

Abstract
Purpose:
Proficient DNA repair by homologous recombination (HR) facilitates resistance to chemo-radiation in glioma stem cells (GSCs). We evaluated whether compromising HR by targeting HSP90, a molecular chaperone required for the function of key HR proteins, using onalespib, a long-acting, brain-penetrant HSP90 inhibitor, would sensitize high-grade gliomas to chemo-radiation in vitro and in vivo.
Experimental Design:
The ability of onalespib to deplete HR client proteins, impair HR repair capacity, and sensitize GBM to chemo-radiation was evaluated in vitro in GSCs, and in vivo using zebrafish and mouse intracranial glioma xenograft models. The effects of HSP90 inhibition on the transcriptome and cytoplasmic proteins was assessed in GSCs and in ex vivo organotypic human glioma slice cultures.
Results:
Treatment with onalespib depleted CHK1 and RAD51, two key proteins of the HR pathway, and attenuated HR repair, sensitizing GSCs to the combination of radiation and temozolomide (TMZ). HSP90 inhibition reprogrammed the transcriptome of GSCs and broadly altered expression of cytoplasmic proteins including known and novel client proteins relevant to GSCs. The combination of onalespib with radiation and TMZ extended survival in a zebra fish and a mouse xenograft model of GBM compared to the standard of care (radiation and TMZ) or onalespib with radiation.
Conclusions:
The results of this study demonstrate that targeting HR by HSP90 inhibition sensitizes GSCs to radiation and chemotherapy and extends survival in zebrafish and mouse intracranial models of GBM. These results provide a preclinical rationale for assessment of HSP90 inhibitors in combination with chemoradiation in GBM patients.
Keywords: DNA repair, Homologous recombination repair, HSP90 inhibition, onalespib, chemoradiation
Introduction
Radiation therapy with concurrent and adjuvant temozolomide (TMZ) following maximum safe surgical resection is the current standard of care treatment for patients with glioblastoma (GBM), a lethal brain tumor with a median survival of less than two years despite aggressive treatment(1). Chemoradiation therapy harnesses the combined DNA damaging effects of fractionated photon radiation therapy and of TMZ, a monofunctional alkylating agent, resulting in irreparable DNA breaks in tumor cells and consequent cell death (2). Ionizing radiation (IR) causes DNA double strand breaks (DSB) by generating high energy free radicals that attack the sugar-phosphate backbone of the DNA strands; this in turn, elicits a DNA damage response (DDR) which initially results in cell cycle arrest in order to attempt strand repair or, in the event of failed repair, leads to activation of apoptotic programs to eliminate the damaged cell. DSB repair is mediated by two major pathways: non-homologous end-joining (NHEJ), a rapid but error prone process or homologous recombination (HR), a slower but high-fidelity repair pathway (3,4). HR is a complex multistep process with various components involved in the sequential recognition, localization and recruitment of partner proteins to double strand breaks in order to orchestrate DNA repair (5). HR initially involves sensing of DS breaks followed by resection and preliminary repair to generate single stranded DNA tails. These DNA ends are recognized by Mre11-Rad50-NBS1 complex which recruits ATM to these foci. In turn, ATM phosphorylates and activates multiple additional substrates including γ-H2AX, a marker of DNA damage as well as BRCA1 and CHK1 which induce cell cycle arrest and assemble the repair complex; the single stranded DNA ends also results in the loading of RAD51 through activation of ATR. Assessment of RAD51 recruitment to the DNA and its resolution in conjunction with the resolution of the γ-H2AX foci hence signals successful HR repair (6). In contrast to IR, TMZ induces aberrant DNA alkylation which through futile mismatch repair cycles requiring a functional MMR system, leads to DSBs and cell death. TMZ-induced DNA methylation is efficiently repaired by the MGMT (O6-methylguanine-DNA methyltransferase) restoring DNA integrity (7). MGMT promoter methylation is known to be a predictor of clinical response to TMZ; however, some subsets of glioma do not respond to TMZ despite MGMT inactivation suggesting the role of additional DNA repair pathways including the base excision repair and the DS break repair pathways that may mediate resistance to TMZ (8). In addition, other proteins known to have roles in HR repair such as ATM, ATR, MRN and RAD51, have been shown to modulate the activity of TMZ in inducing cell cycle arrest and cytotoxicity.
Glioma stem cells (GSCs) are a subpopulation of GBM cells that are postulated to drive resistance to chemo-radiation and lead to tumor recurrence. GSCs have a highly active DNA damage response and a higher DNA repair capacity compared to the normal neural stem cells resulting in greater resistance to radiation (9,10). Several reports demonstrate that targeting HR appears to be sufficient to effectively sensitize GBM cells to DNA damage. (6,9–12). For instance, targeting aspects of the HR pathway with selective inhibitors of ATM (10), CHK1(9) or RAD51 (6) was more effective in radiosensitizing GSCs compared to inhibiting NHEJ (6,11). Similarly, an augmented HR capacity facilitated resistance to TMZ in GSCs (12). These studies were largely conducted using in vitro models and lacked in vivo validation
HSP90 (Heat shock protein 90) is a molecular chaperone which plays a critical role in facilitating the proper folding of various client proteins into their functional forms, stabilizing proteins in the setting of cellular stress and shielding them from proteosome-mediated protein degradation (13). In addition to its role in folding a number of oncogenic survival kinases (13,14), HSP90 also functions as a chaperone for nuclear proteins governing DNA conformation, transcription and DNA repair including those in the HR pathway (15). (16). HSP90 inhibitors such as 17-AAG or NXD30001 have been shown to be radiosensitizers (17–19) or to synergize with TMZ to induce cytotoxicity in GBM cells (20); however, these agents were too toxic for clinical application or not suitable for GBM therapy. Our group first showed the ability of the long acting HSP90 inhibitor, onalespib, to cross the blood brain barrier and synergize with TMZ resulting in improved survival when compared to onalespib or TMZ alone in vivo using both a zebrafish and in a GSC patient derived xenograft model (16). In this study, we establish the mechanistic basis by which HSP90 inhibition by onalespib sensitizes gliomas to chemo-radiation with IR and TMZ in vitro using GSC lines. We demonstrate that exposure to onalespib depletes specific DDR/HR repair client proteins to inhibit HR-mediated DNA repair resulting in the accumulation of unrepaired DNA damage in GSCs, and patient-derived organotypic glioma slice cultures and inducing cell death. These effects of HSP90 inhibition translated into an ability of onalespib to sensitize gliomas to chemoradiation in vivo in two vertebrate models of GBM.
Methods
Cell lines and reagents.
GSC11, GSC23, GSC262, GSC811, GSC214, GSC231 and GSC267, GS2, GS272 and GS711 patient-derived glioma stem cell lines were cultured as neurospheres passaged every 4-7 days in serum-free DMEM/F12 medium containing B-27 Supplement (Life Technologies, Carlsbad, CA), EGF (Gold Bio Technology) and bFGF (FGF2 Gold BioTechnology, St. Louis, MO) (21). U251HF glioma cells were kindly provided by Dr. W. K. Alfred Yung (M. D. Anderson Cancer Center, Houston, TX). The generation and maintenance of the chemiluminescent U251HF-Luc and GSC811-Luc cells were as previously described (16,22). Cell lines were authenticated at the University of Arizona Genetics Core (https://uagc.arl.arizona.edu/services/complete-solutions/cell-line-authentication). Onalespib (AT13387) was purchased from Medkoo Biosciences, Inc. (Morrisville, NC). Ionizing radiation was delivered alone or in combination with onalespib as described in figure legend.
Methylation specific PCR (MSP) analysis of the MGMT promoter.
DNA was extracted from GSC23, GSC214, GSC262, and GSC811 cells and MSP used to analyze positions 118-137 and 174-195 of the MGMT promoter with primers designed to distinguish methylated (met-MGMT) from unmethylated DNA (unmet-MGMT)(5′-3′) using PCR Reagents (Applied Biosystems) as previously described (23) (See Supplementary methods)
Human Glioma Organotypic Slice Culture:
Tissue from resections of recurrent malignant gliomas was obtained in compliance with the Declaration of Helsinki through an institutional review board-approved protocol and written informed consent of the patients undergoing surgery and slice cultures experiments were conducted per our established protocol as described previously (22). Slice cultures were then treated with 0.4μM onalespib or vehicle before being harvested and processed for immunoblots, immunofluorescence, immunohistochemistry or RNA expression analysis.
Immunoblots.
Immunoblotting was performed per manufacturer’s instruction (BIO-RAD, Hercules, CA) with antibodies against RAD51, γ-H2AX, H2AX (EMD Millipore, Temecula, CA), pSer1981ATM, ATM, pThr1989ATR, ATR, pSer345CHK1, CHK1, HSP90, HSP70, pSer308AKT, AKT, pThr202/204ERK1/2, ERK 1/2, pSer240/244S6, S6, p-EGFR, EGFR, cleaved PARP, MSH2, and GAPDH as detailed (Supplementary table 1). The anti-mouse and anti-rabbit IgG-HRP were used as secondary antibodies (GE Healthcare, Piscataway, NJ).
Immunofluorescence Imaging.
GSC262 and GSC811 cells were seeded at density of 4 x 105 on the coated chamber slides, fixed and treated with target-specific primary antibodies and fluorescent secondary antibodies and imaging conducted using confocal microscopy (details provided in Supplementary methods)
Immunohistochemistry and High-Resolution Image Analysis.
