

Abstract
We established a genome-wide compendium of somatic mutation events in 3949 whole cancer genomes representing 19 tumor types. Protein-coding events captured well-established drivers. Noncoding events near tissue-specific genes, such as ALB in the liver or KLK3 in the prostate, characterized localized passenger mutation patterns and may reflect tumor-cell-of-origin imprinting. Noncoding events in regulatory promoter and enhancer regions frequently involved cancer-relevant genes such as BCL6, FGFR2, RAD51B, SMC6, TERT, and XBP1 and represent possible drivers. Unlike most noncoding regulatory events, XBP1 mutations primarily accumulated outside the gene’s promoter, and we validated their effect on gene expression using CRISPR-interference screening and luciferase reporter assays. Broadly, our study provides a blueprint for capturing mutation events across the entire genome to guide advances in biological discovery, therapies, and diagnostics.
INTRODUCTION
A central hallmark of tumor development is that cancer cells acquire somatic mutations in their genomes that are not present in normal tissue. Some mutations are drivers and contribute to the growth of tumor cells, but many others are passengers without apparent effects on tumor biology. Over the past decade, driver mutations have been comprehensively characterized in protein-coding genomic regions by analyzing sequencing data from thousands of tumor-normal pairs. This characterization in protein-coding regions has yielded a wealth of insights into tumor biology, including many genome-inspired drug targets. However, the role of somatic mutations in the other 98% of the cancer genome—the noncoding genome—remains incompletely understood.
RATIONALE
Many statistical approaches detect drivers as recurrent mutation events by comparing the number of mutations with and without effects on protein-coding sequences in each gene. These approaches are therefore inapplicable outside of protein-coding regions, where the roles of somatic mutations remain less well understood. The noncoding genome encompasses a diverse spectrum of elements, including regulatory regions of gene expression that differ in their locations and activities between tumor types. To expand our understanding of mutations beyond protein-coding regions, we designed and implemented a genome-wide, sliding-window approach that detects mutation events irrespective of their locations in regulatory elements or effects on protein-coding sequences.
RESULTS
We developed a composite of three methods to detect recurrent mutation events across the whole genomes of 3949 patients with 19 cancer types and 61.2 million somatic mutations. This approach automatically stratified mutation events into different categories on the basis of their position in the genome. In protein-coding regions, we identified an average of 7.5 events per cancer type and recovered well-established driver mutations. In the noncoding genome, 3.7 events per cancer type occurred adjacent to genes exclusively expressed in specific tissue types (ALB in liver, KLK3 in prostate, SFTPB in lung, SLC5A12 in kidney, TG in thyroid tissue, and many others). These tissue-specific events were unlikely to be prototypical drivers because they stemmed from a mutagenic process that was exclusively active around these genes, instead reflecting possible imprints of the expression programs of the tumor cells of origin. Moreover, we found 3.8 noncoding events per cancer type in regulatory regions of expression, many involving cancer-relevant genes (BCL6, FGFR2, RAD51B, SMC6, TERT, XBP1, and many others). In contrast to most events in regulatory regions, breast cancer mutations near XBP1 mainly accumulated in a regulatory region outside of its promoter. We validated their regulatory effects on gene expression by performing CRISPR-interference screening and luciferase reporter assays, illuminating the potential of genome-wide approaches paired with harmonized sequencing cohorts to comprehensively capture mutation patterns in both known and unknown elements of the noncoding genome.
CONCLUSION
Our study establishes a genome-wide compendium of the diverse mutation patterns that shape the genomes of 19 major cancer types, including events near genes with known roles in tumor biology and some exhibiting experimentally validated effects on gene expression. Our results demonstrate that noncoding mutations are associated with a broad spectrum of different biological processes and that their location in the genome is essential for their accurate interpretation. Broadly, our study provides a blueprint for interpreting whole-genome sequencing data and lays the foundation for future experimental endeavors to implicate noncoding mutations in tumor development, ultimately paving the way for therapies tailored to the noncoding cancer genome.
Graphical Abstract
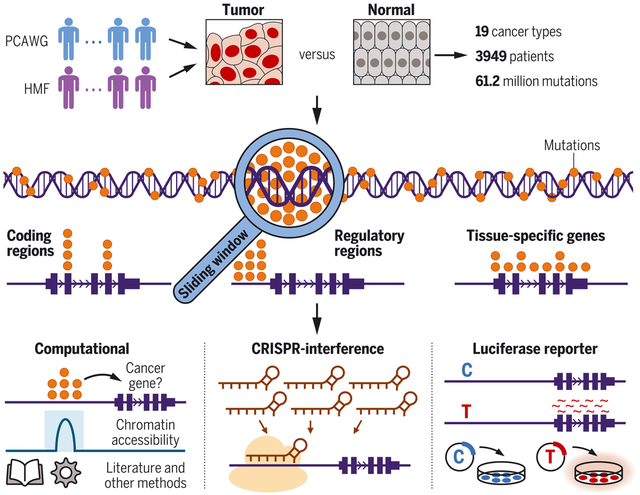
Genome-wide compendium of somatic mutation patterns in human cancer. We analyzed 61.2 million mutations from 3949 patients of 19 cancer types (top). Using a sliding-window approach, we detected mutation events across the entire cancer genome and classified them by their genomic locations (middle). For systematic follow-up, we used both computational and experimental strategies (bottom). PCAWG, Pan-Cancer Analysis of Whole Genomes; HMF, Hartwig Medical Foundation.
Tumors carry different types of somatic mutations in their genomes. Most of these mutations are random “passengers” that are propagated through clonal evolution without contributing to tumor development (1). However, a few are “drivers” that contribute to the uncontrolled growth and proliferation of cancer cells (1) and therefore represent targets for many therapies in precision medicine.
Over the past decade, the characterization of somatic drivers has focused primarily on protein-coding regions (2), where such mutations change the amino acid sequences of oncogenes and tumor suppressor genes. Statistical algorithms have been established to detect drivers as recurrent “mutation events” in large sequencing cohorts of tumor patients (3–5). Applying these algorithms to the sequencing data of thousands of tumor-normal pairs has helped considerably to elucidate which mutations contribute to tumor development in coding regions (2), whereas the role of noncoding somatic mutations in the remaining ~98% of the genome remains less well understood (6).
In the noncoding genome, the detection and interpretation of mutation events are complex. Many algorithms have been established that detect mutation events based on nonsynonymous and synonymous amino acid changes in coding regions (3, 4), rendering them inapplicable to noncoding regions in whole-genome sequencing (WGS) data. Furthermore, the noncoding genome comprises a diverse spectrum of genomic elements, ranging from active regulatory elements of gene expression to inactive heterochromatic regions (7, 8). Therefore, mutation events in different parts of the noncoding genome mirror separate biological processes, as revealed by recent studies such as the Pan-Cancer Analysis of Whole Genomes (PCAWG) (9). Although several mutation events represent possible noncoding drivers, such as those identified in the promoters and enhancers of cancer-relevant genes, others are less likely to be drivers, such as those resulting from mutagenic processes around tissue-specific genes (9, 10).
To address these specific challenges in noncoding regions, we implemented a genome-wide approach that identifies somatic mutation events in point mutations and in short insertions and deletions across the entire cancer genome irrespective of their positions in the genome or their effects on protein-coding sequences. This approach automatically stratifies mutation events based on their genomic locations, thus capturing their different propensities to represent possible drivers or localized passenger mutation patterns. By applying this strategy to a harmonized cohort of 3949 somatic whole cancer genomes and combining it with systematic computational and experimental follow-up, our study establishes a genome-wide compendium of mutation events in 19 major cancer types.
RESULTS
Genome-wide detection of somatic mutation events in whole cancer genomes
For genome-wide detection and classification of somatic mutation events, we proceeded in three steps (Fig. 1, A to C, and fig. S1). First, we tiled the genome with three interval sizes (1, 10, and 100 kb; see illustration in fig. S2) and performed three significance tests in each interval: test 1 to determine whether a genomic region contained more mutations than expected based on its epigenomic signal; test 2 to compare mutation counts between different cancer types in each region; and test 3 to determine whether more mutations clustered together than expected. Second, we integrated P values from these three tests and different interval lengths into a continuous genome-wide signal of significance based on Brown’s method (11), and then adjusted this signal by weighted multiple hypothesis correction based on cancer-specific expression data (12). Third, we identified all statistically significant events in this genome-wide signal [false discovery rate (FDR) < 0.1] and automatically classified them based on their genomic locations into protein-coding regions (mutations in exons of oncogenes and tumor suppressor genes), regulatory regions [promoters and enhancers overlapping with signals of H3K4me3 and H3K27ac histone chromatin immunoprecipitation sequencing (ChIP-seq) (7)], or mutagenic processes around tissue-specific genes (genes exclusively expressed in a specific cancer type). In this way, we captured their different propensities to be possible drivers or passengers building on insights gained from prior studies (9). We excluded events with mutational hotspots in secondary DNA hairpin structures or low genomic mappability; events not meeting any of these criteria were labeled as “other” (Fig. 1, A to C).
FIG. 1.
