

Abstract

The nonclassical extracellular signal-related kinase 5 (ERK5) mitogen-activated protein kinase pathway has been implicated in increased cellular proliferation, migration, survival, and angiogenesis; hence, ERK5 inhibition may be an attractive approach for cancer treatment. However, the development of selective ERK5 inhibitors has been challenging. Previously, we described the development of a pyrrole carboxamide high-throughput screening hit into a selective, submicromolar inhibitor of ERK5 kinase activity. Improvement in the ERK5 potency was necessary for the identification of a tool ERK5 inhibitor for target validation studies. Herein, we describe the optimization of this series to identify nanomolar pyrrole carboxamide inhibitors of ERK5 incorporating a basic center, which suffered from poor oral bioavailability. Parallel optimization of potency and in vitro pharmacokinetic parameters led to the identification of a nonbasic pyrazole analogue with an optimal balance of ERK5 inhibition and oral exposure.
Introduction
Extracellular signal-regulated kinase 5 (ERK5) is a member of the mitogen-activated protein kinase (MAPK) family, which includes ERK1/2, JNK1/2/3, and p38. Activation of the nonclassical MEK5–ERK5 MAPK pathway is associated with increased cellular proliferation, migration, survival, and angiogenesis.1−4 In approximately 50% of hepatocellular carcinomas (HCCs), the MAPK7 gene encoding for ERK5 is amplified.5 ERK5 expression is also upregulated in breast and prostate cancers.6,7 Patients with high levels of ERK5 have a median disease-free survival time of 14 months compared with that of 34 months for patients with low expression.7 Elevated cytoplasmic and nuclear levels of ERK5 serve as independent prognostic markers for advanced prostate cancer, with nuclear ERK5 expression present only in malignant cells.6 Phosphorylated ERK5 associates with, phosphorylates, and activates a number of downstream transcription factors, such as the myocyte enhancer factor (MEF) family, c-Myc, RSK, c-Fos, c-Jun, and Sap1a,8 which are involved in the modulation of apoptosis. ERK5 has also been shown to play a role in cellular invasion and metastatic spread, affecting cell migration and attachment to the extracellular matrix.9 ERK5 activation has also been implicated as a potential resistance mechanism to therapeutics targeting the RAF–MEK1/2–ERK1/2 pathway.10 Selective ERK5 kinase inhibitors will therefore be useful in elucidating the role of this signaling protein in cancer and determining whether they represent potential therapeutics.
There has been significant interest in developing ERK5 inhibitors to interrogate its role in cancer.11,12 Oxindole BIX02189 (Figure 1; 1) was identified as a dual ERK5–MEK5 inhibitor.13 Subsequently, AX15836 (2), a potent and selective ERK5 inhibitor from the pyrimidodiazepinone series, was reported as a useful ERK5 probe14 and BAY-885 (3) was disclosed as a structurally differentiated inhibitor.15
Figure 1.

Published ERK5 inhibitors.
We have described the identification of pyrrole carboxamide-based ERK5 inhibitors (4a,b) with submicromolar potency, excellent kinase selectivity, and encouraging activity in a mouse tumor xenograft model.16 To identify a tool compound from this series, improvement in primary ERK5 inhibitory potency while maintaining the attractive pharmacokinetic properties and selectivity profile was required. The X-ray crystal structure of 4a bound to the adenosine triphosphate (ATP)-binding site of ERK5 (Figure 2) indicates that the ketone and amide carbonyl groups lie coplanar with the pyrrole ring, with the 2,6-disubstituted phenyl ring orthogonal to this plane, occupying a hydrophobic pocket. The pyridyl amide projects toward the solvent-exposed region of the binding pocket, and optimization of this substituent was investigated as a means to improve ERK5 inhibition.
Figure 2.
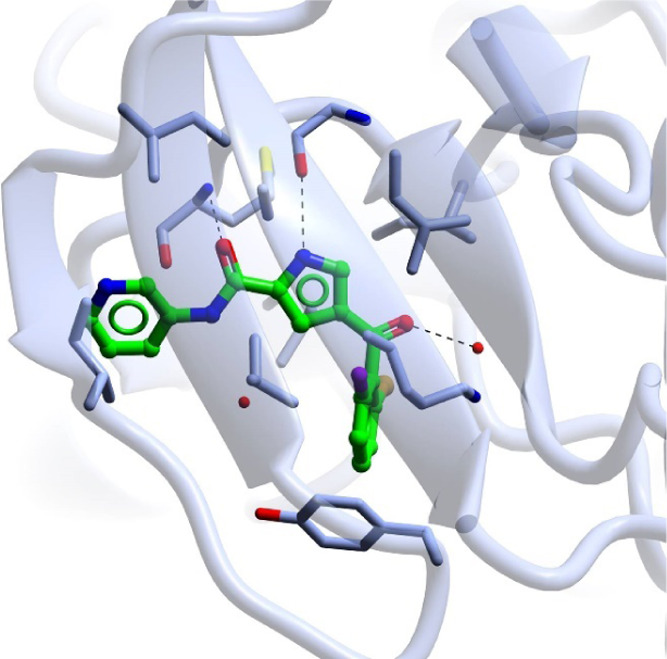
Crystal structure of the ERK5–4a complex determined at a 2.4 Å resolution (PDB ID: 5O7I). H-bonds are shown as dashed lines.
Results and Discussion
Chemistry
5-Pyridyl and 5-pyrimidylamines substituted at the 2-position with O or NH linkers (7a–f and 10a–c) were synthesized from 2-chloro-5-nitro-pyrimidine (5) or 2-chloro-5-nitropyridine (8), respectively, by nucleophilic aromatic substitution with an appropriate amine or alcohol, followed by palladium-catalyzed hydrogenation of the nitro group (Scheme 1). Methylene-linked piperidine 10d was prepared by in situ hydroboration of tert-butyl 4-methylidenepiperidine-1-carboxylate followed by palladium-catalyzed cross-coupling (Scheme 1; method 4).
Scheme 1. Synthesis of 2-Substituted Aminopyrimidines 7a–f and Aminopyridines 10a–d.
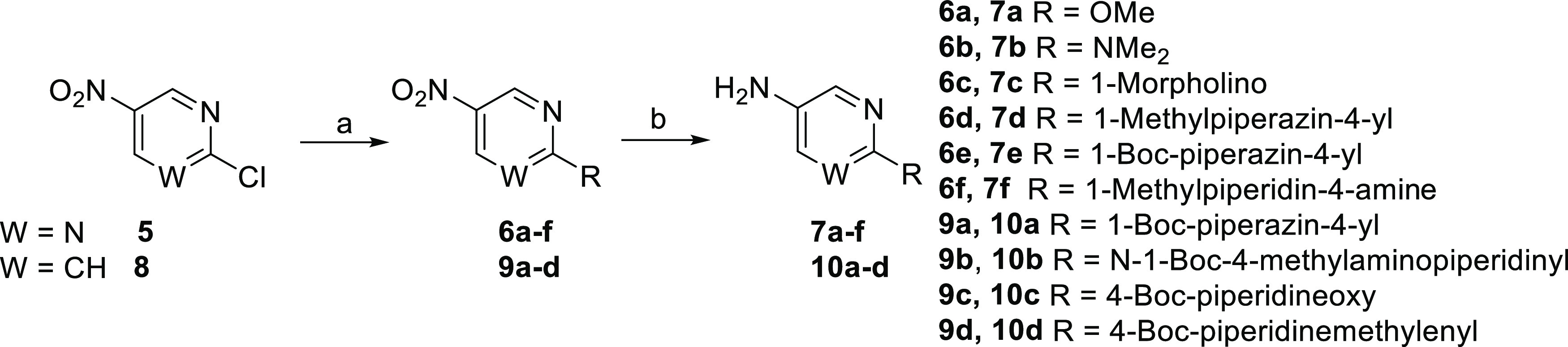
Reagents and conditions: (a) method 1: W = N, R = R1R2N: Et3N, R1R2NH, tetrahydrofuran (THF), room temperature (rt) 18 h, 68–87%; method 2: W = N, R = OMe: Na, MeOH, 65 °C, 1 h, 59%; method 3: W = CH, R = R1R2N: K2CO3, R1R2NH, THF, 80 °C, 0.5–3 h; 78–97%; method 4: (i) tert-butyl 4-methylidenepiperidine-1-carboxylate, 9-BBN (0.5 M in THF), 67 °C, 3 h; (ii) 5, K2CO3, PdCl2dppf, dimethylformamide (DMF)/H2O (10:1), 60 °C, 18 h, 40%; (b) H2, 10% Pd/C, MeOH, CH2Cl2, 92–100%.
A pyrimidine ring synthesis was employed in the synthesis of amine 17 from diethoxyacetonitrile 10 (Scheme 2). Protection of amine 13 as benzyl carbamate 14, hydrolysis of the diethyl acetal, and reductive amination with 1-Boc-piperazine gave 16, which was deprotected to give amine 17.
Scheme 2. Synthesis of tert-Butyl 4-((5-Aminopyrimidin-2-yl)methyl)piperazine-1-carboxylate 17.
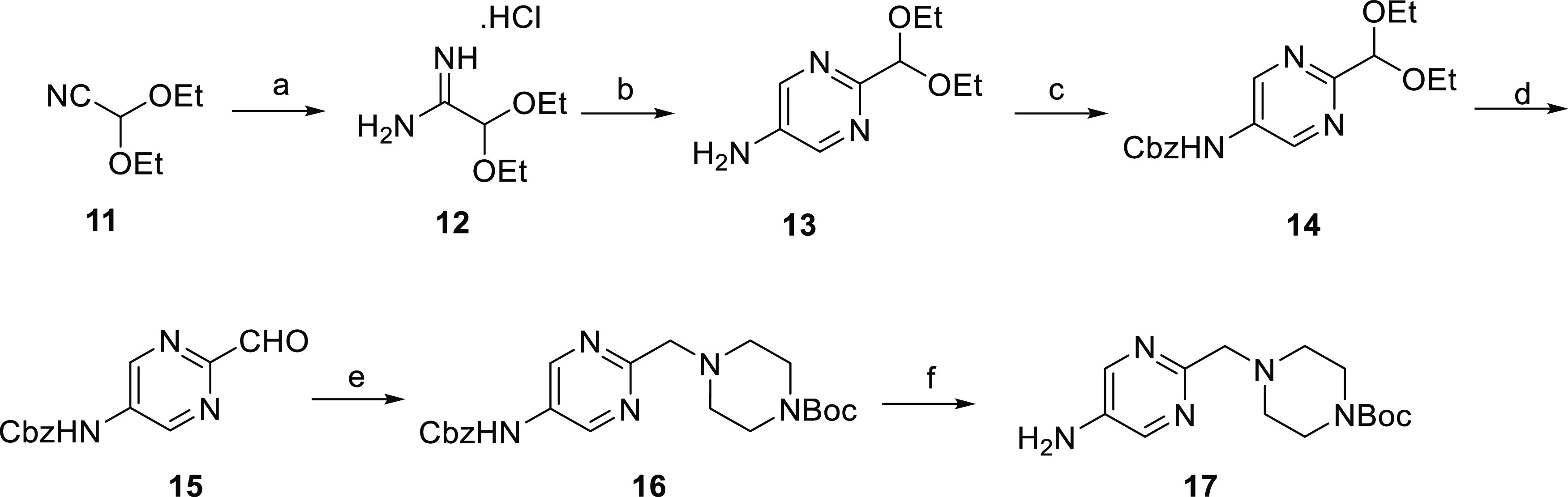
Reagents and conditions: (a) (i) NaOMe, MeOH, RT, 16 h; (ii) NH4Cl, MeOH, RT, 18 h, 89%; (b) (i) N-(3-(dimethylamino)-2-[[(dimethylamino)methylene]amino]prop-2-en-1-ylidene)-N-methylmethanaminium hydrogen dihexafluorophosphate, NaOMe (1 M in MeOH), EtOH, 78 °C, 2.5 h; (ii) 5% aq K2CO3, dioxane, 100 °C, 18 h, 49% (over two steps); (c) benzyl chloroformate, K2CO3, THF/H2O (1:1), RT, 24 h, 80%; (d) HCl (1 M aq), MeCN, RT, 8 h, 89%; (e) (i) tert-butyl piperazine-1-carboxylate, MgSO4, 2,2,2-trifluoroethanol, 1 h, RT; (ii) NaBH4, 2,2,2-trifluoroethanol, 0 °C to RT, 1 h, 42%; and (f) H2, 10% Pd/C, EtOAc, RT, 24 h, 99%.
For the synthesis of substituted 2-pyridylmethylpiperazine 24, the nucleophilic aromatic substitution of 8 with the sodium salt of diethyl malonate followed by double decarboxylation under acidic conditions gave 2-methyl-5-nitropyridine 19 (Scheme 3). N-Oxidation and subsequent rearrangement provided alcohol 21, which was converted to aldehyde 22. Reductive amination with 1-Boc-piperazine followed by reduction of the nitro group gave amine 24. Substituted pyrazolamines were synthesized by Mitsunobu alkylation of 4-nitropyrazole, followed by nitro reduction (Scheme 4).
Scheme 3. Synthesis of tert-Butyl 4-((5-Aminopyridin-2-yl)methyl)piperazine-1-carboxylate 24.
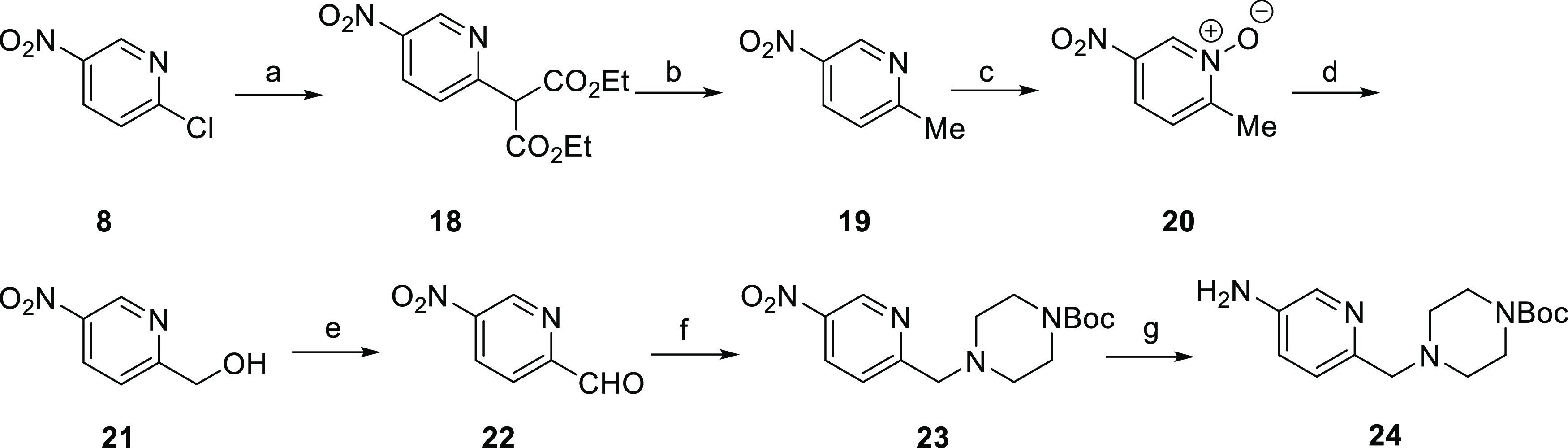
Reagents and conditions: (a) (i) NaH (60% dispersion in mineral oil), diethyl malonate, THF, 0 °C to RT, 1 h; (ii) 2-chloro-5-nitropyridine, 0 °C to RT, 20 h, 64%; (b) 20% aq H2SO4, 100 °C, 2 h, 95%; (c) m-CPBA (74%), dichloromethane (DCM), 0 °C to RT, 16 h, 96%; (d) (i) trifluoroacetic anhydride (TFAA), DCM, 0 °C to RT, 16 h; (ii) MeOH, 0 °C to RT, 8 h, 50%; (e) MnO2, DCM, RT, 16 h, 61%; (f) (i) tert-butyl piperazine-1-carboxylate, MgSO4, 2,2,2-trifluoroethanol, 1 h, 38 °C; (ii) NaBH4, 2,2,2-trifluoroethanol, 0 °C to RT, 1 h, 51%; and (g) H2, 10% Pd/C, MeOH/THF (1:1), 40 °C, 8 h, 95%.
Scheme 4. Synthesis of Substituted Aminopyrazoles 28a–e.
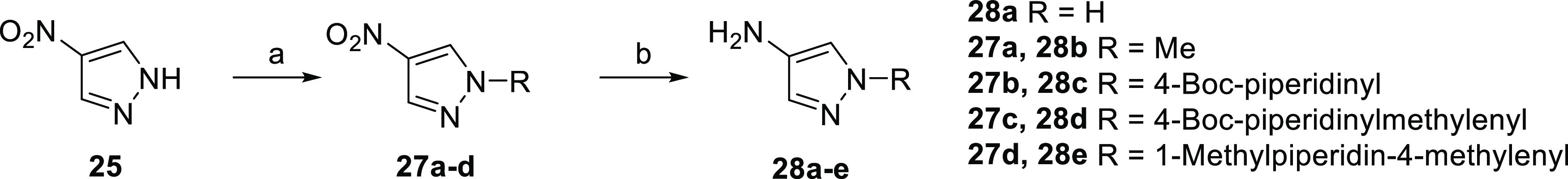
Reagents and conditions: (a) PPh3, diethyl azodicarboxylate (DEAD), THF, R-OH, rt 18 h, 34–82% and (b) H2, 10% Pd/C, MeOH, 95–100%.
Substituted 4-benzoyl-1H-pyrrole-2-carboxylic acids 31a and 31b were synthesized according to Scheme 5. Amines were coupled to the appropriate pyrrole carboxylic acid using cyanuric fluoride, PCl3, or 2-chloro-1-methylpyridinium iodide as activating agents to give targets 32a–k and 33a–f and 34a–i. N-Boc-protected amines were either deprotected under standard acidic conditions (DCM, trifluoroacetic acid, Et3SiH, RT, 2 h) or subjected to direct Eschweiler–Clarke N-methylation [formic acid, formaldehyde (37% wt in water), 95 °C, 3 h].
Scheme 5. Synthesis of Pyrrole Carboxamides 32a–m, 33a–k, and 34a–k.

Reagents and conditions: (a) ArCOCl, AlCl3, 0 °C to RT, 20 h, 89–92%; (b) LiOH, H2O/THF, 67 °C, 18 h, 95–99%; (c) method 1: amine, cyanuric fluoride, pyridine, MeCN, rt, 18 h, 34–76%; method 2: amine, PCl3, MeCN, 155 °C, 5 min, 24–79%; method 3: amine, PyBrOP, pyridine, MeCN, rt, 2 h. 39%; method 4: 2-chloro-1-methylpyridinium iodide, NEt3, DCM, rt, 18 h, 28–49%; (d) TFA, Et3SiH, DCM, rt, 2 h; and (e) HCO2H, HCHO, 100 °C, 3 h.
Replacement of the 3-pyridyl amide of 4a with a 4-pyrimidyl amide (32a) maintained ERK5 inhibitory activity, enabling rapid diversification at the 2-position of the pyrimidine ring (Table 1). Introduction of small alkyl and heteroalkyl substituents (32b–d) and morpholine (32e) at this position led to a reduction in ERK5 potency. However, N-methylpiperazine 32f exhibited a 6-fold increase in potency relative to 32a. The ERK5–4a crystal structure indicated the presence of a small hydrophobic void adjacent to the phenyl ketone, which was targeted through the introduction of a chloro group into the phenyl ketone and resulted in a further improvement in ERK5 inhibition (32g). NH-Piperazine 32k was also tolerated, and pyridinylpiperazine (33f) proved to be equipotent with its pyrimidinyl analogue. 32g was found to be rapidly metabolized in mouse liver microsomes and inhibited the hERG cardiac ion channel. NH-Piperazines 32k and 33f had superior in vitro metabolism and hERG inhibition profiles. In a caco-2 cell permeability assay, both 32k and 33a exhibited poor permeability and high efflux ratios.
Table 1. ERK5 Inhibitory Activity of 2-Substituted Pyrimidine Amides.


ERK5 IC50’s determined using an IMAP FP progressive binding system kit (Molecular Devices #R8127).
μL/min/mg.
Papp 10–6 cm·s–1; - = not determined.
Modeling of the binding pose of 33f was performed by manual ligand building from the crystal structure of the complex of ERK5 and 4a (PDB ID: 5O71). This suggested that the basic center of the piperazine analogues may interact with the acidic side chain of Glu59 in the mouth of the binding pocket (Figure 3). It was speculated that varying the position and basicity of the ionizable center may allow the optimization of potency and ADME properties.
Figure 3.
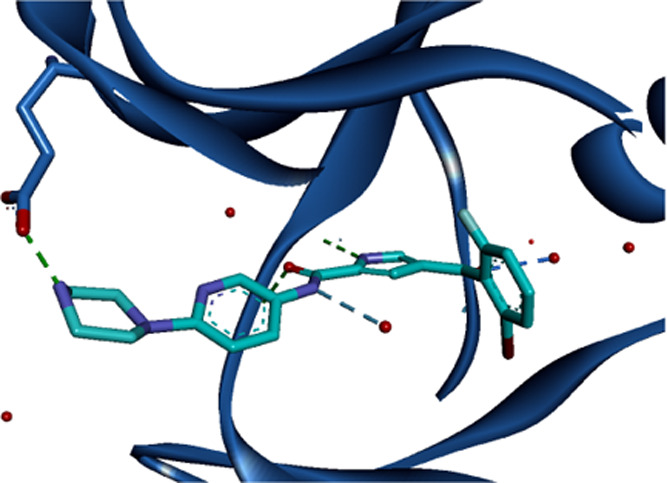
Modeled structure of 33f in the ATP-binding site of ERK5, demonstrating the close proximity of the piperazine basic center with the carboxylate group from the side chain of Glu59.
Structure–activity relationships for the position of the basic center were explored through the introduction of a spacer between the heteroaromatic and aliphatic rings of the amide substituent. Compounds incorporating amine, methylene, and ether linkers all retained low nanomolar ERK5 inhibition in both the pyrimidyl (32) and pyridyl (33) amide series, yielding potent ERK5 inhibitors with improved microsomal clearance (Table 2). Cell-based inhibition of ERK5 autophosphorylation was assessed in HeLa cells using Western blot densitometry of phospho-ERK, with all compounds exhibiting good cellular ERK5 inhibition. However, modulation of efflux pump recognition in the caco-2 permeability assay through variation of the position, linkage, and pKa of the basic center achieved limited success, with 33k having moderate flux and exhibiting an efflux ratio of less than 10.
Table 2. ERK5 Inhibitory Activity and In Vitro ADME Data for 32i–k and 33b–f.


ERK5 IC50’s determined using an IMAP FP progressive binding system kit (Molecular Devices #R8127).
IC50 determined by phospho-ERK5 Western blot densitometry in HeLa cells (1 h incubation with compounds).
μL/min/mg protein.
Papp 10–6 cm·s–1.
In a mouse PK study, compounds 33j and 33k had low clearance, with terminal elimination half-lives of 263 and 80 min, respectively (Table 3). Oral bioavailability was low consistent with the in vitro permeability data.
Table 3. In Vivo Mouse Pharmacokinetic Parameters for Selected Compoundsa.

Dose 10 mg/kg i.v. and p.o.
Five-membered heteroaromatic amides were explored as replacements for the pyridine and pyrimidine amides to determine whether efflux pump recognition could be reduced through subtle changes in size, geometry, and polarity of the amide group. Of the 5-membered heterocycles studied, only 4-pyrazole amides retained low nanomolar ERK5 inhibition (Table 4). NH-Pyrazole 34a was rapidly metabolized in mouse liver microsomes and again suffered from efflux in the caco-2 assay. However, N-methylpyrazole 34b had good permeability and low efflux and was also stable in mouse liver microsomes. Its isomer 34c and other similar 5-membered heterocyclic amides (34d, 34e) incorporating heteroatoms adjacent to the amide linker were significantly less potent.
Table 4. Structures and ERK5 Inhibitory Activity Data for Compounds 34a–k.

ERK5 IC50’s determined using an IMAP FP progressive binding system kit (Molecular Devices #R8127).
IC50 determined by phospho-ERK5 Western blot densitometry in HeLa cells (1 h incubation with compounds).
μL/min/mg protein.
Papp 10–6 cm·s–1.
The pyrazole amides offer an alternative attachment point for the incorporation of a basic center to target the proposed interaction with Glu59. Analogues 34h–k were prepared and provided comparable ERK5 inhibition to their pyrimidyl and pyridyl amide analogues. 34j had the lowest ERK5 IC50 in the binding assay and exhibited good mouse microsomal stability and a caco-2 efflux ratio < 5, a significant reduction in the efflux ratio compared to its pyridyl analogue 33j (ER = 28). However, intrinsic flux remained low. Inhibition of ERK5 autophosphorylation was assessed in HeLa cells. Translation of ERK5 inhibition from the cell-free to the cell-based assay was variable for compounds with poor membrane permeability, with basic pyrazoles 34h and 34j exhibiting 40- and 120-fold lower potencies in the cell-based assay compared to those in the binding assay. In contrast, the cell IC50 of the more membrane-permeable neutral pyrazole 34b was just 3-fold lower than in the binding assay.
Pyrazole amide 34b had low clearance and an oral bioavailability of 42% in the mouse (Table 5). Compound 34b was selective against the closely related MAP3K, p38 (IC50 > 30 μM). No inhibition or the closely related kinases ERK1 and ERK2 and JNKs 1, 2, and 3 were observed in a kinase panel screen, and 34b had an IC50 > 20 μM against BRD4, in contrast to some of the earlier reported ERK5 inhibitors.14 Examination of 394 nonmutant kinases in competition binding assays (DiscoverX KINOMEscan) revealed 34b to inhibit 38 kinases by ≥90% at a concentration of 10 μM (Supporting Information, Table S1). Kds were determined for 10 of these kinases using this assay platform (CSF1R Kd 46 nM, DCLK1 Kd 61 nM, MAPK7 Kd 180 nM, LRRK2 Kd 220 nM, AURKA Kd 290 nM, FGFR1 Kd 380 nM, KIT Kd 420 nM, ABL1 Kd 1.2 μM, JAK3 Kd 1.3 μM, and MEK5 Kd 2.8 μM). Thus, 34b represents a structurally distinct ERK5 inhibitor chemotype, which is a useful addition to the toolkit for interrogating ERK5 signaling. However, the selectivity data should be taken into consideration in biological studies, particularly in vivo where activity against CSF1R and FGFR1 activities could influence host responses (e.g., inflammation or angiogenesis).
Table 5. In Vivo Pharmacokinetic Parameters for 34ba.
| cmpd | Cl (mL/min/kg) | Vd (L/kg) | t1/2 (min) | F (%) |
|---|---|---|---|---|
| 34b | 14 | 0.6 | 80 | 42 |
In vivo studies were performed at a dose of 10 mg/kg i.v. and 10 mg/kg p.o. in mouse.
The structure of 34b bound to ERK5 was solved to a resolution of 2.75 Å, confirming that the binding mode was maintained, with the pyrrole carboxamide forming a bidentate interaction with the hinge region of the ATP-binding site (Figure 4a). The methylpyrrole amide lies in a small channel at the mouth of the binding pocket, lying between the side chain of E146 and the backbone of M140 and the lipophilic side chain on I61 (Figure 4b). The halogenated phenyl ring adopts a conformation orthogonal to the plane of the pyrrole ketone.
Figure 4.

Crystal structure of the ERK5–34b complex determined at 2.75 Å (PDB: 7PUS). (a) Hydrogen-bonding interactions of the pyrrole NH and amide carbonyl to the hinge region of ERK5. (b) Interaction of the pyrazole with the side chains of I61, E146, and the backbone of M140.
The permeability of the compounds within this series varied significantly, with caco-2 Papp values spanning 2 orders of magnitude. Permeability is usually considered to depend primarily on a combination of lipophilicity, molecular size, and hydrogen-bonding potential (or surrogates thereof).17−19 In this series, Papp correlated with molecular weight and hydrogen bond donor count but, interestingly, no relationship with clogP was apparent (Figure 5a–c). Multilinear regression analysis confirmed the significance of molecular weight and hydrogen bond donors and lack of dependence on clogP (p value 0.80 when included in the model; Figure 5d). A multilinear model including molecular weight and donor count alone was able to account for the majority of the variance (RMSE = 0.26). Most significantly, this modeling suggests that it is challenging to achieve a Papp value > 1 in this series with three hydrogen bond donors, highlighting the need to restrict designs to two donors.20 In this case, this effect is likely exacerbated by the presence of two very strong hydrogen-bonding groups (acyl pyrrole and aryl carboxamide) that cannot be readily internally satisfied. It is noteworthy that within this series, molecular weight and basicity are codependent, with the basic compounds also being larger due to the addition of the basic group. The large apparent dependence of permeability on molecular weight within this series may result from the combined effects of both the increased size and basicity as molecular weight increases.
Figure 5.

QSAR modeling of the caco-2 Papp (A to B) data. Correlation with (a) clogP, (b) MWt, (c) distributions against hydrogen bond donor count with paired t-test for significance (Tukey–Kramer method) showing mean diamonds (green) and box plots (red), and (d) multilinear regression model using molecular weight and hydrogen bond donor count. Points are colored by hydrogen bond donor count (green = 2, red = 3); red lines show the line of best fit (solid line) and 95% confidence limits for the fit (dotted curves).
