

SUMMARY
Glycans modify lipids and proteins to mediate inter- and intramolecular interactions across all domains of life. RNA is not thought to be a major target of glycosylation. Here, we challenge this view with evidence that mammals use RNA as a third scaffold for glycosylation. Using a battery of chemical and biochemical approaches, we found that conserved small noncoding RNAs bear sialylated glycans. These “glycoRNAs” were present in multiple cell types and mammalian species, in cultured cells, and in vivo. GlycoRNA assembly depends on canonical N-glycan biosynthetic machinery and results in structures enriched in sialic acid and fucose. Analysis of living cells revealed that the majority of glycoRNAs were present on the cell surface and can interact with anti-dsRNA antibodies and members of the Siglec receptor family. Collectively, these findings suggest the existence of a direct interface between RNA biology and glycobiology, and an expanded role for RNA in extracellular biology.
In brief
Identification of stable mammalian RNAs decorated with glycan structures opens up a new dimension for regulatory control of RNA localization and function by post-transcriptional modification.
Graphical Abstract
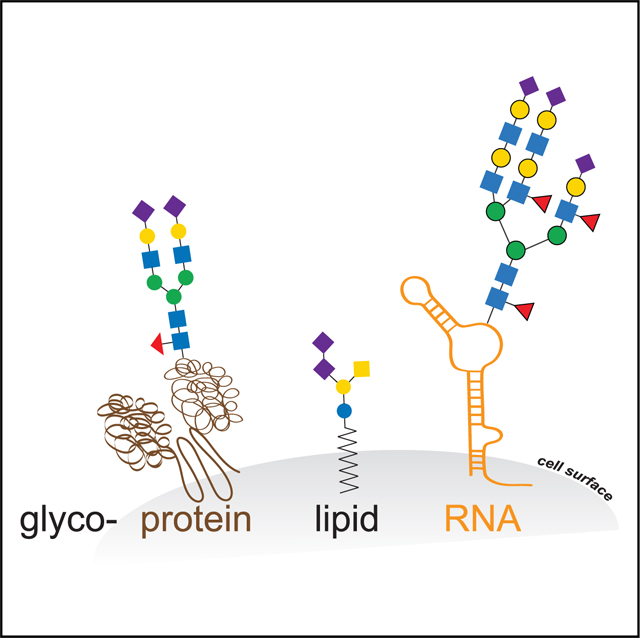
INTRODUCTION
Glycans regulate a myriad of essential cellular functions, especially in the context of cell surface events. For instance, complex glycans facilitate the folding and purposeful trafficking of proteins and lipids for secretion or membrane presentation. Thus, many fundamental processes such as embryogenesis, hostpathogen recognition, and tumor-immune interactions rely on glycosylation (Reily et al., 2019; Varki and Gagneux, 2015). Glycans are present in every cell studied to date across the kingdoms of life (Varki and Gagneux, 2015) and in mammals, are composed of roughly 10 monomeric carbohydrate units.
In a traditionally adjacent field of study, RNA represents another biopolymer that is central to all known life. Although the building blocks of RNA are canonically limited to four bases, post-transcriptional modifications (PTMs) can dramatically expand the chemical diversity of RNA, with >100 PTMs having been identified (Machnicka et al., 2013; Nachtergaele and He, 2017; Frye et al., 2018). It is therefore not surprising that the cellular role for RNA is more complex than that of a simple messenger. For instance, RNAs function as scaffolds, molecular decoys, enzymes, and network regulators across the nucleus and cytosol (Cech and Steitz, 2014; Sharp, 2009; Wang and Chang, 2011). With the exception of a few monosaccharide-based tRNA modifications (Kasai et al., 1976; Okada et al., 1977), there has been no evidence so far of a direct linkage between RNA and glycans in nature.
We previously developed methods for unbiased discovery of protein-associated glycans based on metabolic labeling and bioorthogonal chemistry (Hang et al., 2003; Saxon and Bertozzi, 2000; Woo et al., 2015). In this strategy, we metabolically label cells or animals with precursor sugars functionalized with a clickable azide group. Once incorporated into cellular glycans, the azidosugars enable bioorthogonal reaction with a biotin probe for enrichment, identification, and visualization. Despite the extensive use of these reporter sugars, their implementation appears to have exclusively assayed the protein and lipid compartments of cells. Using an azide-labeled precursor to sialic acid,peracetylated N-azidoacetylmannosamine (Ac4ManNAz), we found that azide reactivity was present on highly purified RNA preparations from labeled cells. Although there was no precedent for a connection between sialoglycans and RNA, either direct or indirect, the fact that RNA is so broadly post-transcriptionally modified in cells motivated us to pursue further investigation.
RESULTS
A glycan metabolic reporter is incorporated into cellular RNA
To explore the possible existence of RNA modified with sialoglycans (hereafter referred to as glycoRNA), we labeled HeLa cells with 100 μM Ac4ManNAz for up to 48 h and then used a rigorous protocol to chemically and enzymatically extract RNA with high purity: RNA is extracted with warm TRIzol (acid phenol and guanidine salts), then ethanol precipitated, desalted via silica columns, stripped of protein contamination via high concentration proteinase K digestion, and repurified over silica columns (Figures 1A and S1A). To visualize azide-labeled components, copper (Cu) free click chemistry (Agard et al., 2004) was used by adding RNA samples to dibenzocyclooctyne-biotin (DBCO-biotin) in denaturing conditions (50% formamide) at 55°C, subsequently separated by denaturing gel electrophoresis and analyzed by blotting (Figures 1B and S1B). In an Ac4ManNAz- and time-dependent manner, we observed biotinylated species in the very high (>10 kb) molecular weight (MW) region. It has recently been reported that high doses of azidosugars can produce non-enzymatic protein labeling (Qin et al., 2018); however, in vitro incubation of total RNA with up to 20 mM Ac4ManNAz did not produce the previously observed biotinylated species on RNAs in the high MW region (Figure S1C). Minor background in vitro labeling was apparent on the 28S rRNA, which can also be seen more variably in some Ac4ManNAz-labeled cellular RNA experiments (e.g., Figure 1B), but no such background labeling was observed in the putative glycoRNA species. Further, treatment of RNA from Ac4ManNAz-labeled HeLa cells with DNase did not affect the glycoRNA signal, whereas treatment with an RNase cocktail (A and T1) efficiently digested the total RNA as well as the biotinylated glycoRNA (Figure S1C and S1D). This effect required RNase enzymatic activity, because pre-blocking of the RNases with an inhibitor, SUPERaseIn, completely rescued the biotinylated glycoRNA (Figure 1C). Thus, cells treated with Ac4ManNAz incorporate the azide label into cellular RNA, which migrates on an agarose gel as a high MW species.
Figure 1. Ac4ManNAz, a glycan reporter, incorporates into mammalian cellular RNA.

(A) Schematic of RNA extraction protocol. Ac4ManNAz, peracetylated N-azidoacetylmannosamine; Prot.K, proteinase K; DBCO, dibenzocyclooctyne.
(B) RNA blotting of RNA from HeLa cells treated with 100 μM Ac4ManNAz for the indicated amount of time. After RNA purification, Ac4ManNAz was conjugated to DBCO-biotin, visualized with streptavidin-IR800 (Strep), and imaged on an infrared scanner. Before RNA transfer to the membrane, total RNA was stained and imaged with SYBR Gold (Sybr) to interrogate quality and loading. All subsequent blots were prepared in this manner, and Ac4ManNAz is always used at 100 μM. The regions where glycoRNAs are present (red text) and non-specific labeling (*) is noted.
(C) RNA Blot of Ac4ManNAz-labeled HeLa RNA treated in vitro with Turbo DNase or RNase cocktail (A/T1) +/− SUPERaseIn (RNase inhibitor).
(D) RNA Blot of murine RNA after in vivo Ac4ManNAz delivery via intraperitoneal injection on indicated days at 300 mg Ac4ManNAz/kg/day. RNA from the liver and spleen were analyzed. Mock (m) mice were injected with DMSO only. RNase treatment was performed on extracted RNA.
See also Figure S1.
Using the same metabolic labeling approach, we looked for the presence of glycoRNA in other cell types and in animals. Human embryonic stem cells (H9), a human myelogenous leukemia line (K562), a human lymphoblastoid cell line (GM12878), a mouse T cell acute lymphoblastic leukemia cell line (T-ALL 4188), and Chinese hamster ovary cells (CHO) all showed evidence of the presence of glycoRNA (Figures S1E and S1F). H9 and 4188 cells showed significantly more labeling with Ac4-ManNAz per mass of total RNA than other cell types (Figures S1E and S1F). Next, we assessed if this labeling could occur in vivo. To this end, we performed intraperitoneal injections of Ac4ManNAz into mice for 2, 4, or 6 days (Chang et al., 2009). In the liver and spleen, the organs that yielded enough total RNA for analysis, we observed dose-dependent and RNase-sensitive Ac4ManNAz labeling of RNAs in the same MW region as glycoRNAs from cultured cells (Figure 1D). These data suggest that glycoRNA is not an artifact of tissue culture and occurs broadly across multiple cell and tissue types and at various abundances.
glycoRNAs are small noncoding RNAs
Across all cell types and organs tested, glycoRNA was found to migrate very slowly by denaturing agarose gel electrophoresis (Figure 1). We hypothesized that if glycoRNAs are indeed large RNAs, they would likely be polyadenylated (poly-A). However, we were consistently unable to purify glycoRNA from extracted RNA via poly-A enrichment (Figure 2A). This was not due to cleavage or degradation of the glycoRNA during the poly-A enrichment procedure (Figure S2A). As an alternative enrichment strategy, we used a commercial fractionation method that leverages length-dependent RNA precipitation and binding to silica columns to separate out “large” (>200 nt) from “small” (<200 nt) transcripts (STAR Methods). To our surprise, the glycoRNA fractionated exclusively with the small RNA population of total RNA (Figure 2B). To validate this observation with an independent fractionation strategy, we applied Ac4ManNAz-labeled RNA to a sucrose gradient and analyzed the distribution of total RNA via SYBR Gold staining and glycoRNA. The sucrose gradient robustly separated the major visible RNAs such as small RNAs/tRNA, 18S rRNA, and 28S rRNA (Figures 2C and S2B). The glycoRNA fractionated with the small RNAs but still demonstrated extremely slow migration (high apparent MW) in the agarose gel (Figure 2C). We speculate that glycoRNA’s anomalous migratory behavior is caused by its associated glycans.
Figure 2. Small, non-polyadenylated, and conserved transcripts comprise the pool of cellular glycoRNA.

(A) Blotting of total or poly-adenylated (poly-A) enriched RNA from HeLa cells treated with Ac4ManNAz.
(B) Blotting of total RNA from HeLa cells treated with Ac4ManNAz after differential precipitation fractionation using silica-based columns.
(C) Blotting of total RNA from H9 human embryonic stem cells (H9) treated with Ac4ManNAz after sucrose density gradient (15%–30% sucrose) fractionation. An input profile is displayed to the right of the gradient.
(D) Scatterplot analysis Ac4ManNAz-enriched RNAs purified from the small RNA fractions of (C) from HeLa and H9 cells. Reads mapping to snRNA, snoRNAs, and Y RNAs are shown. Significance scores (-log10(adjusted p value) are overlaid for HeLa cells as the size of each data point and for H9 cells as the color of each data point.
(E) Representative blot of total RNA from wild-type (WT) or Y5 knockout (KO) 293T cells treated with Ac4ManNAz. Inset: quantification of the blot in (E) from biological triplicates. p value calculated by a paired, two-tailed t test.
See also Figures S2 and S3 and Tables S1 and S2.
A common set of transcripts are glycosylated across diverse cell types
To identify the glycoRNA transcripts, we leveraged the sucrose gradients to isolate only the small RNA fractions from Ac4ManNAz-labeled H9 and HeLa cells. RNA sequencing libraries were generated from small RNAs (input) as well as glycoRNAs that were enriched after streptavidin pulldown. Biological replicates showed high concordance across samples and the bulk of the reads mapped to small, non-polyadenylated RNAs, as expected (Figures S2C and S2D; Table S1). We next asked what RNAs were selectively labeled by Ac4ManNAz treatment. Input expression of tRNA and non-tRNA transcripts were positively correlated between HeLa and H9 cells (Figure S2E). We found a set of Y RNA, small nuclear RNA (snRNA), rRNA, small nucleolar RNAs (snoRNAs), and tRNAs enriched in both H9 and HeLa cells (Figures S2F and S2G). The enrichment values of HeLa and H9 cell glycoRNAs showed strong positive correlation, despite the different lineages of these cell types (Figures 2D and S2H). Thus, Ac4ManNAz enrichment defines 193 RNA transcripts as candidate glycoRNAs (Table S2).
The RNAs we found to be modified have many well-established and critical cellular roles. The Y RNA family stood out, because their binding proteins and ribonucleoproteins (RNPs) (among some other glycoRNAs transcripts identified) are known to be antigens associated with autoimmune diseases such as systemic lupus erythematosus (SLE) (Boccitto and Wolin, 2019; Marshak-Rothstein and Rifkin, 2007). These RNAs are highly conserved in vertebrates and are thought to contribute to cytosolic RNP surveillance, particularly for the 5S rRNA (Köhn et al., 2013; Kowalski and Krude, 2015). Given these features, we sought to validate Y5 as a glycoRNA by gene knockout (KO) via CRISPR/Cas9. A 293T Y5 KO cell line was generated using two single-guide RNAs (sgRNAs) that targeted the 5′ and 3′ regions of the Y5 genomic locus (Figure S3A). Single-cell clones were isolated and a KO was selected for characterization: PCR amplification of the Y5 locus yielded two amplicons corresponding to two different insertion/deletions (Figures S3B and S3C). The KO generated no observable Y5 transcript and had no gross growth defects, consistent with previous reports of Y RNA redundancy (Figures S3D and S3E) (Christov et al., 2006; Gardiner et al., 2009). Ac4ManNAz-labeling of the Y5 KO cells resulted in a significant (~30%, p = 0.033) decrease in the amount of biotin signal compared to wild-type (WT) cells, without any apparent MW changes (Figure 2E). The reduction of glycoRNA signal was consistent with the sequencing data, which identified Y5 as strongly enriched, but among a pool of other candidate glycoRNAs.
Label and label-free detection of sialic acid in glycoRNA
Next, we sought to define the glycan structures on glycoRNAs. The major pathway for Ac4ManNAz metabolism in human cells entails conversion to sialic acid, then to CMP-sialic acid, and finally addition to the termini of glycans (Luchansky et al., 2004). To exclude the possibility that Ac4ManNAz is shunted into unexpected metabolic pathways, we used 9-azido sialic acid (9Az-sialic acid), which is directly converted into CMP-sialic acid (Kosa et al., 1993) as a metabolic label. Consistent with Ac4ManNAz labeling, 9Az-sialic acid produced a similar time-dependent labeling of slowly migrating cellular RNA (Figure 3A). Treatment of Ac4ManNAz-labeled cellular RNA with Vibrio cholerae sialidase (VC-Sia) completely abolished the biotin signal without impacting the integrity of the RNA sample, while a heat-inactivated (HI) VC-Sia was unable to reduce the signal (Figure 3B). We assessed the contribution of canonical sialic acid biosynthesis enzymes through the use of P-3FAX-Neu5Ac, a cell-permeable metabolic inhibitor of sialoside biosynthesis (Rillahan et al., 2012). Treatment of HeLa cells with P-3FAX-Neu5Ac resulted in a dose-dependent reduction in total glycoRNA signal and a concomitant shift toward higher apparent MW on the blot (Figure 3C). This reduced mobility (appearing higher in the gel) of the glycoRNA likely results from less sialic acid, and thus less negative charge per glycoRNA molecule, as has been observed for proteins (Gahmberg and Andersson, 1982).
Figure 3. Glycans modifying RNA contain sialic acid.

(A) Blotting of RNA from HeLa cells treated with 1.75 mM 9-azido sialic acid for indicated times.
(B) Blotting of Ac4ManNAz-labeled HeLa cell RNA treated with Vibrio cholerae (VC) sialidase or heat-inactivated sialidase (VC-sialidase-HI).
(C) Blotting of RNA from HeLa cells treated with Ac4ManNAz and the indicated concentrations of P-3FAX-Neu5Ac.
(D) Unlabeled total RNA from H9 cells was isolated, reacted with the indicated enzyme (no enzymes, RNase cocktail, or Sialidase treatment), cleaned up to remove cleaved metabolites, and processed with the fluorogenic 1,2-diamino-4,5-methylenedioxybenzene (DMB) probe. HPLC analysis quantified the presence and abundance of specific sialic acids. Inset, Sybr gel image of the total RNA for each condition. The main sialic acid peaks are 2 and 3. The identity of peak 1 is unknown, but it is RNase-sensitive.
(E) Quantification of DMB results (D) from 4,188, H9, and HeLa cells from four biological replicates.
See also Figure S3.
To confirm that glycoRNAs are sialylated, we used an independent method not relying on metabolic reporters. The fluorogenic 1,2-diamino-4,5-methylenedioxybenzene (DMB) probe is used to derivatize free sialic acids for detection and quantitation by high-performance liquid chromatography (HPLC)-fluorescence (Bond et al., 2011). We subjected native, total RNA from HeLa, H9, and 4188 cells to the DMB labeling procedure (Figure S3F) and observed the presence of two forms of sialic acid commonly found in animals, Neu5Ac and Neu5Gc (Figures 3D and S3G). These peaks disappeared when the samples were pretreated with VC-Sia or RNase, reinforcing the notion that glycoRNA is modified with sialic acid containing glycans. Notably, we were unable to detect any sialic acid liberated from genomic DNA using the DMB assay (Figure S3H).
