

Abstract
The lack of neuroprotective treatments for retinal ganglion cells (RGCs) and optic nerve (ON) is a central challenge for glaucoma management. Emerging evidence suggests that redox factor NAD+ decline is a hallmark of aging and neurodegenerative diseases. Supplementation with NAD+ precursors and overexpression of NMNAT1, the key enzyme in the NAD+ biosynthetic process, have significant neuroprotective effects. We first profile the translatomes of RGCs in naive mice and mice with silicone oil-induced ocular hypertension (SOHU)/glaucoma by RiboTag mRNA sequencing. Intriguingly, only NMNAT2, but not NMNAT1 or NMNAT3, is significantly decreased in SOHU glaucomatous RGCs, which we confirm by in situ hybridization. We next demonstrate that AAV2 intravitreal injection-mediated overexpression of long half-life NMNAT2 mutant driven by RGC-specific mouse γ-synuclein (mSncg) promoter restores decreased NAD+ levels in glaucomatous RGCs and ONs. Moreover, this RGC-specific gene therapy strategy delivers significant neuroprotection of both RGC soma and axon and preservation of visual function in the traumatic ON crush model and the SOHU glaucoma model. Collectively, our studies suggest that the weakening of NMNAT2 expression in glaucomatous RGCs contributes to a deleterious NAD+ decline, and that modulating RGC-intrinsic NMNAT2 levels by AAV2-mSncg vector is a promising gene therapy for glaucomatous neurodegeneration.
Keywords: NMNAT2, RGC, glaucoma, neuroprotection, gene therapy, NAD+
Graphical abstract
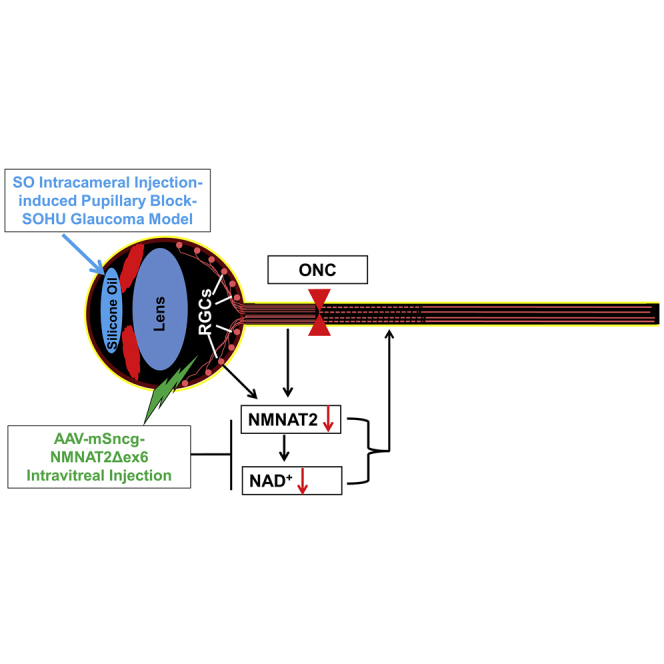
Hu and colleagues discover that NMNAT2 expression decreases significantly in mouse RGCs after IOP elevation, and the NAD+ levels of glaucomatous retina and optic nerves decrease concurrently. They further demonstrate that intravitreal injection of AAV2-mSncg-NMNAT2Δex6 increases the NAD+ levels and achieves significant neuroprotection in a mouse SOHU glaucoma model.
Introduction
Glaucoma is the most common cause of irreversible blindness and will affect more than 100 million people between 40 and 80 years old by 2040.1 It is characterized by optic neuropathy with optic nerve (ON) degeneration followed by progressive retinal ganglion cell (RGC) death.1, 2, 3, 4, 5, 6, 7, 8 Although glaucoma can occur at any intraocular pressure (IOP) level,9 elevated IOP is associated with accelerated progression, probably because of mechanical damage of the ON head.10, 11, 12 The only available treatments act by reducing IOP but fail to completely prevent the progression of glaucomatous neurodegeneration,13, 14, 15, 16 indicating the urgent need for neuroprotection therapies.
It is believed that Wallerian degeneration of RGCs' axons plays a critical role in glaucomatous neurodegeneration,17 and that Wallerian degeneration is closely associated with the reduction of axonal NAD+ level18, 19, 20; an adequate axonal NAD+ level is both necessary and sufficient for axon survival.21, 22, 23 The chimeric mutant protein, slow Wallerian degeneration protein (WldS), contains the full-length NAD+-synthetic enzyme, nicotinamide mononucleotide adenylyltransferase 1 (NMNAT1), and part of the ubiquitin ligase UBE4B. It is translocated to axons, significantly delays Wallerian degeneration,24,25 and rescues glaucomatous neurodegeneration.4,26, 27, 28 Cytoplasmic NMNAT1 mutant (cytNMNAT1) overexpression or feeding of the NAD+ precursor, vitamin B3, has consistently been found to promote substantial RGC soma and axon protection in optic neuropathy models,29,30 indicating that modulating the neuronal NAD+ level through upregulating NMNATs is a promising neuroprotective strategy for glaucoma. However, endogenous NMNAT1 is localized to the nucleus, and NMNAT3 is located in mitochondria; only NMNAT2 is enriched in axons.31,32 This distribution suggests that NMNAT2 plays a predominant role in maintaining axonal integrity.33 Indeed, whereas NMNAT2 knockout mice have truncated ONs,34 NMNAT2 overexpression delays injury-induced axon degeneration both in vitro and in vivo.34, 35, 36, 37 Therefore, NMNAT2 should be a promising choice as a neuronal intrinsic neuroprotective target for axonopathies, but it has not been tested directly in glaucoma.
Taking advantage of our recently developed silicone oil (SO)-induced ocular hypertension (SOHU) mouse glaucoma models38, 39, 40 and RGC-specific adeno-associated virus (AAV) promoter, mouse γ-synuclein (mSncg),41 we here first profiled the translatomes of naive and glaucomatous RGCs. We found that NMNAT2 is the most abundant NMNAT in RGCs, and that NMNAT2, but not NMNAT1 or NMNAT3, is significantly decreased in glaucomatous RGCs prior to significant neurodegeneration. We then forced the expression of a long-life NMNAT2 mutant by AAV intravitreal injection in RGCs and demonstrated that RGC-specific NMNAT2 overexpression increases NAD+ levels in glaucomatous retinas and ONs and has significant neuroprotective effects: it preserves RGC somata and axons and visual function in a clinically relevant mouse SOHU glaucoma model. This readily translatable gene therapy strategy contributes to developing efficient neuroprotective treatments for glaucoma by targeting neuronal intrinsic NMNAT2.
Results
NMNAT2 is significantly decreased in glaucomatous RGCs
To profile RGC transcriptomes, we employed RiboTag mice, which were generated by knocking in the hemagglutinin (HA)-tagged ribosome protein Rpl22 (HA-Rpl22) to the endogenous Rpl22 allele, immediately after the floxed endogenous Rpl22.42 Expression of HA-Rpl22 in RGCs specifically is achieved by intravitreal injection of AAV2-Cre driven by RGC-specific promoter mSncg41 and Cre-mediated deletion of endogenous Rpl22 in RGCs (Figure 1A). We generated the SOHU glaucoma model in one eye and used the contralateral (CL) eye as naive control.38, 39, 40 We then used HA antibody to immunoprecipitate RGC ribosomes from whole-retina lysates directly without the need of retinal cell dissociation and RGC purification. Deep sequencing of translating mRNAs isolated from RGC ribosomes showed high within-group correlation and dramatic differences between naive and glaucomatous RGCs 1 week after SO injection (1 wpi) (Figure 1B). The GO analysis of the differentially expressed genes (DEGs) identified multiple cellular pathways that have been significantly changed in the glaucomatous RGCs (Figure 1C). We focused on genes involved in NAD+ metabolism (Figures 1D and 1E)43: all three isoforms of NMNATs were present in RGCs, but NMNAT2 was the most abundant; NMNAT2, but not NMNAT1 or NMNAT3, was significantly decreased in glaucomatous RGCs, suggesting the primary role of NMNAT2 downregulation in glaucomatous neurodegeneration. We further confirmed the decrease of NMNAT2 in glaucomatous retina by in situ hybridization (ISH; Figure S1). NAPRT (nicotinic acid phosphoribosyltransferase) is the enzyme that generates the substrate of NMNATs by converting nicotinic acid to NAMN, and CD38 is a NAD+-consuming enzyme.43 Both NAPRT and CD38 were upregulated in glaucomatous RGCs (Figure 1E), which may be a compensatory response to the decrease in NMNAT2. In summary, NMNAT2 is the major isoform of NMNAT in RGCs, and its decline is an early event occurring shortly after the onset of ocular hypertension and before significant neurodegeneration.