We examined the activity of 34b in cellular assays to assess the impact on ERK5 kinase and transcriptional activity and proliferation. A recent study, examining the ERK5 kinase inhibitor AX15836 and two derivative compounds, indicated that while these inhibitors suppressed ERK5 kinase activity effectively in HEK293 cells, kinase inhibition also led to a paradoxical activation of ERK5 transcriptional activity by inducing a conformational change in the protein, resulting in the separation of the C-terminal transcriptional activation domain (TAD) from the nuclear localization sequence (NLS), to allow ERK5 nuclear translocation.21,22 To examine whether this paradoxical activation extended to another chemotype, we examined the effect of 34b using the same previously described ERK5:MEF2D luciferase reporter assay.21 When examining a truncated ERK5 construct that lacked both the NLS and TAD (ERK5 ΔTAD), 34b inhibited its kinase activity in cells with an IC50 of 77 ± 4 nM (mean ± SEM, n = 5) (Figure 6a). However, a greater than 13-fold reduction in activity was observed (i.e., IC50 > 1 μM) when 34b was examined against full-length ERK5, suggesting that this compound also induces a paradoxical activation of ERK5 transcriptional activity. The effect of compound treatment on cellular proliferation over a 72 h period was also examined. The concentration of compound 34b that prevented a 50% inhibition of HEK293 growth (GI50) was 19.6 ± 0.5 μM (mean ± SE) (Figure 6b), a value that is 65-fold greater than that required to inhibit the kinase activity of ERK5 ΔTAD in HEK293 cells by 89% (Figure 5a; 0.3 μM, 34b). Comparable GI50 values were obtained with 34b in the human renal cell carcinoma cell line A498 (22.3 ± 1.5 μM), the osteosarcoma cell line SJSA-1 (25.0 ± 0.8), and the breast cancer cell line MDA-MB-231 (26.6 ± 1.4 μM) (mean ± SE, three to five separate experiments). While the ERK5 kinase inhibitor XMD8-92 (5 μM) has been previously shown to inhibit the growth of MDA-MB-231 cells by nearly 40%,23 none of these three tumor cell lines demonstrate a dependency on ERK5 following siRNA gene silencing in publicly accessible data sets (Supporting Information, Figure S50; https://depmap.org/portal/).24 Collectively, these data suggest that the antiproliferative activity of 34b in cells at concentrations of 10 μM and above is unlikely to result from ERK5 kinase inhibition.
Figure 6.

Activity of 34b in HEK293 cellular assays. (a) Activity of 34b in an ERK5:MEF2D luciferase reporter assay examining ERK5 ΔTAD, a truncated form of ERK5 containing the kinase domain but lacking the C-terminal extension, or full-length ERK5 (mean ± SEM, n = 5 separate experiments); (b) growth inhibition following a 72 h incubation with compound 34b (mean ± SEM, n = 8 separate experiments).
Conclusions
Parallel optimization of potency and ADME properties has delivered a compound with balanced potency and oral exposure. Introduction of small lipophilic substituents at the 3-position of the benzoyl group of pyrrole carboxamide ERK5 inhibitors led to improved inhibition. Appending a basic center to the heteroaromatic amide substituent provided nanomolar inhibitors. However, the more potent basic analogues suffered from high efflux ratios in the caco-2 membrane permeability assay that translated to low oral bioavailability in vivo. Smaller, nonbasic analogue 34b provided the best balance of potency and in vitro ADME properties and had good oral bioavailability in mouse.
While 34b (10–300 nM) inhibited the kinase activity of ERK5 without a TAD in cells, its reduced activity against the full-length ERK5 protein suggested that it can also activate ERK5 transcriptional activity in a manner comparable to AX15836 and BAY-885.21,22 Given that this phenomenon has now been observed with three different chemotypes, it highlights a need to evaluate the effect of any new ERK5 kinase inhibitor on ERK5 transcriptional activity. The conformational activation of ERK5 transcriptional activity by compounds may potentially result in a disconnect between the chemical inhibition of ERK5 kinase and phenotypes observed using siRNA-mediated gene silencing or CRISPR/Cas9 gene editing of MAPK7. Nonetheless, such ERK5 kinase inhibitors could find additional utility as ligands for targeted protein degradation strategies that should more closely phenocopy the consequences of ERK5 protein loss following genetic perturbation.
Experimental Section
General Procedures
All commercial reagents were purchased from Sigma-Aldrich Chemical Company, Alfa Aesar, Apollo Scientific, or Tokyo Chemical Industry U.K. Ltd. The chemicals were of the highest available purity. Unless otherwise stated, chemicals were used as supplied without further purification. Anhydrous solvents were obtained from AcroSeal or Aldrich SureSeal bottles and were stored under nitrogen. Petrol refers to the fraction with a boiling point between 40 and 60 °C. Thin-layer chromatography utilized to monitor reaction progress was conducted on plates precoated with silica gel Merck 60F254 or Merck NH2F254S. The eluent was as stated (where this consisted of more than one solvent, the ratio is stated as volume/volume), and visualization was either by short wave (254 nm) ultraviolet light or by treatment with the visualization reagent stated followed by heating. “Flash” medium-pressure liquid chromatography (MPLC) was carried out either on a Biotage SP4 automated purification system or on a Varian 971-FP automated purification system using prepacked Varian or Grace silica or amino-bonded silica cartridges. All reactions carried out in a microwave were performed in a Biotage Initiator with 60 robots. Melting points were determined using a VWR Stuart SMP40 apparatus and are uncorrected. 1H, 13C, and 19F NMR spectra were obtained as either CDCl3, CD3OD, or DMSO-d6 solutions and recorded at 500, 126, and 471 MHz, respectively, on a Bruker Avance III 500 spectrometer. Where 13C NMR data are not quoted, insufficient material was available or problems obtaining high-resolution spectra were encountered. Chemical shifts are quoted in parts per million (δ) referenced to the appropriate deuterated solvent employed. Multiplicities are indicated by s (singlet), d (doublet), t (triplet), q (quartet), quin (quintet), m (multiplet), br (broad), or combinations thereof. Coupling constant values are given in Hz. Homonuclear and heteronuclear two-dimensional NMR experiments were used where appropriate to facilitate the assignment of chemical shifts. Liquid chromatography–mass spectrometry (LC–MS) was carried out on a Waters Acquity UPLC system with PDA and ELSD employing positive or negative electrospray modes as appropriate to the individual compound. High-resolution mass spectrometry was performed by the EPSRC U.K. National Mass Spectrometry Facility, University of Wales Swansea, Singleton Park, Swansea, SA2 8PP. FTIR spectra were recorded on either a Bio-Rad FTS 3000MX diamond ATR or an Agilent Cary 630 FTIR as a neat sample. UV spectra were obtained using a U-2001 Hitachi Spectrophotometer with the sample dissolved in ethanol. All compounds are >95% pure by HPLC.
General Procedure A
To a suspension of AlCl3 (2.5 equiv) in CH2Cl2 (1 mL/mmol AlCl3) at 0 °C was added the relevant acid chloride (2 equiv) followed by methyl-1H-pyrrole-2-carboxylate (1 equiv). The resulting mixture was allowed to reach RT and stirred for 16 h. The reaction was quenched at 0 °C with a 1 M hydrochloric acid (20 mL). The product was extracted with CH2Cl2 (3 × 100 mL) and washed with a saturated aqueous NaHCO3 (2 × 100 mL) and brine (100 mL). The combined organic layers were dried over Na2SO4 and concentrated in vacuo.
General Procedure B
To a solution of pyrrole ester (1.0 equiv) in THF (8 mL/mmol) was added LiOH (20 equiv) in water (13 mL/mmol). The resulting reaction mixture was heated at 60 °C for 18 h, cooled to RT, and acidified to pH 4–5 with 1 M hydrochloric acid. The product was extracted into EtOAc (100 mL/mmol), washed with water (100 mL/mmol) and brine (100 mL/mmol), and dried over Na2SO4. The solvent was removed in vacuo to obtain the product.
General Procedure C
The appropriate carboxylic acid (1.0 equiv) was dissolved in MeCN (5 mL/mmol pyrrole) before the relevant amine (2.5 equiv) was added followed by phosphorus trichloride (1.0 equiv). The mixture was heated using microwave irradiation at 150 °C for 5 min. The reaction was quenched with a few drops of water, and the solvent was removed in vacuo. The residue was dissolved in EtOAc (50 mL/mmol pyrrole) and washed with saturated aqueous NaHCO3 (50 mL/mmol pyrrole) before being extracted with EtOAc (3 × 30 mL/mmol pyrrole). The combined organic extracts were dried over Na2SO4, and the filtrate was concentrated in vacuo to afford the crude product.
General Procedure D
The relevant nitro compound (1 equiv) was dissolved in MeOH (5 mL/mmol) and hydrogenated on a Thales H-cube over a 10% Pd/C CatCart under a full pressure of hydrogen at 40 °C for 2 h with continuous recycling of the reaction mixture at 1 mL/min flow rate. The solvent was removed in vacuo.
General Procedure E
Cyanuric fluoride (0.7 equiv) was added to the relevant carboxylic acid (1 equiv) and pyridine (1 equiv) in MeCN (2 mL/mmol). The relevant amine (2.5 equiv) was added, and the mixture was stirred at RT for 18 h. The reaction was diluted with EtOAc, washed with water and 0.5 M hydrochloric acid, followed by further washes with saturated aqueous NaHCO3 and brine. The organic layer was dried over MgSO4, and the solvent was removed in vacuo.
General Procedure F
The relevant amine (1 equiv), 2-chloro-5-nitropyrimidine (1 equiv), and Et3N (1.1 equiv) were combined in THF (5 mL/mmol) at 0 °C, and the mixture was allowed to stir at RT for 1 h. The solvent was removed in vacuo, and the residue was partitioned between EtOAc (2 × 30 mL) and water (20 mL). The organic layer was washed with brine, dried over MgSO4, and the solvent was removed in vacuo.
General Procedure G
Diethyl azodicarboxylate (1.5 equiv) was added dropwise to a mixture of 4-nitropyrazole (1 equiv), triphenylphosphine (1.73 g, 6.63 mmol, 1.5 equiv), and the substrate alcohol (1 equiv) in THF at 0 °C. The mixture was stirred at 0 °C for 10 min and then allowed to stir at RT for 18 h. The reaction mixture was partitioned between EtOAc (2 × 30 mL) and water (20 mL), washed with brine (20 mL), dried over MgSO4, and the solvent was removed in vacuo.
General Procedure H
Formaldehyde (37% w/v aqueous, 4 equiv) was added to the substrate carbamate (1 equiv) in formic acid (10 mL/mmol), and the mixture was heated to 100 °C for 3 h in a sealed tube. The mixture was allowed to cool, basified with 10% aqueous K2CO3, and extracted with EtOAc (2 × 20 mL). The organic extracts were combined, washed with brine, dried over MgSO4, and the solvent was removed in vacuo.
General Procedure I
Pyrrole acid (1 equiv), Et3N (2.5 equiv), and 2-chloro-1-methylpyridinium iodide (1.1 equiv) were combined in CH2Cl2 (15 mL/mmol) and stirred at RT for 10 min, followed by the addition of the substrate amine (1.25 equiv) in CH2Cl2 (2.5 mL/mmol). The reaction was stirred at RT for 18 h, the solvent was evaporated, and the residue was partitioned between EtOAc (2 × 15 mL) and 10% aqueous K2CO3 (15 mL). The organic layers were combined, washed with brine, dried over MgSO4, and the solvent was removed in vacuo.
General Procedure J
TFA (2 mL/mmol) and Et3SiH (2.5 equiv) were added to the relevant carbamate (1 equiv) in CH2Cl2 (2 mL/mmol), and the mixture was stirred at RT for 2 h. The solvent was removed in vacuo, and the residue was partitioned between EtOAc (5 × 30 mL) and saturated aqueous NaHCO3 (40 mL). The organic extracts were combined, dried over MgSO4, and the solvent was removed in vacuo.
Methyl-4-(2-chloro-6-fluorobenzoyl)-1H-pyrrole-2-carboxylate (30a)
Prepared according to general procedure A, where AlCl3 (2.23 g, 16.8 mmol), CH2Cl2 (17 mL), 2-chloro-6-fluorobenzoyl chloride (1.80 mL, 13.3 mmol), and methyl-1H-pyrrole-2-carboxylate (847 mg, 6.70 mmol) were added. The crude mixture was purified by MPLC on SiO2 with a gradient elution from 0 to 100% EtOAc/petrol to give a white solid (1.74 g, 92%); Rf 0.50 (SiO2, 5% MeOH/CH2Cl2); mp 148–150 °C; λmax (EtOH)/nm 280, 233; IR νmax/cm–1 3226, 1731, 1638, 1604; 1H NMR (500 MHz; DMSO-d6) δH 3.83 (3H, s, OMe), 7.02–7.05 (1H, m, H-3), 7.42 (1H, app td, J = 8.2 and 0.8 Hz, H-5′), 7.49 (1H, d, J = 8.2 Hz, H-3′), 7.57 (1H, dd, J = 1.7 and 3.3 Hz, H-5), 7.61 (1H, td, J = 8.2 and 6.3 Hz, H-4′), 12.94 (1H, br s, NH); 13C NMR (125 MHz; DMSO-d6) δC 51.7 (OMe), 114.6 (CH-pyrrole), 115.0 (d, JCF = 21.8 Hz, C-5′), 124.5 (C-pyrrole), 125.4 (C-pyrrole), 125.9 (d, JCF = 3.2 Hz, C-3′), 127.7 (d, JCF = 23.2 Hz, C-1′), 130.1 (C-pyrrole), 130.3 (d, JCF = 6.4 Hz, C-2′), 131.9 (d, JCF = 9.1 Hz, C-4′), 158.5 (d, JCF = 246.9 Hz, C-6′), 160.3 (CO-NH), 183.7 (CO); 19F NMR (470 MHz; DMSO-d6) δF −114.4; HRMS calcd for C13H1035Cl1F1O3N1 [M + H]+ 282.0328, found 282.0333.
4-(2-Chloro-6-fluorobenzoyl)-1H-pyrrole-2-carboxylic Acid (31a)
Prepared according to general procedure B using LiOH (2.90 g, 121 mmol) in water (80 mL) and ester 30a (1.70 g, 6.05 mmol) in THF (48 mL) to give a white solid (1.60 g, 99%); Rf 0.15 (SiO2, 10% MeOH/CH2Cl2); mp 220–222 °C; λmax (EtOH)/nm 281, 232; IR νmax/cm–1 3313, 1637, 1553; 1H NMR (500 MHz; DMSO-d6) δH 6.98 (1H, br s, H-pyrrole), 7.42 (td, J = 8.2 and 0.6 Hz, H-5′), 7.47–7.51 (2H, m, H-3′ and H-pyrrole), 7.61 (1H, td, J = 8.2 and 6.3 Hz, H-4′), 12.75 (1H, br s, NH-pyrrole), 12.97 (1H, br s, CO2H); 13C NMR (125 MHz; DMSO-d6) δC 114.1 (CH-pyrrole), 114.9 (d, JCF = 21.3 Hz, C-5′), 125.3 (C-pyrrole), 125.8 (d, JCF = 3.2 Hz, C-3′), 125.9 (C-pyrrole), 127.8 (C-pyrrole), 127.9 (d, JCF = 23.2 Hz, C-1′), 129.7 (C-pyrrole), 130.3 (d, JCF = 6.0 Hz, C-2′), 131.8 (d, JCF = 9.1 Hz, C-4′), 158.5 (d, JCF = 247 Hz, C-6′), 161.3 (CO-NH), 183.7 (CO); 19F NMR (470 MHz; DMSO-d6) δF −114.4; HRMS calcd for C12H635Cl1F1N1O3 [M + H]+ 266.0026, found 266.0018.
Methyl-4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxylate (30b)
Prepared according to general procedure A, where AlCl3 (11.9 g, 89.6 mmol) in CH2Cl2 (100 mL), 3,6-dichloro-2-fluorobenzoyl chloride (16.3 mL, 71.7 mmol), and methyl-1H-pyrrole-2-carboxylate (4.48 g, 35.8 mmol) were added. The crude mixture was purified by MPLC on SiO2 with a gradient elution from 0 to 5% EtOAc/petrol to give a white solid (10.1 g, 89%); Rf 0.80 (SiO2, 50:50:0.5 EtOAc/petrol/AcOH); mp 136–138 °C; λmax (EtOH)/nm 282, 229; IR νmax/cm–1 3285, 3230, 1689, 1654; 1H NMR (500 MHz; DMSO-d6) δH 3.84 (3H, s, CH3), 7.12 (1H, app t, J = 1.5 Hz, H-pyrrole), 7.53 (1H, dd, J = 0.9 and 8.5 Hz, H-5′), 7.71 (1H, dd, J = 1.5 and 3.2 Hz, H-pyrrole), 7.80 (1H, app t, J = 8.5 Hz, H-4′), 12.98 (1H, s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 51.7 (CH3), 114.6 (CH-pyrrole), 119.4 (d, JCF = 18.1 Hz, C-3′), 124.8 (C-pyrrole), 124.9 (C-pyrrole), 126.8 (d, JCF = 4.1 Hz, C-5′), 128.9 (d, JCF = 22.7 Hz, C-1′), 129.1 (d, JCF = 5.2 Hz, C-6′), 131.0 (C-pyrrole), 131.9 (C-4′), 153.8 (d, JCF = 248.5 Hz, C-2′), 160.3 (CO2Me), 182.5 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; MS (ES+) m/z 314.2 [M(35Cl) + H]+, 316.1 [M(37Cl) + H]+.
4-(3,6-Dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxylic Acid (31b)
Prepared according to general procedure B using LiOH (26.6 g, 632 mmol) in water (140 mL) and ester 30b (10.0 g, 31.6 mmol) in THF (180 mL) to give a white solid (9.00 g, 95%); Rf 0.25 (SiO2, 50:50:0.5 EtOAc/petrol/AcOH); mp 230–232 °C; λmax (EtOH)/nm 289, 267; IR νmax/cm–1 3264br, 1697, 1646; 1H NMR (500 MHz; DMSO-d6) δH 7.07 (1H, app t, J = 1.6 Hz, H-pyrrole), 7.53 (1H, dd, J = 1.1 and 8.6 Hz, H-5′), 7.71 (1H, dd, J = 1.6 and 3.2 Hz, H-pyrrole), 7.80 (1H, app t, J = 8.6 Hz, H-4′), 12.81 (1H, br s, NH-pyrrole), 13.00 (1H, br s, CO2H); 13C NMR (125 MHz; DMSO-d6) δC 114.1 (CH-pyrrole), 119.3 (d, JCF = 18.1 Hz, C-3′), 124.8 (C-pyrrole), 126.1 (C-pyrrole), 126.8 (d, JCF = 3.6 Hz, C-5′), 129.0 (d, JCF = 23.3 Hz, C-1′), 129.1 (d, JCF = 5.5 Hz, C-6′), 130.6 (C-pyrrole), 131.9 (C-4′), 153.8 (d, JCF = 248.4 Hz, C-2′), 161.3 (CO2H), 182.5 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; MS (ES+) m/z 302.1 [M(35Cl) + H]+, 304.1 [M(37Cl) + H]+.
4-(2-Chloro-6-fluorobenzoyl)-N-(pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32a)
Compound 32a was synthesized according to general procedure C using 4-(2-chloro-6-fluoro-benzoyl)-1H-pyrrole-2-carboxylic acid (31a) (100 mg, 0.37 mmol), MeCN (2 mL), 5-aminopyrimidine (88 mg, 0.93 mmol), and PCl3 (32 μL, 0.37 mmol) to afford the crude product. Purification was achieved using MPLC on SiO2 with a gradient elution from 0 to 10% MeOH/EtOAc to give an orange solid (100 mg, 79%); Rf 0.52 (SiO2, 5% MeOH/EtOAc); mp 227 °C (dec.); λmax (EtOH)/nm 262.0, 292.0; IR 2960, 2862, 1968, 1637 (CO), 1529 (CONH); 1H NMR (500 MHz, DMSO-d6) δ 7.42 (1H, dd, J = 1.0 and 9.0 Hz, H-5′), 7.48–7.50 (2H, m, H-3′ and H-3), 7.55 (1H, s, H-5), 7.60 (1H, ddd, J = 6.3, 8.3 and 8.3 Hz, H-4′), 8.92 (1H, s, N-CH-N-pyrimidine), 9.13 (2H, s, 2 × CH-N-pyrimidine), 10.44 (1H, s, CONH), 12.93 (1H, s, NH); 13C NMR (125 MHz, DMSO-d6) δ 112.1 (C-pyrimidine), 114.9 (C-Ar), 115.1 (d, JCF = 23.4 Hz, C-Ar), 125.3 (C-Ar), 125.9 (C-3), 127.6 (C-2 and C-5), 129.7 (C-4), 130.4 (d, JCF = 22.8 Hz, C-Ar), 131.9 (d, JCF = 8.6 Hz, C-Ar), 134.3 (C-N-pyrimidine), 147.8 (C-Ar), 153.2 (d, JCF = 245.2 Hz, CF), 158.8 (CON), 183.9 (CO); 19F NMR (470 MHz, DMSO-d6) δ −114.3; HRMS m/z calcd for C16H1135ClFN4O2 [M + H]+ 345.0549, found 345.0550.
4-(2-Chloro-6-fluorobenzoyl)-N-(2-methylpyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32b)
Compound 32b was synthesized according to general procedure C using 4-(2-chloro-6-fluorobenzoyl)-1H-pyrrole-2-carboxylic acid (31a) (100 mg, 0.37 mmol), 2-methylpyrimidin-5-amine (102 mg, 0.93 mmol), PCl3 (32 μL, 0.37 mmol), and MeCN (2 mL). The crude mixture was purified by MPLC on SiO2 with a gradient elution from 0 to 8% MeOH/CH2Cl2 to give a yellow solid (53 mg, 0.14 mmol, 40%); Rf 0.45 (SiO2, 5% MeOH/CH2Cl2); mp 270 °C (dec.); λmax (EtOH)/nm 268, 290, 379; IR νmax/cm–1 3320, 1637 (CO), 1516 (CONH); 1H NMR (500 MHz, MeOD) δ 2.68 (3H, s, CH3), 7.25–7.28 (1H, m, H-5′), 7.40 (1H, d, J = 8.5 Hz, H-3′), 7.44 (1H, d, J = 1.5 Hz, H-3), 7.49 (1H, d, J = 1.5 Hz, H-5), 7.53 (1H, ddd, J = 6.1, 8.5 and 8.5 Hz, H-4′), 9.07 (2H, s, CH-pyrimidine); 13C NMR (125 MHz, MeOD) δ 25.0 (CH3), 108.4 (C-pyrimidine), 115.0 (C-Ar), 121.9 (C-Ar), 122.0 (C-3), 126.5 (C-2 and C-5), 127.2 (C-pyrimidine), 131.0 (C-4), 135.6 (C-Ar), 143.2 (C-N-pyridine), 156.7 (N-C-N-pyrimidine), 154.6 (d, JCF = 253.2 Hz, CF), 159.8 (CON), 187.0 (CO); 19F NMR (470 MHz, DMSO-d6) δ −115.3; HRMS m/z calcd for C17H1235ClFN4O2 [M + H]+ 359.0710, found 359.0710.
2-Methoxy-5-nitropyrimidine25,26 (6a)
Sodium (35 mg, 1.50 mmol) was added to MeOH (5 mL), and the mixture was stirred under nitrogen until the sodium had dissolved. 5-Nitro-2-chloropyrimidine (200 mg, 1.25 mmol) was added, and the reaction mixture was heated at reflux for 1 h. The solvent was removed in vacuo, and the residue was purified by MPLC on SiO2 with a gradient elution from 10 to 20% EtOAc/petrol to give a yellow solid (115 mg, 59%); Rf 0.40 (SiO2, 20% EtOAc/petrol); mp 65–67 °C (Lit.25 69–70 °C); λmax (EtOH)/nm 270 nm; IR νmax/cm–1 1567, 1474, 1404, 1315; 1H NMR (500 MHz; DMSO-d6) δH 4.08 (3H, s, OMe), 9.42 (2H, s, H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 56.4 (Me), 138.8 (C-NO2), 156.4 (2 × CH-pyrimidine), 166.7 (C-O-Me); MS (ES+) m/z 156.2 [M + H]+.
2-Methoxypyrimidin-5-amine27 (7a)
Prepared according to general procedure D using nitropyrimidine 6a (140 mg, 0.9 mmol) in MeOH (5 mL) for 90 min. The solvent was removed in vacuo to give a white solid (109 mg, 97%); Rf 0.10 (NH2 SiO2, 5% MeOH/CH2Cl2); mp 113–116 °C (lit.27 119–120 °C); λmax (EtOH)/nm 327, 237; IR νmax/cm–1 3304, 3180, 1648(w), 1565; 1H NMR (500 MHz; DMSO-d6) δH 3.78 (3H, s, OMe), 5.01 (2H, s, NH2), 7.99 (s, 2H, H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 53.9 (OMe), 137.9, (C-NH2), 144.3 (2 × CH-pyrimidine), 157.8 (C-O-Me); MS (ES+) m/z 126.2 [M + H]+.
4-(2-Chloro-6-fluorobenzoyl)-N-(2-methoxypyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32c)
Prepared according to general procedure E using carboxylic acid 31a (86 mg, 0.32 mmol), amine 7a (100 mg, 0.8 mmol), pyridine (26 μL, 0.32 mmol), and cyanuric fluoride (19 μL, 0.22 mmol). Purification by MPLC on SiO2 with a gradient elution from 0 to 3% MeOH/CH2Cl2 gave a white solid (52 mg, 43%); Rf 0.15 (SiO2, 50% EtOAc/petrol); mp 258 °C dec.; λmax (EtOH)/nm 267; IR νmax/cm–1 3337, 2961, 1649, 1616, 1583; 1H NMR (500 MHz; DMSO-d6) δH 3.94 (3H, s, OMe), 7.41–7.48 (2H, m, H-pyrrole and H-5′), 7.51 (1H, d, J = 8.3 Hz, H-3′), 7.54 (1H, br s, H-pyrrole), 7.62 (1H, td, J = 8.3 and 6.3 Hz, H-4′), 8.90 (2H, s, 2 × CH-pyrimidine), 10.34 (1H, s, NH), 12.78 (1H, br s, NH); 19F NMR (470 MHz; DMSO-d6) δF −114.3; 13C NMR (125 MHz; DMSO-d6) δC 54.7 (OMe), 111.6 (CH-pyrrole), 115.0 (d, JCF = 21.3 Hz, C-5′), 125.3 (C-pyrrole), 125.9 (d, JCF = 3.0 Hz, C-3′), 127.8 (C-pyrimidine), 128.0 (d, JCF = 23.2 Hz, C-1′), 128.6 (C-pyrrole), 129.3 (CH-pyrrole), 130.4 (d, JCF = 5.9 Hz, C-2′), 131.9 (d, JCF = 9.1 Hz, C-4′), 151.4 (C-pyrimidine), 156.6 (d, JCF = 248 Hz, C-6′), 158.55 (C-pyrimidine), 161.3 (CO-NH), 183.9 (CO); MS (ES+) m/z 375.3 [M(35Cl) + H]+, 377.3 [M(37Cl) + H]+; HRMS calcd for C17H1235Cl1F1N4O3 [M + H]+ 375.0655, found 375.0648.
N,N-Dimethyl-5-nitropyrimidin-2-amine28 (6b)
Prepared according to general procedure F using 2-chloro-5-nitropyrimidine (300 mg, 1.90 mmol, 1 equiv), Me2NH (1.40 mL, 2.80 mmol, 1.5 equiv, 2.0 M in THF), and Et3N (288 μL, 2.10 mmol, 1.1 equiv) in THF (8 mL) to give a yellow solid (275 mg, 87%); Rf 0.75 (NH2 SiO2, 30% EtOAc/petrol); mp 209–212 °C (lit.28 222 °C); λmax (EtOH)/nm 341, 219; IR νmax/cm–1 1547, 1301; 1H NMR (500 MHz; DMSO-d6) δH 3.31 (6H, s, NMe2), 9.15 (2H, s, 2 × H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 37.4 (NMe2), 133.4 (C-5-pyrimidine), 154.8 (2H, s, 2 × CH-pyrimidine), 161.6 (C-2-pyrimidine); HRMS calcd for C6H9N4O2 [M + H]+ 169.0720, found 169.0720.
N2,N2-Dimethylpyrimidine-2,5-diamine29 (7b)
Prepared according to general procedure D using nitropyrimidine 6b (263 mg, 1.60 mmol) in MeOH (60 mL) and EtOAc (60 mL) for 4 h to give a yellow solid (215 mg, 100%); Rf 0.80 (NH2 SiO2, EtOAc); mp 68–72 °C; λmax (EtOH)/nm 261; IR νmax/cm–1 3206; 1H NMR (500 MHz; DMSO-d6) δH 3.12 (6H, s, NMe2), 4.50 (2H, s, NH2), 7.01 (2H, s, 2 × CH-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 37.2 (NMe2), 133.2 (C-5-pyrimidine), 144.3 (2 × CH-pyrimidine), 156.7 (C-2-pyrimidine); HRMS calcd for C6H11N4 [M + H]+ 139.0978, found 139.0978.
