

Abstract
Elucidating the wiring diagram of the human cell is a central goal of the post-genomic era. We combined genome engineering, confocal live-cell imaging, mass spectrometry and data science to systematically map the localization and interactions of human proteins. Our approach provides a data-driven description of the molecular and spatial networks that organize the proteome. Unsupervised clustering of these networks delineates functional communities that facilitate biological discovery, and uncovers that RNA-binding proteins form a specific sub-group defined by unique interaction and localization properties. Furthermore, we discover that remarkably precise functional information can be derived from protein localization patterns, which often contain enough information to identify molecular interactions. Paired with a fully interactive website (opencell.czbiohub.org), we provide a resource for the quantitative cartography of human cellular organization.
Sequencing the human genome has transformed cell biology by defining the protein parts list that forms the canvas of cellular operation (1, 2). This paves the way for elucidating how the ~20,000 proteins encoded in the genome organize in space and time to define the cell’s functional architecture (3, 4). Where does each protein localize within the cell? Can we comprehensively map how proteins assemble into larger functional communities? A main challenge to answering these fundamental questions is that cellular architecture is organized along multiple scales. Therefore, several approaches need to be combined for its elucidation (5). In a series of pioneering studies, human protein-protein interactions have been mapped using ectopic expression strategies with yeast two-hybrid (Y2H) (6) or epitope tagging coupled to immunoprecipitation-mass spectrometry (IP-MS) (7, 8), while protein localization has been charted using immuno-fluorescence in fixed samples (9). A complementary approach is to directly modify genes in a genome by appending sequences that illuminate specific aspects of the corresponding proteins’ function (commonly referred to as “endogenous tagging” (10)). For example, endogenously tagging a gene with a fluorescent reporter enables to image protein sub-cellular localization in live cells, and supports functional characterization in a native cellular environment (10, 11). The use of endogenous tagging to study the organization of a eukaryotic cell is illustrated by seminal work in the budding yeast S. cerevisiae. There, libraries of tagged strains have enabled the comprehensive mapping of protein localization and molecular interactions across the yeast proteome (12–14). These libraries were made possible by the relative simplicity of homologous recombination and genome engineering in yeast (15). In human cells, earlier work has leveraged alternative strategies including expression from bacterial artificial chromosomes (16) or central-dogma tagging (17) because of the difficulty of site-specific gene editing. CRISPR-mediated genome engineering now allows for homologous recombination-based endogenous tagging to be applied for the interrogation of the human cell (10, 11, 18).
Here, we combine experimental and analytical strategies to create OpenCell, a proteomic map of human cellular architecture. We generated a library of 1,310 CRISPR-edited HEK293T cell lines harboring fluorescent tags on individual proteins, which we characterized by pairing confocal microscopy and mass spectrometry. Our dataset constitutes the most comprehensive live-cell image collection of human protein localization to date. In addition, integration of IP-MS using the fluorescent tags for affinity capture enables measurement of localization and interactions from the same samples. For a quantitative description of cellular architecture, we introduce a data-driven framework to represent protein interactions and localization features, supported by a new machine learning algorithm for image encoding. This approach allows us to delineate communities of functionally related proteins by unsupervised clustering and facilitates the generation of mechanistic hypotheses, including for proteins that had so far remained uncharacterized. We further demonstrate that the localization pattern of each protein is defined by unique and specific features that can be used for functional interpretation, to the point that spatial relationships often contain enough information to predict interactions at the molecular scale. Finally, our analysis enables an unsupervised description of the human proteome’s organization, and highlights in particular that RNA-binding proteins exhibit unique functional signatures that shape the proteome’s network.
Engineered cell library.
Fluorescent protein (FP) fusions are versatile tools that can measure both protein localization by microscopy and protein-protein interactions by acting as affinity handles for IP-MS (18, 19) (Fig. S1A). Here, we constructed a library of fluorescently tagged HEK293T cell lines by targeting human genes with the split-mNeonGreen2 system (20) (Fig. 1A). Split-FPs greatly simplify CRISPR-based genome engineering by circumventing the need for molecular cloning (18), and allowed us to generate endogenous genomic fusions (Fig. 1B) that preserve native expression regulation. A full description of our pipeline is available in the Methods section ((21); summarized in Fig. 1C through E). In brief, FP insertion sites (N- or C-terminus) were chosen on the basis of information from the literature or structural analysis (Fig. S1B; Table S1). For each tagged target we isolated a polyclonal pool of CRISPR-edited cells, which was then characterized by live-cell 3D confocal microscopy, IP-MS, and genotyping of tagged alleles by next-generation sequencing. Open-source software development and advances in instrumentation supported scalability (Fig. 1C). In particular, we developed crispycrunch, a CRISPR design software that enables guide RNA selection and homology donor sequence design (github.com/czbiohub/crispycrunch). We also fully automated the acquisition of data microscopy data in Python for on-the-fly computer vision and selection of desirable fields of view imaged in 96-well plates (github.com/czbiohub/2021-opencell-microscopy-automation). Our mass-spectrometry protocols use the high sensitivity of timsTOF instruments (22) which allowed miniaturization of IP-MS down to 0.8×106 cells of starting material (Fig. S1C; about a tenth of the material required in previous approaches (7, 8)).
Figure 1: the OpenCell library.
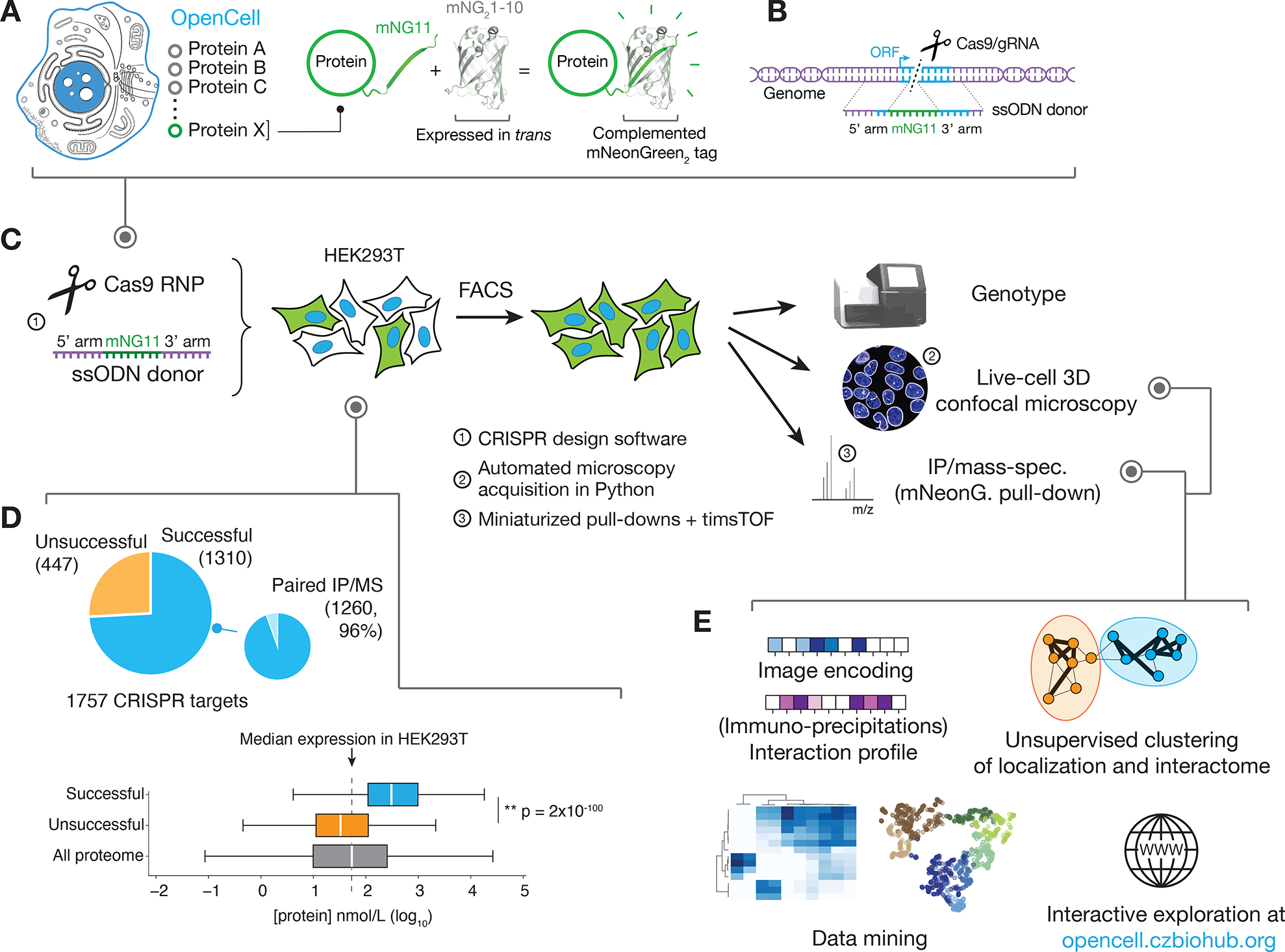
(A) Functional tagging with split-mNeonGreen2. In this system, mNeonGreen2 is separated into two fragments: a short mNG11 fragment, which is fused to a protein of interest, and a large mNG21-10 fragment, which is expressed separately in trans (that is, tagging is done in cells that have been engineered to constitutively express mNG21-10). (B) Endogenous tagging strategy: mNG11 fusion sequences are inserted directly within genomic open reading frames (ORFs) using CRISPR-Cas9 gene editing and homologous recombination with single-stranded oligonucleotides donors (ssODN). (C) The OpenCell experimental pipeline. See text for details. (D) Successful detection of fluorescence in the OpenCell library. Out of 1757 genes that were originally targeted, fluorescent signal was successfully detected for 1310 (top panel). Low protein abundance is the main obstacle to successful detection. Bottom left panel shows the full distribution of abundance for all proteins expressed in HEK293T vs. successfully or unsuccessfully detected OpenCell targets; boxes represent 25th, 50th, and 75th percentiles, and whiskers represent 1.5x interquartile range. Median is indicated by a white line. P-value: Student’s t-test. (E) The OpenCell data analysis pipeline, described in subsequent sections.
