanism favors binding of the proper metal ions in the periplasm, avoiding mismetallation in the cytoplasm. The importance of protein folding in the correct compartment for metal acquisition has been demonstrated for two periplasmic proteins from the cyanobacterium Synechocystis.840 At the moment, no metallochaperones assisting MBL metallation in the periplasm have been identified, and the most accepted hypothesis supports that this process depends on the availability of metal ions in the external milieu (see Section 10). In this regard, the metal contents of purified GOB and L1 enzymes have been shown to be impacted by whether they were recombinantly expressed as periplasmic or cytoplasmic proteins. Expression of GOB-18 in its mature form (without its signal peptide) in the cytoplasm of E. coli, resulted in mixed contents of both Fe(II) and Zn(II),251 while the periplasmic species of GOB-1 and GOB-18 were isolated with only Zn(II) in their active sites.253, 254 Similarly, Crowder and co-workers found that the metal content of L1 depends strongly on the bioavailability of metal ions in the cellular compartment (cytoplasm or periplasm) in which the protein is folded. In rich medium or Zn(II)-supplemented minimal medium, periplasmic L1 bound Zn(II) preferentially as expected.841 In minimal medium with no added metal ions, containing Fe or Mn, the enzyme exhibited different amounts of Zn(II) and Fe, and Mn to a lesser extent. On the other hand, L1 localized in the cytoplasm of E. coli, i.e, lacking the leader sequence, was shown to bind various amounts of Fe and Zn(II) in all media. The metal content of the periplasmic protein was observed to be variable when expressed in minimal medium, as it also bound differing amounts of Fe and Mn(II) upon supplementation with these ions.841 This would reflect the lack of metal homeostasis mechanisms in the bacterial periplasm (see Section 10), leading to the level of these ions in the compartment to be directly ligated to their extracellular concentrations.
The Fe-binding analogues of L1 and GOB-18 were shown to be catalytically inactive,251, 841 casting doubt on the physiologic role of these species. However, Fe(II)-substituted MBLs have recently been shown to be functional, displaying similar catalytic efficiencies although with variabilities in their catalytic mechanism and slight alterations to the active-site coordination with respect to their Zn(II)-bound forms.614 Importantly, these studies were carried out under low-O2 conditions to prevent oxidation to Fe(III), and this factor could explain the previously observed lack of activity. Fe(II) binding could contribute to MBL activity under Zn(II) limitation, although the availability of the former encountered by the enzymes under physiological conditions is unclear.614 Finally, IMP-1 expressed in the E. coli cytoplasm in media supplemented with FeCl3 was reported to be purified bound to Fe(III), although in a form containing a mixed-metal active site with Fe(III) and Zn(II) at the 3H and DCH positions, respectively. The enzyme possessed β-lactamase activity under aerobic conditions.842
According to our analysis of the signal peptide sequences of 671 different MBLs, SignalP 5.0843 predicts that 617 are processed by signal peptidase I, and 47 (all of them lipoproteins) are processed by signal peptidase II. The signal peptides of a small number of enzymes (SFB-1, SLB-1, SPS-1, GOB-P1, GOB-P2, MSI-1, PNGM-1) cannot be accurately predicted, and none of the MBLs is predicted to be exported by the Tat system. Thus, the experimental results on GOB and L1 seem to be of general nature. We conclude that secretion by the Sec system appears as evolutionary favored by enhancing the chances of binding Zn(II) and giving rise to a fully active enzyme.
7.2. Regulation of MBL expression
There is a considerable paucity of information regarding the regulation of MBL expression in their bacterial hosts. Most acquired MBLs are not associated to any regulatory elements, resulting in constitutive expression. However, the characterization of factors controlling expression of intrinsic MBLs is scarce. Here we discuss known cases of regulatory mechanisms operating on MBL expression.
The native enzymes of Stenotrophomonas maltophilia, the MBL L1 and the class A SBL L2, are regulated by a system dependent on the LysR-type transcriptional regulator AmpR.844 Many intrinsic class C SBLs, and also class A enzymes such as Burkholderia spp. PenA, are under control of homologous regulation machinery (see Section 3.1). The general scheme for the system is as follows (further details can be consulted on several excellent recent reviews on the topic30, 845–847). Under normal growth conditions, the AmpR regulator is bound to UDP-N-acetyl-muramyl (UDP-MurNAc) pentapeptide, a biosynthetic intermediary in peptidoglycan production, and in this state acts by repressing the transcription of its regulon (Figure 61). This set of genes includes ampR itself, which is usually located immediately upstream to the gene encoding one of the regulated enzymes but divergently transcribed. When the cell is challenged with β-lactam antibiotics, these inhibit the D,L-transpeptidase and D,D-carboxypeptidase activities of various PBP enzymes, disrupting peptidoglycan (PG) crosslinking and causing secondary damage through the increased activity of lytic transglycosilases which cleave the individual PG strands. This causes the formation of PG degradation products, mainly N-acetyl-glucosamine-1,6-anhydro-N-acetyl-muramyl (GlcNAc-1,6-anhydro-MurNAc) tri-, tetra- and pentapeptides. These compounds enter the cell through the AmpG permease, and the cytoplasmic NagZ glycosylase cleaves their GlcNAc moiety generating the corresponding 1,6-anhydro-MurNAc oligopeptides. These products are also generated during normal cell growth as part of PG turnover, but their cytoplasmic pools are limited by action of the AmpD amidase. This enzyme degrades them into the corresponding peptides and 1,6-anhydro-MurNAc, allowing the products to be recycled into the PG biosynthetic pathway. However, the increased formation of 1,6-anhydro-MurNAc oligopeptides during β-lactam exposure leads to saturation of AmpD, and their accumulation in the cytoplasm. In turn, the increased levels of 1,6-anhydro-MurNAc pentapeptide (or possibly the tripeptide, but recent data supports the former)30, 848 in particular allow this ligand (activating ligand) to replace the UDP-MurNAc-pentapeptide from AmpR, eliciting the transition of the protein from a transcriptional repressor to an activator and triggering an increase of β-lactamase expression. However, not all β-lactams act as equivalent inducers of the AmpR system, as tipically only drugs such as imipenem and cefoxitin trigger enhaced β-lactamase production, probably through increased formation of this activating ligand by inhibition of low molecular mass PBPs such as PBP4 (DacB). These PBPs act as D,D-carboxypeptidases, cleaving the D-Ala-D-Ala bond in the pentapeptide stems to limit the potential PG crosslinking, so their inhibition leads to increased generation of PG degradation products containing intact pentapeptides.
Figure 61.
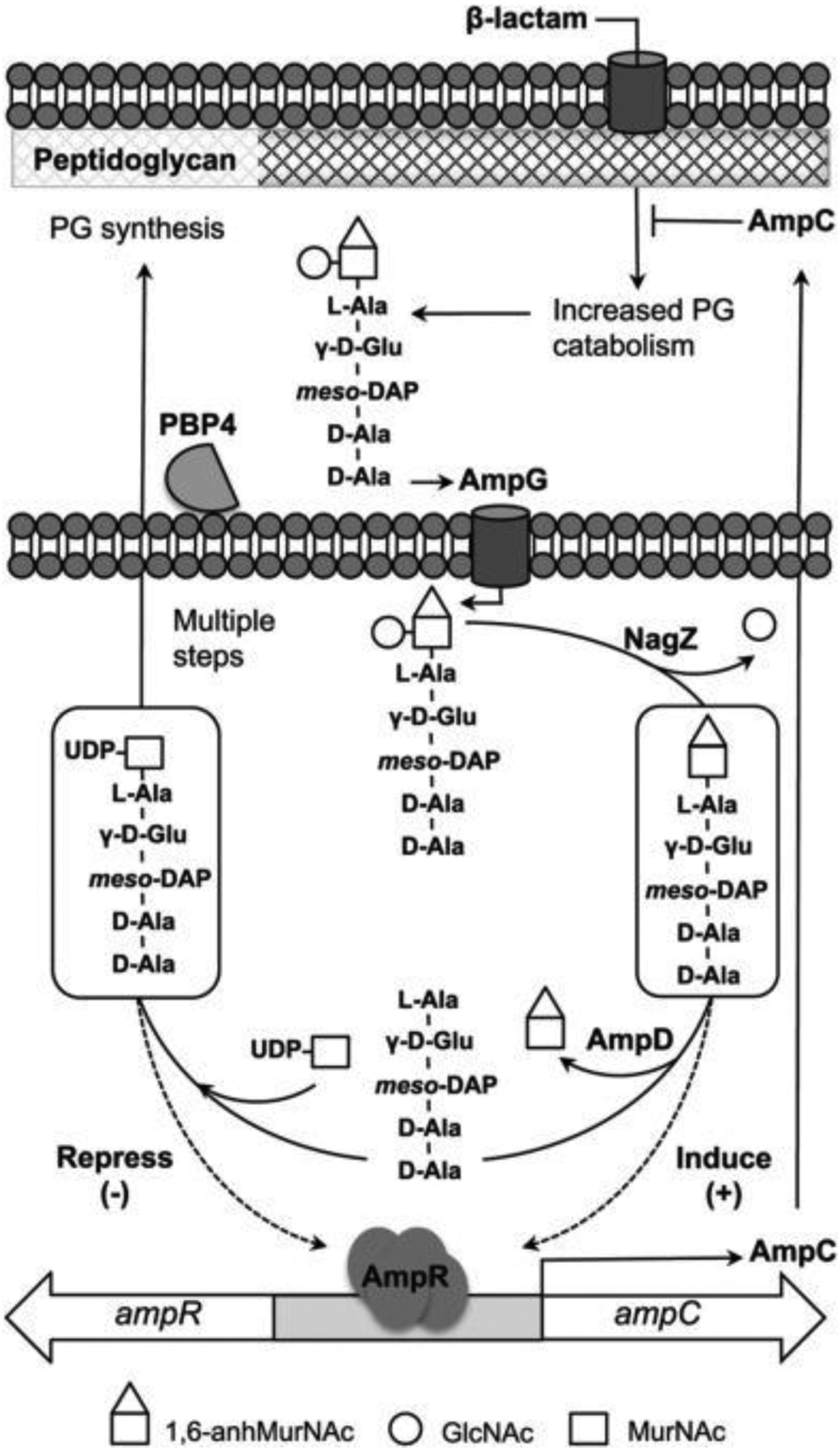
General scheme for the regulation of β-lactamase by AmpR, and its interface with the PG recycling pathway in Gram-negative bacteria. Reproduced with permission from reference848. Copyright 2015 American Society for Biochemistry and Molecular Biology.
S. maltophilia follows the general scheme presented, but with some particularities.845 The organism possesses an additional component of the system, AmpN, which is encoded in the same operon as AmpG. AmpN is a putative membrane protein of yet unidentified function, and both it and AmpG are essential for induction of the intrinsic β-lactamases in S. maltophilia.849 Although both L1 and L2 are regulated, the induction of L1 by AmpR seems to be comparatively weaker, and requires greater activation of AmpR than L2.844 Additionally, induction of β-lactamase expression in this organism can be triggered by a much wider variety of β-lactam drugs, in addition to the typical “inducers” such as cefoxitin and imipenem, which may give rise to the intrinsic resistance to most of these drugs.845 The activation by β-lactams that do not specifically inhibit PBP4 and other D,D-carboxypeptidases suggests that the length of the peptide bound to the activating ligand of AmpR is not as important in S. maltophilia. Furthermore, it has been shown that inactivation of PBP1a (MrcA) leads to overexpression of both β-lactamases in a mechanism that is dependent on AmpR, AmpG and AmpN, but not on the activity of NagZ, suggesting that the activating ligand generated by this alternative AmpR activation pathway differs from the canonical one.850–852
The regulated expression of VarG, an MBL superfamily protein that can confer resistance to β-lactam antibiotics but does not belong to any of the three known MBL subclasses (see Section 4.9), has been recently reported in a Vibrio cholerae strain.600 The expression of this enzyme, together with the VarACDEF drug efflux pump, is under the control of VarR, which belongs to the LysR-type regulator family. The varR gene is located upstream of the gene encoding the MBL, and is transcribed divergently, thus resembling the organization of AmpR-regulated systems. However, the presence of the additional genes encoding the efflux pump, organized in two operators downstream of the varG gene, is a novel feature absent in AmpR regulons. VarG confers resistance to various β-lactam antibiotics when expressed in E. coli, and while its companion efflux pump impacted the MICs of various antibiotics such as macrolides and quinolones, it did not seem to affect β-lactam resistance.600 The expression of this set of genes was found to be repressed by VarR, and is induced by β-lactam antibiotics, but it is still unclear whether the activation mechanism is similar to that of AmpR.
Expression of the Aeromonas spp. chromosomal lactamases is controlled by an alternative regulation scheme.845, 853 All three β-lactamases produced by this genus, namely the CphA/Imi B2 MBL and the SBLs Cep (class C) and AmpH (class D), are under control of the BlrAB two-component system.854, 855 The BlrB sensory kinase is activated by an increase in non-crosslinked disaccharide-pentapeptide units in the peptidoglycan generated as a result of PBP inactivation by β-lactams, mainly of low mass PBPs such as PBP4. However, it is not known whether BlrB interacts directly with its activating ligand or via an unknown intermediary protein. Upon activation, BlrB phosphorylates the BlrA response regulator which in turn induces the production of the Aeromonas native β-lactamases.853 As a consequence of this coordinated regulation of all three enzymes, variants hyperproducing any of the lactamases display simultaneously deregulated production of the others.
Expression of the two intrinsic MBLs from Elizabethkingia meningoseptica, GOB and BlaB, is independently regulated. Both enzymes, together with the native CME class A SBL, are expressed in absence of β-lactams, and none of them seems to be induced by addition of these antibiotics.551 However, BlaB is upregulated under nutrient-limiting conditions, potentially mimicking conditions encountered during infection, with respect to the levels observed in nutrient-rich medium. Expression was also increased in stationary phase cultures, both in rich and minimal medium. Meanwhile, this induction was not observed for GOB.551 This remains the only known example of MBL expression regulated by the growth conditions of the bacterium instead of by the presence of antibiotics. Expression of the chromosomal THIN-B B3 enzyme from Janthinobacterium lividum is also inducible by imipenem, but the regulation mechanism has not been characterized.526
In Gram-positive organisms, the intrinsic β-lactamases from Bacillus anthracis, namely the Bla1 SBL and the Bla2 MBL, were shown to be regulated by an alternative sigma factor termed SigP.856 The closely related species B. cereus and B. thuringiensis also produce MBLs, belonging to the BcII family, and SBLs, under control of an equivalent system. B. anthracis is typically susceptible to penicillin, due both its resistance genes being transcriptionally silent, while certain strains resistant to this antibiotic constitutively express the enzymes.857 It was found that the cause allowing Bla1 and Bla2 production in the resistant strain was the lack of a functional anti-sigma factor (RsiP) that would otherwise abolish the activity of SigP, which is required for transcription of both bla genes.856 RsiP is active in the sensitive strain, and would sequester SigP in the cell membrane, as commonly observed for related anti-sigma factors.858 Homologues of both SigP and RsiP are present in B. cereus and B. thuringiensis. However, β-lactamase expression in these organisms is inducible by β-lactam antibiotics such as ampicillin. The SigP/RsiP pairs of these two organisms were responsive to the signaling pathway triggered by β-lactam antibiotics, which leads to proteolysis of RsiP and the release of SigP, while the proteins from a penicillin-susceptible B. anthracis strain were not.856 The lack of induction of β-lactamase production in B. anthracis would then be caused by the lack of proteolytic degradation of RsiP, preventing the release of SigP and transcription of the corresponding genes. More recently, it was reported that different β-lactams have varying effectiveness in inducing β-lactamase expression in B. thuringiensis, and that the RasP protease was involved in degradation of the anti-sigma factor that allows this regulatory response.859 However, it was found that RasP cleavage of RsiP is not the regulated stage in the degradation of this anti-sigma factor, and a previous proteolysis step carried out by a yet unknown protein is the one initiates the degradation after being triggered by the presence of β-lactams.859
Much less is known about the regulation of MBL expression within mobile genetic elements. An example of such regulation was recently reported for an E. coli isolate harboring a plasmid encoding the IMP-6 enzyme, but which displayed a carbapenem sensitive phenotype and with no detectable cell carbapenemase activity according to the CarbaNP test.860 This was found to be the result of a strong transcriptional repression of the blaIMP-6 gene by the ArdK protein, encoded in the same plasmid, which binds to a sequence upstream of the resistance gene. This transcriptional regulator, part of the Ard alleviation of restriction of DNA system, is ubiquitous in IncN plasmids, acting on the transcription of both Ard genes and of the RepA protein controlling plasmid copy number. The existence of a binding sequence capable of regulating the blaIMP-6 derives from an Is26 insertion sequence introduced upstream of the gene.860 Although the nature of the observed repression of IMP-6 is seemingly fortuitous, with no apparent mechanistic role and with markedly negative impact in the ability to confer resistance, bacteria carrying such plasmids may act as “silent” reservoirs of MBLs in clinical settings, with the presence of the MBL being only detectable through genetic tests instead of rapid activity assays or other phenotypic tests. Finally, the expression of the acquired MBL NDM-1 was reported to be induced by imipenem in a clinical isolate of S. maltophilia, although the underlying mechanism is unknown.861 This raises the possibility that expression of other mobilized MBLs may be affected by regulatory systems either present in the same mobile genetic element or in the chromosome of the host bacterium.
In summary, the mechanisms regulating the production of MBLs warrant further in-depth studies, as most of the knowledge in the topic is restricted to a handful of chromosomal enzymes. It will be especially important to explore whether clinically relevant MBLs, particularly those carried in mobile genetic elements, experience meaningful regulation processes and how this can impact their capability to confer resistance.
8. Membrane-bound MBLs and OMVs
The NDM family of proteins are lipidated, and as a result are bound to the outer membrane (as mentioned in Section 4.7.1.4). This cell localization also results in selective secretion into outer membrane vesicles. These aspects will be discussed in this section, since they represent a novel aspect in the field of MBLs.
8.1. Lipoproteins and protein lipidation machinery
Bacterial lipoproteins are cell envelope proteins that contain a post-translationally modified Cys residue in their N-terminus, which is covalently bound to a set of acyl chains and inserted into one of the lipid membranes of the bacterium, anchoring the polypeptide to the surface of the bilayer.862–867 Protein lipidation is a widely conserved process across bacteria, with a highly homologous biogenesis machinery that is distributed among both Gram-positive and Gram-negative organisms.868
Proteins that undergo this post-translational modification carry out a variety of roles within their microbial hosts.863, 867, 869, 870 The archetypal lipoprotein is E. coli Lpp (also known as Braun’s lipoprotein), which is covalently attached to the peptidoglycan by its C-terminus while its N-terminal end is anchored by lipid groups to the outer membrane.871–873 Lpp, the most abundant lipoprotein in E. coli with around 106 copies per cell, contributes in this way to maintain the structural integrity of the bacterial envelope.863 The lipoprotein Pal carries out a similar role although its attachment to the peptidoglycan is non-covalent. Through its interaction with the Tol system, Pal is able to link the inner membrane, murein sacculus and the outer membrane.874 Other lipoproteins play key roles in the assembly of the outer membrane, such as BamBCDE in the BAM complex required for OM β-barrel protein insertion and folding,875 and LptE in the translocation of lipopolysaccharide units from the inner to the outer membrane.876 In enterobacteria the OM lipoproteins LpoA and LpoB are required for the transpeptidase and transglycosylase activities of PBP1A and PBP1B, respectively, thus playing a key role in PG metabolism.877 Other lipoproteins play a role in sensing, with NlpE and RcsF acting in the relay of signals of cell contact with physical surfaces and envelope stress to the bacteria.878, 879 Additionally, lipoproteins can play important roles in pathogenesis during processes such as cell adhesion and inflammation, with the acylated group of bacterial lipoproteins being a potent inductor of the immune system.869 Furthermore, it is known that loss of function in components of the lipoprotein biogenesis system lead to reduced virulence in organisms such as Listeria monocytogenes and Mycobacterium tuberculosis.869 Lipoproteins possess a particular importance in Gram-positive bacteria, in which the substrate-binding proteins (SBPs) of ABC transport systems are frequently lipoproteins.867, 870 This allows them to be retained in the cell surface in absence of a periplasmic compartment. Similarly, β-lactamases in Gram-positive organisms such as Bacillus cereus and Staphylococcus aureus have been found to be partially or fully processed into lipoproteins, ostensibly to maintain their antibiotic-degrading capabilities associated with the cell.870 Lipidated β-lactamases have also been observed in some Gram-negative bacteria (see Section 8.2). All in all, it is estimated that around 1 to 3% of ORFs in bacterial genomes encode lipoproteins, many of them with a still unknown function.863, 880
The signal peptides of lipoprotein precursors possess a similar structure to other secreted proteins, generally with the presence positively charged residues close to the N-terminus, followed by a stretch of hydrophobic residues.862 However, towards the end of this sequence lipoproteins possess an additional motif with the consensus sequence [LVI]-[ASTVI]-[GAS]-C,880 called a Lipobox. The invariant cysteine residue within the lipobox becomes the N-terminal residue of the mature lipoprotein, binding the lipid groups that anchor it to the membrane surface. There are various tools available to identify possible bacterial lipoproteins, including the specialized online servers LipoP881 and DOLOP.880 The popular SignalP online tool, which detects the presence of leader peptides and transmembrane segments in proteins, can also currently report the presence of a putative lipidation signal.843
Lipoprotein biogenesis takes place mainly in the cytoplasmic membrane.862, 864, 882 After the precursor is exported from the cytoplasm (generally through the Sec pathway, although Tat export of lipoprotein precursors has been observed), it is sequentially processed by three integral membrane enzymes (Figure 62-a). First, Lgt transfers a diacylglycerol group to the free sulfhydryl of the lipobox cysteine. The lipoprotein signal peptidase LspA (signal peptidase II) then cleaves the protein removing the residues preceding the lipidated cysteine, which becomes the new N-terminal amino acid of the protein (Cys+1). Afterwards, the Lnt protein binds an acyl group to the free amino group of this residue. The mature lipoprotein form contains thus a total of three acyl chains bound to its N-terminal Cys, two in an diacylglycerol moiety bound via a thioether bond and an additional amide-linked acyl group (Figure 62-b). These lipid groups are sourced by the Lgt and Lnt proteins from membrane phospholipids, and thus the acyl chains bound to lipoproteins reflect their composition and variation across different species.883 Lgt was observed to use solely phosphatidyl glycerol (PG) as a substrate884 while Lnt uses the -sn1 acyl chain of phosphatidylethanolamine (PE) preferentially.885 Meanwhile, LspA is dependent of the diacylglycerol modification introduced by Lgt for processing of its substrates, being inactive on lipoprotein precursors lacking this moeity.886–888 The crystal structures of Lgt,889 LspA887 and Lnt890 have been recently solved, providing more detailed information regarding their mechanism of action. It should be noted that variations to this general scheme exist in different bacteria, particularly in Gram-positive organisms.867
Figure 62.
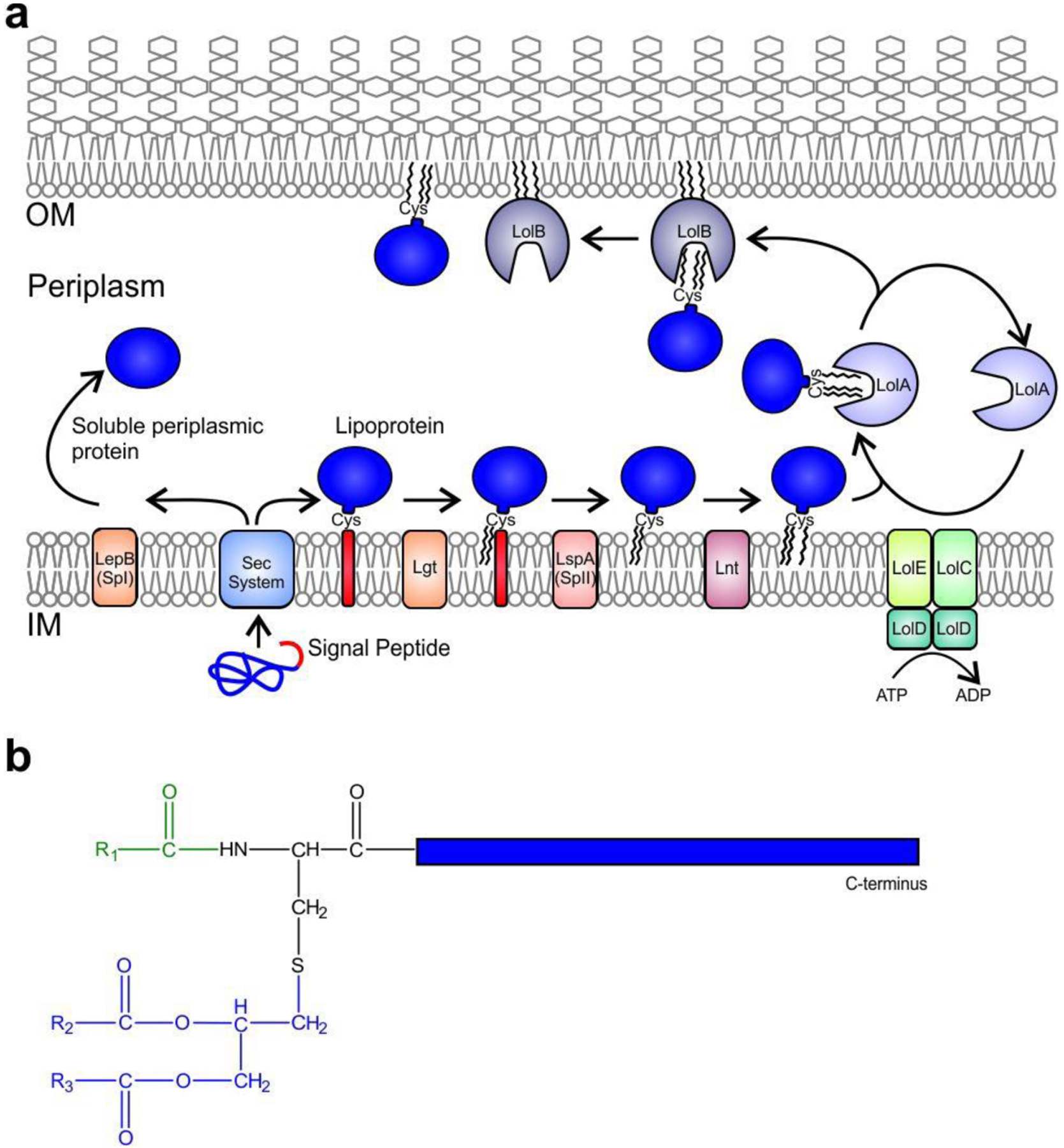
a) Biogenesis pathways for Gram-negative bacterial lipoproteins vs. soluble periplasmic proteins secreted through the Sec system. b) General structure a tri-acylated bacterial lipoprotein. The amide-linked acyl group is highlighted in green, while the S-linked diacylglycerol is shown in blue.
In Gram-negative bacteria, after the attachment of the acyl groups, most lipoproteins (around 90% in E. coli) are transported by the Lol system to the outer membrane (Figure 62-a), although a proportion is retained in the inner bilayer.862, 865, 882 Their fate is largely determined by the identity of the two residues located immediately after the modified cysteine (the +2 and +3 positions).862, 882 The presence of an Asp residue in these locations allows the lipoprotein to bypass the Lol system and remain in the inner membrane, although other residue combinations may also lead to inner membrane retention in E. coli and other bacteria.862, 882, 891 However, most lipoproteins are extracted from this bilayer by LolCDE, an inner membrane ATP binding cassette (ABC) transporter, and are delivered to the soluble periplasmic chaperone LolA.892, 893 Two copies of the cytoplasmic membrane-associated LolD ATPase are present in each LolCDE complex, using ATP hydrolysis to drive the process. LolC and LolE are homologous and present significant structural similarities, forming a transmembrane heterodimer with large loops protruding into the periplasm, but each protein serves a distinct function.894 LolE carries out the recruitment of lipoproteins from the IM while LolC interacts with LolA. The E. coli LolCDE complex binds the diacylated lipoproteins that have not been processed by Lnt with a much lower affinity than triacylated forms,895 and thus is expected to act as a quality control step preventing the export of incompletely modified lipoproteins. Although the lolC and lolE genes are conserved in E. coli and related γ-Proteobacteria, most other Proteobacteria possess a single homologue termed LolF that would carry out both their associated functions.891, 896 LolF forms a homodimer and is considered the precursor of LolC and LolE, which may have arisen by a gene duplication event allowing their subsequent functional specialization.862, 897
When LolA receives the extracted lipoprotein from LolCDE, this periplasmic chaperone shields the lipid groups of its cargo from the aqueous environment during transit from the IM to the OM by accommodating them within a hydrophobic cavity present in its incomplete β-barrel structure. However, this site appears not to be large enough to fit all acyl chains of the lipoprotein, and some of them could instead interact with hydrophobic patches in the LolA surface.898, 899 After crossing the periplasm, the lipoproteins are finally transferred to the outer membrane protein LolB, itself a lipoprotein with a similar structure to LolA (although without significant sequence homology between them),898 which in turns deposits them into the inner bilayer of the OM by a still uncharacterized mechanism.900 LolB has a higher affinity for lipoproteins than LolA, preventing a retrograde traffic of their substrates.901 Notably, it was observed that membrane anchoring is not an absolute requirement for lipoprotein insertion into membranes by LolB, as a soluble mutant could still carry out this activity.902 However, this LolB variant lacking a lipid tether has no membrane preference and deposits lipoproteins indistinctly both into the OM and the IM, but OM lipoproteins incorrectly transferred to the IM can be extracted again by the LolCDE system and end in their correct localization in the steady state.902
Although the inner leaflet of the OM is the final destination for many Gram-negative lipoproteins, some may then cross this bilayer and become either partially or fully exposed to the cell surface, carrying out functions such as nutrient acquisition and cell adhesion and participating in pathogenesis.903, 904 In particular, spirochetes (such as the Lyme disease etiological agent Borrelia burgdorferi) possess numerous surface exposed lipoproteins, that also constitute important antigens.905 The translocation mechanisms used for this process have not been fully characterized yet, although the proteins generally seem to transit to the periplasmic face of the OM via the previously described pathways. A group of the surface lipoproteins (such as E. coli Wza and CsgG) become exposed to cell exterior by assembling into transmembrane oligomers,906, 907 while others cross the OM via the pore in a β-barrel protein assembled by the Bam system. An example of the latter is E. coli RcsF, a sensory OM protein that crosses the membrane by threading through pores in β-barrel outer membrane proteins such as OmpA, OmpC and OmpF.908 Surprisingly, the C-terminus of a subpopulation of Lpp not bound to peptidoglycan is exposed to the cell surface in E. coli, although the mechanism by which it crosses the membrane is unknown.863, 873 A different mechanism for OM exposure is found in pullulanase (PulA) from Klebsiella oxytoca, a lipoprotein that translocates from the inner membrane to the cell surface through a Type-2 secretion system, bypassing the Lol system.904 Finally, a family of dedicated lipoprotein translocators termed Slam was recently reported in Neisseria, with homologues detected in various proteobacteria.909
Gram-positive organisms lack both the outer membrane and the Lol system and as a result, lipoproteins are retained in the cytoplasmic membrane. Some further variations to lipoprotein metabolism exist in low GC content Gram-positive organisms, which lack Lnt and seem to produce diacylated forms instead of the triacylated proteins generated by their high GC content counterparts and Gram-negative organisms.862, 866, 867 However, the detection of triacylated lipoproteins in some low GC content bacteria such as S. aureus indicated the existence of an alternate pathway for acylation of the free N-terminus of the protein. This N-acylation process has been recently reported to be catalyzed by the LnsAB system in S. aureus, which possesses no homology to Lnt.910 Additional lipoprotein forms were found in Gram-positive bacteria, including lyso-form lipoproteins that lack one of the acyl chains (containing instead an S-linked monoacylglycerol and N-acyl group), N-acetylated S-diacylglycerol lipoproteins, and peptidyl lipoproteins in which two additional residues are present in the N-terminus of the protein before the diacylglycerol-bound Cys residue.866 The generation of lyso-forms from diacyl lipoproteins was found to be catalyzed by the integral membrane enzyme Lit, which transfers a fatty acid from the diacylglycerol moiety to the free α-amino group of the N-terminal cysteine.911
The lipidation machinery is essential in various Gram-negative bacteria, since deletion of different genes in the pathway is lethal.864 All proteins in the Lgt-LspA-Lnt triad and those in the Lol system are essential in E. coli, at least in part due to the fact that Lpp accumulation of PG-crosslinked in the inner membrane is lethal coupled with the essentiality of OM lipoproteins such as BamD and LptE.865, 912 However, it was recently found that a combination of mutations, including deletion of lpp, allowed the deletion of lolA and lolB in E. coli (although function of LolCDE remained essential).913 Furthermore, under these conditions some lipoproteins still were being translocated to the outer membrane, implying the existence of a yet uncharacterized alternative to the LolA/LolB tandem for lipoprotein traffic to the OM.913 Supporting this notion, Gram-negative bacteria outside the Proteobacteria phyllum lack LolB (and this protein is absent from α- and ε-Proteobacteria),868 and may thus use an alternate mechanism to deposit lipoproteins in the OM.
The importance of the lipoprotein biogenesis machinery coupled with the lack of homologous proteins in Eukaryotes makes it an appealing molecular target for the development of new classes of antibiotics. Globomycin selectively inhibits LspA, binding the enzyme as an uncleavable substrate analogue,887, 914, 915 and the structure-activity relationships of a series of its derivatives have been explored.916 Antibiotic TA (or myxovirescin) from Myxococcus xanthus also targets this enzyme.917 The Lol system has also been explored as a target. The compound MAC13243 blocks the action of LolA918 while a group of pyridineimidazole drugs inhibits LolCDE.919
8.2. Lipidated β-lactamases
The presence of lipidated β-lactamases has been largely overlooked in the literature. Despite many reports, several lipidated lactamases have been considered as soluble enzymes. Many of them are class A SBLs present in Gram-positive organisms, such as β-lactamase III (also known as BlaZ) from Bacillus cereus,920, 921 PC1/BlaZ from S. aureus922 and PenP from Bacillus licheniformis.923 Homologues of these enzymes carrying a putative lipobox can also be found in related organisms in biological sequence databases. β-Lactamase III and PenP were found to be only partially in their lipidated forms. Indeed, only 50% of the former enzymes were bound to the membrane,921, 923 while a variable proportion of PC1 was membrane anchored in different S. aureus strains.924 The soluble forms of these proteins (termed “exoenzymes”) would be generated by proteolysis of the lipidated form, giving rise to shorter variants due to removal of N-terminal residues.922 This raises the question of the relative advantages of membrane-bound lactamases with respect to extracellular lactamases in terms of resistance.
Although less frequently, lipidated β-lactamases have also been reported in Gram-negative organisms. The best characterized is the class A SBL BRO-1 from M. catarrhalis.925 This enzyme is only partially membrane bound, as 10% was lipidated in its native host, and 45% when expressed recombinantly in E. coli.926 Meanwhile PenA, a class A enzyme from Burkholderia pseudomallei, is fully in the lipidated form and is secreted through the Tat system.927
As previously mentioned in Section 4.7.1.4, the B1 enzyme NDM-1 is a lipoprotein anchored to the outer membrane of Gram-negative bacteria, with a canonical lipobox present within its signal peptide.421, 474 All the protein population is in the membrane associated form, located in the inner leaflet of the outer membrane when expressed either in E. coli or P. aeruginosa.421 This membrane association of the enzyme was also observed in clinical isolates.928 All natural alleles maintain the same lipobox sequence of NDM-1, and membrane anchoring of variants NDM-2 to NDM-16 has been experimentally demonstrated.452
This post-translational modification, unique among all other clinically relevant MBLs, impacts the in vivo properties of the enzyme. Studies carried out in E. coli compared the anchored enzyme and the soluble mutant NDM-1 C26A, in which the Cys26 residue that covalently binds the lipid group is removed.421 Both forms of the enzyme granted similar resistance levels to β-lactam antibiotics in rich medium. Instead, upon metal starvation conditions, membrane-anchored NDM-1 provides higher levels of resistance than the soluble variant. This represents an important adaptation of the enzyme, boosting its capability to grant β-lactam resistance under the low Zn(II) availability that pathogens face during infection as a result of the host’s innate immune response (see Section 10). Additionally, membrane anchoring increases the in vivo stability of the enzyme under Zn(II) starvation: lipidated NDM-1 was proteolyzed at a lower rate than soluble NDM-1 C26A. A similar stabilizing effect for membrane anchoring was observed in the chimeric protein N-VIM, in which the N-terminus of NDM-1 was fused to the globular domain of VIM-2.421 Membrane anchoring also increases secretion of NDM-1 into OMVs (see Section 8.3).
The interaction of NDM-1 with the surface of the OM was recently studied by a combination of computational simulations and experimental methods.476 The protein adopts a stable orientation with respect to the membrane, with the active site exposed to the solvent and patch of the polypeptide establishing specific interactions with the membrane lipids (Figure 63), particularly cardiolipin. Furthermore, it was found that disruption of this interaction via the R39E and R46E mutations (R45E and R52E in the primary sequence numbering of NDM-1) counteracted the advantages provided by membrane anchoring, reducing the capability to confer resistance under Zn(II) depletion and the in vivo stability of the enzyme under such conditions.
Figure 63.
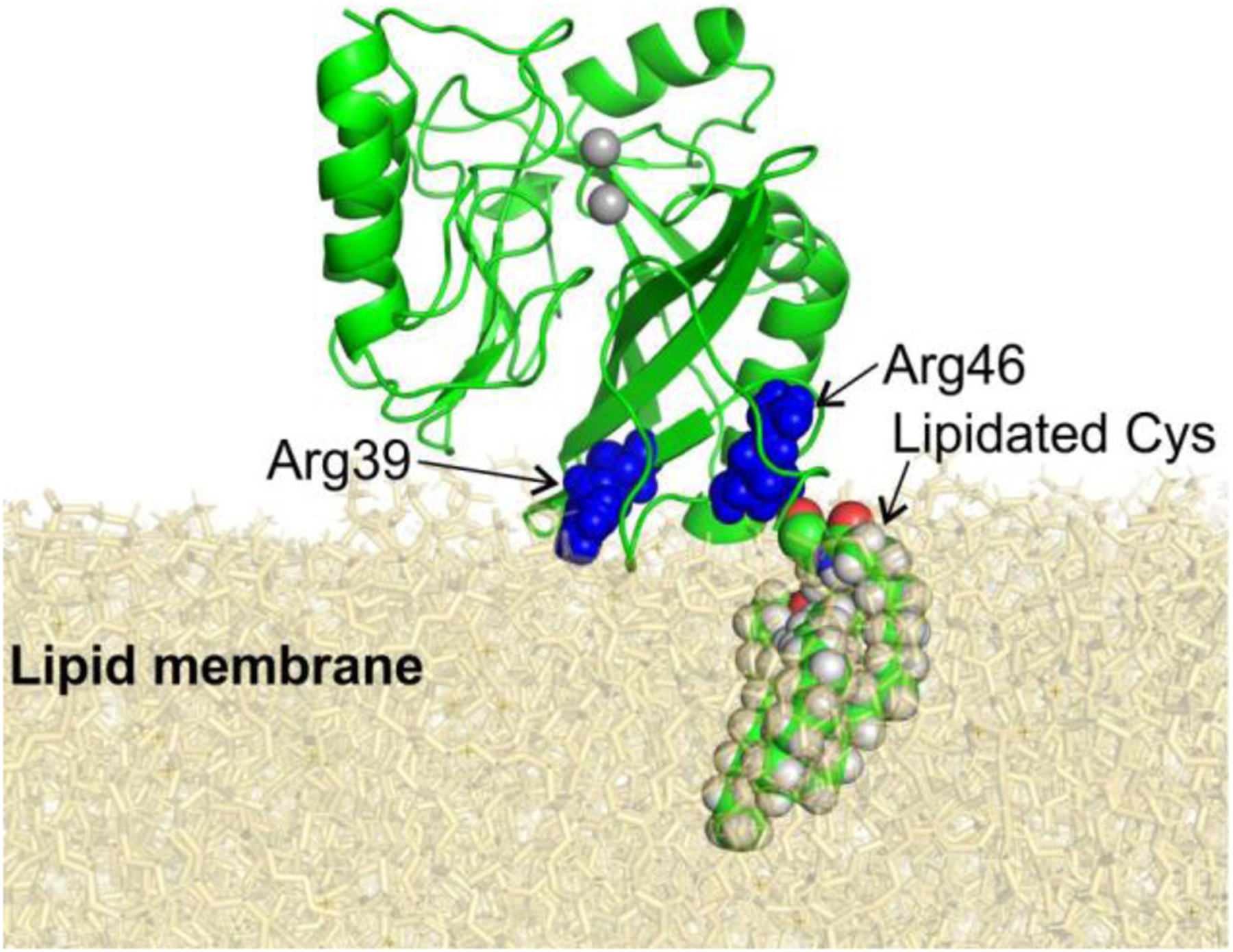
Structural model of the interaction of NDM-1 with the surface of lipid membranes (obtained from MD simulations by Prunotto et al.476). Active-site Zn(II) ions are shown as grey spheres. The N-terminal lipidated Cys and key residues for membrane interaction (Arg 39 and Arg46) are shown as spheres.
8.3. Outer membrane vesicles
Outer membrane vesicles (OMVs) are spherical membrane-enclosed particles released by Gram-negative bacteria into the extracellular milieu.929–934 These vesicles, with a diameter ranging from 20 nm to 250 nm, are formed from protrusions in the bacterial outer membrane (OM), which are then split from the cell and secreted into the environment (Figure 64). The OMVs thus generated are bounded by a lipid bilayer derived from the OM and enclose periplasmic components within their lumen. In recent years, production of similar extracellular vesicles (EVs) has also been observed in Gram-positive bacteria, archaea and eukaryotes, revealing that the formation of EVs is a process common to living cells across all domains of life.935, 936
Figure 64.
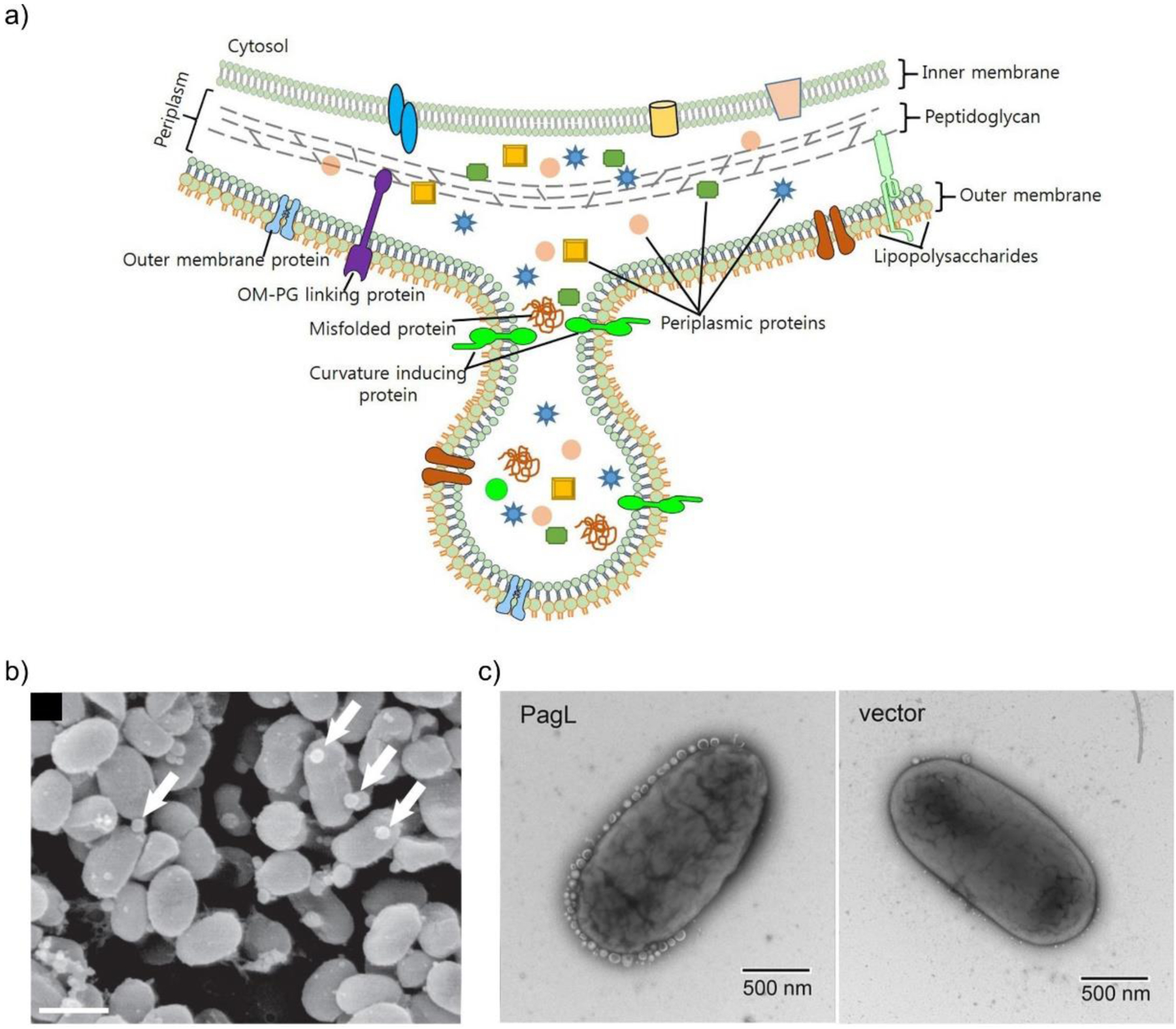
a) Schematic representation of the mechanism of OMV biogenesis. Reproduced with permission from reference933 Jan. Copyright 2017 Markley and Wencewicz under Creative Commons Attribution License (CCBY) https://creativecommons.org/licenses/by/4.0/ b) Scanning electron micrograph of OMVs (indicated by arrows) attached to the surface of Prochlorococcus cells. Scale bar, 1 μm. Reproduced with permission from reference939 Biller et al. Copyright 2014 American Association for the Advancement of Science c) Transmission electron micrographs of S. Typhimurium cells expressing PagL from a vector, or containing the empty vector, showing induction of OMV production by PagL. Adapted with permission from reference940 Elhenawy, Feldman et al. Copyright 2016 American Society for Micrbiology under Creative Commons Attribution 4.0 International License https://creativecommons.org/license/by/4.0/.
The composition of OMVs includes mainly phospholipids, lipopolysaccharide, outer membrane proteins, and periplasmic proteins. Numerous reports indicate the presence of DNA or RNA, but it is unknown how these cellular components are packaged into the vesicles.929, 933 Cytoplasmic and inner membrane proteins were also detected in vesicles, although the presence of these components and possibly that of nucleic acids could be due to the generation of the so-called Outer-inner membrane vesicles (O-IMVs) observed for various organisms, which contain both cellular membranes.937, 938 In particular, the capability of OMVs to transport nucleic acids has important implications regarding their functions (see below).
Although the mechanistic details determining their biogenesis are still controversial, the consensus is that the production of OMVs is a distinct, regulated physiological process that does not implicate cell lysis.929, 932, 933, 941 Furthermore, all Gram-negative bacteria produce OMVs that are released under all growth conditions. Various mutants have been isolated that display either increased (hypervesiculation) or reduced (hypovesiculation) vesicle production, and environmental stimuli can also impact the amount of OMVs released, granting further credence to the notion of a regulated biogenesis process for the vesicles. There are various proposed mechanisms for OMV biogenesis. OMVs may be formed at sites with reduced contacts between the outer membrane and the underlying peptidoglycan layer, as supported by the fact that mutants in proteins that act in tethering these two cell-wall components (such as Lpp, OmpA and the Tol/Pal system) present a hypervesiculation phenotype.942, 943 Although these mutant strains tend to possess a compromised OM integrity, a regulated process may act in a similar fashion during OMV release by locally removing contacts of the OM and peptidoglycan.943 An additional mechanism leading to OMV production may be an asymmetric growth of the outer membrane with respect to the inner bilayer, leading to OM bulging to accommodate an increased amount of membrane lipids. This is supported by the observation of hypervesiculation phenotypes in Haemophilus influenzae and Vibrio cholerae strains that have mutations in the VacJ/Yrb system, a widely conserved ABC transporter among Proteobacteria that was proposed to prevent phospholipid accumulation in the outer leaflet of the OM.944 The observation of a similar phenotype in such diverse organisms points to a potentially widespread mechanism regulating OMV production. An alternative hypothesis posits that an increase in turgor pressure in the periplasmic space by accumulation of peptidoglycan fragments and misfolded proteins may also cause the OM to bulge outwards and lead to OMV biogenesis.930, 945 OMV formation may also be triggered by the accumulation in the outer membrane of curvature-inducing molecules, such as the P. aeruginosa quorum sensing molecule PQS.946 Certain LPS modifications may also contribute to this possible mechanism, as illustrated by the hypervesiculation observed for S. Typhymurium recombinantly expressing the lipid A deacylase PagL940 (Figure 64-c). This enzyme removes acyl chains from the Lipid A core of LPS, producing modified Lipid A species with a reduced hydrophobic cross-section area and an inverted cone shape that could induce the adequate curvature in the outer leaflet of the OM for vesicle biogenesis. PagL expression results in full secretion of the LPS into vesicles and a 4-fold increase in OMV production.940
It has been amply documented that OMVs present differences in composition to the outer membrane and periplasmic compartment from which they are formed. While certain components are enriched in the vesicles, others are excluded, in a process that has been termed “cargo selection”.929, 933 The mechanisms responsible for cargo selection have not been identified, but the existence of this phenomenon supports the conclusion that the formation of these vesicles is a controlled process and not the mere release of fragments of the outer membrane deriving from cell lysis. Examples of cargo selection include the observation that LPS in P. aeruginosa OMVs is almost exclusively of type B LPS, when this is a minor component of the cellular outer bilayer, and the enrichment of extracellular proteases termed gingipains in Porphyromonas gingivalis OMVs while nutrient acquisition proteins RagA/B where excluded.947 Furthermore, various virulence factors have been observed to be enriched in OMVs. In Bacteroides fragilis and B. thetaiotaomicron, proteins enriched in OMVs were found to be predominantly negatively charged (low pI), while most proteins in the OM were positively charged.948 This suggests that an uncharacterized machinery may recruit proteins into OMVs depending on their charge and/or structure.
It is postulated that OMV production constitutes a “Type-0” secretion system, allowing bacteria to release both soluble periplasmic proteins and insoluble components such as membrane proteins into their environment, while protecting their cargo from proteolytic degradation.932 Furthermore, the joint secretion of multiple proteins within OMVs allows their simultaneous delivery in a concentrated form to sites distant from the producing bacteria, which may be important when a set of factors is needed to carry out a given function, such as the combined secretion of adhesins and virulence factors for pathogenesis.930
A wide variety of biological functions has been associated with OMVs.929, 930, 933, 934 Vesicles released by many organisms carry hydrolytic enzymes such as proteases and glycosylases, and thus may contribute to nutrient acquisition through degradation of complex polymers at a distance from the producing organism. OMVs from Neisseria meningitidis and P. gingivalis are enriched in iron-scavenging proteins, and may be involved in acquisition of this metal ion, although it is unknown how the bacterial cells would then obtain the metal from OMVs.949, 950 In this regard, it has been recently shown that P. aeruginosa can capture iron ions from OMVs via a mechanism dependent on the TseF protein, by interacting with the iron-bound PQS contained in the vesicle membrane and with both the siderophore receptor FptA and the porin OprF in the cell surface.951 OMVs are abundant in sea water. Prochlorococcus, the most frequent cyanobacteria in the ocean, was found to produce large amounts of OMVs which can be used as the single carbon source by heterotrophic marine bacteria.939 OMVs may also play a role in predation of other bacteria by Myxococcus xanthus, as vesicles from this organism have been found to contain numerous hydrolytic enzymes and to have the ability to lyse E. coli.952
These vesicles have been posited to be involved in cell-wall remodeling and elimination of toxic material from the bacterial cell. The release of OMVs can increase as part of a cell-wall stress response through the σE pathway,945 and it was shown that the accumulation in misfolded proteins in the periplasm as a result of inactivation of the DegP protease/chaperone also leads to greater production of OMVs.953 Under these conditions, the vesicles may provide a pathway to dispose of protein aggregates and other toxic substances present in the periplasm. Other stress conditions may trigger increased OMV production, as observed for P. aeruginosa under oxygen stress caused by high pO2954 and other external stressors955 and in S. maltophilia upon exposure to the fluoroquinolone ciprofloxacin.956 Additionally, iron limitation was observed to induce a Fur-dependent downregulation of the aforementioned VacJ/Yrb system in H. influenzae, E. coli and V. cholerae,944 leading to hypervesiculation under conditions similar to those that may be encountered by pathogens at infection sites (see Section 10).
It has also been proposed that OMVs can act as “public goods”, not only benefiting the producing organism but the wider community. This is shown by bacteria from the Bacteroides genus, normal components of the human intestinal microbiota, which release OMVs carrying polysaccharide-degrading enzymes that allow the use of the hydrolysis products by organisms that do not express proteins with such activities.957 This collective role of OMVs may also come into play in the sharing of antibiotic resistance enzymes among members of a bacterial community (see below).
OMVs have also been observed to be an important constituent of the extracellular matrix around bacterial cells in biofilms.958–960 The P. aeruginosa quorum sensing compound PQS, which stimulates biofilm formation, is highly hydrophobic and around 80% of it is estimated to be released within OMVs.961 Thus, the PQS quorum sensing response may be mainly mediated by OMVs. Additionally, the vesicles produced by P. gingivalis carry adhesins that aid in the formation of mixed biofilms in dental plaque.962
OMVs also perform important roles during pathogenesis. Bacteria liberate vesicles during infection, and the conditions within the host can lead to an increased production of OMVs. This is exemplified by the increased production of OMVs by Salmonella Typhymurium during intracellular growth, triggered by activation of the expression of PagL as a result of PhoP/PhoQ signalling.940 Additionally, it was reported that the conditions encountered in the human gastrointestinal environment upregulate OMV production by enterohemorragic E. coli (EHEC).963 Large amounts of OMVs were also observed to be released by P. aeruginosa cells infecting the nematode Caenorhabditis elegans.964 This bacterium releases the Cif toxin and other virulence factors within OMVs, which are able to fuse with host airway epithelial cells. This releases the Cif toxin into the cytoplasm where it can trigger the degradation of the CFTR (cystic fibrosis transmembrane conductance regulator) chloride channel, possibly leading to reduced mucus clearance in host airways and helping colonization by P. aeruginosa.965, 966 The VacA toxin from Helicobacter pylori is released both as a free protein and within OMVs, and these vesicles have been shown to be internalized by gastric epithelial cells.967 Additionally, OMVs from E. coli have been shown the ability to deliver virulence factors such as the Shiga toxin and cytolysin A to eukaryotic cells.968, 969 OMVs released during pathogenesis may travel great distances from the infection site, as illustrated by their detection in samples of blood and other biological fluids from patients or animals infected by pathogen Borrelia burgdorferi.970
OMVs from various species transport DNA, both of chromosomal and plasmidic origin, and it has been reported that such vesicles can transfer their genetic material to other bacteria.971 Clinical isolates of A. baumannii release OMVs containing a plasmid harboring the blaOXA-24 gene, which could then transform a different A. baumannii strain. The original plasmid was present in the recipient A. baumannii strain, which presented increased β-lactam resistance.972 Similar OMV mediated transformation was observed in A. baylyi.973 Transfer of chromosomal DNA has been observed among P. gingivalis strains, with the recipient integrating the acquired genes into its chromosome.974 In this case, a reduction in the frequency of transformation was observed when the vesicles were pretreated with DNase, suggesting that part of the DNA carried by the vesicles and transferred to the recipient was located in the surface of these OMVs. In this regard, it was recently reported that OMVs from a range of Gram-negative bacteria carry DNA both in the vesicle lumen and attached to their surface.975 It is still unknown how the recipient strains transfer the genetic material into their cytoplasm. In bacteria capable of natural transformation, such as Acinetobacter, the machinery responsible of this process may also be involved in uptake of DNA from OMVs, as mutation in competence genes leads to decreased efficiency for transformation by vesicles.973 However, DNA transfer by vesicles has also been observed in species not capable of natural transformation, such as E. coli.973, 976 This evidence suggests that OMVs could constitute a novel horizontal gene transfer mechanism, in addition to transformation, transduction and conjugation, which could aid in the dissemination of antibiotic resistance genes. Notably, various recent reports have identified the presence of RNA within OMVs, from organisms such as Prochloroccus, Vibrio chloreae, P. aeruginosa and E. coli.971 This RNA cargo could also contribute to OMV function in processes such as pathogenesis, as the RNA transported by OMVs from P. aeruginosa was found to enter host cells and downregulate their innate immune response.977
There is also considerable evidence supporting the role of OMVs in resistance to antibiotics, phages and other antimicrobial factors. OMVs provide protection towards membrane-active compounds such as the antimicrobial peptide melittin in Pseudomonas syringae or the antibiotics colistin and polymyxin B in E. coli,978 possibly by acting as decoy membranes which titrate the compound and reduce the amount that reaches the actual bacterial cells. OMVs carrying phage receptors may similarly lead to attachment of the viruses to the vesicles and reduce productive infections.978 Moraxella catarrhalis OMVs protect bacteria from the immune system attack by the complement cascade, by capturing and depleting complement factors.979 OMVs may act as carriers of hydrolytic enzymes that inactivate antibiotics, and there are numerous instances of reported secretion of β-lactamases into OMVs. P. aeruginosa isolates obtained from cystic fibrosis patients secreted their intrinsic AmpC lactamase into OMVs.980 The membrane enclosing the enzymes within OMVs can may also act by protecting from inactivation by agents such as extracellular proteases or even host antibodies, as shown in vitro for the BRO-1 SBL secreted into vesicles by Moraxella catarrhalis.981 As previously mentioned, OMVs may act as public goods, and the antibiotic-degrading activity contained within vesicles produced by resistant organisms may benefit other nearby bacteria. OMVs produced by Bacteroides thetaioataomicron carry the chromosomally encoded BtCepA class A SBL, and addition of these purified OMVs to cultures of β-lactam sensitive S. Typhymurium and Bifidobacterium breve strains protects them from the bactericidal action of cephalosporins.982 Furthermore, vesicle production can be influenced by antibiotics, since ceftazidime can lead to an increased OMV production in A. baumannii.983
Secretion of various MBLs into OMVs has also been observed. NDM-1 was found to be secreted into OMVs both in an E. coli lab strain and an Enterobacter cloacae clinical isolate.421 Membrane anchoring was found to increase secretion into the vesicles as the WT enzyme was found at much higher levels in E. coli OMVs compared to soluble variant NDM-1 C26A in spite of similar whole cell expression.
Although expression of NDM-1 did seem to impose a strain on the bacterium, MBL secretion into OMVs may also be linked in certain cases to the stress imposed on the host by the expression of these enzymes. A recent report430 studied the enzymes NDM-1, VIM-2 and SPM-1 produced recombinantly from the same plasmid in three hosts: E. coli, P. aeruginosa and A. baumannii. Both VIM-2 and SPM-1 were secreted in large amounts into OMVs in A. baumannii and E. coli, but not in P. aeruginosa, with no correlation to whole cell expression levels. The secretion of VIM-2 and SPM-1 into OMVs in the former two organisms was found to be triggered by the envelope stressed caused by their production, due to inefficient signal peptide cleavage. This resulted in accumulation of the precursor (unprocessed) forms and impaired cell growth when these MBLs were expressed, in addition to increased OMV production (hypervesiculation). In contrast, no such detrimental effects nor hypervesiculation were observed upon expression of either VIM-2 and SPM-1 in P. aeruginosa, and both enzymes were absent from OMVs released by this organism. As such, OMV secretion of VIM-2 and SPM-1 in the aforementioned bacteria would be caused or boosted by an envelope stress response, which has previously been observed to increase vesiculation, providing a path to eliminate poorly folded proteins from the periplasm. Notably, NDM-1 was secreted into OMVs in all three bacteria while causing neither growth defects nor hypervesiculation.430 This suggests again that membrane anchoring can contribute to release into these vesicles and that NDM-1 is tailored for OMV secretion in a wide range of Gram-negative organisms.
Intrinsic MBLs have also been linked to OMV secretion. Both lactamases produced by Strenotrophomonas maltophilia, the B3 enzyme L1 and class A SBL L2, were found to be released into OMVs. Furthermore, the exposure of this organism to imipenem not only increased the production of these enzymes (see Section 7.2), but also lead to greater liberation of OMVs and abundance of L1 within the vesicles.984 OMVs usually contain porins, allowing the entry of β-lactam antibiotics and other small solutes into the vesicle lumen. Both OMVs purified from β-lactam-exposed S. maltophilia, and vesicles NDM-1 carrying obtained from E. coli possess β-lactase activity.421, 985 Incubation of antibiotic susceptible strains with such vesicles protects them from otherwise lethal β-lactam concentrations.421, 985 Thus, nearby bacterial populations may also benefit from the release of β-lactamase carrying OMVs, and this may be of particular importance in organisms such as S. maltophilia, which is known to co-infect the lungs of cystic fibrosis patients with other species such as P. aeruginosa and Burkholderia cenocepacia. Indeed, a recent study showed that the lactamase activity within OMVs produced by S. maltophilia increases the resistance of strains of the latter two organisms towards imipenem and ticarcillin.985
The potential of OMVs carrying β-lactamases to protect antibiotic sensitive strains during polymicrobial infections was recently validated in the infection model Galleria mellonella (Martinez et al., submitted) During larvae co-infection with a mixture of NDM-1-producing E. coli and carbapenem-sensitive P. aeruginosa, NDM-1 containing vesicles released by E. coli were able to cross-protect P. aeruginosa cells from a meropenem treatment. Furthermore, it was shown that the protective activity of OMVs harboring NDM-1 is stable in the hemolymph of G. mellonella for long periods of time, and that the vesicle scaffold enhances the action of the enzyme.
OMVs have been reported to carry genetic material. A full plasmid harboring blaNDM-1 was observed to be secreted into OMVs by an A. baumannii clinical isolate, and these vesicles have the ability to transform both other A. baumannii strains and E. coli, transmitting the resistance gene.976 The ability of OMVs to carry both active enzymes and their coding genes may contribute to the spread of antibiotic resistance, as the antibiotic degradation activity contributed by such vesicles may grant transient resistance to an otherwise sensitive organism, and increase its chances to permanently acquire such resistance, either by the vesicle bound DNA or via the traditional gene transfer mechanisms.
9. Dissemination and host specificity of MBLs
Bacterial evolution has largely been shaped by the high plasticity of bacterial genomes, leading to their adaptation to most ecosystems as well as to environmental changes generated by human activities, such as the use of antibiotics, industrial contamination and intensive agriculture. Bacteria possess an impressive toolkit that enables them to modify, rearrange and exchange genomic sequences in order to gain new beneficial traits. Bacterial resistance to antibiotics and its continuous evolution are a clear demonstration of this. The acquisition and spread of antibiotic resistance genes among bacteria intimately associated with humans and their domesticated animals are well documented. In the following sections we will describe the general mechanisms that govern the dissemination of antibiotic resistance and the particular case of MBLs.
9.1. Antibiotic resistance genes are ancient
Regardless their evolutionary origin, MBLs, SBLs and many other forms of antibiotic resistance, long predate the discovery and application of antibiotics by humans.986, 987 Microbes in natural environments such as soil and water compete with each other for limited resources, and the production of natural antibiotics as secondary metabolites to hinder the growth of other species could be an effective survival strategy to conquer territory against a large pool of potential competitors.988, 989 As a result, the origin of resistance mechanisms could lie within the very same species producing each antibiotic class to avoid self-destruction in case they possess the biological target of the compound, or in organisms growing in the same environment, as a mechanism ensuring their survival in presence of the antibiotic producer. Alternatively to this microbial warfare hypothesis, natural products regarded as antibiotics have been proposed to act as signalling molecules, and various classes of antibiotics were reported to induce distinct transcriptional responses at concentrations below their MICs, with resistance determinants being actually modulators of these signalling networks.990 Irrespective of their physiological roles in their natural environment, it is well documented that antibiotic resistance genes are ancient in origin. They have been found both in samples predating modern antibiotic chemotherapy and also in remote sites not affected by human activity, such as soil samples obtained from a remote Alaskan site, a deep sea bacterium in the Pacific Ocean or a cave isolated from the outside world for 4 million years.535, 991, 992
9.2. The environmental resistome is a source of antibiotic resistance in clinical settings
Although mutational events contribute to bacterial adaptation, horizontal gene transfer (HGT) seems to be the major cause of rapid proliferation of antibiotic-resistance genes across a wide range of bacteria.993, 994 Apart from cell-to-cell transfer, HGT typically involves assembling or rearrangement of resistance modules containing one or several resistance genes, and the intracellular mobilization of these modules from one DNA molecule to another (e.g., from the chromosome to plasmids or vice versa) (Figure 65-a). At the heart of these processes are mobile genetic elements (MGEs), which include plasmids, bacteriophages, genomic islands (GIs), integrative and conjugative elements (ICEs), integrative and mobilizable elements (IMEs), insertion sequences (ISs), transposons (Tns), integrons and miniature inverted repeat transposable elements (MITEs).994, 995 The success of HGT of resistance genes depends not only on the introduction of DNA into the cytoplasm of the recipient cell, but also on the heritability of the transferred sequences in this microorganism.996–998
Figure 65.
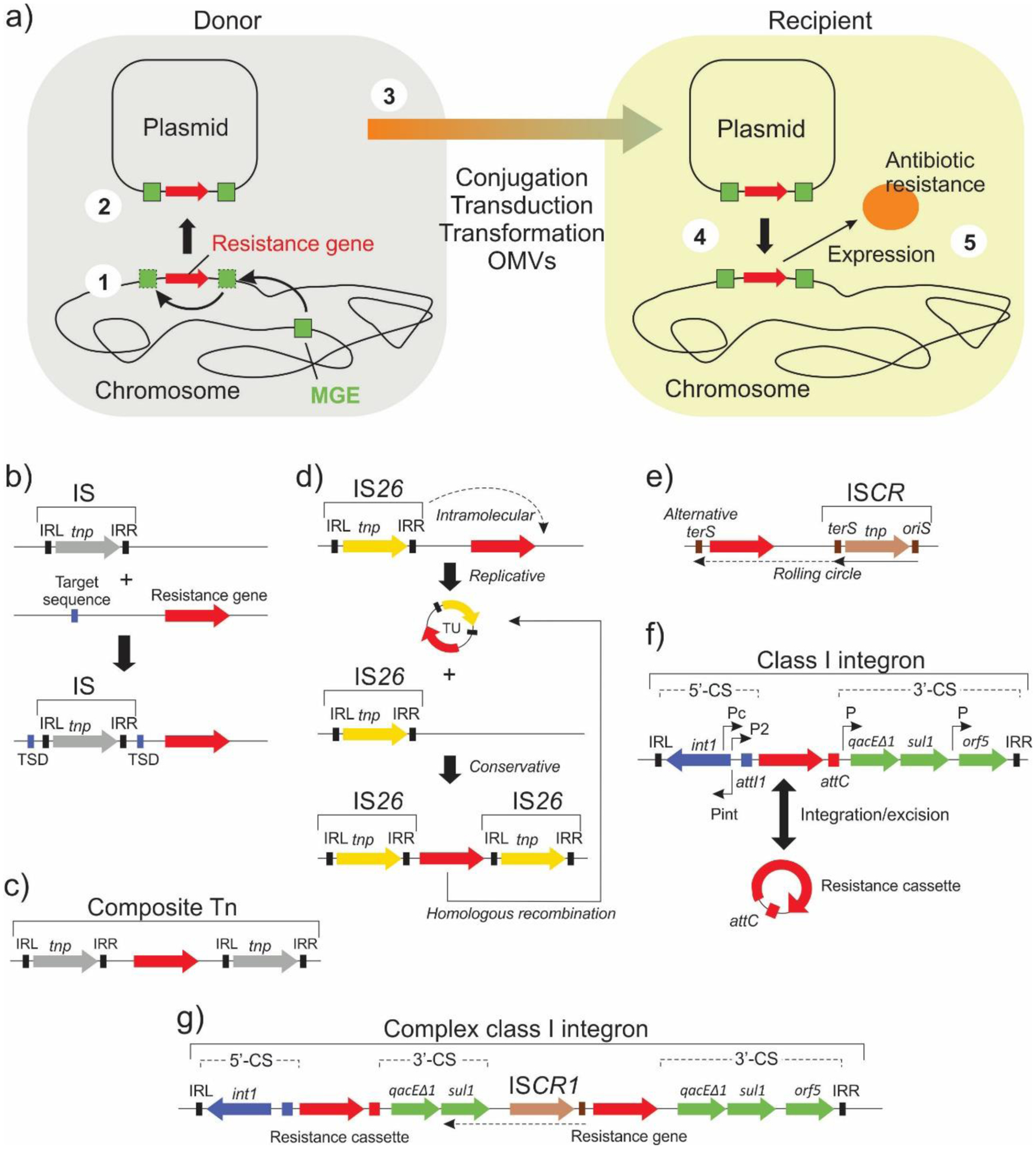
a) Schematic representation of antibiotic resistance gene mobilization and horizontal transfer into a new host. b – g: Structure of commonly encountered mobile genetic elements (MGEs).
There are two types of resistance genes, those typically occurring in clinical environments, which are associated to MGEs and were acquired by HGT (as such, the GC content may be different from that of the host genome), and the intrinsic or endogenous resistance genes which exist in environmental bacteria since ancient times as discussed above.999 Several studies have identified the environmental resistome as a source of resistance genes of clinical interest.1000–1003 In general, the soil and aquatic environments influenced by anthropogenic interventions have been pointed out as the origin and reservoir of resistance genes that further emerge in clinical settings. The resistome of these niches is composed by resistance genes nearly identical or similar to those found in clinics, and harbor a wide range of novel resistance determinants that, if mobilized, may be transferred to pathogens either directly or indirectly via commensal bacteria in humans or animals, leading to infections difficult or impossible to treat. The link between environmental and clinical resistomes has been demonstrated by the finding of organisms from soil samples containing resistance genes with 100% sequence identity to those present in pathogenic bacteria, including SBLs.1004 Additionally, the PER and FOX SBLs have been recently found to have been mobilized from the chromosomes of environmental bacteria.1005, 1006 On the contrary, in minimally impacted environments the resistome is composed by a higher proportion of distant homologs related to intrinsic resistance genes instead of horizontally acquired resistance genes circulating in clinical pathogens.1007–1009
9.3. Generation of acquired resistance genes
A chromosomally-borne intrinsic resistance determinant can gain the characteristics of an acquired gene by means of several genomic rearrangements.994, 995 The first step is the insertion of a MGE, generally an IS, proximal to the resistance gene. Alternatively, the resistance gene may acquire a downstream recombination sequence (attC or 59-be element) and become a gene cassette.1010, 1011 Gene cassettes are small mobile elements (0.5 to 1 kb) consisting of a single gene (occasionally two) and the attC recombination site. These structures, typically lacking a promoter, form non-replicative free circular molecules that insert into integrons (see next).
