

Abstract
INTRODUCTION:
Sleep destabilization is strongly associated with aging and cognitive function decline. Despite sleep fragmentation being central to the most prevalent complaints of sleep problems in elderly populations, the mechanistic underpinnings of sleep instability remain elusive. Fragmented sleep during aging has been observed across species, indicating conserved underlying mechanisms across the phylogenetic tree. Therefore, understanding why the aging brain fails to consolidate sleep may shed light on translational applications for improving the sleep quality of aged individuals.
RATIONALE:
We hypothesized that the decline in sleep quality could be due to malfunction of the neural circuits associated with sleep/wake control. It has been established that hypocretin/orexin (Hcrt/OX) neuronal activity is tightly associated with wakefulness and initiates and maintains the wake state. In this study, we investigated whether the intrinsic excitability of Hcrt neurons is altered, leading to a destabilized control of sleep/wake states during aging.
RESULTS:
Aged mice exhibited sleep fragmentation and a significant loss of Hcrt neurons. Hcrt neurons manifested a more frequent firing pattern, driving wake bouts and disrupting sleep continuity in aged mice. Aged Hcrt neurons were capable of eliciting more prolonged wake bouts upon optogenetic stimulations. These results suggested that hyperexcitability of Hcrt neurons emerges with age. Patch clamp recording in genetically identified Hcrt neurons revealed distinct intrinsic properties between the young and aged groups. Aged Hcrt neurons were hyperexcitable with depolarized membrane potentials (RMPs) and a smaller difference between RMP and the firing threshold. Aged Hcrt neurons expressing ChR2-eYFP were more sensitive to optogenetic stimulations, with a smaller-amplitude attenuation upon repetitive light pulse stimulations. More spikelets were generated in aged Hcrt neurons upon current injections. Recording from non-Hcrt neurons postsynaptic to Hcrt neurons revealed that optogenetic stimulation of Hcrt neurons expressing ChR2-eYFP reliably evoked time-locked postsynaptic currents (PSCs) after optogenetic stimulation more often in the aged group. Aged Hcrt neurons were characterized with a functional impairment of repolarizing M-current mediated by KCNQ2/3 channels and an anatomical loss of KCNQ2, revealed with array tomography at ultrastructural resolution. Single-nucleus RNA-sequencing (snRNA-seq) revealed molecular adaptions, including up-regulated prepro-Hcrt mRNA expression and a smaller fraction of Kcnq family subtypes Kcnq1/2/3/5 in aged Hcrt neurons. CRISPR/SaCas9–mediated disruption of Kcnq2/3 genes selectively in Hcrt neurons was sufficient to recapitulate the aging-associated sleep fragmentation trait in young mice. Pharmacological augmentation of M-current repolarized the RMP, suppressed spontaneous firing activity in aged Hcrt neurons, and consolidated sleep stability in aged mice. Sleep fragmentation in a narcolepsy mouse model with genetic ablation of Hcrt neurons at young ages manifested a mechanism other than hyperexcitable arousal-promoting Hcrt neurons that drives sleep fragmentation during healthy aging.
CONCLUSION:
Our data indicate that emerging hyperexcitability of arousal-promoting Hcrt neurons is strongly associated with fragmented sleep in aged mice, which display a lowered sleep-to-wake transition threshold defined for Hcrt neuronal activity. We have demonstrated that the down-regulation of KCNQ2/3 channels compromising repolarization drives Hcrt neuronal hyperexcitability, which leads to sleep instability during aging. Pharmacological remedy of sleep continuity through targeting KCNQ2/3 channels in aged mice confers a potential translational therapy strategy for improving sleep quality in aged individuals.
Graphical Abstract
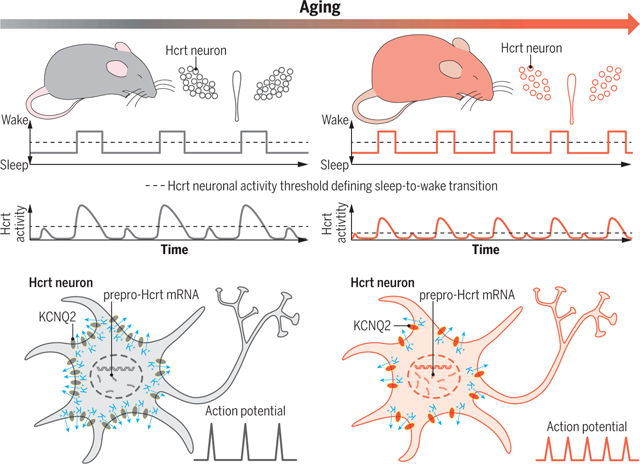
Hyperexcitable Hcrt neurons drive sleep instability during aging. Elevated excitability of Hcrt neurons with depolarized RMPs and adaptive up-regulation of prepro-Hcrt mRNA expression converge to drive sleep/wake instability in the aged brain with substantial Hcrt neuron loss. Hyperexcitable aged Hcrt neurons express functional impairment of KCNQ2/3 channel–mediated M-current and an anatomical loss of KCNQ2, compromising the neurons to repolarize.
Sleep quality declines with age; however, the underlying mechanisms remain elusive. We found that hyperexcitable hypocretin/orexin (Hcrt/OX) neurons drive sleep fragmentation during aging. In aged mice, Hcrt neurons exhibited more frequent neuronal activity epochs driving wake bouts, and optogenetic activation of Hcrt neurons elicited more prolonged wakefulness. Aged Hcrt neurons showed hyperexcitability with lower KCNQ2 expression and impaired M-current, mediated by KCNQ2/3 channels. Single-nucleus RNA-sequencing revealed adaptive changes to Hcrt neuron loss in the aging brain. Disruption of Kcnq2/3 genes in Hcrt neurons of young mice destabilized sleep, mimicking aging-associated sleep fragmentation, whereas the KCNQ-selective activator flupirtine hyperpolarized Hcrt neurons and rejuvenated sleep architecture in aged mice. Our findings demonstrate a mechanism underlying sleep instability during aging and a strategy to improve sleep continuity.
Sleep quality correlates with cognitive function (1, 2), and decline in sleep quality is among the most prevalent complaints during aging in humans (3, 4). Aging is associated with alterations in sleep architecture, prominently sleep fragmentation, which prevents restorative sleep (3, 5). The ability to sustain sleep/wake states during aging is heavily impaired across species (5–7), suggesting that the underlying mechanisms are conserved across the phylogenetic tree. However, the cellular and molecular underpinnings of sleep instability during aging are unknown. A plausible mechanism underlying aging-related sleep fragmentation is that elevated intrinsic excitability of arousal-promoting circuits emerges with age, disrupting sleep stability.
Hypocretin/orexin (Hcrt/OX) neurons (8, 9) in the lateral hypothalamus (LH) play a pivotal role in sleep/wake control (10, 11). Optogenetic stimulation of Hcrt neurons during sleep triggers sleep-to-wake transition (12, 13), whereas optogenetic suppression of Hcrt neuronal activity induces non–rapid eye movement (NREM) sleep (14). Furthermore, genetic Hcrt neuron depletion (15) or Hcrt receptor 2 (HcrtR2) mutation (16) leads to narcolepsy with cataplexy, a condition in which patients suffer sleep and wake fragmentation (17). In vivo electrophysiological recordings demonstrate that Hcrt neuronal activity is correlated with wakefulness and initiates and maintains wake state (18, 19). We thus hypothesized that emerging hyperexcitability of Hcrt neurons drivessleep instability during aging.
Results
Aged mice exhibit fragmented sleep and significant loss of Hcrt neurons
We compared the sleep/wake patterns between young (3 to 5 months) and aged (18 to 22 months) wild-type (WT) mice implanted with electroencephalogram (EEG)–electromyography (EMG) electrodes. Wake and NREM but not REM sleep were more fragmented in aged mice (fig. S1). We next determined the number of Hcrt neurons in these mice and found a significant loss (~38%) of Hcrt neurons in aged mice compared with young mice (fig. S2), indicating a high vulnerability of these neurons in the aging brain.
Fragmented sleep pattern with increased Hcrt neuronal activity in aged mice
We monitored the intrinsic activity of Hcrt neurons using fiber photometry recording of GCaMP6f signals in both young and aged Hcrt∷Cre mice (20) while simultaneously recording EEG-EMG signals (Fig. 1) during the light phase, when mice exhibited a stable sleep/wake pattern (fig. S1). We found scattered Hcrt neuronal GCaMP6f transients during sleep (GS) and GCaMP6f epochs associated with wakefulness (GW) in both young and aged mice. The GCaMP6f amplitude change (Z score) was smaller in the aged group (Fig. 1, C and D), indicating that the threshold of Hcrt neuronal activity that defines sleep-to-wake transition is lower in aged mice. The frequency of GW was significantly higher in aged mice (young, 16.1 ± 0.7 counts/hour versus aged, 22.8 ± 1.4 counts/hour) (Fig. 1D, bottom right). The GW epoch frequencies of Hcrt neurons matched the wake bout counts recorded during the same time window in young and aged WT mice, respectively (fig. S1B). The peak and duration of both GS (Fig. 1C, middle right) and GW (Fig. 1D, middle right) were smaller in aged mice. In the same amount of recording time during the same circadian phase, the mean bout duration of sleep, wake, and sleep-wake (S-W) episodes was shorter in aged mice (Fig. 1E); a fragmented sleep/wake pattern was associated with age. Correlation analysis with a linear fit revealed that sleep bout duration negatively correlates with Hcrt GW epoch frequency (Fig. 1F), suggesting the possibility that sleep bout shortening is driven by Hcrt neuron hyperactivity.
Fig. 1. Spontaneous activity of Hcrt neurons across sleep/wake states in young and aged mice.
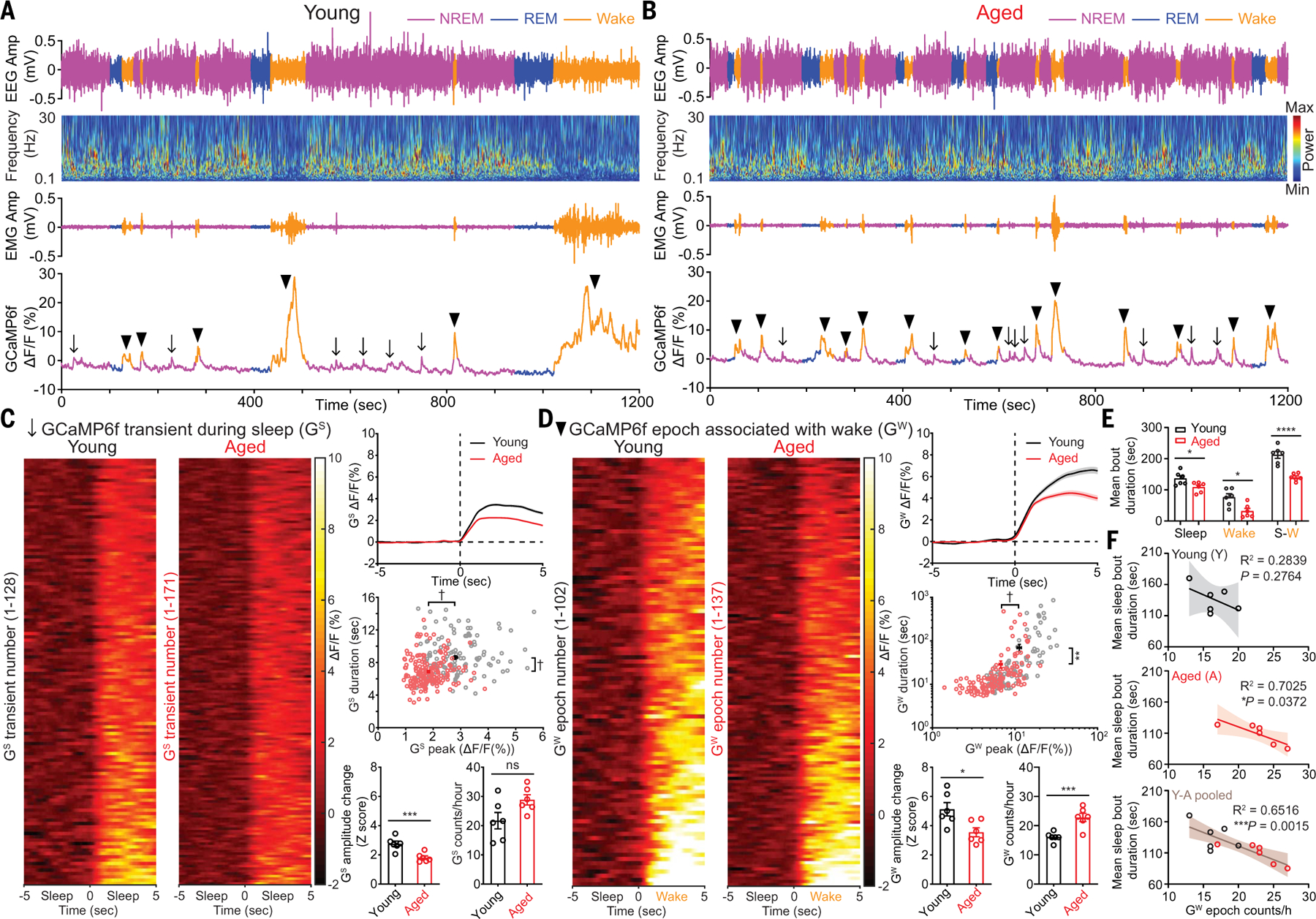
(A and B) Representative EEG, EEG power spectra, EMG, simultaneous Hcrt GCaMP6f signals from (A) young and (B) aged mice. The arrows indicate GCaMP6f transients during sleep (GS), and the black triangles indicate GCaMP6f epochs associated with wakefulness (GW). (C) Staged GS signals during 10 s around the start of GS transients from identical length (6 hours, 1 hour/mouse) of recorded GCaMP6f signals from young and aged mice (n = 6 mice each group), respectively, during light phase, averaged trace plot (right top), scatter plot of individual GS duration against GS peak (young, n = 128; aged, n = 171) (right middle), animal-based comparison of GS signals for Z score and GS frequency (right bottom). (D) Staged GW signals during 10 s around start of GW epochs from identical length (6 hours, 1 hour/mouse) of recorded GCaMP6f signals from young and aged mice, averaged trace plot (right top), scatter plot of individual GW duration against GW peak (young, n = 102; aged, n = 137) (right middle), animal-based comparison of GW signals for Z score and GW frequency (right bottom). (E) Animal-based comparison of mean bout duration for sleep, wake, and entire S-W episodes (n = 6 mice each group). (F) Correlation for mean sleep bout duration against GW bout counts/hour in young, aged, and pooled datasets. Data represent mean ± SEM. In (C) to (E) unpaired t test with Welch’s correction; (F), Spearman correlation, linear fit and 95% confidence band; *P < 0.05, **P < 0.01, ***P < 0.005, ****P < 0.001, †P < 0.0005; statistical details see supplementary text.