Formalin-fixed, paraffin-embedded tissue from normal brain tissue or GBM xenografts were cut into 5 μm thick coronal sections for immunohistochemical staining. Sections were deparaffinized in xylene, dehydrated in ethanol, rinsed with distilled water before incubation with antibodies against EGFR and p-S6 and γ-H2AX (Cell Signaling Technology, Danvers, MA) and visualized using DAB staining kit (Roche-Ventana, Tucson, AZ) according to the manufacturer’s instructions with hematoxylin counterstain. High resolution images were acquired with Aperio ScanScope XT (Aperio, San Diego, CA) at 40x and processed with ImageScope software (Leica Microsystems, Buffalo Grove, IL).
Homologous Recombination Directed Repair Assay (HDR).
HeLa-DR cells were treated with 0.1 μM onalespib for 24 h before being transfected with the pCBASceI plasmid. After 96 h, the number of cells expressing green fluorescence indicating extent of HDR were measured using a Gallios FACSCalibur instrument as described (24).
Comet Assay.
The neutral comet assay, as a means of measuring DNA DSBs, was processed using the CometAssay ESII system (Trevigen) and was performed per manufacturer’s instructions and as described previously (25). Briefly, cell suspensions (1x105 cells/ml) from untreated or onalespib-treated cells were washed with cold PBS, mixed with low melting agarose at 1:10 ration (v/v), and spread onto 2-well CometSlides. The slides were placed in pre-chilled lysis buffer at 4°C for 1h in the dark. After aspiration of the lysis buffer, the slides were placed in TBE at 4°C for 15 minutes, electrophoresed using a CometAssay ESII unit in TBE electrophoresis buffer for 30 minutes at 21V, washed in water and mixed with 70% ethanol for 5 minutes. The slides were air dried at 37°C, stained with SYBR Gold at room temperature for 30 minutes and subsequently imaged by epifluorescence microscopy (Nikon Eclipse 80i) at a magnification of 100x using a FITC filter. Quantification of comet tail moment was scored using Comet Assay IV™ image analysis system (Perceptive Instruments, Suffolk, UK)
Glioma Intracranial Xenograft Zebra Fish and Nude Mouse in vivo Studies.
The animal studies were conducted through a protocol approved by the Institutional Animal Care and Use committee in compliance with the Humane Care and Use of Laboratory Animals Policy. For both the zebrafish and the nude mouse models, intracranial glioma xenografts were generated by implanting U251HF-Luc cells into the forebrain as described previously (16). Briefly, for the zebra fish experiments, after intracerebral glioma cell implantation (day 5), zebrafish (24 animals/group) were randomized to vehicle (DMSO), fractionated radiotherapy (RT) at a dose of 2Gy per day for 5 consecutive days, RT and TMZ at 10μM in fish water (RT+TMZ), RT and onalespib at 0.5μM (RT+AT), or a combination of onalespib and chemoRT for 5 days (from day 5-10). Five days after chemoRT, intracranial tumor burden was determined by imaging the live fish on an Andor spinning disc confocal microscope as described previously (16).
For nude mouse experiments, U251HF-Luc cells were implanted stereotactically (8×105 cells in 2.5μl) into the right frontal lobe of 8-week-old female athymic nude mice (Charles River, Wilmington, MA). After confirmation of tumor formation by chemiluminescence imaging using a IVIS Lumina Series III Pre-clinical In Vivo Imaging System (Perkin Elmer, American Fork, UT), tumor-bearing mice were evenly distributed based on tumor size into four groups (10 mice/group): control (PBS), temozolomide with radiotherapy (RT+TMZ), onalespib (30mg/kg) with radiotherapy (AT+RT), or a combination of onalespib with chemoRT. Temozolomide (1 mg/kg) was administered by oral gavage for 7 days continuously (from day 30-36), RT (2 Gy/day) given for 5 consecutive days (from day 31-35) and onalespib by tail vein injection in two doses (days 32 and 35). Animals were observed daily and euthanized when they showed signs of morbidity (hunched posture and/or weight loss of 20%).
Statistics.
All in vitro experiments were conducted at least three times independently and expressed as mean ± standard error (SEM). Comparisons between groups in vitro and in vivo were performed using two-tailed t-tests, and comparisons between multiple groups were performed using 1-way analysis of variance (ANOVA). In vivo experiments with zebrafish and mouse were analyzed for survival with the Kaplan Meier method and the significance evaluated by log rank test.
Results
DNA damage response (DDR) and HR repair enzymes are expressed in GSCs and are client proteins of HSP90.
MGMT expression status modulates the response of GSCs to both radiation and TMZ (26,27); however, the impact of MGMT expression on tumor cell response to HSP90 inhibitors is unknown. Therefore, we assayed the MGMT promoter methylation status of patient derived GSCs and determined that GSC811, GSC11 and GSC214 were methylated and GSC23, GSC231 GSC262 and GS267 were unmethylated (Fig. 1A).
Figure 1.
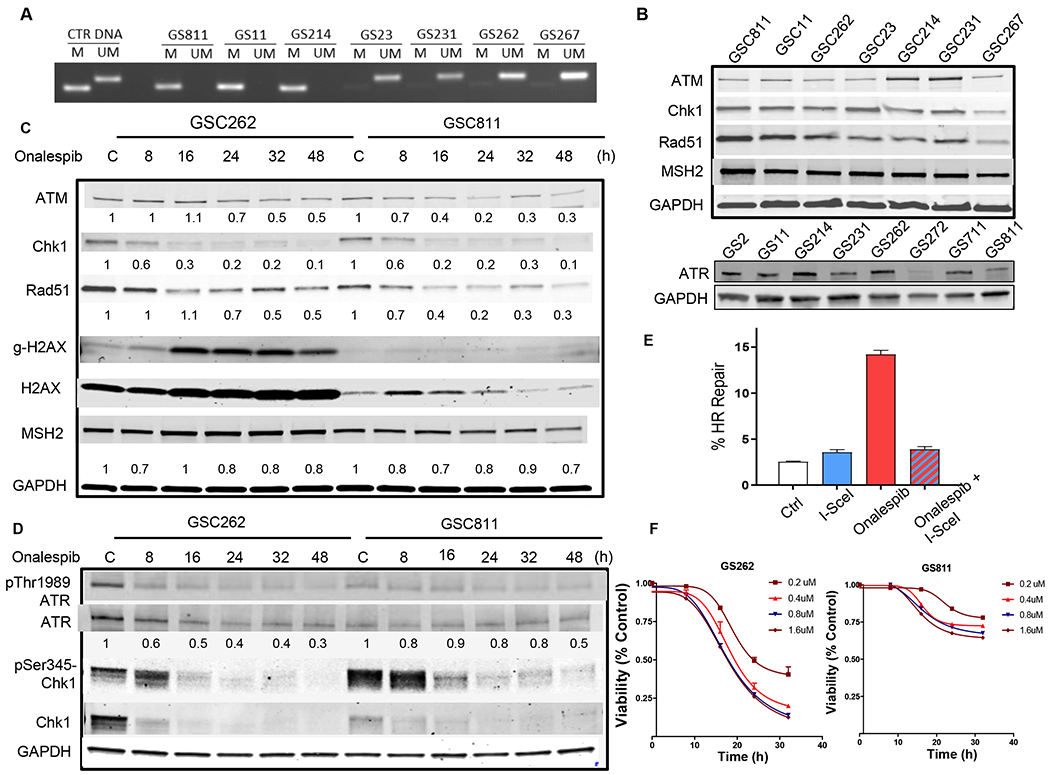
Effect of onalespib treatment on select DNA repair proteins in patient-derived glioma stem cell lines: (A) MGMT promoter methylation status of glioma PDX lines (M, methylated; UM, unmethylated). (B) Expression of ATM, CHK1, RAD51, MSH2 in GSC811, GSC11, GSC262, GSC23, GSC214, GSC231 and GSC267 cells. GAPDH was loaded as a control and the immunoblot is representative of three independent experiment. (C) Effect of onalespib treatment (0.4μM) on levels of ATM, CHK1, RAD51, MSH2 and γ-H2AX in GSC262 and GSC811 cells over time by immunoblotting. GAPDH and total H2AX were evaluated as loading controls. Figure is representative of four independent experiments. (D) Effect of onalespib treatment (0.4μM) over time on the activation of the ATR kinase as measured by levels of p-ATR, ATR, p-CHK1, and CHK1. GAPDH was measured as a loading control. Figure is representative of three independent experiments. (E) Quantitation of percent inhibition of HR in U2OS reporter cells treated with onalespib (0.1for 24h). Graph is representative of three independent experiments in triplicate (p<0.0001, two-tailed p test). (F) Effect of dose and time dependent exposure to onalespib on cell survival in GSC262 and GSC811 cells.
We evaluated the expression of a group of DNA repair proteins selected based on their role in facilitating resistance to both IR and TMZ (11,12,28,29) and found that the HR repair proteins ATM, ATR, CHK1 and RAD51 and MSH2 (a key protein of the MMR pathway) were robustly expressed in all GSCs tested (GSC811, GSC11 and GSC214, GSC23, GSC231, GSC262 and GSC267) irrespective of their MGMT promoter methylation status indicating that GSCs were likely to have proficient DNA repair (Fig. 1B).