Quantitatively, we found that H9, HeLa, and 4,188 cells have approximately 40, 20, and 20 pmol of total sialic acid per μg of total RNA, respectively (Figure 3E). GlycoRNA from 4,188 cells contained more Neu5Gc, whereas H9 cells contained mostly Neu5Ac, and HeLa cells had similar levels of Neu5Ac and Neu5Gc (Figure 3E). Importantly, this quantitative analysis is consistent, with the observed difference in Ac4ManNAz labeling intensity we observed across these cell lines (Figures S1E and S1F). Human cells lack a functional CMAH gene that is responsible for converting Neu5Ac to Neu5Gc, while this pathway exists in mouse cells (Chou et al., 2002). Correspondingly, we found higher Neu5Gc levels in glycoRNA from mouse 4,188 cells as compared to HeLa or H9 cells (Figure 3E). The presence of Neu5Gc in HeLa glycoRNA likely comes from bovine serum in the growth media; H9 cells were grown in serum-free media.
Canonical N-glycan biosynthetic machinery contributes to glycoRNA production
There are two main classes of glycans on proteins, N- and O-glycans, and both can be sialylated. To determine whether glyco-RNA structures were related to glycoprotein-associated glycan structures, we used a combination of genetic, pharmacological, and enzymatic methods. The ldlD mutant CHO cell line lacks the ability to interconvert UDP-glucose(Glc)/GlcNAc into UDP-galactose (Gal)/GalNAc (Figure S4A) (Kingsley et al., 1986). Thus, in minimal growth media, glycoproteins from ldlD CHO cells have stunted N- and O-glycans because the cells cannot produce UDP-Gal (required for N-glycan elongation) and UDP-GalNAc (required to initiate O-glycosylation). We observed very little glycoRNA labeling in Ac4ManNAz-treated ldlD CHO cells (Figure 4A) grown in minimal media. However, supplementation of the media with galactose, but not GalNAc, restored glycoRNA labeling, and supplementation with both galactose and GalNAc further boosted labeling intensity (Figure 4A). This result was reproduced using a human K562 cell line with a CRISPR-Cas9 targeted KO of UDP-galactose-4-epimerase (GALE) (Schumann et al., 2019), which mimics the phenotype of the ldlD CHO cell line (Figure S4B). The pattern of these results were similar to that observed when labeling glycoproteins in these cell types (Möckl et al., 2019), suggesting that glycoRNA glycans are structurally related to those found on proteins.
Figure 4. A distinct set of N-glycans are enriched with glycoRNAs.

(A) Blotting of RNA from ldlD CHO cells labeled with Ac4ManNAz, Galactose (Gal, 10 μM), N-acetylgalactosamine (GalNAc, 100 μM), or all for 24 h.
(B) Blotting of RNA from HeLa cells treated with Ac4ManNAz and indicated concentrations of NGI-1, an inhibitor of OST, for 24 h.
(C) Blotting as in (B) but with the indicated concentrations of kifunensine.
(D) Quantification of Ac4ManNAz signal after treatment of Ac4ManNAz-labeled HeLa cell RNA with the indicated enzymes in vitro each for 1 h at 37°C in biological triplicate.
(E) Schematic of the method used to release glycans from RNA samples and subsequently purify free glycans for mass spectrometry analysis.
(F) Unsupervised clustering analysis of glycans (rows) released from peptide and RNA fractions (columns) of 293, H9, or HeLa cells via PNGaseF cleavage. Glycans had to be found biological replicates of at least one of the six samples to be included.
(G) Principal component analysis of peptide- and RNA PNGaseF-release glycans.
(H) Bar plots of the fraction of glycans containing fucose (red) or sialic acid (purple) modifications that were released from peptides or RNA samples. Numbers below are the absolute numbers of glycans found with each of the modifications from a given dataset.
We next tested the effects of glycosylation inhibitors on glyco-RNA biosynthesis. Oligosaccharyltransferase (OST) mediates protein N-glycosylation by transferring a 14-sugar glycan to asparagine residues on nascent polypeptides during their translocation through the Sec/translocon (Figure S4C) (Mohorko et al., 2011). We tested the effect of NGI-1, a specific and potent small molecule inhibitor of OST (Lopez-Sambrooks et al., 2016), on glycoRNA production. Such treatment caused a dose-dependent loss of glycoRNA labeling with Ac4ManNAz (Figure 4B), suggesting that OST is involved in biosynthesis of glycoRNA-associated glycans. We also perturbed downstream N-glycan processing steps with kifunensine and swainsonine, inhibitors of the N-glycan trimming enzymes a-mannosidase I and II, respectively (Elbein et al., 1990; Tulsiani et al., 1982) (Figure S4C). These treatments also caused a dose-dependent loss of azidosugar labeling (Figures 4C and S4D) accompanied by an increase in apparent MW of the glycoRNA at higher doses, akin to the results seen with P-3FAX-Neu5Ac. We hypothesize that disruption of high-mannose glycan processing produces hyposialylated glycoRNAs with less net negative charge and, therefore, reduced mobility.
To further define the glycan structures on glycoRNA, we employed a panel of endoglycosidases. Purified RNA from Ac4-ManNAz-labeled HeLa cells was first exposed to each enzyme and then reacted with biotin for visualization (Figures 4D and S4E). Treatment of glycoRNA with PNGase F, which cleaves the asparagine side chain amide bond between proteins and N-glycans (Tarentino and Plummer, 1994), strongly abrogated signal from Ac4ManNAz labeling. Endo F2 preferentially cleaves biantennary and high mannose structures, whereas Endo F3 preferentially cleaves fucosylated bi- and triantennary structures, both within the chitobiose core of the glycan (Tarentino and Plummer, 1994). Treatment of glycoRNA with either Endo F2 or F3 resulted in a partial loss of Ac4ManNAz labeling. However, Endo Hf, which is more selective for high-mannose structures, did not affect Ac4ManNAz signal (Figures 4D and S4E). In contrast to these N-glycan digesting enzymes, O-glycosidase (targeting core 1 and core 3 O-glycans) (Koutsioulis et al., 2008) or mucinase (StcE) (Malaker et al., 2019) treatment had no effect on Ac4ManNAz labeling intensity (Figures 4D, S4E, and S4F). As in previous experiments, VC-Sia completely removed the Ac4ManNAz-dependent label (Figures 4D and S4E).
Mass spectrometry defines distinct compositions of glycans on RNA
The above data suggest that glycoRNA are modified with complex-type N-glycans with at least one terminal sialic acid residue. To develop a more precise view of the glycoforms associated with RNA, we optimized a workflow based on PNGaseF-mediated release of glycans from pools of small RNAs, followed by analysis of those glycans by a porous graphitized carbon-based liquid chromatography MS strategy (PGC-LC-MS) (Figure 4D). Glycans were released from small RNA pools from 293T, H9, and HeLa cells and in parallel, peptide samples were similarly processed to compare the glycan profile on the cellular proteins. Two biological replicates were performed for each sample. We found 107 unique glycans present in both replicates in at least one of the six sample types (Figure 4E; STAR Methods) (for all 260 confidently identified glycan, see Table S3). Hierarchical clustering of identified glycans and principal component analysis revealed that glycans released from peptides clustered differentially when compared to those released from RNA. Further, the set of unique glycans found on RNA was smaller and more constrained relative to those on peptides (Figures 4F and 4G). When examining the features that distinguished RNA glycans from peptide glycans, we noticed that both 293T and H9 cells had a higher fraction of glycans modified with fucose on RNA as compared to the peptides from these same cells (Figure 4H). In contrast, glycoRNA glycans from HeLa cells were more likely to contain sialic acid modifications compared to the peptide glycans from HeLa cells (Figure 4H). Overall, the PGC-LC-MS data of PNGaseF-released glycans are in line with Ac4ManNAz labeling and DMB probe experiments. Importantly, because the MS-based approach does not require sialic acid for enrichment or visualization, we were able to reveal an expanded set of glycan compositions that are often fucosylated and sometimes asialylated.
glycoRNAs are associated with cellular membrane
Finally, we assessed the subcellular localization of glycoRNA. The biogenesis of sialylated glycans occurs across many subcellular compartments including the cytosol (processing of ManNAc to Neu5Ac), the nucleus (charging of Neu5Ac with CMP), and the secretory pathway (where sialyltransferases add sialic acid to the termini of glycans) (Varki and Schauer, 2009). The localization of Y RNAs has been reported to be mainly cytoplasmic with a minor fraction in the nucleus (Köhn et al., 2013). Other major classes of glycoRNA transcripts such as tRNAs and sn/snoRNAs are classically localized to the soluble cytosol and nucleus, respectively. To determine where glycoRNAs are distributed inside cells, we used two biochemical strategies: one that isolates nuclei away from membranous organelles and the cytosol (Gagnon et al., 2014) and a second that separates the soluble cytosolic compartment away from membranous organelles (STAR Methods). Nuclear RNA from Ac4ManNAz-labeled HeLa cells yielded no detectable azide-labeled species whereas the membrane fraction exclusively contained the glycoRNA (Figures 5A and 5B). This suggests that glycoRNAs are closely associated with membrane organelles.
Figure 5. glycoRNAs are on the external surface of living cells.

(A) Blotting of RNA and proteins after subcellular fractionation designed to robustly purify nuclei. Non-nuclear proteins GAPDH and β-tubulin and nuclear histone 3 lysine 4 trimethylation (H3K4me3) are visualized by western blot.
(B) Blotting of RNA and proteins after subcellular fractionation designed to separate soluble cytosol from membranous organelles. Membrane proteins RPN1, Sec63, and soluble β-tubulin are visualized by western blot.
(C) Blotting of RNA from HeLa cells labeled with 100 μM Ac4ManNAz for 24 h and then exposed to fresh media containing 100 μM Ac4ManNAz with or without 150 nM VC-Sia for 60 min at 37°C
(D) Quantification of the experiment in (C) across biological triplicates and from 293T or K562 cells treated in the same manner. p value calculated by a paired, two-tailed t test.
(E) Schematic of the lectin-based proximity labeling of RNA on cell surfaces. Living cells are stained with a biotinylated lectin that recruits streptavidin-HRP that is in turn able to generate nitrene radicals from biotin-aniline after the addition of hydrogen peroxide. RNA from these cells is then extracted and analyzed for biotin labeling that reveals if that RNA was in proximity to the lectin.
(F) Blotting of total RNA samples generated as described in (E). Lanes 5 and 6 were processed in vitro (after purifying RNA) with RNase cocktail or VC-Sia to demonstrate any sensitivity of the biotin-aniline signal to these enzymes.
(G) Blotting of total RNA samples similar to (F) however cells were first lysed in a hypotonic buffer, destroying cellular membranes that are normally impermeable to nitrene radicals. Labeling of rRNA is evident here but not in (F).
See also Figure S5.
Because membrane organelles have precise topological configurations, we next assessed if there was a clear topological organization of glycoRNA with respect to the membranes we isolated. Crude cellular membranes and membrane-bound organelles were isolated from Ac4ManNAz-labeled 293T cells and subjected to VC-Sia digestion with or without pre-treatment with Triton X-100 to permeabilize membrane compartments (Figure S5A). If glycoRNAs were topologically confined to the luminal spaces of membrane compartments, VC-Sia would only have access to these species after the addition of Triton X-100. We found that the majority of the glycoRNA signal was sensitive to VC-Sia without Triton X-100, whereas a small but reproducible pool was accessible only after permeabilization (Figure S5B). Thus, although a portion of glycoRNAs appears to reside within the luminal space of membranous organelles, the vast majority seems accessible, or on the surface of membranes in this assay.
glycoRNAs gain access to the surface of living cells
The accessibility to VC-Sia in the experiment above suggests that glycoRNAs do not accumulate in the lumen of intracellular vesicles or membrane organelles, however, it does not precisely define on which membrane surface glycoRNAs may be present. Given the canonical trafficking and localization of glycopolymers, we hypothesized that glycosylation of RNA may afford it the ability to be trafficked to the plasma membrane and be present on the extracellular surface of living cells. We addressed this hypothesis through two orthogonal and complementary approaches.
We first leveraged the robust and specific activity of VC-Sia to cleave sialic acid (including those on glycoRNAs) and its established ability to cleave sialic acids selectively off the surface of living cells (Gray et al., 2020; Xiao et al., 2016). We assayed for changes in Ac4ManNAz signal after adding VC-Sia to culture media on living HeLa cells and in as little as 20 min saw a reduction in the glycoRNA levels (Figures 5C and S5C). Replicating this experiment at 60 min, where the most robust difference was observed (Figure 5D), and performing it on both adherent (HeLa and 293T) and suspension (K562) cells showed that in all cases VC-Sia was able to significantly reduce the levels of glyco-RNA. These data indicate that in a short time frame and in an environment with an intact plasma membrane, VC-Sia has access to >50% of the bulk of glycoRNA purified from cells.
To validate the observation that glycoRNAs localize to live cell surfaces, we required a labeling workflow independent of Ac4ManNAz metabolic incorporation. To achieve this, we combined the Rhee et al. (2013) peroxidase-catalyzed proximity labeling technique with the observation that biotin-aniline has significantly increased reactivity toward RNA relative to biotin phenol, which is favored for protein labeling (Zhou et al., 2019). We took a non-genetic strategy (Bar et al., 2018), leveraging lectins as cell surface affinity tools to bind live cells that could then recruit a peroxidase and deposit biotin-aniline on RNAs near to the bound glycan (Figure 5E). Despite the wide use of lectins as general cell-surface binding reagents, they have specific glycoform binding features and we therefore selected a lectin which, based on our data from Figures 3 and 4, should not bind near glycoRNAs (ConA, specific for high mannose structures) and lectins that should bind directly to glycoRNAs (MAAII, sialic acids; WGA, N-glycans ± sialic acid) (Gao et al., 2019).
Initially we benchmarked this assay against live HeLa cell surface proteins. As expected, all three lectins were capable of recruiting streptavidin-HRP, activating biotin-aniline, and producing specific labeling patterns of cell surface proteins, whereas streptavidin-HRP alone was unable to generate robust labeling (Figure S5D). This and all subsequent experiments were conducted strictly at 4°C to reduce or eliminate vesicular trafficking, membrane recycling, or uptake of extracellular components. We next assayed the RNA from these cells and found specific labeling of a high molecular weight band generated when cells were stained with MAAII or WGA, but not ConA. The signal was partially RNase-sensitive (89% loss) (Figure 5F). On treatment of the purified RNA material with sialidase, we observed a nearly quantitative shift in the biotin signal into the well of the gel without strong reduction in the amount of signal (Figure 5F). This supports the view that biotin-aniline covalently modifies the RNA directly and that the glycan, in particular the sialic acid, contributes to glycoRNA’s dramatically abnormal migration in agarose gels.
Repeating the proximity ligation assay in cell lysate rather than on live cells showed that, without an impermeable (to the activated nitrene radical of biotin-aniline) plasma membrane, MAAII and WGA still labeled glycoRNA. However, all three lectins weakly, but consistently labeled rRNA bands (Figure 5G). The absence of these rRNA bands in the live cell experiment and their intensity relative to the high MW glycoRNA bands suggest once more that the majority of cellular glycoRNAs are on cell surfaces. We note that total RNA exposed to the biotin-aniline label was not fully digested with RNase (Figures 5F and 5G, left, 5th lanes), possibly due to covalent modification of the RNA by the aniline probe or other species. We suggest this as a possible reason that full RNase-sensitivity was not observed in these experiments. In sum, proximity-based labeling of RNAs near complex N-glycans on the surface of living cells detects glycoRNA, consistent with our chemical, genetic, and mass spectrometry results describing RNA glycans (Figures 3 and 4).
Siglec receptors and anti-RNA antibodies recognize cell surface glycoRNAs
Biopolymers localized to the cell surface often participate in molecular interactions with binding partners in cis or in trans at cell-cell junctions. Because glycoRNAs are present on cell surfaces, we hypothesized that they too could engage in these types of interactions. We assessed if existing reagents to study the biology of cell surfaces, such antibody or recombinant protein-based affinity reagents, might interact with cell surface glycoRNA (Figure 6A).
Figure 6. Cell surface glycoRNAs contribute to the binding of select Siglec proteins.

(A) Cartoon model of a glycoRNA on the cells surface depicted with two glycans identified in the PNGaseF release experiment. Prediction locations of binding for the anti-dsRNA antibody (J2) and Siglec-Fc proteins are highlighted.
(B) Fluorescence-activated cell sorting (FACS) analysis of single HeLa cells pre-treated with the indicated enzymes or inhibitors and then stained with the J2 antibody. Gated region (orange) indicates the population shifted toward high J2 binding.
(C) FACS analysis of single HeLa cells pre-treated with the OST inhibitor NGI-1 for 12 h at the indicated concentrations. Dashed vertical line denotes a J2-high population, and for each sample, the fraction of cells within this region is shown as a percentage.
(D) FACS analysis of single HeLa cells pre-treated with RNase then stained with the indicated Siglec-Fc reagents.
See also Figure S6.
Antibodies targeting RNA have been associated with systemic lupus erythematosus (SLE) (Blanco et al., 1991). Additionally, anti-RNA antibodies are used as research tools; for example, the J2 anti-double-stranded RNA (dsRNA) antibody has specificity for ds-regions of RNA (Bonin et al., 2000) (with no cross-reactivity with dsDNA) and is often used to identify cells infected with RNA viruses (Mateer et al., 2019). The J2 antibody is reported to bind dsRNA with a minimum length of ~40 bp (Schönborn et al., 1991). GlycoRNAs are predicted to have duplex RNA regions, however, these are generally <40 bp in length. Nonetheless, we tested whether J2 could bind a small RNA that we find enriched by Ac4ManNAz sequencing (Figure 2), such as the Y5 RNA, using electrophoretic mobility shift assays. As shown in Figure S6A, the J2 antibody was able to shift free Y5 RNA in vitro. This shift was specific to J2 and was not observed using an isotype control antibody. Furthermore, the shift induced by J2 was abrogated in the presence of competing poly-(I:C), which mimics long dsRNA (Figure S6A).
Having confirmed that J2 can bind the RNA component of glycoRNAs like Y5, we next established a flow cytometry assay to probe cell surface RNA. We carried out all experiments on live cells, fixing only after antibodies were bound and background washed away for experimental workflow flexibility. On a technical note, it was critical to use recombinant sources of cell dissociation enzymes. For example, commonly used crude preparations of trypsin contain significant RNase-activity and thus rapidly destroy RNA (Figures S6B and S6C; STAR Methods).
