

SUMMARY
Hematopoietic stem cells (HSCs) rely on complex regulatory networks to preserve stemness. Due to the scarcity of HSCs, technical challenges have limited our insights into the interplay between metabolites, transcription, and the epigenome. In this study, we generated low-input metabolomics, transcriptomics, chromatin accessibility, and chromatin immunoprecipitation data, revealing distinct metabolic hubs that are enriched in HSCs and their downstream multipotent progenitors. Mechanistically, we uncover a non-classical retinoic acid (RA) signaling axis that regulates HSC function. We show that HSCs rely on Cyp26b1, an enzyme conventionally considered to limit RA effects in the cell. In contrast to the traditional view, we demonstrate that Cyp26b1 is indispensable for production of the active metabolite 4-oxo-RA. Further, RA receptor beta (Rarb) is required for complete transmission of 4-oxo-RA-mediated signaling to maintain stem cells. Our findings emphasize that a single metabolite controls stem cell fate by instructing epigenetic and transcriptional attributes.
Graphical abstract
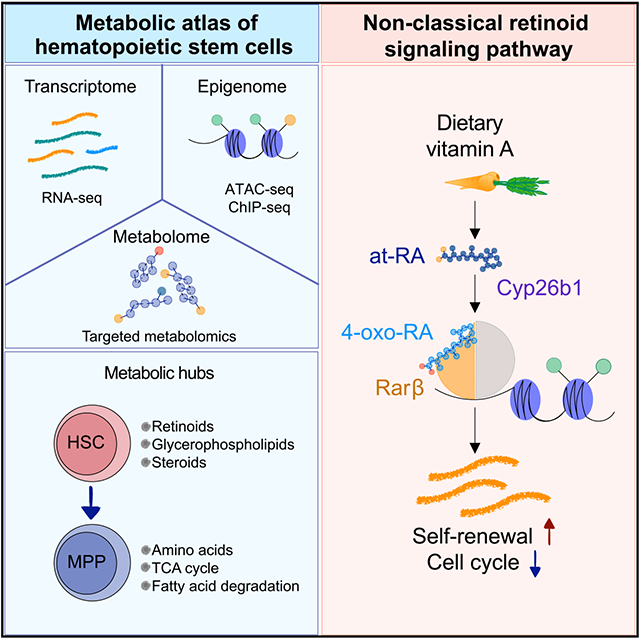
In brief
Schönberger et al. generate and integrate low-input multi-omics data on purified hematopoietic stem cells (HSCs) to identify metabolic and epigenetic signaling hubs relevant to stem cell identity. Mechanistically, the authors uncover a non-classical retinoid signaling axis, Cyp26b1-4-oxo-RA-Rarb, important for maintenance of HSC features.
INTRODUCTION
Hematopoietic stem cells (HSCs) sit at the top of a hierarchically organized system and have the unique capability of long-term reconstitution of the entire blood system (Orkin and Zon, 2008; Till and McCulloch, 1961). HSCs can give rise to multipotent progenitors (MPPs), which differentiate stepwise into lineage-committed progenitors and eventually into effector cells (Ikuta and Weissman, 1992; Okada et al., 1992). Under homeostatic conditions, HSCs are highly quiescent and exhibit low biosynthetic activity (Cabezas-Wallscheid et al., 2017; Wilson et al., 2008). Although currently debated, HSCs are commonly described tas relying on glycolytic ATP production while inhibiting mitochondrial oxidative phosphorylation (OXPHOS) (Chandel et al., 2016; Ito and Suda, 2014; Liang et al., 2020; Vannini et al., 2016). Nevertheless, HSCs must be able to reversibly switch their metabolic program upon stress-induced activation to meet higher energy demands and drive differentiation (Ito and Suda, 2014; Ito et al., 2019; Simsek et al., 2010; Takubo et al., 2013).
Over the last years, it has become evident that metabolites can play an active role in regulating cell fate (Bräutigam et al., 2018; Hinge et al., 2020; Kalaitzidis et al., 2012; Suda et al., 2011; Takubo, 2014). Metabolites can be detected by transcription factors (TFs), modifying gene expression. Furthermore, the metabolic balance of short-chain carbon metabolites is closely linked to the degree of DNA or histone modification and, thus, to transcriptional regulation (Serefidou et al., 2019; Ulrey et al., 2005). Vitamins in particular have been suggested to mediate epigenetic modifications and to play important roles in stem cell function. For instance, vitamin B3 reduces bone marrow (BM) transplantation toxicity (Vannini et al., 2019), and vitamin C suppresses leukemogenesis by changing the DNA methylation status in HSCs (Agathocleous et al., 2017; Cimmino et al., 2017). Previous work has shown that vitamin A/retinoic acid (RA) signaling plays a role in regulating HSCs (Cabezas-Wallscheid et al., 2017; Purton et al., 1999, 2000). In a prior study, we performed proof-of-principle experiments that showed systemic deprivation of vitamin A in mice leads to loss of HSC quiescence (Cabezas-Wallscheid et al., 2017). Treatment with the active vitamin A metabolite all-trans-RA (at-RA) preserves stemness features during stress conditions in vitro and in vivo. Nonetheless, how retinoids regulate hallmarks of HSCs remains largely unknown.
The low frequency of HSCs within the BM leads to technical limitations so far preventing extensive investigation of the interplay between stem cell metabolism and transcriptome/epigenome signaling networks. Significant advances in low-input sampling have been made recently; Agathocleous et al. (2017) detected around 60 metabolites from only 10,000 cells of different stem and progenitor populations. DeVilbiss et al. (2021) were able to identify as many as 160 metabolites in HSCs/MPPs, comparing them with whole BM(WBM) cells. However, the methods applied in these studies did not detect differences between highly purified stem cell populations and immediate downstream progenitor cells. Here we established low-input metabolic and epigenetic methods and showed that HSCs rely on a non-classical RA signaling axis.
RESULTS
Metabolic landscape of HSCs and downstream progenitors
To explore the networks linking metabolism, epigenetics, and transcription in HSCs (Lin−Sca-1+c-Kit+ [LSK] CD150+CD48−CD34−) and their downstream MPPs (LSK CD150−CD48+), we established and employed protocols for low-input metabolomics, RNA sequencing (RNA-seq), chromatin accessibility (assay for transposase-accessible chromatin sequencing; ATAC-seq), and histone chromatin immunoprecipitation sequencing (ChIP-seq) (Figure 1A; Figure S1A).
Figure 1. Metabolic and epigenetic landscape of HSCs and downstream progenitors.

(A) Summary of the experimental setup.
(B) Schematic integration of metabolomics results on HSCs+MPP1 and MPPs. Quantification is depicted as fold change (FC) + SD relative to HSCs+MPP1. Metabolite names and dots (upregulated in HSCs+MPP1)are shown in red (upregulated in MPPs) and blue. RNA-seq expression data of enzymes (padj. < 0.1) are shown as colored lines (red, upregulated in HSCs; blue, upregulated in MPPs; see also Figure S1F). n = 6–12; two to three independent experiments per metabolite; unpaired Student’s t test; *p < 0.05, **p < 0.01, ***p < 0.001.
(C) GSEA of selected Reactome pathways on RNA-seq data enriched in HSCs and MPPs (n = 3).
(D) GSEA of selected Reactome pathways on ranked differential OCRs between HSCs and MPPs in ATAC-seq.
(E) Gene tracks of the Cyp26b1 locus with corresponding RNA-seq, ATAC-seq, and H3K4me3 and H3K27me3 ChIP-seq coverage files.
(F) MTE plots integrating metabolomics, RNA-seq, ATAC-seq, and H3K4me3 and H3K27me3 ChIP-seq data on selected KEGG pathways. Depicted is log2FC between HSCs(+MPP1) and MPPs (red, upregulated in HSCs; blue, upregulated in MPPs). Significance was calculated using an unpaired Student’s t test (metabolomics dataset; *p < 0.05, **p < 0.01, ***p < 0.001) or by DESeq2 padj. value (sequencing datasets; *p < 0.1, **p < 0.05, ***p < 0.01).
Inner circles indicate the average tendencies of the entire KEGG pathway per dataset, and outer circles show individual genes/metabolites. n indicates the number of biological replicates.
First, we aimed to shed light on how the composition of metabolites differs in HSCs and MPPs. We developed two low-input liquid chromatography-tandem mass spectrometry (LC-MS/MS) approaches. One approach detects (semi)polar metabolites such as tricarboxylic acid (TCA) cycle metabolites and amino acids, using as few as 10,000 cells as starting material, and the second approach detects polar lipids, bile acids, and retinoids using 40,000 cells as starting material (STAR Methods). Titration experiments confirmed that metabolites were detected above background levels and showed a signal correlation with the input number (Figure S1B). Because of the scarcity of HSCs, we performed the metabolomics experiments using LSK CD150+CD48− (HSCs and MPP1, hereafter referred to as HSCs+MPP1) and LSK CD150−CD48+ (MPP3+MPP4, hereafter referred to as MPPs) (Cabezas-Wallscheid et al., 2014). We identified 51 metabolites in HSCs+MPP1 and downstream MPPs, of which 23 were found to be significantly different (Figure 1B; Table S2). Compared with previous studies, this represents an advance in the purity of cell populations used and the number of differentially abundant metabolites identified (Figure S1C; Agathocleous et al., 2017; DeVilbiss et al., 2021; Karigane et al., 2016; Takubo et al., 2013).
We found several lipids that were significantly enriched in HSCs+MPP1, such as lyso-phosphocholines (LPCs) and phosphocholines (PCs) (Figure 1B; Table S2). Two of the most highly abundant metabolites in HSCs+MPP1 were RA and its downstream product 4-oxo-RA. This result supports our previous findings and hints at the importance of retinoids in regulation of stem cell function (Figure 1B; Table S2; (Cabezas-Wallscheid et al., 2014, 2017). Further, we detected up to 2-fold higher levels of various amino acids, acyl-carnitines, and TCA cycle-related metabolites in MPPs, reflecting their active metabolic state (Figure 1B; Table S2).
We next performed RNA-seq of HSCs and MPPs, focusing primarily on metabolic genes (Figure S1D). Gene Ontology (GO; Figure S1E; Table S1) and gene set enrichment analysis (GSEA; Figure 1C) revealed that “fatty acid metabolism” and “signaling by RA” were enriched in HSCs, whereas “TCA cycle” and “metabolism of amino acids” were enriched in MPPs, in line with our metabolomics data and previous research (Chandel et al., 2016; Ito and Suda, 2014). Correlation between individual metabolites and their corresponding enzymes was observed (Figure 1B; schematic pathways are highlighted in Figure S1F; Table S1). For example, HSCs showed high expression of glycolytic enzymes and low expression of OXPHOS-related enzymes. By grouping differentially expressed genes (DEGs; adjusted p [padj.] < 0.1) between HSCs and MPPs according to metabolic GO terms (Figure S1G; Table S1), we identified potential upstream regulators (Herrmann et al., 2012; Imrichová et al., 2015). For instance, we found C/EBP homology protein / activating transcription factor 4 (CHOP/ATF4) was the main motif for genes involved in “amino acid metabolism” enriched in MPPs, whereas interferon regulatory factor (IRF) was the main motif for “retinol metabolism” genes highly enriched in HSCs (Figure S1G).
Our results show that, upon transition of HSCs to MPPs, rewiring of the cell metabolic status happens at the transcriptional and metabolic levels.
Epigenetic modulation of metabolic pathways in HSCs
We next explored the chromatin accessibility status in HSCs and MPPs by applying ATAC-seq (Figure S1H). Differences in chromatin accessibility and gene expression correlated significantly, as shown by GSEA (Figures S1I and S1Q). Furthermore, GO analysis of genes annotated to gained or lost open chromatin regions (OCRs) yielded terms such as “cell adhesion” and “positive regulation of transcription” for HSC-specific sites and “intracellular signal transduction” and “activation of GTPase activity” for MPP-specific sites (Figure S1J; Table S1), in line with our previous work and the RNA-seq data (Figures S1D and S1E; Cabezas-Wallscheid et al., 2014). GSEA of genes annotated to ATAC-seq peaks retrieved only a small number of pathways that were significantly enriched in HSCs; these included “retinoic acid metabolism,” indicating epigenetic regulation of this pathway (Figure 1D; Table S1). Differences in binding of TFs enriched for the aforementioned metabolic pathways (Figure S1G) were assessed by digital footprinting analysis of the corresponding motifs. As a control, we used CCCTC-binding factor (CTCF) as a TF generally acting in all cells and Runt-related TFs / ETS-domain TFs (RUNX/ETS) as factors particularly relevant to MPPs (Imperato et al., 2015; Nottingham et al., 2007; Willcockson et al., 2019; Figures S1G and S1K). The analysis revealed higher TF occupancy in MPPs for the motifs ATF4 and CHOP, corresponding to the GO term “amino acid metabolism,” in line with our metabolomics data.
We next compared the promoter chromatin state of HSCs and MPPs by performing low-input ChIP-seq for two histone modifications: histone 3 lysine 4 trimethylation (H3K4me3), associated with active genes, and histone 3 lysine 27 trimethylation (H3K27me3), a repressive mark set by Polycomb repressive complex 2 (PRC2) (Margueron and Reinberg, 2011). To ensure high quality, we performed titration experiments and a comparison with recent publicly available data (Figures S1L and S1M; Zheng et al., 2015). As expected, H3K4me3 and H3K27me3 displayed a global anti-correlation with only a few bivalent promoters (Figure S1N; Wei et al., 2009; Weishaupt et al., 2010).
Consistent with RNA-seq and ATAC-seq analysis, GSEA of the H3K4me3 modification revealed terms like “cell cycle” and “TCA cycle” as enriched in MPPs, whereas “retinoic acid metabolism” and “nuclear receptor transcription” were significantly enriched in HSCs (Figure S1O; Table S1). GSEA of H3K27me3-modified genes highlighted “DNA repair” and “phospholipid metabolism” as enriched in MPPs, suggesting epigenetic repression of these pathways specifically in MPPs (Figure S1P; Table S1). Of note, transcription was positively correlated with OCRs and H3K4me3 promoter marks (Figure S1Q), whereas H3K27me3 showed a weak but significant global anti-correlation, as exemplified by gene loci from different metabolic and cycling-related genes (Figure 1E; Figure S1R).
To globally illustrate metabolic networks governing stemness, we integrated the metabolomics data with the RNA-, ATAC-, and ChIP-seq datasets, focusing on KEGG pathway-annotated enzymes that correspond to the metabolites we detected. For this purpose, we created “metabolome-transcriptome-epigenome” (MTE) plots summarizing all of the assessed layers of regulation (Figure 1F; Figure S1S). For instance, TCA cycle-related enzymes were regulated by H3K4me3, showing a significant average gain of promoter modification in MPPs and, thus, higher gene expression. Further, we identified “sphingo/glycerophospholipid metabolism” and “sterol biosynthesis” as metabolic hubs that previously have not been linked to HSC biology. The most striking outcome of our multi-omics analysis was the strong emphasis on “retinol metabolism” in HSCs. Retinoids were the most highly enriched metabolites in HSCs+MPP1 that strongly correlated with greater chromatin accessibility, higher H3K4me3 but lower H3K27me3 levels, and, consequently, higher levels of expression of enzymes involved in RA metabolism in HSCs (Figures 1B-1F; Figure S1O).
Our integrative multilayer omics approach permits further exploration in future studies to address the role of specific metabolites and enzymes for maintenance of stemness.
Lack of dietary vitamin A leads to loss of the metabolic HSC identity
We next aimed to assess the role of RA metabolism, the most highly enriched metabolic hub detected, in regulating HSC function by specifically disrupting this pathway. Dietary vitamin A (i.e., retinol) serves as the only precursor metabolite for retinoids. Thus, 3-week-old mice were fed control food or a vitamin A-free diet (VAF) for 6 months (Figure 2A). Systemic depletion of vitamin A was confirmed by strongly decreased liver retinol levels after 6 months of VAF diet feeding (Figure S2A). In line with our previous study, flow cytometry analysis of the BM showed a decrease in relative and absolute frequencies of HSCs and a corresponding loss of quiescence (Figures 2B and 2C; Figures S2B and S2C; Cabezas-Wallscheid et al., 2017). Apart from a significant but mild decrease within the erythroid lineage, VAF BM cells did not display any lineage bias (Figure S2D). Moreover, after 48 h of in vitro single-cell cultivation of HSCs, a higher division rate was detected in VAF cells, indicating a cycling-primed state (Figure 2D). Serial colony-forming unit (CFU) assays showed decreased in vitro self-renewal capacity (Figure 2E). In agreement with this finding, serial WBM competitive transplantation assays revealed a significant decrease in peripheral blood (PB) chimerism, displaying a lymphoid bias (Figure 2F; Figures S2E and S2F). Thus, vitamin A deficiency leads to loss of quiescent repopulating HSCs.
Figure 2. Lack of dietary vitamin A leads to loss of metabolic HSC identity and triggers differentiation.

(A) Workflow summarizing analyses performed on VAF HSCs.
(B) Flow cytometry-based analysis of frequencies of control and VAF HSCs. The percentage of single cells is shown. n= 14–19.
(C) Flow cytometry-based analysis of HSC cell cycle control and VAF. The percentage of cell cycle phases (G0, G1, and G2/S/M) is shown. n = 16.
(D) Single cell (SiC) division assay after 48 h in VAF and control HSCs. The percentage of cells is shown. n = 9.
(E) Colony-forming unit assay of HSCs in control and VAF conditions. 1st and 2nd plating. n = 7-8.
