

SUMMARY
Synthetic biology has established powerful tools to precisely control cell function. Engineering these systems to meet clinical requirements has enormous medical implications. Here, we adopted a clinically driven design process to build receptors for the autonomous control of therapeutic cells. We examined the function of key domains involved in regulated intramembrane proteolysis and showed that systematic modular engineering can generate a class of receptors we call SyNthetic Intramembrane Proteolysis Receptors (SNIPRs) that have tunable sensing and transcriptional response abilities. We demonstrate the therapeutic potential of the receptor platform by engineering human primary T cells for multi-antigen recognition and production of dosed, bioactive payloads relevant to the treatment of disease. Our design framework enables the development of fully humanized and customizable transcriptional receptors for the programming of therapeutic cells suitable for clinical translation.
Keywords: cell therapy, synNotch, CAR T cells, cancer immunotherapy, synthetic biology
Graphical Abstract
In brief:
The design framework for fully humanized transcriptional receptors for the programming of therapeutic cells.
INTRODUCTION
Cellular function is influenced by both external and internal stimuli, with responses to these stimuli encoded in the genome. Having control over the cellular transcriptional response to a defined stimulus allows for the development of living, cell-based therapies with programmed therapeutic functions what is natural. In pursuit of this goal, synthetic receptor platforms have been developed, including the Tango (Barnea et al., 2008; Kroeze et al., 2015) and Modular Extracellular Signaling Architecture (MESA) (Daringer et al., 2014) systems, as well as the synthetic Notch receptor (synNotch) (Morsut et al., 2016). Notch and synNotch are type 1 transmembrane proteins that activate through regulated intramembrane proteolysis (RIP), a sequential process that involves ADAM protease-mediated shedding of the extracellular domain (ECD), γ-secretase-mediated cleavage of the transmembrane domain (TMD), and release of an intracellular transcription factor (TF) that traffics to the nucleus (Morsut et al., 2016; Kopan and Ilagan, 2009; Gordon et al., 2009). SynNotch receptors recognize a user-defined membrane-bound antigen via a high-affinity ligand-binding domain (LBD), such as a single-chain variable fragment (scFv) or nanobody and induce custom gene regulation through release of an engineered TF (Morsut et al., 2016).
The first generation synNotch receptor is a powerful tool for engineering cell circuitry for programmed multicellular morphologies (Toda et al. 2018), localized tumor control (Roybal et al., 2016b, Srivastava et al., 2019), multi-antigen tumor recognition (Roybal et al., 2016a, Williams et al., 2020, Hyrenius-Wittsten et al., 2021), and tumor antigen density discrimination (Hernandez-Lopez et al., 2021). Engineered receptors thus hold potential for furthering our understanding of basic biological processes and expanding our therapeutic options in disease. Translating this work into human therapeutic applications is therefore an important engineering goal.
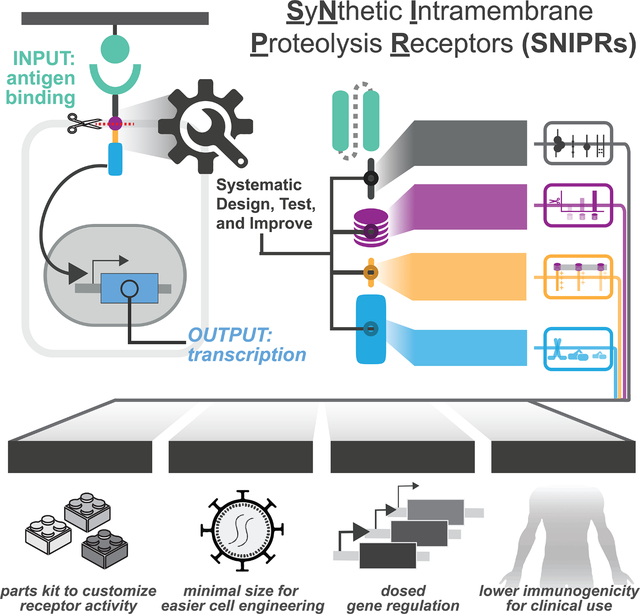
Despite its role in several cell engineering milestones, the original synNotch receptor has known limitations that affect its further advancement to clinical translation. These issues include 1) the use of non-human components that could elicit immune rejection, 2) the lack of clear design rules for building well-expressed receptors with a tunable activity profile, and 3) the large size of the receptor and transcriptional circuit. We originally observed the inflexibility of the original design during our attempts to engineer the human equivalent of synNotch, which is based on murine Notch1 (Fig. S1A, S1B). Receptors built using the human-derived Notch Negative Regulatory Region (NRRs) resulted in poor activation, high ligand-independent signaling, and/or poor expression (Fig. S1B, S1C). Moreover, both human and mouse-derived synNotch were incompatible with multiple transcription factors (TFs) beyond the yeast- and herpesvirus-derived Gal4-VP64 (Fig. S1B). Motivated by these results, we adopted a systematic approach to define functional receptor modules, allowing us to re-engineer the synNotch receptor from the ground up (Fig. 1A).
Figure 1. Design of synthetic RIP receptors for customized antigen-dependent gene regulation in therapeutic cells.
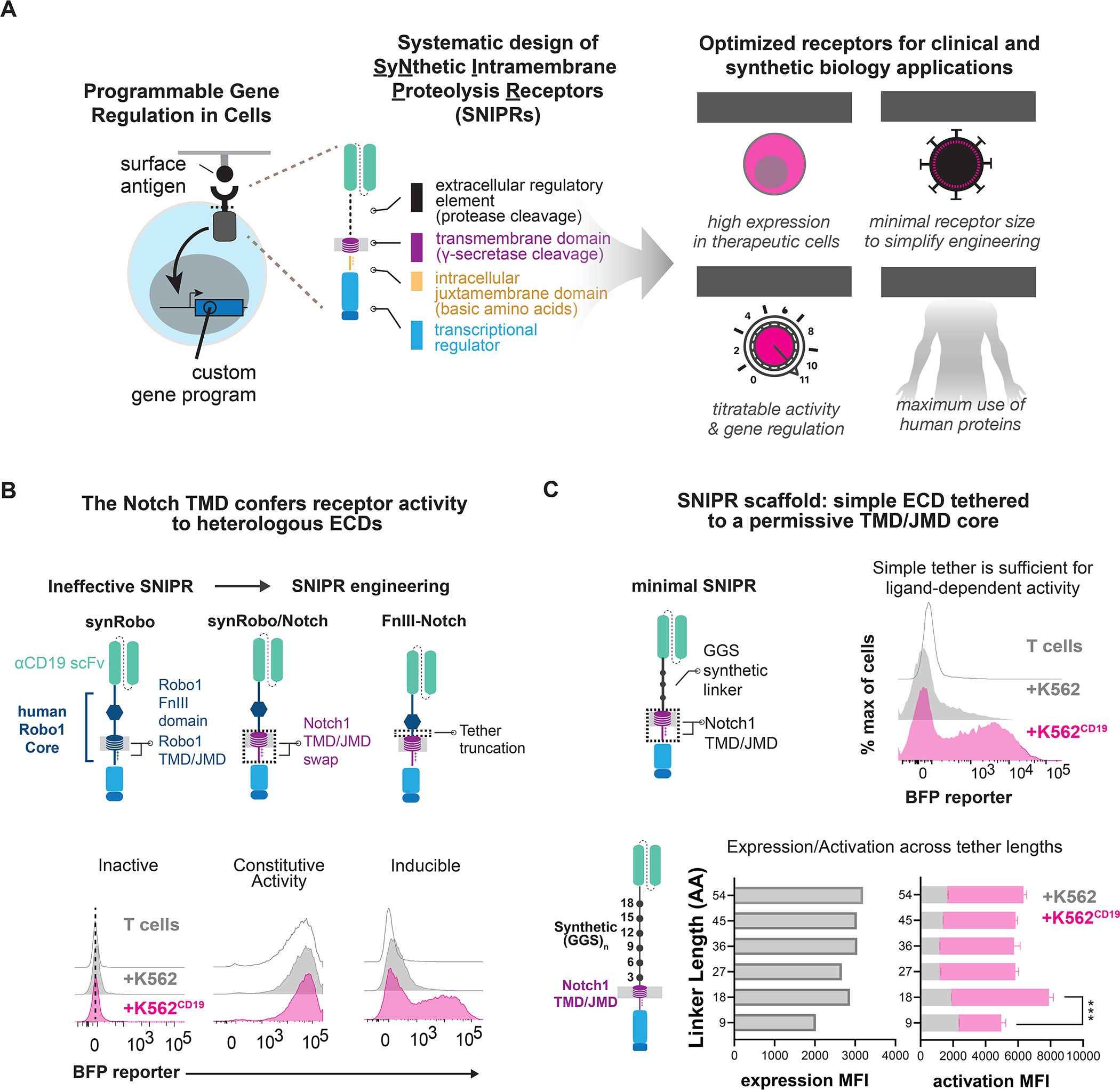
(A) Design of Synthetic Intramembrane Proteolysis Receptors. Receptors are comprised of a ligand binding domain (LBD), an extracellular domain (ECD), a transmembrane domain (TMD), a juxtamembrane domain (JMD), and a transcription factor (TF). Receptor circuits are designed to maximize clinical translation potential (B). A synRobo receptor replaces the Notch1 core with one from human Robo1. Compared to synNotch, a synRobo receptor fails to induce BFP. By replacing the TMD and JMD of Robo1 with those of Notch1, control of BFP production is lost. Deletion of a known ADAM10 cleavage site in the Robo1 ECD rescues ligand-dependent receptor behavior. (C) Same as B, but with minimal SNIPRs constructed using simple (GGS)n ECDs, and the TMD/JMD from Notch1. Statistics were calculated using unpaired T-tests, ***P≤0.001.
Through this approach, we have systematically designed, assembled, and tested a large family of SyNthetic Intramembrane Proteolysis Receptors (SNIPRs). We present design principles of synthetic receptors that undergo RIP and showcase a subset of designs within the larger family that have advantages for synthetic biology and next-generation T cell therapeutics. These optimized SNIPRs are compact in size, well-expressed, compatible with human and humanized synthetic TFs, readily tunable, and are both highly sensitive and specific to their target ligand. We show that these SNIPRs function robustly in SNIPR-chimeric antigen receptor (CAR) dual antigen-sensing circuits in vivo, a therapeutic strategy that enhances tumor specificity and therapeutic efficacy of engineered T cells for solid tumors (Hyrenius-Wittsten et al., 2021, Choe et al., 2021). We also show that we can rationally modify SNIPRs to achieve titratable production of therapeutic payloads such as IL-2, enabling spatially controlled and dosed delivery of therapeutic agents by cells at sites of disease. Though we have focused our efforts on T cells, the toolkit of modular core receptor parts we have characterized can be used for a broad range of applications in synthetic biology, basic biology, and cell therapeutics.
RESULTS
SNIPR development through modular assembly of core receptor domains
To engineer SNIPRs, we took a modular approach for receptor assembly to investigate the role of core domains involved in RIP (Fig. 1A). The ECD of Notch1 and other RIP family proteins contain sites of ADAM protease-mediated shedding (Brou et al., 2000; Mumm et al., 2000), and ECD mutations can impact this regulation (Gordon et al., 2009). The TMD is the site of γ-secretase-mediated cleavage and release of the intracellular domain into the cytosol (De Strooper et al., 1999). While γ-secretase is believed to cleave a diverse peptides (Beel and Sanders, 2008; Haapasalo and Kovacs, 2011), certain TMD mutations are known to negatively impact cleavage efficiency (Huppert et al., 2000). The basic amino acid-rich JMD connects the TMD to the TF, stops translocation of the receptor through the membrane, and interacts with γ-secretase and endocytosis machinery (Le Borgne, 2005).
Through a similar design strategy to that used for synNotch, a prototypical SNIPR, we sought to engineer a second example of a functional SNIPR. We selected human Robo1, a RIP receptor family member known for mediating ligand-directed neuronal pathfinding (Coleman et al., 2010). Like Notch, Robo1 is a type-I transmembrane protein that undergoes ECD shedding upon ligand engagement, followed by γ-secretase-mediated TMD cleavage to free the cytoplasmic tail (Seki et al., 2010). The proteolytic core of Robo1 features an ADAM10 protease-sensitive site that is protected by a type III Fibronectin (Fn-III) domain, as well as the receptor TMD and JMD (Coleman et al., 2010). As with synNotch, we built a synthetic Robo1 receptor (synRobo) against CD19 that contained the Robo1 proteolytic core and the Gal4-VP64 TF, combining it with the cognate Gal4 DNA response element (RE) controlling a BFP reporter (Fig. S1A). Although synRobo expressed at a comparable level to synNotch (Fig. S1D), we observed poor reporter activation when T cells expressing the anti-CD19 synRobo were exposed to K562CD19+ sender cells (Fig. 1B). To determine the cause of this stark difference in activity, we substituted two key domains of synRobo, the TMD and JMD, with the equivalent domains from human Notch1. We found that the resulting Robo1/Notch1 chimeric receptor (RoboNotch) was constitutively active, suggesting that the ECD of synRobo was easily shed, but the Robo1 TMD and JMD were not easily processed (Fig. 1B). We also found that deletion of the putative ADAM10 protease site in the Robo1 ECD of RoboNotch significantly reduced constitutive signaling and restored ligand-dependent activation. Thus, a canonical protease cleavage site in the ECD is not necessary for receptor function, but the receptor activity remains dependent on ADAM protease activity (Fig. 1B, S1D).
The assembly of a second functional SNIPR through engineering of domains from Robo1 and Notch1 prompted us to develop a systematic process to explore the principles of receptor design. We thus built a set of SNIPRs to identify critical features of the ECD, TMD, and JMD that are necessary for optimal receptor function. We began with the ECD, constructing a set of SNIPRs with a series of flexible glycine-glycine-serine repeats ECDs of variable lengths, an anti-CD19 scFv, and the human Notch1 TMD and JMD. These designs were expressed in human T cells and demonstrated ligand-dependent activation across all tested ECD lengths, as well as a dependence on ADAM protease activity (Fig. 1C, S1E). Given that a simple ECD without known protease sites was sufficient for regulated receptor activity, we considered that a broad range of ECDs could be used to assemble functional SNIPRs when paired with a RIP-permissive TMD and JMD. We further hypothesized that additional TMDs and JMDs may be compatible with heterologous ECDs, enabling the modular construction of a family of SNIPRs with diverse activation properties for more customized cellular programming.
ECD engineering controls SNIPR activation parameters
Given our positive results with a Robo-derived ECD and simple linkers, we expanded our survey of ECDs to include synthetic peptides with embedded protease sites, Notch-derived domains, and well-characterized hinge domains sourced from chimeric antigen receptors (CARs). We found that SNIPRs built with synthetic linkers containing exposed ADAM protease sites were constitutively active, while a SNIPR built with a FLAG-tag linker containing an enterokinase cleavage site retained ligand-dependent activity (Fig. 2A). Interestingly, an ECD that incorporated fibroblast activation protein (FAP) cleavage sites demonstrated signaling when co-cultured with K562 cells, an effect that was abrogated with the addition of an LBD, suggesting that ECD shedding is dependent on protease availability and cleavage site accessibility (Fig. 2A, S2A).
Figure 2. The ECD module defines activation triggers and diversifies sensor functions.