Approximately 20% of a population of cultured HeLa cells showed positivity with J2 staining (Figure 6B). This binding was robustly abrogated by pre-treatment of cells with RNase A and was recovered by adding a specific protein inhibitor to the RNase A to block activity (Figure 6B). Similar results were observed using 293T (adherent) and K562 (suspension) cells (Figure S6D). To confirm the distribution of J2 staining the cell surface, we performed confocal imaging of HeLa cells stained with J2 that demonstrated signal in the peripheral edges of cells that was sensitive to RNase A treatment (Figure S6E). We next asked if J2 was detecting glycoRNA on the cell surface by perturbing OST as we previously did in Figure 4. We treated HeLa cells with the OST inhibitor NGI-1 for 12 h and observed a dose-dependent loss of J2 binding to the cell surface (Figure 6C). This is consistent with our whole-cell RNA blotting experiment (Figure 4) and suggests that much of the cell surface RNA recognized by the J2 antibody relies on N-glycosylation for its surface localization.
Finally, we sought to determine whether glycoRNAs can interact with glycan-binding receptors whose ligands have been assumed, based on convention, to be cell-surface glycoproteins and glycolipids. As described above, the N-glycans associated with glycoRNAs are highly sialylated. Thus, we asked whether members of the sialic acid binding-immunoglobulin lectin-type (Siglec) receptor family could recognize glycoRNAs. Notably, with 14 members distributed on all classes of immune cells, the Siglecs are the largest family of sialoside-binding proteins in humans (Duan and Paulson, 2020; Fraschilla and Pillai, 2017). Their roles in immune modulation are well established and include host-pathogen interactions (Chang and Nizet, 2020; Macauley et al., 2014), cancer immune evasion (Barkal et al., 2019; Hudak et al., 2014; Jandus et al., 2014; Stanczak et al., 2018; Xiao et al., 2016), and genetic associations with autoimmune disease (Angata, 2014; Flores et al., 2019). Physiological ligands of individual Siglec family members have been identified in a few settings (Barkal et al., 2019; Läubli et al., 2014), but for the most part, the glycoconjugates that support Siglec binding at immune synapses are not well characterized. All efforts to do so have assumed that Siglec ligands are glycoproteins or glycolipids.
To determine whether human Siglec receptors can bind cell surface glycoRNA, we used soluble Siglec-Fc reagents and probed their binding to cells by flow cytometry. We first determined that 9 of 12 commercial Siglec-Fc reagents were able to bind above background to HeLa cells (Figure S6F). Of these nine, the binding of two Siglec-Fc reagents, Siglec-11 and Siglec-14, was sensitive to RNase A treatment (Figures 6D and S6G). These data support the hypothesis that cell surface glyco-RNAs could be direct Siglec receptor ligands.
DISCUSSION
We found that sialylated N-glycans produced by canonical endoplasmic reticulum (ER)/Golgi-lumen biosynthetic machinery are attached to specific mammalian small noncoding RNAs. These RNAs are consistently modified across several cell types and organisms. This work provides several lines of evidence for the existence of glycoRNA species present at the cell surface. Overall, these findings point to a common strategy for modifying RNA with glycans in mammals. Our discovery opens up further research into the function and chemical structure of these glycoconjugates.
Chemical linkage of RNA to glycan
GlycoRNA was sufficiently robust to withstand stringent protocols to separate RNA from lipids and proteins, including organic phase separation, proteinase K treatment, silica-based RNA purification, and heating in high concentrations of formamide. Although the precise nature of the glycan-RNA linkage has not yet been determined, we speculate that direct glycosylation of native RNA bases is unlikely. The observed sensitivity to PNGase F, which cleaves the glycosidic linkage between asparagine and the proximal GlcNAc of N-glycans, implies an amide bond-containing linker that native nucleobases lack. It is possible that a precursor guanosine modification is necessary to establish an asparagine-like functionality capable of modification by OST, or that a preassembled N-glycan carrier moiety is attached to nucleobases by some other chemistry. These possibilities are consistent with sedimentation of glycoRNAs in the sucrose gradient, which suggests a linker with a relatively small molecular weight.
Precisely defining the chemical and structural features of this linkage will be critical in future studies. A playbook for this endeavor might be found in the process by which multiple laboratories defined the chemical structure of the unusual α-dystroglycan glycans that are associated with various muscular dystrophies (Inamori et al., 2012; Yoshida-Moriguchi et al., 2013; Praissman et al., 2014; Willer et al., 2014; Praissman et al., 2016; Manya et al., 2016). In that story, a combination of analytical approaches, powered by insights from human genetics, ultimately led to the identification of a glycan comprising previously unknown building blocks. Likewise, defining the chemical structure of glycoRNA will benefit from modern genetic tools that might uncover its biosynthetic enzymes.
Regarding glycan structures, our mass spectrometry study provided compositional information and from this we assigned putative structures (Figure S5E). Although future studies will provide unambiguous assignment of these structures, it is interesting to consider the terminal sialyl-Lewis A/X motifs as well as core fucosylation because these are well known to regulate leukocyte trafficking, cancer cell metastasis, and early steps in fertilization (Schneider et al., 2017).
Cell surface glycoRNAs
Glycosylation of proteins and lipids affords these substrate biopolymers novel functions, structures, and localizations. Thus, one might predict a glycan on RNA might similarly control functional aspects of the RNA polymers they modify. A particular hallmark of glycoconjugates is their trafficking through the secretory system for direct secretion or presentation on the cell surface. Our results provide multiple orthogonal lines of evidence that glycoRNAs share this profile. This observation is intriguing because it highlights potentially novel interactions and functions for this class of modified nucleic acids.
We found the majority of cellular glycoRNA is present on the cell surface. However, a number of open and critical questions remain. One concerns the mechanism by which small RNA substrates might gain access to luminal compartments in the secretory pathway. There is precedent for transport of intact RNA transcripts across membranes, such as the C. elegans transporter SID-1 that imports dsRNA for RNA interference (Feinberg and Hunter, 2003) and evidence for similar activity has been found in Drosophila (Saleh et al., 2006). Interestingly, mammals have two SID-1 orthologs that are thought to transport RNAs across intracellular membranes (Aizawa et al., 2016; Nguyen et al., 2019). It is possible that related trafficking systems exist on the ER membrane that would enable N-glycan modification of appropriately functionalized RNA.
Another important question is how glycoRNAs are stably associated with the membranes and in what configuration, and whether these mechanisms are unique to glycoRNA. A recent report has suggested that fragments of specific long coding and noncoding RNAs are present on the surface of cells (Huang et al., 2020). The pathways by which those long RNAs are trafficked and presented on the cell surface have not been established. In our lectin-HRP-aniline experiment (Figure 5), which used lectins with broad glycoform affinities (e.g., ConA and WGA), we detected an entirely non-overlapping set of RNA transcripts, namely small, highly conserved RNAs that are characteristic of glycoRNAs. Examination of the expanding list of proteins which have no canonical RNA-binding domain but are able to directly bind cellular RNAs (Baltz et al., 2012; Beckmann et al., 2015; Castello et al., 2012), some of which are membrane proteins, may provide insights into the mechanism(s) by which (glyco)RNAs localizes to mammalian cell surfaces.
Regulatory implications of cell surface glycoRNAs
The identification of glycoRNA and its localization to the cell surface connects aspects of both glycobiology and RNA biology that had remained isolated from one another. For example, it is interesting to consider disease mechanisms which have classically been connected to RNA but were conceptually incompatible with intact cells. There are a number of RNA and RNA-associated autoantigens associated with autoimmune diseases (Ahlin et al., 2012; Han et al., 2014). However, canonically, cell death is required for these antigens to gain access to the extracellular space (Casciola-Rosen et al., 1994; Golan et al., 1992). Intriguingly, the glycoRNAs we found on HeLa and H9 cells strongly overlap with the small RNAs associated with autoimmune diseases. We speculate that glycosylation is linked to these RNA’s special exposure to the extracellular space and, therefore, to the immune system. Notably, humans have eight secreted RNase A family members that are classically associated with host-defense mechanisms (Koczera et al., 2016; Sorrentino, 2010). There may be an expanded role for these RNases in the metabolism of host-encoded glycoRNAs.
Once a glycoRNA is positioned on the cell surface, direct engagement with and regulation of cell surface receptors is possible. Our exploration of the RNase-sensitivity of Siglec binding to cell surfaces suggests that the ligands of Siglec-11 and Siglec-14 are composed in part of glycoRNAs. Siglecs have been functionally associated with diseases such as autoimmunity and cancer (Eakin et al., 2016; Macauley et al., 2014; Müller et al., 2015). Several Siglec family members function as immune checkpoint receptors that signal through cytosolic immunoreceptor tyrosine-based inhibitory motifs (ITIMs), analogous to the T cell checkpoint receptor PD-1 (Riley, 2009). Intriguingly, monocyte Siglec-14 expression has been correlated with disease severity in SLE patients (Thornhill et al., 2017). Defining the potential intersection of Siglec biology with glycoRNA is an important future pursuit.
In sum, the framework in which glycobiology is presently understood excludes RNA as a substrate for N-glycosylation. Our discovery of glycoRNA suggests the current view is incomplete and points to a new axis of RNA glycobiology, including as of yet undiscovered biosynthetic and trafficking mechanisms. Further, it highlights the possibility that cell surface glycoconjugates, which mediate and regulate important inter-cellular interactions, may now have an expanded template base to include RNA.
Limitations of study
A major focus of the work presented leverages selective metabolic labeling of sialic acid with Ac4ManNAz. Because not all glycans contain sialic acid, it is possible that glycoforms beyond those reported here may also be conjugated to RNAs. Optimization of other glycan labeling strategies is needed to render them compatible with RNA, ideally allowing detection of native, unlabeled RNA-glycan conjugates. Additionally, the precise linkage between the RNA template and carbohydrate remains unknown. Sensitivity to PNGaseF provides hints of the linker’s chemical nature, and sedimentation of glycoRNAs in the sucrose gradient points to a linker with a small molecular weight. However, definitive characterization using unbiased approaches is important to fully define this conjugate, and moreover, may help to map its biosynthetic pathway.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Ryan Flynn (ryan.flynn@childrens.harvard.edu).
Materials availability
All unique/stable reagents generated in this study are available from the Lead Contact with a completed Materials Transfer Agreement.
Data and code availability
All sequencing data have been deposited on the Gene Expression Omnibus (GSE136967; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136967). Sequencing data were analyzed with the FAST-iCLIP pipeline which can be found here (https://github.com/ChangLab/FAST-iCLIP/tree/lite). Glycan release samples were analyzed with GlycoNote (https://github.com/GlycoNote/GlycoNote).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mammalian cell culture
All cells were grown at 37°C and 5% CO2. HeLa and HEK293T cells were cultured in DMEM media supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (P/S). GM12878, K562, K562GALE−/− (Schumann et al., 2019), and MYC T- ALL 4188 cells were cultured in RPMI-1640 media with glutamine supplemented with 10% FBS and 1% P/S. CHO and ldlD-CHO cells were cultured in 1:1 DMEM:F12 media with 3% FBS and 1% P/S. The H9 human embryonic stem cell line was cultured on Matrigel matrix (Corning) coated plates with mTeSR 1 (StemCell Technologies) media.
METHOD DETAILS
Metabolic chemical reporters and inhibitors
Stocks of azide-labeled sugars N-Acetyl-9-azido-9-deoxy-neuraminic acid (9Az sialic acid, Carbosynth) and N-azidoacetylmannosamine-tetraacylated (Ac4ManNAz, Click Chemistry Tools) were made to 500 mM in sterile dimethyl sulfoxide (DMSO). Stocks of unlabeled sugars N-Acetyl-D-galactosamine (GalNAc, Sigma) and D-(+)-Galactose (Gal, Sigma) were made to 500 mM and 50 mM, respectively, in sterile water. In cell experiments ManNAz was used at a final concentration of 100 μM. In vitro experiments with ManNAz used 0, 2, or 20 mM ManNAz (up to 200× the in-cell concentrations) for 2 h at 37°C. The in-cell experiments with 9Az sialic acid used a 1.75 mM final concentration for between 6 and 48 h. Gal and GalNAc were used as media supplements at 10 μM and 100 μM, respectively, and were added simultaneously with ManNAz for labeling.
Working stocks of glycan-biosynthesis inhibitors were all made in DMSO at the following concentrations and stored at −80°C: 10 mM NGI-1 (Sigma), 10 mM Kifunensine (Kif, Sigma), 10 mM Swainsonine (Swain, Sigma), 50 mM P-3FAX-Neu5Ac (Tocris). All compounds were used on cells for 24 h and added simultaneously with ManNAz for labeling.
Metabolic reporters in mouse models
All experiments were performed according to guidelines established by the Stanford University Administrative Panel on Laboratory Animal Care. C57BL/6 mice were crossed and bred in house. ManNAz was prepared by dissolving 100 mg ManNAz in 830 μL 70% DMSO in phosphate buffered saline (PBS), warming to 37°C for 5 min, and then sterile filtering using 0.22 μm Ultrafree MC Centrifugal Filter units (Fisher Scientific); this solution was stored at −20°C. Male C57BL/6 mice (8–12 weeks old) were injected once-daily, intraperitoneally with 100 μL of ManNAz (dosed to 300 mg ManNAz/kg/day), while control mice received the vehicle alone. At 2, 4, and 6 days, mice were euthanized, and their livers and spleens were harvested. The organs were pressed through a nylon cell strainer and resuspended with PBS to create a single cell suspension. RNA was collected as described below.
RNA extraction and purification strategies
A specific series of steps were taken to ensure that RNA analyzed throughout this study was as pure as possible. First TRIzol reagent (Thermo Fisher Scientific) was used as a first step to lyse and denature cells or tissues. After homogenization in TRIzol by pipetting, samples were incubated at 37°C to further denature non-covalent interactions. Phase separation was initiated by adding 0.2× volumes of 100% chloroform, vortexing to mix, and finally spinning at 12,000× g for 15 min at 4°C. The aqueous phase was carefully removed, transferred to a fresh tube and mixed with 2× volumes of 100% ethanol (EtOH). This solution was purified over a Zymo RNA clean and concentrator column (Zymo Research): sample solution was added to Zymo columns and spun at 10,000× g for 20 s and the flow through always discarded. Three separate washes were performed, 1× 400 μL of RNA Prep Buffer (Zymo Research) and 2× 400uL RNA Wash Buffer (Zymo Research) and spun at 10,000x g for 20 s. To elute RNAs, two volumes of pure water were used. Next RNA was subjected to protein digestion by adding 1 μg of Proteinase K (PK, Thermo Fisher Scientific) to 25 μg of purified RNA and incubating it at 37°C for 45 min. After PK digestion, RNA was purified again with a Zymo RNA clean and concentrator as described above. All RNA samples generated in this study were purified at least by these two steps first, with subsequent enzymatic or RNA fractionations occurring in addition to these first two purifications. We found that Zymo-Spin IC and IIICG columns bind up to ~50 and 350 μg of total RNA, respectively; columns in each experiment were selected based on the amount of RNA needed to be purified.
For differential-precipitation of small versus large RNAs, the Zymo RNA clean and concentrator protocol was used as described. Briefly, RNA in an aqueous solution was mixed with 1× volumes of 50% RNA Binding Buffer in 100% EtOH. This mix was applied to the Zymo silica column; the flow through contained small RNAs while the column retained large RNAs. The flow through was mixed with 1× volumes of 100% EtOH, bound to a new Zymo column and purified as described above.
To enrich for poly-adenylated RNA species, RNA initially purified as above was used as the input for the Poly(A)Purist MAG Kit (Thermo Fisher Scientific). Oligo(dT) MagBeads were aliquoted and washed twice in Wash Solution 1. RNA (15 μg total RNA) was brought to 600 ng/μL in 1× Binding Solution, added to washed beads, and heated to 70°C for 5 min. Samples were cooled to 25°C for 60 min, applied to a magnet, supernatant removed, and washed twice with Wash Solution 1 and once with Wash Solution 2. Poly-A enriched RNA was eluted by adding RNA Storage Solution to the beads and heating the samples to 70°C. The elution step was performed twice and the resulting poly-A RNA was cleaned up via the Zymo RNA clean and concentrator as described above.
Enzymatic treatment of RNA samples and cells
Various endo- and exonucleases and glycosidases were used to digest RNA, DNA, or glycans. All digestions were performed on 20 μg of total RNA in a 20 μL at 37°C for 60 min. To digest RNA the following was used: 1 μL of RNase cocktail (0.5U/μL RNaseA and 20U/μL RNase T1, Thermo Fisher Scientific) with 20 mM Tris-HCl (pH 8.0), 100 mM KCl and 0.1 mM MgCl2. To block the RNase activity of the RNase Cocktail, 1 μL of RNase Cocktail was pre-mixed with 8 μL of SUPERaseIn (20U/μL, Thermo Fisher Scientific) for 15 min at 25°C before adding to the RNA solution. To digest DNA, 2 μL of TURBO DNase (2U/μL, Thermo Fisher Scientific) with 1× TURBO DNase buffer (composition not provided by manufacture). To digest glycans: 2 μL of α2–3,6,8 Neuraminidase (50U/μL, New England Biolabs, NEB) with GlycoBuffer 1 (NEB), or 2 μL of Endo-Hf (1,000U/μL, NEB) with GlycoBuffer 3 (NEB), or 2 μL of PNGase F (500U/μL, NEB) with GlycoBuffer 2 (NEB), or 2 mL of Endo-F2 (8U/μL, NEB) with GlycoBuffer 3 (NEB), or 2 mL of Endo-F3 (8U/μL, NEB) with GlycoBuffer 4 (NEB), or 2 μL of O-Glycosidase (40,000U/μL, NEB) with GlycoBuffer 2 (NEB), or 1 μL of StcE (Malaker et al., 2019) at 0.5 μg/μL with or without 20 mM EDTA. For live cell treatments, VC-Sia was expressed and purified as previously described (Gray et al., 2019) and added to cells at 150 nM final concentration in complete growth media for between 20 and 60 min at 37°C.