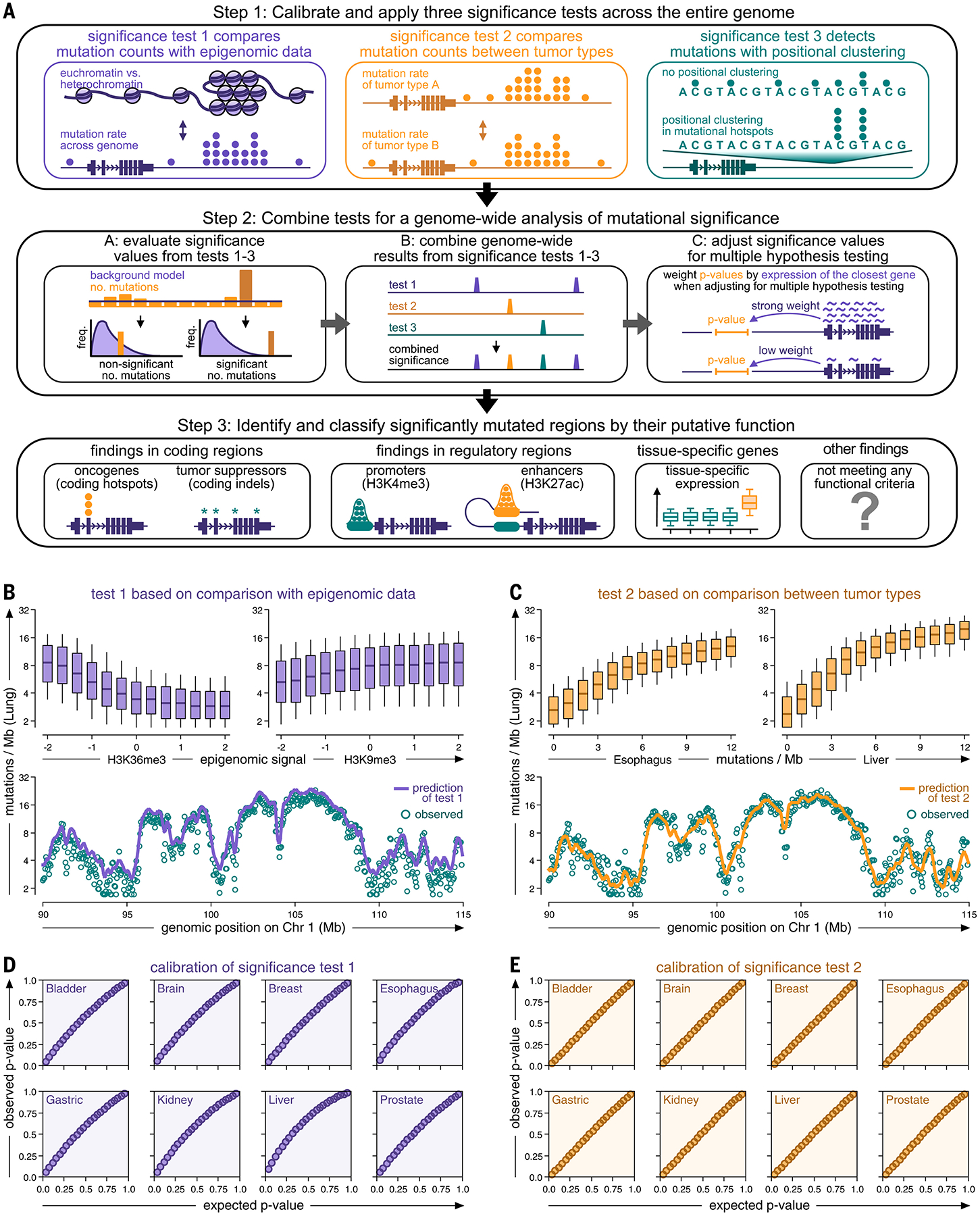
Genome-wide analysis of somatic mutation events in whole cancer genomes. (A) Genome-wide detection of somatic mutation events in whole cancer genome sequencing data. Step 1 combines three complementary test strategies. Step 2 integrates the results of tests 1 to 3 into a joint, genome-wide signal and identifies significant mutation events. Step 3 classifies mutation events according to their genomic location. (B and C) Top: Boxplots comparing mutation rates of a representative cancer type (lung cancer) against epigenomic signals [(B), the rationale of test 1] and mutation rates of other cancer types [(C), the rationale of test 2]. Boxes indicate 25/75% interquartile ranges, vertical lines extend to 10/90% percentiles, and horizontal lines reflect distribution medians. Bottom: Observed (teal dots) and predicted (continuous line) mutation rates (10-kb intervals) plotted against their position on chromosome 1 (function smoothed by Gaussian kernel). (D and E) Q-Q plots comparing observed (y-axis) and expected (x-axis) P values for test 1 (D) and test 2 (E).
Q-Q plots demonstrated that the three significance tests and their combined P values were accurately calibrated to their background signals and exhibited no inflation of low P values (Fig. 1, D and E, and figs. S3 and S4). Histograms revealed that the background models of the three tests matched the observed distributions of mutation rates and positional clustering in the upper distribution tails (fig. S3). These results further suggested that the three tests did not rely on cancer-type-specific assumptions, and that our genome-wide analysis was applicable across a wide range of different cancer types. The materials and methods and supplementary text include a comprehensive explanation of the rationale behind our statistical framework in the context of prior approaches, additional analyses of the performance and accuracy of the three significance tests (figs. S5 to S11), the necessity of combining different tests (fig. S12) and interval sizes (fig. S2) to capture a broad spectrum of mutation events, and a comparison with alternative implementations (figs. S13 to S17).
A genome-wide compendium of mutation events in 19 cancer types
For a harmonized analysis of 6.12 × 107 somatic mutations in 3949 whole genomes from 19 cancer types, we assembled high-confidence samples, regions, mutations, and cancer types from two sequencing consortia, PCAWG (9) and the Hartwig Medical Foundation [HMF (13)]. A detailed description of our filtering criteria and the cancer types included in this study is provided in the materials and methods and figs. S18 to S21. In 19 cancer types, our genome-wide approach detected 142 events in coding regions (average 7.5 per cancer type; 45 in oncogenes and 97 in tumor suppressors), 73 events in regulatory regions (average 3.8 per cancer type; 49 in promoters and 24 in enhancers), 70 events around tissue-specific genes (average 3.7 per cancer type; 70 genes exclusively expressed in a specific cancer type, such as albumin in the liver), and 87 “other” events (average 4.6 per cancer type; the exact role of these findings was less clear) (Fig. 2, A and B; figs. S22 to S24; and tables S1 to 20). To refer to the genomic location of our findings, we annotated them by their closest genes (table S1). For confirmation, we used the activity-by-contact model (14) based on three-dimensional genomic distance, which returned the same genes for 91% of coding, regulatory, and tissue-specific findings (fig. S12, G to I).
FIG. 2.
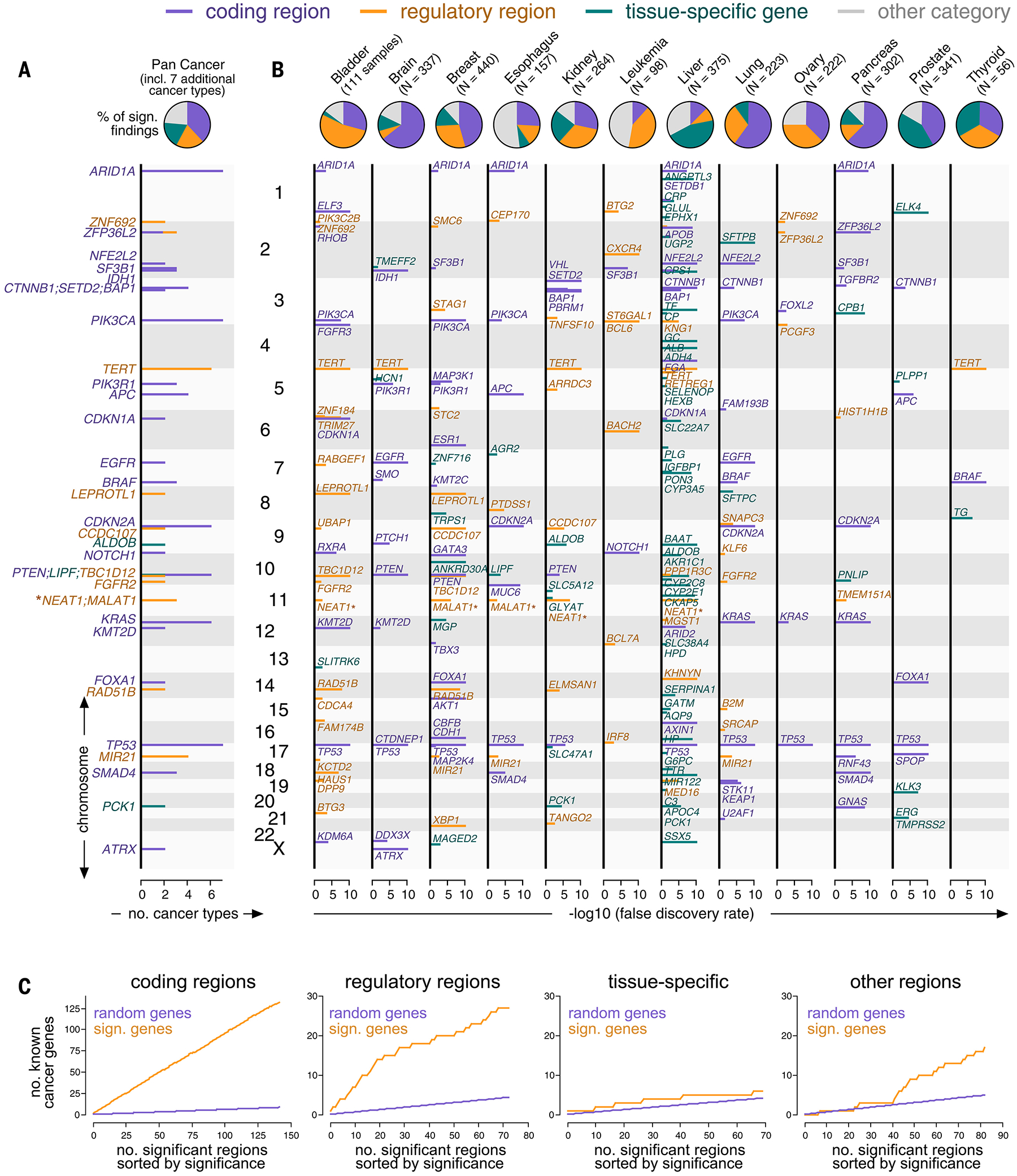
Mutation events identified in a genome-wide analysis of the PCAWG and HMF consortia. (A and B) Top: Pie charts showing the number of mutation events per category (purple: coding, orange: regulatory, teal: tissue-specific, gray: other) in aggregate (A) and individual cancer types (B). Bottom: Genomic positions (y-axis) plotted against their significance in a genome-wide analysis (x-axis) and colored by categories (B). The position (y-axis) of findings recurring in more than one cancer type is plotted against the number of cancer types (x-axis) (A). NEAT1 and MALAT1 are marked by asterisks because their classification was ambiguous. (C) Mutation events sorted by their significance in a genome-wide analysis (x-axis, orange) and plotted against the number of findings involving known cancer genes (y-axis, top). Random overlap between findings and cancer genes serves as a negative control (purple).
Events in protein-coding regions
Findings in protein-coding regions largely captured well-established driver mutations, with 93.0% (132/142) involving canonical cancer genes (Fig. 2C) and 96.5% (137/142) matching the results obtained by two established methods for identifying coding drivers [MutSigCV (3) and dNdScv (4)] (fig. S25, A and B). This low rate of false positives in coding regions supports the robustness of our approach in the entire genome because it uses the same statistics in both coding and noncoding regions. Furthermore, significance values returned by our genome-wide approach in protein-coding regions correlated with the ratio of nonsynonymous to synonymous mutations (fig. S25C), an established marker of positive selection (4). We obtained a similar result in the rest of the genome by predicting the pathogenicity of noncoding mutations based on two bioinformatics scores (15, 16) (fig. S25, D to F).