4-(2-Chloro-6-fluorobenzoyl)-N-(2-(dimethylamino)pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32d)
Prepared according to general procedure E using amine 7b (100 mg, 0.72 mmol, 2.5 equiv), carboxylic acid 31a (77 mg, 0.29 mmol, 1 equiv), cyanuric fluoride (25 μL, 0.20 mmol, 0.7 equiv), pyridine (23 μL, 0.29 mmol, 1 equiv), and MeCN (2 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 50 to 100% EtOAc/petrol gave a white solid (38 mg, 34%); Rf 0.50 (NH2 SiO2, EtOAc); mp 300 °C dec.; λmax (EtOH)/nm 287, 232; IR νmax/cm–1 2951, 1645, 1622; 1H NMR (500 MHz; DMSO-d6) δH 3.15 (6H, s, NMe2), 7.39 (1H, s, H-pyrrole), 7.44 (1H, app t, J = 8.4 Hz, H-5′), 7.49 (1H, s, H-pyrrole), 7.50 (1H, d, J = 8.4 Hz, H-3′), 7.62 (1H, td, J = 8.4 and 6.3 Hz, H-4′), 8.61 (2H, s, 2 × H-pyrimidine), 10.02 (1H, s, CO-NH), 12.70 (1H, s, NH); 19F NMR (470 MHz; DMSO-d6) δF −114.4; 13C NMR (125 MHz; DMSO-d6) δC 36.9 (NMe2), 111.1 (CH-pyrrole), 115.0 (d, JCF = 21.5 Hz, C-5′), 122.9 (C-5-pyrimidine), 125.2 (C-pyrrole), 125.8 (d, JCF = 3.2 Hz, C-3′), 128.1 (d, JCF = 23.2 Hz, C-1′), 128.2 (C-pyrrole), 128.9 (CH-pyrrole), 130.4 (d, JCF = 5.9 Hz, C-2′), 131.8 (d, J = 9.1 Hz, C-4′), 151.2 (2 × CH-pyrimidine), 158.4 (C-2-pyrimidine), 158.6 (d, JCF = 247.0 Hz, C-6′), 159.1 (CO-NH), 183.9 (CO); HRMS calcd for C18H1635Cl1F1N5O2 [M + H]+ 388.0971, found 388.0977.
4-(5-Nitropyrimidin-2-yl)morpholine (6c)
Prepared according to general procedure F using 2-chloro-5-nitropyrimidine (300 mg, 1.88 mmol) morpholine (181 μL, 2.07 mmol), Et3N (288 μL, 2.07 mmol), and THF (12 mL). The residue was purified by MPLC on SiO2 with a gradient elution from 20 to 100% EtOAc/petrol to give a yellow solid (290 mg, 73%); Rf 0.35 (SiO2, 30% EtOAc/petrol); mp 161–164 °C (lit.30 165–168 °C); λmax (EtOH)/nm 339, 221; IR νmax/cm–1 1545 (NO2), 1326 (NO2); 1H NMR (500 MHz; DMSO-d6) δH 3.71–3.75 (4H, m, 2 × CH2-morpholine), 3.94–3.97 (4H, m, 2 × CH2-morpholine), 2.14 (2H, s, 2 × H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 44.5 (2 × CH2-morpholine), 65.8 (2 × CH2-morpholine), 133.4 (C-5-pyrimidine), 155.1 (C-4 and C-6-pyrimidine), 161.0 (C-2-pyrimidine); HRMS calcd for C8H11N4O3 [M + H]+ 211.0826, found 211.0828.
2-Morpholinopyrimidin-5-amine (7c)
Prepared according to general procedure D using nitropyrimidine 6c (278 mg, 1.54 mmol) in MeOH (35 mL) and EtOAc (15 mL) to give a yellow solid (239 mg, 100%); Rf 0.30 (NH2 SiO2, 70% EtOAc/petrol); mp 110–113 °C (lit.30 99 °C); λmax (EtOH)/nm 255; IR νmax/cm–1 3301, 3203 (NH2); 1H NMR (500 MHz; DMSO-d6) δH 3.46–3.50 (4H, m, 2 × CH2-morpholine), 3.63–3.70 (4H, m, 2 × CH2-morpholine), 4.68 (2H, s, NH2), 7.94 (2H, s, 2 × H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 45.0 (2 × CH2-morpholine), 66.0 (2 × CH2-morpholine), 134.8 (C-5-pyrimidine), 143.9 (C-4 and C-6-pyrimidine), 155.9 (C-2-pyrimidine); HRMS calcd for C8H13N4O1 [M + H]+ 181.1084, found 181.1084.
4-(2-Chloro-6-fluorobenzoyl)-N-(2-morpholinopyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32e)
Prepared according to general procedure E using amine 7c (110 mg, 0.61 mmol), carboxylic acid 31a (65 mg, 0.24 mmol), cyanuric fluoride (15 μL, 0.17 mmol), pyridine (20 μL, 0.24 mmol), and MeCN (2 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 50 to 100% EtOAc/petrol gave a white solid (52 mg, 50%); Rf 0.40 (NH2 SiO2, EtOAc); mp 286–287 °C; λmax (EtOH)/nm 292, 232; IR νmax/cm–1 3211, 1656, 1634; 1H NMR (500 MHz; DMSO-d6) δH 3.70 (8H, s, 4 × CH2-morpholine), 7.41 (1H, m, H-pyrrole), 7.44 (1H, app. t, J = 8.3 Hz, H-5′), 7.48–7.52 (2H, m, H-pyrrole and H-3′), 7.62 (1H, td, J = 8.3 and 6.3 Hz, H-4′), 8.68 (2H, s, 2 × H-pyrimidine), 10.10 (CO-NH), 12.72 (NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 44.2 (2 × CH2-morpholine), 65.9 (2 × CH2-morpholine), 111.2 (CH-pyrrole), 115.0 (d, JCF = 21.5 Hz, C-5′), 124.1 (C-pyrimidine), 125.2 (C-pyrrole), 125.8 (JCF = 2.7 Hz, C-3′), 128.7 (d, JCF = 22.7 Hz, C-1′), 128.1 (C-pyrrole), 129.0 (CH-pyrrole), 130.4 (d, JCF = 6.4 Hz, C-2′), 131.8 (d, JCF = 8.6 Hz, C-4′), 151.0 (2 × CH-pyrimidine), 158.4 (CO-NH and C-pyrimidine), 158.6 (d, J = 247.0 Hz, C-6′), 183.9 (CO); 19F NMR (470 MHz; DMSO-d6) δF −114.4; HRMS calcd for C20H1835Cl1F1N5O3 [M + H]+ 430.1077, found 430.1083.
2-(4-Methylpiperazin-1-yl)-5-nitropyrimidine31 (6d)
Prepared according to general procedure F using 1-methylpiperazine (382 μL, 3.45 mmol), 2-chloro-5-nitropyrimidine (500 mg, 3.14 mmol), Et3N (480 μL, 3.45 mmol), and THF (15 mL). The residue was purified by MPLC on SiO2 with a gradient elution from 0 to 13% MeOH/EtOAc to give a yellow solid (573 mg, 82%); Rf 0.60 (NH2 SiO2, EtOAc); mp 149–152 °C; λmax (EtOH)/nm 346, 332, 219; IR νmax/cm–1 1567, 1474; 1H NMR (500 MHz; DMSO-d6) δH 2.26 (3H, s, Me), 2.41–2.46 (4H, m, H-piperazine), 3.92–3.99 (4H, m, H-piperazine), 9.15 (2H, s, H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 44.0 (2 × C-piperazine), 45.5 (NMe), 54.1 (2 × C-piperazine), 133.2 (C-NO2), 155.1 (2 × CH-pyrimidine), 160.8 (C-pyrimidine); MS (ES+) m/z 224.3 [M + H]+; HRMS calcd for C9H14N5O2 [M + H]+ 224.1142, found 224.1136.
2-(4-Methylpiperazin-1-yl)pyrimidin-5-amine32 (7d)
Prepared according to general procedure D using nitropyrimidine 6d (195 mg, 0.87 mmol) and MeOH (5 mL) to give a pale yellow solid (168 mg, 99%); Rf 0.50 (NH2 SiO2, 5% MeOH/CH2Cl2); mp 139–142 °C; λmax (EtOH)/nm 353, 255; IR νmax/cm–1 3347, 3173, 2967, 2920, 1640, 1606; 1H NMR (500 MHz; DMSO-d6) δH 2.22 (3H, s, NMe), 2.31–2.40 (4H, m, H-piperazine), 3.48–3.58 (4H, m, H-piperazine), 4.64 (2H, s, NH2), 7.92 (2H, s, H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 44.3 (2 × C-piperazine), 45.9 (NMe), 54.4 (2 × C-piperazine), 134.3 (C-NH2), 144.0 (2 × CH-pyrimidine), 156.0 (C-pyrimidine); MS (ES+) m/z 194.3 [M + H]+; HRMS calcd for C9H16N5 [M + H]+ 194.1400, found 194.1399.
4-(2-Chloro-6-fluorobenzoyl)-N-(2-(4-methylpiperazin-1-yl)pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32f)
Prepared according to general procedure E using carboxylic acid 31a (150 mg, 0.78 mmol), amine 7d (83 mg, 0.31 mmol), cyanuric fluoride (19 μL, 0.22 mmol), pyridine (25 μL, 0.31 mmol), and MeCN (2 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 0 to 4% MeOH/CH2Cl2 gave a white solid (68 mg, 50%); Rf 0.3 (NH2 SiO2, 5% MeOH/CH2Cl2); mp 246 °C (dec.); λmax (EtOH)/nm 289, 231; IR νmax/cm–1 3186, 1661, 1641, 1606, 1582; 1H NMR (500 MHz; DMSO-d6) δH 2.24 (3H, s, NMe), 2.35–2.42 (4H, m, H-piperazine), 3.68–3.76 (4H, m, H-piperazine), 7.40 (1H, s, H-pyrrole), 7.44 (1H, app t, J = 8.2 Hz, H-5′), 7.48–7.54 (2H, m, H-pyrrole and H-3′), 7.62 (1H, td, J = 8.2 and 6.3 Hz, H-4′), 8.61 (2H, s, 2 × H-pyrimidine), 10.05 (1H, s, CO-NH), 12.69 (1H, s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 43.6 (2 × CH2 piperazine), 45.8 (NMe), 54.3 (2 × CH2 piperazine), 111.1 (CH-pyrrole), 115.0 (d, JCF = 21.5 Hz, C-5′), 125.2 (C-pyrrole), 125.8 (d, JCF = 3.2 Hz, C-3′), 127.9 (C-pyrimidine), 128.0 (d, JCF = 22.7 Hz, C-1′), 128.1 (C-pyrrole), 129.0 (CH-pyrrole), 130.4 (d, JCF = 6.2 Hz, C-2′), 131.8 (d, JCF = 9.1 Hz, C-4′), 151.0 (2 × CH-pyrimidine), 158.4 (CO-NH), 158.4 (C-pyrimidine), 158.5 (d, JCF = 247 Hz, C-6′), 183.9 (CO); 19F NMR (470 MHz; DMSO-d6) δF −114.4; MS (ES+) m/z 443.5 [M(35Cl) + H]+, 445.4 [M(37Cl) + H]+; HRMS calcd for C21H2135Cl1F1N6O2 [M + H]+ 443.193, found 443.193.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(2-(4-methylpiperazin-1-yl)pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32g)
Prepared according to general procedure E using amine 7d (160 mg, 0.83 mmol), carboxylic acid 31b (100 mg, 0.33 mmol), cyanuric fluoride (20 μL, 0.23 mmol), pyridine (27 μL, 0.33 mmol), and MeCN (2 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 0 to 4% MeOH/CH2Cl2 gave a white solid (65 mg, 41%); Rf 0.65 (NH2 SiO2, 5% MeOH/EtOAc); mp 226–228 °C; λmax (EtOH)/nm 287, 227; IR νmax/cm–1 3174, 1663, 1639, 1592; 1H NMR (500 MHz; DMSO-d6) δH 2.25 (3H, s, CH3), 2.37–2.41 (4H, m, 2 × CH2-piperazine), 3.70–3.76 (4H, m, 2 × CH2-piperazine), 7.44 (1H, s, H-pyrrole), 7.56 (1H, dd, J = 1.1 and 8.6 Hz, H-5′), 7.64 (1H, s, H-pyrrole), 7.82 (1H, app t, J = 8.6 Hz, H-4′), 8.64 (2H, s, 2 × H-pyrimidine), 10.07 (1H, s, CO-NH), 12.78 (1H, s, NH); 13C NMR (125 MHz; DMSO-d6) δC 43.6 (2 × CH2-piperazine), 45.8 (CH3), 54.3 (2 × CH2-piperazine), 111.0 (CH-pyrrole), 119.3 (d, JCF = 18.3 Hz, C-3′), 123.7 (C-pyrimidine), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.8 Hz, C-5′), 128.4 (C-pyrrole), 129.1 (d, JCF = 22.3 Hz, C-1′), 129.2 (d, JCF = 5.6 Hz, C-6′), 129.9 (CH-pyrrole), 131.8 (C-4′), 151.1 (2 × CH-pyrimidine), 153.8 (d, JCF = 248.4 Hz, C-2′), 158.4 (C-pyrimidine), 158.4 (CO-NH), 182.6 (NH-pyrrole); 19F NMR (470 MHz; DMSO-d6) δF −116.7; MS (ES+) m/z 477.3 [M(35,35Cl) + H]+, 479.3 [M(35,37Cl) + H]+; HRMS calcd for C21H2035Cl2F1N6O2 [M + H]+ 477.1003, found 477.1008.
tert-Butyl-4-(5-Nitropyrimidin-2-yl)piperazine-1-carboxylate33 (6e)
Prepared according to general procedure F using 2-chloro-5-nitropyrimidine (350 mg, 2.20 mmol), 1-Boc-piperazine (450 mg, 2.40 mmol), Et3N (336 μL, 2.40 mmol), and THF (12 mL). The residue was purified by MPLC on SiO2 with a gradient elution from 15 to 100% EtOAc/petrol to give a yellow solid (460 mg, 68%); Rf 0.40 (SiO2, 20% EtOAc/petrol); mp 196–199 °C; λmax (EtOH)/nm 339, 221; IR νmax/cm–1 1676, 1539, 1325; 1H NMR (500 MHz; DMSO-d6) δH 1.47 (9H, s, C(CH3)3), 3.47–4.57 (4H, m, 2 × CH2-piperazine), 3.92–3.99 (4H, m, 2 × CH2-piperazine), 9.17 (2H, s, 2 × H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 28.0 (C(CH3)3), 42.5 (2 × CH2-piperazine), 43.9 (2 × CH2-piperazine), 79.3 (C(CH3)3), 133.4 (C-5-pyrimidine), 153.8 (CO), 155.1 (C-4 and C-6-pyrimidine), 161.0 (C-2-pyrimidine); HRMS calcd for C13H20N5O4 [M + H]+ 310.1510, found 310.1510.
tert-Butyl-4-(5-aminopyrimidin-2-yl)piperazine-1-carboxylate34 (7e)
Prepared according to general procedure D using nitropyrimidine 6e (440 mg, 1.42 mmol) in MeOH (75 mL) and EtOAc (75 mL) to give a yellow solid (395 mg, 99%); Rf 0.15 (NH2 SiO2, 70% EtOAc/petrol); mp 131–133 °C; λmax (EtOH)/nm 252; IR νmax/cm–1 3336, 1676; 1H NMR (500 MHz; DMSO-d6) δH 1.45 (9H, s, C(CH3)3), 3.38–3.42 (4H, m, 2 × CH2-piperazine), 3.50–3.55 (4H, m, 2 × CH2-piperazine), 4.68 (2H, s, NH2), 7.94 (2H, s, 2 × H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 28.1 (C(CH3)3), 44.2 (2 × CH2-piperazine), 44.7 (2 × CH2-piperazine), 78.9 (C(CH3)3), 134.7 (C-5-pyrimidine), 144.0 (C-4 and C-6-pyrimidine), 154.0 (CO), 155.6 (C-2-pyrimidine); HRMS calcd for C13H20N5O2 [M – H]− 278.1622, found 278.1609.
tert-Butyl-4-(5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyrimidin-2-yl)piperazine-1-carboxylate (32h)
Prepared according to general procedure E using amine 7e (190 mg, 0.68 mmol), carboxylic acid 31b (82 mg, 0.27 mmol), cyanuric fluoride (16 μL, 0.19 mmol), pyridine (22 μL, 0.27 mmol), and MeCN (4 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 40 to 100% EtOAc/petrol gave a white solid (105 mg, 69%); Rf 0.25 (NH2 SiO2, 70% EtOAc/petrol); mp 211–213 °C; λmax (EtOH)/nm 283, 223; IR νmax/cm–1 3199, 1637, 1573; 1H NMR (500 MHz; DMSO-d6) δH 1.46 (9H, s, C(CH3)3), 3.40–3.47 (4H, m, 2 × CH2-piperazine), 3.70–3.76 (4H, m, 2 × CH2-piperazine), 7.44 (1H, s, H-pyrrole), 7.56 (1H, d, J = 8.6 Hz, H-5′), 7.82 (1H, app t, J = 8.6 Hz, H-4′), 8.67 (2H, s, 2 × H-pyrimidine), 10.10 (1H, s, CO-NH), 12.79 (1H, s, NH); 13C NMR (125 MHz; DMSO-d6) δC 28.1 (C(CH3)3), 43.5 (4 × CH2-piperazine), 79.0 (C(CH3)3), 111.1 (CH-pyrrole), 119.3 (d, JCF = 18.0 Hz, C-3′), 124.0 (C-pyrimidine), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.5 Hz, C-5′), 128.3 (C-1′), 128.3 (C-pyrrole), 129.2 (d, JCF = 5.5 Hz, C-6′), 129.2 (CH-pyrrole), 131.8 (C-4′), 151.1 (2 × CH-pyrimidine), 153.8 (d, JCF = 248.4 Hz, C-2′), 158.2 (C-pyrimidine), 158.4 (CO-NH), 182.6 (C–O); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C25H2435Cl2F1N6O4 [M + H]+ 563.1197, found 563.121.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(2-(piperazin-1-yl)pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32k)
Prepared according to general procedure J using Et3SiH (64 μL, 0.40 mmol), TFA (1 mL), CH2Cl2 (1 mL), and carbamate 32h (90 mg, 0.16 mmol) to give a white solid (35 mg, 47%); Rf 0.2 (NH2 SiO2, 5% MeOH/EtOAc); mp 230 °C (dec.); λmax (EtOH)/nm 303, 225; IR νmax/cm–1 3275 (br), 2922, 2847, 1635; 1H NMR (500 MHz; DMSO-d6) δH 2.75–2.79 (4H, m, 2 × CH2-piperazine), 3.64–3.69 (4H, m, 2 × CH2-piperazine), 7.42 (1H, s, H-pyrrole), 7.55 (1H, d, J = 8.6 Hz, H-5′), 7.63 (1H, s, H-pyrrole), 7.82 (1H, app t, J = 8.6 Hz, H-4′), 8.62 (2H, s, 2 × H-pyrimidine), 10.05 (1H, br s, CO-NH); 13C NMR (125 MHz; DMSO-d6) δC 44.9 (2 × C-piperazine), 45.4 (2 × C-piperazine), 111.0 (CH-pyrrole), 119.3 (d, JCF = 18.1 Hz, C-3′), 123.4 (C-pyrimidine), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.6 Hz), 128.4 (C-pyrrole), 129.2 (d, JCF = 23.2 Hz, C-1′), 129.2, (d, JCF = 5.3 Hz, C-6′), 129.9 (CH-pyrrole), 131.8 (C-4′), 151.1 (2 × CH-pyrimidine), 153.8 (d, JCF = 248.7 Hz, C-2′), 158.4 (C-pyrimidine), 158.6 (CO-NH), 182.6 (CO); HRMS calcd for C20H1835Cl2F1N6O2 [M + H]+ 463.0847, found 463.0853.
N-(1-Methylpiperidin-4-yl)-5-nitropyrimidin-2-amine35 (6f)
Prepared according to general procedure F using 2-chloro-5-nitropyrimidine (300 mg, 1.90 mmol), 4-amino-1-methylpiperidine (259 μL, 2.10 mmol), Et3N (288 μL, 2.10 mmol), and THF (10 mL) to give a yellow solid (370 mg, 83%); Rf 0.50 (NH2 SiO2, EtOAc); mp 154–157 °C; λmax (EtOH)/nm 340, 213; IR νmax/cm–1 3242, 1587, 1329; 1H NMR (500 MHz; DMSO-d6) δH 1.61 (2H, qd, J = 3.5 and 11.9 Hz, 2 × H-piperidine), 1.80–1.89 (2H, m, 2 × H-piperidine), 1.93–2.03 (2H, m, 2 × H-piperidine), 2.20 (3H, s, CH3), 2.77–2.84 (2H, m, 2 × H-piperidine), 3.80–3.90 (1H, m, CH-NH), 8.83 (1H, d, J = 8.4 Hz, NH), 9.07 (1H, d, J = 3.4 Hz, H-pyrimidine), 9.13 (1H, d, J = 3.4 Hz, H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 30.9 (2 × CH2-piperidine), 45.9 (N-CH3), 48.4 (CH-NH), 54.1 (2 × CH2-N-piperidine), 133.5 (C-5-pyrimidine), 155.2 (CH-pyrimidine), 155.4 (CH-pyrimidine), 162.1 (C-2-pyrimidine); HRMS calcd for C10H16N5O2 [M + H]+ 238.1299, found 238.1301.
N2-(1-Methylpiperidin-4-yl)pyrimidine-2,5-diamine35 (7f)
Prepared according to general procedure D using nitropyrimidine 6f (360 mg, 1.52 mmol) and MeOH (30 mL) for 2 h to give a pale yellow solid (290 mg, 92%); Rf 0.50 (NH2 SiO2, 5% MeOH/EtOAc); mp 158–161 °C; λmax (EtOH)/nm 248; IR νmax/cm–1 3257, 2967, 2789; 1H NMR (500 MHz; DMSO-d6) δH 1.45 (2H, qd, J = 3.6 and 11.7 Hz, 2 × H-piperidine), 1.78–1.86 (2H, m, 2 × H-piperidine), 1.90–1.99 (2H, m, 2 × H-piperidine), 2.17 (3H, s, CH3), 2.70–2.77 (2H, m, 2 × H-piperidine), 3.46–3.56 (1H, m, CH-NH), 4.41 (2H, s, NH2), 6.02 (1H, d, J = 8.0 Hz, NH), 7.82 (2H, s, 2 × H-pyrimidine); 13C NMR (125 MHz; DMSO-d6) δC 31.8 (2 × CH2-piperidine), 46.1 (N-CH3), 47.5 (CH-NH), 54.6 (2 × CH2N-piperidine), 133.5 (C-5 pyrimidine), 144.6 (2 × CH-pyrimidine), 156.0 (C-2 pyrimidine); HRMS calcd for C10H18N5 [M + H]+ 208.1557, found 208.1559.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(2-((1-methylpiperidin-4-yl)amino)pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32i)
Prepared according to general procedure E using amine 7f (150 mg, 0.72 mmol), carboxylic acid 31b (88 mg, 0.29 mmol), cyanuric fluoride (21 μL, 0.24 mmol), pyridine (23 μL, 0.29 mmol), and MeCN (2 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 1 to 7% MeOH/EtOAc gave a white solid (50 mg, 35%); Rf 0.20 (NH2 SiO2, 5% MeOH/EtOAc); mp 275 °C dec.; λmax (EtOH)/nm 283, 226; IR νmax/cm–1 3163, 1642, 1593; 1H NMR (500 MHz; DMSO-d6) δH 1.54 (2H, qd, J = 3.1 and 11.5 Hz, 2 × H-piperidine), 1.81–1.89 (2H, m, 2 × H-piperidine), 1.92–2.02 (2H, m, 2 × H-piperidine), 2.19 (3H, s, CH3), 2.73–2.81 (2H, m, 2 × H-piperidine), 3.61–3.72 (1H, m, CH-NH-piperidine), 7.02 (1H, d, J = 7.8 Hz, NH-piperidine), 7.42 (1H, s, H-pyrrole), 7.56 (1H, d, J = 8.6 Hz, H-5′), 7.63 (1H, s, H-pyrrole), 7.82 (1H, app t, J = 8.6 Hz, H-4′), 8.51 (2H, s, 2 × H-pyrimidine), 9.98 (1H, s, CO-NH), 12.74 (1H, br s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 31.5 (2 × CH2-piperidine), 46.0 (CH3), 48.6 (CH-NH-piperidine), 54.5 (2 × CH2N-piperidine), 110.9 (CH-pyrrole), 119.3 (d, JCF = 18.0 Hz, C-3′), 123.2 (C-pyrimidine), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.6 Hz, C-5′), 128.5 (C-pyrrole), 128.8 (CH-pyrrole), 129.2 (d, JCF = 22.3 Hz, C-1′), 129.2 (d. JCF = 5.1 Hz, C-6′), 131.8 (C-4′), 151.6 (2 × CH-pyrimidine), 153.8 (d, JCF = 248.9 Hz, C-2′), 158.4 (CO-NH), 158.9 (C-pyrimidine), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C22H2135Cl2F1N6O2 [M + H]+ 491.1160, found 491.1150.
2,2-Diethoxyacetimidamide Hydrochloride36 (12)
Sodium (8 mg, 0.36 mmol) was carefully added to MeOH (5 mL) at RT, and the mixture was stirred under nitrogen until the sodium had dissolved. Diethoxyacetonitrile (11) (1.0 mL, 0.93 g, 7.19 mmol) was added, and the resulting mixture was stirred at RT for 16 h. Solid carbon dioxide was added, and the solvent was removed in vacuo. The resulting oil was dissolved in Et2O (10 mL) and filtered. The filtrate was concentrated in vacuo to afford methyl diethoxyacetimidate as a yellow oil, which was used without further purification. The oil was dissolved in MeOH (5 mL), and ammonium chloride (385 mg, 7.19 mmol) was added in one portion. The resulting solution was stirred at RT overnight before the solvent was concentrated in vacuo. The resulting oil was triturated with Et2O to afford diethoxyacetimidamide hydrochloride as an off-white solid (1.17 g, 89%). The compound was used in the next step without further purification; mp 58.0–60.0 °C (lit. 81.0–82.0 °C);37 IR νmax/cm–1 3259, 3042, 2975, 1692, 1083; 1H NMR (500 MHz, DMSO-d6) δ 1.19 (6H, t, J = 7.0 Hz, OCH2CH3), 3.62 (4H, qd, J = 7.0, 3.0 Hz, OCH2CH3), 5.29 (1H, s, CH(OEt)2), 9.05 (4H, s, NH2 and NH2+); 13C NMR (126 MHz, DMSO-d6) δ 14.9 (OCH2CH3), 63.1 (OCH2CH3), 95.7 (CH(OEt)2), 165.8 (C = NH(NH2)); HRMS (ESI) calcd for C6H15N2O2 [M – Cl]+ 147.1128, found 147.1123; 1H NMR data were identical to literature data.
N-(3-(Dimethylamino)-2-[[(dimethylamino)methylene]amino]prop-2-en-1-ylidene)-N-methylmethanaminium Hydrogen Dihexafluorophosphate38
Phosphorus (V) oxychloride (7.52 mL, 12.4 g, 80.7 mmol) was added dropwise to DMF (16.1 mL) cooled to 10 °C, maintaining the temperature of the solution between 10 and 15 °C during the addition. Once the addition was complete, the reaction was stirred at RT for 20 min. The resulting solution was cooled to 5 °C before powdered glycine hydrochloride (3.00 g, 26.9 mmol) was added in portions; the temperature of the reaction mixture was maintained below 10 °C during the addition. The resulting reaction mixture was heated at 80 °C for 4 h. The hot, dark orange, solution was carefully poured directly into water (43 mL), precooled to 5 °C. The temperature of the solution was kept below 20 °C. Five minutes after the transfer was complete, the reaction mixture was cooled to −5 °C and treated from a plastic vessel with 60% aqueous hexafluorophosphoric acid (7.93 mL, 53.8 mmol). The thick precipitate was collected by filtration and washed with cold EtOH (100 mL) until a pale yellow solid was obtained (5.24 g, 40%); mp 151.0–153.0 °C; λmax (EtOH)/nm 254.6; IR νmax/cm–1 1701, 1611, 1402, 1291; 1H NMR (500 MHz, DMSO-d6) δ 3.19 (9H, s, 3 × NCH3), 3.24 (3H, s, NCH3), 3.29 (6H, s, 2 × NCH3), 7.70 (2H, s, 2 × CH), 8.07 (1H, d, J = 10.4 Hz, CHNH(CH3)2+), 10.74 (1H, d, J = 6.8 Hz, CHNH(CH3)2+); 13C NMR (126 MHz, DMSO-d6) δ 37.0 (NCH3), ca. 40 (overlapping with DMSO) (NCH3), 43.6 (NCH3), 48.8 (NCH3), 100.8 (Cq), 158.1 (CH), 160.7 (CHNH(CH3)2+); 19F NMR (471 MHz, DMSO-d6) δ −70.9 (PF6–), −69.4 (PF6–); MS (ES+) m/z 197.3 [M-HP2F12–]+; HRMS (NSI) calcd for C10H21N4 [M-HP2F12–]+ 197.1761, found 197.1760.