In total, we targeted 1757 genes, of which 1310 (75%) could be detected by fluorescence imaging and form our current dataset (full library details in Table S1). From these, we obtained paired IP-MS measurements for 1260 targets (96%, Fig. 1D). The 1310-protein collection includes a balanced representation of the pathways, compartments and functions of the human proteome (Fig. S1D), with the exception of processes specific to mitochondria, organellar lumen or extracellular matrix. Indeed, the split-FP system tags a gene of interest with a short sequence (mNG11) while a larger FP fragment (mNG21–10) is expressed separately (Fig. 1A). In the version used here, the mNG21–10 fragment is expressed in the nucleo-cytoplasm and prevents access to proteins inside organellar compartments. Membrane proteins can be tagged as long as one terminus extends in the nucleo-cytoplasm. In future iterations, other split systems that contain compartment-specific signal sequences could be used to target organellar lumen (23).
Fluorescent tagging was readily successful for essential genes, suggesting that FP fusions are well tolerated (Fig. S2A). To evaluate other factors contributing to successful fluorescent detection, we measured RNA and protein concentration in HEK293T cells (Fig. S2B; using a 24-fraction scheme for deep proteome quantification; see fully annotated proteome in Table S2). This revealed that protein abundance is the main limitation to detection (Fig. 1D, S2C; see details for unsuccessful targets in Table S3); most successful targets are among the top 50% most abundant (Fig. S2D). Gene-editing efficiency was another important factor: among well-expressed targets, failure was correlated with significantly lower rates of homologous recombination (Fig. S2E), which would impair the selection of edited cells by fluorescence-activated cell sorting (FACS). Training a regression model revealed that the combination of protein abundance and editing efficiency could predict successful detection with 82% accuracy.
To maximize throughput, we used a polyclonal strategy to select genome-edited cells by FACS. Polyclonal pools contain cells with distinct genotypes. HEK293T are pseudo-triploid (24) and a single edited allele is sufficient to confer fluorescence. Moreover, various DNA repair mechanisms compete with homologous recombination for the resolution of CRISPR-induced genomic breaks (25) so that alleles containing non-functional mutations can be present in addition to the desired fusion alleles. However, such alleles do not support fluorescence and are therefore unlikely to impact other measurements, especially in the context of a polyclonal pool. We developed a stringent selection scheme to significantly enrich for fluorescent fusion alleles (Fig. S3A). Our final cell library has a median 61% of mNeonGreen-integrated alleles, 5% wild-type and 26% other non-functional alleles (Fig. S3B, full genotype information in Table S1).
Finally, we verified that our engineering approach maintained the endogenous abundance of the tagged target proteins. For this, we quantified protein expression by Western blotting using antibodies specific to proteins targeted in 12 different cell pools (Fig. S3C), and by single-shot mass spectrometry in 63 tagged lines (Fig. S3D). Both approaches revealed a median abundance of tagged targets in engineered lines at about 80% of untagged HEK293T control, with 5 outliers (8% of total) identified by proteomics (Fig. S3D, all within 3.5-fold of control). Importantly, the overall proteome composition was unchanged in all tagged lines (Fig. S3E–F). Overall, our gene-editing strategy preserves near-endogenous abundances and circumvents the limitations of ectopic overexpression (11, 26, 27), which include aberrant localization, changes in organellar morphology, and masking effects (see the examples of SPTLC1, TOMM20 and MAP1LC3B in Fig. S3G). Therefore, OpenCell supports the functional profiling of tagged proteins in their native cellular context.
Interactome analysis and stoichiometry-driven clustering.
Affinity enrichment coupled to mass spectrometry is an efficient and sensitive method for the systematic mapping of protein interaction networks (28). We isolated tagged proteins (“baits”) from cell lysates solubilized in digitonin, a mild non-ionic detergent that preserves the native structure and properties of membrane proteins (29). Specific protein interactors (“preys”) were identified by proteomics from biological triplicate experiments (see Figure S4A–B and (21) for a detailed description of our statistical analysis, which builds upon established methods (7)). In total, the full interactome from our 1260 OpenCell baits includes 29,922 interactions between 5292 proteins (baits and preys, Fig. 2A, full interactome data in Table S4).
Figure 2: Protein interactome.

(A) Overall description of the interactome. (B) Unsupervised Markov clustering of the interactome graph. (C) Example of community and core cluster definition for the translocon/EMC community. (D) The complete graph of connections between interactome communities. The density of protein-protein interactions between communities is represented by increased edge width. The numbers of targets included in each community is represented by circles of increasing diameters. (E) Distribution of occurrence in PubMed articles vs. RNA expression for all proteins found within interactome communities. The bottom 10th percentile of publication count (poorly characterized proteins) is highlighted. (F) NHSL1/NSHL2/KIAA1522 are part of the SCAR/WAVE community and share amino-acid sequence homology (right panel). (G) DMXL1/2, WDR7 and ROGDI form the human RAVE complex. Heatmaps represent the interaction stoichiometry of preys (lines) in the pull-downs of specific OpenCell targets (columns). See text for details.
To assess the quality of our interactome, we estimated its precision (the fraction of true positive interactions over all interactions) and recall (the fraction of interactions identified compared to a ground truth set) using reference data (Fig. S4B). For recall analysis, we quantified the coverage in our data of interactions included in CORUM (30), a compendium of protein interactions manually curated from the literature. To estimate precision, we quantified how many of our interactions involved protein pairs expected to localize to the same broad cellular compartment (31) (Fig. S4B). To benchmark OpenCell against other large-scale interactomes, we compared its precision and recall to Bioplex (overexpression of HA-tagged baits (8, 32)), the yeast-two-hybrid human reference interactome (HuRI (6)) and our own previous data (GFP fusions expressed from bacterial artificial chromosomes (7)) (Fig. S4C–E). We also calculated compression rates for each dataset as a measure of the overall richness in network patterns and motifs distinguishable from noise, which correlates with overall network quality: real-world networks contain redundant information which can be compressed, while pure noise is not compressible (see (33)) (Fig. S4F). Across all metrics, OpenCell outperformed previous approaches. OpenCell also includes many interactions not reported in previous datasets (Fig. S4E,G). Our interactome may better reflect biological interactions because it preserves near-endogenous protein expression.
A powerful way to interpret interactomes is to identify communities of interactors (8, 13). To this end, we applied unsupervised Markov clustering (MCL) (34) to the graph of interactions defined by our data (5292 baits and preys). We first measured the stoichiometry of each interaction, using a quantitative approach we previously established (7). Interaction stoichiometry measures the abundance of a protein interactor relative to the abundance of the bait in a given immuno-precipitation sample. We have shown that stoichiometry can be interpreted as a proxy for interaction strength, and that interactions can be classified between core (i.e. high) and low stoichiometries (7). In our current data, both high- and low-stoichiometry interactions were significantly enriched for proteins pairs sharing gene ontology annotations (Fig. S4H). Using stoichiometry to assign weights to the edges in the interaction graph (Fig. 2B), a first round of MCL delineated inter-connected protein communities and led to better clustering performance than clustering based on connectivity alone (Fig. S4I). To better delineate stable complexes, we further refined each individual MCL community by additional clustering while removing low-stoichiometry interactions. The resulting sub-clusters outline core interactions within existing communities (Fig. 2B). Figure 2C illustrates how this unsupervised approach enables to delineate functionally related proteins: all subunits of the machinery responsible for the translocation of newly translated proteins at the ER membrane (SEC61/62/63) and of the EMC (ER Membrane Complex) are grouped within respective core interaction clusters, but both are part of the same larger MCL community. This mirrors the recently appreciated cotranslational role of EMC for insertion of transmembrane domains at the ER (35). Additional proteins that have only recently been shown to act cotranslationally are found clustering with translocon or EMC subunits, including ERN1 (IRE1) (36) and CCDC47 (37, 38). Thus, clustering can facilitate mechanistic exploration by grouping proteins involved in related pathways. Overall, we identified 300 communities including a total of 2096 baits and preys (full details in Table S4). Ontology analysis revealed that these communities are significantly enriched for specific cellular functions, supporting their biological relevance (82% of all communities are significantly enriched for specific biological process or molecular function GO ontology terms; see Table S5 for complete analysis). A graph of interactions between communities reveals a richly inter-connected network (Fig. 2D), the structure of which outlines the global architecture of the human interactome (discussed further below).
A direct application of interactome clustering is to help elucidate the cellular roles of the many human proteins that remain poorly characterized (39). We identified poorly characterized proteins by quantifying their occurrence in article titles and abstracts from PubMed (Fig. 2E). Empirically, we determined that proteins in the bottom 10th percentile of publication count (corresponding to less than 10 publications) are very poorly annotated (Fig. 2E). This set encompasses a total of 251 proteins found in interaction communities for which our dataset offers potential mechanistic insights. For example, the proteins NHSL1, NHSL2 and KIAA1522 are all found as part of a community centered around SCAR/WAVE, a large multi-subunit complex nucleating actin polymerization (Fig. 2F). All three proteins share sequence homology and are homologous to NHS (Fig. S5A), a protein mutated in patients with Nance-Horan syndrome. NHS interacts with SCAR/WAVE components to coordinate actin remodeling (40). Thus, NHSL1, NHSL2 and KIAA1522 also act to regulate actin assembly. A recent mechanistic study supports this hypothesis: NHSL1 localizes at the cell’s leading edge and directly binds SCAR/WAVE to negatively regulate its activity, reducing F-actin content in lamellipodia and inhibiting cell migration (41). The authors identified NHSL1’s SCAR/WAVE binding sites, and we find these sequences to be conserved in NSHL2 and KIA1522 (Fig. 2F). Therefore, our data suggests that both NHSL2 and KIAA1522 are also direct SCAR/WAVE binders and possible modulators of the actin cytoskeleton.