Copper-free click conjugation to RNA
Copper-free conditions were used in all experiments to avoid copper in solution during the conjugate of biotin to the azido sugars (ManNAz and 9Az-Sia). All experiments used dibenzocyclooctyne-PEG4-biotin (DBCO-biotin, Sigma) as the alkyne half of the cycloaddition. To perform the SPAAC, RNA in pure water was mixed with 1× volumes of “dye-free” Gel Loading Buffer II (df-GLBII, 95% Formamide, 18mM EDTA, and 0.025% SDS) and 500 μM DBCO-biotin. Typically, these reactions were 10 μL df-GLBII, 9 μL RNA, 1 μL 10mM stock of the DBCO reagent. Samples were conjugated at 55°C for 10 min to denature the RNA and any other possible contaminants. Reactions were stopped by adding 80 μL water, then 2× volumes (200 μL) of RNA Binding Buffer (Zymo), vortexing, and finally adding 3× volumes (300 μL) of 100% EtOH and vortexing. This binding reaction was purified over the Zymo column as described above and analyzed by gel electrophoresis as described below.
RNA gel electrophoresis, blotting, and imaging
Blotting analysis of ManNAz-labeled RNA was performed conceptually similar to a Northern Blot with the following modifications. RNA purified, enriched, or enzymatically digested and conjugated to a DBCO-biotin reagent as a described above was lyophilized dry and subsequently resuspended in 15 μL df-GLBII with 1× SybrGold (Thermo Fisher Scientific). To denature, RNA was incubated at 55°C for 10 min and crashed on ice for 3 min. Samples were then loaded into a 1% agarose-formaldehyde denaturing gel (Northern Max Kit, Thermo Fisher Scientific) and electrophoresed at 110V for 45 min. Total RNA was then visualized in the gel using a UV gel imager. RNA transfer occurred as per the Northern Max protocol for 2 h at 25°C, except 0.45 μm nitrocellulose membrane (NC, GE Life Sciences) was used. This is critical for downstream imaging as most positively charged nylon membranes have strong background in the infrared (IR) spectra. After transfer, RNA was crosslinked to the NC using UV-C light (0.18 J/cm2). NC membranes were then blocked with Odyssey Blocking Buffer, PBS (Li-Cor Biosciences) for 45 min at 25°C. Note that the blocking buffer made with TBS or PBS, both sold from Li-Cor Biosciences, work similarly for this step. After blocking, Streptavidin-IR800 (Li-Cor Biosciences) was diluted to 1:10,000 in Odyssey Blocking Buffer and stained the NC membrane for 30 min at 25°C. Excess streptavidin-IR800 was washed from the membranes by three, serial washes of 0.1% Tween-20 (Sigma) in 1× PBS for 5 min each at 25°C. NC membranes were briefly rinsed in 1× PBS to remove the Tween-20 before scanning on an Odyssey Li-Cor CLx scanner (Li-Cor Biosciences) with the software set to auto-detect the signal intensity for both the 700 and 800 channels. After scanning, images were quantified with the Li-Cor software (when appropriate) in the 800 channel and exported.
DMB assay for sialic acid detection
Unless otherwise noted, all chemicals were supplied by Sigma. Native sialic acids on RNA or DNA were derivatized with 4,5-Methylenedioxy-1,2-phenylenediamine dihydrochloride (DMB) and detected via reverse phase high-performance liquid chromatography (HPLC) according to established methods (Bond et al., 2011). In brief, RNA samples were lyophilized, and 100 μg (or otherwise noted in specific figures) of each sample was dissolved in 2 M acetic acid. Sialic acids were hydrolyzed by incubation at 80°C for 2 h, and then cooled to room temperature before the addition of DMB buffer (7 mM DMB, 0.75 M β-mercaptoethanol, 18 mM Na2SO4, 1.4 M acetic acid). Derivatization was performed at 50°C for 2 h. Following the addition of 0.2 M NaOH, samples were filtered through 10 kDa MWCO filters (Millipore) by centrifugation and stored in the dark at −20°C until use. Separation was performed via reverse phase HPLC using a Poroshell 120 EC-C18 column (Agilent) with a gradient of acetonitrile in water: T(0 min) 2%; T(2 min) 2%; T(5 min) 5%; T(25 min) 10%; T(30 min) 50%; T(31 min) 100%; T(40 min) 100%; T(41 min) 2%; T(45 min) 2%. DMB-derivatized sialic acids were detected by excitation at 373 nm and monitoring emission at 448 nm. Sialic acids standards included N-acetylneuraminic acid (Neu5Ac; Jülich Fine Chemicals), N-glycolylneuraminic acid (Neu5Gc; Carbosynth), 3-deoxy-D-glycero-D-galacto-2-nonulosonic acid (KDN; Carbosynth), and the Glyko Sialic Acid Reference Panel (Prozyme).
Subcellular fractionation
Isolation of highly pure nuclei
Nuclei are intricately entwined with the ER, posing a challenge to biochemically separate nuclei cleanly from the ER without mixing. Gagnon et al. (2014) describe a protocol which cleanly recovers mammalian nuclei after processing without significant residual ER membrane attached. We performed this protocol on adherent ManNAz-labeled HeLa cells without modification to the step-by-step instructions published. Due to the stringent isolation of the nuclei, some fraction of nuclei themselves lyse during the process, contaminating the non-nuclear fraction. Therefore, when examining the fractionation results of this protocol, we consider only the signal left in the nucleus. Signal in the supernatant will be partially mixed ER, Golgi, cytosol, some nuclei, as well as other cellular compartments. After fractionation as per the protocol, TRIzol was used to extract and process the RNA.
Isolation of cytosol and crude membrane fractions
The ProteoExtract® Native Membrane Protein Extraction Kit (EMD Millipore) was used on adherent ManNAz-labeled HeLa cells. This kit uses serial lysis steps: first to gently release soluble cytosol proteins and RNA and second to rupture membranous organelles such as the plasma membrane, Golgi, and ER. Because the lysis buffers are gentle, residual ER/Golgi are left on the nuclear fraction and thus analysis of samples generated from this kit was limited to the efficiently separated soluble cytosolic fractions compared to the membranous fractions. Specifically, cultured HeLa cells first had growth media removed and cells were then washed twice with ice-cold Wash Buffer. Extraction Buffer I (supplemented with protease inhibitor) was added to culture plates and incubated on the cells for 10 min at 4°C, rocking. After the incubation, this buffer was collected as “cytoplasm.” Extraction Buffer II (supplemented with protease inhibitor) was subsequently added to the cells for 30 min at 4°C, rocking. This buffer was collected as “ER/membrane.” These fractions were then extracted with TRIzol and processed as described above.
Membrane protection assay
Large-scale crude membranes were isolated using the Plasma Membrane Protein Extraction Kit (ab65400, Abcam): cultured cells first had growth media removed and cells were then washed twice with ice-cold 1× PBS. In the second PBS wash, cells were scraped off the plate and spun down at 400× g for 4 min at 4°C. Cell pellets were resuspended in 2 mL of Homogenize Buffer Mix per 3× 15cm plates of 80% confluent 293T cells. Cell suspension was Dounce Homogenized on ice for 55 strokes, and this was repeated until all the cell suspension volume was similarly processed. Homogenate was then spun at 700× g for 10 min at 4°C. This pellet was the nuclear fraction and supernatants were transferred to new tubes and spun again at 10,000× g for 30 min at 4°C. The pellets generated from this spin were crude membranes and the supernatant was soluble cytosol. For the protection assay, typically 10× 15cm plates were used for each biological replicate. Crude membranes pellets were resuspended in 800 μL KPBS (136 mM KCl, 10 mM KH2PO4, pH 7.25 was adjusted with KOH) (Abu-Remaileh et al., 2017), 125 mM sucrose, and 2 mM MgCl2, split into 4 reactions, and incubated at 37°C for 1 h with or without 0.1% Triton X-100 or 150 nM VC-Sia (homemade as per above). RNA was extracted with TRIzol and processed as described above for DMB analysis of sialic acid levels.
Protein affinity tools: antibodies and lectins
The following were used for blotting on nitrocellulose membranes at the indicated concentrations: 1:1000 GAPHD (A300–641A, Bethyl), 1:3000 β-tubulin (ab15568, Abcam), 1:5000 H3K4me3 (ab8580, Abcam), 1:1000 RPN1 (A305–026A, Bethyl), 1:1000 Sec63 (A305–084A, Bethyl). Appropriate secondary antibodies conjugated to Li-Cor IR dyes (Li-Cor Biosciences) and used at a final concentration of 0.1 ng/μL. All lectins were bought biotinylated from Vector labs: biotin-wheat germ agglutinin (WGA), biotin-concanavalin A (ConA), and biotin-Maackia Amurensis Lectin II (MAAII). Pierce High Sensitivity Streptavidin-HRP (Strep-HRP, Thermo Fisher Scientific) was used for aniline labeling experiments.
Sucrose gradient fractionation of RNA
RNA used as input for sucrose gradient fractionation was previously extracted, PK treated, and clicked to DBCO-biotin as described above. RNA was sedimented through 15%–30% sucrose gradients following McConkey’s method (McConkey, 1967). Typically, 250–500 μg total RNA was lyophilized and then dissolved in 500 μL buffer containing 50 mM NaCl and 100 mM sodium acetate (pH 5.5). Linear 15%–30% sucrose gradients were prepared in 1×3.5 inch polypropylene tubes (Beckman) using a BioComp 107 Gradient Master. Dissolved RNA was layered on top of pre-chilled gradients, which were then centrifuged using a SW32 Ti rotor at 80,000× g (25,000 rpm) for 18 h in a Beckman Coulter Optima L70-K Ultracentrifuge at 4°C. Gradients were fractionated using a Brandel gradient fractionation system, collecting 0.75 mL fractions. Fractionated RNA was subsequently extracted from the sucrose solution using TRIzol as described above and analyzed by agarose gel electrophoresis or deep sequencing.
Enrichment, deep sequencing, and analysis of ManNAz-labeled RNA
Two rounds of selection performed on RNA samples before sequence analysis to identify transcripts modified with ManNAz-containing glycans. Total RNA from ManNAz-labeled H9 or HeLa cells was extracted, purified, and conjugated to DBCO-biotin as described above. Biological duplicates, at the cell culture level (different passage number), were generated for the purposes of the sequencing experiments. The first enrichment was achieved by sucrose gradient fractionation; after centrifugation fractions containing small RNAs were pooled and TRIzol extracted. The second enrichment was achieved by selective affinity to streptavidin beads as previously published (Duffy and Simon, 2016) with the following specific steps: 10 μL of MyOne C1 Streptavidin beads (Thermo Fisher Scientific), per reaction were blocked with 50 ng/μL glycogen (Thermo Fisher Scientific) in Biotin Wash Buffer (10 mM Tris HCl pH 7.5, 1 mM EDTA, 100 mM NaCl, 0.05% Tween-20) for 1 h at 25°C. Biotinylated small RNAs from H9 and HeLa cells were thawed and 150 ng of each were saved for input library construction. Next, 25 μg of the biotinylated small RNAs were diluted in 750 μL Biotin Wash Buffer (final concentration of ~33 ng/μL) and mixed with the blocked MyOne C1 beads for 2 h at 4°C. Beads were washed to remove non-bound RNAs: twice with 1 mL of ChIRP Wash Buffer (2× SSC, 0.5% SDS), twice with 1 mL of Biotin Wash Buffer, and twice with NT2 Buffer (50 mM Tris HCl, pH 7.5, 150 mM NaCl, 1 mM MgCl2, 0.005% NP-40), all at 25°C for 3 min each.
To construct deep sequencing libraries two approaches were taken using the same enzymes with different steps for the input (Flynn et al., 2016) versus bead-enriched (Zarnegar et al., 2016) samples given that the latter were already conjugated to a bead-support.
Input libraries
The 150 ng of small RNAs isolated before MyOne C1 capture were lyophilized dry and then T4 PNK mix (2 μL 5× buffer (500 mM Tris HCl pH 6.8, 50 mM MgCl2, 50 mM DTT), 1 μL T4 PNK (NEB), 1 μL FastAP (Thermo Fisher Scientific), 0.5 μL SUPERaseIn, and 5.5 μL water) was added for 45 min at 37°C. Next, a pre-adenylated-3′linker was ligated by adding 3′Ligation Mix (1 μL of 3 μM L3-Bio_Linker (Flynn et al., 2016), 1 μL RNA Ligase I (NEB), 1 μL 100 mM DTT, 1 μL 10× RNA Ligase Buffer (NEB) and 6 μL 50% PEG8000 (NEB)) to the T4 PNK reaction and incubating for 4 h at 25°C. Unligated L3-Bio_Linker was digested by adding 2 μL of RecJ (NEB), 1.5 μL 5′ Deadenylase (NEB), 3 μL of 10× NEBuffer 1 (NEB) and incubating the reaction at 37°C for 60 min. Ligated RNA was purified with Zymo columns as described above and lyophilized dry. cDNA synthesis, enrichment of cDNA:RNA hybrids, cDNA elution, cDNA circularization, cDNA cleanup, first-step PCR, PAGE purification, and second-step PCR took place exactly as previously describe (Zarnegar et al., 2016).
Bead-enriched libraries
Washed MyOne C1 beads bounded to ManNAz-labeled small RNAs were processed as described before (Flynn et al., 2016) with the following modifications. For the on-bead ligation step, a non-biotinylated 3′linker oligo was used (L3-Linker) (Flynn et al., 2016) such that all RNAs captured on the beads would be included in the sequencing library. After completing the second-step PCR for both the input and bead-enriched samples, the dsDNA libraries were quantified on a High Sensitivity DNA Bioanalyzer chip (Agilent) and sequenced on a NextSeq 500 instrument (Illumina).
Data analysis
Sequencing data were processed largely as described previously with a pipeline designed to analyze infrared CLIP data (Zarnegar et al., 2016). The specific version of the pipeline used in this work can be found here (https://github.com/ChangLab/FAST-iCLIP/tree/lite). Specifically, the raw reads were removed of PCR duplicates and adaptor sequences trimmed. Next, to address reads mapping to tRNA loci reads were first mapped to a mature tRNA reference using bowtie2 (Langmead and Salzberg, 2012). Mature tRNA reference were obtained from GtRNAdb (Chan and Lowe, 2009) and converted to DNA sequenced in FASTA format. Identical sequences were removed and CCA was added to the 3′ end of each tRNA sequence. Uniquely-mapped reads were extracted using the values of the NM and XS fields of the resulting SAM file (grep -E “@|NM”: *.sam|grep -v “XS”:) (Marchand et al., 2017). Next, reads were mapped to custom sequence indexes of human repetitive RNAs (such as snRNAs and rRNAs) and finally to the human genome reference (GCRh38) (Zarnegar et al., 2016). The number of unique reads for each RNA transcript (e.g., tRNAs, snRNAs, Y RNAs, etc) from each of the two biological replicates was used to calculate fold change between input and enriched samples (ManNAz or EDC capture methods) with the DESeq2 (Love et al., 2014) tool. Statistical analysis was performed using R, and plots generated using ggplot2 (Wickham, 2009) (https://ggplot2.tidyverse.org).
CRISPR/Cas9 knockout of Y5 and characterization
CRISPR gRNA sequences were designed using the CHOPCHOP online webtool (http://chopchop.cbu.uib.no/index.php) (Labun et al., 2016). Guides that flank the Y5 locus were selected (Table S4). Corresponding oligos were ordered from IDT. Oligos were cloned into the Zhang lab generated Cas9 expressing pX458 guide RNA plasmid (Addgene) as previously described (Ran et al., 2013) using Gibson assembly reaction (NEB). Two sgRNAs flanking the human Y5 locus encoded in the pX458 plasmids were co-transfected using Lipofectamine 3000 (Thermo Fisher Scientific) in a 6-well format. Transfected cells were single cell sorted based on GFP expression into 96-well plates using BD influx cell sorter (Stanford FACS facility). Clonal cell lines were allowed to expand, and genomic DNA was isolated for sequenced based genotyping of targeted allele. For this, a 300–500 base-pair region that encompassed the gRNA-targeted site was amplified and the PCR product was Sanger sequenced. Clones with editing events causing large deletions were selected for subsequent experiments and KO loss of expression was confirmed by Northern blotting (below). To evaluate doubling time, 293 WT and KO cells were cultured as described above, initially seeding 20,000 cells per 12-well plate in triplicate. At 24-hour intervals cells were trypsinized and counted using a Countess II FL Automated Cell Counter (Thermo Fisher Scientific).
Small RNA Northern blotting
Detection of small RNAs was achieved by conventional Northern blotting and detection via radiolabeled locked-nucleic acids (LNAs). LNAs (QIAGEN) complementary to the Y5 RNA or 5S rRNA (Table S3) were ordered and 5′end labeled: 200 pmol LNA was added to 3 μL of T4 PNK (NEB), 7 μL 10× T4 PNK buffer, and 1 μL of ATP, [γ−32P]- 3000 Ci/mmol 10 mCi/ml (γ-ATP, Perkin Elmer) in a 70 μL reaction. LNAs were incubated at 37°C for 3 h after which free γ-ATP was purified away using Micro Bio-Spin 6 (Bio-Rad) columns. Columns were brough to 25°C, pre-packed buffer spun out at 1000× g for 2 min. Samples were applied to the dried column matrix and purified by spinning at 1000× g for 4 min. A 12% Urea-PAGE gel (National Diagnostics) was poured and pre-run at 10W for 15 min, after which 2 μg of total RNA from various cell types was separated by running the gel at 15W. After electrophoresis, RNA was transferred to HyBond N+ (GE Life Sciences) using a Semi-Dry transfer apparatus (Bio-Rad) with 0.5× Tris/Borate/EDTA (TBE, Thermo Fisher Scientific) buffer at a constant power of 18V for 90 min at 4°C. Next, RNA was crosslinked to the membrane, and pre-hybridized at 65°C for 60 min in 2 mL of PerfectHyb Plus (Sigma) buffer. Labeled LNA probes were then added to the PerfectHyb Plus buffer (typically 25% of the labeled LNA probe was used for any single membrane hybridization) and incubated at 65°C for 3–16 h (no change in results with longer or shorter hybridizations). Membranes were rinsed 2× 2.5 mL of Low Stringency Northern Buffer (0.1% SDS, 2× SSC (Saline-sodium citrate)) and then washed at 37°C for 2× 5 min in 2.5 mL of High Stringency Northern Buffer (0.1% SDS, 0.5× SSC). Wash membranes were exposed to storage phosphor screens and finally imaged with a GE Typhoon 9410 scanner.