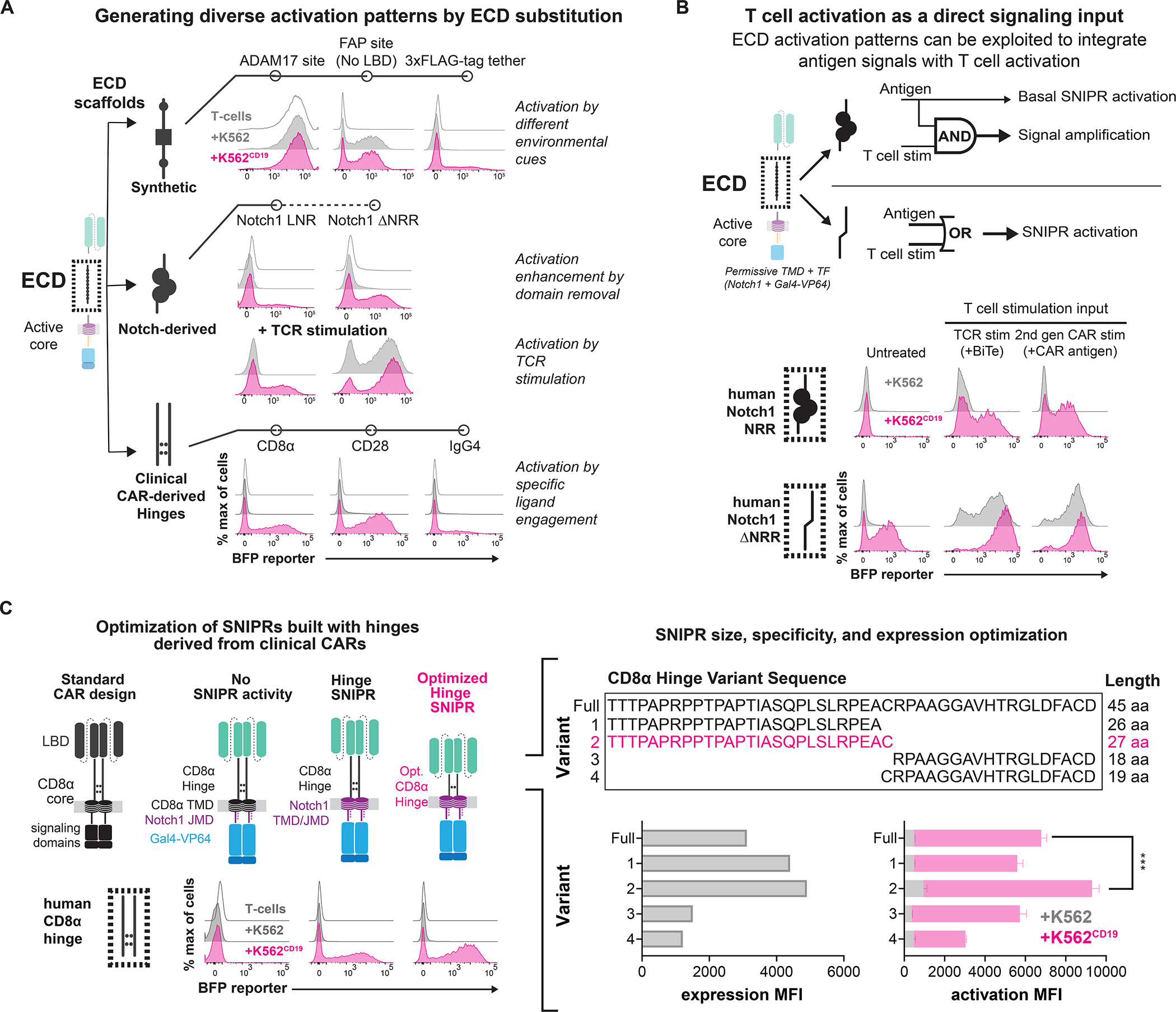
(A) SNIPR ECDs with exposed cleavage sites display ligand-independent signaling. Deleting the NRR from synNotch produces a receptor that is sensitive to both ligand and TCR stimulation. A variety of hinge domains utilized in CARs also demonstrate ligand-dependent signaling. (B) Same as A, but with two methods of T cell stimulation. A SNIPR with the Notch1 NRR core domain displays enhanced activation with a Bi-specific T cell Engager (BiTE) targeting a K562 antigen, and a co-expressed second-generation CAR targeting a separate antigen. A SNIPR with a truncated Notch1 NRR activates with these stimuli independent of the presence of ligand. (C) Same as A, but with variations of the CD8α Hinge. The CD8α Hinge can be optimized to enhance SNIPR expression and activation. Statistics were calculated using unpaired T-tests, ***P≤0.001.
Given the diversity of functional ECDs in SNIPRs, we decided to assess whether a regulatory domain such as the Notch NRR was necessary for receptor function. This SNIPR design with a full deletion of the Notch1 NRR (ΔNRR) exhibited strong ligand-induced signaling but was also triggered by T cell activation alone (Fig. 2A). T cell activation-based receptor activity was observed with several methods of T cell activation, including with the use of a Bi-specific T cell Engager (BiTEs) or a co-expressed second generation CAR (Fig. 2B, S2B). This behavior was not observed in similar truncation variants of the other three human Notch proteins (Fig. S2C). In addition, we observed that T cell activation drove the enhanced activation of SNIPRs, such as synNotch, that appeared insensitive to T cell activation alone (Fig. 2B). These data demonstrate that a spectrum of ECDs is compatible with SNIPR construction and that the choice of ECD can impact the fidelity and sensitivity of the receptor (Fig. 2B, S2C).
Clinically oriented ECD engineering
CARs often include a hinge region derived from immunoglobulin-like domains, such as CD8α or CD28, or from trimeric receptors (e.g. OX-40) in the ECD that affects critical aspects of CAR activity (Guedan et al., 2019). We found that, when used in our SNIPR designs, CD8α and CD28-based hinge ECDs exhibited high expression and receptor activation, with the CD8α hinge exhibiting reduced ligand-independent signaling and thus a better signal-to-noise ratio (Fig. 2A, S2D). However, the full-length CD8α hinge displayed ligand-independent signaling with T cell activation, especially in CD8+ T cells (Fig. S2D, S2E). Given these results, we devised a strategy to improve the functionality of the CD8α hinge ECD through a series of N-terminal and C-terminal truncations. From testing four truncation variants, we found that the 27 amino acid N-terminal region of the CD8α hinge displayed enhanced expression and minimal ligand-independent activity with T cell activation (Fig. 2C, S2F). This optimized CD8α hinge SNIPR was nominated for additional development due to its efficient, high-fidelity activation and compact size, with the full optimized anti-CD19 CD8α hinge SNIPR with Gal4-VP64 being only 1.65kB in length whereas the original synNotch is 2.45kB, a 33% reduction in size.
TMD and JMD engineering can tune receptor activity
The Notch1 TMD and JMD are functional with a broad set of ECDs (Fig. 2A). We next investigated the characteristics of the domains that make them functional, and whether additional TMD or JMD sequences were compatible with SNIPR assembly. To do this, we compiled a list of proteins known to undergo RIP and extracted their TMD and JMD sequences (Haapasalo and Kovacs, 2011) (Fig. 3A, Table S1). We defined the JMD as a stretch of basic amino acids (R/K/H) beginning immediately C-terminal to the TMD and ending before three consecutive non-basic amino acids. Due to the diversity observed in both TMDs and JMDs, we decoupled the TMD-JMD pair into two separate modules for screening and compiled 88 TMDs and 76 JMDs (Table S2, S3). We then inserted them individually into a human synNotch scaffold, replacing the respective Notch1 components, and screened them for receptor activity using a Jurkat reporter cell line in an arrayed format (Fig. 3A, Table S4, S5). We analyzed the TMDs displaying >50% activity to that of the Notch1 TMD or JMD for sequence similarities and identified additional TMDs and JMDs of interest for further testing (Fig. 3B, 3C).
Figure 3. Transmembrane and juxtamembrane domain libraries enable modular assembly of SNIPR architectures.
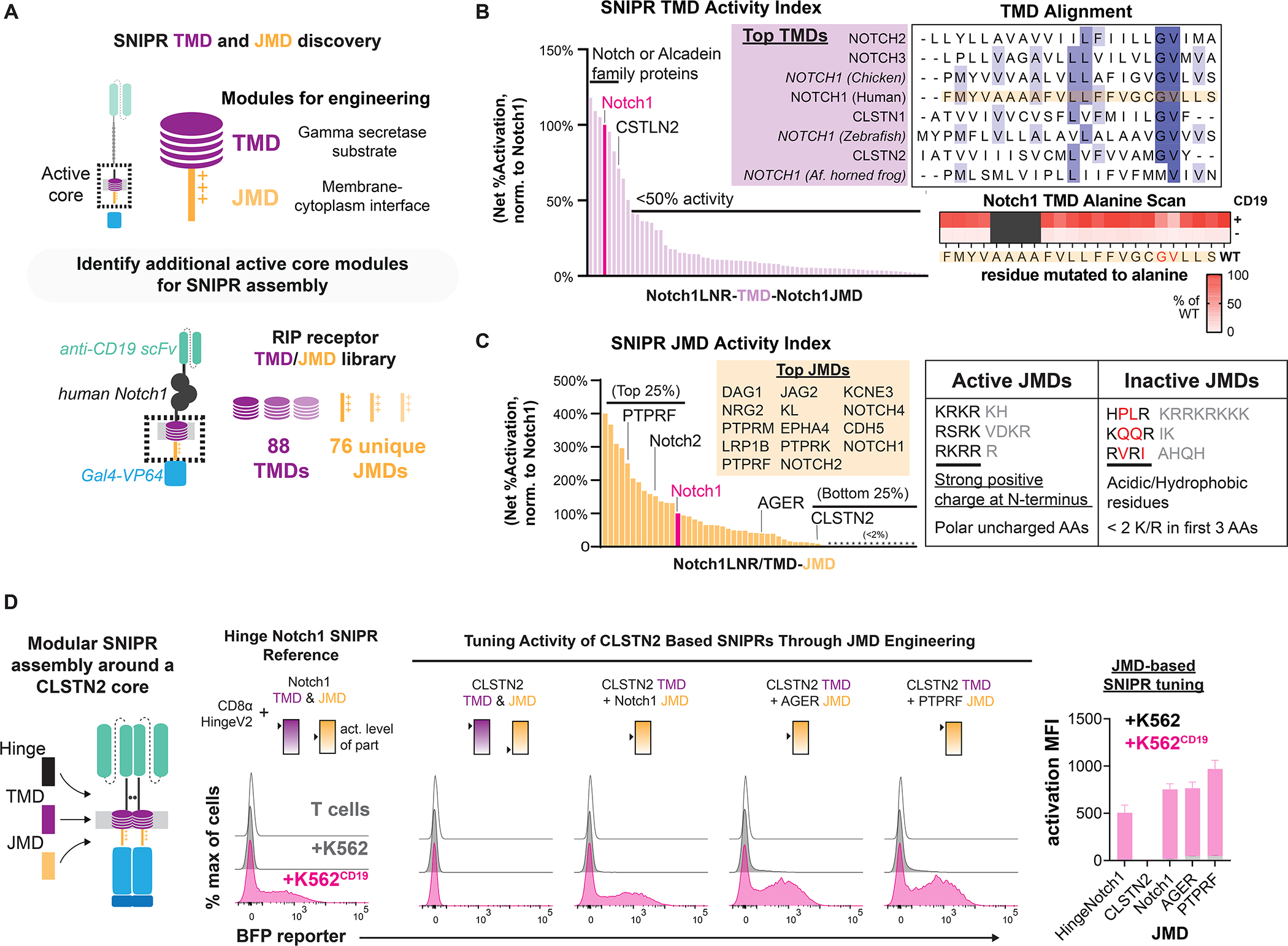
(A) To identify functional receptor TMDs and JMDs for modular assembly, 88 TMDs and 76 JMDs were cloned into a human synNotch scaffold, replacing either the Notch1 TMD or JMD, respectively. Jurkat T cells expressing an inducible BFP reporter were transduced with these SNIPR libraries in an arrayed format. (B) Normalized results of TMD screening in Jurkat T cells. An alignment of the best performing TMDs shows a common Gly-Val motif (Dark blue = >80% agreement with consensus sequence, blue = >60% agreement, light blue = >40% agreement). An alanine scan of the human Notch1 TMD in primary T cells supports the importance of this motif. (C) Same as B, but with the JMD library. High-performing JMDs are strongly basic at their N-termini and may include polar residues but not acidic or hydrophobic residues. (D) Compared to a reference SNIPR containing the Notch1 TMD/JMD, a SNIPR containing the CLSTN2 TMD/JMD is inactive, but receptor function is restored when the CLSTN2 JMD is replaced with the Notch1, AGER or PTPRF JMD.
The top performing TMDs were mainly from the Notch and Calsyntenin (CLSTN) protein families, with the activity of most TMDs below 50% of that of Notch1 (Fig. 3B). Alignment of the Notch and CLSTN TMD sequences reveals a common c-terminal glycine-valine motif associated with γ-secretase cleavage. Previous studies have shown that these sites are essential for efficient intramembrane processing by presenilin (Vooijs et al., 2004; Okochi, 2002). To determine the importance of this motif in SNIPR signaling, we performed an alanine scan within the Notch1 TMD in primary human T cells, using the optimized CD8α hinge Notch ECD and Notch1 JMD (Fig. 3B). Although receptor expression was not reduced (Fig. S3A), we found that substitution of the glycine (G318A) and invariant valine (V319A) reduced receptor activity by 47% and 75%, respectively. Background signaling activity from these receptor variants was also lower, consistent with decreased processing (Fig. 3B, S3B) (Vooijs et al., 2004; Okochi, 2002). In addition, two otherwise non-functional TMDs, from Robo1 and Advanced glycosylation end product-specific receptor (AGER), could be made functional through the addition of a glycine-valine motif and removal of bulky residues near the TMD c-terminus (Fig. S3C).
In contrast to the TMD screen, our results from the JMD screen showed that JMDs sourced from a diverse set of proteins were effective in a SNIPR context (Fig. 3C, Fig. S3D). We found that the top JMD sequences favored basic residues immediately adjacent to the membrane, with basic or polar residues composing the first 4 to 6 amino acids, and at least two R/Ks within the first 3 amino acids. Hydrophobic or acidic residues within this stretch were found to inhibit receptor activation. Replacement of either the Notch1 TMD or JMD did not affect SNIPR sensitivity to T cell activation alone, suggesting that T cell activation affects SNIPR ECD cleavage.
Building non-Notch SNIPRs from functional parts
Thus far, all functional SNIPRs we have studied include sequences derived from the Notch family. To demonstrate the versatility of our modular assembly approach, we engineered a functional ligand-activated receptor without Notch domains, combining the optimized CD8α hinge ECD with the CLSTN2 TMD and functional JMD modules discovered in our screens. Although all receptors expressed (Fig. S3E), the otherwise-active CLSTN2 TMD did not function with its cognate JMD (RVRIAHQH), an expected result given the poor performance of the CLSTN2 JMD in our screen (Fig. 3C). However, receptor functionality was restored by replacing the JMD with that of AGER (RRQRR) or Protein tyrosine phosphatase receptor type F (PTPRF) (KRKRTH), two potent JMDs identified in our screen (Fig. 3D, S3E). Our ability to build functional SNIPRs from a set of functional parts demonstrates that we can tune receptor sensing and activity with modular engineering (Fig. S3E).