(F) Flow cytometry-based analysis of CD45.2 PB chimerism in the competitive transplantation assay of VAF and control WBM cells (CD45.2) versus CD45.1/2 WBM cells. The percentage of CD45.2 chimerism after 1° and 2° transplantation is shown; radio-resistant cells were excluded. n = 9–16.
(G) GSEA of selected Reactome pathways on RNA-seq data of control and VAF HSCs.
(H) Targeted metabolomics results showing significantly different metabolites in VAF and control HSCs+MPP1. Quantification is shown as FC + SD relative to control HSCs+MPP1. n = 9–12.
(I) ATAC-seq digital footprinting analysis showing Tn5 insertions on the forward and reverse strand (red and blue, respectively) and CTCF and RAR motifs in VAF and control HSCs. n = 3.
(J) Cyp26 enzyme expression. Normalized expression counts are shown from VAF RNA-seq. DESeq2 padj.- values: *p < 0.1, **p < 0.05, ***p < 0.01. ND, not detected.
(K) RT-qPCR of Cyp26b1 gene expression in stem and progenitor populations and differentiated cells. Normalized mean relative to Oaz1, B2m, and Tbp expression and relative to HSCs is shown. Significance in HSCs is compared with all other populations. n = 3; one experiment. GE, gene expression.
(B)–(E), (H), (J), and (K), mean + standard deviation (SD); (B), (E), (H), (I), and (K), unpaired Student’s t test; (C), (D), and (F), two-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001; ns, not significant. Significance levels are compared with the control. n indicates the number of biological replicates. For (B)-(F) and (H), two or more independent experiments were performed.
To investigate how retinoids regulate HSC identity, we performed extensive transcriptomic, epigenetic, and metabolic analyses on VAF HSCs. GSEA of RNA-seq data (Figure S2G; Table S1) showed that VAF HSCs exhibit strong downregulation of published HSC signatures such as “dormant HSCs” but enrichment of “MPP” signatures (Cabezas-Wallscheid et al., 2014, 2017; Wilson et al., 2015; Figure S2H; Table S3). Furthermore, Reactome pathways, such as “respiratory electron transport” and “TCA cycle,” were enriched in VAF HSCs (Figure 2G). Next we performed targeted metabolomics and found that vitamin A deficiency led to a significant reduction in the retinoids RA and 4-oxo-RA in HSCs+MPP1 (Figure 2H). Conversely, two TCA cycle metabolites, glutamate and itaconate, were significantly more abundant, suggesting higher metabolic activity of VAF HSCs+MPP1. Most of the metabolites detected in VAF HSCs+MPP1 and VAF MPPs showed similar levels, which is in contrast to control conditions (Figure S2I; Figure 1B; Table S2). We then integrated the metabolomics data with transcriptional changes in enzymes related to the two main metabolically affected pathways, “retinol metabolism” and ”TCA cycle.” Although none of the TCA cycle-related enzymes were transcriptionally deregulated in VAF HSCs, retinol enzymes followed a trend toward downregulation of the pathway (Figure S2J).
To determine the effect of vitamin A deprivation on chromatin accessibility, we applied ATAC-seq (Figure S2K). We integrated the RNA-seq data with genes associated with OCRs specific to control (lost in VAF) and found a significant enrichment of VAF-downregulated genes (Figure S2L). This indicates that vitamin A deficiency leads to loss of OCRs, resulting in reduced expression of the respective genes, as exemplified by two representative gene loci (P2rx7 and Rarb) (Figure S2M). Retinoids can change chromatin accessibility by binding to RA receptors (RARs) (Findley et al., 2019). Thus, we performed digital footprinting analysis for the RAR motif, revealing a significant decrease in VAF HSCs compared with the control (Figure 2I). Additionally, the IRF motif, found previously to be enriched in “retinol metabolism”-annotated genes (Figure S1G), was significantly less occupied in VAF HSCs (Figure S2N), suggesting a potential interplay with interferon regulatory factors.
These data demonstrate that systemic depletion of dietary vitamin A changes the metabolic, transcriptional, and epigenetic profile of HSCs toward a dysfunctional stemness phenotype.
Cyp26b1 is critical for HSC self-renewal and quiescence
To gain deeper insight into the molecular mechanism of RA signaling in HSCs, we screened our omics data for known RA pathway members under homeostatic conditions and upon VAF (Figures 1 and 2). Interestingly, cytochrome P450 family 26 subfamily B member 1 (Cyp26b1) was significantly downregulated under VAF conditions and specifically expressed in HSCs compared with downstream progenitors and differentiated blood cells (Figures 2J and 2K; Figure S2O). In an irreversible reaction, Cyp26 enzymes metabolize at-RA into 4-hydroxy-RA (4-OH-RA), which can be further oxidized to the catabolic metabolite 4-oxo-RA, commonly considered an inactive degradation product (Figure 3A). Thus, the at-RA-Cyp26-4-oxo-RA axis is seen as a system to balance RA levels in a cell (Bowles et al., 2006; Hernandez et al., 2007; Niederreither et al., 2002; Ono et al., 2020; Sakai et al., 2001; Yashiro et al., 2004). Our metabolomics data show that RA and 4-oxo-RA are enriched in HSCs+MPP1, indicating enzymatic activity of Cyp26 in HSCs (Figure 1B). Although Cyp26a1 and Cyp26c1 were not expressed (Figure 2J; Figure S2O), we sought to test whether Cyp26b1 is important for stemness. To this end, we employed two different Cre-driver mouse models to conditionally delete Cyp26b1 in HSCs in the adult hematopoietic system (Mx1Cre x Cyp26b1fl/fl and SclCre x Cyp26b1fl/fl; Figure 3B). In both systems, Cyp26b1 deletion was confirmed in HSCs by quantitative reverse transcription PCR (RT-qPCR), and a potential compensatory upregulation of Cyp26a1 or Cyp26c1 was excluded (Figures S3A and S3J). Four weeks after induction, Mx1Cre x Cyp26b1fl/fl knockout (KO) (Mx-Cyp26b1−/−) mice exhibited a mild but significant decrease in relative and absolute HSC frequencies compared with the Cre (Mx1Cre) or floxed controls (Cyp26b1fl/fl) (Figure 3C; Figures S3B and S3C). Although cell cycle phase distribution was unchanged (Figure S3D), Mx-Cyp26b1−/− HSCs showed a significantly higher in vitro division rate (Figure 3D; Figure S3E). Notably, neither MPPs nor differentiated BM cell frequencies were affected (Figures S3F and S3G). Functionally, HSCs lacking Cyp26b1 showed an impaired in vitro self-renewal capacity (Figure 3E). Competitive BM transplantation experiments showed an overall decrease in CD45.2 chimerism (Figure 3F). Lymphoid lineages were particularly affected (Figure S3H), and a decreased number of donor-derived HSCs after secondary transplants was observed (Figure 3G; Figure S3I). To confirm these findings, we repeated the experiments using the SclCre driver mouse line (Scl-Cyp26b1−/−, which induces adult HSC-specific KO, and observed the same phenotype (Figures S3J-S3S). Additionally, we analyzed mice 7 months after deletion of Cyp26b1 (Figure 3H). This long-term deletion led to an enhanced phenotype of HSC frequencies and function (Figures S3B and S3T-S3W) compared with short-term Cyp26b1 deletion and further resembled the VAF conditions (Figures 2E, 2F, and 3I).
Figure 3. Cyp26b1 is critical for HSC self-renewal and quiescence.

(A) Pathway model.
(B) Workflow summarizing analyses performed on two Cyp26b1 KO mouse models (SclCre and Mx1Cre).
(C) Flow cytometry-based analysis of HSC frequency in Cyp26b1fl/fl (control) and Mx-Cyp26b1−/− (KO). Frequency of single cells is shown. Significance levels are compared with the control. n = 8–10.
(D) Single cell (SiC) division assay after 48h in Cyp26b1fl/fl and Mx-Cyp26b1−/− HSCs. Frequency of cells is shown in percent. n = 10–12.
(E) Colony-forming unit (CFU) assay of Cyp26b1fl/fl and Mx-Cyp26b1−/− HSCs; first, second, and third plating. n = 7–9.
(F) Flow cytometry-based analysis of CD45.2 PB chimerism of 1° and 2° competitive WBM transplantation assays of Cyp26b1fl/fl (CD45.2 control) and Mx-Cyp26b1−/− (CD45.2 KO) cells versus CD45.1/2 cells. Shown is percent CD45.2 chimerism. n = 7–8.
(G) Flow cytometry-based analysis of CD45.2+ HSCs after 16 weeks of 1° and 2° transplantation of HSCs derived from Cyp26b1fl/fl and Mx-Cyp26b1−/− Shown is the percentage of single cells. n = 7.
(H) Workflow summarizing analyses performed on long-term Cyp26b1 KO (Mx-Cyp26b1−/− and Cyp26b1fl/fl 7 months after deletion.
(I) CFU assay of HSCs in 7-month-old Cyp26b1fl/fl and Mx-Cyp26b1−/−; first, second, and third plating. n = 8–10.
(J) GSEA of selected Reactome pathways on RNA-seq of 7-month Mx-Cyp26b1−/− and Cyp26b1fl/fl HSCs.
(K) GSEA of “up-regulated genes in VAF HSCs” on RNA-seq of 7-month Mx-Cyp26b1−/− and Cyp26b1fl/fl HSCs. n = 2.
(L) ATAC-seq digital footprinting analysis on the CTCF and RAR motif in Mx-Cyp26b1−/− and Cyp26b1fl/fl HSCs. Tn5 insertions are shown on positive and negative strands (red and blue, respectively). n = 2.
(M) Targeted metabolomics on retinoids in Scl-Cyp26b1−/− and Cyp26b1fl/fl HSCs+MPP1. Shown is FC compared with Cyp26b1fl/fl HSCs+MPP1. n = 3–12.
(C)–(E), (G), (I), and (M), mean + SD; (C), (E), (G), (I), and (L), unpaired Student’s t test; (D) and (F), two-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. n indicates the number of biological replicates. For (C)–(G), (I), and (M), two or more independent experiments were performed.
We next performed RNA-seq in short-term-deleted (immediately after KO induction) and long-term-deleted (7 months) Mx-Cyp26b1−/− HSCs (Figure 3J; Figure S3X; Table S1). GSEA of both datasets revealed enrichment of “MPP” and “cell cycle” signatures in the absence of Cyp26b1 and downregulation of genes related to “HSC signatures” and, unexpectedly, “RA signaling” after short-term deletion (Figures S3Y-S3Z; Table S3). Furthermore, we found that the GO terms “TCA cycle” and “metabolism of amino acids” were enriched in Mx-Cyp26b1−/− HSCs, whereas “cholesterol metabolism” was depleted, suggesting metabolic priming toward a dysfunctional stemness phenotype similar to VAF conditions (Figure 3J; Figure S3AA).
To see whether loss of Cyp26b1 mimics the transcriptional changes seen in VAF HSCs, we created a gene signature from VAF up- or downregulated DEGs (padj. < 0.1; Table S3) and performed GSEA in Mx-Cyp26b1−/− HSCs. Transcriptional changes observed in short-term Cyp26b1 deletion did not significantly correlate with VAF signatures (data not shown). However, when comparing 7-month Mx-Cyp26b1−/− HSCs with VAF HSCs, up- and downregulated genes, respectively, were significantly enriched (Figure 3K; Figure S3AB; Table S3), suggesting that the phenotype observed in Mx-Cyp26b1−/− HSCs is at least partially established because of the lack of retinoids. We then performed ATAC-seq on Mx-Cyp26b1−/− and Cyp26b1fl/fl HSCs (Figure S3AC). Digital footprinting analysis revealed that the occupancy of the RAR motif was significantly decreased in Mx-Cyp26b1−/− HSCs (Figure 3L), further indicating a role of RA metabolites downstream of Cyp26b1 for RAR activation. To test whether Cyp26b1 is indeed indispensable for 4-oxo-RA production in HSCs, we performed targeted retinoid metabolomics. Importantly, we found that 4-oxo-RA was depleted in Mx-Cyp26b1−/− HSCs+MPP1, whereas RA levels were not affected (Figure 3M).
These data indicate that the enzyme Cyp26b1 plays an important role in maintaining HSC self-renewal and quiescence, presumably by metabolizing at-RA to 4-oxo RA.
4-oxo-RA regulates HSC function
4-oxo-RA is commonly considered a catabolic by-product of processes limiting the effect of at-RA in the cell (Bowles et al., 2006; Ferreira et al., 2020; Ghyselinck and Duester, 2019; Hernandez et al., 2007; Niederreither et al., 2002; Sakai et al., 2001). However, a few studies have shown that 4-oxo-RA can also be an active metabolite (Baron et al., 2005; Idres et al., 2002; Pijnappel et al., 1993; Shealy, 1989; van der Leede et al., 1997). Thus, we aimed to unravel whether and how HSCs respond to 4-oxo-RA. To our surprise, we found that in vitro treatment with 4-oxo-RA maintained stem cell quiescence and prevented HSCs from undergoing cell division to the same extent as at-RA (Figures 4A-4C). Of note, toxicity effects from in vitro treatment were excluded (Figure S4A). When placed into in vitro culture, HSCs start a differentiation process involving increased metabolic activity. We hypothesized that 4-oxo-RA-treatment may prevent HSC activation in vitro, similar to what has been shown for at-RA (Cabezas-Wallscheid et al., 2017; Purton et al., 1999, 2000). Indeed, we observed a significant decrease in nascent protein translation rates and a lower number of reactive oxygen species (ROS)-oxidized HSCs (Figure 4D-E), indicating low use of OXPHOS as an energy source and, thus, maintenance of HSC features (Signer et al., 2014, Ludin et al., 2014; Suda et al., 2011). A mitochondrial stress test on LSK cells revealed significantly lower basal and maximal respiration upon at-RA and 4-oxo-RA treatment (Figure S4B), in line with recent findings that show reduced basal and maximal respiration in HSCs compared with MPPs (Ito et al., 2019). Of note, mitochondrial content was not affected by the treatments (Figure S4C). Functionally, 4-oxo-RA in vitro treatment led to an increased in vitro and in vivo self-renewal capacity (Figures 4F and 4G). To assess whether 4-oxo-RA might protect HSCs in vivo from proliferative activation upon poly(I:C) (pIC) injection, we pre-treated mice with at-RA or 4-oxo-RA (Figure 4H). 4-oxo-RA treatment was associated with higher retention of HSCs in the G0 cell cycle phase, decreased HSC proliferation, and higher self-renewal capacity compared with control conditions after pIC treatment (Figures 4H-4J). The downstream metabolite 4-oxo-RA mimics at-RA effects on HSCs in vitro and in vivo. This indicates that 4-oxo-RA could be crucial for signaling through the RARs in HSCs.
Figure 4. 4-oxo-RA regulates HSC function.

(A) Workflow summarizing in vitro treatments with DMSO, at-RA, and 4-oxo-RA performed on Lin−CD48− BM cells.
(B) Flow cytometry-based analysis of the cell cycle after in vitro treatment, gated on HSCs. The percentage of the cell cycle phase (G0, G1, and G2/S/M) is shown. n = 12.
(C) Single cell division (SiC) assay after 48-h in vitro treatment of HSCs with DMSO, at-RA, and 4-oxo-RA. The percentages of cells are shown. n = 12.
(D) Flow cytometry-based analysis of O-propargyl (OP) puromycin median fluorescence intensity after 72-h in vitro treatment. Gated on HSCs, fold change (FC) normalized to DMSO treatment. n = 6–9.
(E) Flow cytometry-based analysis of ROS in MitoOrp mouse model after 72-h in vitro treatment. Gated on HSCs, shown is the percentage of HSCs. n = 8.
(F) Colony-formung unit (CFU) assay; plating after 72-h in vitro treatment of 1,000 LSK cells with DMSO, at-RA, and 4-oxo-RA; first and second plating. The number of colonies was normalized to the control. n = 7–10.
(G) Flow cytometry-based analysis of CD45.2 PB chimerism over time. Shown is a transplantation assay comparing 72-h in-vitro-treated HSCs with 4-oxo-RA or control (DMSO). Shown is the percentage of CD45.2 chimerism. n = 6–9.
(H) Workflow showing in vivo treatments with DMSO + PBS, DMSO + pIC, at-RA + pIC, and 4-oxo-RA + pIC. Flow cytometry-based analysis of the HSC (LK CD150+ CD48−CD34−) cell cycle. The percentage of the cell cycle phase (G0, G1, and G2/S/M) is shown. Significance levels are compared with DMSO pIC. n = 12.
(I) SiC division assay of 48-h in-vivo-treated HSCs (LK CD150+CD48−CD34−) after DMSO + PBS, DMSO + pIC, at-RA + pIC, and 4-oxo-RA + pIC injections. The percentage of cells is shown. Significance levels are compared with DMSO + pIC. n = 10–12.
(J) CFU assay of HSCs after DMSO + PBS, DMSO + pIC, at-RA +pIC, and 4-oxo-RA+ pIC in vivo injections. The number of colonies is normalized to DMSO after the first and second plating. Significance levels are compared with DMSO + pIC. n = 5–6.
(K) Heatmap of RNA-seq data of 24-h in-vitro-treated HSCs+MPP1. Log2FC threshold ≥ 0.5, padj. < 0.1 in pairwise comparisons. Shown are biological process GO terms and the main associated TF motif for each cluster. NES, normalized enrichment score. n = 3.