Longer wakefulness upon optogenetic activation of aged Hcrt neurons
We then injected adeno-associated virus (AAV) vectors encoding ChR2–enhanced yellow fluorescent protein (eYFP) in the LH of young (3 to 5 months) and aged (18 to 22 months) Hcrt∷ Cre mice (fig. S3, A and B) and implanted fiber optics and EEG-EMG electrodes. After recovery with sufficient virus expression, we stimulated Hcrt neurons in both young and aged mice with a range of blue light intensities (1, 5, 10, 15, and 20 mW) and frequencies (1, 5, 10, 15, and 20 Hz) within 30 s from either NREM onset (Fig. 2, A to F, and fig. S3) or REM sleep onset (Fig. 2, G to L, and fig. S3). Stimulation with high light intensities and frequencies elicited immediate NREM/REM sleep-to-wake transitions in both young and aged mice (Fig. 2, A and G). The sleep-to-wake transition latency is generally shorter in aged mice according to condition-matched comparison (Fig. 2, B and H) and data aggregated for individual mouse (Fig. 2, C and I). Activation of Hcrt neurons evoked significantly longer durations of wakefulness in aged mice as revealed by comparisons based on each stimulation condition (Fig. 2, E and K) and data aggregated for the individual mouse (Fig. 2, F and L) for optogenetic stimulation during either NREM (young, 162.7 ± 5.4 s versus aged, 292.0 ± 8.3 s) (Fig. 2F) or REM sleep (young, 69.0 ± 3.5 s versus aged, 134.0 ± 2.5 s) (Fig. 2L). The surface plots of in vivo optogenetic data demonstrated that compared with the aged mice, the young mice required stronger stimulation to elicit wake bouts with identical lengths, as indicated by the cutaway surface (Fig. 2, D and J) for the mean value of wake duration in aged mice (Fig. 2, F and L). These results demonstrate that the threshold of Hcrt neuronal activity defining the sleep-to-wake transition was lower in aged mice, which is consistent with the hypothesis that aged Hcrt neurons are hyperexcitable.
Fig. 2. More prolonged wake bouts upon optogenetic stimulation of Hcrt neurons expressing ChR2-eYFP in aged mice.
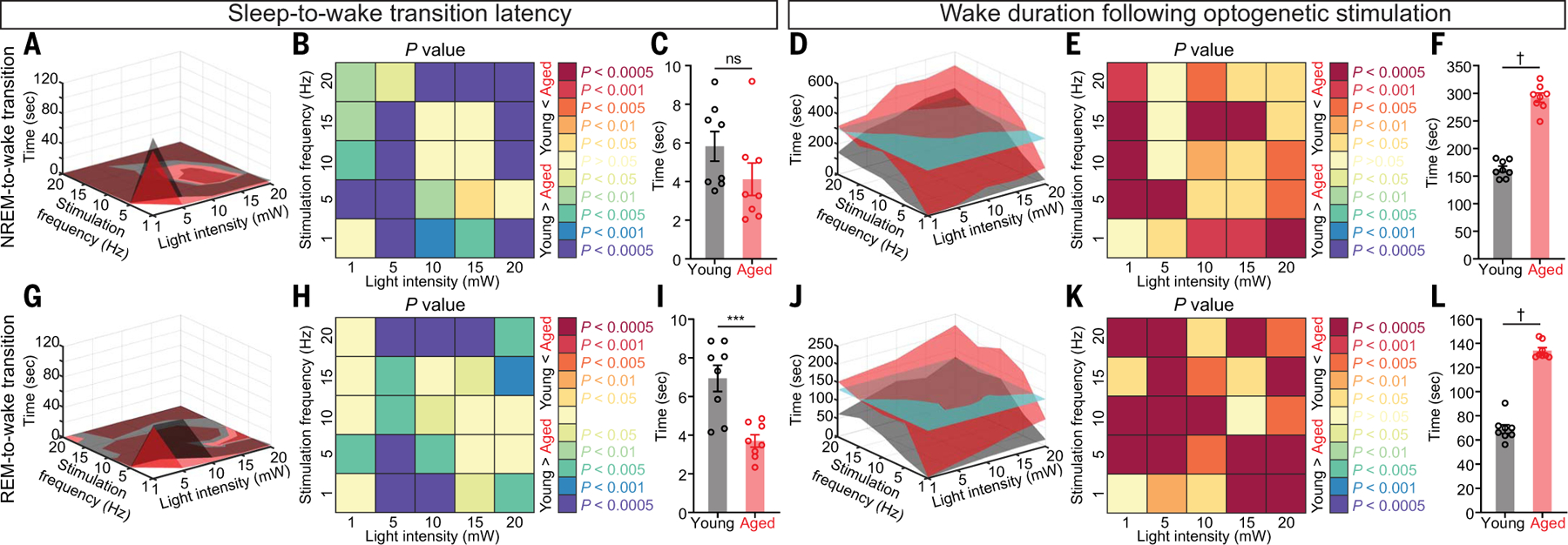
(A) Surface plot of NREM-to-wake transition latency based on the mean value of each stimulation condition. (B and C) Comparison of NREM-to-wake transition latency based on (B) each stimulation condition and (C) the mean value for each animal. (D) Surface plot of wake duration based on the mean value of each stimulation condition. The cyan cutaway surface indicates the mean value for the aged group. (E and F) Comparison of wake duration based on (E) each stimulation condition and (F) the mean value for each animal. (G) Surface plot of REM-to-wake transition latency based on the mean value of each stimulation condition. (H and I) Comparison of REM-to-wake transition latency based on (H) each stimulation condition and (I) the mean value for each animal. (J) Surface plot of wake duration based on the mean value of each stimulation condition. The cyan cutaway surface indicates the mean value for the aged group. (K and L) Comparison of wake duration based on (K) each stimulation condition and (L) the mean value for each animal. In (B), (C), (E), (F), (H), (I), (K), and (L): Mann-Whitney U test; ***P < 0.005, †P < 0.0005. Statistical details are available in the supplementary text.
Elevated intrinsic excitability of aged Hcrt neurons
To determine directly whether the intrinsic excitability of Hcrt neurons differs with age, we recorded spontaneous neuronal activity from ChR2-eYFP–labeled Hcrt neurons with whole-cell patch clamp recording in brain slices from young and aged Hcrt∷Cre mice (Fig. 3). Immunostaining against Hcrt1 confirmed that the recorded neurons infused with biocytin were Hcrt1-positive (Fig. 3A). A higher fraction of aged versus young Hcrt neurons exhibited spontaneous firing (young, 12 of 33 versus aged, 9 of 21) (Fig. 3B). Despite comparable input resistances between young and aged Hcrt neurons (Fig. 3D), the resting membrane potential (RMP) of aged Hcrt neurons was more depolarized than young Hcrt neurons (young, −60.5 ± 1.9 mV versus aged, −51.5 ± 3.1 mV) (Fig. 3E). Ion channels that determine the firing threshold remained unchanged with age because the most negative voltage that must be reached for all-or-none firing to occur (21) was comparable between young and aged Hcrt neurons (young, −34.2 ± 1.6 mV versus aged, −36.3 ± 1.8 mV) (Fig. 3F). The difference between RMP and firing threshold was smaller in aged Hcrt neurons (young, 19.8 ± 2.6 mV versus aged, 11.4 ± 1.5 mV) (Fig. 3G), priming aged Hcrt neurons to fire action potentials (APs) after smaller depolarizations. Young Hcrt neurons also exhibited higher-amplitude APs than those of aged Hcrt neurons (young, 56.2 ± 5.2 mV versus aged, 42.3 ± 1.8 mV) (Fig. 3, B, C and H), although other AP properties did not significantly differ (Fig. 3, I to L).
Fig. 3. Hyperexcitability in aged Hcrt neurons revealed with whole-cell patch clamp recording.
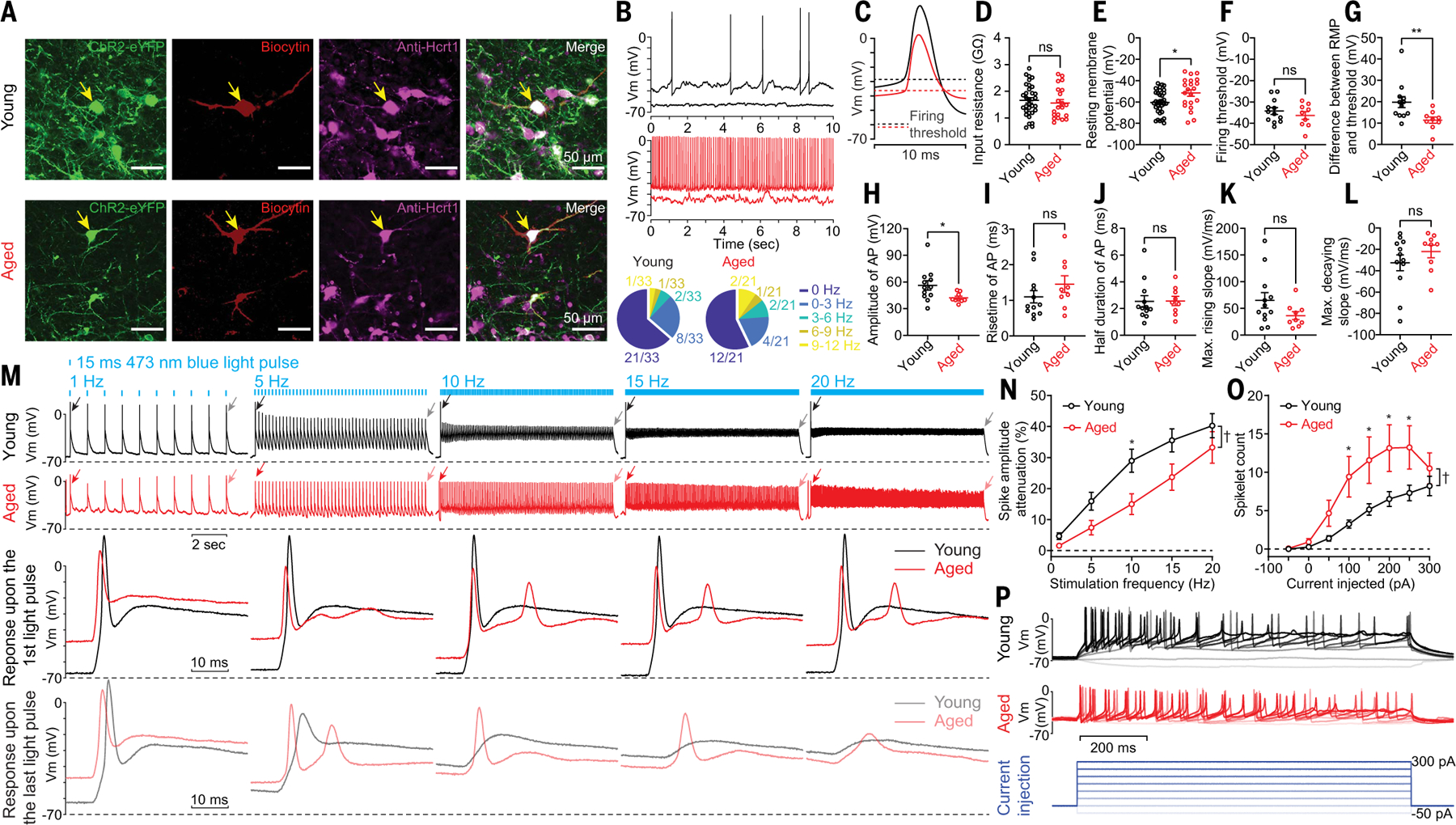
(A) Representative slices containing recorded ChR2-eYFP–labeled Hcrt neurons infused with biocytin. (B) Representative traces and fractions of young and aged Hcrt neurons with and without spontaneous firing activities. (C) Averaged traces of spontaneous APs shown in (B). (D to L) Comparison of basic electrophysiological properties between young and aged Hcrt neurons. (D) Input resistance and (E) RMP of all the recorded young and aged Hcrt neurons (young, n = 33 neurons versus aged, n = 21 neurons from eight mice each group). Comparison of other parameters including (F) firing threshold, (G) difference between RMP and firing threshold, (H) AP peak amplitude, (I) AP rising time, (J) AP half duration, (K) maximum rising slope, and (L) decaying slope of spontaneous APs between young and aged Hcrt neurons (young, n = 12 neurons versus aged, n = 9 neurons from eight mice each group). (M) (Top) Representative traces of young and aged Hcrt neurons expressing ChR2-eYFP upon optogenetic stimulation. (Bottom) The same responses on a slower time base, illustrating the response to the first and last light pulse stimulations at each stimulation frequency (membrane voltage/Vm). (N) Significant reduction of response attenuation calculated based on the first and the last response from trains as in M (young, n = 23 neurons versus aged, n = 21 neurons from eight mice each group). (O and P) Step current injections triggered more spikelets in aged Hcrt neurons than in young Hcrt neurons [(O) young, n = 33 neurons versus aged, n = 26 neurons from eight mice each group; (P) representative traces and current injection protocol]. In (D) to (L): Mann-Whitney U test; (N) and (O): two-way ANOVA followed by post hoc Šidák’s multiple comparisons; *P < 0.05, **P < 0.01, †P < 0.0005. Statistical details are available in the supplementary text.