HSP90 has been shown to function as a molecular chaperone for the HR proteins ATR, CHK1 and RAD51 (11,30), whereas ATM was shown to be an HSP90 client protein in some tumor types but not in others (31,32). Although multiple kinases and proteins are known to be HSP90 clients, efficacy of the HSP90 inhibitors depended on mediating the rapid and sustained destabilization of client proteins. Therefore, in the context of DNA repair, we tested the ability of onalespib, a brain-penetrant long-acting HSP90 inhibitor, to alter the expression of ATM, ATR, CHK1, RAD51 and MSH2 in GSCs. GSC811 (methylated) and GSC262 (unmethylated) cells were exposed to 0.4 μM onalespib for up to 48h, and showed a rapid, time-dependent and sustained loss in the levels of CHK1 and RAD51 by 16h in both lines (Fig. 1C). In contrast, the levels of ATM and MSH2 (Fig. 1C) showed modest and variable declines in these cell lines indicating that the ability of onalespib to target CHK1 and RAD51 were more likely to influence the ability to disrupt DDR and HR repair in GSCs.
Losses in levels of CHK1 and RAD51 were associated with an increase in the levels of γ-H2AX at 16h in the GS262 line and at 32-48h in the GS811 line. γ-H2AX is a highly sensitive marker for free DNA ends generated by DNA damage or during active replication. In general, when γ–H2AX is activated by DNA damage, it is preceded by the activation of ATR and CHK1 as part of the signalosome that mediates the repair of multiple classes of DNA lesions. To determine whether the γ-H2AX activation seen in response to onalespib was a response to DNA damage or active replication, we exposed GSC262 and GSC811 cells to onalespib and evaluated the activation of ATR as measured by phosphorylation on CHK1. We saw that the levels of p-ATR and ATR decreased in GSC262 steadily over time and to a lesser extent in GSC811 (Fig. 1D). However, the kinase activity of ATR was initially activated in both GSC lines early as seen by robust increases in p-CHK1 at 8h following HSP90 inhibition (Fig. 1D, lower panels). However, by 16h the levels of CHK1 showed steep declines with accompanying decreases in p-CHK1 levels indicating that DNA repair checkpoints regulated by the ATR-CHK1 axis were abrogated in both GSC262 and GSC811 due to depletion of the CHK1 protein (Fig. 1D). These changes in the levels of select DNA repair proteins after HSP90 inhibition were also quantitated (Supplemental Fig. 1). We next examined the consequence of HSP90 inhibition on the HR capacity using a previously published HR reporter assay system generated in U2OS cells (33) This reporter system allows measurement of the capacity for HR repair by determining the ability of the cell to repair a double-strand break introduced into a defective green fluorescent protein reporter gene by a transfected I-SceI endonuclease with successful repair leading to reconstitution of a functional GFP yielding fluorescent signal that is detectable and quantifiable by flow cytometry (33). Untreated cells transfected with the I-SceI endonuclease showed an increase in the number of GFP+ cells indicating successful HR. In contrast, exposure to onalespib reduced the number of GFP+ cells almost to basal levels (5%) indicating sustained inhibition of the HR repair process (Fig. 1E).
Next we evaluated the action of onalespib on the viability of GSC262 and GSC811; GSC262 showed a rapid decline in viability as measured by CellTiter-Glo viability assays (Figure 1F) whereas GS811 showed modest toxicity after exposure to onalespib (Fig. 1F). To determine whether this difference in toxicity was caused by cell cycle changes we evaluated the effect of onalespib on the cell cycle transit and saw that GSC262 cells rapidly underwent cell death (Supplemental Fig. 2A) upon exposure to onalespib accounting for the steep decrease in viability seen in Fig. 1F whereas GSC811 underwent an initial G2/M phase cell cycle arrest (Supplemental Fig. 2B) accounting for the modest early cytotoxicity observed for these cells in Fig. 1F, although this was overcome at later time points suggesting delayed but inevitable cytotoxicity.
Taken together, our results show that key proteins of the HR repair pathway such as CHK1 and RAD51 are sensitive and durable clients of HSP90. Consequently, we show that targeting HSP90 compromises HR repair at multiple nodes of the DDR, caused DNA damage and elicits cytotoxicity irrespective of MGMT methylation status.
Onalespib-mediated depletion of DNA damage sensor/repair proteins prevents DDR in response to radiation and sensitizes GSCs to radiation-induced apoptosis.
DNA damage triggers a phosphorylation-dependent signaling cascade in which ATM, and ATR undergo activation by autophosphorylation and in turn, phosphorylate CHK1 and other DNA repair proteins which culminates in the recruitment of RAD51 to the damaged DNA foci. Quantitation of RAD51 foci in conjunction with resolution of γ-H2AX foci provides a measure of successful HR repair (34). To determine whether the onalespib-mediated inhibition of HR capacity sensitized GSC cells of differing MGMT promoter methylation status to radiation or TMZ, we exposed GSC811, GSC11 (MGMT methylated) and GSC262 and GSC23 (MGMT unmethylated) cells to IR (8 Gy) which resulted in the activation of ATM and ATR kinase activity based on the increase in p-CHK1 in both sets of GSC lines indicating the activation of the DDR in GSCs irrespective of MGMT methylation status. In contrast, pre-treatment of cells with onalespib for 72h prior to IR exposure blocked the increase in p-ATM, p-ATR and p-CHK1 in association with variable declines in ATM and ATR and caused a steep and consistent decline in CHK1 levels across all GSC lines indicating an abrogation of radiation-induced DDR signaling (Fig. 2). Further, we observed upregulation of HSP70 in cells exposed to onalespib alone or in combination with IR, which serves as a surrogate marker of successful HSP90 inhibition (Fig. 2) (35).
Figure 2.
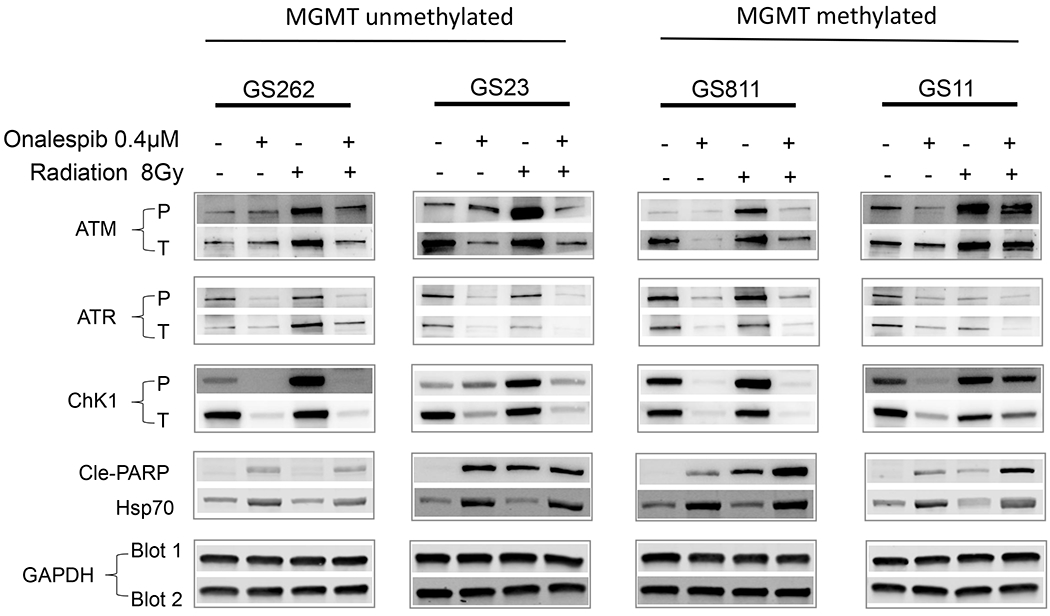
Modulation of IR-mediated DNA damage signaling by onalespib in patient-derived glioma stem cells. Immunoblot analysis of effect on levels of p-ATM, p-ATR and p-CHK1 (as measures of DNA damage signaling) and cleaved PARP (reflecting induction of apoptosis) in GSC811, GSC11, GSC262, and GSC23 cells after IR treatment (8 Gy) and suppression of this effect by pretreatment with onalespib (0.4μM). GAPDH was loaded as a control and HSP70 assayed as a measure of HSP90 inhibition. Figures are representative of three independent experiments.
We then assessed the relationship of onalespib-mediated inhibition of DDR to the accumulation of DNA damage and kinetics of repair by quantitating the appearance and resolution of γ-H2AX foci as a marker of double-strand DNA damage and of RAD51 foci as a marker of active HR. Exposure of GSC262 and GSC811 cells to 3Gy IR caused accumulation of γ-H2AX foci within 30 min of treatment indicating appearance of DNA damage which was largely resolved by 4h post-IR with a concurrent appearance of RAD51 foci indicating active HR repair (Fig. 3A). Exposure of GSCs to onalespib alone resulted in a few foci in both cells. Of note, GSC262 had high levels of γ-H2AX and total H2AX by immunoblot assays at base line (Fig. 1C) which showed a 4-fold induction within 16 h after onalespib treatment (Supplemental Fig. 3). In contrast, γ-H2AX and total H2AX were low in GSC811 but showed a 3-fold induction by 48h (Supplemental Fig. 3) after onalespib treatment; however, the numbers of foci in GSC262 and GSC811 after 24h of onalespib exposure were comparable. These results indicate that formation of foci was a more reliable measure of γ-H2AX at sites of DNA damage compared to immunoblot assessments. However, pretreatment of GSCs with onalespib for 24h prior to IR treatment resulted in a significant increase in the numbers of cells with γ-H2AX foci in both lines, with GSC262 showing a higher number of foci/cell compared to GSC811. Further, these γ-H2AX foci persisted without resolution in both GSC lines indicating unrepaired DNA damage; this was further supported by absence of RAD51 foci in onalespib-treated cells indicating the attenuation of DNA repair (Fig. 3A). These results confirms that HSP90 inhibition abrogates IR-induced dsDNA repair in glioma cells leading to persistent and unrepaired DNA damage.