2-(Diethoxymethyl)pyrimidin-5-amine38 (13)
To a slurry of N-(3-(dimethylamino)-2-[[(dimethylamino)methylene]amino]prop-2-en-1-ylidene)-N-methylmethanaminium hydrogen dihexafluorophosphate (5.70 g, 11.7 mmol) and 2,2-diethoxyacetimidamide hydrochloride (12) (2.56 g, 14.0 mmol) in EtOH (25 mL) was added dropwise a solution of NaOMe in MeOH (2.27 g, 42.0 mmol, 25% w/v); the mixture was heated to reflux halfway through the addition. After refluxing for 2.5 h, the mixture was cooled to 0 °C, the inorganic precipitate was filtered off, washed with cold EtOH (3 × 20 mL), and the filtrate was concentrated in vacuo. The residue was dissolved in CH2Cl2 (50 mL), washed with water (3 × 20 mL), dried over MgSO4, and concentrated in vacuo to give an orange oil. The oil was dissolved in 1,4-dioxane (20 mL), treated with 5% aqueous K2CO3 (30 mL), and heated to reflux overnight. The reaction mixture was cooled to RT and extracted with EtOAc (3 × 40 mL). The organic extracts were washed with water (50 mL) and brine (50 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on SiO2 with a gradient elution from 0 to 5% MeOH/CH2Cl2 to yield an off-white solid (1.12 g, 49%); Rf 0.25 (SiO2, 5% MeOH/CH2Cl2); mp 134.0–136.0 °C; λmax (EtOH)/nm 250.4, 315.4; IR νmax/cm–1 3358, 3324, 3202, 2977, 2929, 2877, 1646, 1583, 1555, 1452; 1H NMR (500 MHz, DMSO-d6) δ 1.09 (6H, t, J = 7.1 Hz, OCH2CH3), 3.49 (2H, dq, J = 9.7, 7.1 Hz, OCH2CH3), 3.61 (2H, dq, J = 9.7, 7.1 Hz, OCH2CH3), 5.27 (1H, s, ArCH(OEt)2), 5.60 (2H, brs, ArNH2), 8.07 (2H, s, H-4, 6); 13C NMR (126 MHz, DMSO-d6) δ 15.2 (OCH2CH3), 61.2 (OCH2CH3), 102.0 (ArCH(OEt)2), 141.1 (C-4, 6), 142.1 (C-5), 153.5 (C-2); MS (ES+) m/z 198.2 [M + H]+; HRMS (NSI) calcd for C9H15N3O2Na [M + Na]+ 220.1056, found 220.1052; 1H and 13C NMR data were identical to literature data.38
Benzyl-(2-(diethoxymethyl)pyrimidin-5-yl)carbamate (14)
To 2-(diethoxymethyl)pyrimidin-5-amine (13) (1.50 g, 7.60 mmol) in THF/H2O (1:1) (20 mL) was added K2CO3 (2.10 g, 15.2 mmol) in one portion, followed by the dropwise addition of benzyl chloroformate (2.17 mL, 15.2 mmol) in THF (5 mL). The resulting reaction mixture was stirred at RT for 24 h. The reaction mixture was diluted with water (20 mL) and extracted with EtOAc (3 × 40 mL). The combined organic extracts were washed with water (40 mL) and brine (40 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified MPLC on SiO2 with a gradient elution from 0 to 40% EtOAc/petrol to yield a clear oil (2.03 g, 80%); Rf 0.32 (SiO2, 40% EtOAc/petrol); λmax (EtOH)/nm 238.0; IR νmax/cm–1 3227, 3032, 2975, 2933, 2882, 1728, 1586, 1525, 1224; 1H NMR (500 MHz, DMSO-d6) δ 1.11 (6H, t, J = 7.0 Hz, 2 × OCH2CH3), 3.54 (2H, dq, J = 9.6, 7.0 Hz, OCH2CH3), 3.65 (2H, dq, J = 9.6, 7.0 Hz, OCH2CH3), 5.20 (2H, s, OCH2Ph), 5.42 (1H, s, ArCH(OEt)2), 7.33–7.47 (5H, m, 5 × ArH), 8.88 (2H, s, H-4, 6), 10.26 (1H, s, ArNHCbz); 13C NMR (126 MHz, DMSO-d6) δ 15.2 (OCH2CH3), 61.6 (OCH2CH3), 66.5 (OCH2Ph), 101.7 (ArCH(OEt)2), 128.2 (CH-Ar), 128.5 (CH-Ar), 133.7 (C-Ar), 136.1 (C-Ar), 146.2 (C-4, 6), 153.4 (ArNHCO2Bn), 159.3 (C-2); MS (ES–) m/z 330.3 [M – H]−; HRMS (NSI) calcd for C17H21N3O4 [M + H]+ 332.1605, found 332.1600.
Benzyl-(2-formylpyrimidin-5-yl)carbamate (15)
To benzyl (2-(diethoxymethyl)pyrimidin-5-yl)carbamate (14) (1.90 g, 5.73 mmol) in MeCN (20 mL) was added 1 M hydrochloric acid (3.50 mL) at RT. The resulting mixture was stirred at RT for 8 h. The solvents were removed in vacuo, and the white residue was dissolved in saturated aqueous NaHCO3 (20 mL). The aqueous layer was extracted with EtOAc (3 × 30 mL), and the organic extracts were washed with water (40 mL) and brine (40 mL), dried over MgSO4, and concentrated in vacuo to give a white solid (1.31 g, 89%). The crude material was used in the next step without further purification; Rf 0.31 (SiO2, 50% petrol/EtOAc); mp 166.5–168.5 °C; λmax (EtOH)/nm 283.4; IR νmax/cm–1 3217, 3062, 3033, 2964, 2876, 1730, 1715, 1586, 1566, 1526; 1H NMR (500 MHz, DMSO-d6) δ 5.24 (2H, s, OCH2Ph), 7.34–7.49 (5H, m, 5 × ArH), 9.10 (2H, s, H-4, 6), 9.89 (1H, s, ArCHO), 10.69 (1H, s, ArNHCbz); 13C NMR (126 MHz, DMSO-d6) δ 66.9 (OCH2Ph), 128.3 (CH), 128.5 (CH), 135.8 (C-Ar), 136.2 (C-Ar), 146.0 (C-4, 6), 153.2, 153.6, 190.4 (ArCHO); MS (ES+) m/z 258.2 [M + H]+; HRMS (NSI) calcd for C13H12N3O3 [M + H]+ 258.0873, found 258.0875.
tert-Butyl-4-((5-(((benzyloxy)carbonyl)amino)pyrimidin-2-yl)methyl)piperazine-1-carboxylate (16)
To benzyl(2-formylpyrimidin-5-yl)carbamate (15) (900 mg, 3.50 mmol) in tetrafluoroethylene (TFE) (25 mL) was added tert-butyl piperazine-1-carboxylate (1.30 g, 7.00 mmol). The resulting solution was stirred at 38 °C for 1 h. The reaction mixture was cooled at 0 °C, and sodium borohydride was added portionwise. The resulting mixture was allowed to warm to RT and stirred for 30 min. The solvent was removed in vacuo, and the crude residue was dissolved in EtOAc (40 mL), neutralized by washing with saturated aqueous NH4Cl (25 mL) and washed with water (20 mL) and brine (20 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 60% EtOAc/petrol to yield a yellow solid (630 mg, 42%); Rf 0.34 (NH2 SiO2, 40% petrol/EtOAc); λmax (EtOH)/nm 236.8 nm; IR νmax/cm–1 2974, 1684, 1591, 1528, 1416; 1H NMR (500 MHz, DMSO-d6) δ 1.38 (9H, s, C(CH3)3), 2.35–2.45 (4H, m, CH2 piperazine), 3.28 (4H, s, CH2 piperazine), 3.65 (2H, s, ArCH2N), 5.19 (2H, s, OCH2Ph), 7.33–7.46 (5H, m, 5 × ArH), 8.83 (2H, s, H-4, 6), 10.17 (1H, s, ArNHCbz); 13C NMR (126 MHz, DMSO-d6) δ 28.0 (C(CH3)3), 43.1 (CH2 piperazine), 52.3 (CH2 piperazine), 63.6 (ArCH2N), 66.4 (OCH2Ph), 78.7 (OC(CH3)3), 128.2 (CH-Ar), 128.2 (CH-Ar), 128.5 (CH-Ar), 132.7 (C-Ar), 136.1 (C-Ar), 146.3 (C-4, 6), 153.4, 153.8, 160.4 (C-2); MS (ES+) m/z 428.5 [M + H]+; HRMS (NSI) calcd for C22H30N5O4 [M + H]+ 428.2292, found 428.2288.
tert-Butyl-4-((5-aminopyrimidin-2-yl)methyl)piperazine-1-carboxylate (17)
tert-Butyl-4-((5-(((benzyloxy)carbonyl)amino)pyrimidin-2-yl)methyl)piperazine-1-carboxylate (16) (600 mg, 1.40 mmol) in EtOAc (28 mL) was subjected to palladium-catalyzed hydrogenation using an H-Cube reactor and a 10% Pd/C CatCart under a full pressure of hydrogen at RT for 24 h with continuous recycling of the reaction mixture at 1 mL/min flow rate. The reaction mixture was concentrated in vacuo to afford a pale yellow solid (407 mg, 99%), which was used in the next step without further purification; Rf 0.26 (NH2 SiO2, 3% MeOH/CH2Cl2); mp 200.5–202.5 °C; λmax (EtOH)/nm 249.0, 318.0; IR νmax/cm–1 3381, 3323, 3197, 2972, 2929, 2894, 2863, 2811, 2775, 1673, 1589, 1554, 1453; 1H NMR (500 MHz, DMSO-d6) δ 1.37 (9H, s, C(CH3)3), 2.35 (4H, t, J = 5.0 Hz, CH2 piperazine), 3.26 (4H, brs, CH2 piperazine), 3.50 (2H, s, ArCH2N), 5.47 (2H, brs, ArNH2), 8.05 (2H, s); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 43.3 (CH2 piperazine), 52.3 (CH2 piperazine), 63.8 (ArCH2N), 78.7 (OC(CH3)3), 141.1, 141.5, 153.8, 154.1; MS (ES+) m/z 294.3 [M + H]+; HRMS (NSI) calcd for C14H24N5O2 [M + H]+ 294.1925, found 294.1926.
tert-Butyl-4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyrimidin-2-yl)methyl)piperazine-1-carboxylate (32j)
Compound 32j was synthesized according to general procedure I using 4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxylic acid 31b (200 mg, 0.66 mmol), triethylamine (231 μL, 167 mg, 1.65 mmol), 2-chloro-1-methylpyridinium iodide (186 mg, 0.73 mmol), tert-butyl 4-((5-aminopyrimidin-2-yl)methyl)piperazine-1-carboxylate (17) (243 mg, 1.65 mmol), and CH2Cl2 (6.60 mL). The crude yellow solid was purified by MPLC on SiO2 with a gradient elution from 0 to 85% EtOAc/petrol to yield a white solid (160 mg, 42%); Rf 0.32 (SiO2, 15% petrol/EtOAc); mp 162.5–164.5 °C; λmax (EtOH)/nm 292.8; IR νmax/cm–1 2967, 2932, 2864, 2815, 1652, 1585, 1555, 1516, 1447, 1423, 1392; 1H NMR (500 MHz, DMSO-d6) δ 1.38 (9H, s, C(CH3)3), 2.43 (4H, t, J = 5.1 Hz, CH2 piperazine), 3.29 (4H, brs, CH2 piperazine), 3.69 (2H, s, ArCH2N), 7.51 (1H, s), 7.53 (1H, dd, J = 8.9, 1.4 Hz), 7.68 (1H, s), 7.79 (1H, dd, J = 8.9, 8.4 Hz), 9.08 (2H, s), 10.43 (1H, s, CONHAr), 12.88 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 43.2 (CH2 piperazine), 52.3 (CH2 piperazine), 63.7 (ArCH2N), 78.7 (OC(CH3)3), 111.9 (C-3), 119.4 (d, J = 18.2 Hz), 124.9, 126.9 (d, J = 3.9 Hz), 128.0, 129.1 (d, J = 23.1 Hz), 129.2 (d, J = 5.1 Hz), 130, 131.9, 132.5, 147.9, 153.8 (CO2N), 153.9 (d, J = 248.5 Hz), 158.7 (CONHAr), 161.1, 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); HRMS (NSI) calcd for C26H28Cl2FN6O4 [M(35Cl35Cl) + H]+ 577.1528, found 577.1521.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(2-(piperazin-1-ylmethyl)pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32l)
Compound 32l was synthesized according to general procedure J using tert-butyl 4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyrimidin-2-yl)methyl)piperazine-1-carboxylate (32j) (60 mg, 0.10 mmol), triethylsilane (41 μL, 30 mg, 0.26 mmol), TFA (0.5 mL), and CH2Cl2 (0.5 mL). The crude yellow solid was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 6% MeOH/CH2Cl2 to yield an off-white solid (35 mg, 70%); Rf 0.29 (NH2 SiO2, 6% MeOH/CH2Cl2); mp 239.5–241.5 °C; λmax (EtOH)/nm 266.0, 293.2; IR νmax/cm–1 3069, 2932, 2812, 1639, 1581, 1558, 1510, 1444, 1389, 1268; 1H NMR (500 MHz, DMSO-d6) δ 2.41 (4H, brs, NCH2 piperazine), 2.70 (4H, t, J = 4.9 Hz, NCH2 piperazine), 3.63 (2H, s, ArCH2N), 7.48 (1H, s, H-3), 7.52 (1H, dd, J = 8.7, 1.4 Hz, H-5′), 7.66 (1H, s, H-5), 7.78 (1H, dd, J = 8.7, 8.4 Hz, H-4′), 9.08 (2H, s, H-4″, 6″), 10.41 (1H, s, CONHAr); 13C NMR (126 MHz, DMSO-d6) δ 45.3 (NCH2 piperazine), 53.6 (NCH2 piperazine), 64.5 (ArCH2N), 112.0 (C-3), 119.3 (d, J = 17.9 Hz, C-3′), 124.9 (C-2 or C-4), 126.9 (d, J = 3.8 Hz, C-5′), 128.3 (C-2 or C-4), 129.2 (d, J = 23.1 Hz, C-1′), 129.2 (d, J = 5.1 Hz, C-6′), 130.7 (C-5), 131.9 (C-4′), 132.4 (C-5″), 147.8 (C-4″, 6″), 153.8 (d, J = 248.5 Hz, C-2′), 158.9 (CONHAr), 161.3 (C-2″), 182.5 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES+) m/z 473.3 [M(35Cl35Cl) + H]+, 475.3 [M(35Cl37Cl) + H]+; HRMS (NSI) calcd for C21H20Cl2FN6O2 [M(35Cl35Cl) + H]+ 477.1003, found 477.0999.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(2-((4-methylpiperazin-1-yl)methyl)pyrimidin-5-yl)-1H-pyrrole-2-carboxamide (32m)
Compound 32m was synthesized according to general procedure H using tert-butyl 4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyrimidin-2-yl)methyl)piperazine-1-carboxylate (32j) (60 mg, 0.10 mmol), formic acid (0.5 mL), and formaldehyde (37% wt in water) (31 μL, 0.42 mmol). The crude yellow solid was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 6% MeOH/CH2Cl2 to yield a white solid (36 mg, 71%); Rf 0.30 (NH2 SiO2, 4% MeOH/CH2Cl2); mp 183.0–185.0 °C; λmax (EtOH)/nm 293.0; IR νmax/cm–1 2935, 2802, 1641, 1581, 1557, 1515, 1447, 1280; 1H NMR (500 MHz, DMSO-d6) δ 2.13 (3H, s, NCH3), 2.29 (4H, brs, NCH2 piperazine), 2.47 (4H, brs, NCH2 piperazine), 3.64 (2H, s, ArCH2N), 7.50 (1H, s, H-3), 7.52 (1H, dd, J = 8.8, 1.4 Hz, H-5′), 7.67 (1H, s, H-5), 7.79 (1H, dd, J = 8.8, 8.4 Hz, H-4′), 9.07 (2H, s, H-4″, 6″), 10.41 (1H, s, CONHAr), 12.85 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 45.7 (NCH3), 52.5 (NCH2 piperazine), 54.7 (NCH2 piperazine), 63.9 (ArCH2N), 111.9 (C-3), 119.4 (d, J = 18.1 Hz, C-3′), 124.8 (C-2 or C-4), 126.9 (d, J = 3.8 Hz, C-5′), 128.0 (C-2 or C-4), 129.1 (d, J = 23.2 Hz, C-1′), 129.2 (d, J = 5.4 Hz, C-6′), 130.5 (C-5), 131.9 (C-4′), 132.4 (C-5″), 147.9 (C-4″, 6″), 153.8 (d, J = 248.7 Hz, C-2′), 158.7 (CONHAr), 161.4 (C-2″), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES+) m/z 491.4 [M(35Cl35Cl) + H]+, 493.4 [M(35Cl37Cl) + H]+; HRMS (NSI) calcd for C22H22Cl2FN6O2 [M(35Cl35Cl) + H]+ 491.1160, found 491.1154.
tert-Butyl-4-(5-nitropyridin-2-yl)piperazine-1-carboxylate39 (9a)
Prepared according to general procedure F using N1-Boc-piperazine (2.35 g, 12.7 mmol), 2-chloro-5-nitropyridine (1.00 g, 6.3 mmol), and K2CO3 (1.75 g, 12.7 mmol) in THF (10 mL) for 72 h to give an orange oil (1.85 g, 97%); Rf 0.5 (SiO2, 50% EtOAc/petrol); mp168–170 °C (Lit.40 169 °C); λmax (EtOH)/nm 358, 228; IR νmax/cm–1 1688, 1594, 1338; 1H NMR (500 MHz; DMSO-d6) δH 1.46 (9H, s, C(CH3)3), 3.46–3.52 (4H, m, 4 × H-piperazine), 3.78–3.83 (4H, m, 4 × H-piperazine), 6.97 (1H, d, J = 9.6 Hz, H-3-pyridine), 8.29 (1H, dd, J = 2.8 and 9.6 Hz, H-4-pyridine), 9.01 (1H, d, J = 2.8 Hz, H-6-pyridine); 13C NMR (125 MHz; DMSO-d6) δC 28.0 (C(CH3)3), 44.1 (C-piperazine), 79.2 (C(CH3)3), 105.7 (C-3-pyridine), 132.9 (C-4-pyridine), 134.4 (C-5-pyridine), 146.0 (C-6-pyridine), 153.8 (C-2-pyridine), 160.1 (CO); HRMS calcd for C14H21N4O4 [M + H]+ 309.1557, found 309.1558.
tert-Butyl-4-(5-aminopyridin-2-yl)piperazine-1-carboxylate41 (10a)
Prepared according to general procedure D using nitropyridine 9a (1.83 g, 5.9 mmol), MeOH (60 mL), and EtOAc (60 mL) for 24 h to give a beige solid (1.65 g, 100%); Rf 0.65 (NH2 SiO2, EtOAc); mp 109 °C (dec.); λmax (EtOH)/nm 255; IR νmax/cm–1 3382, 3321, 2975.8, 2820, 1685; 1H NMR (500 MHz; DMSO-d6) δH 1.45 (9H, s, C(CH3)3), 3.19–3.25 (4H, m, 4 × H-piperazine), 3.40–3.47 (4H, m, 4 × H-piperazine), 4.64 (2H, br s, NH2), 6.68 (1H, d, J = 8.7 Hz, H-3-pyridine), 6.96 (1H, dd, J = 2.9 and 8.7 Hz, H-4-pyridine), 7.64 (1H, d, J = 2.9 Hz, H-6-pyridine); 13C NMR (125 MHz; DMSO-d6) δC 28.1 (C(CH3)3), 43.4 (C-piperazine), 46.3 (C-piperazine), 78.8 (C(CH3)3), 108.8 (C-3-pyridine), 124.4 (C-4-pyridine), 133.3 (C-5-pyridine), 137.6 (C-6-pyridine), 152.0 (C-2-pyridine), 153.9 (CO-carbamate); HRMS calcd for C14H21N4O2 [M – H]− 277.1670, found 277.1666.
tert-Butyl-4-(5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)piperazine-1-carboxylate (33a)
Prepared according to general procedure E using amine 10a (436 mg, 1.57 mmol), carboxylic acid 31b (190 mg, 0.63 mmol), cyanuric fluoride (16 μL, 0.19 mmol), pyridine (51 μL, 0.63 mmol), and MeCN (4 mL) with stirring at 40 °C for 18 h. Purification by MPLC on SiO2 with a gradient elution from 20 to 60% EtOAc/petrol gave a gray solid (160 mg, 45%); Rf 0.5 (NH2 SiO2, EtOAc); mp 159–160 °C; λmax (EtOH)/nm 293, 213; IR νmax/cm–1 1663, 1647; 1H NMR (500 MHz; DMSO-d6) δH 1.46 (9H, s, C(CH3)3), 3.34–3.38 (4H, br s, 8 × H-piperazine), 6.91 (1H, d, J = 9.0 Hz, H-3-pyridine), 7.46 (1H, br s, H-pyrrole), 7.55 (1H, dd, J = 1.3, 8.8 Hz, H-3′), 7.61 (1H, br s, H-pyrrole), 7.82 (1H, app t, J = 8.8 Hz, H-4′), 7.90 (1H, dd, J = 2.5, 9.0 Hz, H-4-pyridine′), 8.46 (1H, d, J = 2.5 Hz, H-6-pyridine), 10.01 (1H, s, CO-NH), 12.72 (1H, s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6, 110 °C) δC 28.7 (C(CH3)3), 43.6 (2 × CH2-piperazine), 45.6 (2 × CH2-piperazine), 79.5 (C(CH3)3), 107.4 (C-3-pyridine), 111.4 (CH-pyrrole), 119.4 (d, JCF = 18.1 Hz), 124.7 (C-pyrrole), 126.4 (C-pyridine), 126.9 (d, JCF = 3.6 Hz), 128.8 (C-pyrrole), 129.2 (d, JCF = 23.2 Hz), 129.2 (d, JCF = 5.0 Hz), 129.8 (CH-pyrrole), 131.0 (C-pyridine), 131.8, 140.0 (C-pyridine), 153.8 (d, JCF = 248.4 Hz), 153.9 (CO-carbamate), 155.6 (C-pyridine), 158.2 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C26H2735Cl2F1N5O4 [M + H]+ 562.1419, found 562.1415.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(6-(piperazin-1-yl)pyridin-3-yl)-1H-pyrrole-2-carboxamide (33f)
Prepared according to general procedure J using carbamate 33a (145 mg, 0.26 mmol), Et3SiH (102 μL, 0.64 mmol), TFA (1.5 mL), and CH2Cl2 (1.5 mL). The reaction was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 4% MeOH/EtOAc to give a yellow solid of 55 mg (46%); Rf 0.4 (NH2 SiO2, 5% MeOH/EtOAc); mp 195 °C (dec.); λmax (EtOH)/nm 293, 213; IR νmax/cm–1 1633; 1H NMR (500 MHz; DMSO-d6) δH 2.78–2.84 (4H, m, 4 × H-piperazine), 3.36–3.41 (4H, m, 4 × H-piperazine), 6.85 (1H, d, J = 9.1 Hz, H-3-pyridine), 7.45 (1H, s, H-pyrrole), 7.55 (1H, dd, J = 1.2, 8.6 Hz), 7.61 (1H, s, H-pyrrole), 7.82 (1H, app t, J = 8.6 Hz), 7.86 (1H, dd, J = 2.6, 9.1 Hz, H-4-pyridine), 8.43 (1H, d, J = 2.6 Hz, H-6-pyridine), 9.98 (1H, s, CO-NH); 13C NMR (125 MHz; DMSO-d6) δC 45.4 (2 × CH2-piperazine), 46.2 (2 × CH2-piperazine), 106.6 (C-3-pyridine), 110.7 (CH-pyrrole), 119.3 (d, JCF = 18.1 Hz, C-3′), 124.7 (C-pyrrole), 125.9 (C-pyridine), 126.9 (d, JCF = 3.7 Hz, C-5′), 128.9 (C-pyrrole), 129.2 (d, JCF = 5.1 Hz), 129.2 (d, JCF = 23.2 Hz), 129.8 (CH-pyrrole), 130.9 (C-2-pyridine), 131.8 (C-4-pyridine), 140.1 (C-6-pyridine), 153.8 (d, JCF = 248.4 Hz), 156.3 (C-5-pyridine), 158.2 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C21H1935Cl2F1N5O2 [M + H]+ 462.0894, found 462.0884.
tert-Butyl-4-(methyl(5-nitropyridin-2-yl)amino)piperidine-1-carboxylate (9b)
Prepared according to general procedure F using 2-chloro-5-nitropyridine (672 mg, 4.24 mmol) in THF (20 mL), triethylamine (650 μL, 472 mg, 4.67 mmol), and tert-butyl 4-(methylamino)piperidine-1-carboxylate (995 μL, 1.00 g, 4.67 mmol). The resulting solution was stirred at reflux overnight. The crude yellow solid was purified by MPLC on SiO2 with a gradient elution from 0 to 25% EtOAc/petrol to yield a yellow solid (1.07 g, 75%); Rf 0.31 (SiO2, 75% petrol/EtOAc); mp 158.5–160.5 °C; λmax (EtOH)/nm 368.6; IR νmax/cm–1 2963, 2926, 1691, 1595, 1571, 1509, 1477, 1410, 1334, 1295, 1241; 1H NMR (500 MHz, DMSO-d6) δ 1.41 (9H, s, C(CH3)3), 1.55–1.74 (4H, m), 2.84 (2H, brs), 2.98 (3H, s, NCH3), 4.06 (2H, brs), 4.79 (1H, brs), 6.81 (1H, d, J = 9.7 Hz), 8.22 (1H, dd, J = 9.7, 2.9 Hz), 8.97 (1H, d, J = 2.9 Hz); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 28, 30.3 (NCH3), 42.7, 78.8 (OC(CH3)3), 105.6, 132.7, 134.1, 146.0, 153.7 (CO2N), 160.1; HRMS (NSI) calcd for C16H25N4O4 [M + H]+ 337.1870, found 337.1871.
tert-Butyl-4-((5-aminopyridin-2-yl)(methyl)amino)piperidine-1-carboxylate (10b)
Prepared according to general procedure D using tert-butyl 4-(methyl(5-nitropyridin-2-yl)amino)piperidine-1-carboxylate (9b) (750 mg, 2.23 mmol), THF (22.5 mL), and MeOH (22.5 mL). The crude pale red solid (650 mg, 95%) was used in the next step without further purification; Rf 0.32 (SiO2, EtOAc); mp 108.0–110.0 °C; λmax (EtOH)/nm 257.0; IR νmax/cm–1 3411, 3339, 3232, 3004, 2945, 1673, 1562, 1494, 1412, 1290, 1270, 1243; 1H NMR (500 MHz, DMSO-d6) δ 1.40 (9H, s, C(CH3)3), 1.46–1.54 (4H, m, H-3′, 5′), 2.66 (3H, s, NCH3), 2.79 (2H, brs, H-2′, 6′), 4.03 (2H, brs, H-2′, 6′), 4.31–4.46 (3H, m, ArNH2 and H-4′), 6.46 (1H, d, J = 8.9 Hz, H-3), 6.91 (1H, dd, J = 8.9, 2.9 Hz, H-4), 7.58 (1H, d, J = 2.9 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 28.4 (C-3′, 5′), 29.9 (NCH3), 43.0 (C-2′, 6′), 52.4 (C-4′), 78.5 (OC(CH3)3), 106.9 (C-3), 125.1 (C-4), 133.5 (C-6), 135.6 (C-5), 151.6 (C-2), 153.8 (CO2N); MS (ES+) m/z 307.4 [M + H]+; HRMS (NSI) calcd for C16H27N4O2 [M + H]+ 307.2129, found 307.2128.