Glycan release from RNA samples
Small RNAs were isolated as described above. RNA samples were sequentially digested with two glycosidases. Typically for experimental samples, 25 μg of small RNA from H9 ES, HeLa, or 293FT cells was resuspended in 10 μL of 1× GlycoBuffer 2 (NEB), 7.5 μL PNGaseF (NEB) and to a final reaction volume of 100 μL with water. PNGaseF cleavage occurred overnight at 37°C. After digestion, released glycans were desalted using PGC SPE columns (Thermo Fisher Scientific). SPE columns were first washed 5× with 80% acetonitrile (ACN) + 0.1% Trifluoroacetic acid (TFA) and then 0.1% TFA. Samples were brought to 500 μL with water and passed over the column twice. SPEs were washed once with 0.1% TFA and finally eluted sequentially in 15% ACN in 0.1% TFA, 35% ACN in 0.1% TFA. ACN was pulled off with a SpeedVac (Labconco), elutions pooled, and dried by lyophilization. After drying, samples were resuspended in 5 μL LC-MS grade water (Thermo Fisher Scientific) for MS analysis.
Glycan release from peptide samples
Peptides were generated from total cell lysate material from H9 ES, HeLa, or 293FT cells. Specifically, 100 μg of protein lysate was processed into tryptic peptides using an S-trap mini column (Protifi). Lysate solutions were brought to 5% SDS and 5 mM DTT final concentration, heated to 95°C for 5 min, cooled to 25°C for 5 min, and then added 25 mM iodoacetamide (Sigma) for alkylation at 25°C for 30 min in the dark. Samples were next acidified by adding phosphoric acid (Sigma) to 1.2% final concentration and then adding 8× volumes of binding buffer (100 mM triethylamonium bicarbonate (TEAB) in 90% methanol), vortexing to mix. Protein samples were next bound the S-trap columns by centrifugation at 4000× g for 10 sections, spins were repeated until all the sample volume had passed over the column matrix. Three washes with binding buffer were performed to rinse the column. Peptides were generated by applying Trypsin (Promega) solution to the column matrix in 50 mM ammonium bicarbonate (Sigma) at a ratio of 1 μg Trypsin to 20 μg protein lysate. Digestion proceeded for 90 min at 47°C. Peptides were eluted by sequentially applying 0.1% formic acid in 50 mM ammonium bicarbonate and 0.1% formic acid in 50% acetonitrile. N-glycans were liberated from the peptide samples as described above for the RNA. After PNGaseF digestion, deglycosylated peptides were removed by bringing the peptide mixture to 500 μL in 0.2% formic acid and passing them over a 10 mg polymeric C-18 SPE (Strata-X) column: free glycans will flow through and were saved. The free glycans were desalted in parallel to the RNA samples with a PGC SPE and finally samples were resuspended in 5 μL LC-MS grade water (Thermo Fisher Scientific) for MS analysis.
Mass Spectrometry
Chromatography
Mass spectrometric data were acquired using the following conditions. Each dried sample was reconstituted in 10 μL of 5 mM ammonium formate and 3 μL of sample were injected onto an UltiMate 3000 RSLCnano UPLC (Thermo Fisher Scientific) system equipped with a 5 μL injection loop. Separation was performed with a capillary column (100 μm ID, 18 cm length) created by hand packing a commercially available fused-silica column (IntegraFrit, New Objective, Woburn, MA) with 5 μm porous graphitic carbon (Hypercarb, PGC, Thermo Fisher Scientific, Waltham, MA) connected to stainless steel emitter (30 um ID, Thermo Fisher Scientific). Mobile phases used were 5 mM ammonium formate (A) and 2:1 isopropanol: acetonitrile (B). The flow rate was 1000 nL/min for 5.5 min at 100% A, then decreased to 300 nL/min over 0.5 min followed by a linear gradient of 15%/min over 1 min., 1.4%/min over 25 min, 6.25%/min over 8 min then followed by a 2 min hold at 100% B, with re-equilibrated at 100% A for 5 min. at 1000 nL/min (including injection time for subsequent injection). The injection valve was switched at the 5.5 min point of the run to remove the sample loop from the flow path during the gradient.
Mass spectrometer
All mass spectrometric data were acquired with a Lumos Orbitrap mass spectrometer (Thermo Fisher Scientific). Positive mode electrospray ionization was performed under nanospray conditions (300 nL/min) using a Thermo Fisher Scientific Nanoflex ion source with a source voltage of 2.2 kV applied to a stainless-steel emitter (30 um ID, Thermo Fisher Scientific), and the capillary temperature was 300°C. The S-Lens RF level setting was 60%.
Free-glycan untargeted screening
Data-dependent fragmentation was performed with full scan (m/z 500 2500) Orbitrap detection at a resolution setting of 120,000, normalized AGC target of 250%, and a maximum ion injection time setting of 50 ms. MS2 spectra were acquired with quadrupole isolation width of m/z 1.6, HCD fragmentation of 25%, Orbitrap detection at a resolution setting of 15000, normalized AGC target of 400%, and maximum ion injection time of 22 ms. Data-dependent parameters were as follows: intensity threshold 2.5 × 104, repeat count of 3 within 30 s, exclusion duration of 20 s, and exclusion mass width of ± 5 ppm with isotopes excluded. A mass exclusion list consisting of previously published endogenous RNA adducts and their 13C isotopologues was used (Table S4). A cycle time of 3 s was used, and data collection was in profile mode.
Analysis
Glycan release samples were analyzed with GlycoNote (https://github.com/MingqiLiu/GlycoNote). Briefly, .raw files were converted to .mgf files and loaded into the GlyoNote GUI. The parameters (Table S5) were used for all glycan release files. GlycoNote output files contained glycan structures and annotated spectra, which were validated manually.
Lectin-proximity labeling of RNA with biotin-aniline
Live Cell Labeling
HeLa cells were cultured as above typically in 10 cm plates. Cells were rinsed twice in ice-cold 1× PBS, discarding after each wash, and blocked in Lectin Blocking Buffer (LBB, 20 mM HEPES, 150 mM NaCl, 1 mM MgCl2, 1 mM MnCl2, 1 mM CaCl2, 2.5% FBS) for 15 min at 4°C. Blocking buffer was then discarded and replaced with LBB+Lectin+Strep-HRP. Typically, 4 mL of this was prepared at a concentration of 5 μg/mL biotinylated lectin and 6 μg/mL Strep-HRP, these components were first mixed together on ice for 30 min prior to the addition of LBB. LBB+Lectin+Strep-HRP staining occurred for 45 min at 4°C, after which the cells were rinsed twice in ice-cold 1× PBS + 1 mM CaCl2 + 1 mM MgCl2 (PBS++). Immediately after this 3 mL of PBS++ with 350 μM biotin-aniline (Iris Biotech GMBH) was added to each plate and incubated on ice for 1 min. Plates were then moved to the bench top and H2O2 was added to a final concentration of 1 mM. This reaction occurred for precisely 2 min, after which plates were brought back on ice, PBS++/biotin-aniline/H2O2 was aspirated, and cells were quickly but gently rinsed twice in Quenching Buffer: 5 mM Trolox, 10 mM sodium ascorbate and 10 mM sodium azide in PBS++ (as described in Fazal et al., 2019). After removing the Quenching Buffer, TRIzol was added directly to the plate and RNA extracted and processed as described above for enzymatic digestions as well as blotting.
In Lysate Labeling
HeLa cells were grown and washed as for the Live Cell Labeling protocol. Cell lysates were generated by adding 500 μL ice-cold 50 mM Tris pH 8 with cOmplete protease inhibitor cocktail (Roche) per 10 cm plate, scraping cells, and pipetting up and down 10 times on ice. Lysate from each 10 cm plate was then incubated with the same pre-complexed ratio of biotinylated lectin and Strep-HRP for 45 min on ice. Lysates were subsequently warmed to 25°C for 2 min, 350 mM biotin-aniline was added to each tube at 25°C for 1 min and then 1 mM H2O2 was added to initiate the reaction. Each reaction was allowed to proceed for exactly 2 min before directly adding 5 mM Trolox, 10 mM sodium ascorbate and 10 mM sodium azide. RNA was extracted from the labeled lysate samples with TRIzol LS (Thermo Fisher Scientific) and processes in parallel with the Live Cell samples.
Optimization of cell-lifting reagents
In the course of establishing a FACS protocol for cell surface glycoRNAs (below), we noticed that standard cell lifting strategies using trypsin resulted in near total destruction of cellular RNA. To understand why this was happening and find an RNA-safe strategy we performed the following quality control experiments:
First, total protein analysis of Trypsin and TrypLE reagents used for tissue culture. Stock Trypsin products from GE Healthcare, Sigma, Stem Cell Technologies, ATCC, and Thermo Fischer Scientific were purchase, separated on an SDS-PAGE gel, stained with Acquastain Protein Gel Stain (Bulldog Bio) and scanned on a Li-Cor to visualize any proteinaceous components of these reagents (Figure S6B). All Trypsin products contained a band that corresponded to the full-length trypsin protein at about 25 kDa, however, every stock also contained a series of lower molecular weight bands of unknown identity.
Second, TrypLE, from the Thermo Fischer Scientific website is an “animal origin-free, recombinant enzyme” that because of its “exceptional purity increases specificity and reduces damage to cells that can be caused by other enzymes present in some trypsin extracts.” We found that TrypLE runs at a very similar molecular weight as trypsin, however, it contains none of these low molecular weight bands (Figure S6B).
Third, we assessed the relative damage these reagents cause to cellular RNA. HeLa cells were grown in 6-well plates as described above, rinsed with 1× PBS, and then incubated at 37°C for 5 min in 250 μL of 1× PBS, Trypsin (GE Healthcare), or TrypLE (Thermo Fischer Scientific). After this incubation, samples were either directly lysed with 750 μL TRIzol LS or resuspended in 750 μL 1× PBS, spun at 300× g for 5 min, supernatant discarded, and then the cell pellet was lysed in TRIzol LS. The results of this experiment are in Figure S6C and show that while PBS and TrypLE cause no RNA degradation, the Trypsin solution completely destroys the RNA if cells are not pre-washed with PBS, and if they are, there is still massive degradation of cellular RNA, even when extracted with TRIzol.
Therefore, it is critical to use TrypLE or other reagents that have been carefully tested for RNase contamination when performing experiments to assay for cell surface glycoRNAs.
Fluorescence-activated cell sorting (FACS) analysis
Cells were grown as described above and if adherent, lifted with TrypLE (Thermo Fisher Scientific) for 4 min at 37°C. Cells were resuspended in FACS Buffer (0.5% bovine serum albumen (Thermo Fisher Scientific) in 1× PBS), counted, and aliquoted to 200,000 cells per 100 μL FACS Buffer, incubating on ice for 30 min to blocking. For RNase digestions, RNase A (Sigma) was added to the blocking buffer at indicated concentrations (typically 2 μM). After blocking, cells were brought to 25°C for 5 min, then spun for 5 min at 4°C and 350× g. Cells were washed once with 150 μL FACS Buffer and spun as above. Two similar approaches were taken to stain for cell surface RNA (1) or cell surface sialic acids (2). In assaying for (1), cells were resuspended in 10 μg/mL anti-J2 antibody (Scicons) in 100 μL FACS Buffer for 30 min on ice, spun as above and washed once as above. Cells were then stained with 8 μg/mL Goat, anti-Mouse-IR680 antibody (Li-Cor Bioscience) in 100 μL FACS Buffer for 30 min on ice and in the dark, spun as above and washed once as above. Finally, cells were fix in 100 μL of FluoroFix Buffer (BioLegend) for 30 min at 25°C in the dark. Cells were finally washed once as above and stored in FACS Buffer at 4°C for analysis. In assaying for (2), recombinant human Siglec-Fc proteins (R&D Systems) were pre-complexed to Alexa Fluor-647 AffiniPure Donkey Anti-Human IgG, Fcγ fragment specific (Jackson Laboratories) both at 1.5 μg/mL in FACS Buffer on ice for 1 h. Cells were resuspended in 100 μL of the pre-complexed Siglec-Fc-Secondary solution and incubated on ice in the dark for 30 min, washed once, and proceeded directly to fixation as described above. FACS data were analyzed and visualized with FloJo software.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq statistics were determined by R package ‘DESeq2.’
Supplementary Material
Figure S1. Ac4ManNAz incorporation into RNA from multiple mammalian cell types, related to Figure 1
(A) SDS-PAGE analysis of Proteinase K digestion of recombinant protein under conditions used to remove protein contaminants from RNA samples.
(B) Blotting of Ac4ManNAz-labeled HeLa RNA with and without Proteinase K treatment of the RNA before copper-free click labeling and visualization. Biological duplicates are shown.
(C) Blotting of in cellulo or in vitro Ac4ManNAz-labeled RNA. Cells were treated with 100 μM Ac4ManNAz for 24 hours while native RNA in vitro was treated with up to 20 mM Ac4ManNAz for 2 hours at 37°C.
(D) Agarose gel analysis of genomic DNA isolated from HeLa cells and subjected to the DNase digestion conditions used to clean up RNA samples.
(E, F) Blotting of RNA from various cell types labeled with Ac4ManNAz including (E) HeLa, GM12878, K562, MYC T-ALL 4188, and CHO cells and (F) HeLa and H9 cells. RNase controls are provided for each cell type assayed.
Figure S2. glycoRNAs are non-polyadenylated, small RNAs, related to Figure 2
(A) Blotting of Ac4ManNAz-labeled HeLa RNA isolated before and at each step of the poly-A enrichment protocol. Three times the amount of RNA was used for the poly-A enrichment (60 μg) such that even low levels of Ac4ManNAz-labeling of poly-A RNA could be detected. FT = flowthrough. 2×-pA = double poly-A enriched RNA.
(B) Representative 254nm signal trace of total RNA signal across the sucrose fractionation. Fractions containing small RNAs, as indicated on the profile with a red bar, were pooled and purified for sequence analysis.
(C) Cross correlation analysis of reads from input and Ac4ManNAz-enriched libraries generated from HeLa and H9 cells.
(D) Pie charts comparing the distribution of mapped reads between input and Ac4ManNAz-enriched libraries generated from HeLa and H9 cells. Colors are used to highlight the common RNA families across libraries.
(E) Correlation analysis at the transcript level of non-tRNA (left) and tRNA (right) transcripts between the HeLa and H9 input sequencing libraries. Error bars are standard deviation of each RNA expressed across the biological duplicate libraries.
(F) volcano plot analysis of Ac4ManNAz-enriched RNAs identified from HeLa small RNA. RNA families are grouped by colors and plotted by their log2-enrichment values in the Ac4ManNAz captured library (x axis) and the confidence of that enrichment value (y axis, -log10(adjusted p value)). Dashed lines are enrichment and p value cutoffs are shown.
(G) Volcano plot analysis as in (F) from H9 cell small RNAs.
(H) Scatterplot analysis Ac4ManNAz-enriched RNAs purified from the small fractions of HeLa and H9 cells. Reads mapping to tRNAs are shown. Significance scores (-log10(adjusted p value) are overlayed for HeLa cells as the size of each datapoint and for H9 cells as the color of each datapoint.
Figure S3. Generation of Y5 knockout 293T cells and DMB detection and quantification of sialic acids on unlabeled cellular RNA, related to Figures 2 and 3
(A) Schematic of the wild-type (WT) locus of the human Y5 transcript. The regions targeted by the two designed sgRNAs are highlighted in orange with the protospacer adjacent motif (PAM) regions in green boxes. The first nucleotide of the Y5 RNA is marked with a green arrow.
(B) PCR analysis of genomic DNA (gDNA) from WT and Y5 KO clones. Primers targeting the Y5 locus (left) showed two new bands in the KO clone.
(C) Schematic of sanger sequencing results from the gDNA PCR in (B). Two specific sequences were isolated by sequencing. A deletion (black) comes from the bottom band in the gel. A separate insertion (red)/deletion (black)/mutation (blue) structure was observed which comes from the top PCR band.
(D) Northern blot of 293T WT and KO total RNA. The KO resulted in a complete loss of the Y5 RNA, with the 5S rRNA serving as a loading control.
(E) Analysis of growth rate between the WT and KO clones across four days of culture. Three independent wells of cells were counted on each day.
(F) Schematic of experimental steps of the DMB assay with associated structures of the two major types of sialic acid (Neu5Ac and Neu5Gc) as well as the DMB reagent.
(G) HPLC-fluorescence traces of DMB modified HeLa (left) and 4188 (right) cellular RNA. The main sialic acid peaks are #2 and 3. Peak 1 is unknown but is sensitive to RNase. Insets are Sybr stained gels of the total RNA for each condition: no enzymes, RNase cocktail, or Sialidase treatment.
(H) HPLC traces of DMB modified 293T (left) and HeLa (right) cellular RNA or DNA. Neu5Ac and Neu5Gc peaks are highlighted across biological duplicates from each cell type with input material run in a denaturing agarose gel for sizes and loading controls (insets). Only cellular RNA produces Neu5Ac and Neu5Gc specific peaks after DMB derivatization.