ISs are small mobile elements that typically carry little more than one transposase (tnp) gene (Figure 65-b).1012 ISs can move almost randomly to new locations in the same or different DNA molecules within a single cell by a cut-and-paste (conservative transposition) or copy-and-paste (replicative transposition) mechanism. Many ISs harbor terminal inverted repeats (IRs) at the ends (IRL and IRR) and after transposition create short flanking direct repeats (DRs) at the target DNA, usually referred to as target site duplications (TSDs). Some ISs do not appear to target specific sequence motifs; others may not have IRs or create target site duplications. While the mobility of some ISs has been shown experimentally, many have been defined only from the transposases that they encode, their IRs, and/or their TSDs. ISs were originally assigned numbers, but now they are assigned with names that include a code for the species in which the IS was first identified and a number (e.g., ISAba1 for A. baumannii). Many ISs include a strong promoter, and insertion upstream of an intrinsic chromosomal gene can influence its expression. Alternatively, an IS may provide a -35 region only, which can combine with an adjacent -10-like sequence to create a hybrid promoter.1012, 1013
Traditionally ISs were not thought of as carrying “passenger” genes, but they can move resistance genes as part of a composite Tn, a region bounded by two copies of the same or related ISs (generally in the same direction) that can move as a single unit (Figure 65-c). Some ISs, however, can move adjacent DNA sequences as single copies. These include IS26, insertion sequence common region elements (ISCRs) and ISEcpI, commonly associated to resistance genes. IS26 can pick up adjacent genes from the chromosome by an intramolecular replicative mechanism, generating free circular molecules termed translocatable units (TUs) that consist of one copy of IS26 and the adjacent gene(s) (Figure 65-d).1014 A TU preferentially inserts next to an existing copy of IS26 in a recipient molecule via a conservative process, generating the typical arrays of resistance genes separated by single copies of IS26 observed in resistance plasmids. New TUs can be generated from these arrays by the same mechanism or by homologous recombination between two copies of IS26. Overall, once a chromosome or plasmid possesses a copy of IS26, it is predisposed to generate and/or acquire further adjacent IS26 TUs. On the other hand, ISCRs are related to an atypical class of ISs designated IS91-like that lack conventional IRs and move by rolling circle replication, without generating target site duplications (and thus being difficult to trace) (Figure 65-e).1015 Replication proceeds from oriS to terS sites (opposite to the direction of transcription of the internal transposase gene) located at the edges of the element. In some cases, ISCRs move adjacent resistance genes when the rolling-circle replication mechanism either misidentifies terS or the terS site has been deleted, as proposed for ISCR1, and proceeds to replicate the DNA adjacent to this site. Replication generates circular ssDNA molecules that can insert into unidentified target sequences or through homologous recombination. ISCR1 has been seemingly responsible for capturing and moving antibiotic resistance genes next to class I integrons, generating the so-called complex class 1 integrons. ISCR1-associated resistance genes are found adjacent to the oriS end, presumably as a result of incorporation of the circular molecule by recombination through the integron 3’-CS (see below). Other ISCRs can be part of GIs, such as ISCR3, or may be included in ICEs, such as ISCR2 or ISCR4. The ISCR elements ISCR3, ISCR4, ISCR5, ISCR14, and ISCR16 all share a similar GC content, and their transposases are related. With the exception of ISCR5, they are all found adjacent to sections of groEL that display the highest identity to the same gene from Xanthomonas spp., indicating that they descend from an ancestral ISCR element in a Xanthomonas-like organism.1016 Finally, ISEcpI, first identified in E. coli, has IRs and creates target site duplications on transposition. ISEcp1 is seemingly able to use IRL in combination with a sequence beyond its IRR end (their promiscuous transposases can recognize a variety of related DNA sequences as a functional IRR) to move an adjacent region, creating target site duplications flanking the whole transposition unit.1017 ISEcp1-like sequences have been involved in mobilization of various blaCTX-M genes.165
Integrons are non-autonomous elements (i.e., incapable of self-transposition) consisting of three components: a gene that encodes an integrase, encoded by the intI gene, needed for site-specific recombination within the integron; an adjacent recombination site (attI) that is recognized by the integrase; and a promoter (Pc), located upstream of the integration site, necessary for efficient transcription of gene cassettes present in the integron.1018, 1019 In general, integrons can incorporate up to five gene cassettes. Different classes of integrons have been defined based on the sequence of the intI gene (called IntI1, IntI2, IntI3, etc., with cognate attI1, attI2, and attI3, etc. sites); integron structures can be found on the INTEGRALL database (http://integrall.bio.ua.pt/). Class I integrons are the most common and widespread in Gram-negative bacteria, especially in clinical settings, being one of the major contributors to the dissemination of antibiotic resistance (Figure 65-f). Class I integrons have three distinct genetic regions two of which are highly conserved, the 5′-conserved segment (5′-CS) and the 3′-conserved segment (3′-CS), that flank the central variable region where the gene cassettes are located. The 5′-CS includes the integrase gene, the recombination site attI1, the promoter Pc located inside the intI1 gene and in some cases a second promoter (P2). The 3′-CS consists of the qacEΔ1 gene, which encodes a functional deletion of a protein that mediates resistance to quaternary ammonium salts, the sulI gene, encoding resistance to sulfonamides, and an open reading frame of unknown function, orf5. Resistance gene cassettes in the variable region are highly mobile as these can be integrated or excised from the integron through the attC site; cassette integration usually occurs at the attI1 site, and the original cassette attC is reformed when the gene cassette is excised from the integron. The last cassette to be integrated is the most proximal to the Pc promoter of the array, and the one exhibiting the highest expression. In addition, the integrase gene carries LexA binding sites close to its promoter region. Upon induction of the SOS response (for instance, by β-lactam antibiotics or during conjugation), the integrase is overexpressed leading to enhanced cassette shuffling and integron diversification. Given that class I integrons incorporate gene cassettes conferring resistance to different types of antibiotics, these structures lead to MDR phenotypes. Class I integrons must embed in other MGEs in order to acquire mobility.1018 Class I integrons can be mobilized as part of composite Tns if they become flanked by two copies of an IS, or in trans by a transposase recognizing the IRs located at the ends of these elements. Alternatively, integrons can exchange resistance cassettes by homologous recombination through the conserved 5’-CS and 3’-CS regions with another integron located in a different genetic structure in the cell. Finally, complex class I integrons generally consist of a class I integron, in which part of the 3’-CS is deleted, immediately followed by an ISCR1 element, one or several non-cassette resistance genes, and a full copy of the 3’-CS (Figure 65-g). In these elements, replication of ISCR1 can mobilize the adjacent integron (see above).
Elemental resistance modules, consisting of one or more resistance genes associated to ISs or as part of integrons or complex class I integrons, can become genetically linked to or embedded in other MGEs, such as miniature inverted repeat transposable elements, ISs, composite Tns, unit Tns or GIs, gaining additional levels of intracellular mobility. These resistance modules, either elemental or “complex”, can move into MGEs capable of performing cell-to-cell transfer, such as ICEs (conjugative Tns), IMEs (mobilizable Tns), plasmids or bacteriophages, favoring the simultaneous horizontal transfer of various resistance genes.994 Miniature inverted repeat transposable elements are non-autonomous derivatives of bacterial IS or transposons that retain the IRs but which have lost central parts, including the transposase gene(s).1020 These elements are transposed when provided a transposase that acts in trans. Unit transposons consist of elements larger than ISs, bounded by IRs rather than by a pair of ISs (unlike composite Tns), and including a transposase gene and an internal passenger gene(s), which may encode antibiotic resistance, acquired by a previous transposition event.995 These elements generally generate TSDs upon transposition. GIs (genomic islands) are clusters of genes within the bacterial chromosome apparently acquired by HGT, given the different GC content and codon usage from the core genome.1021 Resistance islands are genomic islands that contain arrays of several of the elemental resistance modules described above. Genomic islands are generally non-autonomous but can be excised and transferred to a new host.
The transposition events leading to complex resistance modules are difficult to trace, since some MGEs do not leave target site duplications, and intramolecular transposition may generate deletions or sequence inversions (as shown for IS26).994 In addition, since many MGEs, or their associated repeats, are present in multiple copies in different locations of a genome, they can facilitate homologous recombination generating additional exchange, insertion or deletion of sequences.
9.4. Mechanisms of cell-to-cell transfer of resistance genes
Resistance genes can be transferred to recipient cells by several mechanisms including conjugation, transduction, natural transformation or OMV-mediated delivery. Conjugation is the most prevalent one.
Conjugation is a key mechanism of horizontal gene transfer between bacteria and a major contributor of the plasticity and evolution of prokaryotic genomes.1022 Conjugation allows the transfer of DNA between bacterial cells in close contact and is mediated by different types of MGEs. Among them, plasmids are one of the main vectors for the dissemination of antibiotic resistance. Plasmids often carry a variety of antimicrobial resistance genes acquired by transposition or homologous recombination of resistance modules. They have been identified in most bacterial species investigated, which usually harbor multiple plasmids. Plasmids are termed conjugative when they encode the functions needed for self-mobilization, or mobilizable when they rely on the action of other conjugative elements for mobilization.1023, 1024 Conjugative plasmids harbor a transfer (tra) region and an origin of transfer (oriT) which significantly increase the size of the plasmid. The tra region includes two set of genes, those coding for a mating pair formation (MPF) complex, which is a type 4 secretion system (T4SS) providing the mating channel, and the mob genes (also called Dtr, for DNA transfer replication), that encode proteins required for plasmid DNA processing. The latter include a relaxase that specifically nicks the oriT of the DNA strand that is exported to the recipient cell. Mobilizable plasmids contain only a subset of the mob genes and the oriT, thus requiring an MPF of another genetic element present in the same cell for mobilization.
Plasmids conferring multi-drug resistance are usually large (>50 kb), self-conjugative and encode sophisticated mechanisms controlling their copy number, regulating the replication rate.1025 The minimal portion of a plasmid that replicates with the characteristic copy number of the parent plasmid is called the basic replicon.1023, 1024, 1026 Replicons contain the origin of replication (ori) but also encode specific replication initiator proteins (Rep) binding the ori. The strict control of replication implies that two plasmids that share the same replicon cannot be propagated stably in the same cell line (plasmid incompatibility). The relatedness of the sequences controlling replication has been used to classify plasmids in different incompatibility groups, indicated by Inc followed by a letter (e.g. IncF). In this way, closely related plasmids belong to the same Inc group and cannot coexist. Although plasmids encode their own replication initiation, they usually rely on the host’s chromosomally encoded replication machinery for DNA synthesis. The dependence on host-encoded DNA replication proteins as well as systems for mating pair formation, plasmid maintenance or avoidance of host restriction (see next), are among the key factors limiting the host range of plasmids. Narrow-host-range plasmids are efficiently maintained only in closely related bacterial taxa while broad-host-range plasmids have been found or shown to replicate in quite diverse genera. For instance, while plasmids of the IncA/C, IncN and IncL/M groups are broad-host-range, those from the IncX3, IncF, IcnHI1 and IncHI2 are stable only in Enterobacterales. Since conjugation can be an extremely promiscuous process, the transfer of resistance plasmids into hosts in which they cannot replicate is therefore very likely.1027 In these cases, MGEs (e.g., ISs, Tns, and integrons) can provide intracellular mobility mechanisms that give resistance genes an opportunity to move to other functional replicons (the chromosome or other resident plasmids). Thus, even narrow-host-range plasmids can act as suicide vectors for the horizontal spread of resistance genes into divergent hosts.
For small plasmids maintained at a high copy number, efficient inheritance by both daughter cells can be achieved by random segregation.1023, 1024 However, larger resistance plasmids usually exist at a low copy number to minimize the burden on their hosts, which risk being outcompeted by plasmid-free counterparts in the environment. Large low-copy-number plasmids thus usually possess functional modules that contribute to their segregational stability. These include multimer resolution (res) systems,1028 which convert plasmid multimers (arising due to homologous recombination) into monomers that can be segregated independently into daughter cells, partitioning (par) systems,1029 which actively distribute plasmid copies to daughter cells, and addiction systems generally based on toxin–antitoxin factors,1030 which are able to kill daughter cells that do not inherit the plasmid during cell division. In addition, promiscuous broad-host-range plasmids often encode anti-restriction mechanisms to avoid their degradation once incorporated in recipient cells after conjugation, such as the Ard proteins that inhibit type I restriction endonucleases and/or DNA methylases that modify DNA so that it cannot be recognized by these enzymes.1031 Finally, it should be noted that resistance plasmids sharing backbones with similar organizations and functions may differ by their resistance modules. Conversely, plasmids with different backbones may carry similar resistance modules, consequence of module transposition/mobilization events.
Other MGEs transferable by conjugation are the ICEs (integrative and conjugative elements) and IMEs (integrative and mobilizable elements).1032, 1033 ICEs are transposons carrying tra genes and an oriT while IMEs are mobilizable transposons that function as mobilizable plasmids, but without generating target site duplications. In contrast to conjugative plasmids, which are capable of autonomous replication, ICEs generally integrate into the chromosome (without generating target site duplications) and replicate with it. Analyses of bacterial genomes reveal a high prevalence of ICEs and IMEs, suggesting that their abundance exceeds that of conjugative plasmids. In addition, ICEs can promote the transfer of DNA sequences physically linked to the element, leading to the transfer of chromosomal genes or of non-autonomous MGEs. ICEs and IMEs are important vehicles for the acquisition of a broad spectrum of antibiotic resistance genes among bacteria and many other genes that can be advantageous for their host.
Bacteria can also acquire resistance genes through transduction, transformation and OMVs, although these mechanisms are less frequent than conjugation, and occur among bacteria from the same species or closely related species. Bacteriophages, viruses that infect bacteria, are abundant forms of life and co-reside in environments with bacteria.1034 The DNA of bacteriophages can exist in the host cell either in an extrachromosomal form or integrated in the host DNA. During transduction, bacteriophages can transfer fragments of the host DNA (either chromosomal or plasmidic) to the new infected bacterial cell by packaging them into viral particles. As a result, DNA fragments with resistance modules can be horizontally transferred to other cells by this mechanism. The amount of DNA that can be transferred during transduction depends on the size of the phage capsid, that can range from tens to hundreds of kbs. The ability to transduce host DNA seems to be limited to relatively large double-stranded DNA phages.
Natural transformation is the uptake and stabilization of extracellular DNA by competent bacteria. The physiological state of competence, which involves 20 to 50 proteins, is a time-limited response to specific environmental conditions (altered growth conditions, nutrient access, quorum sensing or starvation) and it has been identified in a wide variety of bacteria, including human pathogens.993, 1035 During this process, DNA fragments released from a dead degraded bacterium bind to DNA binding proteins on the surface of a competent living recipient bacterium. Then, depending on the bacterium, either both DNA strands penetrate the recipient, or a nuclease degrades one strand of the fragment and the remaining DNA strand enters the recipient. Although in most cases any fragment of DNA can be taken up (most commonly chromosomal DNA), the lack of sequence similarity between recipient and donor DNA limits the probability of integration by homologous recombination in the new host. However, as with plasmids or transduction, resistance modules can transpose into recipient DNA or integrate by homologous recombination through MGE conserved regions, such as the 5’-CS and 3’-CS of integrons, frequently present in recipient genomes.1018
Finally, in the past few years it has been shown that OMVs carrying resistance plasmids from Acinetobacter spp. are able to transform recipient cells in vitro, although the mechanism by which this occurs remains to be elucidated972, 976 (see Section 8.3).
9.5. horizontal Gene Transfer of blaMBL genes
Resistance to carbapenems in Gram-negative pathogens from the ESKAPE group emerged over the last two decades principally due to the acquisition and spread of plasmid-encoded carbapenemases.1036 This resulted in rapid and extensive worldwide dissemination of carbapenem-resistant bacteria expressing KPC-type, VIM-type, NDM-type and OXA-type carbapenemases, although initially associated to specific bacterial species. Among acquired MBLs of clinical impact, numerous families of the B1 subclass, such as NDMs, VIMs, IMPs, SPM-1, GIMs, SIMs, DIM-1, KHM-1, TMBs, FIM-1, VMB-1,1037 CAM-1,1038 and three members of the B3 subclass, AIM-1, SMB-1 and LMB-1,553 have been identified coded in transferable plasmids or bacterial chromosomes as part of mobile resistance modules (Figure 66). These modules in general contain other genes, some of them conferring resistance to other types of antibiotics.1039 As a result, bacteria expressing acquired MBLs exhibit multi-resistant phenotypes. Similar resistance modules may be found either in different plasmids or in the chromosome, evidencing mobilization of such modules.1040 Also, a given blaMBL gene may be inserted in different immediate contexts,1041 especially when mobilized as gene cassettes, o exist in multiple copies in the same host.1042 Unfortunately, many reports of blaMBL genes lack the characterization of the DNA sequences extending beyond the immediate elemental modules. This results in an incomplete description that does not allow the identification of the plasmid nor the genetic complexity beyond these modules.
Figure 66.
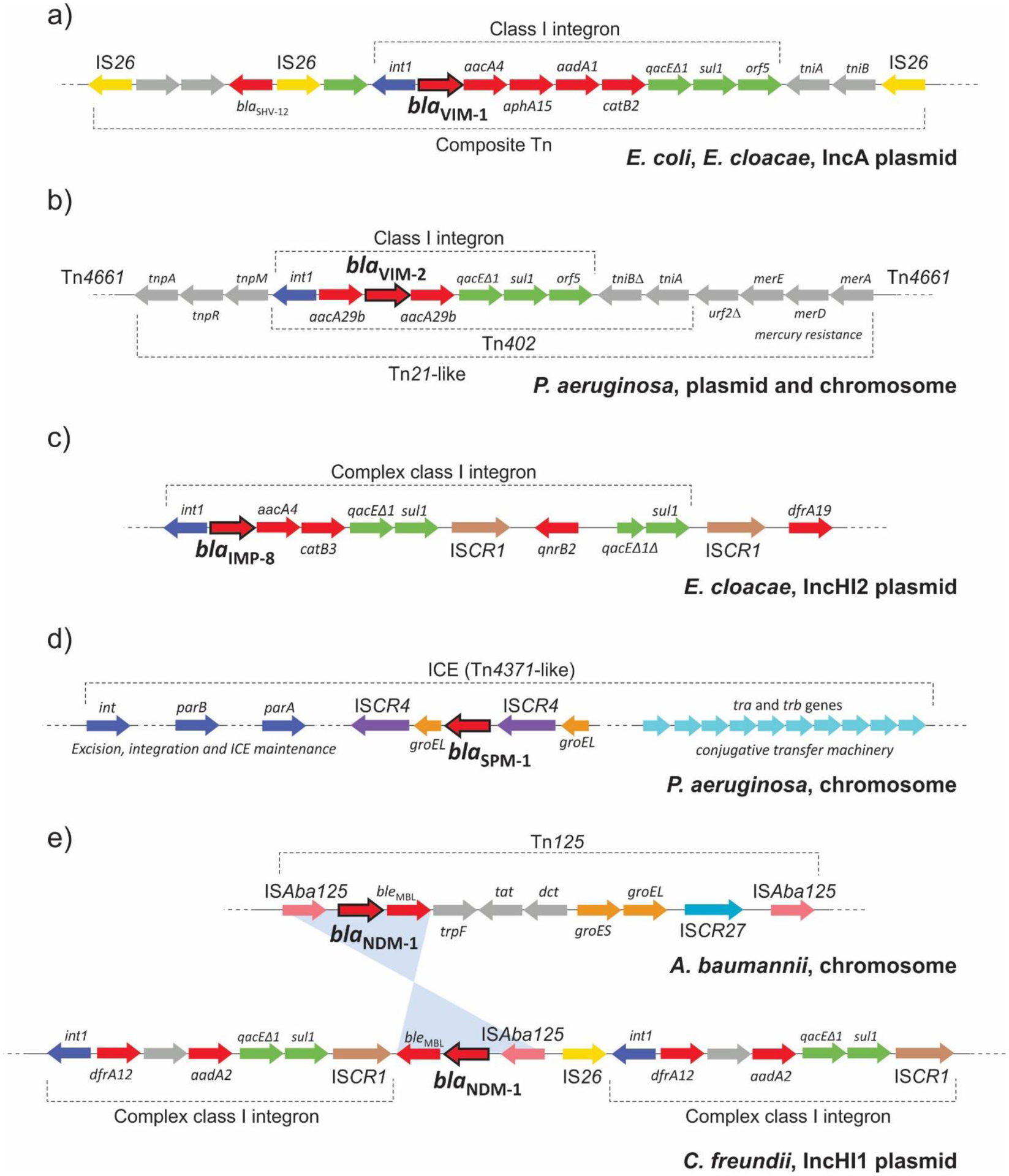
Examples of genetic contexts of blaVIM-1,1043 blaVIM-2,1040 blaIMP-8,1044 blaSPM-1461 and blaNDM-1477, 1045 genes. Resistance genes are indicated as red arrows, and MBL genes are highlighted with bold outlines.
Most clinical blaMBL genes, including those coding for IMPs, VIMs, GIMs, TMB-1, DIM-1, GIMs and DIM-1, are integrated in class I integrons as resistance cassettes, generally in combination with other resistance genes (http://integrall.bio.ua.pt/).1046 The same blaMBL gene may be encountered in different arrays of gene cassettes, and at variable positions with respect to the attI1 site, although in most cases blaMBL genes occupy the first or second position. In the case of VIMs, there are 202 different integron structures deposited at the INTEGRALL database, 48 of which correspond to VIM-1 and 105 to VIM-2. blaMBL genes generally co-exist with aminoglycoside resistance genes (aacA, aadA, aadB, aphA), trimethoprim resistance genes (dfrA, dfrB), chloramphenicol resistance genes (catB3, catB4, catB6) and/or other β-lactamase genes of the OXA type, with aacA4 being the most dominant. Other less frequent genes include blaPSE-1, arr (rifampicin resistance), qac (quaternary ammonium salts resistance) and tnpA (transposase). In many cases orf5 from 3’-CS is absent or there are sequence duplications. ISs have also been found inserted at different integron locations. For instance, an IS26 replacing the 3’-CS region was observed in K. pneumoniae harboring blaVIM-1, and a IS1394 flanked by two aadA1 cassettes was detected in some integrons carrying blaGIM-1. blaIMP-1 is generally associated to class I integrons, but it has also been found in a class 3 integron in S. marcescens. Similarly, blaIMP-27 has been identified in a class 2 integron in P. mirabilis and blaIMP-8 in four complex class I integrons in E. cloacae.417 Another MBL gene associated to complex class I integrons is the B3 blaSMB-1, identified in S. marcescens.557 Integrons carrying blaMBL genes were found in resistance plasmids and to a lesser extent in bacterial chromosomes, in many cases embedded in more complex mobile resistance modules. Indeed, two endemic strains of VIM-2 producing P. aeruginosa from Dutch hospitals were found to carry an almost identical mobile resistance module in the chromosome and in a broad-spectrum conjugative plasmid.1040 In this ~30 kb structure, blaVIM-2 is part of a class I integron (together with aacA) embedded in an immobile Tn402 harboring an incomplete transposition module. This element is in turn embedded in a Tn21-like harboring mercury-resistance genes and lacking the terminal IRR, thus being unable to transpose. The latter is contained within a disrupted Tn4661 harboring a complete Tn4661. Three copies of this module were found at different positions in the chromosome, indicating that it may be a replicative transposable element. blaVIM-2 has also been found in other types of plasmids like pNOR-2000 (not conjugative),423 and in an E. coli plasmid of the IncF group.1047 Similarly, class I integrons containing blaVIM-1 were identified in broad-spectrum plasmids of the IncN and IncA/C groups embedded in other mobilizable MGEs.1043, 1048, 1049 For instance, an IncA/C plasmid from K. pneumoniae was shown to carry a blaVIM-1-containing integron flanked by various copies of IS6, thus forming a composite transposon.1049 Similarly, blaVIM-1-containing integrons were identified embedded in IS26 composite transposons in several IncA plasmids from Enterobacterales.1043 In the case of IMPs, integrons harboring blaIMP-8, blaIMP-4 or blaIMP-6 were identified in IncHI2,1044 IncL/M1050 or IncN1051 plasmids, respectively, among others. As a final example, the blaTMB-1 gene was found in a class 1 integron together with aacA4 and blaOXA-4 located in the chromosome.369
Other acquired blaMBL genes, however, are not associated to integrons. These include those coding for NDMs, KHM-1, SPM-1, AIM-1 and SMB-1.1046 For instance, blaKHM-1 was identified in an IncA/C resistance plasmid from a K. quasipneumoniae isolate (which also harbored other plasmids from different Inc groups) located proximal to ISEc68 and an integrase gene. This plasmid (pTMSNI47-1) also carried blaCTX-M-2, blaDHA-1 and blaOXA-10.1052 The same immediate genetic context of blaKHM-1 was found in a different plasmid from C. freundii (pKHM-1).370
The blaSPM-1 gene, disseminated throughout Brazil by the persistent MDR Pseudomonas aeruginosa clone ST2772 has been identified in the bacterial chromosome embedded in an ICE of the Tn4371 family, which includes broad-host-range ICEs widespread among β- and γ-proteobacteria.461 Although the GC content of the ICE is similar to that of the host, the elemental module containing blaSPM-1 appears to have been incorporated from another source. blaSPM-1 is immediately flanked by 148 bp direct repeats, which, in turn, are flanked by ~2.3 kb DRs composed of ISCR4 and part of the groEL gene (groEL-ISCR4-blaSPM-1-groEL-ISCR4). The groEL sequence upstream of ISCR4 encodes a predicted amino acid sequence with 89% and 87% identity with groEL proteins from Desulfitobacterium hafniense and S. maltophilia, respectively, while the groEL downstream of blaSPM-1 encodes a protein with 73% identity to the groEL protein of Xanthomonas campestris. According to the mechanism of ISCRs replication, this structure suggests that blaSPM-1 was originally mobilized from the chromosome of an environmental bacterium by ISCR4 and introduced by homologous recombination through groEL.1015 Homologous recombination through the direct repeats surrounding blaSPM-1 can lead to gene loss, as observed in one isolate.461
The blaNDM-1 gene has received considerable attention due to its rapid dissemination, and sequences of several plasmids and regions carrying this gene are available.1053 Although blaNDM genes have been found in a variety of genetic contexts, the elemental modules associated to these genes are conserved: an ISAba125 insertion sequence intact or truncated is upstream of blaNDM providing the -35 region of the gene promoter, while a bleomycin resistance gene (bleMBL) is downstream.471 Other genes can be occasionally found downstream of bleMBL, such as trpF (encoding a phosphoribosylanthranilate isomerase), tat (encoding a twin-arginine translocation pathway signal sequence domain protein), dct (encoding a periplasmic divalent cation tolerance protein), and groES-groEL (encoding chaperonin), and an ISCR3-like element, ISCR27. The current hypothesis suggests that the blaNDM-1 gene originated from an unknown environmental bacterium, integrated into the chromosome of A. baumannii, which acted as an intermediate reservoir, to then disseminated by HGT to Enterobacterales and P. aeruginosa.1054 NDM-1-producing A. baumannii harbors another copy of ISAba125 downstream of ISCR27, and the two ISAba125 elements form the composite transposon Tn125. This transposon has been proposed to transfer blaNDM-1 onto broad-host range plasmids responsible of HGT. In fact, its transposition functionality was evidenced in A. baumannii.1055 Initial capture of blaNDM-1 may have been promoted by ISCR27 by the progressive acquisition of DNA segments with different origins at their terIS end, starting with Xanthomonas-like groES-groEL genes (see above). It has been proposed that the segment from blaNDM-1 to ISCR27 was then inserted by rolling-circle inside the aph6 gene (aminoglycoside resistance gene) typically located downstream of ISAba125 in Acinetobacter spp. In fact, the 100 bp region between ISAba125 and blaNDM-1 together with the first 19 bp of blaNDM-1 (encoding the first 6 amino acids of NDM-1) aligned perfectly with the N-terminus and upstream sequence of aphA6.477 Then, the second copy of ISAba125 inserted downstream of ISCR27 to form Tn125. The blaNDM-1-carrying element Tn125 has been interrupted or truncated in Enterobacterales to generate a variety of complex genetic contexts for blaNDM.471 This interruption is largely due to the insertion of many other mobile genetic elements (e.g., IS1, IS5, IS26, IS903, ISEc33, and ISKpn14, etc.) and recombination. The remnants of Tn125 have also been observed in composite transposons formed by two copies of the same insertion sequence, such as IS26 and others, duplicated or mobilized by association with ISCR1 complex class I integrons or MITEs. Other blaNDM variants are associated to similar elemental modules suggesting that they emerged from blaNDM-1 via nucleotide mutations. As with blaSPM-1, MGE or recombination activity can lead to blaNDM-1 loss. Finally, the blaNDM gene has been found principally associated to resistance plasmids, although occasionally found on bacterial chromosomes from P. aeruginosa and Enterobacterales, in addition to A. baumannii.472, 1056, 1057 A wide variety of blaNDM-carrying plasmids have been reported, indicating multiple events of blaNDM-module acquisition and various mechanisms of HGT. These belong to different Inc groups, such as narrow-host-range (Enterobacterales) IncX3 and IncFII, and broad host-range IncC, among the most frequent.471 Besides the traditional HGT mechanisms, it has been recently shown that A. baumannii is capable of intra and inter-species transfer of a resistance plasmid harboring blaNDM-1 gene via OMVs.976 Vesicles released by this strain were able to transform carbapenem-sensitive A. baumannii and E. coli with high transformation frequency. The sizes of the plasmids in the transformants and their restriction digestion patterns were identical to the plasmid in donor A. baumannii.
9.6. Maintenance of blaMBL genes
Acquired blaMBL genes are associated to a wide variety of MGEs and arrays of resistance modules. These genetic structures appear to be similarly efficient in mobilizing resistance genes between different DNA molecules, acquiring other MGEs, etc., as well as in disseminating among different bacteria. However, the extent of dissemination and host specificity displayed by acquired MBLs is highly variable, and this cannot be explained solely based on their genetic contexts. For instance, while blaNDM-1 has rapidly spread all over the world in members of Enterobacterales and non-fermenters, other previously identified blaMBL genes, such as those coding for VIM-2 and SPM-1, remained mostly confined to P. aeruginosa.455, 1058 Similarly, inside the VIM family, although blaVIM-1 and blaVIM-2 are part of class I integrons, and thus exhibit the same potential of mobilization, blaVIM-1 has disseminated over a broader range of hosts including Enterobacterales and non-fermenters.429, 1058, 1059
The success of HGT of resistance genes eventually depends on the heritability of the transferred sequences in the recipient microorganism. Rare events of HGT are unlikely to establish in a larger bacterial population if they do not confer a benefit to the host. In cases where acquisition of a blaMBL gene affects negatively the growth rate of the new host, bacteria harboring the gene will be lost from the larger population as they will be outgrown by members of the population displaying better fitness. However, there may be cases in which the acquired gene and the bacterial host co-adapt by compensatory mutations, so that the initial fitness cost is reduced or even lost.996, 997, 1060 In cases where expression of the blaMBL gene does not result in an initial fitness cost, the maintenance of the gene over time will depend on the frequency with which the cells are exposed to antibiotics, since spontaneous deletions may occur as discussed above for blaNDM-1 and blaSPM-1. In the presence of antibiotics, the blaMBL gene confers an advantage to the new host, and a few HGT events will be sufficient for establishing this resistance determinant in the new host.998 blaMBL-containing cells will rapidly expand their population sizes to become dominant, outcompeting drug susceptible cells.
Protein determinants play a key role in the maintenance of acquired blaMBL genes in a bacterial population, as recently shown by López, Vila and co-workers.430 Indeed, protein expression (instead of MGEs) shapes the dissemination and host-specificity of these enzymes. NDM-1 is widely spread in Enterobacterales and non fermentors, while VIM-2 is preferably expressed by P. aeruginosa, and SPM-1 is almost strictly confined to P. aeruginosa. These distinct host specificities cannot be explained based on the resistance phenotypes, since all three MBLs are able to confer antibiotic resistance when expressed either in E. coli, A. baumannii or P. aeruginosa. However, when expressed under permissive conditions, SPM-1 and VIM-2 elicited a high fitness cost compromising bacterial growth in E. coli and A. baumannii, but not in the frequent host P. aeruginosa.430, 1061 VIM-2 production was also shown to generate a substantial fitness cost in S. Typhimurium.1061 NDM-1, instead, did not induce any fitness cost in all bacterial hosts, revealing that blaNDM-1 is tailored to be successfully maintained in a broad range of hosts. The fitness cost of expressing SPM-1 and VIM-2 in non-frequent hosts is due to an inefficient processing of their signal peptides, resulting in an accumulation of their precursor proteins in the periplasm, which become toxic and cause an envelope stress. The saturation of the protein export machinery is thus responsible for the fitness cost triggered upon expression of these enzymes. This was evidenced by the activation of mechanisms of envelope stress relief, such as a hypervesiculation phenotype (see below) and the activation of the periplasmic housekeeping protease DegP. Recent studies in A. baumannii showed that the global regulator H-NS is also involved in the relief of envelope stress originated by expression of VIM-2 and SPM-1 in this host.1062 Instead, expression of VIM-2 or SPM-1 in the frequent host P. aeruginosa, or NDM-1 in the three bacteria does not lead to precursor accumulation nor envelope stress response.
This phenomenon is not exclusive to MBLs; previous studies have shown that SBLs such as SME-1 or AmpC generates a fitness cost under permissive conditions when expressed in non-frequent hosts.1063, 1064 In the particular case of SME-1 carbapenemase, Marciano et al. reported that the signal peptide of this enzyme is responsible for the fitness cost associated to protein expression.
The importance of the signal peptide sequences in the dissemination of MBLs was further demonstrated by swapping of signal peptides.430 Expression of a VIM-2 variant with the signal peptide of broad-host-range soluble MBLs (such as VIM-1 or IMP-1) abolished the growth defects observed in E. coli and A. baumannii. Similarly, the natural allele VIM-12, which is a VIM-1/VIM-2 hybrid that results from the N-terminal sequence from VIM-1 (including the signal peptide) and the C-terminus from VIM-2, has been reported only in Enterobacterales.1065 Given that VIM-1, VIM-2 and VIM-12 genes share similar genetic contexts, it stems that the signal peptide is what most determines the host specificity. Thus, signal peptides sequence can potentially be used to predict the host specificity and possible spread of new MBL alleles.
VIM-2 has been occasionally found in Enterobacterales, probably as a result of host co-adaptation.1066, 1067 SPM-1, instead, is virtually strictly confined to P. aeruginosa.455 In fact, the substrate profile of SPM-1 is exquisitely adapted to meet the host requirement, i.e., it is optimized for hydrolyzing anti-pseudomonal β-lactam antibiotics.464 However, when expressed at basal levels, SPM-1 was able to confer carbapenem resistance to A. baumannii without affecting bacterial growth. This may explain the rare cases in which SPM-1 can be encountered in Acinetobacter spp.1062
Production of OMVs is a mechanism that helps alleviate envelope stress by eliminating toxic or misfolded proteins from the periplasm of Gram-negative bacteria. This is the case for VIM-2 and SPM-1 in non-frequent hosts, which are released in vesicles as misfolded proteins.430 Instead, in P. aeruginosa, in the absence of stress, these enzymes are not packaged into OMVs. On the contrary, NDM-1 is tailored to be selectively secreted into OMVs of a wide variety of hosts, in a folded active form, without generating an envelope stress. Secretion of NDM-1 into vesicles has been proposed as a mechanism that has contributed to the dissemination of the blaNDM gene. This secretion implies an advantage to the organism under the presence of antibiotics by disseminating the carbapenemase activity beyond the bacterial cell, thus titrating the available antibiotic at the infection site. At the same time, these vesicles act as ‘care packages’ protecting communities of otherwise susceptible bacteria, favoring the opportunity of uptaking the blaNDM gene by any of the HGT mechanisms described above, including the same vesicles transporting NDM protein and plasmidic blaNDM. This could be crucial in polymicrobial infections, resulting in lethal treatment failure. This was recently validated using Galleria mellonella as an infection model (Martinez et al., submitted) (see Section 8.3).
Overall, the faster and wider dissemination of NDM-1, compared to other MBLs, stems from an optimized leader peptide sequence leading to multiple advantages. First, it allows expression of the enzyme in wide variety of hosts without producing a fitness cost under permissive conditions, thus reducing the loss frequency of acquired blaNDM-1 in bacterial populations. Second, the lipidation signal localizes the enzyme in the outer membrane favoring its secretion into OMVs from different hosts. As discussed in Section 10, anchoring to the outer membrane protects the enzyme from degradation by periplasmic proteases in conditions of zinc limitation, as those encountered during infection. Secretion into OMVs contributes to short-term survival of polymicrobial communities exposed to β-lactam antibiotics while augmenting the probability of HGT.
The strict regulation of antibiotic use has been considered as a strategy to reduce antimicrobial resistance, with the assumption that resistance genes impose a fitness cost to the host and the absence of the antibiotic pressure will lead to their loss. In the case of NDMs, however, reverting resistance development is expected to be challenging due to the lack of fitness cost exerted by acquisition and expression of blaNDM, and potential plasmid-host coevolution mechanisms guaranteeing plasmid maintenance.1060
9.7. Origin of acquired blaMBL genes
Acquired MBLs are thought to have emerged from the pool of intrinsic MBLs present in environmental bacteria. Several of these MBLs have been characterized biochemically, including BcII, JOHN-1, BlaB and TUS-1, among others.17, 20, 23 These endogenous enzymes are generally chromosomally encoded and exist in specific bacterial species, most of them belonging to Bacteroidetes or Proteobacteria phylum. The catalytic efficiencies of these enzymes are close to or equal to those reported for acquired MBLs, indicating that these bacteria with intrinsic MBLs are potential reservoirs for future acquired enzymes of clinical impact if mobilized by MGEs. Metagenomic studies on different environmental niches revealed that intrinsic MBLs are much more widespread and diverse than previously expected. Gudeta, Guardabassi and co-workers demonstrated that MBL producers are widespread in soil samples, and that although the MBLs produced by these bacteria are distantly related to MBLs identified in clinical samples, these enzymes can potentially become resistance determinants of clinical relevance if acquired by pathogenic bacteria.530 Reconstruction of resistance genes directly from shotgun metagenomic data also showed that MBLs are abundant in oil-contaminated deep-sea metagenomes and human microbiome.1068 On the other hand, 27 unique MBL genes were identified in hospital wastewaters from the city of Mumbai, India, containing fecal material from a large number of individuals, many of them under antibiotic therapy.1069 In general, these studies revealed the presence of novel MBLs mostly related to previously characterized intrinsic B1 and B3 MBLs, and (to a minor extent) B2 MBLs.
Recent bioinformatics approaches based on Hidden Markov Models were used to search for MBLs within bacterial genomes and plasmids in the NCBI databases as well as metagenome data from human and environmental bacterial communities.221, 467 A set of 2290 unique blaMBL genes were predicted, divided into 817 gene families, most of them previously uncharacterized. Some of these novel MBLs were experimentally confirmed to confer imipenem resistance or reported to be located within a genetic context that suggests they may be mobile.467 Based on phylogenetic analysis, Kristiansson and co-workers proposed an updated classification of MBLs in which subclass B1 and subclass B3 enzymes are divided into five and four monophyletic groups, respectively, where each group contains at least one previously validated MBL (Table 4).221, 467 In agreement with previous studies, only 6 families belonging to subclass B2 were identified, indicating that B2 enzymes are scarce compared to B1 and B3 MBLs. All groups reflect, to a large extent, the taxonomy of the species carrying the MBLs, indicating that the great majority are not mobile or have a restricted host range. Groups B1.1 and B1.2 are dominated by Proteobacteria while B1.3 and B1.4 by Bacteroidetes and Firmicutes to a minor extent in B1.4. In contrast, the groups for B3 do not correlate as well with taxonomy: the first three groups (B3.1–B3.3) all contain genes from multiple phyla although B3.1 is dominated by genes from Proteobacteria, B3.2 by Bacteroidetes and B3.3 by Acidobacteria. The B3.4 group is further divided into six clades (a–f) and most identified species are from the phylum Proteobacteria. Importantly, the vast majority of the mobile MBLs commonly encountered in clinical settings belong to the B1.1, B1.2 and B3.4 groups, suggesting that they have originated from bacterial species within Proteobacteria.
Table 4.
Proposed subdivisions of MBL subclasses B1 and B3 according to Berglund et al.221 The main phyla from which MBLs were isolated and representative enzymes with experimentally confirmed β-lactamase activity are indicated for each group.
| Group | Main phyla (in order of abundance) | Representative MBLs with experimentally confirmed activity |
|---|---|---|
| B1.1 | Proteobacteria | NDM, VIM, FIM |
| B1.2 | Proteobacteria | IMP, GIM, DIM, SIM, KHM, TMB |
| B1.3 | Bacteroidetes | CcrA |
| B1.4 | Bacteroidetes, Firmicutes | BcII, BlaB, JOHN, IND, MUS, TUS |
| B1.5 | Firmicutes, Proteobacteria, Spirochaetes | SPM, SPS |
| B3.1 | Proteobacteria, Firmicutes | CAR |
| B3.2 | Bacteroidetes, Proteobacteria, Acidobacteria | GOB, LRA3, PEDO-1, PEDO-2 |
| B3.3 | Acidobacteria, Proteobacteria | FEZ |
| B3.4a | Proteobacteria | BJP, CAU |
| B3.4b | Proteobacteria | L1 |
| B3.4c | Proteobacteria | AIM, SMB, LMB |
| B3.4d | Proteobacteria | THIN-B |
| B3.4e | Gemmatimonadetes | RM3, LRA2 |
| B3.4f | Proteobacteria | - |
In the case of the widespread B1 enzyme NDM-1, although upon its discovery it showed the greatest similarity to VIM-2, with 32% sequence identity, newer B1 MBLs were identified exhibiting closer homology. The first of them was FIM-1, with 40% sequence identity, which is also an acquired MBL, in this case associated to an ISCR19-like element.367 Intrinsic B1 MBLs related to NDM-1 were later identified from environmental sources, such as MYX-1 from Myxococcus xanthus and ANA-1 from Anaeromyxobacter sp., and most importantly ElBla2-1 from marine bacterium Erythrobacter litoralis, which displays the highest homology to NDM-1 (56% sequence identity).467, 468 Furthermore, ElBla2-1 also displays a putative lipidation signal within its signal peptide. This suggests that the precursor of the NDM family may have already been a lipoprotein, and could have originated in marine bacteria before being mobilized and beginning its spread among clinical pathogens.
The lack of clinically relevant MBLs from phyla different from Proteobacteria is likely a result of the many barriers that prevent genes from being mobilized, transferred and/or maintained within pathogens, including the host-range of MGEs, a lack of ecological connectivity between hosts, non-optimal codons and/or signal peptides generating toxic precursors as demonstrated for VIM-2 and SPM-1.221, 430 In addition to taxonomy, the classification proposed by Kristiansson and co-workers reflect variations in zinc binding sites. For instance, in B1 enzymes, the H116R/C221N substitution is present exclusively in B1.2 (in 2.5% of gene families) while H116G in B1.5 (in 31% of genes families). In B3 enzymes, H116Q substitution is present in B3.2 (75%), B3.3 (19%) and B3.4 (2%) while H116E and H118R/H121Q/H263K in B3.4 (10% and 3% respectively). These variations in active-site ligands might also be related to specific host requirements.
Finally, bioinformatic studies applied on databases from marine microbial communities revealed a high diversity of distant MBL homologs related to the intrinsic B1, B2, and B3 subclasses.1070 This is expected considering that low-impacted/pristine aquatic environments, such as oceans, are unlikely the source of MBLs currently emerging in clinics. The majority of them are related to the chromosomally encoded B3 GOB present in Elizabethkingia genus; only a reduced number are related to acquired MBLs (VIM, SPM-1, and AIM-1). Similar results were obtained in a functional metagenomics analysis of a remote Alaskan soil, considered undisturbed by human activity; all of the recovered MBLs (LRA2, LRA3, LRA7, LRA8, LRA12, LRA17, LRA19) fell into the B3 subclass.535
All in all, MBLs present in the environmental resistome are far more diverse than what is typically encountered in clinical settings, indicating that the pool for the recruitment of novel potentially clinically relevant MBLs is enormous.
10. Zn(II) homeostasis at the host-pathogen interface: Stability and Metal uptake of MBLs in conditions of Zn(II)-limitation
As described in detail in Section 7.1, MBLs are synthesized in the cytoplasm and exported as unfolded polipeptides by the Sec system into the periplasmic space of Gram negative bacteria. Once processed and released into the periplasm as mature proteins, MBLs fold and bind the Zn(II) ions to become fully active carbapenemases. Thus, the activity of MBLs depends on the availability of Zn(II) in the periplasm. Here we will describe the major processes modulating the pool of periplasmic Zn(II) ions, and how they affect the performance and evolution of MBLs. We will discuss in particular the scenario upon a bacterial infection, in which the host immune system sequesters metal ions with the aim of impeding bacterial growth.
10.1. Regulation of metal homeostasis in bacteria
Zn(II) is ubiquitous in life and is required for the structure and/or function of thousands of metalloproteins. Indeed, Zn(II) proteins can represent from 4 to 10% of different proteomes.629, 1071, 1072 In bacteria, Zn(II) is an essential micronutrient required for survival and proliferation. Despite being essential for many bacterial cellular processes, this metal ion is also toxic to the cell if present in high concentrations.1073 In the environment, bacteria may be exposed to quite diverse Zn(II) concentrations, ranging from Zn(II)-limited environments to highly toxic Zn(II) levels. Therefore, to be able to survive in these conditions, both vertebrate hosts and bacterial pathogens have evolved processes to control intracellular Zn(II) homeostasis.
The bacterial response to changes in metal availability is primarily mediated by metalloregulatory proteins, which generally sense changes in cellular metal concentrations and alter the gene expression of metal homeostatic systems.1073–1075 These metalloregulators are widely distributed in bacteria, and they respond to metal limitation, metal intoxication, or both conditions. Homeostatic systems involve mechanisms of Zn(II) acquisition, efflux or intracellular redistribution.
As a result, the levels of available Zn(II) are variable and also difficult to measure. This has led to several misunderstandings that extrapolate the metal loading of MBLs in the test tube to the situation in the periplasm, which offers a different scenario. In addition, comparison of in vitro data is not straightforward since these studies rely on the measurement of affinity constants of the enzymes towards Zn(II) by using different methods which do not provide comparable figures.
10.2. Nutritional immunity
Vertebrate immune systems have evolved sophisticated defense mechanisms to protect themselves against invading pathogens. A primary line of host defense is to sequester and starve invading pathogens from nutrient trace minerals, which are essential for growth within the host.312, 1076–1078 This process, termed nutritional immunity, is a fundamental part of the host innate immune response against many pathogens. The relationship between trace minerals and immunity was first elucidated for iron, in which transferrin, an iron-binding protein in egg whites and human plasma, was found to sequester iron from pathogens preventing microbial growth.1079, 1080 The concept of nutritional immunity has now expanded to host-mediated sequestering of Zn(II) as a defense against invading pathogens.312
Zn(II) is the second most abundant trace metal in humans, with 2–4 g of Zn(II) distributed throughout the body in various tissues.1081, 1082 In the bloodstream, serum Zn(II) (approx. 15 μM) is mostly bound to albumin, transferrin, and α2-macroglobulin, but remains available to transporters to balance Zn(II) levels within cells. The primary regulators of mammalian Zn(II) transport are the Zrt-Irt-like Protein (ZIP) Zn(II) importers and the Zn(II)-transport (ZNT) exporters.1083, 1084 In addition to direct import and export of Zn(II), intracellular Zn(II) sequestration is also required to prevent toxicity. A major component of Zn(II) chelation within eukaryotic cells occurs through the action of metallothionein and glutathione.1085–1087 These are cysteine-containing molecules that link reversible Zn(II) binding with the cellular redox state. reversible Zn(II) binding allows these molecules to serve as components of the intracellular Zn(II) buffering system that includes the potential delivery of Zn(II) to the apo forms of metalloenzymes.
During infection and inflammation, the host limits the availability of Zn(II) in diverse ways. In a more general response to invading pathogens, during the acute phase of inflammation the liver increases the levels of metallothionein and the zinc importer ZIP14, in a process mediated by proinflammatory cytokines such as IL-6, with the aim of reducing the availability of Zn(II) in the plasma.1088 In this case, ZIP14 sequesters circulating zinc in the liver during infection/inflammation and metallothionein binds the resulting intracellular zinc in hepatocytes. In a more localized response, nutritional immunity acts by limiting Zn(II) ions directly at the immediate environment of bacterial pathogens.1073 Indeed, Zn(II) may be actively removed from the lysosome of immune cells as a strategy to limit nutrient Zn(II) availability to pathogens trapped within this cellular compartment. Conversely, extracellular Zn(II) may be mobilized inside immune cells to impart toxicity. However, the most crucial mechanism of Zn(II) limitation involves the sequestration of extracellular Zn(II) by members of the S100 protein family, of which calprotectin (S100A8/S100A9) plays a key role in pathogen clearance during the innate immune response.312, 1073
10.2.1. S100 family of proteins. Calprotectin
The process of acute inflammation (the initial response of the body to harmful stimuli) is initiated by resident immune cells already present in the involved tissue, mainly resident macrophages, dendritic cells and mast cells, among others. These cells possess surface receptors known as pattern recognition receptors (PRRs), which recognize two subclasses of molecules: pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs).1089 PAMPs are compounds that are associated with various pathogens (and OMVs derived from them), but which are distinguishable from host molecules.1089, 1090 These include bacterial cell-wall components such as lipopolysaccharide. DAMPs or alarmins, instead, are endogenous and released upon host-related injury and cell damage. At the onset of an infection, these cells undergo activation (PRRs recognize PAMPs or DAMPs) and release inflammatory mediators responsible for the clinical signs of inflammation, which involves an increased movement of plasma and granulocytes (leukocytes containing granules) from the blood into the injured tissues. Neutrophils are the most abundant type of granulocytes (40% to 70% of the leukocytes in humans) and the major components of acute inflammation (Figure 67-a).1091 These cells play a key role in the first line of host defense against pathogens and are the main phagocytic tool used by the immune system to eliminate pathogens. During an infection, neutrophils rapidly move from the bloodstream to inflamated tissues where they eliminate invading microbes. After initial rolling, contact of blood-circulating neutrophils with activated endothelium close to the site of infection makes these cells arrest and adhere firmly to the vessel wall. Further activation of adherent neutrophils allows them to migrate across the endothelial cell layer and the basal membrane (extravasation), and then through the extracellular matrix thanks to secreted digestive enzymes. Neutrophils infiltrate the connective tissue and are attracted by a gradient of chemotactic factors, created by the local activated immune cells (such as interleukin-8, IL-8, and complement peptides), to the site of inflammation where they kill and degrade invading microorganisms. During this process, neutrophils express and release cytokines, which in turn amplify inflammatory reactions recruiting and activating other cells of the immune system.
Figure 67.
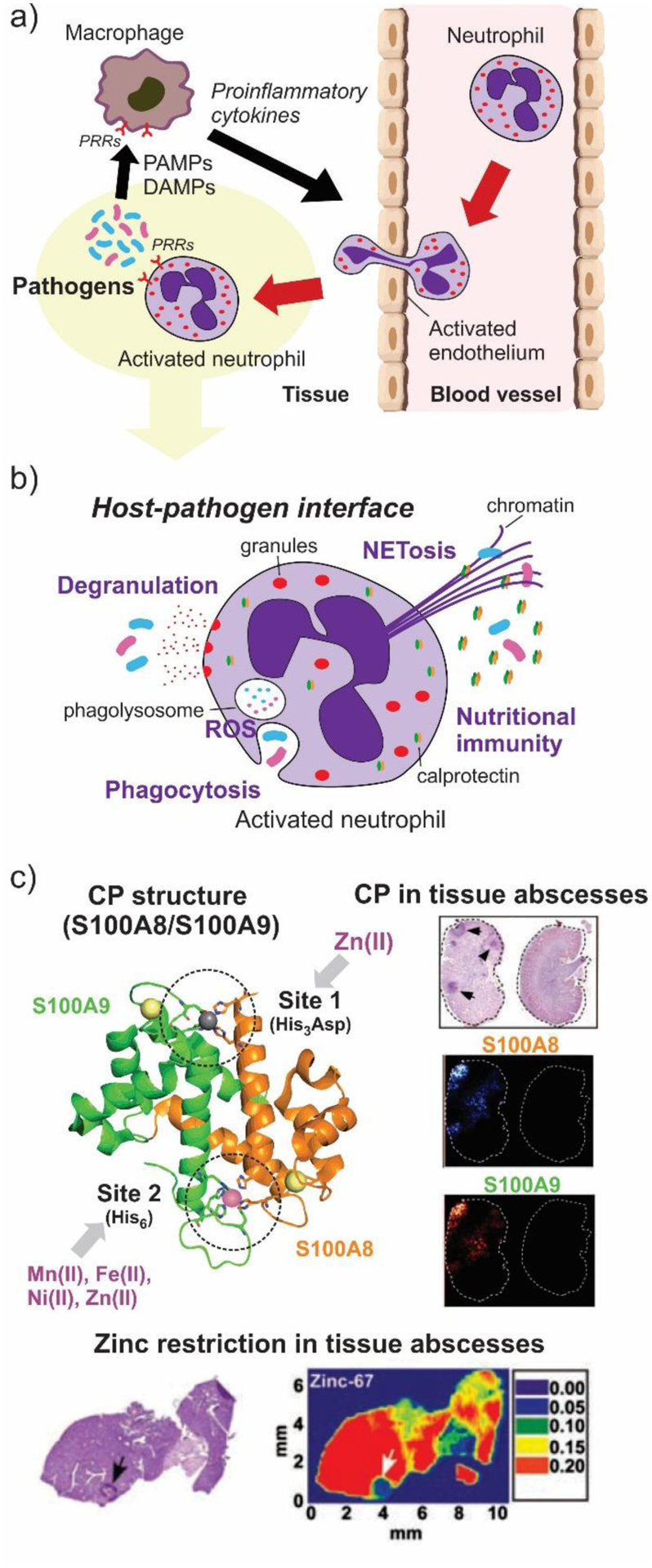
The action of neutrophils in bacterial infections. a) Migration to the sites of infection. b) Mechanisms of pathogen clearance. c) Metal limitation at sites of infection through secretion of calprotectin. Adapted with permission from reference311 Corbin, Skaar et al. Copyright 2008 American Association for the Advancement of Science.
Neutrophils have diverse methods for directly attacking microorganisms: a) phagocytosis, in which neutrophils engulf and destroy small bacteria in phagolysosomes, b) degranulation, in which multiple cytotoxic and proteolytic proteins stored in membrane-enclosed cytoplasmic granules are released extracellularly or into the phagolysosomes, c) NETosis, i.e, generation of neutrophil extracellular traps (NETs) - large extracellular web-like structures composed of cytosolic and granule proteins assembled on a scaffold of decondensed chromatin - aimed to trap, neutralize and kill microorganisms, d) release of reactive oxygen species (ROS) extracellularly or into the phagolysosomes, and e) nutritional immunity by the S100 family of proteins (Figure 67-b).1091, 1092 These processes are generally triggered when pattern recognition receptors and opsonin receptors, located in the surface of neutrophils, recognize PAMPs and opsonins (i.e., plasma-derived antibodies or complement C3b coating the invading microorganism), respectively, in addition to proinflammatory cytokines such as IL-8.
S100A8 (also named calgranulin A; myeloid-related protein 8, MRP8), S100A9 (calgranulin B; MRP14) and S100A12 (calgranulin C; extracellular newly identified RAGE binding protein, EN-RAGE) are among the major proteins secreted by activated neutrophils at the site of infection.1093 These cytosolic proteins, belonging to the calcium-binding S100 protein family, are specifically linked to innate immune functions by their expression in cells of myeloid origin, where they are intensely produced and secreted during infection.1094 While S100 proteins have multiple intracellular functions, most are secreted into the extracellular milieu and bind PRRs receptors which generally results in the initiation and upregulation of an inflammatory response. In contrast to what is considered an ancestral proinflammatory response characteristic of nearly all S100 subgroups, the calgranulin subgroup, composed of S100A7, S100A8, S100A9 and S100A12 in humans, has recently evolved a unique antibacterial activity defined by sequestration of transition metal ions, while also retaining pro-inflammatory activity.312
S100A8 and S100A9 are expressed to high levels in neutrophils, representing close to 40% of the cytoplasmic proteins, and less abundantly in monocytes and early differentiation stages of macrophages.1095 Their expression can also be induced in keratinocytes and epithelial cells under inflammatory conditions. In contrast, S100A12 is more restricted to granulocytes and represents ca. 5% of the cytoplasmic proteins. S100A8 and S100A9 form a heterodimer in the cytosol of neutrophils, known as calprotectin (CP, Figure 67-c), which forms a protease-resistant heterotetramer in the extracellular milieu after secretion or cell necrosis.1096 Extracellular concentrations of CP can be as high as 1 mg/ml at the infection sites, especially in tissue abscesses composed mostly of dead neutrophils. Although the mechanism is not completely understood, it has been proposed that neutrophils secrete CP through phagocyte activation by a Golgi-independent pathway, NETosis (both as a soluble protein or bound to NETs), and/or through neutrophil death in which the cytoplasmic content is released.1093, 1097, 1098 In the extracellular form, (S100A8/S100A9)2, CP acts as a potent metal-chelating host-defense protein, which starves the pathogens from Zn(II) and other first-row divalent cations acting as an extracellular chelator.1096 Similarly, S100A12 homodimers were shown to chelate Zn(II) and Cu(II) although its broader contribution to immunity has been difficult to define since it is not present in mice.1099 Finally, S100A7 (known as psoriasin) is a homodimeric protein produced by keratinocytes which acts as a skin antimicrobial agent by sequestering Zn(II).1100
Early works on CP disclosed a link between metal chelation and its antimicrobial activity. CP is a broad-spectrum inhibitor of bacterial and fungal growth, and its antimicrobial activity can be reversed by supplementation with extra Zn(II) or by mutation of its transition metal-binding sites.1101–1103 Purified CP inhibits the growth of multiple species in vitro, including E. coli, Candida albicans, S. aureus, K. pneumoniae, P. aeruginosa, A. baumannii and S. typhimurium, among others.1095 A unifying feature among S100 proteins is that they form dimers and may form transition metal binding sites at the dimer interface (Figure 67-c).312, 1096, 1104 Both S100A8 and S100A9 in human CP possess two EF-hand Ca(II)-binding domains. Apo CP, without bound Ca(II), is a S100A8/S100A9 heterodimer of low affinity for transition metals. Given the relatively low Ca(II) ion levels in the cytoplasm of neutrophils (nM concentrations in a resting cell), CP heterodimer is likely to be an abundant intracellular species. Calcium binding to apo CP triggers association of two heterodimers to form a (S100A8/S100A9)2 heterotetramer that is resistant to extracellular proteases and exhibits increased affinity for transition metals (from nM or sub-nM in apo-CP to sub-pM in the case of Zn(II) binding to site 1 in Ca(II)-CP). This occurs upon release of CP into the extracellular environment, containing high levels of Ca(II) (~2 mM), which potentiates the growth inhibitory activity of CP. Each CP heterodimer has two transition metal–binding sites that form at the S100A8/S100A9 interface: a His3Asp motif (site 1) selective for Zn(II) (also conserved in S100A12 and S100A7), and a unusual His6 motif (site 2) that endows CP with its functional versatility. This site captures Mn(II) and other divalent metal ions, including Fe(II), Ni(II), and Zn(II).1096 Given its high extracellular levels, CP is usually in excess compared to the concentration of bioavailable metal ions at infection sites. Thus, all bioavailable divalent transition metal ions present at an infection site may be sequestered by CP. In fact, the two metal-binding sites were shown to be important for the antimicrobial activity of CP.
S100A8, S100A9 and S100A12 can also form short-lived homodimers that act as pro-inflammatory cytokines or DAMPs, signaling through two different pattern recognition receptors as endogenous ligands: the receptor for advanced glycation end products (RAGE) and toll-like receptor 4 (TLR4) present in neutrophils and monocytes/macrophages.1095 Extracellular S100A8 and S100A9 have been shown to stimulate the recruitment of neutrophils and other leukocytes to the site of infection, to activate neutrophil functions such as degranulation and phagocytosis, etc. Finally, S100 proteins accomplish different intracellular functions such as tubulin polymerization and cytoskeleton rearrangement, important for enhancing leukocyte migration, phagocytosis and exocytosis, among others.
Imaging mass spectrometry studies have revealed that CP concentration at infection sites correlates with the amount of recruited neutrophils (Figure 67-c).311, 1105 Studies using CP-knockout mice (S100A9−/−, which renders a CP−/− phenotype as S100A8 is destabilized in the absence of S100A9) have demonstrated that S100A8/S100A9 plays a crucial role in the control of bacterial infections.1095 Lack of S100A8/S100A9 leads to a significant increase in the bacterial burden in blood, liver, lungs and spleen. The major mechanism by which CP inhibits pathogen proliferation is through its Zn(II)-chelation activity, especially at the initial stages of infection.1106 Laser ablation with ICP-MS revealed that staphylococcal liver abscesses are devoid of detecTable Zn(II) and Mn(II), unequivocally establishing the abscess as a cation-starved environment due to the presence of CP.311 The lack of immunomodulatory functions associated to S100A8 and S100A9 as DAMPs may not be evident in CP-deficient mice, suggesting that they play a secondary role, in some cases being replaced by immune factors induced to correct for the deficit in CP, or that they act predominantly at a later stage of infection.1095 Corbin et. al. showed that neutrophils extracted from wild-type and CP-deficient mice kill S. aureus in vitro with similar efficiency, suggesting that intracellular CP does not contribute to phagocytic killing.311 In the same line, in vivo studies indicated that recruitment of neutrophils to the sites of infection is unaffected by CP deletion. This was observed in abscessed livers and lungs after infection with S. aureus (96 hour post infection; hpi) and A. baumanni (36 hpi), respectively.311, 1105 Hence, S100A8/S100A9 seems to inhibit the growth of pathogens at infectious sites during the initial phase of infection, allowing time for recruitment and posterior activation of phagocytes, in a process dependent on S100A8 and S100A9, to accelerate the clearance of pathogens. Raquil et. al., however, demonstrated that blockade of S100A8 and/or S100A9 with neutralizing antibodies inhibits phagocyte migration to the alveoli (70% and 80% less neutrophils and macrophages, respectively) in a mouse model of streptococcal pneumonia at 48 hpi, suggesting that S100A8 and S100A9 play a key role in the transmigration of phagocytes from the lung tissue to the alveolar space.1107 In conclusion, the interplay between metal-chelation and immunomodulatory functions of S100 proteins is still a matter of debate and remains to be clarified. In addition, it is important to note that the activities of immune cells such as recruitment or phagocytosis, among others, are also modulated by Zn(II) availability, providing an additional level of complexity.1108
10.3. Zinc homeostasis in Gram-negative bacteria
Under circumstances of Zn(II) deprivation, microorganisms compete for this essential nutrient by increasing the expression of (1) Zn(II) importers or (2) Zn(II)-independent surrogates of essential Zn(II) proteins, with the aim of reutilizing intracellular Zn(II) (Figure 68).1073 The outcome of this competition not only depends on the efficiency of the microbial stress response but also on the tissue where the infection occurs. Kidney abscesses in mice infected with Candida albicans revealed a maximum increase in the expression of Zn(II)-acquisition genes in the fungi 24 h post-infection.1106 The expression levels of these genes were unaltered in CP-deficient mice, pinpointing the key role of CP in this process. At 72 h, the stress response attenuated and the levels of Zn(II) in the kidney restored, even though neutrophil infiltration and CP expression continued to rise. Since the fungal burden in the kidney was mostly unaffected by CP, this reveals how the expression of Zn(II) acquisition systems allows C. albicans to successfully survive the initial Zn(II) restriction exerted by CP while maintaining its intracellular Zn(II) levels. Instead, in lungs, CP-deficient mice were shown to be substantially more susceptible to C. albicans infection.
Figure 68.
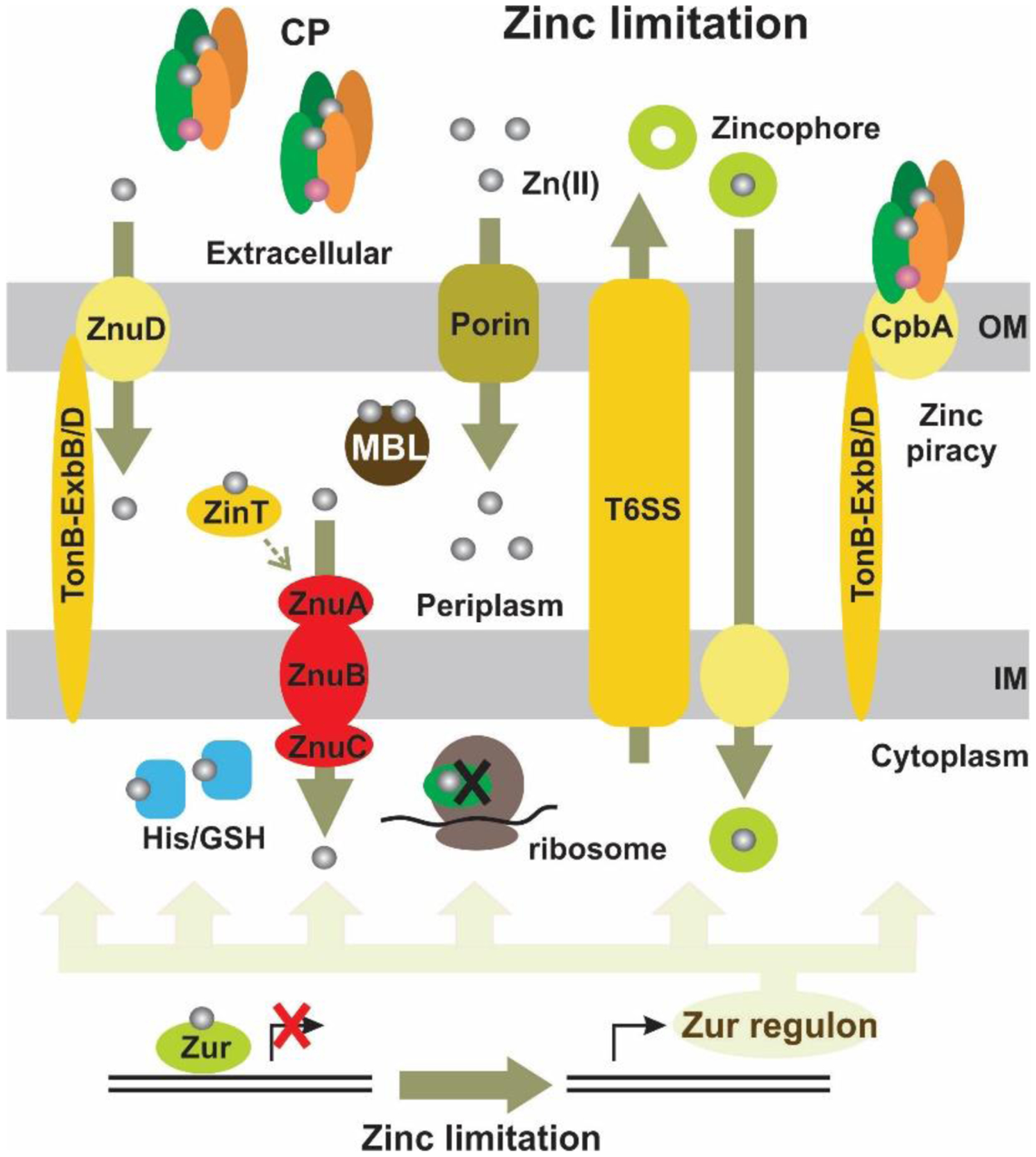
Mechanisms of Zn(II) homeostasis in Gram-negative bacteria, including Zn(II) import by the ZnuABC transporter, replacement of Zn(II)-dependent ribosomal proteins, Zn(II) passive entry into the periplasm through porins and through active transport by the action of ZnuD, and the acquisition of this metal ion by the use of zincophores and zinc piracy.
Bacteria have different strategies to maintain the cytoplasmic Zn(II) levels when challenged by CP (Figure 68). The primary regulator of Zn(II) homeostasis in many bacterial pathogens is the Zn(II) uptake transcriptional regulator (Zur).1109, 1110 Zur is a member of the ferric uptake regulator (Fur) family of metallosensing DNA-binding proteins and is conserved in Proteobacteria, Firmicutes, Cyanobacteria and Actinobacteria.1111 Metal binding to the metalloregulators induces conformational changes and alters the affinity of the regulator for DNA, acting as metal sensor. Zur exhibits sensitivity to fluctuations at femtomolar levels of free intracellular Zn(II).1112 In Zn(II)-replete conditions, Zur is metallated, and binds to conserved palindromic inverted repeat regions (Zur boxes) in the DNA promoter region of its regulon, inhibiting gene transcription. Upon Zn(II) starvation, Zur becomes Zn(II)-depleted and undergoes a conformational change that decreases the affinity to DNA, resulting in derepression of the Zur regulon.1111
In Gram-negative organisms, Zur-regulated derepression involves the induction of genes encoding the high-affinity ZnuABC Zn(II) transporters.1105, 1113–1121 ZnuABC transporters are the major mechanism by which bacteria overcome the action of CP. The ZnuABC complex is a member of the ATP-binding cassette (ABC) transporter protein family located in the inner membrane of Gram-negative bacteria. ZnuA is a periplasmic zinc-binding protein; ZnuB is an integral membrane protein that transports zinc across the cytoplasmic membrane to the cytoplasm; and ZnuC is an ATPase coupling ion transport to ATP hydrolysis.1110, 1122 An auxiliary periplasmic component, ZinT, binds and delivers Zn(II) ions to ZnuA, collaborating in the mechanism of Zn(II) recruitment from the periplasmic environment.1123 This protein, produced by many Gram-negative bacteria, is also regulated by Zur. A strong induction of the A. baumannii znu operon promoter was observed during a pulmonary infection in mice.1105 In line with this, disruption of znuB in A. baumannii or znuA in S. typhimurium increases significantly the sensitivity of bacteria to CP and other Zn(II) chelators.1105, 1118
In addition to the highly conserved ZnuABC (ZinT) transporters, expression of non-Zn(II)-binding ribosomal proteins is also typically increased in response to Zn(II) limitation, with the aim of replacing Zn(II)-requiring isoforms to effectively decrease the total cellular requirement of Zn(II) and promote Zn(II) mobilization (Figure 68).1124
Intracellular Zn(II) levels are relatively high (~0.2 mM for E. coli), but there is essentially no free Zn(II) within the bacterial cell.1112 Thus, Zn(II) is present under different chelated forms that become accessible during Zn(II) starvation. Although bacterial metallothioneins are able to buffer intracellular zinc, these have been identified in a limited number of bacterial genomes.1125, 1126 Instead, E. coli uses glutathione to buffer the concentration of Zn(II) and other divalent cations.1127 A. baumannii utilizes the amino acid L-histidine as a component of its labile zinc pool.1128 During Zn(II) starvation, A. baumannii upregulates the histidine utilization (Hut) system in a Zur-dependent pathway, which catabolizes cellular Zn(II)–His complexes, thereby increasing the levels of bioavailable Zn(II). On the other hand, there are mechanisms to ensure proper metallation of cytoplasmic metalloproteins. In conditions of zinc shortage, this is important for guaranteeing the correct functioning of core cellular processes requiring zinc.1129 G3E GTPases are metallochaperones and/or metal insertases that couple metal (or metal cofactor) delivery to a target protein to GTP binding and hydrolysis. The G3E GTPase superfamily is divided in four subfamilies, one of them denoted COG0523.1130 Members of COG0523 were shown to be Zur-regulated, and may therefore serve as Zn(II) metallochaperones when the metal is limited. These include E. coli YjiA and YeiR and A. baumannii ZigA, which bind Zn(II) and possess GTPase activity.1131, 1132
10.4. Regulation of periplasmic Zn(II)
Most bacterial responses to Zn(II) starvation are aimed to preserve the cytoplasmic pool of this metal ion. Instead, in the periplasmic space of most Gram-negative bacteria, Zn(II) levels are not regulated and depend on the availability of Zn(II) in the extracellular medium. Transport of Zn(II) across the outer membrane is thought to occur by passive diffusion through non-selective porins such as OmpF and OmpC in E. coli (Figure 68).1133, 1134 For this reason, under conditions of Zn(II) limitation, the periplasmic levels of free Zn(II) are expected to be equal to or lower than those in the extracellular milieu due to active import from the periplasm to the cytoplasm by the up-regulated ZnuABC system.
Some bacteria regulate the expression of specific porins in response to Zn(II) availability, such as P. aeruginosa OprD.1135, 1136 Others harbor mechanisms of active Zn(II) transport from the extracellular medium to the periplasm. In N. meningitidis, Zn(II) starvation induces the expression of an outer membrane TonB-dependent Zn(II) transporter named ZnuD (Figure 68).1137 Transport through TonB-dependent receptors requires mechanical energy from the TonB-ExbB-ExbD system, which harnesses energy from the proton motive force generated at the inner membrane to facilitate transport across the outer membrane.1138 ZnuD, conserved in Neisseriaceae, Acinetobacteriaceae, Bordetellaceae and Moraxellaceae families, has been identified as a crucial resistance factor used by N. meningitidis to overcome the NETosis response elicited by neutrophils during the innate host defense.1090 Structural studies on N. meningitides ZnuD demonstrate that large extracellular loops act like a fishing net, facilitating acquisition and sequestration of free Zn(II), although it is possible that ZnuD binds zinc in some chelated form.1139 A. baumannii encodes ZnuD homologs that are directly regulated by Zur: a protein with high homology to N. meningitidis ZnuD1, and a second protein present in some A. baumannii strains, named ZnuD2.1109 The specific contribution of each of these ZnuD transporters to A. baumannii Zn(II) homeostasis is unknown.
Another strategy for acquiring extracellular Zn(II) into the periplasmic space is the secretion of Zn(II)-binding small molecules termed zincophores (Figure 68), which bind the crucial metal ion and deliver it to a specific membrane transporter.1140, 1141 This process is analogous to the iron chelation by secreted siderophores, in which the delivery of iron-loaded siderophores back to the microorganism occurs via specific membrane receptors and transport proteins.