Figure 1.

RGC-specific transcriptome profiling in glaucoma reveals downregulation of NMNAT2
(A) Images of retinal sections and whole mounts of naive Ribo-tag mice 2 weeks after AAV-mSncg-Cre intravitreal injection, showing co-localization of the Ribo-tag (HA-Rpl22) labeled by HA antibody and RGCs labeled by RBPMS antibody. Scale bar of the retinal section, 20 μm; whole mount retina, 50 μm. (B) Heatmap of differentially expressed genes (DEGs) comparing glaucomatous RGCs with naive RGCs. Triplicate samples from each group. (C) Gene Ontology (GO) enrichment analysis of DEGs. Bar plot of the top 10 GO-enriched biological processes of the DEGs in glaucomatous RGCs. (D) GO enrichment analysis of DEGs in nicotinamide nucleotide biosynthetic and metabolic processes. (E) The expression levels and fold changes in glaucomatous and naive RGCs of individual genes involved in NAD+ biosynthesis and metabolism. TPM, transcripts per million. See also Figure S1.
RGC-specific expression of NMNAT2Δex6 and increase of NAD+ in both retina and ON
Because NMNAT2 protein is very labile and rapidly depleted after axotomy, downregulation of axonal NAD+ is known to cause axon degeneration.21,33,44 Two E3 ubiquitin ligases, PHR1/Highwire and SCF, are involved in NMNAT2 degradation and axonal degeneration.45, 46, 47 Our finding that the mRNA level of NMNAT2 in RGCs is decreased by ocular hypertension further supports the notion that the lack of NMNAT2 contributes to glaucomatous neurodegeneration. Therefore, we next examined the effect of RGC-specific NMNAT2 overexpression on RGCs' survival after injury and disease. NMNAT2Δex6 (soluble forms of NMNAT2, lacking exon6) is more stable and has greater axon-protective capacity than wild type (WT) NMNAT2.44,48 We engineered an AAV vector to drive NMNAT2Δex6 under the mSncg promoter and confirmed RGC-specific expression of NMNAT2 after intravitreal injection: HA-tagged NMNAT2 colocalized with RBPMS+ RGCs and Tuj1+ ONs, but not with other layers of retina (Figures S2A, S2B, and S2D). We further confirmed that NMNAT2 overexpression increased NAD+ levels significantly in both naive retina and ON (Figures S2C and S2E). In summary, we established RGC-specific upregulation of NMNAT2, which will enable us to evaluate the RGC autonomous effect of NMNAT2 modulation in glaucomatous neuroprotection.
NMNAT2 overexpression significantly promotes both RGC soma and axon survival after ON crush injury
ON crush (ONC) is extensively used as a convenient optic neuropathy model that injures all RGC axons and causes universal RGC degeneration; it also often serves as an acceptable surrogate glaucoma model. To determine whether a neuronal autonomous effect of NMNATs provides RGC and ON protection, we first injected AAV2-NMNAT2Δex6, AAV2-cytNMNAT1, or AAV2 control into the vitreous of the left eyes of naive mice 2 weeks before ONC. Compared with the CL naive control eyes, ONC caused significant loss of RGC somata and axons (Figure 2A). Consistent with the previous report that the pan-neuronal overexpression of cytNMNAT1 in cytosol and axon protects both RGC somata and axons in optic neuropathy models,30 we found that cytNMNAT1 overexpression also promotes RGC soma and axon survival after a traumatic injury, ONC (Figures 2A and 2B). NMNAT2 overexpression showed similar axon protection as NMNAT1, although NMNAT2 protected RGC somata marginally more effectively than NMNAT1 (Figures 2A and 2B).
Figure 2.

Both NMNAT2 and NMNAT1 overexpression promote neuroprotection of RGC somata and axons after ON crush injury
(A) Upper panel: confocal images of peripheral flat-mounted retinas showing surviving RBPMS-positive (red) RGCs 2 weeks after crush injury. Scale bar, 20 μm. Lower panel: light microscope images of semi-thin transverse sections of ON with PPD staining 2 weeks after crush injury. Scale bar, 10 μm. (B) Quantification of surviving RGC somata and axons represented as percentage of crush-injured eyes compared with the sham contralateral control eyes injected with control AAV2. Data are presented as means ± SD; n = 10 of each group; ∗p < 0.05, ∗∗∗∗p < 0.0001, one-way ANOVA with Tukey's multiple comparisons test. See also Figure S2.
NAD+ levels are significantly decreased in SOHU glaucomatous retinas and ONs and are reversed by NMNAT1 or NMNAT2 overexpression
The downregulation of NMNAT2 mRNA levels in glaucomatous RGCs may result in decrease of NAD+. Indeed, there was a significant decrease in NAD+ levels in retinas and ONs of glaucomatous mice 1 wpi (Figure 3A). Both NMNAT1 and NMNAT2 overexpression increase NAD+ levels in SOHU glaucomatous retinas and ONs (Figure 3A). These data suggest that IOP elevation induces downregulation of NMNAT2 expression and NAD+ production in RGCs and ONs before significant glaucomatous neurodegeneration. Therefore, we next tested the therapeutic effect of NMNAT2 overexpression and NAD+ restoration in the SOHU glaucoma model.
Figure 3.

RGC-specific NMNAT2 overexpression significantly promotes neuronal NAD+ production and survival of both RGC somata and axons in the SOHU glaucoma model
(A) Relative NAD+ levels of retinas and ONs from naive, SOHU glaucoma at 1 week after SO injection (1 wpi), and SOHU glaucoma mice with NMNAT1 or NMNAT2 overexpression, acquired by LC/MS analysis. n = 5 in all the groups. Data are presented as means ± SD; ∗p < 0.05, ∗∗p < 0.01, one-way ANOVA with Dunnett's multiple comparisons test. (B) Representative OCT images of SOHU glaucoma mouse retinas at 3 wpi. Ganglion cell complex (GCC) includes RNFL, GCL, and IPL indicated as double-end arrows. (C) Quantification of GCC thickness measured by OCT at 3 wpi, represented as percentage of GCC thickness in the SOHU eyes compared with the sham contralateral control eyes. n = 20 in both groups. Data are presented as means ± SD; ∗∗∗p < 0.001, two-tailed unpaired t test. (D) Upper panel: representative confocal images of peripheral flat-mounted retinas showing surviving RBPMS-positive (red) RGCs at 3 wpi. Scale bars, 20 μm. Lower panel: light microscopic images of semi-thin transverse sections of ON with PPD staining at 3 wpi. Scale bars, 10 μm. (E) Quantification of surviving RGCs somata and axons at 3 wpi, represented as percentage of glaucomatous eyes compared with the sham contralateral control eyes. n = 19 in both groups. Data are presented as means ± SD; ∗∗∗∗p < 0.0001, two-tailed unpaired t test. See also Figure S3.
NMNAT2 overexpression significantly promotes both RGC soma and axon survival in the SOHU glaucoma model
To determine whether neuronal autonomous NMNAT2 protects in glaucoma, we first performed intravitreal injection of AAV2-mSncg-NMNAT2 or AAV2 control in the left eyes of naive mice 3 weeks before SO injection. Optical coherence tomography (OCT) showed significant thinning of the ganglion cell complex (GCC), including both RGC dendrites and axons, in living animals 3 wpi (Figures 3B and 3C), concurrent with significant IOP elevation (Figure S3A). Histological analysis of postmortem retina whole mounts and ON sections consistently demonstrated a significant loss of RGC somata and axons in SOHU eyes (Figures 3D, 3E, and S3B). RGC-specific expression of NMNAT2 promoted dramatic survival of both RGC somata and axons (Figures 3B–3E and S3B). Taken together, NMNAT2 overexpression in RGCs promotes recovery of NAD+ levels in RGCs and ONs and achieves significant neuroprotection of RGCs and ONs in the SOHU glaucoma model.