Our data also sheds light on the function of ROGDI, whose variants cause Kohlschuetter-Toenz syndrome (a recessive developmental disease characterized by epilepsy and psychomotor regression (42)). ROGDI appears in the literature because of its association with disease, but no study, to our knowledge, specifically determines its molecular function. We first observed that ROGDI’s interaction pattern closely matched that of three other proteins in our dataset: DMXL1, DMXL2 and WDR7 (Fig. 2G). This set exhibited a specific interaction signature with the v-ATPase lysosomal proton pump. All four proteins interact with soluble v-ATPase subunits (ATP6-V1), but not its intra-membrane machinery (ATP6-V0). DMXL1 and WDR7 interact with V1 v-ATPase, and their depletion in cells compromises lysosomal re-acidification (43). Sequence analysis showed that DMXL1 or 2, WDR7 and ROGDI are homologous to proteins from yeast and Drosophila involved in the regulation of assembly of the soluble V1 subunits onto the V0 transmembrane ATPase core (44, 45) (Fig. S5B). In yeast, Rav1 and Rav2 (homologous to DMXL1/2 and ROGDI, respectively) form the stoichiometric RAVE complex, a soluble chaperone that regulates v-ATPase assembly (45). To assess the existence of a human RAVE-like complex, we generated new tagged cell lines for DMXL1 and 2, WDR7, and ROGDI. Because of the low abundance of these proteins, the localization of DMXL2 and ROGDI were not detectable but pull-downs of DMXL1 and WDR7 confirmed a stoichiometric interaction between DMXL1 and 2, WDR7 and ROGDI (Fig. 2G, right panels). No direct interaction between DXML1 and DMXL2 was detected, suggesting that they might nucleate two separate sub-complexes. Therefore, our data reveals a human RAVE-like complex comprising DMXL1 or 2, WDR7 and ROGDI, which we propose acts as a chaperone for v-ATPase assembly based on its yeast homolog. Altogether, these results illustrate how our data can facilitate the generation of new mechanistic hypotheses by combining quantitative analysis and literature curation.
Image dataset: localization annotation and self-supervised machine learning.
A key advantage of our cell engineering approach is to enable the characterization of each tagged protein in live, unperturbed cells. To profile localization, we performed spinning-disk confocal fluorescence microscopy (63× 1.47NA objective) under environmental control (37°C, 5% CO2), and imaged the 3D distribution of proteins in consecutive z-slices. Microscopy acquisition was fully automated in Python to enable scalability (Fig. S6A–B). In particular, we trained a computer vision model to identify fields of view (FOVs) with homogeneous cell density on-the-fly, which reduced experimental variation between images. Our dataset contains a collection of 6375 3D stacks (5 different FOVs for each target) and includes paired imaging of nuclei with live-cell Hoechst 33342 staining.
We manually annotated localization patterns by assigning each protein to one or more of 15 separate cellular compartments such as the nucleolus, centrosome or Golgi apparatus (Fig. 3A). Because proteins often populate multiple compartments at steady-state (9), we graded annotations using a three-tier system: grade 3 identifies prominent localization compartment(s), grade 2 represents less pronounced localizations, and grade 1 annotates weak localization patterns nearing our limit of detection (see Fig. S7A for two representative examples, full annotations in Table S6). Ignoring grade 1 annotations which are inherently less precise, 55% of proteins in our library were detected in multiple locations consistent with known functional relationships. for example, clear connections were observed between secretory compartments (ER, Golgi, vesicles, plasma membrane), or between cytoskeleton and plasma membrane (Fig. S7B, Table S6)). Many proteins are found in both nucleus and cytoplasm (21% of our library), highlighting the importance of the nucleo-cytoplasmic import and export machinery in shaping global cellular function (46, 47). Importantly, because our split-FP system does not enable the detection of proteins in the lumen of organelles, multi-localization involving translocation across an organellar membrane (which is rare but does happen for mitochondrial or peroxisomal proteins) cannot be detected in our data.
Figure 3: live-cell image collection.
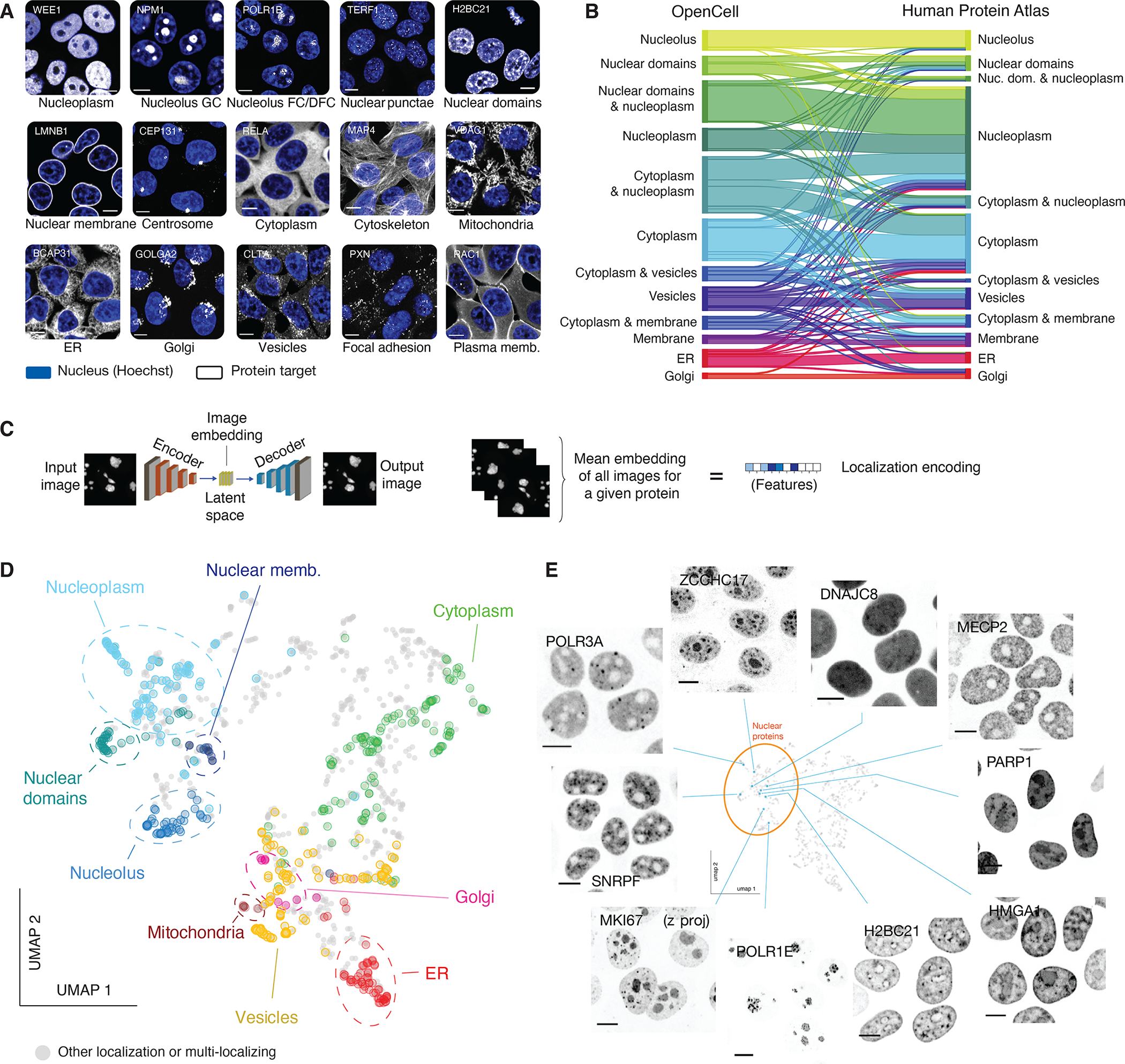
(A) The 15 cellular compartments segregated for annotating localization patterns. The localization of a representative protein belonging to each group is shown (greyscale, gene names in top left corners; scalebar: 10 μm). Nuclear stain (Hoechst) is shown in blue. “Nuclear domains” designate proteins with pronounced non-uniform nucleoplasmic localization, for example chromatin binding proteins. (B) Comparison of annotated localization for proteins included in both OpenCell and Human Protein Atlas datasets. In this flow diagram, colored bands represent groups of proteins that shared the same localization annotation in OpenCell, and the width of the band represents the number of proteins in each group. For readability, only the 12 most common localization groups are shown. Some multi-localization groups are included (e.g. “cytoplasm & nucleoplasm”). (C) Principle of localization encoding by self-supervised machine learning. See text for details. (D) UMAP representation of the OpenCell localization dataset, highlighting targets found to localize to a unique cellular compartment. (E) Representative images for 10 nuclear targets that exemplify the nuanced diversity of localization patterns across the proteome. Scale bars: 10 μm.