Pseudomonas putida secretes the siderophore/zincophore pyridine-2,6-bis(thiocarboxylic acid) (PDTC) that is capable of binding both Fe(III) and Zn(II) ions.1142 Other siderophores/zincophores that were shown to bind Fe, Zn(II) and in some cases other metals, include pyochelin, micacocidin, pyoverdine and yersiniabactin.1143–1146 Bobrov et. al. demonstrated that the Gram-negative pathogen Yersinia pestis uses a dedicated Zn(II)–yersiniabactin importer in the inner membrane, named YbtX, to acquire Zn(II) from the metallophore.1146 In P. aeruginosa, the operon zrmABCD, regulated by Zur, was shown to encode for a metallophore-mediated zinc import system.1147 Type VI secretion systems (T6SSs) have also been implicated in Zn(II) acquisition. Burkholderia thailandensis secretes a Zn(II)-scavenging molecule, TseZ, through a specific type VI secretion system (T6SS4).1148 Zn(II)-bound TseZ is then imported into the bacterial cell, via the heme transporter HmuR, upon sensing extracellular conditions of oxidative stress. Incorporated Zn(II) may be used to metallate Cu/Zn superoxide dismutase enzymes and prevent potential damage from reactive oxygen species. A similar situation occurs in Yersinia pseudotuberculosis, in which T6SS4 secretes a Zn(II)-binding molecule, YezP, that aids in Zn(II) uptake in conditions of oxidative stress.1149 Furthermore, the production of YezP is increased upon zinc limitation. In addition to the production of metal-chelating molecules by bacterial pathogens, some bacteria can utilize host-derived molecules as a metal source. The N. meningitidis TonB-dependent outer membrane receptor protein CpbA is expressed during Zn(II) starvation.1150 CpbA binds human CP, and the presence of CpbA permits N. meningitidis to use CP as its sole Zn(II) source, in a process termed Zn(II) piracy (Figure 68). In addition, the CpbA homolog in Neisseria gonorrhoeae, TdfH, directly binds to CP allowing Zn(II) acquisition from this protein.1150 The Zur-dependent expression of TdfH was shown to enhance bacterial survival after exposure to neutrophil NETs. Finally, changes to the bacterial cell envelope may occur during zinc starvation to accommodate upregulated type VI secretion systems and Zn(II)-uptake machineries. In A. baumannii, one of the most upregulated genes during Zn(II) starvation encodes a cell-wall-modifying enzyme named ZrlA.1151 This Zn(II)-binding peptidase, member of the M15 metallopeptidase family, regulates the abundances of peptidoglycan muropeptides, promotes efficient Zn(II) uptake, and modulates cell envelope permeability and membrane barrier function. In V. cholerae, the M23 family Zn(II)-binding endopeptidase ShyB is involved in peptidoglycan hydrolysis (necessary for cell growth) during Zn(II) limitation.1152 Both ShyB and ZrlA are regulated by Zur, evidencing that bacterial pathogens encode peptidoglycan-modifying enzymes that are important for surviving Zn(II) limitation.
Despite the presence of these mechanisms of active Zn(II) transport to the periplasm, most bacteria lack mechanisms for periplasmic Zn(II) accumulation to respond to metal starvation. Even when these mechanisms are present, bacteria may not accumulate Zn(II) in the periplasm under Zn(II) starvation, except for some cases in which essential periplasmic Zn(II)-enzymes are required. Instead, the periplasmic space may serve as a route of transitory passage to the cytoplasm, driven by receptors for zincophores or the ZnuABC system located in the inner membrane. In this regard, Crowder and co-workers reported that the metal content of periplasmic MBL L1 depends strongly on the bioavailability of metal ions present in the extracellular medium, unlike L1 expressed in the cytoplasm, whose metal content was almost insensitive to changes in extracellular metal composition.841 In fact, metal analyses on the cytoplasmic and periplasmic fractions of E. coli showed that the concentration of metal ions in the periplasm is not tightly controlled and increases with higher concentrations of metal ions in the growth medium. In contrast, the concentration of Zn(II) in the cytoplasm is tightly controlled while that of Fe is less so.
10.5. Effects of Zn(II) limitation on MBLs
MBLs fold and bind one or two Zn(II) ions in the periplasmic space in order to confer resistance against β-lactam antibiotics.838 Exposure of MBL-producing bacteria to sub-inhibitory amounts of metal chelators (EDTA or dipicolinic acid, DPA) renders bacteria sensitive to β-lactam antibiotics, supporting the notion that the levels of periplasmic free Zn(II) are minimally regulated and depend on the availability of extracellular Zn(II).1153 The same effect was observed when using N, N, N′, N′-tetrakis(2-pyridylmethyl)ethylenediamine (TPEN), a specific Zn(II) chelator, or Chelex-treated media, indicating that inhibition of MBLs is due to a reduction in extracellular Zn(II) levels and not to a direct interaction of these enzymes with the metal chelators in the periplasmic space.1154, 1155 In this line, Hood et. al. demonstrated that exposure of A. baumannii to sub-inhibitory concentrations of TPEN activating the Zur regulon renders this bacterium susceptible to carbapenems, despite the up-regulation of ZnuD1 and ZnuD2 located in the outer membrane.1105 Hence, during the initial stages of infection, bacterial resistance to carbapenems is expected to be strongly challenged by the CP-mediated nutritional immunity response elicited by the host. In fact, González et. al. showed that strains of E. coli expressing NDM-1 or VIM-2 become sensitive to carbapenems upon exposure to CP concentrations lower than those typically encountered at infections sites.421 Therefore, nutritional immunity may play a dual action during infections, limiting not only bacterial growth but also MBL-mediated antibiotic resistance.
Classification of MBL producers as carbapenem-resistant is typically based on in vitro susceptibility tests performed on Mueller-Hinton broth (MHB), which contains a concentration of Zn(II) of around 15 μM, largely exceeding the physiological concentration of free Zn(II) in body fluids.1156 Nicolau and co-workers recently proposed that MBL-mediated resistance in Enterobacterales is an artefact of these in vitro methods, suggesting that, unlike SBL carbapenemases, MBLs are not a relevant mechanism of resistance against carbapenems under physiological Zn(II) concentrations.1155 This contention is supported by the finding that the in vivo activity of meropenem against MBL-producing Enterobacterales in murine lung and thigh infection models correlates with the MICs measured in zinc-depleted MHB, which are markedly lower than those measured in regular MHB. The levels of Zn(II) in bronchoalveolar lavage fluid (representing the site of infection in murine lung infection model) drop from 4.5 μM in uninfected mice to undetecTable levels in lung-infected mice, highlighting that Zn(II)-depleted MHB is a better mimic of the physiological conditions encountered during infection. In the same line, there are several reports describing positive clinical outcomes in patients infected with MBL-producing Enterobacterales that were prescribed with β-lactam agents, despite in vitro tests classified these isolates as carbapenem resistant.664, 1157–1159 In addition, animal models of infection with MBL-producing Enterobacterales have demonstrated marked reductions in bacterial counts following administration of simulated human regimens of carbapenems.1160 In contrast, Hobson et. al. showed that the efficacy of MBLs ex vivo in urine from healthy patients is similar to the one observed in regular MHB, due to similar levels of Zn(II) in both media.1161 Although these determinations overlooked the Zn(II) restriction in urine that the immune system imposes in infected patients, collectively these studies evidence the need of improving in vitro susceptibility tests to achieve clinically meaningful MIC values in MBL producers. The Zn(II) content in growth media employed as well as in the sites of infection should be assessed in order to fill the gap between in vivo and in vitro assays, especially given the heterogeneity in Zn(II) levels encountered in different host tissues. In this regard, it has been shown that the modified Hodge test, albeit having an excellent sensitivity for detecting enterobacterial isolates producing Ambler class A (KPC) and class D (OXA-48) carbapenemases, improves its sensitivity for NDM-1 producers from 50% to 85.7% if excess Zn(II) is added to the culture medium.1162 Overall, these studies suggest that MBL-producing Enterobacterales may not exhibit carbapenem resistance in certain host tissues or fluids, casting doubt on the actual role of MBLs as resistance determinants in these bacteria. The situation may differ in other bacteria. For instance, non-fermenters expressing similar levels of MBLs exhibit markedly higher values of carbapenem MICs compared to E. coli, indicating that a stronger Zn(II) limitation is needed to render these bacteria susceptible to carbapenems.430 Hence, although the contribution of MBLs to resistance in Enterobacterales in clinical settings remains to be clarified, it is clear that these pathogens acquire and maintain genes coding for MBLs, which would imply that MBLs are an effective mechanism of resistance. One possibility is that MBLs can be advantageous in conferring resistance to Enterobacterales against penicillins or cephalosporins, whose MIC values are generally higher than those for carbapenems. Despite these controversial issues, it is well established that clinical relevant MBLs are strongly challenged by Zn(II) restriction, and the nutritional immunity response possibly stands as one of the major forces shaping the evolution of these enzymes. In the next section we will describe the mechanisms by which Zn(II) limitation affects MBL performance in the periplasm.
10.6. Zn(II) limitation and MBL stability
In the past few years, a growing body of evidence shed light into how periplasmic Zn(II) mediates MBL resistance, and how these enzymes manage to circumvent conditions of Zn(II) restriction. Initial studies on BcII, an extracellular enzyme secreted by Gram-positive B. cereus, revealed that the metal-binding site is optimized to bind tightly two Zn(II) ions, a condition that is necessary in harsh environments such as the extracellular medium.355 Directed evolution experiments on this enzyme unveiled how substitutions of second-shell residues surrounding the conserved active sites allow MBLs to enhance their Zn(II) binding affinities. Competition experiments of apo-enzymes with chromophoric zinc chelators or EDTA showed that the metal binding affinities of MBLs from the three subclasses increase upon substrate binding.294 Based on this in vitro observation, Wommer et. al. suggested that in Gram-negative bacteria MBLs exist as apo-forms in vivo, increasing their metal affinity and binding one equivalent of Zn(II) in the presence of substrates. This work, however, did not take into account the effect of Zn(II) binding in MBL stability. Different studies on BcII, BlaB, NDM-1, CphA and L1 (i.e., MBLs from all subclasses) have shown that these enzymes are significantly stabilized in vitro by Zn(II).247, 284, 295, 421, 1163 In conclusion, despite these studies provide fruitful insights into the biochemistry and biophysics of MBLs, they do not address the relative stability of the metallated and apo forms in the bacterial periplasm.
The hypothesis suggesting the apo variants as the dominant species in the cell was challenged by different studies. The B3 lactamase L1 does not fold properly in the periplasm of E. coli in the absence of Zn(II), indicating that thermodynamic binding studies with properly folded apo-enzymes cannot be extrapolated to in cell conditions, and that incorporation of Zn(II) may be under kinetic control.1164 In 2016, González et. al. demonstrated that Zn(II) deprivation by DPA or CP from the extracellular medium provokes a marked drop in the levels of clinically relevant periplasmic MBLs in E. coli cells, rendering the bacteria susceptible to β-lactam antibiotics.421 This is due to the fact that soluble apo-MBLs generated under Zn(II)-limiting conditions are unsTable and undergo degradation and/or aggregation in the periplasm (González et al. submitted). Thus, the stability of MBLs in vivo is tightly controlled by the availability of periplasmic Zn(II). This contrasts with the picture of MBLs in vitro: apo-MBLs are well-folded sTable proteins, as revealed by different spectroscopies and the availability of several crystal structures (PDB codes: 6UA1, 3PG4, 3I0V, 3SBL, 2FU6, 3T9M, 3RKJ, 2YNU, 3RKK) while in vivo, these species are much less sTable. Therefore, Zn(II) is not only required for enzyme activity but also for the MBL in cell stability. The lifetime of MBLs in the periplasm depends on an exquisite balance between the kinetics of different processes: metallation, de-metallation, aggregation and degradation (Figure 69), which are not taken into account during in vitro assays. Overall, this contrast reveals that in vitro studies cannot be extrapolated to in vivo, and that specific experiments are required to test in vitro hypotheses.
Figure 69.
Kinetic stability of MBLs in the bacterial periplasm depends on a balance between competing processes of MBL metallation (kon), de-metallation (koff), apo enzyme degradation (kdeg) and aggregation (kagg).
Some MBLs are more fit to endure conditions of nutritional immunity. BcII, SPM-1 and NDM-1, to a lesser extent, are among the most refractory to periplasmic degradation, followed by IMP-1.421 Instead, VIM-2 readily forms insoluble aggregates in the periplasm upon metal starvation. Recently reported clinical alleles from the NDM and VIM families were shown to be more tolerant to metal deprivation.439, 452, 479 Given that these variants exhibit similar resistance profiles when tested under high Zn(II) availability (such as in MHB assays), we conclude that the catalytic activity of the fully metalated enzymes is already optimized, while the clinical evolution of many MBLs seems to be strongly driven by evolutionary constraint of the nutritional immunity response.
According to this model, the performance of an MBL during an infection can be improved by an increase of the Zn(II)-binding affinities (that will avoid the accumulation of unsTable apo-enzymes), and/or by mechanisms leading to stabilization or protection of the apo-enzymes.421 This is the case of the NDM enzymes, since membrane-anchoring to the outer membrane protects NDM-1 from degradation. The soluble variant apo-NDM-1 C26A, instead, is rapidly degraded by periplasmic proteases in vivo. Conversely, an engineered membrane-anchored variant of the otherwise unsTable VIM-2 was more refractory to degradation upon Zn(II) starvation, indicating that lipidation at the N-terminus is a mechanism for stabilizing unsTable apo-MBLs. The stabilities of apo-MBLs cannot be explained solely based on their protein structure. The crystal structures of apo and metal-loaded MBLs are mostly superimposable, indicating that metal removal marginally affects the protein structure (PDB codes: 3S0Z, 3PG4, 1BC2, 3I0V). However, limited proteolysis experiments and NMR studies of holo- and apo-MBLs have revealed that the C-terminus of the protease-sensitive apo forms is much flexible than those of apo-MBLs refractory to degradation (González et al. submitted). In other words, the lifetime of apo-MBLs in the periplasm depends on protein dynamics, which allows the exploration of degradation/aggregation-prone conformations (Figure 70). This mobility at the C-terminus may be transmitted from the active site (L7 loop) through the α3 region, which loses secondary structure upon metal removal. In BcII, although the L7 loop and α3 region increase in flexibility, the effect is more confined and the secondary structure of α3 is marginally affected (González et al. submitted).1163 Metal binding quenches these dynamics, giving rise to more rigid structures, less prone to degradation.
Figure 70.
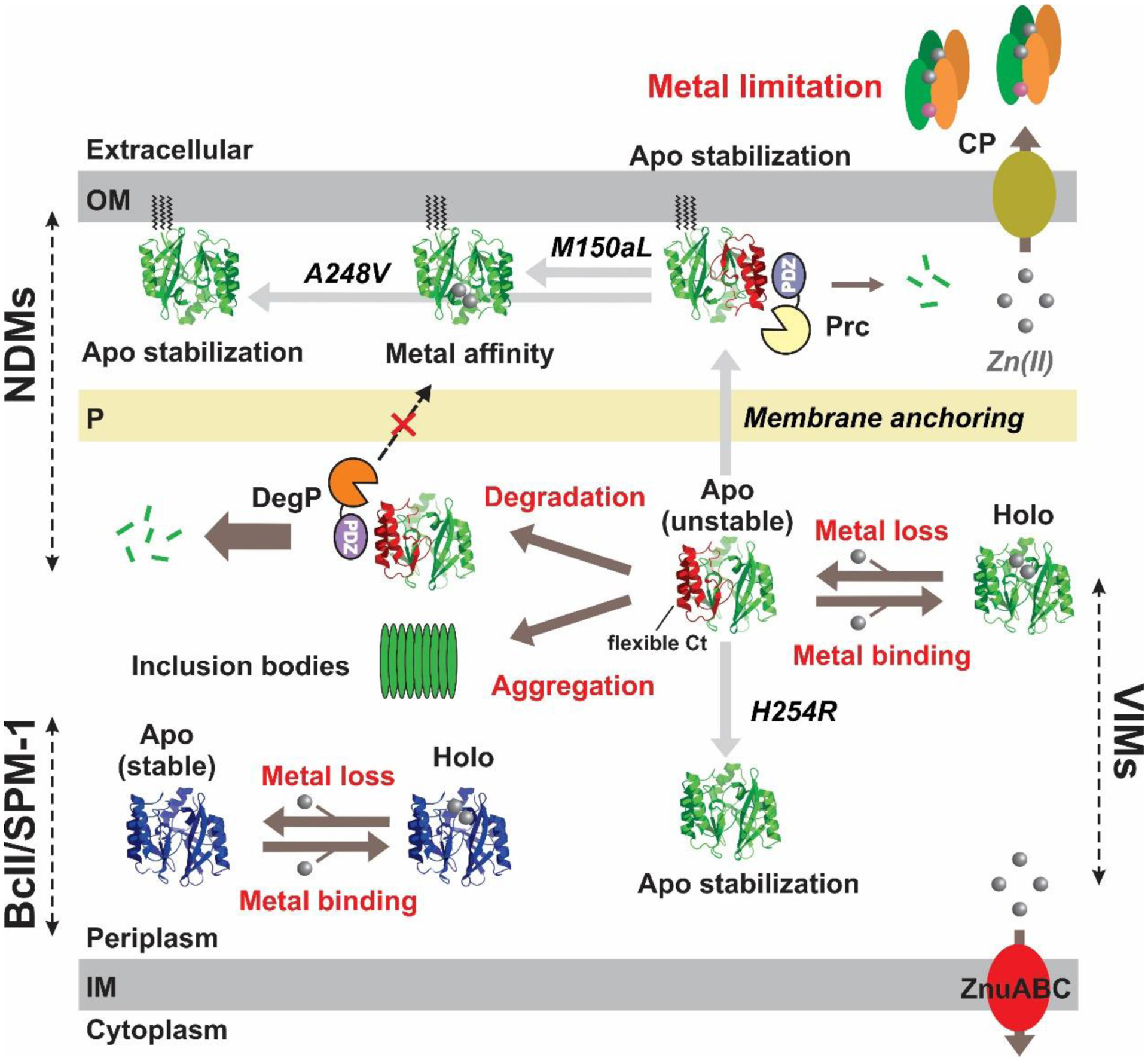
Model of zinc-dependent modulation of MBL stability in vivo for NDMs, VIMs, BcII and SPM-1. Apo enzymes generated during metal limitation are susceptible to degradation and aggregation. Anchoring of NDM-1 to the outer membrane prevents degradation by DegP due to the permeability barrier of the peptidoglycan layer preventing passage of this high molecular weight protease. Mutations H254R (in VIM-2) and M150aL and A248V (in NDM-1) improve tolerance of these MBLs to Zn(II) depletion either by improving stability of the apo enzymes or by increasing their Zn(II) affinity.
DegP and Prc have been identified as the periplasmic proteases responsible for degradation of apo NDM-1 in E. coli (Gonzalez et al. submitted). These proteases, albeit unrelated, harbor a PDZ-domain that selects substrates with flexible or unfolded C-terminal regions, as is the case of soluble apo-NDM-1. Thus, these findings not only provided the structural basis for the degradation/aggregation processes occurring in vivo, but also unveiled a C-terminal degron that tags NDM-1 for degradation upon metal removal from the active site. The L7 loop and α3 region, apart from modulating the flexibility of the C-terminal region, may also be implicated in the delivery of Zn(II) from other periplasmic Zn(II)-proteins to apo-MBLs.1163
The stabilization effect provided by membrane-anchoring is the consequence of a sub-compartmentalization mechanism, since the apo-enzyme cannot be accessed by the main periplasmic protease, DegP, due to restricted permeability of the murein layer (Figure 70) (González et al. submitted). The resting state of soluble Prc is monomeric (74 kDa) while that of DegP hexameric (280 kDa).1165, 1166 Considering that the permeability of the murein layer allows free diffusion of proteins up to 100 kDa,1167 it has been proposed that outer-membrane apo NDM-1 can only be accessed by Prc (González et al. submitted). Soluble NDM-1 (and possibly other soluble periplasmic β-lactamases), instead, is mostly degraded by DegP. Membrane-anchoring stabilizes NDM-1, but does not completely prevent degradation of the apo-enzyme by Prc. As a result, NDM-1 producing Enterobacterales may not resist the action of carbapenems during an infection. Since the first report of NDM-1, new allelic variants have been reported, most of them exhibiting higher tolerance to Zn(II) deprivation.452, 479 This improved fitness has been achieved by accumulating mutations that enhance either the kinetic stability of the apo-enzyme or the Zn(II) binding affinity in vivo without affecting the antibiotic resistance phenotypes.452 In particular, the ubiquitous substitution M150aL increases Zn(II) binding affinity, preventing the formation of apo-enzymes. A248V substitution, located in the C-terminal region in alleles 6, 15, 19 and 27, reduces the flexibility of this region (and α3 region) in the apo-enzyme, thereby limiting proteolysis by Prc (González et al. submitted). The M150aL and A248V substitutions seem act independently of the genetic background: similar Kd values were obtained for NDM-4 (M150aL) and NDM-15 (M150aL, A248V), and a similar increase in tolerance to Zn(II) restriction was observed when comparing NDM-6 (A248V) vs. NDM-1 and NDM-19 (NDM-7 A248V) vs. NDM-7.489 Alleles combining these substitutions, such as NDM-15 (M150aL, A248V), are those showing the best performance under Zn(II) limiting conditions. Crowder and co-workers proposed that NDM-15 has evolved the ability to function as a mono-Zn(II) enzyme with high catalytic efficiency.479 The E152K substitution present in NDM-9 (in the α3 region) was shown to reduce degradation of the apo-enzyme, similar to A248V.452 Crystal structure of NDM-9 shows that a lysine in this position generates a salt-bridge with D236 from loop L10, linking and stabilizing the α3 and C-terminal regions.501
Within the VIM family, initial studies showed that metalated VIM alleles (VIM-1, VIM-2, VIM-4, VIM-5, and VIM-38) exhibiting small variations in the kinetic parameters, display considerable differences in their thermal stabilities, with variations of up to 20°C in Tm.451 A recent study compared the tolerance to Zn(II) deprivation of 45 clinically derived VIM variants from the VIM-1-group, VIM-2-group and VIM-4-group when expressed in E. coli.439 As reported for the NDM family, differences in MIC values were noticeable at limited Zn(II) concentrations. Within the VIM-2-group, VIM-20, harboring the H254R substitution with respect to VIM-2, granted the highest levels of resistance in Zn(II)-depleted medium. The H254R mutation generates a salt bridge between the Glu184 side-chain carboxylate and the Arg254 side-chain guanidino group, increasing the stability of VIM-20 in vitro and in vivo without affecting the catalytic parameters nor the metal-binding affinities. Interestingly, this substitution is located at the C-terminal region of the enzyme, suggesting a similar mechanism of apo-protein stabilization compared to A248V or E149K substitutions in NDMs. The H254R substitution is present in several alleles from different VIM groups (VIM-1, VIM-2 and VIM-7), and it had been previously proposed to accomplish an adaptive role, given variable epistatic effects in antibiotic resistance.425 Given that Glu184 is highly conserved along the entire VIM family, it is expected that a salt-bridge in the C-terminal region will protect various VIM alleles from degradation during Zn(II) scarcity. Similarly, although not characterized structurally, VIM-28 from the VIM-4-group harbors a single substitution proximal to the C-terminus (H224L) compared to VIM-4.439 This variant showed the highest tolerance to metal limitation inside the VIM-4-group. Substitution H224L (and H224Y) is also ubiquitous among different VIM groups.
Finally, in the case of IMP enzymes, a recent work demonstrated that the IMP-1-group (comprising 25% of IMP allelic variants) has evolved towards variants exhibiting higher catalytic activities against newer carbapenems, with no apparent effect coming from zinc limitation conditions.395 Given that IMP-1 exhibits moderate tolerance to zinc deprivation,421 and apparent higher metal-biding affinities compared to VIM-2 and NDM-1,395 these results suggest that IMP enzymes, at least from the IMP-1-group, are already (or partially) optimized to resist conditions of nutritional immunity. However, given the great heterogeneity of the IMP family, further studies are required to evaluate this hypothesis in the remaining IMP groups.
In conclusion, the clinical evolution of the NDM and VIM families evidences how the immune system has forced MBLs to adapt to Zn(II) restriction conditions by increasing either their Zn(II) binding affinities and/or the protein stability in their non metallated form in the periplasm in order to resist the action of periplasmic proteases or to avoid protein aggregation. Zn(II) binding may not only protect already folded MBLs but may also assist in protein folding of newly synthesized MBLs. While metal limitation has been the major force guiding the evolution of the more recent NDM family, diversification of the broader VIM family, which begun earlier in the late 80s, has also been impacted by exposure to antibiotics, in particular ceftazidime, which selected variants exhibiting higher resistance against this antibiotic.425 VIM-2 is an enigmatic case. VIM-2 is the most commonly found VIM variant in clinical settings despite being highly unsTable in conditions of Zn(II) limitation.421 However, this enzyme is mostly found in P. aeruginosa isolates, in which the levels of resistance granted by MBLs are higher compared to other microorganisms, due to the lower permeability of the outer membrane towards carbapenems.123, 1058 It is likely that higher levels of Zn(II) limitation are required to sensitize this bacterium. Finally, considering that the nutritional immunity response and bacterial homeostatic systems are in constant co-evolution,312 it is expected that MBLs will be progressively challenged by Zn(II) restriction in the near future.
10.7. Metallation of MBLs from Gram-positives
A somewhat different situation is expected for MBLs produced by Gram positive bacteria, namely the BcII family. Intrinsic to B. cereus, B. thuringiensis and B. anthracis, the BcII family comprises 195 closely-related allelic variants that are produced as extracellular enzymes (http://bldb.eu/).262, 373, 1168 Similar to MBLs from Gram negatives, BcII enzymes harbor a signal peptide for secretion through the Sec system, indicating that these enzymes are synthesized as precursors in the cytoplasm, exported across the cell membrane as unfolded polypeptides, and processed by type I signal peptidases to be finally released as mature enzymes into the extracellular medium.1169 Based on this, it is possible that BcII variants fold and bind Zn(II) ions in the space between the cell membrane and the cell wall, before diffusing away from the cell. Zn(II)-homeostasis systems in Bacillus species are similar to those from Gram negative organisms: under Zn(II) limitation, Zur de-represses the expression of ZnuABC, which imports Zn(II) ions from the extracellular medium to the cytoplasm.1073 Therefore, in conditions of Zn(II) scarcity, metallation of BcII in the surroundings of the cell membrane will be challenged not only by the low availability of extracellular Zn(II) ions but also by the action of the ZnuABC importer. However, the high thermostability and rigidity of apo-BcII, together with its optimized metal-dissociation constants, suggests that these enzymes are adapted to thrive as sTable extracellular apo-enzymes, capable of binding Zn(II) ions efficiently upon different stringent circumstances, and being resistant to extracellular proteases (González et al. submitted).355 MBLs from Gram-negatives, instead, require the shield of outer membrane vesicle scaffolds for being secreted into the extracellular medium.421
11. Detection of MBL producers
In a context of increasingly prevalent antibiotic resistance among clinical pathogens, the ability to detect the resistance mechanisms present in a specific isolate is of crucial importance both for defining therapeutic options for the affected patient and for maintaining resistance surveillance and updated epidemiologic information to prevent and control outbreaks. There is a very large variety of methods that allow detection of MBL production (or more generally, β-lactamase production) within bacteria. These detection strategies, and their relative diagnostic merits, have been reviewed extensively elsewhere.471, 1156, 1170–1176 Here we present a summary of the main methods currently available for detection of organisms producing MBLs.
Prior to a detailed characterization of their resistance mechanisms, clinical isolates are generally subjected to antimicrobial susceptibility testing (AST) methods to identify their resistance profile towards a panel of antibiotics. These range from simple disc diffusion tests and minimum inhibitory concentration (MIC) determinations to automated systems that allow combined microorganism identification and AST such as Vitek2 (bioMérieux) or Phoenix (BD Diagnostics).1177 If reduced susceptibility to certain antibiotics is detected, e.g. reduced diameter of the inhibition zone in a disk diffusion test or MICs above the established breakpoints, subsequent tests may be carried out to determine the underlying resistance mechanisms. There are two general types of methods: those evidencing the presence or activity of the resistance mechanism itself (phenotypic methods) and those detecting the coding gene (genotypic or molecular methods).1170, 1171, 1173 Multiple factors influence the choice of detection method to be carried out, including its performance (i.e. sensitivity and specificity) and the possibility that these may vary among different types of isolates, the turnaround time required to obtain the final result, cost, equipment requirements and the level of expertise needed to perform it and analyze the results. It is unlikely that any test procedure will be optimal across all these criteria.1156 Furthermore, it should be noted that not all tests may have been approved as clinical diagnostic assays, most of them being for research use only. The standard workflow of AST followed by identification of resistance mechanisms may take several days,1174 but due to the value of rapid resistance diagnosis some newer phenotypic and molecular tests allow the direct use of patient specimens.
An unifying characteristic of MBLs is their capability to degrade carbapenems. Many phenotypic detection methods are based in the hydrolytic activity presented by these enzymes, together with other carbapenemases, using it to differentiate their occurrence from other forms of resistance carbapenem. Although most of these phenotypic tests lack by themselves the capability to identify the type of carbapenemase involved, they can provide this information when combined with appropriate inhibitors (such as EDTA or DPA for MBLs) that abolish activity for specific carbapenemases.
The hydrolysis reaction carried out by β-lactamases generates protons as a side product, and can thus be detected via the resulting acidification of the reaction medium. This principle is exploited in the Carba-NP test (Figure 71-b),1178 in which an imipenem solution is incubated with bacterial extracts in presence of the pH indicator phenol red, that visually confirms the presence of carbapenemase activity by a color shift (red to yellow). The test presents relatively high sensitivity in Enterobacterales, although it was found to have issues detecting OXA-48 type carbapenemases and with mucoid isolates presenting false negatives.1179 Low sensitivity was also observed for detection of carbapenemases in A. baumannii isolates, although the CarbAcineto NP variant was developed to overcome these limitations using altered lysis conditions and a larger inoculum.1180 Although these tests do not discriminate between carbapenemase classes, the Carba-NP test II grants this capability, via the use of assays in presence of tazobactam to detect KPC and EDTA to infer the presence of MBLs.1181
Figure 71.

Methods for carbapenemase detection. a) Modified Hodge Test (MHT). b) Carba NP positive result, showing tube with no imipenem added (red) and with imipenem added (yellow). c-d) Modified carbapenem inactivation method (mCIM) positive (c) and negative (d) results. e-f) mCIM and EDTA-mCIM (eCIM) for a SBL producer (e) and for an MBL producer (f). g) double disk synergy test for detection of MBLs, using imipenem (IMP) and EDTA disks. h) NG-Test Carba 5 (NG Biotech) lateral flow immunoassay (LFIA) showing the different carbapenemase detected. Panels a), b), c), d), e), f) and h) adapted with permission from refence1175 Tamma et al. Copyright 2018 American Society for Microbiology. Panel g) adapted with permission from reference1185 Lee et al. Copyright 2003 American Society for Microbiology.
Various tests use solid medium in which the isolate to be tested has been spread, and are performed by placing a disc or strip containing the drug within the plate. The diffusion of the antibiotic creates a concentration gradient and results in an inhibition zone in which the bacterium cannot grow, and whose size informs of the resistance level (i.e. growth closer to the source implies lower susceptibility). The popular eTest uses a manufactured strip that contains a longitudinal antibiotic gradient (imipenem or meropenem in the case of carbapenemase detection), and antibiotic MICs can be directly read from a grading on the strip according to the point in which the edge of the inhibition zone contacts it.1182 A version of the test allows specific detection of MBLs by incorporating a duplicate antibiotic gradient on the opposite half of the strip with EDTA.1183 Differences in resistance under both conditions reveal the presence of MBLs. The combined disc test (CDT) works by placing two discs, one containing a carbapenem alone and the same drug plus a metal chelator (or other MBL inhibitor), on a plate streaked with the bacterial isolate.1184 After growth, the inhibition zones around both discs are compared and a sufficiently larger zone around the disc containing the inhibitor indicates the presence of MBL-mediated resistance. Similarly, the double disc synergy test (DDST) also uses two discs on a plate in which the isolate to be tested has been spread, but one disc contains the carbapenem and the other the inhibitor alone (Figure 71-g).1184, 1185 The discs are placed in proximity to each other, and the synergy between the two compounds (revealing the presence of an MBL) is indicated by a larger inhibition zone in the space between the two discs.
Mass spectrometry is widely used in clinical microbiology for the identification of genus and species of bacterial isolates, but it can also be applied to detect carbapenemase activity. MALDI-TOF protocols allow very sensitive and specific detection of carbapenemases through the observation of mass peaks corresponding to hydrolyzed antibiotic molecules after incubation with bacterial extracts.1186, 1187 Similarly to other methods, comparison of assays ran in presence or absence of metal chelators can add the ability to distinguish the presence of these enzymes from other carbapenemases.1188
Other phenotypic methods such as the carbapenem inactivation method (CIM) and the modified Hodge test (MHT) are growth-based.1156, 1175 In these cases, detection of carbapenemase activity depends on an antibiotic sensitive indicator strain being able to grow under specific conditions.
The CIM tests the hydrolysis of carbapenems by a bacterial strain, by incubating a suspension of the isolate to be assayed for 2 h with a meropenem-containing disk.1189 After incubation, the disc is placed in a plate streaked with a carbapenem sensitive E. coli strain and, following overnight growth, the diameter of the inhibition zone around the disc is measured. In a positive test, a small inhibition area is observed, since it implies that the tested isolate possessed carbapenemase activity and hydrolyzed the antibiotic in the disc during the previous incubation. The most widely used variant of this test in the modified CIM (mCIM, Figure 71-c, d), in which the incubation period is extended to 4 h and the bacterial isolate is resuspended in tryptic soy broth instead of water, leading to a very high sensitivity (around 97%) in Enterobacterales while maintaining specificity.1190 For non-glucose-fermenting organisms, although the mCIM presents similar detection parameters in P. aeruginosa, the sensitivity and specificity are considerably lower for A. baumannii. A variant (CIMTris) which used 0.5 M TrisHCl for extraction was found to possess high sensitivity and specificity for detection of carbapenemases in both P. aeruginosa and A. baumannii.1191 While the CIM-type tests do not identify the particular type of carbapenemase, the EDTA-mCIM (eCIM) test allows discrimination of MBLs from carbapenem-hydrolyzing SBLs by means of a duplicate assay in which the disc is incubated with the bacterial isolate in presence of EDTA.1192 Thus, the production of an MBL is revealed by an increase in the radius of the inhibition zone in the plate that received the disc from the assay with added chelator with respect to that which followed the standard mCIM procedure (Figure 71-e, f). However, coproduction of MBL and SBL type carbapenemases may lead to false negative results.
In the MHT (Figure 71-a), the carbapenemase activity is detected by monitoring the protective effect towards a β-lactam sensitive E. coli strain used as an indicator. In the MHT, the carbapenemase activity is detected through the protection it grants to a β-lactam sensitive E. coli strain used as an indicator.1162 A lawn of the indicator strain is spread on the surface of a plate, upon which a disk containing a carbapenem is placed in the center, while each tested isolate is streaked in a radial stripe from the center towards the edge. The stripes containing resistant bacteria will grow towards the center further than the indicator strain, for which an inhibition zone will be visible. However, if the resistance is caused by a β-lactamase, the degradation of the antibiotic by the cells and the enzyme they release into the medium will generate a zone of lower concentration of the drug near the corresponding stripe in which the indicator strain may grow. Thus, for positive results the indicator strain will grow along the corresponding stripe into the inhibition zone, giving rise to a distinct clover-like shape. The modified Hodge test is known to display low sensitivity for detection of NDM-1 producing bacteria (although this can be partially remedied by the addition of ZnSO4 to the growth media).1162 A variation, termed the Triton Hodge Test (THT), greatly improves the detection of strains producing this enzyme, also increasing sensitivity for other enzymes without resulting in a greater false positive rate.928 The assay is carried out as the MHT, but with the addition to the solid medium of the non-ionic detergent Triton X-100. This inexpensive modification aids in dissolving the membrane-anchored NDM-1 from the producer bacterium membranes, allowing it to diffuse into the surrounding medium. Due to its difficulties in detecting NDM-1 and other enzymes, the MHT was removed from the recommended tests in the 2018 CLSI guidelines, although it is still widely used due to its simplicity and low cost.1173, 1175
Lateral flow immunoassays (LFIA) are immunochromatographic methods that allow the detection of MBL proteins (or other β-lactamases) within cell lysates using specific antibodies.1193, 1194 These assays can be multiplexed, enabling the simultaneous detection of a predetermined set of enzymes. Furthermore, the tests can be very rapid, offer extremely high sensitivity and specificity, and do not require technical expertise nor additional equipment. However, detection of allelic variants of an enzyme may be impaired if the mutations differentiating them affect the epitope recognized by the antibody. A commercial LFIA test, termed NG-Test Carba 5 (Figure 71-h), was developed to detect the 5 main carbapenemase families (KPC, VIM, NDM, IMP, OXA-48).1193 Starting from a single colony of a clinical isolate resuspended in extraction buffer, the test offered reliable detection at 15 min of incubation, with 100% sensitivity paired with very high specificity. The LFIA test RESIST-4 K-SeT also offers similar performance characteristics,1194 detecting the same set of enzymes with the exception of IMPs, which remain infrequent in Western countries.
Various molecular/genetic methods allow the direct detection of blaMBL genes and are generally considered the reference methods due to their high sensitivity and specificity.1170, 1171, 1174 Furthermore, these methods generally allow for a higher degree of information regarding the detected resistance determinant, i.e. identification of the particular enzyme or enzyme family instead of a general detection of the type of resistance mechanism involved, and can be considerably faster than phenotypic methods requiring bacterial growth. It should also be noted, however, that most of these methods require prior knowledge of the genes to be detected, and will not be able to identify novel resistance determinants. Additionally, some molecular methods require greater technical expertise and specialized equipment which may only be available at reference labs.
PCR-based methodologies are widely applied. These include simple end-point PCR with a single pair of primers followed by gel electrophoresis to detect the amplification products, but can also be multiplexed to allow parallel detection of multiple resistance genes.1195, 1196 This requires a greater technical effort to develop compatible primer sets to simultaneously amplify a set of distinct resistance genes, and generating products of sufficiently different molecular weight to allow detection of each amplicon. Amplification products can also be sequenced to obtain nucleotide-level data on each detected gene.
Real-time PCR can also be used to detect resistance genes. It has the distinct advantage that amplification and result visualization is performed by the thermocycler, which typically monitors a fluorescent signal from a probe informing the amount of generated product along the reaction, instead of requiring a subsequent step of gel electrophoresis. Real-time PCR detection tests exist in single1197–1199 and multiplex1200, 1201 variants, with commercial platforms also available. Multiplex real-time PCR may use probes labeled with different fluorophores to increase specificity in detection with respect to end-point PCR. The automated commercial test Xpert Carba-R can detect the most common carbapenemases (blaKPC, blaNDM, blaVIM, blaIMP, blaOXA-48) with a sensitivity of 100% and 97% specificity, and can perform detection both from cultured isolates or directly from rectal swab specimens.1202, 1203 Similar capabilities are found in the BD MAX Check-points Check CPO1204 and the GenePOC Carba1205 automated assays.
Loop-mediated isothermal amplification (LAMP) methods have also been reported for carbapenemase detection.1206, 1207 The LAMP technique is based on DNA amplification by a DNA polymerase with strand-displacement activity (such as Bst polymerase), using a set of 4 or 6 specially designed primers binding to the region to be copied.1208, 1209 Methods using LAMP offer comparative advantages with respect to PCR detection in that they do not need a thermocycler, as the amplification is carried out at a constant temperature in a thermal bath, and their tolerance to typical polymerase inhibitors. Furthermore, they are faster to perform (reaction times can be below 1h) and do not require gel electrophoresis, as visualization can be performed by turbidity reading or using fluorescent dyes.1206, 1207 The commercial assay Eazyplex SuperBug CRE is based on LAMP coupled to detection via fluorescent probes, and allows detection of VIM, NDM, KPC and OXA-48 family carbapenemases with high sensitivity and specificity.1210
DNA microarrays also offer capabilities for detection of antibiotic resistance, either coupled to PCR amplification or directly using DNA from bacterial sources.1171, 1174 They offer advantages particularly in parallel screening for a large range of different genes, potentially offering the simultaneous detection of hundreds of targets with nearly 100% sensitivity. Furthermore, appropriately design oligonucleotide probes on the DNA array can allow detection of different allelic variants, which can be challenging with other genotypic tests without sequencing. Commercial microarray-based tests include Verigene1211, 1212 and Check-MDR CT103,1213 with the former reporting a turnaround time of <3 h for detection.
The growing adoption of advanced DNA sequencing platforms and the continuous reduction in their operative costs, have led to whole-genome sequencing (WGS) of microorganisms constituting a possible alternative for the detection of resistance genes.1214–1217 Albeit at a higher technical complexity and requirement of computational power for data analysis than other procedures, WGS techniques allow for the direct identification of allelic variants that may not be detected by other molecular methods, in addition to permitting the simultaneous characterization of their genetic environment, organism identification, and detection of additional resistance mechanisms.1216 WGS methods have shown sensitivities >87% and specificities >98% in the detection of antimicrobial resistance.1215, 1216 These techniques are being increasingly used for antimicrobial resistance surveillance, and are able to predict accurately phenotypic resistance profiles.1214 Currently, the minimum estimated cost for sequencing of a bacterial genome is around US$ 80, with a 1 week turnaround time (although sequencing is possible in 24h with higher associated costs).1216 While current per-genome costs may still prevent use for routine diagnosis, WGS approaches offer enormous future potential for detection of antibiotic resistance and other clinical microbiology applications.1217
12. β-Lactamases as an unique model system to study protein evolution
This review has focused on the current and potential future impact of β-lactamases on human health, and chemical strategies to limit it. There is, however, a different facet to the scientific study of these enzymes, less directly related to their clinical role as mechanisms of resistance and focused instead on the proteins themselves. β-Lactamases provide a unique model to study protein evolution, in which the fitness and survival of the host bacteria in presence of antibiotics is directly tied to the function of the enzyme. β-Lactamases are also one of the most intensively studied enzyme families, many thousands of known proteins and a wealth of available information regarding their capability to confer resistance and in vitro biochemical properties.355, 1218–1224 As such, the natural adaptation of β-lactamases to different selective pressures, originating from the introduction of the various classes of β-lactam drugs and by the impact of Zn(II) starvation, can be observed and analyzed along the timeline of first detection of variants within the largest enzyme families, as we have reviewed in Section 4.7. Within the laboratory, this same characteristic can be exploited to perform in vitro evolution studies. These involve challenging bacteria expressing libraries of variants, generated either by random mutagenesis or site-saturation approaches, with conditions that surpass the resistance granted by the original enzyme. Both with the natural variants and those obtained from in vitro experiments, important insights can be obtained into the molecular mechanisms allowing adaptation of the enzyme to challenges such as the hydrolysis of a poor substrate.
Mutations have pleiotropic effects, being able to alter catalytic efficiency, affinity to inhibitors, stability, dynamics, folding rates, expression levels, processing and secreation to the periplasm and metal binding affinities (which are relevant for MBLs).1225, 1226 In addition, many mutations are deleterious for the activity or stability of the proteins, severely impairing their fitness in a biological context. However, some deleterious mutations can be advantageous when combined with others. This evidences the existence of epistatic interactions in evolution355, 1227–1231 and partially accounts for the observation that different families of B1 MBLs evolve by accumulation of different mutations, as discussed in Section 4.7. Thus, the understanding of epistatic interaction between mutations together with their impact in different protein features helps to describe the adaptive landscape of a protein under certain external, sometimes challenging circumstances (concentration of antibiotic, temperature, presence of an inhibitor, Zn(II) starvation, etc). Directed evolution also allow to select the optimal variants in their physiological environment, providing lessons that may help understanding the evolution in the clinics, provided the environment for selection is adequately chosen in the laboratory.
These type of studies in MBLs can be classified into two categories: (1) evolution experiments aimed to improve or study the evolutionary features of existing MBLs or (2) generation of MBL activity in a protein scaffold which is not able to hydrolyze β-lactams.
12.1. Evolution of MBLs
In 2004, Hall reported an in vitro evolution study of IMP-1to explore its ability to further improve its ability to confer imipenem resistance.1232 Different libraries with ca. 107 transformants each were generated with an average of 1.2 mutations per gene by using an error-prone polymerase and screened for imipenem resistance. Since none of the libraries resulted in variants with improved MIC, it was concluded that IMP-1 did not have the potential to evolve further resistance to imipenem. These results are in line with the resistance profile of the different clinical IMP alleles discussed in Section 4.7: practically none of the 80 IMP alleles provides more resistance to imipenem compared to IMP-1. Intriguingly, several alleles have evolved more resistance to other, more modern carbapenems, as recently highlighted for the subgroup IMP-1 by Crowder, Bonomo and coworkers.395 This may be due to the evolutionary pressure imposed by the new carbapenems or to the possibility that the IMP scaffold has exhausted its capacity to evolve further resistance to imipenem.
Directed evolution of BcII by DNA shuffling towards a poorly hydrolyzed substrate, cephalexin, resulted in an enzyme which increased the catalytic efficiency against this cephalosporin by one order of magnitude.274 This gain of function was not in detriment of the hydrolytic capabilities towards other substrates, therefore resulting in an enzyme with a broadened substrate spectrum. The evolved variant (M5) included four mutations outside the active site, one of which corresponds to a second sphere residue (G262S) (Figure 72). The single G262S mutation increases the activity in vitro by changing the hydrogen bond network at the active site and the flanking loops,354 showing a high epistatic interaction with mutation N70S. These two mutations have been shown to increase the flexibility of the active-site loops L3 and L10 by NMR spectroscopy,347 accounting for the expansion of the substrate profile of the evolved enzyme and revealing the high plasticity of the MBL scaffold to evolve new functions. Mutation G262S is also relevant in shaping the substrate spectrum of the IMP family1233 (Section 4.7). Reconstruction of the possible evolutionary pathways reveals that the accumulation of mutations cannot occur in a random order, but is highly conditioned by strong epistatic interactions.355 This study also revealed that the accumulation of some mutations resulted in increased resistance but in spite of these variants being equally active in vitro. In other words, the catalytic efficiency cannot account for the MIC data in E. coli. Instead, measurement of enzymatic activity in an environment resembling bacterial periplasm could provide an accurate description of the resistance profile. This gap between in vitro and in cell studies is due to the different Zn(II) binding affinities of the enzymes. In vitro assays are always performed in excess Zn(II) conditions that ensure that the active site of the MBLs are fully metallated, but in the periplasm the amount of metallated enzyme depends on the Zn(II) availability. As a result, enzyme variants with impaired Zn(II) efficiency provide less resistance since the actual concentration of active, metallated enzyme is lower. This work also showed that the evolved M5 variant had an improved affinity for Zn(II) with respect to BcII.
Figure 72.
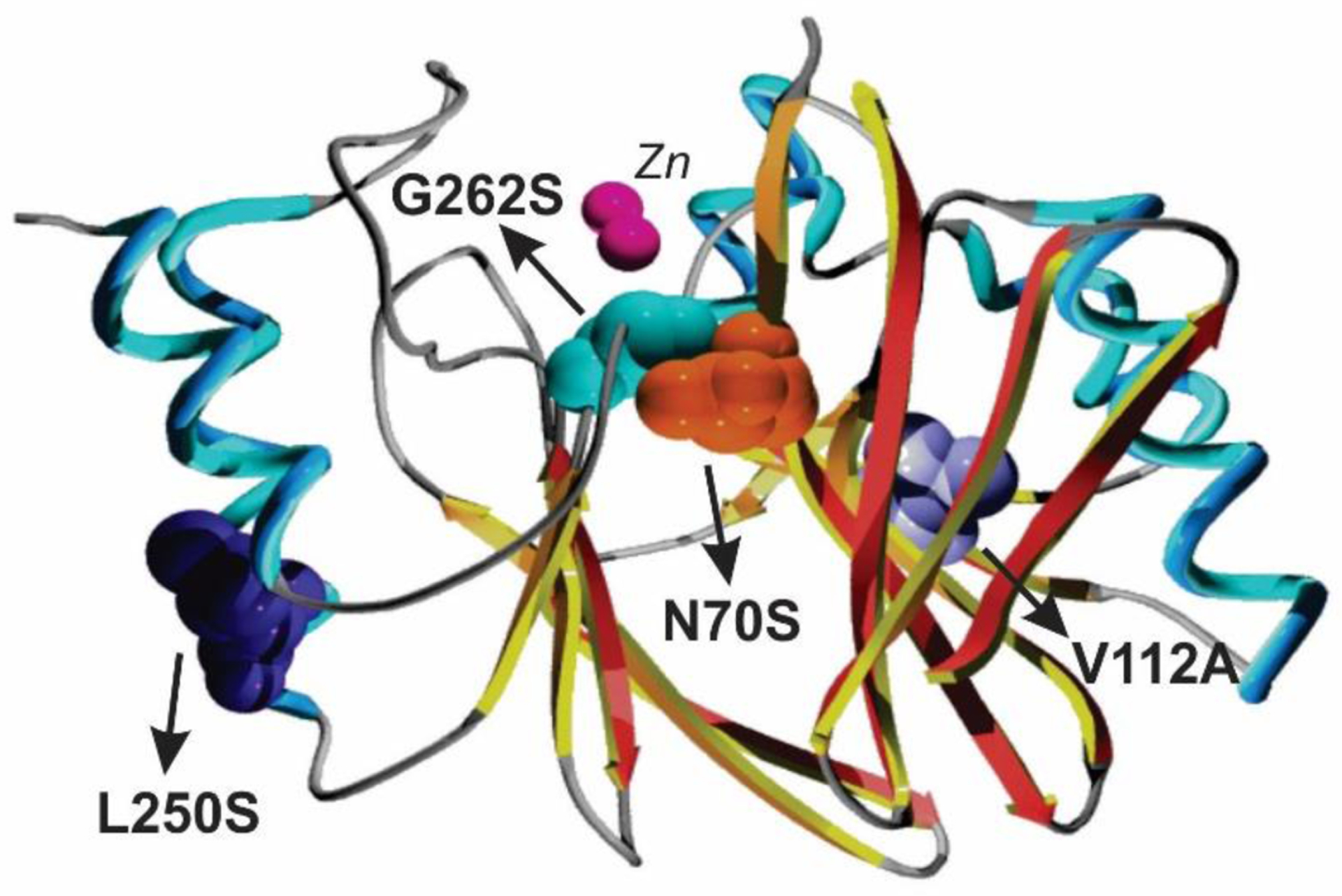
Structure of evolved variant of BcII, M5 (PDB 3FCZ). Mutations with respect to wild type BcII are indicated as spheres. Zinc ions are shown as magenta spheres.
Phage display technology was also applied to perform directed evolution on BcII by Galleni and Fastrez, which resulted in different variants with increased activity against benzylpenicillin distributed in the protein structure, all of them being destabilizing with respect to wt BcII.1234
Schenk and Ollis performed directed evolution on the B3 enzyme AIM-1 by error-prone PCR using cefoxitin as the antibiotic for selection. Initial rounds of selection were performed by sublethal concentrations of cefoxitin to induce neutral drift, and then higher concentrations of the antibiotic were employed.556 This resulted in a large number of mutations, but F114L turned to be the most relevant, broadening the resistance profile of AIM-1 by improving the catalytic efficiency towards antibiotic from different families.
There are some important lessons that can be learned from these directed evolution experiments: (1) they can predict the limits of evolution of a given MBL scaffold towards specific antibiotics, since epistatic effects limit the number of mutations that can accumulate and provide fitness at the same time, (2) they are useful in discovering mutational hotspots that are hard (or impossible) to identify rationally as drivers leading to optimized resistance levels.
12.2. in vitro generation of MBL activity
There have been several attemps to generate MBL activity into scaffolds unable to hydrolyze β-lactams. The first successful report relied on exploiting one member of the MBL superfamily, glyoxalase II, that shares the same protein fold and a metal binding motif. Kim, Benkovic and coworkers1235 redesigned rationally the active site of glyoxalase II by removing the substrate recognition domain and by grafting the metal ligand set and the loops of IMP-1. This protein scaffold was the starting point for directed evolution using cefotaxime as selector antibiotic. The evolved protein (evMBL8) displayed kcat/KM values of 102 against cefotaxime.
The Tezcan lab developed MBL activity on a different protein scaffold by using a supramolecular assembly approach.1236 This was pursued by assembling a four-helix bundle scaffold (based on cytochrome cb562) that can self-assemble into a tetrameric arrangement with Zn(II) binding sites with His and Glu ligands at the interfaces between monomer and a solvent molecule. Engineering this site resulted in an artificial metalloenzyme able to confer resistance to ampicillin in E. coli cells. Directed evolution experiments on this protein1237 revealed that structural dynamics in the adjacencies of the metal site are essential for the improvement of β-lactamase activity, in line with previous findings on natural MBLs.347
13. Can we fill the gap between in vitro and in vivo experiments?
As discussed along this review, the study of β-lactamases is of interest to different fields, from clinical microbiology to biological inorganic chemistry, and as a consequence, there are different languages involved and communication barriers that have not been necessarily sorted out despite many years of research. In addition, there are different observables that need to be correlated or at least compared on a similar basis. Clinicians are interested in knowing the minimum inhibitory concentration (MIC) of an antibiotic able to kill a bacterial strain expressing a β-lactamase, while biochemists are interested in determining kinetic parameters in the test tube and the structural determinant of function. Unfortunately, the most active enzymes are not necessarily those able to provide the largest resistance. There are many issues that need to be taken into account for this comparison. The first aspect to be considered is the impact of the permeability barrier of the outer membrane in Gram-negative bacteria, that slows down the diffusion of the different antibiotics to reach the target enzyme. This aspect has been addressed by Zimmermann and Rosselet, who combined for the first time the Michaelis-Menten enzymatic treatment with the Fick diffusion model to predict the behavior in bacteria.1238 Later, Nikaido and Normark1239 refined this model and proposed a new parameter, the “target access index”, that was later analyzed and improved by Frère, Joris and coworkers.1240
A second aspect that deserves attention is the impact of the physiological environment on the β-lactamase itself. This aspect has been addressed in general by Pielak, Selenko and others,1241–1243 which involves considering the effect of macromolecular crowding, ionic strength, the interaction with other molecules not present in the in vitro studies, among others. These differences have an impact not only on the enzymatic activity, but also on the protein stability (which may be exposed to the action of proteases, highly abundant in the periplasmic space). In the particular case of MBLs, in order to be active, the enzymes need to be properly metallated, and this represents an additional factor to be considered which may ultimately determine the fate of the enzyme in this cellular compartment. As already discussed in Section 10, apo MBLs are sTable in solution in vitro, while they are degraded by periplasmic proteases in the cell. This inconsistency claims for parallel studies in vitro and in the cell.
To address this aspect, Vila, Gonzalez and coworkers have developed a simple experimental approach to measure the activity of MBL in periplasmic extracts (in periplasma) and retrieve quantitative biochemical and biophysical parameters.355, 464, 1228 This approach also enables examining the impact of expression in different bacteria. This was relevant in the case of SPM-1, expressed only in P. aeruginosa. Activities measured in periplasmic extracts from P. aeruginosa normalized relative to the concentration of enzyme determined by immunoblotting could account for the MIC levels measured in the same bacterial host.464 This enables an estimation of apparent kcat and KM values. These values were shown to correlate better with MIC values than those determined in vitro.355 In the case of evolved BcII variants, the differences could be accounted for by changes in the Zn(II) binding affinity and by the impact of available Zn(II) under physiological conditions. The amount of Zn(II) rich MHB medium, used for MIC determinations, ranges between 15 and 25 μM, depending on the vendor.355, 928, 1162, 1244 This is a source of discrepancy that poses a dilemma: which is the adequate Zn(II) concentration to be used? Without a simple and unifying answer, and given that Zn(II) concentrations are decreased during an infection due to the process of nutritional immunity,311 it is mandatory to test different Zn(II) concentrations in these studies.452 An alternative approach involves measuring the metal concentration in the tissue or body fluid in which the infection occurs and performing in vitro susceptibility tests with similar levels of Zn(II). Noteworthy, allelic variants or MBL mutants obtained in the laboratory granting considerable levels of resistance under canonical Zn(II)-replete conditions, may exhibit a differential behavior in zinc-scarce media, leading to misinterpretations.300, 452, 464 Finally, enzymes with high catalytic efficiencies in vitro may not confer adequate resistance levels if the expression of the enzyme generates a fitness cost to the host, is not correctly processed, exported, etc.430, 1061, 1063
There are two approaches to determine the stability of MBLs under physiological conditions. Thermodynamic stability can be determined in periplasmic extracts by measuring the activity of the enzyme after incubation at different temperatures for a fixed period of time. This enables the quantitation of an apparent TM in periplasma that can be compared with the in vitro TM.355 More relevant to the resistance is the measurement of the kinetic stability, that can be quantitated by measuring the periplasmic degradation of the apo MBL by immunoblotting after treatment with a chelating agent.421, 452 The degradation process can be fit to a single exponential curve, providing a t1/2 that provides information about the lifetime of an MBL upon Zn(II) starvation conditions, that is a critical factor for these enzymes to confer resistance.
In the case of membrane-anchored lactamases, such as NDM variants, a similar approach has been devised to study these proteins in a medium mimicking as close as possible their physiological environment. Bacterial spheroplasts are devoid of soluble, periplasmic lactamases, but retain the membrane-bound NDM-1.421 These preparations can be exploited to retrieve reliable biochemical and biophysical parameters, especially when the protein-membrane interactions are expected to affect the enzyme performance.1245 An alternative, more controlled, approach includes the insertion of recombinant lipidated MBLs in artificial liposomes. This strategy has allowed the identification of specific interactions between the globular domain of NDM-1 and the bacterial outer membrane.476
The field of biochemistry under physiological conditions is blooming now, and there will be more biophysical tools to address these aspects in the coming years. We envisage that these approaches will have a great impact on the study of MBL activity and evolution in the near future.
14. Future Perspectives and Concluding Remarks
The challenge of understanding the diverse facets underlying the action of MBLs as a powerful machinery conferring resistance to antibiotics has been fascinating, and has claimed for efforts from different disciplines. The initial report of an MBL, BcII, dates back to 1956, while its characterization as such from Sabath and Abraham was achieved in 1966. During these six decades of research, the three latest decades have witnessed the largest grow on MBL research accompanying their clinical dissemination. At this moment, the structural determinants of function of main MBLs are well known, as well as the essential features of their mechanism of action against different antibiotics. Among the thousands of tested inhibitors, the boronate-based compounds offer a promising future, as well as the possibility of using cefiderocol. However, it is clear that the IMP, VIM, and NDM enzymes continue to evolve with different driving forces, providing different scaffolds that not only increase resistance, but could also provide future escape mechanisms to the newly developed antibiotics. There is an urgent need to improve our understanding of the evolutionary landscapes of these clinically relevant MBL families, as well as to keep an eye alert on the environmental niches of related resistance genes that may offer surprises, as was the case of the membrane-bound NDM.
The understanding of the MBL physiology in the bacterial cell requires solid efforts, from the behavior under metal-starvation conditions, their adaptation to different bacterial hosts and the dissemination mediated by Outer Membrane Vesicles. This aspect has been mostly overlooked, and the knowledge gained on the biochemistry of these enzymes should be reinforced by going deeper on the birth, life and death of these enzymes within and outside the bacterial cell. This knowledge could provide alternative to conventional inhibitors to thwart the scourge that these enzymes represent to maintain the potential of antibiotics to save lives.
Figure 5.
a) Structural comparison of the global fold of the transpeptidase domain of a PBP enzyme (PBP2a – PDB 3ZG0) and SBLs from classes A (KPC-2 – PDB 20V5), C (PDC-3 – PDB 4GZB) and D (OXA-10 – PDB 1K55), and a class B enzyme, i.e., an MBL (NDM-1 – PDB 4EYL). The Ω-loop region in SBLs are highlighted in yellow. b) Active sites of class A, C and D SBLs, with key catalytic residues displayed as sticks.
Figure 24.
Structures of VIM-1 (a, PDB 5N5G) and VIM-2 (b, PDB 1KO3) highlighting the positions presenting sequence variation within the VIM-1 and VIM-2 enzyme groups. The positions are colored in the cartoon representation according to increasing absolute frequency of mutation within VIM enzymes belonging to each group. The side chains of those positions with a frequency of substitution in the top 4 categories are shown as sticks.
Figure 34.
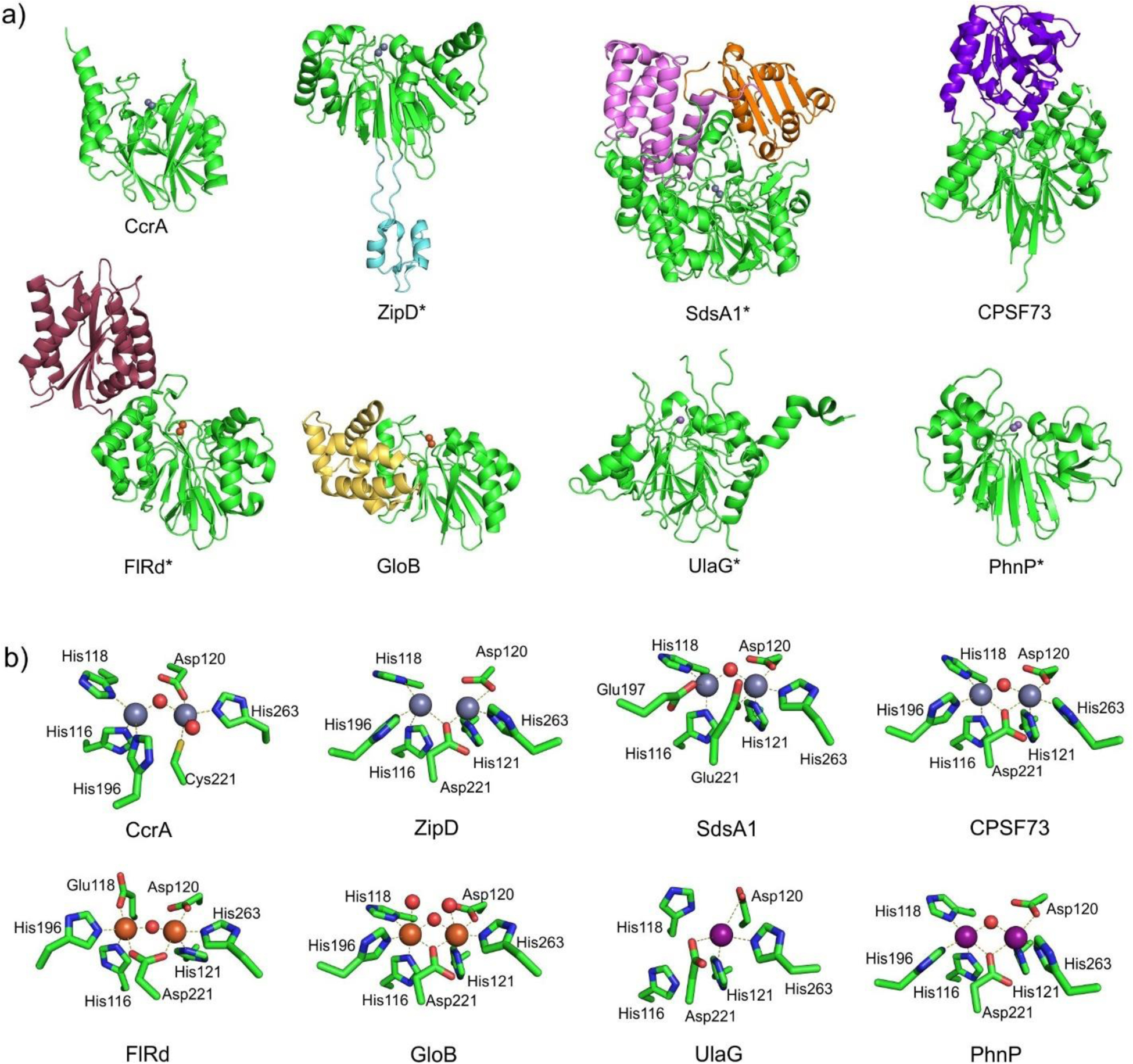
a) Comparison of the general structures of various members of the MBL superfamily. For each protein, the characteristic MBL fold domain is indicated in green, while additional domains present in some MBL superfamily members are highlighted in other colors. Metal ions within the active site are shown as spheres, and the Zn2 (or equivalent) site is oriented towards the front. Proteins names ending in an asterisk indicate that the physiological form of protein comprises homodimers or other homooligomers, which are not shown. b) Active-site metal coordination spheres for the MBL superfamily members indicated in panel a). Zn(II), Fe(II)/Fe(III) and Mn(II) ions are represented as grey, orange and light violet spheres, respectively, while water molecules / hydroxide ions are shown as red spheres. PDB codes for the structures are: CcrA – 1ZNB, ZipD – 2CBN, SdsA1 – 2CFU, CPSF73 – 2I7V, FlRd – 4D02, GloB – 2QED, UlaG – 2WYM, PhnP – 3P2U.
Figure 42.
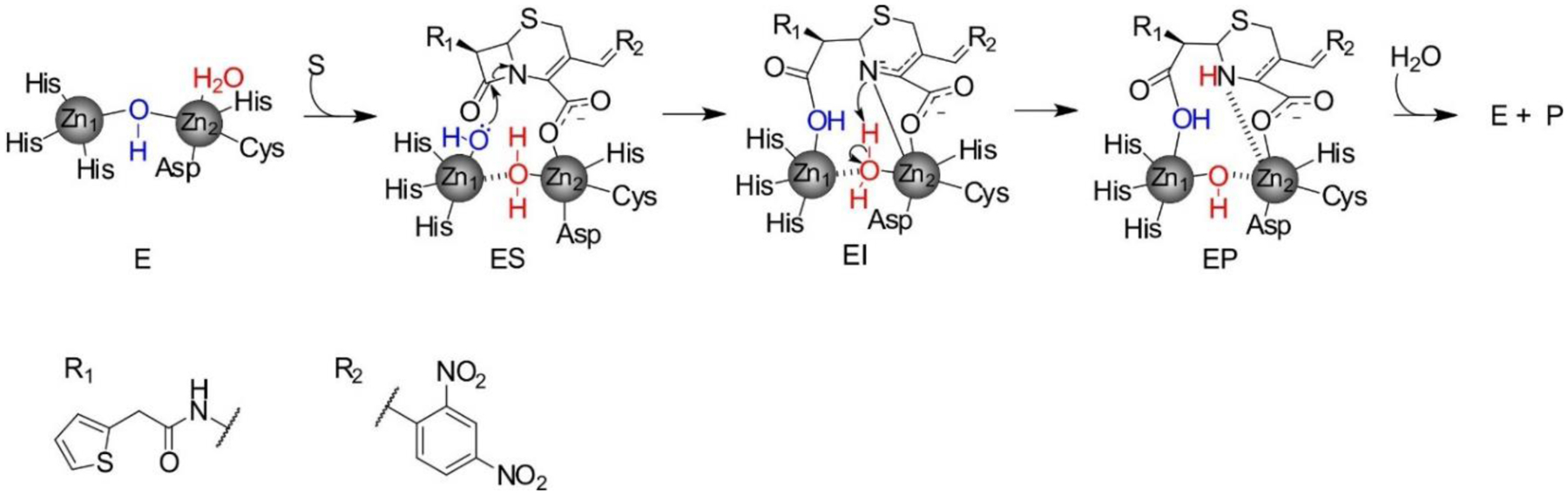
Mechanism of nitrocefin hydrolysis by bi-Zn(II) MBLs. Based on the work from Wang, Fast and Benkovic242.
Figure 46.
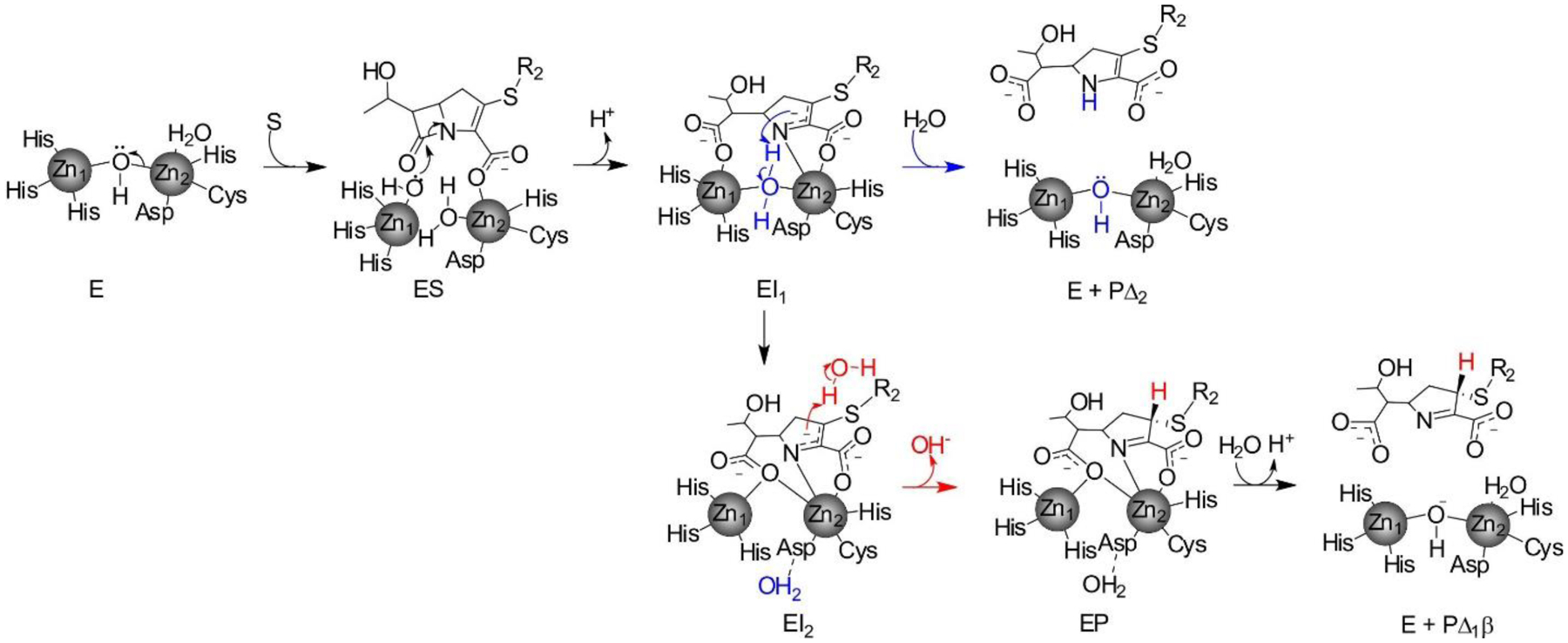
Mechanism of carbapenem hydrolysis by di-Zn(II) MBLs. Reproduced with permission from reference241 Lisa, Palacios and coworkers. Copyright 2017 Springer Nature under Creative Commons Attribution 4.0 International License https://creativecommons.org/license/by/4.0/.
Figure 48.
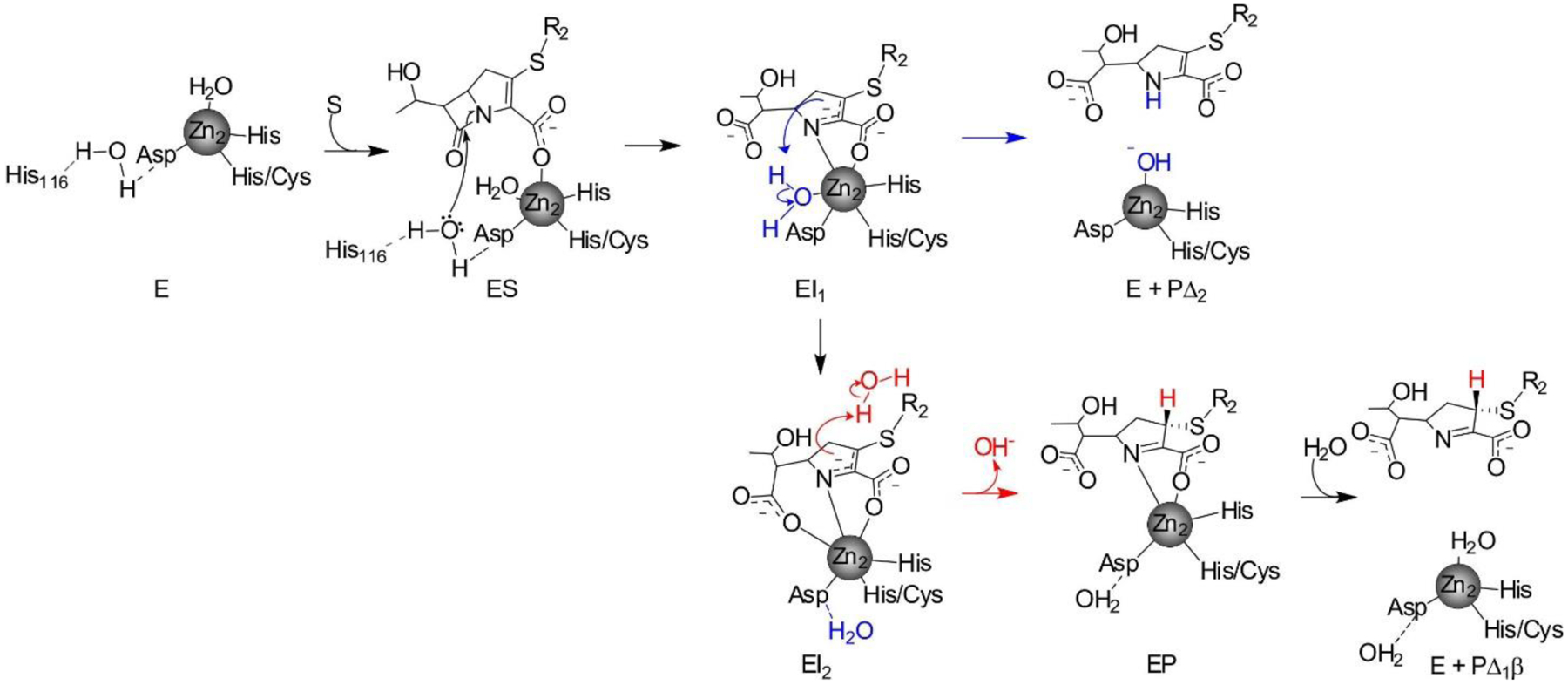
Mechanism of carbapenem hydrolysis by mono-Zn(II) MBLs. Reproduced with permission from reference241 Lisa, Palacios and coworkers. Copyright 2017 Springer Nature under Creative Commons Attribution 4.0 International License https://creativecommons.org/license/by/4.0/.