NMNAT2 overexpression preserves visual functions of glaucomatous mice
In addition to morphological protection, we also investigated whether RGC-specific expression of NMNAT2 preserved visual function in glaucomatous mice. The pattern electroretinogram (PERG), a sensitive electrophysiological assay of general RGC function, is obtained in response to a visual stimulus consisting of contrast reversal patterned gratings at constant mean luminance. This stimulus differs from the uniform flashes of light used in flash electroretinogram (ERG).49 Because our PERG system can measure both eyes at the same time, there is an internal control to serve as a reference and normalization to minimize variations.50 We previously employed PERG to examine the changes of RGC function in the SOHU glaucoma model.38,40 The peak-to-trough (P1-N2) amplitude ratio of the SOHU eyes to CL eyes increased significantly after NMNAT2 overexpression in RGCs (Figure 4A). Optokinetic tracking response (OKR) is a natural reflex that objectively assesses mouse visual acuity.51,52 The mouse eye will only track a grating stimulus that is moving from the temporal to nasal visual field, which allows both eyes to be measured independently.52,53 Consistent with our postmortem histological and in vivo morphological and electrophysiological results, NMNAT2 significantly preserved visual acuity of the glaucomatous eyes (Figure 4B).
Figure 4.

NMNAT2 overexpression preserves visual functions of glaucomatous mice
(A) Left: representative waveforms of PERG at 3 wpi. Right: quantification of P1-N2 amplitude of PERG at 3 wpi, represented as a percentage of glaucomatous eyes compared with the sham contralateral control eyes. n = 20 in both groups. Data are presented as means ± SD; ∗∗∗∗p < 0.0001, two-tailed unpaired t test. (B) Visual acuity measured by OKR at 3 wpi, represented as percentage of glaucomatous eyes compared with the sham contralateral control eyes. Data are presented as means ± SD; ∗∗∗∗p < 0.0001, two-tailed unpaired t test.
Discussion
In this study, we determined the expression levels of the genes involved in NAD+ metabolism in both naive and SOHU glaucomatous RGCs. Intriguingly, NMNAT2 is the dominant form of NMNATs in RGCs, and its mRNA level, but not that of NMNAT1 or NMNAT3, is significantly decreased in glaucomatous RGCs. This decrease occurs concurrently in glaucomatous retinas and ONs with a decrease in NAD+. Two other genes that are also involved in NAD+ metabolism, NAPRT and CD38, are also upregulated in glaucomatous RGCs, which may be a compensatory response to the decrease of NMNAT2. We next demonstrated that intravitreal injection of AAV2-mSncg-NMNAT2Δex6 increases NMNAT2 expression and NAD+ levels specifically in RGCs and ONs. We then tested this gene therapy strategy in two optic neuropathy models, traumatic ONC and ocular hypertension glaucoma. RGC-specific NMNAT2 overexpression significantly promotes survival of RGC somata and axons in both models and preserves visual function in SOHU glaucoma eyes. These results contrast dramatically with that of another study showing that NMNAT2 overexpression fails to provide neuroprotection in an experimental autoimmune encephalomyelitis/optic neuritis model,54 suggesting that NMNAT2 dysfunction is uniquely associated with traumatic and glaucomatous retina/ON injuries.
The axonal NAD+ level declines rapidly in injured axons, primarily because of depletion of the axonal NAD+-synthetic enzyme NMNAT2 and activation of the NAD+-consuming enzyme SARM1 (sterile alpha and TIR motif-containing protein 1), a downstream acting pro-degeneration factor for NMNAT2.18, 19, 20 Both the short half-life of NMNAT2 protein identified before44 and decreased NMNAT2 transcription in the SOHU glaucoma model identified by this study (Figure 1E) may contribute to the NMNAT2 depletion and therefore cause axon degeneration. An additional factor that may also contribute is the decreased axonal transport of NMNAT2 found with aging, a common risk factor in chronic neurodegenerative diseases.55 Significantly reduced levels of NMNAT2 mRNA and protein have been identified consistently in Alzheimer disease,37 and loss-of-function NMNAT2 mutations have been detected in rare neurological diseases.56,57 A causative NMNAT2 mutation has not been found yet for glaucoma, but germline deletion of NMNAT2 causes ON truncation in mice.58 The observations that NMNAT2 is the predominant NMNAT isoform in RGCs and that it provides significant neuroprotection of RGCs and ONs suggest that NMNAT2 would offer an excellent therapeutic target for glaucoma neuroprotection. Multiple NMNAT1 mutations cause Leber congenital amaurosis type 9 (LCA9), an autosomal recessive photoreceptor degenerative disease.59, 60, 61, 62, 63 The mutations in LCA9 probably act through photoreceptor-autonomous effects because pan-neuronal NMNAT1 deletion specifically causes degeneration of photoreceptors, but not of RGCs.64,65 The cytosolic form of NMNAT1 also protects RGCs and ONs in the ONC model, however, although NMNAT2 protection of the RGC soma is slightly better. Levels of NAD+ in glaucomatous retinas and ONs induced by NMNAT1 are comparable with those of NMNAT2, suggesting that they may provide similar neuroprotection in glaucoma. Taken together, our evidence indicates that a low NMNAT2 level is pro-neurodegenerative, and that strategies boosting neuronal intrinsic NMNAT2 and NAD+ levels represent a promising therapeutic treatment for glaucoma and other axonopathies. The retina is readily accessible for administration of a virus, which is applied by a localized injection that is confined to the eye and requires minimal virus load and causes minimal, if any, systemic effects. Considered along with these other advantages, our demonstration that AAV-mediated RGC-specific promoter mSncg-driven NMNAT2 overexpression provides significant neuroprotection of glaucomatous RGCs/ONs and preservation of visual functions establishes a firm proof of concept for a translatable gene therapy approach to glaucoma.
SARM1 is a Toll-like receptor adapter protein but with intrinsic NAD+ hydrolase activity that causes axon degeneration by degrading axonal NAD+ significantly after injury-induced activation.23,66, 67, 68 Importantly, SARM1 deletion rescues NMNAT2-deficient axons,58 indicating that SARM1 works downstream of NMNAT2 depletion. Although we did not detect a significant increase of SARM1 transcription in glaucomatous RGCs, blocking SARM1 activity is another promising strategy for neuroprotection, as demonstrated in many, but not all, neurodegenerative disease models.18 Germline deletion of SARM1 in mouse delays ON degeneration significantly after traumatic crush injury but has no effect on RGC survival.69 However, SARM1 deletion blocks TNF-α-induced RGC and ON degeneration.70 We are testing whether RGC-specific SARM1 inhibition protects against glaucomatous neurodegeneration, which will help to determine whether NMNAT2 activation or SARM1 inhibition is the better therapeutic strategy for glaucoma.
In summary, consistent with the notion that a decline in neuronal NAD+ is a hallmark of many neurodegenerative diseases,43,71 including models of photoreceptor degenerative diseases72,73 and the aging DBA2/J genetic glaucoma mouse line,29 we found decreased NMNAT2 expression in glaucomatous RGCs, which may lead to NAD+ decline in RGCs and ONs. We further demonstrated that modulating RGC-intrinsic levels of NMNAT2 by an AAV2-mSncg vector represents a potent gene therapy strategy for protecting both RGC somata and axons in traumatic ON injury and the SOHU glaucoma model.
Materials and methods
Animals
C57BL/6J WT (#000664) and RiboTag (#011029) mice (7–9 weeks old) were purchased from Jackson Laboratories (Bar Harbor, ME, USA) and housed in standard cages on a 12-h light/dark cycle. All experimental procedures were performed in compliance with animal protocols approved by the IACUC at Stanford University School of Medicine.
Constructs
The AAV2 vector containing the mSncg promoter, NMNAT2Δex6, and cytNMNAT1 has been described before.30,41,44,48 The 3HA-NMNAT2Δex6 is driven by the mSncg promoter, and the cytNMNAT1 is driven by the universal CAG promoter.
AAV production and intravitreal injection
The detailed procedure of AAV production has been described previously.41,74, 75, 76 The AAV titers were determined by real-time PCR and diluted to 1.5 × 1012 vector genomes (vg)/mL. For intravitreal injection, mice were anesthetized with xylazine and ketamine based on their body weight (0.01 mg xylazine/g + 0.08 mg ketamine/g). A pulled and polished microcapillary needle was inserted into the peripheral retina just behind the ora serrata. Approximately 2 μL of the vitreous was removed to allow injection of 2 μL AAV into the vitreous chamber to achieve 3 × 109 vg/retina. The CL eyes were injected with 2 μL AAV2 as control.