We next compared light-evoked firing activity between the young and aged Hcrt neurons expressing ChR2-eYFP. We tested different stimulation frequencies and compared the response attenuation, which is defined as the amplitude difference between the first and last light pulse–evoked responses after a train of blue light stimulations. The response attenuation was significantly smaller in aged Hcrt neurons than in young Hcrt neurons (Fig. 3, M and N). Recordings from non-Hcrt neurons postsynaptic to Hcrt neurons (fig. S4A) demonstrated that optogenetic stimulation of Hcrt neurons expressing ChR2-eYFP reliably evoked time-locked postsynaptic currents (PSCs) after optogenetic stimulation more often in the aged group than in the young group (young, 3 of 15 versus aged, 6 of 18) (fig. S4B). More neurons in slices from young mice exhibited PSC failures compared with those from aged mice (fig. S4, C and D). To test whether differences in excitability could be attributed to differential expression of ChR2-eYFP, we performed step-current injection in both young and aged Hcrt neurons. More spikelets were evoked in aged Hcrt neurons by the same current injection protocol (Fig. 3, O and P), again indicating hyperexcitability of aged Hcrt neurons.
Impaired M-current with loss of KCNQ2 channels in aged Hcrt neurons
Hyperexcitability in aged Hcrt neurons with depolarized RMPs suggested a change in ionic conductance such as K+ conductance mediated by voltage-gated potassium channels (KCNQ). There are five mammalian subtypes named KCNQ1 to −5 (Kv7.1 to Kv7.5) (22, 23), and KCNQ2 pairs with the KCNQ3 subunit to form KCNQ2/3 heterotetramers, constituting primarily the molecular substrate of the M-current (IM) (24), which plays a critical role in governing neuronal subthreshold excitability, repolarization, and sensitivity to synaptic inputs (23–25). To test this idea, we applied KCNQ2/3 channel-selective modulators to Hcrt neurons expressing eYFP in brain slices from either young or aged mice (Fig. 4, A–F). Perfusion of a KCNQ2/3-selective blocker, XE991 (50 μM), significantly depolarized the RMP and increased the firing frequency in young Hcrt neurons (Fig. 4, A to C). Reciprocally, application of a KCNQ2/3-selective activator, flupirtine (50 μM), hyperpolarized the RMP and reduced the firing frequency in aged Hcrt neurons (Fig. 4, D to F). XE991 reduced IM in young Hcrt neurons (before, −11.2 ± 1.3 pA versus after, −5.2 ± 0.8 pA) (Fig. 4G, top). Conversely, flupirtine increased IM in both young (before, −16.9 ± 3.8 pA versus after, −22.6 ± 5.2 pA) (Fig. 4G, bottom) and aged (before, −14.6 ± 1.1 pA versus after: −18.0 ± 1.5 pA) (Fig. 4H, bottom) Hcrt neurons. The basal IM in aged Hcrt neurons was significantly smaller than in young Hcrt neurons (young, −17.1 ± 1.9 pA versus aged, −11.8 ± 1.2 pA) (Fig. 4I). These results were validated with array tomography at ultrastructural resolution (26), which revealed a significant reduction in KCNQ2 immunoreactivity in aged Hcrt neurons (Fig. 4J).
Fig. 4. Impaired IM associated with loss of KCNQ2 in aged Hcrt neurons.
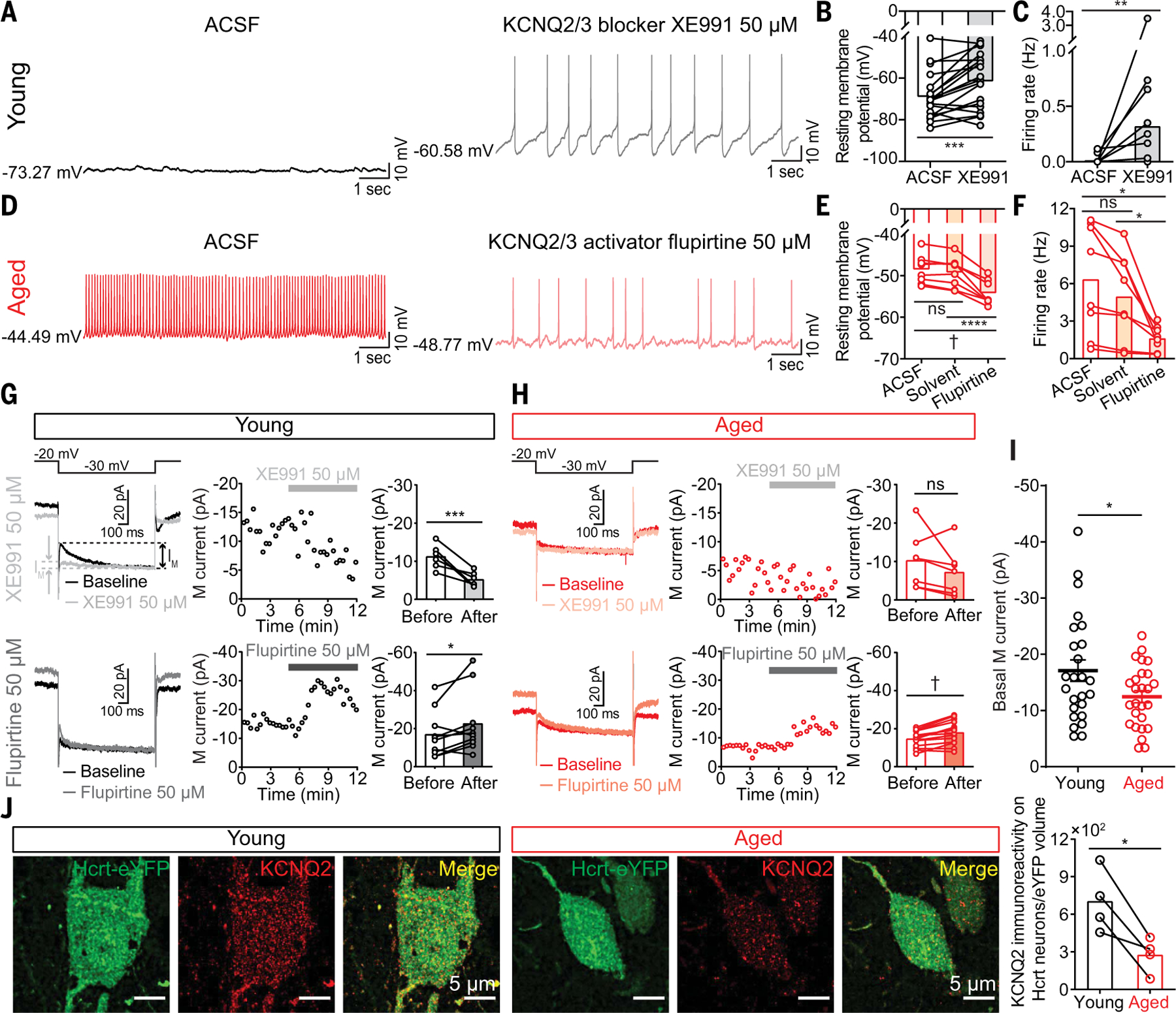
(A) Representative traces from a young Hcrt neuron (left) before or (right) in the presence of a KCNQ2/3 blocker XE991 (50 μM). (B) XE991 depolarized RMP and (C) increased firing frequency in young Hcrt neurons (n = 19 neurons from seven mice). (D) Representative traces from an aged Hcrt neuron (left) before or (right) in the presence of a KCNQ2/3 activator flupirtine (50 μM). (E) Flupirtine hyperpolarized RMP and (F) decreased firing frequency in aged Hcrt neurons (n = 8 neurons from five mice). (G) IM in young Hcrt neurons modulated by XE991 (top; n = 6 neurons from three mice) and flupirtine (bottom; n = 10 neurons from five mice). (H) IM in aged Hcrt neurons modulated by XE991 (top; n = 7 neurons from three mice) and flupirtine (bottom; n = 15 neurons from five mice). (I) Basal IM in young Hcrt neurons (n = 25 neurons from nine mice) versus in aged Hcrt neurons (n = 26 neurons from nine mice). (J) Array tomography revealed reduced KCNQ2 expression in aged Hcrt neurons (n = 4 mice/group). In (B) and (C), Wilcoxon matched-pairs signed rank test; (E) and (F), RM one-way ANOVA followed by post hoc Tukey’s multiple comparisons; (G), (H), and (J), paired t test; (I) unpaired t test with Welch’s correction; statistical details are available in the supplementary text.
To determine the extent of differences between young and aged Hcrt neurons at the transcriptomic level, we performed single-nucleus RNA-sequencing (snRNA-seq). Aged Hcrt neurons developed adaptive up-regulation of prepro-Hcrt mRNA expression in both male (fig. S5) and female (fig. S6) mice, suggesting potential compensatory syntheses of Hcrt neuropeptides by individual Hcrt neurons. Although the normalized expression level of Kcnq subtypes was not significantly different between the young and aged groups, the percentage of aged Hcrt neurons actively expressing Kcnq1/2/3/5 mRNAs, the dominant subtypes, was lower (figs. S5E and S6E) and expected to contribute to the hyperexcitability of aged Hcrt neurons.
CRISPR/SaCas9–mediated disruption of Kcnq2/3 genes in young Hcrt neurons destabilizes NREM sleep
Given the broad expression of KCNQ channels in the brain (25), what is the contribution of IM in Hcrt neurons to the overall sleep architecture? To answer this question, we used CRISPR/SaCas9–mediated disruption (27) of Kcnq2/3 genes specifically in Hcrt neurons in young mice to mirror the impaired IM observed in aged Hcrt neurons. We designed AAV vectors for Cre-dependent expression of SaCas9 (28), sgControl, and sgKcnq2/3 targeting Kcnq2/3 genes in Hcrt neurons (Fig. 5). Young Hcrt∷Cre mice from different litters were randomly separated into two groups. We delivered a viral mixture of SaCas9 and sgControl to the Hcrt field bilaterally in the control group, whereas the other group received bilateral injection of a SaCas9 and sgKcnq2/3 viral mixture for disruption of Kcnq2/3 genes (Fig. 5A). All the mice were implanted with EEG-EMG electrodes for sleep/wake pattern monitoring for a 2-day consecutive recording of EEG-EMG signals weekly for 8 weeks. CRISPR/SaCas9–mediated deactivation of Kcnq2/3 genes in Hcrt neurons was sufficient to generate NREM sleep fragmentation in young mice (NREM mean bout length at 8 weeks after virus injection, 48 hours; sgControl, 2.1 ± 0.04 min versus sgKcnq2/3, 1.7 ± 0.1 min; light phase, sgControl, 2.2 ± 0.1 min versus sgKcnq2/3, 1.9 ± 0.1 min; dark phase, sgControl, 2.0 ± 0.1 min versus sgKcnq2/3, 1.6 ± 0.1 min) (Fig. 5B). We then performed whole-cell recording from individual mCherry-labeled Hcrt neurons (Fig. 5C) after EEG-EMG recording at 8 weeks after virus injection and found that the Hcrt neurons with Kcnq2/3 gene disruptions showed a depolarized RMP (sgControl, −67.0 ± 2.4 mV versus sgKcnq2/3, −55.9 ± 3.3 mV) (Fig. 5D) and spontaneous firing activity in higher proportion (Fig. 5E), mimicking the aged Hcrt neurons. EEG-EMG recording up to 12 weeks after virus infection in a subset of these mice (fig. S7) further validated our observations at 8 weeks after virus injection. Analyses of the basic electrophysiological properties of the spontaneous APs revealed that artificial disruption of KCNQ2/3 channels in young Hcrt neurons (fig. S7, E and F) mimicked some features observed in the aged Hcrt neurons, including a trend of reduction in the maximum rising/decaying slope of spontaneous APs (Fig. 3, D to L).
Fig. 5. CRISPR/SaCas9–mediated disruption of Kcnq2/3 genes in Hcrt neurons leads to NREM sleep fragmentation in young mice.