Events in regulatory regions
Events in regulatory regions were significantly enriched for canonical cancer genes (P < 0.001, Fisher’s exact test), with 37.0% (27/73) of the findings linked to genes in the Cancer Gene Census (17) or the Oncology Knowledge Base (18), compared with the 4.1% (the percentage of cancer genes among all genes) that would be expected to occur by chance (Fig. 2C). Because of the link between these regions and gene expression, some findings in this category have been discussed as plausible noncoding drivers in the literature (6, 9, 10, 19). This includes mutation events in the TERT promoter (telomere regulation), which we identified in bladder, brain, head and neck, kidney, liver, and thyroid cancer, and mutations at MIR21 (cancer-promoting microRNA gene), which we detected in breast, esophagus, gastric, and lung cancer. Furthermore, consistent with these prior studies (6, 9, 10, 19), we found noncoding mutations upstream of FOXA1 in breast cancer and downstream of FOXA1 in prostate cancer, in addition to many coding mutations in the same gene.
Our study expanded this category by 46 additional findings in promoters and enhancers of genes potentially relevant to cancer (Figs. 2, A and B, and 3A and figs. S22 and S23). For example, we identified recurrent events in the promoters of leukemia-related genes, including BACH2, BTG2, CXCR4, BCL6, BCL7A, and IRF8. Other mutations accumulated in promoters of the cancer-associated genes FGFR2 in bladder and lung cancer; B2M, KLF6, and SRCAP (chromatin remodeling complex) in lung cancer; and MDM4, PIK3C2B, CDCA4 (cell cycle gene), and BTG3 (antiproliferation factor) in bladder cancer. We found additional events in the promoters of MED16 (coactivator of RNA polymerase II transcription) in liver cancer, as well as STAG1 (cohesion of sister chromatids during the S-phase), SMC6 (maintenance of telomere length), and GEN1 (double-strand break repair) in breast cancer.
FIG. 3.
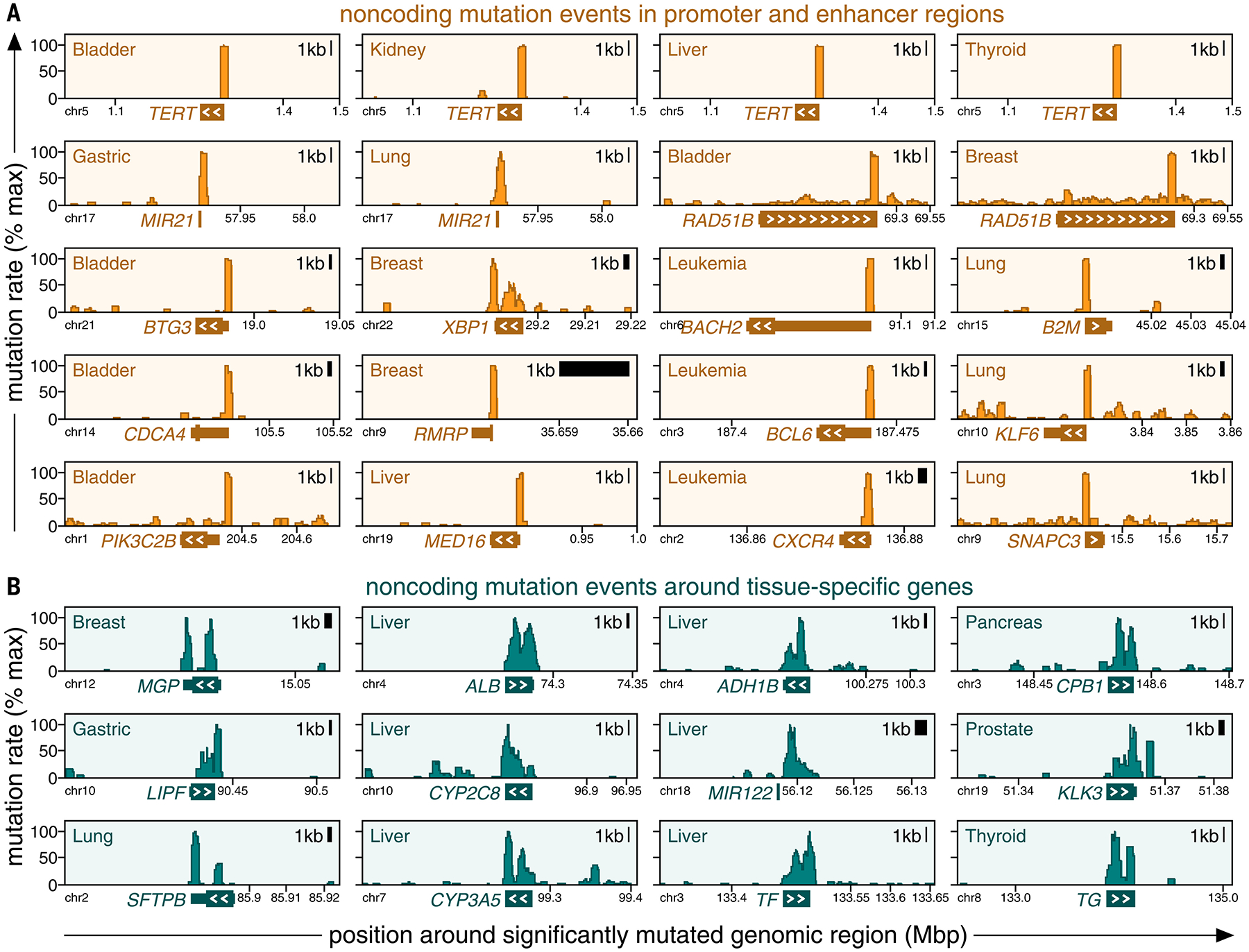
Categories of mutation events exhibit different mutation patterns. Positional clustering of mutations (y-axis, percentage of maximum) is plotted against genomic positions (x-axis) around mutation events that fall into regulatory regions [(A), orange] or overlap with tissue-specific genes [(B), teal]. Genomic boundaries of the closest gene are marked at the bottom of each plot, and white arrowheads mark the direction of its transcription.
Other additional findings were in enhancers, including RAD51B (canonical cancer gene involved in double-strand break repair) in bladder and breast cancer, ETS2 (transcription factor related to proliferation, apoptosis, and telomere maintenance) in colorectal cancer, ST6GAL1 (glycosyltransferase inducing an invasive phenotype) in leukemia, and XBP1 (established function as an estrogen-induced transcription factor) in breast cancer. Some mutations in this category recurred as hotspots in the same genomic position, including BTG3, FGFR2, MED16, PIK3C2B, SMC6, STAG1, and TERT (fig. S26A and table S21), although the occurrence of this mutation pattern was rare in noncoding regulatory regions compared with its high frequency in coding regions.
Events near tissue-specific genes
In contrast to protein-coding and regulatory regions, findings around tissue-specific genes are unlikely to represent candidate driver events themselves because of their reported link to localized mutagenic processes (9, 10) and lack of enrichment for known cancer genes (Fig. 2C). However, according to the MalaCards database (20), 42.9% (30/70) of tissue-specific genes linked to mutation events exhibited physiological roles in their associated normal tissues, compared with the 3.9% (the percentage of genes included in the MalaCards database) that would be expected to occur by chance (fig. S26, B and C). Therefore, mutation events in this category were significantly enriched around genes with reported physiological roles independent of cancer signaling (P < 0.001, Fisher’s exact test), concordant with their unique expression in a specific tissue type. Some of our findings near tissue-specific genes have been observed in previous studies, either as primary results (10) or as incidental findings annotated as nondrivers (9). These included LIPF in gastroesophageal cancer, ALDOB in kidney and liver cancer, SFTPB and SFTPC in lung cancer, CPB1 and PNLIP in pancreatic cancer, TG in thyroid cancer, and 12 tissue-specific genes in liver cancer (including ALB, CYP3A5, FGA, and MIR122).
Our study expanded this category by 54 additional findings (Figs. 2, A and B, and 3B and figs. S22 and S23), including TMEFF2 (survival factor for neurons) and HCN1 (hyperpolarization-activated cation channel in neurons) in brain tumors, as well as STC2 (glycoprotein induced by estrogen), TRPS1 (repressor of GATA-regulated genes), ANKRD30A (serologically defined breast cancer antigen), and MGP (estrogen-regulated matrix protein involved in cellular differentiation) in breast cancer. Other additional events in this category included KLK3 (prostate-specific antigen, a serum marker for prostate cancer), PLPP1 (androgen-regulated phosphatase expressed on the cell surface), and TMPRSS2 (androgen-regulated serine protease) in prostate cancer, and GCG (glucagon, a pancreatic hormone) in neuroendocrine tumors. Furthermore, we identified tissue-specific events around SLC5A12 (lactate reabsorption in proximal tubules), KCNJ15 (potassium channel in the kidney), GLYAT (glycine-acyltransferase), and PCK1 (gluconeogenesis) in kidney cancer, as well as MUC6 (mucin; protects epithelium from gastric acid) and AGR2 (expressed in mucus-secreting tissues and overexpressed in Barrett’s esophagus) in gastroesophageal tumors. Moreover, liver cancer exhibited the largest number of additional mutation events in the tissue-specific category, including 18 genes encoding liver-specific proteins (including C3, CRP, and TF) and 17 genes associated with liver metabolism and detoxification (including AKR1C1, BAAT, CYP2E1, G6PC, and HEXB).
Other events
For some events, the status remained less clear. For example, in agreement with the prior literature, we identified events at the neighboring genes NEAT1 and NEAT2 in breast, bladder, esophagus, kidney, and liver cancer. Our genome-wide approach placed them in the regulatory category (fig. S27), whereas PCAWG interpreted them as being the result of a transcription-related mutational process (9), and other studies arrived at different conclusions regarding their relevance in tumor signaling (19, 21).
Furthermore, some noncoding events did not fall into the protein-coding, regulatory, or tissue-specific categories. This “other” category exhibited mild enrichment for canonical cancer genes (Fig. 2C) and included MAD1L1 and MAD2L1 (mitotic spindle assembly checkpoint) in brain and ovarian tumors; NF1 (tumor suppressor) in breast tumors; DCC (known cancer gene) in esophageal cancer; KCNJ15 (potassium channel) in kidney cancer; TCL1A, BCR, and NFKBIE (known cancer genes) in leukemia; as well as ABHD5 (lipid binding), LIPG (lipase), FN1 (fibronectin), HNF4A (hepatocyte nuclear factor), MAP2K6 (mitogen-activated kinase), and ERRFI1 (ERBB receptor feedback inhibitor) in liver cancer. In addition, APC and SMAD4 in colorectal cancer harbored noncoding splice site mutations outside of canonical exon-intron boundaries (fig. S22D).