Precision control and customization of T cell therapeutics with SNIPRs
Based on the design principles of SNIPRs we uncovered, we next assembled receptors from a menu of ECDs, TMDs, and JMDs with a range of activation characteristics. Our design criteria included robust expression, a range of ligand-dependent activation levels, and low ligand-independent activation (Fig. 4A, S4A). For the ECD, we selected the optimized CD8α hinge, due to its strong expression, compact size, and selective response to ligand. We then screened through a selection of high-performing TMDs and JMDs, using a constant Notch1 JMD or TMD, respectively. From this process, we decided to keep the Notch1 TMD due to its robust activation and best-in-class levels of ligand-independent signaling, along with the ready availability of mutants for tunability (Fig. 4A). We screened this ECD-TMD combination against a panel of JMDs, choosing a set of SNIPRs with a range of activation levels. Although SNIPR expression levels varied between TMDs and JMDs, these differences did not always correlate with SNIPR activation, supporting a role for the JMD in affecting activity beyond impacting expression. The set of SNIPRs remained sensitive to ADAM protease and γ-secretase inhibition, suggesting a continued role for these proteases in SNIPR activation (Fig. S4B).
Figure 4. Enhanced sensitivity and tunable gene regulation through SNIPR engineering.
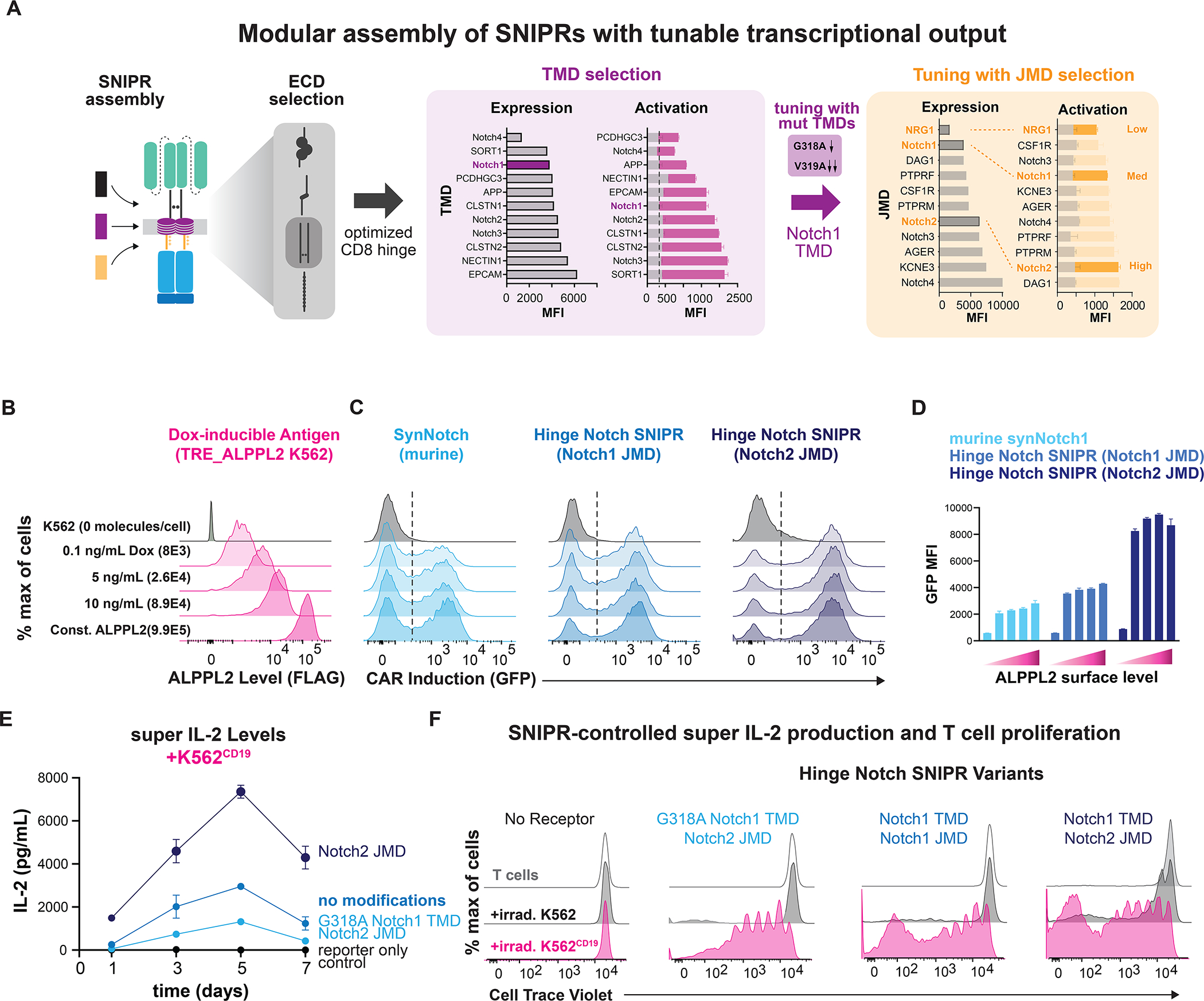
(A) From analyzing activity of high-performing SNIPR-BFP circuits, the Notch1 TMD was selected for further testing. Three JMDs and two TMD alanine mutants were selected to produce a wide output range. (B) K562 cells transduced with a doxycycline-inducible FLAG-tagged ALPPL2 cassette express ALPPL2 in a dose-dependent manner. (C) CD4+ T cells expressing anti-ALPPL2 SNIPR-MCAM CAR circuits were co-incubated with sender cells for 48 hours and CAR output was measured using a t2a GFP system. (D) Graphical representation of C. (E) Supernatant IL-2 concentration was assayed using ELISA. (F) T cells stained with Cell Trace Violet were co-incubated with irradiated sender cells in media without IL-2 for 9 days. T cell proliferation was measured using flow cytometry.
Having extensively investigated the range of domains that can be used to build functional SNIPRs, we determined how to utilize these receptors to control the therapeutic function of T cells. Many cancers adapt to CAR T cell therapy through antigen escape, downregulating their levels of surface CAR antigen (Majzner and Mackall, 2018). Having observed that our SNIPRs exhibited improved activation to CD19, we decided to test their ability to sense a range of surface antigen levels. To do this, we activated T cells engineered with alkaline phosphatase placental-like 2 (ALPPL2) targeted SNIPRs with a K562 cell line that expresses the tumor-specific antigen ALPPL2 in response to Doxycycline (Hyrenius-Wittsten, 2021) (Fig. 4B, S4C, S4D). We were able to achieve expression levels from 8×103 to 9.9×105 molecules of ALPPL2 per K562 (Fig. 4B). The optimized CD8α Hinge Notch SNIPR is more sensitive to lower ligand levels than synNotch without an increase in basal activity and that use of the Notch2 JMD further boosts sensitivity (Fig. 4C, 4D). These data suggest that SNIPRs could be useful in a wider array of immunotherapeutic applications where antigen density is heterogenous across the tumor mass.
Immune cell function is regulated by cytokines in a dose-dependent fashion, and side effects occur when a high dose of cytokines is given systemically as an immunotherapeutic (Pachella et al., 2015). Given that SNIPR activity is readily tuned through the TMD and JMD, we wanted to show how SNIPRs can be used to drive defined levels of the engineered T cell growth factor, super IL-2 (Levin et al., 2012). To do this, we built single viral vector constructs containing SNIPRs with a range of activity levels and an inducible super IL-2 cassette (Fig. S4E). We observed that CD4+ T cells expressing a SNIPR with the enhancing Notch2 JMD secreted higher amounts of super IL-2 in response to ligand expressed on K562 sender cells that had been irradiated to prevent culture overgrowth, while those with the additional dampening TMD mutation G318A secreted lower amounts of super IL-2 (Fig. 4E). The different amounts of induced super IL-2 produced by each SNIPR circuit correlated with T cell proliferation rates, exhibiting our ability to tune therapeutic T cell activity, and did not correlate purely with SNIPR expression levels (Fig. 4F, S4F). While we use the example of super IL-2 to demonstrate the capabilities of the SNIPR platform, this principle of receptor tuning can be applied toward a broad range of therapeutic programs (Roybal et al., 2016a). Indeed, we have found high-performing SNIPRs to be compatible with a wide array of LBDs (Fig. S4G).
Development of humanized SNIPRs with potential for clinical translation
We have explored the core regulatory domains (ECD, TMD, and JMD) that control the ligand-dependent cleavage of SNIPRs and have identified optimized cores. However, the synthetic Gal4-VP64 TF is a design liability for clinical translation as it is derived from yeast (Gal4) and herpesvirus proteins (VP64). To engineer a humanized receptor, we constructed TFs comprised of DNA-binding domains (DBD) fused to the transactivation domain of human NF-κB p65. We examined both DBDs sourced from human proteins, as well as engineered orthogonal synthetic zinc fingers (synTFs), for their ability to function in the SNIPR context (Fig 5A). Human protein-derived DBDs are advantageous for minimizing immunogenicity, whereas synTFs minimize off-target effects as verified by RNA-seq (Israni et al., 2021). Human protein-derived DBDs were chosen based on size and lack of expression in T cells (Uhlen et al., 2015), and included the eye development-associated paired box protein Pax-6 (Pax6) (Xu et al., 1999) and the liver-specific protein hepatocyte-nuclear factor 1-alpha (HNF1A) (Roscilli et al., 2002). SynTF candidates were selected for their orthogonality and potent transcriptional activity (Israni et al., 2021). RE cassettes for these TFs were constructed by tandem assembly of cognate binding motifs upstream to a minimal promoter. These RE cassettes proved to be orthogonal in T cells, as they were not activated in the absence of target cells (Fig. 5B). Humanized SNIPRs activated to target cells, and activation varied across TFs, suggesting that circuit function is subject to the efficiency of each TF in driving transcriptional activation (Fig. 5B). To examine whether TF compatibility extends to the original synNotch receptor, we tested the two TFs with the highest activation, HNF1A and ZF10, with the mouse synNotch and humanized receptor variant, and found that neither expressed nor activated as efficiently as the equivalent CD8α Hinge Notch SNIPRs (Fig. 5C, S5A). Compared to the Gal4-based receptor, Pax6 and HNF1A-based SNIPRs are comprised of peptide sequences with predicted immunogenicity on par with current clinical CAR designs, greatly reducing the risk of rejection in therapeutic applications (Fig. 5D). We found that peptides predicted to be most immunogenic are at junctions and thus could be eliminated by adjusting linker sequences without affecting receptor function. We examined whether a humanized anti-CD19 SNIPR→CAR circuit can eliminate target cells, an important benchmark for clinical utility. We found that HNF1A-based SNIPRs induced expression of an anti-BCMA CAR at a slower rate than Gal4-based SNIPRs, but at sufficient levels to clear in vitro tumor targets (Fig. S5B, S5C, S5D). We also found that functional SNIPR circuits could be integrated at low vector copy numbers (VCN), meeting requirements for clinical manufacturing (Fig. S5E). Finally, to further assess clinical viability, we performed RNA-sequencing to examine the transcriptomes of SNIPR-T cells utilizing HNF1A, our strongest human TF. We found that SNIPR circuit activation resulted in strong expression of circuit components, with minimal off-target changes in gene expression (Fig. 5E, S5F). Together, these data demonstrate that optimized SNIPR designs compatible with a broad range of TFs can overcome major obstacles to clinical translation.
Figure 5. Humanization of SNIPRs to reduce immunogenicity potential for cell-based therapies.
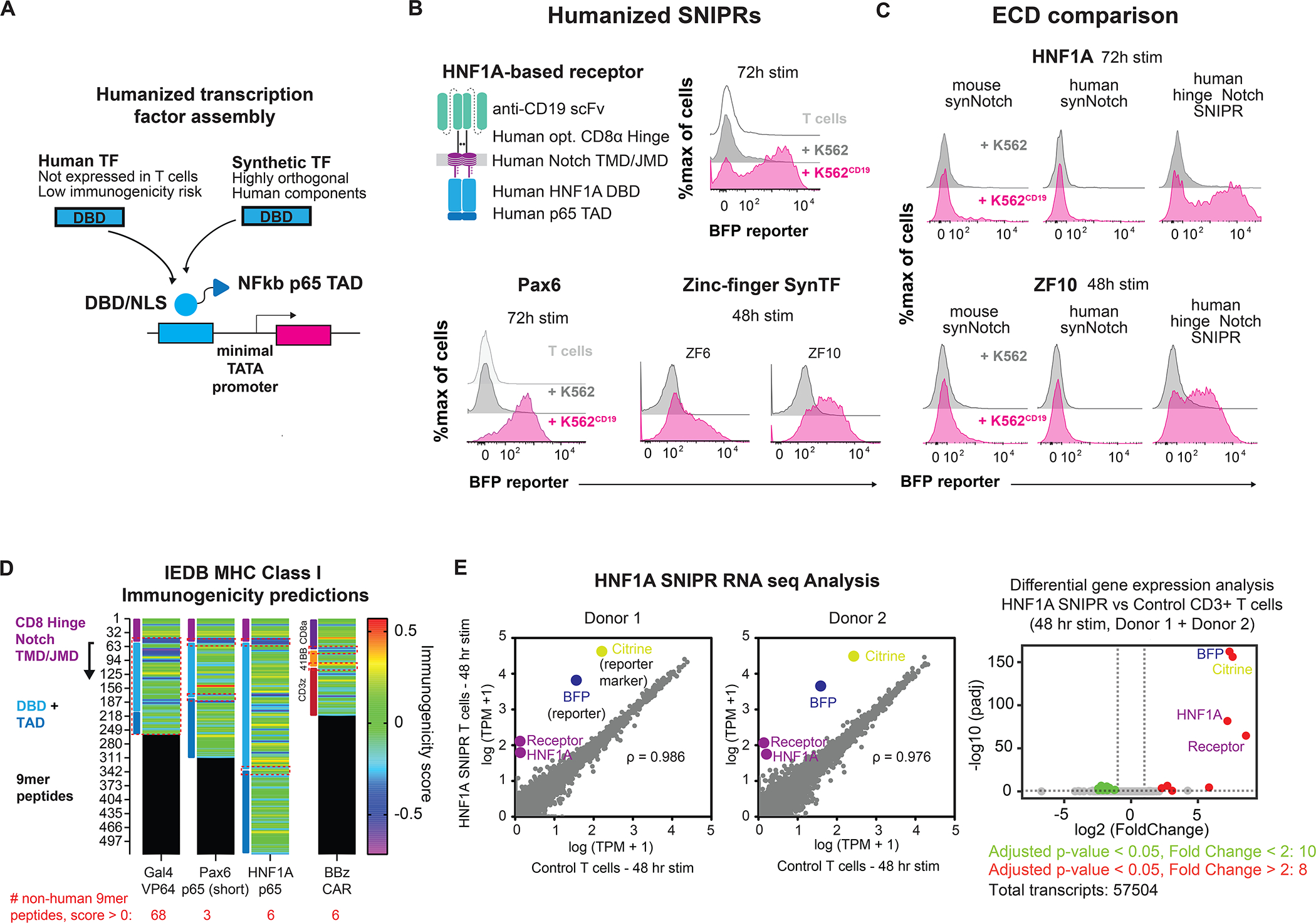
(A) Humanized TF and RE construction. (B) Activity of fully humanized SNIPRs. (C) SNIPR receptor scaffold compatibility with humanized TFs. (D) Assessing SNIPR immunogenicity. 9mer peptide sequences for SNIPRs with Gal4-VP64, Pax6, and HNF1A transcription factors were assessed for MHC I immunogenicity. Relative immunogenic potential across receptors was examined by comparing immunogenicity scores in regions derived from non-contiguous human protein sources (highlighted in red dashed boxes). Average scores: Pax6 0.039, HNF1A 0.156, BBz 0.102. (E) HNF1A SNIPR RNA-sequencing analysis. HNF1A SNIPR T cells were induced with target cells for 48 hours and sorted to remove targets for RNA-sequencing analysis. Correlation of transcriptomes against non-SNIPR T cells in two donors show few differences apart from SNIPR circuit components. Pearson correlation coefficients (left panel) were calculated for native transcripts (gray). Differential gene analysis shows few upregulated or downregulated genes compared to control cells following circuit induction (right panel).