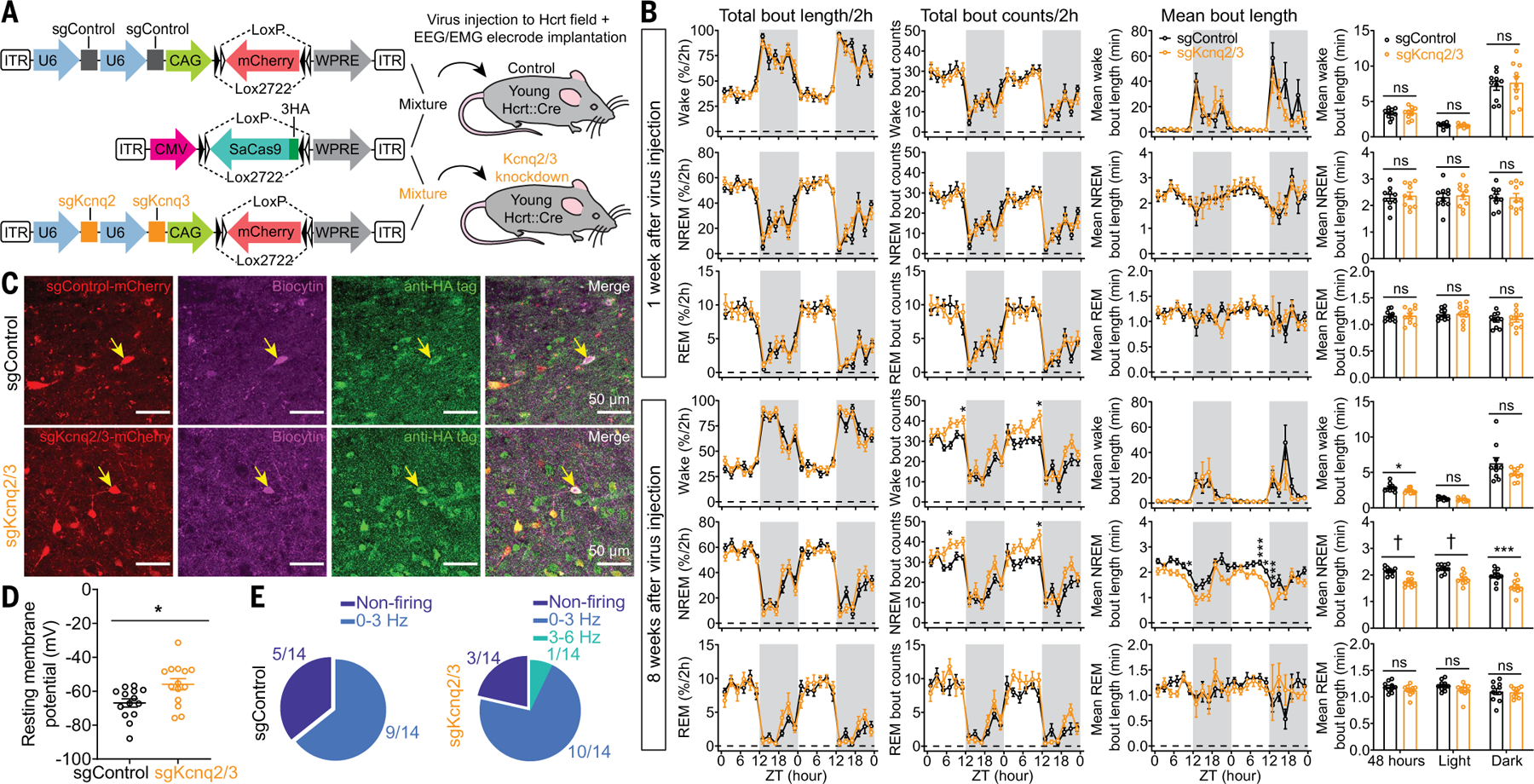
(A) Schematic of AAV sgControl, AAV SaCas9, AAV sgKcnq2/3 vector design, and bilateral viral infection of Hcrt neurons in young Hcrt∷Cre mice. (B) Two-hour (left) binned percentage, (middle left) bout counts, (middle right) mean bout length and (right) mean bout length based on circadian phase for wake, NREM, and REM sleep at 1 week (top) and 8 weeks (bottom) after injection of a virus mixture, as illustrated in (A) (n = 10 mice/group, dark phase indicated by gray shielding). (C) Representative slices expressing sgRNA with fluorescent mCherry flag and SaCas9–3HA for sgControl and sgKcnq2/3 group, respectively. Patch clamp recorded cells were labeled with biocytin, and post hoc antibody staining against HA tag confirmed the cells expressing SaCas9 for data analyses. (D) Comparison of RMPs between sgControl and sgKcnq2/3 group (n = 14 neurons from three mice each group). (E) Fractions of neurons with different firing frequencies in the (left) sgControl and (right) sgKcnq2/3 groups. Data indicate mean ± SEM [(B) left to middle right, two-way RM ANOVA followed by Šidák’s multiple comparisons; (B) right, Holm-Šidák; (D) Mann-Whitney U test; *P < 0.05, ***P < 0.005, †P < 0.0005; statistical details are avialable in the supplementary text].
In vivo evaluation of KCNQ2/3-selective ligands
To further test the role of KCNQ2/3 channels in sleep modulation, we administered (intraperitoneally)either the KCNQ2/3-selective blocker XE991 (2 mg/kg) or the saline vehicle as control to young (3 to 5 months) WT mice at the beginning of the light phase, when there is a strong sleep pressure. XE991 significantly increased the wake amount during the first hour after injection compared with that of the control group (Fig. 6A) without affecting the power spectra (Fig. 6, C, E, and G). Reciprocally, we injected the KCNQ2/3–selective activator, flupirtine (intraperitoneally, 20 mg/kg) or 0.3% dimethyl sulfoxide (DMSO) in saline (v/v) as vehicle to aged (18 to 22 months) WT mice at the beginning of the light phase. Flupirtine significantly increased the amount and stability of NREM sleep compared with the control group (Fig. 6B). Flupirtine also increased the theta band power during NREM sleep and the initial REM sleep segment after drug administration (Fig. 6, D, F and H). Sleep quality is correlated with cognitive functions (1, 2), and flupirtine administered to aged mice at the beginning of light phase after the familiarization session in the object-recognition task also improved the subsequent exploration of the new object (fig. S8).
Fig. 6. Pharmacological manipulation of sleep/wake states with KCNQ2/3 ligands.
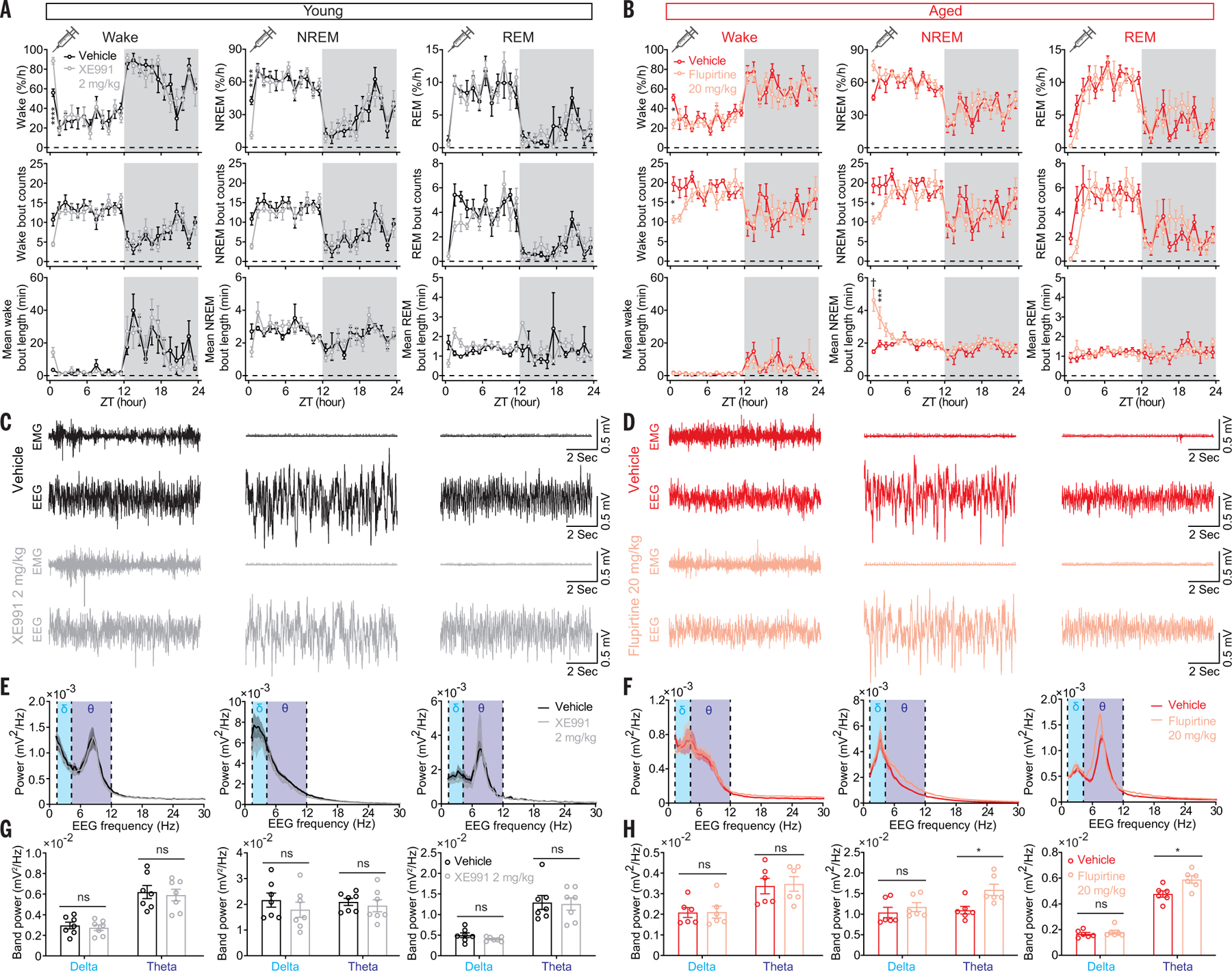
(A) Significantly increased wake amount by the KCNQ2/3 blocker XE991 (2 mg/kg) in young mice. (B) Significantly increased NREM amount and mean bout length by the KCNQ2/3 activator flupirtine (20 mg/kg) in aged mice. (C) Representative EMG-EEG raw traces from vehicle- and XE991-treated (2 mg/kg) young mice. (D) Representative EMG-EEG raw traces from vehicle- and flupirtine-treated (20 mg/kg) aged mice. (E) Power spectra of EEG for vehicle- and XE991-treated young mice and (F) power spectra of EEG for vehicle- and flupirtine-treated aged mice. (G) Comparison of delta, theta band power between vehicle- and XE991-treated young mice and (H) between vehicle- and flupirtine-treated aged mice. Data indicate mean ± SEM [young, n = 7 mice each group; aged, n = 6 mice each group; (A) and (B) two-way liner mixed-effects model followed by Šidák’s multiple comparisons, dark phase indicated by gray shielding; (G) and (H) Holm-Šidák, *P < 0.05, ***P < 0.005, ****P < 0.001, †P < 0.0005; statistical details are available in the supplementary text].
Sleep instability in a narcolepsy mouse model with genetic ablation of Hcrt neurons
Abrupt Hcrt neuron loss in pathological conditions at young ages causes type I narcolepsy, a condition characterized by excessive daytime sleepiness and a sudden loss of muscle tone with EEG pattern resembling REM sleep, known as cataplexy, as well as sleep fragmentation (17). We crossed OX(Hcrt)–enhanced green fluorescent protein (eGFP) mice (29) with OX(Hcrt)-ataxin3 mice, a narcolepsy mouse model (15), to generate mice with Hcrt neurons that express both eGFP and ataxin3, allowing us to examine the activity of these neurons while monitoring EEG-EMG patterns of their litter-mates (figs. S9 and S10). EEG-EMG recording from 5-week-old mice expressing ataxin3 exhibited mild NREM sleep fragmentation, with three of six mice showing cataplexy-like EEGEMG epochs compared with their control group with intact Hcrt neurons (fig. S9, A to C), according to established criteria (30). In recordings from brain slices, although Hcrt cells from the ataxin3-expressing group had a more depolarized RMP—a smaller difference between RMP and firing threshold—as well as a higher fraction of spontaneously firing neurons compared with that of age-matched controls, other basic electrophysiological properties were similar (fig. S9, D to G). Ataxin3-expressing mice at an age of 12 weeks showed obvious NREM sleep fragmentation with a severe cataplexy-like EEG-EMG pattern (six of six mice) (fig. S10A). Immunostaining against Hcrt1 showed a near complete Hcrt neuron degeneration at the age of 12 weeks in mice expressing ataxin3 (fig. S10B), confirming Hcrt neuron loss around a similar age in this narcolepsy mouse model (15). Thus, although both healthy aged WT mice and young mice expressing ataxin3 show sleep fragmentation, the underlying mechanisms are not identical.
Discussion
Sleep quality decline—in particular, fragmented nocturnal sleep during aging—impairs daytime well-being in elderly individuals (3). However, the mechanistic underpinnings of aging-related sleep fragmentation are unknown. In this study, we report a mechanism underlying sleep instability during aging. We found a generally fragmented sleep/wake pattern in aged mice with reduced wakefulness during their active phase (fig. S1), replicating the sleep traits in aged populations (3). We further observed more frequent Hcrt GCaMP6f activity epochs driving the sleep fragmentation in aged mice (Fig. 1). Despite a reduction in the number of wake-promoting Hcrt neurons in aged mice (fig. S2), these mice paradoxically manifested significantly longer wake bouts in response to Hcrt neuron optogenetic stimulation (Fig. 2 and fig. S3). Immunostaining against Hcrt1 confirmed that the majority of the virus-infected neurons are hypocretinergic (fig. S3, B and C), and the small fraction of non-Hcrt neurons may be assigned to the ~30 cell types described in the LH (31), none of which are likely to affect bout length directly (32–34). Aged Hcrt neurons exhibited increased intrinsic excitability, with more depolarized RMPs (Fig. 3), more spikelets per unit current (Fig. 3, O and P), and up-regulated prepro-Hcrt nuclear mRNA expression (figs. S5 and S6).
To understand the molecular mechanisms underlying the phenotypes described above, we focused on the ion channels that regulate the intrinsic excitability of neurons. Potassium channels play an important role in governing the excitability of neurons through repolarizing action potentials (22, 25). Specifically, KCNQ2/3 channels are expressed broadly in brain regions controlling neuronal network oscillations and synchronization (35), and a moderate loss of function of these channels causes epilepsy in humans (36, 37) and mice (38). In aged Hcrt neurons, we discovered impaired KCNQ2/3–mediated IM associated with lower KCNQ2 channel density (Fig. 4, I and J). The loss of KCNQ2 may be due to oxidation, a known factor for potassium conductance impairment during aging (39). Decreased levels of transcription factor specificity protein 1 (sp1) in senescent cells (40) might be another reason for impaired IM in aged Hcrt neurons because sp1 has been shown to activate the expression of KCNQ2/3 and augment IM (41). Our data support the hypothesis that the arousal circuitry, and particularly the Hcrt system, becomes hyperexcitable during aging. Selective disruption of Kcnq2/3 genes in young Hcrt neurons was sufficient to depolarize these neurons and cause sleep fragmentation (Fig. 5 and fig. S7), mimicking the sleep instability observed in aged mice (fig. S1). Further advancing these findings, systemic administration of a KCNQ2/3 blocker increased wakefulness (Fig. 6A), whereas a KCNQ2/3 activator consolidated NREM sleep (Fig. 6B). Specifically targeting the Hcrt system with a pharmacological tool, application of a dual Hcrt/OX receptor antagonist MK6096 (filorexant, 20 mg/kg, intraperitoneally) also increased the amount of NREM sleep and mean NREM bout length within 6 hours after drug administration (fig. S11). Our pharmacological data may open a new approach for conquering sleep quality decline during aging.
On the basis of our scRNA-seq data (figs. S5 and S6), KCNQ1/5 mRNA are expressed in Hcrt neurons and are expected to have a lower KCNQ subunit expression level in aged Hcrt neurons both in male (fig. S5E) and female mice (fig. S6E). Factors other than impaired M-current with a key role in neuron repolarization may also contribute to increased excitability of arousal-promoting circuits during aging. Together with KCNQ family, G protein–gated inward rectifying K+ channels (Girks) are expressed in the brain and mediate outward potassium current in hyperpolarizing neurons and decreasing intrinsic excitability (42).