Altogether, our study establishes a genome-wide compendium of somatic mutation events for 19 cancer types, categorized by their genomic locations and different biology, including many findings from recent studies and several additional results (see table S1 for literature references). A complete list of our findings in each cancer type is provided in tables S2 to S20, annotated by their genomic locations, mutation frequencies, status as known cancer genes, and significance values returned by our genome-wide approach.
Systematic follow-up on mutation events identified in our genome-wide analysis
We performed three systematic follow-up analyses to examine the ability of our approach to detect mutation events in the noncoding genome and evaluate the plausibility of our results.
Inspection of the genomic territory around mutation events
Although our genome-wide approach examined the entire genome, 76.6% (285/372) of the mutation events occurred in coding, regulatory, or tissue-specific regions (Fig. 2, A and B, and figs. S22 and S23), which account for 10.2% of the genome. Furthermore, they accumulated in regulatory and transcribed regions based on ChIP-seq data from normal tissue (7) (fig. S28A), and this enrichment was even more pronounced in chromatin accessibility data [assay for transposase-accessible chromatin using sequencing (ATAC-seq)] from the same type of tumor tissue, when available (8) (fig. S28B). Moreover, mutation events exhibited strong enrichment around the following four markers (figs. S29 and S30 and tables S2 to S20): (i) ATAC-seq peaks that existed in tumor but not in normal tissue (fig. S29, A and B), (ii) ATAC-seq peaks that correlated with the expression of their closest gene (fig. S29, C and D), (iii) methylation markers that correlated negatively with the expression of their associated genes (fig. S29, E and F), and (iv) genome-wide association study (GWAS) peaks from germline data (fig. S29, G and H).
The accumulation of events around these four markers prompted us to investigate whether the performance of our genome-wide analysis could be improved by restricting it to regions around these four markers. However, this restricted version missed a substantial number of findings (fig. S30H), including many events associated with known cancer genes. Furthermore, the applicability of the four markers varied between cancer types, depending on the availability of ATAC-seq data (8). Similar results were obtained when restricting our analysis to five databases of established promoter and enhancer regions (22–26) (fig. S30, C and D), illuminating the potential of a genome-wide approach.
Compatibility with prior findings and methods
Previous studies, including PCAWG, reported 30.1% (43/143) of the noncoding mutation events in the tissue-specific and regulatory categories observed herein (6, 9, 10, 19), compared with the 1.47% (the percentage of genes for which noncoding findings had been reported previously) that would be expected by chance (P < 0.001, Fisher’s exact test). Conversely, our genome-wide analysis identified 39 of the noncoding findings from prior work (39/65 previous findings; 30/39 previous findings with an FDR < 10−4) (tables S22 and S23). Tissue-specific events in this comparison were interpreted differently in prior studies that either reported them as primary results (10) or incidental, nondriver findings (9). Furthermore, our WGS dataset overlapped with that of previous studies, so that shared findings affirm the general compatibility of our genome-wide approach in regions evaluated by both our study and prior work.
For further comparison, we ran four existing and available methods [DriverPower (27), Larva (28), MutSpot (29), and OncodriveFML (5)] on the entire WGS dataset. This revealed that our genome-wide approach identified nearly all the noncoding events detected by these four methods in the genomic territory included in our analysis (figs. S31 and S32). This comparison further highlighted the importance of excluding low-quality mutations and low-coverage regions from our genome-wide analysis for technical considerations (figs. S18 and S32), given that not all parts of the genome are amenable to WGS.
Analysis of the statistical power of our genome-wide approach to detect mutation events
This analysis demonstrated that the power of our approach varied substantially between cancer types, depending on their background mutation rates, the available number of samples, and the size of the genomic territory included in the analysis (fig. S33). Additional technical factors beyond those captured in this model may interfere with the statistical power (9). Although combining the HMF and PCAWG consortia increased the statistical power of our study considerably, the amount of whole-genome data was still smaller than the amount of whole-exome data generated over more than a decade and used to characterize mutations in coding regions (2). Therefore, there may be noncoding events in addition to those identifiable in the available data (fig. S33), as was concordantly concluded in a power analysis by the PCAWG study (9).
Characterization of mutation and expression patterns of tissue-specific genes
We next studied the pattern of mutation events near or within tissue-specific genes in more detail (fig. S34). We first focused on liver cancer, which contained the largest number of events in this category. Consistent with previous studies connecting this category of mutations with localized mutagenic processes (9, 10), noncoding regions around tissue-specific genes were enriched for insertions and deletions (“indels”) (Fig. 4A). These indels were longer than those in the rest of the genome (83.2 versus 22.4% of deletions had target lengths >1 bp; 30.1 versus 15.5% for insertions) (Fig. 4, B and C, and fig. S34A). In addition, we observed that indels around tissue-specific genes accumulated in A/T-rich nucleotide contexts and resembled Catalogue of Somatic Mutations in Cancer (COSMIC) indel signatures ID4 and ID8 (30), a pattern that rarely occurred in the rest of the genome (fig. S34, B to H). Comparison of mutations around tissue-specific versus highly expressed genes yielded the same differences (fig. S34, I and J), suggesting that mutation events in this category only occurred around genes exhibiting unique expression in a particular tissue type and not around highly expressed genes in general. Concordantly, expression and mutation rates exhibited positive correlation in noncoding regions around tissue-specific genes, the opposite of their relationship in the rest of the genome (fig. S35, A and B). In addition to mutations, other recurrent events accumulated in proximity to tissue-specific genes, including hypermethylation (fig. S35, C and D) and copy number loss (fig. S35, E to H). We obtained similar results in cancer types other than liver (fig. S34K).
FIG. 4.
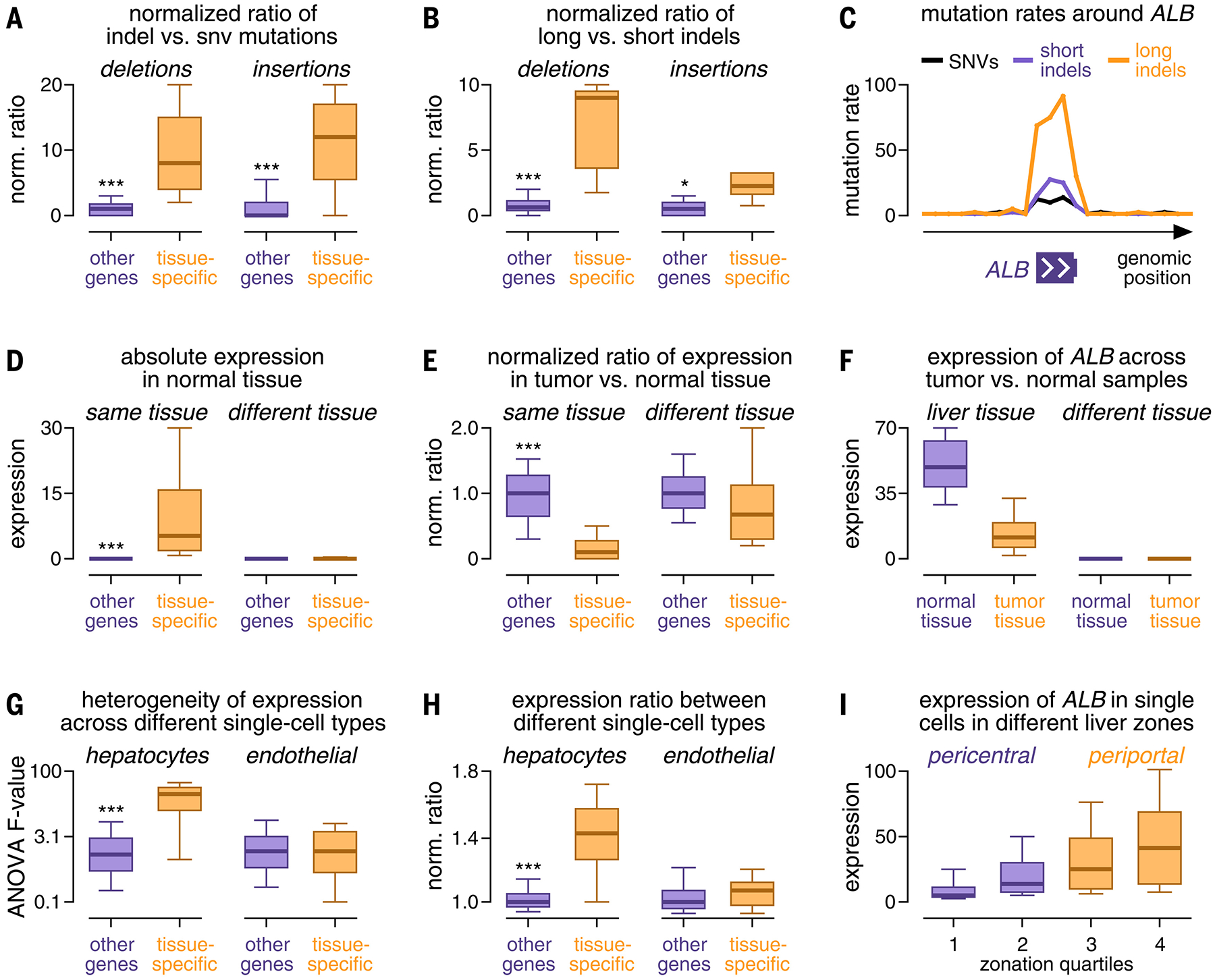
Characterization of the expression and mutation patterns of tissue-specific genes. (A and B) Box plots comparing the ratio of the number of indels to single-nucleotide variants (SNVs) (A) and the ratio of the number of long to short indels (B) between tissue-specific genes (orange) and other genes (purple). (C) Mutation rates of SNVs (black), short indels (purple), and long indels (orange) (y-axis, percentage of maximum) plotted against their genomic position around ALB (x-axis). (D and E) Box plots comparing the expression (D) and expression ratio in tumor versus normal tissue (E) of tissue-specific genes (orange) and other genes (purple). (F) Box plots comparing ALB expression (y-axis) between samples from tumor tissue (orange) and normal tissue (purple). (G and H) Box plots comparing heterogeneous expression of tissue-specific genes (orange) and other genes (purple) in single-cell data of hepatocytes (left) and endothelial cells (right) based on an analysis of variance (ANOVA) test (G) and the expression ratio between cell types (H). (I) Box plots comparing ALB expression in cells from different histological zones of the liver (x-axis). Boxes in (A) to (I) indicate the 25/75% interquartile range, vertical lines extend to 10/90% percentiles, and horizontal lines reflect distribution medians. Significant differences (Mann-Whitney U test) are marked with asterisks: *P < 0.05, **P < 0.01, ***P < 0.001.