Figure 3.
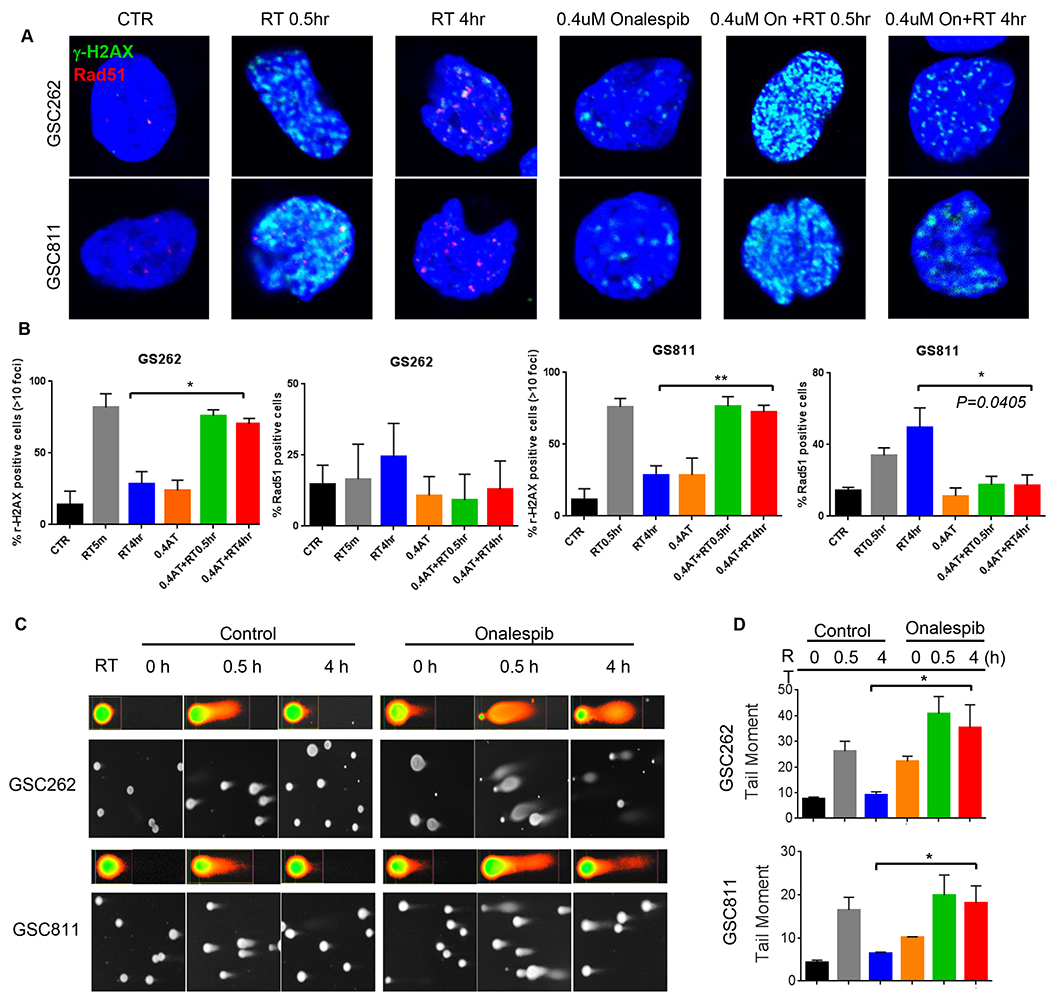
Effect of onalespib on IR-induced DNA damage and subsequent repair in GSC811 and GSC262 cells. (A) Immunofluorescence images (upper panels) of time-course of appearance and resolution of γ-H2AX (marker of DNA damage) and RAD51 foci (marker of ongoing HR repair) after IR-induced DNA damage alone or in combination with onalespib in GSC (upper panels). (B) Quantitation of immunofluorescent γ-H2AX foci and Rad-51 foci in GSCs treated with IR alone or IR+onalespib. Graph represents at least three independent experiments (*=p<0.05, **=p<0.005, two-tailed p test, error bars=SEM). (C) Comet assay imaging of GSC811 and GSC262 cells at 0.5h or 4h after treatment with IR alone or IR+onalespib (0.4μM) showing persistence of comet tails in the onalespib treated cells at 4h post-treatment indicating failure to repair IR-induced DNA strand breaks. (D) Quantification of comet tail moment indicating onalespib-mediated abrogation of cellular DNA repair after IR. Graphs represent triplicate experiments (*=p< 0.05). (RT=radiation therapy; AT: onalespib)
To further confirm the effects of onalespib on DNA repair, we utilized the neutral comet assay, which allows visualization and quantitation of double strand DNA damage. Nuclei with DNA strand breaks form comets under an electrical field whereas repaired DNA results in a regression in the comet tail length. GSC cells exposed to IR for 30 min showed an increase in comet tail length and moment which subsequently decreased by 4h post IR indicating successful DNA repair. In contrast, GSCs pretreated with 0.4 μM onalespib for 24h before IR treatment showed persistent comets indicating unresolved DNA damage confirming lack of active DNA repair (Fig. 3Cand 3D).
Similar to IR, exposure of GSC811, GSC11, GSC262 and GSC23 to TMZ activated ATM and ATR based on the phosphorylation of p-CHK1with GS23 having the maximal induction in p-CHK1 followed by GS811, GS11 and GS262. Activation of ATR, in particular, is linked to resistance to TMZ (36). Pre-exposure to onalespib prevented the ATM- and ATR-driven activation of CHK1 (Fig. 4A). Correspondingly, GSC811, GSC11, GSC262 and GSC23 cells exposed to either onalespib alone or onalespib plus TMZ activated apoptosis as measured by the appearance of cleaved PARP with GSC811 and GSC23 also showing sensitivity to TMZ as a single agent (Fig. 4A).
Figure 4.
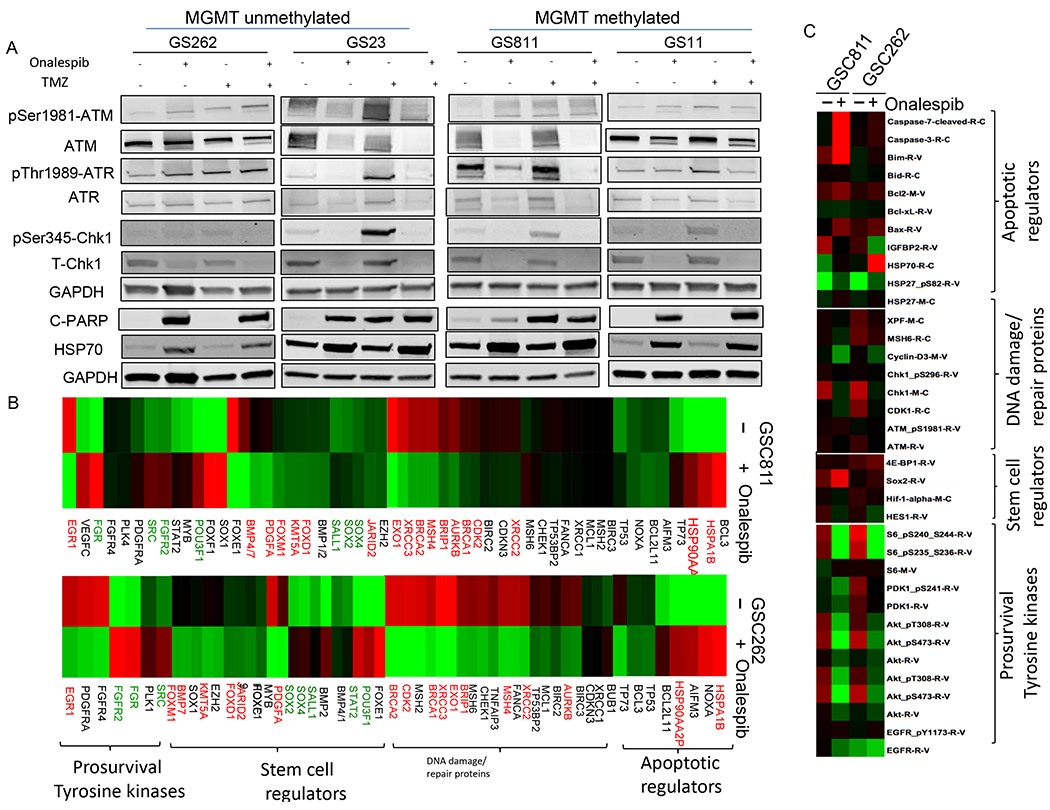
Modulation of TMZ-mediated DNA damage signaling by onalespib in patient-derived glioma stem cells and the effect of onalespib on transcriptome and proteome in patient-derived glioma cells. (A) Immunoblot analysis of effect on levels of p-ATM, p-ATR, and p-CHK1 (as measures of DNA damage signaling) and cleaved PARP (reflecting induction of apoptosis) in GSC811, GSC11, GSC262, and GSC23 cells after TMZ treatment and suppression of this effect by pretreatment with onalespib (0.4μM). GAPDH was loaded as a control and HSP70 assayed as a measure of HSP90 inhibition. Figures are representative of three independent experiments. (B) RNA-seq data analysis of expression of various classes of genes on GSC811 and GSC262 exposed to onalespib (0.4 μM for 24h) compared with untreated control cells. Upregulated (red) and down regulated (green) genes are indicated. (C) Changes in select set of proteins in GSCs exposed to onalespib assessed by the RPPA assay.