(L) Log2FC of all cluster 2 genes depicted from 3 RNA-seq datasets (4-oxo-RA 24-h in vitro treatment of HSCs+MPP1, VAF HSCs, and 7-month Cyp26b1 KO HSCs). Genes shown in bold contain the RAR motif. Grey indicates not detected.
(B)–(F), (H), and (J), mean + SD; (D), (F), and (J), unpaired Student’s t test; (B), (C), (E), (G), and (I), two-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. n indicates the number of biological replicates. For (B)-(J), two or more independent experiments were performed.
Next we performed RNA-seq on 24-h in-vitro-cultured HSCs+MPP1 treated with dimethyl sulfoxide (DMSO), at-RA, or 4-oxo-RA (Figure S4D). GSEA of 4-oxo-RA-treated versus DMSO-treated HSCs+MPP1 revealed a strong correlation for the signatures “dormant HSCs” and “HSCs” (Figure S4E). Although the overall transcriptional response to at-RA and 4-oxo-RA was very similar (Figure S4D; Table S1), 4-oxo-RA treatment led to additional enrichment for HSC-specific signatures as well as metabolic pathways such as “signaling by GPCR” and “Netrin-1 signaling” (Renders et al., 2021; Figure S4F and S4G). In contrast, at-RA-treated HSCs+MPP1 were further enriched for “amino acid metabolism” and “TCA cycle,” indicating that HSC hallmarks are even better preserved by the metabolite 4-oxo-RA.
An unbiased clustering analysis identified 4 clusters for which we performed GO analysis and a regulatory motif prediction (Figure 4K; Tables S1 and S4). Interestingly, cluster 1 was dominated by genes associated with “DNA replication,” showing strong downregulation by both RA metabolites and enrichment for the E2F motif, which is known to be involved in cell cycle regulation (Dimova and Dyson, 2005; Helin, 1998). Genes in cluster 2, which included Cyp26b1 and Rarb, were highly upregulated by both RAs and linked to “inflammatory response.” This cluster showed enrichment for RAR motifs, suggesting that these are likely direct target genes of 4-oxo-RA and at-RA, in contrast to genes in clusters 3 and 4. Additionally, RT-qPCR analysis showed cluster 2 genes were already induced after 2 h of treatment, further indicating a direct response (Figure S4H). Similarly, genes induced by 4-oxo-RA are also expressed at significantly higher levels in HSCs compared with downstream MPP populations, whereas 4-oxo-RA-repressed genes are strongly induced upon differentiation (Figure S4I). Thus, 4-oxo-RA-treatment leads to direct and indirect transcriptional changes, inducing HSC-specific and repressing MPP-specific genes.
We showed that 4-oxo-RA is a potent metabolite that regulates gene expression in HSCs and plays an essential role in the retention of HSC quiescence and self-renewal capacity.
4-oxo-RA acts downstream of Cyp26b1 and is required for HSC maintenance
To unravel whether the lack of 4-oxo-RA causes the transcriptional changes observed with VAF diet feeding or upon Cyp26b1 deletion, we compared DEGs of in vitro 4-oxo-RA treatment with gene expression fold changes found in VAF and Mx-Cyp26b1−/− HSCs (Figure 4L; Figures S4J and S4K; Table S4). Globally, transcriptome data of VAF HSCs, short-term as well as long-term deletion of Cyp26b1 in HSCs, and, interestingly, MPPs resembled the DMSO control condition upon HSC+MPP1 in vitro treatment (Figure S4K). Importantly, VAF signatures for up- and downregulated genes showed a significant and strong anti-correlation compared with 4-oxo-RA-induced and 4-oxo-RA-repressed genes, respectively (Figure S4J). Cluster 2 genes in particular, approximately 80% of which contain a RAR motif, were not only strongly downregulated in VAF HSCs but also reduced in 7-month Mx-Cyp26b1−/− HSCs (Figure 4L). Thus, 4-oxo-RA is likely the metabolic signal modulating core signature genes that were found to be deregulated in VAF and Cyp26b1 KO HSCs; however, an at-RA-mediated effect cannot be excluded.
To address whether the positive effects of at-RA on HSC function are conferred by binding of at-RA to RARs or by the Cyp26b1-downstream metabolite 4-oxo-RA, we performed RNA-seq on Mx-Cyp26b1−/− HSCs+MPP1 that were treated in vitro with DMSO, at-RA, or 4-oxo-RA (Figure 5A; Table S1). GSEA of the DMSO condition revealed a strong correlation with the MPP signature when comparing Mx-Cyp26b1−/− HSCs+MPP1 against Cyp26b1fl/fl HSCs+MPP1, consistent with our previous findings (Figures S3E, S3P, and S5A; Table S3). Although at-RA treatment exerted a transcriptional response in Mx-Cyp26b1−/− HSCs+MPP1, we found significant enrichment of the MPP signature that, in contrast, was lost upon 4-oxo-RA treatment (Figure S5A). This suggests that 4-oxo-RA, but not at-RA, can rescue the loss of Cyp26b1 in Mx-Cyp26b1−/− HSCs. To evaluate a functional rescue, we treated Cyp26b1fl/fl and Mx-Cyp26b1−/− or Scl-Cyp26b1−/− HSCs with at-RA, 4-oxo-RA, and their upstream metabolite retinol (Figures 5B and 5C; Figure S5B). Upon treatment with retinol, at-RA, and 4-oxo-RA, Cyp26b1fl/fl HSCs retained their quiescence and showed enhanced self-renewal capability. In contrast, in Scl-Cyp26b1−/− HSCs, only 4-oxo-RA led to enhanced stem cell quiescence and function, whereas retinol and at-RA treatments had no effect (Figures 5B and 5C). To validate these findings in vivo, we pre-treated Cyp26b1fl/fl and Scl-Cy-p26b1−/− mice with at-RA or 4-oxo-RA before challenging them with pIC. Strikingly, 4-oxo-RA, but not at-RA, retained quiescence in Scl-Cyp26b1−/− HSCs and led to increased in vitro self-renewal capacity compared with DMSO pIC conditions (Figures 5D and 5E).
Figure 5. The Cyp26b1-downstream metabolite 4-oxo-RA is required for HSC maintenance.

(A) Workflow showing rescue experiments performed on the different Cyp26b1 KO mouse models.
(B) Workflow showing the single cell (SiC) division assay after 48 h in vitro treatment of Scl-Cyp26b1−/− compared with Cyp26b1fl/fl HSCs with DMSO, retinol, at-RA, and 4-oxo-RA. The percentage of cells is shown. n = 12.
(C) Workflow depicting the colony-forming unit (CFU) assay after 72 h in vitro treatment of Scl-Cyp26b1−/− compared with Cyp26b1fl/fl HSCs with DMSO, retinol, at-RA, and 4-oxo-RA. The number of colonies was normalized to control DMSO treatment of each corresponding plating. n = 6–8.
(D) Workflow showing in vivo treatments with DMSO + PBS, DMSO + poly I:C (pIC), at-RA + pIC, and 4-oxo-RA + pIC of Scl-Cyp26b1−/− compared with Cyp26b1fl/fl mice and flow cytometry-based analysis of the HSC cell cycle. The percentage of HSCs in the G2/S/M cell cycle phase is shown. Significance levels are compared with DMSO pIC. n = 6–7.
(E) HSC CFU assay after 72 h in vivo treatment of Scl-Cyp26b1−/− and Cyp26b1fl/fl mice with DMSO + PBS, DMSO + pIC, at-RA + pIC, and 4-oxo-RA + pIC. The number of colonies was normalized to DMSO + PBS treatment of each corresponding plating. n = 4–8.
(B)–(E), mean + SD; (C)–(E), unpaired Student’s t test; (B), two-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. n indicates the number of biological replicates. Significance levels are compared with the control. For (B)–(E), two or more independent experiments were performed.
These data indicate that 4-oxo-RA, produced in a cell-autonomous manner by Cyp26b1 in HSCs, is the main active RA metabolite required to convey the complete transcriptional response to maintain HSC properties.
Rarb is required to mediate 4-oxo-RA-dependent maintenance of HSC function
Next we aimed to investigate which TF may be responsible for transmitting 4-oxo-RA-dependent effects on HSC function. RA metabolites enter the nucleus to bind to RARs; to modulate gene expression, RARs dimerize with retinoid X receptors (RXRs) and bind to RA response elements (RAREs) on genomic DNA (McKenna, 2012). RARs as well as RXRs consist of three different subtypes (α, β, and γ) that show cell-type-specific expression patterns, creating platforms for distinct RA effects in various tissues (Bushue and Wan, 2010; Conserva et al., 2019; Tang and Gudas, 2011). Our gene expression data revealed that Rarb (in contrast to Rarg and Rara) was specifically expressed in HSCs but not in downstream progenitors or differentiated cells and showed significant downregulation upon VAF conditions (Figures 6A and 6B; Figures S6A and S6B). Moreover, some studies report that 4-oxo-RA is a potent ligand and activator of Rarb specifically (Faria et al., 1999; Idres et al., 2002; Pijnappel et al., 1993; Sonneveld et al., 1999), raising the possibility that 4-oxo-RA’s effects in HSCs could be mediated through Rarb.
Figure 6. Rarb is required to mediate 4-oxo-RA-dependent maintenance of HSC self-renewal.

(A) RT-qPCR of Rarb GE in stem and progenitor populations and differentiated cells. Normalized mean relative to Oaz1, B2m, and Tbp expression and relative to HSCs is shown. Significance in HSCs is compared with all other populations. n = 3, one experiment.
(B) Normalized expression counts (DESeq2) of RAR and RXR genes from VAF RNA-seq. DESeq2 padj. values: *p < 0.1, **p < 0.05, ***p < 0.01.
(C) Workflow showing in vitro Rarb KD.
(D) Flow cytometry-based analysis of the frequency of HSCs+MPP1 (LSK CD150+CD48−). The percentage of single cells is shown. n = 3, one experiment.
(E) Colony-forming unit (CFU) assay of GFP+ LSK cells after in vitro Rarb KD; first and second plating. The number of colonies was normalized to the control. n = 12.
(F) Workflow showing competitive transplantation of GFP+LSK shRarb (CD45.2) and GFP+LSK (CD45.1) and flow cytometry-based analysis of PB chimerism over time. Shown is the percentage of chimerism. n = 12–16.
(G) Workflow showing the single cell (SiC) division assay after 48-h in vitro treatment of control HSCs compared with Rarb KO with DMSO, retinol, at-RA, and 4-oxo-RA. The percentage of single cells is shown. n = 12.
(H) Workflow depicting CFUs after 72-h in vitro treatment of control HSCs compared with Rarb KO with DMSO, retinol, at-RA, and 4-oxo-RA. The number of colonies was normalized to control DMSO treatment of each corresponding plating. n = 6–8.
(I) Transplantation assay comparing 72-h in-vitro-treated control and Rarb KO HSCs with at-RA, 4-oxo-RA, or DMSO. Shown is the percentage of CD45.2 chimerism over time. n = 6–9.
(J) Boxplots comparing 24-h in vitro 4-oxo-RA effects on GE in control and Rarb KO HSCs+MPP1 RNA-seq versus DMSO treatment. All genes of heatmap clusters 1–4 from RNA-seq of in vitro treatments after 24 h (Figure 4K) are shown. n = 2–3.
(A), (B), (D), (E), (G), and (I), mean + SD; (A), (D), (E), and (H), unpaired Student’s t test; (F), (G), and (I), two-way ANOVA. *p < 0.05, **p < 0.01, ***p < 0.001. n indicates the number of biological replicates. For (B)–(F), two or more independent experiments were performed.
To test Rarb function in HSCs, we first established an in vitro retroviral knockdown (KD) system (Figure 6C; Figure S6C). Rarb KD was validated by RT-qPCR (Figure S6D) and led to decreased HSC frequencies and reduced in vitro self-renewal capacity (Figures 6D and 6E). In vivo competitive transplantation of Rarb KD and control LSK cells showed that silencing of Rarb leads to a severe decrease in PB CD45.2 chimerism (Figure 6F; Figure S6E). Additionally, we performed in vivo transplantation assays using a constitutive Rarb KO mouse model. Rarb KO was confirmed by RT-qPCR, whereas compensatory upregulation of other RARs was excluded (Figure S6F). 16 weeks after transplantation of Rarb KO HSCs, CD45.2+ BM cells revealed a lower frequency of HSCs compared with the control condition (Figure S6G). These data show that Rarb plays an important role in maintenance of stem cell features.
To unravel whether Rarb is required for mediation of 4-oxo-RA’s effects on stemness, we treated Rarb KO HSCs with retinol, at-RA, or 4-oxo-RA in vitro and analyzed their functional response. Interestingly, none of the metabolites had an effect on proliferation or on in vitro or in vivo self-renewal capacity in Rarb KO HSCs (Figures 6G-6I). A previous study has described the role of Rarg in mediating RA signaling in HSCs (Purton et al., 2006). To investigate a potential involvement of Rarg in (4-oxo-) RA-mediated signaling, we used a constitutive Rarg KO mouse model and treated Rarg KO and control HSCs with DMSO, at-RA, or 4-oxo-RA in vitro (Figures S6H and S6I). Interestingly, Rarg KO HSCs responded to at-RA and 4-oxo-RA treatment with enhanced quiescence and increased in vitro self-renewal capacity, similar to control HSCs (Figures S6H and S6I). Of note, Rarg KO HSCs did not show expression of any RAR except Rarb (Figure S6F). These data suggest that Rarb, but not Rarg, is essential for transmitting RA signaling in HSCs.
To evaluate whether Rarb is involved in transcriptional regulation of 4-oxo-RA response genes (Figure 4K; Table S4), we performed RNA-seq in Rarb KO HSCs+MPP1 (Figure 6J; Table S1). Although Rarb KO HSCs+MPP1 remained responsive to at-RA and 4-oxo-RA treatment, the overall effect was significantly inferior compared with wild-type cells.
These results indicate that Rarb is required to establish a critical gene dosage for proper and complete transmission of RA signaling in HSCs and, ultimately, for maintenance of stem cell identity. We showed that a single metabolite controls stem cell fate by instructing epigenetic and transcriptional attributes. Mechanistically, we dissected an unexpected and non-classical retinoid signaling axis, showing that Cyp26b1 catalyzes production of 4-oxo-RA, which subsequently mediates signaling via Rarb.
DISCUSSION
In this study, we established and applied low-input targeted metabolomics approaches to uncover differences between purified HSCs+MPP1 and their downstream MPPs. Joint analysis of global data on gene expression, chromatin accessibility, and histone modifications emphasized metabolic players that have so far not been linked to HSC biology. For instance, we observed a high abundance of various lipids in HSCs+MPP1. Several studies have previously highlighted changing lipid compositions in relation to stem cell aging or dietary intake (Hermetet et al., 2019; Lu et al., 2019). There is evidence that specific lipid species may have a direct effect on transcription; e.g., by modulating DNA polymerase activity or binding to nuclear receptors (Ardah et al., 2018; Clémot et al., 2020; Kamath-Loeb et al., 2014; Novello et al., 1975). However, further lipidomics will be required to illuminate differential use of signaling or membrane lipids in HSCs and downstream progenitors. The distinct metabolic properties of both populations are further characterized by enrichment of various amino acids in MPPs. The involvement of glutaminolysis in stress-mediated activation of different stem cell systems as well as in a leukemic context has been emphasized previously (Gallipoli et al., 2018; Kim et al., 2020; Sommerkamp et al., 2020). Our metabolomics data revealed higher levels of glutamine and higher expression of its synthesizing enzyme, glutamine synthetase, in HSCs(+MPP1) compared with downstream MPPs but lower levels of glutamate, suggesting that MPPs may rely on energy production supported by glutaminolysis.
The most highly enriched metabolic hub in HSCs was retinol metabolism, as indicated by metabolic, transcriptional, and epigenetic analyses. It is currently widely accepted that vitamin A is metabolized to bioactive at-RA via a classical RA pathway (Cañete et al., 2017). Here we show that HSCs rely on a non-classical RA signaling axis, Cyp26b1-4-oxo-RA-Rarb. Cyp26 enzymes are mostly described as preventing excess levels of intracellular at-RA via its degradation to inactive metabolites such as 4-oxo-RA (Bowles et al., 2006; Ghyselinck and Duester, 2019; Hernandez et al., 2007; Niederreither et al., 2002; Sakai et al., 2001). KO studies of Cyp26 enzymes have shown their relevance in various tissues during development and in adulthood (Ono et al., 2020; Snyder et al., 2020; Yashiro et al., 2004). A previous publication suggested that CYP26B1 expression is exclusively found and required in human BM niche cells to inactivate at-RA in stem and progenitor cells (Ghiaur et al., 2013). In mice, we showed that Cyp26b1 was expressed in HSCs and that its deletion led to a deficit in 4-oxo-RA levels and loss of HSC function. To our knowledge, Cyp26b1 has not yet been considered essential for production of a bioactive retinoid. Strikingly, 4-oxo-RA treatment resulted in rapid and strong upregulation of genes and enhanced stemness features. Although few studies have previously demonstrated that 4-oxo-RA can act as a bioactive metabolite (Baron et al., 2005; Gaemers et al., 1996; Pijnappel et al., 1993; Satre et al., 1989; Shealy, 1989), so far its role has not been studied in the hematopoietic context. Mechanistically, we hypothesize that 4-oxo-RA induces expression of a small group of genes in HSCs that are deregulated in VAF and Cyp26b1−/− HSCs and many of which code for membrane-associated proteins (e.g., Clec1a/b and P2rx7). This, in turn, may lead to the ability to respond to BM niche signals and, thus, to maintain HSC self-renewal and quiescence. Even though we observed a discrepancy between the lineage biases in VAF and Cyp26b1−/− transplantations, we nevertheless identified a common core retinoid signaling signature, supporting our pathway hypothesis. We suspect that systemic depletion of vitamin A might have additional effects on the hematopoietic system.