However, mutation events did not occur ubiquitously around all tissue-specific genes, with most cancer types harboring >100 tissue-specific genes but five or fewer tissue-specific events (fig. S36A). Furthermore, the number of events in this category differed greatly between cancer types (Fig. 2B and fig. S22, A and B), and the fraction of indels and their lengths varied considerably between individual tissue-specific genes (fig. S36, B and C). These observations suggest that some but not all tissue-specific genes harbor a mutation pattern in their surrounding noncoding territory that deviates from the rest of the genome. These differences manifested as mutation events detected by our genome-wide approach and characterized the specific genomic regions and genes where this localized mutation pattern occurred.
Finally, we explored whether characterizing mutation events around tissue-specific genes could offer insights into tumor biology. We hypothesized that these events might be connected to the cell of origin from which a tumor developed, given that these genes exhibited (i) tissue-specific expression (Fig. 4D), (ii) lower expression in tumor cells than in normal cells (Fig. 4, E and F, and fig. S36, D to F), and (iii) physiological roles in their respective tissues (fig. S26, B and C). Consistent with this hypothesis, many tissue-specific genes were heterogeneously expressed in single-cell data from normal tissues (fig. S37, A and B), particularly those harboring mutation events (fig. S37, C and D). For instance, in single-cell expression data for liver (31), most tissue-specific genes with mutation events were differentially expressed (87.5%; 35/40) between cells from different histological zones (Fig. 4, G to I, and fig. S38, A to D) compared with 15.5% for arbitrary genes expressed in the liver (P < 0.001, Fisher’s exact test). Similarly, in single-cell expression data for kidney (32), all tissue-specific genes with mutation events were expressed in a specific cell type (proximal tubule cells, 100%; 5/5) (fig. S38, E and F) compared with 26.4% for arbitrary, heterogeneously expressed genes (P = 0.001, Fisher’s exact test). Likewise, papillary and clear-cell kidney tumors, which originate from proximal tubule cells, carried mutations around tissue-specific genes more frequently than chromophobe kidney tumors that originate from collecting-duct epithelial cells (33) (60.9 versus 14.0%; P < 0.001, Fisher’s exact test) (fig. S38G).
Our analyses thus established a general, reciprocal link among a localized mutation pattern in tumor genomes, tissue-specific expression in bulk expression data, and heterogeneous expression in single-cell data of the related normal tissue. Therefore, the localized mutation pattern around tissue-specific genes may reflect a potential imprint of the characteristic expression program of the cell type from which a tumor originated (Fig. 4, G to I, and figs. S37 and S38), which could be of use in diagnostics.
Evaluation of mutation events in promoter and enhancer regions
We next used the following analyses to further assess the noncoding mutation events in regulatory promoter and enhancer regions.
Transcription factor binding sites
We used a permutation test to identify recurrent mutations that changed transcription factor binding motifs in the JASPAR database (34) (see the materials and methods). This test revealed that mutations changed binding motifs in 15.1% (11/73) of our findings in the regulatory category (fig. S39A), mainly in two binding motifs (81.8%; 9/11): Mutations in the ELK4 motif produced two binding sites in the TERT promoter in many cancer types (35) (fig. S39A), whereas mutations in the EGR1 motif (36) removed transcription factor binding sites from the promoters of antiproliferative genes such as BTG3 or STAG1 (fig. S39, A and B). We found an additional hotspot in the FOXA1 promoter that produced a binding site for E2F1 (19) (fig. S39A). In addition to these single-gene analyses, we analyzed mutations across regulatory regions in aggregate and detected additional changes to transcription factor binding sites in regulatory regions (fig. S40).
Differential expression
Differential expression analysis required matched mutation and expression data from the same tumor samples, and the limited availability of such data restricted our search to 12 cancer types (fig. S41, A to C, and materials and methods). In addition, we identified potential confounders of differential expression (fig. S41, D to G), including copy number, methylation, and the positive correlation between expression and mutation rates around tissue-specific genes, which was opposite to their negative correlation in the rest of the genome (fig. S35, A and B). Keeping these intrinsic limitations in mind, the genes linked to 49 mutation events (23 coding, seven regulatory, 19 tissue specific) were associated with differential expression between mutated and nonmutated samples after multiple hypothesis correction (fig. S42). For seven of 12 cancer types, the number of differentially expressed genes was higher than would be expected by chance (fig. S43, A to D). In addition to evaluating differential expression for each mutation event separately, we performed two aggregate analyses and detected additional potential associations between noncoding mutations and differential expression (fig. S43, E and F).
Physical interactions
Noncoding mutation events involved many genes that exhibited direct physical interactions with established driver genes identified from analyses of coding regions, suggesting that they targeted the same pathway (37) (fig. S44A and materials and methods).
Differences in survival
We tested whether findings in the regulatory category were associated with differences in the survival of mutated and nonmutated cancer patients. Using a log-rank test, we detected significant differences for TERT in brain (P = 3 × 10−5) and thyroid (P = 5 × 10−2) cancer, B2M and FGFR2 in lung cancer (P = 9 × 10−4 and 1 × 10−2, respectively), ARRDC3 in kidney cancer (P = 4 × 10−2), PIK3C2B in bladder cancer (P = 8 × 10−3), BCL6 in leukemia (P = 1 × 10−2), and XBP1 in breast cancer (P = 8 × 10−4) (fig. S44B). These analyses provide additional support for the plausibility of some of the mutation events in this category, in addition to their location in regulatory regions and enrichment for canonical cancer genes (Fig. 2C).
Experimental evaluation of regulatory regions and noncoding mutations around XBP1
Although many events in the regulatory category fell into the promoter regions of known cancer genes (Figs. 2 and 3A), some events occurred outside of canonical regulatory regions. For example, XBP1 mutations, which were present in ~6% of the breast cancer patients in our WGS cohort, did not primarily target the XBP1 promoter but rather clustered in a narrow, noncoding region downstream of XBP1 (Fig. 3A and fig. S45A), a pattern unlikely to occur by random chance (fig. S45B).
Previous studies have connected XBP1 to breast cancer (38, 39) and estrogen receptor signaling (40, 41). Concordantly, Gene Set Enrichment Analysis showed estrogen receptor–dependent signaling to be the most differentially expressed pathway (FDR = 7 × 10−4) between breast cancer samples with high versus low XBP1 expression (Fig. 5 and fig. S46). Furthermore, XBP1 was only expressed in prediction analysis of microarray 50 [PAM50 (42)] expression types related to hormone receptor signaling (luminal A/B, HER2-enriched types) but not in other breast tumors (basal-like type) (fig. S47). In addition, the average ATAC-seq signal around XBP1 was 1.83-fold higher in receptor-positive versus receptor-negative breast tumors (P < 0.001, basal-like versus non–basal-like PAM50 subtype, Mann-Whitney U test) (fig. S46, D and E), suggesting that regulatory regions around XBP1 exhibited primary activity in the hormone receptor–related subtype. We confirmed somatic mutations around XBP1 using Sanger sequencing in breast tumors from our WGS cohort (fig. S48).
FIG. 5.
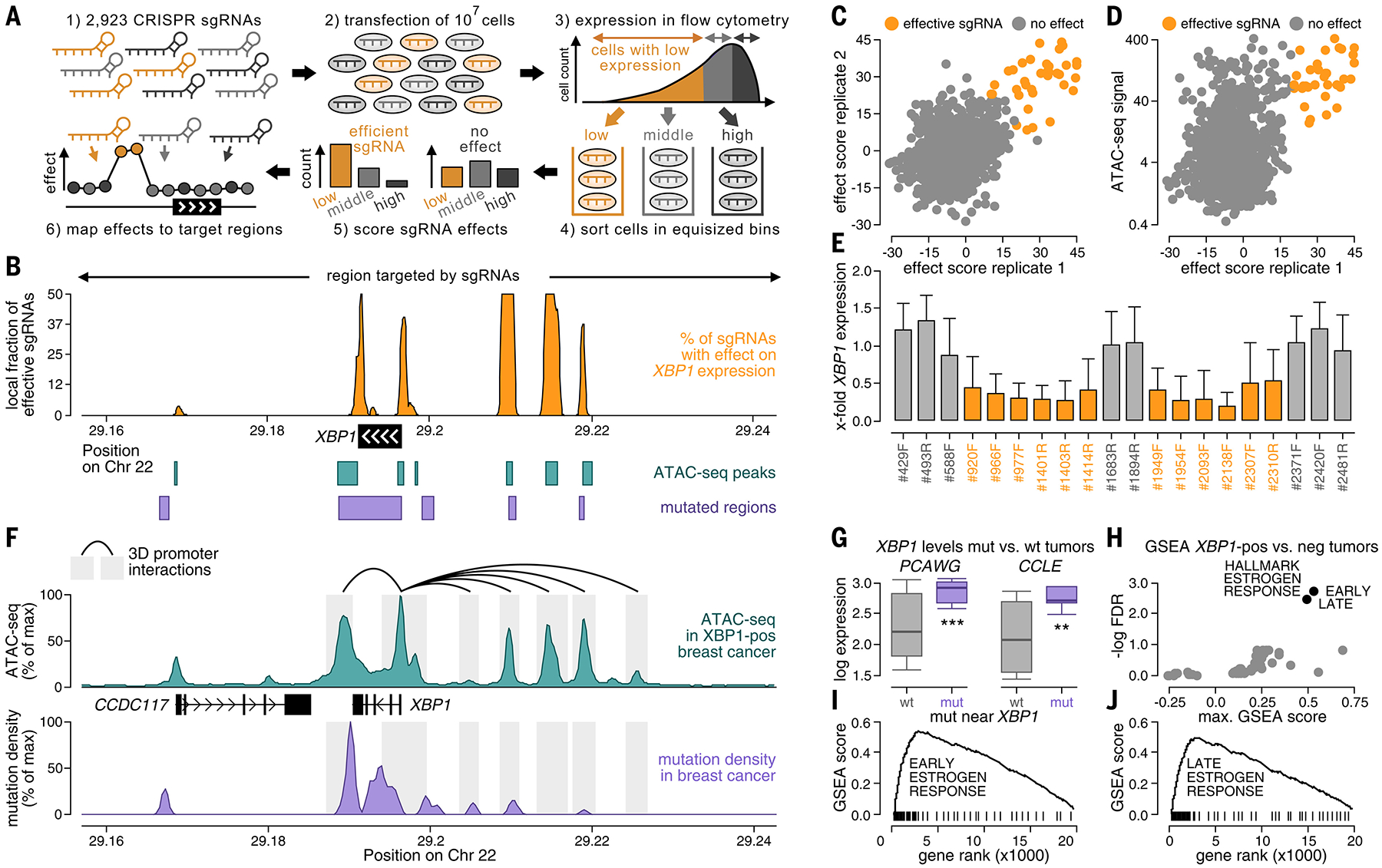
Noncoding somatic mutations occur in regulatory regions around XBP1. (A) CRISPRi screening of regions around XBP1 using a library of 2923 sgRNAs in breast cancer cells (CAMA1). Regulatory regions were localized based on sgRNAs, for which KRAB-mediated silencing of their target region led to decreased XBP1 expression in flow cytometry (orange). (B) Fractions of effective sgRNAs (y-axis) plotted against their position around XBP1 (x-axis). Positions of ATAC-seq peaks (teal, bottom), noncoding mutations (purple, bottom), and target regions of the sgRNAs (top) are annotated. (C and D) Efficacies of sgRNAs (sliding window of 10 adjacent sgRNAs) compared between experimental replicates [x-axis versus y-axis (C)] and the ATAC-seq signal of their target regions in breast cancer [y-axis (D)]. (E) Bar graphs displaying the XBP1 expression ratio before and after CRISPRi in regulatory regions (orange) and nonregulatory regions (gray) for individual sgRNAs. Error bars reflect the SD across cells. (F) Mutation densities (purple), ATAC-seq signals (teal), and three-dimensional interactions in the breast cancer genome of MCF7 (ChIA-PET, black) plotted against their genomic position around XBP1 (x-axis). (G) XBP1 expression compared between breast tumors with [purple, mutated (mut)] and without [gray, wild-type (wt)] mutations around XBP1 in PCAWG (left) and CCLE (right). Boxes indicate the 25/75% interquartile range, vertical lines extend to 10/90% percentiles, and horizontal lines reflect distribution medians of XBP1 expression. Significant differences (Mann-Whitney U test) are annotated with asterisks: *P < 0.05, **P < 0.01, ***P < 0.001. (H) Gene Set Enrichment Analysis analyzing expression differences in tumors with high versus low XBP1 expression by computing an enrichment score (x-axis) and a significance value (y-axis) for each hallmark signature. For (I) and (J), gene ranks (x-axis) are plotted against enrichment scores (y-axis) for early (I) and late (J) estrogen response signatures (black).