To benchmark our dataset, we compared our localization annotations against the Human Protein Atlas (HPA), the reference antibody-based compendium of human protein localization (9). This revealed significant agreement between datasets: 75% of proteins share at least one localization annotation in common (Fig. 3B; this includes 25% of all proteins that share the exact same set of annotations, see full description in Table S7A). Because HPA mostly reports on cell lines other than HEK293T, a perfect overlap is not expected as proteins might differentially localize between related compartments in different cell types. However, the annotations for 147 proteins (11% of our data) were fully inconsistent between the two datasets (Fig. S7C). An extensive curation of the literature on the localization of those proteins allowed us to resolve discrepancies for 115 proteins (i.e., 78% of that set; full curation in Table S8). Of these, existing literature evidence supported the OpenCell results for 113 (98.3%) of the 115 cases (Fig. S7D). This validates that endogenous tagging can help refine the curation of localization in the human proteome. Finally, our dataset includes 350 targets that have orthologs in S. cerevisiae. Comparison between OpenCell and yeast localization annotations (48) revealed a high degree of concordance (Fig. S7E; Table S7B; 81% of proteins share at least one annotation in common, including 36% perfect matches).
While expert annotation remains the best performing strategy to curate protein localization (49, 50), the low-dimensional description it allows is not well suited for quantitative comparisons. Recent developments in image analysis and machine learning offer new opportunities to extract high-dimensional features from microscopy images (50, 51). Therefore, we developed a deep learning model to quantitatively represent the localization pattern of each protein in our dataset (52). Briefly, our model is a variant of an autoencoder (Fig. 3C): a form of neural network that learns to vectorize an image through paired tasks of encoding (from an input image to a vector in a latent space) and decoding (from the latent space vector to a new output image). After training, a consensus representation for a given protein can be obtained from the average of the encodings from all its associated images. This generates a high-dimensional “localization encoding” (Fig. 3C) that captures the complex set of features that define the spatial distribution of a protein at steady state and across many individual cells. One of the main advantages of this approach is that it is self-supervised. Therefore, as opposed to supervised machine learning strategies that are trained to recognize pre-annotated patterns (for example, manual annotations of protein localization (50)), our method extracts localization signatures from raw images without any a priori assumptions or manually assigned labels. To visualize the relationships between these high-dimensional encodings, we embedded the encodings for all 1,310 OpenCell targets in two dimensions using UMAP, an algorithm that reduces high-dimensional datasets to two dimensions (UMAP 1 and UMAP 2) while attempting to preserve the global and local structures of the original data (53). The resulting map is organized in distinct territories that closely match manual annotations (Fig. 3D, highlighting mono-localizing proteins). This validates that the encoding approach yields a quantitative representation of the biologically relevant information in our microscopy data. The separation of different protein clusters in the UMAP embedding (further discussed below) mirrors the fascinating diversity of localization patterns across the full proteome. Images from nuclear proteins offer compelling illustrative examples of this diversity and reveal how fine-scale details can define the localization of proteins within the same organelle (Fig. 3E).
Functional specificity of protein localization in the human cell.
Extracting functional insights directly from cellular images is a major goal of modern cell biology and data science (54). In this context, our image library and associated machine learning encodings enable us to explore what degree of functional relationship can be inferred between proteins solely based on their localization. For this, we first employed an unsupervised Leiden clustering strategy commonly used to identify cell types in single-cell RNA sequencing datasets (55). Clusters group proteins that share similar localization properties (every protein in the dataset is included in a cluster); these groups can then be analyzed for how well they match different sets of ground-truth annotations (Fig. 4A). The average size of clusters is controlled by varying a hyper-parameter called resolution (Fig. S8A). Systematically varying clustering resolution in our dataset revealed that not only did low-resolution clusters delineate proteins belonging to the same organelles (Fig. 4A–B), clustering at higher resolution also enabled to delineate functional pathways and even molecular complexes of interacting proteins (Fig. 4A–C). This demonstrates that the spatial distribution of each protein in the cell is highly specific, to the point that proteins sharing closely related functions can be identified on the sole basis of the similarity between their spatial distributions. This is further illustrated by how finely high-resolution clusters encapsulate proteins specialized in defined cellular functions (Fig. 4C). For example, our analysis not only separated P-body proteins (cluster #83) from other forms of punctated cytoplasmic structures, but also unambiguously differentiated vesicular trafficking pathways despite their very similar localization patterns: the endosomal machinery (#40), plasma membrane endocytic pits (#117) or COP-II vesicles (#143) were all delineated with high precision (Fig. 4C). Among ER proteins, the translocon clusters with the SRP receptor, EMC subunits and the OST glycosylation complex, all responsible for cotranslational operations (#9). This performance extends to cytoplasmic (Fig. S8A) and nuclear clusters (Fig. S8B), revealing that spatial patterning is not limited to membrane-bound organelles and that sub-compartments exist also in the nucleo-cytoplasm. An illustrative example is a cytoplasmic cluster (#17) formed by a group of RNA-binding proteins (including ATXN2L, NUFIP2 or FXR1, Fig. 4C) that separate into granules upon stress conditions (56–59). Stress granules are not formed under the standard growth conditions used in our experiments, but the ability of our analysis to cluster these proteins together reveals an underlying specificity to their cytoplasmic localization (i.e., “texture”) even in the absence of stress.
Figure 4: protein functional features derived from unsupervised image analysis.
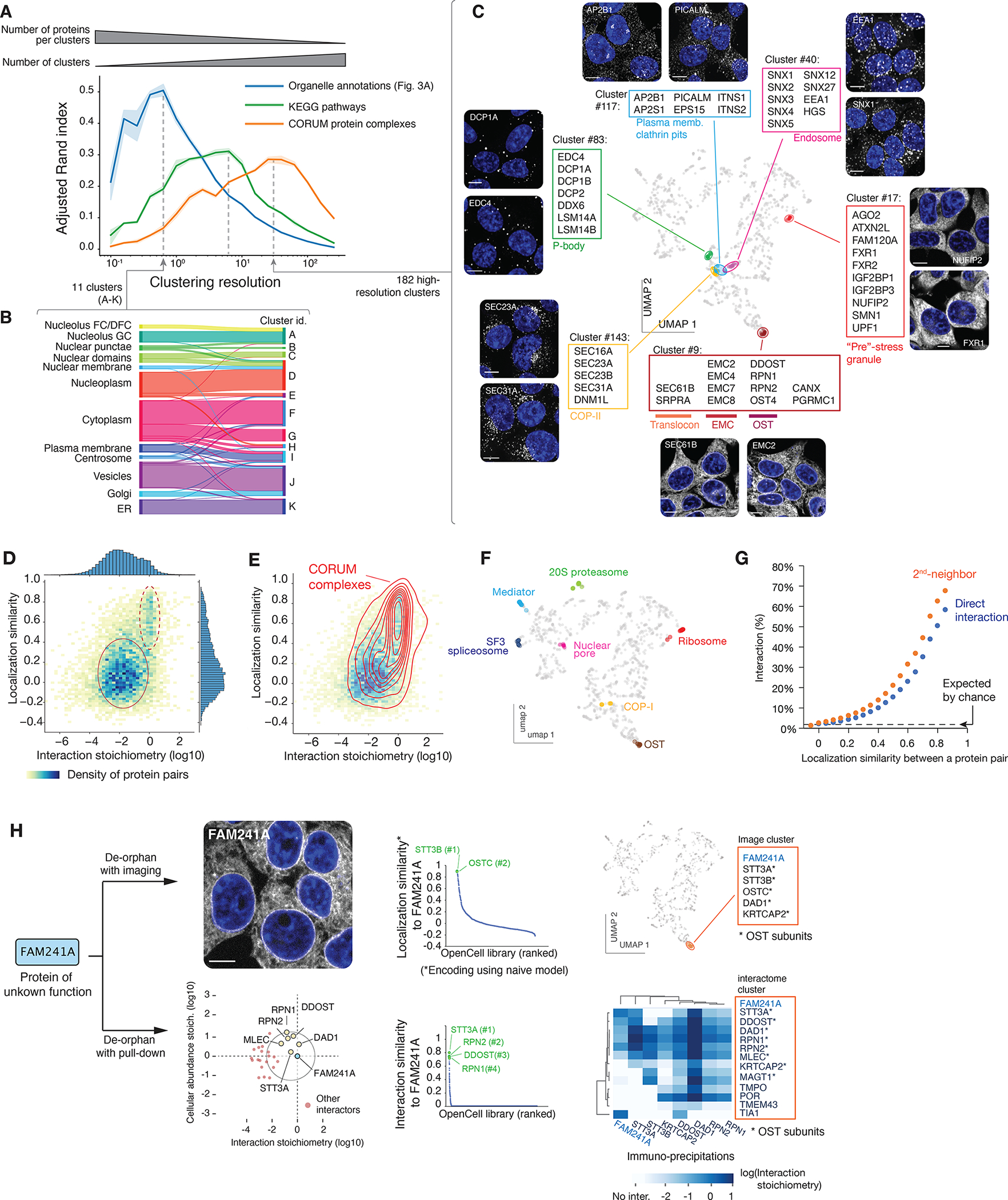
(A) Comparison of image-based Leiden clusters with ground-truth annotations. The Adjusted Rand Index (ARI, (86)) of clusters relative to three ground-truth datasets is plotted as a function of the Leiden clustering resolution. ARI (a metric between 0 and 1, see Materials and Methods) measures how well the groups from a given partition (in our case, the groups of proteins delineated at different clustering resolutions) match groups defined in a reference set. The amplitude of the ARI curves is approximately equal to the number of pairs of elements that partition similarly between sets; the resolution at which each curve reaches its maximum corresponds to the resolution that best captures the information in each ground-truth dataset. At a low resolution, Leiden clustering delineates groups that recapitulate about half of the organellar localization annotations, while at increasing resolutions, clustering recapitulates about a third of pathways annotated in KEGG, or molecular protein complexes annotated in CORUM. Shaded regions show standard deviations calculated from 9 separate repeat rounds of clustering, and average values are shown as a solid line. (B) High correspondence between low-resolution image clusters and cellular organelles. (C) Examples of functional groups delineated by high-resolution image clusters, highlighted on the localization UMAP. (D) Heatmap distribution of localization similarity (defined as the Pearson correlation between two deep learning-derived encoding vectors) vs. interaction stoichiometry between all interacting pairs of OpenCell targets. Two discrete sub-groups are outlined: low stoichiometry/low localization similarity pairs (solid line) and high stoichiometry/high localization similarity pairs (dashed line). (E) Probability density distribution of CORUM interactions mapped on the graph from (D). Contours correspond to iso-proportions of density thresholds for each 10th percentile. (F) Localization patterns of different subunits from example stable protein complexes, represented on the localization UMAP. (G) Frequency of direct (1st-neighbor) or once-removed (2nd neighbor, having a direct interactor in common) protein-protein interactions between any two pairs of OpenCell targets sharing localization similarities above a given threshold (x-axis). (H) Parallel identification of FAM241A as a new OST subunit by imaging or mass-spectrometry. See text for details.