Our findings in aged animals may extend to neurodegenerative conditions such as Alzheimer’s disease (AD). The level of amyloid-β (Aβ, a marker of AD) in brain interstitial fluid correlates with wakefulness and increases with elevated hypocretinergic tone in AD mouse models (43). Aβ reduces gene expression of Kcnq2/3 and Girk2/3/4 in hippocampal slices incubated with Aβ fragment Aβ25–35 (44) in line with a deleterious cycle of hyperactivation, with multiple excitatory elements implicating Aβ-induced hyperexcitation in hippocampal neurons (45), which links AD pathology–mediated down-regulation of K+ channels to neuronal excitability. Loss of hyperexcitable arousal-promoting neurons destabilizing sleep during aging could be drastically exacerbated by AD pathology, as evidenced by a study of post mortem brains from AD patients that demonstrate a ~32% Hcrt neuron loss compared with their age-matched controls without AD (46).
Among the main downstream targets of Hcrt neurons (47, 48), locus coeruleus noradrenergic (LC NA) neurons displayed a milder cell count loss (~15%) (fig. S12) compared with Hcrt neuron loss (~38%) (fig. S2) in the same group of aged mice. Optogenetic activation of LC NA neurons elicited sleep-to-wake transitions and maintained wakefulness more robustly, as indicated by shorter sleep-to-wake transition latencies and longer wake bout durations in aged mice with the same optogenetic stimulations in young and aged mice (fig. S13). Electrophysiological recordings revealed a more depolarized RMP and a higher proportion of neurons with spontaneous firing activity in aged LC NA neurons (fig. S14). These results suggested a potential up-regulation of excitability in other arousal-promoting brain nuclei during aging.
Sleep instability and fragmentation not only appears in the elderly but also have been observed in type I narcolepsy patients with loss of Hcrt neurons at young ages, yet the pathology may develop gradually (17), which raises the question that whether the spontaneous activity of the remaining Hcrt neurons are up-regulated to compensate for the rapid Hcrt neuron loss in the narcolepsy pathological condition. With a narcolepsy mouse model, we found that the remaining Hcrt neurons exhibited depolarized RMPs and a higher fraction with spontaneous firing activity (fig. S9, D to G), despite other AP properties being similar to those of age-matched controls around 5 weeks of age. Yet because the 12-week-old ataxin3+ mice have almost completely lost their Hcrt neurons, the even more fragmented sleep features in these mice (compared with the sleep pattern around 5 weeks) (figs. S9 and S10) cannot be attributed to alteration of Hcrt/OX neuronal excitability. Increased depolarization of the remaining Hcrt neurons in ataxin3-expressing mice may result from toxicity introduced by ataxin3 expression or neural circuit reorganization or malfunction during rapid Hcrt neuron loss, whereas elevated depolarization in aged mice is the result of an altered complement of membrane ion channels. Although it is possible that there is also neural circuit reorganization or malfunction accompanying chronic Hcrt neuron loss during natural healthy aging, the absence of a cataplexy-like EEG-EMG pattern in healthy aged WT mice highlighted that the mechanisms underlying sleep fragmentation in young OX(Hcrt)-ataxin3 mice and healthy aged WT mice are not identical.
Collectively, our study delineates that elevated Hcrt neuron excitability is associated with sleep fragmentation during aging. A lower threshold defining sleep-to-wake transitions driven by hyperexcitable Hcrt neurons accounts for sleep fragmentation and yields longer wake bouts upon optogenetic stimulation of these neurons in aged mice. The restoration of sleep stability in aged mice upon manipulation of KCNQ channels may lead to the development of potential therapeutic strategies in aged individuals whose sleep fragmentation contributes to aging-related neurodegeneration.
Materials and methods
Animals
Experiments with mice were performed following the protocols approved by the Stanford University Animal Care and Use Committee in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Discomfort, distress, and pain were minimized with anesthesia and analgesic medications. Mice were housed in a temperature- and humidity-controlled animal facility with a 12-hours/12-hours light/dark cycle (9 am light on, Zeitgeber time 0/ZT 0), unless otherwise specified. Mice had ad libitum access to standard laboratory mouse food pellets and water. 2–3 month young male adult wild type (WT) mice were acquired from Jackson Laboratory (Jax) and 18 month old WT male mice were acquired from National Institute on Aging (NIA). OX(Hcrt)-ataxin3 heterozygotes (15), Hcrt-IRES-Cre knock-in (Hcrt∷Cre) heterozygotes (20), OX(Hcrt)-eGFP heterozygotes (29) and tyrosine hydroxylase (TH)-IRES-Cre knock-in (TH∷Cre) heterozygotes (European Mouse Mutant Archive; EMMA ID: EM:00254) (49) were backcrossed onto C57BL/6J background. Male mice were used in the experiments, unless otherwise specified. Mice at an age younger than 5 months belonged to the young group, whereas mice older than 18 months were considered as aged. Animals from multiple litters were randomly assigned to control or experimental group under each experimental paradigm. Group sizes were determined based on earlier publications (13, 50, 51).
EEG-EMG electrode preparation and implantation
Mini-screw (US Micro Screw) was soldered to one tip of an insulated mini-wire with two tips exposed, and the other tip of the mini-wire was soldered to a golden pin aligned in an electrode socket. A micro-ring was made on one side of an insulated mini-wire with the other end soldered to a separate golden pin in the electrode socket. Each electrode socket contained 4 channels with 2 mini-screw channels for EEG recording and 2 micro-ring channels for EMG recording as described in earlier work from our lab (12, 47, 50). The resistance of all the channels was controlled with a digital Multimeter (Fluke) to be lower than 1.5 ohms for ideal conductance. Mice were mounted onto an animal stereotaxic frame (David Kopf Instruments) under anesthesia with intraperitoneal injection of a mixture of ketamine (100 mg/kg) and xylazine (20 mg/kg). Two mini-screws were placed in the skull above the frontal (AP: −2 mm; ML: ± 1 mm) and temporal (AP: 3 mm, ML: ± 2.5 mm) cortices for EEG signal sampling and two micro-rings were placed in the neck muscles for EMG signal acquisition. Electrode socket was secured with Metabond (Parkell, Japan) and dental acrylic on skull for recording in freely moving mice. Buprenorphine SR (0.5 mg/kg) was administered subcutaneously to mice before and after surgery for pain relief. After surgery, revertidine (5 mg/kg) was administered (intraperitoneally) to mice to facilitate recovery from anesthesia.
Virus injection with and without fiber optic implantation
Optogenetic experiments
0.3 μl AAV-DJ-EF1α-DIO-hChR2(H134R)-eYFP viruses (ChR2-eYFP, 6.5 × 1012 gc/ml, Stanford Virus Core, Lot no. 4176) was delivered to LH (AP: −1.35 mm, ML: ± 0.95 mm, DV: −5.15 mm) of anesthetized young (3 to 5 months) or aged (18 to 22 months) Hcrt∷Cre mice with a 5 μl Hamilton microsyringe according to stereotaxic coordinates determined on a Kopf stereotaxic frame. AAV-DJ-EF1α-DIO-eYFP viruses (eYFP, 6.9 × 1012 gc/ml, Stanford Virus Core, Lot no. 3010) was used as control or for in vitro pharmacology experiments. A glass fiber (200 μm core diameter, Doric Lenses, Franquet, Québec, Canada) was implanted with the tip right above the injection site for optogenetic stimulations later on. After fixing the glass fiber with Metabond, the EEG/EMG electrodes were implanted with dental acrylic fixation. Similar procedure was performed for virus injection in TH∷Cre mice targeting LC NA neurons (AP: −5.46 mm, ML: ± 1.2 mm, DV: −3.6 mm). Mice were allowed to recover for at least 2 weeks to get sufficient virus expression before connecting to the EEG/EMG recording cables and optical stimulation patch cord. EEG/EMG electrode and fiber optic implantation were omitted in the mice infected with ChR2-eYFP or eYFP viruses used for in vitro electrophysiology experiments.
Fiber photometry
For fiber photometry, 0.3 μl AAV vectors encoding GCaMP6f (AAV-DJ-EF1α-DIO-GCaMP6f, 1.1 × 1012 gc/ml, Stanford Virus Core, Lot no. 3725) were delivered to LH (AP: −1.35 mm, ML: ± 0.95 mm, DV: −5.15 mm) of young (3 to 5 months) or aged (18 to 22 months) Hcrt∷Cre mice with a 5 μl Hamilton micro-syringe. A glass fiber (400 μm core diameter, Doric Lenses) was implanted with the tip at the injection site for GCaMP6f signal acquisition afterwards. EEG/EMG electrodes were implanted following fixation of fiber optic and secured with Metabond and dental acrylic. Mice were allowed to recover for at least 2 weeks to get sufficient virus expression before connecting to the EEG-EMG recording cables and fiber photometry recording patch cord.
Single-nucleus RNA-sequencing (snRNA-seq)
To label telomeres in the nuclei, 0.3 μl AAV vectors encoding Cre-dependent DsRed-hTRF2 (52) (AAV-DJ-DIO-DsRed-hTRF2, 1.95 × 1012 gc/ml, customer viruses packaged at Stanford Virus Core, Lot no. 4422) were bilaterally injected to the LH (AP: −1.35 mm, ML: ± 0.95 mm, DV: −5.15 mm) of young (3 months) and aged (18 months) male and female Hcrt∷Cre mice [3 mice per condition (young/aged male/female)].
EEG/EMG recording and analysis
Mice were singly-housed after surgery and allowed to recover for 1 week with access to food and water ad libitum before EEG/EMG recording. EEG/EMG signals were amplified through a multiple channel amplifier (Grass Instruments) and acquired with VitalRecorder (Kissei Comtec Co.) with a sampling frequency of 256 Hz followed by offline signal analysis. The bandpass was set between 0.1 and 120 Hz. Raw EEG/EMG data were exported to Matlab (MathWorks, Natick, MA, USA) and analyzed with custom scripts and Matlab built- in tools based on described criteria (12) to determine behavioral states. Cataplexy-like EEG/EMG pattern was determined based on the criteria described in the original publication reporting the OX(Hcrt)-ataxin3 narcolepsy mouse model (15) and the consensus definition of cataplexy in mouse models of narcolepsy: (i) ≥ 10 s of EMG atonia; (ii) EEG with theta band domination; (iii) behavioral immobility preceded by ≥ 40 s of wakefulness (30). For optogenetic and fiber photometry recording experiments, simultaneous EEG/EMG signals were recorded to determine behavioral states. The latency of sleep-to-wake transition and the duration of wakefulness following optogenetic stimulation during sleep were determined in SleepSign (Kissei Comtec Co.) with indication of stimulation timestamps on the raw EEG/EMG signals. EEG power spectral analysis was performed with the same method as described earlier (13). EEG band power calculation was based on: delta (1 to 4 Hz); theta (4 to 12 Hz). EEG band power comparison between vehicle- and KCNQ2/3 ligand-treated groups was conducted based on signals during 1 hour (for vehicle versus XE991) and 3 hours (for vehicle versus flupirtine) following injection for wakefulness and NREM sleep based on the dynamic of drug’s effect. As both XE991 and flupirtine postponed REM sleep onset, EEG band power was computed based on the initial REM sleep epoch after injection of vehicle/drug. The investigator was blind to the group information while conducting the EEG/EMG data analysis.
In vivo optogenetic stimulation
After recovery and sufficient virus expression (>2 weeks), mice injected with viruses expressing Cre- dependent ChR2-eYFP were connected to EEG/EMG recording cables and fiber optic patch cords (200 μm core diameter, Doric Lenses) for one week acclimation in special cages with open top which allowed mice to move freely. Following acclimation, optogenetic stimulation with a range of frequencies (1, 5, 10, 15, and 20 Hz, controlled by A.M.P.I. Master 8) and a range of blue light (473 nm) intensities (1, 5, 10, 15, and 20 mW, Laser-glow Technologies, calibrated with Thorlabs light meter) was performed. Each stimulation train consisted of 15 ms light pulses for 10 s with a given light intensity and frequency according to a randomized 5 (light intensities) × 5 (frequencies) matrix generated in Matlab. Sleep-to-wake transition experiments were performed between ZT5-ZT9 of their inactive phase when mice have a strong sleep pressure. Light stimulations were delivered to mice within 30 s of NREM or REM sleep onset to determine the latency of sleep-to-wake transition and duration of wake bout following optogenetic stimulation. The onset of light stimulation was time-stamped during recording for offline analysis afterwards.
Fiber photometry signal acquisition and analysis
After recovery, sufficient virus expression (>2 weeks), and habituation to EEG/EMG cable and fiber optic patch cord (400 μm core diameter, Doric Lenses), mice injected with AAV viruses expressing Cre-dependent GCaMP6f were connected to EEG/EMG recording setup and a custom-built fiber photometry setup (50). Briefly, a 470-nm LED (M470D3, Thorlabs, NJ, USA) was sinusoidally modulated at 211 Hz and passed through a GFP excitation filter followed by a dichroic mirror (MD 498, ThorLabs) for reflection. The light stream was sent through a high NA (0.48), large core (400 μm) optical fiber patch cord (Doric Lenses, Québec, Canada), which was connected with a zirconia connector (Doric Lenses, Québec, Canada) to the dental acrylic-secured fiber optic implant (0.48NA, 400 μm, Doric Lenses, Québec, Canada) with the tip on the injection site. Separately, a 405-nm LED was modulated at 531 Hz and filtered by a 405-nm bandpass filter and sent through the optical fiber patch cord to mouse brain to evoke reference fluorescence, which was independent of Ca2+ release. GCaMP6f fluorescence and reference fluorescence were sampled by the same fiber patch cord through a GFP emission filter (MF525-39, ThorLabs), and center-aligned to a photodetector (Model 2151, Newport, Irvine, CA, USA) with a lens (LA1540-A, ThorLabs). The analog signals were amplified by two lock-in amplifiers for GFP fluorescence and reference fluorescence respectively (30 ms time constant, model SR380, Stanford Research Systems, Sunnyvale, CA, USA). Matlab-based custom software was used to control the LEDs and sample both the GFP fluorescence and reference fluorescence through a multifunction data acquisition device (National Instruments, Austin, TX, USA) with 256 Hz sampling frequency in a real-time manner. ΔF/F was obtained by subtracting the reference fluorescence signal from the 470-nm excited GFP emission signal to remove the system interference. The optical fiber patch cord was photobleached to minimize autofluorescence prior to recording according to the user manual (Doric Lenses, Québec, Canada). The recording was conducted between ZT5-ZT9 of their inactive phase when mice have a strong sleep pressure.