ONC model
ONC was performed 2 weeks following AAV injection.75, 76, 77, 78 After anesthetization by intraperitoneal injection of Avertin (0.3 mg/g), the ON was exposed intraorbitally, while care was taken not to damage the underlying ophthalmic artery, and crushed with a jeweler's forceps (Dumont #5; Fine Science Tools, Foster City, CA, USA) for 5 s approximately 0.5 mm behind the eyeball. Eye ointment containing neomycin (Akorn, Somerset, NJ, USA) was applied to protect the cornea after surgery.
SOHU glaucoma model and IOP measurement
SOHU mouse models and IOP measurement have been detailed before.38, 39, 40 In brief, mice were anesthetized by an intraperitoneal injection of Avertin (0.3 mg/g) and received the SO (1,000 mPa/s; Alcon Laboratories, Fort Worth, TX, USA) injection at 9–10 weeks of age. Prior to injection, one drop of 0.5% proparacaine hydrochloride (Akorn) was applied to the cornea to reduce its sensitivity during the procedure. A 32G needle was tunneled through the layers of the cornea at the superotemporal side close to the limbus to reach the anterior chamber without injuring the lens or iris. Following this entry, ∼2 μL SO (1,000 mPa/s; Silikon; Alcon Laboratories) was injected slowly into the anterior chamber using a homemade sterile glass micropipette, until the oil droplet expanded to cover most areas of the iris (diameter ∼1.8–2.2 mm). After the injection, veterinary antibiotic ointment (BNP ophthalmic ointment; Vetropolycin; Dechra, Overland Park, KS, USA) was applied to the surface of the injected eye. The CL control eyes received mock injection with 2 μL normal saline to the anterior chamber. Throughout the procedure, artificial tears (Systane Ultra Lubricant Eye Drops; Alcon Laboratories) were applied to keep the cornea moist.
The detailed procedure for IOP measurement has been described before.38,39 The IOP of both eyes was measured by the TonoLab tonometer (Colonial Medical Supply, Espoo, Finland) according to product instructions under a sustained flow of isoflurane (3% isoflurane at 2 L/min mixed with oxygen) delivered to the nose by a special rodent nose cone (Xenotec, Rolla, MO, USA). 1% Tropicamide Sterile Ophthalmic Solution (Akorn) was applied three times at 3-min intervals to fully dilate the pupils (about 10 min) before taking measurements. During this procedure, artificial tears were applied to keep the cornea moist. Because IOP measurement requires pupil dilation, which essentially relieves ocular hypertension during the period of pupil dilation, we measure only IOP 3 weeks after SO injection immediately before sacrificing the animals in the acute and severe ND (no dilation) SOHU model that we described before.40
RiboTag immunoprecipitation (Ribo-IP), RNA extraction, RNA sequencing (RNA-seq), and data analysis
RiboTag mice (B6N.129-Rpl22tm1.1Psam; Jackson Laboratory) were intravitreally injected with 2 μL AAV2-mSncg-Cre (1.5 × 1012 vg/mL) to achieve RGC ribosome labeling. Three weeks after the AAV injection, SOHU model mice were prepared as previously described.38, 39, 40 Freshly isolated retinas (10–12 retinas/condition) at 1 wpi in SOHU eyes and CL control eyes were homogenized with a pestle in homogenization buffer (50 mM Tris HCl, 100 mM KCl, 12 mM MgCl2, 1% Nonidet P-40 [NP-40], 1 mM DTT, 1× protease inhibitor [Sigma], 200 U/mL RNasin [Promega, Madison, WI, USA], 100 μg/mL cycloheximide, and 1 mg/mL heparin). Samples were then centrifuged at 12,000 × g for 10 min and the supernatant used for IP. A total of 10 μL anti-HA antibody (BioLegend, San Diego, CA, USA) was added into each sample and incubated for 4 h in a cold room with rotation before incubation with protein G magnetic beads (prewashed with homogenization buffer; Thermo Fisher Scientific, South San Francisco, CA, USA) overnight at 4°C with rotation. Sample tubes were placed in a magnetic adaptor to aggregate the magnetic beads, and the supernatant was discarded. The beads were washed for 10 min three times in a high-salt buffer (50 mM Tris HCl, 300 mM KCl, 12 mM MgCl2, 1% NP-40, 1 mM DTT, 100 μg/mL cycloheximide) before resuspension in TRIzol (Thermo Fisher Scientific) for RNA isolation with PureLink RNA Mini Kit (Thermo Fisher Scientific) following the manufacturer's protocol. RNA integrity was analyzed using a 2100 Bioanalyzer (Agilent, Santa Clara, CA, USA) with the RNA Pico chip; the samples with RNA integrity number (RIN) greater than 6 were used for library preparation. Library preparation and sequencing were performed using the ultra-low-input RNA-seq service from GENEWIZ (South Plainfield, NJ, USA). In brief, the cDNA was generated using the SMART-Seq v4 Ultra Low Input RNA kit (Takara, Mountain View, CA). Libraries were then constructed using the Illumina Nextera XT kit (Illumina, San Diego, CA), and sequencing was performed on the Illumina HiSeq 4000 sequencer with 2 × 150-bp paired-end configuration. Three biological replicate samples were prepared and sequenced for each condition.
RNA-seq raw data were trimmed by trim-galore to remove adaptor sequences and aligned with Hisat2 to the mouse reference genome (version mm10). The aligned reads ranges are from 59.7 million (93.36%) to 67.1 million (94.25%). MultiQC was used to assess the quality of the sequence data. The count matrix of all the genes in the individual samples was determined by featureCounts. TPMs (transcripts per million) of each gene were calculated; TPM = (reads mapped to transcript/transcript length)/sum (reads mapped to transcript/transcript length) × 106. Samples were further processed by DESeq2 package working in R to determine DEGs between SOHU RGCs versus naive RGCs. A total of 1,132 DEGs in SOHU were detected with the cutoff, >1.2-fold change upregulated or <0.7-fold downregulated, and adjusted p < 0.05. A heatmap was generated using heatmap package to illustrate the DEGs of different groups. We used clusterProfiler package working in R to perform enrichment analysis of gene clusters and visualize the top enriched Gene Ontology (GO) terms. The log10 (adjusted p value) of the enriched biological process was plotted to indicate the significance of the enrichment of each item. All the raw data and processed data have been submitted to the Gene Expression Omnibus (GEO); the accession number is GEO: GSE182483.
Fluorescent ISH (FISH) with retina cross sections
FISH was performed by using the RNAscope Multiplex Fluorescent Detection Reagents V2 (Advanced Cell Diagnostics [ACD], Hayward, CA, USA) according to the manufacturer's instructions. RNAscope probe Mm-NMNAT2-C2 was purchased from ACD. Adult mice were perfused with ice-cold 4% paraformaldehyde (PFA)/PBS, and eyes were dissected out and fixed in 4% PFA/PBS at 4°C overnight. The eyes were dehydrated with increasing concentrations of sucrose solution (10%, 20%, and 30%) overnight before embedding in OCT on dry ice. Serial cross sections (12 μm) were cut with a Leica cryostat and collected on Superfrost Plus Slides. The sections were pretreated with protease and then subjected to ISH with RNAscope Multiplex Fluorescent Detection Reagents V2 according to the manufacturer's instruction (ACD). In brief, sections were hybridized with the probe solution, followed by amplification and probe detection using TSA plus fluorophores (AKOYA, Marlborough, MA, USA). The sections were mounted with Fluoromount G (Southern Biotech, Birmingham, AL, USA). Images were captured by a Zeiss LSM 880 confocal laser-scanning microscope with 40×/1.0 Oil DIC (Carl Zeiss Microscopy, Thornwood, NY, USA). The quantification of NMNAT2 fluorescence intensity was measured by NIH ImageJ. The results were calculated as the mean gray value (integrated density/area).