tert-Butyl-4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)(methyl)amino)piperidine-1-carboxylate (33b)
Compound 33b was synthesized according to general procedure I using 4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxylic acid (31b) (300 mg, 0.99 mmol), triethylamine (346 μL, 251 mg, 2.48 mmol), 2-chloro-1-methylpyridinium iodide (279 mg, 1.09 mmol), tert-butyl 4-((5-aminopyridin-2-yl)(methyl)amino)piperidine-1-carboxylate (10b) (380 mg, 1.24 mmol), and CH2Cl2 (9.9 mL). The crude yellow solid was purified by MPLC on SiO2 with a gradient elution from 0 to 50% EtOAc/petrol to yield a pale orange solid (285 mg, 49%); Rf 0.31 (SiO2, 50% petrol/EtOAc); mp 173.0–175.0 °C; λmax (EtOH)/nm 290.0; IR νmax/cm–1 3223, 2958, 2931, 2865, 1656, 1638, 1527, 1494, 1448, 1421, 1393, 1365, 1291; 1H NMR (500 MHz, DMSO-d6) δ 1.41 (9H, s, C(CH3)3), 1.52–1.63 (4H, m, CH2CH2NBoc), 2.79 (3H, s, NCH3), 2.81 (2H, brs, CH2CH2NBoc), 4.06 (2H, brs, CH2CH2NBoc), 4.58 (1H, tt, J = 10.5, 5.9 Hz, ArN (CH3)CH), 6.67 (1H, d, J = 9.1 Hz, H-3″), 7.41 (1H, s, H-3), 7.51 (1H, dd, J = 8.7, 1.4 Hz, H-5′), 7.56 (1H, s, H-5), 7.71–7.83 (2H, m, H-4′ and H-4″), 8.36 (1H, d, J = 2.7 Hz, H-6″), 9.91 (1H, s, CONHAr), 12.67 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 28.6 (CH2CH2NBoc), 29.6 (NCH3), 43.4 (CH2CH2NBoc), 51.9 (ArN (CH3)CH), 78.6 (OC(CH3)3), 105.6 (C-3″), 110.6 (C-3), 119.3 (d, J = 18.2 Hz, C-3′), 124.7 (C-2 or C-4 and C-5″), 126.9 (d, J = 3.3 Hz, C-5′), 128.9 (C-2 or C-4), 129.2 (d, J = 5.2 Hz, C-6′), 129.2 (d, J = 23.0 Hz, C-1′), 129.6 (C-5), 131.4 (C-4′ or C-4″), 131.8 (C-4′ or C-4″), 140.4 (C-6″), 153.8 (CO2N or C-2″), 153.8 (d, J = 248.4 Hz, C-2′), 155.2 (CO2N or C-2″), 158.2 (CONHAr), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7; MS (ES–) m/z 588.3 [M(35Cl35Cl)-H]−, 590.3 [M(35Cl37Cl)-H]−; HRMS (NSI) calcd for C28H31Cl2FN5O4 [M(35Cl35Cl) + H]+ 590.1732, found 590.1725.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(6-(methyl(1-methylpiperidin-4-yl)amino)pyridin-3-yl)-1H-pyrrole-2-carboxamide (33g)
Prepared according to general procedure H using tert-butyl 4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)(methyl)amino)piperidine-1-carboxylate (33b) (100 mg, 0.17 mmol), formic acid (0.85 mL), and formaldehyde (37% wt in water) (50 μL, 0.68 mmol). The crude yellow solid was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 3% MeOH/CH2Cl2 to yield a white solid (63 mg, 74%); Rf 0.32 (NH2 SiO2, 3% MeOH/CH2Cl2); mp 217.5–219.5 °C; λmax (EtOH)/nm 291.4; IR νmax/cm–1 3313, 2981, 2971, 2782, 1646, 1584, 1532, 1500, 1448, 1394, 1296, 1281, 1264; 1H NMR (500 MHz, DMSO-d6) δ 1.42 −1.57 (2H, m, CH2CH2NMe), 1.75 (2H, dddd, J = 12.2, 12.2, 12.2 and 3.8 Hz, CH2CH2NMe), 1.99 (2H, ddd, J = 12.2, 12.2 and 2.5 Hz, CH2CH2NMe), 2.17 (3H, s, NCH3), 2.81 (3H, s, NCH3), 2.82–2.85 (2H, m, CH2CH2NMe), 4.34 (1H, tt, J = 12.2, 3.8 Hz, ArN (CH3)CH), 6.64 (1H, d, J = 9.1 Hz, H-5″), 7.41 (1H, s, H-3), 7.51 (1H, dd, J = 8.8, 1.4 Hz, H-5′), 7.56 (1H, s, H-5), 7.75 (1H, dd, J = 9.1, 2.7 Hz, H-4″), 7.77 (1H, dd, J = 8.8, 8.5 Hz, H-4′), 8.35 (1H, d, J = 2.7 Hz, H-2″), 9.89 (1H, s, CONHAr), 12.66 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 28.4 (CH2CH2NMe), 29.6 (NCH3), 46.0 (NCH3), 51.8 (ArN (CH3)CH), 55.1 (CH2CH2NMe), 105.5 (C-5″), 110.6 (C-3), 119.3 (d, J = 18.1 Hz, C-3′), 124.5 (C-3″), 124.7 (C-2 or C-4), 126.9 (d, J = 3.8 Hz, C-5′), 128.9 (C-2 or C-4), 129.2 (d, J = 5.5 Hz, C-6′), 129.2 (d, J = 23.1 Hz, C-1′), 129.7 (C-5), 131.3 (C-4′ or C-4″), 131.8 (C-4′ or C-4″), 140.4 (C-2″), 153.8 (d, J = 247.2 Hz, C-2′), 155.3 (C-6″), 158.2 (CONHAr), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES–) m/z 502.3 [M(35Cl35Cl)-H]−, 504.3 [M(35Cl37Cl)-H]−; HRMS (NSI) calcd for C24H25Cl2FN5O2 [M(35Cl35Cl) + H]+ 504.1364, found 504.1352.
Diethyl-2-(5-nitropyridin-2-yl)malonate (18)
To a suspension of sodium hydride (60% dispersion in mineral oil, 4.04 g, 101 mmol) in THF (60 mL), cooled in an ice bath, was added diethyl malonate (7.66 mL, 8.08 g, 50.5 mmol). The resulting solution was stirred at 0 °C for 10 min and allowed to warm to RT. After 1 h, the reaction mixture was cooled in an ice bath and a solution of 2-chloro-5-nitropyridine (8.0 g, 50.5 mmol) in THF (20 mL) was added dropwise. The resulting mixture was then stirred overnight at RT. Upon completion, the mixture was diluted with EtOAc (40 mL), quenched by the cautious addition of 1 M hydrochloric acid (20 mL), and extracted with EtOAc (3 × 60 mL). The pooled organic extracts were washed with water and brine (40 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on SiO2 with a gradient elution from 0 to 15% EtOAc/petrol to yield a yellow solid (9.05 g, 64%); Rf 0.32 (SiO2, 15% EtOac/petrol); mp 86.5–88.5 °C (lit. 97.0–99.0 °C);42 λmax (EtOH)/nm 247.6, 272.4; IR νmax/cm–1 3129, 3074, 2986, 2939, 1662, 1637, 1588, 1529, 1509, 1341; 1H NMR (500 MHz, DMSO-d6) δ 1.19 (6H, t, J = 7.1 Hz, OCH2CH3), 4.19 (4H, qd, J = 7.1, 1.9 Hz, OCH2CH3), 5.41 (1H, s, ArCH(CO2Et)2), 7.77 (1H, d, J = 8.6 Hz, H-3), 8.64 (1H, dd, J = 8.6, 2.7 Hz, H-4), 9.33 (1H, d, J = 2.7 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 13.9 (OCH2CH3), 59.2 (ArCH(CO2Et)2), 61.8 (OCH2CH3), 125.0 (C-3), 132.4 (C-4), 143.7 (C-5), 144.3 (C-6), 158.9 (C-2), 166.5 (ArCH(CO2Et)2); MS (ES+) m/z 283.3 [M + H]+; HRMS (ESI) calcd for C12H15N2O6 [M + H]+ 283.0925, found 283.0917.
2-Methyl-5-nitropyridine (19)
To diethyl-2-(5-nitropyridin-2-yl)malonate (18) (8.0 g, 28.3 mmol) was added cold 20% aqueous sulfuric acid (80 mL). The resulting solution was stirred at 100 °C for 2 h. Upon completion, the mixture was cooled in an ice bath, neutralized by the cautious addition of 2 M aqueous sodium hydroxide until pH 8–9, and extracted with CH2Cl2 (3 × 100 mL). The pooled organic extracts were washed with water (100 mL) and brine (100 mL), dried over MgSO4, and concentrated in vacuo to give a pale yellow solid (3.71 g, 95%), which was used without further purification; Rf 0.32 (10% EtOAc/petrol); mp 109.5–110.5 °C (lit. 110–111);43 λmax (EtOH)/nm 252.8, 276.4; IR νmax/cm–1 3039, 3019, 2950, 2854, 1600, 1572, 1512, 1469; 1H NMR (500 MHz, DMSO-d6) δ 2.62 (3H, s, ArCH3), 7.57 (1H, d, J = 8.6 Hz, H-3), 8.48 (1H, dd, J = 8.6, 2.8 Hz, H-4), 9.24 (1H, d, J = 2.8 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 24.3 (ArCH3), 123.8 (C-3), 131.6 (C-4), 142.5 (C-5), 144.1 (C-6), 165.2 (C-2); MS (ES+) m/z 139.1 [M + H]+; HRMS (APCI) calcd for C6H7N2O2 [M + H]+ 139.0502, found 139.0500; 1H NMR, 13C NMR and IR data were identical to literature data.44,45
2-Methyl-5-nitropyridine-1-oxide (20)
To 2-methyl-5-nitropyridine (19) (2.0 g, 14.5 mmol) in CH2Cl2 (50 mL), cooled in an ice bath, was added 3-chloroperbenzoic acid (74%, 5.06 g, 21.7 mmol) in portions. The resulting solution was stirred in an ice bath for 1 h and allowed to warm to RT. After 16 h, the reaction was quenched by the addition of saturated aqueous NaHCO3 (50 mL) and stirred for 30 min. The aqueous layer was extracted with CH2Cl2 (3 × 40 mL). The pooled organic extracts were washed with water (50 mL) and brine (50 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on SiO2 with a gradient elution from 0 to 100% EtOAc/petrol to yield a yellow solid (2.15 g, 96%); Rf 0.27 (100% EtOAc); mp 149.5–151.5 °C; λmax (EtOH)/nm 247.8, 278.2; IR νmax/cm–1 3126, 3102, 3036, 2920, 1564, 1519, 1491, 1350, 1285; 1H NMR (500 MHz, DMSO-d6) δ 2.44 (3H, s, ArCH3), 7.76 (1H, d, J = 8.7 Hz, H-3), 8.05 (1H, dd, J = 8.7, 2.2 Hz, H-4), 9.01 (1H, d, J = 2.2 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 17.4 (ArCH3), 119.2 (C-4), 126.4 (C-3), 134.6 (C-6), 145.0 (C-5), 154.7 (C-2); MS (ES+) m/z 155.2 [M + H]+; HRMS (NSI) calcd for C6H7N2O3 [M + H]+ 155.0451, found 155.0448.
(5-Nitropyridin-2-yl)methanol (21)
To 2-methyl-5-nitropyridine 1-oxide (20) (1.0 g, 6.49 mmol) in CH2Cl2 (20 mL), cooled in an ice bath, was added dropwise trifluoroacetic anhydride (1.80 mL, 2.73 g, 13.0 mmol). The resulting solution was stirred in an ice bath for 1 h and allowed to warm to RT. After 16 h, the reaction was cooled in an ice bath, quenched by the addition of MeOH (15 mL), and stirred for 8 h. The volatiles were concentrated in vacuo. The crude residue was dissolved in EtOAc (30 mL), washed with saturated aqueous NaHCO3 (30 mL), and extracted with EtOAc (2 × 35 mL). The combined organic extracts were washed with water (40 mL) and brine (40 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on SiO2 with a gradient elution from 0 to 45% EtOAc/petrol to yield a yellow solid (500 mg, 50%); Rf 0.31 (45% EtOAc/petrol); mp 95.0–97.0 °C; λmax (EtOH)/nm 253.2, 276.2; IR νmax/cm–1 3161, 3040, 2917, 2852, 1596, 1575, 1515, 1451, 1434, 1345; 1H NMR (500 MHz, DMSO-d6) δ 4.69 (2H, s, ArCH2OH), 5.77 (1H, brs, ArCH2OH), 7.75 (1H, d, J = 8.7 Hz, H-3), 8.61 (1H, dd, J = 8.7, 2.7 Hz, H-4), 9.28 (1H, d, J = 2.7 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 64.0 (ArCH2OH), 120.4 (C-3), 132.1 (C-4), 143.0 (C-5), 143.9 (C-6), 168.9 (C-2); MS (ES+) m/z 155.2 [M + H]+; HRMS (APCI) calcd for C6H7N2O3 [M + H]+ 155.0451, found 155.0450.
5-Nitropicolinaldehyde (22)
To (5-nitropyridin-2-yl)methanol (21) (1.2 g, 7.79 mmol) in CH2Cl2 (30 mL) was added manganese oxide (6.77 g, 77.9 mmol). The resulting solution was stirred at RT for 16 h. Upon completion, the heterogeneous mixture was filtered through celite and washed with CH2Cl2 (15 mL). The filtrate was concentrated in vacuo to give a yellow solid. The crude solid was purified by MPLC on SiO2 with a gradient elution from 0 to 20% EtOAc/petrol to yield an orange solid (720 mg, 61%); Rf 0.28 (20% EtOAc/petrol); mp 65.0–67.0 °C (lit. 55 °C);44 λmax (EtOH)/nm 248.2, 273.6; IR νmax/cm–1 3102, 2981, 2889, 2845, 1712, 1598, 1528, 1349; 1H NMR (500 MHz, DMSO-d6) δ 8.16 (1H, dd, J = 8.5, 0.7 Hz, H-3), 8.80 (1H, dd, J = 8.5, 2.5 Hz, H-4), 9.56 (1H, d, J = 2.5 Hz, H-6), 10.08 (1H, d, J = 0.7 Hz, ArCHO); 13C NMR (126 MHz, DMSO-d6) δ 122.4 (C-3), 133.4 (C-4), 145.3 (C-6), 146.1 (C-5), 155.2 (C-2), 192.0 (ArCHO); MS (ES+) m/z 153.2 [M + H]+; HRMS (APCI) calcd for C6H5N2O3 [M + H]+ 153.0295, found 153.0292; 1H and 13C NMR data were identical to literature data.44
tert-Butyl-4-((5-nitropyridin-2-yl)methyl)piperazine-1-carboxylate (23)
To 5-nitropicolinaldehyde (22) (300 mg, 1.97 mmol) in TFE (10 mL) was added tert-butyl piperazine-1-carboxylate (367 mg, 1.97 mmol). The resulting solution was stirred at 38 °C for 1 h. Once cooled at 0 °C, sodium borohydride was carefully added. The resulting mixture was allowed to warm to RT and then stirred for 30 min. Upon completion, the solvent was removed in vacuo. The crude residue was dissolved in EtOAc (30 mL), neutralized by washing with saturated aqueous NH4Cl (20 mL), washed with water (20 mL) and brine (20 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on SiO2 with a gradient elution from 0 to 50% EtOAc/petrol to yield a white solid (325 mg, 51%); Rf 0.34 (petrol/EtOAc, 1:1); mp 107.5–109.5 °C; λmax (EtOH)/nm 246.4, 305.4; IR νmax/cm–1 2981, 2941, 2881, 2820, 1686, 1601, 1580, 1523, 1420, 1356, 1345; 1H NMR (500 MHz, DMSO-d6) δ 1.39 (9H, s, C(CH3)3), 2.40 (4H, t, J = 5.0 Hz, CH2 piperazine), 3.34 (4H, t, J = 5.0 Hz, CH2 piperazine), 3.76 (2H, brs, ArCH2N), 7.75 (1H, d, J = 8.6 Hz, H-3), 8.57 (1H, dd, J = 8.6, 2.7 Hz, H-4), 9.29 (1H, d, J = 2.7 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 43.0 (CH2 piperazine), 52.5 (CH2 piperazine), 63.0 (ArCH2N), 78.8 (OC(CH3)3), 123.1 (C-3), 132.0 (C-4), 143.2 (C-5), 144.1 (C-6), 153.8 (CO2N), 165.2 (C-2); MS (ES–) m/z 321.2 [M – H]−; HRMS (NSI) calcd for C15H23N4O4 [M + H]+ 323.1714, found 323.1712.
tert-Butyl-4-((5-aminopyridin-2-yl)methyl)piperazine-1-carboxylate (24)
Compound 24 was synthesized according to general procedure D using tert-butyl 4-((5-nitropyridin-2-yl)methyl)piperazine-1-carboxylate (23) (300 mg, 0.93 mmol), THF (9.3 mL), and MeOH (9.3 mL). The crude colorless oil (258 mg, 95%) was used in the next step without further purification; Rf 0.32 (100%, EtOAc); λmax (EtOH)/nm 246.4, 305.4; IR νmax/cm–1 3410, 3327, 3204, 2980, 2925, 2891, 2808, 2769, 1671, 1598, 1574, 1495, 1455, 1423; 1H NMR (500 MHz, DMSO-d6) δ 1.38 (9H, s, C(CH3)3), 2.29 (4H, t, J = 5.1 Hz, CH2 piperazine), 3.28 (4H, t, J = 5.1 Hz, CH2 piperazine), 3.39 (2H, s, ArCH2N), 5.18 (2H, brs, ArNH2), 6.88 (1H, dd, J = 8.3, 2.8 Hz, H-4), 7.03 (1H, d, J = 8.3 Hz, H-3), 7.83 (1H, d, J = 2.8 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 28.0 (C(CH3)3), 43.5 (CH2 piperazine), 52.4 (CH2 piperazine), 63.3 (ArCH2N), 78.7 (OC(CH3)3), 120.4 (C-4), 123.1 (C-3), 135.2 (C-6), 143.5 (C-2 or C-5), 144.6 (C-2 or C-5), 153.8 (CO2N); MS (ES+) m/z 293.5 [M + H]+; HRMS (NSI) calcd for C15H25N4O2 [M + H]+ 293.1972, found 293.1971.
tert-Butyl-4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)methyl)piperazine-1-carboxylate (33c)
Prepared according to general procedure I using 4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxylic acid (31b) (211 mg, 0.70 mmol), triethylamine (243 μL, 177 mg, 1.75 mmol), 2-chloro-1-methylpyridinium iodide (196 mg, 0.77 mmol), tert-butyl 4-((5-aminopyridin-2-yl)methyl)piperazine-1-carboxylate (24) (255 mg, 0.87 mmol), and CH2Cl2 (7 mL). The crude yellow solid was purified by MPLC on SiO2 with a gradient elution from 0 to 80% EtOAc/petrol to yield a pale orange solid (113 mg, 28%); Rf 0.28 (petrol/EtOAc, 8:2); mp 147.0–149.0 °C; λmax (EtOH)/nm 295.8; IR νmax/cm–1 3238, 2964, 2932, 2871, 2819, 1652, 1531, 1448, 1423, 1393; 1H NMR (500 MHz, DMSO-d6) δ 1.39 (9H, s, C(CH3)3), 2.32–2.42 (4H, m, CH2 piperazine), 3.33 (4H, brs, CH2 piperazine), 3.56 (2H, s, ArCH2N), 7.42 (1H, d, J = 8.5 Hz, H-3″), 7.48–7.54 (2H, m, H-3 and H-5′), 7.62 (1H, s, H-5), 7.78 (1H, dd, J = 8.7, 8.2 Hz, H-4′), 8.11 (1H, dd, J = 8.5, 2.6 Hz, H-4″), 8.80 (1H, d, J = 2.6 Hz, H-6″), 10.23 (1H, s, CONHAr), 12.77 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 43.2 (CH2 piperazine), 52.5 (CH2 piperazine), 63.2 (ArCH2N), 78.7 (OC(CH3)3), 111.4 (C-5), 119.3 (d, J = 18.0 Hz, C-3′), 122.7 (C-3″), 124.8 (C-2 or C-4), 126.9 (d, J = 3.7 Hz, C-5′), 127.6 (C-4″), 128.5 (C-2 or C-4), 129.1 (d, J = 22.9 Hz, C-1′), 129.2 (d, J = 5.3 Hz, C-6′), 130.3 (C-5), 131.9 (C-4′), 134.1 (C-5″), 140.7 (C-6″), 152.8 (CO2N or C-2″), 153.8 (CO2N or C-2″), 153.9 (d, J = 248.7 Hz, C-2′), 158.6 (CONHAr), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES+) m/z 576.5 [M(35Cl35Cl) + H]+, 578.5 [M(35Cl37Cl) + H]+; HRMS (NSI) calcd for C27H29Cl2FN5O4 [M(35Cl35Cl) + H]+ 576.1575, found 576.1568.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(6-((4-methylpiperazin-1-yl)methyl)pyridin-3-yl)-1H-pyrrole-2-carboxamide (33i)
Prepared according to general procedure H using tert-butyl 4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)methyl)piperazine-1-carboxylate (33c) (45 mg, 0.08 mmol), formic acid (0.4 mL), and formaldehyde (37% wt in water) (23 μL, 0.31 mmol). The crude yellow solid was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 5% MeOH/CH2Cl2 to yield a white solid (25 mg, 66%); Rf 0.28 (NH2 SiO2, 0–5% MeOH/CH2Cl2); mp 161.5–163.5 °C; λmax (EtOH)/nm 296.0; IR νmax/cm–1 3215, 2933, 2802, 1639, 1590, 1527, 1491, 1446, 1391; 1H NMR (500 MHz, DMSO-d6) δ 2.16 (3H, s, NCH3), 2.22–2.49 (8H, m, NCH2 piperazine), 3.53 (2H, s, ArCH2N), 7.39 (1H, d, J = 8.5 Hz, H-5″), 7.47–7.55 (2H, m, H-3 and H-5′), 7.61 (1H, s, H-5), 7.78 (1H, dd, J = 8.8, 8.4 Hz, H-4′), 8.10 (1H, dd, J = 8.5, 2.5 Hz, H-4″), 8.79 (1H, d, J = 2.5 Hz, H-2″), 10.22 (1H, s, CONHAr), 12.76 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 45.7 (NCH3), 52.6 (NCH2 piperazine), 54.7 (NCH2 piperazine), 63.3 (ArCH2N), 111.4 (C-3), 119.4 (d, J = 18.0 Hz), 122.6 (C-5″), 124.8 (C-2 or C-4), 126.9 (d, J = 3.7 Hz, C-5′), 127.6 (C-4″), 128.5 (C-2 or C-4), 129.1 (d, J = 23.2 Hz, C-1′), 129.2 (d, J = 4.9 Hz, C-6′), 130.2 (C-5), 131.9 (C-4′), 134.0 (C-3″), 140.7 (C-2″), 153.2 (C-6″), 153.9 (d, J = 248.5 Hz, C-2′), 158.6 (CONHAr), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES+) m/z 490.4 [M(35Cl35Cl) + H]+, 492.5 [M(35Cl37Cl) + H]+; HRMS (NSI) calcd for C23H23Cl2FN5O2 [M(35Cl35Cl) + H]+ 490.1207, found 490.1195.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(6-(piperazin-1-ylmethyl)pyridin-3-yl)-1H-pyrrole-2-carboxamide (33h)
Prepared according to general procedure J using tert-butyl 4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)methyl)piperazine-1-carboxylate (33c) (50 mg, 0.09 mmol), triethylsilane (35 μL, 25 mg, 0.22 mmol), TFA (0.45 mL), and CH2Cl2 (0.45 mL). The crude yellow solid was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 10% MeOH/CH2Cl2 to yield a white solid (30 mg, 73%); Rf 0.25 (NH2 SiO2, 0–10% MeOH/CH2Cl2); mp 179.5–181.5 °C; λmax (EtOH)/nm 296.0; IR νmax/cm–1 3105, 2921, 2812, 1637, 1589, 1525, 1490, 1445, 1389; 1H NMR (500 MHz, DMSO-d6) δ 2.33 (4H, brs, NCH2 piperazine), 2.70 (4H, t, J = 4.8 Hz, NCH2 piperazine), 3.51 (2H, s, ArCH2N), 7.40 (1H, d, J = 8.5 Hz, H-5″), 7.49 (1H, s, H-3), 7.52 (1H, dd, J = 8.8, 0.6 Hz, H-5′), 7.60 (1H, s, H-5), 7.78 (1H, dd, J = 8.8, 8.3 Hz, H-4′), 8.09 (1H, dd, J = 8.5, 2.6 Hz, H-4″), 8.79 (1H, d, J = 2.6 Hz, H-2″), 10.20 (1H, s, CONHAr); 13C NMR (126 MHz, DMSO-d6) δ 45.5 (NCH2 piperazine), 54.0 (NCH2 piperazine), 64.1 (ArCH2N), 111.4 (C-3), 119.3 (d, J = 17.9 Hz, C-3′), 122.6 (C-5″), 124.8 (C-2 or C-4), 126.9 (d, J = 3.7 Hz, C-5′), 127.5 (C-4″), 128.7 (C-2 or C-4), 129.2 (d, J = 23.0 Hz, C-1′), 129.2 (d, J = 5.2 Hz, C-6′), 130.4 (C-5), 131.8 (C-4′), 134.0 (C-3″), 140.6 (C-2″), 153.2 (C-6″), 154.8 (d, J = 248.8 Hz, C-2′), 158.7 (CONHAr), 182.5 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.6 (ArF); MS (ES+) m/z 476.5 [M(35Cl35Cl) + H]+, 478.5 [M(35Cl37Cl) + H]+; HRMS (NSI) calcd for C22H21Cl2FN5O2 [M(35Cl35Cl) + H]+ 476.1051, found 476.1040.
tert-Butyl-4-((5-nitropyridin-2-yl)methyl)piperidine-1-carboxylate (9d)
To a degassed sample of tert-butyl 4-methylidenepiperidine-1-carboxylate (750 mg, 3.80 mmol) was added 9-BBN (0.5 M in THF) (7.60 mL, 3.80 mmol). The resulting solution was sparged with nitrogen for 15 min and then reflux for 3 h. After cooling to RT, N,N-dimethylformamide (7 mL) and water (0.7 mL) were added and the resulting solution was sparged with nitrogen for 15 min. To the degassed mixture were added 2-chloro-5-nitropyridine (1.20 g, 7.60 mmol), potassium carbonate (788 mg, 5.70 mmol), and [1,1′-bis(diphenylphosphino)ferrocene]dichloropalladium(II) complex with CH2Cl2 (233 mg, 0.28 mmol). The resulting mixture was heated at 60 °C overnight. Upon completion, the heterogeneous mixture was filtered through celite and the solvent was removed in vacuo. The crude residue was dissolved in a mixture of EtOAc (30 mL) and water (30 mL) and extracted with EtOAc (3 × 30 mL). The combined organic extracts were washed with water (40 mL) and brine (40 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on SiO2 with a gradient elution from 0 to 25% EtOAc/petrol to yield a yellow solid (485 mg, 40%); Rf 0.31 (25% EtOAc/petrol/EtOAc); mp 80.5–82.5 °C; λmax (EtOH)/nm 254.8, 278.4; IR νmax/cm–1 3044, 2972, 2922, 2851, 1683, 1597, 1576, 1515, 1468, 1423, 1354, 1287, 1238; 1H NMR (500 MHz, DMSO-d6) δ 1.04–1.15 (2H, m, H-3′, 5′), 1.38 (9H, s, C(CH3)3), 1.48–1.56 (2H, m, H-3′, 5′), 1.91–2.03 (1H, m, H-4′), 2.66 (2H, brs, H-2′, 6′), 2.82 (2H, d, J = 7.1 Hz, ArCH2), 3.89 (2H, d, J = 13.2 Hz, H-2′, 6′), 7.56 (1H, d, J = 8.6 Hz, H-3), 8.50 (1H, dd, J = 8.6, 2.7 Hz, H-4), 9.29 (1H, d, J = 2.7 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 31.3 (C-3′, 5′), 35.8 (C-4′), 43.1 (C-2′, 6′), 44.0 (ArCH2), 78.4 (OC(CH3)3), 124.2 (C-3), 131.6 (C-4), 142.6 (C-5), 144.2 (C-6), 153.8 (CO2N), 167.0 (C-2); MS (ES+) m/z 320.3 [M + H]+; HRMS (NSI) calcd for C16H24N3O4 [M + H]+ 322.1761, found 322.1763.
tert-Butyl-4-((5-aminopyridin-2-yl)methyl)piperidine-1-carboxylate (10d)
Prepared according to general procedure D using tert-butyl 4-((5-nitropyridin-2-yl)methyl)piperidine-1-carboxylate (9d) (300 mg, 0.93 mmol), THF (9.3 mL), and MeOH (9.3 mL). The crude orange solid (261 mg, 96%) was used in the next step without further purification; Rf 0.26 (NH2 SiO2, 50% EtOAc/petrol); mp 107.5–109.5 °C; λmax (EtOH)/nm 243.6, 307.4; IR νmax/cm–1 3398, 3323, 3219, 2981, 2917, 2852, 1668, 1573, 1493, 1425, 1366; 1H NMR (500 MHz, DMSO-d6) δ 1.00 (2H, dddd, J = 12.3, 12.3, 12.3 and 4.3 Hz, H-3′, 5′), 1.38 (9H, s, C(CH3)3), 1.47–1.54 (2H, m, H-3′, 5′), 1.76 (1H, ttt, J = 12.3, 7.2 and 4.3 Hz, H-4′), 2.44 (2H, d, J = 7.2 Hz, ArCH2CH), 2.64 (2H, brs, H-2′, 6′), 3.88 (2H, d, J = 13.1 Hz, H-2′, 6′), 5.03 (2H, brs, ArNH2), 6.81–6.84 (2H, m, H-3 and H-4), 7.84 (1H, d, J = 1.7 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 31.5 (C-3′, 5′), 36.2 (C-4′), 42.9 (C-2′, 6′), 43.3 (ArCH2), 78.3 (OC(CH3)3), 120.4 (C-3 or C-4), 123.0 (C-3 or C-4), 135.7 (C-6), 142.5 (C-5), 146.8 (C-2), 153.8 (CO2N); MS (ES+) m/z 292.4 [M + H]+; HRMS (NSI) calcd for C16H26N3O2 [M + H]+ 292.2020, found 292.2019.