Our results highlighted RARs as potential regulators for the majority of 4-oxo-RA target genes. Depending on RAR and RXR expression patterns, different cell types may react differently to various RA metabolites (Bushue and Wan, 2010; Michaille et al., 1994, 1995; Rowe et al., 1992). For example, Idres et al. (2002) demonstrated that at-RA has a higher binding affinity to Rarg, whereas 4-oxo-RA binds more strongly to Rarb. Indeed, HSCs exhibited high and specific expression of Rarb (Cabezas-Wallscheid et al., 2014, 2017; Dahlin et al., 2018; Qian et al., 2018). When we treated Rarb KO HSCs with at-RA and 4-oxo-RA, functional responses on cell division and self-renewal were completely abolished. Although we measured an effect on gene expression, it was significantly dampened. Although a previous study has shown that Rarg plays an essential role in mediating at-RA signaling in LSK cells (Purton et al., 2006), we found that Rarg KO HSCs still functionally responded to 4-oxo-RA and at-RA, although to a slightly lower extent. This discrepancy may be explained by use of different populations (LSK in Purton et al., 2006 versus HSCs used here) and the fact that Rarb is HSC specific, whereas Rarg is also expressed in progenitor populations. However, because we did not perform transplantation experiments, the role of Rarg in mediating 4-oxo-RA signaling in HSCs requires further examination.
Our results suggest that binding of 4-oxo-RA to Rarb is required in HSCs to establish a critical gene dosage for proper and complete transmission of RA signaling to maintain stem cell features. A possible explanation for the remaining transcriptional effects in Rarb KO HSCs may be that these are conferred by other TFs. Previous publications have shown that retinoids can also cause a transcriptional response via unconventional RAR or RXR heterodimers; e.g., via RXR-peroxisome proliferator-activated receptors (Ppars) (Dawson and Xia, 2012; Xu et al., 2001). Additionally, at-RA has been shown to mediate gene expression independent of RARs via other TFs, such as Irf1 (Coyle et al., 2017). Indeed, our data revealed potential crosstalk between RA and IRF signaling in HSCs, as observed previously in other cell types (Bernardo et al., 2017; Luo and Ross, 2006).
This alternative non-classical RA pathway may also have implications for leukemia therapies. At present, at-RA therapy is exquisitely effective, in combination with arsenic trioxide, as a differentiation therapy in acute promyelocytic leukemia with the PML-RARA fusion protein but is generally not potent for treatment of other leukemia subtypes (Chen et al., 1991; Grimwade et al., 2010). It is possible that at-RA therapy not only promotes differentiation of leukemic cells but also supports leukemic stem cell quiescence via its downstream metabolite 4-oxo-RA and, thus, increases the risk of relapse. Hence, recent advances in the design of Cyp26-enzyme inhibitors and Cyp26-resistant synthetic at-RA-derivatives (Hernandez et al., 2020; Nelson et al., 2013; Teodorescu et al., 2020) may help to improve prognosis.
Limitations of the study
For technical reasons, the HSC+MPP1 metabolic profiles are based on targeted approaches and were generated using a less stringent sorting scheme compared with the highly pure HSC populations used for RNA-, ATAC-, and ChIP-seq. Thus, further development of metabolomics tools might increase the number of detected metabolites. Further, the discrepancy in HSC purity may lead to underestimation of metabolic differences between HSCs and MPPs compared with the other genome-wide approaches.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents can be directed to and are available from the lead contact, Nina Cabezas-Wallscheid (cabezas@ie-freiburg.mpg.de).
Materials availability
Plasmids generated in this study are available upon request from the lead contact, but a completed Materials Transfer Agreement is required.
Data and code availability
The raw data were deposited in ArrayExpress (https://www.ebi.ac.uk/arrayexpress) and are available under the accession numbers [E-MTAB-9729, E-MTAB-9745, E-MTAB-10659, E-MTAB-9752, E-MTAB-9749, E-MTAB-9779, E-MTAB-9780, E-MTAB-9778, E-MTAB-10661]. Further information can be found in the STAR Methods section.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mouse models
All mice were bred in-house in the animal facility at the MPIE in individually ventilated cages (IVCs), except for the Rarg KO mouse model, which was imported from our collaborator in France (animal facility licensed by agreement #C6721837; N.B.G and B.F. qualified in compliance with the European Community guidelines for laboratory animal care and use - 2010/63/UE). Mice were euthanized by cervical dislocation according to German guidelines. Animal procedures were performed according to protocols approved by the German authorities and the Regierungspräsidium Freiburg (the sacrificing of animals for scientific purposes according to §4 (3) of the German Animal Protection Act; animal protocol numbers G-17/97, G-17/147, G-20/58).
Vitamin A-free diet
3- to 4-week old C57BL/6J female mice were kept in a vitamin A-free diet (0 IU/g vitamin A) or control food (25 IU/g vitamin A) (ssniff Spezialdiäaten GmbH) for 16 to 24 weeks and analyzed the same day.
Cyp26b1 knockout
Conditional deletion of Cyp26b1 was performed using two independent Cre-driver mice, the Mx1Cre line (full name B6. Cg-Tg(Mx1-cre)1Cgn/J) (Kühn et al., 1995) or SclCre line (full name B6. Tg(Tal1-cre)42-056Jrg) (Göthert et al., 2005). These were then crossed with the Cyp26b1 fl/fl (full name B6-Cyp26b1tm2Hmd) (MacLean et al., 2007) mouse line and induced at 7 to 8 weeks of age. KO of Cyp26b1 using the Mx1Cre driver was performed by intraperitoneal injection of 100μg poly I:C /mouse every other day over 10 days (for a total of 5 injections), followed by a recovery phase of 4 weeks before analysis. Knockout using the SclCre driver was achieved by administering tamoxifen food (400mg/kg tamoxifen-citrate food, ssniff Spezialdiäten GmbH) over a period of 3 to 4 weeks before analysis. Male and female mice were used for analysis. Induction treatments were also administered to Cyp26b1 fl/fl, MxCre and SclCre littermate controls.
Rarb knockout
Rarb straight KO mice were purchased from Jax (Stock Rarbtm1Vg1/HsvJ, stock No 022999) and analyzed at 7 to 8 weeks of age together with either wild-type (WT) littermate controls or C57BL/6J mice of the same age. Male and female mice were used for analysis.
Rarg knockout
Rarg straight KO mice (Chapellier et al., 2002) were imported from N.B.G., kept for 2 weeks under quarantine conditions, and analyzed at 8 weeks of age together with either WT littermate controls or C57BL/6J mice of the same age. Male and female mice were used for analysis.
MitoOrp mouse model
The MitoOrp mouse model (full name B6-Gt(ROSA)26Sor(mito-Orp-roGFP2)Tg(CMV-cre)1Cgn) (Fujikawa et al., 2016) was used to measure ROS levels after in vitro treatments of HSPCs (specified elsewhere). Male and female mice were used for experiments at the age of 8 to 20 weeks.
Transplantation models
B6Ly5.1 (CD45.1) or C57BL/6J x B6Ly5.1(CD45.1/2) female mice were used as transplantation recipients and/or supportive/competitive donors.
METHOD DETAILS
HSC/MPP isolation and fluorescence-activated cell sorting (FACS)
Murine BM cells were isolated from pooled femurs, tibias, ilia, and vertebrae by gentle crushing in PBS using a mortar and pestle. Erythrocyte lysis was performed using ACK Lysing Buffer (Thermo Fisher Scientific). To enrich for lineage negative (Lin−) cells, we used the Dynabeads Untouched Mouse CD4 Cells Kit (Invitrogen). Briefly, the erylysed BM was stained for 30 to 45 minutes with a 1:4 dilution of the Lineage Cocktail provided in the Dynabeads Untouched Mouse CD4 Cells Kit (Invitrogen). Labeled cells were then incubated for 20 minutes with washed polyclonal sheep anti-rat IgG coated Dynabeads (as provided in the kit) at 400 uL/mouse. Depletion of Lin+ cells was performed using a magnet. To achieve further purification of HSCs and progenitor cells we used fluorescence- activated cell sorting (FACS). Cell populations were defined as follows: HSCs (LSK CD150+ CD48− CD34−), MPP1 (LSK CD150+ CD48− CD34+), MPP2 (LSK CD150+ CD48+ CD34+), MPP3 (LSK CD150− CD48+ CD34+ CD135−), MPP4 (LSK CD150− CD48+ CD34+ CD135+), B cells (B220+), myeloid cells (CD11b+, GR1+) and T cells (CD4+CD8+). Therefore, the depleted cell fraction was stained for 30 to 60 minutes using the following monoclonal antibodies: anti-lineage (anti-CD4 [clone GK1.5], anti-CD8a [53-6.7], anti-CD11b [M1/70], anti- B220 [RA3-6B2], anti-GR1 [RB6-8C5] and anti-TER119 [Ter-119]) all PE-Cy7 or BV650 or FITC; anti-CD117/c-Kit (2B8)-PE or BV711; anti-Ly6a/Sca-1 (D7)-APCCy7; anti-CD34 (RAM34)-FITC or AF700; anti-CD150 (TC15-12F12.2)-BV605 or PECy5; anti-CD48 (HM48-1)-BV421 or PECy7. Monoclonal antibody conjugates were purchased from eBioscience or BioLegend. Sorting was then performed using a FACS Aria II, III or FACSymphony (Becton Dickinson). Subsequently, cells were collected into ice-cold PBS for reconstitution assays and ChIP-seq, Stem Pro®-34 SFM (Life Technologies) without cytokines for targeted metabolomics, Complete Stem Cell Medium (specified elsewhere) for in vitro treatments, single cell division assays and in vitro treatment transplants, PBS with 1% BSA for ATAC-seq, RNA lysis buffer (ARCTURUS PicoPure RNA Isolation Kit (Life Technologies, Invitrogen)) for population RNA-seq, RT-qPCR and stored at −80°C.
Bulk RNA-seq
Generation
Population RNA-seq data were generated as previously described (Cabezas-Wallscheid et al., 2014). RNA of freshly sorted cells was first extracted using the PicoPURE Arcturus kit (Applied Biosystems). For in vitro treatment experiments, cells were harvested after 24 hours and directly transferred into SMARTseq v4 reaction buffer. Subsequently, cDNA libraries were prepared using SMARTseq v4 (Takara Bio) chemicals with 12 cycles of amplification. Further, the NEBNext Ultra II FS DNA library kit (NEB) was employed to generate uniquely- and dually-barcoded sequencing libraries from cDNA libraries. To this end, 3 to 10ng of cDNA library was fragmented for 22.5 minutes, adaptors were ligated and libraries were amplified using cycle numbers according to input material. RNA-seq libraries were sequenced at 55 million reads depth, 100bp paired end on the Illumina NovaSeq platform.
Low-level processing
Raw FASTQ files were mapped against the mm10 reference genome using the mRNA-seq tool from the bioinformatics pipeline snakePipes (Bhardwaj et al., 2019). In this tool, Alignment mode was used for the mapping of sequenced reads with STAR (STAR_2.7.4a) (Dobin et al., 2013), followed by expression counts quantification by featureCounts (Liao et al., 2014). The quality of the data was checked by running Deeptools QC (3.3.2) (Ramírez et al., 2016). Read coverages for track visualization were obtained via deepTools bamCoverage using RPKM normalization. Genes with an average expression higher than 100 counts in any condition were selected for further analysis. DESeq2 was used to test for differential expression (Love et al., 2014); results were considered significant at a false discovery rate (FDR) equal to 0.1.
Downstream analysis
Principal component analysis (PCA) visualization was performed based on variance stabilized read counts using the DESeq2 package. The expression values of DEGs were used for the visualization of the effect on metabolic pathways and the generation of custom summary metabolic diagrams. For evaluation of expression of the previously published gene signatures of HSC/MPP (Cabezas-Wallscheid et al., 2014), Activation/Dormancy HSCs (Cabezas-Wallscheid et al., 2017), MolO/NoMo HSCs (Wilson et al., 2015) and Retinoic acid (Cabezas-Wallscheid et al., 2017) in the pairwise comparisons, GSEA was performed with the fgseaMultilevel(minSize = 15, maxSize = 500, eps = 0) function from the fgsea package (Sergushichev, 2016), considering significant pathways at a FDR equal to 0.1. We also performed enrichment of the mouse Reactome pathways (Jassal et al., 2020) as well as our herein-described VAF RNA-seq upregulated or downregulated genes and gene sets (obtained from the GO terms of the metabolic pathways of "fatty acid metabolic process" [GO:0006631], "glycolytic process" [GO:0006096], "nucleotide metabolic process" [GO:0009117], "reactive oxygen species metabolic process" [GO:0072593], "tricarboxylic acid cycle" [GO:0006099], "cellular amino acid metabolic process" [GO:0006520], "cholesterol metabolic process" [GO:0008203] and "retinoic acid metabolism" [including "retinoic acid metabolic process" (GO:0042573), "cellular response to retinoic acid" (GO:0071300), "retinoic acid receptor binding" (GO:0042974) and "retinoic acid receptor signaling pathway" (GO:0048384)]). The GSEA enrichment profile of specific signatures were plotted using gseaplot2 function (Yu, 2020). Lists of expression levels of the genes involved in the already mentioned GO terms in our datasets were manually generated and represented as barplots using Prism. For in vitro treatments RNA-seq datasets, the expression values of the resulting 2077 DEGs from the pairwise comparisons of wildtype 24-hour, 4-oxo-RA and at-RA treatments were VST-transformed and relativized across samples, showing the amount by which each gene deviated in a specific sample from the gene’s average (Love et al., 2015). The Euclidean distances between these genes were calculated with the dist function, and then sorted into 4 clusters that covary across samples of the same condition using the hclust function with the ward.D2 clustering criteria and cutree(k = 4) (Murtagh and Legendre, 2014). The resulting tree was represented using the pheatmap package (Kolde, 2019). We tested for over-representation of the clustered genes on GO enrichment analysis using Biological Processes of DAVID 6.8 (Huang et al., 2009). Finally, the log2FC values for the clustered genes in different pairwise comparisons were visualized in boxplots.
The raw data were deposited in ArrayExpress (https://www.ebi.ac.uk/arrayexpress) and are available under the accession numbers [E-MTAB-9729, E-MTAB-9745, E-MTAB-10659, E-MTAB-9752, E-MTAB-9749].
ATAC-seq
Generation
ATAC-seq was generated according to Corces and colleagues (Corces et al., 2017) with some variations. In brief, 1,000 to 3,000 cells were sorted according to surface markers and DAPI into PBS 1% BSA in order to ensure viability. Cells were spun down at 500 x g for 10 minutes at 4°C, supernatant was removed and 20μl of complete transposition buffer (1ml Tn5, 10μl 2x TD buffer (Illumina), 0.01% digitonin, 0.3 x PBS) was carefully added to the invisible cell pellet. The transposition reaction was subsequently carried out at 37°C for 30 minutes on a Thermomixer at 700 rpm. DNA was purified using QIAGEN MinElute Reaction Cleanup and eluted in 20μl EB buffer. Libraries were amplified initially for 5 cycles and further amplification was adjusted to input material as assessed by qPCR. Libraries were cleaned up using AMPure XP beads (Beckman Coulter), double-sided size selection was performed by removing large fragments that precipitate with 0.5 x volume and purifying fragments that precipitate with 1.8 x volume ratio. Clean-up was done twice in order to efficiently remove primers and adaptor dimers. ATAC-seq libraries were sequenced at 75 million reads depth per sample, 100bp paired end on an Illumina NovaSeq platform.
Low-level processing
Reads were aligned in paired-end mode to the mm10 genome using Bowtie2 (Langmead and Salzberg, 2012). Normalization of resulting bam files was obtained using deepTools multiBamSummary (Ramírez et al., 2014). Read coverages for visualization were obtained via deeptools bamCoverage using RPKM normalization and scaling factors obtained above. Peak detection was performed via MACS2 callpeak (Zhang et al., 2008) with a q-value of 0.001.
Downstream analysis
Clustering was performed as previously described (Cauchy et al., 2016) with modifications. The union of peaks was obtained via bedtools merge (Quinlan and Hall, 2010) from ATAC summits. Regions showing log2 fold changes versus control either < −1 or > +1 were considered specific; those between −1 and +1 were considered shared. ATAC matrices were obtained using deepTools computeMatrix ± 3 Kbp around summits. Heatmaps were generated using deeptools plotHeatmap. For GSEA of ATAC-specific sites in RNA-seq log2FC ranked genes (Subramanian et al., 2005), gene sets were created by annotating the top 300 specific peaks to the nearest gene using bedtools closest. GSEAs were run on log2 transformed expression data, using weighted as the enrichment statistic, difference of classes as the ranking metric and gene set as the permutation type. For digital genomic footprinting analyses, pyDNase wellington_footprints was used (Piper et al., 2013) in ATAC mode, shifting coordinates by +4bp and −5bp for the positive and negative strand, respectively. Average Tn5 insertion profiles and footprinting occupancy scores were obtained using pyDNase dnase_average_profiles and wellington_score_heatmap, respectively, also in ATAC mode.