Figure 49.
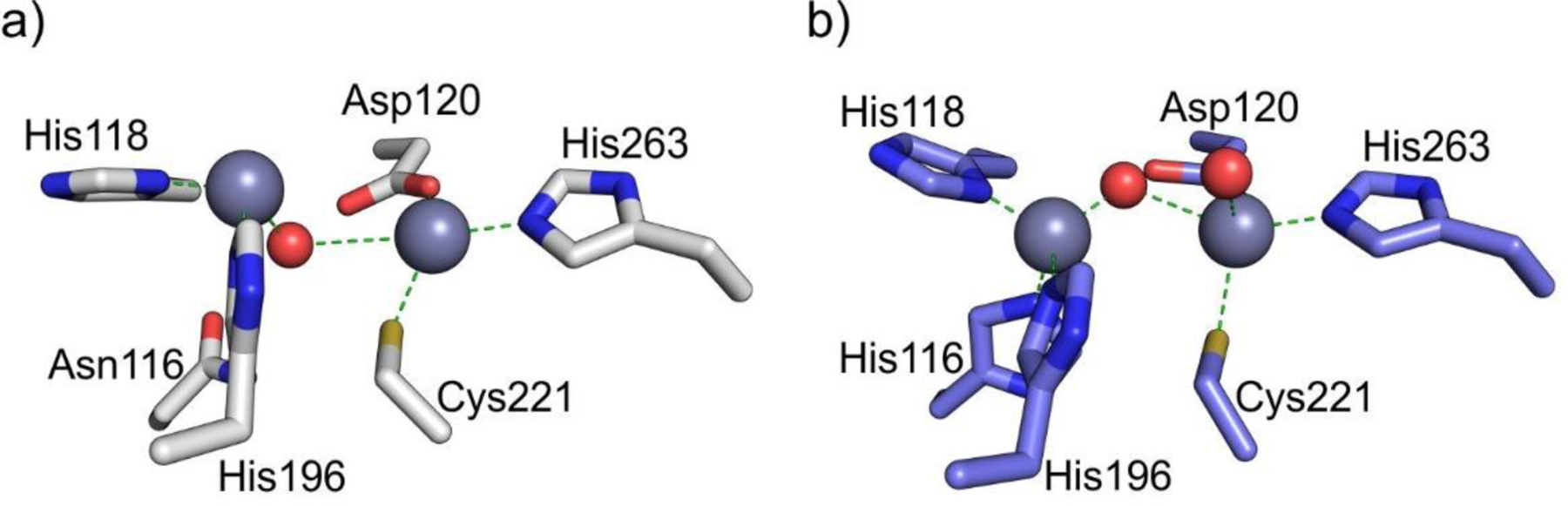
a) Structure of the bi-Zn(II) form of CphA (PDB 3F9O). b) Structure of NDM-1 (PDB 3SPU, chain B). Zn(II) ions and water molecules / hydroxide ions are shown as grey and red spheres, respectively, while protein residues are shown as sticks. Interactions between the Zn(II) ions and their coordination residues are shown as green dashed lines.
Table 1.
Zn(II)-ligand, Zn(II)-Zn(II) and Zn(II)-water distances in the active site of MBLs in the unbound forms obtained from X-ray crystallographic structures.
| PDB code | Enzyme (subclass) | Resolution (Å) | Reference | Chain | Zn1-Zn2 | Wat1-Zn1 | Wat1-Zn2 | Wat2-Zn2 |
|---|---|---|---|---|---|---|---|---|
| 1BC2 | BcII (B1) | 1.90 | 226 | A | 3.8 | 1.9 | 2.5 | 2.6 |
| B | 4.4 | 1.9 | 3.1 | 2.7 | ||||
| 5ZGZ | NDM-1 (B1) | 0.95 | 258 | A | 3.6 | 2.0 | 2.0 | 2.2 |
| 5N5G | VIM-1 (B1) | 1.29 | 259 | A | 3.6 | 1.9 | 2.2 | 2.1 |
| 6CQS | SPS-1 (B1) | 1.70 | 230 | A | 3.5 | - | - | 2.7 |
| 2FM6 | L1 (B3) | 1.75 | 260 | A | 3.5 | 1.9 | 1.9 | 2.5 |
| B | 3.5 | 1.8 | 2.1 | 2.5 | ||||
| 3LVZ | BJP(B3) | 1.4 | 261 | A | 3.4 | 1.9 | 2.1 | 2.5 |
| B | 3.4 | 2.0 | 2.0 | 2.4 | ||||
| 5K0W | GOB-18 (B3) | 2.61 | 254 | A | 3.5 | 2.1 | 2.1 | - |
| B | 3.8 | 2.3 | 2.2 |
Table 3.
Relevant inter-Zn(II), Zn(II)-water, amino acid side chains-water, and hydrolysis product-enzyme distances in the active site of MBLs obtained from X-ray crystallographic structures of EP(a) or EI(b) complexes. Res. : crystal structure resolution (in Å); ch. : crystal structure chain; ref. : reference publication for the crystal structure.
| PDB code | Enzyme | Res.(Å) | Hydrolyzed β-lactam (adduct) | Ref. | Ch. | Zn1-Zn2 | Wat1-Zn1 | Wat1-Zn2 | Wat2-Zn2 | C7O-Zn1 | C7O-Zn2 | C3O-Zn2 | N4-Zn2 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 3Q6X | NDM-1a (B1) | 1.3 | ampicillin (EP) | 258 | A | 4.6 | 2.1 | 3.0 | - | 2.4 | 4.3 | 2.2 | 2.2 |
| B | 4.6 | 2.0 | 3.0 | - | 2.5 | 4.3 | 2.2 | 2.2 | |||||
| 4EYF | NDM-1a (B1) | 1.8 | benzylpenicillin (EP) | 229 | A | 4.6 | 1.9 | 3.1 | - | 2.5 | 4.4 | 2.2 | 2.2 |
| B | 4.6 | 2.0 | 3.1 | - | 2.4 | 4.4 | 2.1 | 2.2 | |||||
| 4EY2 | NDM-1a (B1) | 1.2 | methicillin (EP) | 229 | A | 4.6 | 2.0 | 3.0 | - | 2.5 | 4.4 | 2.2 | 2.1 |
| B | 4.6 | 2.0 | 3.0 | - | 2.5 | 4.4 | 2.2 | 2.2 | |||||
| 4EYB | NDM-1a (B1) | 1.2 | oxacillin (EP) | 229 | A | 4.5 | 2.0 | 2.9 | - | 2.5 | 4.4 | 2.2 | 2.1 |
| B | 4.6 | 2.0 | 3.0 | - | 2.5 | 4.4 | 2.1 | 2.1 | |||||
| PDB code | Enzyme | Res.(Å) | Hydrolyzed β-lactam (adduct) | Ref. | Ch. | Zn1-Zn2 | Wat1-Zn1 | Wat1-Zn2 | Wat2-Zn2 | C8O-Zn1 | C8O-Zn2 | C4O-Zn2 | N5-Zn2 |
| 2AIO | L1 (B3)a | 1.7 | moxalactam (EP) | 330 | A | 3.7 | 2.0 | 2.2 | - | 2.4 | 4.2 | 2.3 | 2.4 |
| 4RL0 | NDM-1b (B1) | 1.3 | cefuroxime (EI) | 329 | A | 3.8 | 2.0 | 2.2 | - | 2.8 | 4.2 | 2.1 | 2.4 |
| B | 3.8 | 2.0 | 2.2 | - | 2.9 | 4.2 | 2.2 | 2.3 | |||||
| 4RL2 | NDM-1a (B1) | 2.0 | cephalexin (EP) | 329 | A | 4.5 | 1.8 | 3.0 | - | 2.5 | 4.3 | 2.3 | 2.4 |
| B | 4.5 | 2.0 | 2.8 | - | 2.4 | 4.3 | 2.3 | 2.4 | |||||
| PDB code | Enzyme | Res.(Å) | Hydrolyzed β-lactam (adduct) | Ref. | Ch. | Zn1-Zn2 | Wat1-Zn1 | Wat1-Zn2 | Wat2-Zn2 | C7O-Zn1 | C7O-Zn2 | C3O-Zn2 | N4-Zn2 |
| 5N5I | VIM-1a (B1) | 2.2 | meropenem (EP) | 259 | A | 3.5 | 1.8 | 2.3 | - | 6.0; 6.4 | 6.3; 5.8 | 2.6 | 3.0 |
| 4EYL | NDM-1a (B1) | 1.9 | meropenem (EP) | 229 | A | 4.0 | - | - | - | 2.3 | 2.5 | 3.0 | 2.2 |
| B | 3.9 | - | - | - | 2.2 | 2.7 | 2.9 | 2.3 | |||||
| 4RBS | NDM-1a (B1) | 2.4 | meropenem (EP) | To be published (Kim, Y. et al) | A | 4.0 | - | - | - | 2.0 | 3.3 | 3.0 | 2.1 |
| B | 4.0 | 2.2 | 3.0 | 3.1 | 2.1 | ||||||||
| 5YPN | NDM-1 (B1) b | 2.12 | meropenem (EI2) | 328 | A | 4.1 | - | - | - | 2.0 | 2.6 | 3.0 | 2.0 |
| B | 4.3 | - | - | - | 2.2 | 2.9 | 2.7 | 2.0 | |||||
| 5YPM | NDM-1 (B1) b | 2.15 | meropenem (EI1) | 328 | A | 4.2 | - | - | - | 2.1 | 3.2 | 2.6 | 2.0 |
| B | 4.2 | - | - | - | 2.1 | 3.0 | 2.4 | 2.0 | |||||
| C | 4.2 | - | - | - | 2.1 | 3.1 | 2.5 | 1.9 | |||||
| D | 4.2 | - | - | - | 2.0 | 3.2 | 2.3 | 2.0 | |||||
| E | 4.2 | - | - | - | 2.2 | 3.1 | 2.4 | 2.0 | |||||
| F | 4.3 | - | - | - | 2.1 | 3.2 | 2.4 | 2.0 | |||||
| G | 4.2 | - | - | - | 2.3 | 2.8 | 2.3 | 1.9 | |||||
| H | 3.9 | - | - | - | 2.3 | 3.0 | 2.6 | 2.0 | |||||
| Wat1-H118 | Wat1-D120 | Wat1-H196 | Wat1-Zn2 | Wat2-Zn2 | C3O-Zn2 | N4-Zn2 | |||||||
| 1X8I | CphA (B2)a | 1.9 | biapenem (EP) | 245 | A | 2.7 | 2.8 | 4.0 | 3.4 | - | 2.4 | 2.2 |
Acknowledgments
We want to thank all members of the Metal Team at Rosario who contributed for more than 20 years to the study of metallo-β-lactamases, as well as to all coworkers throughout the world, particularly to Robert Bonomo, who has been provided friendship and a critical perspective along the years. We are deeply thankful to the support of our home institutions, CONICET and the University of Rosario, and the following funding agencies that supported our research: Fundacion Antorchas, ANPCyT, MinCYT, the Howard Hughes Medical Institute, the John Simon Guggenheim Foundation and the US National Institutes of Health (NIAID grant 2R01AI100560-06A1).
Biographies
Guillermo Bahr was born in Chajarí, Entre Ríos, Argentina in 1988. He obtained his B.S. degree in Biotechnology from the National University of Rosario (UNR) in 2013, and his PhD degree in Biological Sciences from UNR in 2018, under the supervision of Prof. Alejandro J. Vila at the Institute of Molecular and Cell Biology of Rosario (IBR). He is currently a postdoctoral researcher at the Vila lab, and a TA in the Biophysics department in the Faculty of Pharmacy and Biochemistry at the UNR. His research is focused on the biochemical and biophysical characterization of metallo-β-lactamases, and he is also interested in bioinformatics and the applications of computer programming in science.
Lisandro J. González was born in Rosario, Santa Fé, Argentina in 1979. After his B.S. degree in Biotechnology from the National University of Rosario (UNR) in 2006, he graduated as a PhD in Biological Sciences (UNR, 2012) and held a postdoctoral position, both under the supervision of Prof. Alejandro J. Vila at the Institute of Molecular and Cell Biology of Rosario (IBR). He currently holds a tenured position as an associate researcher (CONICET), developing his activities at IBR. With more than ten years of experience in the field of metallo-β-lactamases, Lisandro’s major interests in the field are focused to fill the gap between in vivo and in vitro observations, aimed to broaden the understanding of bacterial resistance in more complex contexts such as the bacterial host or upon conditions of pathogenesis.
Alejandro J. Vila received his B.S. degree in chemistry in 1986, and his PhD degree in synthetic organic chemistry from the National University of Rosario (UNR) in 1990 under the supervision of Professor Manuel González-Sierra. He was a postdoctoral fellow at the Department of Chemistry in the University of Florence, Italy with Ivano Bertini, where he started working in NMR of metalloproteins. In 1993 he started his independent career as Assistant Professor of Biophysics at the University of Rosario, studying the electronic structure of copper proteins. In 2001 he joined the Institute of Molecular and Cellular Biology of Rosario (IBR). He is the leader of the Metalloprotein lab, head of the Protein NMR Facility, Full Professor of Biophysics at the University of Rosario and Superior Researcher of CONICET. His lab studies metalloproteins (mostly zinc and copper), and metal-ion homeostasis and trafficking. He has received numerous awards, including the Howard Hughes Medical Institute International Research Fellowship, the John Simon Guggenheim Memorial Foundation, the E.P.Abraham Award, and the Ivano Bertini Award, for scientific achievements in copper research. Alejandro is not only passionate about metalloproteins, but also about literature, cooking, red wine and music.
References
- 1.Fleming A On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to Their Use in the Isolation of B. Influenzæ. British Journal of Experimental Pathology 1929, 10, 226–236. [Google Scholar]
- 2.Fleming A Streptococcal Meningitis Treated with Penicillin.: Measurement of Bacteriostatic Power of Blood and Cerebrospinal Fluid. The Lancet 1943, 242, 434–438. [Google Scholar]
- 3.Abraham EP History of B-Lactam Antibiotics. In Antibiotics: Containing the Beta-Lactam Structure, Demain AL; Solomon NA, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 1983; pp 1–14. [Google Scholar]
- 4.Bennett JW; Chung KT Alexander Fleming and the Discovery of Penicillin. Adv. Appl. Microbiol 2001, 49, 163–184. [DOI] [PubMed] [Google Scholar]
- 5.Bush K; Bradford PA Beta-Lactams and Beta-Lactamase Inhibitors: An Overview. Cold Spring Harb Perspect Med 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fisher JF; Meroueh SO; Mobashery S Bacterial Resistance to Beta-Lactam Antibiotics: Compelling Opportunism, Compelling Opportunity. Chem. Rev 2005, 105, 395–424. [DOI] [PubMed] [Google Scholar]
- 7.Bonomo RA Beta-Lactamases: A Focus on Current Challenges. Cold Spring Harb Perspect Med 2017, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Llarrull LI; Testero SA; Fisher JF; Mobashery S The Future of the Beta-Lactams. Curr. Opin. Microbiol 2010, 13, 551–557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.O’Neill J Tackling a Crisis for the Health and Wealth of Nations. Review on Antimicrobial Resistance 2014. [Google Scholar]
- 10.Langford BJ; So M; Raybardhan S; Leung V; Soucy JR; Westwood D; Daneman N; MacFadden DR Antibiotic Prescribing in Patients with Covid-19: Rapid Review and Meta-Analysis. Clin. Microbiol. Infect 2021, 27, 520–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Abraham EP; Newton GG A Comparison of the Action of Penicillinase on Benzylpenicillin and Cephalosporin N and the Competitive Inhibition of Penicillinase by Cephalosporin C. Biochem. J 1956, 63, 628–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Crompton B; Jago M; Crawford K; Newton GG; Abraham EP Behaviour of Some Derivatives of 7-Aminocephalosporanic Acid and 6-Aminopenicillanic Acidas Substrates, Inhibitors and Inducers of Penicillinases. Biochem. J 1962, 83, 52–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sabath LD; Abraham EP Zinc as a Cofactor for Cephalosporinase from Bacillus cereus 569. Biochem. J 1966, 98, 11C–13C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Boyd SE; Livermore DM; Hooper DC; Hope WW Metallo-Beta-Lactamases: Structure, Function, Epidemiology, Treatment Options, and the Development Pipeline. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Palacios AR; Rossi MA; Mahler GS; Vila AJ Metallo-Beta-Lactamase Inhibitors Inspired on Snapshots from the Catalytic Mechanism. Biomolecules 2020, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bush K; Bradford PA Epidemiology of Beta-Lactamase-Producing Pathogens. Clin. Microbiol. Rev 2020, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tooke CL; Hinchliffe P; Bragginton EC; Colenso CK; Hirvonen VHA; Takebayashi Y; Spencer J Beta-Lactamases and Beta-Lactamase Inhibitors in the 21st Century. J. Mol. Biol 2019, 431, 3472–3500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Linciano P; Cendron L; Gianquinto E; Spyrakis F; Tondi D Ten Years with New Delhi Metallo-Beta-Lactamase-1 (NDM-1): From Structural Insights to Inhibitor Design. ACS Infect Dis 2019, 5, 9–34. [DOI] [PubMed] [Google Scholar]
- 19.Ju LC; Cheng Z; Fast W; Bonomo RA; Crowder MW The Continuing Challenge of Metallo-Beta-Lactamase Inhibition: Mechanism Matters. Trends Pharmacol. Sci 2018, 39, 635–647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bebrone C Metallo-Beta-Lactamases (Classification, Activity, Genetic Organization, Structure, Zinc Coordination) and Their Superfamily. Biochem. Pharmacol 2007, 74, 1686–1701. [DOI] [PubMed] [Google Scholar]
- 21.Crowder MW; Spencer J; Vila AJ Metallo-Beta-Lactamases: Novel Weaponry for Antibiotic Resistance in Bacteria. Acc. Chem. Res 2006, 39, 721–728. [DOI] [PubMed] [Google Scholar]
- 22.Shi C; Chen J; Kang X; Shen X; Lao X; Zheng H Approaches for the Discovery of Metallo-Beta-Lactamase Inhibitors: A Review. Chem Biol Drug Des 2019, 94, 1427–1440. [DOI] [PubMed] [Google Scholar]
- 23.Palzkill T Metallo-Beta-Lactamase Structure and Function. Ann. N. Y. Acad. Sci 2013, 1277, 91–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Walsh TR; Toleman MA; Poirel L; Nordmann P Metallo-Beta-Lactamases: The Quiet before the Storm? Clin. Microbiol. Rev 2005, 18, 306–325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Silver LL; Bush K Antibiotics and Antibiotic Resistance p xi, 404 pages.
- 26.Walsh C Antibiotics That Act on Cell Wall Biosynthesis. Antibiotics: actions, origins, resistance ASM Press, Washington, DC: 2003, 23–49. [Google Scholar]
- 27.King DT; Sobhanifar S; Strynadka NC One Ring to Rule Them All: Current Trends in Combating Bacterial Resistance to the Beta-Lactams. Protein Sci 2016, 25, 787–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Papp-Wallace KM; Endimiani A; Taracila MA; Bonomo RA Carbapenems: Past, Present, and Future. Antimicrob. Agents Chemother 2011, 55, 4943–4960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Testero SA; Fisher JF; Mobashery S B-Lactam Antibiotics. In Burger’s Medicinal Chemistry and Drug Discovery, pp 257–402. [Google Scholar]
- 30.Dik DA; Fisher JF; Mobashery S Cell-Wall Recycling of the Gram-Negative Bacteria and the Nexus to Antibiotic Resistance. Chem. Rev 2018, 118, 5952–5984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cho H; Uehara T; Bernhardt TG Beta-Lactam Antibiotics Induce a Lethal Malfunctioning of the Bacterial Cell Wall Synthesis Machinery. Cell 2014, 159, 1300–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tomasz A The Mechanism of the Irreversible Antimicrobial Effects of Penicillins: How the Beta-Lactam Antibiotics Kill and Lyse Bacteria. Annu. Rev. Microbiol 1979, 33, 113–137. [DOI] [PubMed] [Google Scholar]
- 33.Baquero F; Levin BR Proximate and Ultimate Causes of the Bactericidal Action of Antibiotics. Nat. Rev. Microbiol 2021, 19, 123–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Neu HC Aztreonam Activity, Pharmacology, and Clinical Uses. Am. J. Med 1990, 88, 2S–6S; discussion 38S-42S. [DOI] [PubMed] [Google Scholar]
- 35.Retzlaff CL; Kussrow A; Schorkopf T; Saetear P; Bornhop DJ; Hardaway JA; Sturgeon SM; Wright J; Blakely RD Metallo-Beta-Lactamase Domain-Containing Protein 1 (MBLAC1) Is a Specific, High-Affinity Target for the Glutamate Transporter Inducer Ceftriaxone. ACS Chem Neurosci 2017, 8, 2132–2138. [DOI] [PubMed] [Google Scholar]
- 36.Rothstein JD; Patel S; Regan MR; Haenggeli C; Huang YH; Bergles DE; Jin L; Dykes Hoberg M; Vidensky S; Chung DS, et al. Beta-Lactam Antibiotics Offer Neuroprotection by Increasing Glutamate Transporter Expression. Nature 2005, 433, 73–77. [DOI] [PubMed] [Google Scholar]
- 37.Fleming A On the Antibacterial Action of Cultures of a Penicillium, with Special Reference to Their Use in the Isolation of B. influenzae. 1929. Bull World Health Organ 2001, 79, 780–790. [PMC free article] [PubMed] [Google Scholar]
- 38.Page MGP Beta-Lactam Antibiotics. In Antibiotic Discovery and Development, Dougherty TJ; Pucci MJ, Eds. Springer US: Boston, MA, 2012; pp 79–117. [Google Scholar]
- 39.Bush K B-Lactam Antibiotics: Penicillins. In Antibiotic and Chemotherapy, Finch RG; Greenwood D; Whitley RJ; Norrby SR, Eds. Elsevier Health Sciences: 2010; pp 200–225. [Google Scholar]
- 40.Abraham EP Cephalosporins 1945–1986. Drugs 1987, 34 Suppl 2, 1–14. [DOI] [PubMed] [Google Scholar]
- 41.Chaudhry SB; Veve MP; Wagner JL Cephalosporins: A Focus on Side Chains and Beta-Lactam Cross-Reactivity. Pharmacy (Basel) 2019, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Neu HC Relation of Structural Properties of Beta-Lactam Antibiotics to Antibacterial Activity. Am. J. Med 1985, 79, 2–13. [DOI] [PubMed] [Google Scholar]
- 43.Kahan JS; Kahan FM; Goegelman R; Currie SA; Jackson M; Stapley EO; Miller TW; Miller AK; Hendlin D; Mochales S, et al. Thienamycin, a New Beta-Lactam Antibiotic. I. Discovery, Taxonomy, Isolation and Physical Properties. J Antibiot (Tokyo) 1979, 32, 1–12. [DOI] [PubMed] [Google Scholar]
- 44.Kahan FM; Kropp H; Sundelof JG; Birnbaum J Thienamycin: Development of Imipenen-Cilastatin. J. Antimicrob. Chemother 1983, 12 Suppl D, 1–35. [DOI] [PubMed] [Google Scholar]
- 45.Weaver SS; Bodey GP; LeBlanc BM Thienamycin: New Beta-Lactam Antibiotic with Potent Broad-Spectrum Activity. Antimicrob. Agents Chemother 1979, 15, 518–521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kropp H; Gerckens L; Sundelof JG; Kahan FM Antibacterial Activity of Imipenem: The First Thienamycin Antibiotic. Rev Infect Dis 1985, 7 Suppl 3, S389–410. [DOI] [PubMed] [Google Scholar]
- 47.El-Gamal MI; Oh CH Current Status of Carbapenem Antibiotics. Curr. Top. Med. Chem 2010, 10, 1882–1897. [DOI] [PubMed] [Google Scholar]
- 48.Zhanel GG; Wiebe R; Dilay L; Thomson K; Rubinstein E; Hoban DJ; Noreddin AM; Karlowsky JA Comparative Review of the Carbapenems. Drugs 2007, 67, 1027–1052. [DOI] [PubMed] [Google Scholar]
- 49.Sykes RB; Bonner DP; Bush K; Georgopapadakou NH Azthreonam (SQ 26,776), a Synthetic Monobactam Specifically Active against Aerobic Gram-Negative Bacteria. Antimicrob. Agents Chemother 1982, 21, 85–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Breuer H; Cimarusti CM; Denzel T; Koster WH; Slusarchyk WA; Treuner UD Monobactams--Structure-Activity Relationships Leading to SQ 26,776. J. Antimicrob. Chemother 1981, 8 Suppl E, 21–28. [DOI] [PubMed] [Google Scholar]
- 51.Sykes RB; Cimarusti CM; Bonner DP; Bush K; Floyd DM; Georgopapadakou NH; Koster WM; Liu WC; Parker WL; Principe PA, et al. Monocyclic Beta-Lactam Antibiotics Produced by Bacteria. Nature 1981, 291, 489–491. [DOI] [PubMed] [Google Scholar]
- 52.Sykes RB; Bonner DP; Bush K; Georgopapadakou NH; Wells JS Monobactams--Monocyclic Beta-Lactam Antibiotics Produced by Bacteria. J. Antimicrob. Chemother 1981, 8 Suppl E, 1–16. [DOI] [PubMed] [Google Scholar]
- 53.Aoki H; Sakai H; Kohsaka M; Konomi T; Hosoda J Nocardicin A, a New Monocyclic Beta-Lactam Antibiotic. I. Discovery, Isolation and Characterization. J Antibiot (Tokyo) 1976, 29, 492–500. [DOI] [PubMed] [Google Scholar]
- 54.Imada A; Kitano K; Kintaka K; Muroi M; Asai M Sulfazecin and Isosulfazecin, Novel Beta-Lactam Antibiotics of Bacterial Origin. Nature 1981, 289, 590–591. [DOI] [PubMed] [Google Scholar]
- 55.Tanaka SK; Summerill RA; Minassian BF; Bush K; Visnic DA; Bonner DP; Sykes RB In Vitro Evaluation of Tigemonam, a Novel Oral Monobactam. Antimicrob. Agents Chemother 1987, 31, 219–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McNulty CA; Garden GM; Ashby J; Wise R Pharmacokinetics and Tissue Penetration of Carumonam, a New Synthetic Monobactam. Antimicrob. Agents Chemother 1985, 28, 425–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Wright AJ The Penicillins. Mayo Clin Proc 1999, 74, 290–307. [DOI] [PubMed] [Google Scholar]
- 58.Giamarellou H; Antoniadou A Antipseudomonal Antibiotics. Med Clin North Am 2001, 85, 19–42, v. [DOI] [PubMed] [Google Scholar]
- 59.Greenwood D B-Lactam Antibiotics: Cephalosporins. In Antibiotic and Chemotherapy, Finch RG; Greenwood D; Whitley RJ; Norrby SR, Eds. Elsevier Health Sciences: 2010; pp 170–199. [Google Scholar]
- 60.Marshall WF; Blair JE The Cephalosporins. Mayo Clin Proc 1999, 74, 187–195. [DOI] [PubMed] [Google Scholar]
- 61.Zhanel GG; Sniezek G; Schweizer F; Zelenitsky S; Lagace-Wiens PR; Rubinstein E; Gin AS; Hoban DJ; Karlowsky JA Ceftaroline: A Novel Broad-Spectrum Cephalosporin with Activity against Meticillin-Resistant Staphylococcus Aureus. Drugs 2009, 69, 809–831. [DOI] [PubMed] [Google Scholar]
- 62.Barbour A; Schmidt S; Rand KH; Derendorf H Ceftobiprole: A Novel Cephalosporin with Activity against Gram-Positive and Gram-Negative Pathogens, Including Methicillin-Resistant Staphylococcus Aureus (Mrsa). Int. J. Antimicrob. Agents 2009, 34, 1–7. [DOI] [PubMed] [Google Scholar]
- 63.Lovering AL; Gretes MC; Safadi SS; Danel F; de Castro L; Page MG; Strynadka NC Structural Insights into the Anti-Methicillin-Resistant Staphylococcus aureus (MRSA) Activity of Ceftobiprole. J. Biol. Chem 2012, 287, 32096–32102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Llarrull LI; Fisher JF; Mobashery S Molecular Basis and Phenotype of Methicillin Resistance in Staphylococcus aureus and Insights into New Beta-Lactams That Meet the Challenge. Antimicrob. Agents Chemother 2009, 53, 4051–4063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhanel GG; Chung P; Adam H; Zelenitsky S; Denisuik A; Schweizer F; Lagace-Wiens PR; Rubinstein E; Gin AS; Walkty A, et al. Ceftolozane/Tazobactam: A Novel Cephalosporin/Beta-Lactamase Inhibitor Combination with Activity against Multidrug-Resistant Gram-Negative Bacilli. Drugs 2014, 74, 31–51. [DOI] [PubMed] [Google Scholar]
- 66.Brook I Treatment of Anaerobic Infection. Expert Rev Anti Infect Ther 2007, 5, 991–1006. [DOI] [PubMed] [Google Scholar]
- 67.Zhanel GG; Golden AR; Zelenitsky S; Wiebe K; Lawrence CK; Adam HJ; Idowu T; Domalaon R; Schweizer F; Zhanel MA, et al. Cefiderocol: A Siderophore Cephalosporin with Activity against Carbapenem-Resistant and Multidrug-Resistant Gram-Negative Bacilli. Drugs 2019, 79, 271–289. [DOI] [PubMed] [Google Scholar]
- 68.Ito A; Nishikawa T; Matsumoto S; Yoshizawa H; Sato T; Nakamura R; Tsuji M; Yamano Y Siderophore Cephalosporin Cefiderocol Utilizes Ferric Iron Transporter Systems for Antibacterial Activity against Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2016, 60, 7396–7401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Ito-Horiyama T; Ishii Y; Ito A; Sato T; Nakamura R; Fukuhara N; Tsuji M; Yamano Y; Yamaguchi K; Tateda K Stability of Novel Siderophore Cephalosporin S-649266 against Clinically Relevant Carbapenemases. Antimicrob. Agents Chemother 2016, 60, 4384–4386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Dobias J; Denervaud-Tendon V; Poirel L; Nordmann P Activity of the Novel Siderophore Cephalosporin Cefiderocol against Multidrug-Resistant Gram-Negative Pathogens. Eur. J. Clin. Microbiol. Infect. Dis 2017, 36, 2319–2327. [DOI] [PubMed] [Google Scholar]
- 71.Riera E; Cabot G; Mulet X; Garcia-Castillo M; del Campo R; Juan C; Canton R; Oliver A Pseudomonas aeruginosa Carbapenem Resistance Mechanisms in Spain: Impact on the Activity of Imipenem, Meropenem and Doripenem. J. Antimicrob. Chemother 2011, 66, 2022–2027. [DOI] [PubMed] [Google Scholar]
- 72.El-Gamal MI; Brahim I; Hisham N; Aladdin R; Mohammed H; Bahaaeldin A Recent Updates of Carbapenem Antibiotics. Eur. J. Med. Chem 2017, 131, 185–195. [DOI] [PubMed] [Google Scholar]
- 73.Jain A; Utley L; Parr TR; Zabawa T; Pucci MJ Tebipenem, the First Oral Carbapenem Antibiotic. Expert Rev Anti Infect Ther 2018, 16, 513–522. [DOI] [PubMed] [Google Scholar]
- 74.Kattan JN; Villegas MV; Quinn JP New Developments in Carbapenems. Clin. Microbiol. Infect 2008, 14, 1102–1111. [DOI] [PubMed] [Google Scholar]
- 75.Patel G; Bonomo RA “Stormy Waters Ahead”: Global Emergence of Carbapenemases. Front Microbiol 2013, 4, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bush K Proliferation and Significance of Clinically Relevant Beta-Lactamases. Ann. N. Y. Acad. Sci 2013, 1277, 84–90. [DOI] [PubMed] [Google Scholar]
- 77.Nordmann P; Dortet L; Poirel L Carbapenem Resistance in Enterobacteriaceae: Here Is the Storm! Trends Mol. Med 2012, 18, 263–272. [DOI] [PubMed] [Google Scholar]
- 78.Blais J; Lopez S; Li C; Ruzin A; Ranjitkar S; Dean CR; Leeds JA; Casarez A; Simmons RL; Reck F In Vitro Activity of Lys228, a Novel Monobactam Antibiotic, against Multidrug-Resistant Enterobacteriaceae. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Theuretzbacher U; Bush K; Harbarth S; Paul M; Rex JH; Tacconelli E; Thwaites GE Critical Analysis of Antibacterial Agents in Clinical Development. Nat. Rev. Microbiol 2020, 18, 286–298. [DOI] [PubMed] [Google Scholar]
- 80.Bush K; Bradford PA Interplay between Beta-Lactamases and New Beta-Lactamase Inhibitors. Nat. Rev. Microbiol 2019, 17, 295–306. [DOI] [PubMed] [Google Scholar]
- 81.Jacoby GA AmpC Beta-Lactamases. Clin. Microbiol. Rev 2009, 22, 161–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Juan C; Macia MD; Gutierrez O; Vidal C; Perez JL; Oliver A Molecular Mechanisms of Beta-Lactam Resistance Mediated by AmpC Hyperproduction in Pseudomonas aeruginosa Clinical Strains. Antimicrob. Agents Chemother 2005, 49, 4733–4738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Corvec S; Caroff N; Espaze E; Giraudeau C; Drugeon H; Reynaud A Ampc Cephalosporinase Hyperproduction in Acinetobacter baumannii Clinical Strains. J. Antimicrob. Chemother 2003, 52, 629–635. [DOI] [PubMed] [Google Scholar]
- 84.Karaiskos I; Lagou S; Pontikis K; Rapti V; Poulakou G The “Old” and the “New” Antibiotics for MDR Gram-Negative Pathogens: For Whom, When, and How. Front Public Health 2019, 7, 151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Eichenberger EM; Thaden JT Epidemiology and Mechanisms of Resistance of Extensively Drug Resistant Gram-Negative Bacteria. Antibiotics (Basel) 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Kirby WM Extraction of a Highly Potent Penicillin Inactivator from Penicillin Resistant Staphylococci. Science 1944, 99, 452–453. [DOI] [PubMed] [Google Scholar]
- 87.Lowy FD Antimicrobial Resistance: The Example of Staphylococcus aureus. J. Clin. Invest 2003, 111, 1265–1273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Fisher JF; Mobashery S Beta-Lactam Resistance Mechanisms: Gram-Positive Bacteria and Mycobacterium tuberculosis. Cold Spring Harb Perspect Med 2016, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Zapun A; Contreras-Martel C; Vernet T Penicillin-Binding Proteins and Beta-Lactam Resistance. FEMS Microbiol. Rev 2008, 32, 361–385. [DOI] [PubMed] [Google Scholar]
- 90.Fisher JF; Mobashery S Beta-Lactams against the Fortress of the Gram-Positive Staphylococcus aureus Bacterium. Chem. Rev 2021, 121, 3412–3463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Parker MT; Hewitt JH Methicillin Resistance in Staphylococcus aureus. Lancet 1970, 1, 800–804. [DOI] [PubMed] [Google Scholar]
- 92.Peacock SJ; Paterson GK Mechanisms of Methicillin Resistance in Staphylococcus aureus. Annu. Rev. Biochem 2015, 84, 577–601. [DOI] [PubMed] [Google Scholar]
- 93.Lim D; Strynadka NC Structural Basis for the Beta Lactam Resistance of PBP2a from Methicillin-Resistant Staphylococcus aureus. Nat. Struct. Biol 2002, 9, 870–876. [DOI] [PubMed] [Google Scholar]
- 94.Fuda C; Hesek D; Lee M; Morio K; Nowak T; Mobashery S Activation for Catalysis of Penicillin-Binding Protein 2a from Methicillin-Resistant Staphylococcus aureus by Bacterial Cell Wall. J. Am. Chem. Soc 2005, 127, 2056–2057. [DOI] [PubMed] [Google Scholar]
- 95.Otero LH; Rojas-Altuve A; Llarrull LI; Carrasco-Lopez C; Kumarasiri M; Lastochkin E; Fishovitz J; Dawley M; Hesek D; Lee M, et al. How Allosteric Control of Staphylococcus aureus Penicillin Binding Protein 2a Enables Methicillin Resistance and Physiological Function. Proc Natl Acad Sci U S A 2013, 110, 16808–16813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Fontana R; Cerini R; Longoni P; Grossato A; Canepari P Identification of a Streptococcal Penicillin-Binding Protein That Reacts Very Slowly with Penicillin. J. Bacteriol 1983, 155, 1343–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Sauvage E; Kerff F; Fonze E; Herman R; Schoot B; Marquette JP; Taburet Y; Prevost D; Dumas J; Leonard G, et al. The 2.4-a Crystal Structure of the Penicillin-Resistant Penicillin-Binding Protein Pbp5fm from Enterococcus faecium in Complex with Benzylpenicillin. Cell. Mol. Life Sci 2002, 59, 1223–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Rice LB; Bellais S; Carias LL; Hutton-Thomas R; Bonomo RA; Caspers P; Page MG; Gutmann L Impact of Specific PBP5 Mutations on Expression of Beta-Lactam Resistance in Enterococcus Faecium. Antimicrob. Agents Chemother 2004, 48, 3028–3032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Klare I; Rodloff AC; Wagner J; Witte W; Hakenbeck R Overproduction of a Penicillin-Binding Protein Is Not the Only Mechanism of Penicillin Resistance in Enterococcus faecium. Antimicrob. Agents Chemother 1992, 36, 783–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Montealegre MC; Roh JH; Rae M; Davlieva MG; Singh KV; Shamoo Y; Murray BE Differential Penicillin-Binding Protein 5 (Pbp5) Levels in the Enterococcus faecium Clades with Different Levels of Ampicillin Resistance. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Hackbarth CJ; Kocagoz T; Kocagoz S; Chambers HF Point Mutations in Staphylococcus aureus PBP2 Gene Affect Penicillin-Binding Kinetics and Are Associated with Resistance. Antimicrob. Agents Chemother 1995, 39, 103–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Hakenbeck R; Bruckner R; Denapaite D; Maurer P Molecular Mechanisms of Beta-Lactam Resistance in Streptococcus pneumoniae. Future Microbiol 2012, 7, 395–410. [DOI] [PubMed] [Google Scholar]
- 103.Moya B; Beceiro A; Cabot G; Juan C; Zamorano L; Alberti S; Oliver A Pan-Beta-Lactam Resistance Development in Pseudomonas aeruginosa Clinical Strains: Molecular Mechanisms, Penicillin-Binding Protein Profiles, and Binding Affinities. Antimicrob. Agents Chemother 2012, 56, 4771–4778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Diaz Caballero J; Clark ST; Coburn B; Zhang Y; Wang PW; Donaldson SL; Tullis DE; Yau YC; Waters VJ; Hwang DM, et al. Selective Sweeps and Parallel Pathoadaptation Drive Pseudomonas aeruginosa Evolution in the Cystic Fibrosis Lung. mBio 2015, 6, e00981–00915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Clark ST; Sinha U; Zhang Y; Wang PW; Donaldson SL; Coburn B; Waters VJ; Yau YCW; Tullis DE; Guttman DS, et al. Penicillin-Binding Protein 3 Is a Common Adaptive Target among Pseudomonas aeruginosa Isolates from Adult Cystic Fibrosis Patients Treated with Beta-Lactams. Int. J. Antimicrob. Agents 2019, 53, 620–628. [DOI] [PubMed] [Google Scholar]
- 106.Matic V; Bozdogan B; Jacobs MR; Ubukata K; Appelbaum PC Contribution of Beta-Lactamase and PBP Amino Acid Substitutions to Amoxicillin/Clavulanate Resistance in Beta-Lactamase-Positive, Amoxicillin/Clavulanate-Resistant Haemophilus influenzae. J. Antimicrob. Chemother 2003, 52, 1018–1021. [DOI] [PubMed] [Google Scholar]
- 107.Matteo MJ; Granados G; Olmos M; Wonaga A; Catalano M Helicobacter pylori Amoxicillin Heteroresistance Due to Point Mutations in Pbp-1a in Isogenic Isolates. J. Antimicrob. Chemother 2008, 61, 474–477. [DOI] [PubMed] [Google Scholar]
- 108.Zapun A; Morlot C; Taha MK Resistance to Beta-Lactams in Neisseria ssp Due to Chromosomally Encoded Penicillin-Binding Proteins. Antibiotics (Basel) 2016, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Delcour AH Outer Membrane Permeability and Antibiotic Resistance. Biochim. Biophys. Acta 2009, 1794, 808–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nikaido H Molecular Basis of Bacterial Outer Membrane Permeability Revisited. Microbiol. Mol. Biol. Rev 2003, 67, 593–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dam S; Pages JM; Masi M Stress Responses, Outer Membrane Permeability Control and Antimicrobial Resistance in Enterobacteriaceae. Microbiology (Reading) 2018, 164, 260–267. [DOI] [PubMed] [Google Scholar]
- 112.Pages JM; James CE; Winterhalter M The Porin and the Permeating Antibiotic: A Selective Diffusion Barrier in Gram-Negative Bacteria. Nat. Rev. Microbiol 2008, 6, 893–903. [DOI] [PubMed] [Google Scholar]
- 113.Vergalli J; Bodrenko IV; Masi M; Moynie L; Acosta-Gutierrez S; Naismith JH; Davin-Regli A; Ceccarelli M; van den Berg B; Winterhalter M, et al. Porins and Small-Molecule Translocation across the Outer Membrane of Gram-Negative Bacteria. Nat. Rev. Microbiol 2020, 18, 164–176. [DOI] [PubMed] [Google Scholar]
- 114.Ferenci T Maintaining a Healthy SPANC Balance through Regulatory and Mutational Adaptation. Mol. Microbiol 2005, 57, 1–8. [DOI] [PubMed] [Google Scholar]
- 115.Shen Z; Ding B; Ye M; Wang P; Bi Y; Wu S; Xu X; Guo Q; Wang M High Ceftazidime Hydrolysis Activity and Porin Ompk35 Deficiency Contribute to the Decreased Susceptibility to Ceftazidime/Avibactam in Kpc-Producing Klebsiella pneumoniae. J. Antimicrob. Chemother 2017, 72, 1930–1936. [DOI] [PubMed] [Google Scholar]
- 116.Humphries RM; Hemarajata P Resistance to Ceftazidime-Avibactam in Klebsiella pneumoniae Due to Porin Mutations and the Increased Expression of Kpc-3. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Garcia-Sureda L; Juan C; Domenech-Sanchez A; Alberti S Role of Klebsiella pneumoniae LamB Porin in Antimicrobial Resistance. Antimicrob. Agents Chemother 2011, 55, 1803–1805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Acosta-Gutierrez S; Ferrara L; Pathania M; Masi M; Wang J; Bodrenko I; Zahn M; Winterhalter M; Stavenger RA; Pages JM, et al. Getting Drugs into Gram-Negative Bacteria: Rational Rules for Permeation through General Porins. ACS Infect Dis 2018, 4, 1487–1498. [DOI] [PubMed] [Google Scholar]
- 119.De E; Basle A; Jaquinod M; Saint N; Mallea M; Molle G; Pages JM A New Mechanism of Antibiotic Resistance in Enterobacteriaceae Induced by a Structural Modification of the Major Porin. Mol. Microbiol 2001, 41, 189–198. [DOI] [PubMed] [Google Scholar]
- 120.Lou H; Chen M; Black SS; Bushell SR; Ceccarelli M; Mach T; Beis K; Low AS; Bamford VA; Booth IR, et al. Altered Antibiotic Transport in Ompc Mutants Isolated from a Series of Clinical Strains of Multi-Drug Resistant E. coli. PLoS One 2011, 6, e25825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Garcia-Fernandez A; Miriagou V; Papagiannitsis CC; Giordano A; Venditti M; Mancini C; Carattoli A An Ertapenem-Resistant Extended-Spectrum-Beta-Lactamase-Producing Klebsiella pneumoniae Clone Carries a Novel OmpK36 Porin Variant. Antimicrob. Agents Chemother 2010, 54, 4178–4184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Chevalier S; Bouffartigues E; Bodilis J; Maillot O; Lesouhaitier O; Feuilloley MGJ; Orange N; Dufour A; Cornelis P Structure, Function and Regulation of Pseudomonas aeruginosa Porins. FEMS Microbiol. Rev 2017, 41, 698–722. [DOI] [PubMed] [Google Scholar]
- 123.Hancock RE Resistance Mechanisms in Pseudomonas aeruginosa and Other Nonfermentative Gram-Negative Bacteria. Clin. Infect. Dis 1998, 27 Suppl 1, S93–99. [DOI] [PubMed] [Google Scholar]
- 124.Trias J; Nikaido H Outer Membrane Protein D2 Catalyzes Facilitated Diffusion of Carbapenems and Penems through the Outer Membrane of Pseudomonas aeruginosa. Antimicrob. Agents Chemother 1990, 34, 52–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rodriguez-Martinez JM; Poirel L; Nordmann P Molecular Epidemiology and Mechanisms of Carbapenem Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2009, 53, 4783–4788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wolter DJ; Hanson ND; Lister PD Insertional Inactivation of OprD in Clinical Isolates of Pseudomonas aeruginosa Leading to Carbapenem Resistance. FEMS Microbiol. Lett 2004, 236, 137–143. [DOI] [PubMed] [Google Scholar]
- 127.Quale J; Bratu S; Gupta J; Landman D Interplay of Efflux System, AmpC, and OprD Expression in Carbapenem Resistance of Pseudomonas aeruginosa Clinical Isolates. Antimicrob. Agents Chemother 2006, 50, 1633–1641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Perez FJ; Gimeno C; Navarro D; Garcia-de-Lomas J Meropenem Permeation through the Outer Membrane of Pseudomonas aeruginosa Can Involve Pathways Other Than the OprD Porin Channel. Chemotherapy 1996, 42, 210–214. [DOI] [PubMed] [Google Scholar]
- 129.Vila J; Marti S; Sanchez-Cespedes J Porins, Efflux Pumps and Multidrug Resistance in Acinetobacter baumannii. J. Antimicrob. Chemother 2007, 59, 1210–1215. [DOI] [PubMed] [Google Scholar]
- 130.Sugawara E; Nikaido H Ompa Is the Principal Nonspecific Slow Porin of Acinetobacter baumannii. J. Bacteriol 2012, 194, 4089–4096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Moran-Barrio J; Cameranesi MM; Relling V; Limansky AS; Brambilla L; Viale AM The Acinetobacter Outer Membrane Contains Multiple Specific Channels for Carbapenem Beta-Lactams as Revealed by Kinetic Characterization Analyses of Imipenem Permeation into Acinetobacter baylyi Cells. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.del Mar Tomas M; Beceiro A; Perez A; Velasco D; Moure R; Villanueva R; Martinez-Beltran J; Bou G Cloning and Functional Analysis of the Gene Encoding the 33- to 36-Kilodalton Outer Membrane Protein Associated with Carbapenem Resistance in Acinetobacter baumannii. Antimicrob. Agents Chemother 2005, 49, 5172–5175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Masi M; Refregiers M; Pos KM; Pages JM Mechanisms of Envelope Permeability and Antibiotic Influx and Efflux in Gram-Negative Bacteria. Nat Microbiol 2017, 2, 17001. [DOI] [PubMed] [Google Scholar]
- 134.Du D; Wang-Kan X; Neuberger A; van Veen HW; Pos KM; Piddock LJV; Luisi BF Multidrug Efflux Pumps: Structure, Function and Regulation. Nat. Rev. Microbiol 2018, 16, 523–539. [DOI] [PubMed] [Google Scholar]
- 135.Blair JM; Richmond GE; Piddock LJ Multidrug Efflux Pumps in Gram-Negative Bacteria and Their Role in Antibiotic Resistance. Future Microbiol 2014, 9, 1165–1177. [DOI] [PubMed] [Google Scholar]
- 136.Li XZ; Nikaido H Efflux-Mediated Drug Resistance in Bacteria: An Update. Drugs 2009, 69, 1555–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Du D; Wang Z; James NR; Voss JE; Klimont E; Ohene-Agyei T; Venter H; Chiu W; Luisi BF Structure of the AcrAB-TolC Multidrug Efflux Pump. Nature 2014, 509, 512–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Poirel L; Nordmann P Carbapenem Resistance in Acinetobacter baumannii: Mechanisms and Epidemiology. Clin. Microbiol. Infect 2006, 12, 826–836. [DOI] [PubMed] [Google Scholar]
- 139.Coyne S; Courvalin P; Perichon B Efflux-Mediated Antibiotic Resistance in Acinetobacter spp. Antimicrob. Agents Chemother 2011, 55, 947–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Padilla E; Llobet E; Domenech-Sanchez A; Martinez-Martinez L; Bengoechea JA; Alberti S Klebsiella pneumoniae AcrAB Efflux Pump Contributes to Antimicrobial Resistance and Virulence. Antimicrob. Agents Chemother 2010, 54, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Pages JM; Lavigne JP; Leflon-Guibout V; Marcon E; Bert F; Noussair L; Nicolas-Chanoine MH Efflux Pump, the Masked Side of Beta-Lactam Resistance in Klebsiella pneumoniae Clinical Isolates. PLoS One 2009, 4, e4817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Dreier J; Ruggerone P Interaction of Antibacterial Compounds with RND Efflux Pumps in Pseudomonas aeruginosa. Front Microbiol 2015, 6, 660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ambler RP The Structure of Beta-Lactamases. Philosophical transactions of the Royal Society of London. Series B, Biological sciences 1980, 289, 321–331. [DOI] [PubMed] [Google Scholar]
- 144.Bush K The ABCD’s of Beta-Lactamase Nomenclature. J. Infect. Chemother 2013, 19, 549–559. [DOI] [PubMed] [Google Scholar]
- 145.Fisher J; Belasco JG; Khosla S; Knowles JR Beta-Lactamase Proceeds Via an Acyl-Enzyme Intermediate. Interaction of the Escherichia coli RTEM Enzyme with Cefoxitin. Biochemistry 1980, 19, 2895–2901. [DOI] [PubMed] [Google Scholar]
- 146.Cartwright SJ; Waley SG Cryoenzymology of Beta-Lactamases. Biochemistry 1987, 26, 5329–5337. [DOI] [PubMed] [Google Scholar]
- 147.Strynadka NC; Adachi H; Jensen SE; Johns K; Sielecki A; Betzel C; Sutoh K; James MN Molecular Structure of the Acyl-Enzyme Intermediate in Beta-Lactam Hydrolysis at 1.7 A Resolution. Nature 1992, 359, 700–705. [DOI] [PubMed] [Google Scholar]
- 148.Fisher JF; Mobashery S Three Decades of the Class a Beta-Lactamase Acyl-Enzyme. Curr Protein Pept Sci 2009, 10, 401–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Philippon A; Dusart J; Joris B; Frere JM The Diversity, Structure and Regulation of Beta-Lactamases. Cell. Mol. Life Sci 1998, 54, 341–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Galleni M; Lamotte-Brasseur J; Raquet X; Dubus A; Monnaie D; Knox JR; Frere JM The Enigmatic Catalytic Mechanism of Active-Site Serine Beta-Lactamases. Biochem. Pharmacol 1995, 49, 1171–1178. [DOI] [PubMed] [Google Scholar]
- 151.Frere JM Beta-Lactamases and Bacterial Resistance to Antibiotics. Mol. Microbiol 1995, 16, 385–395. [DOI] [PubMed] [Google Scholar]
- 152.Massova I; Mobashery S Kinship and Diversification of Bacterial Penicillin-Binding Proteins and Beta-Lactamases. Antimicrob. Agents Chemother 1998, 42, 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Herzberg O; Moult J Bacterial Resistance to Beta-Lactam Antibiotics: Crystal Structure of Beta-Lactamase from Staphylococcus aureus PC1 at 2.5 A Resolution. Science 1987, 236, 694–701. [DOI] [PubMed] [Google Scholar]
- 154.Hall BG; Barlow M Evolution of the Serine Beta-Lactamases: Past, Present and Future. Drug Resist Updat 2004, 7, 111–123. [DOI] [PubMed] [Google Scholar]
- 155.Urbach C; Fastrez J; Soumillion P A New Family of Cyanobacterial Penicillin-Binding Proteins. A Missing Link in the Evolution of Class A Beta-Lactamases. J. Biol. Chem 2008, 283, 32516–32526. [DOI] [PubMed] [Google Scholar]
- 156.Tripathi LP; Sowdhamini R Genome-Wide Survey of Prokaryotic Serine Proteases: Analysis of Distribution and Domain Architectures of Five Serine Protease Families in Prokaryotes. BMC Genomics 2008, 9, 549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Wilke MS; Hills TL; Zhang HZ; Chambers HF; Strynadka NC Crystal Structures of the Apo and Penicillin-Acylated Forms of the BlaR1 Beta-Lactam Sensor of Staphylococcus aureus. J. Biol. Chem 2004, 279, 47278–47287. [DOI] [PubMed] [Google Scholar]
- 158.Keckesova Z; Donaher JL; De Cock J; Freinkman E; Lingrell S; Bachovchin DA; Bierie B; Tischler V; Noske A; Okondo MC, et al. LACTB Is a Tumour Suppressor That Modulates Lipid Metabolism and Cell State. Nature 2017, 543, 681–686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Meini MR; Llarrull LI; Vila AJ Overcoming Differences: The Catalytic Mechanism of Metallo-Beta-Lactamases. FEBS Lett. 2015, 589, 3419–3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Palzkill T Structural and Mechanistic Basis for Extended-Spectrum Drug-Resistance Mutations in Altering the Specificity of TEM, CTX-M, and KPC Beta-Lactamases. Front Mol Biosci 2018, 5, 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Rammelkamp CH; Maxon T Resistance of Staphylococcus aureus to the Action of Penicillin. Proc. Soc. Exp. Biol. Med 1942, 51, 386–389. [Google Scholar]
- 162.Bush K Past and Present Perspectives on Beta-Lactamases. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Datta N; Kontomichalou P Penicillinase Synthesis Controlled by Infectious R Factors in Enterobacteriaceae. Nature 1965, 208, 239–241. [DOI] [PubMed] [Google Scholar]
- 164.Chaves J; Ladona MG; Segura C; Coira A; Reig R; Ampurdanes C SHV-1 Beta-Lactamase Is Mainly a Chromosomally Encoded Species-Specific Enzyme in Klebsiella pneumoniae. Antimicrob. Agents Chemother 2001, 45, 2856–2861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Bonnet R Growing Group of Extended-Spectrum Beta-Lactamases: The CTX-M Enzymes. Antimicrob. Agents Chemother 2004, 48, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Canton R; Gonzalez-Alba JM; Galan JC CTX-M Enzymes: Origin and Diffusion. Front Microbiol 2012, 3, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Poirel L; Le Thomas I; Naas T; Karim A; Nordmann P Biochemical Sequence Analyses of GES-1, a Novel Class a Extended-Spectrum Beta-Lactamase, and the Class 1 Integron In52 from Klebsiella pneumoniae. Antimicrob. Agents Chemother 2000, 44, 622–632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Walther-Rasmussen J; Hoiby N Class A Carbapenemases. J. Antimicrob. Chemother 2007, 60, 470–482. [DOI] [PubMed] [Google Scholar]
- 169.Rapp RP; Urban C Klebsiella pneumoniae Carbapenemases in Enterobacteriaceae: History, Evolution, and Microbiology Concerns. Pharmacotherapy 2012, 32, 399–407. [DOI] [PubMed] [Google Scholar]
- 170.Vourli S; Giakkoupi P; Miriagou V; Tzelepi E; Vatopoulos AC; Tzouvelekis LS Novel GES/IBC Extended-Spectrum Beta-Lactamase Variants with Carbapenemase Activity in Clinical Enterobacteria. FEMS Microbiol. Lett 2004, 234, 209–213. [DOI] [PubMed] [Google Scholar]
- 171.Rasmussen BA; Bush K; Keeney D; Yang Y; Hare R; O’Gara C; Medeiros AA Characterization of IMI-1 Beta-Lactamase, a Class a Carbapenem-Hydrolyzing Enzyme from Enterobacter cloacae. Antimicrob. Agents Chemother 1996, 40, 2080–2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Naas T; Vandel L; Sougakoff W; Livermore DM; Nordmann P Cloning and Sequence Analysis of the Gene for a Carbapenem-Hydrolyzing Class a Beta-Lactamase, SME-1, from Serratia marcescens S6. Antimicrob. Agents Chemother 1994, 38, 1262–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Hackbarth CJ; Chambers HF BlaI and BlaR1 Regulate Beta-Lactamase and PBP2a Production in Methicillin-Resistant Staphylococcus aureus. Antimicrob. Agents Chemother 1993, 37, 1144–1149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Datz M; Joris B; Azab EA; Galleni M; Van Beeumen J; Frere JM; Martin HH A Common System Controls the Induction of Very Different Genes. The Class-A Beta-Lactamase of Proteus vulgaris and the Enterobacterial Class-C Beta-Lactamase. Eur. J. Biochem 1994, 226, 149–157. [DOI] [PubMed] [Google Scholar]
- 175.Naas T; Nordmann P Analysis of a Carbapenem-Hydrolyzing Class A Beta-Lactamase from Enterobacter cloacae and of Its Lysr-Type Regulatory Protein. Proc Natl Acad Sci U S A 1994, 91, 7693–7697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Trepanier S; Prince A; Huletsky A Characterization of the PenA and PenR Genes of Burkholderia cepacia 249 Which Encode the Chromosomal Class A Penicillinase and Its LysR-Type Transcriptional Regulator. Antimicrob. Agents Chemother 1997, 41, 2399–2405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Aubron C; Poirel L; Ash RJ; Nordmann P Carbapenemase-Producing Enterobacteriaceae, U.S. Rivers. Emerg Infect Dis 2005, 11, 260–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Ambler RP; Coulson AF; Frere JM; Ghuysen JM; Joris B; Forsman M; Levesque RC; Tiraby G; Waley SG A Standard Numbering Scheme for the Class a Beta-Lactamases. Biochem. J 1991, 276 (Pt 1), 269–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Chen Y; Bonnet R; Shoichet BK The Acylation Mechanism of CTX-M Beta-Lactamase at 0.88 a Resolution. J. Am. Chem. Soc 2007, 129, 5378–5380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Tomanicek SJ; Wang KK; Weiss KL; Blakeley MP; Cooper J; Chen Y; Coates L The Active Site Protonation States of Perdeuterated Toho-1 Beta-Lactamase Determined by Neutron Diffraction Support a Role for Glu166 as the General Base in Acylation. FEBS Lett. 2011, 585, 364–368. [DOI] [PubMed] [Google Scholar]
- 181.Meroueh SO; Fisher JF; Schlegel HB; Mobashery S Ab Initio QM/MM Study of Class A Beta-Lactamase Acylation: Dual Participation of Glu166 and Lys73 in a Concerted Base Promotion of Ser70. J. Am. Chem. Soc 2005, 127, 15397–15407. [DOI] [PubMed] [Google Scholar]
- 182.Tremblay LW; Xu H; Blanchard JS Structures of the Michaelis Complex (1.2 a) and the Covalent Acyl Intermediate (2.0 a) of Cefamandole Bound in the Active Sites of the Mycobacterium tuberculosis Beta-Lactamase K73A and E166A Mutants. Biochemistry 2010, 49, 9685–9687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Golemi-Kotra D; Meroueh SO; Kim C; Vakulenko SB; Bulychev A; Stemmler AJ; Stemmler TL; Mobashery S The Importance of a Critical Protonation State and the Fate of the Catalytic Steps in Class A Beta-Lactamases and Penicillin-Binding Proteins. J. Biol. Chem 2004, 279, 34665–34673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Vandavasi VG; Langan PS; Weiss KL; Parks JM; Cooper JB; Ginell SL; Coates L Active-Site Protonation States in an Acyl-Enzyme Intermediate of a Class A Beta-Lactamase with a Monobactam Substrate. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Vandavasi VG; Weiss KL; Cooper JB; Erskine PT; Tomanicek SJ; Ostermann A; Schrader TE; Ginell SL; Coates L Exploring the Mechanism of Beta-Lactam Ring Protonation in the Class A Beta-Lactamase Acylation Mechanism Using Neutron and X-Ray Crystallography. J. Med. Chem 2016, 59, 474–479. [DOI] [PubMed] [Google Scholar]
- 186.Pan X; He Y; Lei J; Huang X; Zhao Y Crystallographic Snapshots of Class A Beta-Lactamase Catalysis Reveal Structural Changes That Facilitate Beta-Lactam Hydrolysis. J. Biol. Chem 2017, 292, 4022–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Pratt RF Beta-Lactamases: Why and How. J. Med. Chem 2016, 59, 8207–8220. [DOI] [PubMed] [Google Scholar]
- 188.Abraham EP; Chain E An Enzyme from Bacteria able to Destroy Penicillin. Nature 1940, 146, 837–837. [PubMed] [Google Scholar]
- 189.Burman LG; Park JT; Lindstrom EB; Boman HG Resistance of Escherichia coli to Penicillins: Identification of the Structural Gene for the Chromosomal Penicillinase. J. Bacteriol 1973, 116, 123–130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Walther-Rasmussen J; Hoiby N Plasmid-Borne AmpC Beta-Lactamases. Can. J. Microbiol 2002, 48, 479–493. [DOI] [PubMed] [Google Scholar]
- 191.Mack AR; Barnes MD; Taracila MA; Hujer AM; Hujer KM; Cabot G; Feldgarden M; Haft DH; Klimke W; van den Akker F, et al. A Standard Numbering Scheme for Class C Beta-Lactamases. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Oefner C; D’Arcy A; Daly JJ; Gubernator K; Charnas RL; Heinze I; Hubschwerlen C; Winkler FK Refined Crystal Structure of Beta-Lactamase from Citrobacter freundii Indicates a Mechanism for Beta-Lactam Hydrolysis. Nature 1990, 343, 284–288. [DOI] [PubMed] [Google Scholar]
- 193.Kato-Toma Y; Iwashita T; Masuda K; Oyama Y; Ishiguro M pKa Measurements from Nuclear Magnetic Resonance of Tyrosine-150 in Class C Beta-Lactamase. Biochem. J 2003, 371, 175–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Lobkovsky E; Moews PC; Liu H; Zhao H; Frere JM; Knox JR Evolution of an Enzyme Activity: Crystallographic Structure at 2-A Resolution of Cephalosporinase from the ampC Gene of Enterobacter cloacae P99 and Comparison with a Class a Penicillinase. Proc Natl Acad Sci U S A 1993, 90, 11257–11261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Beadle BM; Trehan I; Focia PJ; Shoichet BK Structural Milestones in the Reaction Pathway of an Amide Hydrolase: Substrate, Acyl, and Product Complexes of Cephalothin with AmpC Beta-Lactamase. Structure 2002, 10, 413–424. [DOI] [PubMed] [Google Scholar]
- 196.Tripathi R; Nair NN Mechanism of Acyl-Enzyme Complex Formation from the Henry-Michaelis Complex of Class C Beta-Lactamases with Beta-Lactam Antibiotics. J. Am. Chem. Soc 2013, 135, 14679–14690. [DOI] [PubMed] [Google Scholar]
- 197.Honore N; Nicolas MH; Cole ST Inducible Cephalosporinase Production in Clinical Isolates of Enterobacter Cloacae Is Controlled by a Regulatory Gene That Has Been Deleted from Escherichia coli. EMBO J 1986, 5, 3709–3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Juan C; Moya B; Perez JL; Oliver A Stepwise Upregulation of the Pseudomonas aeruginosa Chromosomal Cephalosporinase Conferring High-Level Beta-Lactam Resistance Involves Three AmpD Homologues. Antimicrob. Agents Chemother 2006, 50, 1780–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Kaneko K; Okamoto R; Nakano R; Kawakami S; Inoue M Gene Mutations responsible for Overexpression of AmpC Beta-Lactamase in Some Clinical Isolates of Enterobacter cloacae. J. Clin. Microbiol 2005, 43, 2955–2958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Bulychev A; Mobashery S Class C Beta-Lactamases Operate at the Diffusion Limit for Turnover of Their Preferred Cephalosporin Substrates. Antimicrob. Agents Chemother 1999, 43, 1743–1746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Leonard DA; Bonomo RA; Powers RA Class D Beta-Lactamases: A Reappraisal after Five Decades. Acc. Chem. Res 2013, 46, 2407–2415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Poirel L; Naas T; Nordmann P Diversity, Epidemiology, and Genetics of Class D BetaLactamases. Antimicrob. Agents Chemother 2010, 54, 24–38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Evans BA; Amyes SG OXA Beta-Lactamases. Clin. Microbiol. Rev 2014, 27, 241–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Pitout JDD; Peirano G; Kock MM; Strydom KA; Matsumura Y The Global Ascendency of OXA-48-Type Carbapenemases. Clin. Microbiol. Rev 2019, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Couture F; Lachapelle J; Levesque RC Phylogeny of LCR-1 and OXA-5 with Class a and Class D Beta-Lactamases. Mol. Microbiol 1992, 6, 1693–1705. [DOI] [PubMed] [Google Scholar]
- 206.Golemi D; Maveyraud L; Vakulenko S; Samama JP; Mobashery S Critical Involvement of a Carbamylated Lysine in Catalytic Function of Class D Beta-Lactamases. Proc Natl Acad Sci U S A 2001, 98, 14280–14285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Maveyraud L; Golemi-Kotra D; Ishiwata A; Meroueh O; Mobashery S; Samama JP High-Resolution X-Ray Structure of an Acyl-Enzyme Species for the Class D OXA-10 Beta-Lactamase. J. Am. Chem. Soc 2002, 124, 2461–2465. [DOI] [PubMed] [Google Scholar]
- 208.Antunes NT; Lamoureaux TL; Toth M; Stewart NK; Frase H; Vakulenko SB Class D Beta-Lactamases: Are They All Carbapenemases? Antimicrob. Agents Chemother 2014, 58, 2119–2125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Bush K; Jacoby GA; Medeiros AA A Functional Classification Scheme for Beta-Lactamases and Its Correlation with Molecular Structure. Antimicrob. Agents Chemother 1995, 39, 1211–1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Bush K; Jacoby GA Updated Functional Classification of Beta-Lactamases. Antimicrob. Agents Chemother 2010, 54, 969–976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Thai QK; Bos F; Pleiss J The Lactamase Engineering Database: A Critical Survey of TEM Sequences in Public Databases. BMC Genomics 2009, 10, 390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Naas T; Oueslati S; Bonnin RA; Dabos ML; Zavala A; Dortet L; Retailleau P; Iorga BI Beta-Lactamase Database (BLDB) - Structure and Function. J. Enzyme Inhib. Med. Chem 2017, 32, 917–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Daiyasu H; Osaka K; Ishino Y; Toh H Expansion of the Zinc Metallo-Hydrolase Family of the Beta-Lactamase Fold. FEBS Lett. 2001, 503, 1–6. [DOI] [PubMed] [Google Scholar]
- 214.Gonzalez JM Visualizing the Superfamily of Metallo-Beta-Lactamases through Sequence Similarity Network Neighborhood Connectivity Analysis. Heliyon 2021, 7, e05867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Baier F; Tokuriki N Connectivity between Catalytic Landscapes of the Metallo-Beta-Lactamase Superfamily. J. Mol. Biol 2014, 426, 2442–2456. [DOI] [PubMed] [Google Scholar]
- 216.Pettinati I; Brem J; Lee SY; McHugh PJ; Schofield CJ The Chemical Biology of Human Metallo-Beta-Lactamase Fold Proteins. Trends Biochem. Sci 2016, 41, 338–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Carfi A; Pares S; Duee E; Galleni M; Duez C; Frere JM; Dideberg O The 3-D Structure of a Zinc Metallo-Beta-Lactamase from Bacillus cereus Reveals a New Type of Protein Fold. EMBO J 1995, 14, 4914–4921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Galleni M; Lamotte-Brasseur J; Rossolini GM; Spencer J; Dideberg O; Frere JM; Metallo-beta-lactamases Working G Standard Numbering Scheme for Class B Beta-Lactamases. Antimicrob. Agents Chemother 2001, 45, 660–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Garau G; Garcia-Saez I; Bebrone C; Anne C; Mercuri P; Galleni M; Frere JM; Dideberg O Update of the Standard Numbering Scheme for Class B Beta-Lactamases. Antimicrob. Agents Chemother 2004, 48, 2347–2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Rasmussen BA; Bush K Carbapenem-Hydrolyzing Beta-Lactamases. Antimicrob. Agents Chemother 1997, 41, 223–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 221.Berglund F; Johnning A; Larsson DGJ; Kristiansson E An Updated Phylogeny of the Metallo-Beta-Lactamases. J. Antimicrob. Chemother 2021, 76, 117–123. [DOI] [PubMed] [Google Scholar]
- 222.Hall BG; Salipante SJ; Barlow M The Metallo-Beta-Lactamases Fall into Two Distinct Phylogenetic Groups. J. Mol. Evol 2003, 57, 249–254. [DOI] [PubMed] [Google Scholar]
- 223.Hall BG; Salipante SJ; Barlow M Independent origins of Subgroup Bl + B2 and Subgroup B3 Metallo-Beta-Lactamases. J. Mol. Evol 2004, 59, 133–141. [DOI] [PubMed] [Google Scholar]
- 224.Hall BG; Barlow M Revised Ambler Classification of Beta-Lactamases. J. Antimicrob. Chemother 2005, 55, 1050–1051. [DOI] [PubMed] [Google Scholar]
- 225.Frere JM; Galleni M; Bush K; Dideberg O Is It Necessary to Change the Classification of Beta-Lactamases? J. Antimicrob. Chemother 2005, 55, 1051–1053. [DOI] [PubMed] [Google Scholar]
- 226.Fabiane SM; Sohi MK; Wan T; Payne DJ; Bateson JH; Mitchell T; Sutton BJ Crystal Structure of the Zinc-Dependent Beta-Lactamase from Bacillus cereus at 1.9 A Resolution: Binuclear Active Site with Features of a Mononuclear Enzyme. Biochemistry 1998, 37, 12404–12411. [DOI] [PubMed] [Google Scholar]
- 227.Concha NO; Rasmussen BA; Bush K; Herzberg O Crystal Structure of the Wide-Spectrum Binuclear Zinc Beta-Lactamase from Bacteroides fragilis. Structure 1996, 4, 823–836. [DOI] [PubMed] [Google Scholar]
- 228.Carfi A; Duee E; Paul-Soto R; Galleni M; Frere JM; Dideberg O X-Ray Structure of the ZnII Beta-Lactamase from Bacteroides fragilis in an Orthorhombic Crystal Form. Acta Crystallogr D Biol Crystallogr 1998, 54, 45–57. [DOI] [PubMed] [Google Scholar]
- 229.King DT; Worrall LJ; Gruninger R; Strynadka NC New Delhi Metallo-Beta-Lactamase: Structural Insights into Beta-Lactam Recognition and Inhibition. J. Am. Chem. Soc 2012, 134, 11362–11365. [DOI] [PubMed] [Google Scholar]
- 230.Cheng Z; VanPelt J; Bergstrom A; Bethel C; Katko A; Miller C; Mason K; Cumming E; Zhang H; Kimble RL, et al. A Noncanonical Metal Center Drives the Activity of the Sediminispirochaeta smaragdinae Metallo-Beta-Lactamase SPS-1. Biochemistry 2018, 57, 5218–5229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.de Seny D; Prosperi-Meys C; Bebrone C; Rossolini GM; Page MI; Noel P; Frere JM; Galleni M Mutational Analysis of the Two Zinc-Binding Sites of the Bacillus cereus 569/H/9 Metallo-Beta-Lactamase. Biochem. J 2002, 363, 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 232.Yang Y; Keeney D; Tang X; Canfield N; Rasmussen BA Kinetic Properties and Metal Content of the Metallo-Beta-Lactamase CcrA harboring Selective Amino Acid Substitutions. J. Biol. Chem 1999, 274, 15706–15711. [DOI] [PubMed] [Google Scholar]
- 233.Haruta S; Yamaguchi H; Yamamoto ET; Eriguchi Y; Nukaga M; O’Hara K; Sawai T Functional Analysis of the Active Site of a Metallo-Beta-Lactamase Proliferating in Japan. Antimicrob. Agents Chemother 2000, 44, 2304–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Vanhove M; Zakhem M; Devreese B; Franceschini N; Anne C; Bebrone C; Amicosante G; Rossolini GM; Van Beeumen J; Frere JM, et al. Role of Cys221 and Asn116 in the Zinc-Binding Sites of the Aeromonas hydrophila Metallo-Beta-Lactamase. Cell. Mol. Life Sci 2003, 60, 2501–2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Materon IC; Palzkill T Identification of Residues Critical for Metallo-Beta-Lactamase Function by Codon Randomization and Selection. Protein Sci 2001, 10, 2556–2565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 236.Sun Z; Hu L; Sankaran B; Prasad BVV; Palzkill T Differential Active Site Requirements for NDM-1 Beta-Lactamase Hydrolysis of Carbapenem Versus Penicillin and Cephalosporin Antibiotics. Nat Commun 2018, 9, 4524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Chen JZ; Fowler DM; Tokuriki N Comprehensive Exploration of the Translocation, Stability and Substrate Recognition Requirements in VIM-2 Lactamase. Elife 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Wang Z; Benkovic SJ Purification, Characterization, and Kinetic Studies of a Soluble Bacteroides fragilis Metallo-Beta-Lactamase That Provides Multiple Antibiotic Resistance. J. Biol. Chem 1998, 273, 22402–22408. [DOI] [PubMed] [Google Scholar]
- 239.Rasia RM; Vila AJ Structural Determinants of Substrate Binding to Bacillus cereus Metallo-Beta-Lactamase. J. Biol. Chem 2004, 279, 26046–26051. [DOI] [PubMed] [Google Scholar]
- 240.Tioni MF; Llarrull LI; Poeylaut-Palena AA; Marti MA; Saggu M; Periyannan GR; Mata EG; Bennett B; Murgida DH; Vila AJ Trapping and Characterization of a Reaction Intermediate in Carbapenem Hydrolysis by B. cereus Metallo-Beta-Lactamase. J. Am. Chem. Soc 2008, 130, 15852–15863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Lisa MN; Palacios AR; Aitha M; Gonzalez MM; Moreno DM; Crowder MW; Bonomo RA; Spencer J; Tierney DL; Llarrull LI, et al. A General Reaction Mechanism for Carbapenem Hydrolysis by Mononuclear and Binuclear Metallo-Beta-Lactamases. Nat Commun 2017, 8, 538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Wang Z; Fast W; Benkovic SJ On the Mechanism of the Metallo-Beta-Lactamase from Bacteroides fragilis. Biochemistry 1999, 38, 10013–10023. [DOI] [PubMed] [Google Scholar]
- 243.Abboud MI; Kosmopoulou M; Krismanich AP; Johnson JW; Hinchliffe P; Brem J; Claridge TDW; Spencer J; Schofield CJ; Dmitrienko GI Cyclobutanone Mimics of Intermediates in Metallo-Beta-Lactamase Catalysis. Chemistry 2018, 24, 5734–5737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 244.Tripathi R; Nair NN Mechanism of Meropenem Hydrolysis by New Delhi Metallo B-Lactamase. ACS Catalysis 2015, 5, 2577–2586. [Google Scholar]
- 245.Garau G; Bebrone C; Anne C; Galleni M; Frere JM; Dideberg O A Metallo-Beta-Lactamase Enzyme in Action: Crystal Structures of the Monozinc Carbapenemase Cpha and Its Complex with Biapenem. J. Mol. Biol 2005, 345, 785–795. [DOI] [PubMed] [Google Scholar]
- 246.Fonseca F; Bromley EH; Saavedra MJ; Correia A; Spencer J Crystal Structure of Serratia fonticola Sfh-I: Activation of the Nucleophile in Mono-Zinc Metallo-Beta-Lactamases. J. Mol. Biol 2011, 411, 951–959. [DOI] [PubMed] [Google Scholar]