Onalespib-mediated reprogramming of the transcriptome and proteome sensitizes GSCs to chemo and radiation therapy
While most of the cellular HSP90 resides in the cytoplasm to regulate the folding, maturation and chaperoning of cytoplasmic proteins, a small fraction of HSP90 translocates to the nucleus and chaperones nuclear proteins including several DNA repair proteins and transcription factors.(37) We examined the consequence of HSP90 inhibition on nuclear client proteins and its effect on gene expression profiles using RNA Seq and on the cellular proteome using reverse phase proteomic arrays (RPPA). GSC811 and GSC262 cells were exposed to 0.4μM onalespib for 24h after which GSCs were harvested and analyzed by RNA Seq. HSP90 inhibition resulted in a reciprocal upregulation of HSP70 (HSPA1B) and HSP90AA2P (HSP90 pseudogene) in both cell lines which are expected markers confirming HSP90 inhibition. We observed a downregulation in the expression of BRCA1/2 and XRCC2 (key regulators of the HR pathway), XRCC3 (replication fork repair), and MSH4 and EXO1 (mismatch repair) in both GSC lines (Fig. 4B, in red). Similarly, expression levels of the KMT5A and JARID2 (repressive chromatin remodelers) and FoxM1 (transcription factor that promotes radio-resistance) were downregulated (Fig. 4B).
Next, we investigated the consequence of HSP90 inhibition on the proteome of GSC cells using RPPA assay in order to identify changes in client protein levels or their downstream target proteins as a consequence of onalespib treatments. We noted the expected increase in Hsp70 and Hsp27 protein levels confirming HSP90 inhibition. With reference to DNA repair pathways relevant to the current project, HSP90 inhibition resulted in decreased levels of several client proteins including key DNA damage and repair factors such as ATM, XPF, CHK1, γH2AX and CDC25 suggesting a broad effect on DDR at a protein level (Fig. 4C). In addition, in concordance with our previous findings (16) we found that HSP90 inhibition depletes both total and phospho protein levels of key survival kinases such as EGFR, AKT and the downstream signaling kinase such as S6 in glioma cell lines and GSCs indicating complete shutdown of these crucial survival signaling pathways in gliomas. The RPPA array also showed increases in pro-apoptotic Bcl2 family proteins such as BIM (Bcl2L11, also seen in GSC262 at the mRNA levels) and BAX, which are effectors of intrinsic apoptosis pathway components including Caspase 3 and Caspase-7. In addition, the RPPA array showed consistent increases in the levels of the anti-apoptotic protein, Bcl2, possibly as a measure of defense against the strong pro-apoptotic signal induced by HSP90.
HSP90 inhibition depletes HR DNA repair proteins and survival kinases in primary GBM organotypic slice cultures.
To examine whether the changes seen in RNA and protein expression profile in GSC due to HSP90 inhibition in GSCs were relevant to human gliomas, we examined the effects of onalespib in an ex-vivo human glioma organotypic slice culture model. Using our previously published protocol, surgically resected human GBM samples were rapidly transferred from the operating room to the lab and live organotypic human glioma slices were generated using a vibratome (22). These slices also maintain the microenvironment in GBM comprised of stromal cells, vascular endothelial cells, astrocytes, and myeloid cells which often secrete factors that facilitate chemo- and radioresistance in GBM (38). Expression analysis of primary GBM slices exposed to onalespib showed the expected upregulation of HSP70 and HSP90AA (HSP90 pseudogene), a class effect of HSP90 inhibitors. Similar to our findings in GSC, there was a significant decrease in the expression levels of several DNA damage factors including CHK1 (HR), FANCA, XRCC1/3 (HR and replication fork repair), MSH2, MSH4, MSH6 (MMR repair), BRCA1 (HR) and EXO1 (BER). In addition, there was a downregulation of certain stem cell, tyrosine and angiogenic kinases including the FOX group of transcription factors, and survival kinases such as FGR, EGR1, FGFR4, FGFR2, PDGFRA and VEGFC. Conversely, several stem cell factors such as BMP4/7, POU3F1, SOX1, SOX2 and SOX4 were upregulated in response to HSP90 inhibition (Fig. 5A) which could potentially represent cellular attempts to enter into a more resistant stem-like state or could be a consequence of changes in transcription. At a protein level, immunoblot analysis of organotypic GBM slice cultures showed a strong decrease in the levels of the HR proteins, ATM, CHK1 and RAD51, after HSP90 inhibition (Fig. 5B). These results in human GBM patient tissue samples further confirm that the in vitro results in patient derived GSCs may be potentially predictive of similar biological responses in human GBMs in a clinical setting at a gene expression and protein level.
Figure 5.
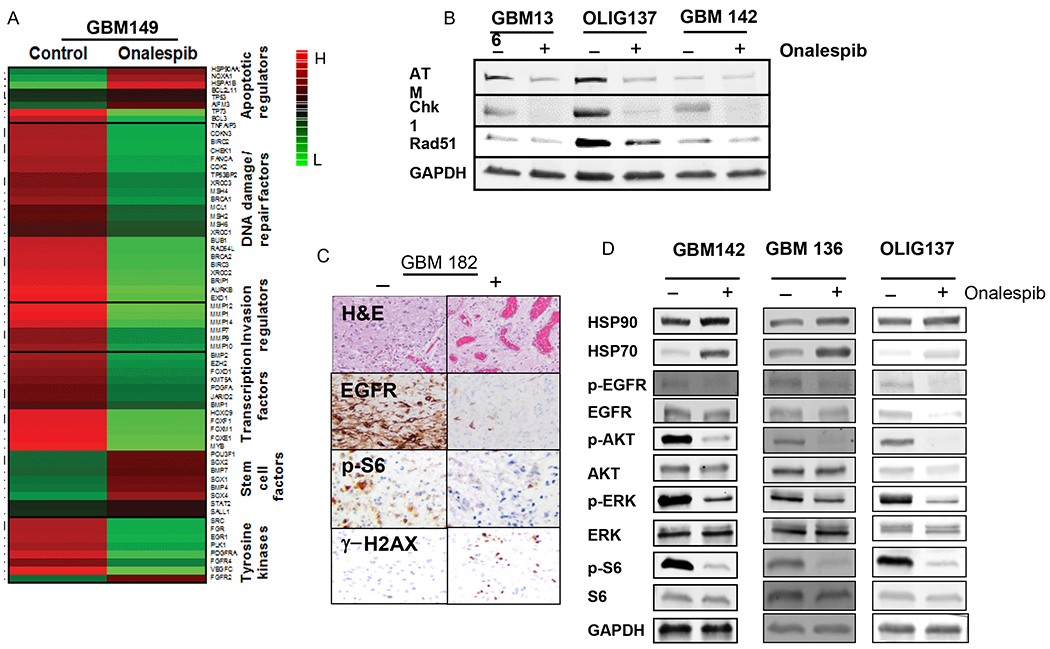
Effects of onalespib (0.4μM) in an ex-vivo patient-derived organotypic slice culture model (A) Changes in gene expression upon treatment of a human glioma slice culture specimen (GBM149) with onalespib as determined by RNA-Seq analysis. (B) Immunoblot analysis of levels of DNA-repair proteins, ATM, CHK1, and RAD51 in human glioma slice culture specimens (GBM136, OLIG137 and GBM142) upon treatment with onalespib. GAPDH was assayed as a loading control.(C) Immunohistochemical assessment of tissue levels of EGFR, p-S6 and γH2AX in a patient-derived tumor slice specimen (GBM182). (D) Immunoblot analysis of human tumor tissue exposed to onalespib or DMSO for levels of HSP90, HSP70, AKT, p-AKT, ERK, p-ERK, S6, p-S6, GAPDH (as loading control) levels.
We have previously shown that exposure to onalespib attenuates EGFR and its downstream survival signaling pathway components in GSCs (16). To determine whether onalespib had similar inhibitory effects in primary brain tumor tissue, we treated organotypic human GBM slices with 0.4μM onalespib for 24 h and examined changes in EGFR pathways components. We observed that untreated GBM slices, as assessed by immunohistochemistry, showed robust expression of EGFR and its downstream signaling mediator, p-S6; however, slices from the same tumor treated with onalespib 0.4μM for 24h showed dramatic reduction of these two proteins suggesting that both upstream and downstream components of this crucial pathway were robustly inhibited by onalespib. Further, an increase in γ-H2AX was noted after exposure to onalespib suggesting induction of DNA damage in these live human tumor slices (Fig. 5C). These results were further confirmed by western blot analysis of human GBM slices treated with onalespib which showed a rapid attenuation of EGFR and its downstream signaling mediators AKT, p-ERK and p-S6 (Fig. 5D). These data from patient-derived tumor tissue further confirm that in addition to depletion of DNA repair related client proteins of HSP90, onalespib can strongly downregulate pro-survival pathways downstream of EGFR, the most prevalent of aberrantly expression receptor tyrosine kinases in GBM.