To assess differential accessibility in pairwise comparisons at the gene level, mapped read counts on the peaks located along the gene body of all replicates filtered with bedtools window (Quinlan and Hall, 2010) were quantified by featureCounts (Liao et al., 2014) using the ‘-O’ option to allow multi-overlapping reads. DESeq2 was used to test for differential accessibility (Love et al., 2014); results were considered significant at an FDR = 0.1. As described in the RNA-seq methods above, the DESeq2 output was fed into the fgsea package (Sergushichev, 2016) for performing GSEA of the mouse Reactome pathways (Jassal et al., 2020) and gene sets of the selected metabolic GO terms, considering significant pathways at an FDR equal to 0.1.
Gene ontology analysis for genes annotated to HSC- or MPP-specific ATAC-seq peaks was done using the Biological Processes database of DAVID (Huang et al., 2009).
The raw data were deposited in ArrayExpress (https://www.ebi.ac.uk/arrayexpress) and are available under the accession numbers [E-MTAB-9778, E-MTAB-9779, E-MTAB-9780].
ChIP-seq
Generation
For low-input ChIP-seq we followed the TAF ChIP-seq protocol as described by Akhtar et al. (2019) with few modifications. In brief, cells were stained for FACS sorting with respective antibodies and subsequently crosslinked for 10 minutes at room temperature using PBS with 1% formaldehyde. The reaction was quenched by 125mM glycine and cells were washed twice with ice-cold PBS. 4,000 DAPI-negative cells were then sorted according to surface markers into 200 μl RIPA buffer. Nuclei were broken by mild sonication (Diagenode Bioruptor, 3 cycles, low power, 30 s on/off) and chromatin was incubated with antibody-coupled protein G dynabeads (Thermo Fisher Scientific) overnight at 4°C. Chromatin was then fragmented by incubation with 1μl Tn5 enzyme in 20μl 1x transposition buffer (Illumina) for 40 minutes at 37°C. Stringent washing steps with low salt (once) and high-salt RIPA (four times) as well as TE buffer (twice) were performed to remove all fragmented chromatin that was not associated with the antibody-bead-complex. Reverse crosslinking was performed with proteinase K at 60°C overnight before DNA was isolated using phenol-chloroform extraction and subsequent ethanol precipitation overnight at −80°C. Sequencing libraries were then amplified using unique dual barcode primers (Illumina) and 14 PCR cycles. Libraries were cleaned up using AMPure XP beads (Beckman Coulter). Double-sided size selection was performed by removing large fragments that precipitate with 0.2 x volume and purifying fragments that precipitate with 1.0 x volume ratio. Clean-up was done twice in order to efficiently remove primers and adaptor dimers. ChIP-seq libraries were sequenced at 50 million reads depth per sample, 100bp paired end on an Illumina NovaSeq platform.
Low-level processing
ChIP-Seq datasets were aligned to the mm10 genome and processed as described above for ATAC-Seq datasets. Additionally, to correct for amplification of simple repeat elements due to low starting material, coverage files were subsequently masked for those using bedtools intersect. H3K4me3 enriched regions intersecting with gene TSSs (±1 Kbp) were called via MACS2 callpeak (Zhang et al., 2008) with a q-value of 0.01, using–nomodel–nolambda parameters. H3K27me3 TSS enriched regions were identified using the same method, except adding the –broad parameter for broad peak calling, since H3K27me3 histone mark tends to spread along broad regions of enrichment as suggested in the literature (Adriaens et al., 2016; Chokeshaiusaha et al., 2018; Nakato and Sakata, 2021). In addition, the Pearson correlation coefficient between the H3K4me3 ChIP-seq datasets performed on 1,000 and 10,000 cells were estimated based on the read coverage matrix resulting from the multiBamsummary step.
Downstream analyses
To assess differential histone mark enrichment in pairwise comparisons at gene promoter level, the reads in the TSSs detected peaks were quantified by featureCounts (Liao et al., 2014) as described in the ATAC-seq methods. DESeq2 was again used to test for differential enrichment (FDR = 0.1) and fgsea package was used for the GSEA of the pathways of interest as described above. For the visualization of the overall differences in TSS enrichment of H3K4me3 and H3K27me3 histone marks, genes with an average quantified enrichment higher than 100 counts in any condition were selected. Row z-scores of the average enrichment per condition in the resulting gene promoter regions for relativization across samples were obtained in R via double transposition of center-scaled: t(scale(t(x)). The Euclidean distances between the enrichment patterns of the genes were calculated with the dist function, and then sorted into 5 clusters using the hclust function with the ward.D2 clustering criteria and cutree(k = 5) (Murtagh and Legendre, 2014). The resulting tree was represented using the pheatmap package (Kolde, 2019).
The raw data were deposited in ArrayExpress (https://www.ebi.ac.uk/arrayexpress) and are available under the accession number [E-MTAB-10661].
Targeted metabolomics: (semi-)polar metabolites
For low-input targeted metabolomics on polar metabolites 10,000 HSCs+MPP1 (LSK CD150+)CD48−) and MPPs (LSK CD150−CD48+) were sorted using flow cytometry into ice-cold Stem Pro-34 SFM (Life Technologies) without cytokines. For extraction of polar metabolites cells were then washed with 2.8% glycerol solution and metabolites were extracted with 100μl 80% MeOH extraction buffer containing 13C yeast extract as internal standard. Samples were subsequently vacuum-concentrated (EZ2 elite, Genevac) and stored at −80°C until further processing. Targeted metabolite quantification by LC-MS was carried out using an Agilent 1290 Infinity II UHPLC in line with an Agilent 6495 QQQ-MS operating in MRM mode. MRM settings were optimized separately for all compounds using pure standards. LC separation was done on a Phenomenex Luna propylamine column (50 × 2 mm, 3 mm particles) using a solvent gradient of 100% buffer B (5 mM ammonium carbonate in 90% acetonitrile) to 90% buffer A (10 mM NH4 in water). Flow rate was adjusted from 1000 to 750 μl/min. Autosampler temperature was 5°C and the injection volume was 2 μl. Data processing was performed by Agilent MassHunter (Version B.08.00). At least two negative controls were measured for each experiment to evaluate metabolite background levels. Metabolites were only included if they were detected above background levels and retention time was identical to the 13C yeast standard qualifier peak. To evaluate metabolic differences, the area under the curve (AUC) was determined. Data were normalized to the mean of every metabolite in the HSC population per experiment. HSC+MPP1 replicates represent either a single biological replicate or a pool of 2 biological replicates, MPP replicates represent only 1 biological replicate of the corresponding mice.
Targeted metabolomics: retinoids and polar lipids
Low-input targeted metabolomics on retinoids, bile acids, and polar lipids were performed on 30,000 to 50,000 FACS-sorted HSCs+MPP1 (LSK CD150+CD48−) and MPPs (LSK CD150−CD48+). Cells were directly sorted into 100μl 100% acetonitrile (ACN) reaching a final concentration of roughly 80% ACN due to contamination of flow-PBS. Samples were then vacuum-concentrated (EZ2 elite, Genevac) and stored at −80°C until further processing. Targeted quantification of these metabolites by LC-MS was carried out using an Agilent 1290 Infinity II UHPLC in combination with a ProLab Zirconium Ultra microLC pump in line with an Agilent 6495 QQQ-MS operating in MRM mode. ESI coupling was achieved with a prototype microLC ESI source (ProLab). MRM settings were optimized separately for selected retinoids, bile acids, and polar lipids using pure standards and transferred for the detection of their respective isomers. LC separation was performed on a 100 × 0.3 mm column with 1.8μm Zorbax Eclipse plus C18 (Agilent) packed by Dr. Maisch GmbH. The solvent gradient was 100% buffer A (10 mM ammonium formiate in 90:10 water:methanol) to 100% buffer B (10 mM ammonium formiate in 90:10-propanol:acetonitrile). Flow rate was adjusted to 5 μl/min. Autosampler temperature was 5°C and the injection volume was 5 μl. Data processing was performed using Agilent MassHunter Software. At least two negative controls were measured for each experiment to evaluate metabolite background levels. Metabolites were only included if they were detected above background levels and retention time was similar to the standard qualifier peak. To evaluate metabolic differences, the area under the curve (AUC) was determined. Data were normalized to the mean of every metabolite in the HSC population per experiment and the mean of negative controls was subtracted (resulting negative values were set to 0). Whereas each HSC+MPP1 replicate consisted of a pool of biological replicates (4 to 8 mice), MPP replicates represented only 1 biological replicate of the corresponding mice.
Integration of omics datasets and conditions
To study the overall integration between the different omics datasets in the pairwise comparisons between HSC and MPP cell types we generated a swarm plot with the ranked log2FC expression in the RNA-seq genes with a FDR threshold of 0.1. Swarm plots of the corresponding ATAC-seq gene body and ChIP-seq TSS peaks counts, as described above, were also generated, and pairwise Pearson’s correlation tests were performed with respect to the RNA expression swarm plot. In addition, MTE (Metabolome-Transcriptome-Epigenome)-plots were custom-generated using the “Metabologram” plot as previously described (Hakimi et al., 2016) in order to integrate the effects on the different omics datasets at a metabolic pathway level in an unbiased way. The metabolites involved in each targeted-metabolomics pathway and the individual enzymatic genes detected in RNA-seq from the respective KEGG pathway are plotted as slices in the periphery. The color scale represents the log2FC metabolite abundance, as well as, the individual enzyme log2FC value in RNA-seq expression, ATAC accessibility (gene body peaks), and histone marks H3K4me3 and H3K27me3 enrichment assessed with ChIP-seq (TSS peaks) in the comparison between MPP and HSC cell types. The individual significances of metabolites log2FC are calculated using an unpaired Student’s t test (*p < 0.05; **p < 0.01; ***p < 0.001) whereas the represented significance in the RNA-seq, ATAC-seq and ChIP-seq bars are determined by the DESeq2 adjusted p value (*p < 0.1; **p < 0.05; ***p < 0.01). Moreover, the average log2FC values in the different enzymes and metabolites perturbations are plotted in the center, and a Wilcoxon test was used to calculate the significance of the change based on the individual measurements (*p < 0.1; **p < 0.05; ***p < 0.01). Only the pathways involving at least 5 detected enzymes in RNA-seq and at least 1 measured metabolite are shown. In the case of the pairwise comparison between VAF and control HSCs conditions, only metabolites abundance and the enzyme RNA-seq expression in selected pathways are assessed in the plots. All figures were generated with ggplot2 package (Wickham, 2016)
For the visualization of the RNA-, ATAC-, and ChIP-seq data, we used the Integrative Genomics Viewer (IGV) browser and represented merged coverage tracks of individual replicates.
For the integration of the RNA-seq expression perturbations in different conditions, we took the in vitro WT 24-hour 4-oxo-RA and at-RA treatments RNA-seq heatmap as reference and generated VST-transformed heatmaps as previously described for the same genes and clusters in the other pairwise comparisons. Additionally, the expression log2FC of the genes corresponding to cluster 2 were shown individually for different pairwise comparisons, and the presence/absence of a RAR motif in their regulatory cistrome, as retrieved using i-cis-target (Herrmann et al., 2012) was indicated.
In vivo experiments
In vivo treatments
8-to 16-week old C57BL/6J, Cyp26b1 KO, or Cyp26b1fl/fl, mice were injected intraperitoneally 24 hours before sacrificing with either 30mg/kg at-RA (Sigma-Aldrich) or 30mg/kg 4-oxo-RA (Sigma-Aldrich), or with the respective amount of DMSO in PBS. After 8 hours, 100μg poly I:C per mouse was injected. Mice were sacrificed 16 hours later and BM cells were subjected to further analysis such as cell cycle, single cell division assays or colony-forming unit assays (described below).
Characterization of in vivo mouse models
Cell cycle analysis.
HSC surface staining defined as (LSK CD150+CD48−CD34− was performed on erylysed BM cells. Cells were fixed for 10 minutes at 4°C with BD Cytofix/Cytoperm Buffer (Becton Dickinson and Company). Subsequently, intracellular Ki-67 (BD Biosciences) staining was performed using PermWash solution for at least 45 minutes at 4°C (Becton Dickinson and Company). Prior to flow cytometry analysis cells were stained with DAPI (Sigma-Aldrich) at RT for at least 30 minutes.
Single cell division assay
Single HSCs (LSK CD150+CD48−CD34−) were sorted into 72-well Terasaki plates (Greiner Bio-One) containing Complete Stem Cell Medium (specified in Enrichment of HSPCs and cultivation). After 48 hours, each well was checked manually for the number of cell divisions: 1 cell indicated no division, 2 cells indicated 1 division, > 2 cells indicated more than 1 division.
Serial colony-forming-unit assays (CFU)s of HSCs
200-300 HSCs (LSK CD150+CD48−CD34−) from the various mouse models and their corresponding controls were sorted into and cultured in MethoCult M3434 (StemCell Technologies). 7 Days after plating of CFUs, colonies were quantified (a colony was defined as > 300 cells). In each plating, 10,000 cells of each replicate were used for second and third platings and cultured in MethoCult M3434 (StemCell Technologies). Colonies were quantified 3 and 5 days after the second and third re-platings respectively.
Competitive transplantation experiments
1.5 × 106 WBM cells from CD45.2+ KO or VAF diet models, as well as their corresponding control condition were transplanted into fully irradiated (4.5 Gray +5 Gray) CD45.1 (Ly5.1) mice in competition to 1.5 × 106 WBM cells from 3- to 6-month-old CD45.1/2 mice (i.e., the same age as competitor) by tail vein injection within 24 hours after irradiation. As quality control, a 50:50 ratio of CD45.2 versus CD45.1/2 was confirmed prior to transplantation by flow cytometry. Contribution of CD45.2-donor cells was monitored in PB at 4, 8, 12 and 16 weeks post-transplantation. CD45.2 chimerism was determined by flow cytometry using the following monoclonal antibodies: anti-CD45.1 (A20)–PECy7 or FITC, anti-CD45.2 (104)–PB, anti-CD11b (M1/70)-APCCy7, anti-GR1 (RB6.8C5)-APC, anti-CD8a (53.6.7)- PECy5, anti-CD4 (GK1.5)-PECy5, anti-B220 (RA3.6B2)-AF700. Radioresistant cells (CD45.1) were included to evaluate overall potency of WBM cells.
HSC transplantation experiments
500 HSCs (LSK CD150+CD48−CD34−) from CD45.2+ KO or VAF diet models and control conditions were transplanted into fully irradiated (4.5 Gray +5 Gray) CD45.1 (Ly5.1) mice together with 105 supportive WBM cells from 3- to 6-month-old CD45.1/2 mice (the same age as donor) by tail vein injection within 24 hours after irradiation. Contribution of CD45.2-donor cells was monitored in PB at 4, 8, 12, and 16 weeks post-transplantation. CD45.2 chimerism was determined by flow cytometry using the following monoclonal antibodies: anti-CD45.1 (A20)–PECy7 or FITC, anti-CD45.2 (104)–PB, anti-CD11b (M1/70)-APCCy7, anti-GR1 (RB6.8C5)-APC, anti-CD8a (53.6.7)- PECy5, anti-CD4 (GK1.5)-PECy5, anti-B220 (RA3.6B2)-AF700. Radioresistant cells (CD45.1) were included to evaluate overall potency of HSCs.
Gene expression by RT-qPCR analysis
1,000-2,000 HSCs, MPPs or differentiated cells were sorted using FACS into RNA extraction buffer. Total RNA was isolated using the ARCTURUS Pico Pure Isolation Kit, and reverse transcription was conducted using SuperScript VILO cDNA Synthesis Kit (Invitrogen) according to manufacturer’s guidelines. For RT-qPCR analysis, a Fast SYBR Green Master Mix (Thermo Fisher Scientific) was used with a StepONE Real-Time PCR System (Applied Biosystems). RNA expression was normalized to the geometric mean of Oaz1, B2m and Tbp housekeeping gene expression and represented as relative quantification (ratio: 2 DDCT). For the full list of primers see Table S5. The analysis of amplification curves was carried out using the StepONE software (Applied Biosystems).
In vitro experiments
Enrichment of HSPCs and cultivation
To enrich hematopoietic stem and progenitor cells (HSPCs) for in vitro assays, lineage marker (CD4, CD8a, B220, Gr-1, CD11b, Ter119)-positive and CD48-positive cells were depleted from BM using biotin-labeled antibodies and biotin binder streptavidin Dynabeads (Invitrogen). For BM preparation and depletion process, see above. HSPCs were subsequently cultured in Complete Stem Cell Medium (StemPro-34 SFM, LifeTechnologies containing 50ng/ml SCF, 25ng/ml TPO, 30ng/ml Flt3-Ligand [all Preprotech], 100ml/ml Penicillin/Streptomycin, and 2mM L-Glutamine [both Gibco]). Cells were cultured in 24-, 96- or 384-well ultra-low attachment plates and were treated with either at-RA (5μM final) (Sigma-Aldrich), retinol (10μM final) (Sigma-Aldrich), or 4-oxo-RA (5μM final) (Santa Cruz Biotechnology) or the respective amount of DMSO.
Caspase-3/DAPI staining
For active Caspase 3/DAPI staining, in vitro-treated cells (5μM at-RA, 10μM retinol, 5μM 4-oxo-RA or DMSO) were harvested after 72 hours and subsequently stained with HSC/MPP surface markers and DAPI (1μg/ml) in PBS for 30 minutes. Cells were then fixed for 10 minutes at 4°C and permeabilized using Cytofix/Cytoperm (Becton Dickinson and Company). Intracellular staining with the antiactive Caspase 3-PE antibody (BD) was performed for at least 4 hours at 4°C in PermWash solution (Becton Dickinson and Company).