We used two experimental assays to further assess mutations near XBP1 and to provide proof-of-principle support for the possible biological relevance of mutation events outside of canonical regulatory regions (Fig. 5 and figs. S49 to S55).
As a first experiment, we performed a CRISPR interference (CRISPRi) screen to localize positive regulatory regions around XBP1 (Fig. 5A). We tiled the genomic region around XBP1 with a library of 2923 single-guide RNAs (sgRNAs), including the territory outside of canonical promoters and enhancers, and repressed the target regions of these sgRNAs through Krüppel associated box (KRAB)–mediated silencing in breast cancer cells (CAMA1). We then used flow cytometry [CRISPRi-Flow fluorescence in situ hybridization (CRISPRi-FlowFISH)] to quantify to what extent repression of a candidate regulatory region down-regulated XBP1 expression (14) (Fig. 5A and fig. S49). This screen identified five positive regulatory regions (four upstream and one downstream of XBP1) in which KRAB-mediated repression down-regulated XBP1 expression (Fig. 5B). These regulatory regions were consistent between experimental replicates (Fig. 5, C to E), and CRISPRi-FlowFISH screening results correlated with an independent experimental assay (quantitative polymerase chain reaction, R = 0.59; 29 sgRNAs tested in both assays) (fig. S50). In particular, many breast cancer mutations accumulated in the regulatory region that this experiment identified downstream of XBP1.
Companion analysis of ATAC-seq data from 74 breast tumors (8) confirmed the five regulatory regions from our screening experiment at a higher resolution, where they colocalized with five distinct ATAC-seq peaks around XBP1 (Fig. 5F). These peaks were exclusive to breast tumors with high XBP1 expression (Fig. 5F and fig. S46E), and their ATAC-seq signals correlated with XBP1 expression (fig. S51, A to C), with the highest correlation being observed in the ATAC-seq peak downstream of XBP1 (R = 0.80). In addition, regulatory regions physically interacted with the XBP1 promoter in the three-dimensional structure of the MCF7 breast cancer genome (43) (Fig. 5F), and breast cancer–specific transcription factors bound to upstream regulatory regions of XBP1 in breast cancer ChIP-seq data (fig. S51, D and E). Thus, our first experimental strategy demonstrated that important noncoding mutation events can occur outside of canonical regulatory regions, illuminating the potential of a genome-wide approach to capture somatic mutation events in both known and unknown elements of the noncoding genome.
As a second experiment, we used a luciferase reporter assay to examine the effect of mutations observed in breast cancer genomes near XBP1 on transcriptional activity directly (figs. S52 and S53A). For this purpose, we cloned the mutated and nonmutated 193-bp sequences around 10 mutations near XBP1 that were observed in our WGS cohort into the regulatory region of a luciferase reporter plasmid. We measured their luciferase signal in breast cancer cells (CAMA1) as a marker of their effect on transcriptional activity. For five of 10 mutations, we obtained significantly higher luciferase activity (P < 0.05; Mann-Whitney U test) for mutated sequences compared with their corresponding nonmutated sequences (fig. S52, A and B). For three mutations, we measured a >1.5-fold higher luciferase signal, which was similar to that reported for established noncoding mutations, including those in the TERT and FOXA1 promoters (~2-fold) (19, 35). Furthermore, despite variation between independent experiments, results correlated robustly between replicates (fig. S52C).
Differential expression analysis concordantly revealed that breast tumors with mutations around XBP1 were associated with elevated XBP1 expression relative to that observed in nonmutated tumors, both in tumor patients [PCAWG (9)] and in the Cancer Cell Line Encyclopedia [CCLE (44)] (Fig. 5, G to J, and fig. S42). Likewise, analysis of matched RNA sequencing (RNA-seq) and ATAC-seq data from two samples (three XBP1 mutations) in our WGS cohort revealed that XBP1 mutations correlated with increased fractions of mutated reads in RNA-seq and ATAC-seq data compared with their corresponding WGS data (two of three mutations examined) (fig. S53, B and C). In addition, mutations near XBP1 exhibited differential pathogenicity compared with mutations in the rest of the genome based on two bioinformatics scores (15, 16) (fig. S53, D and E). Thus, the second experimental strategy confirmed that specific mutations observed in breast cancer patients near XBP1 were associated with increased expression and activity of their downstream regulatory region.
The supplementary materials contain additional analyses related to the phenotypes associated with XBP1 mutations, including tumor cell proliferation (fig. S54), drug efficacy (fig. S55, A and B), the activity of related pathways (fig. S55, C and D), and patient survival (fig. S55E).
Discussion
Our study establishes a genome-wide compendium of somatic mutation events in 19 major cancer types and advances the field related to four major challenges.
First, noncoding regions comprise a heterogeneous spectrum of genomic elements, and mutation events in different parts of the noncoding genome relate to diverse aspects of tumor biology. To capture these biological differences, our approach automatically stratified mutation events based on their genomic location: Events in protein-coding regions corresponded to established coding drivers that alter protein structures of cancer-related genes. Some mutations in regulatory regions have been discussed as plausible noncoding drivers that could change protein levels of cancer-related genes with low expression in normal tissue to recruit them for oncogenesis (6, 9, 10, 19). Events near tissue-specific genes characterized localized passenger mutation patterns linked to characteristic expression programs and physiological processes in the tumor cell of origin and are unlikely to represent prototypical oncogenic drivers. Some noncoding events could not be associated with any of these categories, so their status remains less clear. In addition, although our classification was guided by the insights from prior studies (9, 10), the exact terminology and criteria differed between studies: Our category of tissue-specific genes (based on their expression pattern) was largely equivalent to PCAWG’s annotation of “transcriptional processes” (based on a review of their fraction of long indels), our category of regulatory regions was mostly labeled as “candidate drivers” by PCAWG, and our upfront filter of low-quality mutations and regions was consistent with the “technical artifacts” filter used by PCAWG. Despite broad overall consistency, these classifications diverged for individual results observed in both our study and prior work. Therefore, careful follow-up is required to determine the biology of individual mutation events in detail beyond their genomic location and capture the multifaceted functional effects of somatic mutations in noncoding regions.
Our second challenge was that the current understanding of regulatory regions and other functional elements in the noncoding cancer genome is likely incomplete given that their activity and location can vary between cell types, between tumor and normal tissue, and even between patients with the same tumor type (8, 45). Therefore, databases of regulatory regions (22–26) and ChIP-seq signals from normal tissue (7) may not capture the full diversity and versatility of functional elements in noncoding cancer genomes, and differences in the epigenomic structure of tumor and normal cells may be critical for characterizing mutation events in tumor-specific regulatory regions. Several analyses in our study, including experimental evaluation of XBP1 mutations, highlighted that important noncoding mutation events can occur outside of canonical regulatory elements. Although tumor-specific ATAC-seq and methylation data improved the enrichment for putative functional events, many mutation events linked to cancer genes still fell outside of these regions. To address this challenge, our genome-wide analysis locates mutation events across the entire genome instead of restricting its search to canonical functional regions. In contrast to previous annotation-unbiased approaches (9), our approach tiles the genome with multiple interval sizes. This proved critical for its use and performance in the noncoding genome, which harbors no predefined genomic boundaries and is ~50-fold larger than exons in coding regions. Our results may inform future experimental and clinical characterizations of tumor-specific regulatory elements, prioritize regions for hybrid-capture sequencing, and enable profiling of these mutation events at a higher read coverage.
The third challenge was that detecting somatic mutation events is technically more challenging in noncoding than in coding regions. To detect mutation events based on mutational excess, many established statistical concepts use synonymous mutations as a control of the regional background mutation rate in coding regions (3, 4). These concepts are inapplicable to the noncoding genome because synonymous mutations are available in coding regions only. Therefore, methods for identifying mutation events in the noncoding genome are required to use epigenomic features to calibrate their statistical models and detect mutational excess, which is a statistically more complex problem. Furthermore, the search for activating mutations in coding regions has been guided by hotspots of mutations that recur in the same position, and these are less frequently observed in noncoding regions (9), possibly because noncoding mutations might converge on similar biological effects in independent genomic positions. The statistical power to detect noncoding mutation events is further limited by the large number of hypotheses resulting from the size of the noncoding genome and its lack of predefined genomic regions. In addition, although thousands of whole cancer genomes have been sequenced, the amount of WGS data that captures noncoding somatic mutations is still smaller than that available for mutations in protein-coding regions. To account for these technical difficulties, we harmonized data from two WGS consortia (9, 13) and implemented a statistical approach allowing us to detect mutation events irrespective of their effects on protein-coding sequences or location within predefined genomic regions. Our approach incorporates established principles from other fields and methods (4, 9–12, 46, 47) but differs in critical aspects from many existing methods. For example, instead of negative binomial regression, our genome-wide analysis is based on a segmented statistical model, which gives it greater flexibility to account for overdispersion of mutation counts and complex relationships between epigenomic and mutation data. Furthermore, instead of using synonymous mutations in coding regions for comparison, our analysis compares mutation counts of the tumor type being studied with epigenomics data and sequencing data from unrelated tumor types. Prospective histone modification ChIP-seq data from large cohorts of tumor samples could be integrated into our approach and might improve its calibration to tumor-specific background mutation rates.