A direct comparison between imaging and interactome data allows us to further examine the extent to which molecular-level relationships (that is, protein interactions) can be derived from a comparison of localization patterns. For OpenCell targets that directly interact, we compared the correlation between their localization encodings derived from machine learning (defining a “localization similarity”) and the stoichiometry of their interaction. This “localization similarity” measures the similarity between the global steady-state distributions of two proteins, as opposed to a direct measure of co-localization. We find that most proteins interact with low stoichiometry (as we previously described (7)) and without strong similarities in their spatial distribution (Fig 4D, solid oval). This means that while low-stoichiometry interactors co-localize at least partially to interact, their global distribution within the cell is different at steady state. On the other hand, high stoichiometry interactors share very similar localization signatures (Fig 4D, dashed oval). Indeed, proteins interacting within stable complexes annotated in CORUM fall into this category (Fig 4E), and the localization signatures of different subunits from large complexes are positioned very closely in UMAP embedding (Fig. 4F). In an important correlate, we found that a high similarity of spatial distribution is a strong predictor of molecular interaction. Across the entire set of target pairs (predicted to interact or not), proteins that share high localization similarities are also very likely to interact (Fig. 4G). For example, target pairs with a localization similarity greater than 0.85 have a 58% chance of being direct interactors, and a 68% chance of being second-neighbors (i.e., sharing a direct interactor in common). This suggests that protein-protein interactions could be identified from a quantitative comparison of spatial distribution alone. To test this, we focused on FAM241A (C4orf32), a protein of unknown function that was not part of our original library and asked whether we could predict its interactions using imaging data alone, compared to the classical de-orphaning approach that uses interaction proteomics. We thus generated a FAM241A endogenous fusion that was analyzed with live imaging and IP-MS separately. Encoding its localization pattern using a “naïve” machine learning model that was never trained with images of this new target revealed a very high localization similarity with two subunits of the ER oligo-saccharyl transferase OST (>0.85 similarity to STT3B and OSTC), and high-resolution Leiden clustering placed FAM241A in an image cluster containing only OST subunits (Fig 4H, top). This analysis suggested that FAM241A is a high-stoichiometry interactor of OST. IP-MS identified that FAM241A was indeed a stoichiometric subunit of the OST complex (Fig. 4H, bottom). While the specific function of FAM241A in protein glycosylation remains to be fully elucidated, this proof-of-concept example establishes that live-cell imaging can be used as a specific readout to predict molecular interactions.
Collectively, our analyses establish that the spatial distribution of a given protein contains highly specific information from which precise functional attributes can be extracted by modern machine learning algorithms. In addition, we show that while high-stoichiometry interactors share very similar localization patterns, most proteins interact with low stoichiometry and share different localization signatures. This reinforces the importance of low-stoichiometry interactions for defining the overall structure of the cellular network, not only providing the “glue” that holds the interactome network together (7) but also connecting different cellular compartments.
RNA-binding proteins form a unique group in both interactome and spatial networks.
To gain insight into global signatures that organize the proteome, we further examined the structures of our imaging and interactome datasets. First, we reduced the dimensionality of each dataset by grouping proteins into their respective spatial clusters (as defined by the high-resolution localization-based clusters in Figs. 4A, 4C) or interaction communities (as defined in Fig. 2B). We then separately clustered these spatial groups (Fig. S9A) and interaction communities (Fig. S9B) to formalize paired hierarchical descriptions of the human proteome organization. These hierarchies are highly structured and delineate clear groups of proteins (see comparison to hierarchies expected by chance, Fig. S9C). In both hierarchies, groups isolated at an intermediate hierarchical layer outline “modules” which are enriched for specific cellular functions or compartments (Fig. S9A–B; full ontology analysis in Suppl. Tables 5 & 9). At a higher layer, each dataset is partitioned into three “branches”, which represent core signatures that shape the proteome’s architecture from a molecular or spatial perspective (Fig. S9A–B). The structure of the localization-based hierarchy (Fig. S9A) recapitulates the human cell’s architecture across its three key compartments (nucleus, cytoplasm, membrane-bound organelles, Fig. S10A–B), which validates the relevance of our unsupervised hierarchical analysis. This motivated a deeper examination of the hierarchical architecture of the interactome (Fig. S9B, ontology analysis in Table S5). We found that intermediate-layer modules of the interactome delineate specific cellular functions such as transcription or vesicular transport (Fig. S9B), reflecting as expected that functional pathways are formed by groups of proteins that physically interact (60, 61). More strikingly, the highest-layer structure showed that two of the three interactome branches were defined by clear functional signatures (Fig. S10C–E): branch B is significantly enriched in proteins that reside in or interact with lipid membranes, while branch C is significantly enriched in RNA-binding proteins (RNA-BPs) (Fig. 5B). This indicates that both membrane-related proteins and RNA-BPs interact more preferentially with each other than with other kinds of proteins in the cell.
Figure 5: segregation of RNA-BPs in both interactome and imaging datasets.

(A) Hierarchical structure of the interactome dataset, see full description in Figure S9B. (B) Distribution of membrane-related (transmembrane or membrane-binding) and RNA-BPs within the three interactome branches. (C) Distribution of intrinsic disorder in the RNA-BP branch of the interactome hierarchy (related to Figure S10). Two separate scores are shown for completeness: IUPRED2 (87), and metapredict (88), a new aggregative disorder scoring algorithm. Boxes represent 25th, 50th, and 75th percentiles, and whiskers represent 1.5x inter-quartile range. Median is represented by a white line. ** p < 10–4 (Student’s t-test), exact p-values are shown. (D) Distribution of RNA-BP percentage across spatial clusters, comparing our data to a control in which the membership of proteins across clusters was randomized 1,000 times. Lines indicate parts of the distribution over-represented in our data vs control (**: p < 2×10−3, Fisher’s exact t-test). (E) Distribution of disorder score (IUPRED2) across spatial clusters, comparing our data to a control in which the membership of proteins across clusters was randomized 1,000 times. Lines indicate parts of the distribution over-represented in our data vs control (**: p < 2×10−3, Fisher’s exact t-test). (F) Ontology enrichment analysis of proteins contained in high-disorder spatial clusters (average disorder score > 0.45). Enrichment compares to the whole set of OpenCell targets (p-value: Fisher’s exact test). (G) Prevalence of proteins annotated to be involved in biomolecular condensation in high-disorder vs. other spatial clusters. Boxes represent 25th, 50th, and 75th percentiles, and whiskers represent 1.5x inter-quartile range. Median is represented by a white line. Note that for both distributions, the median is zero. (H) Distribution of high-disorder spatial clusters in the UMAP embedding from Fig. 3D. Individual nuclear clusters are not outlined for readability. Multiple high-disorder spatial clusters include compartments or proteins known to be characterized by biomolecular condensation behaviors, which are marked by an asterisk.
That membrane-related proteins form a specific interaction group is perhaps not surprising as the membrane surfaces that sequester them within the three-dimensional cell will be partially maintained upon detergent solubilization. On the other hand, the fact that RNA-BPs also form a specific interaction group is unexpected, since our protein interactions were measured in nuclease-treated samples (21) in which most RNAs are degraded. This suggests that protein features beyond binding to RNAs themselves might drive the preferential interactions of RNA-BPs with each other. Therefore, we reasoned that the biophysical properties of proteins within each interactome branch might underly their segregation. Indeed, an analysis of protein sequence features revealed a separation of different biophysical properties in each branch (Fig. S10F–G). Branch B was enriched for hydrophobic sequences (Fig. 5C), consistent with its enrichment for membrane-related proteins, while branch C was enriched for intrinsic disorder (Fig. 5C). This is consistent with the fact that RNA-BPs are significantly more disordered than other proteins in the proteome (Fig. S11A, (62)). RNA-BPs are also among the most abundant in the cell (Fig. S11B), and form a higher number of interactions than other proteins (Fig. S11C–D).
IP-MS measures protein interactions in vitro after lysis and therefore does not directly address the spatial relationship between interacting proteins. Thus, we sought to further examine how RNA-BPs distribute in our live-cell imaging data. If RNA-BPs segregate into interacting groups in vivo, this should also manifest at the level of their intracellular localization: they should enrich in the same spatial clusters derived from our unsupervised machine learning analysis. Indeed, the distribution of RNA-BP content within spatial clusters revealed a significant over-representation of clusters that are either strongly enriched or depleted for RNA-BPs (Fig. 5D). Since spatial clusters can be interpreted as defining “micro-compartments” within the cell, both enrichment and depletion have functional implications: not only are RNA-BPs enriched within the same micro-compartments, they tend to also be excluded from others. 16 out of the 26 spatial clusters (62%) that are highly enriched in RNA-BPs include at least one protein involved in biomolecular condensation (as curated in PhaSepDB (63)), which might reflect a prevalent role for biomolecular condensation in shaping the RNA-BP proteome. Collectively, both interactome and imaging data underscore that RNA-BPs (a prevalent group of proteins that represents 13% of proteins expressed in HEK293T cells, see Table S2) form a distinct sub-group within the proteome characterized by unique properties.