ROS measurement
The Mito-roGFP2-Orp1 probe allows qualitative measurements of H2O2 levels in the mitochondrial matrix of intact living cells (Fujikawa et al., 2016). Fluorescence ratios (405/488 nm) of enriched HSPCs expressing mito-roGFP2-Orp1 were measured by flow cytometry after 72 hours of in vitro cultivation (described in Enrichment of HSPCs and cultivation). As control, samples were treated with either 2mM diamide (Sigma-Aldrich) (fully oxidized) or 10mM DTT (Santa Cruz Biotechnology) (fully reduced).
Measurement of protein de novo synthesis by O-propargyl-puromycin (OP-Puro). Analysis of OP-Puro incorporation was performed using the Click-iT® PlusOPP Protein Synthesis Assay Kit (Thermo Fisher Scientific). To measure de novo protein synthesis, in vitro-treated HSPCs (5μM at-RA, 5μM 4-oxo-RA or DMSO) were incubated for 30 minutes in medium supplemented with OP-Puro (Thermo Fisher Scientific, 20 μM final concentration) at 37°C. After harvesting, cells were surface-stained for HSC markers LSK CD150+ CD48− CD34− cells at 4°C. The cells were then fixed and permeabilized for 10 minutes using the Cytofix/Cytoperm Buffer (Becton Dickinson and Company). The copper-catalyzed azide-alkyne cycloaddition (CuAAC) was performed according to the manufacturer’s instructions.
Mito Tracker Green staining on in vitro-treated cells
MitoTracker Green (MTG) localizes to the mitochondria of living cells independent of mitochondrial membrane potential. Surface staining was performed after harvesting of in vitro-treated, lineage-depleted cells (described in Enrichment of HSPCs and Treatment) and incubated for 30 minutes at 4°C. Cells were then incubated with MTG (60nM, Invitrogen) and verapamil (50μM, Sigma-Aldrich) at 37°C for 20 to 30 minutes according to de Almeida et al. (2017). Cells were subsequently resuspended in PBS supplemented with verapamil and subjected to flow cytometry-based analysis.
Seahorse assays of in vitro-treated cells
To measure oxygen consumption rates after in vitro treatments we performed mitochondrial stress tests using the Agilent Seahorse XF Analyzer (Agilent) according to the manufacturer’s instructions. Briefly, 50,000 LSK cells were sorted into 96-well ultra-low attachment plates and immediately treatedwith at-RA(5μM), 4-oxo-RA(5μM), or the respective amount of DMSO for 24 hours. Cells were then transferred to a poly-D-lysine (Sigma-Aldrich) coated 96-well seahorse plate and cultivated for 1 hour in a CO2-free incubator in XF Seahorse medium(XF DMEM Medium, 10mMglucose, 1mMpyruvate, 2mMglutamine) (all Agilent). The measurement protocol included injections of 10μM Oligomycin (Sigma-Aldrich), 15 μM FCCP (Sigma-Aldrich) and Rotenone 1μM (Sigma-Aldrich) + Antimycin A 10μM (Sigma-Aldrich) conducted in sequence.
Single cell division assays of in vitro-treated cells
Single HSCs (LSK CD150+CD48−CD34−) were sorted into 72-well Terasaki plates (Greiner Bio-One) containing Complete Stem Cell Medium (described in Enrichment of HSPCs and cultivation) and immediately treated with retinol (5μM final), at-RA (5μM final), 4-oxo-RA (5μM final), or the respective amount of DMSO. After 48 hours, each well was checked manually for the number of cell divisions; 1 cell indicated no division, 2 cells indicated 1 division, and more than 2 cells indicated more than 1 division.
Colony-forming-unit assays (CFUs) of in vitro-treated cells
1,000 FACS-sorted LSK cells from 8-to 12-week-old mice or 200 FACS-sorted HSCs (LSK CD150+CD48−CD34−) were cultured in 100μl Complete Stem Cell Medium (described in Enrichment of HSPCs and cultivation) in 96-well ultra-low attachment plates, and were immediately treated with retinol (10μM), at-RA (5μM), 4-oxo-RA (5μM), or the respective amount of DMSO. 72 hours after treatment, all cells of a single well were harvested and cultured in MethoCult M3434 (StemCell Technologies). 3 to 5 days after plating colony numbers were quantified. 10,000 cells of each replicate were used for second and third replating and cultured in MethoCult M3434 (StemCell Technologies). Three days after second and third re-platings respectively, colonies were quantified and replated.
Serial transplantation experiments of in vitro-treated cells
100HSCs (LSKCD150+CD48−CD34−) from 8-16-week-old mice were FACS sorted and cultured in 100μl Complete Stem Cell Medium (described in Enrichment of HSPCs and cultivation) in 384-well plates and immediately treated with at-RA(5μM), 4-oxo-RA(5μM), or the respective amount of DMSO. 72 hours after treatment, all cells from a single well were harvested and transplanted into fully irradiated (4.5 +5 Gray) B6 Ly5.1 mice (CD45.1), together with 105 supportive spleen cells (CD45.1/2). Contribution of CD45.2-donor cells was monitored in PB at 4, 8, 12, and 16 weeks post-transplantation in primary recipients. CD45.2 chimerism was analyzed by flow cytometry using the following monoclonal antibodies: anti-CD45.1 (A20)–PECy7 or FITC; anti-CD45.2 (104)–PB.
RNA-seq/RT-qPCR on in vitro-treated cells
1,000 HSCs (LSK CD150+CD48−) were sorted and cultured in 100μl Complete Stem Cell Medium (described in Enrichment of HSPCs and cultivation) in 96-well low attachment plates. These cells were subsequently harvested after 0 hours, 2 hours or 24 hours to check for gene expression as described above.
Rarb in vitro-knockdown
Knock-down (KD) of Rarb was achieved by retroviral transduction of BM cells enriched for HSPCs, with a shRNA targeting Rarb or renilla-luciferin-2-monooxygenase as a control. The retroviral miR-E system was used (Sauvergeau laboratory, Montreal, Canada). Briefly, shRNAs were designed using a genome-wide sensor-based shRNA ranked prediction list (Fellmann et al., 2013) and subsequently cloned into a plasmid. This plasmid contained a MSCV promoter including a GFP-IRES and an ampicillin resistance gene. Transformation of the plasmid was performed in competent DH5α E.coli where the ampicilin resistance gene enabled selection of successfully transformed plasmids. Single colonies were selected and expanded in lysogeny broth (LB) medium with ampicillin. In order to confirm the correct plasmid sequence, plasmid DNA was extracted with the Monarch Plasmid Miniprep Kit (NEB) according to the manufacturer’s instructions. Validation of the MSCV-shRNA-Rarb-GFP-IRES construct was then carried out by gel electrophoresis and Sanger sequencing. For virus production, we transfected PlatinumE cells with the MSCV-shRNA-Rarb plasmids using jetPRIME (Polyplus transfection). Virus-containing supernatant was collected and filtered after 48 hours and 72 hours post-transfection and stored at 4°C. HSPC enriched BM cells (see HSC/MPP isolation and flow cytometry-based sorting) were then transduced by spinoculation. We then transferred the cells to a 6-well plate with Complete StemPro Media (specified in Enrichment of HSPCs and cultivation) adding Polybrene (Sigma-Aldrich) and filtered virus particles (multiplicity of infection approximately 50). Transduced LSK GFP+ cells were collected 48 hours after transduction via flow cytometry-based sorting and subjected to further analysis.
For competitive transplantation assays we transduced CD45.1 HSPCs with the control shRNA (shRenilla) and CD45.2 with shRarb. After 48 hours 5,000 LSK GFP+ cells were sorted from each condition, pooled and transplanted into fully irradiated (4.5+5 Gray) CD45.1/2 mice together with 105 supportive WBM cells (CD45.1/2). Contribution of CD45.1 versus CD45.2-donor cells was monitored in PB at 4, 8, 12, and 16 weeks post-transplantation in primary recipients. Chimerism was analyzed by flow cytometry using the following monoclonal antibodies: anti-CD45.1 (A20)–PECy7 or FITC; anti-CD45.2 (104)–PB.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical analysis was performed using unpaired Student’s t test or two-way ANOVA with correction for multiple comparison (by either Tukey test or Sidak’s correction). All data are presented as mean ± SD. Significance levels were set at p* < 0.05, p** < 0.01, and p*** < 0.001. GraphPad Prism was used for statistical analyses.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| CD8a-BV650 | BioLegend | 100742; RRID:AB_2563056 |
| CD11b-BV650 | BioLegend | 101259; RRID:AB_2566568 |
| Gr1-BV650 | BioLegend | 108442; RRID:AB_2686974 |
| TER119-BV650 | BioLegend | 116235; RRID:AB_11204244 |
| B220-BV650 | BioLegend | 103241; RRID:AB_11204069 |
| CD4-BV650 | BioLegend | 563232; RRID:AB_2738083 |
| CD8a-PE/Cy7 | BioLegend | 100722; RRID:AB_312761 |
| CD11b-PE/Cy7 | BioLegend | 101216; RRID:AB_312799 |
| Gr1-PE/Cy7 | BioLegend | 108416; RRID:AB_313381 |
| TER119-PE/Cy7 | BioLegend | 116221; RRID:AB_2137789 |
| B220-PE/Cy7 | BioLegend | 103222; RRID:AB_313005 |
| CD4-PE/Cy7 | BioLegend | 100422; RRID:AB_2660860 |
| cKit-BV711 | BioLegend | 105835; RRID:AB_2565956 |
| cKit-BV421 | BioLegend | 105828; RRID:AB_11204256 |
| cKit-PE | BioLegend | 105808; RRID:AB_313217 |
| Sca1-APC/Cy7 | BioLegend | 108126; RRID:AB_10645327 |
| CD150-PE/Cy5 | BioLegend | 115912; RRID:AB_493598 |
| CD48-Pacific Blue | BioLegend | 103418; RRID:AB_756140 |
| Ki67-PE | Invitrogen | 12-5698-80; RRID:AB_11149672 |
| Gr1-APC | BioLegend | 108412; RRID:AB_313377 |
| CD11b-APC/Cy7 | BioLegend | 101226; RRID:AB_830642 |
| CD150-BV605 | BioLegend | 115927; RRID:AB_11204248 |
| CD48-BV421 | BioLegend | 103428; RRID:AB_2650894 |
| CD34-FITC | BD Biosciences | 553733; RRID:AB_395017 |
| CD34-AF700 | BD Biosciences | 560518; RRID:AB_1727471 |
| CD45.1-FITC | BioLegend | 110706; RRID:AB_313495 |
| CD45.2-Pacific Blue | BioLegend | 109820; RRID:AB_492872 |
| CD45.1-PE/Cy7 | BioLegend | 110730; RRID:AB_1134168 |
| CD127-PE | BioLegend | 121112; RRID:AB_493509 |
| CD16/32-APC | BioLegend | 101326; RRID:AB_1953273 |
| CD8a-FITC | BioLegend | 100706; RRID:AB_312745 |
| CD11b-FITC | BioLegend | 101206; RRID:AB_312789 |
| Gr1-FITC | BioLegend | 108406; RRID:AB_313371 |
| Ter119-FITC | BioLegend | 116206; RRID:AB_313707 |
| B220-FITC | BioLegend | 103206; RRID:AB_312991 |
| CD4-FITC | BioLegend | 100406; RRID:AB_312691 |
| CD48-PE/Cy7 | BioLegend | 103424; RRID:AB_2075049 |
| B220-AF700 | BioLegend | 103232; RRID:AB_493717 |
| Active Caspase3-PE | BD Biosciences | 561011; RRID:AB_2033931 |
| CD4-PE/Cy5 | BioLegend | 100410; RRID:AB_312695 |
| CD8a-PE/Cy5 | BioLegend | 100710; RRID:AB_312749 |
| H3K4me3 | Millipore | 07-473; RRID:AB_1977252 |
| H3K27me3 | Cell Signaling | C36B11; RRID:AB_11220433 |
| Chemicals, peptides, and recombinant proteins | ||
| BD Cytofix/Cytoperm Kit | BD | 554722 |
| Dynabeads Untouched Mouse CD4 Kit | Life Technologies | 11415D |
| FastSYBR Green Master Mix | Thermo Fisher Scientific | 4385610 |
| OneComp eBeads | eBioscience | 01-1111-41 |
| AMPure/RNAClean XP beads | Beckman Coulter | A63881 |
| Deoxynucleotide Solution, Mix | NEB | N0447S |
| ProtoScript II Reverse Transcriptase | NEB | M0368L |
| Murine Rnase Inhibitor | NEB | M0314L |
| Rnase H, recombinant | NEB | M0297L |
| DNA Polymerase I | NEB | M0209L |
| DAPI | Sigma-Aldrich | D9542 |
| All-trans-retinoic acid (at-RA) | Sigma-Aldrich | R2625 |
| 13-cis-4oxo-Retinoic acid (in vivo) | Sigma-Aldrich | 42073 |
| 4-Oxo-(9-cis,13-cis)-Retinoic acid (in vitro) | Santa Cruz Biotechnology | sc-477908 |
| Retinol | Sigma-Aldrich | R7632 |
| Rotenone | Sigma-Aldrich | R8875 |
| Antimycin A | Sigma-Aldrich | A8674 |
| Oligomycin | Sigma-Aldrich | O4876 |
| FCCP | Sigma-Aldrich | C2920 |
| Recombinant Human Flt3 | PeproTech | 300-19 |
| Recombinant Murine Stem Cell Factor | PeproTech | 250-03 |
| Recombinant Murine Thrombopoietin | PeproTech | 315-14 |
| ACK Lysing Buffer | Lonza | 10-548E |
| Poly-D-Lysine | Sigma-Aldrich | P6407 |
| StemPro-34 SFM (1X) | Life Technologies | 10639011 |
| Methocult Mouse M3434 | Stem Cell Technologies | M3434 |
| XF DMEM medium | Agilent | 103575-100 |
| XF 1.0 M Glucose Solution | Agilent | 103577-100 |
| XF 100 mM Pyruvate Solution | Agilent | 103578-100 |
| XF 200 mM Glutamine Solution | Agilent | 103579-100 |
| MitoTracker Green | Thermo Fisher Scientific | M7514 |
| Protein G Dynabeads | Invitrogen | 10003D |
| Q5® High-Fidelity 2X Master Mix | NEB | M0492S |
| NEBNext® Multiplex Oligos for Illumina® (96 Unique Dual Index Primer Pairs) | NEB | E6440 |
| Formaldehyde 16% | Pierce | 28906 |
| IDT unique dual indexing primers | Illumina | 20027213 |
| Digitonin | Promega | G9441 |
| BSA solution | Sigma-Aldrich | A8577 |
| Critical commercial assays | ||
| SuperScript VILO cDNA Synthesis Kit | Invitrogen | 11754050 |
| QIAGEN Plasmid Maxi Kit | QIAGEN | 12163 |
| PicoPure® RNA Isolation Kit | Thermo Fisher Scientific | KIT0204 |
| Click-iT® Plus OPP Protein Synthesis Assay Kit *Alexa Fluor® 594 picolyl azide | Life Technologies | C10457 |
| MEGAscript T7 Transcription Kit | Thermo FFisher Scientific | AM1334 |
| QIAGEN MinElute PCR clean up kit | QIAGEN | 28204 |
| Tagment DNA TDE1 Enzyme and Buffer Kits | Illumina | 20034197 |
| NEBNext Ultra II FS DNA library kit | NEB | E7805 |
| SMARTseq v4 | Takare Bio | 634889 |
| NEBNext Secondary Strand Synthesis Reaction Buffer | NEB | B6117S |
| Nextera XT DNA Sample Preparation Kits | Illumina | 1502354 |
| Deposited data | ||
| RNA Seq of control and Vitamin A-deficient HSCs and MPPs | This paper | ArrayExpress: E-MTAB-9729 |
| RNA Seq of control and Cyp26b1KO HSCs | This paper | ArrayExpress: E-MTAB-9745 |
| RNA Seq of 7-month-old control and Cyp26b1KO HSCs | This paper | ArrayExpress: E-MTAB-10659 |
| RNA Seq of control and Cyp26b1KO HSCs+MPP1 after 24h of in vitro culture and treatment | This paper | ArrayExpress: E-MTAB-9752 |
| RNA Seq of wildtype and Rarb KO HSCs after 24h of in vitro culture and treatment | This paper | ArrayExpress: E-MTAB-9749 |
| ATAC-seq of HSCs and MPPs | This paper | ArrayExpress: E-MTAB-9779 |
| ATAC-seq of HSCs in Vitamin A-free diet | This paper | ArrayExpress: E-MTAB-9780 |
| ATAC-seq of control and Cyp26b1KO HSCs | This paper | ArrayExpress: E-MTAB-9778 |
| ChIP-seq for H3K4me3 and H3K27me3 in HSCs and MPPs | This paper | ArrayExpress: E-MTAB-10661 |
| Experimental models: Organisms/strains | ||
| C57BL/6J (CD45.2) | MPI-IE | RRID:IMSR_JAX:002014 |
| B6Ly5.1 (CD45.1) | MPI-IE | RRID:IMSR_JAX:000664 |
| B6Ly5.1(CD45.1/2) | MPI-IE | N/A |
| Mx1Cre (B6. Cg-Tg(Mx1-cre)1Cgn/J) | Kühn et al., 1995 | RRID:IMSR_JAX:003556 |
| SclCre (B6. Tg(Tal1-cre)42-056Jrg) | Göthert et al., 2005 | MGI: 3579158 |
| Cyp26b1 fl/fl (B6-Cyp26b1tm2Hmd) | MacLean et al., 2007 | RRID:IMSR_RBRC04333 |
| RarbKO (Stock Rarbtm1Vg1/HsvJ) | The Jackson Laboratory | RRID:IMSR_JAX:022999 |
| MitoOrp (B6-Gt(ROSA)26Sor(mito-Orp-roGFP2) Tg(CMV-cre)1Cgn) | Fujikawa et al., 2016 | N/A |
| RargKO | Chapellier et al., 2002; Provided by N.B. Ghyselinck | N/A |
| Oligonucleotides | ||
| shRNA Rarb TGCTGTTGACAGTGAGCGCCCGCAGA AGATGTATTCTTCTGAATACTTCTGCG GATGCCTA CTGCCTCGGA | Sigma-Aldrich | N/A |
| shRNA Renilla AAGGTATATTGCTGTTGACAGTGAGC GCAGGAATTATAATGCTTATCTATAGTGAAGCCACA GATGTATAGATAAGCATTATAATTCCTATGCCTACTG CCTCGGACTTCAAGGGGCTA | Sigma-Aldrich | N/A |
| Software and algorithms | ||
| FACSDiva | BD | RRID:SCR_001456 |
| FlowJo10 | FlowJo LLC | RRID:SCR_008520 |
| Prism8 | Graphpad Software | RRID:SCR_002798 |
| Integrative Genomics Viewer (IGV) | N/A | RRID:SCR_011793 |
| DAVID v6.8 | Huang et al., 2009 | https://david.ncifcrf.gov |
| snakePipes (2.5.1) | Bhardwaj et al., 2019 | https://github.com/maxplanck-ie/snakepipes |
| deepTools (3.4.0) | Ramírez et al., 2014 | https://github.com/deeptools/deepTools/ |
| STAR v2.7.4a | Dobin et al., 2013 | https://github.com/alexdobin/STAR |
| Bowtie2 (2.4.4) | Langmead and Salzberg, 2012 | https://github.com/BenLangmead/bowtie2 |
| MACS2 (2.2.7.1) | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| bedtools (2.30.0) | Quinlan and Hall, 2010 | https://github.com/arq5x/bedtools2 |
| pyDNase (0.3.0) | Piper et al., 2013 | https://github.com/jpiper/pyDNase |
| Subread (2.0.3) | Liao et al., 2014 | https://sourceforge.net/projects/subread/ |
| GSEA_4.1.0 | Subramanian et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
Highlights.