In vivo testing of SNIPR-CAR circuits
Current challenges in CAR immunotherapy include the difficulty in defining a tumor with a single antigen. Systemic and unintended toxicity through on-target, off-tumor CAR activity has limited the clinical development of CARs and potent cytokine therapies (Ellis et al., 2021; Morgan et al., 2010). A multi-antigen recognition platform where a SNIPR binds a primary tumor antigen and drives expression of a CAR to a secondary antigen helps to mitigate risk of toxicity through more precise tumor recognition, and our humanized SNIPRs reduce the chance for immune rejection (Roybal et al., 2016b).
Humanized SNIPR→CAR circuits performed with high-fidelity during in vitro testing, but the question remained of their performance in vivo, where they would be exposed to a more diverse set of proteases and other environmental factors. To assess the performance and specificity of the optimized CD8α hinge SNIPRs in vivo, we examined the ability of SNIPR circuit T cells to control tumor growth in dual-antigen xenograft models. To test our benchmark anti-CD19 SNIPR→anti-BCMA CAR circuit in vivo, CD19+/BCMA+ K562 tumors were implanted in the left flank, and BCMA+ K562 tumors were implanted in the right flank of NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice. Four days after implantation, these mice were treated with untransduced T cells, anti-BCMA CAR T cells, or anti-CD19 SNIPR circuit T cells containing either a Gal4-VP64- or HNF1A-p65-driven anti-BCMA CAR (Fig. S6A, S6B). The anti-BCMA CAR and anti-CD19 SNIPR→anti-BCMA CAR circuit T cells selectively controlled dual-positive tumor growth. These data support the potency and specificity of SNIPR→CAR circuits in an in vivo setting and represent a successful demonstration of a humanized circuit in vivo. As a further assessment of specificity of the SNIPR→CAR circuit T cells, we harvested the tumors and spleen and observed BCMA CAR expression only in the dual antigen target tumor (Fig. S6F–H).
We next examined whether our humanized SNIPR circuits can specifically recognize clinically relevant antigen pairs for solid tumors. We recently reported ALPPL2 as a tumor specific antigen that can be targeted in combination with the tumor-associated antigens mesothelin or HER2 in mesothelioma or ovarian cancer, respectively (Hyrenius-Wittsten et al., 2021). We examined the ability of humanized anti-ALPPL2 SNIPR→anti-HER2 CAR circuits to eliminate dual-antigen ovarian cancer SK-OV-3 cells, both in vitro and in vivo. Using live cell imaging assays, we observed that SNIPR→CAR T cells cleared ALPPL2+/HER2+ SK-OV-3 but not HER2+ only cells (Fig 6A). Kinetic analysis showed that SNIPR circuit-mediated cytotoxicity was slower than CAR-T cell killing, due to the time needed for CAR induction as previously observed (Fig 6B) (Hyrenius-Wittsten et al., 2021). To examine in vivo efficacy, ALPPL2+/HER2+ and ALPPL2-/HER2+ SK-OV-3 cells were implanted in the left and right flanks, respectively, of NSG mice. Eight days after implantation, these mice were treated with untransduced T cells, anti-HER2 CAR T cells, or anti-ALPPL2 SNIPR→anti-HER2 CAR circuit T cells (Fig 6C). Anti-HER2 CAR-T cells and anti-ALPPL2 SNIPR→anti-HER2 CAR circuit T cells controlled tumor growth in the dual-positive tumor, but only the HER2 CAR controlled tumor growth in the single-positive tumor. Analysis of SNIPR T cell activation and CAR expression in the spleen, dual-positive, and single-positive tumors in a repeat experiment found specific SNIPR-T cell infiltration and CAR expression in the dual-positive tumor (Fig. 6D–G). Similarly, we observed that SNIPR circuits could specifically clear ALPPL2+/Mesothelin+ M28 epithelioid mesothelioma (Fig S6I, S6J). Together, these data demonstrate the clinical potential of humanized SNIPR→CAR circuits for more precise recognition of solid tumor targets.
Figure 6. Humanized SNIPR – CAR circuits exhibit precise dual antigen targeting in preclinical in vivo models of solid tumors.
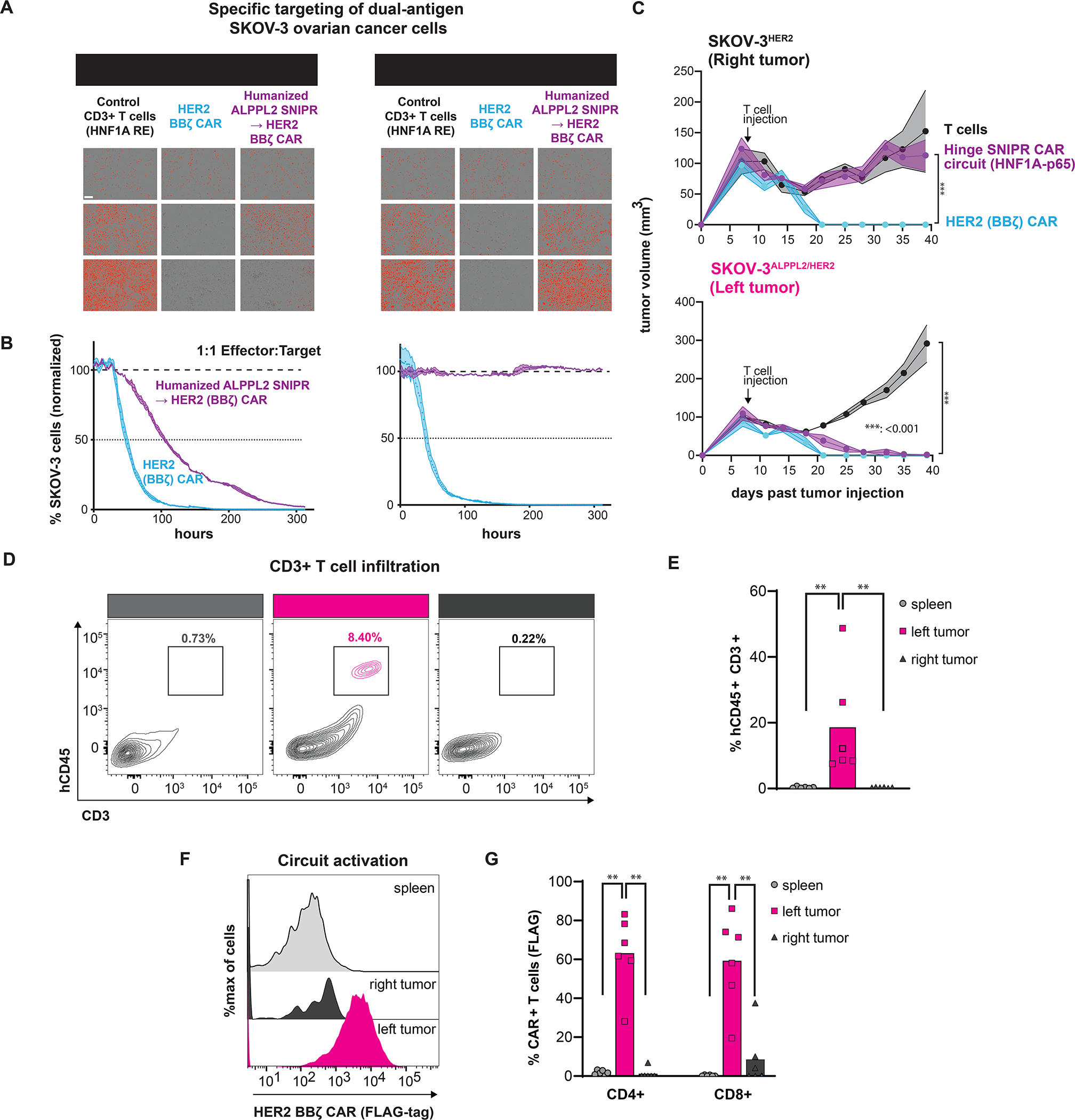
(A) Incucyte live cell imaging showing killing kinetics and specificity of humanized anti-ALPPL2 SNIPR –> anti-HER2 CAR circuits against SK-OV-3 ovarian tumor cells. (B) Quantitation of incucyte assay killing in (A). (C) Humanized SNIPR → CAR circuits clear dual positive ALPPL2+/HER2+ SK-OV-3 tumors in vivo. Statistics calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc comparing anti-HER2 CAR-T cells to circuit T cells (top) and untransduced T cells to anti-HER2 CAR and Circuit T cells (bottom). ***P ≤ 0.001. (D) In vivo assessment of fully human SNIPR circuit activation and trafficking. (E) Quantitation of T cells in the spleen and tumors. (F) Circuit activation of humanized SNIPR circuits in the spleen and tumors. (G) Quantitation of CAR surface expression in (F). Statistics were calculated using Mann-Whitney U-test (E and G), **P ≤ 0.01.
DISCUSSION
From our investigations into the ECDs, TMDs, and JMDs of RIP receptors, we have constructed a large set of receptors that function like Notch and have begun to define the guidelines for the synthetic assembly of these receptors we call SNIPRs (Fig. 7, Tables S2, S3). Overactive, inactive, and suboptimal core domains that control RIP all significantly reduce SNIPR performance, even when assembled with functional domains at other positions, suggesting that all three domains must be optimized for maximum ligand-dependent cleavage. We find that ECD specificity can be optimized by avoiding exposed protease sites and minimizing length, although SNIPR activity in response to alternative stimuli, such as T cell activation, may require direct observation to discover. We also find that additional TMDs and JMDs can be used to construct SNIPRs, and that TMDs and JMDs can be tuned through point mutations to meet individual clinical requirements, such as improving specificity or regulating levels of a delivered therapeutic. In addition, we find that SNIPRs containing suboptimal modules, such as the human synNotch ECD, can be improved through either direct ECD engineering, such as deletion of the NRR (Fig. 2A, 2B), or increasing activity in another module, such as the JMD. All three core SNIPR components, along with the LBD and TF, can impact receptor expression. Our systematic exploration of SNIPR parts identified receptors that are well-expressed and activate with high fidelity, two key features for cell therapy.
Figure 7. Design framework for next generation synthetic receptors for custom transcriptional regulation in therapeutic cells.
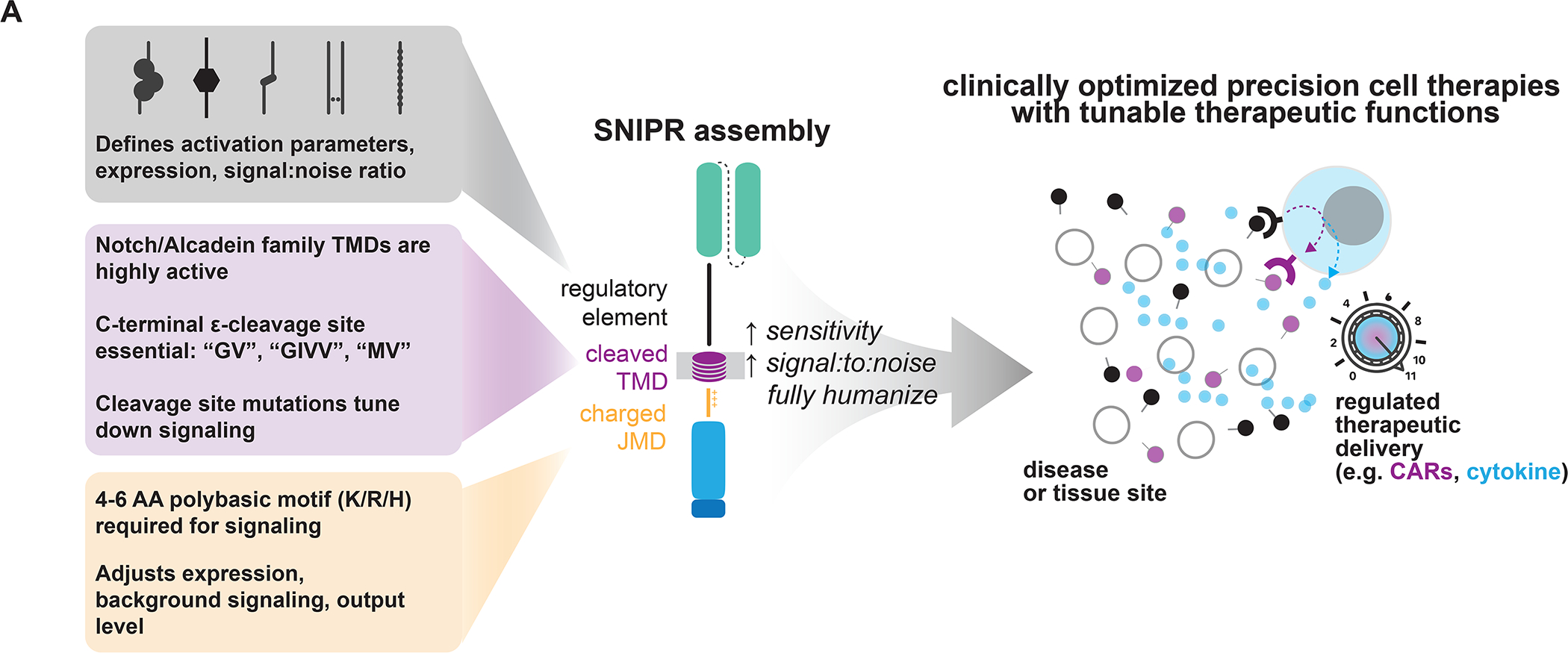
SNIPRs can be built through design of the receptor ECD, TMD, and JMD. The receptor ECD represents the first regulatory site and affects receptor activation parameters, expression, and stringency for ligand. Several known C-terminal motifs in the receptor TMD, commonly found in the Notch and Calsyntenin families, appear to be important for receptor signaling. Highly basic residues in the receptor JMD are required for signaling, and the choice of JMD can strongly affect receptor expression and output levels. By combining these elements, clinically relevant SNIPRs can be built that utilize fully human proteins and are compact, highly expressed, and regulatable. Our SNIPR design framework opens the possibility to build customized precision cellular therapeutics.