The final challenge was that there is currently no consensus on which events in the noncoding genome represent genuine drivers (6). In coding regions, many statistical tools detect mutation events based on established markers of positive selection (such as the ratio of nonsynonymous to synonymous mutations or equivalent measures), and their findings thus uniformly harbor signs of positive selection by design (3, 4). In noncoding regions, positive selection markers have not been established, and mutation events are identified based on their deviations from a careful statistical background model, including events resulting from positive selection or localized mutagenic processes. Therefore, the performance of statistical models in noncoding regions cannot be evaluated by classifying findings into true versus false positives, which is a common procedure used in coding regions (2, 4). Furthermore, experimental validation of the “driverness” of mutation events identified by statistical methods remains a general limitation of the field, particularly in noncoding regions, because experimental assays to capture the oncogenic effects of noncoding mutations beyond expression changes are limited. To address these challenges, our study included multiple pan-cancer follow-up strategies, including literature support of the genes linked to noncoding mutation events, comparison with other methods, and analysis of statistical power. Furthermore, we benchmarked mutation events against orthogonal ChIP-seq, ATAC-seq, RNA-seq, drug response, transcription factor binding, protein interaction, and patient survival data. We also established four markers to identify events in candidate regulatory regions outside of traditional ChIP-seq signals and databases. In addition to these computational strategies, our study combined two experimental assays to further assess XBP1 by characterizing regulatory regions of gene expression (CRISPRi screen) and assessing the effects of noncoding mutations in these regions on expression (luciferase reporter assay). These assays gauge orthogonal effects because point mutations in luciferase reporter experiments change only a few nucleotides, whereas sgRNAs in CRISPRi experiments can affect up to several kilobases around their target regions through KRAB-mediated silencing (48) and thus do not mimic the effect of point mutations. In particular, this combined strategy enables experimental follow-up irrespective of the location of mutations in canonical regulatory regions, and could therefore guide future experimental endeavors.
Moving forward, our findings could be further evaluated in prospective multiomics datasets derived from the same patients as mutation sequencing data. These data would allow a deeper characterization of our findings in the context of differential expression (matched expression data), tumor-specific, long-distance promoter-enhancer interactions (matched chromosome conformation capture data), and changes in transcription factor binding (matched transcription factor ChIP-seq data). Furthermore, some of our noncoding findings may be of direct clinical interest because they converge on genes that have been previously explored as direct or indirect targets of cancer therapies, such as TERT and imetelstat, FOXA1 and fulvestrant, FGFR2 and infigratinib, BCR and ibrutinib, or RAD51B, GEN1, or STAG1 and olaparib. Additionally, our study revealed that XBP1 mutations potentially created additional therapeutic avenues. However, many other noncoding findings were linked to genes that have not been nominated as drug targets. These could provide critical starting points for the development of personalized therapies based on noncoding cancer genomes, particularly for patients with resistance to primary treatment or no druggable options in protein-coding regions.
Broadly, given the growing use of somatic WGS in the clinical setting and in biobank-scale datasets, our study establishes a critical step toward expanding our understanding of somatic mutations from protein-coding regions to the remaining ~98% of the genome. It also provides a blueprint for prioritizing noncoding mutations for translational investigation and therapeutic development.
Materials and methods summary
We combined three complementary significance tests for the genome-wide detection of somatic mutation events, which are local accumulations or clusters of somatic mutations that deviate from the pattern observed in the rest of the genome. These three tests integrated and extended principles established in other fields or methods (4, 9–12, 46, 47), as outlined below.
Significance test 1 models the mutational background based on epigenomic signals, taking into account differences in mutation rates between euchromatic and heterochromatic regions (47) (see section 1.2 of the materials and methods). Using this background model, test 1 identifies genomic regions with larger numbers of mutations than would be expected by chance. A similar principle to that of test 1 had been applied in some previous studies that accounted for epigenomic signals by using negative binomial regression to detect mutational significance in coding (4) or noncoding (10) regions. Significance test 1 generalizes these approaches by using a four-component mixture model [H3K4me1, H3K9me3, H3K27me3, and H3K36me3 histone ChIP-seq data (7)] that allows for nonexponential relationships between mutation rates and epigenomic signals.
Significance test 2 compares the number of mutations per genomic interval between unrelated cancer types and identifies genomic regions with an unusually large number of mutations in a particular cancer type (see section 1.2 of the materials and methods). In this way, test 2 detects accumulations of mutations that are specific to a certain cancer type and could reflect a specific biology in that type of tumor tissue. To take into consideration nonlinear dependencies of mutation counts between cancer types, test 2 uses a segmented statistical model to arrange genomic regions into bins and estimate the background mutation rate within each bin separately. Furthermore, it accounts for differences in mutation rates between tumor types using regional distribution variance. Although test 1 used epigenomic data from normal tissue, test 2 serves as a proxy for tumor-specific epigenomic data given that the epigenomic structure differs between tumor and normal tissue. The importance of these differences has been highlighted in the context of somatic mutations by previous studies (8, 45).
Significance test 3 detects positional clustering of mutations around biologically relevant positions in the cancer genome (see section 1.3 of the materials and methods). In addition to the biological function of genomic positions, other factors, including nucleotide contexts, coverage fluctuation, read mappability, and kataegis events, affect positional clustering. Concepts similar to those of test 3 have been used in other methods for analyzing coding and noncoding regions (9, 29). Therefore, test 3 examines whether mutations occur in different positions than expected by chance, but it does not analyze whether the total number of mutations deviates from the expectation and thus does not require calibration against regional fluctuations of the background mutation rates.
To combine signals from tests 1 through 3, we tiled the genome into 1-, 10-, and 100-kb intervals with 25% overlap and performed the three tests in each of these intervals (all mutations and indels only). This strategy of an unbiased, genome-wide analysis builds on established principles from noncancer germline studies (46) and an annotation-unbiased strategy in PCAWG that analyzes 2-kb intervals (9). For each 10- and 100-kb interval, we obtained multiple P values from the interval and its subintervals (linked P values of its consecutive, nonoverlapping 1- and 10-kb subintervals; see sections 1.2 and 1.4 of the materials and methods). We then combined them using Brown’s method (11), which was also used in previous cancer genomics studies, including PCAWG (9), and then adjusted them using weighted multiple hypothesis correction (12). To derive a genome-wide signal of significance, we selected maximally significant, nonoverlapping intervals, as described previously (10), and favored 10- over 100-kb intervals because they allowed us to optimize the resolution of our signal (see section 1.4 of the materials and methods). In this genome-wide signal, we identified mutation events as significant regions with an FDR < 0.1 (peak value < 0.05).
To classify mutation events, we annotated them based on their closest gene and their putative function (see section 1.5 of the materials and methods): coding regions [regions with the most mutations in exons or splice sites in exon-intron boundaries and findings detected by MutSigCV (3) or dNdScv (4)]; regulatory regions [regions with the most mutations in H3K4me3 or H3K27ac ChIP-seq peaks from Roadmap (7)]; tissue-specific genes (mutations around genes that are expressed in a particular tumor type); and “other” findings (mutations with unclear functions that fit no other criteria). We excluded regions with low-alignability mutations or hotspots in DNA loops (see section 1.5 of the materials and methods).
A more detailed description of the significance tests and statistical framework can be found in the materials and methods.
Supplementary Material
ACKNOWLEDGMENTS
We thank M. Meyerson, M. Brown, G. Getz, E. Rheinbay, P. Priestley, S. Chen, X. Zhou, H. Cao, M. Lupien, J. Kreisberg, and J. Ma for valuable feedback and suggestions; B. Reardon, S. Camp, B. Jiang, C. Ricker, and K. Mandl for proofreading our manuscript and improving its readability; C. Otis, P. Rogers, and the Flow Cytometry Facility of the Broad Institute for technical assistance; and J. Hyle and Y. Zhang from the St. Jude Children’s Research Hospital for experimental support. This publication and the underlying study have been made possible partly through data requested (DR-050) and made available by HMF and the Center of Personalized Cancer Treatment (CPCT). Furthermore, the results presented in this study are in part based on data generated by the The Cancer Genome Atlas (TCGA) Research Network (https://www.cancer.gov/tcga) and the PCAWG and International Cancer Genome Consortium (ICGC) networks through the Data Access Compliance Office (DACO-1078465). We further acknowledge the contributions of the many clinical networks across ICGC and TCGA in providing samples and data to the PCAWG Consortium and the contributions of the Technical Working Group and the Germline Working Group of the PCAWG Consortium for the collation, realignment, and harmonized variant-calling of the cancer genomes used in this study. Finally, we thank the patients and their families for their participation in the individual CPCT, HMF, ICGC, and TCGA projects.
Funding:
F.D. was supported by the National Institutes of Health (grant no. K99 CA262152), the Claudia Adams Barr Program for Innovative Cancer Research, the EMBO Long-Term Fellowship Program (grant no. ALTF 502-2016), and the AWS Cloud Credits for Research Program. J.T.N. was supported by a Merkin Institute Fellowship. E.M.V.A. was supported by the National Institutes of Health (grant nos. R01 CA227388 and R21 CA242861). F.D., J.T.N., and E.M.V.A. were supported by ASPIRE Awards from The Mark Foundation for Cancer Research.
Competing interests:
E.M.V.A. is a consultant for Tango Therapeutics, Genome Medical, Invitae, Foresite Capital, Enara Bio, Monte Rosa Therapeutics, Manifold Bio, Janssen, Dynamo, and Illumina; received research support from Novartis and Bristol-Myers Squibb and travel support from Roche and Genentech; and is an equity holder of Syapse, Tango Therapeutics, Syapse, Enara Bio, Monte Rosa Therapeutics, and Genome Medical. J.T.N. is a consultant for AbbVie Inc. The other authors declare no competing interests.