- 247.Hernandez Valladares M; Felici A; Weber G; Adolph HW; Zeppezauer M; Rossolini GM; Amicosante G; Frere JM; Galleni M Zn(Ii) Dependence of the Aeromonas hydrophila AE036 Metallo-Beta-Lactamase Activity and Stability. Biochemistry 1997, 36, 11534–11541. [DOI] [PubMed] [Google Scholar]
- 248.Bebrone C; Delbruck H; Kupper MB; Schlomer P; Willmann C; Frere JM; Fischer R; Galleni M; Hoffmann KM The Structure of the Dizinc Subclass B2 Metallo-Beta-Lactamase CphA Reveals That the Second Inhibitory Zinc Ion Binds in the Histidine Site. Antimicrob. Agents Chemother 2009, 53, 4464–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Simona F; Magistrato A; Vera DM; Garau G; Vila AJ; Carloni P Protonation State and Substrate Binding to B2 Metallo-Beta-Lactamase CphA from Aeromonas hydrofila. Proteins 2007, 69, 595–605. [DOI] [PubMed] [Google Scholar]
- 250.Ullah JH; Walsh TR; Taylor IA; Emery DC; Verma CS; Gamblin SJ; Spencer J The Crystal Structure of the L1 Metallo-Beta-Lactamase from Stenotrophomonas maltophilia at 1.7 A Resolution. J. Mol. Biol 1998, 284, 125–136. [DOI] [PubMed] [Google Scholar]
- 251.Moran-Barrio J; Gonzalez JM; Lisa MN; Costello AL; Peraro MD; Carloni P; Bennett B; Tierney DL; Limansky AS; Viale AM, et al. The Metallo-Beta-Lactamase GOB Is a Mono-Zn(II) Enzyme with a Novel Active Site. J. Biol. Chem 2007, 282, 18286–18293. [DOI] [PubMed] [Google Scholar]
- 252.Lisa MN; Hemmingsen L; Vila AJ Catalytic Role of the Metal Ion in the Metallo-Beta-Lactamase GOB. J. Biol. Chem 2010, 285, 4570–4577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Horsfall LE; Izougarhane Y; Lassaux P; Selevsek N; Lienard BM; Poirel L; Kupper MB; Hoffmann KM; Frere JM; Galleni M, et al. Broad Antibiotic Resistance Profile of the Subclass B3 Metallo-Beta-Lactamase GOB-1, a Di-Zinc Enzyme. FEBS J. 2011, 278, 1252–1263. [DOI] [PubMed] [Google Scholar]
- 254.Moran-Barrio J; Lisa MN; Larrieux N; Drusin SI; Viale AM; Moreno DM; Buschiazzo A; Vila AJ Crystal Structure of the Metallo-Beta-Lactamase GOB in the Periplasmic Dizinc Form Reveals an Unusual Metal Site. Antimicrob. Agents Chemother 2016, 60, 6013–6022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Paton R; Miles RS; Amyes SG Biochemical Properties of Inducible Beta-Lactamases Produced from Xanthomonas Maltophilia. Antimicrob. Agents Chemother 1994, 38, 2143–2149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 256.Crowder MW; Walsh TR; Banovic L; Pettit M; Spencer J Overexpression, Purification, and Characterization of the Cloned Metallo-Beta-Lactamase L1 from Stenotrophomonas Maltophilia. Antimicrob. Agents Chemother 1998, 42, 921–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 257.Simm AM; Higgins CS; Carenbauer AL; Crowder MW; Bateson JH; Bennett PM; Clarke AR; Halford SE; Walsh TR Characterization of Monomeric L1 Metallo-Beta-Lactamase and the Role of the N-Terminal Extension in Negative Cooperativity and Antibiotic Hydrolysis. J. Biol. Chem 2002, 277, 24744–24752. [DOI] [PubMed] [Google Scholar]
- 258.Zhang H; Ma G; Zhu Y; Zeng L; Ahmad A; Wang C; Pang B; Fang H; Zhao L; Hao Q Active-Site Conformational Fluctuations Promote the Enzymatic Activity of NDM-1. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Salimraj R; Hinchliffe P; Kosmopoulou M; Tyrrell JM; Brem J; van Berkel SS; Verma A; Owens RJ; McDonough MA; Walsh TR, et al. Crystal Structures of VIM-1 Complexes Explain Active Site Heterogeneity in Vim-Class Metallo-Beta-Lactamases. FEBS J. 2019, 286, 169–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Nauton L; Kahn R; Garau G; Hernandez JF; Dideberg O Structural Insights into the Design of Inhibitors for the L1 Metallo-Beta-Lactamase from Stenotrophomonas maltophilia. J. Mol. Biol 2008, 375, 257–269. [DOI] [PubMed] [Google Scholar]
- 261.Docquier JD; Benvenuti M; Calderone V; Stoczko M; Menciassi N; Rossolini GM; Mangani S High-Resolution Crystal Structure of the Subclass B3 Metallo-Beta-Lactamase BJP-1: Rational Basis for Substrate Specificity and Interaction with Sulfonamides. Antimicrob. Agents Chemother 2010, 54, 4343–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Kuwabara S; Abraham EP Some Properties of Two Extracellular Beta-Lactamases from Bacillus cereus 569/H. Biochem. J 1967, 103, 27C–30C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Davies RB Comparison of Beta-Lactamase II from Bacillus cereus 569/H/9 with a Beta-Lactamase from Bacillus cereus 5/B/6. Biochem. J 1975, 145, 409–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Kuwabara S Purification and Properties of Two Extracellular Beta-Lactamases from Bacillus cereus 569-H. Biochem. J 1970, 118, 457–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Davies RB; Abraham EP Metal Cofactor Requirements of Beta-Lactamase II. Biochem. J 1974, 143, 129–135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Baldwin GS; Galdes A; Hill HA; Smith BE; Waley SG; Abraham EP Histidine Residues of Zinc Ligands in Beta-Lactamase II. Biochem. J 1978, 175, 441–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Baldwin GS; Waley SG; Abraham EP Identification of Histidine Residues That Act as Zinc Ligands in Beta-Lactamase II by Differential Tritium Exchange. Biochem. J 1979, 179, 459–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Galdes A; Hill HA; Baldwin GS; Waley SG; Abraham EP The 1H Nuclear-Magnetic-Resonance Spectroscopy of Cobalt(II)-Beta-Lactamase II. Biochem. J 1980, 187, 789–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Baldwin GS; Galdes A; Hill HA; Waley SG; Abraham EP A Spectroscopic Study of Metal Ion and Ligand Binding to Beta-Lactamase II. J. Inorg. Biochem 1980, 13, 189–204. [DOI] [PubMed] [Google Scholar]
- 270.Bicknell R; Waley SG Cryoenzymology of Bacillus cereus Beta-Lactamase II. Biochemistry 1985, 24, 6876–6887. [DOI] [PubMed] [Google Scholar]
- 271.Bicknell R; Schaffer A; Waley SG; Auld DS Changes in the Coordination Geometry of the Active-Site Metal During Catalysis of Benzylpenicillin Hydrolysis by Bacillus cereus Beta-Lactamase II. Biochemistry 1986, 25, 7208–7215. [DOI] [PubMed] [Google Scholar]
- 272.Ambler RP; Daniel M; Fleming J; Hermoso JM; Pang C; Waley SG The Amino Acid Sequence of the Zinc-Requiring Beta-Lactamase II from the Bacterium Bacillus cereus 569. FEBS Lett. 1985, 189, 207–211. [DOI] [PubMed] [Google Scholar]
- 273.Hussain M; Carlino A; Madonna MJ; Lampen JO Cloning and Sequencing of the Metallothioprotein Beta-Lactamase II Gene of Bacillus cereus 569/H in Escherichia Coli. J. Bacteriol 1985, 164, 223–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Tomatis PE; Rasia RM; Segovia L; Vila AJ Mimicking Natural Evolution in Metallo-Beta-Lactamases through Second-Shell Ligand Mutations. Proc Natl Acad Sci U S A 2005, 102, 13761–13766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Lim HM; Pene JJ Mutations Affecting the Catalytic Activity of Bacillus cereus 5/B/6 Beta-Lactamase II. J. Biol. Chem 1989, 264, 11682–11687. [PubMed] [Google Scholar]
- 276.Lim HM; Iyer RK; Pene JJ Site-Directed Mutagenesis of Dicarboxylic Acids near the Active Site of Bacillus cereus 5/B/6 Beta-Lactamase II. Biochem. J 1991, 276 (Pt 2), 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Sutton BJ; Artymiuk PJ; Cordero-Borboa AE; Little C; Phillips DC; Waley SG An X-Ray-Crystallographic Study of Beta-Lactamase II from Bacillus cereus at 0.35 nm Resolution. Biochem. J 1987, 248, 181–188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Carfi A; Duee E; Galleni M; Frere JM; Dideberg O 1.85 A Resolution Structure of the Zinc(II) Beta-Lactamase from Bacillus cereus. Acta Crystallogr D Biol Crystallogr 1998, 54, 313–323. [DOI] [PubMed] [Google Scholar]
- 279.Orellano EG; Girardini JE; Cricco JA; Ceccarelli EA; Vila AJ Spectroscopic Characterization of a Binuclear Metal Site in Bacillus cereus Beta-Lactamase II. Biochemistry 1998, 37, 10173–10180. [DOI] [PubMed] [Google Scholar]
- 280.Davies AM; Rasia RM; Vila AJ; Sutton BJ; Fabiane SM Effect of pH on the Active Site of an Arg121Cys Mutant of the Metallo-Beta-Lactamase from Bacillus cereus: Implications for the Enzyme Mechanism. Biochemistry 2005, 44, 4841–4849. [DOI] [PubMed] [Google Scholar]
- 281.Murphy TA; Catto LE; Halford SE; Hadfield AT; Minor W; Walsh TR; Spencer J Crystal Structure of Pseudomonas aeruginosa SPM-1 Provides Insights into Variable Zinc Affinity of Metallo-Beta-Lactamases. J. Mol. Biol 2006, 357, 890–903. [DOI] [PubMed] [Google Scholar]
- 282.Garcia-Saez I; Docquier JD; Rossolini GM; Dideberg O The Three-Dimensional Structure of VIM-2, a Zn-Beta-Lactamase from Pseudomonas aeruginosa in Its Reduced and Oxidised Form. J. Mol. Biol 2008, 375, 604–611. [DOI] [PubMed] [Google Scholar]
- 283.Kim Y; Tesar C; Mire J; Jedrzejczak R; Binkowski A; Babnigg G; Sacchettini J; Joachimiak A Structure of Apo- and Monometalated Forms of NDM-1 --a Highly Potent Carbapenem-Hydrolyzing Metallo-Beta-Lactamase. PLoS One 2011, 6, e24621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Kim Y; Maltseva N; Wilamowski M; Tesar C; Endres M; Joachimiak A Structural and Biochemical Analysis of the Metallo-Beta-Lactamase L1 from Emerging Pathogen Stenotrophomonas maltophilia Revealed the Subtle but Distinct Di-Metal Scaffold for Catalytic Activity. Protein Sci 2020, 29, 723–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Breece RM; Llarrull LI; Tioni MF; Vila AJ; Tierney DL X-Ray Absorption Spectroscopy of Metal Site Speciation in the Metallo-Beta-Lactamase BcII from Bacillus cereus. J. Inorg. Biochem 2012, 111, 182–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Llarrull LI; Tioni MF; Kowalski J; Bennett B; Vila AJ Evidence for a Dinuclear Active Site in the Metallo-Beta-Lactamase BcII with Substoichiometric Co(II). A New Model for Metal Uptake. J. Biol. Chem 2007, 282, 30586–30595. [DOI] [PubMed] [Google Scholar]
- 287.Llarrull LI; Tioni MF; Vila AJ Metal Content and Localization During Turnover in B. cereus Metallo-Beta-Lactamase. J. Am. Chem. Soc 2008, 130, 15842–15851. [DOI] [PubMed] [Google Scholar]
- 288.Hemmingsen L; Damblon C; Antony J; Jensen M; Adolph HW; Wommer S; Roberts GC; Bauer R Dynamics of Mononuclear Cadmium Beta-Lactamase Revealed by the Combination of NMR and PAC Spectroscopy. J. Am. Chem. Soc 2001, 123, 10329–10335. [DOI] [PubMed] [Google Scholar]
- 289.Paul-Soto R; Zeppezauer M; Adolph HW; Galleni M; Frere JM; Carfi A; Dideberg O; Wouters J; Hemmingsen L; Bauer R Preference of Cd(II) and Zn(II) for the Two Metal Sites in Bacillus cereus Beta-Lactamase II: A Perturbed Angular Correlation of Gamma-Rays Spectroscopic Study. Biochemistry 1999, 38, 16500–16506. [DOI] [PubMed] [Google Scholar]
- 290.de Seny D; Heinz U; Wommer S; Kiefer M; Meyer-Klaucke W; Galleni M; Frere JM; Bauer R; Adolph HW Metal Ion Binding and Coordination Geometry for Wild Type and Mutants of Metallo-Beta -Lactamase from Bacillus cereus 569/H/9 (BcII): A Combined Thermodynamic, Kinetic, and Spectroscopic Approach. J. Biol. Chem 2001, 276, 45065–45078. [DOI] [PubMed] [Google Scholar]
- 291.Hawk MJ; Breece RM; Hajdin CE; Bender KM; Hu Z; Costello AL; Bennett B; Tierney DL; Crowder MW Differential Binding of Co(II) and Zn(II) to Metallo-Beta-Lactamase Bla2 from Bacillus anthracis. J. Am. Chem. Soc 2009, 131, 10753–10762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Hu Z; Periyannan G; Bennett B; Crowder MW Role of the Zn1 and Zn2 Sites in Metallo-Beta-Lactamase L1. J. Am. Chem. Soc 2008, 130, 14207–14216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Yang H; Aitha M; Marts AR; Hetrick A; Bennett B; Crowder MW; Tierney DL Spectroscopic and Mechanistic Studies of Heterodimetallic Forms of Metallo-Beta-Lactamase NDM-1. J. Am. Chem. Soc 2014, 136, 7273–7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 294.Wommer S; Rival S; Heinz U; Galleni M; Frere JM; Franceschini N; Amicosante G; Rasmussen B; Bauer R; Adolph HW Substrate-Activated Zinc Binding of Metallo-Beta -Lactamases: Physiological Importance of Mononuclear Enzymes. J. Biol. Chem 2002, 277, 24142–24147. [DOI] [PubMed] [Google Scholar]
- 295.Jacquin O; Balbeur D; Damblon C; Marchot P; De Pauw E; Roberts GC; Frere JM; Matagne A Positively Cooperative Binding of Zinc Ions to Bacillus cereus 569/H/9 Beta-Lactamase II Suggests That the Binuclear Enzyme Is the Only Relevant Form for Catalysis. J. Mol. Biol 2009, 392, 1278–1291. [DOI] [PubMed] [Google Scholar]
- 296.Badarau A; Page MI Enzyme Deactivation Due to Metal-Ion Dissociation During Turnover of the Cobalt-Beta-Lactamase Catalyzed Hydrolysis of Beta-Lactams. Biochemistry 2006, 45, 11012–11020. [DOI] [PubMed] [Google Scholar]
- 297.Badarau A; Damblon C; Page MI The Activity of the Dinuclear Cobalt-Beta-Lactamase from Bacillus cereus in Catalysing the Hydrolysis of Beta-Lactams. Biochem. J 2007, 401, 197–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 298.Karsisiotis AI; Damblon CF; Roberts GC A Variety of Roles for Versatile Zinc in Metallo-Beta-Lactamases. Metallomics 2014, 6, 1181–1197. [DOI] [PubMed] [Google Scholar]
- 299.Griffin DH; Richmond TK; Sanchez C; Moller AJ; Breece RM; Tierney DL; Bennett B; Crowder MW Structural and Kinetic Studies on Metallo-Beta-Lactamase IMP-1. Biochemistry 2011, 50, 9125–9134. [DOI] [PubMed] [Google Scholar]
- 300.Gonzalez JM; Meini MR; Tomatis PE; Medrano Martin FJ; Cricco JA; Vila AJ Metallo-Beta-Lactamases Withstand Low Zn(II) Conditions by Tuning Metal-Ligand Interactions. Nat. Chem. Biol 2012, 8, 698–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 301.Eriksson AE; Jones TA; Liljas A Refined Structure of Human Carbonic Anhydrase II at 2.0 A Resolution. Proteins 1988, 4, 274–282. [DOI] [PubMed] [Google Scholar]
- 302.Lindskog S Structure and Mechanism of Carbonic Anhydrase. Pharmacol. Ther 1997, 74, 1–20. [DOI] [PubMed] [Google Scholar]
- 303.Lipscomb WN; Strater N Recent Advances in Zinc Enzymology. Chem. Rev 1996, 96, 2375–2434. [DOI] [PubMed] [Google Scholar]
- 304.Karlin S; Zhu ZY Classification of Mononuclear Zinc Metal Sites in Protein Structures. Proc Natl Acad Sci U S A 1997, 94, 14231–14236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Mitic N; Smith SJ; Neves A; Guddat LW; Gahan LR; Schenk G The Catalytic Mechanisms of Binuclear Metallohydrolases. Chem. Rev 2006, 106, 3338–3363. [DOI] [PubMed] [Google Scholar]
- 306.Mazzei L; Musiani F; Ciurli S The Structure-Based Reaction Mechanism of Urease, a Nickel Dependent Enzyme: Tale of a Long Debate. J. Biol. Inorg. Chem 2020, 25, 829–845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Breece RM; Hu Z; Bennett B; Crowder MW; Tierney DL Motion of the Zinc Ions in Catalysis by a Dizinc Metallo-Beta-Lactamase. J. Am. Chem. Soc 2009, 131, 11642–11643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Dudev T; Lim C Metal Binding Affinity and Selectivity in Metalloproteins: Insights from Computational Studies. Annu Rev Biophys 2008, 37, 97–116. [DOI] [PubMed] [Google Scholar]
- 309.Dudev T; Lim C Principles Governing Mg, Ca, and Zn Binding and Selectivity in Proteins. Chem. Rev 2003, 103, 773–788. [DOI] [PubMed] [Google Scholar]
- 310.Horton LB; Shanker S; Mikulski R; Brown NG; Phillips KJ; Lykissa E; Venkataram Prasad BV; Palzkill T Mutagenesis of Zinc Ligand Residue Cys221 Reveals Plasticity in the IMP-1 Metallo-Beta-Lactamase Active Site. Antimicrob. Agents Chemother 2012, 56, 5667–5677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Corbin BD; Seeley EH; Raab A; Feldmann J; Miller MR; Torres VJ; Anderson KL; Dattilo BM; Dunman PM; Gerads R, et al. Metal Chelation and Inhibition of Bacterial Growth in Tissue Abscesses. Science 2008, 319, 962–965. [DOI] [PubMed] [Google Scholar]
- 312.Antelo GT; Vila AJ; Giedroc DP; Capdevila DA Molecular Evolution of Transition Metal Bioavailability at the Host-Pathogen Interface. Trends Microbiol 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Bebrone C; Anne C; Kerff F; Garau G; De Vriendt K; Lantin R; Devreese B; Van Beeumen J; Dideberg O; Frere JM, et al. Mutational Analysis of the Zinc- and Substrate-Binding Sites in the CphA Metallo-Beta-Lactamase from Aeromonas hydrophila. Biochem. J 2008, 414, 151–159. [DOI] [PubMed] [Google Scholar]
- 314.Sun Z; Mehta SC; Adamski CJ; Gibbs RA; Palzkill T Deep Sequencing of Random Mutant Libraries Reveals the Active Site of the Narrow Specificity CphA Metallo-Beta-Lactamase Is Fragile to Mutations. Sci Rep 2016, 6, 33195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Yanchak MP; Taylor RA; Crowder MW Mutational Analysis of Metallo-Beta-Lactamase CcrA from Bacteroides Fragilis. Biochemistry 2000, 39, 11330–11339. [DOI] [PubMed] [Google Scholar]
- 316.Crisp J; Conners R; Garrity JD; Carenbauer AL; Crowder MW; Spencer J Structural Basis for the Role of Asp-120 in Metallo-Beta-Lactamases. Biochemistry 2007, 46, 10664–10674. [DOI] [PubMed] [Google Scholar]
- 317.Garrity JD; Carenbauer AL; Herron LR; Crowder MW Metal Binding Asp-120 in Metallo-Beta-Lactamase L1 from Stenotrophomonas maltophilia Plays a Crucial Role in Catalysis. J. Biol. Chem 2004, 279, 920–927. [DOI] [PubMed] [Google Scholar]
- 318.Llarrull LI; Fabiane SM; Kowalski JM; Bennett B; Sutton BJ; Vila AJ Asp-120 Locates Zn2 for Optimal Metallo-Beta-Lactamase Activity. J. Biol. Chem 2007, 282, 18276–18285. [DOI] [PubMed] [Google Scholar]
- 319.Kim Y; Cunningham MA; Mire J; Tesar C; Sacchettini J; Joachimiak A NDM-1, the Ultimate Promiscuous Enzyme: Substrate Recognition and Catalytic Mechanism. FASEB J. 2013, 27, 1917–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Raczynska JE; Shabalin IG; Minor W; Wlodawer A; Jaskolski M A Close Look onto Structural Models and Primary Ligands of Metallo-Beta-Lactamases. Drug Resist Updat 2018, 40, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Das CK; Nair NN Hydrolysis of Cephalexin and Meropenem by New Delhi Metallo-Beta-Lactamase: The Substrate Protonation Mechanism Is Drug Dependent. Phys. Chem. Chem. Phys 2017, 19, 13111–13121. [DOI] [PubMed] [Google Scholar]
- 322.Spencer J; Clarke AR; Walsh TR Novel Mechanism of Hydrolysis of Therapeutic Beta-Lactams by Stenotrophomonas maltophilia L1 Metallo-Beta-Lactamase. J. Biol. Chem 2001, 276, 33638–33644. [DOI] [PubMed] [Google Scholar]
- 323.Sharma NP; Hajdin C; Chandrasekar S; Bennett B; Yang KW; Crowder MW Mechanistic Studies on the Mononuclear ZnII-Containing Metallo-Beta-Lactamase ImiS from Aeromonas sobria. Biochemistry 2006, 45, 10729–10738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Garrity JD; Pauff JM; Crowder MW Probing the Dynamics of a Mobile Loop above the Active Site of L1, a Metallo-Beta-Lactamase from Stenotrophomonas maltophilia, Via Site-Directed Mutagenesis and Stopped-Flow Fluorescence Spectroscopy. J. Biol. Chem 2004, 279, 39663–39670. [DOI] [PubMed] [Google Scholar]
- 325.Christianson DW; Mangani S; Shoham G; Lipscomb WN Binding of D-Phenylalanine and D-Tyrosine to Carboxypeptidase A. J. Biol. Chem 1989, 264, 12849–12853. [PubMed] [Google Scholar]
- 326.Dal Peraro M; Vila AJ; Carloni P; Klein ML Role of Zinc Content on the Catalytic Efficiency of B1 Metallo Beta-Lactamases. J. Am. Chem. Soc 2007, 129, 2808–2816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Suarez D; Diaz N; Merz KM Jr. Molecular Dynamics Simulations of the Dinuclear Zinc-Beta-Lactamase from Bacteroides fragilis Complexed with Imipenem. J. Comput. Chem 2002, 23, 1587–1600. [DOI] [PubMed] [Google Scholar]
- 328.Feng H; Liu X; Wang S; Fleming J; Wang DC; Liu W The Mechanism of NDM-1-Catalyzed Carbapenem Hydrolysis Is Distinct from That of Penicillin or Cephalosporin Hydrolysis. Nat Commun 2017, 8, 2242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Feng H; Ding J; Zhu D; Liu X; Xu X; Zhang Y; Zang S; Wang DC; Liu W Structural and Mechanistic Insights into NDM-1 Catalyzed Hydrolysis of Cephalosporins. J. Am. Chem. Soc 2014, 136, 14694–14697. [DOI] [PubMed] [Google Scholar]
- 330.Spencer J; Read J; Sessions RB; Howell S; Blackburn GM; Gamblin SJ Antibiotic Recognition by Binuclear Metallo-Beta-Lactamases Revealed by X-Ray Crystallography. J. Am. Chem. Soc 2005, 127, 14439–14444. [DOI] [PubMed] [Google Scholar]
- 331.Wockel S; Galezowska J; Dechert S; Meyer F Binding of Beta-Lactam Antibiotics to a Bioinspired Dizinc Complex Reminiscent of the Active Site of Metallo-Beta-Lactamases. Inorg. Chem 2012, 51, 2486–2493. [DOI] [PubMed] [Google Scholar]
- 332.Borgianni L; Vandenameele J; Matagne A; Bini L; Bonomo RA; Frere JM; Rossolini GM; Docquier JD Mutational Analysis of VIM-2 Reveals an Essential Determinant for Metallo-Beta-Lactamase Stability and Folding. Antimicrob. Agents Chemother 2010, 54, 3197–3204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Moali C; Anne C; Lamotte-Brasseur J; Groslambert S; Devreese B; Van Beeumen J; Galleni M; Frere JM Analysis of the Importance of the Metallo-Beta-Lactamase Active Site Loop in Substrate Binding and Catalysis. Chem. Biol 2003, 10, 319–329. [DOI] [PubMed] [Google Scholar]
- 334.Huntley JJ; Fast W; Benkovic SJ; Wright PE; Dyson HJ Role of a Solvent-Exposed Tryptophan in the Recognition and Binding of Antibiotic Substrates for a Metallo-Beta-Lactamase. Protein Sci 2003, 12, 1368–1375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Materon IC; Beharry Z; Huang W; Perez C; Palzkill T Analysis of the Context Dependent Sequence Requirements of Active Site Residues in the Metallo-Beta-Lactamase IMP-1. J. Mol. Biol 2004, 344, 653–663. [DOI] [PubMed] [Google Scholar]
- 336.LaCuran AE; Pegg KM; Liu EM; Bethel CR; Ai N; Welsh WJ; Bonomo RA; Oelschlaeger P Elucidating the Role of Residue 67 in IMP-Type Metallo-Beta-Lactamase Evolution. Antimicrob. Agents Chemother 2015, 59, 7299–7307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Palacios AR; Mojica MF; Giannini E; Taracila MA; Bethel CR; Alzari PM; Otero LH; Klinke S; Llarrull LI; Bonomo RA, et al. The Reaction Mechanism of Metallo-Beta-Lactamases Is Tuned by the Conformation of an Active-Site Mobile Loop. Antimicrob. Agents Chemother 2019, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Lohans CT; Brem J; Schofield CJ New Delhi Metallo-Beta-Lactamase 1 Catalyzes Avibactam and Aztreonam Hydrolysis. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Poeylaut-Palena AA; Tomatis PE; Karsisiotis AI; Damblon C; Mata EG; Vila AJ A Minimalistic Approach to Identify Substrate Binding Features in B1 Metallo-Beta-Lactamases. Bioorg. Med. Chem. Lett 2007, 17, 5171–5174. [DOI] [PubMed] [Google Scholar]
- 340.Yuan Q; He L; Ke H A Potential Substrate Binding Conformation of Beta-Lactams and Insight into the Broad Spectrum of NDM-1 Activity. Antimicrob. Agents Chemother 2012, 56, 5157–5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Gonzalez JM; Buschiazzo A; Vila AJ Evidence of Adaptability in Metal Coordination Geometry and Active-Site Loop Conformation among B1 Metallo-Beta-Lactamases. Biochemistry 2010, 49, 7930–7938. [DOI] [PubMed] [Google Scholar]
- 342.Bebrone C; Anne C; De Vriendt K; Devreese B; Rossolini GM; Van Beeumen J; Frere JM; Galleni M Dramatic Broadening of the Substrate Profile of the Aeromonas hydrophila CphA Metallo-Beta-Lactamase by Site-Directed Mutagenesis. J. Biol. Chem 2005, 280, 28195–28202. [DOI] [PubMed] [Google Scholar]
- 343.Scrofani SD; Chung J; Huntley JJ; Benkovic SJ; Wright PE; Dyson HJ NMR Characterization of the Metallo-Beta-Lactamase from Bacteroides fragilis and Its Interaction with a Tight-Binding Inhibitor: Role of an Active-Site Loop. Biochemistry 1999, 38, 14507–14514. [DOI] [PubMed] [Google Scholar]
- 344.Huntley JJ; Scrofani SD; Osborne MJ; Wright PE; Dyson HJ Dynamics of the Metallo-Beta-Lactamase from Bacteroides fragilis in the Presence and Absence of a Tight-Binding Inhibitor. Biochemistry 2000, 39, 13356–13364. [DOI] [PubMed] [Google Scholar]
- 345.Karsisiotis AI; Damblon CF; Roberts GC Solution Structures of the Bacillus cereus Metallo-Beta-Lactamase BcII and Its Complex with the Broad Spectrum Inhibitor R-Thiomandelic Acid. Biochem. J 2013, 456, 397–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 346.Aitha M; Moller AJ; Sahu ID; Horitani M; Tierney DL; Crowder MW Investigating the Position of the Hairpin Loop in New Delhi Metallo-Beta-Lactamase, NDM-1, During Catalysis and Inhibitor Binding. J. Inorg. Biochem 2016, 156, 35–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Gonzalez MM; Abriata LA; Tomatis PE; Vila AJ Optimization of Conformational Dynamics in an Epistatic Evolutionary Trajectory. Mol. Biol. Evol 2016, 33, 1768–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Brem J; Struwe WB; Rydzik AM; Tarhonskaya H; Pfeffer I; Flashman E; van Berkel SS; Spencer J; Claridge TD; McDonough MA, et al. Studying the Active-Site Loop Movement of the Sao Paolo Metallo-Beta-Lactamase-1. Chem Sci 2015, 6, 956–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 349.Abboud MI; Hinchliffe P; Brem J; Macsics R; Pfeffer I; Makena A; Umland KD; Rydzik AM; Li GB; Spencer J, et al. (19) F-NMR Reveals the Role of Mobile Loops in Product and Inhibitor Binding by the Sao Paulo Metallo-Beta-Lactamase. Angew. Chem. Int. Ed. Engl 2017, 56, 3862–3866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Aitha M; Moritz L; Sahu ID; Sanyurah O; Roche Z; McCarrick R; Lorigan GA; Bennett B; Crowder MW Conformational Dynamics of Metallo-Beta-Lactamase CcrA During Catalysis Investigated by Using Deer Spectroscopy. J. Biol. Inorg. Chem 2015, 20, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Sharma N; Hu Z; Crowder MW; Bennett B Conformational Changes in the Metallo-Beta-Lactamase ImiS During the Catalytic Reaction: An EPR Spectrokinetic Study of Co(II)-Spin Label Interactions. J. Am. Chem. Soc 2008, 130, 8215–8222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 352.Lancaster KM Biological Outer-Sphere Coordination. In Molecular Electronic Structures of Transition Metal Complexes I, Mingos DMP; Day P; Dahl JP, Eds. Springer Berlin Heidelberg: Berlin, Heidelberg, 2012; pp 119–153. [Google Scholar]
- 353.Cox JD; Hunt JA; Compher KM; Fierke CA; Christianson DW Structural Influence of Hydrophobic Core Residues on Metal Binding and Specificity in Carbonic Anhydrase Ii. Biochemistry 2000, 39, 13687–13694. [DOI] [PubMed] [Google Scholar]
- 354.Tomatis PE; Fabiane SM; Simona F; Carloni P; Sutton BJ; Vila AJ Adaptive Protein Evolution Grants Organismal Fitness by Improving Catalysis and Flexibility. Proc Natl Acad Sci U S A 2008, 105, 20605–20610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 355.Meini MR; Tomatis PE; Weinreich DM; Vila AJ Quantitative Description of a Protein Fitness Landscape Based on Molecular Features. Mol. Biol. Evol 2015, 32, 1774–1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 356.Oelschlaeger P; Mayo SL; Pleiss J Impact of Remote Mutations on Metallo-Beta-Lactamase Substrate Specificity: Implications for the Evolution of Antibiotic Resistance. Protein Sci 2005, 14, 765–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 357.Liu EM; Pegg KM; Oelschlaeger P The Sequence-Activity Relationship between Metallo-Beta-Lactamases IMP-1, IMP-6, and IMP-25 Suggests an Evolutionary Adaptation to Meropenem Exposure. Antimicrob. Agents Chemother 2012, 56, 6403–6406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 358.Iyobe S; Kusadokoro H; Ozaki J; Matsumura N; Minami S; Haruta S; Sawai T; O’Hara K Amino Acid Substitutions in a Variant of IMP-1 Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2000, 44, 2023–2027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Cuchural GJ Jr.; Malamy MH; Tally FP Beta-Lactamase-Mediated Imipenem Resistance in Bacteroides fragilis. Antimicrob. Agents Chemother 1986, 30, 645–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 360.Rasmussen BA; Gluzman Y; Tally FP Cloning and Sequencing of the Class B Beta-Lactamase Gene (CcrA) from Bacteroides Fragilis Tal3636. Antimicrob. Agents Chemother 1990, 34, 1590–1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 361.Sato K; Fujii T; Okamoto R; Inoue M; Mitsuhashi S Biochemical Properties of Beta-Lactamase Produced by Flavobacterium odoratum. Antimicrob. Agents Chemother 1985, 27, 612–614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Bellais S; Naas T; Nordmann P Genetic and Biochemical Characterization of CGB-1, an Ambler Class B Carbapenem-Hydrolyzing Beta-Lactamase from Chryseobacterium gleum. Antimicrob. Agents Chemother 2002, 46, 2791–2796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 363.Saino Y; Kobayashi F; Inoue M; Mitsuhashi S Purification and Properties of Inducible Penicillin Beta-Lactamase Isolated from Pseudomonas maltophilia. Antimicrob. Agents Chemother 1982, 22, 564–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Watanabe M; Iyobe S; Inoue M; Mitsuhashi S Transferable Imipenem Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 1991, 35, 147–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 365.Poirel L; Magalhaes M; Lopes M; Nordmann P Molecular Analysis of Metallo-Beta-Lactamase Gene bla(SPM-1)-Surrounding Sequences from Disseminated Pseudomonas aeruginosa Isolates in Recife, Brazil. Antimicrob. Agents Chemother 2004, 48, 1406–1409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Poirel L; Rodriguez-Martinez JM; Al Naiemi N; Debets-Ossenkopp YJ; Nordmann P Characterization of DIM-1, an Integron-Encoded Metallo-Beta-Lactamase from a Pseudomonas stutzeri Clinical Isolate in the Netherlands. Antimicrob. Agents Chemother 2010, 54, 2420–2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 367.Pollini S; Maradei S; Pecile P; Olivo G; Luzzaro F; Docquier JD; Rossolini GM FIM-1, a New Acquired Metallo-Beta-Lactamase from a Pseudomonas aeruginosa Clinical Isolate from Italy. Antimicrob. Agents Chemother 2013, 57, 410–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 368.Pfennigwerth N; Lange F; Belmar Campos C; Hentschke M; Gatermann SG; Kaase M Genetic and Biochemical Characterization of HMB-1, a Novel Subclass B1 Metallo-Beta-Lactamase Found in a Pseudomonas aeruginosa Clinical Isolate. J. Antimicrob. Chemother 2017, 72, 1068–1073. [DOI] [PubMed] [Google Scholar]
- 369.El Salabi A; Borra PS; Toleman MA; Samuelsen O; Walsh TR Genetic and Biochemical Characterization of a Novel Metallo-Beta-Lactamase, TMB-1, from an Achromobacter xylosoxidans Strain Isolated in Tripoli, Libya. Antimicrob. Agents Chemother 2012, 56, 2241–2245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 370.Sekiguchi J; Morita K; Kitao T; Watanabe N; Okazaki M; Miyoshi-Akiyama T; Kanamori M; Kirikae T KHM-1, a Novel Plasmid-Mediated Metallo-Beta-Lactamase from a Citrobacter freundii Clinical Isolate. Antimicrob. Agents Chemother 2008, 52, 4194–4197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 371.Castanheira M; Toleman MA; Jones RN; Schmidt FJ; Walsh TR Molecular Characterization of a Beta-Lactamase Gene, blaGIM-1, Encoding a New Subclass of Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2004, 48, 4654–4661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Lee K; Yum JH; Yong D; Lee HM; Kim HD; Docquier JD; Rossolini GM; Chong Y Novel Acquired Metallo-Beta-Lactamase Gene, bla(SIM-1), in a Class 1 Integron from Acinetobacter baumannii Clinical Isolates from Korea. Antimicrob. Agents Chemother 2005, 49, 4485–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 373.Chen Y; Succi J; Tenover FC; Koehler TM Beta-Lactamase Genes of the Penicillin-Susceptible Bacillus anthracis Sterne Strain. J. Bacteriol 2003, 185, 823–830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 374.Rossolini GM; Franceschini N; Riccio ML; Mercuri PS; Perilli M; Galleni M; Frere JM; Amicosante G Characterization and Sequence of the Chryseobacterium (Flavobacterium) meningosepticum Carbapenemase: A New Molecular Class B Beta-Lactamase Showing a Broad Substrate Profile. Biochem. J 1998, 332 (Pt 1), 145–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Bellais S; Leotard S; Poirel L; Naas T; Nordmann P Molecular Characterization of a Carbapenem-Hydrolyzing Beta-Lactamase from Chryseobacterium (Flavobacterium) indologenes. FEMS Microbiol. Lett 1999, 171, 127–132. [DOI] [PubMed] [Google Scholar]
- 376.Naas T; Bellais S; Nordmann P Molecular and Biochemical Characterization of a Carbapenem-Hydrolysing Beta-Lactamase from Flavobacterium johnsoniae. J. Antimicrob. Chemother 2003, 51, 267–273. [DOI] [PubMed] [Google Scholar]
- 377.Mammeri H; Bellais S; Nordmann P Chromosome-Encoded Beta-Lactamases TUS-1 and MUS-1 from Myroides odoratus and Myroides odoratimimus (formerly Flavobacterium odoratum), New Members of the Lineage of Molecular Subclass B1 Metalloenzymes. Antimicrob. Agents Chemother 2002, 46, 3561–3567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 378.Bellais S; Girlich D; Karim A; Nordmann P EBR-1, a Novel Ambler Subclass B1 Beta-Lactamase from Empedobacter brevis. Antimicrob. Agents Chemother 2002, 46, 3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 379.Osano E; Arakawa Y; Wacharotayankun R; Ohta M; Horii T; Ito H; Yoshimura F; Kato N Molecular Characterization of an Enterobacterial Metallo Beta-Lactamase Found in a Clinical Isolate of Serratia marcescens That Shows Imipenem Resistance. Antimicrob. Agents Chemother 1994, 38, 71–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 380.Pal A; Tripathi A An in Silico Approach for Understanding the Molecular Evolution of Clinically Important Metallo-Beta-Lactamases. Infect Genet Evol 2013, 20, 39–47. [DOI] [PubMed] [Google Scholar]
- 381.Laraki N; Franceschini N; Rossolini GM; Santucci P; Meunier C; de Pauw E; Amicosante G; Frere JM; Galleni M Biochemical Characterization of the Pseudomonas aeruginosa 101/1477 Metallo-Beta-Lactamase IMP-1 Produced by Escherichia Coli. Antimicrob. Agents Chemother 1999, 43, 902–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Concha NO; Janson CA; Rowling P; Pearson S; Cheever CA; Clarke BP; Lewis C; Galleni M; Frere JM; Payne DJ, et al. Crystal Structure of the IMP-1 Metallo Beta-Lactamase from Pseudomonas aeruginosa and Its Complex with a Mercaptocarboxylate Inhibitor: Binding Determinants of a Potent, Broad-Spectrum Inhibitor. Biochemistry 2000, 39, 4288–4298. [DOI] [PubMed] [Google Scholar]
- 383.Docquier JD; Riccio ML; Mugnaioli C; Luzzaro F; Endimiani A; Toniolo A; Amicosante G; Rossolini GM IMP-12, a New Plasmid-Encoded Metallo-Beta-Lactamase from a Pseudomonas putida Clinical Isolate. Antimicrob. Agents Chemother 2003, 47, 1522–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 384.Santella G; Docquier JD; Gutkind G; Rossolini GM; Radice M Purification and Biochemical Characterization of IMP-13 Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2011, 55, 399–401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 385.Mendes RE; Toleman MA; Ribeiro J; Sader HS; Jones RN; Walsh TR Integron Carrying a Novel Metallo-Beta-Lactamase Gene, blaIMP-16, and a Fused Form of Aminoglycoside-Resistant Gene Aac(6’)-30/Aac(6’)-Ib’: Report from the Sentry Antimicrobial Surveillance Program. Antimicrob. Agents Chemother 2004, 48, 4693–4702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 386.Borgianni L; Prandi S; Salden L; Santella G; Hanson ND; Rossolini GM; Docquier JD Genetic Context and Biochemical Characterization of the IMP-18 Metallo-Beta-Lactamase Identified in a Pseudomonas aeruginosa Isolate from the United States. Antimicrob. Agents Chemother 2011, 55, 140–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 387.Jeannot K; Poirel L; Robert-Nicoud M; Cholley P; Nordmann P; Plesiat P IMP-29, a Novel IMP-Type Metallo-Beta-Lactamase in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2012, 56, 2187–2190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 388.Pfennigwerth N; Geis G; Gatermann SG; Kaase M Description of IMP-31, a Novel Metallo-Beta-Lactamase Found in an ST235 Pseudomonas aeruginosa Strain in Western Germany. J. Antimicrob. Chemother 2015, 70, 1973–1980. [DOI] [PubMed] [Google Scholar]
- 389.Kubota H; Suzuki Y; Okuno R; Uchitani Y; Ariyoshi T; Takemura N; Mihara F; Mezaki K; Ohmagari N; Matsui M, et al. IMP-68, a Novel Imp-Type Metallo-Beta-Lactamase in Imipenem-Susceptible Klebsiella pneumoniae. mSphere 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 390.Yamaguchi Y; Kuroki T; Yasuzawa H; Higashi T; Jin W; Kawanami A; Yamagata Y; Arakawa Y; Goto M; Kurosaki H Probing the Role of Asp-120(81) of Metallo-Beta-Lactamase (IMP-1) by Site-Directed Mutagenesis, Kinetic Studies, and X-Ray Crystallography. J. Biol. Chem 2005, 280, 20824–20832. [DOI] [PubMed] [Google Scholar]
- 391.Widmann M; Pleiss J Protein Variants Form a System of Networks: Microdiversity of IMP Metallo-Beta-Lactamases. PLoS One 2014, 9, e101813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 392.Yano H; Kuga A; Okamoto R; Kitasato H; Kobayashi T; Inoue M Plasmid-Encoded Metallo-Beta-Lactamase (IMP-6) Conferring Resistance to Carbapenems, Especially Meropenem. Antimicrob. Agents Chemother 2001, 45, 1343–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 393.Oelschlaeger P; Aitha M; Yang H; Kang JS; Zhang AL; Liu EM; Buynak JD; Crowder MW Meropenem and Chromacef Intermediates Observed in IMP-25 Metallo-Beta-Lactamase-Catalyzed Hydrolysis. Antimicrob. Agents Chemother 2015, 59, 4326–4330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 394.Tada T; Nhung PH; Miyoshi-Akiyama T; Shimada K; Phuong DM; Anh NQ; Ohmagari N; Kirikae T IMP-51, a Novel Imp-Type Metallo-Beta-Lactamase with Increased Doripenem- and Meropenem-Hydrolyzing Activities, in a Carbapenem-Resistant Pseudomonas aeruginosa Clinical Isolate. Antimicrob. Agents Chemother 2015, 59, 7090–7093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 395.Cheng Z; Bethel CR; Thomas PW; Shurina BA; Alao JP; Thomas CA; Yang K; Zhang H; Sturgill AM; Kravats AN, et al. Carbapenem Use Is Driving the Evolution of Imipenemase (IMP)-1 Variants. Antimicrob. Agents Chemother 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 396.Oelschlaeger P; Schmid RD; Pleiss J Modeling Domino Effects in Enzymes: Molecular Basis of the Substrate Specificity of the Bacterial Metallo-Beta-Lactamases IMP-1 and IMP-6. Biochemistry 2003, 42, 8945–8956. [DOI] [PubMed] [Google Scholar]
- 397.Oelschlaeger P; Mayo SL Hydroxyl Groups in the BetaBeta Sandwich of Metallo-Beta-Lactamases Favor Enzyme Activity: A Computational Protein Design Study. J. Mol. Biol 2005, 350, 395–401. [DOI] [PubMed] [Google Scholar]
- 398.Pegg KM; Liu EM; George AC; LaCuran AE; Bethel CR; Bonomo RA; Oelschlaeger P Understanding the Determinants of Substrate Specificity in IMP Family Metallo-Beta-Lactamases: The Importance of Residue 262. Protein Sci 2014, 23, 1451–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 399.Oelschlaeger P; Pleiss J Hydroxyl Groups in the Betabeta Sandwich of Metallo-Beta-Lactamases Favor Enzyme Activity: Tyr218 and Ser262 Pull Down the Lid. J. Mol. Biol 2007, 366, 316–329. [DOI] [PubMed] [Google Scholar]
- 400.Softley CA; Zak KM; Bostock MJ; Fino R; Zhou RX; Kolonko M; Mejdi-Nitiu R; Meyer H; Sattler M; Popowicz GM Structure and Molecular Recognition Mechanism of IMP-13 Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 401.Zhang CJ; Faheem M; Dang P; Morris MN; Kumar P; Oelschlaeger P Mutation S115T in IMP-Type Metallo-Beta-Lactamases Compensates for Decreased Expression Levels Caused by Mutation S119G. Biomolecules 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 402.Oelschlaeger P; Schmid RD; Pleiss J Insight into the Mechanism of the IMP-1 Metallo-Beta-Lactamase by Molecular Dynamics Simulations. Protein Eng 2003, 16, 341–350. [DOI] [PubMed] [Google Scholar]
- 403.Haruta S; Yamamoto ET; Eriguchi Y; Sawai T Characterization of the Active-Site Residues Asparagine 167 and Lysine 161 of the IMP-1 Metallo Beta-Lactamase. FEMS Microbiol. Lett 2001, 197, 85–89. [DOI] [PubMed] [Google Scholar]
- 404.Brown NG; Horton LB; Huang W; Vongpunsawad S; Palzkill T Analysis of the Functional Contributions of Asn233 in Metallo-Beta-Lactamase IMP-1. Antimicrob. Agents Chemother 2011, 55, 5696–5702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 405.Yamaguchi Y; Matsueda S; Matsunaga K; Takashio N; Toma-Fukai S; Yamagata Y; Shibata N; Wachino J; Shibayama K; Arakawa Y, et al. Crystal Structure of IMP-2 Metallo-Beta-Lactamase from Acinetobacter spp.: Comparison of Active-Site Loop Structures between IMP-1 and IMP-2. Biol. Pharm. Bull 2015, 38, 96–101. [DOI] [PubMed] [Google Scholar]
- 406.Furuyama T; Nonomura H; Ishii Y; Hanson ND; Shimizu-Ibuka A Structural and Mutagenic Analysis of Metallo-Beta-Lactamase IMP-18. Antimicrob. Agents Chemother 2016, 60, 5521–5526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 407.Iwata S; Tada T; Hishinuma T; Tohya M; Oshiro S; Kuwahara-Arai K; Ogawa M; Shimojima M; Kirikae T Emergence of Carbapenem-Resistant Providencia rettgeri and Providencia stuartii Producing IMP-Type Metallo-Beta-Lactamase in Japan. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 408.Iyobe S; Kusadokoro H; Takahashi A; Yomoda S; Okubo T; Nakamura A; O’Hara K Detection of a Variant Metallo-Beta-Lactamase, IMP-10, from Two Unrelated Strains of Pseudomonas aeruginosa and an Alcaligenes xylosoxidans Strain. Antimicrob. Agents Chemother 2002, 46, 2014–2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 409.Tada T; Miyoshi-Akiyama T; Shimada K; Shimojima M; Kirikae T IMP-43 and IMP-44 Metallo-Beta-Lactamases with Increased Carbapenemase Activities in Multidrug-Resistant Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2013, 57, 4427–4432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 410.Tada T; Nhung PH; Miyoshi-Akiyama T; Shimada K; Tsuchiya M; Phuong DM; Anh NQ; Ohmagari N; Kirikae T Multidrug-Resistant Sequence Type 235 Pseudomonas aeruginosa Clinical Isolates Producing IMP-26 with Increased Carbapenem-Hydrolyzing Activities in Vietnam. Antimicrob. Agents Chemother 2016, 60, 6853–6858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 411.Riccio ML; Franceschini N; Boschi L; Caravelli B; Cornaglia G; Fontana R; Amicosante G; Rossolini GM Characterization of the Metallo-Beta-Lactamase Determinant of Acinetobacter baumannii Ac-54/97 Reveals the Existence of bla(IMP) Allelic Variants Carried by Gene Cassettes of Different Phylogeny. Antimicrob. Agents Chemother 2000, 44, 1229–1235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 412.Hinchliffe P; Gonzalez MM; Mojica MF; Gonzalez JM; Castillo V; Saiz C; Kosmopoulou M; Tooke CL; Llarrull LI; Mahler G, et al. Cross-Class Metallo-Beta-Lactamase Inhibition by Bisthiazolidines Reveals Multiple Binding Modes. Proc Natl Acad Sci U S A 2016, 113, E3745–3754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 413.Pegg KM; Liu EM; Lacuran AE; Oelschlaeger P Biochemical Characterization of IMP-30, a Metallo-Beta-Lactamase with Enhanced Activity toward Ceftazidime. Antimicrob. Agents Chemother 2013, 57, 5122–5126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 414.Samuelsen O; Castanheira M; Walsh TR; Spencer J Kinetic Characterization of VIM-7, a Divergent Member of the Vim Metallo-Beta-Lactamase Family. Antimicrob. Agents Chemother 2008, 52, 2905–2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 415.Murphy TA; Simm AM; Toleman MA; Jones RN; Walsh TR Biochemical Characterization of the Acquired Metallo-Beta-Lactamase SPM-1 from Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2003, 47, 582–587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 416.Yong D; Toleman MA; Giske CG; Cho HS; Sundman K; Lee K; Walsh TR Characterization of a New Metallo-Beta-Lactamase Gene, bla(NDM-1), and a Novel Erythromycin Esterase Gene Carried on a Unique Genetic Structure in Klebsiella pneumoniae Sequence Type 14 from India. Antimicrob. Agents Chemother 2009, 53, 5046–5054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 417.Dixon N; Fowler RC; Yoshizumi A; Horiyama T; Ishii Y; Harrison L; Geyer CN; Moland ES; Thomson K; Hanson ND IMP-27, a Unique Metallo-Beta-Lactamase Identified in Geographically Distinct Isolates of Proteus mirabilis. Antimicrob. Agents Chemother 2016, 60, 6418–6421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 418.Perez-Llarena FJ; Fernandez A; Zamorano L; Kerff F; Beceiro A; Aracil B; Cercenado E; Miro E; Oliver A; Oteo J, et al. Characterization of a Novel IMP-28 Metallo-Beta-Lactamase from a Spanish Klebsiella oxytoca Clinical Isolate. Antimicrob. Agents Chemother 2012, 56, 4540–4543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 419.Deshpande LM; Davies TA; Blandino G; Nicoletti G; Jones RN; Castanheira M IMP-33, a New IMP Variant Detected in Pseudomonas aeruginosa from Sicily. Antimicrob. Agents Chemother 2013, 57, 6401–6403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 420.Shakibaie MR; Azizi O; Shahcheraghi F Insight into Stereochemistry of a New IMP Allelic Variant (IMP-55) Metallo-Beta-Lactamase Identified in a Clinical Strain of Acinetobacter baumannii. Infect Genet Evol 2017, 51, 118–126. [DOI] [PubMed] [Google Scholar]
- 421.Gonzalez LJ; Bahr G; Nakashige TG; Nolan EM; Bonomo RA; Vila AJ Membrane Anchoring Stabilizes and Favors Secretion of New Delhi Metallo-Beta-Lactamase. Nat. Chem. Biol 2016, 12, 516–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 422.Lauretti L; Riccio ML; Mazzariol A; Cornaglia G; Amicosante G; Fontana R; Rossolini GM Cloning and Characterization of blaVIM, a New Integron-Borne Metallo-Beta-Lactamase Gene from a Pseudomonas aeruginosa Clinical Isolate. Antimicrob. Agents Chemother 1999, 43, 1584–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 423.Poirel L; Naas T; Nicolas D; Collet L; Bellais S; Cavallo JD; Nordmann P Characterization of VIM-2, a Carbapenem-Hydrolyzing Metallo-Beta-Lactamase and Its Plasmid- and Integron-Borne Gene from a Pseudomonas aeruginosa Clinical Isolate in France. Antimicrob. Agents Chemother 2000, 44, 891–897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 424.Pournaras S; Tsakris A; Maniati M; Tzouvelekis LS; Maniatis AN Novel Variant (bla(VIM-4)) of the Metallo-Beta-Lactamase Gene bla(VIM-1) in a Clinical Strain of Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2002, 46, 4026–4028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 425.Martinez-Garcia L; Gonzalez-Alba JM; Baquero F; Canton R; Galan JC Ceftazidime Is the Key Diversification and Selection Driver of VIM-Type Carbapenemases. mBio 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 426.Toleman MA; Rolston K; Jones RN; Walsh TR blaVIM-7, an Evolutionarily Distinct Metallo-Beta-Lactamase Gene in a Pseudomonas aeruginosa Isolate from the United States. Antimicrob. Agents Chemother 2004, 48, 329–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 427.Kazmierczak KM; Rabine S; Hackel M; McLaughlin RE; Biedenbach DJ; Bouchillon SK; Sahm DF; Bradford PA Multiyear, Multinational Survey of the Incidence and Global Distribution of Metallo-Beta-Lactamase-Producing Enterobacteriaceae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2016, 60, 1067–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 428.Nordmann P; Poirel L The Difficult-to-Control Spread of Carbapenemase Producers among Enterobacteriaceae Worldwide. Clin. Microbiol. Infect 2014, 20, 821–830. [DOI] [PubMed] [Google Scholar]
- 429.Tato M; Coque TM; Baquero F; Canton R Dispersal of Carbapenemase blaVIM-1 Gene Associated with Different Tn402 Variants, Mercury Transposons, and Conjugative Plasmids in Enterobacteriaceae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2010, 54, 320–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 430.Lopez C; Ayala JA; Bonomo RA; Gonzalez LJ; Vila AJ Protein Determinants of Dissemination and Host Specificity of Metallo-Beta-Lactamases. Nat Commun 2019, 10, 3617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 431.Docquier JD; Lamotte-Brasseur J; Galleni M; Amicosante G; Frere JM; Rossolini GM On Functional and Structural Heterogeneity of VIM-Type Metallo-Beta-Lactamases. J. Antimicrob. Chemother 2003, 51, 257–266. [DOI] [PubMed] [Google Scholar]
- 432.Franceschini N; Caravelli B; Docquier JD; Galleni M; Frere JM; Amicosante G; Rossolini GM Purification and Biochemical Characterization of the VIM-1 Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2000, 44, 3003–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 433.Lassaux P; Traore DA; Loisel E; Favier A; Docquier JD; Sohier JS; Laurent C; Bebrone C; Frere JM; Ferrer JL, et al. Biochemical and Structural Characterization of the Subclass B1 Metallo-Beta-Lactamase VIM-4. Antimicrob. Agents Chemother 2011, 55, 1248–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 434.Juan C; Beceiro A; Gutierrez O; Alberti S; Garau M; Perez JL; Bou G; Oliver A Characterization of the New Metallo-Beta-Lactamase VIM-13 and Its Integron-Borne Gene from a Pseudomonas aeruginosa Clinical Isolate in Spain. Antimicrob. Agents Chemother 2008, 52, 3589–3596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 435.Merino M; Perez-Llarena FJ; Kerff F; Poza M; Mallo S; Rumbo-Feal S; Beceiro A; Juan C; Oliver A; Bou G Role of Changes in the L3 Loop of the Active Site in the Evolution of Enzymatic Activity of VIM-Type Metallo-Beta-Lactamases. J. Antimicrob. Chemother 2010, 65, 1950–1954. [DOI] [PubMed] [Google Scholar]
- 436.Rodriguez-Martinez JM; Nordmann P; Fortineau N; Poirel L VIM-19, a Metallo-Beta-Lactamase with Increased Carbapenemase Activity from Escherichia coli and Klebsiella pneumoniae. Antimicrob. Agents Chemother 2010, 54, 471–476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 437.Mojica MF; Mahler SG; Bethel CR; Taracila MA; Kosmopoulou M; Papp-Wallace KM; Llarrull LI; Wilson BM; Marshall SH; Wallace CJ, et al. Exploring the Role of Residue 228 in Substrate and Inhibitor Recognition by Vim Metallo-Beta-Lactamases. Biochemistry 2015, 54, 3183–3196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 438.Castanheira M; Deshpande LM; Mendes RE; Rodriguez-Noriega E; Jones RN; Morfin-Otero R Comment On: Role of Changes in the L3 Loop of the Active Site in the Evolution of Enzymatic Activity of VIM-Type Metallo-Beta-Lactamases. J. Antimicrob. Chemother 2011, 66, 684–685; author reply 686. [DOI] [PubMed] [Google Scholar]
- 439.Cheng Z; Shurina BA; Bethel CR; Thomas PW; Marshall SH; Thomas CA; Yang K; Kimble RL; Montgomery JS; Orischak MG, et al. A Single Salt Bridge in VIM-20 Increases Protein Stability and Antibiotic Resistance under Low-Zinc Conditions. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 440.Hishinuma T; Tada T; Uchida H; Shimojima M; Kirikae T A Novel VIM-Type Metallo-Beta-Lactamase Variant, VIM-60, with Increased Hydrolyzing Activity against Fourth-Generation Cephalosporins in Pseudomonas aeruginosa Clinical Isolates in Japan. Antimicrob. Agents Chemother 2019, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 441.Leiros HK; Skagseth S; Edvardsen KS; Lorentzen MS; Bjerga GE; Leiros I; Samuelsen O His224 Alters the R2 Drug Binding Site and Phe218 Influences the Catalytic Efficiency of the Metallo-Beta-Lactamase VIM-7. Antimicrob. Agents Chemother 2014, 58, 4826–4836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 442.Borra PS; Leiros HK; Ahmad R; Spencer J; Leiros I; Walsh TR; Sundsfjord A; Samuelsen O Structural and Computational Investigations of VIM-7: Insights into the Substrate Specificity of VIM Metallo-Beta-Lactamases. J. Mol. Biol 2011, 411, 174–189. [DOI] [PubMed] [Google Scholar]
- 443.Schneider I; Keuleyan E; Rasshofer R; Markovska R; Queenan AM; Bauernfeind A VIM-15 and VIM-16, Two New VIM-2-Like Metallo-Beta-Lactamases in Pseudomonas aeruginosa Isolates from Bulgaria and Germany. Antimicrob. Agents Chemother 2008, 52, 2977–2979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 444.Leiros HK; Edvardsen KS; Bjerga GE; Samuelsen O Structural and Biochemical Characterization of VIM-26 Shows That Leu224 Has Implications for the Substrate Specificity of VIM Metallo-Beta-Lactamases. FEBS J. 2015, 282, 1031–1042. [DOI] [PubMed] [Google Scholar]
- 445.Papagiannitsis CC; Pollini S; De Luca F; Rossolini GM; Docquier JD; Hrabak J Biochemical Characterization of VIM-39, a VIM-1-Like Metallo-Beta-Lactamase Variant from a Multidrug-Resistant Klebsiella pneumoniae Isolate from Greece. Antimicrob. Agents Chemother 2015, 59, 7811–7814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 446.Bogaerts P; Bebrone C; Huang TD; Bouchahrouf W; Degheldre Y; Deplano A; Hoffmann K; Glupczynski Y Detection and Characterization of VIM-31, a New Variant of VIM-2 with Tyr224His and His252Arg Mutations, in a Clinical Isolate of Enterobacter cloacae. Antimicrob. Agents Chemother 2012, 56, 3283–3287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 447.Kupper MB; Herzog K; Bennink S; Schlomer P; Bogaerts P; Glupczynski Y; Fischer R; Bebrone C; Hoffmann KM The Three-Dimensional Structure of VIM-31--a Metallo-Beta-Lactamase from Enterobacter cloacae in Its Native and Oxidized Form. FEBS J. 2015, 282, 2352–2360. [DOI] [PubMed] [Google Scholar]
- 448.Koh TH; Yamaguchi K; Ishii Y Characterisation of the Metallo-Beta-Lactamase VIM-6 and Its Genetic Support. Int. J. Antimicrob. Agents 2008, 32, 446–449. [DOI] [PubMed] [Google Scholar]
- 449.Marchiaro P; Tomatis PE; Mussi MA; Pasteran F; Viale AM; Limansky AS; Vila AJ Biochemical Characterization of Metallo-Beta-Lactamase VIM-11 from a Pseudomonas aeruginosa Clinical Strain. Antimicrob. Agents Chemother 2008, 52, 2250–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 450.Papagiannitsis CC; Kotsakis SD; Petinaki E; Vatopoulos AC; Tzelepi E; Miriagou V; Tzouvelekis LS Characterization of Metallo-Beta-Lactamase VIM-27, an A57S Mutant of VIM-1 Associated with Klebsiella pneumoniae St147. Antimicrob. Agents Chemother 2011, 55, 3570–3572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 451.Makena A; Duzgun AO; Brem J; McDonough MA; Rydzik AM; Abboud MI; Saral A; Cicek AC; Sandalli C; Schofield CJ Comparison of Verona Integron-Borne Metallo-Beta-Lactamase (VIM) Variants Reveals Differences in Stability and Inhibition Profiles. Antimicrob. Agents Chemother 2015, 60, 1377–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 452.Bahr G; Vitor-Horen L; Bethel CR; Bonomo RA; Gonzalez LJ; Vila AJ Clinical Evolution of New Delhi Metallo-Beta-Lactamase (NDM) Optimizes Resistance under Zn(II) Deprivation. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 453.Kontou M; Pournaras S; Kristo I; Ikonomidis A; Maniatis AN; Stathopoulos C Molecular Cloning and Biochemical Characterization of VIM-12, a Novel Hybrid VIM-1/VIM-2 Metallo-Beta-Lactamase from a Klebsiella pneumoniae Clinical Isolate, Reveal Atypical Substrate Specificity. Biochemistry 2007, 46, 13170–13178. [DOI] [PubMed] [Google Scholar]
- 454.Liu Z; Zhang R; Li W; Yang L; Liu D; Wang S; Shen J; Wang Y Amino Acid Changes at the VIM-48 C-Terminus Result in Increased Carbapenem Resistance, Enzyme Activity and Protein Stability. J. Antimicrob. Chemother 2019, 74, 885–893. [DOI] [PubMed] [Google Scholar]
- 455.Toleman MA; Simm AM; Murphy TA; Gales AC; Biedenbach DJ; Jones RN; Walsh TR Molecular Characterization of SPM-1, a Novel Metallo-Beta-Lactamase Isolated in Latin America: Report from the Sentry Antimicrobial Surveillance Programme. J. Antimicrob. Chemother 2002, 50, 673–679. [DOI] [PubMed] [Google Scholar]
- 456.Silva FM; Carmo MS; Silbert S; Gales AC SPM-1-Producing Pseudomonas aeruginosa: Analysis of the Ancestor Relationship Using Multilocus Sequence Typing, Pulsed-Field Gel Electrophoresis, and Automated Ribotyping. Microb. Drug Resist 2011, 17, 215–220. [DOI] [PubMed] [Google Scholar]
- 457.Hopkins KL; Meunier D; Findlay J; Mustafa N; Parsons H; Pike R; Wright L; Woodford N SPM-1 Metallo-Beta-Lactamase-Producing Pseudomonas aeruginosa ST277 in the UK. J. Med. Microbiol 2016, 65, 696–697. [DOI] [PubMed] [Google Scholar]
- 458.Salabi AE; Toleman MA; Weeks J; Bruderer T; Frei R; Walsh TR First Report of the Metallo-Beta-Lactamase SPM-1 in Europe. Antimicrob. Agents Chemother 2010, 54, 582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 459.Rossi F The Challenges of Antimicrobial Resistance in Brazil. Clin. Infect. Dis 2011, 52, 1138–1143. [DOI] [PubMed] [Google Scholar]
- 460.Shahcheraghi F; Abbasalipour M; Feizabadi M; Ebrahimipour G; Akbari N Isolation and Genetic Characterization of Metallo-Beta-Lactamase and Carbapenamase Producing Strains of Acinetobacter baumannii from Patients at Tehran Hospitals. Iran J Microbiol 2011, 3, 68–74. [PMC free article] [PubMed] [Google Scholar]
- 461.Fonseca EL; Marin MA; Encinas F; Vicente AC Full Characterization of the Integrative and Conjugative Element Carrying the Metallo-Beta-Lactamase blaSPM-1 and Bicyclomycin Bcr1 Resistance Genes Found in the Pandemic Pseudomonas aeruginosa Clone SP/ST277. J. Antimicrob. Chemother 2015, 70, 2547–2550. [DOI] [PubMed] [Google Scholar]
- 462.Nascimento AP; Ortiz MF; Martins WM; Morais GL; Fehlberg LC; Almeida LG; Ciapina LP; Gales AC; Vasconcelos AT Intraclonal Genome Stability of the Metallo-Beta-Lactamase SPM-1-Producing Pseudomonas aeruginosa ST277, an Endemic Clone Disseminated in Brazilian Hospitals. Front Microbiol 2016, 7, 1946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 463.Gonzalez LJ; Stival C; Puzzolo JL; Moreno DM; Vila AJ Shaping Substrate Selectivity in a Broad-Spectrum Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 464.Gonzalez LJ; Moreno DM; Bonomo RA; Vila AJ Host-Specific Enzyme-Substrate Interactions in SPM-1 Metallo-Beta-Lactamase Are Modulated by Second Sphere Residues. PLoS Pathog 2014, 10, e1003817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 465.Kieffer N; Poirel L; Fournier C; Haltli B; Kerr R; Nordmann P Characterization of PAN-1, a Carbapenem-Hydrolyzing Class B Beta-Lactamase from the Environmental Gram-Negative Pseudobacteriovorax antillogorgiicola. Front Microbiol 2019, 10, 1673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 466.Castanheira M; Deshpande LM; Mathai D; Bell JM; Jones RN; Mendes RE Early Dissemination of NDM-1- and OXA-181-Producing Enterobacteriaceae in Indian Hospitals: Report from the Sentry Antimicrobial Surveillance Program, 2006–2007. Antimicrob. Agents Chemother 2011, 55, 1274–1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 467.Berglund F; Marathe NP; Osterlund T; Bengtsson-Palme J; Kotsakis S; Flach CF; Larsson DGJ; Kristiansson E Identification of 76 Novel B1 Metallo-Beta-Lactamases through Large-Scale Screening of Genomic and Metagenomic Data. Microbiome 2017, 5, 134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 468.Jiang XW; Cheng H; Huo YY; Xu L; Wu YH; Liu WH; Tao FF; Cui XJ; Zheng BW Biochemical and Genetic Characterization of a Novel Metallo-Beta-Lactamase from Marine Bacterium Erythrobacter litoralis HTCC 2594. Sci Rep 2018, 8, 803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 469.Dortet L; Poirel L; Nordmann P Worldwide Dissemination of the NDM-Type Carbapenemases in Gram-Negative Bacteria. Biomed Res Int 2014, 2014, 249856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 470.Nordmann P; Poirel L; Walsh TR; Livermore DM The Emerging NDM Carbapenemases. Trends Microbiol 2011, 19, 588–595. [DOI] [PubMed] [Google Scholar]
- 471.Wu W; Feng Y; Tang G; Qiao F; McNally A; Zong Z NDM Metallo-Beta-Lactamases and Their Bacterial Producers in Health Care Settings. Clin. Microbiol. Rev 2019, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 472.Poirel L; Dortet L; Bernabeu S; Nordmann P Genetic Features of blaNDM-1-Positive Enterobacteriaceae. Antimicrob. Agents Chemother 2011, 55, 5403–5407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 473.Dortet L; Nordmann P; Poirel L Association of the Emerging Carbapenemase NDM-1 with a Bleomycin Resistance Protein in Enterobacteriaceae and Acinetobacter baumannii. Antimicrob. Agents Chemother 2012, 56, 1693–1697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 474.King D; Strynadka N Crystal Structure of New Delhi Metallo-Beta-Lactamase Reveals Molecular Basis for Antibiotic Resistance. Protein Sci 2011, 20, 1484–1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 475.Zalucki YM; Jen FE; Pegg CL; Nouwens AS; Schulz BL; Jennings MP Evolution for Improved Secretion and Fitness May Be the Selective Pressures Leading to the Emergence of Two NDM Alleles. Biochem. Biophys. Res. Commun 2020, 524, 555–560. [DOI] [PubMed] [Google Scholar]
- 476.Prunotto A; Bahr G; Gonzalez LJ; Vila AJ; Dal Peraro M Molecular Bases of the Membrane Association Mechanism Potentiating Antibiotic Resistance by New Delhi Metallo-Beta-Lactamase 1. ACS Infect Dis 2020, 6, 2719–2731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 477.Toleman MA; Spencer J; Jones L; Walsh TR blaNDM-1 Is a Chimera Likely Constructed in Acinetobacter baumannii. Antimicrob. Agents Chemother 2012, 56, 2773–2776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 478.Makena A; Brem J; Pfeffer I; Geffen RE; Wilkins SE; Tarhonskaya H; Flashman E; Phee LM; Wareham DW; Schofield CJ Biochemical Characterization of New Delhi Metallo-BetaLactamase Variants Reveals Differences in Protein Stability. J. Antimicrob. Chemother 2015, 70, 463–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 479.Cheng Z; Thomas PW; Ju L; Bergstrom A; Mason K; Clayton D; Miller C; Bethel CR; VanPelt J; Tierney DL, et al. Evolution of New Delhi Metallo-Beta-Lactamase (NDM) in the Clinic: Effects of Ndm Mutations on Stability, Zinc Affinity, and Mono-Zinc Activity. J. Biol. Chem 2018, 293, 12606–12618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 480.Hornsey M; Phee L; Wareham DW A Novel Variant, NDM-5, of the New Delhi Metallo-Beta-Lactamase in a Multidrug-Resistant Escherichia coli ST648 Isolate Recovered from a Patient in the United Kingdom. Antimicrob. Agents Chemother 2011, 55, 5952–5954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 481.Gottig S; Hamprecht AG; Christ S; Kempf VA; Wichelhaus TA Detection of NDM-7 in Germany, a New Variant of the New Delhi Metallo-Beta-Lactamase with Increased Carbapenemase Activity. J. Antimicrob. Chemother 2013, 68, 1737–1740. [DOI] [PubMed] [Google Scholar]