We have found that many SNIPR ECDs that lack regulatory domains such as the NRR remain functional. This result adds to previous screens of ECDs in a Notch context, which found that proteolytic switches with homology to Notch could substitute for the Notch NRR, albeit with a reduced signal-to-noise ratio (Hayward et al., 2019). In contrast, we find that ECDs with no homology with Notch outperform it in the context of a synthetic receptor. One commonality between functional ECDs is a relative lack of known protease cleavage sites. While a simple glycine-serine linker is a sufficient ECD, we find the addition of ADAM or MMP9 cleavage sites to this inherently unstructured linker leads to uncontrolled SNIPR activation. In addition, removal of known ADAM10 cleavage sites in the Robo1 and Notch1 ECDs improved the signal-to-noise ratios for SNIPRs utilizing these components. This discovery suggests that there is a large realm of permissive ECDs with a common mechanism of activation. Although most SNIPRs exclude known sites for ADAM protease cleavage, we find that all tested SNIPRs continue to rely on ADAM protease and γ-secretase activity. Our finding of enhanced SNIPR signaling during T cell activation may be explained by higher ADAM10 and ADAM17 activity (Li et al., 2007, Lambrecht et al., 2018), but the mechanism for either ligand-dependent or -independent activation for the SNIPRs requires further study. The mechanism of Notch activation through RIP is well-investigated, but the roles of other cellular processes, such as ubiquitination (Moretti et al., 2013), receptor endocytosis (Kandachar and Roegiers, 2012), and receptor trafficking (Yamamoto et al., 2009) remain unclarified and could regulate SNIPR activity.
We were surprised to observe a lack of diversity in high-performing TMDs, having selected candidate TMDs from reported γ-secretase substrates (Haapasalo and Kovacs, 2011). This finding may be specific for SNIPRs expressed in human T cells, and SNIPRs containing non-functional TMDs from our screen may be more active when expressed in other tissue or cell types.
In conclusion, our systematic engineering of SNIPRs has allowed us to build customizable receptor cores that provide not only the spatial discrimination afforded by previous synthetic receptors, such as synNotch, but also sensitivity at a range of antigen levels and greater compatibility with humanized components, thereby lowering the risk for immunogenicity. This added functionality is of clear benefit to current immunotherapies, such as CAR T cells, and should help provide a titrated therapeutic response while mitigating known issues of these technologies, such as premature T cell exhaustion and on-target/off-tumor systemic toxicity. For example, local titrated delivery of a potent cytokine, such as IL-12 (Lasek et al., 2014), to a tumor site using therapeutic cells may significantly improve efficacy and clinical outcomes as compared to the severe toxicity observed during systemic IV administration. These receptors should provide biomedical research with a comprehensive toolkit for directing a range of cell-based therapies to their intended targets combined with programmed localized therapeutic activity.
Limitations of the Study
Limitations of the study include our focus on applying SNIPR technology in human primary T cells. Guidelines that define optimal SNIPR performance, such as TMD and JMD selection, may vary between tissue types and organisms, and a screen of SNIPR performance between a wide array of cell types would be helpful in determining the universality of our SNIPR engineering rules. In addition, while we expect that enhanced SNIPR activity in the context of T cell activation would be beneficial in the context of an induced CAR, where activation of the induced CAR boosts the initial SNIPR signal, any TCR/CAR-mediated boost in SNIPR signaling may be unwanted in situations where only small amounts of an otherwise toxic therapeutic payload are desired. Additional engineering strategies such as TRAC KO (Eyquem et al., 2017) should be considered in these situations where SNIPR-TCR interactions are of concern. Finally, care should be taken to reduce unintended CAR activation at co-localized normal tissues (Srivastava et al., 2019) through the selection of target antigens for SNIPR-CAR circuits and the choice of SNIPR core.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Kole Roybal (kole.roybal@ucsf.edu).
Material availability
Plasmids generated in this study will be deposited to Addgene.
Data and code availability
All raw data reported in this paper will be shared by the Lead Contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice were used for all in vivo experiments. All mice were between 8 and 12 weeks of age at start of experimentation and were not involved in previous procedures. Females were used for all experiments using K562 and SK-OV-3 as target cells, and males were used for all experiments using M28 as target cells. All mice were group housed with a maximum of 5 mice per cage. For experimentation, littermates of the same sex were randomly assigned to experimental groups. All mice were supplemented with amoxicillin/clavulanate (Zoetis) during experimentation for infection prophylaxis. All experimentation was performed in accordance with the IACUC guidelines present at UCSF.
Cell lines
Lenti-X 293T cells originate from female fetal tissue. U87 cells originate from a male patient with malignant glioma, likely glioblastoma. MDA-MB-468 cells originate from a female with breast adenocarcinoma. SKBR3 cells originate from a female with breast adenocarcinoma. Lenti-X 293T, U87, MDA-MB-468, and SKBR3 cells were all cultured in DMEM supplemented with 10% FBS and 50 U/mL penicillin-streptomycin. K562 cells originate from a female with chronic myelogenous leukemia. They were cultured in IMEM supplemented with 10% FBS and 50 U/mL penicillin-streptomycin. Jurkat T cells originate from a male T cell leukemia patient, and M28 cells originate from a human mesothelioma culture. Both were cultured in RPMI-1640 supplemented with 10% FBS, 50 U/mL penicillin-streptomycin, and 1X Glutamax (ThermoFisher). SK-OV-3 cells originate from a female with ovarian cancer. They were cultured in McCoy’s 5A media supplemented with 10% FBS and 50 U/mL penicillin-streptomycin. All cell lines are cultured in a 37°C incubator with 5% CO2.
Primary Human T cell Isolation and Culture
Primary CD4+ and CD8+ T cells were isolated from anonymous donor blood after apheresis by negative selection (STEMCELL Technologies #15062 & 15063). Blood was obtained from Blood Centers of the Pacific (San Francisco, CA) as approved by the University Institutional Review Board. T cells were cryopreserved in RPMI-1640 (Thermo Fisher #11875093) with 20% human AB serum (Valley Biomedical Inc., #HP1022) and 10% DMSO. After thawing, T cells were cultured in human T cell medium consisting of X-VIVO 15 (Lonza #04-418Q), 5% Human AB serum and 10 mM neutralized N-acetyl L-Cysteine (Sigma-Aldrich #A9165) supplemented with 30 units/mL IL-2 (NCI BRB Preclinical Repository) for most experiments. For experiments involving the induction of super IL-2, primary T cells were maintained in human T cell media supplemented with IL-2 until experimentation, whereupon media was replaced with media without supplemental IL-2.
Bacteria
Stellar chemically competent E. coli, a HST08 strain, (Takara) were used for molecular cloning. They were cultured in LB broth supplemented with a selection antibiotic at 37°C in a shaker set to 250 rpm or on LB-agar plates supplemented with a selection antibiotic at 37°C in a plate incubator.
METHOD DETAILS
Receptor and Response Element Construct Design
Receptors were built by fusing the CD19 scFV (Porter et al., 2011), ALPPL2 M25FYIA scFV (Hyrenius-Wittsten et al., 2021), HER2 4D5-8 scFv (Carter et al., 1992), EGFRviii 139 scFv (Morgan et al., 2012), or LaG17 nanobody (Fridy et al., 2014) to an extracellular domain comprised of: the human Notch1 (P46531) minimal regulatory region (Ile1427 to His1735), a truncated human Notch1 Notch Regulatory Region (Ile1427 to Glu1447, Thr1725 to His1735), a CD8α (P01732) hinge region (Thr138 to Asp182), a CD28 (P10747) hinge region (Ile114 to Pro152), an IgG4 hinge region, an OX40 hinge region, (the type III fibronectin domain from Robo1 (Q9Y6N7, Lys769 to Pro897), truncated CD8α hinges and fibronectin domains (as described), or Gly-Gly-Ser linkers of variable length (as described). All extracellular domains were fused to a transmembrane domain and intracellular juxtamembrane domain (as described), and a transcriptional element composed of Gal4 DBD VP64, Pax6(M1 to Ala139)-p65(Pro428 to Ser551), HNF1A(Met1 to Met283 with Thr-Cys-Arg linker)-p65(Asp361 to Ser551), or ZF-p65 (14). All receptors contain an N-terminal CD8α signal peptide (MALPVTALLLPLALLLHAARP) for membrane targeting and a myc-tag (EQKLISEEDL) for easy determination of surface expression with α-myc AF647 (Cell-Signaling #2233). The receptors were cloned into a modified pHR’SIN:CSW vector containing a PGK promoter for all primary T cell experiments. The pHR’SIN:CSW vector was also modified to make the response element plasmids. Five copies of the Gal4 DNA binding domain target sequence (GGAGCACTGTCCTCCGAACG), or four copies of the Pax6 consensus DBD recognition motif (ATTTTCACGCATGAGTGCACAG) and HNF1A DBD recognition motif (GTTAATNATTAAC) were cloned 5′ to a minimal synthetic pybTATA promoter. Also included in the response element plasmids is a PGK promoter that either constitutively drives expression of a fluorophore (mCitrine or mCherry) to easily identify transduced T cells or a SNIPR for single vector experimentation. Inducible CAR vectors contained CARs tagged N-terminally with FLAG-tag, and in some cases C-terminally with a t2a GFP system. All induced elements were cloned via a BamHI site in the multiple cloning site 3′ to the Gal4 response elements. All constructs were cloned via In-Fusion cloning (Takara # 638951).
Lentiviral Transduction of Human T cells
Pantropic VSV-G pseudotyped lentivirus was produced via transfection of Lenti-X 293T cells (Clontech #11131D) with a pHR’SIN:CSW transgene expression vector and the viral packaging plasmids pCMVdR8.91 and pMD2.G using Mirus Trans-IT Lenti (Mirus #MIR6606). Primary T cells were thawed the same day, and after 24 hours in culture, were stimulated with Human T-Activator CD3/CD28 Dynabeads (Life Technologies #11131D) at a 1:3 cell:bead ratio. At 48 hours, viral supernatant was harvested, and the primary T cells were exposed to the virus for 24 hours. At day 5 post T cell stimulation, the Dynabeads were removed, and the T cells were sorted for assays with a Beckton Dickinson (BD) FACs ARIA II. Sorted T cells were expanded until day 10 for in vivo assays and until day 14 for in vitro assays.
Generation of Receptor Jurkat cells for Screening
E6-1 Jurkat T cells (ATCC# TIB-152) were lentivirally transduced with a reporter plasmid encoding a Gal4 driven tagBFP response element and a constitutively expressed mCitrine cassette. Reporter positive cells were sorted for mCitrine positivity and expanded. Individual cultures of reporter positive Jurkat T cells were lentivirally transduced in a 96 well plate with myc-tagged α-CD19 human SynNotch1 receptors with modified transmembrane or juxtamembrane domains. After viral transduction, the receptor transduction efficiency for each Jurkat cell population was measured with a BD FACSymphony Fortessa X-50 following staining with anti-myc AF647 (Cell-Signaling #2233).
Cancer Cell Lines for Screening
The cancer cell lines used were K562 myelogenous leukemia cells (ATCC #CCL-243). K562s were lentivirally transduced to stably express either human CD19 at equivalent levels as Daudi tumors (ATCC #CCL-213), BCMA, or both BCMA and CD19. CD19 levels were determined by staining the cells with α-CD19 APC (Biolegend #302212). BCMA levels were determined by staining the cells with α-BCMA APC (Biolegend #357505). All cell lines were sorted for expression of the transgenes.
MCAM BiTE Production
MCAM BiTE was produced from transfecting LentiX-293T cells with a pHR’SIN:CSW transgene expression vector. 293T media was replaced with T cell media 24 hours after transfection. MCAM BiTE was harvested 48 hours post-media replacement by collecting supernatant and removing 293T cells via centrifugation.
Doxycycline inducible ALPPL2
A clonal line of K562 cells expressing a doxycycline-inducible FLAG-tagged ALPPL2 cassette was treated with doxycycline (Abcam) at doses ranging between 0.1–100 ng/mL for 24 hours prior to co-incubation with T cells. Surface expression levels were assessed by flow cytometry using an PE-conjugated anti-FLAG antibody (Biolegend) prior to assay. Surface ligand levels were quantified using a Quantibrite PE Phycoerythrin Fluorescence Quantitation Kit (BD Biosciences).
In vitro SNIPR Activation Assays
For in vitro SNIPR activation assays, 1×105 T cells or Jurkat T cells were co-cultured with the indicated target cells at a 1:1 ratio. Co-cultures were performed in 96 well round bottom plates for K562 target cells (VWR) and 96 well flat bottom plates (VWR) for all other target cell lines. Adherent target cells were plated 1 day prior to co-culture with T cells. To exogenously activate T cells, MCAM BiTEs were added to co-cultures when indicated. When activating a co-expressed ALPPL2 CAR, ALPPL2+ K562 cells were added to the co-culture in a 1:1 ratio with T cells. The cultures were analyzed at the time points indicated for reporter activation using a BD FACSymphony Fortessa X-50. All flow cytometry analysis was performed in FlowJo software (BD). TMD sequence alignment was performed using ClustalX and visualized using Jalview.
Incucyte killing assay
For in vitro engineered T cell killing assays, ALPPL2+/HER2+ or ALPPL2-/HER2+ SK-OV-3 tumor cells expressing nuclear mKate2 were seeded in 96-well flat-bottom plates. After 24 hours, engineered T cells were added at an expected effector:target ratio of 1:1. Plates were imaged every 2 hours using the IncuCyte S3 Live-Cell Analysis System (Essen Bioscience) for a duration of 14 days. Three images per well at ×10 magnification were collected. Experiments were performed in RPMI 1640 with 10% FBS, penicillin (100 U/ml) and streptomycin (100 μg/ml), and 1× GlutaMAX supplemented with IL-2 (30 U/ml). Media was replaced every 5 days.
Super IL-2 Induction Assays
For proliferation assays, primary CD4+ were stained with Cell Trace Violet (Thermo Fisher #C34557) and stimulated with irradiated K562 or CD19+ K562 target cells in human T cell media without IL-2 supplementation. T cell division was assayed using flow cytometry at the indicated time points using a BD FACSymphony Fortessa X-50. For induced super IL-2 quantification, CD4+ T cells were stimulated with irradiated K562 or CD19+ K562 target cells in human T cell media without IL-2 supplementation and supernatant was harvested at the indicated timepoints. Super IL-2 levels in the supernatant were measured using an IL-2 Human Instant ELISA kit (Thermo Fisher #BMS221INST).
Vector Copy Number Measurements
Vector copy number was measured using a previously described droplet digital PCR method (Wang et. al., 2008). Genomic DNA was extracted from T cells transduced with SNIPR receptor circuits with a NucleoSpin Tissue kit (Macherey-Nagel) and digested with 40 IU of BamHI-HF and EcoRI-HF (New England Biolabs) at 37°C for 1 hour. ddPCRs amplifying HIV-1 Ψ and human TERT (hTERT) in duplex were set up and analyzed using PCR primers and protocols described previously (Wang et. al., 2008). The hTERT primers were 5’-GGCACACGTGGCTTTTCG-3’ and 5’-GGTGAACCTCGTAAGTTTATGCAA-3’. The HIV-1 Ψ primers were 5’-TACTGACGCTCTCGCACC-3’ and 5’-TCTCGACGCAGGACTCG-3’. The hTERT probe was 5’-TCAGGACGTCGAGTGGACACGGTG-3’ with SUN dye and ZEN/IBFQ double quenchers. The HIV-1 Ψ probe was 5’-ATCTCTCTCCTTCTAGCCTC-3’ with a 6-FAM dye and ZEN/IBFQ double quenchers. PCRs were set up in accordance with ddPCR Supermix for Probes (No dUTP) guidelines (Bio-Rad) and droplets were generated using a QX200 Droplet Generator (Bio-Rad). The PCR program was one cycle at 95°C for 10 minutes, followed by 40 cycles at 94°C for 30 minutes and at 60°C for 1 minute, then one cycle at 98°C for 10 minutes. Samples were immediately analyzed by a QX100 Droplet reader using QuantaSoft software (Bio-Rad). Vector copy number was calculated using the formula: 2 × (copies of HIV-1 Ψ) / (copies of hTERT).