To reveal the Hcrt neuronal activity in driving behavioral pattern changes, we used a bottom-up analysis strategy, i.e., GCaMP6f data were staged independent of simultaneous EEG/EMG signals. We then separated the increased GCaMP6f into two categories: GCaMP6f transients during sleep (GS) and GCaMP6f epochs associated with wakefulness (GW) (Fig. 1). All the GS and GW were staged from the same amount of recording conducted during ZT5-ZT9 from equal group size (1 hour/each mouse, n = 6 mice each group) for comparison of Hcrt neuronal activity between young and aged mice. All the GCaMP ΔF/F transients with a Z score >1% (equals GCaMP6f ΔF/F value ~0.3–0.6 for individual animal) of the highest ΔF/F value of the entire trace were staged. After data staging, each GCaMP6f epoch was normalized to its own 5 s baseline with time 0 defined for the beginning of GCaMP6f rising phase. Heatmaps were generated for each category based on 10 s of normalized GCaMP6f epochs with 5 s prior to and 5 s after time 0. A Z score was calculated by subtracting the mean value of GCaMP6f trace prior to time 0 from the mean value of GCaMP6f after time 0 and an averaged Z score based on each animal was used for statistical comparisons. As the GS Z score was generally small, only the GS transients with Z score > mean (GS Z score) – 3 × SEM (GS Z score) were included with ideal signal-to-noise ratio for subsequent analyses. By definition, all the GW epochs were qualified for analyses. GS scatter plot was generated with the duration of GS against its peak value, and GW scatter plot was generated with the duration of wake-associated GW epoch against its maximum peak value (maximum GCaMP6f ΔF/F, if given epoch appeared with multi-peaks). Animal-based frequencies of GS and GW were compared between the young and aged groups. Durations of sleep, wake, and S-W epochs were compared. Spearman correlation analysis with a linear fit was perform between GW frequency (counts/hour) and mean sleep bout duration. The investigator was blind to the group information while conducting the GCaMP6f data staging.
Chemical preparation and application
XE991 dihydrochloride (Cat. no. 2000, referred to as XE991) and flupirtine maleate (Cat. no. 2867, referred to as flupirtine) were purchased from Tocris. XE991 was prepared in saline with a concentration of 50 μM for in vitro electrophysiology and prepared in saline with a concentration of 0.2 mg/ml for in vivo experiment with a dosage of 2 mg/kg (0.1 ml/10 g, intraperitoneally). 5 mM flupirtine stock solution (0.9% saline containing 0.3% dimethyl sulfoxide/DMSO, v/v) was added to artificial cerebrospinal fluid (ACSF) to reach a concentration of 50 μM for in vitro electrophysiology. Flupirtine was prepared at a concentration of 2 mg/ml in 0.9% saline containing 0.3% DMSO (v/v, vehicle) for in vivo experiments with a dosage of 20 mg/kg (0.1 ml/10 g, intraperitoneally). Flupirtine solution was ultrasonicated prior to application. Counterbalanced crossover design was used for in vivo pharmacology experiments to reveal the drug’s effect. Two rounds of drug administrations were separated by at least one week for a complete wash-out of drug’s effect. 4-Aminopyridine (4-AP) was purchased from Sigma-Aldrich (Cat. no. 275875). 100 mM 4-AP stock solution (0.9% sodium chloride/saline as vehicle) was added to ACSF to reach a concentration of 50 μM for in vitro electrophysiology of IM experiment. MK6096 (Merck) was prepared at a concentration of 2 mg/ml in a mixture (v/v, vehicle) of 50% 0.9% saline and 50% Poly ethylene glycol (average Mn 400, PEG400, Sigma-Aldrich Cat. no. 202398) for in vivo pharmacology experiment as previously described (13).
In vitro electrophysiology
All the in vitro electrophysiology experiments were performed during the light phase (ZT3-ZT9). 3–9 mice were used each group. Slices were randomly assigned to groups examining effects of XE991 or flupirtine on M current in the in vitro pharmacology experiments.
Slice preparation
Mice from both groups were decapitated after anesthesia with sevoflurane or perfused with ice-cold slicing solution under anesthesia. To increase the chances of acquiring a healthy slice, we used a sucrose-based or choline-based ACSF for brain slice preparation to reduce the cell excitotoxicity and loss during slice preparation (53). After decapitation, the brain was rapidly dissected and immersed in ice-cold sucrose/choline-based ACSF slicing solution (pH 7.4, 95% O2 / 5% CO2). 300 μm-thick coronal brain slices containing Hcrt neurons with eYFP fluorescence were sectioned using a VT1200s vibratome (Leica Microsystems). After ~20 min incubation at ~35°C, the slices were stored at room temperature. Slices were used for maximally 5 hours after dissection. Experiments were performed at room temperature 21° to 24°C.
Recording and data analysis
During experiments, slices were superfused with a physiological extracellular solution containing: 125 mM NaCl, 2.5 mM KCl, 25 mM NaHCO3, 1.25 mM NaH2PO4, 25 mM D-glucose, 2 mM CaCl2, and 1 mM MgCl2 (pH 7.4 in 95% O2/5% CO2, ~325 mOsm). Neurons were chosen based on eYFP expression and visualized with an Olympus BX51WI with Nomarski optics connected to a camera (Q-imaging). Thick wall borosilicate pipettes (1B150F-4, World Precision Instruments Inc.) were pulled using a P-97 puller (Sutter Instruments) and electrodes with a resistance of 3–6 megohms were used for recording. Intracellular solution used for whole-cell recording contained: 120 mM K-methyl-sulfonate, 10 mM NaCl, 10 mM EGTA, 1 mM CaCl2, 10 mM HEPES, 0.5 mM NaGTP, 5 mM MgATP, pH adjusted to 7.2 with KOH, osmolarity adjusted to 305 mOsm with sucrose; 0.2% biocytin was added for post-hoc staining. Neurons were recorded under current-clamp to examine excitability, or under voltage-clamp to examine PSCs. 1 s step currents from −50 pA to 300 pA were used to evoke AP firing. For optogenetic stimuli, a 15-ms blue light pulse (3.4 mW, calibrated with Thorlab light meter) was given at 1 Hz, 5 Hz, 10 Hz, 15 Hz, and 20 Hz in a randomized manner for 10 s to compare light-induced activity between the young and aged groups, and the interval between sweeps was 20 s. Data were acquired with a Multiclamp 700B amplifier (Axon Instruments, USA), and sampled at 10 kHz. Stimulus generation and data acquisition were performed using pClamp 10. Data were analyzed using Stimfit 0.14.9 (www.stimfit.org) and R 3.5.1 (the R project for statistical computing). RMP values were measured and averaged from temporal windows (at least 50 ms prior to the peak of a given AP for spontaneously firing neurons) with minimal membrane potential variance (54). The RMPs were determined without predicted/measured junction potential correction. All the amplitudes of APs and spikelets were calculated from RMPs. Depolarization events with a peak value above −20 mV, and with a half width shorter than 6 ms were qualified for spikelet analyses. PSC recording from non-fluorescent neuron innervated by fluorescent Hcrt neuron expressing ChR2-eYFP was performed as illustrated in fig. S4A. For the PSC failure analysis, a success PSC was considered when a current deflection bigger than 10 pA occurred time-locked to the light pulse. The investigators were blind to the group information while conducting the data analyses.
LC neurons were recorded in slices prepared from WT young (2 to 3 months) and aged (18 to 21 months) mice, infused with biocytin, followed by antibody staining against tyrosine hydroxylase (TH). Only the neurons positive for both biocytin and TH were included for data analyses.
IM recording
For recording of the slowly deactivating M-current (IM) mediated by KCNQ2/3, perforated patch recordings were used to maintain the integrity of second messenger signaling cascades and minimize current rundown. The pore-forming antibiotic nystatin was dissolved in DMSO at 50 mg/ml. This stock solution was diluted in an internal pipette solution and vortexed and ultrasonicated for a final concentration of 100 to 200 μg/ml. Pipette tips were prefilled by brief immersion into antibiotic-free solution and then pipettes were back filled with nystatin. After the cell-attached configuration was attained, the access resistance was periodically monitored with hyperpolarizing voltage steps (10 mV, 20 ms) and capacitative transients were cancelled. After 10 to 20 min, recording was started once the access resistance stabilized. The recording was terminated if a sudden change in access resistance occurred. Extracellular solution contained 4-AP (5 mM) to minimize contamination by other potassium currents, and AMPARs, glycine receptors and GABAA receptors were blocked by 6,7-dinitroquinoxaline-2,3-dione (DNQX, Tocris Cat. no. 0189, 10 μM), strychnine (Sigma-Aldrich Cat. no. S0532, 1 μM), (2R)-amino-5-phosphonopentanoate (APV, Tocris Cat. no. 0106, 100 μM) and bicuculline (Sigma-Aldrich Cat. no. 285269, 10 μM). IM was recorded using a standard deactivation protocol (1000 ms hyperpolarizing steps to −30 mV from a holding potential of −20 mV every 10 s, intersweep holding potential −20 mV). IM did not inactivate with this protocol, while contamination by other voltage-gated currents was minimized. IM was measured as the inward relaxation current caused by deactivation of IM during this voltage step (Fig. 4, G and H). After obtaining at least a stable 5 min baseline, XE991 (50 μM) or flupirtine (50 μM) was applied. The effect of XE991 or flupirtine was determined by comparing averaged IM amplitudes over a 5 min period just before XE991 or flupirtine application with averaged IM amplitudes during the 5 to 10 min period after XE991 or flupirtine application.
Array tomography (AT)
Tissue preparation
Array creation and immunohistochemistry were described in detail in a previous publication (55). In short, a small piece of tissue (~2 mm high by 1 mm wide by 1 mm deep), covering the LH containing eYFP-labeled Hcrt neurons, was microwave-fixed in 4% Para-formaldehyde (PFA). The fixed tissue was then dehydrated in graded steps of ethanol, and then embedded in LR White resin overnight at 50°C. The embedded tissue was sectioned on an ultramicrotome at a thickness of 70 nm and placed as a ribbon array directly on gelatin or carbon coated glass coverslips. The ultrathin physical sectioning allows AT to achieve true isotropic voxels of ~100 nm. To assure that the brain tissue from animals were prepared and imaged in as similar conditions as possible, all samples were paired starting at the tissue preparation step. Thus, young and aged animals were prepared in tandem, placed on the same coverslip, stained together and imaged together. Furthermore, to minimize the impact of locational differences in the gathered tissue, multiple blocks were generated from LH of each mouse, and screened at 20× using 4′,6-diamidino-2-phenylindole (DAPI) fluorescence. Then similar tissue blocks were used for further analysis.
Immunohistochemistry
Immunohistochemistry was then carried out on the arrays using primary antibody against KCNQ2 (Alomone Cat. no. AGP-065). The primary antibodies were visualized via fluorescence-labeled secondary antibody (Alexa 594, Invitrogen Cat. no. A11076), and mounted in SlowFade Gold antifade with DAPI (Invitrogen Cat. no. S36938).
Microscopy
Wide-field imaging of ribbons were accomplished on a Zeiss Axio Imager.Z1 Upright Fluorescence Microscope with motorized stage and Axiocam HR Digital Camera as previously described (56). A position list was generated for each ribbon array of ultrathin sections using custom software modules written for Axiovision. Single fields of view were imaged for each position in the position list using a Zeiss 63×/1.4 NA Plan-Apochromat objective.
Image registration and processing
Image stacks from AT were imported into Fiji (ImageJ) and aligned using both rigid and affine transformations with the Register Virtual Stacks plugin. The aligned image stacks were further registered across image sessions using Fiji and TrackEM. The aligned and registered image stacks were imported into Matlab and deconvolved using the native implementation of Richardson-Lucy deconvolution with empirical or theoretical PSFs with 10 iterations (56). Custom functions were written to automate and facilitate this workflow.
eYFP segmentation
eYFP delimited protein amount was calculated using custom Matlab software. eYFP volumes were slightly dilated via morphological operations and used to segment protein data in image space. Segmentation custom functions were used to quantify the number and amount of proteins encapsulated by eYFP.