Immunohistochemistry of whole-mount and cross sections of retina
The detailed procedures have been published before.38, 39, 40, 41,77,79 In brief, after perfusion fixation with 4% PFA in PBS, mice eyeballs and ONs were dissected out and post-fixed with 4% PFA for 2 h at room temperature. 30% sucrose was then used for cryoprotection of the tissues. Retinas were dissected out for whole-mount retina immunostaining. For cross sections of retina, the eyeballs were embedded in Tissue-Tek OCT on dry ice for subsequent cryo-section with a Leica cryostat. The primary antibodies used for immunostaining are anti-RBPMS at 1:4,000 (custom-made at ProSci), anti-HA at 1:200 (11867423001; Roche), and anti-Tuj1 at 1:200 (845502; BioLegend). Secondary antibodies were then applied (1:200; Jackson ImmunoResearch, West Grove, PA, USA) and incubated for 1 h at room temperature before mounting.
RGC counting
The detailed procedures have been published before.38, 39, 40, 41,77 For peripheral RGC counting, whole-mount retinas were immunostained with the RBPMS antibody, eight fields sampled from peripheral regions of each retina using a 40× lens with a Zeiss M2 epifluorescence microscope, and RBPMS + RGCs counted by Volocity software (Quorum Technologies). The percentage of RGC survival was calculated as the ratio of surviving RGC numbers in injured eyes compared with CL uninjured eyes. The investigators who counted the cells were masked to the treatment of the samples.
ON semi-thin sections and quantification of surviving axons
The detailed procedure of ON semi-thin section preparation and paraphenylenediamine (PPD) staining has been described previously.38,40,41,77,79 In brief, ONs were post-fixed in situ with 2% glutaraldehyde and 2% PFA in 0.1 M PBS. Semi-thin (1 μm) cross sections of the ON 2 mm distal to the eye (globe) were collected. After PPD staining, four sections of each ON were imaged through a 100× lens of a Zeiss M2 epifluorescence microscope to cover the entire area of the ON without overlap. Two areas of 21.4 μm × 29.1 μm were cropped from the center of each image, and the surviving axons within the designated areas were counted manually. After counting all the images taken from a single nerve, the mean of the surviving axon number was calculated for each ON. The mean of the surviving axon number in the injured ON was compared with that in the CL control ON to yield a percentage of axon survival value. The investigators who counted the axons were masked to the treatment of the samples.
NAD+ measurement with NAD+/NADH Assay Kit
The NAD+ levels of naive retinas 2 weeks after AAV intravitreal injection were measured according to the manufacturer's protocol with the NAD+/NADH Assay Kit (KA1657; Abnova). Mice were sacrificed by cervical dislocation, and retinas and ONs were collected gently and quickly. One retina per sample or two ONs per sample were homogenized in NAD+ extraction buffer and then heated at 60°C for 5 min. The homogenates added with an assay buffer were centrifuged at 14,000 rpm for 5 min to remove cellular debris. After adding reagents to 40 μL supernatants and standard solutions, the absorbance was determined at 565 nm by TECAN SPARK Plate Reader (Tecan, Switzerland). The results were normalized to a microgram of protein concentration.
NAD+ measurement with liquid chromatography mass spectrometry (LC/MS)
One week after SO intracameral injection, mice were sacrificed by cervical dislocation, and retinas and ONs were dissected out immediately. The tissues were rinsed with ice-cold PBS before snap freezing in liquid nitrogen. One retina per sample or two ONs per sample were homogenized in 400 μL extraction buffer, including 40:40:20 of methanol/acetonitrile/H2O by the “bead beating” method with FastPrep-24 grinder (MP Biomedicals) at 4°C (ONs: 6.5 m/s, 60 s/pulse, 5 pulses with 30-s interval; retina: 6.5 m/s, 60 s/pulse, 1 pulse). Samples were then centrifuged at 4°C for 15,000 rpm × 15 min, and 150 μL of the supernatant was transferred to 9-mm plastic screw thread vial (Thermo Scientific) and kept at −80°C before LC/MS analysis. Details about MS analysis have been described in a previous publication.80 In brief, 10 μL of the lysates was injected into an Agilent 1290 Infinity LC system coupled to a Q-TOF 6545 mass spectrometer (Agilent). Data were collected in negative ionization mode. A hydrophilic interaction chromatography method (HILIC) with a BEH amide column (100 × 2.1 mm inner diameter [i.d.], 1.7 μm; Waters) was used for compound separation at 35°C with a flow rate of 0.3 mL/min. The mobile phase A consisted of 25 mM ammonium acetate and 25 mM ammonium hydroxide in water, and mobile phase B was acetonitrile. The gradient elution was 0–1 min, 85% B; 1–12 min, 85% B → 65% B; 12–12.2 min, 65% B → 40% B; 12.2–15 min, 40% B. After the gradient, the column was re-equilibrated at 85% B for 5 min. The overall run time was 20 min. NAD+ was quantified as area under the curve of the MS peak and normalized with the total protein levels in each sample. NAD+/protein ratio was then normalized to average NAD+/protein value in the naive group for each sample to generate the “NAD+ relative level to naive” for comparison.
Spectral-domain OCT (SD-OCT) imaging
The detailed procedure has been published previously.38,40,77 In brief, after anesthetization (0.01 mg xylazine/g + 0.08 mg ketamine/g) and pupil dilation, the retina fundus images were captured with the Heidelberg Spectralis SLO/OCT system (Heidelberg Engineering, Germany). The mouse retina was scanned with the ring scan mode centered by the ON head under high-resolution mode (each B-scan consisted of 1,536 A-scans). The scanning ring had a fixed diameter of 160 μm, and the focal length was fixed at scale 37 D. The ON head was always placed in the center of the ring, which allowed scanning the same area and same distance from the ON head to the ring of each eye. The GCC includes retinal nerve fiber layer (RNFL), ganglion cell layer (GCL), and inner plexiform layer (IPL). The average thickness of GCC around the ON head was measured manually with the aid of Heidelberg software. The investigators who measured the thickness of GCC were masked to the treatment of the samples.
PERG recording
The detailed procedure has been published previously.38,40,49,50,77 In brief, after anesthetization (0.01 mg xylazine/g + 0.08 mg ketamine/g) and pupil dilation, PERG of both eyes was recorded simultaneously with the Miami PERG system (Intelligent Hearing Systems, Miami, FL, USA) according to the manufacturer's instructions. The pattern remained at a contrast of 100% and a luminance of 800 cd/m2 and consisted of four cycles of black-gray elements, with a spatial frequency of 0.052 c/d. Two consecutive recordings of 200 traces were averaged to achieve one readout; each trace recorded up to 1,020 ms. The first positive peak in the waveform was designated as P1, and the second negative peak as N2. The amplitude was measured from P1 to N2.
OKR
The detailed procedure has been published previously.38,51,52 In brief, mice were placed on a platform in the center of four 17-inch LCD computer monitors (Dell, Phoenix, AZ, USA), with a video camera above the platform to capture the movement of the mouse. A rotating cylinder with vertical sine-wave grating was computed and projected to the four monitors by OptoMotry software (CerebralMechanics, Lethbridge, AB, Canada). The sine-wave grating, settled at 100% contrast and speed of 12°/s, provides a virtual reality environment to measure the spatial acuity (cycles/degree) of the left eye when rotated clockwise and the right eye when rotated counterclockwise. The maximum frequency (cycles/degree) that the mouse could track was identified and recorded by investigators masked to treatment.
Statistical analysis
GraphPad Prism 7 was used to generate graphs and for statistical analyses. Data are presented as means ± SD. Student's t test was used for two-group comparison, and one-way ANOVA with post hoc test was used for multiple comparisons.
Acknowledgments
We are grateful to Drs. Michael Coleman and Jon Gilley for the generous gift of the NMNAT2Δex6 construct, and Drs. Jeff Milbrandt and Yo Sasaki for the generous gift of the cytNMNAT1 construct. We thank Hu lab members and Dr. Alan Tessler for the critical discussion and reading of the manuscript. Y.H. was supported by NIH grants EY024932, EY023295, EY028106, and EY031063 and grants from the Glaucoma Research Foundation (CFC3), BrightFocus Foundation, and Chan Zuckerberg Initiative Neurodegeneration Collaborative Pairs Pilot Projects. F.F. was supported by Natural Science Foundation of Hunan Province (Reference: 2018JJ3742). Portions of this work were supported by NIH grants R01EY025295 and R01EY032159, VA merit CX001298, Stanford Children's Health Research Institute Award to Y.S., and American Cancer Society Research Scholar Grant (RSG-20-036-01) and a Stanford Maternal and Child Health Research Institute Research Scholar Award (2020) to J.Y. We are grateful for an unrestricted grant from Research to Prevent Blindness and National Eye Institute (NEI) P30 EY026877 to the Department of Ophthalmology at Stanford University.