RNA-seq sample preparation
4 ×106 primary human CD3+ T cells with and without SNIPR circuits were co-cultured 1:1 with K562CD19+ target cells for 48 hours. Following circuit induction, 2×106 T cells T cells were sorted, washed with PBS, flash frozen, and submitted to Genewiz for mRNA extraction (polyA selection), library preparation, and next-generation sequencing (Illumina, 2×150bp, ~350M PE reads).
In vivo assays
For anti-ALPPL2 SNIPR→anti-HER2 CAR circuit testing, NOD.Cg-PrkdcscidIl2rgtm1Wjl/SzJ (NSG) mice between 8–12 weeks of age were injected subcutaneously with 4×106 SK-OV-3ALPPL2/HER2 target cells into the left flank and 4×106 SK-OV-3HER2 control cells into the right flank. 8 days post tumor injection, 1.5×106 of untransduced bulk CD3+ T cells, anti-HER2 CAR-T cells, or anti-ALPPL2 SNIPR→anti-HER2 CAR circuit T cells were injected retro-orbitally, and tumor volume was measured by caliper regularly. For SK-OV-3 tumor T cell analysis, NSG mice were injected subcutaneously with 2.5×106 SK-OV-3ALPPL2/HER2 target cells into the left flank and 2.5×106 SK-OV-3HER2 control cells into the right flank. 33 days post tumor injection, 1.5×106 of anti-ALPPL2 SNIPR-anti-HER2 CAR circuit T cells were injected retro-orbitally. 16 days post T cell injection, mice were sacrificed and T cell presence in the spleen, left, and right tumors was measured using flow cytometry.
For anti-ALPPL2 SNIPR→anti-Mesothelin CAR circuit testing, NSG mice between 8–12 weeks of age were injected subcutaneously with 4×106 M28ALPPL2/MESO target cells into the left flank and 4×106 M28ALPPL2KO/MESO control cells into the right flank. 10 days post tumor injection, 3×106 of untransduced bulk CD3+ T cells, anti-MESO CAR-T cells, or anti-ALPPL2 SNIPR-anti-MESO CAR single vector circuit T cells were injected retro-orbitally and tumor volume was measured by caliper every few days.
For anti-CD19 SNIPR→anti-BCMA CAR circuit testing, NSG mice between 8–12 weeks of age were implanted with either 1×106 K562CD19+/BCMA+ tumor cells subcutaneously in the left flank alone or with an additional 1×106 K562BCMA+ tumor cells subcutaneously in the right flank. Four days after tumor implantation, 2.5 or 3×106 engineered primary human CD4+ and CD8+ T cells (total of 5 or 6×106 T cells) were intravenously infused through tail vein injection. Tumor volume was monitored via caliper regularly.
QUANTIFICATION AND STATISTICAL ANALYSIS
RNA-seq analysis
RNA-seq analysis was performed by Genewiz as follows. Reads were trimmed (Trimmomatic v.0.36) and mapped to the 30-574668504_GRCh38 reference genome available on ENSEMBL plus SNIPR circuit sequences using STAR aligner v.2.5.2b. Unique gene hit counts were calculated by using featureCounts from the Subread package v.1.5.2, TPM plots were generated using R, and gene expression analysis was performed using DESeq2.
In vivo and in vitro statistical analysis
Statistics for tumor measurements were calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc. Tumor takedown MFI statistics were calculated using Mann-Whitney U-test. In vitro reporter induction MFI statistics were calculated using unpaired T tests. P-values less than 0.05 were considered significant. Incucyte images were analyzed using the IncuCyte S3 Software (Essen Bioscience) to detect and count the number of mKate2+ nuclei per image.
Supplementary Material
Supplemental Figure 1. Related to Figure 1. Construct designs, expression of Robo/FnIII receptors, and drug inhibition studies. (A) Schematic of SNIPRs and reporter constructs. All SNIPRs tested are expressed under a constitutive pGK promoter and contain an N-terminal CD8α signal peptide for membrane trafficking and myc-tag for measuring expression. Expression of tagBFP is regulated by a 5x Gal4 UAS response element and a minimal pybTATA promoter. For identifying reporter+ cells, mCitrine is expressed constitutively under a pGK promoter. Right, receptor expression of murine and human synNotch receptors. (B) The original synNotch receptor is composed of a ligand-binding domain (LBD) fused to a proteolysis-regulated core from murine Notch1 and a Gal4-VP64 transcriptional unit. T cells expressing a CD19 synNotch receptor, constructed with either a murine or human Notch1 core, were tested with various transcriptional factors for their ability to transmit ligand-dependent signaling. SynNotch T cells were co-incubated with either K562 or K562CD19 sender cells for 24 hours and BFP output was measured using flow cytometry proteins. Compared to the original murine synNotch design, synNotch receptors utilizing human components fail to efficiently induce BFP. (C) SynNotch receptors using cores from human Notch 1, Notch 2, and Notch 3 do not display strong ligand-dependent activation. Human synNotch 4 did not express. (D) Representative expression MFIs for synNotch/synRobo. Expression MFI does not correlate with receptor signaling ability. (E) Receptor activity is dependent on ADAM/γ-secretase activity. SNIPR activation is inhibited by the ADAM inhibitor GI254023X and the gamma secretase inhibitor DAPT.
Supplemental Figure 2. Related to Figure 2. Protease site regulation, reporter constructs with CAR, hinge engineering. (A) Protease site exposure can regulate SNIPR signaling. Addition of a fibroblast activation protein (FAP) protease site into a synthetic linker ECD confers SNIPR activity in the presence of K562 sender cells. Addition of a ligand binding domain restores ligand specificity. (B) Diagram of reporter construct with constitutively co-expressed 2nd generation ALPPL2 CAR (41BB-zeta). (C) Behavior of ΔNRR variants of human Notch1–4. Notch1 ΔNRR demonstrates sensitivity to T cell activation through activation of a co-expressed anti-ALPPL2 CAR. Notch2 ΔNRR demonstrates constitutive signaling. Notch3 ΔNRR demonstrates significant ligand-independent signaling. Notch4 ΔNRR demonstrates weak ligand-dependent signaling but no sensitivity to T cell activation. (D) Activation of anti-CD19 CD8α Hinge SNIPR variants. 1st row: expression of SNIPR and reporter construct in CD8+ T cells. 2nd row: Activation of the CD8α Hinge SNIPR variants with K562CD19. 3rd row: Activation of the CD8α Hinge SNIPR variants with K562CD19 in the presence of MCAM BiTEs. 4th row: Activation of the CD8α Hinge SNIPR variants with K562CD19 in the presence of a co-expressed 2nd generation ALPPL2 CAR. Superimposed bar graphs displaying ligand-independent (K562) and ligand-dependent (K562CD19) activation are shown. (E) Same as C, but with the full length CD8α Hinge Notch1 expressed in CD4+ T cells. The CD8α-based SNIPR is not as sensitive to T cell activation in CD4+ T cells as in CD8+ cells. (F) Same as B, but with CD8α Hinge SNIPR variants.
Supplemental Figure 3. Related to Figure 3. Alanine scan, fixing non-functional TMDs, non-Notch receptors. (A) Expression of Notch1 TMD Alanine scan mutants. (B) Results from Alanine scan in terms of %BFP+. (C) Fixing a non-functional TMD through residue substitution. The performance of the Robo1 TMD in a SNIPR setting can be improved by inserting a Gly-Val motif at an equivalent position to the Notch1 TMD and replacing the Robo1 JMD with that of Notch1. The performance of the AGER TMD in a SNIPR setting can be improved by replacing the residues C-terminal to its Gly-Val motif with those from the highest performing TMD, Notch1 from G. gallus. T cells expressing a SNIPR-BFP circuit were co-incubated with the indicated K562 cells for 48 hours and the percentage of BFP+ cells was measured using flow cytometry. (D) Human synNotch expression and activation improvement through JMD substitution identified by a screen. Replacing the human Notch 1 JMD with the Notch 2 JMD increases receptor expression and activation in primary T cells. (E) Activation of anti-CD19 SNIPRs with non-Notch components. 1st row: expression of SNIPR and reporter construct in CD8+ T cells. 2nd row: Activation of the SNIPR variants with K562CD19. 3rd row: Activation of the CD8α Hinge SNIPR variants in the presence of MCAM BiTEs. 4th row: Activation of the CD8α Hinge SNIPR variants in the presence of a co-expressed 2nd generation anti-ALPPL2 CAR. Superimposed bars displaying ligand-independent (K562) and ligand-dependent (K562CD19) activation are shown.
Supplemental Figure 4. Related to Figure 4. Hinge SNIPR TMD and JMD, drug inhibition studies, construct designs, expression data, testing with additional LBDs. (A) Superimposed bar graphs displaying activation of Hinge SNIPRs with variable TMDs and JMDs with an activated co-expressed ALPPL2 CAR at 48 hours. (B) SNIPR activation is dependent on ADAM and gamma-secretase activity. T cells expressing a CD19 Hinge Notch SNIPR-BFP circuit and co-expressed ALPPL2 CAR were co-incubated with the indicated conditions for 48 hours. BFP output was measured using flow cytometry. (C) Design of inducible ALPPL2 cassette and MCAM CAR response element. rtTA3 is expressed under an SFFV promoter and induces ALPPL2 expression in the presence of doxycycline. MCAM CAR is expressed under an inducible minimal TATA promoter and 5XGal4 UAS enhancer with a constitutively expressed mCherry. (D) Expression of ALPPL2 SNIPR was measured using myc-staining. (E) Design of an induced Super IL-2 single vector. Super IL-2 is expressed under an inducible minimal TATA promoter and 5XGal4 UAS enhancer with a constitutively expressed SNIPR. (F) Expression of SNIPR-super IL-2 circuits were measured using myc-staining. (G) Activation of Hinge SNIPR with additional LBDs. Hinge SNIPR with Notch2 JMD is effective against membrane bound GFP, EGFRviii, and HER2 when expressing an anti-GFP nanobody, anti-EGFRviii scFv, or anti-HER2 scFv, respectively.
Supplemental Figure 5. Related to Figure 5. Constructs, expression of humanized and non-humanized receptors, and individual tumor growth curves. (A) Comparison of humanized TFs with SNIPR ECDs. Expression profile of optimized CD8α Hinge SNIPRs and synNotch receptors using the human transcription factor HNF1A and synTF ZF10 was determined by surface staining of the myc-tagged receptors. (B) Design of BCMA CAR, CD19 SNIPR, and induced BCMA CAR response vector. BCMA CAR is expressed under a constitutive SFFV promoter. CD19 SNIPR is expressed under a constitutive pGK promoter. BCMA CAR is expressed under an inducible minimal TATA promoter and 5XGal4 UAS enhancer with a constitutively expressed mCherry. (C) Receptor expression of optimized CD8α Hinge SNIPRs in CD4+ and CD8+ T cells used for in vivo testing of circuit function. (D) Vector copy number (VCN) measurements of SNIPR circuits in both single-vector and dual-vector format. Genomic DNA was isolated from SNIPR T cells and the number of copies of HIV and a TERT2 reference gene was assayed using a duplex ddPCR reaction done in triplicate. The VCN was calculated using the formula 2 * (#HIV copies) / (#TERT2 copies).
(E) Target cell killing by a fully humanized SNIPR circuit. T cells expressing a SNIPR-CAR circuit were co-incubated with K562 target cells for 72 hours. Circuit induction was measured by FLAG-tag staining for the BCMA-CAR and flow cytometry (left panel). Target cells were cleared by 48 hours, as measured by DRAQ7 staining and flow cytometry (right panel).
(F) Differential gene analysis of transcriptomes from fully HNF1A SNIPR Circuits following induction, versus control cells. Analysis was performed with two different human T cell donors, and genes with greater than two-fold difference and p-value < 0.01 are listed here.
Supplemental Figure 6. Additional in vivo data. Related to Figure 6. (A) NSG mice (5 per experimental group) were injected subcutaneously with 1×106 K562CD19/BCMA target cells into the left flank and 1×106 K562BCMA control cells into the right flank. 4 days post tumor injection, 6×106 untransduced, BCMA CAR, or CD19 SNIPR-BCMA CAR circuit T cells (3×106 each CD4+ and CD8+) were injected via tail vein, and tumor volume was measured by caliper every few days. Statistics were calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc comparing untransduced T cells to BCMA CAR (top, *) and Circuit T cells (top, **) on Day 21, and comparing BCMA CAR T to circuit T cells (bottom). *P ≤ 0.05, **P ≤ 0.01. (B) Individual tumor growth curves for data shown in (A). (C) NSG mice were injected with 1×106 K562CD19+/BCMA+ target cells into the left flank. 4 days post tumor injection, 5×106 BCMA CAR or CD19 SNIPR-BCMA CAR circuit T cells (2.5×106 each CD4+ and CD8+) or PBS control were injected, and tumor volume was measured over time. (D) Survival curve for mice in C. (E) Same as C, but with NOD mice injected with 1×106 K562CD19+/BCMA+ target cells into the left flank and 1×106 K562BCMA target cells into the right flank. (F) Human T cell presence in the spleen, left, and right tumors was measured using flow cytometry. (G, H) T cell abundance (G) and CAR induction (H) of human T cells in the spleen, left, and right tumors was measured using a t2a system. Statistics were calculated using one-way ANOVA with Dunnett’s test post hoc. *P ≤ 0.05; **P ≤ 0.01; ns, not significant. (I) NSG mice were injected subcutaneously with 4×106 M28ALPPL2/MESO target cells into the left flank and 4×106 M28ALPPL2KO/MESO control cells into the right flank. 10 days post tumor injection, 3×106 of untransduced T cells, anti-MESO CAR-T cells, or anti-ALPPL2 SNIPR-anti-MESO CAR single vector circuit T cells (G4VP64) were injected retro-orbitally, and tumor volume was measured by caliper every few days. Statistics were calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc comparing untransduced T cells to anti-MESO CAR and Circuit T cells (top, **) and untransduced T cells to circuit T cells (bottom) ns = not significant, **P ≤ 0.01. (J) NSG mice were injected subcutaneously with 4×106 M28ALPPL2/MESO target cells into the left flank and 4×106 M28ALPPL2KO/MESO control cells into the right flank. 9 days post tumor injection, 3×106 of untransduced T cells, anti-MESO CAR-T cells, or anti-ALPPL2 SNIPR-anti-MESO CAR circuit T cells (HNF1A-p65) were injected retro-orbitally, and tumor volume was measured by caliper every few days. Statistics were calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc comparing untransduced T cells to anti-HER2 CAR and Circuit T cells (top) and anti-MESO CAR-T cells to circuit T cells (bottom). ****P ≤ 0.0001.