Single-nucleus isolation, FACS sorting, RNA library preparation and sequencing
3 weeks after virus injection, mice were deeply anesthetized using isoflurane and perfused with 1× PBS. The brains were rapidly dissected and transferred to a chilled metal Brain Slicer Matrix (Zivic Instruments, 500 μm coronal slice intervals), and the brain sections containing Hcrt neurons (AP: −1.0 ~ −2.0 mm) were sliced and transferred to 1× PBS on ice. Bilateral hypothalamic areas (LH) were identified and dissected under a stereoscope. LH tissue blocks were then transferred to a glass dounce homogenizer (Sigma-Aldrich) on ice and homogenized in 1 ml Homogenization Buffer (57) containing Tris (pH 8.0, 10 mM), sucrose (250 mM), KCl (25 mM), MgCl2 (5 mM), Triton-X100 (0.1%), RNasin Plus RNase Inhibitor (0.5%, Promega Cat. no. N2615), SUPERase·In RNase Inhibitor (0.5%, Thermo-Fisher Cat. no. AM2694), Protease Inhibitor Cocktail (1×, Promega Cat. no. G6521), DTT (0.1 mM) and DAPI (1:1000, Invitrogen Cat. no. D3571). LH tissue blocks from 3 mice per condition (young/aged male/female) were pooled each condition for isolation of nuclei. The nuclei were released by sequentially applying 10 to 12 strokes of the loose dounce pestle and 10 to 12 strokes of the tight dounce pestle on ice, followed by filtering the suspension through a 35 μm cell strainer (Falcon). The nuclei were then spun down by centrifugation (10 min, 900× g at 4°C) and resus-pended in the Wash Buffer (1× PBS containing 0.8% BSA, 0.5% RNasin Plus RNase Inhibitor and 0.5% SUPERase·In RNase Inhibitor). The single-nucleus suspension was further washed twice in Wash Buffer by centrifugation (10 min, 900× g at 4°C). Fluorescence activated cell sorting (FACS) was performed using the 70-μm nozzle and optimal gates collecting the DsRed/DAPI double positive events and excluding debris and doublets. Sorted DsRed+ single nuclei were confirmed using a fluorescence microscope, and manually counted using a hemocytometer. snRNA-seq libraries were prepared using 10x Genomics Chromium Single Cell 3′ Reagents v3 following manufacturer’s instructions. Briefly, the concentration of single nuclei solution prepared from dissected LH tissue was determined using DAPI staining and Trypan Blue staining. The nuclei solution was loaded onto a Chromium Chip B to capture seven to ten thousand nuclei in droplets containing the reverse transcription reagents. After reverse transcription, the now barcoded cDNA was recovered and amplified for 12 polymerase chain reaction (PCR) cycles. After qualitative and quantitative control of the cDNA, the final libraries were constructed by fragmenting the cDNA, End Repair, and A-Tailing. After adapter ligation, the libraries were amplified for 11 PCR cycles. The libraries were sequenced using an Illumina MiSeq v3 150-cycle kit to check library quality and confirm the number of captured nuclei. Then all the barcoded samples were mixed and deep sequenced on an Illumina HiSeqX sequencing machine across 4 different lanes to avoid lane variability and potential lane failure.
snRNA-seq data analysis
Illumina fastq files were processed through the 10x Genomics cellranger pipeline according to the manufacturer’s instructions. Briefly, reads were aligned to the mm10 mouse genome using a custom gtf annotation file which labeled all “transcripts” as “exons,” thus allowing to count intronic as well as exonic reads. The four libraries were then combined using cellranger aggr command to match sequencing depth per cell across libraries. All further processing of the genes X cells count matrix was performed in Seurat V3 (58) using scTransform normalization (59). First, the population of Hcrt+ neurons were identified out of all sequenced cells by coarse Louvian clustering of the entire sequencing dataset. Only one cluster showed Hcrt expression. This cluster was then separately subclustered, and all doublet clusters were removed. No large batch effects were observed at this level. A core set of three clusters, all of which expressed Hcrt at high levels, served as the basis for the analysis of age related effects.
CRISPR/SaCas9-mediated Kcnq2/3 gene disruption in Hcrt neurons
The target sites of Kcnq2/3 genes for Staphylococcus aureus CRISPR/Cas9 (CRISPR/SaCas9) were designed by CHOPCHOP (http://chopchop.cbu.uib.no) (60). The target sequences were as follows: sgKcnq2: 5′-CGCGTGTGGAGTCGGGCGCGC-3′, sgKcnq3: 5′-GCGGCCACCGCCCTCCAGCAG-3′.
Oligonucleotides encoding guide sequences were purchased from Sigma-Aldrich and cloned individually into BsaI fragment of pX601 (Addgene plasmid 61591). U6-sgKcnq2 and U6-sgKcnq3 fragments were PCR-amplified, respectively using pX601-sgKcnq as a template. Amplified fragments were cloned tandemly into MluI-digested pAAV CAG FLEX mCherry by Gibson assembly method. The primers used were as follows; Gibson1-F: 5′-TAGGGGTTCCTGCGGCCGCAGAGGGCCTATTTCCCATG-3′, Gibson1-R: 5′-ATAGGCCCTCTCTAGAAAAAATCTCGCCAAC-3′, Gibson2-F: 5′-TTTTTCTAGAGAGGGCCTATTTCCCATG-3′, Gibson2-R: 5′-TCATTATTGACGTCAATGGAAAAAATCTCGCCAACAAGTTG-3′. AAV constructs carrying nontargeting guide sequences (5′-GCGAGGTATTCGGCTCCGCGT-3′) were used as control. For the Cre- dependent SaCas9 construct, SaCas9 fused with 3× HA tag was PCR amplified using pX601 as a template. Amplified fragment was cloned into AscI/NheI-double digested pAAV-U6-SaCas9gRNA(SapI)-CMV-SaCas9-DIO-pA (Addgene plasmid 113691). Next, the plasmid was digested by MluI and applied to self- ligation to remove U6 promoter and single-guide RNA (sgRNA) scaffold sequences. pAAV CMV-DIO-SaCas9–3HA (SaCas9), pAAV U6 sgKcnq2-U6 sgKcnq3 CAG FLEX mCherry (sgKcnq2/3) and pAAV U6 sgControl-U6 sgControl CAG FLEX mCherry (sgControl) were packaged into AAV-DJ by the Wu Tsai Neurosciences Institute Gene Vector and Virus Core at Stanford University.
20 young (6 to 8 weeks old) Hcrt∷Cre mice were separated into two groups in a random manner (n = 10/group). Under anesthetics and analgesic, according to the Hcrt neuron field coordinates as described above, one group received bilateral stereotaxic injection of a 0.6 μl (each side, 0.3 mm apart in depth) mixture of SaCas9 (2.4 × 1013 gc/ml) and sgControl (6.24 × 1012 gc/ml) and implanted with EEG/EMG electrodes to serve as the control group. The other group received bilateral stereotaxic injection of a 0.6 μl mixture of SaCas9 and sgKcnq2/3 (2.97 × 1012 gc/ml) and implanted with EEG/EMG electrodes to monitor the effect of Hcrt neuron- selective Kcnq2/3 gene disruption on sleep architecture. After surgery, mice were connected to EEG/EMG recording cables and singly-housed with food and water ad libitum to recover, and for EEG/EMG signal recording. EEG/EMG signals were recorded continuously on day 6 and day 7 weekly up to 8 weeks (EEG/EMG recording lasted until 12 weeks in half of each group) after surgery. Following recording in week 8/12 after virus injection, slices were prepared from each group for in vitro electrophysiology experiment to determine RMP and firing property of the Hcrt neurons labeled by mCherry flag. Patch clamp recorded cells were infused with biocytin for subsequent immunostaining. The data were used for statistical analysis only if the recorded neurons were stained to co-express biocytin and HA tag.
Histology
For in vivo experiments, upon accomplishment of recordings, mice were perfused under anesthesia described above with ice-cold 1× PBS and followed by 4% PFA for immunostaining against Hcrt1/OXA for Hcrt neurons, and TH for LC NA neurons. Brains were rapidly extracted, postfixed with 4% PFA at 4°C overnight, and equilibrated in 30% sucrose in 1× PBS containing 0.1% NaN3. Then, brains were sectioned at −22°C with a cryostat (Leica Microsystems) at a thickness of 35 μm. Slices were collected from anterior to posterior consecutively to 24-well plates containing PBS with 0.1% NaN3, covered with aluminum foil, and stored at 4°C until immunostaining and imaging. Primary antibody against OXA/Hcrt1 (SC-8070, Lot no. A2915, Goat polyclonal IgG) was purchased from Santa Cruz Biotechnology. Primary antibody against TH (Chicken polyclonal anti-peptide, Cat. TYH, Lot no. TYH1897983) was purchased from Avēs. Primary antibody against HA tag (Rabbit Anti-HA tag pAb, Item no. 561, Lot no. 067) was purchased from MBL InternationalCorporation.Secondary antibodies: Alexa Fluor 488 Goat anti-chicken IgG (H+L, Ref. no. A11039, Lot no. 1094413), Alexa Fluor 488 donkey anti-goat IgG (H+L, Ref. no. A11055, Lot no. 1869589), Alexa Fluor 488 donkey anti-rabbit IgG (H+L, Ref. no. A21206, Lot no. 1910751), Alexa Fluor 647 donkey anti-goat IgG (H+L, Ref. no. A21447, Lot no. 2175459), were purchased from Invitrogen (Manufacturer: Life Technologies). Alexa Fluor 594 streptavidin conjugate (Ref. no. S11227, Lot no. 1991448) and Alexa Fluor 647 streptavidin conjugate (Ref. no. S32357, Lot no. 1738557) to label neurons infused with biocytin were purchased from Invitrogen. For the WT mice used for comparison of sleep patterns, sections around LH and LC were washed in 1× PBS for 5 min, 3 times and incubated in a blocking solution of PBS with 0.3% Triton X-100 (PBST) and 4% bovine serum albumin (BSA) for 1 hour. Following that, OXA/Hcrt1 primary antibody was added to the blocking solution (1:800) overnight. On the second day, sections were washed in 1× PBS for 3 times (5 min/time), and incubated in blocking buffer for 2 hours. After blocking, secondary antibody was added to the blocking buffer for 2 hours (dilution 1:800). After 3 times of 5-min 1× PBS washing, brain sections were mounted onto gelatin-coated slides, covered with Fluoroshield containing DAPI mounting media (Sigma-Aldrich, F6057) and cover glass for imaging with wild field microscope (Zeiss AxioImager, Germany) for entire section or LSM710 Confocal Microscope for enlarged visualization (Zeiss, Germany). For brain slices infected with Cre-dependent viruses, slices around the injection site were collected and stained with appropriate antibodies as described above. Alexa Fluor 594 streptavidin conjugate or Alexa Fluor 647 streptavidin conjugate for staining of biocytin was added together with the secondary fluorescent antibody for Hcrt1, TH or HA tag on the second staining day for in vitro experiment slices.
Object recognition test
Aged mice (~20 months, singly-housed with a reversed 12 hours/12 hours light/dark cycle, 9 pm light on, Zeitgeber time 0/ZT 0) were used to evaluate flupirtine’s effect on memory ability in the object recognition task. The object recognition task was performed according to a protocol described by Leger et al. (61). The protocol consisted of habituation, familiarization and test sessions (fig. S8). During each habituation session, an individual mouse was released to the arena (34 cm × 17 cm, non-transparent open field filled with Sani-Chip pine bedding floor) for habituation of 5 min. Each mouse underwent two habituation sessions conducted during ZT16-18 and ZT22-24 for 3 consecutive days. During the familiarization session (Day 4: ZT22-24), each mouse was allowed to explore two identical objects for a total of 5 min. Each object was placed at the same distance from the walls and corners of the field without spatial or odor cues (bedding was changed; arena and objects were cleaned with 70% ethanol before each exposure). After the familiarization session, mice were intraperitoneally injected with either vehicle or flupirtine (20 mg/kg) at the beginning of the following light phase. During the test session (Day 5: ZT22-24), mice were placed in the same arena with one of the familiar objects from the familiarization session replaced by a similar size novel object. The position of the novel object (left or right) was randomized for each mouse and each group tested. Time spent facing away from object within the 7 cm radius or climbing on object was not qualified as exploration. Mice were randomly assigned to control/flupirtine group through a counterbalanced crossover design. Two rounds of object recognition task (with two sets of familiar and novel objects) were separated by one week for a complete drug wash-out. Animal-based averaged value of two rounds of familiarization was presented. Mouse with over 65% preference for either object during the familiarization session was not qualified to proceed to the next session.
Statistics
One/two hour-binned sleep comparisons were analyzed by two-way repeated measure (RM) analysis of variance (ANOVA) (linear mixed-effects model for counterbalanced crossover design) followed by Šidák’s multiple comparisons. Holm-Šidák was used for comparison based on 24 hours/light/dark phase. Unpaired t-test with Welch’s correction was used for GCaMP6f data and in vivo optogenetic data analyses. For slice electrophysiology, Mann-Whitney U test, RM one-way ANOVA, two-way ANOVA were used to analyze appropriate datasets. Paired test was used for data analyses of experiments with paired design. Spearman correlation with a linear fit was performed for 2-demensional data correlation analysis. For snRNA-seq data, differentially expressed genes across ages were determined using the Wilcoxon rank-sum test, considering only those genes with a Bonferroni adjusted P < 0.05. Differences with P < 0.05 were considered significant for all experiments. In figures, *, **, ***, ****, and † indicate P < 0.05, P <0.01, P <0.005, P < 0.001, and P < 0.0005, respectively, and ns indicates not significant. Data with error bars were reported as mean ± SEM. Details on statistical analyses have been described in the supplementary text.
Supplementary Material
ACKNOWLEDGMENTS
We thank T. Sakurai and X. (Simon) Xie for providing OX(Hcrt)-eGFP mice and OX(Hcrt)-ataxin3 mice. We thank L. Luo for facilitating snRNA-seq data analysis. We thank Stanford Wu Tsai Neuroscience Microscopy Service for imaging.
Funding:
This work was supported in part by National Institutes of Health grants R01AG047671 (L.d.L.), R01MH116470 (L.d.L.), R01NS104950 (L.d.L. and P.M.), K01AG061230 (G.X.W.), P30EY026877 (G.X.W. and P.M.), R01DA011289 (J.A.K.), R01NS106301 (G.S.), Sleep Research Society Foundation Career Development Award 030-JP-21 (S.-B.L.), Stanford Alzheimer’s Disease Center-Scully Family Seed Grant P50AG047366 (L.d.L. and P.M.), and the New York Stem Cell Foundation (G.S.). G.S. is a New York Stem Cell Foundation-Robertson Investigator.
Footnotes
Competing interests: L.d.L. and S.-B.L. are co-inventors on a WIPO (World Intellectual Property Organization) patent WO/2021/119018 related to the work described in this manuscript. L.d.L. is a Chief Scientific Advisor to and receives consulting fees from Attune Neurosciences. L.d.L. is a paid consultant to Eisai Pharmaceuticals and Idorsia Pharmaceuticals. G.S. is a cofounder of Epiodyne, a drug discovery company, an inventor on a patent application (US application no. 62/962,581) related to imaging of neural dynamics to discover analgesics, and a member of the NIH PSPP (Preclinical Screening Platform for Pain) External Consulting Board. The remaining authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
science.org/doi/10.1126/science.abh3021
Materials and Methods
MDAR Reproducibility Checklist
Data and materials availability:
The sequencing reads and preprocessed data are available from NCBI Gene Expression Omnibus (GSE160581). Code is freely available on Github (https://github.com/justuskebschull/Hyprocretin_Li2020). All other data are available in the main text or the supplementary materials. All materials are available from the corresponding author upon reasonable request.