Figure S4. Cellular and in vitro assays define the requirement of the N-glycan pathway for glycoRNA, related to Figure 4
(A) Diagram of the main cellular pathway for interconversion of glucose (Glc) to galactose (Gal). Normal cell culture media contains Glu without Gal. The enzyme GALE is responsible for converting uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc) to UDP-GalNAc and UDP-Glc to UDP-Gal. ldlD CHO cells contain a loss-of-function mutation in the GALE pathway rendering them deficient in the production of N- and O-linked glycans when grown in normal media. If supple- mented with Gal or GalNAc, these cells can selectively elaborate N- and O-linked glycans, respectively. Importantly, biosynthesis of CMP-sialic acid is unaffected by GALE mutation.
(B) Blotting of RNA from K562-GALE-KO cells labeled with Ac4ManNAz, Gal (10 μM), or GalNAc (100 μM) for 24 hours.
(C) An overview of major steps in the ER- and Golgi-localized steps of N-glycan biosynthesis. Dolichol-linked precursor glycans are transferred to ER-lumen- facing peptides (X) by OST. These glycans are sequentially modified by enzymes that first trim and then elaborate the glycan tree. Specific chemical inhibitorssuch as NGI-1 (inhibiting OST), Kifunesine (inhibiting a-mannosidase I) and Swainsonine (inhibiting α-mannosidase II) are employed to interrogate the importance of each of these enzymes to the accumulation of Ac4ManNAz-labeling of RNA.
(D) Blotting of Ac4ManNAz-labeled HeLa cell total RNA after treatment with the indicated concentrations of Swainsonine.
(E) Blotting of Ac4ManNAz-labeled HeLa cell total RNA after in vitro treatment with the indicated enzymes; representative of three independent experiments and quantified results are presented in Figure 4D.
(F) Blotting of Ac4ManNAz-labeled HeLa cell total RNA after in vitro treatment with the mucinase StcE. Addition of EDTA inhibits the mucinase as a control.
Figure S5. Membrane isolation and live-cell assays define the cell surface as a major reservoir for glycoRNAs, related to Figure 5
(A) Schematic of isolated cellular membranes treated with or without Triton X-100 or VC-Sia for 1 hour at 37°C before isolating RNA and assaying sialic acid levels via DMB.
(B) Quantification of membrane protection assay performed in biological quadruplicates from 293T cells. Significance calculated with an unpaired t test.
(C) Blotting of RNA from HeLa cells labeled with 100 μM Ac4ManNAz for 24 hours and then expose to fresh media containing 100 μM Ac4ManNAz with or without 150 nM VC-Sia for 20, 40, or 60 minutes at 37°C.
(D) SDS-PAGE analysis followed by blotting and visualization of biotin signal with StrepIR800. Samples were generated from cells in Figure 5F, here protein lysate was analyzed revealing that all three biotinylated lectins are capable of activating biotin-aniline and labeling cell surface proteins.
Figure S6. RNA and sialic acid affinity reagents stain cell surfaces in a manner depended on RNA, related to Figure 6
(A) Native PAGE of synthetic Y5 RNA 3′ end labeled with Cy-7 (Y5-Cy7) bound to antibodies, J2 or a non-targeting IgG, with or without competing dsRNA (poly- (I:C)). Quantification of the shifted (red) and free (black) Y5-Cy7 is plotted blow each corresponding lane.
(B) SDS-PAGE analysis of trypsin and TrypLE products stained for total protein content. Regions of each lane are annotated with presumptive identity of bands.
(C) Agarose gel analysis of total cellular RNA stained with Sybr for visualization. To live cells, PBS, Trypsin, or TrypLE was added and incubated for 5 minutes at 37°C. RNA was directly extracted from this or extracted after rinsing the cells once in PBS. RNA stability was significantly impacted with the trypsin reagent with or without PBS washing.
(D) FACS analysis of single 293T (left) or K562 (right) cells pre-treated or not with RNase A (red outline) and stained with the J2 antibody (orange).
(E) Representative confocal images of HeLa cells stained with the J2 antibody with or without prior treatment with RNase A. Image acquired at 40× magnification and scale is as indicated on the image.
(F) FACS analysis of single HeLa cells stained with a panel of commercially available recombinant human Siglec-Fc reagents. Siglec-2, −3, and -Mag did not stain HeLa cells above background.
(G) FACS analysis of single HeLa cells stained with the 9 Siglec-Fc reagents that were above background in (F). Red outlines indicate cells were pre-treated with RNase A.
KEY RESOURCES TABLE
| Reagent or resource | Source | Identifier |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| anti-RPN1 | Bethyl Laboratories | Cat# A305–026A; RRID: AB_2621220 |
| anti-GAPDH | Bethyl Laboratories | Cat# A300–641A; RRID: AB_513619 |
| anti-b-tubulin | Abcam | Cat# ab15568; RRID: AB_2210952 |
| anti-H3K4me3 | Abcam | Cat# ab8580; RRID: AB_306649 |
| anti-Sec63 | Bethyl Laboratories | Cat# A305–084A; RRID: AB_2631479 |
| IRDye 800CW Goat anti-Mouse IgG Secondary Antibody | Li-Cor Biosciences | Cat# 926–32210; RRID: AB_621842 |
| IRDye 800CW Goat anti-Rabbit IgG Secondary Antibody | Li-Cor Biosciences | Cat# 926–32211; RRID: AB_621843 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| RPMI 1640 Medium | Thermo Fisher Scientific | Cat# 11875085 |
| mTeSR 1 Media | STEMCELL Technologies | Cat# 85850 |
| Ham’s F-12 Nutrient Mix Media | Thermo Fisher Scientific | Cat# 11765062 |
| DMEM media, high glucose | Thermo Fisher Scientific | Cat# 11965092 |
| Penicillin/streptomycin | Sigma | Cat# 15140122 |
| Matrigel matrix | Corning | Cat# 354234 |
| Fetal Bovine Serum | Thermo Fisher Scientific | Cat# 16000036 |
| N-Acetyl-9-azido-9-deoxy-neuraminic acid; 9Az sialic acid | Carbosynth | Cat# MA30919 |
| N-azidoacetylmannosamine-tetraacylated; Ac4ManNAz | Click Chemistry Tools | Cat# 1084 |
| Dimethyl sulfoxide, anhydrous | Sigma | Cat# 276855 |
| N-Acetyl-D-galactosamine; GalNAc | Sigma | Cat# A2795 |
| D-(+)-Galactose; Gal | Sigma | Cat# G0750 |
| NGI-1 | Sigma | Cat# SML1620 |
| Kifunensine | Sigma | Cat# K1140 |
| Swainsonine | Sigma | Cat# S9263 |
| P-3FAX-Neu5Ac | Torcis | Cat# 5760 |
| TRIzol | Thermo Fisher Scientific | Cat# 15596026 |
| Chloroform | Sigma | Cat# C2432 |
| Proteinase K | Thermo Fisher Scientific | Cat# AM2546 |
| TURBO DNase | Thermo Fisher Scientific | Cat# AM2238 |
| RNase cocktail | Thermo Fisher Scientific | Cat# AM2286 |
| a2–3,6,8,9 Neuraminidase A | New England Biolabs | Cat# P0722 |
| Endo Hf | New England Biolabs | Cat# P0703 |
| PNGaseF | New England Biolabs | Cat# P0704 |
| Endo F2 | New England Biolabs | Cat# P0772 |
| Endo F3 | New England Biolabs | Cat# P0771 |
| O-Glycosidase | New England Biolabs | Cat# P0733 |
| StcE | Malaker et al., 2019 | N/A |
| Vibrio cholerae Sialidase; VC-Sia | Gray et al., 2020 | N/A |
| dibenzocyclooctyne-PEG4-biotin; DBCO-biotin | Sigma | Cat# 760749 |
| UltraPure Formamide | Thermo Fisher Scientific | Cat# 15515026 |
| UltraPure EDTA | Thermo Fisher Scientific | Cat# 15575020 |
| UltraPure SDS | Thermo Fisher Scientific | Cat# 15553027 |
| SYBR Gold Nucleic Acid Gel Stain | Thermo Fisher Scientific | Cat# S11494 |
| 0.45 um nitrocellulose membrane | Cytiva Life Sciences | Cat# 10600002 |
| Odyssey Blocking Buffer | Li-Cor Biosciences | Cat# 927–40000 |
| IRDye 800CW Streptavidin | Li-Cor Biosciences | Cat# 926–32230 |
| Tween-20 | Sigma | Cat# P7949 |
| 4,5-methylenedioxy-1,2-phenylenediamine dihydrochloride; DMB | Sigma | Cat# 66807 |
| Acetic Acid | Sigma | Cat# 695092 |
| 2-Mercaptoethanol | Sigma | Cat# M6250 |
| Sodium Sulfate | Sigma | Cat# 239313 |
| N-acetylneuraminic acid; Neu5Ac | Carbosynth | Cat# MA00746 |
| N-glycolylneuraminic acid; Neu5Gc | Carbosynth | Cat# MG05324 |
| 3-deoxy-D-glycero-D-galacto-2-nonulosonic acid; KDN | Carbosynth | Cat# MD04703 |
| Glyko Sialic Acid Reference Panel | Agilent | Cat# GKRP-2503 |
| biotin-wheat germ agglutinin; WGA | Vector Laboratories | Cat# B-1025 |
| biotin-concanavalin A; ConA | Vector Laboratories | Cat# B-1005 |
| biotin-Maackia Amurensis Lectin II; MAAII | Vector Laboratories | Cat# B-1265 |
| Pierce High Sensitivity Streptavidin-HRP; Strep-HRP | Thermo Fisher Scientific | Cat# 21130 |
| Sucrose | Sigma | Cat# S0389 |
| Dithiothreitol; DTT | Thermo Fisher Scientific | Cat# R0861 |
| MyOne C1 Streptavidin beads | Thermo Fisher Scientific | Cat# 65001 |
| Glycogen | Thermo Fisher Scientific | Cat# AM9510 |
| T4 PNK | New England Biolabs | Cat# M0201 |
| FastAP | Thermo Fisher Scientific | Cat# EF0651 |
| RNA Ligase I | New England Biolabs | Cat# M0204 |
| 5′ Deadenylase | New England Biolabs | Cat# M0331 |
| RecJf | New England Biolabs | Cat# M0264 |
| gamma-32p-ATP | Perkin Elmer | Cat# NEG502Z250UC |
| UltraPure TBE Buffer | Thermo Fisher Scientific | Cat# 15581044 |
| Hybond-N+ Membrane | VWR | Ca# 95038–376 |
| PerfectHyb Plus | Sigma | Cat# H7033 |
| UltraPure SSC | Thermo Fisher Scientific | Cat# 15557044 |
| Acetonitrile, Optima LC/MS Grade | Fisher Scientific | Cat# A955–500 |
| Trifluoroacetic acid for HPLC, ≥ 99.0% | Sigma | Cat# 302031 |
| Formic Acid, 99.0+%, Optima LC/MS Grade | Fisher Scientific | Cat# A117–50 |
| Ammonium bicarbonate | Sigma | Cat# A6141 |
| Ammonium formate | Sigma | Cat# 70221 |
| Water, Optima LC/MS Grade | Fisher Scientific | Cat# W6500 |
| Isopropanol, Optima LC/MS Grade | Fisher Scientific | Cat# A461–500 |
| Sodium ascorbate | Sigma | Cat# A7631 |
| Trolox | Sigma | Cat# 238813 |
| Sodium Azide | Sigma | Cat# S2002 |
| Manganese(II) chloride | Sigma | Cat# 244589 |
| Calcium chloride | Sigma | Cat# 499609 |
| Magnesium chloride | Sigma | Cat# 63069 |
| Biotin-aniline | Iris Biotech GMBH | Cat# LS-3970 |
| TRIzol LS | Thermo Fisher Scientific | Cat# 10296028 |
| rec-hSiglec-2 | R&D Systems | Cat# 1968-SL-050 |
| rec-hSiglec-3 | R&D Systems | Cat# 1137-SL-050 |
| rec-hSiglec-Mag | R&D Systems | Cat# 8940-MG-050 |
| rec-hSiglec-5 | R&D Systems | Cat# 1072-SL-050 |
| rec-hSiglec-6 | R&D Systems | Cat# 2859-SL-050 |
| rec-hSiglec-7 | R&D Systems | Cat# 1138-SL-050 |
| rec-hSiglec-8 | R&D Systems | Cat# 9045-SL-050 |
| rec-hSiglec-9 | R&D Systems | Cat# 1139-SL-050 |
| rec-hSiglec-10 | R&D Systems | Cat# 2130-SL-050 |
| rec-hSiglec-11 | R&D Systems | Cat# 3258-SL-050 |
| rec-hSiglec-14 | R&D Systems | Cat# 4905-SL-050 |
| rec-hSiglec-15 | R&D Systems | Cat# 9227-SL-050 |
| rec-hIgG1 | R&D Systems | Cat# 110-HG-100 |
| Bovine Serum Albumin | Sigma | Cat# A9418 |
| IRDye 680 RD Goat anti-Mouse IgG | Li-Cor Biosciences | Cat# 926–68070 |
| AffiniPure Mouse Anti-Human IgG, Fcγ fragment specific | Jackson ImmunoResearch | Cat# 209–605-098 |
| J2 antibody | Scicons | Cat# J2 |
| FluoroFix Buffer | BioLegend | Cat# 422101 |
| TrypLE | Thermo Fisher Scientific | Cat# 12604021 |
| Trypsin LC-MS grade | Promega | Cat# V5117 |
| Trypsin | HyClone | Cat# SH3004201 |
| Trypsin | Thermo Fisher Scientific | Cat# 25300054 |
| Trypsin | Sigma | Cat# T4174 |
| Trypsin | Sigma | Cat# T4549 |
| Trypsin | Stem Cell Technologies | Cat# 07901 |
| Trypsin | ATCC | Cat# 30–2101 |
|
| ||
| Critical commercial assays | ||
|
| ||
| ProteoExtract Native Membrane Protein Extraction Kit |
EMD Millipore | Cat# 444810 |
| Plasma Membrane Protein Extraction Kit | Abcam | Cat# ab65400 |
| RNA Clean and Concentrator 5 | Zymo Research | Cat# R1013 |
| Poly(A)Purist MAG Kit | Thermo Fisher Scientific | Cat# AM1922 |
|
| ||
| Deposited data | ||
|
| ||
| Input- and ManNAz-seq from HeLa cells | This Study | Gene Expression Omnibus: GSE136967 |
| Input- and ManNAz-seq from H9 ES cells | This Study | Gene Expression Omnibus: GSE136967 |
|
| ||
| Experimental models: cell lines | ||
|
| ||
| HeLa | ATCC | Cat# ATCC-CCL-2 |
| HEK293T | ATCC | Cat# ATCC-CRL-3216 |
| K562 | ATCC | Cat# ATCC-CCL-243 |
| T-ALL 4188 | Lab of Dean Felsher (Stanford); Felsher and Bishop, 1999 | N/A |
| H9 ES cells | WiCell | Cat# WA09 |
| GM12878 | Lab of Howard Chang (Stanford) | N/A |
| K562GALE−/− | Lab of Carolyn Bertozzi (Stanford); Schumann et al., 2020 | N/A |
| CHO-K1 | ATCC | Cat# ATCC-CCL-61 |
| ldlD-CHO | Lab of Monty Krieger (MIT); Kingsley et al., 1986 | N/A |
|
| ||
| Experimental models: organisms/strains | ||
|
| ||
| Mouse: C57BL/6 | Jackson Laboratories | N/A |
|
| ||
| Oligonucleotides | ||
|
| ||
| RNY5_gDNA_PCR_F: GTTGATTAACATAATTCTTAC |
This paper | N/A |
| RNY5_gDNA_PCR_R: CATTTTGAAGGTTAATACTTC |
This paper | N/A |
| sgRNA_RNY5–1_px458_F: CACCGCACAGTTGGTCCGAGTGTTG |
This paper | N/A |
| HS_Y5_LNA: aa+cag+caa+gct+agt+caa+gcg |
This paper | N/A |
| HS_5S_LNA: cga+ccc+tgc+tta+gct+tcc+ga |
This paper | N/A |
| sgRNA_RNY5–1_px458_R: AAACCAACACTCGGACCAACTGTGC |
This paper | N/A |
| sgRNA_RNY5–2_px458_F: CACCGAAGCTAGTCAAGCGCGGTTG |
This paper | N/A |
| sgRNA_RNY5–2_px458_R: AAACCAACCGCGCTTGACTAGCTTC |
This paper | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| R | R Project | https://www.r-project.org/ |
| ggplot2 | https://www.tidyverse.org/ | https://ggplot2.tidyverse.org |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| FAST-iCLIP | Flynn et al., 2015 | https://github.com/ChangLab/FAST-iCLIP/tree/lite |
| CHOPCHOP | Labun et al., 2019 | https://chopchop.cbu.uib.no/ |
| Adobe Illustrator CC | Adobe | https://www.adobe.com |
| GlycoNote | Dr. Ming Qi Liu | https://github.com/MingqiLiu/GlycoNote |
Highlights.