Supplemental Table S1. Sequences of screened TMDs and JMDs, related to Figure 3. TMDs and JMDs were sourced from known γ-secretase substrates (19), and Notch1 homologs. TMD sequences were extracted from Uniprot annotations and were extended to the first basic residue (R, K, or H). Two TMDs from proteins not believed to be γ-secretase substrates were selected for negative control. JMD sequences began immediately c-terminal to the TMD and ended immediately prior to 3 non-basic residues.
Supplemental Table S2. Amino acid sequences of highlighted SNIPR components, related to Figures 1–6, supplemental figures S1–S6.
Supplemental Table S3. LBD, ECD, TMD, JMD, and TFs of highlighted SNIPR designs, related to Figures 1–6, supplemental figures S1–S6.
Supplemental Table S4. Jurkat TMD screen expression and activation data, related to Figure 3.
Supplemental Table S5. Jurkat JMD screen expression and activation data, related to Figure 3.
Key resources table.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Myc-Tag (9B11) Mouse mAb | Cell-Signaling Technology | Cat#2233 |
| PE anti-DYKDDDDK Tag Antibody | Biolegend | Cat#637310 |
| Chemicals, peptides, and recombinant proteins | ||
| In-Fusion Snap Assembly Master Mix with Competent Cells | Takara | Cat#638951 |
| N-Acetyl-L-cysteine (NAC) | Sigma-Aldrich | Cat#A9165 |
| Human AB Serum | Valley Biomedical Inc. | Cat#HP1022 |
| Recombinant human IL-2 protein | NCI BRB Preclinical Repository | https://ncifrederick.cancer.gov/research/brb/ |
| Mirus Trans-IT Lenti Transfection Reagent | Mirus | Cat#MIR6606 |
| ddPCR Supermix for Probes (No dUTP) | Bio-rad | Cat#1863024 |
| Critical commercial assays | ||
| RosetteSep Human CD4+ T Cell Enrichment Cocktail | STEMCELL Technologies | Cat#15062 |
| RosetteSep Human CD8+ T Cell Enrichment Cocktail | STEMCELL Technologies | Cat#15063 |
| IL-2 Human Instant ELISA Kit | Thermo Fisher | Cat#BMS221INST |
| Experimental models: Cell lines | ||
| LentiX 293T | Clontech | Cat#11131D |
| K562 myelogenous leukemia cells | ATCC | Cat#CCL-243 |
| E6-1 Jurkat T cells | ATCC | Cat#T1B-152 |
| K562 CD19 (CD19 extracellular domain_myctag_hPDGFRtransmembrane) | Roybal et al., 2016 | N/A |
| K562 GFP | Roybal et al., 2016 | N/A |
| K562 ALPPL2 | Hyrenius-Wittsten et al, 2021 | N/A |
| K562 TRE ALPPL2 | Hyrenius-Wittsten et al, 2021 | N/A |
| MDA-MB-468 | ATCC | Cat#HTB-132 |
| U87 MG | ATCC | Cat#HTB-14 |
| U87EGFRviii | Choe et al., 2021 | N/A |
| M28MESO | Hyrenius-Wittsten et al, 2021 | N/A |
| M28MESO/ALPPL2KO | Hyrenius-Wittsten et al, 2021 | N/A |
| SK-OV-3 | UCSF Cell and Genome Engineering Core | Cat#CCLZR377 |
| SK-OV-3ALPPL2 | Hyrenius-Wittsten et al, 2021 | N/A |
| Experimental models: Organisms/strains | ||
| NOD scid IL-2Rγ−/− (NSG) (8–12 weeks) | Jackson Laboratories | Cat#005557 |
| Oligonucleotides | ||
| hTERT Probe: 5′-(SUN)-TCAGGACGTCGAGTGGACACGGTG-(ZEN/IBFQ)-3′ | IDT | N/A |
| HIV-1 Ψ Probe 5′-(FAM)-ATCTCTCTCCTTCTAGCCTC-(ZEN/IBFQ)-3′ | IDT | N/A |
| hTERT Forward Primer: 5′-GGCACACGTGGCTTTTCG-3′ | IDT | N/A |
| hTERT Reverse Primer: 5′-GGTGAACCTCGTAAGTTTATGCAA-3′ | IDT | N/A |
| HIV-1 Ψ Forward Primer: 5′-TACTGACGCTCTCGCACC-3′ | IDT | N/A |
| HIV-1 Ψ Reverse Primer: 5′-TCTCGACGCAGGACTCG-3′ | IDT | N/A |
| Recombinant DNA | ||
| pHR_SFFV | Addgene | ID#79121 |
| pHR_PGK | Addgene | ID#79120 |
| pHR_Gal4UAS_PGK_mCherry | Addgene | ID#79124 |
| pHR_Gal4UAS_tBFP_PGK_mCherry | Addgene | ID#79130 |
| Software and algorithms | ||
| Prism Version 9 | Graphpad | N/A |
| FlowJo V10.7.1 | FlowJo LLC | N/A |
| Jalview | Jalview | N/A |
| Incucyte Software v2018B | Sartorius | N/A |
| QuantaSoft | Bio-Rad | N/A |
HIGHLIGHTS.
SyNthetic Intermembrane Proteolysis Receptors exhibit modularity
SNIPRs can be rationally optimized for sensitivity and strength of gene regulation
SNIPRs are compatible with a range of human and programmable transcription factors
Humanized SNIPR→CAR-T cells exhibit potent and precise anti-tumor activity in vivo
ACKNOWLEDGEMENTS
I.Z. is funded by an F30 fellowship from the National Cancer Institute (5F30CA250247-02). R.L. was supported by a T32 training grant (5T32AI07334-29). K.T.R. acknowledges funding from the Parker Institute for Cancer Immunotherapy and an NIH Director’s New Innovator Award (DP2 CA239143). This work was also supported by NIH grant R01EB029483 (K.T.R., A.S.K.). A.S.K. acknowledges funding from a DARPA Young Faculty Award (D16AP00142), NIH Director’s New Innovator Award (1DP2AI131083), and DoD Vannevar Bush Faculty Fellowship (N00014-20-1-2825). We acknowledge the PFCC supported in part by Grant NIH P30 DK063720 and by the NIH S10 Instrumentation Grant S10 1S10OD021822-01.
Footnotes
DECLARATION OF INTERESTS
I.Z., R.L., and K.T.R. are co-inventors on patents for synthetic receptors (PRV 62/905,258, 62/905,262, 62/905,266, 62/905,268, 62/905,251, 62/905,263). R.L. and K.T.R. are co-inventors on patents for synthetic receptors PRV 62/007,807. R.L., I.Z., D.P., D.V.I., A.S.K. and K.T.R. are co-inventors for synthetic receptors PRV 63/007,795. K.T.R. is a cofounder of Arsenal Biosciences, consultant, SAB member, and stockholder. K.T.R. is an inventor on patents for synthetic Notch receptors (WO2016138034A1, PRV/2016/62/333,106) and receives licensing fees and royalties. The patents were licensed by Cell Design Labs and are now part of Gilead. He was a founding scientist/consultant and stockholder in Cell Design Labs, now a Gilead Company. K.T.R. holds stock in Gilead. K.T.R. is on the SAB of Ziopharm Oncology and an Advisor to Venrock. A.S.K. is a scientific advisor for and holds equity in Senti Biosciences and Chroma Medicine and is a co-founder of Fynch Biosciences and K2 Biotechnologies. B.L. is an inventor on patents (WO2017095823A1 and US20180369409A1) held by University of California that cover ALPPL2-targeted anticancer therapy and ALPPL2-targeting antibodies. Unrelated to this work, B.L. is a founder and stockholder of Fortis Therapeutics and Vivace Therapeutics, and a consultant for Merck Sharpe & Dohme.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Barnea G, Strapps W, Herrada G, Berman Y, Ong J, Kloss B, Axel R, and Lee KJ (2008). The genetic design of signaling cascades to record receptor activation. Proc Natl Acad Sci U S A 105, 64–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beel AJ, and Sanders CR (2008). Substrate specificity of gamma-secretase and other intramembrane proteases. Cell Mol Life Sci 65, 1311–1334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, and Israël A (2000). A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell 5, 207–216. [DOI] [PubMed] [Google Scholar]
- Carter P, Presta L, Gorman CM, Ridgway JB, Henner D, Wong WL, Rowland AM, Kotts C, Carver ME, and Shepard HM (1992). Humanization of an anti-p185HER2 antibody for human cancer therapy. Proceedings of the National Academy of Sciences 89, 4285–4289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choe JH, Watchmaker PB, Simic MS, Gilbert RD, Li AW, Krasnow NA, Downey KM, Yu W, Carrera DA, Celli A, et al. (2021). SynNotch-CAR T cells overcome challenges of specificity, heterogeneity, and persistence in treating glioblastoma. Sci. Transl. Med. 13, eabe7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coleman HA, Labrador J-P, Chance RK, and Bashaw GJ (2010). The Adam family metalloprotease Kuzbanian regulates the cleavage of the roundabout receptor to control axon repulsion at the midline. Development 137, 2417–2426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daringer NM, Dudek RM, Schwarz KA, and Leonard JN (2014). Modular Extracellular Sensor Architecture for Engineering Mammalian Cell-based Devices. ACS Synth. Biol. 3, 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, et al. (1999). A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain. Nature 398, 518–522. [DOI] [PubMed] [Google Scholar]
- Ellis GI, Sheppard NC, and Riley JL (2021). Genetic engineering of T cells for immunotherapy. Nature Reviews Genetics 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eyquem J, Mansilla-Soto J, Giavridis T, van der Stegen SJC, Hamieh M, Cunanan KM, Odak A, Gönen M, and Sadelain M (2017). Targeting a CAR to the TRAC locus with CRISPR/Cas9 enhances tumour rejection. Nature 543, 113–117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fridy PC, Li Y, Keegan S, Thompson MK, Nudelman I, Scheid JF, Oeffinger M, Nussenzweig MC, Fenyö D, Chait BT, et al. (2014). A robust pipeline for rapid production of versatile nanobody repertoires. Nat Methods 11, 1253–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon WR, Roy M, Vardar-Ulu D, Garfinkel M, Mansour MR, Aster JC, and Blacklow SC (2009). Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood 113, 4381–4390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedan S, Calderon H, Posey AD, and Maus MV (2019). Engineering and Design of Chimeric Antigen Receptors. Molecular Therapy - Methods & Clinical Development 12, 145–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haapasalo A, and Kovacs DM (2011). The many substrates of presenilin/γ-secretase. J Alzheimers Dis 25, 3–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayward AN, Aird EJ, and Gordon WR (2019). A toolkit for studying cell surface shedding of diverse transmembrane receptors. ELife 8, e46983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hernandez-Lopez RA, Yu W, Cabral KA, Creasey OA, Lopez Pazmino M. del P., Tonai Y, De Guzman A, Mäkelä A, Saksela K, Gartner ZJ, et al. (2021). T cell circuits that sense antigen density with an ultrasensitive threshold. Science 371, 1166–1171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, and Kopan R (2000). Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature 405, 966–970. [DOI] [PubMed] [Google Scholar]
- Hyrenius-Wittsten A, Su Y, Park M, Garcia JM, Alavi J, Perry N, Montgomery G, Liu B, and Roybal KT (2021). SynNotch CAR circuits enhance solid tumor recognition and promote persistent antitumor activity in mouse models. Sci. Transl. Med. 13, eabd8836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Israni DV, Li H-S, Gagnon KA, Sander JD, Roybal KT, Joung JK, Wong WW, and Khalil AS (2021). Clinically-driven design of synthetic gene regulatory programs in human cells (Synthetic Biology). [Google Scholar]
- Kandachar V, and Roegiers F (2012). Endocytosis and control of Notch signaling. Current Opinion in Cell Biology 24, 534–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klebanoff CA, and Restifo NP (2016). Customizing Functionality and Payload Delivery for Receptor-Engineered T Cells. Cell 167, 304–306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopan R, and Ilagan MXG (2009). The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137, 216–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroeze WK, Sassano MF, Huang X-P, Lansu K, McCorvy JD, Giguère PM, Sciaky N, and Roth BL (2015). PRESTO-Tango as an open-source resource for interrogation of the druggable human GPCRome. Nat Struct Mol Biol 22, 362–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lambrecht BN, Vanderkerken M, and Hammad H (2018). The emerging role of ADAM metalloproteinases in immunity. Nat Rev Immunol 18, 745–758. [DOI] [PubMed] [Google Scholar]
- Lasek W, Zagożdżon R, and Jakobisiak M (2014). Interleukin 12: still a promising candidate for tumor immunotherapy? Cancer Immunol Immunother 63, 419–435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Borgne R (2005). The roles of receptor and ligand endocytosis in regulating Notch signaling. Development 132, 1751–1762. [DOI] [PubMed] [Google Scholar]
- Levin AM, Bates DL, Ring AM, Krieg C, Lin JT, Su L, Moraga I, Raeber ME, Bowman GR, Novick P, et al. (2012). Exploiting a natural conformational switch to engineer an interleukin-2 “superkine.” Nature 484, 529–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, Hartmann D, Black RA, Rossi JJ, Blobel CP, et al. (2007). Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J 26, 494–504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majzner RG, and Mackall CL (2018). Tumor Antigen Escape from CAR T-cell Therapy. Cancer Discov 8, 1219–1226. [DOI] [PubMed] [Google Scholar]
- Moretti J, and Brou C (2013). Ubiquitinations in the Notch Signaling Pathway. IJMS 14, 6359–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Yang JC, Kitano M, Dudley ME, Laurencot CM, and Rosenberg SA (2010). Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther 18, 843–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Johnson LA, Davis JL, Zheng Z, Woolard KD, Reap EA, Feldman SA, Chinnasamy N, Kuan C-T, Song H, et al. (2012). Recognition of Glioma Stem Cells by Genetically Modified T Cells Targeting EGFRvIII and Development of Adoptive Cell Therapy for Glioma. Human Gene Therapy 23, 1043–1053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morsut L, Roybal KT, Xiong X, Gordley RM, Coyle SM, Thomson M, and Lim WA (2016). Engineering Customized Cell Sensing and Response Behaviors Using Synthetic Notch Receptors. Cell 164, 780–791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, and Kopan R (2000). A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell 5, 197–206. [DOI] [PubMed] [Google Scholar]
- Okochi M (2002). Presenilins mediate a dual intramembranous gamma-secretase cleavage of Notch-1. The EMBO Journal 21, 5408–5416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pachella LA, Madsen LT, and Dains JE (2015). The Toxicity and Benefit of Various Dosing Strategies for Interleukin-2 in Metastatic Melanoma and Renal Cell Carcinoma. J Adv Pract Oncol 6, 212–221. [PMC free article] [PubMed] [Google Scholar]
- Porter DL, Levine BL, Kalos M, Bagg A, and June CH (2011). Chimeric antigen receptor-modified T cells in chronic lymphoid leukemia. N Engl J Med 365, 725–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roscilli G, Rinaudo CD, Cimino M, Sporeno E, Lamartina S, Ciliberto G, and Toniatti C (2002). Long-term and tight control of gene expression in mouse skeletal muscle by a new hybrid human transcription factor. Mol Ther 6, 653–663. [PubMed] [Google Scholar]
- Roybal KT, Williams JZ, Morsut L, Rupp LJ, Kolinko I, Choe JH, Walker WJ, McNally KA, and Lim WA (2016a). Engineering T Cells with Customized Therapeutic Response Programs Using Synthetic Notch Receptors. Cell 167, 419–432.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roybal KT, Rupp LJ, Morsut L, Walker WJ, McNally KA, Park JS, and Lim WA (2016b). Precision Tumor Recognition by T Cells With Combinatorial Antigen-Sensing Circuits. Cell 164, 770–779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seki M, Watanabe A, Enomoto S, Kawamura T, Ito H, Kodama T, Hamakubo T, and Aburatani H (2010). Human ROBO1 is cleaved by metalloproteinases and gamma-secretase and migrates to the nucleus in cancer cells. FEBS Lett 584, 2909–2915. [DOI] [PubMed] [Google Scholar]