Author contributions
Y.H., F.F., and H.H. designed the experiments; F.F., P.Z., and X.F. performed Ribo-IP and RNA-seq analysis; F.F., P.L., H.H., L. Li, and W.C. generated animal models and acquired histology and function analysis; L. Liu provided AAV production; F.F., D.L., H.J., and J.Y. performed LC/MS analysis; Y.S. provided reagents; Y.H., F.F., H.H., and D.L. prepared the manuscript.
Declaration of interests
A patent application has been submitted through Stanford Office of Technology Licensing for neuroprotection of the gene therapy identified in this manuscript. The authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.ymthe.2022.01.035.
Supplemental information
References
- 1.Tham Y.C., Li X., Wong T.Y., Quigley H.A., Aung T., Cheng C.Y. Global prevalence of glaucoma and projections of glaucoma burden through 2040: a systematic review and meta-analysis. Ophthalmology. 2014;121:2081–2090. doi: 10.1016/j.ophtha.2014.05.013. [DOI] [PubMed] [Google Scholar]
- 2.Quigley H.A., Nickells R.W., Kerrigan L.A., Pease M.E., Thibault D.J., Zack D.J. Retinal ganglion cell death in experimental glaucoma and after axotomy occurs by apoptosis. Invest. Ophthalmol. Vis. Sci. 1995;36:774–786. [PubMed] [Google Scholar]
- 3.Libby R.T., Li Y., Savinova O.V., Barter J., Smith R.S., Nickells R.W., John S.W. Susceptibility to neurodegeneration in a glaucoma is modified by Bax gene dosage. PLoS Genet. 2005;1:17–26. doi: 10.1371/journal.pgen.0010004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Howell G.R., Libby R.T., Jakobs T.C., Smith R.S., Phalan F.C., Barter J.W., Barbay J.M., Marchant J.K., Mahesh N., Porciatti V., et al. Axons of retinal ganglion cells are insulted in the optic nerve early in DBA/2J glaucoma. J. Cell Biol. 2007;179:1523–1537. doi: 10.1083/jcb.200706181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nickells R.W., Howell G.R., Soto I., John S.W. Under pressure: cellular and molecular responses during glaucoma, a common neurodegeneration with axonopathy. Annu. Rev. Neurosci. 2012;35:153–179. doi: 10.1146/annurev.neuro.051508.135728. [DOI] [PubMed] [Google Scholar]
- 6.Jonas J.B., Aung T., Bourne R.R., Bron A.M., Ritch R., Panda-Jonas S. Glaucoma. Lancet. 2017;390:2183–2193. doi: 10.1016/S0140-6736(17)31469-1. [DOI] [PubMed] [Google Scholar]
- 7.Weinreb R.N., Leung C.K., Crowston J.G., Medeiros F.A., Friedman D.S., Wiggs J.L., Martin K.R. Primary open-angle glaucoma. Nat. Rev. Dis. Primers. 2016;2:16067. doi: 10.1038/nrdp.2016.67. [DOI] [PubMed] [Google Scholar]
- 8.Calkins D.J. Adaptive responses to neurodegenerative stress in glaucoma. Prog. Retin. Eye Res. 2021:100953. doi: 10.1016/j.preteyeres.2021.100953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Iyer J., Vianna J.R., Chauhan B.C., Quigley H.A. Toward a new definition of glaucomatous optic neuropathy for clinical research. Curr. Opin. Ophthalmol. 2020;31:85–90. doi: 10.1097/ICU.0000000000000644. [DOI] [PubMed] [Google Scholar]
- 10.Mafwiri M., Bowman R.J., Wood M., Kabiru J. Primary open-angle glaucoma presentation at a tertiary unit in Africa: intraocular pressure levels and visual status. Ophthalmic Epidemiol. 2005;12:299–302. doi: 10.1080/09286580500180572. [DOI] [PubMed] [Google Scholar]
- 11.Burgoyne C.F. A biomechanical paradigm for axonal insult within the optic nerve head in aging and glaucoma. Exp. Eye Res. 2011;93:120–132. doi: 10.1016/j.exer.2010.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Stowell C., Burgoyne C.F., Tamm E.R., Ethier C.R., Lasker I.I.o.A., Glaucomatous Neurodegeneration P. Biomechanical aspects of axonal damage in glaucoma: a brief review. Exp. Eye Res. 2017;157:13–19. doi: 10.1016/j.exer.2017.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Heijl A., Leske M.C., Bengtsson B., Hyman L., Bengtsson B., Hussein M., Early Manifest Glaucoma Trial, G. Reduction of intraocular pressure and glaucoma progression: results from the Early Manifest Glaucoma Trial. Arch. Ophthalmol. 2002;120:1268–1279. doi: 10.1001/archopht.120.10.1268. [DOI] [PubMed] [Google Scholar]
- 14.Garway-Heath D.F., Crabb D.P., Bunce C., Lascaratos G., Amalfitano F., Anand N., Azuara-Blanco A., Bourne R.R., Broadway D.C., Cunliffe I.A., et al. Latanoprost for open-angle glaucoma (UKGTS): a randomised, multicentre, placebo-controlled trial. Lancet. 2015;385:1295–1304. doi: 10.1016/S0140-6736(14)62111-5. [DOI] [PubMed] [Google Scholar]
- 15.Anderson D.R., Normal Tension Glaucoma S. Collaborative normal tension glaucoma study. Curr. Opin. Ophthalmol. 2003;14:86–90. doi: 10.1097/00055735-200304000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Wormald R., Virgili G., Azuara-Blanco A. Systematic reviews and randomised controlled trials on open angle glaucoma. Eye (Lond) 2020;34:161–167. doi: 10.1038/s41433-019-0687-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Howell G.R., Soto I., Libby R.T., John S.W. Intrinsic axonal degeneration pathways are critical for glaucomatous damage. Exp. Neurol. 2013;246:54–61. doi: 10.1016/j.expneurol.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Figley M.D., DiAntonio A. The SARM1 axon degeneration pathway: control of the NAD(+) metabolome regulates axon survival in health and disease. Curr. Opin. Neurobiol. 2020;63:59–66. doi: 10.1016/j.conb.2020.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sambashivan S., Freeman M.R. SARM1 signaling mechanisms in the injured nervous system. Curr. Opin. Neurobiol. 2021;69:247–255. doi: 10.1016/j.conb.2021.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Coleman M.P., Hoke A. Programmed axon degeneration: from mouse to mechanism to medicine. Nat. Rev. Neurosci. 2020;21:183–196. doi: 10.1038/s41583-020-0269-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wang J., Zhai Q., Chen Y., Lin E., Gu W., McBurney M.W., He Z. A local mechanism mediates NAD-dependent protection of axon degeneration. J. Cell Biol. 2005;170:349–355. doi: 10.1083/jcb.200504028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Wang J.T., Medress Z.A., Vargas M.E., Barres B.A. Local axonal protection by WldS as revealed by conditional regulation of protein stability. Proc. Natl. Acad. Sci. U S A. 2015;112:10093–10100. doi: 10.1073/pnas.1508337112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gerdts J., Brace E.J., Sasaki Y., DiAntonio A., Milbrandt J. SARM1 activation triggers axon degeneration locally via NAD(+) destruction. Science. 2015;348:453–457. doi: 10.1126/science.1258366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mack T.G., Reiner M., Beirowski B., Mi W., Emanuelli M., Wagner D., Thomson D., Gillingwater T., Court F., Conforti L., et al. Wallerian degeneration of injured axons and synapses is delayed by a Ube4b/Nmnat chimeric gene. Nat. Neurosci. 2001;4:1199–1206. doi: 10.1038/nn770. [DOI] [PubMed] [Google Scholar]
- 25.Sasaki Y., Vohra B.P., Baloh R.H., Milbrandt J. Transgenic mice expressing the Nmnat1 protein manifest robust delay in axonal degeneration in vivo. J. Neurosci. 2009;29:6526–6534. doi: 10.1523/JNEUROSCI.1429-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Beirowski B., Babetto E., Coleman M.P., Martin K.R. The WldS gene delays axonal but not somatic degeneration in a rat glaucoma model. Eur. J. Neurosci. 2008;28:1166–1179. doi: 10.1111/j.1460-9568.2008.06426.x. [DOI] [PubMed] [Google Scholar]