Synthetic, clickable sugars that label glycoproteins and glycolipids also label RNAs
RNA-glycan conjugates, glycoRNAs, are conserved, small, noncoding RNAs
GlycoRNAs possess N-glycans that are highly sialylated and fucosylated
GlycoRNAs are displayed on the cell surface and can bind Siglec receptors
ACKNOWLEDGMENTS
We thank Phillip Sharp, Robert Spitale, Eliezer Calo, Steven Banik, and the Bertozzi Group members for critical discussion. We also thank Hannah Long, Ulla Gerling-Driessen, and Cheen Ang for help with human ES cell culture, Caitlyn Miller for synthesis of 9Az sialic acid, Dean Felsher for the T-ALL 4188 cells, Melissa Gray for expression and purification of VC-Sialidase, Sam Wattrus for help with confocal imaging, and Edgar Henriquez for help doing the cell sorting and FACS analysis. Cell sorting and FACS analysis was performed on an instrument in the Shared FACS Facility obtained using NIH S10 Shared Instrument Grants S10RR025518-01 and 1S10OD026831-01. Mass spectrometry was performed in the Analytical Biochemistry Shared Resource, Masonic Cancer Center, University of Minnesota, supported in part by NCI (P30CA077598). This work utilized the computational resources of the NIH HPC Biowulf cluster (https://hpc.nih.gov/). This work was supported by grants from Damon Runyon Cancer Research Foundation (DRG-2286-17 to R.A.F.), Burroughs Wellcome Fund Career Award for Medical Scientists (to R.A.F.), NIH (R01 CA200423-17 to C.R.B. and R01 AI141970 to J.E.C.), Stanford Medical Scientist Training Program and NIH F30 Predoctoral Fellowship (F30-CA232541 to B.A.H.S), National Institute of General Medical Sciences F32 Postdoctoral Fellowship (F32-GM126663-01 to S.A.M.), National Science Foundation Graduate Research Fellowship (NSF GRF) (DGE-114747 to A.G.J.), and NSF-GRF/Stanford Graduate Fellowship/Stanford ChEM-H Chemistry/Biology Interface Predoctoral Training Program (to K.P.). P.J.B. is supported by the Intramural Research Program of the NIH, National Cancer Institute, and Center for Cancer Research of the United States of America.
DECLARATION OF INTERESTS
C.R.B. is co-founder and SAB member of Redwood Bioscience (a subsidiary of Catalent), Enable Biosciences, Palleon Pharmaceuticals, InterVenn Bio, Oli-Lux Bio, and Lycia Therapeutics and a member of the Board of Directors of Eli Lilly.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.cell.2021.04.023.
REFERENCES
- Abu-Remaileh M, Wyant GA, Kim C, Laqtom NN, Abbasi M, Chan SH, Freinkman E, and Sabatini DM (2017). Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 358, 807–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agard NJ, Prescher JA, and Bertozzi CR (2004). A strain-promoted [3 + 2] azide-alkyne cycloaddition for covalent modification of biomolecules in living systems. J. Am. Chem. Soc 126, 15046–15047. [DOI] [PubMed] [Google Scholar]
- Ahlin E, Mathsson L, Eloranta M-L, Jonsdottir T, Gunnarsson I, Rönnblom L, and Rönnelid J. (2012). Autoantibodies associated with RNA are more enriched than anti-dsDNA antibodies in circulating immune complexes in SLE. Lupus 21, 586–595. [DOI] [PubMed] [Google Scholar]
- Aizawa S, Fujiwara Y, Contu VR, Hase K, Takahashi M, Kikuchi H, Kabuta C, Wada K, and Kabuta T. (2016). Lysosomal putative RNA transporter SIDT2 mediates direct uptake of RNA by lysosomes. Autophagy 12, 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Angata T. (2014). Associations of genetic polymorphisms of Siglecs with human diseases. Glycobiology 24, 785–793. [DOI] [PubMed] [Google Scholar]
- Baltz AG, Munschauer M, Schwanhäusser B, Vasile A, Murakawa Y, Schueler M, Youngs N, Penfold-Brown D, Drew K, Milek M, et al. (2012). The mRNA-bound proteome and its global occupancy profile on protein-coding transcripts. Mol. Cell 46, 674–690. [DOI] [PubMed] [Google Scholar]
- Bar DZ, Atkatsh K, Tavarez U, Erdos MR, Gruenbaum Y, and Collins FS (2018). Biotinylation by antibody recognition-a method for proximity labeling. Nat. Methods 15, 127–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barkal AA, Brewer RE, Markovic M, Kowarsky M, Barkal SA, Zaro BW, Krishnan V, Hatakeyama J, Dorigo O, Barkal LJ, and Weissman IL (2019). CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckmann BM, Horos R, Fischer B, Castello A, Eichelbaum K, Alleaume A-M, Schwarzl T, Curk T, Foehr S, Huber W, et al. (2015). The RNA-binding proteomes from yeast to man harbour conserved enigmRBPs. Nat. Commun 6, 10127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanco F, Kalsi J, and Isenberg DA (1991). Analysis of antibodies to RNA in patients with systemic lupus erythematosus and other autoimmune rheumatic diseases. Clin. Exp. Immunol 86, 66–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boccitto M, and Wolin SL (2019). Ro60 and Y RNAs: structure, functions, and roles in autoimmunity. Crit. Rev. Biochem. Mol. Biol 54, 133–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bond MR, Zhang H, Kim J, Yu S-H, Yang F, Patrie SM, and Kohler JJ (2011). Metabolism of diazirine-modified N-acetylmannosamine analogues to photo-cross-linking sialosides. Bioconjug. Chem 22, 1811–1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonin M, Oberstrass J, Lukacs N, Ewert K, Oesterschulze E, Kassing R, and Nellen W. (2000). Determination of preferential binding sites for anti-dsRNA antibodies on double-stranded RNA by scanning force microscopy. RNA 6, 563–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casciola-Rosen LA, Anhalt G, and Rosen A. (1994). Autoantigens targeted in systemic lupus erythematosus are clustered in two populations of surface structures on apoptotic keratinocytes. J. Exp. Med 179, 1317–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castello A, Fischer B, Eichelbaum K, Horos R, Beckmann BMB, Strein C, Davey NEN, Humphreys DTD, Preiss T, Steinmetz LML, et al. (2012). Insights into RNA biology from an atlas of mammalian mRNA-binding proteins. Cell 149, 1393–1406. [DOI] [PubMed] [Google Scholar]
- Cech TR, and Steitz JA (2014). The noncoding RNA revolution-trashing old rules to forge new ones. Cell 157, 77–94. [DOI] [PubMed] [Google Scholar]
- Chan PP, and Lowe TM (2009). GtRNAdb: a database of transfer RNA genes detected in genomic sequence. Nucleic Acids Res. 37, D93–D97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang Y-C, and Nizet V. (2020). Siglecs at the Host-Pathogen Interface. Adv. Exp. Med. Biol 1204, 197–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang PV, Chen X, Smyrniotis C, Xenakis A, Hu T, Bertozzi CR, and Wu P. (2009). Metabolic labeling of sialic acids in living animals with alkynyl sugars. Angew. Chem. Int. Ed. Engl 48, 4030–4033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou H-H, Hayakawa T, Diaz S, Krings M, Indriati E, Leakey M, Paabo S, Satta Y, Takahata N, and Varki A. (2002). Inactivation of CMP-N-acetylneuraminic acid hydroxylase occurred prior to brain expansion during human evolution. Proc. Natl. Acad. Sci. USA 99, 11736–11741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christov CP, Gardiner TJ, Szüts D, and Krude T. (2006). Functional requirement of noncoding Y RNAs for human chromosomal DNA replication. Mol. Cell. Biol 26, 6993–7004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duan S, and Paulson JC (2020). Siglecs as Immune Cell Checkpoints in Disease. Annu. Rev. Immunol 38, 365–395. [DOI] [PubMed] [Google Scholar]
- Duffy EE, and Simon MD (2016). Enriching s4 U-RNA Using Methane Thiosulfonate (MTS) Chemistry. Curr. Protoc. Chem. Biol 8, 234–250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eakin AJ, Bustard MJ, McGeough CM, Ahmed T, Bjourson AJ, and Gibson DS (2016). Siglec-1 and −2 as potential biomarkers in autoimmune disease. Proteomics Clin. Appl 10, 635–644. [DOI] [PubMed] [Google Scholar]
- Elbein AD, Tropea JE, Mitchell M, and Kaushal GP (1990). Kifunensine, a potent inhibitor of the glycoprotein processing mannosidase I. J. Biol. Chem 265, 15599–15605. [PubMed] [Google Scholar]
- Fazal FM, Han S, Parker KR, Kaewsapsak P, Xu J, Boettiger AN, Chang HY, and Ting AY (2019). Atlas of Subcellular RNA Localization Revealed by APEX-Seq. Cell 178, 473–490.e26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feinberg EH, and Hunter CP (2003). Transport of dsRNA into cells by the transmembrane protein SID-1. Science 301, 1545–1547. [DOI] [PubMed] [Google Scholar]
- Felsher DW, and Bishop JM (1999). Reversible tumorigenesis by MYC in hematopoietic lineages. Mol. Cell 4, 199–207. [DOI] [PubMed] [Google Scholar]
- Flores R, Zhang P, Wu W, Wang X, Ye P, Zheng P, and Liu Y. (2019). Siglec genes confer resistance to systemic lupus erythematosus in humans and mice. Cell. Mol. Immunol 16, 154–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Martin L, Spitale RC, Do BT, Sagan SM, Zarnegar B, Qu K, Khavari PA, Quake SR, Sarnow P, et al. (2015). Dissecting noncoding and pathogen RNA-protein interactomes. RNA 21, 135–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flynn RA, Zhang QC, Spitale RC, Lee B, Mumbach MR, and Chang HY (2016). Transcriptome-wide interrogation of RNA secondary structure in living cells with icSHAPE. Nat. Protoc 11, 273–290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fraschilla I, and Pillai S. (2017). Viewing Siglecs through the lens of tumor immunology. Immunol. Rev 276, 178–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frye M, Harada BT, Behm M, and He C. (2018). RNA modifications modulate gene expression during development. Science 361, 1346–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gagnon KT, Li L, Janowski BA, and Corey DR (2014). Analysis of nuclear RNA interference in human cells by subcellular fractionation and Argonaute loading. Nat. Protoc 9, 2045–2060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gahmberg CG, and Andersson LC (1982). Role of sialic acid in the mobility of membrane proteins containing O-linked oligosaccharides on polyacrylamide gel electrophoresis in sodium dodecyl sulfate. Eur. J. Biochem 122, 581–586. [DOI] [PubMed] [Google Scholar]
- Gao C, Hanes MS, Byrd-Leotis LA, Wei M, Jia N, Kardish RJ, McKitrick TR, Steinhauer DA, and Cummings RD (2019). Unique Binding Specificities of Proteins toward Isomeric Asparagine-Linked Glycans. Cell Chem. Biol 26, 535–547.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardiner TJ, Christov CP, Langley AR, and Krude T. (2009). A conserved motif of vertebrate Y RNAs essential for chromosomal DNA replication. RNA 15, 1375–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golan TD, Elkon KB, Gharavi AE, and Krueger JG (1992). Enhanced membrane binding of autoantibodies to cultured keratinocytes of systemic lupus erythematosus patients after ultraviolet B/ultraviolet A irradiation. J. Clin. Invest 90, 1067–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gray M, Stanczak MA, Xiao H, Pijnenborg JFA, Malaker SA, Weidenbacher PA, Tanzo JT, Ahn G, Woods EC, Läubli H, et al. (2019). Targeted Desialylation Overcomes Glyco-Immune Checkpoints and Potentiates the Anticancer Immune Response in Vivo. ChemRxiv. https://doi.org/10. 26434/chemrxiv.8187146.v2. [Google Scholar]
- Gray MA, Stanczak MA, Mantuano NR, Xiao H, Pijnenborg JFA, Malaker SA, Miller CL, Weidenbacher PA, Tanzo JT, Ahn G, et al. (2020). Targeted glycan degradation potentiates the anticancer immune response in vivo. Nat. Chem. Biol 16, 1376–1384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han J-H, Umiker BR, Kazimirova AA, Fray M, Korgaonkar P, Selsing E, and Imanishi-Kari T. (2014). Expression of an anti-RNA autoantibody in a mouse model of SLE increases neutrophil and monocyte numbers as well as IFN-I expression. Eur. J. Immunol 44, 215–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang HC, Yu C, Kato DL, and Bertozzi CR (2003). A metabolic labeling approach toward proteomic analysis of mucin-type O-linked glycosylation. Proc. Natl. Acad. Sci. USA 100, 14846–14851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N, Fan X, Zaleta-Rivera K, Nguyen TC, Zhou J, Luo Y, Gao J, Fang RH, Yan Z, Chen ZB, et al. (2020). Natural display of nuclear-encoded RNA on the cell surface and its impact on cell interaction. Genome Biol. 21, 225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudak JE, Canham SM, and Bertozzi CR (2014). Glycocalyx engineering reveals a Siglec-based mechanism for NK cell immunoevasion. Nat. Chem. Biol 10, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Inamori K, Yoshida-Moriguchi T, Hara Y, Anderson ME, Yu L, and Campbell KP (2012). Dystroglycan function requires xylosyl- and glucuronyltransferase activities of LARGE. Science 335, 93–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jandus C, Boligan KF, Chijioke O, Liu H, Dahlhaus M, Démoulins T, Schneider C, Wehrli M, Hunger RE, Baerlocher GM, et al. (2014). Interactions between Siglec-7/9 receptors and ligands influence NK cell-dependent tumor immunosurveillance. J. Clin. Invest 124, 1810–1820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kasai H, Nakanishi K, Macfarlane RD, Torgerson DF, Ohashi Z, McCloskey JA, Gross HJ, and Nishimura S. (1976). Letter: The structure of Q* nucleoside isolated from rabbit liver transfer ribonucleic acid. J. Am. Chem. Soc 98, 5044–5046. [DOI] [PubMed] [Google Scholar]
- Kingsley DM, Kozarsky KF, Hobbie L, and Krieger M. (1986). Reversible defects in O-linked glycosylation and LDL receptor expression in a UDP-Gal/UDP-GalNAc 4-epimerase deficient mutant. Cell 44, 749–759. [DOI] [PubMed] [Google Scholar]
- Koczera P, Martin L, Marx G, and Schuerholz T. (2016). The Ribonuclease A Superfamily in Humans: Canonical RNases as the Buttress of Innate Immunity. Int. J. Mol. Sci 17, 1278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Köhn M, Pazaitis N, and Hüttelmaier S. (2013). Why YRNAs? About Versatile RNAs and Their Functions. Biomolecules 3, 143–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosa RE, Brossmer R, and Gross HJ (1993). Modification of cell surfaces by enzymatic introduction of special sialic acid analogues. Biochem. Biophys. Res. Commun 190, 914–920. [DOI] [PubMed] [Google Scholar]
- Koutsioulis D, Landry D, and Guthrie EP (2008). Novel endo-alpha-N-acetylgalactosaminidases with broader substrate specificity. Glycobiology 18, 799–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowalski MP, and Krude T. (2015). Functional roles of non-coding Y RNAs. Int. J. Biochem. Cell Biol 66, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Gagnon JA, Thyme SB, and Valen E. (2016). CHOPCHOP v2: a web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res. 44 (W1), W272–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labun K, Montague TG, Krause M, Torres Cleuren YN, Tjeldnes H, and Valen E. (2019). CHOPCHOP v3: expanding the CRISPR web toolbox beyond genome editing 47. 10.1093/nar/gkz365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Läubli H, Alisson-Silva F, Stanczak MA, Siddiqui SS, Deng L, Verhagen A, Varki N, and Varki A. (2014). Lectin galactoside-binding soluble 3 binding protein (LGALS3BP) is a tumor-associated immunomodulatory ligand for CD33-related Siglecs. J. Biol. Chem 289, 33481–33491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Sambrooks C, Shrimal S, Khodier C, Flaherty DP, Rinis N, Charest JC, Gao N, Zhao P, Wells L, Lewis TA, et al. (2016). Oligosaccharyltransferase inhibition induces senescence in RTK-driven tumor cells. Nat. Chem. Biol 12, 1023–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luchansky SJ, Argade S, Hayes BK, and Bertozzi CR (2004). Metabolic functionalization of recombinant glycoproteins. Biochemistry 43, 12358–12366. [DOI] [PubMed] [Google Scholar]
- Macauley MS, Crocker PR, and Paulson JC (2014). Siglec-mediated regulation of immune cell function in disease. Nat. Rev. Immunol 14, 653–666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machnicka MA, Milanowska K, Osman Oglou O, Purta E, Kurkowska M, Olchowik A, Januszewski W, Kalinowski S, Dunin-Horkawicz S, Rother KM, et al. (2013). MODOMICS: a database of RNA modification pathways–2013 update. Nucleic Acids Res. 41, D262–D267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malaker SA, Pedram K, Ferracane MJ, Bensing BA, Krishnan V, Pett C, Yu J, Woods EC, Kramer JR, Westerlind U, et al. (2019). The mucin-selective protease StcE enables molecular and functional analysis of human cancer-associated mucins. Proc. Natl. Acad. Sci. USA 116, 7278–7287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manya H, Yamaguchi Y, Kanagawa M, Kobayashi K, Tajiri M, Akasaka-Manya K, Kawakami H, Mizuno M, Wada Y, Toda T, and Endo T. (2016). The Muscular Dystrophy Gene TMEM5 Encodes a Ribitol b1,4-Xylosyltransferase Required for the Functional Glycosylation of Dystroglycan. J. Biol. Chem 291, 24618–24627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchand V, Pichot F, Thüring K, Ayadi L, Freund I, Dalpke A, Helm M, and Motorin Y. (2017). Next-Generation Sequencing-Based RiboMethSeq Protocol for Analysis of tRNA 2′-O-Methylation. Biomolecules 7, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshak-Rothstein A, and Rifkin IR (2007). Immunologically active autoantigens: the role of toll-like receptors in the development of chronic inflammatory disease. Annu. Rev. Immunol 25, 419–441. [DOI] [PubMed] [Google Scholar]
- Mateer E, Paessler S, and Huang C. (2019). Confocal Imaging of Double-Stranded RNA and Pattern Recognition Receptors in Negative-Sense RNA Virus Infection. J. Vis. Exp (143), 10.3791/59095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McConkey EH (1967). The fractionation of RNA’s by sucrose gradient centrifugation. Methods Enzymol. 12, 620–634. [Google Scholar]