These results motivated a broader analysis of the contribution of intrinsic disorder to the spatial organization of the proteome in our dataset. Plotting the distribution of mean intrinsic disorder within spatial clusters revealed a significant over-representation of clusters both enriched and depleted in disordered proteins (Fig. 5E). 26 out of 182 total spatial clusters were enriched for disordered proteins, covering 13% of the proteins in our imaging dataset. Overall, the extent to which disordered proteins segregate spatially is similar to the degree of segregation found for hydrophobic proteins: an analogous analysis revealed that 10% of proteins in our dataset are found within clusters significantly enriched for high hydrophobicity (Fig. S12E), which map to membrane-bound organelles (Fig. S12F). This supports the hypothesis that intrinsic disorder is as important a feature as hydrophobicity in organizing the spatial distribution of the human proteome. Consistent with our previous analysis, high-disorder clusters were enriched for RNA-BPs (Fig. 5F), with 15 out of these 26 clusters containing over 50% of RNA-BPs. High-disorder clusters were also enriched for proteins annotated to participate in biomolecular condensation (Fig. 5G), and were predominantly found in the nucleus (19 clusters, 73% of total, Fig. 5H). 5 out of 7 high-disorders clusters found in the cytosol delineate compartments for which biomolecular condensation has been proposed to play an important role (Fig. 5G), namely P-bodies (64), stress granules (59), centrosome (65), cell junctions (66) and the interface between cell surface and actin cytoskeleton (67).
Interactive data sharing at opencell.czbiohub.org
To enable widespread access to the OpenCell datasets, we built an interactive web application that provides side-by-side visualizations of the 3D confocal images and of the interaction network for each tagged protein, together with RNA and protein abundances for the whole proteome (Fig. 6). Our web interface is fully described in Suppl. Fig S12.
Figure 6: the OpenCell website.
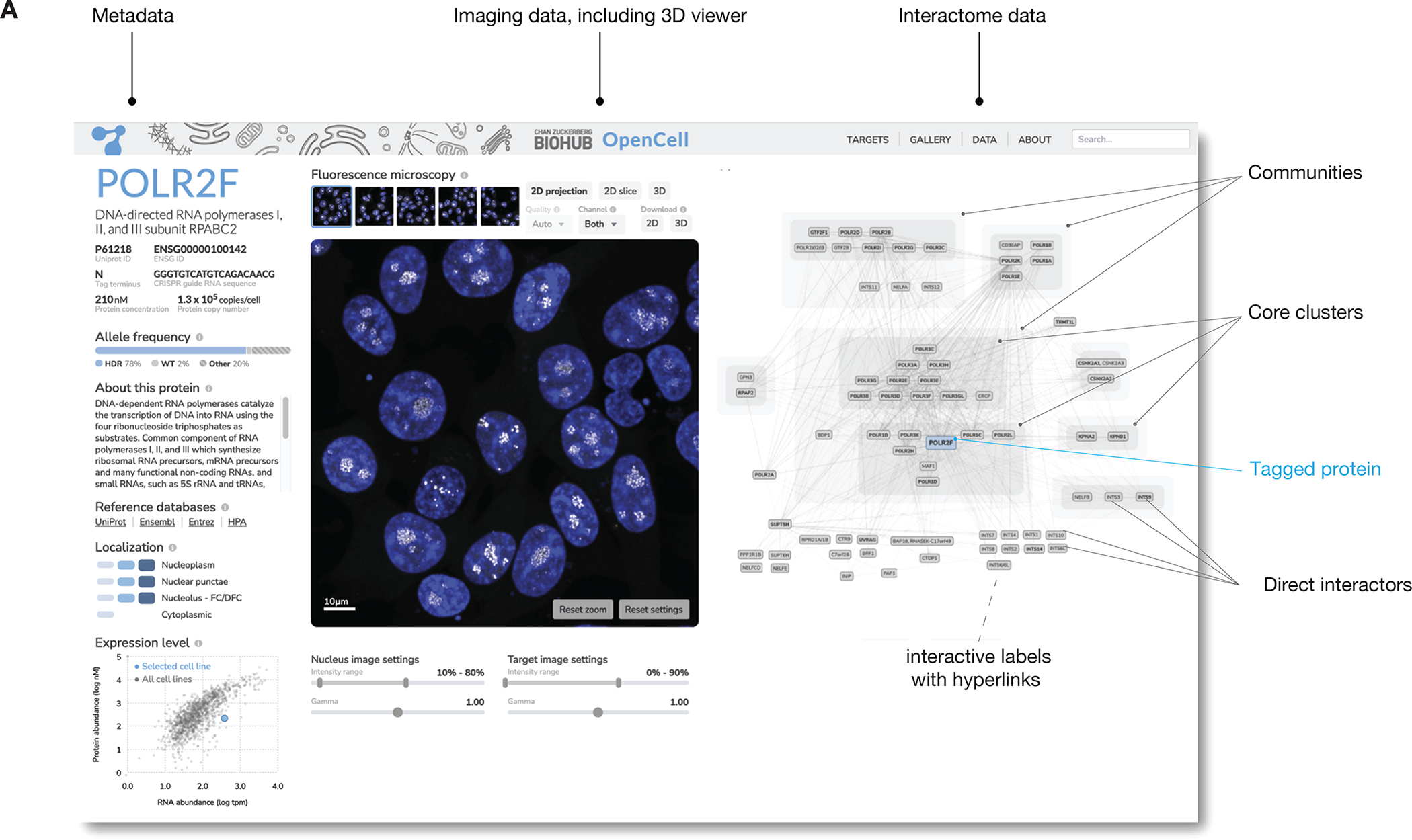
Shown is an annotated screenshot from our web-app at http://opencell.czbiohub.org, which is described in more details in Suppl Fig. S12.
Discussion
OpenCell combines three strategies to augment the description of human cellular architecture. First, we present an integrated experimental pipeline for high-throughput cell biology, fueled by scalable methods for genome engineering, live-cell microscopy and IP-MS. Second, we provide an open-source resource of well-curated localization and interactome measurements, easily accessible through an interactive web interface at opencell.czbiohub.org. And third, we developed an analytical framework for the representation and comparison of interaction or localization signatures (including a self-supervised machine learning approach for image encoding). Finally, we demonstrate how our dataset can be used both for fine-grained mechanistic exploration (to explore the function of multiple proteins that were previously uncharacterized), as well as for investigating the core organizational principles of the proteome.
Our current strategy that combines split-FPs and HEK293T – a cell line that is heavily transformed but easily manipulatable – is mostly constrained by scalability considerations. Excitingly, technological advances are quickly broadening the set of cellular systems that can be engineered and profiled at scale. Advances in stem cell technologies enable the generation of libraries that can be differentiated in multiple cell types (11), while innovations in genome engineering (for example, by modulating DNA repair (68)) pave the way for the scalable insertion of gene-sized payload, for the combination of multiple edits in the same cell, or for increased homozygosity in polyclonal pools. In addition, recent developments in high-throughput light-sheet microscopy (69) might soon enable the systematic description of 4D intracellular dynamics (70).
A central feature of our approach is to use endogenous fluorescent tags to study protein function. Genome-edited cells enable to examine protein function at near-native expression levels (which can circumvent some limitations of over-expression (71)), and to measure protein localization in live cells (which can avoid artefacts caused by fixation or antibody labeling (72)). Comparing our data to the current reference datasets of protein-proteins interactions (Fig. S4C–F) or localization (Fig. S7C–D) highlights the performance of our strategy. In addition, our high success rate tagging essential genes (Fig. S2A; see also (73) in yeast) and the successful tagging of the near-complete yeast proteome (14, 73) support that fluorescent tagging generally preserves normal protein physiology. However, limitations exist for specific protein targets. FPs are as big as an average human protein and their insertion can impair function or localization, for example by occluding important interaction interfaces or impairing sub-cellular targeting sequences. In other cases, tags can affect expression or degradation rates, which might explain why we find tagged proteins being expressed at 80% of their endogenous abundance, and 8% of targets in our dataset having outlier abundances at steady-state (Fig. S3D). Further, tagging often cannot discriminate between different isoforms of a protein (such as splicing or post-translationally modified variants). Finally, relying on endogenous expression can be an obstacle given the low concentration of most proteins in the human cell: even using a very bright FP like mNeonGreen (74), detecting proteins in the bottom 50% percentile of abundance is difficult (Fig. S2D). Solutions to this obstacle include using FP repeats to increase signal (18, 23) or using tags that bind chemical fluorophores (e.g., HaloTag (75)), which can be brighter than FPs or operate at wavelengths where cellular auto-fluorescence is decreased (76). Overall, the full description of human cellular architecture remains a formidable challenge which will require complementary methods being applied in parallel. The diversity of large-scale cell biology approaches is a solution to this problem (6, 8, 9, 11, 31, 70, 77–80). Mirroring the advances in genomics following the human genome sequence (2), open-source systematic datasets will likely play an important role in how the growth of cell biology measurements can be transformed into fundamental discoveries by an entire community (81).