tert-Butyl-4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)methyl)piperidine-1-carboxylate (33d)
Prepared according to general procedure I using 4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxylic acid (31b) (250 mg, 0.83 mmol), triethylamine (288 μL, 209 mg, 2.07 mmol), 2-chloro-1-methylpyridinium iodide (233 mg, 0.91 mmol), tert-butyl 4-((5-aminopyridin-2-yl)methyl)piperidine-1-carboxylate (10d) (301 mg, 1.03 mmol), and CH2Cl2 (8.30 mL). The crude yellow solid was purified by MPLC on SiO2 with a gradient elution from 0 to 100% EtOAc/petrol to yield an orange solid (201 mg, 42%); Rf 0.29 (50% EtOAc/petrol); mp 132.0–134.0 °C; λmax (EtOH)/nm 295.6; IR νmax/cm–1 3238, 2926, 2853, 1651, 1592, 1531, 1448, 1424, 1394, 1366; 1H NMR (500 MHz, DMSO-d6) δ 1.01–1.10 (2H, m, CH2CH2NBoc), 1.38 (9H, s, C(CH3)3), 1.53 (2H, dd, J = 12.6, 3.6 Hz, CH2CH2NBoc), 1.83–1.90 (1H, m, ArCH2CH), 2.62 (2H, d, J = 7.1 Hz, ArCH2CH), 2.66 (2H, brs, CH2CH2NBoc), 3.90 (2H, d, J = 13.1 Hz, CH2CH2NBoc), 7.21 (1H, d, J = 8.4 Hz, H-3″), 7.50 (1H, s, H-3), 7.52 (1H, dd, J = 8.8, 1.2 Hz, H-5′), 7.61 (1H, s, H-5), 7.78 (1H, dd, J = 8.8, 8.4 Hz, H-4′), 8.02 (1H, dd, J = 8.4, 2.5 Hz, H-4″), 8.78 (1H, d, J = 2.5 Hz, H-6″), 10.18 (1H, s, CONHAr), 12.75 (1H, s, NH-pyrrole);13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 31.5 (CH2CH2NBoc), 36.0 (ArCH2CH), 43.6 (ArCH2CH), 43.9 (CH2CH2NBoc), 78.4 (OC(CH3)3), 111.3 (C-3), 119.3 (d, J = 17.8 Hz, C-3′), 123.1 (C-3″), 124.8 (C-2 or C-4), 126.9 (d, J = 3.7 Hz, C-5′), 127.5 (C-4″), 128.5 (C-2 or C-4), 129.1 (d, J = 22.8 Hz, C-1′), 129.2 (d, J = 5.4 Hz, C-6′), 130.2 (C-5), 131.9 (C-4′), 133.2 (C-5″), 141.1 (C-6″), 153.8 (CO2N or C-2″), 153.9 (d, J = 248.6 Hz, C-2′), 154.8 (CO2N or C-2″), 158.5 (CONHAr), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES+) m/z 573.4 [M(35Cl35Cl) + H]+, 575.4 [M(35Cl37Cl) + H]+; HRMS (NSI) calcd for C28H30Cl2FN4O4 [M(35Cl35Cl) + H]+ 575.1623, found 575.1616.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(6-((1-methylpiperidin-4-yl)methyl)pyridin-3-yl)-1H-pyrrole-2-carboxamide (33j)
Prepared according to general procedure H using tert-butyl 4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)methyl)piperidine-1-carboxylate (33d) (100 mg, 0.17 mmol), formic acid (0.85 mL), and formaldehyde (37% wt in water) (52 μL, 0.69 mmol). The crude yellow solid was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 4% MeOH/CH2Cl2 to yield an off-white solid (64 mg, 75%); Rf 0.26 (NH2 SiO2, 4% MeOH/CH2Cl2); mp 148.0–150.0 °C; λmax (EtOH)/nm 261.4, 296.0; IR νmax/cm–1 3286, 2926, 2846, 2788, 1646, 1592, 1525, 1493, 1447, 1392, 1282, 1237, 1224; 1H NMR (500 MHz, DMSO-d6) δ 1.11–1.31 (2H, m, CH2CH2NMe), 1.50 (2H, d, J = 12.0 Hz, CH2CH2NMe), 1.57–1.70 (1H, m, ArCH2CH), 1.77 (2H, dd, J = 12.0, 12.0 Hz, CH2CH2NMe), 2.11 (3H, s, NCH3), 2.59 (2H, d, J = 7.0 Hz, ArCH2CH), 2.70 (2H, d, J = 10.9 Hz, CH2CH2NMe), 7.20 (1H, d, J = 8.4 Hz, H-5″), 7.49 (1H, s, H-3), 7.52 (1H, d, J = 8.8 Hz, H-5′), 7.61 (1H, s, H-5), 7.78 (1H, dd, J = 8.8, 8.3 Hz, H-4′), 8.02 (1H, d, J = 8.4 Hz, H-4″), 8.78 (1H, s, H-2″), 10.17 (1H, s, CONHAr), 12.72 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 31.7 (CH2CH2NMe), 35.6 (ArCH2CH), 43.8 (ArCH2CH), 46.2 (NCH3), 55.3 (CH2CH2NMe), 111.3 (C-3), 119.3 (d, J = 18.1 Hz, C-3′), 123.0 (C-5″), 124.8 (C-2 or C-4), 126.9 (d, J = 3.7 Hz, C-5′), 127.4 (C-4″), 128.6 (C-2 or C-4), 129.2 (d, J = 23.0 Hz, C-1′), 129.2 (d, J = 5.0 Hz, C-3′), 130.2 (C-5), 131.8 (C-4′), 133.1 (C-3″), 141.0 (C-2″), 153.9 (d, J = 248.9 Hz, C-2′), 155.1 (CONHAr or C-6″), 158.6 (CONHAr or C-6″), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.6 (ArF); MS (ES–) m/z 487.3 [M(35Cl35Cl)-H]−, 489.3 [M(35Cl37Cl)-H]−; HRMS (NSI) calcd for C24H24Cl2FN4O2 [M(35Cl35Cl) + H]+ 489.1255, found 489.1243.
tert-Butyl-4-((5-nitropyridin-2-yl)oxy)piperidine-1-carboxylate46 (9c)
To a suspension of sodium hydride (60% dispersion in mineral oil, 246 mg, 6.15 mmol) in THF (20 mL), cooled in an ice bath, was added 1-Boc-4-hydroxypiperidine (1.24 g, 6.15 mmol). The resulting solution was stirred at 0 °C for 10 min and allowed to warm to RT. After 1 h, the reaction mixture was cooled in an ice bath and 2-chloro-5-nitropyridine (650 mg, 4.10 mmol) was added in small portions. The resulting mixture was then stirred overnight at RT. Upon completion, the mixture was diluted with EtOAc (20 mL), quenched by the cautious addition of saturated aqueous NaHCO3 (20 mL), and extracted with EtOAc (3 × 20 mL). The combined organic extracts were washed with water (40 mL) and brine (40 mL), dried over MgSO4, and concentrated in vacuo. The crude product was purified by MPLC on SiO2 with a gradient elution from 0 to 100% EtOAc/petrol to yield an off-white solid (1.03 g, 78%); Rf 0.30 (10% petrol/EtOAc); mp 109.5–111.5 °C; λmax (EtOH)/nm 295.0; IR νmax/cm–1 2961, 2924, 2873, 1675, 1605, 1579, 1513, 1473, 1425, 1349, 1318, 1273; 1H NMR (500 MHz, DMSO-d6) δ 1.41 (9H, s, C(CH3)3), 1.60 (2H, dddd, J = 12.9, 8.8, 8.8 and 4.0 Hz, H-3′, 5′axial), 2.03–1.90 (2H, m, H-3′, 5′equ), 3.20 (2H, brs, H-2′, 6′axial), 3.69 (2H, ddd, J = 12.9, 4.6 and 4.6 Hz, H-2′, 6′equ), 5.32 (1H, tt, J = 8.8, 4.0 Hz, H-4′), 7.02 (1H, d, J = 9.1 Hz, H-3), 8.47 (1H, dd, J = 9.1, 2.9 Hz, H-4), 9.07 (1H, d, J = 2.9 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 28.0 (C(CH3)3), 30.2 (C-3′, 5′), 40.6 (C-2′, 6′) 72.3 (C-4′), 78.8 (OC(CH3)3), 111.8 (C-3), 134.9 (C-4), 139.3 (C-5), 144.6 (C-6), 153.9 (CO2N), 165.9 (C-2); HRMS (NSI) calcd for C15H22N3O5 [M + H]+ 324.1554, found 324.1553; 1H NMR data were identical to literature data.
tert-Butyl-4-((5-aminopyridin-2-yl)oxy)piperidine-1-carboxylate (10c)
Prepared according to general procedure D using tert-butyl 4-((5-nitropyridin-2-yl)oxy)piperidine-1-carboxylate (9c) (800 mg, 2.47 mmol), THF (24.7 mL), and MeOH (24.7 mL). The crude pale yellow solid (700 mg, 96%) was used in the next step without further purification; Rf 0.29 (100% EtOAc); mp 160.0–162.0 °C; λmax (EtOH)/nm 236.6, 313.8; IR νmax/cm–1 3384, 3335, 2980, 2961, 2925, 2865, 1681, 1485, 1418, 1369, 1270, 1249, 1236; 1H NMR (500 MHz, DMSO-d6) δ 1.40 (9H, s, C(CH3)3), 1.46 (2H, dddd, J = 13.2, 9.2, 9.1 and 4.1 Hz, H-3′, 5′axial), 1.87 (2H, ddd, J = 13.2, 5.8 and 3.3 Hz, H-3′, 5′equ), 3.12 (2H, brs, H-2′, 6′axial), 3.66 (2H, ddd, J = 13.2, 4.9 and 4.9 Hz, H-2′, 6′equ), 4.74 (2H, brs, ArNH2), 4.93 (1H, tt, J = 8.3, 3.8 Hz, H-4′), 6.51 (1H, d, J = 8.6 Hz, H-3), 6.98 (1H, dd, J = 8.6, 2.9 Hz, H-4), 7.48 (1H, d, J = 2.9 Hz, H-6); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 30.7 (C-3′, 5′), 40.6 (C-2′, 6′), 69.3 (C-4′), 78.6 (OC(CH3)3), 110.9 (C-3), 126.3 (C-4), 131.1 (C-6), 139.4 (C-5), 153.9 (C-2 or CO2N), 154.3 (C-2 or CO2N); MS (ES+) m/z 294.4 [M + H]+; HRMS (ESI) calcd for C15H24N3O3 [M + H]+ 294.1812, found 294.1811.
tert-Butyl-4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)oxy)piperidine-1-carboxylate (33e)
Prepared according to general procedure J using 4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxylic acid (31b) (300 mg, 0.99 mmol), triethylamine (346 μL, 251 mg, 2.48 mmol), 2-chloro-1-methylpyridinium iodide (279 mg, 1.09 mmol), tert-butyl 4-((5-aminopyridin-2-yl)oxy)piperidine-1-carboxylate (10e) (364 mg, 1.24 mmol), and CH2Cl2 (9.9 mL). The crude yellow solid was purified by MPLC on SiO2 with a gradient elution from 0 to 40% EtOAc/petrol to yield a pale orange solid (275 mg, 48%); Rf 0.30 (60% petrol/EtOAc); mp 226.5–228.5 °C; λmax (EtOH)/nm 261.6; IR νmax/cm–1 3166, 2980, 2936, 2857, 1658, 1647, 1593, 1556, 1538, 1484, 1433, 1365, 1263;1H NMR (500 MHz, DMSO-d6) δ 1.41 (9H, s, C(CH3)3), 1.53 (2H, dddd, J = 13.0, 9.1, 9.0 and 4.0 Hz, CH2CH2NBoc), 1.94 (2H, ddd, J = 13.0, 5.9 and 3.4 Hz, CH2CH2NBoc), 3.16 (2H, brs, CH2CH2NBoc), 3.70 (2H, ddd, J = 13.0, 4.8 and 4.8 Hz, CH2CH2NBoc), 5.13 (1H, tt, J = 8.3, 3.8 Hz, ArOCH), 6.81 (1H, d, J = 8.9 Hz, H-3″), 7.45 (1H, s, H-3), 7.52 (1H, dd, J = 8.7, 1.3 Hz, H-5′), 7.59 (1H, s, H-5), 7.78 (1H, dd, J = 8.7, 8.5 Hz, H-4′), 7.98 (1H, dd, J = 8.9, 2.7 Hz, H-4″), 8.44 (1H, d, J = 2.7 Hz, H-6″), 10.10 (1H, s, CONHAr), 12.73 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 28.1 (C(CH3)3), 30.6 (CH2CH2NBoc), 40.7 (CH2CH2NBoc), 70.0 (ArOCH), 78.7 (OC(CH3)3), 110.8 (C-3″), 111.0 (C-3), 119.3 (d, J = 18.1 Hz, C-3′), 124.7 (C-2 or C-4), 126.9 (d, J = 3.4 Hz, C-5′), 128.6 (C-2 or C-4), 129.2 (d, J = 23.3 Hz, C-1′), 129.2 (d, J = 5.3 Hz, C-6′), 129.5 (C-5″), 130.0 (C-5), 131.8 (C-4′), 132.5 (C-4″), 138.6 (C-6″), 153.8 (d, J = 248.3 Hz, C-2′), 153.9 (CO2N), 158.4 (CONHAr or C-2″), 158.7 (CONHAr or C-2″), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES–) m/z 575.3 [M(35Cl35Cl)-H]−, 577.3 [M(35Cl37Cl)-H]−; HRMS (NSI) calcd for C27H28Cl2FN4O5 [M(35Cl35Cl) + H]+ 577.1415, found 577.1408.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(6-((1-methylpiperidin-4-yl)oxy)pyridin-3-yl)-1H-pyrrole-2-carboxamide (33k)
Prepared according to general procedure H using tert-butyl 4-((5-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)pyridin-2-yl)oxy)piperidine-1-carboxylate (33e) (100 mg, 0.17 mmol), formic acid (0.85 mL), and formaldehyde (37% wt in water) (52 μL, 0.69 mmol). The crude yellow solid was purified by MPLC on NH2 SiO2 with a gradient elution from 0 to 3% MeOH/CH2Cl2 to yield a white solid (60 mg, 71%); Rf 0.28 (NH2 SiO2, 3% MeOH/CH2Cl2); mp 189.5–191.5 °C; λmax (EtOH)/nm 263.0; IR νmax/cm–1 3357, 2981, 2971, 1670, 1635, 1588, 1528, 1485, 1450, 1289, 1275, 1231; 1H NMR (500 MHz, DMSO-d6) δ 1.64 (2H, dddd, J = 12.9, 9.3, 9.3 and 3.6 Hz, CH2CH2NMe), 1.89–2.00 (2H, m, CH2CH2NMe), 2.14 (2H, dd, J = 12.9, 12.9 Hz, CH2CH2NMe), 2.17 (3H, s, NCH3), 2.63 (2H, ddd, J = 12.9, 4.5 and 4.5 Hz, CH2CH2NMe), 4.92 (1H, tt, J = 8.6, 3.9 Hz, ArOCH), 6.79 (1H, d, J = 8.9 Hz, H-5″), 7.45 (1H, d, J = 1.9 Hz, H-3), 7.52 (1H, dd, J = 8.8, 1.4 Hz, H-5′), 7.59 (1H, d, J = 1.9 Hz, H-5), 7.78 (1H, dd, J = 8.8, 8.4 Hz, H-4′), 7.96 (1H, dd, J = 8.9, 2.7 Hz, H-4″), 8.43 (1H, d, J = 2.7 Hz, H-2″), 10.09 (1H, s, CONHAr), 12.72 (1H, s, NH-pyrrole); 13C NMR (126 MHz, DMSO-d6) δ 30.7 (CH2CH2NMe), 45.8 (NCH3), 52.8 (CH2CH2NMe), 70.2 (ArOCH), 110.8 (C-5″), 111.0 (C-3), 119.3 (d, J = 18.2 Hz, C-3′), 124.7 (C-2 or C-4), 126.9 (d, J = 3.8 Hz, C-5′), 128.6 (C-2 or C-4), 129.2 (d, J = 24.0 Hz, C-1′ and C-3″), 129.2 (d, J = 5.2 Hz, C-6′), 130.0 (C-5), 131.8 (C-4′), 132.5 (C-4″), 138.6 (C-2″), 153.8 (d, J = 248.6 Hz, C-2′), 158.4 (CONHAr or C-6″), 158.9 (CONHAr or C-6″), 182.6 (ArCO); 19F NMR (471 MHz, DMSO-d6) δ −116.7 (ArF); MS (ES–) m/z 489.3 [M(35Cl35Cl)-H]−, 491.2 [M(35Cl37Cl)-H]−; HRMS (NSI) calcd for C23H22Cl2FN4O3 [M(35Cl35Cl) + H]+ 491.1048, found 491.1038.
1H-Pyrazol-4-amine47 (28a)
Prepared according to general procedure D using 4-nitropyrazole (500 mg, 4.40 mmol) and MeOH (30 mL) to give a red gum (360 mg, 98%); λmax (EtOH)/nm 238; IR νmax/cm–1 3374, 3114, 2955, 2891, 2842, 1585; 1H NMR (500 MHz; DMSO-d6) δH 7.03 (2H, s, 2 × H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 122.5, 130.0; MS: No mass ion detected.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1H-pyrazol-4-yl)-1H-pyrrole-2-carboxamide (34a)
Prepared according to general procedure E using amine 28a (69 mg, 0.83 mmol), carboxylic acid 31b (100 mg, 0.33 mmol), cyanuric fluoride (20 μL, 0.24 mmol), pyridine (27 μL, 0.34 mmol), and MeCN (2 mL) with stirring at RT for 18 h. Purification by MPLC on SiO2 with a gradient elution from 2 to 8% MeOH/EtOAc gave a yellow solid (78 mg, 64%); Rf 0.35 (NH2 SiO2, 5% MeOH/EtOAc); mp 300–304 °C; λmax (EtOH)/nm 255, 223; IR νmax/cm–1 3361.0, 3241.5 br, 2971.9, 1636.3, 1586.2; 1H NMR (500 MHz; DMSO-d6) δH 7.36 (1H, s, H-pyrrole), 7.56 (1H, dd, J = 1.1 & 8.5 Hz, H-5′), 7.60 (1H, s, H-pyrrole), 7.57 (1H, s, H-pyrazole), 7.82 (1H, app t, J = 8.5 Hz, H-4′), 7.97 (1H, s, H-pyrazole), 10.29 (1H, s, CO-NH), 12.60–12.73 (2H, m, NH-pyrrole and NH-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 110.1 (CH-pyrrole), 119.3 (d, JCF = 18.1 Hz, C-3′), 120.8 (CH-pyrazole), 124.7 (C-pyrrole), 126.9 (d, JCF = 4.1 Hz, C-5′), 128.8 (C-pyrrole), 129.2 (d, JCF = 5.2 Hz, C-6′), 129.2 (d, JCF = 22.7 Hz, C-1′), 129.5 (C-pyrrole), 131.0 (CH-pyrazole), 131.8 (C-4′), 153.8 (d, JCF = 248.8 Hz, C-2′), 156.9 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C15H1035Cl2F1N4O2 [M + H]+ 367.0159, found 367.0158.
1-Methyl-4-nitro-1H-pyrazole48 (27a)
Prepared according to general procedure G using MeOH (11 μL, 2.65 mmol), PPh3 (1.04 g, 4.0 mmol), 4-nitropyrazole (300 mg, 2.7 mmol), and DEAD (626 μL, 4.0 mmol) in THF (5 mL) with stirring at RT for 18 h. The residue was purified by MPLC on SiO2 with a gradient elution from 20 to 50% EtOAc/petrol to give an impure product, which was repurified by MPLC on SiO2 with a gradient elution from 10 to 40% EtOAc/petrol to give a white solid (227 mg, 67%); Rf 0.80 (SiO2, EtOAc); mp 90–94 °C (Lit.48 91–92 °C); λmax (EtOH)/nm 266; IR νmax/cm–1 1504, 1310; 1H NMR (500 MHz; DMSO-d6) δH 3.92 (3H, s, CH3), 8.25 (1H, s, H-pyrazole), 8.86 (1H, s, H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 39.6 (CH3), 131.0 (CH-pyrazole), 134.8 (CH-pyrazole), 135.5 (C-NO2). MS (ES+) 128.1 [M + H]+.
1-Methyl-1H-pyrazol-4-amine (28b)
Prepared according to general procedure D using nitropyrazole 27a (200 mg, 1.57 mmol) and MeOH (10 mL) for 2 h to give an orange oil (150 mg, 98%); Rf 0.10 (SiO2, 100% EtOAc); λmax (EtOH)/nm 244; IR νmax/cm–1 3322, 3111; 1H NMR (500 MHz; DMSO-d6) δH 3.68 (3H, s, CH3), 3.82 (2H, br s, NH2), 6.90 (1H, d, J = 0.6 Hz, H-pyrazole), 7.01 (1H, d, J = 0.6 Hz, H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 38.3 (CH3), 117.2 (CH-pyrazole), 128.9 (CH-pyrazole), 131.0 (C-pyrazole); MS: No mass ion detected.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1-methyl-1H-pyrazol-4-yl)-1H-pyrrole-2-carboxamide (34b)
Prepared according to general procedure E using amine 28b (130 mg, 1.3 mmol) and carboxylic acid 31b (162 mg, 0.54 mmol), cyanuric fluoride (32 μL, 0.37 mmol), pyridine (43 μL, 0.50 mmol), and MeCN (2 mL) with stirring at RT for 18 h. Purification by MPLC on SiO2 with a gradient elution from 30 to 60% EtOAc/petrol gave a yellow solid (120 mg, 59%); Rf 0.10 (SiO2, 50% EtOAc/petrol); mp 216–219 °C; λmax (EtOH)/nm 252, 225; IR νmax/cm–1 3195, 3121, 2938, 1630; 1H NMR (500 MHz; DMSO-d6) δH 3.85 (3H, s, CH3), 7.36 (1H, s, H-pyrrole), 7.54 (1H, s, H-pyrazole), 7.56 (1H, dd, J = 1.2 and 8.5 Hz, H-5′), 7.59 (1H, s, H-pyrrole), 7.82 (1H, app t, J = 8.5 Hz, H-4′), 7.98 (1H, s, H-pyrazole), 10.28 (1H, s, CO-NH), 12.68 (NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 38.7 (CH3), 110.2 (CH-pyrrole), 119.3 (d, JCF = 18.2 Hz, C-3′), 121.2 (CH-pyrazole), 121.4 (C-pyrazole), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.7 Hz, C-5′), 128.7 (C-pyrrole), 129.2 (d, JCF = 23.2 Hz, C-1′), 129.2 (d, JCF = 5.2 Hz, C-6′), 129.5 (C-pyrrole), 129.9 (CH-pyrazole), 131.8 (C-4′), 153.8 (d, JCF = 248.4 Hz, C-2′), 156.8 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C16H1235Cl2F1N4O2 [M + H]+ 381.0319, found 381.0318.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1-methyl-1H-pyrazol-3-yl)-1H-pyrrole-2-carboxamide (34c)
Prepared according to general procedure C using carboxylic acid 31b (100 mg, 0.33 mmol), 3-amino-5-methylpyrazole (113 mg, 1.16 mmol), PCl3 (29 μL, 0.33 mmol), and MeCN (1.50 mL). Purification by MPLC on SiO2 with a gradient elution from 50 to 80% EtOAc/petrol gave a white solid, which was repurified by MPLC on NH2 SiO2 with a gradient elution from 50 to 80% EtOAc/petrol to give a white solid (85 mg, 67%); Rf 0.30 (SiO2, 75% EtOAc/petrol); mp 240–242 °C; λmax (EtOH)/nm 254sh; IR νmax/cm–1 3200, 3131, 1636, 1574; 1H NMR (500 MHz; DMSO-d6) δH 3.81 (3H, s, CH3), 6.57 (1H, d, J = 2.2 Hz, H-pyrazole), 7.55 (1H, dd, J = 1.4 and 8.6 Hz, H-5′), 7.56 (1H, br s, H-pyrrole), 7.60 (1H, br s, H-pyrrole), 7.62 (1H, d, J = 2.2 Hz, H-pyrazole), 7.81 (1H, app t, J = 8.6 Hz, H-4′), 10.75 (1H, s, CO-NH), 12.64 (1H, br s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 38.3 (CH3), 97.2 (C-4-pyrazole), 111.5 (CH-pyrrole), 119.2 (d, JCF = 18.2 Hz, C-3′), 124.7 (C-pyrrole), 126.8 (d, JCF = 3.6 Hz, C-5′), 128.5 (CH-pyrrole), 129.2 (d, JCF = 5.1 Hz, C-6′), 129.3 (d, JCF = 23.2 Hz, C-1′), 129.6 (C-pyrrole), 130.9 (C-5-pyrazole), 131.7 (C-4′), 146.6 (C-3-pyrazole), 153.8 (d, JCF = 248.4 Hz, C-2′), 157.4 (CO-NH), 170.3, 182.5 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.6; HRMS calcd for C16H1235Cl2F1N4O2 [M + H]+ 381.0316, found 381.0313.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(3-methylisoxazol-5-yl)-1H-pyrrole-2-carboxamide (34d)
Prepared according to general procedure C using carboxylic acid 31b (75 mg, 0.25 mmol), 5-amino-3-methylisoxazole (85 mg, 0.87 mmol), PCl3 (22 μL, 0.25 mmol), and MeCN (1 mL). Purification by MPLC on SiO2 with a gradient elution from 10 to 40% EtOAc/petrol gave a white solid (32 mg, 34%); Rf 0.60 (SiO2, 40% EtOAc/petrol); mp 228 °C dec.; λmax (EtOH)/nm 294, 240; IR νmax/cm–1 3253, 3126, 3055, 1680, 1640; 1H NMR (500 MHz; DMSO-d6) δH 2.25 (3H, s, CH3), 6.29 (1H, s, H-isoxazole), 7.57 (1H, dd, J = 1.3 and 8.4 Hz, H-3′), 7.62 (1H, br s, H-pyrrole), 7.74 (1H, s, H-pyrrole), 7.83 (1H, app t, J = 8.6 Hz, H-4′), 11.80 (1H, s, CO-NH), 12.94 (1H, s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 11.3 (CH3), 89.2 (CH-isoxazole), 113.0 (CH-pyrrole), 119.3 (d, JCF = 18.0 Hz, C-3′), 124.9 (C-pyrrole), 126.9 (d, JCF = 3.8 Hz, C-5′), 127.1 (C-pyrrole), 129.0 (d, JCF = 23.0 Hz, C-1′), 129.2 (d, JCF = 5.1 Hz, C-6′), 130.7 (C-pyrrole), 131.9 (C-4′), 153.8 (d, JCF = 248.4 Hz, C-2′), 156.3 (CO-NH), 160.7 (C-isoxazole), 161.1 (C-isoxazole), 182.6, (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.6; HRMS calcd for C16H1135Cl2F1N3O3 [M + H]+ 382.0156, found 382.0153.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1-methyl-1H-imidazol-4-yl)-1H-pyrrole-2-carboxamide (34e)
Prepared according to general procedure C using carboxylic acid 31b (125 mg, 0.33 mmol), 1-methyl-1H-imidazol-4-amine (100 mg, 1.03 mmol), PCl3 (29 μL, 0.33 mmol), and MeCN (1.50 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 0 to 4% MeOH/EtOAc gave a white solid (30 mg, 24%); Rf 0.70 (5% MeOH/EtOAc); mp 258–260 °C; λmax (EtOH)/nm 227, 250 sh; IR νmax/cm–1 3262, 3122, 1657, 1637; 1H NMR (500 MHz; DMSO-d6) δH 3.68 (3H, CH3), 7.33 (1H, s, H-imidazole), 7.47 (1H, s, H-imidazole), 7.52–7.60 (3H, m, H-5′ and 2 × H-pyrrole), 7.81 (1H, app t, J = 8.3 Hz, H-4′), 10.67 (1H, s, CO-NH), 12.61 (1H, s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 33.1 (CH3), 108.2 (CH-imidazole), 111.2 (CH-pyrrole), 119.2 (d, JCF = 18.3 Hz, C-3′), 124.7 (C-pyrrole), 126.8 (d, JCF = 3.6 Hz, C-5′), 128.5 (CH-pyrrole), 129.2 (d, JCF = 5.4 Hz, C-6′), 129.3 (d, JCF = 22.7 Hz, C-1′), 131.7 (C-4′), 133.9 (CH-imidazole), 137.6 (C-imidazole), 153.8 (d, JCF = 248.4 Hz, C-2′), 156.6 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.2; HRMS calcd for C16H1235Cl2F1N4O2 [M + H]+ 381.0316, found 381.0315.