Onalespib Sensitizes Gliomas in vivo to Chemoradiation in Vertebrate Models of GBM
We have previously demonstrated the utility of zebrafish as a reproducible and efficient vertebrate intracranial xenograft model to screen for efficacy of preclinical therapeutic agents in gliomas (16,39); using this model, we reported that the combination of onalespib and TMZ was superior to onalespib or TMZ alone in decreasing tumor volume and extending survival in glioma-bearing zebrafish. Here, we extend these findings to determine the effect of onalespib in combination with chemoRT in the zebrafish model. U251HF cells expressing green fluorescent protein (U251HF-GFP, 50 cells/animal) were implanted into the midbrain of zebrafish embryos and the resultant tumor growth confirmed 5 days post-transplant (dpt) using fluorescent imaging (Fig. 6A and 6B, 40x magnification). Animals with confirmed intracranial tumors were randomized to the following treatment groups: vehicle (DMSO), radiation alone, TMZ+RT, Onalespib+RT, Onalespib+TMZ+ RT. RT was administered at 2Gy as a single dose; TMZ (10μM), onalespib (0.4μM) and DMSO (1%) were added to fish water from 5 to 10 days post-transplantation. Fish exposed to DMSO alone had the shortest survival; those exposed to Onalespib + RT or TMZ + RT had a longer survival than those exposed to RT alone. Zebrafish bearing intracranial gliomas and exposed to the combination of onalespib, TMZ and RT had the longest survival (Fig. 6C) compared to controls suggesting efficacy of this combination in this in-vivo vertebrate model of GBM.
Figure 6.
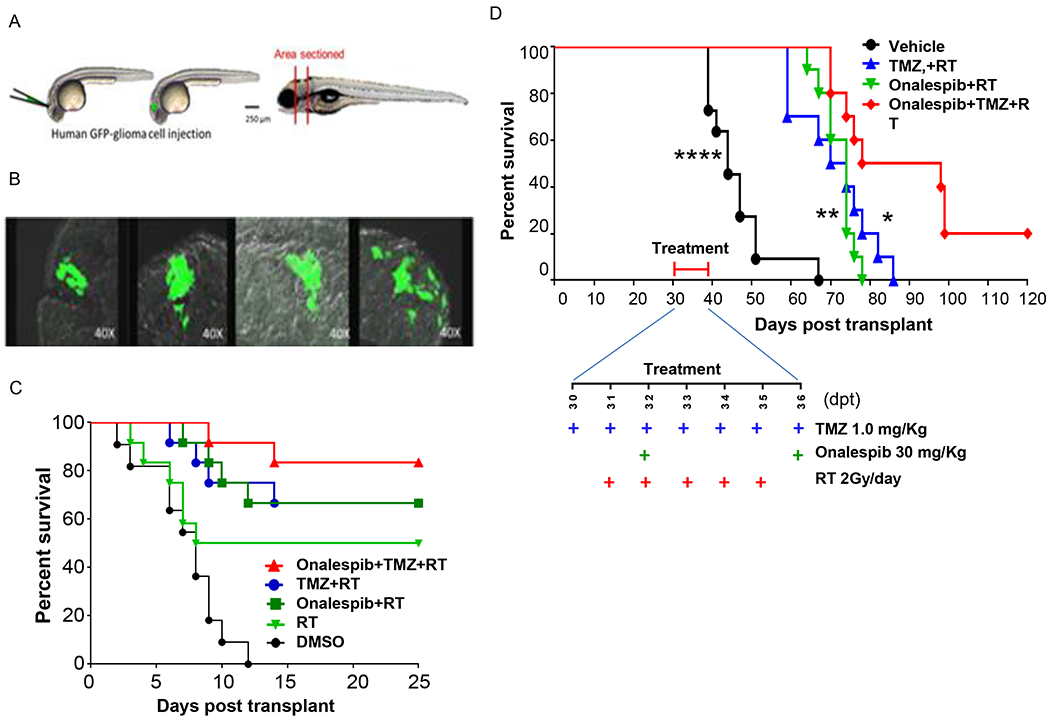
Onalespib-mediated effects on chemoRT therapy and effects on survival in a zebrafish and nude mouse intracranial glioma xenograft models. (A) Schematic diagram of intracranial human U251HF-GFP glioma-xenotransplanted zebrafish model. (B) GFP-expressing diffusely infiltrative tumor cells in intracranial glioma xenograft seen in transverse sections at various levels in the zebrafish brain. (C) Kaplan-Meier survival curves for zebrafish glioma (n=11-12 animals/group). U251HF-GFP xenotransplanted zebrafishes treated with DMSO, RT (2 Gy/day for 5 days), TMZ (10 μmol/L) plus RT, onalespib (0.5 μmol/L) plus RT, or a combination of TMZ (10 μmol/L) and onalespib (0.5 μmol/L) with RT. (D) Kaplan-Meier survival curves for U251HF-Luc xenotransplanted nude mice (n=10-12 mice/group) treated with vehicle (PBS), TMZ (1.0 mg/Kg) + RT (2 Gy/day for 5 days), onalespib (20 mg/Kg) + RT, or a combination of onalespib and TMZ+RT; differences in survival were assessed by the log-rank test. (combination vs. vehicle, ****, P < 0.0001; combination vs. onalespib + RT, **, P < 0.01; combination vs. TMZ + RT, *, P < 0.05) (TMZ=temozolomide; RT= radiotherapy).
To confirm these results in a second in vivo model, we utilized a mammalian mouse intracranial orthotopic glioma model in which mice were implanted with the infiltrative and highly angiogenic U251HF-Luc cells expressing luciferase, in the mouse forebrain allowing monitoring of tumor growth using bioluminescent imaging. Upon confirmation of tumor formation by IVIS imaging, mice were equally assigned based on tumor size to the following treatment groups: PBS, TMZ+RT, onalespib+RT and onalespib+TMZ+RT. Again, while the combination of TMZ+RT and onalespib+RT elicited moderate increases in survival, mice receiving the combination of onalespib+TMZ+RT had the best improvement in median survival (Fig. 6D). These results confirm the efficacy of onalespib against gliomas in vivo and provide support for the potential for the combination of HSP90 inhibition with DNA-damaging treatments such as TMZ and RT against human gliomas.
Discussion
Resistance to chemotherapy and radiation remains a significant barrier to the successful treatment of GBM; this has been attributed to several factors including the presence of the subset of GSCs within the bulk tumor, the adaptive upregulation of survival pathways in tumor cells in response to therapy and tumor heterogeneity that allows resistant clones to emerge during therapy (9,10,40–42). Consequently, pathway-specific targeted therapies based on known molecular pathways have largely failed to improve outcome in patients with GBM.
Activation of evolutionally conserved stress response and DNA repair pathways has been shown to mediate treatment resistance in various malignancies including gliomas regardless of their genetic and biological characteristics (43,44). The current first line standard of care therapy for GBMs and anaplastic astrocytomas consists of a combination of RT with temozolomide which exerts its by inducing DNA damage and apoptosis. Signal transducers such as AKT which are highly active in GBMs, enhance DNA repair and radio-resistance mechanisms (45). Further, proteins involved in HR such as ATM (29), CHK1 (9), BRCA1(46) and RAD51(6) have also been implicated in radio-resistance of gliomas and other tumors. In particular, GSCs exhibit increased HR in comparison to normal neural progenitor cells which is proposed to be one of the major mechanisms by which they exert their effect (6,47). Given that HSP90 serves as a molecular chaperone for several protein components of the HR pathway, it has been noted that high levels of HSP90 are strongly correlated with radio-resistance in various malignancies; conversely, HSP90 inhibition can sensitize cells to radiation (48). We previously showed that onalespib, which has favorable brain pharmacokinetics, decreases the levels of oncogenic survival kinases such as EGFR and its downstream effectors phospho-AKT, ERK and S6 which are highly relevant to radio-resistance. We also reported that HSP90 inhibition enhances the effects of monofunctional alkylator, temozolomide, in vitro and in vivo in gliomas.(16) Extending these results to the effects radiation or chemotherapy induced DNA damage and repair mechanisms in gliomas, we demonstrate that HSP90 inhibition caused a steep decline in the levels of CHK1 and RAD51 protein in patient derived GSCs. This resulted in abolishing HR-mediated repair in a U2OS cell-based HR reporter assay system which may not represent the best reporter system to shed light on the effect of onalespib on the HR repair capacity of glioma cells. However, based on our results where onalespib-treated GSC cells depleted levels of CHK1 and RAD51, were unable to sustain phosphorylation on CHK1 in response to IR or TMZ, demonstrated an absence of RAD51 repair foci and accumulated unrepaired DNA fragments which were reflected by the persistence of γ-H2AX foci, and well as unrepaired DNA Comet tails after DNA damage, suggest that it is very likely that conducting the HR reporter assay in glioma cells would demonstrate a similar strong attenuation in repair capacity.