In addition to presenting a resource of measurements and protocols, we also demonstrate how our data can be used to study the global signatures that pattern the proteome. Our analysis reveals that RNA-binding proteins, which form one of the biggest functional family in the cell, are characterized by a unique set of properties and segregate from other proteins in term of both interactions and spatial distribution. It would be fascinating to explore to which extent RNA itself might act as a structural organizer of the cellular proteome (62, 82). This is for example the case for some non-coding RNAs whose main function is to template protein interactions to form nuclear bodies (83). High intrinsic disorder is one of the distinguishing features of RNA-BPs, which likely contributes to their unique properties. Beyond RNA-BPs, our data supports a general role for intrinsic disorder in shaping the spatial distribution of human proteins. For example, 13% of proteins in our dataset are found in spatial clusters that are significantly enriched for disordered proteins. This adds to the growing appreciation that intrinsic disorder, which is much more prevalent in eukaryotic vs. prokaryotic proteomes (84, 85), plays a key role in the functional sub-compartmentalization of the eukaryotic nucleo- and cytoplasm in the context of biomolecular condensation (86).
Lastly, we show that the spatial distribution of each human protein is very specific, to the point that remarkably detailed functional relationships can be inferred on the sole basis of similarities between localization patterns – including the prediction of molecular interactions (which complements other studies (87)). This highlights that intracellular organization is defined by fine-grained features that go beyond membership to a given organelle. Our demonstration that self-supervised deep learning models can identify complex but deterministic signatures from light microscopy images opens exciting avenues for the use of imaging as an information-rich method for deep phenotyping and functional genomics (51). Because light microscopy is easily scalable, can be performed live and enables measurements at the single-cell level, this should offer rich opportunities for the full quantitative description of cellular diversity in normal physiology and disease.
Supplementary Material
Acknowledgements.
We thank N. Neff, M. Tan, R. Sit and A. Detweiler for help with high-throughput sequencing; G. Margulis, A. Sellas, E. Ho and J. Mann for operational support; A. McGeever for help with web application architecture and deployment; and S. Schmid for critical feedback. M.D.L. thanks C. L. Tan for continuous discussions. H.K. was supported by an International Research Fellowship from the Japan Society for the Promotion of Science. R.M.B. was supported by a NIH pre-doctoral fellowship (F31 HL143882). B.H. was supported by NIH (R01GM131641) and is a Chan Zuckerberg Biohub Investigator. J.S.W. was supported by NIH (1RM1HG009490) and is an investigator with the Howard Hughes Medical Institute.
Footnotes
Competing interests. J.S.W. declares outside interest in Chroma Therapeutics, KSQ Therapeutics, Maze Therapeutics, Amgen, Tessera Therapeutics and 5 AM Ventures. M. M. is an indirect shareholder in EvoSep Biosystems.
Material and Methods are available as a supplementary file (see below).
Data and materials availability.
Mass spectrometry raw data and associated MaxQuant output tables are deposited to the ProteomeXchange Consortium via the PRIDEpartner repository (accession PXD024909 for interactome data, and accession PXD029191 for whole-cell abundance data). Bulk RNA-seq raw data and associated kallisto transcript abundance tables are available on GEO (accession GSE186192). Raw microscopy images are hosted by AWS’s Open Datasets Program at https://registry.opendata.aws/czb-opencell/.
References
- 1.Consortium IHGS, Finishing the euchromatic sequence of the human genome. Nature. 431, 931–945 (2004). [DOI] [PubMed] [Google Scholar]
- 2.Hood L, Rowen L, The Human Genome Project: big science transforms biology and medicine. Genome Med. 5, 79 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nurse P, Hayles J, The Cell in an Era of Systems Biology. Cell. 144, 850–854 (2011). [DOI] [PubMed] [Google Scholar]
- 4.Mast FD, Ratushny AV, Aitchison JD, Systems cell biology. The Journal of Cell Biology. 206, 695–706 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Lundberg E, Borner GHH, Spatial proteomics: a powerful discovery tool for cell biology. Nature Reviews Molecular Cell Biology. 20, 285–302 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Luck K, Kim D-K, Lambourne L, Spirohn K, Begg BE, Bian W, Brignall R, Cafarelli T, Campos-Laborie FJ, Charloteaux B, Choi D, Coté AG, Daley M, Deimling S, Desbuleux A, Dricot A, Gebbia M, Hardy MF, Kishore N, Knapp JJ, Kovács IA, Lemmens I, Mee MW, Mellor JC, Pollis C, Pons C, Richardson AD, Schlabach S, Teeking B, Yadav A, Babor M, Balcha D, Basha O, Bowman-Colin C, Chin S-F, Choi SG, Colabella C, Coppin G, D’Amata C, Ridder DD, Rouck SD, Duran-Frigola M, Ennajdaoui H, Goebels F, Goehring L, Gopal A, Haddad G, Hatchi E, Helmy M, Jacob Y, Kassa Y, Landini S, Li R, van Lieshout N, MacWilliams A, Markey D, Paulson JN, Rangarajan S, Rasla J, Rayhan A, Rolland T, San-Miguel A, Shen Y, Sheykhkarimli D, Sheynkman GM, Simonovsky E, Taşan M, Tejeda A, Tropepe V, Twizere J-C, Wang Y, Weatheritt RJ, Weile J, Xia Y, Yang X, Yeger-Lotem E, Zhong Q, Aloy P, Bader GD, Rivas JDL, Gaudet S, Hao T, Rak J, Tavernier J, Hill DE, Vidal M, Roth FP, Calderwood MA, A reference map of the human binary protein interactome. Nature. 580, 1–7 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hein MY, Hubner NC, Poser I, Cox J, Nagaraj N, Toyoda Y, Gak IA, Weisswange I, Mansfeld J, Buchholz F, Hyman AA, Mann M, A Human Interactome in Three Quantitative Dimensions Organized by Stoichiometries and Abundances. Cell. 163, 712–723 (2015). [DOI] [PubMed] [Google Scholar]
- 8.Huttlin EL, Bruckner RJ, Paulo JA, Cannon JR, Ting L, Baltier K, Colby G, Gebreab F, Gygi MP, Parzen H, Szpyt J, Tam S, Zarraga G, Pontano-Vaites L, Swarup S, White AE, Schweppe DK, Rad R, Erickson BK, Obar RA, Guruharsha KG, Li K, Artavanis-Tsakonas S, Gygi SP, Harper JW, Architecture of the human interactome defines protein communities and disease networks. Nature. 545 (2017), doi: 10.1038/nature22366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thul PJ, Åkesson L, Wiking M, Mahdessian D, Geladaki A, Blal HA, Alm T, Asplund A, Björk L, Breckels LM, Bäckström A, Danielsson F, Fagerberg L, Fall J, Gatto L, Gnann C, Hober S, Hjelmare M, Johansson F, Lee S, Lindskog C, Mulder J, Mulvey CM, Nilsson P, Oksvold P, Rockberg J, Schutten R, Schwenk JM, Sivertsson Å, Sjöstedt E, Skogs M, Stadler C, Sullivan DP, Tegel H, Winsnes C, Zhang C, Zwahlen M, Mardinoglu A, Pontén F, von Feilitzen K, Lilley KS, Uhlén M, Lundberg E, A subcellular map of the human proteome. Science. 356, eaal3321 (2017). [DOI] [PubMed] [Google Scholar]
- 10.Bukhari H, Müller T, Endogenous Fluorescence Tagging by CRISPR. Trends Cell Biol. 29, 912–928 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Roberts B, Haupt A, Tucker A, Grancharova T, Arakaki J, Fuqua MA, Nelson A, Hookway C, Ludmann SA, Mueller IA, Yang R, Horwitz AR, Rafelski SM, Gunawardane RN, Molecular biology of the cell, in press, doi: 10.1091/mbc.e17-03-0209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghaemmaghami S, Ghaemmaghami S, Huh W-K, Bower K, Bower K, Howson RW, Belle A, Belle A, Dephoure N, Dephoure N, O’Shea EK, Weissman JS, Global analysis of protein expression in yeast. Nature. 425, 737–741 (2003). [DOI] [PubMed] [Google Scholar]
- 13.Collins SR, Kemmeren P, Zhao X-C, Zhao X-C, Greenblatt JF, Spencer F, Spencer F, Holstege FCP, Holstege FCP, Weissman JS, Krogan NJ, Toward a comprehensive atlas of the physical interactome of Saccharomyces cerevisiae. Molecular & cellular proteomics : MCP. 6, 439–450 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Weill U, Yofe I, Sass E, Stynen B, Davidi D, Natarajan J, Ben-Menachem R, Avihou Z, Goldman O, Harpaz N, Chuartzman S, Kniazev K, Knoblach B, Laborenz J, Boos F, Kowarzyk J, Ben-Dor S, Zalckvar E, Herrmann JM, Rachubinski RA, Pines O, Rapaport D, Michnick SW, Levy ED, Schuldiner M, Genome-wide SWAp-Tag yeast libraries for proteome exploration. Nature Methods. 15 (2018), doi: 10.1038/s41592-018-0044-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Baudin A, Ozier-Kalogeropoulos O, Denouel A, Lacroute F, Cullin C, A simple and efficient method for direct gene deletion in Saccharomyces cerevisiae. Nucleic acids research. 21, 3329–3330 (1993). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Poser I, Sarov M, Hutchins JRA, Hériché J-K, Toyoda Y, Pozniakovsky A, Weigl D, Nitzsche A, Hegemann B, Bird AW, Pelletier L, Kittler R, Hua S, Naumann R, Augsburg M, Sykora MM, Hofemeister H, Zhang Y, Nasmyth K, White KP, Dietzel S, Mechtler K, Durbin R, Stewart AF, Peters J-M, Buchholz F, Hyman AA, BAC TransgeneOmics: a high-throughput method for exploration of protein function in mammals. Nature methods. 5, 409–415 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sigal A, Danon T, Cohen A, Milo R, Geva-Zatorsky N, Lustig G, Liron Y, Alon U, Perzov N, Generation of a fluorescently labeled endogenous protein library in living human cells. Nature protocols. 2, 1515–1527 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Leonetti MD, Sekine S, Kamiyama D, Weissman JS, Huang B, A scalable strategy for high-throughput GFP tagging of endogenous human proteins. Proceedings of the National Academy of Sciences of the United States of America. 113, E3501–8 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hubner NC, Bird AW, Cox J, Splettstoesser B, Bandilla P, Poser I, Hyman A, Mann M, Quantitative proteomics combined with BAC TransgeneOmics reveals in vivo protein interactions. The Journal of cell biology. 189, 739–754 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Feng S, Sekine S, Pessino V, Li H, Leonetti MD, Huang B, Improved split fluorescent proteins for endogenous protein labeling. Nature communications. 8, 370 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.See supplementary Materials and Methods online.