REFERENCES AND NOTES
- 1.Scullin MK, Bliwise DL, Sleep, cognition, and normal aging: Integrating a half century of multidisciplinary research. Perspect. Psychol. Sci 10, 97–137 (2015). doi: 10.1177/1745691614556680; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Yaffe K, Falvey CM, Hoang T, Connections between sleep and cognition in older adults. Lancet Neurol 13, 1017–1028 (2014). doi: 10.1016/S1474-4422(14)70172-3; [DOI] [PubMed] [Google Scholar]
- 3.Carskadon MA, Brown ED, Dement WC, Sleep fragmentation in the elderly: Relationship to daytime sleep tendency. Neurobiol. Aging 3, 321–327 (1982). doi: 10.1016/0197-4580(82)90020-3; [DOI] [PubMed] [Google Scholar]
- 4.Mander BA, Winer JR, Walker MP, Sleep and human aging. Neuron 94, 19–36 (2017). doi: 10.1016/j.neuron.2017.02.004; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rodriguez JC, Dzierzewski JM, Alessi CA, Sleep problems in the elderly. Med. Clin. North Am 99, 431–439 (2015). doi: 10.1016/j.mcna.2014.11.013; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koh K, Evans JM, Hendricks JC, Sehgal A, A Drosophila model for age-associated changes in sleep:wake cycles. Proc. Natl. Acad. Sci. U.S.A 103, 13843–13847 (2006). doi: 10.1073/pnas.0605903103; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wimmer ME et al. , Aging in mice reduces the ability to sustain sleep/wake states. PLOS ONE 8, e81880 (2013). doi: 10.1371/journal.pone.0081880; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.de Lecea L et al. , The hypocretins: Hypothalamus-specific peptides with neuroexcitatory activity. Proc. Natl. Acad. Sci. U.S.A 95, 322–327 (1998). doi: 10.1073/pnas.95.1.322; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sakurai T et al. , Orexins and orexin receptors: A family of hypothalamic neuropeptides and G protein–coupled receptors that regulate feeding behavior. Cell 92, 573–585 (1998). doi: 10.1016/S0092-8674(00)80949-6; [DOI] [PubMed] [Google Scholar]
- 10.Mahoney CE, Cogswell A, Koralnik IJ, Scammell TE, The neurobiological basis of narcolepsy. Nat. Rev. Neurosci 20, 83–93 (2019). doi: 10.1038/s41583-018-0097-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sakurai T, The neural circuit of orexin (hypocretin): Maintaining sleep and wakefulness. Nat. Rev. Neurosci 8, 171–181 (2007). doi: 10.1038/nrn2092; [DOI] [PubMed] [Google Scholar]
- 12.Adamantidis AR, Zhang F, Aravanis AM, Deisseroth K, de Lecea L, Neural substrates of awakening probed with optogenetic control of hypocretin neurons. Nature 450, 420–424 (2007). doi: 10.1038/nature06310; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Li SB, Nevárez N, Giardino WJ, de Lecea L, Optical probing of orexin/hypocretin receptor antagonists. Sleep 41, (2018). doi: 10.1093/sleep/zsy141; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tsunematsu T et al. , Acute optogenetic silencing of orexin/hypocretin neurons induces slow-wave sleep in mice. J. Neurosci 31, 10529–10539 (2011). doi: 10.1523/JNEUROSCI.0784-11.2011; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hara J et al. , Genetic ablation of orexin neurons in mice results in narcolepsy, hypophagia, and obesity. Neuron 30, 345–354 (2001). doi: 10.1016/S0896-6273(01)00293-8; [DOI] [PubMed] [Google Scholar]
- 16.Lin L et al. , The sleep disorder canine narcolepsy is caused by a mutation in the hypocretin (orexin) receptor 2 gene. Cell 98, 365–376 (1999). doi: 10.1016/S0092-8674(00)81965-0; [DOI] [PubMed] [Google Scholar]
- 17.Scammell TE, Narcolepsy. N. Engl. J. Med 373, 2654–2662 (2015). doi: 10.1056/NEJMra1500587; [DOI] [PubMed] [Google Scholar]
- 18.Lee MG, Hassani OK, Jones BE, Discharge of identified orexin/hypocretin neurons across the sleep-waking cycle. J. Neurosci 25, 6716–6720 (2005). doi: 10.1523/JNEUROSCI.1887-05.2005; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mileykovskiy BY, Kiyashchenko LI, Siegel JM, Behavioral correlates of activity in identified hypocretin/orexin neurons. Neuron 46, 787–798 (2005). doi: 10.1016/j.neuron.2005.04.035; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Giardino WJ et al. , Parallel circuits from the bed nuclei of stria terminalis to the lateral hypothalamus drive opposing emotional states. Nat. Neurosci 21, 1084–1095 (2018). doi: 10.1038/s41593-018-0198-x; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bean BP, The action potential in mammalian central neurons. Nat. Rev. Neurosci 8, 451–465 (2007). doi: 10.1038/nrn2148; [DOI] [PubMed] [Google Scholar]
- 22.Greene DL, Hoshi N, Modulation of Kv7 channels and excitability in the brain. Cell. Mol. Life Sci 74, 495–508 (2017). doi: 10.1007/s00018-016-2359-y; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Howard RJ, Clark KA, Holton JM, Minor DL Jr., Structural insight into KCNQ (Kv7) channel assembly and channelopathy. Neuron 53, 663–675 (2007). doi: 10.1016/j.neuron.2007.02.010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wang HS et al. , KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 282, 1890–1893 (1998). doi: 10.1126/science.282.5395.1890; [DOI] [PubMed] [Google Scholar]
- 25.Jentsch TJ, Neuronal KCNQ potassium channels: Physiology and role in disease. Nat. Rev. Neurosci 1, 21–30 (2000).doi: 10.1038/35036198; [DOI] [PubMed] [Google Scholar]
- 26.Wang GX, Smith SJ, Mourrain P, Fmr1 KO and fenobam treatment differentially impact distinct synapse populations of mouse neocortex. Neuron 84, 1273–1286 (2014). doi: 10.1016/j.neuron.2014.11.016; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ran FA et al. , In vivo genome editing using Staphylococcus aureus Cas9. Nature 520, 186–191 (2015). doi: 10.1038/nature14299; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kumar N et al. , The development of an AAV-based CRISPR SaCas9 genome editing system that can be delivered to neurons in vivo and regulated via doxycycline and cre-recombinase. Front. Mol. Neurosci 11, 413 (2018). doi: 10.3389/fnmol.2018.00413; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamanaka A et al. , Hypothalamic orexin neurons regulate arousal according to energy balance in mice. Neuron 38, 701–713 (2003). doi: 10.1016/S0896-6273(03)00331-3; [DOI] [PubMed] [Google Scholar]
- 30.Scammell TE, Willie JT, Guilleminault C, Siegel JM, International Working Group on Rodent Models of Narcolepsy, A consensus definition of cataplexy in mouse models of narcolepsy. Sleep 32, 111–116 (2009). doi: 10.5665/sleep/32.1.111; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mickelsen LE et al. , Single-cell transcriptomic analysis of the lateral hypothalamic area reveals molecularly distinct populations of inhibitory and excitatory neurons. Nat. Neurosci 22, 642–656 (2019). doi: 10.1038/s41593-019-0349-8; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Izawa S et al. , REM sleep-active MCH neurons are involved in forgetting hippocampus-dependent memories. Science 365, 1308–1313 (2019). doi: 10.1126/science.aax9238; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kosse C, Schöne C, Bracey E, Burdakov D, Orexin-driven GAD65 network of the lateral hypothalamus sets physical activity in mice. Proc. Natl. Acad. Sci. U.S.A 114, 4525–4530 (2017). doi: 10.1073/pnas.1619700114; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Naganuma F et al. , Lateral hypothalamic neurotensin neurons promote arousal and hyperthermia. PLOS Biol 17, e3000172 (2019). doi: 10.1371/journal.pbio.3000172; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cooper EC, Harrington E, Jan YN, Jan LY, M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J. Neurosci 21, 9529–9540 (2001). doi: 10.1523/JNEUROSCI.21-24-09529.2001; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Biervert C et al. , A potassium channel mutation in neonatal human epilepsy. Science 279, 403–406 (1998). doi: 10.1126/science.279.5349.403; [DOI] [PubMed] [Google Scholar]
- 37.Schroeder BC, Kubisch C, Stein V, Jentsch TJ, Moderate loss of function of cyclic-AMP-modulated KCNQ2/KCNQ3 K+ channels causes epilepsy. Nature 396, 687–690 (1998). doi: 10.1038/25367; [DOI] [PubMed] [Google Scholar]
- 38.Singh NA et al. , Mouse models of human KCNQ2 and KCNQ3 mutations for benign familial neonatal convulsions show seizures and neuronal plasticity without synaptic reorganization. J. Physiol 586, 3405–3423 (2008). doi: 10.1113/jphysiol.2008.154971; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sesti F, Oxidation of K+ channels in aging and neurodegeneration. Aging Dis 7, 130–135 (2016). doi: 10.14336/AD.2015.0901; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Kim SY, Kang HT, Han JA, Park SC, The transcription factor Sp1 is responsible for aging-dependent altered nucleocytoplasmic trafficking. Aging Cell 11, 1102–1109 (2012). doi: 10.1111/acel.12012; [DOI] [PubMed] [Google Scholar]
- 41.Mucha M et al. , Transcriptional control of KCNQ channel genes and the regulation of neuronal excitability. J. Neurosci 30, 13235–13245 (2010). doi: 10.1523/JNEUROSCI.1981-10.2010; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lüscher C, Slesinger PA, Emerging roles for G protein–gated inwardly rectifying potassium (GIRK) channels in health and disease. Nat. Rev. Neurosci 11, 301–315 (2010). doi: 10.1038/nrn2834; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kang JE et al. , Amyloid-β dynamics are regulated by orexin and the sleep-wake cycle. Science 326, 1005–1007 (2009). doi: 10.1126/science.1180962; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mayordomo-Cava J, Yajeya J, Navarro-López JD, Jiménez-Díaz L, Amyloid-β(25–35) modulates the expression of GirK and KCNQ channel genes in the hippocampus. PLOS ONE 10, e0134385 (2015). doi: 10.1371/journal.pone.0134385; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zott B et al. , A vicious cycle of β amyloid-dependent neuronal hyperactivation. Science 365, 559–565 (2019). doi: 10.1126/science.aay0198; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oh J et al. , Profound degeneration of wake-promoting neurons in Alzheimer’s disease. Alzheimers Dement 15, 1253–1263 (2019). doi: 10.1016/j.jalz.2019.06.3916; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Carter ME et al. , Tuning arousal with optogenetic modulation of locus coeruleus neurons. Nat. Neurosci 13, 1526–1533 (2010). doi: 10.1038/nn.2682; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Peyron C et al. , Neurons containing hypocretin (orexin) project to multiple neuronal systems. J. Neurosci 18, 9996–10015 (1998). doi: 10.1523/JNEUROSCI.18-23-09996.1998; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lindeberg J et al. , Transgenic expression of Cre recombinase from the tyrosine hydroxylase locus. Genesis 40, 67–73 (2004). doi: 10.1002/gene.20065; [DOI] [PubMed] [Google Scholar]
- 50.Eban-Rothschild A, Rothschild G, Giardino WJ, Jones JR, de Lecea L, VTA dopaminergic neurons regulate ethologically relevant sleep-wake behaviors. Nat. Neurosci 19, 1356–1366 (2016). doi: 10.1038/nn.4377; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Li SB et al. , Hypothalamic circuitry underlying stress-induced insomnia and peripheral immunosuppression. Sci. Adv 6, eabc2590 (2020). doi: 10.1126/sciadv.abc2590; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Zada D, Bronshtein I, Lerer-Goldshtein T, Garini Y, Appelbaum L, Sleep increases chromosome dynamics to enable reduction of accumulating DNA damage in single neurons. Nat. Commun 10, 895 (2019). doi: 10.1038/s41467-019-08806-w; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aghajanian GK, Rasmussen K, Intracellular studies in the facial nucleus illustrating a simple new method for obtaining viable motoneurons in adult rat brain slices. Synapse 3, 331–338 (1989). doi: 10.1002/syn.890030406; [DOI] [PubMed] [Google Scholar]
- 54.Pernía-Andrade AJ, Jonas P, Theta-gamma-modulated synaptic currents in hippocampal granule cells in vivo define a mechanism for network oscillations. Neuron 81, 140–152 (2014). doi: 10.1016/j.neuron.2013.09.046; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Micheva KD, Smith SJ, Array tomography: A new tool for imaging the molecular architecture and ultrastructure of neural circuits. Neuron 55, 25–36 (2007). doi: 10.1016/j.neuron.2007.06.014; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Wang G, Smith SJ, Sub-diffraction limit localization of proteins in volumetric space using Bayesian restoration of fluorescence images from ultrathin specimens. PLOS Comput. Biol 8, e1002671 (2012). doi: 10.1371/journal.pcbi.1002671; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bakken TE et al. , Single-nucleus and single-cell transcriptomes compared in matched cortical cell types. PLOS ONE 13, e0209648 (2018). doi: 10.1371/journal.pone.0209648; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Stuart T et al. , Comprehensive integration of single-cell data. Cell 177, 1888–1902.e21 (2019). doi: 10.1016/j.cell.2019.05.031; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). doi: 10.1186/s13059-019-1874-1; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Labun K, Montague TG, Gagnon JA, Thyme SB, Valen E, CHOPCHOP v2: A web tool for the next generation of CRISPR genome engineering. Nucleic Acids Res 44 (W1), W272–6 (2016). doi: 10.1093/nar/gkw398; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Leger M et al. , Object recognition test in mice. Nat. Protoc 8, 2531–2537 (2013). doi: 10.1038/nprot.2013.155; [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The sequencing reads and preprocessed data are available from NCBI Gene Expression Omnibus (GSE160581). Code is freely available on Github (https://github.com/justuskebschull/Hyprocretin_Li2020). All other data are available in the main text or the supplementary materials. All materials are available from the corresponding author upon reasonable request.