- 482.Zou D; Huang Y; Zhao X; Liu W; Dong D; Li H; Wang X; Huang S; Wei X; Yan X, et al. A Novel New Delhi Metallo-Beta-Lactamase Variant, NDM-14, Isolated in a Chinese Hospital Possesses Increased Enzymatic Activity against Carbapenems. Antimicrob. Agents Chemother 2015, 59, 2450–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 483.Liu Z; Wang Y; Walsh TR; Liu D; Shen Z; Zhang R; Yin W; Yao H; Li J; Shen J Plasmid-Mediated Novel blaNDM-17 Gene Encoding a Carbapenemase with Enhanced Activity in a Sequence Type 48 Escherichia coli Strain. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 484.Liu L; Feng Y; McNally A; Zong Z blaNDM-21, a New Variant of blaNDM in an Escherichia coli Clinical Isolate Carrying blaCTX-M-55 and rmtB. J. Antimicrob. Chemother 2018, 73, 2336–2339. [DOI] [PubMed] [Google Scholar]
- 485.Liu Z; Li J; Wang X; Liu D; Ke Y; Wang Y; Shen J Novel Variant of New Delhi Metallo-Beta-Lactamase, NDM-20, in Escherichia coli. Front Microbiol 2018, 9, 248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 486.Liu Z; Piccirilli A; Liu D; Li W; Wang Y; Shen J Deciphering the Role of V88l Substitution in NDM-24 Metallo-B-Lactamase. Catalysts 2019, 9, 744. [Google Scholar]
- 487.Cuzon G; Bonnin RA; Nordmann P First Identification of Novel NDM Carbapenemase, NDM-7, in Escherichia coli in France. PLoS One 2013, 8, e61322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 488.Rahman M; Shukla SK; Prasad KN; Ovejero CM; Pati BK; Tripathi A; Singh A; Srivastava AK; Gonzalez-Zorn B Prevalence and Molecular Characterisation of New Delhi Metallo-Beta-Lactamases NDM-1, NDM-5, NDM-6 and NDM-7 in Multidrug-Resistant Enterobacteriaceae from India. Int. J. Antimicrob. Agents 2014, 44, 30–37. [DOI] [PubMed] [Google Scholar]
- 489.Mancini S; Keller PM; Greiner M; Bruderer V; Imkamp F Detection of NDM-19, a Novel Variant of the New Delhi Metallo-Beta-Lactamase with Increased Carbapenemase Activity under Zinc-Limited Conditions, in Switzerland. Diagn. Microbiol. Infect. Dis 2019, 95, 114851. [DOI] [PubMed] [Google Scholar]
- 490.Nordmann P; Boulanger AE; Poirel L NDM-4 Metallo-Beta-Lactamase with Increased Carbapenemase Activity from Escherichia coli. Antimicrob. Agents Chemother 2012, 56, 2184–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 491.Tada T; Miyoshi-Akiyama T; Shimada K; Kirikae T Biochemical Analysis of Metallo-Beta-Lactamase NDM-3 from a Multidrug-Resistant Escherichia coli Strain Isolated in Japan. Antimicrob. Agents Chemother 2014, 58, 3538–3540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 492.Tada T; Miyoshi-Akiyama T; Dahal RK; Sah MK; Ohara H; Kirikae T; Pokhrel BM NDM-8 Metallo-Beta-Lactamase in a Multidrug-Resistant Escherichia coli Strain Isolated in Nepal. Antimicrob. Agents Chemother 2013, 57, 2394–2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 493.Tada T; Shrestha B; Miyoshi-Akiyama T; Shimada K; Ohara H; Kirikae T; Pokhrel BM NDM-12, a Novel New Delhi Metallo-Beta-Lactamase Variant from a Carbapenem-Resistant Escherichia coli Clinical Isolate in Nepal. Antimicrob. Agents Chemother 2014, 58, 6302–6305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 494.Shrestha B; Tada T; Miyoshi-Akiyama T; Shimada K; Ohara H; Kirikae T; Pokhrel BM Identification of a Novel NDM Variant, NDM-13, from a Multidrug-Resistant Escherichia coli Clinical Isolate in Nepal. Antimicrob. Agents Chemother 2015, 59, 5847–5850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 495.Khajuria A; Praharaj AK; Kumar M; Grover N Presence of a Novel Variant NDM-10, of the New Delhi Metallo-Beta-Lactamase in a Klebsiella pneumoniae Isolate. Indian J Med Microbiol 2016, 34, 121–123. [DOI] [PubMed] [Google Scholar]
- 496.Wang X; Li H; Zhao C; Chen H; Liu J; Wang Z; Wang Q; Zhang Y; He W; Zhang F, et al. Novel NDM-9 Metallo-Beta-Lactamase Identified from a ST107 Klebsiella pneumoniae Strain Isolated in China. Int. J. Antimicrob. Agents 2014, 44, 90–91. [DOI] [PubMed] [Google Scholar]
- 497.Stewart AC; Bethel CR; VanPelt J; Bergstrom A; Cheng Z; Miller CG; Williams C; Poth R; Morris M; Lahey O, et al. Clinical Variants of New Delhi Metallo-Beta-Lactamase Are Evolving to Overcome Zinc Scarcity. ACS Infect Dis 2017, 3, 927–940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 498.Chen J; Chen H; Zhu T; Zhou D; Zhang F; Lao X; Zheng H Asp120Asn Mutation Impairs the Catalytic Activity of NDM-1 Metallo-Beta-Lactamase: Experimental and Computational Study. Phys. Chem. Chem. Phys 2014, 16, 6709–6716. [DOI] [PubMed] [Google Scholar]
- 499.Liang Z; Li L; Wang Y; Chen L; Kong X; Hong Y; Lan L; Zheng M; Guang-Yang C; Liu H, et al. Molecular Basis of NDM-1, a New Antibiotic Resistance Determinant. PLoS One 2011, 6, e23606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 500.Chiou J; Leung TY; Chen S Molecular Mechanisms of Substrate Recognition and Specificity of New Delhi Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2014, 58, 5372–5378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 501.Raczynska JE; Imiolczyk B; Komorowska M; Sliwiak J; Czyrko-Horczak J; Brzezinski K; Jaskolski M flexible Loops of New Delhi Metallo-Beta-Lactamase Modulate Its Activity Towards Different Substrates. Int. J. Biol. Macromol 2020, 158, 104–115. [DOI] [PubMed] [Google Scholar]
- 502.Khan AU; Rehman MT Role of Non-Active-Site Residue Trp-93 in the Function and Stability of New Delhi Metallo-Beta-Lactamase 1. Antimicrob. Agents Chemother 2016, 60, 356–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 503.Marcoccia F; Mercuri PS; Galleni M; Celenza G; Amicosante G; Perilli M A Kinetic Study of the Replacement by Site Saturation Mutagenesis of Residue 119 in NDM-1 Metallo-Beta-Lactamase. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 504.Guo Y; Wang J; Niu G; Shui W; Sun Y; Zhou H; Zhang Y; Yang C; Lou Z; Rao Z A Structural View of the Antibiotic Degradation Enzyme NDM-1 from a Superbug. Protein Cell 2011, 2, 384–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 505.Chen J; Chen H; Shi Y; Hu F; Lao X; Gao X; Zheng H; Yao W Probing the Effect of the Non-Active-Site Mutation Y229W in New Delhi Metallo-Beta-Lactamase-1 by Site-Directed Mutagenesis, Kinetic Studies, and Molecular Dynamics Simulations. PLoS One 2013, 8, e82080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 506.Marcoccia F; Leiros HS; Aschi M; Amicosante G; Perilli M Exploring the Role of L209 Residue in the Active Site of NDM-1 a Metallo-Beta-Lactamase. PLoS One 2018, 13, e0189686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 507.Piccirilli A; Brisdelli F; Aschi M; Celenza G; Amicosante G; Perilli M Kinetic Profile and Molecular Dynamic Studies Show That Y229W Substitution in an NDM-1/L209F Variant Restores the Hydrolytic Activity of the Enzyme toward Penicillins, Cephalosporins, and Carbapenems. Antimicrob. Agents Chemother 2019, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 508.Segatore B; Massidda O; Satta G; Setacci D; Amicosante G High Specificity of CphA-Encoded Metallo-Beta-Lactamase from Aeromonas hydrophila AE036 for Carbapenems and Its Contribution to Beta-Lactam Resistance. Antimicrob. Agents Chemother 1993, 37, 1324–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 509.Felici A; Amicosante G; Oratore A; Strom R; Ledent P; Joris B; Fanuel L; Frere JM An Overview of the Kinetic Parameters of Class B Beta-Lactamases. Biochem. J 1993, 291 (Pt 1), 151–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 510.Hernandez Valladares M; Kiefer M; Heinz U; Soto RP; Meyer-Klaucke W; Nolting HF; Zeppezauer M; Galleni M; Frere JM; Rossolini GM, et al. Kinetic and Spectroscopic Characterization of Native and Metal-Substituted Beta-Lactamase from Aeromonas hydrophila AE036. FEBS Lett. 2000, 467, 221–225. [DOI] [PubMed] [Google Scholar]
- 511.Crawford PA; Sharma N; Chandrasekar S; Sigdel T; Walsh TR; Spencer J; Crowder MW Over-Expression, Purification, and Characterization of Metallo-Beta-Lactamase ImiS from Aeromonas veronii Bv. Sobria. Protein Expr Purif 2004, 36, 272–279. [DOI] [PubMed] [Google Scholar]
- 512.Massidda O; Rossolini GM; Satta G The Aeromonas hydrophila CphA Gene: Molecular Heterogeneity among Class B Metallo-Beta-Lactamases. J. Bacteriol 1991, 173, 4611–4617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 513.Zervosen A; Valladares MH; Devreese B; Prosperi-Meys C; Adolph HW; Mercuri PS; Vanhove M; Amicosante G; van Beeumen J; Frere JM, et al. Inactivation of Aeromonas hydrophila Metallo-Beta-Lactamase by Cephamycins and Moxalactam. Eur. J. Biochem 2001, 268, 3840–3850. [DOI] [PubMed] [Google Scholar]
- 514.Bottoni C; Perilli M; Marcoccia F; Piccirilli A; Pellegrini C; Colapietro M; Sabatini A; Celenza G; Kerff F; Amicosante G, et al. Kinetic Studies on CphA Mutants Reveal the Role of the P158-P172 Loop in Activity Versus Carbapenems. Antimicrob. Agents Chemother 2016, 60, 3123–3126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 515.Periyannan GR; Costello AL; Tierney DL; Yang KW; Bennett B; Crowder MW Sequential Binding of Cobalt(II) to Metallo-Beta-Lactamase CcrA. Biochemistry 2006, 45, 1313–1320. [DOI] [PubMed] [Google Scholar]
- 516.Costello AL; Sharma NP; Yang KW; Crowder MW; Tierney DL X-Ray Absorption Spectroscopy of the Zinc-Binding Sites in the Class B2 Metallo-Beta-Lactamase ImiS from Aeromonas veronii Bv. Sobria. Biochemistry 2006, 45, 13650–13658. [DOI] [PubMed] [Google Scholar]
- 517.Saavedra MJ; Peixe L; Sousa JC; Henriques I; Alves A; Correia A Sfh-I, a Subclass B2 Metallo-Beta-Lactamase from a Serratia fonticola Environmental Isolate. Antimicrob. Agents Chemother 2003, 47, 2330–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 518.Poirel L; Palmieri M; Brilhante M; Masseron A; Perreten V; Nordmann P PFM-Like Enzymes Are a Novel Family of Subclass B2 Metallo-Beta-Lactamases from Pseudomonas synxantha Belonging to the Pseudomonas fluorescens Complex. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 519.Mercuri PS; Esposito R; Bletard S; Di Costanzo S; Perilli M; Kerff F; Galleni M Mutational Effects on Carbapenem Hydrolysis of YEM-1, a New Subclass B2 Metallo-Beta-Lactamase from Yersinia mollaretii. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 520.Walsh TR; Hall L; Assinder SJ; Nichols WW; Cartwright SJ; MacGowan AP; Bennett PM Sequence Analysis of the L1 Metallo-Beta-Lactamase from Xanthomonas maltophilia. Biochim. Biophys. Acta 1994, 1218, 199–201. [DOI] [PubMed] [Google Scholar]
- 521.Bellais S; Aubert D; Naas T; Nordmann P Molecular and Biochemical Heterogeneity of Class B Carbapenem-Hydrolyzing Beta-Lactamases in Chryseobacterium meningosepticum. Antimicrob. Agents Chemother 2000, 44, 1878–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 522.Stoczko M; Frere JM; Rossolini GM; Docquier JD Postgenomic Scan of Metallo-Beta-Lactamase Homologues in Rhizobacteria: Identification and Characterization of BJP-1, a Subclass B3 Ortholog from Bradyrhizobium japonicum. Antimicrob. Agents Chemother 2006, 50, 1973–1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 523.Boschi L; Mercuri PS; Riccio ML; Amicosante G; Galleni M; Frere JM; Rossolini GM The Legionella (Fluoribacter) gormanii Metallo-Beta-Lactamase: A New Member of the Highly Divergent Lineage of Molecular-Subclass B3 Beta-Lactamases. Antimicrob. Agents Chemother 2000, 44, 1538–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 524.Simm AM; Higgins CS; Pullan ST; Avison MB; Niumsup P; Erdozain O; Bennett PM; Walsh TR A Novel Metallo-Beta-Lactamase, MBL1b, Produced by the Environmental Bacterium Caulobacter crescentus. FEBS Lett. 2001, 509, 350–354. [DOI] [PubMed] [Google Scholar]
- 525.Docquier JD; Pantanella F; Giuliani F; Thaller MC; Amicosante G; Galleni M; Frere JM; Bush K; Rossolini GM CAU-1, a Subclass B3 Metallo-Beta-Lactamase of Low Substrate Affinity Encoded by an Ortholog Present in the Caulobacter crescentus Chromosome. Antimicrob. Agents Chemother 2002, 46, 1823–1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 526.Rossolini GM; Condemi MA; Pantanella F; Docquier JD; Amicosante G; Thaller MC Metallo-Beta-Lactamase Producers in Environmental Microbiota: New Molecular Class B Enzyme in Janthinobacterium lividum. Antimicrob. Agents Chemother 2001, 45, 837–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 527.Thaller MC; Borgianni L; Di Lallo G; Chong Y; Lee K; Dajcs J; Stroman D; Rossolini GM Metallo-Beta-Lactamase Production by Pseudomonas otitidis: A Species-Related Trait. Antimicrob. Agents Chemother 2011, 55, 118–123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 528.Stoczko M; Frere JM; Rossolini GM; Docquier JD Functional Diversity among Metallo-Beta-Lactamases: Characterization of the CAR-1 Enzyme of Erwinia carotovora. Antimicrob. Agents Chemother 2008, 52, 2473–2479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 529.Vella P; Miraula M; Phelan E; Leung EW; Ely F; Ollis DL; McGeary RP; Schenk G; Mitic N Identification and Characterization of an Unusual Metallo-Beta-Lactamase from Serratia proteamaculans. J. Biol. Inorg. Chem 2013, 18, 855–863. [DOI] [PubMed] [Google Scholar]
- 530.Gudeta DD; Bortolaia V; Amos G; Wellington EM; Brandt KK; Poirel L; Nielsen JB; Westh H; Guardabassi L The Soil Microbiota Harbors a Diversity of Carbapenem-Hydrolyzing Beta-Lactamases of Potential Clinical Relevance. Antimicrob. Agents Chemother 2016, 60, 151–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 531.Girlich D; Poirel L; Nordmann P Diversity of Naturally Occurring Ambler Class B Metallo-Beta-Lactamases in Erythrobacter spp. J. Antimicrob. Chemother 2012, 67, 2661–2664. [DOI] [PubMed] [Google Scholar]
- 532.Salimraj R; Zhang L; Hinchliffe P; Wellington EM; Brem J; Schofield CJ; Gaze WH; Spencer J Structural and Biochemical Characterization of Rm3, a Subclass B3 Metallo-Beta-Lactamase Identified from a Functional Metagenomic Study. Antimicrob. Agents Chemother 2016, 60, 5828–5840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 533.Zhang L; Calvo-Bado L; Murray AK; Amos GCA; Hawkey PM; Wellington EM; Gaze WH Novel Clinically Relevant Antibiotic Resistance Genes Associated with Sewage Sludge and Industrial Waste Streams Revealed by Functional Metagenomic Screening. Environ. Int 2019, 132, 105120. [DOI] [PubMed] [Google Scholar]
- 534.Lau CH; van Engelen K; Gordon S; Renaud J; Topp E Novel Antibiotic Resistance Determinants from Agricultural Soil Exposed to Antibiotics Widely Used in Human Medicine and Animal Farming. Appl. Environ. Microbiol 2017, 83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 535.Allen HK; Moe LA; Rodbumrer J; Gaarder A; Handelsman J Functional Metagenomics Reveals Diverse Beta-Lactamases in a Remote Alaskan Soil. ISME J 2009, 3, 243–251. [DOI] [PubMed] [Google Scholar]
- 536.Pedroso MM; Selleck C; Enculescu C; Harmer JR; Mitic N; Craig WR; Helweh W; Hugenholtz P; Tyson GW; Tierney DL, et al. Characterization of a Highly Efficient Antibiotic-Degrading Metallo-Beta-Lactamase Obtained from an Uncultured Member of a Permafrost Community. Metallomics 2017, 9, 1157–1168. [DOI] [PubMed] [Google Scholar]
- 537.Bicknell R; Emanuel EL; Gagnon J; Waley SG The Production and Molecular Properties of the Zinc Beta-Lactamase of Pseudomonas maltophilia IID 1275. Biochem. J 1985, 229, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 538.Brooke JS Stenotrophomonas maltophilia: An Emerging Global Opportunistic Pathogen. Clin. Microbiol. Rev 2012, 25, 2–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 539.Sanchez MB Antibiotic Resistance in the Opportunistic Pathogen Stenotrophomonas maltophilia. Front Microbiol 2015, 6, 658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 540.Carenbauer AL; Garrity JD; Periyannan G; Yates RB; Crowder MW Probing Substrate Binding to Metallo-Beta-Lactamase L1 from Stenotrophomonas maltophilia by Using Site-Directed Mutagenesis. BMC Biochem 2002, 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 541.Leiros HK; Borra PS; Brandsdal BO; Edvardsen KS; Spencer J; Walsh TR; Samuelsen O Crystal Structure of the Mobile Metallo-Beta-Lactamase AIM-1 from Pseudomonas aeruginosa: Insights into Antibiotic Binding and the Role of Gln157. Antimicrob. Agents Chemother 2012, 56, 4341–4353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 542.Wachino J; Yamaguchi Y; Mori S; Kurosaki H; Arakawa Y; Shibayama K Structural Insights into the Subclass B3 Metallo-Beta-Lactamase SMB-1 and the Mode of Inhibition by the Common Metallo-Beta-Lactamase Inhibitor Mercaptoacetate. Antimicrob. Agents Chemother 2013, 57, 101–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 543.Garcia-Saez I; Mercuri PS; Papamicael C; Kahn R; Frere JM; Galleni M; Rossolini GM; Dideberg O Three-Dimensional Structure of FEZ-1, a Monomeric Subclass B3 Metallo-Beta-Lactamase from Fluoribacter gormanii, in Native Form and in Complex with D-Captopril. J. Mol. Biol 2003, 325, 651–660. [DOI] [PubMed] [Google Scholar]
- 544.Rodriguez MM; Herman R; Ghiglione B; Kerff F; D’Amico Gonzalez G; Bouillenne F; Galleni M; Handelsman J; Charlier P; Gutkind G, et al. Crystal Structure and Kinetic Analysis of the Class B3 Di-Zinc Metallo-Beta-Lactamase LRA-12 from an Alaskan Soil Metagenome. PLoS One 2017, 12, e0182043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 545.Mojica MF; Rutter JD; Taracila M; Abriata LA; Fouts DE; Papp-Wallace KM; Walsh TJ; LiPuma JJ; Vila AJ; Bonomo RA Population Structure, Molecular Epidemiology, and Beta-Lactamase Diversity among Stenotrophomonas maltophilia Isolates in the United States. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 546.Avison MB; Higgins CS; von Heldreich CJ; Bennett PM; Walsh TR Plasmid Location and Molecular Heterogeneity of the L1 and L2 Beta-Lactamase Genes of Stenotrophomonas maltophilia. Antimicrob. Agents Chemother 2001, 45, 413–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 547.Sanschagrin F; Dufresne J; Levesque RC Molecular Heterogeneity of the L-1 Metallo-Beta-Lactamase Family from Stenotrophomonas maltophilia. Antimicrob. Agents Chemother 1998, 42, 1245–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 548.Crossman LC; Gould VC; Dow JM; Vernikos GS; Okazaki A; Sebaihia M; Saunders D; Arrowsmith C; Carver T; Peters N, et al. The Complete Genome, Comparative and Functional Analysis of Stenotrophomonas maltophilia Reveals an Organism Heavily Shielded by Drug Resistance Determinants. Genome Biol 2008, 9, R74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 549.Hu R; Zhang Q; Gu Z Molecular Diversity of Chromosomal Metallo-Beta-Lactamase Genes in Elizabethkingia Genus. Int. J. Antimicrob. Agents 2020, 56, 105978. [DOI] [PubMed] [Google Scholar]
- 550.Lin JN; Lai CH; Yang CH; Huang YH Elizabethkingia Infections in Humans: From Genomics to Clinics. Microorganisms 2019, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 551.Gonzalez LJ; Vila AJ Carbapenem Resistance in Elizabethkingia meningoseptica Is Mediated by Metallo-Beta-Lactamase BlaB. Antimicrob. Agents Chemother 2012, 56, 1686–1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 552.Lange F; Pfennigwerth N; Hartl R; Kerschner H; Achleitner D; Gatermann SG; Kaase M LMB-1, a Novel Family of Class B3 MBLs from an Isolate of Enterobacter cloacae. J. Antimicrob. Chemother 2018, 73, 2331–2335. [DOI] [PubMed] [Google Scholar]
- 553.Dabos L; Rodriguez CH; Nastro M; Dortet L; Bonnin RA; Famiglietti A; Iorga BI; Vay C; Naas T Lmb-1 Producing Citrobacter freundii from Argentina, a Novel Player in the Field of MBLs. Int. J. Antimicrob. Agents 2020, 55, 105857. [DOI] [PubMed] [Google Scholar]
- 554.Yong D; Toleman MA; Bell J; Ritchie B; Pratt R; Ryley H; Walsh TR Genetic and Biochemical Characterization of an Acquired Subgroup B3 Metallo-Beta-Lactamase Gene, blaAIM-1, and Its Unique Genetic Context in Pseudomonas aeruginosa from Australia. Antimicrob. Agents Chemother 2012, 56, 6154–6159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 555.Zhou H; Guo W; Zhang J; Li Y; Zheng P; Zhang H Draft Genome Sequence of a Metallo-Beta-Lactamase (blaAIM-1)-Producing Klebsiella pneumoniae ST1916 Isolated from a Patient with Chronic Diarrhoea. J Glob Antimicrob Resist 2019, 16, 165–167. [DOI] [PubMed] [Google Scholar]
- 556.Hou CD; Liu JW; Collyer C; Mitic N; Pedroso MM; Schenk G; Ollis DL Insights into an Evolutionary Strategy Leading to Antibiotic Resistance. Sci Rep 2017, 7, 40357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 557.Wachino J; Yoshida H; Yamane K; Suzuki S; Matsui M; Yamagishi T; Tsutsui A; Konda T; Shibayama K; Arakawa Y SMB-1, a Novel Subclass B3 Metallo-Beta-Lactamase, Associated with ISCR1 and a Class 1 Integron, from a Carbapenem-Resistant Serratia marcescens Clinical Isolate. Antimicrob. Agents Chemother 2011, 55, 5143–5149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 558.Pedroso MM; Waite DW; Melse O; Wilson L; Mitic N; McGeary RP; Antes I; Guddat LW; Hugenholtz P; Schenk G Broad Spectrum Antibiotic-Degrading Metallo-Beta-Lactamases Are Phylogenetically Diverse. Protein Cell 2020, 11, 613–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 559.Alderson RG; Barker D; Mitchell JB One Origin for Metallo-Beta-Lactamase Activity, or Two? An Investigation Assessing a Diverse Set of Reconstructed Ancestral Sequences Based on a Sample of Phylogenetic Trees. J. Mol. Evol 2014, 79, 117–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 560.Hagelueken G; Adams TM; Wiehlmann L; Widow U; Kolmar H; Tummler B; Heinz DW; Schubert WD The Crystal Structure of SdsA1, an Alkylsulfatase from Pseudomonas aeruginosa, Defines a Third Class of Sulfatases. Proc Natl Acad Sci U S A 2006, 103, 7631–7636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 561.Dong YJ; Bartlam M; Sun L; Zhou YF; Zhang ZP; Zhang CG; Rao Z; Zhang XE Crystal Structure of Methyl Parathion Hydrolase from Pseudomonas Sp. Wbc-3. J. Mol. Biol 2005, 353, 655–663. [DOI] [PubMed] [Google Scholar]
- 562.Magotti P; Bauer I; Igarashi M; Babagoli M; Marotta R; Piomelli D; Garau G Structure of Human N-Acylphosphatidylethanolamine-Hydrolyzing Phospholipase D: Regulation of Fatty Acid Ethanolamide Biosynthesis by Bile Acids. Structure 2015, 23, 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 563.van den Bosch TJM; Tan K; Joachimiak A; Welte CU Functional Profiling and Crystal Structures of Isothiocyanate Hydrolases Found in Gut-Associated and Plant-Pathogenic Bacteria. Appl. Environ. Microbiol 2018, 84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 564.Momb J; Wang C; Liu D; Thomas PW; Petsko GA; Guo H; Ringe D; Fast W Mechanism of the Quorum-Quenching Lactonase (AiiA) from Bacillus thuringiensis. 2. Substrate Modeling and Active Site Mutations. Biochemistry 2008, 47, 7715–7725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 565.Liu D; Thomas PW; Momb J; Hoang QQ; Petsko GA; Ringe D; Fast W Structure and Specificity of a Quorum-Quenching Lactonase (AiiB) from Agrobacterium tumefaciens. Biochemistry 2007, 46, 11789–11799. [DOI] [PubMed] [Google Scholar]
- 566.Li de la Sierra-Gallay I; Pellegrini O; Condon C Structural Basis for Substrate Binding, Cleavage and Allostery in the tRNA Maturase RNase Z. Nature 2005, 433, 657–661. [DOI] [PubMed] [Google Scholar]
- 567.Ma M; Li de la Sierra-Gallay I; Lazar N; Pellegrini O; Durand D; Marchfelder A; Condon C; van Tilbeurgh H The Crystal Structure of Trz1, the Long Form Rnase Z from Yeast. Nucleic Acids Res 2017, 45, 6209–6216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 568.Dorleans A; Li de la Sierra-Gallay I; Piton J; Zig L; Gilet L; Putzer H; Condon C Molecular Basis for the Recognition and Cleavage of RNA by the Bifunctional 5′-3′ Exo/Endoribonuclease RNase J. Structure 2011, 19, 1252–1261. [DOI] [PubMed] [Google Scholar]
- 569.Karim MF; Liu S; Laciak AR; Volk L; Koszelak-Rosenblum M; Lieber MR; Wu M; Curtis R; Huang NN; Carr G, et al. Structural Analysis of the Catalytic Domain of Artemis Endonuclease/SNM1C Reveals Distinct Structural Features. J. Biol. Chem 2020, 295, 12368–12377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 570.Mandel CR; Kaneko S; Zhang H; Gebauer D; Vethantham V; Manley JL; Tong L Polyadenylation Factor CPSF-73 Is the pre-mRNA 3′-End-Processing Endonuclease. Nature 2006, 444, 953–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 571.Garau G; Lemaire D; Vernet T; Dideberg O; Di Guilmi AM Crystal Structure of Phosphorylcholine Esterase Domain of the Virulence Factor Choline-Binding Protein E from Streptococcus pneumoniae: New Structural Features among the Metallo-Beta-Lactamase Superfamily. J. Biol. Chem 2005, 280, 28591–28600. [DOI] [PubMed] [Google Scholar]
- 572.Frazao C; Silva G; Gomes CM; Matias P; Coelho R; Sieker L; Macedo S; Liu MY; Oliveira S; Teixeira M, et al. Structure of a Dioxygen Reduction Enzyme from Desulfovibrio gigas. Nat. Struct. Biol 2000, 7, 1041–1045. [DOI] [PubMed] [Google Scholar]
- 573.Romao CV; Vicente JB; Borges PT; Victor BL; Lamosa P; Silva E; Pereira L; Bandeiras TM; Soares CM; Carrondo MA, et al. Structure of Escherichia coli Flavodiiron Nitric Oxide Reductase. J. Mol. Biol 2016, 428, 4686–4707. [DOI] [PubMed] [Google Scholar]
- 574.Pettinati I; Brem J; McDonough MA; Schofield CJ Crystal Structure of Human Persulfide Dioxygenase: Structural Basis of Ethylmalonic Encephalopathy. Hum. Mol. Genet 2015, 24, 2458–2469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 575.Sattler SA; Wang X; Lewis KM; DeHan PJ; Park CM; Xin Y; Liu H; Xian M; Xun L; Kang C Characterizations of Two Bacterial Persulfide Dioxygenases of the Metallo-Beta-Lactamase Superfamily. J. Biol. Chem 2015, 290, 18914–18923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 576.Puehringer S; Metlitzky M; Schwarzenbacher R The Pyrroloquinoline Quinone Biosynthesis Pathway Revisited: A Structural Approach. BMC Biochem 2008, 9, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 577.Koehn EM; Latham JA; Armand T; Evans RL 3rd; Tu X; Wilmot CM; Iavarone AT; Klinman JP Discovery of Hydroxylase Activity for Pqqb Provides a Missing Link in the Pyrroloquinoline Quinone Biosynthetic Pathway. J. Am. Chem. Soc 2019, 141, 4398–4405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 578.Makris TM; Knoot CJ; Wilmot CM; Lipscomb JD Structure of a Dinuclear Iron Cluster-Containing Beta-Hydroxylase Active in Antibiotic Biosynthesis. Biochemistry 2013, 52, 6662–6671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 579.Garces F; Fernandez FJ; Montella C; Penya-Soler E; Prohens R; Aguilar J; Baldoma L; Coll M; Badia J; Vega MC Molecular Architecture of the Mn2+-Dependent Lactonase UlaG Reveals an RNase-Like Metallo-Beta-Lactamase Fold and a Novel Quaternary Structure. J. Mol. Biol 2010, 398, 715–729. [DOI] [PubMed] [Google Scholar]
- 580.He SM; Wathier M; Podzelinska K; Wong M; McSorley FR; Asfaw A; Hove-Jensen B; Jia Z; Zechel DL Structure and Mechanism of PhnP, a Phosphodiesterase of the Carbon-Phosphorus Lyase Pathway. Biochemistry 2011, 50, 8603–8615. [DOI] [PubMed] [Google Scholar]
- 581.Podzelinska K; He SM; Wathier M; Yakunin A; Proudfoot M; Hove-Jensen B; Zechel DL; Jia Z Structure of PhnP, a Phosphodiesterase of the Carbon-Phosphorus Lyase Pathway for Phosphonate Degradation. J. Biol. Chem 2009, 284, 17216–17226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 582.Rabbani N; Xue M; Thornalley PJ Activity, Regulation, Copy Number and Function in the Glyoxalase System. Biochem. Soc. Trans 2014, 42, 419–424. [DOI] [PubMed] [Google Scholar]
- 583.Schilling O; Wenzel N; Naylor M; Vogel A; Crowder M; Makaroff C; Meyer-Klaucke W flexible Metal Binding of the Metallo-Beta-Lactamase Domain: Glyoxalase II Incorporates Iron, Manganese, and Zinc in Vivo. Biochemistry 2003, 42, 11777–11786. [DOI] [PubMed] [Google Scholar]
- 584.Marasinghe GP; Sander IM; Bennett B; Periyannan G; Yang KW; Makaroff CA; Crowder MW Structural Studies on a Mitochondrial Glyoxalase II. J. Biol. Chem 2005, 280, 40668–40675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 585.Campos-Bermudez VA; Leite NR; Krog R; Costa-Filho AJ; Soncini FC; Oliva G; Vila AJ Biochemical and Structural Characterization of Salmonella Typhimurium Glyoxalase II: New Insights into Metal Ion Selectivity. Biochemistry 2007, 46, 11069–11079. [DOI] [PubMed] [Google Scholar]
- 586.Vogel A; Schilling O; Niecke M; Bettmer J; Meyer-Klaucke W ElaC Encodes a Novel Binuclear Zinc Phosphodiesterase. J. Biol. Chem 2002, 277, 29078–29085. [DOI] [PubMed] [Google Scholar]
- 587.Levy S; Allerston CK; Liveanu V; Habib MR; Gileadi O; Schuster G Identification of LACTB2, a Metallo-Beta-Lactamase Protein, as a Human Mitochondrial Endoribonuclease. Nucleic Acids Res 2016, 44, 1813–1832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 588.Baker JA; Simkovic F; Taylor HM; Rigden DJ Potential DNA Binding and Nuclease Functions of ComEC Domains Characterized in Silico. Proteins 2016, 84, 1431–1442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 589.Fernández-Moreno MA; Martínez E; Boto L; Hopwood DA; Malpartida F Nucleotide Sequence and Deduced Functions of a Set of Cotranscribed Genes of Streptomyces Coelicolor A3(2) Including the Polyketide Synthase for the Antibiotic Actinorhodin. J. Biol. Chem 1992, 267, 19278–19290. [PubMed] [Google Scholar]
- 590.Taguchi T; Awakawa T; Nishihara Y; Kawamura M; Ohnishi Y; Ichinose K Bifunctionality of Activ as a Cyclase-Thioesterase Revealed by in Vitro Reconstitution of Actinorhodin Biosynthesis in Streptomyces Coelicolor A3(2). ChemBioChem 2017, 18, 316–323. [DOI] [PubMed] [Google Scholar]
- 591.Awakawa T; Yokota K; Funa N; Doi F; Mori N; Watanabe H; Horinouchi S Physically Discrete Beta-Lactamase-Type Thioesterase Catalyzes Product Release in Atrochrysone Synthesis by Iterative Type I Polyketide Synthase. Chem. Biol 2009, 16, 613–623. [DOI] [PubMed] [Google Scholar]
- 592.Li Y; Chooi YH; Sheng Y; Valentine JS; Tang Y Comparative Characterization of Fungal Anthracenone and Naphthacenedione Biosynthetic Pathways Reveals an Alpha-Hydroxylation-Dependent Claisen-Like Cyclization Catalyzed by a Dimanganese Thioesterase. J. Am. Chem. Soc 2011, 133, 15773–15785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 593.Baier F; Copp JN; Tokuriki N Evolution of Enzyme Superfamilies: Comprehensive Exploration of Sequence-Function Relationships. Biochemistry 2016, 55, 6375–6388. [DOI] [PubMed] [Google Scholar]
- 594.Miraula M; Schenk G; Mitic N Promiscuous Metallo-Beta-Lactamases: MIM-1 and MIM-2 May Play an Essential Role in Quorum Sensing Networks. J. Inorg. Biochem 2016, 162, 366–375. [DOI] [PubMed] [Google Scholar]
- 595.Baier F; Hong N; Yang G; Pabis A; Miton CM; Barrozo A; Carr PD; Kamerlin SC; Jackson CJ; Tokuriki N Cryptic Genetic Variation Shapes the Adaptive Evolutionary Potential of Enzymes. Elife 2019, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 596.Park KS; Kim TY; Kim JH; Lee JH; Jeon JH; Karim AM; Malik SK; Lee SH PNGM-1, a Novel Subclass B3 Metallo-Beta-Lactamase from a Deep-Sea Sediment Metagenome. J Glob Antimicrob Resist 2018, 14, 302–305. [DOI] [PubMed] [Google Scholar]
- 597.Park YS; Kim TY; Park H; Lee JH; Nguyen DQ; Hong MK; Lee SH; Kang LW Structural Study of Metal Binding and Coordination in Ancient Metallo-Beta-Lactamase PNGM-1 Variants. Int J Mol Sci 2020, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 598.Lee JH; Takahashi M; Jeon JH; Kang LW; Seki M; Park KS; Hong MK; Park YS; Kim TY; Karim AM, et al. Dual Activity of PNGM-1 Pinpoints the Evolutionary Origin of Subclass B3 Metallo-Beta-Lactamases: A Molecular and Evolutionary Study. Emerg Microbes Infect 2019, 8, 1688–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 599.Diene SM; Pinault L; Keshri V; Armstrong N; Khelaifia S; Chabriere E; Caetano-Anolles G; Colson P; La Scola B; Rolain JM, et al. Human Metallo-Beta-Lactamase Enzymes Degrade Penicillin. Sci Rep 2019, 9, 12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 600.Lin HV; Massam-Wu T; Lin CP; Wang YA; Shen YC; Lu WJ; Hsu PH; Chen YH; Borges-Walmsley MI; Walmsley AR The Vibrio cholerae Var Regulon Encodes a Metallo-Beta-Lactamase and an Antibiotic Efflux Pump, Which Are Regulated by VarR, a LysR-Type Transcription Factor. PLoS One 2017, 12, e0184255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 601.Page MI; Badarau A The Mechanisms of Catalysis by Metallo Beta-Lactamases. Bioinorg Chem Appl 2008, 576297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 602.Cricco JA; Orellano EG; Rasia RM; Ceccarelli EA; Vila AJ Metallo-B-Lactamases: Does It Take Two to Tango? Coord. Chem. Rev 1999, 190–192, 519–535. [Google Scholar]
- 603.Wang Z; Fast W; Valentine AM; Benkovic SJ Metallo-Beta-Lactamase: Structure and Mechanism. Curr. Opin. Chem. Biol 1999, 3, 614–622. [DOI] [PubMed] [Google Scholar]
- 604.Bounaga S; Laws AP; Galleni M; Page MI The Mechanism of Catalysis and the Inhibition of the Bacillus cereus Zinc-Dependent Beta-Lactamase. Biochem. J 1998, 331 (Pt 3), 703–711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 605.Badarau A; Page MI The Variation of Catalytic Efficiency of Bacillus cereus Metallo-Beta-Lactamase with Different Active Site Metal Ions. Biochemistry 2006, 45, 10654–10666. [DOI] [PubMed] [Google Scholar]
- 606.Badarau A; Page MI Loss of Enzyme Activity During Turnover of the Bacillus cereus Beta-Lactamase Catalysed Hydrolysis of Beta-Lactams Due to Loss of Zinc Ion. J. Biol. Inorg. Chem 2008, 13, 919–928. [DOI] [PubMed] [Google Scholar]
- 607.Wang Z; Fast W; Benkovic SJ Direct Observation of an Enzyme-Bound Intermediate in the Catalytic Cycle of the Metallo-B-Lactamase from Bacteroides fragilis. J. Am. Chem. Soc 1998, 120, 10788–10789. [Google Scholar]
- 608.Bertini I; Luchinat C Cobalt(II) as a Probe of the Structure and Function of Carbonic Anhydrase. Acc. Chem. Res 1983, 16, 272–279. [Google Scholar]
- 609.Vila AJ; Fernández CO Structure of the Metal Site in Rhus Vernicifera Stellacyanin: A Paramagnetic NMR Study on Its Co(Ii) Derivative. J. Am. Chem. Soc 1996, 118, 7291–7298. [Google Scholar]
- 610.Fernandez CO; Sannazzaro AI; Vila AJ Alkaline Transition of Rhus vernicifera Stellacyanin, an Unusual Blue Copper Protein. Biochemistry 1997, 36, 10566–10570. [DOI] [PubMed] [Google Scholar]
- 611.Selleck C; Larrabee JA; Harmer J; Guddat LW; Mitic N; Helweh W; Ollis DL; Craig WR; Tierney DL; Monteiro Pedroso M, et al. AIM-1: An Antibiotic-Degrading Metallohydrolase That Displays Mechanistic Flexibility. Chemistry 2016, 22, 17704–17714. [DOI] [PubMed] [Google Scholar]
- 612.Damblon C; Jensen M; Ababou A; Barsukov I; Papamicael C; Schofield CJ; Olsen L; Bauer R; Roberts GC The Inhibitor Thiomandelic Acid Binds to Both Metal Ions in Metallo-Beta-Lactamase and Induces Positive Cooperativity in Metal Binding. J. Biol. Chem 2003, 278, 29240–29251. [DOI] [PubMed] [Google Scholar]
- 613.Djoko KY; Achard MES; Phan MD; Lo AW; Miraula M; Prombhul S; Hancock SJ; Peters KM; Sidjabat HE; Harris PN, et al. Copper Ions and Coordination Complexes as Novel Carbapenem Adjuvants. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 614.Cahill ST; Tarhonskaya H; Rydzik AM; Flashman E; McDonough MA; Schofield CJ; Brem J Use of Ferrous Iron by Metallo-Beta-Lactamases. J. Inorg. Biochem 2016, 163, 185–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 615.Fast W; Sutton LD Metallo-Beta-Lactamase: Inhibitors and Reporter Substrates. Biochim. Biophys. Acta 2013, 1834, 1648–1659. [DOI] [PubMed] [Google Scholar]
- 616.Baldwin JE; Schofield C 1 the Biosynthesis of ß-Lactams. In The Chemistry of B-Lactams, Page MI, Ed. Springer Netherlands: Dordrecht, 1992; pp 1–78. [Google Scholar]
- 617.Dal Peraro M; Vila AJ; Carloni P Structural Determinants and Hydrogen-Bond Network of the Mononuclear Zinc(II)-Beta-Lactamase Active Site. J. Biol. Inorg. Chem 2002, 7, 704–712. [DOI] [PubMed] [Google Scholar]
- 618.Dal Peraro M; Vila AJ; Carloni P Protonation State of Asp120 in the Binuclear Active Site of the Metallo-Beta-Lactamase from Bacteroides Fragilis. Inorg. Chem 2003, 42, 4245–4247. [DOI] [PubMed] [Google Scholar]
- 619.Dal Peraro M; Vila AJ; Carloni P Substrate Binding to Mononuclear Metallo-Beta-Lactamase from Bacillus cereus. Proteins 2004, 54, 412–423. [DOI] [PubMed] [Google Scholar]
- 620.Dal Peraro M; Llarrull LI; Rothlisberger U; Vila AJ; Carloni P Water-Assisted Reaction Mechanism of Monozinc Beta-Lactamases. J. Am. Chem. Soc 2004, 126, 12661–12668. [DOI] [PubMed] [Google Scholar]
- 621.Simona F; Magistrato A; Dal Peraro M; Cavalli A; Vila AJ; Carloni P Common Mechanistic Features among Metallo-Beta-Lactamases: A Computational Study of Aeromonas hydrophila CphA Enzyme. J. Biol. Chem 2009, 284, 28164–28171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 622.Suarez D; Merz KM Jr. Molecular Dynamics Simulations of the Mononuclear Zinc-Beta-Lactamase from Bacillus cereus. J. Am. Chem. Soc 2001, 123, 3759–3770. [DOI] [PubMed] [Google Scholar]
- 623.Diaz N; Suarez D; Merz KM Jr. Molecular Dynamics Simulations of the Mononuclear Zinc-Beta-Lactamase from Bacillus cereus Complexed with Benzylpenicillin and a Quantum Chemical Study of the Reaction Mechanism. J. Am. Chem. Soc 2001, 123, 9867–9879. [DOI] [PubMed] [Google Scholar]
- 624.Suarez D; Brothers EN; Merz KM Jr. Insights into the Structure and Dynamics of the Dinuclear Zinc Beta-Lactamase Site from Bacteroides fragilis. Biochemistry 2002, 41, 6615–6630. [DOI] [PubMed] [Google Scholar]
- 625.Park H; Brothers EN; Merz KM Jr. Hybrid Qm/Mm and Dft Investigations of the Catalytic Mechanism and Inhibition of the Dinuclear Zinc Metallo-Beta-Lactamase CcrA from Bacteroides fragilis. J. Am. Chem. Soc 2005, 127, 4232–4241. [DOI] [PubMed] [Google Scholar]
- 626.Xu D; Guo H; Cui Q Antibiotic Deactivation by a Dizinc B-Lactamase: Mechanistic Insights from QM/MM and DFT Studies. J. Am. Chem. Soc 2007, 129, 10814–10822. [DOI] [PubMed] [Google Scholar]
- 627.Zheng M; Xu D New Delhi Metallo-Beta-Lactamase I: Substrate Binding and Catalytic Mechanism. J. Phys. Chem. B 2013, 117, 11596–11607. [DOI] [PubMed] [Google Scholar]
- 628.Khrenova MG; Nemukhin AV Modeling the Transient Kinetics of the L1 Metallo-Beta-Lactamase. J. Phys. Chem. B 2018, 122, 1378–1386. [DOI] [PubMed] [Google Scholar]
- 629.Andreini C; Bertini I; Rosato A Metalloproteomes: A Bioinformatic Approach. Acc. Chem. Res 2009, 42, 1471–1479. [DOI] [PubMed] [Google Scholar]
- 630.Maret W; Li Y Coordination Dynamics of Zinc in Proteins. Chem. Rev 2009, 109, 4682–4707. [DOI] [PubMed] [Google Scholar]
- 631.Brem J; Cain R; Cahill S; McDonough MA; Clifton IJ; Jimenez-Castellanos JC; Avison MB; Spencer J; Fishwick CW; Schofield CJ Structural Basis of Metallo-Beta-Lactamase, Serine-Beta-Lactamase and Penicillin-Binding Protein Inhibition by Cyclic Boronates. Nat Commun 2016, 7, 12406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 632.Ippolito JA; Baird TT Jr.; McGee SA; Christianson DW; Fierke CA Structure-Assisted Redesign of a Protein-Zinc-Binding Site with Femtomolar Affinity. Proc Natl Acad Sci U S A 1995, 92, 5017–5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 633.Voordouw G; Milo C; Roche RS The Determination of the Binding Constant of Metalloenzymes for Their Active Site Metal Ion from Ligand Inhibition Data. Theoretical Analysis and Application to the Inhibition of Thermolysin by 1,10-Phenanthroline. Anal. Biochem 1976, 70, 313–326. [DOI] [PubMed] [Google Scholar]
- 634.Vallee BL; Riordan JF; Coleman JE Carboxypeptidase A: Approaches to the Chemical Nature of the Active Center and the Mechanisms of Action. Proc Natl Acad Sci U S A 1963, 49, 109–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 635.Rasia RM; Vila AJ Exploring the Role and the Binding Affinity of a Second Zinc Equivalent in B. cereus Metallo-Beta-Lactamase. Biochemistry 2002, 41, 1853–1860. [DOI] [PubMed] [Google Scholar]
- 636.Goto M; Yasuzawa H; Higashi T; Yamaguchi Y; Kawanami A; Mifune S; Mori H; Nakayama H; Harada K; Arakawa Y Dependence of Hydrolysis of Beta-Lactams with a Zinc(II)-Beta-Lactamase Produced from Serratia Marcescens (IMP-1) on pH and Concentration of Zinc(II) Ion: Dissociation of Zn(II) from IMP-1 in Acidic Medium. Biol. Pharm. Bull 2003, 26, 589–594. [DOI] [PubMed] [Google Scholar]
- 637.Schenk G; Mitic N; Gahan LR; Ollis DL; McGeary RP; Guddat LW Binuclear Metallohydrolases: Complex Mechanistic Strategies for a Simple Chemical Reaction. Acc. Chem. Res 2012, 45, 1593–1603. [DOI] [PubMed] [Google Scholar]
- 638.Kaminskaia NV; He C; Lippard SJ Reactivity of Mu-Hydroxodizinc(II) Centers in Enzymatic Catalysis through Model Studies. Inorg. Chem 2000, 39, 3365–3373. [DOI] [PubMed] [Google Scholar]
- 639.Fast W; Wang Z; Benkovic SJ Familial Mutations and Zinc Stoichiometry Determine the Rate-Limiting Step of Nitrocefin Hydrolysis by Metallo-Beta-Lactamase from Bacteroides fragilis. Biochemistry 2001, 40, 1640–1650. [DOI] [PubMed] [Google Scholar]
- 640.McManus-Munoz S; Crowder MW Kinetic Mechanism of Metallo-Beta-Lactamase L1 from Stenotrophomonas maltophilia. Biochemistry 1999, 38, 1547–1553. [DOI] [PubMed] [Google Scholar]
- 641.Cook PF Enzyme Mechanism from Isotope Effects. CRC Press: Boca Raton, 1991; p 500 p. [Google Scholar]
- 642.Anderson VE Isotope Effects on Enzyme-Catalyzed Reactions. Curr. Opin. Struct. Biol 1992, 2, 757–764. [Google Scholar]
- 643.Page MI The Mechanisms of Reactions Of .Beta.-Lactam Antibiotics. Acc. Chem. Res 1984, 17, 144–151. [Google Scholar]
- 644.Hu Z; Periyannan GR; Crowder MW Folding Strategy to Prepare Co(II)-Substituted Metallo-Beta-Lactamase L1. Anal. Biochem 2008, 378, 177–183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 645.Garrity JD; Bennett B; Crowder MW Direct Evidence That the Reaction Intermediate of Metallo-Beta-Lactamase L1 Is Metal Bound. Biochemistry 2005, 44, 1078–1087. [DOI] [PubMed] [Google Scholar]
- 646.Crawford PA; Yang KW; Sharma N; Bennett B; Crowder MW Spectroscopic Studies on Cobalt(II)-Substituted Metallo-Beta-Lactamase Imis from Aeromonas veronii Bv. Sobria. Biochemistry 2005, 44, 5168–5176. [DOI] [PubMed] [Google Scholar]
- 647.Zhang H; Hao Q Crystal Structure of NDM-1 Reveals a Common Beta-Lactam Hydrolysis Mechanism. FASEB J. 2011, 25, 2574–2582. [DOI] [PubMed] [Google Scholar]
- 648.Boyd DB; Herron DK; Lunn WHW; Spitzer WA Electronic Structures of Cephalosporins and Penicillins. 11. Parabolic Relationships between Antibacterial Activity of Cephalosporins And Beta-Lactam Reactivity Predicted from Molecular Orbital Calculations. J. Am. Chem. Soc 1980, 102, 1812–1814. [Google Scholar]
- 649.Hamilton-Miller JMT; Newton GGF; Abraham EP Products of Aminolysis and Enzymic Hydrolysis of the Cephalosporins. Biochem. J 1970, 116, 371–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 650.O’Callaghan CH; Morris A; Kirby SM; Shingler AH Novel Method for Detection of Beta-Lactamases by Using a Chromogenic Cephalosporin Substrate. Antimicrob. Agents Chemother 1972, 1, 283–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 651.Bebrone C; Moali C; Mahy F; Rival S; Docquier JD; Rossolini GM; Fastrez J; Pratt RF; Frere JM; Galleni M CENTA as a Chromogenic Substrate for Studying Beta-Lactamases. Antimicrob. Agents Chemother 2001, 45, 1868–1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 652.Jones RN; Wilson HW; Novick WJ Jr.; Barry AL; Thornsberry C In Vitro Evaluation of CENTA, a New Beta-Lactamase-susceptible Chromogenic Cephalosporin Reagent. J. Clin. Microbiol 1982, 15, 954–958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 653.Yu S; Vosbeek A; Corbella K; Severson J; Schesser J; Sutton LD A Chromogenic Cephalosporin for Beta-Lactamase Inhibitor Screening Assays. Anal. Biochem 2012, 428, 96–98. [DOI] [PubMed] [Google Scholar]
- 654.Kaminskaia NV; Spingler B; Lippard SJ Intermediate in Beta-Lactam Hydrolysis Catalyzed by a Dinuclear Zinc(Ii) Complex: Relevance to the Mechanism of Metallo-Beta-Lactamase. J. Am. Chem. Soc 2001, 123, 6555–6563. [DOI] [PubMed] [Google Scholar]
- 655.Thomas PW; Zheng M; Wu S; Guo H; Liu D; Xu D; Fast W Characterization of Purified New Delhi Metallo-Beta-Lactamase-1. Biochemistry 2011, 50, 10102–10113. [DOI] [PubMed] [Google Scholar]
- 656.Aitha M; Marts AR; Bergstrom A; Moller AJ; Moritz L; Turner L; Nix JC; Bonomo RA; Page RC; Tierney DL, et al. Biochemical, Mechanistic, and Spectroscopic Characterization of Metallo-Beta-Lactamase VIM-2. Biochemistry 2014, 53, 7321–7331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 657.Rasia R; Vila A Mechanistic Study of the Hydrolysis of Nitrocefin Mediated by B. cereus Metallo-beta-Lactamase. Arkivoc 2003, 2003. [Google Scholar]
- 658.Charnas RL; Knowles JR Inhibition of the RTEM Beta-Lactamase from Escherichia coli. Interaction of Enzyme with Derivatives of Olivanic Acid. Biochemistry 1981, 20, 2732–2737. [DOI] [PubMed] [Google Scholar]
- 659.Easton CJ; Knowles JR Inhibition of the RTEM Beta-Lactamase from Escherichia coli. Interaction of the Enzyme with Derivatives of Olivanic Acid. Biochemistry 1982, 21, 2857–2862. [DOI] [PubMed] [Google Scholar]
- 660.Ratcliffe RW; Wildonger KJ; Di Michele L; Douglas AW; Hajdu R; Goegelman RT; Springer JP; Hirshfield J Studies on the Structures of Imipenem, Dehydropeptidase I-Hydrolyzed Imipenem, and Related Analogs. The Journal of Organic Chemistry 1989, 54, 653–660. [Google Scholar]
- 661.Wachino J; Yamaguchi Y; Mori S; Jin W; Kimura K; Kurosaki H; Arakawa Y Structural Insights into Recognition of Hydrolyzed Carbapenems and Inhibitors by Subclass B3 Metallo-Beta-Lactamase SMB-1. Antimicrob. Agents Chemother 2016, 60, 4274–4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 662.Lohans CT; Freeman EI; Groesen EV; Tooke CL; Hinchliffe P; Spencer J; Brem J; Schofield CJ Mechanistic Insights into Beta-Lactamase-Catalysed Carbapenem Degradation through Product Characterisation. Sci Rep 2019, 9, 13608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 663.Reddy N; Shungube M; Arvidsson PI; Baijnath S; Kruger HG; Govender T; Naicker TA 2018–2019 Patent Review of Metallo Beta-Lactamase Inhibitors. Expert Opin Ther Pat 2020, 30, 541–555. [DOI] [PubMed] [Google Scholar]
- 664.Zmarlicka MT; Nailor MD; Nicolau DP Impact of the New Delhi Metallo-Beta-Lactamase on Beta-Lactam Antibiotics. Infect Drug Resist 2015, 8, 297–309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 665.King DT; Strynadka NC Targeting Metallo-Beta-Lactamase Enzymes in Antibiotic Resistance. Future Med Chem 2013, 5, 1243–1263. [DOI] [PubMed] [Google Scholar]
- 666.McGeary RP; Tan DT; Schenk G Progress toward Inhibitors of Metallo-Beta-Lactamases. Future Med Chem 2017, 9, 673–691. [DOI] [PubMed] [Google Scholar]
- 667.Perez-Llarena FJ; Bou G Beta-Lactamase Inhibitors: The Story So Far. Curr. Med. Chem 2009, 16, 3740–3765. [DOI] [PubMed] [Google Scholar]
- 668.Krajnc A; Lang PA; Panduwawala TD; Brem J; Schofield CJ Will Morphing Boron-Based Inhibitors Beat the Beta-Lactamases? Curr. Opin. Chem. Biol 2019, 50, 101–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 669.Bonomo RA Cefiderocol: A Novel Siderophore Cephalosporin Defeating Carbapenem-Resistant Pathogens. Clin. Infect. Dis 2019, 69, S519–S520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 670.Ito A; Kohira N; Bouchillon SK; West J; Rittenhouse S; Sader HS; Rhomberg PR; Jones RN; Yoshizawa H; Nakamura R, et al. In Vitro Antimicrobial Activity of S-649266, a Catechol-Substituted Siderophore Cephalosporin, When Tested against Non-Fermenting Gram-Negative Bacteria. J. Antimicrob. Chemother 2016, 71, 670–677. [DOI] [PubMed] [Google Scholar]
- 671.Kohira N; West J; Ito A; Ito-Horiyama T; Nakamura R; Sato T; Rittenhouse S; Tsuji M; Yamano Y In Vitro Antimicrobial Activity of a Siderophore Cephalosporin, S-649266, against Enterobacteriaceae Clinical Isolates, Including Carbapenem-Resistant Strains. Antimicrob. Agents Chemother 2016, 60, 729–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 672.Neu HC; Fu KP Clavulanic Acid, a Novel Inhibitor of Beta-Lactamases. Antimicrob. Agents Chemother 1978, 14, 650–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 673.Reading C; Cole M Clavulanic Acid: A Beta-Lactamase-Inhiting Beta-Lactam from Streptomyces clavuligerus. Antimicrob. Agents Chemother 1977, 11, 852–857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 674.Fisher J; Charnas RL; Knowles JR Kinetic Studies on the Inactivation of Escherichia coli RTEM Beta-Lactamase by Clavulanic Acid. Biochemistry 1978, 17, 2180–2184. [DOI] [PubMed] [Google Scholar]
- 675.English AR; Retsema JA; Girard AE; Lynch JE; Barth WE CP-45,899, a Beta-Lactamase Inhibitor That Extends the Antibacterial Spectrum of Beta-Lactams: Initial Bacteriological Characterization. Antimicrob. Agents Chemother 1978, 14, 414–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 676.Aronoff SC; Jacobs MR; Johenning S; Yamabe S Comparative Activities of the Beta-Lactamase Inhibitors Ytr 830, Sodium Clavulanate, and Sulbactam Combined with Amoxicillin or Ampicillin. Antimicrob. Agents Chemother 1984, 26, 580–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 677.Vena A; Castaldo N; Bassetti M The Role of New Beta-Lactamase Inhibitors in Gram-Negative Infections. Curr. Opin. Infect. Dis 2019, 32, 638–646. [DOI] [PubMed] [Google Scholar]
- 678.Ehmann DE; Jahic H; Ross PL; Gu RF; Hu J; Kern G; Walkup GK; Fisher SL Avibactam Is a Covalent, reversible, Non-Beta-Lactam Beta-Lactamase Inhibitor. Proc Natl Acad Sci U S A 2012, 109, 11663–11668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 679.Coleman K Diazabicyclooctanes (DBOs): A Potent New Class of Non-Beta-Lactam Beta-Lactamase Inhibitors. Curr. Opin. Microbiol 2011, 14, 550–555. [DOI] [PubMed] [Google Scholar]
- 680.Kaye KS; Boucher HW; Brown ML; Aggrey A; Khan I; Joeng HK; Tipping RW; Du J; Young K; Butterton JR, et al. Comparison of Treatment Outcomes between Analysis Populations in the RESTORE-IMI 1 Phase 3 Trial of Imipenem-Cilastatin-Relebactam Versus Colistin Plus Imipenem-Cilastatin in Patients with Imipenem-Nonsusceptible Bacterial Infections. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 681.Tooke CL; Hinchliffe P; Lang PA; Mulholland AJ; Brem J; Schofield CJ; Spencer J Molecular Basis of Class A Beta-Lactamase Inhibition by Relebactam. Antimicrob. Agents Chemother 2019, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 682.Monogue ML; Giovagnoli S; Bissantz C; Zampaloni C; Nicolau DP In Vivo Efficacy of Meropenem with a Novel Non-Beta-Lactam-Beta-Lactamase Inhibitor, Nacubactam, against Gram-Negative Organisms Exhibiting Various Resistance Mechanisms in a Murine Complicated Urinary Tract Infection Model. Antimicrob. Agents Chemother 2018, 62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 683.Livermore DM; Mushtaq S; Warner M; Vickers A; Woodford N In Vitro Activity of Cefepime/Zidebactam (WCK 5222) against Gram-Negative Bacteria. J. Antimicrob. Chemother 2017, 72, 1373–1385. [DOI] [PubMed] [Google Scholar]
- 684.Mushtaq S; Vickers A; Woodford N; Haldimann A; Livermore DM Activity of Nacubactam (RG6080/OP0595) Combinations against MBL-Producing Enterobacteriaceae. J. Antimicrob. Chemother 2019, 74, 953–960. [DOI] [PubMed] [Google Scholar]
- 685.Durand-Reville TF; Guler S; Comita-Prevoir J; Chen B; Bifulco N; Huynh H; Lahiri S; Shapiro AB; McLeod SM; Carter NM, et al. ETX2514 Is a Broad-Spectrum Beta-Lactamase Inhibitor for the Treatment of Drug-Resistant Gram-Negative Bacteria Including Acinetobacter baumannii. Nat Microbiol 2017, 2, 17104. [DOI] [PubMed] [Google Scholar]
- 686.Lomovskaya O; Sun D; Rubio-Aparicio D; Nelson K; Tsivkovski R; Griffith DC; Dudley MN Vaborbactam: Spectrum of Beta-Lactamase Inhibition and Impact of Resistance Mechanisms on Activity in Enterobacteriaceae. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 687.Castanheira M; Rhomberg PR; Flamm RK; Jones RN Effect of the Beta-Lactamase Inhibitor Vaborbactam Combined with Meropenem against Serine Carbapenemase-Producing Enterobacteriaceae. Antimicrob. Agents Chemother 2016, 60, 5454–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 688.Rotondo CM; Wright GD Inhibitors of Metallo-Beta-Lactamases. Curr. Opin. Microbiol 2017, 39, 96–105. [DOI] [PubMed] [Google Scholar]
- 689.Ganta SR; Perumal S; Pagadala SR; Samuelsen O; Spencer J; Pratt RF; Buynak JD Approaches to the Simultaneous Inactivation of Metallo- and Serine-Beta-Lactamases. Bioorg. Med. Chem. Lett 2009, 19, 1618–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 690.Buynak JD; Chen H; Vogeti L; Gadhachanda VR; Buchanan CA; Palzkill T; Shaw RW; Spencer J; Walsh TR Penicillin-Derived Inhibitors That Simultaneously Target Both Metallo- and Serine-Beta-Lactamases. Bioorg. Med. Chem. Lett 2004, 14, 1299–1304. [DOI] [PubMed] [Google Scholar]
- 691.Murphy BP; Pratt RF A Thiono-Beta-Lactam Substrate for the Beta-Lactamase II of Bacillus cereus. Evidence for Direct Interaction between the Essential Metal Ion and Substrate. Biochem. J 1989, 258, 765–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 692.Tsang WY; Dhanda A; Schofield CJ; Frere JM; Galleni M; Page MI The Inhibition of Metallo-Beta-Lactamase by Thioxo-Cephalosporin Derivatives. Bioorg. Med. Chem. Lett 2004, 14, 1737–1739. [DOI] [PubMed] [Google Scholar]
- 693.Nagano R; Adachi Y; Imamura H; Yamada K; Hashizume T; Morishima H Carbapenem Derivatives as Potential Inhibitors of Various Beta-Lactamases, Including Class B Metallo-Beta-Lactamases. Antimicrob. Agents Chemother 1999, 43, 2497–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 694.Nagano R; Adachi Y; Hashizume T; Morishima H In Vitro Antibacterial Activity and Mechanism of Action of J-111,225, a Novel 1beta-Methylcarbapenem, against Transferable IMP-1 Metallo-Beta-Lactamase Producers. J. Antimicrob. Chemother 2000, 45, 271–276. [DOI] [PubMed] [Google Scholar]
- 695.Zaengle-Barone JM; Jackson AC; Besse DM; Becken B; Arshad M; Seed PC; Franz KJ Copper Influences the Antibacterial Outcomes of a Beta-Lactamase-Activated Prochelator against Drug-Resistant Bacteria. ACS Infect Dis 2018, 4, 1019–1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 696.Jackson AC; Zaengle-Barone JM; Puccio EA; Franz KJ A Cephalosporin Prochelator Inhibits New Delhi Metallo-Beta-Lactamase 1 without Removing Zinc. ACS Infect Dis 2020, 6, 1264–1272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 697.Gonzalez MM; Kosmopoulou M; Mojica MF; Castillo V; Hinchliffe P; Pettinati I; Brem J; Schofield CJ; Mahler G; Bonomo RA, et al. Bisthiazolidines: A Substrate-Mimicking Scaffold as an Inhibitor of the NDM-1 Carbapenemase. ACS Infect Dis 2015, 1, 544–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 698.Bounaga S; Galleni M; Laws AP; Page MI Cysteinyl Peptide Inhibitors of Bacillus cereus Zinc Beta-Lactamase. Bioorg. Med. Chem 2001, 9, 503–510. [DOI] [PubMed] [Google Scholar]
- 699.Mollard C; Moali C; Papamicael C; Damblon C; Vessilier S; Amicosante G; Schofield CJ; Galleni M; Frere JM; Roberts GC Thiomandelic Acid, a Broad Spectrum Inhibitor of Zinc Beta-Lactamases: Kinetic and Spectroscopic Studies. J. Biol. Chem 2001, 276, 45015–45023. [DOI] [PubMed] [Google Scholar]
- 700.Heinz U; Bauer R; Wommer S; Meyer-Klaucke W; Papamichaels C; Bateson J; Adolph HW Coordination Geometries of Metal Ions in D- or L-Captopril-Inhibited Metallo-Beta-Lactamases. J. Biol. Chem 2003, 278, 20659–20666. [DOI] [PubMed] [Google Scholar]
- 701.Sun Q; Law A; Crowder MW; Geysen HM Homo-Cysteinyl Peptide Inhibitors of the L1 Metallo-Beta-Lactamase, and SAR as Determined by Combinatorial Library Synthesis. Bioorg. Med. Chem. Lett 2006, 16, 5169–5175. [DOI] [PubMed] [Google Scholar]
- 702.Meng Z; Tang ML; Yu L; Liang Y; Han J; Zhang C; Hu F; Yu JM; Sun X Novel Mercapto Propionamide Derivatives with Potent New Delhi Metallo-Beta-Lactamase-1 Inhibitory Activity and Low Toxicity. ACS Infect Dis 2019, 5, 903–916. [DOI] [PubMed] [Google Scholar]
- 703.Tehrani K; Martin NI Thiol-Containing Metallo-Beta-Lactamase Inhibitors Resensitize Resistant Gram-Negative Bacteria to Meropenem. ACS Infect Dis 2017, 3, 711–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 704.Lassaux P; Hamel M; Gulea M; Delbruck H; Mercuri PS; Horsfall L; Dehareng D; Kupper M; Frere JM; Hoffmann K, et al. Mercaptophosphonate Compounds as Broad-Spectrum Inhibitors of the Metallo-Beta-Lactamases. J. Med. Chem 2010, 53, 4862–4876. [DOI] [PubMed] [Google Scholar]
- 705.Wang YL; Liu S; Yu ZJ; Lei Y; Huang MY; Yan YH; Ma Q; Zheng Y; Deng H; Sun Y, et al. Structure-Based Development of (1-(3’-Mercaptopropanamido)Methyl)Boronic Acid Derived Broad-Spectrum, Dual-Action Inhibitors of Metallo- and Serine-Beta-Lactamases. J. Med. Chem 2019, 62, 7160–7184. [DOI] [PubMed] [Google Scholar]
- 706.Brem J; van Berkel SS; Zollman D; Lee SY; Gileadi O; McHugh PJ; Walsh TR; McDonough MA; Schofield CJ Structural Basis of Metallo-Beta-Lactamase Inhibition by Captopril Stereoisomers. Antimicrob. Agents Chemother 2016, 60, 142–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 707.Rossi M-A; Martinez V; Hinchliffe P; Mojica MF; Castillo V; Moreno DM; Smith R; Spellberg B; Drusano GL; Banchio C, et al. 2-Mercaptomethyl-Thiazolidines Use Conserved Aromatic–S Interactions to Achieve Broad-Range Inhibition of Metallo-B-Lactamases. Chemical Science 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 708.Siemann S; Clarke AJ; Viswanatha T; Dmitrienko GI Thiols as Classical and Slow-Binding Inhibitors of IMP-1 and Other Binuclear Metallo-Beta-Lactamases. Biochemistry 2003, 42, 1673–1683. [DOI] [PubMed] [Google Scholar]
- 709.Klingler FM; Wichelhaus TA; Frank D; Cuesta-Bernal J; El-Delik J; Muller HF; Sjuts H; Gottig S; Koenigs A; Pos KM, et al. Approved Drugs Containing Thiols as Inhibitors of Metallo-Beta-Lactamases: Strategy to Combat Multidrug-Resistant Bacteria. J. Med. Chem 2015, 58, 3626–3630. [DOI] [PubMed] [Google Scholar]
- 710.Buttner D; Kramer JS; Klingler FM; Wittmann SK; Hartmann MR; Kurz CG; Kohnhauser D; Weizel L; Bruggerhoff A; Frank D, et al. Challenges in the Development of a Thiol-Based Broad-Spectrum Inhibitor for Metallo-Beta-Lactamases. ACS Infect Dis 2018, 4, 360–372. [DOI] [PubMed] [Google Scholar]
- 711.Cushman DW; Ondetti MA Design of Angiotensin Converting Enzyme Inhibitors. Nat. Med 1999, 5, 1110–1113. [DOI] [PubMed] [Google Scholar]
- 712.Antony J; Gresh N; Olsen L; Hemmingsen L; Schofield CJ; Bauer R Binding of D- and L-Captopril Inhibitors to Metallo-Beta-Lactamase Studied by Polarizable Molecular Mechanics and Quantum Mechanics. J. Comput. Chem 2002, 23, 1281–1296. [DOI] [PubMed] [Google Scholar]
- 713.Garcia-Saez I; Hopkins J; Papamicael C; Franceschini N; Amicosante G; Rossolini GM; Galleni M; Frere JM; Dideberg O The 1.5-A Structure of Chryseobacterium meningosepticum Zinc Beta-Lactamase in Complex with the Inhibitor, D-Captopril. J. Biol. Chem 2003, 278, 23868–23873. [DOI] [PubMed] [Google Scholar]
- 714.Li N; Xu Y; Xia Q; Bai C; Wang T; Wang L; He D; Xie N; Li L; Wang J, et al. Simplified Captopril Analogues as NDM-1 Inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 386–389. [DOI] [PubMed] [Google Scholar]
- 715.Yusof Y; Tan DTC; Arjomandi OK; Schenk G; McGeary RP Captopril Analogues as Metallo-Beta-Lactamase Inhibitors. Bioorg. Med. Chem. Lett 2016, 26, 1589–1593. [DOI] [PubMed] [Google Scholar]
- 716.Ma G; Wang S; Wu K; Zhang W; Ahmad A; Hao Q; Lei X; Zhang H Structure-Guided Optimization of D-Captopril for Discovery of Potent NDM-1 Inhibitors. Bioorg. Med. Chem 2021, 29, 115902. [DOI] [PubMed] [Google Scholar]
- 717.Toney JH; Hammond GG; Fitzgerald PM; Sharma N; Balkovec JM; Rouen GP; Olson SH; Hammond ML; Greenlee ML; Gao YD Succinic Acids as Potent Inhibitors of Plasmid-Borne IMP-1 Metallo-Beta-Lactamase. J. Biol. Chem 2001, 276, 31913–31918. [DOI] [PubMed] [Google Scholar]
- 718.Moloughney JG; J DT; Toney JH Novel IMP-1 Metallo-Beta-Lactamase Inhibitors Can Reverse Meropenem Resistance in Escherichia coli Expressing IMP-1. FEMS Microbiol. Lett 2005, 243, 65–71. [DOI] [PubMed] [Google Scholar]
- 719.Hiraiwa Y; Saito J; Watanabe T; Yamada M; Morinaka A; Fukushima T; Kudo T X-Ray Crystallographic Analysis of IMP-1 Metallo-Beta-Lactamase Complexed with a 3-Aminophthalic Acid Derivative, Structure-Based Drug Design, and Synthesis of 3,6-Disubstituted Phthalic Acid Derivative Inhibitors. Bioorg. Med. Chem. Lett 2014, 24, 4891–4894. [DOI] [PubMed] [Google Scholar]
- 720.Hiraiwa Y; Morinaka A; Fukushima T; Kudo T Metallo-Beta-Lactamase Inhibitory Activity of 3-Alkyloxy and 3-Amino Phthalic Acid Derivatives and Their Combination Effect with Carbapenem. Bioorg. Med. Chem 2013, 21, 5841–5850. [DOI] [PubMed] [Google Scholar]
- 721.Hiraiwa Y; Morinaka A; Fukushima T; Kudo T Metallo-Beta-Lactamase Inhibitory Activity of Phthalic Acid Derivatives. Bioorg. Med. Chem. Lett 2009, 19, 5162–5165. [DOI] [PubMed] [Google Scholar]
- 722.Ishii Y; Eto M; Mano Y; Tateda K; Yamaguchi K In Vitro Potentiation of Carbapenems with Me1071, a Novel Metallo-Beta-Lactamase Inhibitor, against Metallo-Beta-Lactamase- Producing Pseudomonas aeruginosa Clinical Isolates. Antimicrob. Agents Chemother 2010, 54, 3625–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 723.Livermore DM; Mushtaq S; Morinaka A; Ida T; Maebashi K; Hope R Activity of Carbapenems with Me1071 (Disodium 2,3-Diethylmaleate) against Enterobacteriaceae and Acinetobacter Spp. With Carbapenemases, Including NDM Enzymes. J. Antimicrob. Chemother 2013, 68, 153–158. [DOI] [PubMed] [Google Scholar]
- 724.Yamada K; Yanagihara K; Kaku N; Harada Y; Migiyama Y; Nagaoka K; Morinaga Y; Nakamura S; Imamura Y; Miyazaki T, et al. In Vivo Efficacy of Biapenem with Me1071, a Novel Metallo-Beta-Lactamase (MBL) Inhibitor, in a Murine Model Mimicking Ventilator-Associated Pneumonia Caused by MBL-Producing Pseudomonas aeruginosa. Int. J. Antimicrob. Agents 2013, 42, 238–243. [DOI] [PubMed] [Google Scholar]
- 725.Feng L; Yang KW; Zhou LS; Xiao JM; Yang X; Zhai L; Zhang YL; Crowder MW N-Heterocyclic Dicarboxylic Acids: Broad-Spectrum Inhibitors of Metallo-Beta-Lactamases with Co-Antibacterial Effect against Antibiotic-Resistant Bacteria. Bioorg. Med. Chem. Lett 2012, 22, 5185–5189. [DOI] [PubMed] [Google Scholar]
- 726.Chen AY; Thomas CA; Thomas PW; Yang K; Cheng Z; Fast W; Crowder MW; Cohen SM Iminodiacetic Acid as a Novel Metal-Binding Pharmacophore for New Delhi Metallo-Beta-Lactamase Inhibitor Development. ChemMedChem 2020, 15, 1272–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 727.Jackson AC; Pinter TBJ; Talley DC; Baker-Agha A; Patel D; Smith PJ; Franz KJ Benzimidazole and Benzoxazole Zinc Chelators as Inhibitors of Metallo-Beta-Lactamase NDM-1. ChemMedChem 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 728.Maveyraud L; Pratt RF; Samama JP Crystal Structure of an Acylation Transition-State Analog of the TEM-1 Beta-Lactamase. Mechanistic Implications for Class A Beta-Lactamases. Biochemistry 1998, 37, 2622–2628. [DOI] [PubMed] [Google Scholar]