Although HSP90 inhibitions successfully depletes DNA repair and survival client proteins in vitro across multiple tumor cell lines, prior studies with earlier generation HSP90 inhibitors reported limited success in trials likely due to insufficient inhibition of target client proteins in human tumor tissue. Using onalespib which is a long-acting HSP90 inhibitor, we tested whether HSP90 inhibition could target DNA repair proteins and survival pathways in primary human glioma tissue using an ex vivo organotypic glioma slice culture model. We noted a rapid decline of not only CHK1 and RAD51 in glioma tissue but also of the survival kinase, EGFR and its downstream effectors AKT and ERK, confirming the potential of HSP90 inhibition in abrogating radio-resistance pathways directly in primary GBM tissue.
Assessment of the genome wide effect of HSP90 inhibition in gliomas showed that exposure to onalespib downregulated several additional transcripts including components of other DNA repair pathways such as BER and MMR and several resistance-facilitating chromatin modulators and transcription factors. These results were further confirmed at the protein level by the RPPA analysis demonstrating decreases in several survival kinases that are known HSP90 client proteins. Although many DNA repair proteins which decreased at the transcript level were not represented in the RPPA panel, decreases in specific proteins such as XRCC3 and XPF suggested that HSP90 inhibition can targets multiple additional DNA repair pathways which may otherwise provide a resistance mechanism for the glioma cells by activating compensatory DNA repair pathways. These cell and tumor level data were further confirmed in an in vivo setting in which we utilized a schedule for chemoRT similar to that used in the clinical setting (fractionated RT in combination with temozolomide). Exposure to onalespib strongly sensitized gliomas to chemoRT both a zebrafish and a mouse intracranial xenograft model of GBM. Of particular note, these results were seen in a panel of glioma cell lines and PDX lines which were varied in their MGMT promoter methylation status and other genetic characteristics suggesting that this strategy has the potential to be active across heterogeneous glioma subtypes regardless of their genetic or epigenetic characteristics.
There have been no clinical studies of HSP90 inhibition in human gliomas to date. While single agent activity of HSP90 inhibitors may serve to target multiple relevant signaling pathways in gliomas, this may not provide an adequate cytotoxic signal in these tumors given their extensive tumor heterogeneity which may circumvent such inhibition. Here, we examined the possibility that the most effective strategy to utilize HSP90 inhibitors in this setting would be as sensitizers of radiation therapy and alkylating agents, the current standards of care for malignant gliomas. In this context, our results provide a strong pre-clinical mechanistic and efficacy rationale for combining HSP90 inhibitors with radiation and alkylator therapy in first line and recurrent setting in patients with GBM. A NCI-funded phase I/II trial of HSP90 inhibitors in combination with chemoRT is currently being planned and will test this therapeutic concept in adult patients with newly diagnosed GBM.
Supplementary Material
Translational Relevance.
De novo upregulation of DNA repair pathway components, tumor heterogeneity, and the intrinsic resistance of glioma stem-like cells (GSCs) are mediators of resistance of glioblastoma (GBM) to ionizing radiation (IR) and temozolomide (TMZ). Overcoming DNA repair in a tumor selective manner can hence represent a strategy to restore sensitivity to chemo-radiation therapy. Several key DNA repair and survival proteins in GBM are client proteins of heat shock protein 90 (HSP90), a molecular chaperone required for their proper folding and function. We demonstrate that HSP90 inhibition with onalespib, a long-acting brain penetrant HSP90 inhibitor, promotes degradation of these client proteins, augmenting the efficacy of chemo-radiation in vitro and in vivo. HSP90 inhibition also reprograms the transcriptome and proteome, modifying known and novel client proteins, which may serve as additional therapeutic targets for combination therapies. Our findings provide a strong rationale for assessment of HSP90 inhibitors in combination with chemoradiation therapy in patients with high grade gliomas.
Acknowledgments
We gratefully acknowledge the assistance of the OSU tissue procurement team for patient consenting, tissue collection and data management.
Funding:
This study was supported by NCI grant R01CA235673 (VP, DS), the Salvino Family & Accenture Brain Cancer Research Fund, Lisa B, Landes Brain Cancer Fund, the Roc on Research Fund for Neuro-oncology, The Glioblastoma Foundation and the M.D. Anderson GBM Moonshot fund.
Conflict of Interest:
VP reports grant funding from the National Cancer Institute, the Salvino Family & Accenture Brain Cancer Research Fund, Lisa B, Landes Brain Cancer Fund, the Roc on Research Fund for Neuro-oncology, and The Glioblastoma Foundation to support this research project, personal consultancy relationship with Bayer pharmaceuticals, Orbus Therapeutics, Forma Therapeutics, Prelude Therapeutics, Ziopharm and Novocure, and equity in Amarin and Gilead Pharmaceuticals. DS reports grant funding from the National Cancer Institute and consultancy and equity with Newave Pharma; AW reports current employment with Abbvie; CT reports current employment with Incyte Corporation. The remaining authors report no relevant conflicts of interest
Data Availability
RNA-seq data for GSC lines and primary GBM experiments reported in this article are deposited to the Gene Expression Omnibus under accession number: GSE193635 at https://urldefense.com/v3/__https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE193635__;!!KGKeukY!kZlj9RkmtpFePsOvn0itBI_LvC1dY_IO7o26GeplxvOhTzeioboniGTXm5gtCvWEXZA$ and is accessible with token ufuzkowovnidtof.
References:
- 1.Johnson DR, O’Neill BP. Glioblastoma survival in the United States before and during the temozolomide era. Journal of neuro-oncology 2012;107(2):359–64 doi 10.1007/s11060-011-0749-4. [DOI] [PubMed] [Google Scholar]
- 2.Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. The New England journal of medicine 2005;352(10):987–96 doi 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- 3.Cannan WJ, Pederson DS. Mechanisms and Consequences of Double-Strand DNA Break Formation in Chromatin. Journal of cellular physiology 2016;231(1):3–14 doi 10.1002/jcp.25048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bee L, Fabris S, Cherubini R, Mognato M, Celotti L. The efficiency of homologous recombination and non-homologous end joining systems in repairing double-strand breaks during cell cycle progression. PLoS One 2013;8 doi 10.1371/journal.pone.0069061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Marechal A, Zou L. DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 2013;5(9) doi 10.1101/cshperspect.a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.King HO, Brend T, Payne HL, Wright A, Ward TA, Patel K, et al. RAD51 Is a Selective DNA Repair Target to Radiosensitize Glioma Stem Cells. Stem cell reports 2017;8(1):125–39 doi 10.1016/j.stemcr.2016.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hegi ME, Diserens AC, Gorlia T, Hamou MF, de Tribolet N, Weller M, et al. MGMT gene silencing and benefit from temozolomide in glioblastoma. The New England journal of medicine 2005;352(10):997–1003 doi 10.1056/NEJMoa043331. [DOI] [PubMed] [Google Scholar]
- 8.Yoshimoto K, Mizoguchi M, Hata N, Murata H, Hatae R, Amano T, et al. Complex DNA repair pathways as possible therapeutic targets to overcome temozolomide resistance in glioblastoma. Front Oncol 2012;2:186 doi 10.3389/fonc.2012.00186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB, et al. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444(7120):756–60 doi 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 10.Lim YC, Roberts TL, Day BW, Harding A, Kozlov S, Kijas AW, et al. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Molecular cancer therapeutics 2012;11(9):1863–72 doi 10.1158/1535-7163.MCT-11-1044. [DOI] [PubMed] [Google Scholar]
- 11.Balbous A, Cortes U, Guilloteau K, Rivet P, Pinel B, Duchesne M, et al. A radiosensitizing effect of RAD51 inhibition in glioblastoma stem-like cells. BMC cancer 2016;16:604 doi 10.1186/s12885-016-2647-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gil Del Alcazar CR, Todorova PK, Habib AA, Mukherjee B, Burma S. Augmented HR Repair Mediates Acquired Temozolomide Resistance in Glioblastoma. Molecular cancer research : MCR 2016;14(10):928–40 doi 10.1158/1541-7786.MCR-16-0125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schopf FH, Biebl MM, Buchner J. The HSP90 chaperone machinery. Nature reviews Molecular cell biology 2017;18(6):345–60 doi 10.1038/nrm.2017.20. [DOI] [PubMed] [Google Scholar]