- Srivastava S, Salter AI, Liggitt D, Yechan-Gunja S, Sarvothama M, Cooper K, Smythe KS, Dudakov JA, Pierce RH, Rader C, et al. (2019). Logic-Gated ROR1 Chimeric Antigen Receptor Expression Rescues T Cell-Mediated Toxicity to Normal Tissues and Enables Selective Tumor Targeting. Cancer Cell 35, 489–503.e8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toda S, Blauch LR, Tang SKY, Morsut L, and Lim WA (2018). Programming self-organizing multicellular structures with synthetic cell-cell signaling. Science eaat0271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Fagerberg L, Hallstrom BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson A, Kampf C, Sjostedt E, Asplund A, et al. (2015). Tissue-based map of the human proteome. Science 347, 1260419–1260419. [DOI] [PubMed] [Google Scholar]
- Vooijs M, Schroeter EH, Pan Y, Blandford M, and Kopan R (2004). Ectodomain shedding and intramembrane cleavage of mammalian Notch proteins is not regulated through oligomerization. J Biol Chem 279, 50864–50873. [DOI] [PubMed] [Google Scholar]
- Wang Y, Bergelson S, and Feschenko M (2018). Determination of Lentiviral Infectious Titer by a Novel Droplet Digital PCR Method. Human Gene Therapy Methods 29, 96–103. [DOI] [PubMed] [Google Scholar]
- Weber EW, Parker KR, Sotillo E, Lynn RC, Anbunathan H, Lattin J, Good Z, Belk JA, Daniel B, Klysz D, et al. (2021). Transient rest restores functionality in exhausted CAR-T cells through epigenetic remodeling. Science 372, eaba1786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams JZ, Allen GM, Shah D, Sterin IS, Kim KH, Garcia VP, Shavey GE, Yu W, Puig-Saus C, Tsoi J, et al. (2020). Precise T cell recognition programs designed by transcriptionally linking multiple receptors. Science 370, 1099–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu HE, Rould MA, Xu W, Epstein JA, Maas RL, and Pabo CO (1999). Crystal structure of the human Pax6 paired domain-DNA complex reveals specific roles for the linker region and carboxy-terminal subdomain in DNA binding. Genes Dev 13, 1263–1275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto S, Charng W-L, and Bellen HJ (2010). Endocytosis and Intracellular Trafficking of Notch and Its Ligands. In Current Topics in Developmental Biology, (Elsevier; ), pp. 165–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure 1. Related to Figure 1. Construct designs, expression of Robo/FnIII receptors, and drug inhibition studies. (A) Schematic of SNIPRs and reporter constructs. All SNIPRs tested are expressed under a constitutive pGK promoter and contain an N-terminal CD8α signal peptide for membrane trafficking and myc-tag for measuring expression. Expression of tagBFP is regulated by a 5x Gal4 UAS response element and a minimal pybTATA promoter. For identifying reporter+ cells, mCitrine is expressed constitutively under a pGK promoter. Right, receptor expression of murine and human synNotch receptors. (B) The original synNotch receptor is composed of a ligand-binding domain (LBD) fused to a proteolysis-regulated core from murine Notch1 and a Gal4-VP64 transcriptional unit. T cells expressing a CD19 synNotch receptor, constructed with either a murine or human Notch1 core, were tested with various transcriptional factors for their ability to transmit ligand-dependent signaling. SynNotch T cells were co-incubated with either K562 or K562CD19 sender cells for 24 hours and BFP output was measured using flow cytometry proteins. Compared to the original murine synNotch design, synNotch receptors utilizing human components fail to efficiently induce BFP. (C) SynNotch receptors using cores from human Notch 1, Notch 2, and Notch 3 do not display strong ligand-dependent activation. Human synNotch 4 did not express. (D) Representative expression MFIs for synNotch/synRobo. Expression MFI does not correlate with receptor signaling ability. (E) Receptor activity is dependent on ADAM/γ-secretase activity. SNIPR activation is inhibited by the ADAM inhibitor GI254023X and the gamma secretase inhibitor DAPT.
Supplemental Figure 2. Related to Figure 2. Protease site regulation, reporter constructs with CAR, hinge engineering. (A) Protease site exposure can regulate SNIPR signaling. Addition of a fibroblast activation protein (FAP) protease site into a synthetic linker ECD confers SNIPR activity in the presence of K562 sender cells. Addition of a ligand binding domain restores ligand specificity. (B) Diagram of reporter construct with constitutively co-expressed 2nd generation ALPPL2 CAR (41BB-zeta). (C) Behavior of ΔNRR variants of human Notch1–4. Notch1 ΔNRR demonstrates sensitivity to T cell activation through activation of a co-expressed anti-ALPPL2 CAR. Notch2 ΔNRR demonstrates constitutive signaling. Notch3 ΔNRR demonstrates significant ligand-independent signaling. Notch4 ΔNRR demonstrates weak ligand-dependent signaling but no sensitivity to T cell activation. (D) Activation of anti-CD19 CD8α Hinge SNIPR variants. 1st row: expression of SNIPR and reporter construct in CD8+ T cells. 2nd row: Activation of the CD8α Hinge SNIPR variants with K562CD19. 3rd row: Activation of the CD8α Hinge SNIPR variants with K562CD19 in the presence of MCAM BiTEs. 4th row: Activation of the CD8α Hinge SNIPR variants with K562CD19 in the presence of a co-expressed 2nd generation ALPPL2 CAR. Superimposed bar graphs displaying ligand-independent (K562) and ligand-dependent (K562CD19) activation are shown. (E) Same as C, but with the full length CD8α Hinge Notch1 expressed in CD4+ T cells. The CD8α-based SNIPR is not as sensitive to T cell activation in CD4+ T cells as in CD8+ cells. (F) Same as B, but with CD8α Hinge SNIPR variants.
Supplemental Figure 3. Related to Figure 3. Alanine scan, fixing non-functional TMDs, non-Notch receptors. (A) Expression of Notch1 TMD Alanine scan mutants. (B) Results from Alanine scan in terms of %BFP+. (C) Fixing a non-functional TMD through residue substitution. The performance of the Robo1 TMD in a SNIPR setting can be improved by inserting a Gly-Val motif at an equivalent position to the Notch1 TMD and replacing the Robo1 JMD with that of Notch1. The performance of the AGER TMD in a SNIPR setting can be improved by replacing the residues C-terminal to its Gly-Val motif with those from the highest performing TMD, Notch1 from G. gallus. T cells expressing a SNIPR-BFP circuit were co-incubated with the indicated K562 cells for 48 hours and the percentage of BFP+ cells was measured using flow cytometry. (D) Human synNotch expression and activation improvement through JMD substitution identified by a screen. Replacing the human Notch 1 JMD with the Notch 2 JMD increases receptor expression and activation in primary T cells. (E) Activation of anti-CD19 SNIPRs with non-Notch components. 1st row: expression of SNIPR and reporter construct in CD8+ T cells. 2nd row: Activation of the SNIPR variants with K562CD19. 3rd row: Activation of the CD8α Hinge SNIPR variants in the presence of MCAM BiTEs. 4th row: Activation of the CD8α Hinge SNIPR variants in the presence of a co-expressed 2nd generation anti-ALPPL2 CAR. Superimposed bars displaying ligand-independent (K562) and ligand-dependent (K562CD19) activation are shown.
Supplemental Figure 4. Related to Figure 4. Hinge SNIPR TMD and JMD, drug inhibition studies, construct designs, expression data, testing with additional LBDs. (A) Superimposed bar graphs displaying activation of Hinge SNIPRs with variable TMDs and JMDs with an activated co-expressed ALPPL2 CAR at 48 hours. (B) SNIPR activation is dependent on ADAM and gamma-secretase activity. T cells expressing a CD19 Hinge Notch SNIPR-BFP circuit and co-expressed ALPPL2 CAR were co-incubated with the indicated conditions for 48 hours. BFP output was measured using flow cytometry. (C) Design of inducible ALPPL2 cassette and MCAM CAR response element. rtTA3 is expressed under an SFFV promoter and induces ALPPL2 expression in the presence of doxycycline. MCAM CAR is expressed under an inducible minimal TATA promoter and 5XGal4 UAS enhancer with a constitutively expressed mCherry. (D) Expression of ALPPL2 SNIPR was measured using myc-staining. (E) Design of an induced Super IL-2 single vector. Super IL-2 is expressed under an inducible minimal TATA promoter and 5XGal4 UAS enhancer with a constitutively expressed SNIPR. (F) Expression of SNIPR-super IL-2 circuits were measured using myc-staining. (G) Activation of Hinge SNIPR with additional LBDs. Hinge SNIPR with Notch2 JMD is effective against membrane bound GFP, EGFRviii, and HER2 when expressing an anti-GFP nanobody, anti-EGFRviii scFv, or anti-HER2 scFv, respectively.
Supplemental Figure 5. Related to Figure 5. Constructs, expression of humanized and non-humanized receptors, and individual tumor growth curves. (A) Comparison of humanized TFs with SNIPR ECDs. Expression profile of optimized CD8α Hinge SNIPRs and synNotch receptors using the human transcription factor HNF1A and synTF ZF10 was determined by surface staining of the myc-tagged receptors. (B) Design of BCMA CAR, CD19 SNIPR, and induced BCMA CAR response vector. BCMA CAR is expressed under a constitutive SFFV promoter. CD19 SNIPR is expressed under a constitutive pGK promoter. BCMA CAR is expressed under an inducible minimal TATA promoter and 5XGal4 UAS enhancer with a constitutively expressed mCherry. (C) Receptor expression of optimized CD8α Hinge SNIPRs in CD4+ and CD8+ T cells used for in vivo testing of circuit function. (D) Vector copy number (VCN) measurements of SNIPR circuits in both single-vector and dual-vector format. Genomic DNA was isolated from SNIPR T cells and the number of copies of HIV and a TERT2 reference gene was assayed using a duplex ddPCR reaction done in triplicate. The VCN was calculated using the formula 2 * (#HIV copies) / (#TERT2 copies).
(E) Target cell killing by a fully humanized SNIPR circuit. T cells expressing a SNIPR-CAR circuit were co-incubated with K562 target cells for 72 hours. Circuit induction was measured by FLAG-tag staining for the BCMA-CAR and flow cytometry (left panel). Target cells were cleared by 48 hours, as measured by DRAQ7 staining and flow cytometry (right panel).
(F) Differential gene analysis of transcriptomes from fully HNF1A SNIPR Circuits following induction, versus control cells. Analysis was performed with two different human T cell donors, and genes with greater than two-fold difference and p-value < 0.01 are listed here.
Supplemental Figure 6. Additional in vivo data. Related to Figure 6. (A) NSG mice (5 per experimental group) were injected subcutaneously with 1×106 K562CD19/BCMA target cells into the left flank and 1×106 K562BCMA control cells into the right flank. 4 days post tumor injection, 6×106 untransduced, BCMA CAR, or CD19 SNIPR-BCMA CAR circuit T cells (3×106 each CD4+ and CD8+) were injected via tail vein, and tumor volume was measured by caliper every few days. Statistics were calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc comparing untransduced T cells to BCMA CAR (top, *) and Circuit T cells (top, **) on Day 21, and comparing BCMA CAR T to circuit T cells (bottom). *P ≤ 0.05, **P ≤ 0.01. (B) Individual tumor growth curves for data shown in (A). (C) NSG mice were injected with 1×106 K562CD19+/BCMA+ target cells into the left flank. 4 days post tumor injection, 5×106 BCMA CAR or CD19 SNIPR-BCMA CAR circuit T cells (2.5×106 each CD4+ and CD8+) or PBS control were injected, and tumor volume was measured over time. (D) Survival curve for mice in C. (E) Same as C, but with NOD mice injected with 1×106 K562CD19+/BCMA+ target cells into the left flank and 1×106 K562BCMA target cells into the right flank. (F) Human T cell presence in the spleen, left, and right tumors was measured using flow cytometry. (G, H) T cell abundance (G) and CAR induction (H) of human T cells in the spleen, left, and right tumors was measured using a t2a system. Statistics were calculated using one-way ANOVA with Dunnett’s test post hoc. *P ≤ 0.05; **P ≤ 0.01; ns, not significant. (I) NSG mice were injected subcutaneously with 4×106 M28ALPPL2/MESO target cells into the left flank and 4×106 M28ALPPL2KO/MESO control cells into the right flank. 10 days post tumor injection, 3×106 of untransduced T cells, anti-MESO CAR-T cells, or anti-ALPPL2 SNIPR-anti-MESO CAR single vector circuit T cells (G4VP64) were injected retro-orbitally, and tumor volume was measured by caliper every few days. Statistics were calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc comparing untransduced T cells to anti-MESO CAR and Circuit T cells (top, **) and untransduced T cells to circuit T cells (bottom) ns = not significant, **P ≤ 0.01. (J) NSG mice were injected subcutaneously with 4×106 M28ALPPL2/MESO target cells into the left flank and 4×106 M28ALPPL2KO/MESO control cells into the right flank. 9 days post tumor injection, 3×106 of untransduced T cells, anti-MESO CAR-T cells, or anti-ALPPL2 SNIPR-anti-MESO CAR circuit T cells (HNF1A-p65) were injected retro-orbitally, and tumor volume was measured by caliper every few days. Statistics were calculated using one-way analysis of variance (ANOVA) with Dunnet’s test post hoc comparing untransduced T cells to anti-HER2 CAR and Circuit T cells (top) and anti-MESO CAR-T cells to circuit T cells (bottom). ****P ≤ 0.0001.
Supplemental Table S1. Sequences of screened TMDs and JMDs, related to Figure 3. TMDs and JMDs were sourced from known γ-secretase substrates (19), and Notch1 homologs. TMD sequences were extracted from Uniprot annotations and were extended to the first basic residue (R, K, or H). Two TMDs from proteins not believed to be γ-secretase substrates were selected for negative control. JMD sequences began immediately c-terminal to the TMD and ended immediately prior to 3 non-basic residues.
Supplemental Table S2. Amino acid sequences of highlighted SNIPR components, related to Figures 1–6, supplemental figures S1–S6.
Supplemental Table S3. LBD, ECD, TMD, JMD, and TFs of highlighted SNIPR designs, related to Figures 1–6, supplemental figures S1–S6.
Supplemental Table S4. Jurkat TMD screen expression and activation data, related to Figure 3.
Supplemental Table S5. Jurkat JMD screen expression and activation data, related to Figure 3.
Data Availability Statement
All raw data reported in this paper will be shared by the Lead Contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.