- 729.Chen CC; Rahil J; Pratt RF; Herzberg O Structure of a Phosphonate-Inhibited Beta-Lactamase. An Analog of the Tetrahedral Transition State/Intermediate of Beta-Lactam Hydrolysis. J. Mol. Biol 1993, 234, 165–178. [DOI] [PubMed] [Google Scholar]
- 730.Yang KW; Feng L; Yang SK; Aitha M; LaCuran AE; Oelschlaeger P; Crowder MW New Beta-Phospholactam as a Carbapenem Transition State Analog: Synthesis of a Broad-Spectrum Inhibitor of Metallo-Beta-Lactamases. Bioorg. Med. Chem. Lett 2013, 23, 5855–5859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 731.Skagseth S; Akhter S; Paulsen MH; Muhammad Z; Lauksund S; Samuelsen O; Leiros HS; Bayer A Metallo-Beta-Lactamase Inhibitors by Bioisosteric Replacement: Preparation, Activity and Binding. Eur. J. Med. Chem 2017, 135, 159–173. [DOI] [PubMed] [Google Scholar]
- 732.Hinchliffe P; Tanner CA; Krismanich AP; Labbe G; Goodfellow VJ; Marrone L; Desoky AY; Calvopina K; Whittle EE; Zeng F, et al. Structural and Kinetic Studies of the Potent Inhibition of Metallo-Beta-Lactamases by 6-Phosphonomethylpyridine-2- Carboxylates. Biochemistry 2018, 57, 1880–1892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 733.Pemberton OA; Jaishankar P; Akhtar A; Adams JL; Shaw LN; Renslo AR; Chen Y Heteroaryl Phosphonates as Noncovalent Inhibitors of Both Serine- and Metallocarbapenemases. J. Med. Chem 2019, 62, 8480–8496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 734.Dembitsky VM; Al Quntar AA; Srebnik M Natural and Synthetic Small Boron-Containing Molecules as Potential Inhibitors of Bacterial and Fungal Quorum Sensing. Chem. Rev 2011, 111, 209–237. [DOI] [PubMed] [Google Scholar]
- 735.Diaz DB; Yudin AK The Versatility of Boron in Biological Target Engagement. Nat Chem 2017, 9, 731–742. [DOI] [PubMed] [Google Scholar]
- 736.Liu B; Trout REL; Chu GH; McGarry D; Jackson RW; Hamrick JC; Daigle DM; Cusick SM; Pozzi C; De Luca F, et al. Discovery of Taniborbactam (VNRX-5133): A Broad-Spectrum Serine- and Metallo-Beta-Lactamase Inhibitor for Carbapenem-Resistant Bacterial Infections. J. Med. Chem 2020, 63, 2789–2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 737.Krajnc A; Brem J; Hinchliffe P; Calvopina K; Panduwawala TD; Lang PA; Kamps J; Tyrrell JM; Widlake E; Saward BG, et al. Bicyclic Boronate VNRX-5133 Inhibits Metallo- and Serine-Beta-Lactamases. J. Med. Chem 2019, 62, 8544–8556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 738.Hecker SJ; Reddy KR; Lomovskaya O; Griffith DC; Rubio-Aparicio D; Nelson K; Tsivkovski R; Sun D; Sabet M; Tarazi Z, et al. Discovery of Cyclic Boronic Acid QPX7728, an Ultrabroad-Spectrum Inhibitor of Serine and Metallo-Beta-Lactamases. J. Med. Chem 2020, 63, 7491–7507. [DOI] [PubMed] [Google Scholar]
- 739.Hamrick JC; Docquier JD; Uehara T; Myers CL; Six DA; Chatwin CL; John KJ; Vernacchio SF; Cusick SM; Trout REL, et al. VNRX-5133 (Taniborbactam), a Broad-Spectrum Inhibitor of Serine- and Metallo-Beta-Lactamases, Restores Activity of Cefepime in Enterobacterales and Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 740.Tsivkovski R; Totrov M; Lomovskaya O Biochemical Characterization of QPX7728, a New Ultrabroad-Spectrum Beta-Lactamase Inhibitor of Serine and Metallo-Beta-Lactamases. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 741.Richter MF; Drown BS; Riley AP; Garcia A; Shirai T; Svec RL; Hergenrother PJ Predictive Compound Accumulation Rules Yield a Broad-Spectrum Antibiotic. Nature 2017, 545, 299–304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 742.Lomovskaya O; Tsivkovski R; Nelson K; Rubio-Aparicio D; Sun D; Totrov M; Dudley MN Spectrum of Beta-Lactamase Inhibition by the Cyclic Boronate QPX7728, an Ultrabroad-Spectrum Beta-Lactamase Inhibitor of Serine and Metallo-Beta-Lactamases: Enhancement of Activity of Multiple Antibiotics against Isogenic Strains Expressing Single Beta-Lactamases. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 743.Lomovskaya O; Nelson K; Rubio-Aparicio D; Tsivkovski R; Sun D; Dudley MN Impact of Intrinsic Resistance Mechanisms on Potency of QPX7728, a New Ultrabroad-Spectrum Beta-Lactamase Inhibitor of Serine and Metallo-Beta-Lactamases in Enterobacteriaceae, Pseudomonas aeruginosa, and Acinetobacter baumannii. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 744.Nelson K; Rubio-Aparicio D; Sun D; Dudley M; Lomovskaya O In Vitro Activity of the Ultrabroad-Spectrum-Beta-Lactamase Inhibitor QPX7728 against Carbapenem-Resistant Enterobacterales with Varying Intrinsic and Acquired Resistance Mechanisms. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 745.Sabet M; Tarazi Z; Griffith DC In Vivo Activity of QPX7728, an Ultrabroad-Spectrum Beta-Lactamase Inhibitor, in Combination with Beta-Lactams against Carbapenem-Resistant Klebsiella pneumoniae. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 746.Cahill ST; Cain R; Wang DY; Lohans CT; Wareham DW; Oswin HP; Mohammed J; Spencer J; Fishwick CW; McDonough MA, et al. Cyclic Boronates Inhibit All Classes of Beta-Lactamases. Antimicrob. Agents Chemother 2017, 61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 747.Parkova A; Lucic A; Krajnc A; Brem J; Calvopina K; Langley GW; McDonough MA; Trapencieris P; Schofield CJ Broad Spectrum Beta-Lactamase Inhibition by a Thioether Substituted Bicyclic Boronate. ACS Infect Dis 2020, 6, 1398–1404. [DOI] [PubMed] [Google Scholar]
- 748.Olsen L; Jost S; Adolph HW; Pettersson I; Hemmingsen L; Jorgensen FS New Leads of Metallo-Beta-Lactamase Inhibitors from Structure-Based Pharmacophore Design. Bioorg. Med. Chem 2006, 14, 2627–2635. [DOI] [PubMed] [Google Scholar]
- 749.Shi C; Chen J; Xiao B; Kang X; Lao X; Zheng H Discovery of NDM-1 Inhibitors from Natural Products. J Glob Antimicrob Resist 2019, 18, 80–87. [DOI] [PubMed] [Google Scholar]
- 750.Kang JS; Zhang AL; Faheem M; Zhang CJ; Ai N; Buynak JD; Welsh WJ; Oelschlaeger P Virtual Screening and Experimental Testing of B1 Metallo-Beta-Lactamase Inhibitors. J Chem Inf Model 2018, 58, 1902–1914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 751.Brindisi M; Brogi S; Giovani S; Gemma S; Lamponi S; De Luca F; Novellino E; Campiani G; Docquier JD; Butini S Targeting Clinically-Relevant Metallo-Beta-Lactamases: From High-Throughput Docking to Broad-Spectrum Inhibitors. J. Enzyme Inhib. Med. Chem 2016, 31, 98–109. [DOI] [PubMed] [Google Scholar]
- 752.Spyrakis F; Celenza G; Marcoccia F; Santucci M; Cross S; Bellio P; Cendron L; Perilli M; Tondi D Structure-Based Virtual Screening for the Discovery of Novel Inhibitors of New Delhi Metallo-Beta-Lactamase-1. ACS Med Chem Lett 2018, 9, 45–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 753.Wang X; Lu M; Shi Y; Ou Y; Cheng X Discovery of Novel New Delhi Metallo-Beta-Lactamases-1 Inhibitors by Multistep Virtual Screening. PLoS One 2015, 10, e0118290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 754.Yu ZJ; Liu S; Zhou S; Li H; Yang F; Yang LL; Wu Y; Guo L; Li GB Virtual Target Screening Reveals Rosmarinic Acid and Salvianolic Acid a Inhibiting Metallo- and Serine-Beta-Lactamases. Bioorg. Med. Chem. Lett 2018, 28, 1037–1042. [DOI] [PubMed] [Google Scholar]
- 755.Wang X; Yang Y; Gao Y; Niu X Discovery of the Novel Inhibitor against New Delhi Metallo-Beta-Lactamase Based on Virtual Screening and Molecular Modelling. Int J Mol Sci 2020, 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 756.Gilpin ML; Fulston M; Payne D; Cramp R; Hood I Isolation and Structure Determination of Two Novel Phenazines from a Streptomyces with Inhibitory Activity against Metallo-Enzymes, Including Metallo-Beta-Lactamase. J Antibiot (Tokyo) 1995, 48, 1081–1085. [DOI] [PubMed] [Google Scholar]
- 757.Payne DJ; Hueso-Rodriguez JA; Boyd H; Concha NO; Janson CA; Gilpin M; Bateson JH; Cheever C; Niconovich NL; Pearson S, et al. Identification of a Series of Tricyclic Natural Products as Potent Broad-Spectrum Inhibitors of Metallo-Beta-Lactamases. Antimicrob. Agents Chemother 2002, 46, 1880–1886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 758.Denny BJ; Lambert PA; West PW The Flavonoid Galangin Inhibits the L1 Metallo-Beta-Lactamase from Stenotrophomonas maltophilia. FEMS Microbiol. Lett 2002, 208, 21–24. [DOI] [PubMed] [Google Scholar]
- 759.King AM; Reid-Yu SA; Wang W; King DT; De Pascale G; Strynadka NC; Walsh TR; Coombes BK; Wright GD Aspergillomarasmine a Overcomes Metallo-Beta-Lactamase Antibiotic Resistance. Nature 2014, 510, 503–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 760.Toney JH; Fitzgerald PM; Grover-Sharma N; Olson SH; May WJ; Sundelof JG; Vanderwall DE; Cleary KA; Grant SK; Wu JK, et al. Antibiotic Sensitization Using Biphenyl Tetrazoles as Potent Inhibitors of Bacteroides fragilis Metallo-Beta-Lactamase. Chem. Biol 1998, 5, 185–196. [DOI] [PubMed] [Google Scholar]
- 761.Toney JH; Cleary KA; Hammond GG; Yuan X; May WJ; Hutchins SM; Ashton WT; Vanderwall DE Structure-Activity Relationships of Biphenyl Tetrazoles as Metallo-Beta-Lactamase Inhibitors. Bioorg. Med. Chem. Lett 1999, 9, 2741–2746. [DOI] [PubMed] [Google Scholar]
- 762.Lienard BM; Selevsek N; Oldham NJ; Schofield CJ Combined Mass Spectrometry and Dynamic Chemistry Approach to Identify Metalloenzyme Inhibitors. ChemMedChem 2007, 2, 175–179. [DOI] [PubMed] [Google Scholar]
- 763.Lienard BM; Huting R; Lassaux P; Galleni M; Frere JM; Schofield CJ Dynamic Combinatorial Mass Spectrometry Leads to Metallo-Beta-Lactamase Inhibitors. J. Med. Chem 2008, 51, 684–688. [DOI] [PubMed] [Google Scholar]
- 764.Rydzik AM; Brem J; van Berkel SS; Pfeffer I; Makena A; Claridge TD; Schofield CJ Monitoring Conformational Changes in the NDM-1 Metallo-Beta-Lactamase by 19F NMR Spectroscopy. Angew. Chem. Int. Ed. Engl 2014, 53, 3129–3133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 765.Li GB; Abboud MI; Brem J; Someya H; Lohans CT; Yang SY; Spencer J; Wareham DW; McDonough MA; Schofield CJ NMR-Filtered Virtual Screening Leads to Non-Metal Chelating Metallo-Beta-Lactamase Inhibitors. Chem Sci 2017, 8, 928–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 766.Ma J; McLeod S; MacCormack K; Sriram S; Gao N; Breeze AL; Hu J Real-Time Monitoring of New Delhi Metallo-Beta-Lactamase Activity in Living Bacterial Cells by 1H NMR Spectroscopy. Angew. Chem. Int. Ed. Engl 2014, 53, 2130–2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 767.Brem J; van Berkel SS; Aik W; Rydzik AM; Avison MB; Pettinati I; Umland KD; Kawamura A; Spencer J; Claridge TD, et al. Rhodanine Hydrolysis Leads to Potent Thioenolate Mediated Metallo-Beta-Lactamase Inhibition. Nat Chem 2014, 6, 1084–1090. [DOI] [PubMed] [Google Scholar]
- 768.Grant EB; Guiadeen D; Baum EZ; Foleno BD; Jin H; Montenegro DA; Nelson EA; Bush K; Hlasta DJ The Synthesis and SAR of Rhodanines as Novel Class C Beta-Lactamase Inhibitors. Bioorg. Med. Chem. Lett 2000, 10, 2179–2182. [DOI] [PubMed] [Google Scholar]
- 769.Zervosen A; Lu WP; Chen Z; White RE; Demuth TP Jr.; Frere JM Interactions between Penicillin-Binding Proteins (PBPs) and Two Novel Classes of PBP Inhibitors, Arylalkylidene Rhodanines and Arylalkylidene Iminothiazolidin-4-Ones. Antimicrob. Agents Chemother 2004, 48, 961–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 770.Spicer T; Minond D; Enogieru I; Saldanha SA; Allais C; Liu Q; Mercer BA; Roush WR; Hodder P Ml302, a Novel Beta-Lactamase (Bla) Inhibitor. In Probe Reports from the Nih Molecular Libraries Program, Bethesda (MD), 2010. [PubMed] [Google Scholar]
- 771.Zhang D; Markoulides MS; Stepanovs D; Rydzik AM; El-Hussein A; Bon C; Kamps J; Umland KD; Collins PM; Cahill ST, et al. Structure Activity Relationship Studies on Rhodanines and Derived Enethiol Inhibitors of Metallo-Beta-Lactamases. Bioorg. Med. Chem 2018, 26, 2928–2936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 772.Xiang Y; Chen C; Wang WM; Xu LW; Yang KW; Oelschlaeger P; He Y Rhodanine as a Potent Scaffold for the Development of Broad-Spectrum Metallo-Beta-Lactamase Inhibitors. ACS Med Chem Lett 2018, 9, 359–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 773.Nichols DA; Renslo AR; Chen Y Fragment-Based Inhibitor Discovery against Beta-Lactamase. Future Med Chem 2014, 6, 413–427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 774.Christopeit T; Carlsen TJ; Helland R; Leiros HK Discovery of Novel Inhibitor Scaffolds against the Metallo-Beta-Lactamase VIM-2 by Surface Plasmon Resonance (SPR) Based Fragment Screening. J. Med. Chem 2015, 58, 8671–8682. [DOI] [PubMed] [Google Scholar]
- 775.Christopeit T; Leiros HK Fragment-Based Discovery of Inhibitor Scaffolds Targeting the Metallo-Beta-Lactamases NDM-1 and VIM-2. Bioorg. Med. Chem. Lett 2016, 26, 1973–1977. [DOI] [PubMed] [Google Scholar]
- 776.Christopeit T; Albert A; Leiros HS Discovery of a Novel Covalent Non-Beta-Lactam Inhibitor of the Metallo-Beta-Lactamase NDM-1. Bioorg. Med. Chem 2016, 24, 2947–2953. [DOI] [PubMed] [Google Scholar]
- 777.Klingler FM; Moser D; Buttner D; Wichelhaus TA; Lohr F; Dotsch V; Proschak E Probing Metallo-Beta-Lactamases with Molecular Fragments Identified by Consensus Docking. Bioorg. Med. Chem. Lett 2015, 25, 5243–5246. [DOI] [PubMed] [Google Scholar]
- 778.Cain R; Brem J; Zollman D; McDonough MA; Johnson RM; Spencer J; Makena A; Abboud MI; Cahill S; Lee SY, et al. In Silico Fragment-Based Design Identifies Subfamily B1 Metallo-Beta-Lactamase Inhibitors. J. Med. Chem 2018, 61, 1255–1260. [DOI] [PubMed] [Google Scholar]
- 779.Vella P; Hussein WM; Leung EW; Clayton D; Ollis DL; Mitic N; Schenk G; McGeary RP The Identification of New Metallo-Beta-Lactamase Inhibitor Leads from Fragment-Based Screening. Bioorg. Med. Chem. Lett 2011, 21, 3282–3285. [DOI] [PubMed] [Google Scholar]
- 780.Jiang L; Yuan C; Huang M A General Strategy to Inhibit Serine Protease by Targeting Its Autolysis Loop. FASEB J. 2021, 35, e21259. [DOI] [PubMed] [Google Scholar]
- 781.Leroux AE; Biondi RM Renaissance of Allostery to Disrupt Protein Kinase Interactions. Trends Biochem. Sci 2020, 45, 27–41. [DOI] [PubMed] [Google Scholar]
- 782.Lu X; Smaill JB; Ding K New Promise and Opportunities for Allosteric Kinase Inhibitors. Angew. Chem. Int. Ed. Engl 2020, 59, 13764–13776. [DOI] [PubMed] [Google Scholar]
- 783.Sohier JS; Laurent C; Chevigne A; Pardon E; Srinivasan V; Wernery U; Lassaux P; Steyaert J; Galleni M Allosteric Inhibition of VIM Metallo-Beta-Lactamases by a Camelid Nanobody. Biochem. J 2013, 450, 477–486. [DOI] [PubMed] [Google Scholar]
- 784.Kim SK; Sims CL; Wozniak SE; Drude SH; Whitson D; Shaw RW Antibiotic Resistance in Bacteria: Novel Metalloenzyme Inhibitors. Chem Biol Drug Des 2009, 74, 343–348. [DOI] [PubMed] [Google Scholar]
- 785.Khan NH; Bui AA; Xiao Y; Sutton RB; Shaw RW; Wylie BJ; Latham MP A DNA Aptamer Reveals an Allosteric Site for Inhibition in Metallo-Beta-Lactamases. PLoS One 2019, 14, e0214440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 786.Rotondo CM; Marrone L; Goodfellow VJ; Ghavami A; Labbe G; Spencer J; Dmitrienko GI; Siemann S Arginine-Containing Peptides as Potent Inhibitors of VIM-2 Metallo-Beta-Lactamase. Biochim. Biophys. Acta 2015, 1850, 2228–2238. [DOI] [PubMed] [Google Scholar]
- 787.Ouyang X; Chang YN; Yang KW; Wang WM; Bai JJ; Wang JW; Zhang YJ; Wang SY; Xie BB; Wang LL A DNA Nanoribbon as a Potent Inhibitor of Metallo-Beta-Lactamases. Chem Commun (Camb) 2017, 53, 8878–8881. [DOI] [PubMed] [Google Scholar]
- 788.Huang PJ; Pautler R; Shanmugaraj J; Labbe G; Liu J Inhibiting the VIM-2 Metallo-Beta-Lactamase by Graphene Oxide and Carbon Nanotubes. ACS Appl Mater Interfaces 2015, 7, 9898–9903. [DOI] [PubMed] [Google Scholar]
- 789.Tummino PJ; Harvey PJ; McQuade T; Domagala J; Gogliotti R; Sanchez J; Song Y; Hupe D The Human Immunodeficiency Virus Type 1 (HIV-1) Nucleocapsid Protein Zinc Ejection Activity of Disulfide Benzamides and Benzisothiazolones: Correlation with Anti-Hiv and Virucidal Activities. Antimicrob. Agents Chemother 1997, 41, 394–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 790.Maret W Oxidative Metal Release from Metallothionein Via Zinc-Thiol/Disulfide Interchange. Proc Natl Acad Sci U S A 1994, 91, 237–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 791.Payne DJ; Bateson JH; Gasson BC; Proctor D; Khushi T; Farmer TH; Tolson DA; Bell D; Skett PW; Marshall AC, et al. Inhibition of Metallo-Beta-Lactamases by a Series of Mercaptoacetic Acid Thiol Ester Derivatives. Antimicrob. Agents Chemother 1997, 41, 135–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 792.Boerzel H; Koeckert M; Bu W; Spingler B; Lippard SJ Zinc-Bound Thiolate-Disulfide Exchange: A Strategy for Inhibiting Metallo-Beta-Lactamases. Inorg. Chem 2003, 42, 1604–1615. [DOI] [PubMed] [Google Scholar]
- 793.Badarau A; Llinas A; Laws AP; Damblon C; Page MI Inhibitors of Metallo-Beta-Lactamase Generated from Beta-Lactam Antibiotics. Biochemistry 2005, 44, 8578–8589. [DOI] [PubMed] [Google Scholar]
- 794.Chen C; Yang KW; Wu LY; Li JQ; Sun LY Disulfiram as a Potent Metallo-Beta-Lactamase Inhibitor with Dual Functional Mechanisms. Chem Commun (Camb) 2020, 56, 2755–2758. [DOI] [PubMed] [Google Scholar]
- 795.Chiou J; Wan S; Chan KF; So PK; He D; Chan EW; Chan TH; Wong KY; Tao J; Chen S Ebselen as a Potent Covalent Inhibitor of New Delhi Metallo-Beta-Lactamase (NDM-1). Chem Commun (Camb) 2015, 51, 9543–9546. [DOI] [PubMed] [Google Scholar]
- 796.Chen C; Xiang Y; Yang KW; Zhang Y; Wang WM; Su JP; Ge Y; Liu Y A Protein Structure-Guided Covalent Scaffold Selectively Targets the B1 and B2 Subclass Metallo-Beta-Lactamases. Chem Commun (Camb) 2018, 54, 4802–4805. [DOI] [PubMed] [Google Scholar]
- 797.Su J; Liu J; Chen C; Zhang Y; Yang K Ebsulfur as a Potent Scaffold for Inhibition and Labelling of New Delhi Metallo-Beta-Lactamase-1 in Vitro and in Vivo. Bioorg. Chem 2019, 84, 192–201. [DOI] [PubMed] [Google Scholar]
- 798.Chen C; Liu Y; Zhang YJ; Ge Y; Lei JE; Yang KW The Assemblage of Covalent and Metal Binding Dual Functional Scaffold for Cross-Class Metallo-Beta-Lactamases Inhibition. Future Med Chem 2019, 11, 2381–2394. [DOI] [PubMed] [Google Scholar]
- 799.Jin WB; Xu C; Cheung Q; Gao W; Zeng P; Liu J; Chan EWC; Leung YC; Chan TH; Wong KY, et al. Bioisosteric Investigation of Ebselen: Synthesis and in Vitro Characterization of 1,2-Benzisothiazol-3(2H)-One Derivatives as Potent New Delhi Metallo-Beta-Lactamase Inhibitors. Bioorg. Chem 2020, 100, 103873. [DOI] [PubMed] [Google Scholar]
- 800.Kurosaki H; Yamaguchi Y; Higashi T; Soga K; Matsueda S; Yumoto H; Misumi S; Yamagata Y; Arakawa Y; Goto M Irreversible Inhibition of Metallo-Beta-Lactamase (IMP-1) by 3-(3-Mercaptopropionylsulfanyl)Propionic Acid Pentafluorophenyl Ester. Angew. Chem. Int. Ed. Engl 2005, 44, 3861–3864. [DOI] [PubMed] [Google Scholar]
- 801.Thomas PW; Cammarata M; Brodbelt JS; Monzingo AF; Pratt RF; Fast W A Lysine-Targeted Affinity Label for Serine-Beta-Lactamase Also Covalently Modifies New Delhi Metallo-Beta-Lactamase-1 (NDM-1). Biochemistry 2019, 58, 2834–2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 802.Lambert RJ; Hanlon GW; Denyer SP The Synergistic Effect of EDTA/Antimicrobial Combinations on Pseudomonas aeruginosa. J. Appl. Microbiol 2004, 96, 244–253. [DOI] [PubMed] [Google Scholar]
- 803.Amaral KF; Rogero MM; Fock RA; Borelli P; Gavini G Cytotoxicity Analysis of EDTA and Citric Acid Applied on Murine Resident Macrophages Culture. Int Endod J 2007, 40, 338–343. [DOI] [PubMed] [Google Scholar]
- 804.Azumah R; Dutta J; Somboro AM; Ramtahal M; Chonco L; Parboosing R; Bester LA; Kruger HG; Naicker T; Essack SY, et al. In Vitro Evaluation of Metal Chelators as Potential Metallo-Beta -Lactamase Inhibitors. J. Appl. Microbiol 2016, 120, 860–867. [DOI] [PubMed] [Google Scholar]
- 805.Aoki N; Ishii Y; Tateda K; Saga T; Kimura S; Kikuchi Y; Kobayashi T; Tanabe Y; Tsukada H; Gejyo F, et al. Efficacy of Calcium-EDTA as an Inhibitor for Metallo-Beta-Lactamase in a Mouse Model of Pseudomonas aeruginosa Pneumonia. Antimicrob. Agents Chemother 2010, 54, 4582–4588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 806.Yoshizumi A; Ishii Y; Livermore DM; Woodford N; Kimura S; Saga T; Harada S; Yamaguchi K; Tateda K Efficacies of Calcium-EDTA in Combination with Imipenem in a Murine Model of Sepsis Caused by Escherichia coli with NDM-1 Beta-Lactamase. J. Infect. Chemother 2013, 19, 992–995. [DOI] [PubMed] [Google Scholar]
- 807.Kimura S; Ishii Y; Yamaguchi K Evaluation of Dipicolinic Acid for Detection of IMP- or VIM-Type Metallo-Beta-Lactamase-Producing Pseudomonas aeruginosa Clinical Isolates. Diagn. Microbiol. Infect. Dis 2005, 53, 241–244. [DOI] [PubMed] [Google Scholar]
- 808.Horsfall LE; Garau G; Lienard BM; Dideberg O; Schofield CJ; Frere JM; Galleni M Competitive Inhibitors of the CphA Metallo-Beta-Lactamase from Aeromonas hydrophila. Antimicrob. Agents Chemother 2007, 51, 2136–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 809.Chen AY; Thomas PW; Stewart AC; Bergstrom A; Cheng Z; Miller C; Bethel CR; Marshall SH; Credille CV; Riley CL, et al. Dipicolinic Acid Derivatives as Inhibitors of New Delhi Metallo-Beta-Lactamase-1. J. Med. Chem 2017, 60, 7267–7283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 810.Chen AY; Thomas PW; Cheng Z; Xu NY; Tierney DL; Crowder MW; Fast W; Cohen SM Investigation of Dipicolinic Acid Isosteres for the Inhibition of Metallo-Beta-Lactamases. ChemMedChem 2019, 14, 1271–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 811.Thomas CA; Cheng Z; Yang K; Hellwarth E; Yurkiewicz CJ; Baxter FM; Fullington SA; Klinsky SA; Otto JL; Chen AY, et al. Probing the Mechanisms of Inhibition for Various Inhibitors of Metallo-Beta-Lactamases VIM-2 and NDM-1. J. Inorg. Biochem 2020, 210, 111123. [DOI] [PubMed] [Google Scholar]
- 812.Sosibo SC; Somboro AM; Amoako DG; Osei Sekyere J; Bester LA; Ngila JC; Sun DD; Kumalo HM Impact of Pyridyl Moieties on the Inhibitory Properties of Prominent Acyclic Metal Chelators against Metallo-Beta-Lactamase-Producing Enterobacteriaceae: Investigating the Molecular Basis of Acyclic Metal Chelators’ Activity. Microb. Drug Resist 2019, 25, 439–449. [DOI] [PubMed] [Google Scholar]
- 813.Somboro AM; Tiwari D; Bester LA; Parboosing R; Chonco L; Kruger HG; Arvidsson PI; Govender T; Naicker T; Essack SY Nota: A Potent Metallo-Beta-Lactamase Inhibitor. J. Antimicrob. Chemother 2015, 70, 1594–1596. [DOI] [PubMed] [Google Scholar]
- 814.Falconer SB; Reid-Yu SA; King AM; Gehrke SS; Wang W; Britten JF; Coombes BK; Wright GD; Brown ED Zinc Chelation by a Small-Molecule Adjuvant Potentiates Meropenem Activity in Vivo against NDM-1-Producing Klebsiella pneumoniae. ACS Infect Dis 2015, 1, 533–543. [DOI] [PubMed] [Google Scholar]
- 815.Haenni AL; Robert M; Vetter W; Roux L; Barbier M; Lederer E [Chemical Structure of Aspergellomarasmines A and B]. Helv. Chim. Acta 1965, 48, 729–750. [DOI] [PubMed] [Google Scholar]
- 816.Mikami Y; Suzuki T Novel Microbial Inhibitors of Angiotensin-Converting Enzyme, Aspergillomarasmines A and B. Agric. Biol. Chem 1983, 47, 2693–2695. [Google Scholar]
- 817.Rotondo CM; Sychantha D; Koteva K; Wright GD Suppression of Beta-Lactam Resistance by Aspergillomarasmine A Is Influenced by Both the Metallo-Beta-Lactamase Target and the Antibiotic Partner. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 818.Liao D; Yang S; Wang J; Zhang J; Hong B; Wu F; Lei X Total Synthesis and Structural Reassignment of Aspergillomarasmine A. Angew. Chem. Int. Ed. Engl 2016, 55, 4291–4295. [DOI] [PubMed] [Google Scholar]
- 819.Koteva K; King AM; Capretta A; Wright GD Total Synthesis and Activity of the Metallo-Beta-Lactamase Inhibitor Aspergillomarasmine A. Angew. Chem. Int. Ed. Engl 2016, 55, 2210–2212. [DOI] [PubMed] [Google Scholar]
- 820.Albu SA; Koteva K; King AM; Al-Karmi S; Wright GD; Capretta A Total Synthesis of Aspergillomarasmine A and Related Compounds: A Sulfamidate Approach Enables Exploration of Structure-Activity Relationships. Angew. Chem. Int. Ed. Engl 2016, 55, 13259–13262. [DOI] [PubMed] [Google Scholar]
- 821.Zhang J; Wang S; Wei Q; Guo Q; Bai Y; Yang S; Song F; Zhang L; Lei X Synthesis and Biological Evaluation of Aspergillomarasmine A Derivatives as Novel NDM-1 Inhibitor to Overcome Antibiotics Resistance. Bioorg. Med. Chem 2017, 25, 5133–5141. [DOI] [PubMed] [Google Scholar]
- 822.Bergstrom A; Katko A; Adkins Z; Hill J; Cheng Z; Burnett M; Yang H; Aitha M; Mehaffey MR; Brodbelt JS, et al. Probing the Interaction of Aspergillomarasmine A with Metallo-Beta-Lactamases NDM-1, VIM-2, and Imp-7. ACS Infect Dis 2018, 4, 135–145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 823.Li H; Wang R; Sun H Systems Approaches for Unveiling the Mechanism of Action of Bismuth Drugs: New Medicinal Applications Beyond Helicobacter pylori Infection. Acc. Chem. Res 2019, 52, 216–227. [DOI] [PubMed] [Google Scholar]
- 824.Wang H; Zhou Y; Xu X; Li H; Sun H Metalloproteomics in Conjunction with Other Omics for Uncovering the Mechanism of Action of Metallodrugs: Mechanism-Driven New Therapy Development. Curr. Opin. Chem. Biol 2020, 55, 171–179. [DOI] [PubMed] [Google Scholar]
- 825.Wang R; Lai TP; Gao P; Zhang H; Ho PL; Woo PC; Ma G; Kao RY; Li H; Sun H Bismuth Antimicrobial Drugs Serve as Broad-Spectrum Metallo-Beta-Lactamase Inhibitors. Nat Commun 2018, 9, 439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 826.Sun H; Zhang Q; Wang R; Wang H; Wong YT; Wang M; Hao Q; Yan A; Kao RY; Ho PL, et al. Resensitizing Carbapenem- and Colistin-Resistant Bacteria to Antibiotics Using Auranofin. Nat Commun 2020, 11, 5263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 827.Kumar NG; Kumar G; Mallick S; Ghosh SK; Pramanick P; Ghosh AS Bio-Surfactin Stabilised Silver Nanoparticles Exert Inhibitory Effect over New-Delhi Metallo-Beta-Lactamases (NDMs) and the Cells Harbouring NDMs. FEMS Microbiol. Lett 2019, 366. [DOI] [PubMed] [Google Scholar]
- 828.Chen C; Yang K Ruthenium Complexes as Prospective Inhibitors of Metallo-Beta-Lactamases to Reverse Carbapenem Resistance. Dalton Trans 2020, 49, 14099–14105. [DOI] [PubMed] [Google Scholar]
- 829.Chen C; Sun LY; Gao H; Kang PW; Li JQ; Zhen JB; Yang KW Identification of Cisplatin and Palladium(II) Complexes as Potent Metallo-Beta-Lactamase Inhibitors for Targeting Carbapenem-Resistant Enterobacteriaceae. ACS Infect Dis 2020, 6, 975–985. [DOI] [PubMed] [Google Scholar]
- 830.Pradel N; Delmas J; Wu LF; Santini CL; Bonnet R Sec- and Tat-Dependent Translocation of Beta-Lactamases across the Escherichia coli Inner Membrane. Antimicrob. Agents Chemother 2009, 53, 242–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 831.Denks K; Vogt A; Sachelaru I; Petriman NA; Kudva R; Koch HG The Sec Translocon Mediated Protein Transport in Prokaryotes and Eukaryotes. Mol. Membr. Biol 2014, 31, 58–84. [DOI] [PubMed] [Google Scholar]
- 832.du Plessis DJ; Nouwen N; Driessen AJ The Sec Translocase. Biochim. Biophys. Acta 2011, 1808, 851–865. [DOI] [PubMed] [Google Scholar]
- 833.Frain KM; Robinson C; van Dijl JM Transport of Folded Proteins by the Tat System. Protein J 2019, 38, 377–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 834.McDonough JA; Hacker KE; Flores AR; Pavelka MS Jr.; Braunstein M The Twin-Arginine Translocation Pathway of Mycobacterium smegmatis Is Functional and Required for the Export of Mycobacterial Beta-Lactamases. J. Bacteriol 2005, 187, 7667–7679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 835.Rholl DA; Papp-Wallace KM; Tomaras AP; Vasil ML; Bonomo RA; Schweizer HP Molecular Investigations of PenA-Mediated Beta-Lactam Resistance in Burkholderia pseudomallei. Front Microbiol 2011, 2, 139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 836.Balder R; Shaffer TL; Lafontaine ER Moraxella catarrhalis Uses a Twin-Arginine Translocation System to Secrete the Beta-Lactamase BRO-2. BMC Microbiol 2013, 13, 140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 837.Schriefer EM; Hoffmann-Thoms S; Schmid FX; Schmid A; Heesemann J Yersinia enterocolitica and Photorhabdus asymbiotica Beta-Lactamases BlaA Are Exported by the Twin-Arginine Translocation Pathway. Int. J. Med. Microbiol 2013, 303, 16–24. [DOI] [PubMed] [Google Scholar]
- 838.Moran-Barrio J; Limansky AS; Viale AM Secretion of GOB Metallo-Beta-Lactamase in Escherichia coli Depends Strictly on the Cooperation between the Cytoplasmic DnaK Chaperone System and the Sec Machinery: Completion of Folding and Zn(II) Ion Acquisition Occur in the Bacterial Periplasm. Antimicrob. Agents Chemother 2009, 53, 2908–2917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 839.Brambilla L; Moran-Barrio J; Viale AM Low-Molecular-Mass Penicillin Binding Protein 6b (DacD) Is Required for Efficient GOB-18 Metallo-Beta-Lactamase Biogenesis in Salmonella enterica and Escherichia coli. Antimicrob. Agents Chemother 2014, 58, 205–211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 840.Tottey S; Waldron KJ; Firbank SJ; Reale B; Bessant C; Sato K; Cheek TR; Gray J; Banfield MJ; Dennison C, et al. Protein-Folding Location Can Regulate Manganese-Binding Versus Copper- or Zinc-Binding. Nature 2008, 455, 1138–1142. [DOI] [PubMed] [Google Scholar]
- 841.Hu Z; Gunasekera TS; Spadafora L; Bennett B; Crowder MW Metal Content of Metallo-Beta-Lactamase L1 Is Determined by the Bioavailability of Metal Ions. Biochemistry 2008, 47, 7947–7953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 842.Carruthers TJ; Carr PD; Loh CT; Jackson CJ; Otting G Iron(III) Located in the Dinuclear Metallo-Beta-Lactamase IMP-1 by Pseudocontact Shifts. Angew. Chem. Int. Ed. Engl 2014, 53, 14269–14272. [DOI] [PubMed] [Google Scholar]
- 843.Almagro Armenteros JJ; Tsirigos KD; Sonderby CK; Petersen TN; Winther O; Brunak S; von Heijne G; Nielsen H SignalP 5.0 Improves Signal Peptide Predictions Using Deep Neural Networks. Nat. Biotechnol 2019, 37, 420–423. [DOI] [PubMed] [Google Scholar]
- 844.Okazaki A; Avison MB Induction of L1 and L2 Beta-Lactamase Production in Stenotrophomonas maltophilia Is Dependent on an AmpR-Type Regulator. Antimicrob. Agents Chemother 2008, 52, 1525–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 845.Juan C; Torrens G; Gonzalez-Nicolau M; Oliver A Diversity and Regulation of Intrinsic Beta-Lactamases from Non-Fermenting and Other Gram-Negative Opportunistic Pathogens. FEMS Microbiol. Rev 2017, 41, 781–815. [DOI] [PubMed] [Google Scholar]
- 846.Fisher JF; Mobashery S The Sentinel Role of Peptidoglycan Recycling in the Beta-Lactam Resistance of the Gram-Negative Enterobacteriaceae and Pseudomonas aeruginosa. Bioorg. Chem 2014, 56, 41–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 847.Zeng X; Lin J Beta-Lactamase Induction and Cell Wall Metabolism in Gram-Negative Bacteria. Front Microbiol 2013, 4, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 848.Vadlamani G; Thomas MD; Patel TR; Donald LJ; Reeve TM; Stetefeld J; Standing KG; Vocadlo DJ; Mark BL The Beta-Lactamase Gene Regulator AmpR Is a Tetramer That Recognizes and Binds the D-Ala-D-Ala Motif of Its Repressor UDP-N-Acetylmuramic Acid (MurNAc)-Pentapeptide. J. Biol. Chem 2015, 290, 2630–2643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 849.Huang YW; Lin CW; Hu RM; Lin YT; Chung TC; Yang TC AmpN-AmpG Operon Is Essential for Expression of L1 and L2 Beta-Lactamases in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother 2010, 54, 2583–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 850.Talfan A; Mounsey O; Charman M; Townsend E; Avison MB Involvement of Mutation in ampD I, mrcA, and at Least One Additional Gene in Beta-Lactamase Hyperproduction in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother 2013, 57, 5486–5491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 851.Lin CW; Lin HC; Huang YW; Chung TC; Yang TC Inactivation of mrcA Gene Derepresses the Basal-Level Expression of L1 and L2 Beta-Lactamases in Stenotrophomonas maltophilia. J. Antimicrob. Chemother 2011, 66, 2033–2037. [DOI] [PubMed] [Google Scholar]
- 852.Huang YW; Hu RM; Lin CW; Chung TC; Yang TC NagZ-Dependent and NagZ-Independent Mechanisms for Beta-Lactamase Expression in Stenotrophomonas maltophilia. Antimicrob. Agents Chemother 2012, 56, 1936–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 853.Tayler AE; Ayala JA; Niumsup P; Westphal K; Baker JA; Zhang L; Walsh TR; Wiedemann B; Bennett PM; Avison MB Induction of Beta-Lactamase Production in Aeromonas hydrophila Is Responsive to Beta-Lactam-Mediated Changes in Peptidoglycan Composition. Microbiology (Reading) 2010, 156, 2327–2335. [DOI] [PubMed] [Google Scholar]
- 854.Alksne LE; Rasmussen BA Expression of the AsbA1, OXA-12, and AsbM1 Beta-Lactamases in Aeromonas jandaei AER 14 Is Coordinated by a Two-Component Regulon. J. Bacteriol 1997, 179, 2006–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 855.Niumsup P; Simm AM; Nurmahomed K; Walsh TR; Bennett PM; Avison MB Genetic Linkage of the Penicillinase Gene, Amp, and BlrAB, Encoding the Regulator of Beta-Lactamase Expression in Aeromonas Spp. J. Antimicrob. Chemother 2003, 51, 1351–1358. [DOI] [PubMed] [Google Scholar]
- 856.Ross CL; Thomason KS; Koehler TM An Extracytoplasmic Function Sigma Factor Controls Beta-Lactamase Gene Expression in Bacillus anthracis and Other Bacillus cereus Group Species. J. Bacteriol 2009, 191, 6683–6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 857.Chen Y; Tenover FC; Koehler TM Beta-Lactamase Gene Expression in a Penicillin-Resistant Bacillus anthracis Strain. Antimicrob. Agents Chemother 2004, 48, 4873–4877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 858.Paget MS Bacterial Sigma Factors and Anti-Sigma Factors: Structure, Function and Distribution. Biomolecules 2015, 5, 1245–1265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 859.Ho TD; Nauta KM; Muh U; Ellermeier CD Activation of the Extracytoplasmic Function Sigma Factor Sigma(P) by Beta-Lactams in Bacillus thuringiensis Requires the Site-2 Protease RasP. mSphere 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 860.Segawa T; Sekizuka T; Suzuki S; Shibayama K; Matsui M; Kuroda M The Plasmid-Encoded Transcription Factor ArdK Contributes to the Repression of the IMP-6 Metallo-Beta-Lactamase Gene blaIMP-6, Leading to a Carbapenem-susceptible Phenotype in the blaIMP-6-Positive Escherichia coli Strain A56–1S. PLoS One 2018, 13, e0208976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 861.Liu W; Zou D; Wang X; Li X; Zhu L; Yin Z; Yang Z; Wei X; Han L; Wang Y, et al. Proteomic Analysis of Clinical Isolate of Stenotrophomonas maltophilia with blaNDM-1, blaL1 and blaL2 Beta-Lactamase Genes under Imipenem Treatment. J Proteome Res 2012, 11, 4024–4033. [DOI] [PubMed] [Google Scholar]
- 862.Narita SI; Tokuda H Bacterial Lipoproteins; Biogenesis, Sorting and Quality Control. Biochim. Biophys. Acta 2017, 1862, 1414–1423. [DOI] [PubMed] [Google Scholar]
- 863.Braun V; Hantke K Lipoproteins: Structure, Function, Biosynthesis. Subcell Biochem 2019, 92, 39–77. [DOI] [PubMed] [Google Scholar]
- 864.Buddelmeijer N The Molecular Mechanism of Bacterial Lipoprotein Modification--How, When and Why? FEMS Microbiol. Rev 2015, 39, 246–261. [DOI] [PubMed] [Google Scholar]
- 865.Grabowicz M Lipoproteins and Their Trafficking to the Outer Membrane. EcoSal Plus 2019, 8. [DOI] [PubMed] [Google Scholar]
- 866.Nakayama H; Kurokawa K; Lee BL Lipoproteins in Bacteria: Structures and Biosynthetic Pathways. FEBS J. 2012, 279, 4247–4268. [DOI] [PubMed] [Google Scholar]
- 867.Hutchings MI; Palmer T; Harrington DJ; Sutcliffe IC Lipoprotein Biogenesis in Gram-Positive Bacteria: Knowing When to Hold ‘Em, Knowing When to Fold ‘Em. Trends Microbiol 2009, 17, 13–21. [DOI] [PubMed] [Google Scholar]
- 868.Sutcliffe IC; Harrington DJ; Hutchings MI A Phylum Level Analysis Reveals Lipoprotein Biosynthesis to Be a Fundamental Property of Bacteria. Protein Cell 2012, 3, 163–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 869.Kovacs-Simon A; Titball RW; Michell SL Lipoproteins of Bacterial Pathogens. Infect. Immun 2011, 79, 548–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 870.Sutcliffe IC; Russell RR Lipoproteins of Gram-Positive Bacteria. J. Bacteriol 1995, 177, 1123–1128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 871.Braun V; Rehn K Chemical Characterization, Spatial Distribution and Function of a Lipoprotein (Murein-Lipoprotein) of the E. coli Cell Wall. The Specific Effect of Trypsin on the Membrane Structure. Eur. J. Biochem 1969, 10, 426–438. [DOI] [PubMed] [Google Scholar]
- 872.Hantke K; Braun V Covalent Binding of Lipid to Protein. Diglyceride and Amide-Linked Fatty Acid at the N-Terminal End of the Murein-Lipoprotein of the Escherichia coli Outer Membrane. Eur. J. Biochem 1973, 34, 284–296. [DOI] [PubMed] [Google Scholar]
- 873.Asmar AT; Collet JF Lpp, the Braun Lipoprotein, Turns 50-Major Achievements and Remaining Issues. FEMS Microbiol. Lett 2018, 365. [DOI] [PubMed] [Google Scholar]
- 874.Godlewska R; Wisniewska K; Pietras Z; Jagusztyn-Krynicka EK Peptidoglycan-Associated Lipoprotein (Pal) of Gram-Negative Bacteria: Function, Structure, Role in Pathogenesis and Potential Application in Immunoprophylaxis. FEMS Microbiol. Lett 2009, 298, 1–11. [DOI] [PubMed] [Google Scholar]
- 875.Ricci DP; Silhavy TJ The Bam Machine: A Molecular Cooper. Biochim. Biophys. Acta 2012, 1818, 1067–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 876.Qiao S; Luo Q; Zhao Y; Zhang XC; Huang Y Structural Basis for Lipopolysaccharide Insertion in the Bacterial Outer Membrane. Nature 2014, 511, 108–111. [DOI] [PubMed] [Google Scholar]
- 877.Lupoli TJ; Lebar MD; Markovski M; Bernhardt T; Kahne D; Walker S Lipoprotein Activators Stimulate Escherichia coli Penicillin-Binding Proteins by Different Mechanisms. J. Am. Chem. Soc 2014, 136, 52–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 878.May KL; Lehman KM; Mitchell AM; Grabowicz M A Stress Response Monitoring Lipoprotein Trafficking to the Outer Membrane. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 879.Kimkes TEP; Heinemann M How Bacteria Recognise and Respond to Surface Contact. FEMS Microbiol. Rev 2020, 44, 106–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 880.Babu MM; Priya ML; Selvan AT; Madera M; Gough J; Aravind L; Sankaran K A Database of Bacterial Lipoproteins (DOLOP) with Functional Assignments to Predicted Lipoproteins. J. Bacteriol 2006, 188, 2761–2773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 881.Juncker AS; Willenbrock H; Von Heijne G; Brunak S; Nielsen H; Krogh A Prediction of Lipoprotein Signal Peptides in Gram-Negative Bacteria. Protein Sci 2003, 12, 1652–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 882.Zuckert WR Secretion of Bacterial Lipoproteins: Through the Cytoplasmic Membrane, the Periplasm and Beyond. Biochim. Biophys. Acta 2014, 1843, 1509–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 883.Lai SH; Philbrick WM; Wu HC Acyl Moieties in Phospholipids Are the Precursors for the Fatty Acids in Murein Lipoprotein of Escherichia coli. J. Biol. Chem 1980, 255, 5384–5387. [PubMed] [Google Scholar]
- 884.Sankaran K; Wu HC Lipid Modification of Bacterial Prolipoprotein. Transfer of Diacylglyceryl Moiety from Phosphatidylglycerol. J. Biol. Chem 1994, 269, 19701–19706. [PubMed] [Google Scholar]
- 885.Hillmann F; Argentini M; Buddelmeijer N Kinetics and Phospholipid Specificity of Apolipoprotein N-Acyltransferase. J. Biol. Chem 2011, 286, 27936–27946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 886.Tokunaga M; Tokunaga H; Wu HC Post-Translational Modification and Processing of Escherichia coli Prolipoprotein in Vitro. Proc Natl Acad Sci U S A 1982, 79, 2255–2259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 887.Vogeley L; El Arnaout T; Bailey J; Stansfeld PJ; Boland C; Caffrey M Structural Basis of Lipoprotein Signal Peptidase II Action and Inhibition by the Antibiotic Globomycin. Science 2016, 351, 876–880. [DOI] [PubMed] [Google Scholar]
- 888.Kitamura S; Wolan DW Probing Substrate Recognition of Bacterial Lipoprotein Signal Peptidase Using FRET Reporters. FEBS Lett. 2018, 592, 2289–2296. [DOI] [PubMed] [Google Scholar]
- 889.Mao G; Zhao Y; Kang X; Li Z; Zhang Y; Wang X; Sun F; Sankaran K; Zhang XC Crystal Structure of E. coli Lipoprotein Diacylglyceryl Transferase. Nat Commun 2016, 7, 10198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 890.Noland CL; Kattke MD; Diao J; Gloor SL; Pantua H; Reichelt M; Katakam AK; Yan D; Kang J; Zilberleyb I, et al. Structural Insights into Lipoprotein N-Acylation by Escherichia coli Apolipoprotein N-Acyltransferase. Proc Natl Acad Sci U S A 2017, 114, E6044–E6053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 891.Grabowicz M Lipoprotein Transport: Greasing the Machines of Outer Membrane Biogenesis: Re-Examining Lipoprotein Transport Mechanisms among Diverse Gram-Negative Bacteria While Exploring New Discoveries and Questions. Bioessays 2018, 40, e1700187. [DOI] [PubMed] [Google Scholar]
- 892.Yakushi T; Masuda K; Narita S; Matsuyama S; Tokuda H A New ABC Transporter Mediating the Detachment of Lipid-Modified Proteins from Membranes. Nat. Cell Biol 2000, 2, 212–218. [DOI] [PubMed] [Google Scholar]
- 893.Ito Y; Kanamaru K; Taniguchi N; Miyamoto S; Tokuda H A Novel Ligand Bound ABC Transporter, LolCDE, Provides Insights into the Molecular Mechanisms Underlying Membrane Detachment of Bacterial Lipoproteins. Mol. Microbiol 2006, 62, 1064–1075. [DOI] [PubMed] [Google Scholar]
- 894.Yasuda M; Iguchi-Yokoyama A; Matsuyama S; Tokuda H; Narita S Membrane Topology and Functional Importance of the Periplasmic Region of ABC Transporter LolCDE. Biosci Biotechnol Biochem 2009, 73, 2310–2316. [DOI] [PubMed] [Google Scholar]
- 895.Fukuda A; Matsuyama S; Hara T; Nakayama J; Nagasawa H; Tokuda H Aminoacylation of the N-Terminal Cysteine Is Essential for Lol-Dependent Release of Lipoproteins from Membranes but Does Not Depend on Lipoprotein Sorting Signals. J. Biol. Chem 2002, 277, 43512–43518. [DOI] [PubMed] [Google Scholar]
- 896.LoVullo ED; Wright LF; Isabella V; Huntley JF; Pavelka MS Jr. Revisiting the Gram-Negative Lipoprotein Paradigm. J. Bacteriol 2015, 197, 1705–1715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 897.Mizutani M; Mukaiyama K; Xiao J; Mori M; Satou R; Narita S; Okuda S; Tokuda H Functional Differentiation of Structurally Similar Membrane Subunits of the ABC Transporter LolCDE Complex. FEBS Lett. 2013, 587, 23–29. [DOI] [PubMed] [Google Scholar]
- 898.Takeda K; Miyatake H; Yokota N; Matsuyama S; Tokuda H; Miki K Crystal Structures of Bacterial Lipoprotein Localization Factors, LolA and LolB. EMBO J 2003, 22, 3199–3209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 899.Remans K; Pauwels K; van Ulsen P; Buts L; Cornelis P; Tommassen J; Savvides SN; Decanniere K; Van Gelder P Hydrophobic Surface Patches on LolA of Pseudomonas aeruginosa Are Essential for Lipoprotein Binding. J. Mol. Biol 2010, 401, 921–930. [DOI] [PubMed] [Google Scholar]
- 900.Okuda S; Tokuda H Model of Mouth-to-Mouth Transfer of Bacterial Lipoproteins through Inner Membrane LolC, Periplasmic LolA, and Outer Membrane LolB. Proc Natl Acad Sci U S A 2009, 106, 5877–5882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 901.Taniguchi N; Matsuyama S; Tokuda H Mechanisms Underlying Energy-Independent Transfer of Lipoproteins from LolA to LolB, Which Have Similar Unclosed Beta-Barrel Structures. J. Biol. Chem 2005, 280, 34481–34488. [DOI] [PubMed] [Google Scholar]
- 902.Tsukahara J; Mukaiyama K; Okuda S; Narita S; Tokuda H Dissection of LolB Function--Lipoprotein Binding, Membrane Targeting and Incorporation of Lipoproteins into Lipid Bilayers. FEBS J. 2009, 276, 4496–4504. [DOI] [PubMed] [Google Scholar]
- 903.Hooda Y; Moraes TF Translocation of Lipoproteins to the Surface of Gram Negative Bacteria. Curr. Opin. Struct. Biol 2018, 51, 73–79. [DOI] [PubMed] [Google Scholar]
- 904.Konovalova A; Silhavy TJ Outer Membrane Lipoprotein Biogenesis: Lol Is Not the End. Philos Trans R Soc Lond B Biol Sci 2015, 370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 905.Dowdell AS; Murphy MD; Azodi C; Swanson SK; Florens L; Chen S; Zuckert WR Comprehensive Spatial Analysis of the Borrelia burgdorferi Lipoproteome Reveals a Compartmentalization Bias toward the Bacterial Surface. J. Bacteriol 2017, 199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 906.Dong C; Beis K; Nesper J; Brunkan-Lamontagne AL; Clarke BR; Whitfield C; Naismith JH Wza the Translocon for E. coli Capsular Polysaccharides Defines a New Class of Membrane Protein. Nature 2006, 444, 226–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 907.Goyal P; Krasteva PV; Van Gerven N; Gubellini F; Van den Broeck I; Troupiotis-Tsailaki A; Jonckheere W; Pehau-Arnaudet G; Pinkner JS; Chapman MR, et al. Structural and Mechanistic Insights into the Bacterial Amyloid Secretion Channel CsgG. Nature 2014, 516, 250–253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 908.Konovalova A; Perlman DH; Cowles CE; Silhavy TJ Transmembrane Domain of Surface-Exposed Outer Membrane Lipoprotein RcsF Is Threaded through the Lumen of Beta-Barrel Proteins. Proc Natl Acad Sci U S A 2014, 111, E4350–4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 909.Hooda Y; Lai CC; Judd A; Buckwalter CM; Shin HE; Gray-Owen SD; Moraes TF Slam Is an Outer Membrane Protein That Is Required for the Surface Display of Lipidated Virulence Factors in Neisseria. Nat Microbiol 2016, 1, 16009. [DOI] [PubMed] [Google Scholar]
- 910.Gardiner J. H. t.; Komazin G; Matsuo M; Cole K; Gotz F; Meredith TC Lipoprotein N-Acylation in Staphylococcus aureus Is Catalyzed by a Two-Component Acyl Transferase System. mBio 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 911.Armbruster KM; Komazin G; Meredith TC Bacterial Lyso-Form Lipoproteins Are Synthesized Via an Intramolecular Acyl Chain Migration. J. Biol. Chem 2020, 295, 10195–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 912.Yakushi T; Tajima T; Matsuyama S; Tokuda H Lethality of the Covalent Linkage between Mislocalized Major Outer Membrane Lipoprotein and the Peptidoglycan of Escherichia coli. J. Bacteriol 1997, 179, 2857–2862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 913.Grabowicz M; Silhavy TJ Redefining the Essential Trafficking Pathway for Outer Membrane Lipoproteins. Proc Natl Acad Sci U S A 2017, 114, 4769–4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 914.Hussain M; Ichihara S; Mizushima S Accumulation of Glyceride-Containing Precursor of the Outer Membrane Lipoprotein in the Cytoplasmic Membrane of Escherichia coli Treated with Globomycin. J. Biol. Chem 1980, 255, 3707–3712. [PubMed] [Google Scholar]
- 915.Dev IK; Harvey RJ; Ray PH Inhibition of Prolipoprotein Signal Peptidase by Globomycin. J. Biol. Chem 1985, 260, 5891–5894. [PubMed] [Google Scholar]
- 916.Kiho T; Nakayama M; Yasuda K; Miyakoshi S; Inukai M; Kogen H Structure-Activity Relationships of Globomycin Analogues as Antibiotics. Bioorg. Med. Chem 2004, 12, 337–361. [DOI] [PubMed] [Google Scholar]
- 917.Xiao Y; Gerth K; Muller R; Wall D Myxobacterium-Produced Antibiotic TA (Myxovirescin) Inhibits Type II Signal Peptidase. Antimicrob. Agents Chemother 2012, 56, 2014–2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 918.Pathania R; Zlitni S; Barker C; Das R; Gerritsma DA; Lebert J; Awuah E; Melacini G; Capretta FA; Brown ED Chemical Genomics in Escherichia coli Identifies an Inhibitor of Bacterial Lipoprotein Targeting. Nat. Chem. Biol 2009, 5, 849–856. [DOI] [PubMed] [Google Scholar]
- 919.McLeod SM; Fleming PR; MacCormack K; McLaughlin RE; Whiteaker JD; Narita S; Mori M; Tokuda H; Miller AA Small-Molecule Inhibitors of Gram-Negative Lipoprotein Trafficking Discovered by Phenotypic Screening. J. Bacteriol 2015, 197, 1075–1082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 920.Pollock MR The Cell-Bound Penicillinase of Bacillus cereus. J. Gen. Microbiol 1956, 15, 154–169. [DOI] [PubMed] [Google Scholar]
- 921.Nielsen JB; Lampen JO Beta-Lactamase III of Bacillus cereus 569: Membrane Lipoprotein and Secreted Protein. Biochemistry 1983, 22, 4652–4656. [DOI] [PubMed] [Google Scholar]
- 922.Nielsen JB; Lampen JO Membrane-Bound Penicillinases in Gram-Positive Bacteria. J. Biol. Chem 1982, 257, 4490–4495. [PubMed] [Google Scholar]
- 923.Nielsen JB; Caulfield MP; Lampen JO Lipoprotein Nature of Bacillus licheniformis Membrane Penicillinase. Proc Natl Acad Sci U S A 1981, 78, 3511–3515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 924.East AK; Dyke KG Cloning and Sequence Determination of Six Staphylococcus aureus Beta-Lactamases and Their Expression in Escherichia coli and Staphylococcus aureus. J. Gen. Microbiol 1989, 135, 1001–1015. [DOI] [PubMed] [Google Scholar]
- 925.Bootsma HJ; Aerts PC; Posthuma G; Harmsen T; Verhoef J; van Dijk H; Mooi FR Moraxella (Branhamella) catarrhalis BRO Beta-Lactamase: A Lipoprotein of Gram-Positive Origin? J. Bacteriol 1999, 181, 5090–5093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 926.Bootsma HJ; van Dijk H; Verhoef J; Fleer A; Mooi FR Molecular Characterization of the BRO Beta-Lactamase of Moraxella (Branhamella) catarrhalis. Antimicrob. Agents Chemother 1996, 40, 966–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 927.Randall LB; Dobos K; Papp-Wallace KM; Bonomo RA; Schweizer HP Membrane-Bound PenA Beta-Lactamase of Burkholderia pseudomallei. Antimicrob. Agents Chemother 2016, 60, 1509–1514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 928.Pasteran F; Gonzalez LJ; Albornoz E; Bahr G; Vila AJ; Corso A Triton Hodge Test: Improved Protocol for Modified Hodge Test for Enhanced Detection of NDM and Other Carbapenemase Producers. J. Clin. Microbiol 2016, 54, 640–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 929.Schwechheimer C; Kuehn MJ Outer-Membrane Vesicles from Gram-Negative Bacteria: Biogenesis and Functions. Nat. Rev. Microbiol 2015, 13, 605–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 930.Kulp A; Kuehn MJ Biological Functions and Biogenesis of Secreted Bacterial Outer Membrane Vesicles. Annu. Rev. Microbiol 2010, 64, 163–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 931.Haurat MF; Elhenawy W; Feldman MF Prokaryotic Membrane Vesicles: New Insights on Biogenesis and Biological Roles. Biol. Chem 2015, 396, 95–109. [DOI] [PubMed] [Google Scholar]
- 932.Guerrero-Mandujano A; Hernandez-Cortez C; Ibarra JA; Castro-Escarpulli G The Outer Membrane Vesicles: Secretion System Type Zero. Traffic 2017, 18, 425–432. [DOI] [PubMed] [Google Scholar]
- 933.Jan AT Outer Membrane Vesicles (OMVs) of Gram-Negative Bacteria: A Perspective Update. Front Microbiol 2017, 8, 1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 934.Beveridge TJ Structures of Gram-Negative Cell Walls and Their Derived Membrane Vesicles. J. Bacteriol 1999, 181, 4725–4733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 935.Deatherage BL; Cookson BT Membrane Vesicle Release in Bacteria, Eukaryotes, and Archaea: A Conserved yet Underappreciated Aspect of Microbial Life. Infect. Immun 2012, 80, 1948–1957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 936.Gill S; Catchpole R; Forterre P Extracellular Membrane Vesicles in the Three Domains of Life and Beyond. FEMS Microbiol. Rev 2019, 43, 273–303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 937.Perez-Cruz C; Delgado L; Lopez-Iglesias C; Mercade E Outer-Inner Membrane Vesicles Naturally Secreted by Gram-Negative Pathogenic Bacteria. PLoS One 2015, 10, e0116896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 938.Toyofuku M; Nomura N; Eberl L Types and origins of Bacterial Membrane Vesicles. Nat. Rev. Microbiol 2019, 17, 13–24. [DOI] [PubMed] [Google Scholar]
- 939.Biller SJ; Schubotz F; Roggensack SE; Thompson AW; Summons RE; Chisholm SW Bacterial Vesicles in Marine Ecosystems. Science 2014, 343, 183–186. [DOI] [PubMed] [Google Scholar]
- 940.Elhenawy W; Bording-Jorgensen M; Valguarnera E; Haurat MF; Wine E; Feldman MF LPS Remodeling Triggers Formation of Outer Membrane Vesicles in Salmonella. MBio 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 941.McBroom AJ; Johnson AP; Vemulapalli S; Kuehn MJ Outer Membrane Vesicle Production by Escherichia coli Is Independent of Membrane Instability. J. Bacteriol 2006, 188, 5385–5392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 942.Bernadac A; Gavioli M; Lazzaroni JC; Raina S; Lloubes R Escherichia coli Tol-Pal Mutants Form Outer Membrane Vesicles. J. Bacteriol 1998, 180, 4872–4878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 943.Deatherage BL; Lara JC; Bergsbaken T; Rassoulian Barrett SL; Lara S; Cookson BT Biogenesis of Bacterial Membrane Vesicles. Mol. Microbiol 2009, 72, 1395–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 944.Roier S; Zingl FG; Cakar F; Durakovic S; Kohl P; Eichmann TO; Klug L; Gadermaier B; Weinzerl K; Prassl R, et al. A Novel Mechanism for the Biogenesis of Outer Membrane Vesicles in Gram-Negative Bacteria. Nat Commun 2016, 7, 10515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 945.McBroom AJ; Kuehn MJ Release of Outer Membrane Vesicles by Gram-Negative Bacteria Is a Novel Envelope Stress Response. Mol. Microbiol 2007, 63, 545–558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 946.Mashburn-Warren L; Howe J; Garidel P; Richter W; Steiniger F; Roessle M; Brandenburg K; Whiteley M Interaction of Quorum Signals with Outer Membrane Lipids: Insights into Prokaryotic Membrane Vesicle Formation. Mol. Microbiol 2008, 69, 491–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 947.Haurat MF; Aduse-Opoku J; Rangarajan M; Dorobantu L; Gray MR; Curtis MA; Feldman MF Selective Sorting of Cargo Proteins into Bacterial Membrane Vesicles. J. Biol. Chem 2011, 286, 1269–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 948.Elhenawy W; Debelyy MO; Feldman MF Preferential Packing of Acidic Glycosidases and Proteases into Bacteroides Outer Membrane Vesicles. mBio 2014, 5, e00909–00914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 949.Lappann M; Otto A; Becher D; Vogel U Comparative Proteome Analysis of Spontaneous Outer Membrane Vesicles and Purified Outer Membranes of Neisseria Meningitidis. J. Bacteriol 2013, 195, 4425–4435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 950.Veith PD; Chen YY; Gorasia DG; Chen D; Glew MD; O’Brien-Simpson NM; Cecil JD; Holden JA; Reynolds EC Porphyromonas gingivalis Outer Membrane Vesicles Exclusively Contain Outer Membrane and Periplasmic Proteins and Carry a Cargo Enriched with Virulence Factors. J Proteome Res 2014, 13, 2420–2432. [DOI] [PubMed] [Google Scholar]
- 951.Lin J; Zhang W; Cheng J; Yang X; Zhu K; Wang Y; Wei G; Qian PY; Luo ZQ; Shen X A Pseudomonas T6SS Effector Recruits PQS-Containing Outer Membrane Vesicles for Iron Acquisition. Nat Commun 2017, 8, 14888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 952.Berleman JE; Allen S; Danielewicz MA; Remis JP; Gorur A; Cunha J; Hadi MZ; Zusman DR; Northen TR; Witkowska HE, et al. The Lethal Cargo of Myxococcus xanthus Outer Membrane Vesicles. Front Microbiol 2014, 5, 474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 953.Schwechheimer C; Kuehn MJ Synthetic Effect between Envelope Stress and Lack of Outer Membrane Vesicle Production in Escherichia coli. J. Bacteriol 2013, 195, 4161–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 954.Sabra W; Lunsdorf H; Zeng AP Alterations in the Formation of Lipopolysaccharide and Membrane Vesicles on the Surface of Pseudomonas aeruginosa PAO1 under Oxygen Stress Conditions. Microbiology (Reading) 2003, 149, 2789–2795. [DOI] [PubMed] [Google Scholar]
- 955.Macdonald IA; Kuehn MJ Stress-Induced Outer Membrane Vesicle Production by Pseudomonas aeruginosa. J. Bacteriol 2013, 195, 2971–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 956.Devos S; Van Putte W; Vitse J; Van Driessche G; Stremersch S; Van Den Broek W; Raemdonck K; Braeckmans K; Stahlberg H; Kudryashev M, et al. Membrane Vesicle Secretion and Prophage Induction in Multidrug-Resistant Stenotrophomonas maltophilia in Response to Ciprofloxacin Stress. Environ. Microbiol 2017, 19, 3930–3937. [DOI] [PubMed] [Google Scholar]
- 957.Rakoff-Nahoum S; Coyne MJ; Comstock LE An Ecological Network of Polysaccharide Utilization among Human Intestinal Symbionts. Curr. Biol 2014, 24, 40–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 958.Schooling SR; Beveridge TJ Membrane Vesicles: An Overlooked Component of the Matrices of Biofilms. J. Bacteriol 2006, 188, 5945–5957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 959.Yonezawa H; Osaki T; Kurata S; Fukuda M; Kawakami H; Ochiai K; Hanawa T; Kamiya S Outer Membrane Vesicles of Helicobacter pylori TK1402 Are Involved in Biofilm Formation. BMC Microbiol 2009, 9, 197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 960.Wang W; Chanda W; Zhong M The Relationship between Biofilm and Outer Membrane Vesicles: A Novel Therapy Overview. FEMS Microbiol. Lett 2015, 362, fnv117. [DOI] [PubMed] [Google Scholar]
- 961.Mashburn LM; Whiteley M Membrane Vesicles Traffic Signals and Facilitate Group Activities in a Prokaryote. Nature 2005, 437, 422–425. [DOI] [PubMed] [Google Scholar]
- 962.Zhu Y; Dashper SG; Chen YY; Crawford S; Slakeski N; Reynolds EC Porphyromonas gingivalis and Treponema denticola Synergistic Polymicrobial Biofilm Development. PLoS One 2013, 8, e71727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 963.Bauwens A; Kunsmann L; Marejkova M; Zhang W; Karch H; Bielaszewska M; Mellmann A Intrahost Milieu Modulates Production of Outer Membrane Vesicles, Vesicle-Associated Shiga Toxin 2A and Cytotoxicity in Escherichia coli O157:H7 and O104:H4. Environ Microbiol Rep 2017, 9, 626–634. [DOI] [PubMed] [Google Scholar]
- 964.Irazoqui JE; Troemel ER; Feinbaum RL; Luhachack LG; Cezairliyan BO; Ausubel FM Distinct Pathogenesis and Host Responses During Infection of C. Elegans by P. aeruginosa and S. aureus. PLoS Pathog 2010, 6, e1000982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 965.Bomberger JM; Maceachran DP; Coutermarsh BA; Ye S; O’Toole GA; Stanton BA Long-Distance Delivery of Bacterial Virulence Factors by Pseudomonas aeruginosa Outer Membrane Vesicles. PLoS Pathog 2009, 5, e1000382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 966.Bomberger JM; Ye S; Maceachran DP; Koeppen K; Barnaby RL; O’Toole GA; Stanton BA A Pseudomonas aeruginosa Toxin That Hijacks the Host Ubiquitin Proteolytic System. PLoS Pathog 2011, 7, e1001325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 967.Fiocca R; Necchi V; Sommi P; Ricci V; Telford J; Cover TL; Solcia E Release of Helicobacter pylori Vacuolating Cytotoxin by Both a Specific Secretion Pathway and Budding of Outer Membrane Vesicles. Uptake of Released Toxin and Vesicles by Gastric Epithelium. J. Pathol 1999, 188, 220–226. [DOI] [PubMed] [Google Scholar]
- 968.Wai SN; Lindmark B; Soderblom T; Takade A; Westermark M; Oscarsson J; Jass J; Richter-Dahlfors A; Mizunoe Y; Uhlin BE Vesicle-Mediated Export and Assembly of Pore-Forming Oligomers of the Enterobacterial ClyA Cytotoxin. Cell 2003, 115, 25–35. [DOI] [PubMed] [Google Scholar]
- 969.Kunsmann L; Ruter C; Bauwens A; Greune L; Gluder M; Kemper B; Fruth A; Wai SN; He X; Lloubes R, et al. Virulence from Vesicles: Novel Mechanisms of Host Cell Injury by Escherichia coli O104:H4 Outbreak Strain. Sci Rep 2015, 5, 13252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 970.Dorward DW; Schwan TG; Garon CF Immune Capture and Detection of Borrelia burgdorferi Antigens in Urine, Blood, or Tissues from Infected Ticks, Mice, Dogs, and Humans. J. Clin. Microbiol 1991, 29, 1162–1170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 971.Domingues S; Nielsen KM Membrane Vesicles and horizontal Gene Transfer in Prokaryotes. Curr. Opin. Microbiol 2017, 38, 16–21. [DOI] [PubMed] [Google Scholar]
- 972.Rumbo C; Fernandez-Moreira E; Merino M; Poza M; Mendez JA; Soares NC; Mosquera A; Chaves F; Bou G horizontal Transfer of the Oxa-24 Carbapenemase Gene Via Outer Membrane Vesicles: A New Mechanism of Dissemination of Carbapenem Resistance Genes in Acinetobacter baumannii. Antimicrob. Agents Chemother 2011, 55, 3084–3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 973.Fulsundar S; Harms K; Flaten GE; Johnsen PJ; Chopade BA; Nielsen KM Gene Transfer Potential of Outer Membrane Vesicles of Acinetobacter baylyi and Effects of Stress on Vesiculation. Appl. Environ. Microbiol 2014, 80, 3469–3483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 974.Ho MH; Chen CH; Goodwin JS; Wang BY; Xie H Functional Advantages of Porphyromonas gingivalis Vesicles. PLoS One 2015, 10, e0123448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 975.Bitto NJ; Chapman R; Pidot S; Costin A; Lo C; Choi J; D’Cruze T; Reynolds EC; Dashper SG; Turnbull L, et al. Bacterial Membrane Vesicles Transport Their DNA Cargo into Host Cells. Sci Rep 2017, 7, 7072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 976.Chatterjee S; Mondal A; Mitra S; Basu S Acinetobacter baumannii Transfers the blaNDM-1 Gene Via Outer Membrane Vesicles. J. Antimicrob. Chemother 2017, 72, 2201–2207. [DOI] [PubMed] [Google Scholar]
- 977.Koeppen K; Hampton TH; Jarek M; Scharfe M; Gerber SA; Mielcarz DW; Demers EG; Dolben EL; Hammond JH; Hogan DA, et al. A Novel Mechanism of Host-Pathogen Interaction through sRNA in Bacterial Outer Membrane Vesicles. PLoS Pathog 2016, 12, e1005672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 978.Manning AJ; Kuehn MJ Contribution of Bacterial Outer Membrane Vesicles to Innate Bacterial Defense. BMC Microbiol 2011, 11, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 979.Tan TT; Morgelin M; Forsgren A; Riesbeck K Haemophilus Influenzae Survival During Complement-Mediated Attacks Is Promoted by Moraxella catarrhalis Outer Membrane Vesicles. J. Infect. Dis 2007, 195, 1661–1670. [DOI] [PubMed] [Google Scholar]
- 980.Ciofu O; Beveridge TJ; Kadurugamuwa J; Walther-Rasmussen J; Hoiby N Chromosomal Beta-Lactamase Is Packaged into Membrane Vesicles and Secreted from Pseudomonas aeruginosa. J. Antimicrob. Chemother 2000, 45, 9–13. [DOI] [PubMed] [Google Scholar]
- 981.Schaar V; Paulsson M; Morgelin M; Riesbeck K Outer Membrane Vesicles Shield Moraxella catarrhalis Beta-Lactamase from Neutralization by Serum IgG. J. Antimicrob. Chemother 2013, 68, 593–600. [DOI] [PubMed] [Google Scholar]