Metabolic and epigenetic hubs that regulate stemness programs
Lack of dietary vitamin A leads to loss of the metabolic HSC identity
Non-classical retinoid signaling axis maintains HSC function: Cyp26b1–4-oxo-RA-Rarb
Rarb is required to establish the critical dosage of stemness genes
ACKNOWLEDGMENTS
We thank the MPI-IE Core Facilities (Flow Cytometry, Deep Sequencing, Laboratory Animal) for assistance and excellent work. This work was supported by the Max Planck Society, the ERC-Stg-2017 (VitASTEM, 759206), the Behrens-Weise-Foundation, the German Research Foundation (DFG) under the German Excellence Strategy (CIBSS-EXC-2189, project ID 390939984), SFB1425 (Project #422681845), SFB992 (Project #192904750; B07), and the European Union’s Horizon 2020 Research and Innovation Programme under the Marie Skłodowska-Curie Actions Grant (agreement 813091). We thank S. Renders and M. Derecka for carefully reading the manuscript.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.stem.2021.10.002.
REFERENCES
- Adriaens ME, Prickaerts P, Chan-Seng-Yue M, van den Beucken T, Dahlmans VEH, Eijssen LM, Beck T, Wouters BG, Voncken JW, and Evelo CTA (2016). Quantitative analysis of ChIP-seq data uncovers dynamic and sustained H3K4me3 and H3K27me3 modulation in cancer cells under hypoxia. Epigenetics Chromatin 9, 48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agathocleous M, Meacham CE, Burgess RJ, Piskounova E, Zhao Z, Crane GM, Cowin BL, Bruner E, Murphy MM, Chen W, et al. (2017). Ascorbate regulates haematopoietic stem cell function and leukaemogenesis. Nature 549, 476–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akhtar J, More P, Albrecht S, Marini F, Kaiser W, Kulkarni A, Wojnowski L, Fontaine JF, Andrade-Navarro MA, Silies M, and Berger C (2019). TAF-ChIP: an ultra-low input approach for genome-wide chromatin immunoprecipitation assay. Life Sci. Alliance 2, e201900318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ardah MT, Parween S, Varghese DS, Emerald BS, and Ansari SA (2018). Saturated fatty acid alters embryonic cortical neurogenesis through modulation of gene expression in neural stem cells. J. Nutr. Biochem 62, 230–246. [DOI] [PubMed] [Google Scholar]
- Baron JM, Heise R, Blaner WS, Neis M, Joussen S, Dreuw A, Marquardt Y, Saurat JH, Merk HF, Bickers DR, and Jugert FK (2005). Retinoic acid and its 4-oxo metabolites are functionally active in human skin cells in vitro. J. Invest. Dermatol 125, 143–153. [DOI] [PubMed] [Google Scholar]
- Bernardo AR, Cosgaya JM, Aranda A, and Jiménez-Lara AM (2017). Pro-apoptotic signaling induced by Retinoic acid and dsRNA is under the control of Interferon Regulatory Factor-3 in breast cancer cells. Apoptosis 22, 920–932. [DOI] [PubMed] [Google Scholar]
- Bhardwaj V, Heyne S, Sikora K, Rabbani L, Rauer M, Kilpert F, Richter AS, Ryan DP, and Manke T (2019). snakePipes: facilitating flexible, scalable and integrative epigenomic analysis. Bioinformatics 35, 4757–4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bowles J, Knight D, Smith C, Wilhelm D, Richman J, Mamiya S, Yashiro K, Chawengsaksophak K, Wilson MJ, Rossant J, et al. (2006). Retinoid signaling determines germ cell fate in mice. Science 312, 596–600. [DOI] [PubMed] [Google Scholar]
- Bräutigam L, Zhang J, Dreij K, Spahiu L, Holmgren A, Abe H, Tew KD, Townsend DM, Kelner MJ, Morgenstern R, and Johansson K (2018). MGST1, a GSH transferase/peroxidase essential for development and hematopoietic stem cell differentiation. Redox Biol. 17, 171–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bushue N, and Wan Y-JY (2010). Retinoid pathway and cancer therapeutics. Adv. Drug Deliv. Rev 62, 1285–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Klimmeck D, Hansson J, Lipka DB, Reyes A, Wang Q, Weichenhan D, Lier A, von Paleske L, Renders S, et al. (2014). Identification of regulatory networks in HSCs and their immediate progeny via integrated proteome, transcriptome, and DNA methylome analysis. Cell Stem Cell 15, 507–522. [DOI] [PubMed] [Google Scholar]
- Cabezas-Wallscheid N, Buettner F, Sommerkamp P, Klimmeck D, Ladel L, Thalheimer FB, Pastor-Flores D, Roma LP, Renders S, Zeisberger P, et al. (2017). Vitamin A-Retinoic Acid Signaling Regulates Hematopoietic Stem Cell Dormancy. Cell 169, 807–823.e19. [DOI] [PubMed] [Google Scholar]
- Cañete A, Cano E, Muñoz-Chápuli R, and Carmona R (2017). Role of Vitamin A/Retinoic Acid in Regulation of Embryonic and Adult Hematopoiesis. Nutrients 9, 159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cauchy P, Maqbool MA, Zacarias-Cabeza J, Vanhille L, Koch F, Fenouil R, Gut M, Gut I, Santana MA, Griffon A, et al. (2016). Dynamic recruitment of Ets1 to both nucleosome-occupied and -depleted enhancer regions mediates a transcriptional program switch during early T-cell differentiation. Nucleic Acids Res. 44, 3567–3585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandel NS, Jasper H, Ho TT, and Passegué E (2016). Metabolic regulation of stem cell function in tissue homeostasis and organismal ageing. Nat. Cell Biol 18, 823–832. [DOI] [PubMed] [Google Scholar]
- Chapellier B, Mark M, Bastien J, Dierich A, LeMeur M, Chambon P, and Ghyselinck NB (2002). A conditional floxed (loxP-flanked) allele for the retinoic acid receptor beta (RARbeta) gene. Genesis 32, 91–94. [DOI] [PubMed] [Google Scholar]
- Chen SJ, Zhu YJ, Tong JH, Dong S, Huang W, Chen Y, Xiang WM, Zhang L, Li XS, Qian GQ, et al. (1991). Rearrangements in the second intron of the RARA gene are present in a large majority of patients with acute promyelocytic leukemia and are used as molecular marker for retinoic acidinduced leukemic cell differentiation. Blood 78, 2696–2701. [PubMed] [Google Scholar]
- Chokeshaiusaha K, Puthier D, Nguyen C, and Sananmuang T (2018). A demonstration of the H3 trimethylation ChIP-seq analysis of galline follicular mesenchymal cells and male germ cells. Asian-Australas. J. Anim. Sci 31, 791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cimmino L, Dolgalev I, Wang Y, Yoshimi A, Martin GH, Wang J, Ng V, Xia B, Witkowski MT, Mitchell-Flack M, et al. (2017). Restoration of TET2 Function Blocks Aberrant Self-Renewal and Leukemia Progression. Cell 170, 1079–1095.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clémot M, Sênos Demarco R, and Jones DL (2020). Lipid Mediated Regulation of Adult Stem Cell Behavior. Front. Cell Dev. Biol 8, 115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conserva MR,Anelli L, Zagaria A, Specchia G, and Albano F (2019). The Pleiotropic Role of Retinoic Acid/Retinoic Acid Receptors Signaling: From Vitamin A Metabolism to Gene Rearrangements in Acute Promyelocytic Leukemia. Int. J. Mol. Sci 20, 2921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corces MR, Trevino AE, Hamilton EG, Greenside PG, Sinnott-Armstrong NA, Vesuna S, Satpathy AT, Rubin AJ, Montine KS, Wu B, et al. (2017). An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nat. Methods 14, 959–962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coyle KM, Maxwell S, Thomas ML, and Marcato P (2017). Profiling of the transcriptional response to all-trans retinoic acid in breast cancer cells reveals RARE-independent mechanisms of gene expression. Sci. Rep 7, 16684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlin JS, Hamey FK, Pijuan-Sala B, Shepherd M, Lau WWY, Nestorowa S, Weinreb C, Wolock S, Hannah R, Diamanti E, et al. (2018). A single-cell hematopoietic landscape resolves 8 lineage trajectories and defects in Kit mutant mice. Blood 131, e1–e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson MI, and Xia Z (2012). The retinoid X receptors and their ligands. Biochim. Biophys. Acta 1821, 21–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Almeida MJ, Luchsinger LL, Corrigan DJ, Williams LJ, and Snoeck HW (2017). Dye-Independent Methods Reveal Elevated Mitochondrial Mass in Hematopoietic Stem Cells. Cell Stem Cell 21>, 725–729 e724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeVilbiss AW, Zhao Z, Martin-Sandoval MS, Ubellacker JM, Tasdogan A, Agathocleous M, Mathews TP, and Morrison SJ (2021). Metabolomic profiling of rare cell populations isolated by flow cytometry from tissues. eLife 10, e61980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimova DK, and Dyson NJ (2005). The E2F transcriptional network: old acquaintances with new faces. Oncogene 24, 2810–2826. [DOI] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2013). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faria TN, Mendelsohn C, Chambon P, and Gudas LJ (1999). The targeted disruption of both alleles of RARbeta(2) in F9 cells results in the loss of retinoic acid-associated growth arrest. J. Biol. Chem 274, 26783–26788. [DOI] [PubMed] [Google Scholar]
- Fellmann C, Hoffmann T, Sridhar V, Hopfgartner B, Muhar M, Roth M, Lai DY, Barbosa IA, Kwon JS, Guan Y, et al. (2013). An optimized microRNA backbone for effective single-copy RNAi. Cell Rep. 5, 1704–1713. [DOI] [PubMed] [Google Scholar]
- Ferreira R, Napoli J, Enver T, Bernardino L, and Ferreira L (2020). Advances and challenges in retinoid delivery systems in regenerative and therapeutic medicine. Nat. Commun 11, 4265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findley AS, Richards AL, Petrini C, Alazizi A, Doman E, Shanku AG, Davis GO, Hauff N, Sorokin Y, Wen X, et al. (2019). Interpreting Coronary Artery Disease Risk Through Gene-Environment Interactions in Gene Regulation. Genetics 213, 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujikawa Y, Roma LP, Sobotta MC, Rose AJ, Diaz MB, Locatelli G, Breckwoldt MO, Misgeld T, Kerschensteiner M, Herzig S, et al. (2016). Mouse redox histology using genetically encoded probes. Sci. Signal 9, rs1. [DOI] [PubMed] [Google Scholar]
- Gaemers IC, van Pelt AM, van der Saag PT, and de Rooij DG (1996). All-trans-4-oxo-retinoic acid: a potent inducer of in vivo proliferation of growth-arrested A spermatogonia in the vitamin A-deficient mouse testis. Endocrinology 137, 479–485. [DOI] [PubMed] [Google Scholar]
- Gallipoli P, Giotopoulos G, Tzelepis K, Costa ASH, Vohra S, Medina-Perez P, Basheer F, Marando L, Di Lisio L, Dias JML, et al. (2018). Glutaminolysis is a metabolic dependency in FLT3ITD acute myeloid leukemia unmasked by FLT3 tyrosine kinase inhibition. Blood 131, 1639–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiaur G, Yegnasubramanian S, Perkins B, Gucwa JL, Gerber JM, and Jones RJ (2013). Regulation of human hematopoietic stem cell self-renewal by the microenvironment’s control of retinoic acid signaling. Proc. Natl. Acad. Sci. USA 110, 16121–16126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghyselinck NB, and Duester G (2019). Retinoic acid signaling pathways. Development 146, dev167502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göthert JR, Gustin SE, Hall MA, Green AR, Göttgens B, Izon DJ, and Begley CG (2005). In vivo fate-tracing studies using the Scl stem cell enhancer: embryonic hematopoietic stem cells significantly contribute to adult hematopoiesis. Blood 105, 2724–2732. [DOI] [PubMed] [Google Scholar]
- Grimwade D, Mistry AR, Solomon E, and Guidez F (2010). Acute promyelocytic leukemia: a paradigm for differentiation therapy. Cancer Treat. Res 145, 219–235. [DOI] [PubMed] [Google Scholar]
- Hakimi AA, Reznik E, Lee CH, Creighton CJ, Brannon AR, Luna A, Aksoy BA, Liu EM, Shen R, Lee W, et al. (2016). An Integrated Metabolic Atlas of Clear Cell Renal Cell Carcinoma. Cancer Cell 29, 104–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helin K(1998). Regulation of cell proliferation by the E2F transcription factors. Curr. Opin. Genet. Dev 8, 28–35. [DOI] [PubMed] [Google Scholar]
- Hermetet F, Buffière A, Aznague A, Pais de Barros JP, Bastie JN, Delva L, and Quéré R (2019). High-fat diet disturbs lipid raft/TGF-β signaling-mediated maintenance of hematopoietic stem cells in mouse bone marrow. Nat. Commun 10, 523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez RE, Putzke AP, Myers JP, Margaretha L, and Moens CB (2007). Cyp26 enzymes generate the retinoic acid response pattern necessary for hindbrain development. Development 134, 177–187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez D, Palau L, Norsworthy K, Anders NM, Alonso S, Su M, Petkovich M, Chandraratna R, Rudek MA, Smith BD, et al. (2020). Overcoming microenvironment-mediated protection from ATRA using CYP26-resistant retinoids. Leukemia 34, 3077–3081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrmann C, Van de Sande B, Potier D, and Aerts S (2012). i-cisTarget: an integrative genomics method for the prediction of regulatory features and cis-regulatory modules. Nucleic Acids Res. 40, e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hinge A, He J, Bartram J, Javier J, Xu J, Fjellman E, Sesaki H, Li T, Yu J, Wunderlich M, et al. (2020). Asymmetrically Segregated Mitochondria Provide Cellular Memory of Hematopoietic Stem Cell Replicative History and Drive HSC Attrition. Cell Stem Cell 26, 420–430.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang W, Sherman BT, and Lempicki RA (2009). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Idres N, Marill J, Flexor MA, and Chabot GG (2002). Activation of retinoic acid receptor-dependent transcription by all-trans-retinoic acid metabolites and isomers. J. Biol. Chem 277, 31491–31498. [DOI] [PubMed] [Google Scholar]
- Ikuta K, and Weissman IL (1992). Evidence that hematopoietic stem cells express mouse c-kit but do not depend on steel factor for their generation. Proc. Natl. Acad. Sci. USA 89, 1502–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imperato MR, Cauchy P, Obier N, and Bonifer C (2015). The RUNX1-PU.1 axis in the control of hematopoiesis. Int. J. Hematol 101, 319–329. [DOI] [PubMed] [Google Scholar]
- Imrichová H, Hulselmans G, Atak ZK, Potier D, and Aerts S (2015). i-cisTarget 2015 update: generalized cis-regulatory enrichment analysis in human, mouse and fly. Nucleic Acids Res. 43 (W1), W57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, and Suda T (2014). Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol 15, 243–256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito K, Bonora M, and Ito K (2019). Metabolism as master of hematopoietic stem cell fate. Int. J. Hematol 109, 18–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jassal B, Matthews L, Viteri G, Gong C, Lorente P, Fabregat A, Sidiropoulos K, Cook J, Gillespie M, Haw R, et al. (2020). The reactome pathway knowledgebase. Nucleic Acids Res. 48 (D1), D498–D503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaitzidis D, Sykes SM, Wang Z, Punt N, Tang Y, Ragu C, Sinha AU, Lane SW, Souza AL, Clish CB, et al. (2012). mTOR complex 1 plays critical roles in hematopoiesis and Pten-loss-evoked leukemogenesis. Cell Stem Cell 11, 429–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath-Loeb AS, Balakrishna S, Whittington D, Shen JC, Emond MJ, Okabe T, Masutani C, Hanaoka F, Nishimura S, and Loeb LA (2014). Sphingosine, a modulator of human translesion DNA polymerase activity. J. Biol. Chem 289, 21663–21672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karigane D, Kobayashi H, Morikawa T, Ootomo Y, Sakai M, Nagamatsu G, Kubota Y, Goda N, Matsumoto M, Nishimura EK, et al. (2016). p38α Activates Purine Metabolism to Initiate Hematopoietic Stem/Progenitor Cell Cycling in Response to Stress. Cell Stem Cell 19, 192–204. [DOI] [PubMed] [Google Scholar]