tert-Butyl-4-(4-nitro-1H-pyrazol-1-yl)piperidine-1-carboxylate49,50 (27b)
Prepared according to general procedure G using Boc-4-piperidinol (889 mg, 4.4 mmol), PPh3 (1.73 g, 6.6 mmol), 4-nitropyrazole (500 mg, 4.4 mmol), and DEAD (1.04 mL, 5.3 mmol) in THF (10 mL). The residue was purified by MPLC on SiO2 with a gradient elution from 20 to 40% EtOAc/petrol to give a white solid (880 mg, 67%); Rf 0.50 (SiO2, 20% EtOAc/petrol); mp 116–118 °C; λmax (EtOH)/nm 274; IR νmax/cm–1 3099, 2971, 2875, 1671, 1301; 1H NMR (500 MHz; DMSO-d6) δH 1.45 (9H, s, C(CH3)3), 1.85 (2H, qd, J = 4.2 and 11.6 Hz, 2 × H-piperidine), 2.04–2.11 (2H, m, 2 × H-piperidine), 2.80–3.07 (2H, m, 2 × H-piperidine), 3.99–4.19 (2H, m, 2 × H-piperidine), 4.50 (1H, tt, J = 4.2 and 11.6 Hz, CH2CHCCH2-piperidine), 8.32 (1H, s, H-pyrazole), 9.00 (1H, s, H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 28.0 (C(CH3)3), 31.3 (2 × CH2-piperidine), 42.4 (2 × CH2-piperidine), 59.5 (CH-N-piperidine), 78.9 (C(CH3)3), 128.9 (C-pyrazole), 134.8 (C-pyrazole), 135.3 (C-pyrazole), 153.7 (CO); HRMS calcd for C13H19N4O4 [M – H]− 295.1412, found 295.1408.
tert-Butyl-4-(4-amino-1H-pyrazol-1-yl)piperidine-1-carboxylate49,50 (28c)
Prepared according to general procedure D using nitropyrazole 27b (310 mg, 1.05 mmol) in MeOH (15 mL) and EtOAc (15 mL) for 3 h to give a white solid (279 mg, 100%); Rf 0.40 (NH2 SiO2, 100% EtOAc); mp 86–89 °C dec.; λmax (EtOH)/nm 248; IR νmax/cm–1 3238, 2972, 2930, 2865, 1697, 1669; 1H NMR (500 MHz; DMSO-d6) δH 1.45 (9H, s, C(CH3)3), 1.71 (2H, qd, J = 4.2 and 11.7 Hz, 2 × H-piperidine), 1.90–1.97 (2H, m, 2 × H-piperidine), 2.75–3.04 (2H, m, 2 × H-piperidine), 3.80 (2H, br s, NH2), 3.96–4.09 (2H, m, 2 × H-piperidine), 4.16 (1H, tt, J = 4.2 and 11.7 Hz, CH2CHCCH2-piperidine), 6.94 (1H, s, H-pyrazole), 7.10 (1H, s, H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 28.0 (C(CH3)3), 31.9 (2 × CH2-piperidine), 43.2 (2 × CH2-piperidine), 57.6 (CHN-piperidine), 78.7 (C(CH3)3), 114.4 (CH-pyrazole), 128.9 (CH-pyrazole), 131.1 (C-pyrazole), 153.8 (CO); HRMS calcd for C13H23N4O2 [M + H]+ 267.1816, found 267.1815.
tert-Butyl-4-(4-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)-1H-pyrazol-1-yl)piperidine-1-carboxylate (34f)
Prepared according to general procedure E using amine 28c (200 mg, 0.75 mmol), carboxylic acid 31b (91 mg, 0.30 mmol), cyanuric fluoride (18 μL, 0.21 mmol), pyridine (24 μL, 0.30 mmol), and MeCN (2 mL). Purification by MPLC on SiO2 with a gradient elution from 40 to 70% EtOAc/petrol gave a yellow oil (125 mg, 76%); Rf 0.35 (SiO2, 70% EtOAc/petrol); mp 144 °C dec.; IR νmax/cm–1 3123 br, 2975, 1637; 1H NMR (500 MHz; DMSO-d6) δH 1.46 (9H, s, C(CH3)3), 1.80 (2H, app qd, J = 4.1 and 11.6 Hz, 2 × H-piperidine), 1.97–2.04 (2H, m, 2 × H-piperidine), 2.82–3.02 (2H, m, 2 × H-piperidine), 4.01–4.12 (2H, m, 2 × H-piperidine), 4.38 (1H, tt, J = 3.9 and 11.5 Hz, CH2CHCCH2-piperidine), 7.36 (1H, s, CH-pyrrole), 7.55 (1H, dd, J = 1.1 and 8.6 Hz, H-5′), 7.58–7.62 (2H, m, H-pyrrole and H-pyrazole), 7.82 (1H, app t, J = 8.6 Hz, H-4′), 8.02 (1H, s, H-pyrazole), 10.30 (1H, s, CO-NH), 12.67 (1H, s, NH-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 28.0 (C(CH3)3), 31.9 (2 × CH2-piperidine), 42.8 (2 × CH2-piperidine), 58.0 (CHN-piperidine), 78.8 (C(CH3)3), 110.2 (CH-pyrrole), 118.9 (CH-pyrazole), 119.3 (d, JCF = 18.1 Hz, C-3′), 120.9 (C-pyrazole), 124.7 (C-pyrrole), 126.9 (d, JCF = 4.1 Hz, C-5′), 128.7 (C-pyrrole), 129.2 (JCF = 23.1 Hz, C-1′), 129.2 (JCF = 5.1 Hz, C-6′), 129.6 (C-pyrrole), 130.0 (CH-pyrazole), 131.8 (C-4′), 153.8 (JCF = 248.4 Hz, C-2′), 153.8 (CO-carbamate), 156.8 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C25H2735Cl2F1N5O4 [M + H]+ 550.1419, found 550.1414.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1-(piperidin-4-yl)-1H-pyrazol-4-yl)-1H-pyrrole-2-carboxamide (34h)
Prepared according to general procedure J using carbamate 34f (110 mg, 0.20 mmol), Et3SiH (80 μL, 0.50 mmol), TFA (1 mL), and CH2Cl2 (1 mL). The residue was purified by MPLC on NH2 SiO2 with a gradient elution from 1 to 4% MeOH/CH2Cl2 to give a white solid (62 mg, 69%); Rf 0.30 (NH2 SiO2, 10% MeOH/CH2Cl2); mp 199 °C dec.; λmax (EtOH)/nm 255, 223; IR νmax/cm–1 3122 br, 2949, 1633, 1592; 1H NMR (500 MHz; DMSO-d6) δH 1.78 (2H, app qd, J = 3.9 and 11.8 Hz, 2 × H-piperidine), 1.91–1.98 (2H, m, 2 × H-piperidine), 2.57–2.66 (2H, m, 2 × H-piperidine), 3.01–3.11 (2H, m, 2 × H-piperidine), 4.21 (1H, tt, J = 4.1 and 11.7 Hz, N-CH-piperidine), 7.35 (1H, s, H-pyrrole), 7.56 (1H, d, J = 8.6 Hz, H-5′), 7.57–7.61 (2H, m, H-pyrrole and H-pyrazole), 7.82 (1H, app t, J = 8.6 Hz, H-4′), 7.98 (1H, s, H-pyrazole), 10.27 (1H, s, CO-NH); 13C NMR (125 MHz; DMSO-d6) δC 33.5 (2 × CH2-piperidine), 45.1 (2 × CH2-piperidine), 59.0 (CHN-piperidine), 110.2 (CH-pyrrole), 118.4 (CH-pyrazole), 119.3 (d, JCF = 18.0 Hz, C-3′), 120.8 (C-pyrazole), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.6 Hz, C-5′), 128.8 (CH-pyrrole), 129.2 (JCF = 22.9 Hz, C-1′), 129.2 (JCF = 4.8 Hz, C-6′), 129.7 (C-pyrrole), 130.0 (CH-pyrazole), 131.8 (C-4′), 153.8 (JCF = 248.5 Hz, C-2′), 156.9 (CO-NH), 182.5 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C20H1935Cl2F1N5O2 [M + H]+ 450.0894, found 450.0888.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1-(1-methylpiperidin-4-yl)-1H-pyrazol-4-yl)-1H-pyrrole-2-carboxamide (34i)
Prepared according to general procedure H using carbamate 34f (75 mg, 0.137 mmol), formic acid (1.5 mL), and formaldehyde (44 μL, 0.55 mmol). The residue was purified by MPLC on NH2 SiO2 with a gradient elution from 2 to 6% MeOH/EtOAc to give a white solid (63 mg, 100%); Rf 0.25 (5% MeOH/EtOAc); mp 220–222 °C; λmax (EtOH)/nm 252; IR νmax/cm–1 3121, 2938, 2788, 1631; 1H NMR (500 MHz; DMSO-d6) δH 1.93–2.03 (4H, m, 4 × H-piperidine), 2.09–2.21 (2H, m, 2 × H-piperidine), 2.28 (3H, s, NCH3), 2.88–2.98 (2H, m, 2 × H-piperidine), 4.10–4.211 (1H, m, N-CH-piperidine), 7.36 (1H, s, H-pyrrole), 7.56 (1H, dd, J = 1.3 and 8.6 Hz, H-5′), 7.58–7.61 (2H, m, H-pyrrole and H-pyrazole), 7.82 (1H, app t, J = 8.6 Hz, H-4′), 8.01 (1H, s, H-pyrazole), 10.29 (1H, s, CO-NH), 12.67 (1H, s, NH-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 31.8 (2 × CH2-piperidine), 45.5 (N-CH3), 54.0 (2 × CH2-piperidine), 57.8 (CHN-piperidine), 110.1 (CH-pyrrole), 118.8 (CH-pyrazole), 119.3 (d, JCF = 18.2 Hz, C-3′), 120.8 (C-pyrazole), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.7 Hz, C-5′), 128.7 (CH-pyrrole), 129.2 (d, JCF = 23.1 Hz, C-1′), 129.2 (d, JCF = 4.8 Hz, C-6′), 129.5 (C-pyrrole), 129.9 (CH-pyrazole), 131.8 (C-4′), 154.8 (d, JCF = 248.5 Hz, C-2′), 156.8 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C21H2135Cl2F1N5O2 [M + H]+ 464.1051, found 464.1043.
tert-Butyl-4-((4-nitro-1H-pyrazol-1-yl)methyl)piperidine-1-carboxylate (27c)
Prepared according to general procedure G using Boc-4-piperidinemethanol (951 mg, 4.4 mmol), PPh3 (1.74 g, 6.6 mmol), 4-nitropyrazole (500 mg, 4.4 mmol), and DEAD (1.04 mL, 6.6 mmol) in THF (10 mL). The residue was purified by MPLC on SiO2 with a gradient elution from 10 to 60% EtOAc/petrol to give a white solid (1.135 g, 82%); Rf 0.35 (SiO2, 40% EtOAc/petrol); mp 158–160 °C; λmax (EtOH)/nm 271; IR νmax/cm–1 1665, 1506, 1312.; 1H NMR (500 MHz; DMSO-d6) δH 1.10 (2H, qd, J = 4.1 and 12.6 Hz, 2 × H-piperidine), 1.42 (9H, s, C(CH3)3), 1.45–1.53 (2H, m, 2 × H-piperidine), 2.02–2.12 (1H, m, H-piperidine), 2.61–2.82 (m, 2 × CH-N-piperidine), 3.88–4.01 (2H, m, 2 × CH-N-piperidine), 4.13 (2H, d, J = 7.2 Hz, N-CH2-CH), 8.31 (H-pyrazole), 8.92 (H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 28.0 (C(CH3)3), 28.7 (C-piperidine), 35.9 (2 × C-piperidine), 42.8 (2 × C-piperidine), 57.2 (N-CH2-CH), 78.5 (C(CH3)3), 130.8 (CH-pyrazole), 134.7 (C-4-pyrazole), 135.6 (CH-pyrazole), 153.8 (CO); HRMS calcd for C14H21O4N4 [M – H]− 309.1568, found 309.1565.
tert-Butyl-4-((4-amino-1H-pyrazol-1-yl)methyl)piperidine-1-carboxylate (28d)
Prepared according to general procedure D using nitropyrazole 27c (1.1 g, 3.5 mmol) in MeOH (40 mL) and EtOAc (40 mL) for 5 h to give an orange solid (945 mg, 95%); Rf 0.35 (NH2 SiO2, 100% EtOAc); mp 104–107 °C; λmax (EtOH)/nm 247; IR νmax/cm–1 2966, 2929, 1672; 1H NMR (500 MHz; DMSO-d6) δH 1.04 (2H, qd, J = 4.3 and 12.3 Hz, 2 × H-piperidine), 1.42 (9H, s, C(CH3)3), 1.42–1.49 (2H, m, 2 × H-piperidine), 1.85–1.97 (1H, m, H-piperidine), 2.58–2.82 (m, 2 × CH-N-piperidine), 3.79–3.87 (4H, m, N-CH2-CH and NH2), 3.88–3.99 (2H, m, 2 × H-N-piperidine), 6.92 (H-pyrazole), 7.03 (H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 28.1 (C(CH3)3), 29.0 (C-piperidine), 36.7 (2 × C-piperidine), 42.5 (2 × C-piperidine), 56.2 (N-CH2-CH), 78.4 (C(CH3)3), 117.0 (C-2-pyrazole), 129.2 (C-4-pyrazole), 130.5 (C-3-pyrazole), 153.8 (CO); HRMS calcd for C14H25N4O4 [M + H]+ 281.1972, found 281.1972.
tert-Butyl-4-((4-(4-(3,6-dichloro-2-fluorobenzoyl)-1H-pyrrole-2-carboxamido)-1H-pyrazol-1-yl)methyl)piperidine-1-carboxylate (34g)
Prepared according to general procedure E using amine 28d (450 mg, 1.6 mmol), carboxylic acid 31a (195 mg, 0.64 mmol), cyanuric fluoride (18 μL, 0.21 mmol), and pyridine (52 μL, 0.64 mmol) in MeCN (2 mL) with stirring at RT for 18 h. Purification by MPLC on SiO2 with a gradient elution from 50 to 100% EtOAc/petrol gave a yellow solid (214 mg, 59%); Rf 0.10 (NH2 SiO2, 100% EtOAc); mp 211 °C dec.; λmax (EtOH)/nm 254; IR νmax/cm–1 3126, 2977, 2932, 2860, 1645, 1592; 1H NMR (500 MHz; DMSO-d6) δH 1.09 (2H, qd, J = 3.6 and 12.3 Hz, 2 × CH-piperidine), 1.42 (9H, s, C(CH3)3), 1.44–1.52 (2H, m, 2 × H-piperidine), 1.94–2.07 (1H, m, H-piperidine), 2.60–2.80 (m, 2 × CH-N-piperidine), 3.87–4.01 (2H, m, 2 × CH-N-piperidine), 4.02 (2H, d, J = 7.1 Hz, N-CH2-CH), 7.36 (1H, s, H-pyrrole), 7.56 (1H, dd, J = 1.3 and 8.6 Hz, H-4′), 7.58 (s, H-pyrazole), 7.60 (1H, s, H-pyrrole), 7.82 (1H, app t, J = 8.6 Hz, H-3′), 7.99 (1H, s, H-pyrazole), 10.29 (1H, s, CO-NH), 12.66 (1H, s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 28.1 (C(CH3)3), 30.0 (C-piperidine), 36.6 (2 × C-piperidine), 42.8 (2 × C-piperidine), 56.3 (N-CH2-CH), 78.5 (C(CH3)3), 110.1 (CH-pyrrole), 119.3 (d, JCF = 18.0 Hz, C-3′), 120.9 (C-pyrazole), 121.2 (C-pyrazole), 124.7 (C-pyrrole), 126.9 (d, JCF = 3.8 Hz, C-5′), 128.7 (C-pyrrole), 129.2 (d, JCF = 23.0 Hz, C-1′), 129.2 (d, JCF = 5.1 Hz, C-6′), 129.5 (C-pyrrole), 130.2 (C-3-pyrazole), 131.8 (C-4′), 153.8 (CO-carbamate), 153.8 (d, JCF = 248.5 Hz, C-2′), 156.8 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C26H2935Cl2F1N5O4 [M + H]+ 564.1575, found 564.1566.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1-(piperidin-4-ylmethyl)-1H-pyrazol-4-yl)-1H-pyrrole-2-carboxamide (34j)
Prepared according to general procedure J using carbamate 34y (200 mg, 0.35 mmol), Et3SiH (283 μL, 1.77 mmol), TFA (1.5 mL), and CH2Cl2 (1.5 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 3 to 15% MeOH/EtOAc gave a white solid (106 mg, 64%); Rf 0.05 (SiO2, 5% MeOH/EtOAc); mp 147 °C dec.; λmax (EtOH)/nm 254; IR νmax/cm–1 2926, 1633, 1592; 1H NMR (500 MHz; DMSO-d6) δH 1.09 (2H, qd, J = 3.6 and 12.0 Hz, 2 × H-piperidine), 1.39–1.47 (2H, m, 2 × H-piperidine), 1.82–1.94 (1H, m, H-piperidine), 2.43 (td, J = 12.0 and 2.2 Hz, 2 × CH-N-piperidine), 2.90–2.97 (2H, m, 2 × CH-N-piperidine), 3.97 (2H, d, J = 7.1 Hz, pyrazole-CH2-piperidine), 7.33 (1H, s, H-pyrrole), 7.55 (1H, dd, J = 1.3 and 8.7 Hz, H-4′), 7.57 (1H, s, H-pyrazole), 7.58 (1H, s, H-pyrrole), 7.82 (1H, app t, J = 8.7 Hz, H-3′), 7.97 (1H, s, H-pyrazole), 10.26 (1H, s, CO-NH); 13C NMR (125 MHz; DMSO-d6) δC 30.2 (C-piperidine), 37.3 (2 × C-piperidine), 45.5 (2 × C-piperidine), 57.2 (pyrazole-CH2-piperidine), 110.2 (CH-pyrrole), 119.3 (d, JCF = 18.2 Hz, C-3′), 120.9 (C-pyrazole), 121.1 (C-pyrazole), 124.7 (C-pyrrole), 126.8 (d, JCF = 3.8 Hz, C-5′), 129.1 (C-pyrrole), 129.2 (d, JCF = 11.6 Hz, C-6′), 129.2 (CH-pyrrole), 129.3 (d, JCF = 20.5 Hz, C-1′), 130.0 (C-pyrazole), 131.7 (C-4′), 153.8 (d, JCF = 248.86 Hz, C-2′), 157.0 (CO-NH), 182.4 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.6; HRMS calcd for C21H2135Cl2F1N5O2 [M + H]+ 464.1051, found 464.1038.
1-Methyl-4-((4-nitro-1H-pyrazol-1-yl)methyl)piperidine (27d)
Prepared according to general procedure G using 1-methyl-4-piperidinemethanol (349 μL, 2.7 mmol), PPh3 (1.04 g, 4.0 mmol), 4-nitropyrazole (300 mg, 2.7 mmol), and DEAD (626 μL, 4.0 mmol) in THF (6 mL). Purification by MPLC on NH2 SiO2 with a gradient elution from 30 to 80% CH2Cl2/petrol gave a white solid. This solid was dissolved in 5% MeOH/CH2Cl2 and passed through an SCX ion-exchange column, eluting with 5% MeOH/CH2Cl2 followed by 80:20:2 CH2Cl2/MeOH/NH4OH to give a beige solid (200 mg, 34%). Rf 0.30 (NH2 SiO2, 100% CH2Cl2); mp 70–73 °C; λmax (EtOH)/nm 274; IR νmax/cm–1 3067, 2943, 2903, 2797, 1511, 1312; 1H NMR (500 MHz; DMSO-d6) δH 1.24 (2H, qd, J = 3.6 and 12.5 Hz, 2 × H-piperidine), 1.42–1.50 (2H, m, 2 × H-piperidine), 1.77–1.89 (3H, m, 3 × H-piperidine), 2.16 (3H, s, CH3), 2.72–2.80 (2H, m, 2 × H-piperidine), 4.11 (2H, d, J = 7.3 Hz, pyrazole-CH2-piperidine), 8.30 (1H, s, H-pyrazole), 8.93(1H, s, H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 28.9 (2 × CH2-piperidine), 35.5 (CH-piperidine), 46.0 (CH3), 54.6 (2 × CH2-piperidine), 57.5 (N-CH2-CH), 130.8 (CH-pyrazole), 134.7 (C-pyrazole), 135.6 (CH-pyrazole); HRMS calcd for C10H17N4O2 [M + H]+ 225.1346, found 225.1341.
1-((1-Methylpiperidin-4-yl)methyl)-1H-pyrazol-4-amine (28e)
Prepared according to general procedure D using nitropyrazole 27d (190 mg, 0.85 mmol) and MeOH (20 mL) for 2 h to give a pale brown oil (150 mg, 91%); Rf 0.40 (NH2 SiO2, 7% MeOH/CH2Cl2); mp 50–55 °C; λmax (EtOH)/nm 246; IR νmax/cm–1 3304, 3127, 2919, 2794; 1H NMR (500 MHz; DMSO-d6) δH 1.18 (2H, qd, J = 12.5 and 3.7 Hz, 2 × H-piperidine), 1.39–1.47 (2H, m, 2 × H-piperidine), 1.61–1.72 (1H, m, H-piperidine), 1.74–1.84 (2H, m, 2 × H-piperidine), 2.15 (3H, s, CH3), 2.70–2.78 (2H, m, 2 × H-piperidine), 3.80 (2H, d, J = 7.3 Hz, N-CH2-CH), 6.91 (1H, s, H-pyrazole), 7.02 (1H, s, H-pyrazole); 13C NMR (125 MHz; DMSO-d6) δC 29.3 (2 × CH2-piperidine), 36.3 (2 × CH2-piperidine), 46.1 (CH3), 54.9 (2 × CH2-piperidine), 56.5 (N-CH2-CH), 116.9 (CH-pyrazole), 129.0 (CH-pyrazole), 130.5 (C-pyrazole); HRMS calcd for C10H18N4 [M + H]+ 195.1604, found 195.1600.
4-(3,6-Dichloro-2-fluorobenzoyl)-N-(1-((1-methylpiperidin-4-yl)methyl)-1H-pyrazol-4-yl)-1H-pyrrole-2-carboxamide (34k)
Amine 28e (140 mg, 0.43 mmol) was added to carboxylic acid 31b (87 mg, 0.29 mmol), pyridine (23 μL, 0.29 mmol), and bromotripyrrolidinophosphonium hexafluorophosphate (PyBrOP) (200 mg, 0.43 mmol) in MeCN (2 mL). The mixture was stirred at RT for 1 h and then partitioned between EtOAc (2 × 30 mL) and H2O (20 mL). The organic layers were combined, washed with brine, dried (MgSO4), and the solvent was removed in vacuo. Purification by MPLC on NH2 SiO2 with gradient elution from 1 to 4% MeOH/CH2Cl2 gave a beige solid (53 mg, 39%); Rf 0.40 (NH2 SiO2, 7% MeOH/CH2Cl2); mp 124–128 °C; λmax (EtOH)/nm 252; IR νmax/cm–1 2935, 1639, 1592; 1H NMR (500 MHz; DMSO-d6) δH 1.23 (2H, qd, J = 3.0 and 12.5 Hz, 2 × H-piperidine), 1.41–1.51 (2H, m, 2 × H-piperidine), 1.70–1.85 (3H, m, 3 × H-piperidine), 2.15 (CH3), 2.72–2.80 (2H, m, 2 × H-piperidine), 4.00 (2H, d, J = 7.3 Hz, N-CH2-CH), 7.35 (1H, s, H-pyrrole), 7.53–7.61 (3H, m, H-4′, H-pyrrole and H-pyrazole), 7.82 (1H, app t, J = 8.4 Hz, H-3′), 7.97 (1H, s, H-pyrazole), 10.28 (1H, s, CO-NH), 12.52 (1H, br s, NH-pyrrole); 13C NMR (125 MHz; DMSO-d6) δC 29.2 (2 × C-piperidine), 36.2 (C-piperidine), 46.1 (CH3), 54.8 (2 × C-piperidine), 56.7 (N-CH2-CH), 110.5 (CH-pyrrole), 121.0 (d, JCF = 29.7 Hz, C-3′), 121.3 (CH-pyrazole), 124.7 (C-pyrrole), 126.9 (d, JCF = 4.1 Hz, C-5′), 128.2 (C-pyrrole), 128.7 (CH-pyrrole), 129.2 (d, JCF = 21.8 Hz, C-1′), 129.2 (d, JCF = 5.4 Hz, C-6′), 130.1 (C-1′), 130.5 (C-pyrazole), 131.8 (C-4′), 153.8 (d, JCF = 248.2 Hz, C-2′), 156.8 (CO-NH), 182.6 (CO); 19F NMR (470 MHz; DMSO-d6) δF −116.7; HRMS calcd for C22H2335Cl2F1N5O2 [M + H]+ 478.1207, found 478.1195.
Biological Assay Protocols
ERK5 IMAP Assay Protocol
Preparation of Assay Buffer (1×)
The 0.01% Tween-20 5× stock was supplied as part of the IMAP FP progressive binding system kit (Molecular Devices R7436) and was diluted to 1× using Milli-Q H2O. One microliter of a 1 M dithiothreitol (DTT) stock was added for every 1 mL of a 1× assay buffer to give a final concentration of 1 mM DTT. Preparation of ERK5 working solution. The final dilution was dependent on the activity of the enzyme batches. The initial batch (08/08/08) was used as a 1 in 1 in a 350 final dilution in assay buffer. A 1:175 dilution of ERK5 stock was performed in a 1× assay buffer. For 1 plate, 13 μL of ERK5 stock was added to 2262 μL of a 1× assay buffer. Aliquots were stored at −80 °C. Batch PO080808 was used at a stock concentration of 73.4 ng/μL.
Preparation of ATP/Substrate Working Solution
For one plate, ATP disodium salt (90 μL, 20 mM) (Sigma-Aldrich A7699) and FAM-EGFR-derived peptide (15 μL, 100 μM) (LVEPLTPSGEAPNQ(K-5FAM)-COOH) (Molecular Devices RP7129; reconstituted in Milli-Q H2O to a stock concentration of 100 μL; stored at −20°C) were added to 2295 μL of a 1× assay buffer.
Preparation of IMAP Binding Solution
For one plate, 20.5 μL of IMAP binding reagent stock, 1476 μL of 1× binding buffer A (60%), and 984 μL of binding buffer B (40%) [IMAP FP Progressive screening express kit (Molecular Devices R8127)] were added to 9819.5 μL of Milli-Q H2O.
Assay Procedure
One microliter of compound (in 60:40 H2O/DMSO) or 60:40 H2O/DMSO (for controls and blanks) was dry-spotted into the relevant wells of a 384-well assay plate using a MATRIX PlateMate Plus. Five microliters of ERK5 working solution was added to test and control wells, and 5 μL of a 1× assay buffer was added to blanks; 4 μL of ATP/substrate working solution was added to all wells using a Matrix multichannel pipette. The plate was sealed using DMSO-resistant 205 clear seal and incubated for 2 h at 37 °C. One microliter of the kinase reaction mixture from the first plate was dry-spotted into a second 384-well assay plate using the MATRIX PlateMate Plus. Nine microliters of assay buffer was added, followed by 30 μL of IMAP binding solution using a multichannel pipette. The plate was incubated at RT in darkness for 2 h. The assay plate was then read on an Analyst HT plate reader (Molecular Devices) using the settings described below; measurement mode = fluorescence polarization; method ID = ERK5; integration time = 100 ms; excitation filter = fluorescein 485–20; emission filter = 530–25; dichroic mirror = 505 nm; plate definition file = Corning 384 black fb; Z-height = 5.715 mm (middle); G-factor = 1; attenuator = out; detector counting = Smartread+; and sensitivity = 2.
p38α LANCE Assay Protocol
Preparation of Assay Buffer (1×)
A 1× assay buffer was prepared freshly from the following reagents: 250 mM tris(hydroxymethyl)aminomethane (Tris) pH 7.5, 25 mM MgCl2, 2.5 mM ethylene glycol tetraacetic acid (EGTA), 10 mM dithiothreitol (DTT), and 0.05% Triton X100 in Milli-Q H2O (NB: 1× buffer final assay concentrations were 5× lower than stated above).
Preparation of p38α/SAPK2 Working Solution
The p38α/SAPK2, active N-terminal GST-tagged recombinant full-length protein (Millipore 14–251) was supplied as a 10 μg/4 μL stock. This was diluted to a 10 μg/40 μL (1 μM) concentration by the addition of 156 μL of tris/HCl (pH 7.5, 50 mM), NaCl (150 mM), EGTA (0.1 mM), Brij-35 surfactant (0.03%), glycerol (50%), and 0.1% 2-mercaptoethanol (0.1%). The final dilution was dependent on the activity of the enzyme batches. The p38α concentration used in the assay was 1 nM. A 2× working stock solution (2 nM, 500-fold dilution of 1 μM stock) in a 1× assay buffer was prepared. For one plate, 9.4 μL of p38α (1 μM) was added to 1870.6 μL of Milli-Q H2O.
Preparation of ATP/Substrate Working Solution
For one plate, ATP disodium salt (17.5 μL, 200 mM stock) (Sigma-Aldrich A7699) and Ulight-MBP Peptide (50 μL, 5 μM stock) (Perkin Elmer TRF0109) were added to 400 μL of 5× assay buffer and 1532.5 μL of Milli-Q H2O.
Preparation of Ethylenediaminetetraacetic Acid (EDTA)/Antibody Detection Reagent
For one plate, 84 μL of ethylenediaminetetraacetic acid (EDTA) (0.5 M) (Sigma-Aldrich E4378-100G) and 27 μL of Europium-anti-phospho-MBP antibody (0.625 μM) (Perkin Elmer) were added to 420 μL of LANCE detection buffer (1×) and 3669 of Milli-Q H2O.