- 14.Genest O, Wickner S, Doyle SM. Hsp90 and Hsp70 chaperones: Collaborators in protein remodeling. The Journal of biological chemistry 2019;294(6):2109–20 doi 10.1074/jbc.REV118.002806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sottile ML, Nadin SB. Heat shock proteins and DNA repair mechanisms: an updated overview. Cell stress & chaperones 2018;23(3):303–15 doi 10.1007/s12192-017-0843-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Canella A, Welker AM, Yoo JY, Xu J, Abas FS, Kesanakurti D, et al. Efficacy of Onalespib, a Long-Acting Second-Generation HSP90 Inhibitor, as a Single Agent and in Combination with Temozolomide against Malignant Gliomas. Clinical cancer research : an official journal of the American Association for Cancer Research 2017;23(20):6215–26 doi 10.1158/1078-0432.CCR-16-3151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sauvageot CM, Weatherbee JL, Kesari S, Winters SE, Barnes J, Dellagatta J, et al. Efficacy of the HSP90 inhibitor 17-AAG in human glioma cell lines and tumorigenic glioma stem cells. Neuro Oncol 2009;11(2):109–21 doi 10.1215/15228517-2008-060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Stingl L, Stühmer T, Chatterjee M, Jensen MR, Flentje M, Djuzenova CS. Novel HSP90 inhibitors, NVP-AUY922 and NVP-BEP800, radiosensitise tumour cells through cell-cycle impairment, increased DNA damage and repair protraction. British Journal of Cancer 2010;102(11):1578–91 doi 10.1038/sj.bjc.6605683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen H, Gong Y, Ma Y, Thompson RC, Wang J, Cheng Z, et al. A Brain-Penetrating Hsp90 Inhibitor NXD30001 Inhibits Glioblastoma as a Monotherapy or in Combination With Radiation. Front Pharmacol 2020;11:974-doi 10.3389/fphar.2020.00974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Choi EJ, Cho BJ, Lee DJ, Hwang YH, Chun SH, Kim HH, et al. Enhanced cytotoxic effect of radiation and temozolomide in malignant glioma cells: targeting PI3K-AKT-mTOR signaling, HSP90 and histone deacetylases. BMC cancer 2014;14(1):17 doi 10.1186/1471-2407-14-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Mascarenhas R, Pietrzak M, Smith RM, Webb A, Wang D, Papp AC, et al. Allele-Selective Transcriptome Recruitment to Polysomes Primed for Translation: Protein-Coding and Noncoding RNAs, and RNA Isoforms. PLoS One 2015;10(9):e0136798 doi 10.1371/journal.pone.0136798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xu J, Sampath D, Lang FF, Prabhu S, Rao G, Fuller GN, et al. Vorinostat modulates cell cycle regulatory proteins in glioma cells and human glioma slice cultures. Journal of neuro-oncology 2011;105(2):241–51 doi 10.1007/s11060-011-0604-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Esteller M, Hamilton SR, Burger PC, Baylin SB, Herman JG. Inactivation of the DNA repair gene O6-methylguanine-DNA methyltransferase by promoter hypermethylation is a common event in primary human neoplasia. Cancer research 1999;59(4):793–7. [PubMed] [Google Scholar]
- 24.Kotian S, Liyanarachchi S, Zelent A, Parvin JD. Histone deacetylases 9 and 10 are required for homologous recombination. The Journal of biological chemistry 2011;286(10):7722–6 doi 10.1074/jbc.C110.194233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Olive PL, Banath JP. The comet assay: a method to measure DNA damage in individual cells. Nature protocols 2006;1(1):23–9 doi 10.1038/nprot.2006.5. [DOI] [PubMed] [Google Scholar]
- 26.Kitange GJ, Carlson BL, Schroeder MA, Grogan PT, Lamont JD, Decker PA, et al. Induction of MGMT expression is associated with temozolomide resistance in glioblastoma xenografts. Neuro Oncol 2009;11(3):281–91 doi 10.1215/15228517-2008-090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Rivera AL, Pelloski CE, Gilbert MR, Colman H, De La Cruz C, Sulman EP, et al. MGMT promoter methylation is predictive of response to radiotherapy and prognostic in the absence of adjuvant alkylating chemotherapy for glioblastoma. Neuro Oncol 2010;12(2):116–21 doi 10.1093/neuonc/nop020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bao S, Wu Q, McLendon RE, Hao Y, Shi Q, Hjelmeland AB. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006;444 doi 10.1038/nature05236. [DOI] [PubMed] [Google Scholar]
- 29.Lim YC, Roberts TL, Day BW, Harding A, Kozlov S, Kijas AW. A role for homologous recombination and abnormal cell-cycle progression in radioresistance of glioma-initiating cells. Molecular cancer therapeutics 2012;11 doi 10.1158/1535-7163.mct-11-1044. [DOI] [PubMed] [Google Scholar]
- 30.Arlander SJ, Eapen AK, Vroman BT, McDonald RJ, Toft DO, Karnitz LM. Hsp90 inhibition depletes Chk1 and sensitizes tumor cells to replication stress. The Journal of biological chemistry 2003;278(52):52572–7 doi 10.1074/jbc.M309054200. [DOI] [PubMed] [Google Scholar]
- 31.Elaimy AL, Ahsan A, Marsh K, Pratt WB, Ray D, Lawrence TS, et al. ATM is the primary kinase responsible for phosphorylation of Hsp90alpha after ionizing radiation. Oncotarget 2016;7(50):82450–7 doi 10.18632/oncotarget.12557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Pennisi R, Antoccia A, Leone S, Ascenzi P, di Masi A. Hsp90α regulates ATM and NBN functions in sensing and repair of DNA double-strand breaks. The FEBS Journal 2017;284(15):2378–95 doi 10.1111/febs.14145. [DOI] [PubMed] [Google Scholar]
- 33.Lai TH, Ewald B, Zecevic A, Liu C, Sulda M, Papaioannou D, et al. HDAC Inhibition Induces MicroRNA-182, which Targets RAD51 and Impairs HR Repair to Sensitize Cells to Sapacitabine in Acute Myelogenous Leukemia. Clinical cancer research : an official journal of the American Association for Cancer Research 2016;22(14):3537–49 doi 10.1158/1078-0432.CCR-15-1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000;10(15):886–95 doi 10.1016/s0960-9822(00)00610-2. [DOI] [PubMed] [Google Scholar]
- 35.Jhaveri K, Chandarlapaty S, Iyengar N, Morris PG, Corben AD, Patil S, et al. Biomarkers That Predict Sensitivity to Heat Shock Protein 90 Inhibitors. Clinical breast cancer 2016;16(4):276–83 doi 10.1016/j.clbc.2015.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Eich M, Roos WP, Nikolova T, Kaina B. Contribution of ATM and ATR to the resistance of glioblastoma and malignant melanoma cells to the methylating anticancer drug temozolomide. Molecular cancer therapeutics 2013;12(11):2529–40 doi 10.1158/1535-7163.MCT-13-0136. [DOI] [PubMed] [Google Scholar]
- 37.Calderwood SK, Neckers L. Hsp90 in Cancer: Transcriptional Roles in the Nucleus. Advances in cancer research 2016;129:89–106 doi 10.1016/bs.acr.2015.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vaira V, Fedele G, Pyne S, Fasoli E, Zadra G, Bailey D, et al. Preclinical model of organotypic culture for pharmacodynamic profiling of human tumors. Proceedings of the National Academy of Sciences 2010;107(18):8352–6 doi 10.1073/pnas.0907676107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Welker AM, Jaros BD, Puduvalli VK, Imitola J, Kaur B, Beattie CE. Standardized orthotopic xenografts in zebrafish reveal glioma cell-line-specific characteristics and tumor cell heterogeneity. Disease models & mechanisms 2016;9(2):199–210 doi 10.1242/dmm.022921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Santivasi WL, Xia F. The role and clinical significance of DNA damage response and repair pathways in primary brain tumors. Cell Biosci 2013;3(1):10-doi 10.1186/2045-3701-3-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim J, Lee IH, Cho HJ, Park CK, Jung YS, Kim Y, et al. Spatiotemporal Evolution of the Primary Glioblastoma Genome. Cancer cell 2015;28(3):318–28 doi 10.1016/j.ccell.2015.07.013. [DOI] [PubMed] [Google Scholar]
- 42.Osuka S, Van Meir EG. Overcoming therapeutic resistance in glioblastoma: the way forward. The Journal of clinical investigation 2017;127(2):415–26 doi 10.1172/JCI89587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Camphausen K, Tofilon PJ. Inhibition of Hsp90: a multitarget approach to radiosensitization. Clinical cancer research : an official journal of the American Association for Cancer Research 2007;13(15 Pt 1):4326–30 doi 10.1158/1078-0432.CCR-07-0632. [DOI] [PubMed] [Google Scholar]
- 44.Dote H, Burgan WE, Camphausen K, Tofilon PJ. Inhibition of hsp90 compromises the DNA damage response to radiation. Cancer research 2006;66(18):9211–20 doi 10.1158/0008-5472.CAN-06-2181. [DOI] [PubMed] [Google Scholar]
- 45.Turner KM, Sun Y, Ji P, Granberg KJ, Bernard B, Hu L, et al. Genomically amplified Akt3 activates DNA repair pathway and promotes glioma progression. Proceedings of the National Academy of Sciences 2015;112(11):3421–6 doi 10.1073/pnas.1414573112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kan C, Zhang J. BRCA1 Mutation: A Predictive Marker for Radiation Therapy? International journal of radiation oncology, biology, physics 2015;93(2):281–93 doi 10.1016/j.ijrobp.2015.05.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lim YC, Roberts TL, Day BW, Stringer BW, Kozlov S, Fazry S. Increased sensitivity to ionizing radiation by targeting the homologous recombination pathway in glioma initiating cells. Mol Oncol 2014;8 doi 10.1016/j.molonc.2014.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ernst A, Anders H, Kapfhammer H, Orth M, Hennel R, Seidl K, et al. HSP90 inhibition as a means of radiosensitizing resistant, aggressive soft tissue sarcomas. Cancer letters 2015;365(2):211–22 doi 10.1016/j.canlet.2015.05.024. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data for GSC lines and primary GBM experiments reported in this article are deposited to the Gene Expression Omnibus under accession number: GSE193635 at https://urldefense.com/v3/__https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE193635__;!!KGKeukY!kZlj9RkmtpFePsOvn0itBI_LvC1dY_IO7o26GeplxvOhTzeioboniGTXm5gtCvWEXZA$ and is accessible with token ufuzkowovnidtof.