Data and materials availability:
Code used to perform our analyses is available on Zenodo (49). Access to controlled data from the PCAWG project in the ICGC was obtained through DACO-1078465. Clinical annotations and somatic whole-genome sequencing data from the CPCT and the HMF were obtained through a data access request (DR-050). Access requests for these data can be submitted under https://www.hartwigmedicalfoundation.nl/en/applying-for-data (HMF, CPCT) and https://daco.icgc.org (PCAWG, ICGC). All other data used in this study are publicly available without restrictions.
REFERENCES AND NOTES
- 1.Stratton MR, Campbell PJ, Futreal PA, The cancer genome. Nature 458, 719–724 (2009). doi: 10.1038/nature07943; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bailey MH et al. , Comprehensive characterization of cancer driver genes and mutations. Cell 174, 1034–1035 (2018). doi: 10.1016/j.cell.2018.07.034; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lawrence MS et al. , Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 499, 214–218 (2013). doi: 10.1038/nature12213; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Martincorena I et al. , Universal patterns of selection in cancer and somatic tissues. Cell 171, 1029–1041.e21 (2017). doi: 10.1016/j.cell.2017.09.042; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mularoni L, Sabarinathan R, Deu-Pons J, Gonzalez-Perez A, López-Bigas N, OncodriveFML: A general framework to identify coding and non-coding regions with cancer driver mutations. Genome Biol. 17, 128 (2016). doi: 10.1186/s13059-016-0994-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Elliott K, Larsson E, Non-coding driver mutations in human cancer. Nat. Rev. Cancer 21, 500–509 (2021). doi: 10.1038/s41568-021-00371-z; [DOI] [PubMed] [Google Scholar]
- 7.Bernstein BE et al. , The NIH Roadmap Epigenomics Mapping Consortium. Nat. Biotechnol 28, 1045–1048 (2010). doi: 10.1038/nbt1010-1045; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Corces MR et al. , The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898 (2018). doi: 10.1126/science.aav1898; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rheinbay E et al. , Analyses of non-coding somatic drivers in 2,658 cancer whole genomes. Nature 578, 102–111 (2020). doi: 10.1038/s41586-020-1965-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Imielinski M, Guo G, Meyerson M, Insertions and deletions target lineage-defining genes in human cancers. Cell 168, 460–472.e14 (2017). doi: 10.1016/j.cell.2016.12.025; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Brown MB, 400: A method for combining non-independent, one-sided tests of significance. Biometrics 31, 987–992 (1975). doi: 10.2307/2529826 [DOI] [Google Scholar]
- 12.Ignatiadis N, Klaus B, Zaugg JB, Huber W, Data-driven hypothesis weighting increases detection power in genome- scale multiple testing. Nat. Methods 13, 577–580 (2016). doi: 10.1038/nmeth.3885; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Priestley P et al. , Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216 (2019). doi: 10.1038/s41586-019-1689-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fulco CP et al. , Activity-by-contact model of enhancer- promoter regulation from thousands of CRISPR perturbations. Nat. Genet 51, 1664–1669 (2019). doi: 10.1038/s41588-019-0538-0; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kircher M et al. , A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet 46, 310–315 (2014). doi: 10.1038/ng.2892; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shihab HA et al. , An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 31, 1536–1543 (2015). doi: 10.1093/bioinformatics/btv009; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Futreal PA et al. , A census of human cancer genes. Nat. Rev. Cancer 4, 177–183 (2004). doi: 10.1038/nrc1299; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Chakravarty D et al. , OncoKB: A precision oncology knowledge base. JCO Precis. Oncol 2017, PO.17.00011 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rheinbay E et al. , Recurrent and functional regulatory mutations in breast cancer. Nature 547, 55–60 (2017). doi: 10.1038/nature22992; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rappaport N et al. , MalaCards: An integrated compendium for diseases and their annotation. Database 2013, bat018 (2013). doi: 10.1093/database/bat018; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fujimoto A et al. , Whole-genome mutational landscape and characterization of noncoding and structural mutations in liver cancer. Nat. Genet 48, 500–509 (2016). doi: 10.1038/ng.3547; [DOI] [PubMed] [Google Scholar]
- 22.Moore JE et al. , Expanded encyclopaedias of DNA elements in the human and mouse genomes. Nature 583, 699–710 (2020). doi: 10.1038/s41586-020-2493-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fishilevich S et al. , GeneHancer: Genome-wide integration of enhancers and target genes in GeneCards. Database 2017, bax028 (2017). doi: 10.1093/database/bax028; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Andersson R et al. , An atlas of active enhancers across human cell types and tissues. Nature 507, 455–461 (2014). doi: 10.1038/nature12787; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zerbino DR, Wilder SP, Johnson N, Juettemann T, Flicek PR, The ensembl regulatory build. Genome Biol. 16, 56 (2015). doi: 10.1186/s13059-015-0621-5; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Visel A, Minovitsky S, Dubchak I, Pennacchio LA, VISTA Enhancer Browser—A database of tissue-specific human enhancers. Nucleic Acids Res. 35 (Database), D88–D92 (2007). doi: 10.1093/nar/gkl822; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Shuai S, Gallinger S, Stein L; PCAWG Drivers and Functional Interpretation Working Group; PCAWG Consortium, Combined burden and functional impact tests for cancer driver discovery using DriverPower. Nat. Commun 11, 734 (2020). doi: 10.1038/s41467-019-13929-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lochovsky L, Zhang J, Fu Y, Khurana E, Gerstein M, LARVA: An integrative framework for large-scale analysis of recurrent variants in noncoding annotations. Nucleic Acids Res. 43, 8123–8134 (2015). doi: 10.1093/nar/gkv803; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Guo YA, Chang MM, Skanderup AJ, MutSpot: Detection of non-coding mutation hotspots in cancer genomes. NPJ Genom. Med 5, 26 (2020). doi: 10.1038/s41525-020-0133-4; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Alexandrov LB et al. , The repertoire of mutational signatures in human cancer. Nature 578, 94–101 (2020). doi: 10.1038/s41586-020-1943-3; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Aizarani N et al. , A human liver cell atlas reveals heterogeneity and epithelial progenitors. Nature 572, 199–204 (2019). doi: 10.1038/s41586-019-1373-2; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liao J et al. , Single-cell RNA sequencing of human kidney. Sci. Data 7, 4 (2020). doi: 10.1038/s41597-019-0351-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kawano N et al. , Composite distal nephron-derived renal cell carcinoma with chromophobe and collecting duct carcinomatous elements. Pathol. Int 55, 360–365 (2005). doi: 10.1111/j.1440-1827.2005.01837.x; [DOI] [PubMed] [Google Scholar]
- 34.Sandelin A, Alkema W, Engström P, Wasserman WW, Lenhard B, JASPAR: An open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32, D91–D94 (2004). doi: 10.1093/nar/gkh012; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Huang FW et al. , Highly recurrent TERT promoter mutations in human melanoma. Science 339, 957–959 (2013). doi: 10.1126/science.1229259; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Krones-Herzig A et al. , Early growth response 1 acts as a tumor suppressor in vivo and in vitro via regulation of p53. Cancer Res. 65, 5133–5143 (2005). doi: 10.1158/0008-5472.CAN-04-3742; [DOI] [PubMed] [Google Scholar]
- 37.Szklarczyk D et al. , The STRING database in 2017: Quality-controlled protein-protein association networks, made broadly accessible. Nucleic Acids Res. 45 (D1), D362–D368 (2017). doi: 10.1093/nar/gkw937; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gupta A et al. , NCOA3 coactivator is a transcriptional target of XBP1 and regulates PERK-eIF2a-ATF4 signalling in breast cancer. Oncogene 35, 5860–5871 (2016). doi: 10.1038/onc.2016.121; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen S et al. , The emerging role of XBP1 in cancer. Biomed. Pharmacother 127, 110069 (2020). doi: 10.1016/j.biopha.2020.110069; [DOI] [PubMed] [Google Scholar]
- 40.Sengupta S, Sharma CG, Jordan VC, Estrogen regulation of X-box binding protein-1 and its role in estrogen induced growth of breast and endometrial cancer cells. Horm. Mol. Biol. Clin. Investig 2, 235–243 (2010). doi: 10.1515/hmbci.2010.025; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wang C et al. , Estrogen receptor antagonist fulvestrant inhibits proliferation and promotes apoptosis of prolactinoma cells by regulating the IRE1/XBP1 signaling pathway. Mol. Med. Rep 18, 4037–4041 (2018). doi: 10.3892/mmr.2018.9379; [DOI] [PubMed] [Google Scholar]
- 42.Parker JS et al. , Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol 27, 1160–1167 (2009). doi: 10.1200/JCO.2008.18.1370; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Li G et al. , Extensive promoter-centered chromatin interactions provide a topological basis for transcription regulation. Cell 148, 84–98 (2012). doi: 10.1016/j.cell.2011.12.014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barretina J et al. , The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607 (2012). doi: 10.1038/nature11003; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sur I, Taipale J, The role of enhancers in cancer. Nat. Rev. Cancer 16, 483–493 (2016). doi: 10.1038/nrc.2016.62; [DOI] [PubMed] [Google Scholar]
- 46.Tam V et al. , Benefits and limitations of genome-wide association studies. Nat. Rev. Genet 20, 467–484 (2019). doi: 10.1038/s41576-019-0127-1; [DOI] [PubMed] [Google Scholar]
- 47.Schuster-Böckler B, Lehner B, Chromatin organization is a major influence on regional mutation rates in human cancer cells. Nature 488, 504–507 (2012). doi: 10.1038/nature11273; [DOI] [PubMed] [Google Scholar]
- 48.Groner AC et al. , KRAB-zinc finger proteins and KAP1 can mediate long-range transcriptional repression through heterochromatin spreading. PLOS Genet. 6, e1000869 (2010). doi: 10.1371/journal.pgen.1000869; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Dietlein F, Wang AB, Fagre C, Tang A, Besselink N, Cuppen E, Li C, Sunyaev SR, Neal JT, Van Allen EM, Source code for: Genome-wide analysis of somatic noncoding mutation patterns in cancer. Zenodo (2022); doi: 10.5281/zenodo.5913867 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Code used to perform our analyses is available on Zenodo (49). Access to controlled data from the PCAWG project in the ICGC was obtained through DACO-1078465. Clinical annotations and somatic whole-genome sequencing data from the CPCT and the HMF were obtained through a data access request (DR-050). Access requests for these data can be submitted under https://www.hartwigmedicalfoundation.nl/en/applying-for-data (HMF, CPCT) and https://daco.icgc.org (PCAWG, ICGC). All other data used in this study are publicly available without restrictions.