Assay Procedure
One microliter of compound (in 80:20 H2O/DMSO) or 80:20 H2O/DMSO was dry-spotted into the relevant wells of a 384-well assay plate using the MATRIX PlateMate Plus. Five microliters of p38α working solution was added to test and control wells, and 5 μL of assay buffer was added to blanks; 4 μL of the ATP/substrate working solution was added to all wells using a Thermo Multidrop Combi or Matrix multichannel pipette. The plate was sealed using DMSO-resistant clear seal and incubated for 1 h at 37 °C. Ten microliters of the EDTA/antibody working solution was added to all wells using a Thermo Multidrop Combi or Matrix multichannel pipette. The plate was incubated at RT in darkness for 2 h. The assay plate was then read on a PheraStar microplate reader using the settings described below; Pherastar: measurement mode = TRF; method ID = LANCE HTRF ERK5; optic module: 337, 665, 620 nm. Focal height = 6.0, positioning delay, 0.1 s, number of flashes per well = 100, integration start = 60.
Cell Growth Inhibition Assays
Human cell lines were obtained from the American Type Culture Collection (ATCC) and maintained at 37 °C in 5% CO2 with 95% humidity. Cells were cultured in RPMI-1640 medium containing 2 mM l-glutamine and 10% (v/v) fetal bovine serum (Life Technologies). Cell lines were authenticated by short tandem repeat profiling (LGC Standards) and routinely tested for mycoplasma contamination at 3 monthly intervals. Cell proliferation was assessed using a previously described sulforhodamine B (SRB) assay51 following a 72 h incubation with compound.
Western Blot Densitometry Cell-Based Assay in Hela Cells
Protocol
HeLa cells were serum-starved overnight followed by treatment with ERK5 inhibitors for 1 h. Cells were then stimulated with 100 ng/mL EGF for 10 min. The cells were harvested and lysed at 4 °C for 5–10 min in Laemmli buffer containing Halt protease and phosphate inhibitors (Pierce). The lysates were boiled for 10 min at 100 °C. A 20 μm sample was run on a 6% tris–glycine gel and transferred to nitrocellulose. Western blotting was done with ERK5 antibody (Cell Signaling #3372S). The IC50 was calculated from densitometry of the top (phospho-ERK) bands. Values represent single determinations or the mean ± standard deviation (SD) (n = 3–5).
BRD4 Expression, Purification, and Surface Plasmon Resonance Protocols
Expression and Purification of Recombinant BRD4 Bromodomain 1
Harvested bacterial cells were resuspended in lysis buffer comprising 50 mM HEPES (pH 7.4), 200 mM NaCl, 10 mM imidazole, 0.5 mg mL–1 lysozyme, and 0.2 mg mL–1 DNAse at 4 °C for 1 h. After sonication and centrifugation (1 h at 35,000g), the supernatant was purified by immobilized Ni2+ ion affinity chromatography. The peak fractions were pooled and incubated with GST-tagged HRV 3C protease (50:1) at 4 °C overnight. The cleaved His-tag was separated from BRD4 by size exclusion chromatography using a Superdex 75 (26/60) column (GE Healthcare), equilibrated, and run in 50 mM HEPES (pH 7.4), 200 mM NaCl, and 1 mM DTT. All purification steps were performed using an ÄKTA Pure (GE Healthcare) at 4 °C.
Surface Plasmon Resonance
SPR-based ligand binding assays were performed using a BIAcore S200 (GE Healthcare) at 25 °C using single cycle affinity. Immobilization of BRD4 was achieved using standard amine coupling on a CM5 chip surface. The surface was prepared through activation with 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide/N-hydroxy succinimide (EDC/NHS), followed by injection of 10 μg mL–1 BRD4 until a target level of 8000 RU was reached. The surface was then quenched using 1 M ethanolamine and washed with running buffer (10 mM HEPES, 150 mM NaCl, 0.01% (v/v) Tween-20, 0.5 mM TCEP, and 1% (v/v) DMSO) at a flow rate of 30 μL min–1. XMD8-92 and 46 were injected in a dose–response manner (nine points ranging from 0 to 20 μM) with a contact time of 30 s and a dissociation time of 160 s in series across the reference and BRD4-immobilized flow cells using solvent correction to account for bulk refractive index changes. The reference channel response was subtracted from the BRD4-immobilized channel response, and dose–response data were fitted using an affinity steady-state 1:1 binding model to determine the Kd.
ERK5-Dependent Cellular Reporter Assay
To examine the inhibition of ERK5 kinase and transcriptional activity in cells, a previously described ERK5:MEF2D reporter assay was used.21 Using Lipofectamine 2000 (ThermoFisher Scientific), HEK293 cells in 96-well plates were transfected with a constitutively active form of MEK5 (pEGFR–MEK5D), HA-tagged ERK5 (either full-length or a.a. 1–492, which lacked the NLS and C-terminal TAD), a GAL4-activated DNA-binding domain fused to the ERK5 substrate MEF2D (rat, a.a. 87–428), a 5XGAL4–luciferase reporter construct, and a CMV–renilla luciferase reporter construct. Compound 34b was added 4 h after transfection, and cells were incubated (37 °C, 5% CO2) for a further 20 h, prior to the determination of firefly and renilla luciferase using a dual luciferase reporter assay kit (Promega). The firefly luciferase activity was normalized to the renilla luciferase signal to quantify the ERK5-driven transcriptional activity.
Crystallographic Protocols
Preparation of ERK5–34b Complex Crystals
The purified unphosphorylated ERK5 kinase domain (residues 46–402) was purchased from Proteros Biostructures GmbH, and cocrystals with compound were prepared in a similar manner as described.52 ERK5 (46–402) at 11.5 mg mL–1 in storage buffer (50 mM HEPES (pH 6.5), 150 mM NaCl, 10% (v/v) glycerol, 2 mM DTT) was mixed with 34b (100 mM in 100% DMSO) to give a final concentration of 1 mM 34b and 1% (v/v) DMSO. Complex formation was allowed to proceed for 2 h on ice. The sample was then clarified by centrifugation (5 min, 16,000g, 4 °C) immediately before use in crystallization. Crystals were grown by sitting drop vapor diffusion at 20 °C in 96-well MRC plates by mixing the protein: compound complex with crystallization buffer comprising 5% (v/v) PEG 6000, 0.1 M 2-(N-morpholino)ethanesulfonic acid (MES) (pH 6.0), 5 mM DTT in a 1:1 ratio to give a 0.8 μL drop. Drops were immediately streak-seeded with a seed stock prepared from crystals of ERK5 with an indazole ERK5 inhibitor (in house series—unpublished structure). The seed stock was prepared by looping two crystals of the ERK5: indazole complex into a 2 μL crystallization buffer [5% (v/v) PEG 6000, 0.1 M MES (pH 6.0), 5 mM DTT]. The buffer and crystals were transferred into a microcentrifuge tube containing a stabilization buffer [20 μL 5% (v/v) PEG 6000, 0.1 M MES (pH 6.0), 5 mM DTT plus 80 μL 6% (v/v) PEG 6000, 0.1 M MES (pH 6.25), 5 mM DTT], and the crystals were crushed by vortexing with a Teflon bead. The seed stock was aliquoted into cryotubes, flash-frozen in liquid nitrogen, and stored at −80 °C until use.
X-ray Diffraction Data Collection, Structure Solution, and Refinement for the Complex of ERK5 with 34b
Crystals were passed briefly through a cryoprotectant solution comprising 4.9% (v/v) PEG 6000, 70 mM MES (pH 6.0), 3.5 mM DTT, 30% (v/v) glycerol, 1% (v/v) DMSO, and 10 mM 34b before flash cooling in liquid nitrogen. Data were collected at 100K on beamline I04 at the DIAMOND Light Source (Oxford, U.K.). Data processing was carried out using XDS, POINTLESS/AIMLESS (PMID: 21460446), and other programs of the CCP4i suite (PMID: 15299374) run through the CCP4i2 gui. Structures were solved by molecular replacement using PHASER (PMID: 19461840) and pdb 5BYZ as a starting model. REFMAC (PMID: 15299926) was employed for refinement, and model building was performed using COOT (PMID: 20383002). PDB was deposited within the protein database www.pdb.org using accession code: 7PUS. The authors will release the atomic coordinates upon article publication.
In vitro pharmacokinetic profiling was performed at Cyprotex. Assay protocols can be found at https://www.cyprotex.com/admepk.
In Vivo Pharmacokinetic Methods
Mice were treated intravenously with 10 mg/kg of compound in a vehicle of 10% N-methyl pyrrolidone (NMP) in saline. Blood samples were collected via the tail vein at 15, 30, and 60 min and by cardiac puncture under terminal anesthesia at 120, 240, and 360 min (nine mice in total; three per time point with serial sampling). Oral pharmacokinetic studies were performed in an analogous manner following the administration of a 10 mg/kg compound by oral gavage in the same vehicle. Drug levels were determined by liquid chromatography–mass spectrometry (LC–MS) analysis against a standard curve prepared in control plasma. All in vivo experiments were reviewed and approved by institutional animal welfare committees.
Acknowledgments
The authors thank Cancer Research U.K. (Grant References C2115/A21421 and DRCDDRPGMApr2020\100002) and the Medical Research Council (Grant Reference MR/K007580/1) for financial support. Astex Pharmaceuticals is also gratefully acknowledged for financial support under a strategic alliance agreement with Newcastle University. The authors would also like to thank Diamond Light Source for beamtime (proposals mx9948-60 and mx13587-7), the staff of beamlines I04 and I24, and Dr. Arnaud Baslé for assistance with crystal testing and data collection. The authors gratefully acknowledge friends and collaborators at Astex Pharmaceuticals for helpful discussions during the course of this work and particularly thank Tom Davies. The use of the EPSRC U.K. National Mass Spectrometry Facility at the University of Wales (Swansea) is also gratefully acknowledged.
Glossary
Abbreviations Used
- ERK5
extracellular regulated kinase 5
- ER
efflux ratio
- MLM
mouse liver microsomes
- MPLC
medium-pressure liquid chromatography
- LC–MS
liquid chromatography–mass spectrometry
- IC50
half-maximal inhibitory concentration
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jmedchem.1c01756.
Accession Codes
PDB was deposited within the protein database at www.pdb.org using accession code: 7PUS. The authors will release the atomic coordinates upon article publication.
Author Contributions
# D.C.M. and T.R. contributed equally to this work.
The authors declare no competing financial interest.
Author Status
∇ Deceased 24 September 2014.
Supplementary Material
References
- Perez-Madrigal D.; Finegan K. G.; Paramo B.; Tournier C. The extracellular-regulated protein kinase 5 (erk5) promotes cell proliferation through the down-regulation of inhibitors of cyclin dependent protein kinases (cdks). Cell. Signalling 2012, 24, 2360–2368. 10.1016/j.cellsig.2012.08.001. [DOI] [PubMed] [Google Scholar]
- Carvajal-Vergara X.; Tabera S.; Montero J. C.; Esparís-Ogando A.; López-Pérez R.; Mateo G.; Gutiérrez N.; Parmo-Cabañas M.; Teixidó J.; San Miguel J. F.; Pandiella A. Multifunctional role of erk5 in multiple myeloma. Blood 2005, 105, 4492–4499. 10.1182/blood-2004-08-2985. [DOI] [PubMed] [Google Scholar]
- Stecca B.; Rovida E. Impact of erk5 on the hallmarks of cancer. Int J. Mol. Sci. 2019, 20, 1426 10.3390/ijms20061426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang V. T.; Yan T. J.; Cavanaugh J. E.; Flaherty P. T.; Beckman B. S.; Burow M. E. Oncogenic signaling of mek5-erk5. Cancer Lett. 2017, 392, 51–59. 10.1016/j.canlet.2017.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zen K.; Yasui K.; Nakajima T.; Zen Y.; Zen K.; Gen Y.; Mitsuyoshi H.; Minami M.; Mitsufuji S.; Tanaka S.; Itoh Y.; Nakanuma Y.; Taniwaki M.; Arii S.; Okanoue T.; Yoshikawa T. Erk5 is a target for gene amplification at 17p11 and promotes cell growth in hepatocellular carcinoma by regulating mitotic entry. Genes, Chromosomes Cancer 2009, 48, 109–120. 10.1002/gcc.20624. [DOI] [PubMed] [Google Scholar]
- McCracken S. R.; Ramsay A.; Heer R.; Mathers M. E.; Jenkins B. L.; Edwards J.; Robson C. N.; Marquez R.; Cohen P.; Leung H. Y. Aberrant expression of extracellular signal-regulated kinase 5 in human prostate cancer. Oncogene 2008, 27, 2978–2988. 10.1038/sj.onc.1210963. [DOI] [PubMed] [Google Scholar]
- Montero J. C.; Ocana A.; Abad M.; Ortiz-Ruiz M. J.; Pandiella A.; Esparis-Ogando A. Expression of erk5 in early stage breast cancer and association with disease free survival identifies this kinase as a potential therapeutic target. PLoS One 2009, 4, e5565 10.1371/journal.pone.0005565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishimoto S.; Nishida E. Mapk signalling: Erk5 versus erk1/2. EMBO Rep. 2006, 7, 782–786. 10.1038/sj.embor.7400755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nithianandarajah-Jones G. N.; Wilm B.; Goldring C. E. P.; Müller J.; Cross M. J. Erk5: Structure, regulation and function. Cell. Signalling 2012, 24, 2187–2196. 10.1016/j.cellsig.2012.07.007. [DOI] [PubMed] [Google Scholar]
- Tubita A.; Tusa I.; Rovida E. Playing the whack-a-mole game: Erk5 activation emerges among the resistance mechanisms to raf-mek1/2-erk1/2-targeted therapy. Front. Cell Dev. Biol. 2021, 9, 518 10.3389/fcell.2021.647311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J.; Erazo T.; Ferguson F. M.; Buckley D. L.; Gomez N.; Muñoz-Guardiola P.; Diéguez-Martínez N.; Deng X.; Hao M.; Massefski W.; Fedorov O.; Offei-Addo N. K.; Park P. M.; Dai L.; DiBona A.; Becht K.; Kim N. D.; McKeown M. R.; Roberts J. M.; Zhang J.; Sim T.; Alessi D. R.; Bradner J. E.; Lizcano J. M.; Blacklow S. C.; Qi J.; Xu X.; Gray N. S. Structural and atropisomeric factors governing the selectivity of pyrimido-benzodiazipinones as inhibitors of kinases and bromodomains. ACS Chem. Biol. 2018, 13, 2438–2448. 10.1021/acschembio.7b00638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pereira D. M.; Rodrigues C. M. P. Targeted avenues for cancer treatment: The mek5/erk5 signaling pathway. Trends Mol. Med. 2020, 26, 394–407. 10.1016/j.molmed.2020.01.006. [DOI] [PubMed] [Google Scholar]
- Tatake R. J.; O’Neill M. M.; Kennedy C. A.; Wayne A. L.; Jakes S.; Wu D.; Kugler S. Z.; Kashem M. A.; Kaplita P.; Snow R. J. Identification of pharmacological inhibitors of the mek5/erk5 pathway. Biochem. Biophys. Res. Commun. 2008, 377, 120–125. 10.1016/j.bbrc.2008.09.087. [DOI] [PubMed] [Google Scholar]
- Lin E. C. K.; Amantea C. M.; Nomanbhoy T. K.; Weissig H.; Ishiyama J.; Hu Y.; Sidique S.; Li B.; Kozarich J. W.; Rosenblum J. S. Erk5 kinase activity is dispensable for cellular immune response and proliferation. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 11865–11870. 10.1073/pnas.1609019113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen D.; Lemos C.; Wortmann L.; Eis K.; Holton S. J.; Boemer U.; Moosmayer D.; Eberspaecher U.; Weiske J.; Lechner C.; Prechtl S.; Suelzle D.; Siegel F.; Prinz F.; Lesche R.; Nicke B.; Nowak-Reppel K.; Himmel H.; Mumberg D.; von Nussbaum F.; Nising C. F.; Bauser M.; Haegebarth A. Discovery and characterization of the potent and highly selective (piperidin-4-yl)pyrido[3,2-d]pyrimidine based in vitro probe bay-885 for the kinase erk5. J. Med. Chem. 2019, 62, 928–940. 10.1021/acs.jmedchem.8b01606. [DOI] [PubMed] [Google Scholar]
- Myers S. M.; Miller D. C.; Molyneux L.; Arasta M.; Bawn R. H.; Blackburn T. J.; Cook S. J.; Edwards N.; Endicott J. A.; Golding B. T.; Griffin R. J.; Hammonds T.; Hardcastle I. R.; Harnor S. J.; Heptinstall A. B.; Lochhead P. A.; Martin M. P.; Martin N. C.; Newell D. R.; Owen P. J.; Pang L. C.; Reuillon T.; Rigoreau L. J. M.; Thomas H. D.; Tucker J. A.; Wang L.-Z.; Wong A.-C.; Noble M. E. M.; Wedge S. R.; Cano C. Identification of a novel orally bioavailable erk5 inhibitor with selectivity over p38α and brd4. Eur J. Med. Chem. 2019, 178, 530–543. 10.1016/j.ejmech.2019.05.057. [DOI] [PubMed] [Google Scholar]
- Waring M. J. Defining optimum lipophilicity and molecular weight ranges for drug candidates—molecular weight dependent lower logd limits based on permeability. Bioorg. Med. Chem. Lett. 2009, 19, 2844–2851. 10.1016/j.bmcl.2009.03.109. [DOI] [PubMed] [Google Scholar]
- Lipinski C. A.; Lombardo F.; Dominy B. W.; Feeney P. J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Delivery Rev. 2001, 46, 3–26. 10.1016/S0169-409X(96)00423-1. [DOI] [PubMed] [Google Scholar]
- Veber D. F.; Johnson S. R.; Cheng H.-Y.; Smith B. R.; Ward K. W.; Kopple K. D. Molecular properties that influence the oral bioavailability of drug candidates. J. Med. Chem. 2002, 45, 2615–2623. 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Waring M. J.; Clarke D. S.; Fenwick M. D.; Godfrey L.; Groombridge S. D.; Johnstone C.; McKerrecher D.; Pike K. G.; Rayner J. W.; Robb G. R.; Wilson I. Property based optimisation of glucokinase activators—discovery of the phase iib clinical candidate azd1656. MedChemComm 2012, 3, 1077–1081. 10.1039/c2md20077e. [DOI] [Google Scholar]
- Lochhead P. A.; Tucker J. A.; Tatum N. J.; Wang J.; Oxley D.; Kidger A. M.; Johnson V. P.; Cassidy M. A.; Gray N. S.; Noble M. E. M.; Cook S. J. Paradoxical activation of the protein kinase-transcription factor erk5 by erk5 kinase inhibitors. Nat. Commun. 2020, 11, 1383 10.1038/s41467-020-15031-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cook S. J.; Tucker J. A.; Lochhead P. A. Small molecule erk5 kinase inhibitors paradoxically activate erk5 signalling: Be careful what you wish for.... Biochem. Soc. Trans. 2020, 48, 1859–1875. 10.1042/BST20190338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Q.; Deng X.; Lu B.; Cameron M.; Fearns C.; Patricelli M. P.; Yates J. R.; Gray N. S.; Lee J.-D. Pharmacological inhibition of bmk1 suppresses tumor growth through promyelocytic leukemia protein. Cancer Cell 2010, 18, 258–267. 10.1016/j.ccr.2010.08.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McFarland J. M.; Ho Z. V.; Kugener G.; Dempster J. M.; Montgomery P. G.; Bryan J. G.; Krill-Burger J. M.; Green T. M.; Vazquez F.; Boehm J. S.; Golub T. R.; Hahn W. C.; Root D. E.; Tsherniak A. Improved estimation of cancer dependencies from large-scale rnai screens using model-based normalization and data integration. Nat. Commun. 2018, 9, 4610 10.1038/s41467-018-06916-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. J.; Sugimoto T. Aza-analogues of pteridine. Part ii. The novel use of silver oxide in transetherification of alkoxy-1,2,4,6,8-penta-azanaphthalenes, alkoxynitropyrimidines, and related systems. J. Chem. Soc. C 1970, 2661–2666. 10.1039/j39700002661. [DOI] [Google Scholar]
- Cherkasov V. M.; Remennikov G. Y.; Kisilenko A. A.; Romanenko E. A. Sigma complexes in the pyrimidine series. 2. ∑ complexes of 5-nitropyrimidine and methoxy-substituted 5-nitropyrimidines with the acetone anion. Chem. Heterocycl. Compd. 1980, 16, 179–182. 10.1007/BF02401696. [DOI] [Google Scholar]
- Krchňák V.; Arnold Z. Novel pyrimidine derivatives, reactions and ultraviolet spectra. Collect. Czech. Chem. Commun. 1975, 40, 1396–1402. 10.1135/cccc19751396. [DOI] [Google Scholar]
- Rahamin Y.; Sharvit J.; Mandelbaum A.; Sprecher M. Electron impact-induced methyl migration in dimethylaminoheteroaromatic systems. J. Org. Chem. 1967, 32, 3856–3859. 10.1021/jo01287a030. [DOI] [Google Scholar]
- Nara S. J.; Jha M.; Brinkhorst J.; Zemanek T. J.; Pratt D. A. A simple cu-catalyzed coupling approach to substituted 3-pyridinol and 5-pyrimidinol antioxidants. J. Org. Chem. 2008, 73, 9326–9333. 10.1021/jo801501e. [DOI] [PubMed] [Google Scholar]
- Hurst D. T.; Christophides J. The synthesis of some 2-substituted 5-nitropyrimidines. Heterocycles 1977, 6, 1999–2004. 10.3987/R-1977-12-1999. [DOI] [Google Scholar]
- Axten J. M.; Betancourt J. R.; Johnson N. W.; Semones M.. Anthraniliamides. WO Patent WO20081478312008.
- Tsou H.-R.; Otteng M.; Tran T.; Floyd M. B.; Reich M.; Birnberg G.; Kutterer K.; Ayral-Kaloustian S.; Ravi M.; Nilakantan R.; Grillo M.; McGinnis J. P.; Rabindran S. K. 4-(phenylaminomethylene)isoquinoline-1,3(2h,4h)-diones as potent and selective inhibitors of the cyclin-dependent kinase 4 (cdk4). J. Med. Chem. 2008, 51, 3507–3525. 10.1021/jm800072z. [DOI] [PubMed] [Google Scholar]
- Quattropani A.; Kulkarni S. S.; Giri A. G.. Glycosidase Inhibitors. WO Patent WO20160304432016.
- Qian Y.; Wertheimer S. J.; Ahmad M.; Cheung A. W.-H.; Firooznia F.; Hamilton M. M.; Hayden S.; Li S.; Marcopulos N.; McDermott L.; Tan J.; Yun W.; Guo L.; Pamidimukkala A.; Chen Y.; Huang K.-S.; Ramsey G. B.; Whittard T.; Conde-Knape K.; Taub R.; Rondinone C. M.; Tilley J.; Bolin D. Discovery of orally active carboxylic acid derivatives of 2-phenyl-5-trifluoromethyloxazole-4-carboxamide as potent diacylglycerol acyltransferase-1 inhibitors for the potential treatment of obesity and diabetes. J. Med. Chem. 2011, 54, 2433–2446. 10.1021/jm101580m. [DOI] [PubMed] [Google Scholar]
- DiMauro E. F.; Newcomb J.; Nunes J. J.; Bemis J. E.; Boucher C.; Chai L.; Chaffee S. C.; Deak H. L.; Epstein L. F.; Faust T.; Gallant P.; Gore A.; Gu Y.; Henkle B.; Hsieh F.; Huang X.; Kim J. L.; Lee J. H.; Martin M. W.; McGowan D. C.; Metz D.; Mohn D.; Morgenstern K. A.; Oliveira-dos-Santos A.; Patel V. F.; Powers D.; Rose P. E.; Schneider S.; Tomlinson S. A.; Tudor Y.-Y.; Turci S. M.; Welcher A. A.; Zhao H.; Zhu L.; Zhu X. Structure-guided design of aminopyrimidine amides as potent, selective inhibitors of lymphocyte specific kinase: Synthesis, structure–activity relationships, and inhibition of in vivo t cell activation. J. Med. Chem. 2008, 51, 1681–1694. 10.1021/jm7010996. [DOI] [PubMed] [Google Scholar]
- Hattori Y.; Nishikawa M.; Kusamoto T.; Kume S.; Nishihara H. Steric interference on the redox-conjugated pyrimidine ring rotation of mono- and dinuclear copper complexes with (4-methyl-2-pyrimidinyl)imine ligands. Chem. Lett. 2014, 43, 1037–1039. 10.1246/cl.140238. [DOI] [Google Scholar]
- Schaefer F. C. Synthesis of the s-triazine system. Preparation of unsymmetrically substituted s-triazines by reaction of amidine salts with imidates. J. Org. Chem. 1962, 27, 3608–3613. 10.1021/jo01057a052. [DOI] [Google Scholar]
- Dousson C. B.; Heron N. M.; Hill G. B. Valuable synthetic building blocks: Useful 2-substituted 5-aminopyrimidines from a stable precursor. Synthesis 2005, 11, 1817–1821. 10.1055/s-2005-865338. [DOI] [Google Scholar]
- Adjabeng G.; Bifulco N.; Davis-Ward R. G.; Dickerson S. H.; Donaldson K. H.; Harris P. A.; Hornberger K.; Petrov K.; Rheaul T. R.; Schaaf G.; Stellwagen J.; Uehling D. E.; Waterson A. G.. Thiazole and Oxazole Kinase Inhibitors. WO Patent WO20090326672009.
- Dodic N.Bioisosteric Benzamide Derivatives and Their Use as apob-100 Secretion Inhibitors. U.S. Patent US20040099882004.
- Spencer J.; Patel H.; Callear S. K.; Coles S. J.; Deadman J. J. Synthesis and solid state study of pyridine- and pyrimidine-based fragment libraries. Tetrahedron Lett. 2011, 52, 5905–5909. 10.1016/j.tetlet.2011.07.147. [DOI] [Google Scholar]
- Gruber W.; Schlögl K. Lithiumreaktionen an α-picolinen. Monatsh. Chem. 1950, 81, 473–479. 10.1007/BF00906435. [DOI] [Google Scholar]
- Hosaan A.; Fadda A. A. Enamine rearrangement of pyridinium salts to indole ring: A combined experimental and molecular modeling study. J. Heterocycl. Chem. 2013, 50, 638–644. 10.1002/jhet.1629. [DOI] [Google Scholar]
- Mundinger S.; Jakob U.; Bichovski P.; Bannwarth W. Modification and optimization of the bis-picolylamide-based relay protection for carboxylic acids to be cleaved by unusual complexation with cu2+ salts. J. Org. Chem. 2012, 77, 8968–8979. 10.1021/jo301349t. [DOI] [PubMed] [Google Scholar]
- Tohda Y.; Eiraku M.; Nakagawa T.; Usami Y.; Ariga M.; Kawashima T.; Tani K.; Watanabe H.; Mori Y. Nucleophilic reaction upon electron-deficient pyridone derivatives. One-pot synthesis of 3-nitropyridines by ring transformation of 1-methyl-3,5-dinitro-2-pyridone with ketones or aldehydes in the presence of ammonia. Bull. Chem. Soc. Jpn. 1990, 63, 2820–2827. 10.1246/bcsj.63.2820. [DOI] [Google Scholar]
- Ohtake N.; Mizutani S.; Yoshimoto R.; Okita S.; Kanatani A.. Novel Piperidine Derivative. WO Patent WO20050284382005.
- Park S.; Lee I. S.; Park J. A magnetically separable gold catalyst for chemoselective reduction of nitro compounds. Org. Biomol. Chem. 2013, 11, 395–399. 10.1039/C2OB27025K. [DOI] [PubMed] [Google Scholar]
- Ravi P.; Gore G. M.; Sikder A. K.; Tewari S. P. Silica–sulfuric acid catalyzed nitrodeiodination of iodopyrazoles. Synth. Commun. 2012, 42, 3463–3471. 10.1080/00397911.2011.584261. [DOI] [Google Scholar]
- Zabierek A. A.; Konrad K. M.; Haidle A. M. A practical, two-step synthesis of 1-alkyl-4-aminopyrazoles. Tetrahedron Lett. 2008, 49, 2996–2998. 10.1016/j.tetlet.2008.02.169. [DOI] [Google Scholar]
- Barlamm B. C.; Foote K. M.; Ple P.. Pyridine Compounds. WO Patent WO20091535892009.
- Skehan P.; Storeng R.; Scudiero D.; Monks A.; McMahon J.; Vistica D.; Warren J. T.; Bokesch H.; Kenney S.; Boyd M. R. New colorimetric cytotoxicity assay for anticancer-drug screening. J. Natl. Cancer Inst. 1990, 82, 1107–1112. 10.1093/jnci/82.13.1107. [DOI] [PubMed] [Google Scholar]
- Chen H.; Tucker J.; Wang X.; Gavine P. R.; Phillips C.; Augustin M. A.; Schreiner P.; Steinbacher S.; Preston M.; Ogg D. Discovery of a novel allosteric inhibitor-binding site in erk5: Comparison with the canonical kinase hinge atp-binding site. Acta Crystallogr., Sect. D: Struct. Biol. 2016, 72, 682–693. 10.1107/S2059798316004502. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.