- 982.Stentz R; Horn N; Cross K; Salt L; Brearley C; Livermore DM; Carding SR Cephalosporinases Associated with Outer Membrane Vesicles Released by Bacteroides Spp. Protect Gut Pathogens and Commensals against Beta-Lactam Antibiotics. J. Antimicrob. Chemother 2015, 70, 701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 983.Koning RI; de Breij A; Oostergetel GT; Nibbering PH; Koster AJ; Dijkshoorn L Cryo-Electron Tomography Analysis of Membrane Vesicles from Acinetobacter baumannii ATCC19606 T. Res. Microbiol 2013, 164, 397–405. [DOI] [PubMed] [Google Scholar]
- 984.Devos S; Van Oudenhove L; Stremersch S; Van Putte W; De Rycke R; Van Driessche G; Vitse J; Raemdonck K; Devreese B The Effect of Imipenem and Diffusible Signaling Factors on the Secretion of Outer Membrane Vesicles and Associated Ax21 Proteins in Stenotrophomonas maltophilia. Front Microbiol 2015, 6, 298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 985.Devos S; Stremersch S; Raemdonck K; Braeckmans K; Devreese B Intra- and Interspecies Effects of Outer Membrane Vesicles from Stenotrophomonas maltophilia on Beta-Lactam Resistance. Antimicrob. Agents Chemother 2016, 60, 2516–2518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 986.D’Costa VM; King CE; Kalan L; Morar M; Sung WW; Schwarz C; Froese D; Zazula G; Calmels F; Debruyne R, et al. Antibiotic Resistance Is Ancient. Nature 2011, 477, 457–461. [DOI] [PubMed] [Google Scholar]
- 987.Perron GG; Whyte L; Turnbaugh PJ; Goordial J; Hanage WP; Dantas G; Desai MM Functional Characterization of Bacteria Isolated from Ancient Arctic Soil Exposes Diverse Resistance Mechanisms to Modern Antibiotics. PLoS One 2015, 10, e0069533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 988.Abrudan MI; Smakman F; Grimbergen AJ; Westhoff S; Miller EL; van Wezel GP; Rozen DE Socially Mediated Induction and Suppression of Antibiosis During Bacterial Coexistence. Proc Natl Acad Sci U S A 2015, 112, 11054–11059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 989.Raaijmakers JM; Mazzola M Diversity and Natural Functions of Antibiotics Produced by Beneficial and Plant Pathogenic Bacteria. Annu. Rev. Phytopathol 2012, 50, 403–424. [DOI] [PubMed] [Google Scholar]
- 990.Yim G; Wang HH; Davies J Antibiotics as Signalling Molecules. Philos Trans R Soc Lond B Biol Sci 2007, 362, 1195–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 991.Bhullar K; Waglechner N; Pawlowski A; Koteva K; Banks ED; Johnston MD; Barton HA; Wright GD Antibiotic Resistance Is Prevalent in an Isolated Cave Microbiome. PLoS One 2012, 7, e34953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 992.Toth M; Smith C; Frase H; Mobashery S; Vakulenko S An Antibiotic-Resistance Enzyme from a Deep-Sea Bacterium. J. Am. Chem. Soc 2010, 132, 816–823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 993.Thomas CM; Nielsen KM Mechanisms of, and Barriers to, horizontal Gene Transfer between Bacteria. Nat. Rev. Microbiol 2005, 3, 711–721. [DOI] [PubMed] [Google Scholar]
- 994.Partridge SR; Kwong SM; Firth N; Jensen SO Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev 2018, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 995.Siguier P; Gourbeyre E; Chandler M Known Knowns, Known Unknowns and Unknown Unknowns in Prokaryotic Transposition. Curr. Opin. Microbiol 2017, 38, 171–180. [DOI] [PubMed] [Google Scholar]
- 996.Bjorkman J; Andersson DI The Cost of Antibiotic Resistance from a Bacterial Perspective. Drug Resist Updat 2000, 3, 237–245. [DOI] [PubMed] [Google Scholar]
- 997.Andersson DI Persistence of Antibiotic Resistant Bacteria. Curr. Opin. Microbiol 2003, 6, 452–456. [DOI] [PubMed] [Google Scholar]
- 998.Enne VI; Delsol AA; Davis GR; Hayward SL; Roe JM; Bennett PM Assessment of the Fitness Impacts on Escherichia coli of Acquisition of Antibiotic Resistance Genes Encoded by Different Types of Genetic Element. J. Antimicrob. Chemother 2005, 56, 544–551. [DOI] [PubMed] [Google Scholar]
- 999.Arzanlou M; Chai WC; Venter H Intrinsic, Adaptive and Acquired Antimicrobial Resistance in Gram-Negative Bacteria. Essays Biochem 2017, 61, 49–59. [DOI] [PubMed] [Google Scholar]
- 1000.Forsberg KJ; Patel S; Gibson MK; Lauber CL; Knight R; Fierer N; Dantas G Bacterial Phylogeny Structures Soil Resistomes across Habitats. Nature 2014, 509, 612–616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1001.Dantas G; Sommer MO Context Matters - the Complex Interplay between Resistome Genotypes and Resistance Phenotypes. Curr. Opin. Microbiol 2012, 15, 577–582. [DOI] [PubMed] [Google Scholar]
- 1002.Figueira V; Vaz-Moreira I; Silva M; Manaia CM Diversity and Antibiotic Resistance of Aeromonas Spp. In Drinking and Waste Water Treatment Plants. Water Res 2011, 45, 5599–5611. [DOI] [PubMed] [Google Scholar]
- 1003.Pruden A; Arabi M; Storteboom HN Correlation between Upstream Human Activities and Riverine Antibiotic Resistance Genes. Environ. Sci. Technol 2012, 46, 11541–11549. [DOI] [PubMed] [Google Scholar]
- 1004.Forsberg KJ; Reyes A; Wang B; Selleck EM; Sommer MO; Dantas G The Shared Antibiotic Resistome of Soil Bacteria and Human Pathogens. Science 2012, 337, 1107–1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1005.Ebmeyer S; Kristiansson E; Larsson DGJ The Mobile FOX AmpC Beta-Lactamases Originated in Aeromonas allosaccharophila. Int. J. Antimicrob. Agents 2019, 54, 798–802. [DOI] [PubMed] [Google Scholar]
- 1006.Ebmeyer S; Kristiansson E; Larsson DGJ PER Extended-Spectrum Beta-Lactamases Originate from Pararheinheimera Spp. Int. J. Antimicrob. Agents 2019, 53, 158–164. [DOI] [PubMed] [Google Scholar]
- 1007.Storteboom H; Arabi M; Davis JG; Crimi B; Pruden A Tracking Antibiotic Resistance Genes in the South Platte River Basin Using Molecular Signatures of Urban, Agricultural, and Pristine Sources. Environ. Sci. Technol 2010, 44, 7397–7404. [DOI] [PubMed] [Google Scholar]
- 1008.Hatosy SM; Martiny AC The Ocean as a Global Reservoir of Antibiotic Resistance Genes. Appl. Environ. Microbiol 2015, 81, 7593–7599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1009.Chen B; Yang Y; Liang X; Yu K; Zhang T; Li X Metagenomic Profiles of Antibiotic Resistance Genes (ARGs) between Human Impacted Estuary and Deep Ocean Sediments. Environ. Sci. Technol 2013, 47, 12753–12760. [DOI] [PubMed] [Google Scholar]
- 1010.Hall RM; Collis CM Mobile Gene Cassettes and Integrons: Capture and Spread of Genes by Site-Specific Recombination. Mol. Microbiol 1995, 15, 593–600. [DOI] [PubMed] [Google Scholar]
- 1011.Recchia GD; Hall RM origins of the Mobile Gene Cassettes Found in Integrons. Trends Microbiol 1997, 5, 389–394. [DOI] [PubMed] [Google Scholar]
- 1012.Siguier P; Gourbeyre E; Varani A; Ton-Hoang B; Chandler M Everyman’s Guide to Bacterial Insertion Sequences. In Mobile DNA Iii, 2015; pp 555–590. [DOI] [PubMed] [Google Scholar]
- 1013.Vandecraen J; Chandler M; Aertsen A; Van Houdt R The Impact of Insertion Sequences on Bacterial Genome Plasticity and Adaptability. Crit. Rev. Microbiol 2017, 43, 709–730. [DOI] [PubMed] [Google Scholar]
- 1014.Harmer CJ; Pong CH; Hall RM Structures Bounded by Directly-Oriented Members of the IS26 Family Are Pseudo-Compound Transposons. Plasmid 2020, 111, 102530. [DOI] [PubMed] [Google Scholar]
- 1015.Toleman MA; Bennett PM; Walsh TR ISCR Elements: Novel Gene-Capturing Systems of the 21st Century? Microbiol. Mol. Biol. Rev 2006, 70, 296–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1016.Toleman MA; Walsh TR Evolution of the ISCR3 Group of ISCR Elements. Antimicrob. Agents Chemother 2008, 52, 3789–3791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1017.Boyd DA; Tyler S; Christianson S; McGeer A; Muller MP; Willey BM; Bryce E; Gardam M; Nordmann P; Mulvey MR Complete Nucleotide Sequence of a 92-Kilobase Plasmid harboring the CTX-M-15 Extended-Spectrum Beta-Lactamase Involved in an Outbreak in Long-Term-Care Facilities in Toronto, Canada. Antimicrob. Agents Chemother 2004, 48, 3758–3764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1018.Domingues S; da Silva GJ; Nielsen KM Integrons: Vehicles and Pathways for horizontal Dissemination in Bacteria. Mob Genet Elements 2012, 2, 211–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1019.Stalder T; Barraud O; Casellas M; Dagot C; Ploy MC Integron Involvement in Environmental Spread of Antibiotic Resistance. Front Microbiol 2012, 3, 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1020.Delihas N Small Mobile Sequences in Bacteria Display Diverse Structure/Function Motifs. Mol. Microbiol 2008, 67, 475–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1021.Rodriguez-Valera F; Martin-Cuadrado AB; Lopez-Perez M flexible Genomic Islands as Drivers of Genome Evolution. Curr. Opin. Microbiol 2016, 31, 154–160. [DOI] [PubMed] [Google Scholar]
- 1022.Banuelos-Vazquez LA; Torres Tejerizo G; Brom S Regulation of Conjugative Transfer of Plasmids and Integrative Conjugative Elements. Plasmid 2017, 91, 82–89. [DOI] [PubMed] [Google Scholar]
- 1023.Smillie C; Garcillan-Barcia MP; Francia MV; Rocha EP; de la Cruz F Mobility of Plasmids. Microbiol. Mol. Biol. Rev 2010, 74, 434–452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1024.Virolle C; Goldlust K; Djermoun S; Bigot S; Lesterlin C Plasmid Transfer by Conjugation in Gram-Negative Bacteria: From the Cellular to the Community Level. Genes (Basel) 2020, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1025.Carattoli A Plasmids and the Spread of Resistance. Int. J. Med. Microbiol 2013, 303, 298–304. [DOI] [PubMed] [Google Scholar]
- 1026.Thomas CM Paradigms of Plasmid Organization. Mol. Microbiol 2000, 37, 485–491. [DOI] [PubMed] [Google Scholar]
- 1027.Lacroix B; Citovsky V Transfer of DNA from Bacteria to Eukaryotes. mBio 2016, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1028.LeBard RJ; Jensen SO; Arnaiz IA; Skurray RA; Firth N A Multimer Resolution System Contributes to Segregational Stability of the Prototypical Staphylococcal Conjugative Multiresistance Plasmid Psk41. FEMS Microbiol. Lett 2008, 284, 58–67. [DOI] [PubMed] [Google Scholar]
- 1029.Baxter JC; Funnell BE Plasmid Partition Mechanisms. Microbiol Spectr 2014, 2. 1030. [DOI] [PubMed] [Google Scholar]
- 1030.Hayes F Toxins-Antitoxins: Plasmid Maintenance, Programmed Cell Death, and Cell Cycle Arrest. Science 2003, 301, 1496–1499. [DOI] [PubMed] [Google Scholar]
- 1031.Tock MR; Dryden DT The Biology of Restriction and Anti-Restriction. Curr. Opin. Microbiol 2005, 8, 466–472. [DOI] [PubMed] [Google Scholar]
- 1032.Botelho J; Schulenburg H The Role of Integrative and Conjugative Elements in Antibiotic Resistance Evolution. Trends Microbiol 2021, 29, 8–18. [DOI] [PubMed] [Google Scholar]
- 1033.Johnson CM; Grossman AD Integrative and Conjugative Elements (ICEs): What They Do and How They Work. Annu. Rev. Genet 2015, 49, 577–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1034.Frost LS; Leplae R; Summers AO; Toussaint A Mobile Genetic Elements: The Agents of Open Source Evolution. Nat. Rev. Microbiol 2005, 3, 722–732. [DOI] [PubMed] [Google Scholar]
- 1035.Overballe-Petersen S; Harms K; Orlando LA; Mayar JV; Rasmussen S; Dahl TW; Rosing MT; Poole AM; Sicheritz-Ponten T; Brunak S, et al. Bacterial Natural Transformation by Highly Fragmented and Damaged DNA. Proc Natl Acad Sci U S A 2013, 110, 19860–19865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1036.Hammoudi Halat D; Ayoub Moubareck C The Current Burden of Carbapenemases: Review of Significant Properties and Dissemination among Gram-Negative Bacteria. Antibiotics (Basel) 2020, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1037.Zheng Z; Cheng Q; Chan EW; Chen S Genetic and Biochemical Characterization of VMB-1, a Novel Metallo-Beta-Lactamase Encoded by a Conjugative, Broad-Host Range IncC Plasmid from Vibrio Spp. Adv Biosyst 2020, 4, e1900221. [DOI] [PubMed] [Google Scholar]
- 1038.Boyd DA; Lisboa LF; Rennie R; Zhanel GG; Dingle TC; Mulvey MR Identification of a Novel Metallo-Beta-Lactamase, CAM-1, in Clinical Pseudomonas aeruginosa Isolates from Canada. J. Antimicrob. Chemother 2019, 74, 1563–1567. [DOI] [PubMed] [Google Scholar]
- 1039.Bush K Carbapenemases: Partners in Crime. J Glob Antimicrob Resist 2013, 1, 7–16. [DOI] [PubMed] [Google Scholar]
- 1040.van der Zee A; Kraak WB; Burggraaf A; Goessens WHF; Pirovano W; Ossewaarde JM; Tommassen J Spread of Carbapenem Resistance by Transposition and Conjugation among Pseudomonas aeruginosa. Front Microbiol 2018, 9, 2057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1041.Bashar S; Sanyal SK; Sultana M; Hossain MA Emergence of IntI1 Associated blaVIM-2 Gene Cassette-Mediated Carbapenem Resistance in Opportunistic Pathogen Pseudomonas stutzeri. Emerg Microbes Infect 2017, 6, e29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1042.Jovcic B; Lepsanovic Z; Begovic J; Filipic B; Kojic M Two Copies of blaNDM-1 Gene Are Present in NDM-1 Producing Pseudomonas aeruginosa Isolates from Serbia. Antonie Van Leeuwenhoek 2014, 105, 613–618. [DOI] [PubMed] [Google Scholar]
- 1043.Arcari G; Di Lella FM; Bibbolino G; Mengoni F; Beccaccioli M; Antonelli G; Faino L; Carattoli A A Multispecies Cluster of VIM-1 Carbapenemase-Producing Enterobacterales Linked by a Novel, Highly Conjugative, and Broad-Host-Range IncA Plasmid Forebodes the Reemergence of VIM-1. Antimicrob. Agents Chemother 2020, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1044.Chen YT; Liao TL; Liu YM; Lauderdale TL; Yan JJ; Tsai SF Mobilization of QnrB2 and ISCR1 in Plasmids. Antimicrob. Agents Chemother 2009, 53, 1235–1237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1045.Dolejska M; Villa L; Poirel L; Nordmann P; Carattoli A Complete Sequencing of an IncHI1 Plasmid Encoding the Carbapenemase NDM-1, the ArmA 16s Rna Methylase and a Resistance-Nodulation-Cell Division/Multidrug Efflux Pump. J. Antimicrob. Chemother 2013, 68, 34–39. [DOI] [PubMed] [Google Scholar]
- 1046.Zhao WH; Hu ZQ Acquired Metallo-Beta-Lactamases and Their Genetic Association with Class 1 Integrons and ISCR Elements in Gram-Negative Bacteria. Future Microbiol 2015, 10, 873–887. [DOI] [PubMed] [Google Scholar]
- 1047.Paul D; Dhar D; Maurya AP; Mishra S; Sharma GD; Chakravarty A; Bhattacharjee A Occurrence of Co-Existing blaVIM-2 and blaNDM-1 in Clinical Isolates of Pseudomonas aeruginosa from India. Ann Clin Microbiol Antimicrob 2016, 15, 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1048.Miriagou V; Papagiannitsis CC; Kotsakis SD; Loli A; Tzelepi E; Legakis NJ; Tzouvelekis LS Sequence of pNL194, a 79.3-Kilobase IncN Plasmid Carrying the blaVIM-1 Metallo-Beta-Lactamase Gene in Klebsiella pneumoniae. Antimicrob. Agents Chemother 2010, 54, 4497–4502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1049.Esposito EP; Gaiarsa S; Del Franco M; Crivaro V; Bernardo M; Cuccurullo S; Pennino F; Triassi M; Marone P; Sassera D, et al. A Novel IncA/C1 Group Conjugative Plasmid, Encoding VIM-1 Metallo-Beta-Lactamase, Mediates the Acquisition of Carbapenem Resistance in ST104 Klebsiella pneumoniae Isolates from Neonates in the Intensive Care Unit of V. Monaldi Hospital in Naples. Front Microbiol 2017, 8, 2135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1050.Partridge SR; Ginn AN; Paulsen IT; Iredell JR pEI1573 Carrying blaIMP-4, from Sydney, Australia, Is Closely Related to Other IncL/M Plasmids. Antimicrob. Agents Chemother 2012, 56, 6029–6032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1051.Kayama S; Shigemoto N; Kuwahara R; Oshima K; Hirakawa H; Hisatsune J; Jove T; Nishio H; Yamasaki K; Wada Y, et al. Complete Nucleotide Sequence of the IncN Plasmid Encoding IMP-6 and CTX-M-2 from Emerging Carbapenem-Resistant Enterobacteriaceae in Japan. Antimicrob. Agents Chemother 2015, 59, 1356–1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1052.Suzuki Y; Ida M; Kubota H; Ariyoshi T; Murakami K; Kobayashi M; Kato R; Hirai A; Suzuki J; Sadamasu K Multiple Beta-Lactam Resistance Gene-Carrying Plasmid Harbored by Klebsiella quasipneumoniae Isolated from Urban Sewage in Japan. mSphere 2019, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1053.Partridge SR; Iredell JR Genetic Contexts of blaNDM-1. Antimicrob. Agents Chemother 2012, 56, 6065–6067; author reply 6071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1054.Bonnin RA; Poirel L; Nordmann P New Delhi Metallo-Beta-Lactamase-Producing Acinetobacter baumannii: A Novel Paradigm for Spreading Antibiotic Resistance Genes. Future Microbiol 2014, 9, 33–41. [DOI] [PubMed] [Google Scholar]
- 1055.Bontron S; Nordmann P; Poirel L Transposition of Tn125 Encoding the NDM-1 Carbapenemase in Acinetobacter baumannii. Antimicrob. Agents Chemother 2016, 60, 7245–7251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1056.Rahman M; Prasad KN; Pathak A; Pati BK; Singh A; Ovejero CM; Ahmad S; Gonzalez-Zorn B RmtC and RmtF 16S rRNA Methyltransferase in NDM-1-Producing Pseudomonas aeruginosa. Emerg Infect Dis 2015, 21, 2059–2062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1057.Shen P; Yi M; Fu Y; Ruan Z; Du X; Yu Y; Xie X Detection of an Escherichia coli Sequence Type 167 Strain with Two Tandem Copies of blaNDM-1 in the Chromosome. J. Clin. Microbiol 2017, 55, 199–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1058.Hong DJ; Bae IK; Jang IH; Jeong SH; Kang HK; Lee K Epidemiology and Characteristics of Metallo-Beta-Lactamase-Producing Pseudomonas aeruginosa. Infect Chemother 2015, 47, 81–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1059.Vatopoulos A High Rates of Metallo-Beta-Lactamase-Producing Klebsiella pneumoniae in Greece--a Review of the Current Evidence. Euro Surveill 2008, 13. [PubMed] [Google Scholar]
- 1060.Stalder T; Rogers LM; Renfrow C; Yano H; Smith Z; Top EM Emerging Patterns of Plasmid-Host Coevolution That Stabilize Antibiotic Resistance. Sci Rep 2017, 7, 4853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1061.Cordeiro NF; Chabalgoity JA; Yim L; Vignoli R Synthesis of Metallo-Beta-Lactamase VIM-2 Is Associated with a Fitness Reduction in Salmonella enterica Serovar Typhimurium. Antimicrob. Agents Chemother 2014, 58, 6528–6535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1062.Huang F; Fitchett N; Razo-Gutierrez C; Le C; Martinez J; Ra G; Lopez C; Gonzalez LJ; Sieira R; Vila AJ, et al. The H-NS Regulator Plays a Role in the Stress Induced by Carbapenemase Expression in Acinetobacter baumannii. mSphere 2020, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1063.Marciano DC; Karkouti OY; Palzkill T A Fitness Cost Associated with the Antibiotic Resistance Enzyme SME-1 Beta-Lactamase. Genetics 2007, 176, 2381–2392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1064.Morosini MI; Ayala JA; Baquero F; Martinez JL; Blazquez J Biological Cost of AmpC Production for Salmonella enterica Serotype Typhimurium. Antimicrob. Agents Chemother 2000, 44, 3137–3143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1065.Pournaras S; Ikonomidis A; Tzouvelekis LS; Tokatlidou D; Spanakis N; Maniatis AN; Legakis NJ; Tsakris A VIM-12, a Novel Plasmid-Mediated Metallo-Beta-Lactamase from Klebsiella pneumoniae That Resembles a VIM-1/VIM-2 Hybrid. Antimicrob. Agents Chemother 2005, 49, 5153–5156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1066.Galani I; Souli M; Chryssouli Z; Katsala D; Giamarellou H First Identification of an Escherichia coli Clinical Isolate Producing Both Metallo-Beta-Lactamase VIM-2 and Extended-Spectrum Beta-Lactamase IBC-1. Clin. Microbiol. Infect 2004, 10, 757–760. [DOI] [PubMed] [Google Scholar]
- 1067.Ghaith DM; Zafer MM; Ismail DK; Al-Agamy MH; Bohol MFF; Al-Qahtani A; Al-Ahdal MN; Elnagdy SM; Mostafa IY First Reported Nosocomial Outbreak of Serratia marcescens Harboring bla IMP-4 and bla VIM-2 in a Neonatal Intensive Care Unit in Cairo, Egypt. Infect Drug Resist 2018, 11, 2211–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1068.Berglund F; Osterlund T; Boulund F; Marathe NP; Larsson DGJ; Kristiansson E Identification and Reconstruction of Novel Antibiotic Resistance Genes from Metagenomes. Microbiome 2019, 7, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1069.Marathe NP; Berglund F; Razavi M; Pal C; Droge J; Samant S; Kristiansson E; Larsson DGJ Sewage Effluent from an Indian Hospital Harbors Novel Carbapenemases and Integron-Borne Antibiotic Resistance Genes. Microbiome 2019, 7, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1070.Fonseca EL; Andrade BGN; Vicente ACP The Resistome of Low-Impacted Marine Environments Is Composed by Distant Metallo-Beta-Lactamases Homologs. Front Microbiol 2018, 9, 677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1071.Andreini C; Banci L; Bertini I; Rosato A Zinc through the Three Domains of Life. J Proteome Res 2006, 5, 3173–3178. [DOI] [PubMed] [Google Scholar]
- 1072.Andreini C; Banci L; Bertini I; Rosato A Counting the Zinc-Proteins Encoded in the Human Genome. J Proteome Res 2006, 5, 196–201. [DOI] [PubMed] [Google Scholar]
- 1073.Lonergan ZR; Skaar EP Nutrient Zinc at the Host-Pathogen Interface. Trends Biochem. Sci 2019, 44, 1041–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1074.Fillat MF The Fur (Ferric Uptake Regulator) Superfamily: Diversity and Versatility of Key Transcriptional Regulators. Arch. Biochem. Biophys 2014, 546, 41–52. [DOI] [PubMed] [Google Scholar]
- 1075.Hobman JL; Wilkie J; Brown NL A Design for Life: Prokaryotic Metal-Binding MerR Family Regulators. BioMetals 2005, 18, 429–436. [DOI] [PubMed] [Google Scholar]
- 1076.Hood MI; Skaar EP Nutritional Immunity: Transition Metals at the Pathogen-Host Interface. Nat. Rev. Microbiol 2012, 10, 525–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1077.Lopez CA; Skaar EP The Impact of Dietary Transition Metals on Host-Bacterial Interactions. Cell Host Microbe 2018, 23, 737–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1078.Weiss G; Carver PL Role of Divalent Metals in Infectious Disease Susceptibility and Outcome. Clin. Microbiol. Infect 2018, 24, 16–23. [DOI] [PubMed] [Google Scholar]
- 1079.Schade AL; Caroline L Raw Hen Egg White and the Role of Iron in Growth Inhibition of Shigella dysenteriae, Staphylococcus aureus, Escherichia coli and Saccharomyces cerevisiae. Science 1944, 100, 14–15. [DOI] [PubMed] [Google Scholar]
- 1080.Schade AL; Caroline L An Iron-Binding Component in Human Blood Plasma. Science 1946, 104, 340–341. [DOI] [PubMed] [Google Scholar]
- 1081.Eggleton WG The Zinc and Copper Contents of the Organs and Tissues of Chinese Subjects. Biochem. J 1940, 34, 991–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1082.King JC; Shames DM; Woodhouse LR Zinc Homeostasis in Humans. J. Nutr 2000, 130, 1360S–1366S. [DOI] [PubMed] [Google Scholar]
- 1083.Murakami M; Hirano T Intracellular Zinc Homeostasis and Zinc Signaling. Cancer Sci 2008, 99, 1515–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1084.Gaither LA; Eide DJ Eukaryotic Zinc Transporters and Their Regulation. BioMetals 2001, 14, 251–270. [DOI] [PubMed] [Google Scholar]
- 1085.Petering DH; Mahim A Proteomic High Affinity Zn(2+) Trafficking: Where Does Metallothionein Fit In? Int J Mol Sci 2017, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1086.Babula P; Masarik M; Adam V; Eckschlager T; Stiborova M; Trnkova L; Skutkova H; Provaznik I; Hubalek J; Kizek R Mammalian Metallothioneins: Properties and Functions. Metallomics 2012, 4, 739–750. [DOI] [PubMed] [Google Scholar]
- 1087.Rana U; Kothinti R; Meeusen J; Tabatabai NM; Krezoski S; Petering DH Zinc Binding Ligands and Cellular Zinc Trafficking: Apo-Metallothionein, Glutathione, TPEN, Proteomic Zinc, and Zn-Sp1. J. Inorg. Biochem 2008, 102, 489–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1088.Liuzzi JP; Lichten LA; Rivera S; Blanchard RK; Aydemir TB; Knutson MD; Ganz T; Cousins RJ Interleukin-6 Regulates the Zinc Transporter Zip14 in Liver and Contributes to the Hypozincemia of the Acute-Phase Response. Proc Natl Acad Sci U S A 2005, 102, 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1089.O’Neill LA; Golenbock D; Bowie AG The History of Toll-Like Receptors - Redefining Innate Immunity. Nat. Rev. Immunol 2013, 13, 453–460. [DOI] [PubMed] [Google Scholar]
- 1090.Lappann M; Danhof S; Guenther F; Olivares-Florez S; Mordhorst IL; Vogel U In Vitro Resistance Mechanisms of Neisseria meningitidis against Neutrophil Extracellular Traps. Mol. Microbiol 2013, 89, 433–449. [DOI] [PubMed] [Google Scholar]
- 1091.Witko-Sarsat V; Rieu P; Descamps-Latscha B; Lesavre P; Halbwachs-Mecarelli L Neutrophils: Molecules, Functions and Pathophysiological Aspects. Lab. Invest 2000, 80, 617–653. [DOI] [PubMed] [Google Scholar]
- 1092.Papayannopoulos V Neutrophil Extracellular Traps in Immunity and Disease. Nat. Rev. Immunol 2018, 18, 134–147. [DOI] [PubMed] [Google Scholar]
- 1093.Boussac M; Garin J Calcium-Dependent Secretion in Human Neutrophils: A Proteomic Approach. Electrophoresis 2000, 21, 665–672. [DOI] [PubMed] [Google Scholar]
- 1094.Gonzalez LL; Garrie K; Turner MD Role of S100 Proteins in Health and Disease. Biochim Biophys Acta Mol Cell Res 2020, 1867, 118677. [DOI] [PubMed] [Google Scholar]
- 1095.Wang S; Song R; Wang Z; Jing Z; Wang S; Ma J S100A8/A9 in Inflammation. Front Immunol 2018, 9, 1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1096.Zygiel EM; Nolan EM Transition Metal Sequestration by the Host-Defense Protein Calprotectin. Annu. Rev. Biochem 2018, 87, 621–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1097.Urban CF; Ermert D; Schmid M; Abu-Abed U; Goosmann C; Nacken W; Brinkmann V; Jungblut PR; Zychlinsky A Neutrophil Extracellular Traps Contain Calprotectin, a Cytosolic Protein Complex Involved in Host Defense against Candida albicans. PLoS Pathog 2009, 5, e1000639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1098.Achouiti A; Vogl T; Urban CF; Rohm M; Hommes TJ; van Zoelen MA; Florquin S; Roth J; van ť Veer C; de Vos AF, et al. Myeloid-Related Protein-14 Contributes to Protective Immunity in Gram-Negative Pneumonia Derived Sepsis. PLoS Pathog 2012, 8, e1002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1099.Cunden LS; Gaillard A; Nolan EM Calcium Ions Tune the Zinc-Sequestering Properties and Antimicrobial Activity of Human S100A12. Chem Sci 2016, 7, 1338–1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1100.Cunden LS; Brophy MB; Rodriguez GE; Flaxman HA; Nolan EM Biochemical and Functional Evaluation of the Intramolecular Disulfide Bonds in the Zinc-Chelating Antimicrobial Protein Human S100A7 (Psoriasin). Biochemistry 2017, 56, 5726–5738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1101.Sohnle PG; Collins-Lech C; Wiessner JH The Zinc-reversible Antimicrobial Activity of Neutrophil Lysates and Abscess Fluid Supernatants. J. Infect. Dis 1991, 164, 137–142. [DOI] [PubMed] [Google Scholar]
- 1102.Sohnle PG; Hunter MJ; Hahn B; Chazin WJ Zinc-reversible Antimicrobial Activity of Recombinant Calprotectin (Migration Inhibitory Factor-Related Proteins 8 and 14). J. Infect. Dis 2000, 182, 1272–1275. [DOI] [PubMed] [Google Scholar]
- 1103.Kehl-Fie TE; Chitayat S; Hood MI; Damo S; Restrepo N; Garcia C; Munro KA; Chazin WJ; Skaar EP Nutrient Metal Sequestration by Calprotectin Inhibits Bacterial Superoxide Defense, Enhancing Neutrophil Killing of Staphylococcus aureus. Cell Host Microbe 2011, 10, 158–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1104.Gilston BA; Skaar EP; Chazin WJ Binding of Transition Metals to S100 Proteins. Sci China Life Sci 2016, 59, 792–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1105.Hood MI; Mortensen BL; Moore JL; Zhang Y; Kehl-Fie TE; Sugitani N; Chazin WJ; Caprioli RM; Skaar EP Identification of an Acinetobacter baumannii Zinc Acquisition System That Facilitates Resistance to Calprotectin-Mediated Zinc Sequestration. PLoS Pathog 2012, 8, e1003068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1106.Besold AN; Gilston BA; Radin JN; Ramsoomair C; Culbertson EM; Li CX; Cormack BP; Chazin WJ; Kehl-Fie TE; Culotta VC Role of Calprotectin in Withholding Zinc and Copper from Candida albicans. Infect. Immun 2018, 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1107.Raquil MA; Anceriz N; Rouleau P; Tessier PA Blockade of Antimicrobial Proteins S100A8 and S100A9 Inhibits Phagocyte Migration to the Alveoli in Streptococcal Pneumonia. J. Immunol 2008, 180, 3366–3374. [DOI] [PubMed] [Google Scholar]
- 1108.Rink L; Haase H Zinc Homeostasis and Immunity. Trends Immunol 2007, 28, 1–4. [DOI] [PubMed] [Google Scholar]
- 1109.Mortensen BL; Rathi S; Chazin WJ; Skaar EP Acinetobacter baumannii Response to Host-Mediated Zinc Limitation Requires the Transcriptional Regulator Zur. J. Bacteriol 2014, 196, 2616–2626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1110.Patzer SI; Hantke K The ZnuABC High-Affinity Zinc Uptake System and Its Regulator Zur in Escherichia coli. Mol. Microbiol 1998, 28, 1199–1210. [DOI] [PubMed] [Google Scholar]
- 1111.Lee JW; Helmann JD Functional Specialization within the Fur Family of Metalloregulators. BioMetals 2007, 20, 485–499. [DOI] [PubMed] [Google Scholar]
- 1112.Outten CE; O’Halloran TV Femtomolar Sensitivity of Metalloregulatory Proteins Controlling Zinc Homeostasis. Science 2001, 292, 2488–2492. [DOI] [PubMed] [Google Scholar]
- 1113.Lewis DA; Klesney-Tait J; Lumbley SR; Ward CK; Latimer JL; Ison CA; Hansen EJ Identification of the ZnuA-Encoded Periplasmic Zinc Transport Protein of Haemophilus ducreyi. Infect. Immun 1999, 67, 5060–5068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1114.Campoy S; Jara M; Busquets N; Perez De Rozas AM; Badiola I; Barbe J Role of the High-Affinity Zinc Uptake ZnuABC System in Salmonella enterica Serovar Typhimurium Virulence. Infect. Immun 2002, 70, 4721–4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1115.Garrido ME; Bosch M; Medina R; Llagostera M; Perez de Rozas AM; Badiola I; Barbe J The High-Affinity Zinc-Uptake System ZnuACB Is under Control of the Iron-Uptake Regulator (Fur) Gene in the Animal Pathogen Pasteurella multocida. FEMS Microbiol. Lett 2003, 221, 31–37. [DOI] [PubMed] [Google Scholar]
- 1116.Kim S; Watanabe K; Shirahata T; Watarai M Zinc Uptake System (ZnuA Locus) of Brucella abortus Is Essential for Intracellular Survival and Virulence in Mice. J. Vet. Med. Sci 2004, 66, 1059–1063. [DOI] [PubMed] [Google Scholar]
- 1117.Yang X; Becker T; Walters N; Pascual DW Deletion of ZnuA Virulence Factor Attenuates Brucella abortus and Confers Protection against Wild-Type Challenge. Infect. Immun 2006, 74, 3874–3879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1118.Ammendola S; Pasquali P; Pistoia C; Petrucci P; Petrarca P; Rotilio G; Battistoni A High-Affinity Zn2+ Uptake System ZnuABC Is Required for Bacterial Zinc Homeostasis in Intracellular Environments and Contributes to the Virulence of Salmonella enterica. Infect. Immun 2007, 75, 5867–5876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1119.Davis LM; Kakuda T; DiRita VJ A Campylobacter jejuni ZnuA Orthologue Is Essential for Growth in Low-Zinc Environments and Chick Colonization. J. Bacteriol 2009, 191, 1631–1640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1120.Sabri M; Houle S; Dozois CM Roles of the Extraintestinal Pathogenic Escherichia coli ZnuACB and ZupT Zinc Transporters During Urinary Tract Infection. Infect. Immun 2009, 77, 1155–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1121.Corbett D; Wang J; Schuler S; Lopez-Castejon G; Glenn S; Brough D; Andrew PW; Cavet JS; Roberts IS Two Zinc Uptake Systems Contribute to the Full Virulence of Listeria monocytogenes During Growth in Vitro and in Vivo. Infect. Immun 2012, 80, 14–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1122.Yatsunyk LA; Easton JA; Kim LR; Sugarbaker SA; Bennett B; Breece RM; Vorontsov II; Tierney DL; Crowder MW; Rosenzweig AC Structure and Metal Binding Properties of ZnuA, a Periplasmic Zinc Transporter from Escherichia coli. J. Biol. Inorg. Chem 2008, 13, 271–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1123.Petrarca P; Ammendola S; Pasquali P; Battistoni A The Zur-Regulated ZinT Protein Is an Auxiliary Component of the High-Affinity ZnuABC Zinc Transporter That Facilitates Metal Recruitment During Severe Zinc Shortage. J. Bacteriol 2010, 192, 1553–1564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1124.Pederick VG; Eijkelkamp BA; Begg SL; Ween MP; McAllister LJ; Paton JC; McDevitt CA ZnuA and Zinc Homeostasis in Pseudomonas aeruginosa. Sci Rep 2015, 5, 13139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1125.Blindauer CA Bacterial Metallothioneins: Past, Present, and Questions for the Future. J. Biol. Inorg. Chem 2011, 16, 1011–1024. [DOI] [PubMed] [Google Scholar]
- 1126.Robinson NJ; Whitehall SK; Cavet JS Microbial Metallothioneins. Adv. Microb. Physiol 2001, 44, 183–213. [DOI] [PubMed] [Google Scholar]
- 1127.Helbig K; Bleuel C; Krauss GJ; Nies DH Glutathione and Transition-Metal Homeostasis in Escherichia coli. J. Bacteriol 2008, 190, 5431–5438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1128.Nairn BL; Lonergan ZR; Wang J; Braymer JJ; Zhang Y; Calcutt MW; Lisher JP; Gilston BA; Chazin WJ; de Crecy-Lagard V, et al. The Response of Acinetobacter baumannii to Zinc Starvation. Cell Host Microbe 2016, 19, 826–836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1129.Capdevila DA; Edmonds KA; Giedroc DP Metallochaperones and Metalloregulation in Bacteria. Essays Biochem 2017, 61, 177–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1130.Haas CE; Rodionov DA; Kropat J; Malasarn D; Merchant SS; de Crecy-Lagard V A Subset of the Diverse COG0523 Family of Putative Metal Chaperones Is Linked to Zinc Homeostasis in All Kingdoms of Life. BMC Genomics 2009, 10, 470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1131.Sydor AM; Jost M; Ryan KS; Turo KE; Douglas CD; Drennan CL; Zamble DB Metal Binding Properties of Escherichia coli YjiA, a Member of the Metal Homeostasis-Associated COG0523 Family of GTPases. Biochemistry 2013, 52, 1788–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1132.Jordan MR; Wang J; Weiss A; Skaar EP; Capdevila DA; Giedroc DP Mechanistic Insights into the Metal-Dependent Activation of Zn(II)-Dependent Metallochaperones. Inorg. Chem 2019, 58, 13661–13672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1133.Easton JA; Thompson P; Crowder MW Time-Dependent Translational Response of E. coli to Excess Zn(II). J Biomol Tech 2006, 17, 303–307. [PMC free article] [PubMed] [Google Scholar]
- 1134.Sigdel TK; Cilliers R; Gursahaney PR; Thompson P; Easton JA; Crowder MW Probing the Adaptive Response of Escherichia coli to Extracellular Zn(II). BioMetals 2006, 19, 461–471. [DOI] [PubMed] [Google Scholar]
- 1135.Conejo MC; Garcia I; Martinez-Martinez L; Picabea L; Pascual A Zinc Eluted from Siliconized Latex Urinary Catheters Decreases OprD Expression, Causing Carbapenem Resistance in Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2003, 47, 2313–2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1136.Lim CK; Hassan KA; Penesyan A; Loper JE; Paulsen IT The Effect of Zinc Limitation on the Transcriptome of Pseudomonas protegens Pf-5. Environ. Microbiol 2013, 15, 702–715. [DOI] [PubMed] [Google Scholar]
- 1137.Stork M; Bos MP; Jongerius I; de Kok N; Schilders I; Weynants VE; Poolman JT; Tommassen J An Outer Membrane Receptor of Neisseria meningitidis Involved in Zinc Acquisition with Vaccine Potential. PLoS Pathog 2010, 6, e1000969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1138.Noinaj N; Guillier M; Barnard TJ; Buchanan SK TonB-Dependent Transporters: Regulation, Structure, and Function. Annu. Rev. Microbiol 2010, 64, 43–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1139.Calmettes C; Ing C; Buckwalter CM; El Bakkouri M; Chieh-Lin Lai C; Pogoutse A; Gray-Owen SD; Pomes R; Moraes TF The Molecular Mechanism of Zinc Acquisition by the Neisserial Outer-Membrane Transporter ZnuD. Nat Commun 2015, 6, 7996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1140.Johnstone TC; Nolan EM Beyond Iron: Non-Classical Biological Functions of Bacterial Siderophores. Dalton Trans 2015, 44, 6320–6339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1141.Watly J; Potocki S; Rowinska-Zyrek M Zinc Homeostasis at the Bacteria/Host Interface-from Coordination Chemistry to Nutritional Immunity. Chemistry 2016, 22, 15992–16010. [DOI] [PubMed] [Google Scholar]
- 1142.Leach LH; Morris JC; Lewis TA The Role of the Siderophore Pyridine-2,6-Bis (Thiocarboxylic Acid) (PDTC) in Zinc Utilization by Pseudomonas putida DSM 3601. BioMetals 2007, 20, 717–726. [DOI] [PubMed] [Google Scholar]
- 1143.Braud A; Geoffroy V; Hoegy F; Mislin GL; Schalk IJ Presence of the Siderophores Pyoverdine and Pyochelin in the Extracellular Medium Reduces Toxic Metal Accumulation in Pseudomonas aeruginosa and Increases Bacterial Metal Tolerance. Environ Microbiol Rep 2010, 2, 419–425. [DOI] [PubMed] [Google Scholar]
- 1144.Kobayashi S; Nakai H; Ikenishi Y; Sun WY; Ozaki M; Hayase Y; Takeda R Micacocidin A, B and C, Novel Antimycoplasma Agents from Pseudomonas Sp. II. Structure Elucidation. J Antibiot (Tokyo) 1998, 51, 328–332. [DOI] [PubMed] [Google Scholar]
- 1145.Braud A; Hoegy F; Jezequel K; Lebeau T; Schalk IJ New Insights into the Metal Specificity of the Pseudomonas aeruginosa Pyoverdine-Iron Uptake Pathway. Environ. Microbiol 2009, 11, 1079–1091. [DOI] [PubMed] [Google Scholar]
- 1146.Bobrov AG; Kirillina O; Fetherston JD; Miller MC; Burlison JA; Perry RD The Yersinia pestis Siderophore, Yersiniabactin, and the ZnuABC System Both Contribute to Zinc Acquisition and the Development of Lethal Septicaemic Plague in Mice. Mol. Microbiol 2014, 93, 759–775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1147.Mastropasqua MC; D’Orazio M; Cerasi M; Pacello F; Gismondi A; Canini A; Canuti L; Consalvo A; Ciavardelli D; Chirullo B, et al. Growth of Pseudomonas aeruginosa in Zinc Poor Environments Is Promoted by a Nicotianamine-Related Metallophore. Mol. Microbiol 2017, 106, 543–561. [DOI] [PubMed] [Google Scholar]
- 1148.Si M; Wang Y; Zhang B; Zhao C; Kang Y; Bai H; Wei D; Zhu L; Zhang L; Dong TG, et al. The Type VI Secretion System Engages a Redox-Regulated Dual-Functional Heme Transporter for Zinc Acquisition. Cell Rep 2017, 20, 949–959. [DOI] [PubMed] [Google Scholar]
- 1149.Wang T; Si M; Song Y; Zhu W; Gao F; Wang Y; Zhang L; Zhang W; Wei G; Luo ZQ, et al. Type VI Secretion System Transports Zn2+ to Combat Multiple Stresses and Host Immunity. PLoS Pathog 2015, 11, e1005020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1150.Stork M; Grijpstra J; Bos MP; Manas Torres C; Devos N; Poolman JT; Chazin WJ; Tommassen J Zinc Piracy as a Mechanism of Neisseria meningitidis for Evasion of Nutritional Immunity. PLoS Pathog 2013, 9, e1003733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1151.Lonergan ZR; Nairn BL; Wang J; Hsu YP; Hesse LE; Beavers WN; Chazin WJ; Trinidad JC; VanNieuwenhze MS; Giedroc DP, et al. An Acinetobacter baumannii, Zinc-Regulated Peptidase Maintains Cell Wall Integrity During Immune-Mediated Nutrient Sequestration. Cell Rep 2019, 26, 2009–2018 e2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1152.Murphy SG; Alvarez L; Adams MC; Liu S; Chappie JS; Cava F; Dorr T Endopeptidase Regulation as a Novel Function of the Zur-Dependent Zinc Starvation Response. mBio 2019, 10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1153.Shin KS; Son BR; Hong SB; Kim J Dipicolinic Acid-Based Disk Methods for Detection of Metallo-Beta-Lactamase-Producing Pseudomonas Spp. And Acinetobacter Spp. Diagn. Microbiol. Infect. Dis 2008, 62, 102–105. [DOI] [PubMed] [Google Scholar]
- 1154.Principe L; Vecchio G; Sheehan G; Kavanagh K; Morroni G; Viaggi V; di Masi A; Giacobbe DR; Luzzaro F; Luzzati R, et al. Zinc Chelators as Carbapenem Adjuvants for Metallo-Beta-Lactamase-Producing Bacteria: In Vitro and in Vivo Evaluation. Microb. Drug Resist 2020, 26, 1133–1143. [DOI] [PubMed] [Google Scholar]
- 1155.Asempa TE; Abdelraouf K; Nicolau DP Metallo-Beta-Lactamase Resistance in Enterobacteriaceae Is an Artefact of Currently Utilized Antimicrobial Susceptibility Testing Methods. J. Antimicrob. Chemother 2020, 75, 997–1005. [DOI] [PubMed] [Google Scholar]
- 1156.Workneh M; Yee R; Simner PJ Phenotypic Methods for Detection of Carbapenemase Production in Carbapenem-Resistant Organisms: What Method Should Your Laboratory Choose? Clin. Microbiol. Newsl 2019, 41, 11–22. [Google Scholar]
- 1157.Chibabhai V; Nana T; Bosman N; Thomas T; Lowman W Were All Carbapenemases Created Equal? Treatment of NDM-Producing Extensively Drug-Resistant Enterobacteriaceae: A Case Report and Literature Review. Infection 2018, 46, 1–13. [DOI] [PubMed] [Google Scholar]
- 1158.Peirano G; Ahmed-Bentley J; Woodford N; Pitout JD New Delhi Metallo-Beta-Lactamase from Traveler Returning to Canada. Emerg Infect Dis 2011, 17, 242–244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1159.Zhang X; Li X; Wang M; Yue H; Li P; Liu Y; Cao W; Yao D; Liu L; Zhou X, et al. Outbreak of NDM-1-Producing Klebsiella pneumoniae Causing Neonatal Infection in a Teaching Hospital in Mainland China. Antimicrob. Agents Chemother 2015, 59, 4349–4351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1160.Wiskirchen DE; Nordmann P; Crandon JL; Nicolau DP Efficacy of Humanized Carbapenem Exposures against New Delhi Metallo-Beta-Lactamase (NDM-1)-Producing Enterobacteriaceae in a Murine Infection Model. Antimicrob. Agents Chemother 2013, 57, 3936–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1161.Hobson CA; Cointe A; Bidet P; Poupon J; Bonacorsi S; Birgy A Urine Zinc Concentrations Allow Proper Expression of Metallo-Beta-Lactamases in Enterobacteriaceae. J. Antimicrob. Chemother 2020, 75, 3077–3079. [DOI] [PubMed] [Google Scholar]
- 1162.Girlich D; Poirel L; Nordmann P Value of the Modified Hodge Test for Detection of Emerging Carbapenemases in Enterobacteriaceae. J. Clin. Microbiol 2012, 50, 477–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1163.Selevsek N; Rival S; Tholey A; Heinzle E; Heinz U; Hemmingsen L; Adolph HW Zinc Ion-Induced Domain Organization in Metallo-Beta-Lactamases: A flexible “Zinc Arm” for Rapid Metal Ion Transfer? J. Biol. Chem 2009, 284, 16419–16431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1164.Periyannan G; Shaw PJ; Sigdel T; Crowder MW In Vivo Folding of Recombinant Metallo-Beta-Lactamase L1 Requires the Presence of Zn(Ii). Protein Sci 2004, 13, 2236–2243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1165.Su MY; Som N; Wu CY; Su SC; Kuo YT; Ke LC; Ho MR; Tzeng SR; Teng CH; Mengin-Lecreulx D, et al. Structural Basis of Adaptor-Mediated Protein Degradation by the Tail-Specific PDZ-Protease Prc. Nat Commun 2017, 8, 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1166.Krojer T; Sawa J; Schafer E; Saibil HR; Ehrmann M; Clausen T Structural Basis for the Regulated Protease and Chaperone Function of DegP. Nature 2008, 453, 885–890. [DOI] [PubMed] [Google Scholar]
- 1167.Vazquez-Laslop N; Lee H; Hu R; Neyfakh AA Molecular Sieve Mechanism of Selective Release of Cytoplasmic Proteins by Osmotically Shocked Escherichia coli. J. Bacteriol 2001, 183, 2399–2404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1168.Materon IC; Queenan AM; Koehler TM; Bush K; Palzkill T Biochemical Characterization of Beta-Lactamases Bla1 and Bla2 from Bacillus anthracis. Antimicrob. Agents Chemother 2003, 47, 2040–2042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1169.Lim HM; Pene JJ; Shaw RW Cloning, Nucleotide Sequence, and Expression of the Bacillus cereus 5/B/6 Beta-Lactamase Ii Structural Gene. J. Bacteriol 1988, 170, 2873–2878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1170.Matsumura Y; Pitout JD Recent Advances in the Laboratory Detection of Carbapenemase-Producing Enterobacteriaceae. Expert Rev Mol Diagn 2016, 16, 783–794. [DOI] [PubMed] [Google Scholar]
- 1171.Osei Sekyere J; Govinden U; Essack SY Review of Established and Innovative Detection Methods for Carbapenemase-Producing Gram-Negative Bacteria. J. Appl. Microbiol 2015, 119, 1219–1233. [DOI] [PubMed] [Google Scholar]
- 1172.Viau R; Frank KM; Jacobs MR; Wilson B; Kaye K; Donskey CJ; Perez F; Endimiani A; Bonomo RA Intestinal Carriage of Carbapenemase-Producing Organisms: Current Status of Surveillance Methods. Clin. Microbiol. Rev 2016, 29, 1–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1173.Lutgring JD; Limbago BM The Problem of Carbapenemase-Producing-Carbapenem-Resistant-Enterobacteriaceae Detection. J. Clin. Microbiol 2016, 54, 529–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1174.Lupo A; Papp-Wallace KM; Sendi P; Bonomo RA; Endimiani A Non-Phenotypic Tests to Detect and Characterize Antibiotic Resistance Mechanisms in Enterobacteriaceae. Diagn. Microbiol. Infect. Dis 2013, 77, 179–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1175.Tamma PD; Simner PJ Phenotypic Detection of Carbapenemase-Producing Organisms from Clinical Isolates. J. Clin. Microbiol 2018, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1176.Bonomo RA; Burd EM; Conly J; Limbago BM; Poirel L; Segre JA; Westblade LF Carbapenemase-Producing Organisms: A Global Scourge. Clin. Infect. Dis 2018, 66, 1290–1297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1177.van Belkum A; Dunne WM Jr. Next-Generation Antimicrobial Susceptibility Testing. J. Clin. Microbiol 2013, 51, 2018–2024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1178.Nordmann P; Poirel L; Dortet L Rapid Detection of Carbapenemase-Producing Enterobacteriaceae. Emerg Infect Dis 2012, 18, 1503–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1179.Tijet N; Boyd D; Patel SN; Mulvey MR; Melano RG Evaluation of the Carba NP Test for Rapid Detection of Carbapenemase-Producing Enterobacteriaceae and Pseudomonas aeruginosa. Antimicrob. Agents Chemother 2013, 57, 4578–4580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1180.Dortet L; Poirel L; Errera C; Nordmann P Carbacineto NP Test for Rapid Detection of Carbapenemase-Producing Acinetobacter Spp. J. Clin. Microbiol 2014, 52, 2359–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1181.Dortet L; Poirel L; Nordmann P Rapid Identification of Carbapenemase Types in Enterobacteriaceae and Pseudomonas Spp. By Using a Biochemical Test. Antimicrob. Agents Chemother 2012, 56, 6437–6440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1182.Citron DM; Ostovari MI; Karlsson A; Goldstein EJ Evaluation of the E Test for Susceptibility Testing of Anaerobic Bacteria. J. Clin. Microbiol 1991, 29, 2197–2203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1183.Walsh TR; Bolmstrom A; Qwarnstrom A; Gales A Evaluation of a New ETest for Detecting Metallo-Beta-Lactamases in Routine Clinical Testing. J. Clin. Microbiol 2002, 40, 2755–2759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1184.Picao RC; Andrade SS; Nicoletti AG; Campana EH; Moraes GC; Mendes RE; Gales AC Metallo-Beta-Lactamase Detection: Comparative Evaluation of double-Disk Synergy Versus Combined Disk Tests for IMP-, GIM-, SIM-, SPM-, or VIM-Producing Isolates. J. Clin. Microbiol 2008, 46, 2028–2037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1185.Lee K; Lim YS; Yong D; Yum JH; Chong Y Evaluation of the Hodge Test and the Imipenem-EDTA double-Disk Synergy Test for Differentiating Metallo-Beta-Lactamase-Producing Isolates of Pseudomonas Spp. And Acinetobacter Spp. J. Clin. Microbiol 2003, 41, 4623–4629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1186.Hrabak J; Studentova V; Walkova R; Zemlickova H; Jakubu V; Chudackova E; Gniadkowski M; Pfeifer Y; Perry JD; Wilkinson K, et al. Detection of NDM-1, VIM-1, KPC, OXA-48, and OXA-162 Carbapenemases by Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry. J. Clin. Microbiol 2012, 50, 2441–2443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1187.Burckhardt I; Zimmermann S Using Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry to Detect Carbapenem Resistance within 1 to 2.5 Hours. J. Clin. Microbiol 2011, 49, 3321–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1188.Oviano M; Barba MJ; Fernandez B; Ortega A; Aracil B; Oteo J; Campos J; Bou G Rapid Detection of OXA-48-Producing Enterobacteriaceae by Matrix-Assisted Laser Desorption Ionization-Time of Flight Mass Spectrometry. J. Clin. Microbiol 2016, 54, 754–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1189.van der Zwaluw K; de Haan A; Pluister GN; Bootsma HJ; de Neeling AJ; Schouls LM The Carbapenem Inactivation Method (CIM), a Simple and Low-Cost Alternative for the Carba NP Test to Assess Phenotypic Carbapenemase Activity in Gram-Negative Rods. PLoS One 2015, 10, e0123690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1190.Pierce VM; Simner PJ; Lonsway DR; Roe-Carpenter DE; Johnson JK; Brasso WB; Bobenchik AM; Lockett ZC; Charnot-Katsikas A; Ferraro MJ, et al. Modified Carbapenem Inactivation Method for Phenotypic Detection of Carbapenemase Production among Enterobacteriaceae. J. Clin. Microbiol 2017, 55, 2321–2333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1191.Uechi K; Tada T; Shimada K; Kuwahara-Arai K; Arakaki M; Tome T; Nakasone I; Maeda S; Kirikae T; Fujita J A Modified Carbapenem Inactivation Method, Cimtris, for Carbapenemase Production in Acinetobacter and Pseudomonas Species. J. Clin. Microbiol 2017, 55, 3405–3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1192.Sfeir MM; Hayden JA; Fauntleroy KA; Mazur C; Johnson JK; Simner PJ; Das S; Satlin MJ; Jenkins SG; Westblade LF EDTA-Modified Carbapenem Inactivation Method: A Phenotypic Method for Detecting Metallo-Beta-Lactamase-Producing Enterobacteriaceae. J. Clin. Microbiol 2019, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1193.Boutal H; Vogel A; Bernabeu S; Devilliers K; Creton E; Cotellon G; Plaisance M; Oueslati S; Dortet L; Jousset A, et al. A Multiplex Lateral Flow Immunoassay for the Rapid Identification of NDM-, KPC-, IMP- and VIM-Type and OXA-48-Like Carbapenemase-Producing Enterobacteriaceae. J. Antimicrob. Chemother 2018, 73, 909–915. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1194.Glupczynski Y; Evrard S; Huang TD; Bogaerts P Evaluation of the Resist-4 K-Set Assay, a Multiplex Immunochromatographic Assay for the Rapid Detection of OXA-48-Like, KPC, VIM and NDM Carbapenemases. J. Antimicrob. Chemother 2019, 74, 1284–1287. [DOI] [PubMed] [Google Scholar]
- 1195.Poirel L; Walsh TR; Cuvillier V; Nordmann P Multiplex PCR for Detection of Acquired Carbapenemase Genes. Diagn. Microbiol. Infect. Dis 2011, 70, 119–123. [DOI] [PubMed] [Google Scholar]
- 1196.Doyle D; Peirano G; Lascols C; Lloyd T; Church DL; Pitout JD Laboratory Detection of Enterobacteriaceae That Produce Carbapenemases. J. Clin. Microbiol 2012, 50, 3877–3880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1197.Naas T; Ergani A; Carrer A; Nordmann P Real-Time PCR for Detection of NDM-1 Carbapenemase Genes from Spiked Stool Samples. Antimicrob. Agents Chemother 2011, 55, 4038–4043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1198.Manchanda V; Rai S; Gupta S; Rautela RS; Chopra R; Rawat DS; Verma N; Singh NP; Kaur IR; Bhalla P Development of Taqman Real-Time Polymerase Chain Reaction for the Detection of the Newly Emerging Form of Carbapenem Resistance Gene in Clinical Isolates of Escherichia coli, Klebsiella pneumoniae, and Acinetobacter baumannii. Indian J Med Microbiol 2011, 29, 249–253. [DOI] [PubMed] [Google Scholar]
- 1199.Diene SM; Bruder N; Raoult D; Rolain JM Real-Time PCR Assay Allows Detection of the New Delhi Metallo-Beta-Lactamase (NDM-1)-Encoding Gene in France. Int. J. Antimicrob. Agents 2011, 37, 544–546. [DOI] [PubMed] [Google Scholar]
- 1200.Bisiklis A; Papageorgiou F; Frantzidou F; Alexiou-Daniel S Specific Detection of blaVIM and blaIMP Metallo-Beta-Lactamase Genes in a Single Real-Time PCR. Clin. Microbiol. Infect 2007, 13, 1201–1203. [DOI] [PubMed] [Google Scholar]
- 1201.Monteiro J; Widen RH; Pignatari AC; Kubasek C; Silbert S Rapid Detection of Carbapenemase Genes by Multiplex Real-Time PCR. J. Antimicrob. Chemother 2012, 67, 906–909. [DOI] [PubMed] [Google Scholar]
- 1202.Traczewski MM; Carretto E; Canton R; Moore NM; Carba RST Multicenter Evaluation of the Xpert Carba-R Assay for Detection of Carbapenemase Genes in Gram-Negative Isolates. J. Clin. Microbiol 2018, 56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1203.Moore NM; Canton R; Carretto E; Peterson LR; Sautter RL; Traczewski MM; Carba RST Rapid Identification of Five Classes of Carbapenem Resistance Genes Directly from Rectal Swabs by Use of the Xpert Carba-R Assay. J. Clin. Microbiol 2017, 55, 2268–2275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1204.Girlich D; Oueslati S; Bernabeu S; Langlois I; Begasse C; Arangia N; Creton E; Cotellon G; Sauvadet A; Dortet L, et al. Evaluation of the BD Max Check-Points CPO Assay for the Detection of Carbapenemase Producers Directly from Rectal Swabs. J Mol Diagn 2020, 22, 294–300. [DOI] [PubMed] [Google Scholar]
- 1205.Lucena Baeza L; Pfennigwerth N; Hamprecht A Rapid and Easy Detection of Carbapenemases in Enterobacterales in the Routine Laboratory Using the New Genepoc Carba/Revogene Carba C Assay. J. Clin. Microbiol 2019, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1206.Liu W; Zou D; Li Y; Wang X; He X; Wei X; Shao C; Li X; Shang W; Yu K, et al. Sensitive and Rapid Detection of the New Delhi Metallo-Beta-Lactamase Gene by Loop-Mediated Isothermal Amplification. J. Clin. Microbiol 2012, 50, 1580–1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1207.Cheng C; Zheng F; Rui Y Rapid Detection of blaNDM, blaKPC, blaIMP, and blaVIM Carbapenemase Genes in Bacteria by Loop-Mediated Isothermal Amplification. Microb. Drug Resist 2014, 20, 533–538. [DOI] [PubMed] [Google Scholar]
- 1208.Notomi T; Okayama H; Masubuchi H; Yonekawa T; Watanabe K; Amino N; Hase T Loop-Mediated Isothermal Amplification of DNA. Nucleic Acids Res 2000, 28, E63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1209.Mori Y; Notomi T Loop-Mediated Isothermal Amplification (LAMP): A Rapid, Accurate, and Cost-Effective Diagnostic Method for Infectious Diseases. J. Infect. Chemother 2009, 15, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1210.Garcia-Fernandez S; Morosini MI; Marco F; Gijon D; Vergara A; Vila J; Ruiz-Garbajosa P; Canton R Evaluation of the Eazyplex(R) Superbug Cre System for Rapid Detection of Carbapenemases and ESBLs in Clinical Enterobacteriaceae Isolates Recovered at Two Spanish Hospitals. J. Antimicrob. Chemother 2015, 70, 1047–1050. [DOI] [PubMed] [Google Scholar]
- 1211.Mancini N; Infurnari L; Ghidoli N; Valzano G; Clementi N; Burioni R; Clementi M Potential Impact of a Microarray-Based Nucleic Acid Assay for Rapid Detection of Gram-Negative Bacteria and Resistance Markers in Positive Blood Cultures. J. Clin. Microbiol 2014, 52, 1242–1245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1212.Dodemont M; De Mendonca R; Nonhoff C; Roisin S; Denis O Performance of the Verigene Gram-Negative Blood Culture Assay for Rapid Detection of Bacteria and Resistance Determinants. J. Clin. Microbiol 2014, 52, 3085–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1213.Cuzon G; Naas T; Bogaerts P; Glupczynski Y; Nordmann P Evaluation of a DNA Microarray for the Rapid Detection of Extended-Spectrum Beta-Lactamases (TEM, SHV and CTX-M), Plasmid-Mediated Cephalosporinases (CMY-2-Like, DHA, FOX, ACC-1, ACT/MIR and CMY-1-Like/MOX) and Carbapenemases (KPC, OXA-48, VIM, IMP and NDM). J. Antimicrob. Chemother 2012, 67, 1865–1869. [DOI] [PubMed] [Google Scholar]
- 1214.Neuert S; Nair S; Day MR; Doumith M; Ashton PM; Mellor KC; Jenkins C; Hopkins KL; Woodford N; de Pinna E, et al. Prediction of Phenotypic Antimicrobial Resistance Profiles from Whole Genome Sequences of Non-Typhoidal Salmonella enterica. Front Microbiol 2018, 9, 592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1215.Hendriksen RS; Bortolaia V; Tate H; Tyson GH; Aarestrup FM; McDermott PF Using Genomics to Track Global Antimicrobial Resistance. Front Public Health 2019, 7, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1216.Su M; Satola SW; Read TD Genome-Based Prediction of Bacterial Antibiotic Resistance. J. Clin. Microbiol 2019, 57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1217.Motro Y; Moran-Gilad J Next-Generation Sequencing Applications in Clinical Bacteriology. Biomol Detect Quantif 2017, 14, 1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1218.Davidi D; Longo LM; Jablonska J; Milo R; Tawfik DS A Bird’s-Eye View of Enzyme Evolution: Chemical, Physicochemical, and Physiological Considerations. Chem. Rev 2018, 118, 8786–8797. [DOI] [PubMed] [Google Scholar]
- 1219.Weinreich DM; Delaney NF; Depristo MA; Hartl DL Darwinian Evolution Can Follow Only Very Few Mutational Paths to Fitter Proteins. Science 2006, 312, 111–114. [DOI] [PubMed] [Google Scholar]
- 1220.Thomas VL; McReynolds AC; Shoichet BK Structural Bases for Stability-Function Tradeoffs in Antibiotic Resistance. J. Mol. Biol 2010, 396, 47–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1221.Meroueh SO; Minasov G; Lee W; Shoichet BK; Mobashery S Structural Aspects for Evolution of Beta-Lactamases from Penicillin-Binding Proteins. J. Am. Chem. Soc 2003, 125, 9612–9618. [DOI] [PubMed] [Google Scholar]
- 1222.Wang X; Minasov G; Shoichet BK Evolution of an Antibiotic Resistance Enzyme Constrained by Stability and Activity Trade-Offs. J. Mol. Biol 2002, 320, 85–95. [DOI] [PubMed] [Google Scholar]
- 1223.Petrosino J; Cantu C 3rd; Palzkill T Beta-Lactamases: Protein Evolution in Real Time. Trends Microbiol 1998, 6, 323–327. [DOI] [PubMed] [Google Scholar]
- 1224.Pabis A; Risso VA; Sanchez-Ruiz JM; Kamerlin SC Cooperativity and Flexibility in Enzyme Evolution. Curr. Opin. Struct. Biol 2018, 48, 83–92. [DOI] [PubMed] [Google Scholar]
- 1225.Abriata LA; Palzkill T; Dal Peraro M How Structural and Physicochemical Determinants Shape Sequence Constraints in a Functional Enzyme. PLoS One 2015, 10, e0118684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1226.Abriata LA; Salverda ML; Tomatis PE Sequence-Function-Stability Relationships in Proteins from Datasets of Functionally Annotated Variants: The Case of TEM Beta-Lactamases. FEBS Lett. 2012, 586, 3330–3335. [DOI] [PubMed] [Google Scholar]
- 1227.Poelwijk FJ; Kiviet DJ; Weinreich DM; Tans SJ Empirical Fitness Landscapes Reveal accessible Evolutionary Paths. Nature 2007, 445, 383–386. [DOI] [PubMed] [Google Scholar]
- 1228.Knies JL; Cai F; Weinreich DM Enzyme Efficiency but Not Thermostability Drives Cefotaxime Resistance Evolution in TEM-1 Beta-Lactamase. Mol. Biol. Evol 2017, 34, 1040–1054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1229.Ortlund EA; Bridgham JT; Redinbo MR; Thornton JW Crystal Structure of an Ancient Protein: Evolution by Conformational Epistasis. Science 2007, 317, 1544–1548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1230.Starr TN; Thornton JW Epistasis in Protein Evolution. Protein Sci 2016, 25, 1204–1218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1231.Lunzer M; Miller SP; Felsheim R; Dean AM The Biochemical Architecture of an Ancient Adaptive Landscape. Science 2005, 310, 499–501. [DOI] [PubMed] [Google Scholar]
- 1232.Hall BG In Vitro Evolution Predicts That the IMP-1 Metallo-Beta-Lactamase Does Not Have the Potential to Evolve Increased Activity against Imipenem. Antimicrob. Agents Chemother 2004, 48, 1032–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1233.Oelschlaeger P Outsmarting Metallo-Beta-Lactamases by Mimicking Their Natural Evolution. J. Inorg. Biochem 2008, 102, 2043–2051. [DOI] [PubMed] [Google Scholar]
- 1234.Ponsard I; Galleni M; Soumillion P; Fastrez J Selection of Metalloenzymes by Catalytic Activity Using Phage Display and Catalytic Elution. ChemBioChem 2001, 2, 253–259. [DOI] [PubMed] [Google Scholar]
- 1235.Park HS; Nam SH; Lee JK; Yoon CN; Mannervik B; Benkovic SJ; Kim HS Design and Evolution of New Catalytic Activity with an Existing Protein Scaffold. Science 2006, 311, 535–538. [DOI] [PubMed] [Google Scholar]
- 1236.Song WJ; Tezcan FA A Designed Supramolecular Protein Assembly with in vivo Enzymatic Activity. Science 2014, 346, 1525–1528. [DOI] [PubMed] [Google Scholar]
- 1237.Song WJ; Yu J; Tezcan FA Importance of Scaffold Flexibility/Rigidity in the Design and Directed Evolution of Artificial Metallo-Beta-Lactamases. J. Am. Chem. Soc 2017, 139, 16772–16779. [DOI] [PubMed] [Google Scholar]
- 1238.Zimmermann W; Rosselet A Function of the Outer Membrane of Escherichia coli as a Permeability Barrier to Beta-Lactam Antibiotics. Antimicrob. Agents Chemother 1977, 12, 368–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1239.Nikaido H; Normark S Sensitivity of Escherichia coli to Various Beta-Lactams Is Determined by the Interplay of Outer Membrane Permeability and Degradation by Periplasmic Beta-Lactamases: A Quantitative Predictive Treatment. Mol. Microbiol 1987, 1, 29–36. [DOI] [PubMed] [Google Scholar]
- 1240.Frere JM; Joris B; Crine M; Martin HH Quantitative Relationship between Sensitivity to Beta-Lactam Antibiotics and Beta-Lactamase Production in Gram-Negative Bacteria--II. Non-Steady-State Treatment and Progress Curves. Biochem. Pharmacol 1989, 38, 1427–1433. [DOI] [PubMed] [Google Scholar]
- 1241.Cohen RD; Pielak GJ A Cell Is More Than the Sum of Its (Dilute) Parts: A Brief History of Quinary Structure. Protein Sci 2017, 26, 403–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1242.Theillet FX; Binolfi A; Frembgen-Kesner T; Hingorani K; Sarkar M; Kyne C; Li C; Crowley PB; Gierasch L; Pielak GJ, et al. Physicochemical Properties of Cells and Their Effects on Intrinsically Disordered Proteins (IDPs). Chem. Rev 2014, 114, 6661–6714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1243.Rivas G; Minton AP Macromolecular Crowding in vitro, in vivo, and in between. Trends Biochem. Sci 2016, 41, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1244.Nordmann P; Girlich D; Poirel L Detection of Carbapenemase Producers in Enterobacteriaceae by Use of a Novel Screening Medium. J. Clin. Microbiol 2012, 50, 2761–2766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 1245.Giannini E; Gonzalez LJ; Vila AJ A Simple Protocol to Characterize Bacterial Cell-Envelope Lipoproteins in a Native-Like Environment. Protein Sci 2019, 28, 2004–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]