- Möckl L, Pedram K, Roy AR, Krishnan V, Gustavsson A-K, Dorigo O, Bertozzi CR, and Moerner WE (2019). Quantitative Super-Resolution Microscopy of the Mammalian Glycocalyx. Dev. Cell 50, 57–72.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohorko E, Glockshuber R, and Aebi M. (2011). Oligosaccharyltransferase: the central enzyme of N-linked protein glycosylation. J. Inherit. Metab. Dis 34, 869–878. [DOI] [PubMed] [Google Scholar]
- Müller J, Lunz B, Schwab I, Acs A, Nimmerjahn F, Daniel C, and Nitschke L. (2015). Siglec-G Deficiency Leads to Autoimmunity in Aging C57BL/6 Mice. J. Immunol 195, 51–60. [DOI] [PubMed] [Google Scholar]
- Nachtergaele S, and He C. (2017). The emerging biology of RNA post-transcriptional modifications. RNA Biol. 14, 156–163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen TA, Smith BRC, Elgass KD, Creed SJ, Cheung S, Tate MD, Belz GT, Wicks IP, Masters SL, and Pang KC (2019). SIDT1 Localizes to Endolysosomes and Mediates Double-Stranded RNA Transport into the Cytoplasm. J. Immunol 202, 3483–3492. [DOI] [PubMed] [Google Scholar]
- Okada N, Shindo-Okada N, and Nishimura S. (1977). Isolation of mammalian tRNAAsp and tRNATyr by lectin-Sepharose affinity column chromatography. Nucleic Acids Res. 4, 415–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praissman JL, Live DH, Wang S, Ramiah A, Chinoy ZS, Boons G-J, Moremen KW, and Wells L. (2014). B4GAT1 is the priming enzyme for the LARGE-dependent functional glycosylation of α-dystroglycan. eLife 3, e03943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Praissman JL, Willer T, Sheikh MO, Toi A, Chitayat D, Lin Y-Y, Lee H, Stalnaker SH, Wang S, Prabhakar PK, et al. (2016). The functional O-mannose glycan on α-dystroglycan contains a phospho-ribitol primed for matriglycan addition. eLife 5, e14473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qin W, Qin K, Fan X, Peng L, Hong W, Zhu Y, Lv P, Du Y, Huang R, Han M, et al. (2018). Artificial Cysteine S-Glycosylation Induced by Per-O-Acetylated Unnatural Monosaccharides during Metabolic Glycan Labeling. Angew. Chem. Int. Ed. Engl 57, 1817–1820. [DOI] [PubMed] [Google Scholar]
- Ran FA, Hsu PD, Wright J, Agarwala V, Scott DA, and Zhang F. (2013). Genome engineering using the CRISPR-Cas9 system. Nat. Protoc 8, 2281–2308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reily C, Stewart TJ, Renfrow MB, and Novak J. (2019). Glycosylation in health and disease. Nat. Rev. Nephrol 15, 346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee H-W, Zou P, Udeshi ND, Martell JD, Mootha VK, Carr SA, and Ting AY (2013). Proteomic mapping of mitochondria in living cells via spatially restricted enzymatic tagging. Science 339, 1328–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riley James L. (2009). PD-1 signaling in primary T cells. Immunological Reviews 229, 114–125. 10.1111/j.1600-065X.2009.00767.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rillahan CD, Antonopoulos A, Lefort CT, Sonon R, Azadi P, Ley K, Dell A, Haslam SM, and Paulson JC (2012). Global metabolic inhibitors of sialyl- and fucosyltransferases remodel the glycome. Nat. Chem. Biol 8, 661–668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saleh M-C, van Rij RP, Hekele A, Gillis A, Foley E, O’Farrell PH, and Andino R. (2006). The endocytic pathway mediates cell entry of dsRNA to induce RNAi silencing. Nat. Cell Biol 8, 793–802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saxon E, and Bertozzi CR (2000). Cell surface engineering by a modified Staudinger reaction. Science 287, 2007–2010. [DOI] [PubMed] [Google Scholar]
- Schneider M, Al-Shareffi E, and Haltiwanger RS (2017). Biological functions of fucose in mammals. Glycobiology 27, 601–618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schönborn J, Oberstrass J, Breyel E, Tittgen J, Schumacher J, and Lukacs N. (1991). Monoclonal antibodies to double-stranded RNA as probes of RNA structure in crude nucleic acid extracts. Nucleic Acids Res. 19, 2993–3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schumann B, Malaker SA, Wisnovsky SP, Debets MF, Agbay AJ, Fernandez D, Wagner LJS, Lin L, Choi J, Fox DM, et al. (2019). Chemical precision glyco-mutagenesis by glycosyltransferase engineering in living cells. bioRxiv. 10.1101/669861. [DOI] [Google Scholar]
- Schumann B, Malaker SA, Wisnovsky SP, Debets MF, Agbay AJ, Fernandez D, Wagner LJS, Lin L, Li Z, Choi J, et al. (2020). Bump-and-Hole Engineering Identifies Specific Substrates of Glycosyltransferases in Living Cells. Mol. Cell 78, 824–834.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp PA (2009). The centrality of RNA. Cell 136, 577–580. [DOI] [PubMed] [Google Scholar]
- Sorrentino S. (2010). The eight human “canonical” ribonucleases: molecular diversity, catalytic properties, and special biological actions of the enzyme proteins. FEBS Lett. 584, 2194–2200. [DOI] [PubMed] [Google Scholar]
- Stanczak MA, Siddiqui SS, Trefny MP, Thommen DS, Boligan KF, von Gunten S, Tzankov A, Tietze L, Lardinois D, Heinzelmann-Schwarz V, et al. (2018). Self-associated molecular patterns mediate cancer immune evasion by engaging Siglecs on T cells. J. Clin. Invest 128, 4912–4923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarentino AL, and Plummer TH Jr. (1994). Enzymatic deglycosylation of asparagine-linked glycans: purification, properties, and specificity of oligosac-charide-cleaving enzymes from Flavobacterium meningosepticum. Methods Enzymol. 230, 44–57. [DOI] [PubMed] [Google Scholar]
- Thornhill SI, Mak A, Lee B, Lee HY, Poidinger M, Connolly JE, and Fairhurst A-M (2017). Monocyte Siglec-14 expression is upregulated in patients with systemic lupus erythematosus and correlates with lupus disease activity. Rheumatology (Oxford) 56, 1025–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tulsiani DR, Harris TM, and Touster O. (1982). Swainsonine inhibits the biosynthesis of complex glycoproteins by inhibition of Golgi mannosidase II. J. Biol. Chem 257, 7936–7939. [PubMed] [Google Scholar]
- Varki A, and Gagneux P. (2015). Biological Functions of Glycans. In Essentials of Glycobiology, Varki A, Cummings RD, Esko JD, Stanley P, Hart GW, Aebi M, Darvill AG, Kinoshita T, Packer NH, and Prestegard JH, et al. , eds. (Cold Spring Harbor Laboratory Press; ). [PubMed] [Google Scholar]
- Varki A, and Schauer R. (2009). Sialic Acids. In Essentials of Glycobiology, Varki A, Cummings RD, Esko JD, Freeze HH, Stanley P, Bertozzi CR, Hart GW, and Etzler ME, eds. (Cold Spring Harbor Laboratory Press; ). [PubMed] [Google Scholar]
- Wang KC, and Chang HY (2011). Molecular mechanisms of long noncoding RNAs. Mol. Cell 43, 904–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H. (2009). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
- Willer T, Inamori K, Venzke D, Harvey C, Morgensen G, Hara Y, Beltrán Valero de Bernabé D, Yu L, Wright KM, and Campbell KP (2014). The glucuronyltransferase B4GAT1 is required for initiation of LARGE-mediated a-dystroglycan functional glycosylation. eLife 3, e03941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woo CM, Iavarone AT, Spiciarich DR, Palaniappan KK, and Bertozzi CR (2015). Isotope-targeted glycoproteomics (IsoTaG): a mass-independent platform for intact N- and O-glycopeptide discovery and analysis. Nat. Methods 12, 561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao H, Woods EC, Vukojicic P, and Bertozzi CR (2016). Precision glycocalyx editing as a strategy for cancer immunotherapy. Proc. Natl. Acad. Sci. USA 113, 10304–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida-Moriguchi T, Willer T, Anderson ME, Venzke D, Whyte T, Muntoni F, Lee H, Nelson SF, Yu L, and Campbell KP (2013). SGK196 is a glycosylation-specific O-mannose kinase required for dystroglycan function. Science 341, 896–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarnegar BJ, Flynn RA, Shen Y, Do BT, Chang HY, and Khavari PA (2016). irCLIP platform for efficient characterization of protein-RNA interactions. Nat. Methods 13, 489–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Y, Wang G, Wang P, Li Z, Yue T, Wang J, and Zou P. (2019). Expanding APEX2 Substrates for Proximity-Dependent Labeling of Nucleic Acids and Proteins in Living Cells. Angew. Chem. Int. Ed. Engl 58, 11763–11767. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Ac4ManNAz incorporation into RNA from multiple mammalian cell types, related to Figure 1
(A) SDS-PAGE analysis of Proteinase K digestion of recombinant protein under conditions used to remove protein contaminants from RNA samples.
(B) Blotting of Ac4ManNAz-labeled HeLa RNA with and without Proteinase K treatment of the RNA before copper-free click labeling and visualization. Biological duplicates are shown.
(C) Blotting of in cellulo or in vitro Ac4ManNAz-labeled RNA. Cells were treated with 100 μM Ac4ManNAz for 24 hours while native RNA in vitro was treated with up to 20 mM Ac4ManNAz for 2 hours at 37°C.
(D) Agarose gel analysis of genomic DNA isolated from HeLa cells and subjected to the DNase digestion conditions used to clean up RNA samples.
(E, F) Blotting of RNA from various cell types labeled with Ac4ManNAz including (E) HeLa, GM12878, K562, MYC T-ALL 4188, and CHO cells and (F) HeLa and H9 cells. RNase controls are provided for each cell type assayed.
Figure S2. glycoRNAs are non-polyadenylated, small RNAs, related to Figure 2
(A) Blotting of Ac4ManNAz-labeled HeLa RNA isolated before and at each step of the poly-A enrichment protocol. Three times the amount of RNA was used for the poly-A enrichment (60 μg) such that even low levels of Ac4ManNAz-labeling of poly-A RNA could be detected. FT = flowthrough. 2×-pA = double poly-A enriched RNA.
(B) Representative 254nm signal trace of total RNA signal across the sucrose fractionation. Fractions containing small RNAs, as indicated on the profile with a red bar, were pooled and purified for sequence analysis.
(C) Cross correlation analysis of reads from input and Ac4ManNAz-enriched libraries generated from HeLa and H9 cells.
(D) Pie charts comparing the distribution of mapped reads between input and Ac4ManNAz-enriched libraries generated from HeLa and H9 cells. Colors are used to highlight the common RNA families across libraries.
(E) Correlation analysis at the transcript level of non-tRNA (left) and tRNA (right) transcripts between the HeLa and H9 input sequencing libraries. Error bars are standard deviation of each RNA expressed across the biological duplicate libraries.
(F) volcano plot analysis of Ac4ManNAz-enriched RNAs identified from HeLa small RNA. RNA families are grouped by colors and plotted by their log2-enrichment values in the Ac4ManNAz captured library (x axis) and the confidence of that enrichment value (y axis, -log10(adjusted p value)). Dashed lines are enrichment and p value cutoffs are shown.
(G) Volcano plot analysis as in (F) from H9 cell small RNAs.
(H) Scatterplot analysis Ac4ManNAz-enriched RNAs purified from the small fractions of HeLa and H9 cells. Reads mapping to tRNAs are shown. Significance scores (-log10(adjusted p value) are overlayed for HeLa cells as the size of each datapoint and for H9 cells as the color of each datapoint.
Figure S3. Generation of Y5 knockout 293T cells and DMB detection and quantification of sialic acids on unlabeled cellular RNA, related to Figures 2 and 3
(A) Schematic of the wild-type (WT) locus of the human Y5 transcript. The regions targeted by the two designed sgRNAs are highlighted in orange with the protospacer adjacent motif (PAM) regions in green boxes. The first nucleotide of the Y5 RNA is marked with a green arrow.
(B) PCR analysis of genomic DNA (gDNA) from WT and Y5 KO clones. Primers targeting the Y5 locus (left) showed two new bands in the KO clone.
(C) Schematic of sanger sequencing results from the gDNA PCR in (B). Two specific sequences were isolated by sequencing. A deletion (black) comes from the bottom band in the gel. A separate insertion (red)/deletion (black)/mutation (blue) structure was observed which comes from the top PCR band.
(D) Northern blot of 293T WT and KO total RNA. The KO resulted in a complete loss of the Y5 RNA, with the 5S rRNA serving as a loading control.
(E) Analysis of growth rate between the WT and KO clones across four days of culture. Three independent wells of cells were counted on each day.
(F) Schematic of experimental steps of the DMB assay with associated structures of the two major types of sialic acid (Neu5Ac and Neu5Gc) as well as the DMB reagent.
(G) HPLC-fluorescence traces of DMB modified HeLa (left) and 4188 (right) cellular RNA. The main sialic acid peaks are #2 and 3. Peak 1 is unknown but is sensitive to RNase. Insets are Sybr stained gels of the total RNA for each condition: no enzymes, RNase cocktail, or Sialidase treatment.
(H) HPLC traces of DMB modified 293T (left) and HeLa (right) cellular RNA or DNA. Neu5Ac and Neu5Gc peaks are highlighted across biological duplicates from each cell type with input material run in a denaturing agarose gel for sizes and loading controls (insets). Only cellular RNA produces Neu5Ac and Neu5Gc specific peaks after DMB derivatization.
Figure S4. Cellular and in vitro assays define the requirement of the N-glycan pathway for glycoRNA, related to Figure 4
(A) Diagram of the main cellular pathway for interconversion of glucose (Glc) to galactose (Gal). Normal cell culture media contains Glu without Gal. The enzyme GALE is responsible for converting uridine 5′-diphospho-N-acetylglucosamine (UDP-GlcNAc) to UDP-GalNAc and UDP-Glc to UDP-Gal. ldlD CHO cells contain a loss-of-function mutation in the GALE pathway rendering them deficient in the production of N- and O-linked glycans when grown in normal media. If supple- mented with Gal or GalNAc, these cells can selectively elaborate N- and O-linked glycans, respectively. Importantly, biosynthesis of CMP-sialic acid is unaffected by GALE mutation.
(B) Blotting of RNA from K562-GALE-KO cells labeled with Ac4ManNAz, Gal (10 μM), or GalNAc (100 μM) for 24 hours.
(C) An overview of major steps in the ER- and Golgi-localized steps of N-glycan biosynthesis. Dolichol-linked precursor glycans are transferred to ER-lumen- facing peptides (X) by OST. These glycans are sequentially modified by enzymes that first trim and then elaborate the glycan tree. Specific chemical inhibitorssuch as NGI-1 (inhibiting OST), Kifunesine (inhibiting a-mannosidase I) and Swainsonine (inhibiting α-mannosidase II) are employed to interrogate the importance of each of these enzymes to the accumulation of Ac4ManNAz-labeling of RNA.
(D) Blotting of Ac4ManNAz-labeled HeLa cell total RNA after treatment with the indicated concentrations of Swainsonine.
(E) Blotting of Ac4ManNAz-labeled HeLa cell total RNA after in vitro treatment with the indicated enzymes; representative of three independent experiments and quantified results are presented in Figure 4D.
(F) Blotting of Ac4ManNAz-labeled HeLa cell total RNA after in vitro treatment with the mucinase StcE. Addition of EDTA inhibits the mucinase as a control.
Figure S5. Membrane isolation and live-cell assays define the cell surface as a major reservoir for glycoRNAs, related to Figure 5
(A) Schematic of isolated cellular membranes treated with or without Triton X-100 or VC-Sia for 1 hour at 37°C before isolating RNA and assaying sialic acid levels via DMB.
(B) Quantification of membrane protection assay performed in biological quadruplicates from 293T cells. Significance calculated with an unpaired t test.
(C) Blotting of RNA from HeLa cells labeled with 100 μM Ac4ManNAz for 24 hours and then expose to fresh media containing 100 μM Ac4ManNAz with or without 150 nM VC-Sia for 20, 40, or 60 minutes at 37°C.
(D) SDS-PAGE analysis followed by blotting and visualization of biotin signal with StrepIR800. Samples were generated from cells in Figure 5F, here protein lysate was analyzed revealing that all three biotinylated lectins are capable of activating biotin-aniline and labeling cell surface proteins.
Figure S6. RNA and sialic acid affinity reagents stain cell surfaces in a manner depended on RNA, related to Figure 6
(A) Native PAGE of synthetic Y5 RNA 3′ end labeled with Cy-7 (Y5-Cy7) bound to antibodies, J2 or a non-targeting IgG, with or without competing dsRNA (poly- (I:C)). Quantification of the shifted (red) and free (black) Y5-Cy7 is plotted blow each corresponding lane.
(B) SDS-PAGE analysis of trypsin and TrypLE products stained for total protein content. Regions of each lane are annotated with presumptive identity of bands.
(C) Agarose gel analysis of total cellular RNA stained with Sybr for visualization. To live cells, PBS, Trypsin, or TrypLE was added and incubated for 5 minutes at 37°C. RNA was directly extracted from this or extracted after rinsing the cells once in PBS. RNA stability was significantly impacted with the trypsin reagent with or without PBS washing.
(D) FACS analysis of single 293T (left) or K562 (right) cells pre-treated or not with RNase A (red outline) and stained with the J2 antibody (orange).
(E) Representative confocal images of HeLa cells stained with the J2 antibody with or without prior treatment with RNase A. Image acquired at 40× magnification and scale is as indicated on the image.
(F) FACS analysis of single HeLa cells stained with a panel of commercially available recombinant human Siglec-Fc reagents. Siglec-2, −3, and -Mag did not stain HeLa cells above background.
(G) FACS analysis of single HeLa cells stained with the 9 Siglec-Fc reagents that were above background in (F). Red outlines indicate cells were pre-treated with RNase A.
Data Availability Statement
All sequencing data have been deposited on the Gene Expression Omnibus (GSE136967; https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136967). Sequencing data were analyzed with the FAST-iCLIP pipeline which can be found here (https://github.com/ChangLab/FAST-iCLIP/tree/lite). Glycan release samples were analyzed with GlycoNote (https://github.com/GlycoNote/GlycoNote).