- Kim CS, Ding X, Allmeroth K, Biggs LC, Kolenc OI, L’Hoest N, Chacon-Martinez CA, Edlich-Muth C, Giavalisco P, Quinn KP, et al. (2020). Glutamine Metabolism Controls Stem Cell Fate Reversibility and Long-Term Maintenance in the Hair Follicle. Cell Metab. 32, 629–642 e628. [DOI] [PubMed] [Google Scholar]
- Kolde R (2019). Pheatmap: pretty heatmaps, R Package Version 1.0.12. https://rdrr.io/cran/pheatmap/. [Google Scholar]
- Kühn R, Schwenk F, Aguet M, and Rajewsky K (1995). Inducible gene targeting in mice. Science 269, 1427–1429. [DOI] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang R, Arif T, Kalmykova S, Kasianov A, Lin M, Menon V, Qiu J, Bernitz JM, Moore K, Lin F, et al. (2020). Restraining Lysosomal Activity Preserves Hematopoietic Stem Cell Quiescence and Potency. Cell Stem Cell 26, 359–376.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2014). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Anders S, Kim V, and Huber W (2015). RNA-Seq workflow: gene-level exploratory analysis and differential expression. F1000Res. 4, 1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, Chen Y, Wang H, Bai Y, Zhao J, Zhang X, Liang L, Chen Y, Ye C, Li Y, et al. (2019). Integrated Lipidomics and Transcriptomics Characterization upon Aging-Related Changes of Lipid Species and Pathways in Human Bone Marrow Mesenchymal Stem Cells. J. Proteome Res 18, 2065–2077. [DOI] [PubMed] [Google Scholar]
- Ludin A, Gur-Cohen S, Golan K, Kaufmann KB, Itkin T, Medaglia C, Lu XJ, Ledergor G, Kollet O, and Lapidot T (2014). Reactive oxygen species regulate hematopoietic stem cell self-renewal, migration and development, as well as their bone marrow microenvironment. Antioxid. Redox Signal 21, 1605–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo XM, and Ross AC (2006). Retinoic acid exerts dual regulatory actions on the expression and nuclear localization of interferon regulatory factor-1. Exp. Biol. Med. (Maywood) 231, 619–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacLean G, Li H, Metzger D, Chambon P, and Petkovich M (2007). Apoptotic extinction of germ cells in testes of Cyp26b1 knockout mice. Endocrinology 148, 4560–4567. [DOI] [PubMed] [Google Scholar]
- Margueron R, and Reinberg D (2011). The Polycomb complex PRC2 and its mark in life. Nature 469, 343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKenna NJ (2012). EMBO Retinoids 2011: Mechanisms, biology and pathology of signaling by retinoic acid and retinoic acid receptors. Nucl. Recept. Signal 10, e003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaille JJ, Blanchet S, Kanzler B, Garnier JM, and Dhouailly D (1994). Characterization of cDNAs encoding the chick retinoic acid receptor gamma 2 and preferential distribution of retinoic acid receptor gamma transcripts during chick skin development. Dev. Dyn 201, 334–343. [DOI] [PubMed] [Google Scholar]
- Michaille JJ, Kanzler B, Blanchet S, Garnier JM, and Dhouailly D (1995). Characterization of cDNAs encoding two chick retinoic acid receptor alpha isoforms and distribution of retinoic acid receptor alpha, beta and gamma transcripts during chick skin development. Int. J. Dev. Biol 39, 587–596 [PubMed] [Google Scholar]
- Murtagh F, and Legendre P (2014). Ward’s Hierarchical Clustering Method: Clustering Criterion and Agglomerative Algorithm. Journal of Classification 31, 274–295. [Google Scholar]
- Nakato R, and Sakata T (2021). Methods for ChIP-seq analysis: A practical workflow and advanced applications. Methods 187, 44–53. [DOI] [PubMed] [Google Scholar]
- Nelson CH, Buttrick BR, and Isoherranen N (2013). Therapeutic potential of the inhibition of the retinoic acid hydroxylases CYP26A1 and CYP26B1 by xenobiotics. Curr. Top. Med. Chem 13, 1402–1428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niederreither K, Abu-Abed S, Schuhbaur B, Petkovich M, Chambon P, and Dollé P (2002). Genetic evidence that oxidative derivatives of retinoic acid are not involved in retinoid signaling during mouse development. Nat. Genet 31, 84–88. [DOI] [PubMed] [Google Scholar]
- Nottingham WT, Jarratt A, Burgess M, Speck CL, Cheng JF, Prabhakar S, Rubin EM, Li PS, Sloane-Stanley J, Kong-A-San J, and de Bruijn MF (2007). Runx1-mediated hematopoietic stem-cell emergence is controlled by a Gata/Ets/SCL-regulated enhancer. Blood 110, 4188–4197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novello F, Muchmore JH, Bonora B, Capitani S, and Manzoli FA (1975). Effect of phospholipids on the activity of DNA polymerase I from E. coli. Ital. J. Biochem 24, 325–334. [PubMed] [Google Scholar]
- Okada S, Nakauchi H, Nagayoshi K, Nishikawa S, Miura Y, and Suda T (1992). In vivo and in vitro stem cell function of c-kit-and Sca-1-positive murine hematopoietic cells. Blood 80, 3044–3050. [PubMed] [Google Scholar]
- Ono K, Keller J, López Ramírez O, González Garrido A, Zobeiri OA, Chang HHV, Vijayakumar S, Ayiotis A, Duester G, Della Santina CC, et al. (2020). Retinoic acid degradation shapes zonal development of vestibular organs and sensitivity to transient linear accelerations. Nat. Commun 11, 63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orkin SH, and Zon LI (2008). Hematopoiesis: an evolving paradigm for stem cell biology. Cell 132, 631–644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pijnappel WW, Hendriks HF, Folkers GE, van den Brink CE, Dekker EJ, Edelenbosch C, van der Saag PT, and Durston AJ (1993). The retinoid ligand 4-oxo-retinoic acid is a highly active modulator of positional specification. Nature 366, 340–344. [DOI] [PubMed] [Google Scholar]
- Piper J, Elze MC, Cauchy P, Cockerill PN, Bonifer C, and Ott S (2013). Wellington: a novel method for the accurate identification of digital genomic footprints from DNase-seq data. Nucleic Acids Res. 41, e201. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purton LE, Bernstein ID, and Collins SJ (1999). All-trans retinoic acid delays the differentiation of primitive hematopoietic precursors (lin-c-kit+Sca-1(+)) while enhancing the terminal maturation of committed granulocyte/monocyte progenitors. Blood 94, 483–495. [PubMed] [Google Scholar]
- Purton LE, Bernstein ID, and Collins SJ (2000). All-trans retinoic acid enhances the long-term repopulating activity of cultured hematopoietic stem cells. Blood 95, 470–477. [PubMed] [Google Scholar]
- Purton LE, Dworkin S, Olsen GH, Walkley CR, Fabb SA, Collins SJ, and Chambon P (2006). RARgamma is critical for maintaining a balance between hematopoietic stem cell self-renewal and differentiation. J. Exp. Med 203,1283–1293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qian P, De Kumar B, He XC, Nolte C, Gogol M, Ahn Y, Chen S, Li Z, Xu H, Perry JM, et al. (2018). Retinoid-Sensitive Epigenetic Regulation of the Hoxb Cluster Maintains Normal Hematopoiesis and Inhibits Leukemogenesis. Cell Stem Cell 22, 740–754.e7. [DOI] [PubMed] [Google Scholar]
- Quinlan AR, and Hall IM (2010). BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Dündar F, Diehl S, Grüning BA, and Manke T (2014). deepTools: a flexible platform for exploring deep-sequencing data. Nucleic Acids Res. 42, W187–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44 (W1), W160–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Renders S, Svendsen AF, Panten J, Rama N, Maryanovich M, Sommerkamp P, Ladel L, Redavid AR, Gibert B, Lazare S, et al. (2021). Niche derived netrin-1 regulates hematopoietic stem cell dormancy via its receptor neogenin-1. Nat. Commun 12, 608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowe A, Richman JM, and Brickell PM (1992). Development of the spatial pattern of retinoic acid receptor-beta transcripts in embryonic chick facial primordia. Development 114, 805–813. [DOI] [PubMed] [Google Scholar]
- Sakai Y, Meno C, Fujii H, Nishino J, Shiratori H, Saijoh Y, Rossant J, and Hamada H (2001). The retinoic acid-inactivating enzyme CYP26 is essential for establishing an uneven distribution of retinoic acid along the anterioposterior axis within the mouse embryo. Genes Dev. 15, 213–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satre MA, Penner JD, and Kochhar DM (1989). Pharmacokinetic assessment of teratologically effective concentrations of an endogenous retinoic acid metabolite. Teratology 39, 341–348. [DOI] [PubMed] [Google Scholar]
- Serefidou M, Venkatasubramani AV, and Imhof A (2019). The Impact of One Carbon Metabolism on Histone Methylation. Front. Genet 10, 764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sergushichev AA (2016). An algorithm for fast preranked gene set enrichment analysis using cumulative statistic calculation. bioRxiv. 10.1101/060012. [DOI] [Google Scholar]
- Shealy YF (1989). Synthesis and evaluation of some new retinoids for cancer chemoprevention. Prev. Med 18, 624–645. [DOI] [PubMed] [Google Scholar]
- Signer RA, Magee JA, Salic A, and Morrison SJ (2014). Haematopoietic stem cells require a highly regulated protein synthesis rate. Nature 509, 49–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simsek T, Kocabas F, Zheng J, Deberardinis RJ, Mahmoud AI, Olson EN, Schneider JW, Zhang CC, and Sadek HA (2010). The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 7, 380–390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snyder JM, Zhong G, Hogarth C, Huang W, Topping T, LaFrance J, Palau L, Czuba LC, Griswold M, Ghiaur G, and Isoherranen N (2020). Knockout of Cyp26a1 and Cyp26b1 during postnatal life causes reduced lifespan, dermatitis, splenomegaly, and systemic inflammation in mice. FASEB J. 34, 15788–15804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sommerkamp P, Altamura S, Renders S, Narr A, Ladel L, Zeisberger P, Eiben PL, Fawaz M, Rieger MA, Cabezas-Wallscheid N, et al. (2020). Differential Alternative Polyadenylation Landscapes Mediate Hematopoietic Stem Cell Activation and Regulate Glutamine Metabolism. Cell Stem Cell 26, 722–738 e727. [DOI] [PubMed] [Google Scholar]
- Sonneveld E, van den Brink CE, van der Leede BJ, Maden M, and van der Saag PT (1999). Embryonal carcinoma cell lines stably transfected with mRARbeta2-lacZ: sensitive system for measuring levels of active retinoids. Exp. Cell Res 250, 284–297. [DOI] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suda T, Takubo K, and Semenza GL (2011). Metabolic regulation of hematopoietic stem cells in the hypoxic niche. Cell Stem Cell 9, 298–310. [DOI] [PubMed] [Google Scholar]
- Takubo K (2014). [Homeostatic regulation of hematopoiesis by the hematopoietic stem cell niche]. Seikagaku 86, 755–765. [PubMed] [Google Scholar]
- Takubo K, Nagamatsu G, Kobayashi CI, Nakamura-Ishizu A, Kobayashi H, Ikeda E, Goda N, Rahimi Y, Johnson RS, Soga T, et al. (2013). Regulation of glycolysis by Pdk functions as a metabolic checkpoint for cell cycle quiescence in hematopoietic stem cells. Cell Stem Cell 12, 49–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang XH, and Gudas LJ (2011). Retinoids, retinoic acid receptors, and cancer. Annu. Rev. Pathol 6, 345–364. [DOI] [PubMed] [Google Scholar]
- Teodorescu P, Pasca S, Dima D, Tomuleasa C, and Ghiaur G (2020). Targeting the Microenvironment in MDS: The Final Frontier. Front. Pharmacol 11, 1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Till JE, and McCULLOCH EA (1961). A direct measurement of the radiation sensitivity of normal mouse bone marrow cells. Radiat. Res 14, 213–222. [PubMed] [Google Scholar]
- Ulrey CL, Liu L, Andrews LG, and Tollefsbol TO (2005). The impact of metabolism on DNA methylation. Hum. Mol. Genet 14, R139–R147 [DOI] [PubMed] [Google Scholar]
- van der Leede BM, van den Brink CE, Pijnappel WW, Sonneveld E, van der Saag PT, and van der Burg B (1997). Autoinduction of retinoic acid metabolism to polar derivatives with decreased biological activity in retinoic acid-sensitive, but not in retinoic acid-resistant human breast cancer cells. J. Biol. Chem 272, 17921–17928. [DOI] [PubMed] [Google Scholar]
- Vannini N, Girotra M, Naveiras O, Nikitin G, Campos V, Giger S, Roch A, Auwerx J, and Lutolf MP (2016). Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun 7, 13125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vannini N, Campos V, Girotra M, Trachsel V, Rojas-Sutterlin S, Tratwal J, Ragusa S, Stefanidis E, Ryu D, Rainer PY, et al. (2019). The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 24, 405–418.e7. [DOI] [PubMed] [Google Scholar]
- Wei G, Wei L, Zhu J, Zang C, Hu-Li J, Yao Z, Cui K, Kanno Y, Roh TY, Watford WT, et al. (2009). Global mapping of H3K4me3 and H3K27me3 reveals specificity and plasticity in lineage fate determination of differentiating CD4+ T cells. Immunity 30, 155–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weishaupt H, Sigvardsson M, and Attema JL (2010). Epigenetic chromatin states uniquely define the developmental plasticity of murine hematopoietic stem cells. Blood 115, 247–256. [DOI] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer). [Google Scholar]
- Willcockson MA, Taylor SJ, Ghosh S, Healton SE, Wheat JC, Wilson TJ, Steidl U, and Skoultchi AI (2019). Runx1 promotes murine erythroid progenitor proliferation and inhibits differentiation by preventing Pu.1 downregulation. Proc. Natl. Acad. Sci. USA 116, 17841–17847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson A, Laurenti E, Oser G, van der Wath RC, Blanco-Bose W, Jaworski M, Offner S, Dunant CF, Eshkind L, Bockamp E, et al. (2008). Hematopoietic stem cells reversibly switch from dormancy to self-renewal during homeostasis and repair. Cell 135, 1118–1129. [DOI] [PubMed] [Google Scholar]
- Wilson NK, Kent DG, Buettner F, Shehata M, Macaulay IC, Calero-Nieto FJ, Sánchez Castillo M, Oedekoven CA, Diamanti E, Schulte R, et al. (2015). Combined Single-Cell Functional and Gene Expression Analysis Resolves Heterogeneity within Stem Cell Populations. Cell Stem Cell 16, 712–724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HE, Lambert MH, Montana VG, Plunket KD, Moore LB, Collins JL, Oplinger JA, Kliewer SA, Gampe RT Jr., McKee DD, et al. (2001). Structural determinants of ligand binding selectivity between the peroxisome proliferator-activated receptors. Proc. Natl. Acad. Sci. USA 98, 13919–13924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yashiro K, Zhao X, Uehara M, Yamashita K, Nishijima M, Nishino J, Saijoh Y, Sakai Y, and Hamada H (2004). Regulation of retinoic acid distribution is required for proximodistal patterning and outgrowth of the developing mouse limb. Dev. Cell 6, 411–422. [DOI] [PubMed] [Google Scholar]
- Yu G (2020). enrichplot: Visualization of Functional Enrichment Result, R Package Version 1.8.1. https://bioconductor.org/packages/release/bioc/html/enrichplot.html. [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, and Liu XS (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng X, Yue S, Chen H, Weber B, Jia J, and Zheng Y (2015). Low-Cell-Number Epigenome Profiling Aids the Study of Lens Aging and Hematopoiesis. Cell Rep. 13, 1505–1518. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data were deposited in ArrayExpress (https://www.ebi.ac.uk/arrayexpress) and are available under the accession numbers [E-MTAB-9729, E-MTAB-9745, E-MTAB-10659, E-MTAB-9752, E-MTAB-9749, E-MTAB-9779, E-MTAB-9780, E-MTAB-9778, E-MTAB-10661]. Further information can be found in the STAR Methods section.