- 27.Williams P.A., Harder J.M., Foxworth N.E., Cardozo B.H., Cochran K.E., John S.W.M. Nicotinamide and WLD(S) act together to prevent neurodegeneration in glaucoma. Front. Neurosci. 2017;11:232. doi: 10.3389/fnins.2017.00232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Risner M.L., Pasini S., McGrady N.R., D'Alessandro K.B., Yao V., Cooper M.L., Calkins D.J. Neuroprotection by Wld(S) depends on retinal ganglion cell type and age in glaucoma. Mol. Neurodegener. 2021;16:36. doi: 10.1186/s13024-021-00459-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Williams P.A., Harder J.M., Foxworth N.E., Cochran K.E., Philip V.M., Porciatti V., Smithies O., John S.W. Vitamin B3 modulates mitochondrial vulnerability and prevents glaucoma in aged mice. Science. 2017;355:756–760. doi: 10.1126/science.aal0092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhu Y., Zhang L., Sasaki Y., Milbrandt J., Gidday J.M. Protection of mouse retinal ganglion cell axons and soma from glaucomatous and ischemic injury by cytoplasmic overexpression of Nmnat1. Invest. Ophthalmol. Vis. Sci. 2013;54:25–36. doi: 10.1167/iovs.12-10861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Berger F., Lau C., Dahlmann M., Ziegler M. Subcellular compartmentation and differential catalytic properties of the three human nicotinamide mononucleotide adenylyltransferase isoforms. J. Biol. Chem. 2005;280:36334–36341. doi: 10.1074/jbc.M508660200. [DOI] [PubMed] [Google Scholar]
- 32.Cahoy J.D., Emery B., Kaushal A., Foo L.C., Zamanian J.L., Christopherson K.S., Xing Y., Lubischer J.L., Krieg P.A., Krupenko S.A., et al. A transcriptome database for astrocytes, neurons, and oligodendrocytes: a new resource for understanding brain development and function. J. Neurosci. 2008;28:264–278. doi: 10.1523/JNEUROSCI.4178-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gilley J., Coleman M.P. Endogenous Nmnat2 is an essential survival factor for maintenance of healthy axons. PLoS Biol. 2010;8:e1000300. doi: 10.1371/journal.pbio.1000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gilley J., Adalbert R., Yu G., Coleman M.P. Rescue of peripheral and CNS axon defects in mice lacking NMNAT2. J. Neurosci. 2013;33:13410–13424. doi: 10.1523/JNEUROSCI.1534-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Feng Y., Yan T., Zheng J., Ge X., Mu Y., Zhang Y., Wu D., Du J.L., Zhai Q. Overexpression of Wld(S) or Nmnat2 in mauthner cells by single-cell electroporation delays axon degeneration in live zebrafish. J. Neurosci. Res. 2010;88:3319–3327. doi: 10.1002/jnr.22498. [DOI] [PubMed] [Google Scholar]
- 36.Yan T., Feng Y., Zheng J., Ge X., Zhang Y., Wu D., Zhao J., Zhai Q. Nmnat2 delays axon degeneration in superior cervical ganglia dependent on its NAD synthesis activity. Neurochem. Int. 2010;56:101–106. doi: 10.1016/j.neuint.2009.09.007. [DOI] [PubMed] [Google Scholar]
- 37.Ali Y.O., Allen H.M., Yu L., Li-Kroeger D., Bakhshizadehmahmoudi D., Hatcher A., McCabe C., Xu J., Bjorklund N., Taglialatela G., et al. NMNAT2:HSP90 complex mediates proteostasis in proteinopathies. PLoS Biol. 2016;14:e1002472. doi: 10.1371/journal.pbio.1002472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhang J., Li L., Huang H., Fang F., Webber H.C., Zhuang P., Liu L., Dalal R., Tang P.H., Mahajan V.B., et al. Silicone oil-induced ocular hypertension and glaucomatous neurodegeneration in mouse. Elife. 2019;8:e45881. doi: 10.7554/eLife.45881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang J., Fang F., Li L., Huang H., Webber H.C., Sun Y., Mahajan V.B., Hu Y. A reversible silicon oil-induced ocular hypertension model in mice. J. Vis. Exp. 2019;153 doi: 10.3791/60409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Fang F., Zhang J., Zhuang P., Liu P., Li L., Huang H., Webber H.C., Xu Y., Liu L., Dalal R., et al. Chronic mild and acute severe glaucomatous neurodegeneration derived from silicone oil-induced ocular hypertension. Scientific Rep. 2021;11:9052. doi: 10.1038/s41598-021-88690-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang Q., Zhuang P., Huang H., Li L., Liu L., Webber H.C., Dalal R., Siew L., Fligor C.M., Chang K.C., et al. Mouse gamma-synuclein promoter-mediated gene expression and editing in mammalian retinal ganglion cells. J. Neurosci. 2020;40:3896–3914. doi: 10.1523/JNEUROSCI.0102-20.2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sanz E., Yang L., Su T., Morris D.R., McKnight G.S., Amieux P.S. Cell-type-specific isolation of ribosome-associated mRNA from complex tissues. Proc. Natl. Acad. Sci. U S A. 2009;106:13939–13944. doi: 10.1073/pnas.0907143106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350:1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 44.Milde S., Gilley J., Coleman M.P. Subcellular localization determines the stability and axon protective capacity of axon survival factor Nmnat2. PLoS Biol. 2013;11:e1001539. doi: 10.1371/journal.pbio.1001539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Babetto E., Beirowski B., Russler E.V., Milbrandt J., DiAntonio A. The Phr1 ubiquitin ligase promotes injury-induced axon self-destruction. Cell Rep. 2013;3:1422–1429. doi: 10.1016/j.celrep.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Xiong X., Hao Y., Sun K., Li J., Li X., Mishra B., Soppina P., Wu C., Hume R.I., Collins C.A. The Highwire ubiquitin ligase promotes axonal degeneration by tuning levels of Nmnat protein. PLoS Biol. 2012;10:e1001440. doi: 10.1371/journal.pbio.1001440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yamagishi Y., Tessier-Lavigne M. An atypical SCF-like ubiquitin ligase complex promotes Wallerian degeneration through regulation of axonal Nmnat2. Cell Rep. 2016;17:774–782. doi: 10.1016/j.celrep.2016.09.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Milde S., Fox A.N., Freeman M.R., Coleman M.P. Deletions within its subcellular targeting domain enhance the axon protective capacity of Nmnat2 in vivo. Scientific Rep. 2013;3:2567. doi: 10.1038/srep02567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Porciatti V. Electrophysiological assessment of retinal ganglion cell function. Exp. Eye Res. 2015;141:164–170. doi: 10.1016/j.exer.2015.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chou T.H., Bohorquez J., Toft-Nielsen J., Ozdamar O., Porciatti V. Robust mouse pattern electroretinograms derived simultaneously from each eye using a common snout electrode. Invest. Ophthalmol. Vis. Sci. 2014;55:2469–2475. doi: 10.1167/iovs.14-13943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Prusky G.T., Alam N.M., Beekman S., Douglas R.M. Rapid quantification of adult and developing mouse spatial vision using a virtual optomotor system. Invest. Ophthalmol. Vis. Sci. 2004;45:4611–4616. doi: 10.1167/iovs.04-0541. [DOI] [PubMed] [Google Scholar]
- 52.Douglas R.M., Alam N.M., Silver B.D., McGill T.J., Tschetter W.W., Prusky G.T. Independent visual threshold measurements in the two eyes of freely moving rats and mice using a virtual-reality optokinetic system. Vis. Neurosci. 2005;22:677–684. doi: 10.1017/S0952523805225166. [DOI] [PubMed] [Google Scholar]
- 53.Douglas R.M., Neve A., Quittenbaum J.P., Alam N.M., Prusky G.T. Perception of visual motion coherence by rats and mice. Vis. Res. 2006;46:2842–2847. doi: 10.1016/j.visres.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 54.Liu P., Huang H., Fang F., Liu L., Li L., Feng X., Chen W., Dalal R., Sun Y., Hu Y. Neuronal NMNAT2 overexpression does not achieve significant neuroprotection in experimental autoimmune encephalomyelitis/optic neuritis. Front. Cell. Neurosci. 2021;15:754651. doi: 10.3389/fncel.2021.754651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Milde S., Adalbert R., Elaman M.H., Coleman M.P. Axonal transport declines with age in two distinct phases separated by a period of relative stability. Neurobiol. Aging. 2015;36:971–981. doi: 10.1016/j.neurobiolaging.2014.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Huppke P., Wegener E., Gilley J., Angeletti C., Kurth I., Drenth J.P.H., Stadelmann C., Barrantes-Freer A., Bruck W., Thiele H., et al. Homozygous NMNAT2 mutation in sisters with polyneuropathy and erythromelalgia. Exp. Neurol. 2019;320:112958. doi: 10.1016/j.expneurol.2019.112958. [DOI] [PubMed] [Google Scholar]
- 57.Lukacs M., Gilley J., Zhu Y., Orsomando G., Angeletti C., Liu J., Yang X., Park J., Hopkin R.J., Coleman M.P., et al. Severe biallelic loss-of-function mutations in nicotinamide mononucleotide adenylyltransferase 2 (NMNAT2) in two fetuses with fetal akinesia deformation sequence. Exp. Neurol. 2019;320:112961. doi: 10.1016/j.expneurol.2019.112961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gilley J., Orsomando G., Nascimento-Ferreira I., Coleman M.P. Absence of SARM1 rescues development and survival of NMNAT2-deficient axons. Cell Rep. 2015;10:1974–1981. doi: 10.1016/j.celrep.2015.02.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Falk M.J., Zhang Q., Nakamaru-Ogiso E., Kannabiran C., Fonseca-Kelly Z., Chakarova C., Audo I., Mackay D.S., Zeitz C., Borman A.D., et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012;44:1040–1045. doi: 10.1038/ng.2361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Perrault I., Hanein S., Zanlonghi X., Serre V., Nicouleau M., Defoort-Delhemmes S., Delphin N., Fares-Taie L., Gerber S., Xerri O., et al. Mutations in NMNAT1 cause Leber congenital amaurosis with early-onset severe macular and optic atrophy. Nat. Genet. 2012;44:975–977. doi: 10.1038/ng.2357. [DOI] [PubMed] [Google Scholar]
- 61.Koenekoop R.K., Wang H., Majewski J., Wang X., Lopez I., Ren H., Chen Y., Li Y., Fishman G.A., Genead M., et al. Mutations in NMNAT1 cause Leber congenital amaurosis and identify a new disease pathway for retinal degeneration. Nat. Genet. 2012;44:1035–1039. doi: 10.1038/ng.2356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chiang P.W., Wang J., Chen Y., Fu Q., Zhong J., Chen Y., Yi X., Wu R., Gan H., Shi Y., et al. Exome sequencing identifies NMNAT1 mutations as a cause of Leber congenital amaurosis. Nat. Genet. 2012;44:972–974. doi: 10.1038/ng.2370. [DOI] [PubMed] [Google Scholar]
- 63.Kaplan J., Perrault I., Hanein S., Dollfus H., Rozet J.M. [Mutations in NMNAT1 cause Leber congenital amaurosis with severe macular and optic atrophy] Med. Sci. (Paris) 2013;29:26–27. doi: 10.1051/medsci/2013291008. [DOI] [PubMed] [Google Scholar]
- 64.Sasaki Y., Kakita H., Kubota S., Sene A., Lee T.J., Ban N., Dong Z., Lin J.B., Boye S.L., DiAntonio A., et al. SARM1 depletion rescues NMNAT1-dependent photoreceptor cell death and retinal degeneration. Elife. 2020;9:e62027. doi: 10.7554/eLife.62027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Eblimit A., Zaneveld S.A., Liu W., Thomas K., Wang K., Li Y., Mardon G., Chen R. NMNAT1 E257K variant, associated with Leber Congenital Amaurosis (LCA9), causes a mild retinal degeneration phenotype. Exp. Eye Res. 2018;173:32–43. doi: 10.1016/j.exer.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Essuman K., Summers D.W., Sasaki Y., Mao X., DiAntonio A., Milbrandt J. The SARM1 toll/interleukin-1 receptor domain possesses intrinsic NAD(+) cleavage activity that promotes pathological axonal degeneration. Neuron. 2017;93:1334–1343 e1335. doi: 10.1016/j.neuron.2017.02.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Osterloh J.M., Yang J., Rooney T.M., Fox A.N., Adalbert R., Powell E.H., Sheehan A.E., Avery M.A., Hackett R., Logan M.A., et al. dSarm/Sarm1 is required for activation of an injury-induced axon death pathway. Science. 2012;337:481–484. doi: 10.1126/science.1223899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gerdts J., Summers D.W., Sasaki Y., DiAntonio A., Milbrandt J. Sarm1-mediated axon degeneration requires both SAM and TIR interactions. J. Neurosci. 2013;33:13569–13580. doi: 10.1523/JNEUROSCI.1197-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Fernandes K.A., Mitchell K.L., Patel A., Marola O.J., Shrager P., Zack D.J., Libby R.T., Welsbie D.S. Role of SARM1 and DR6 in retinal ganglion cell axonal and somal degeneration following axonal injury. Exp. Eye Res. 2018;171:54–61. doi: 10.1016/j.exer.2018.03.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ko K.W., Milbrandt J., DiAntonio A. SARM1 acts downstream of neuroinflammatory and necroptotic signaling to induce axon degeneration. J. Cell Biol. 2020;219:e201912047. doi: 10.1083/jcb.201912047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lautrup S., Sinclair D.A., Mattson M.P., Fang E.F. NAD(+) in brain aging and neurodegenerative disorders. Cell Metab. 2019;30:630–655. doi: 10.1016/j.cmet.2019.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Lin J.B., Kubota S., Ban N., Yoshida M., Santeford A., Sene A., Nakamura R., Zapata N., Kubota M., Tsubota K., et al. NAMPT-mediated NAD(+) biosynthesis is essential for vision in mice. Cell Rep. 2016;17:69–85. doi: 10.1016/j.celrep.2016.08.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Lin J.B., Apte R.S. NAD(+) and sirtuins in retinal degenerative diseases: a look at future therapies. Prog. Retin. Eye Res. 2018;67:118–129. doi: 10.1016/j.preteyeres.2018.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang L., Li S., Miao L., Huang H., Liang F., Teng X., Xu L., Wang Q., Xiao W., Ridder W.H., 3rd, et al. Rescue of glaucomatous neurodegeneration by differentially modulating neuronal endoplasmic reticulum stress molecules. J. Neurosci. 2016;36:5891–5903. doi: 10.1523/JNEUROSCI.3709-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yang L., Miao L., Liang F., Huang H., Teng X., Li S., Nuriddinov J., Selzer M.E., Hu Y. The mTORC1 effectors S6K1 and 4E-BP play different roles in CNS axon regeneration. Nat. Commun. 2014;5:5416. doi: 10.1038/ncomms6416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Miao L., Yang L., Huang H., Liang F., Ling C., Hu Y. mTORC1 is necessary but mTORC2 and GSK3beta are inhibitory for AKT3-induced axon regeneration in the central nervous system. Elife. 2016;5:e14908. doi: 10.7554/eLife.14908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li L., Huang H., Fang F., Liu L., Sun Y., Hu Y. Longitudinal morphological and functional assessment of RGC neurodegeneration after optic nerve crush in mouse. Front. Cell. Neurosci. 2020;14:109. doi: 10.3389/fncel.2020.00109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Huang H., Miao L., Yang L., Liang F., Wang Q., Zhuang P., Sun Y., Hu Y. AKT-dependent and -independent pathways mediate PTEN deletion-induced CNS axon regeneration. Cell Death Dis. 2019;10:203. doi: 10.1038/s41419-018-1289-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Huang H., Miao L., Liang F., Liu X., Xu L., Teng X., Wang Q., Ridder W.H., 3rd, Shindler K.S., Sun Y., Hu Y. Neuroprotection by eIF2alpha-CHOP inhibition and XBP-1 activation in EAE/optic neuritiss. Cell Death Dis. 2017;8:e2936. doi: 10.1038/cddis.2017.329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Li Y., Gruber J.J., Litzenburger U.M., Zhou Y., Miao Y.R., LaGory E.L., Li A.M., Hu Z., Yip M., Hart L.S., et al. Acetate supplementation restores chromatin accessibility and promotes tumor cell differentiation under hypoxia. Cell Death Dis. 2020;11:102. doi: 10.1038/s41419-020-2303-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.