- 22.Meier F, Beck S, Grassl N, Lubeck M, Park MA, Raether O, Mann M, Parallel Accumulation–Serial Fragmentation (PASEF): Multiplying Sequencing Speed and Sensitivity by Synchronized Scans in a Trapped Ion Mobility Device. J Proteome Res. 14, 5378–5387 (2015). [DOI] [PubMed] [Google Scholar]
- 23.Kamiyama D, Sekine S, Barsi-Rhyne B, Hu J, Chen B, Gilbert LA, Ishikawa H, Leonetti MD, Marshall WF, Weissman JS, Huang B, Versatile protein tagging in cells with split fluorescent protein. Nature communications. 7, 11046 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin Y-C, Boone M, Meuris L, Lemmens I, Roy NV, Soete A, Reumers J, Moisse M, Plaisance S, Drmanac R, Chen J, Speleman F, Lambrechts D, de Peer YV, Tavernier J, Callewaert N, Genome dynamics of the human embryonic kidney 293 lineage in response to cell biology manipulations. Nature communications. 5, 4767 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lin S, Staahl B, Alla RK, Doudna JA, Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery. eLife. 3 (2014), doi: 10.7554/elife.04766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Doyon JB, Zeitler B, Cheng J, Cheng AT, Cherone JM, Santiago Y, Lee AH, Vo TD, Doyon Y, Miller JC, Paschon DE, Zhang L, Rebar EJ, Gregory PD, Urnov FD, Drubin DG, Rapid and efficient clathrin-mediated endocytosis revealed in genome-edited mammalian cells. Nature cell biology. 13, 331–337 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gibson TJ, Seiler M, Veitia RA, The transience of transient overexpression. Nat Methods. 10, 715–721 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Keilhauer EC, Hein MY, Mann M, Accurate protein complex retrieval by affinity enrichment mass spectrometry (AE-MS) rather than affinity purification mass spectrometry (AP-MS). Mol Cell Proteom Mcp. 14, 120–35 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thomas JA, Tate CG, Quality Control in Eukaryotic Membrane Protein Overproduction. J Mol Biol. 426, 4139–4154 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giurgiu M, Reinhard J, Brauner B, Dunger-Kaltenbach I, Fobo G, Frishman G, Montrone C, Ruepp A, CORUM: the comprehensive resource of mammalian protein complexes—2019. Nucleic Acids Res. 47, gky973- (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Itzhak DN, Tyanova S, Cox J, Borner GH, Global, quantitative and dynamic mapping of protein subcellular localization. eLife. 5, 570 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Huttlin EL, Bruckner RJ, Navarrete-Perea J, Cannon JR, Baltier K, Gebreab F, Gygi MP, Thornock A, Zarraga G, Tam S, Szpyt J, Panov A, Parzen H, Fu S, Golbazi A, Maenpaa E, Stricker K, Thakurta SG, Rad R, Pan J, Nusinow DP, Paulo JA, Schweppe DK, Vaites LP, Harper JW, Gygi SP, Biorxiv, in press, doi: 10.1101/2020.01.19.905109. [DOI] [Google Scholar]
- 33.Royer L, Reimann M, Stewart AF, Schroeder M, Network Compression as a Quality Measure for Protein Interaction Networks. PLoS ONE. 7, e35729 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Enright AJ, Dongen SV, Ouzounis CA, An efficient algorithm for large-scale detection of protein families. Nucleic Acids Research. 30, 1575–1584 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shurtleff MJ, Itzhak DN, Hussmann JA, Oakdale NTS, Costa EA, Jonikas M, Weibezahn J, Popova KD, Jan CH, Sinitcyn P, Vembar SS, Hernandez H, Cox J, Burlingame AL, Brodsky JL, Frost A, Borner GH, Weissman JS, The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. Elife. 7, e37018 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Acosta-Alvear D, Karagöz GE, Fröhlich F, Li H, Walther TC, Walter P, The unfolded protein response and endoplasmic reticulum protein targeting machineries converge on the stress sensor IRE1. Elife. 7, e43036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McGilvray PT, Anghel SA, Sundaram A, Zhong F, Trnka MJ, Fuller JR, Hu H, Burlingame AL, Keenan RJ, An ER translocon for multi-pass membrane protein biogenesis. Elife. 9, e56889 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chitwood PJ, Hegde RS, An intramembrane chaperone complex facilitates membrane protein biogenesis. Nature. 584, 630–634 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Stoeger T, Gerlach M, Morimoto RI, Amaral LAN, Large-scale investigation of the reasons why potentially important genes are ignored. PLoS biology. 16, e2006643 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brooks SP, Coccia M, Tang HR, Kanuga N, Machesky LM, Bailly M, Cheetham ME, Hardcastle AJ, The Nance–Horan syndrome protein encodes a functional WAVE homology domain (WHD) and is important for co-ordinating actin remodelling and maintaining cell morphology. Hum Mol Genet. 19, 2421–2432 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Law A-L, Jalal S, Mosis F, Pallett T, Guni A, Brayford S, Yolland L, Marcotti S, Levitt JA, Poland SP, Rowe-Sampson M, Jandke A, Köchl R, Pula G, Ameer-Beg SM, Stramer BM, Krause M, Biorxiv, in press, doi: 10.1101/2020.05.11.083030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schossig A, Wolf NI, Fischer C, Fischer M, Stocker G, Pabinger S, Dander A, Steiner B, Tönz O, Kotzot D, Haberlandt E, Amberger A, Burwinkel B, Wimmer K, Fauth C, Grond-Ginsbach C, Koch MJ, Deichmann A, von Kalle C, Bartram CR, Kohlschütter A, Trajanoski Z, Zschocke J, Mutations in ROGDI Cause Kohlschütter-Tönz Syndrome. Am J Hum Genetics. 90, 701–707 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Merkulova M, Păunescu TG, Azroyan A, Marshansky V, Breton S, Brown D, Mapping the H+ (V)-ATPase interactome: identification of proteins involved in trafficking, folding, assembly and phosphorylation. Scientific Reports. 5 (2015), doi: 10.1038/srep14827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yan Y, Denef N, Schüpbach T, The Vacuolar Proton Pump, V-ATPase, Is Required for Notch Signaling and Endosomal Trafficking in Drosophila. Dev Cell. 17, 387–402 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Vasanthakumar T, Rubinstein JL, Structure and Roles of V-type ATPases. Trends Biochem Sci. 45, 295–307 (2020). [DOI] [PubMed] [Google Scholar]
- 46.Görlich D, Kutay U, TRANSPORT BETWEEN THE CELL NUCLEUS AND THE CYTOPLASM. Annu Rev Cell Dev Bi. 15, 607–660 (1999). [DOI] [PubMed] [Google Scholar]
- 47.Lusk CP, King MC, The nucleus: keeping it together by keeping it apart. Curr Opin Cell Biol. 44, 44–50 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Breker M, Gymrek M, Schuldiner M, A novel single-cell screening platform reveals proteome plasticity during yeast stress responsesYeast proteome plasticity. J Cell Biology. 200, 839–850 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Sullivan DP, Winsnes CF, Åkesson L, Hjelmare M, Wiking M, Schutten R, Campbell L, Leifsson H, Rhodes S, Nordgren A, Smith K, Revaz B, Finnbogason B, Szantner A, Lundberg E, Deep learning is combined with massive-scale citizen science to improve large-scale image classification. Nat Biotechnol. 36, 820–828 (2018). [DOI] [PubMed] [Google Scholar]
- 50.Ouyang W, Winsnes CF, Hjelmare M, Cesnik AJ, Åkesson L, Xu H, Sullivan DP, Dai S, Lan J, Jinmo P, Galib SM, Henkel C, Hwang K, Poplavskiy D, Tunguz B, Wolfinger RD, Gu Y, Li C, Xie J, Buslov D, Fironov S, Kiselev A, Panchenko D, Cao X, Wei R, Wu Y, Zhu X, Tseng K-L, Gao Z, Ju C, Yi X, Zheng H, Kappel C, Lundberg E, Analysis of the Human Protein Atlas Image Classification competition. Nat Methods. 16, 1254–1261 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chandrasekaran SN, Ceulemans H, Boyd JD, Carpenter AE, Image-based profiling for drug discovery: due for a machine-learning upgrade? Nat Rev Drug Discov. 20, 145–159 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kobayashi H, Cheveralls KC, Leonetti MD, Royer LA, Self-Supervised Deep-Learning Reveals High-Resolution Functional Features from Protein Localization Microscopy. in preparation. [DOI] [PMC free article] [PubMed]
- 53.McInnes L, Healy J, Melville J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. Arxiv (2018). [Google Scholar]
- 54.Meijering E, Carpenter AE, Peng H, Hamprecht FA, Olivo-Marin J-C, Imagining the future of bioimage analysis. Nat Biotechnol. 34, 1250–1255 (2016). [DOI] [PubMed] [Google Scholar]
- 55.Traag VA, Waltman L, van Eck NJ, From Louvain to Leiden: guaranteeing well-connected communities. Sci Rep-uk. 9, 5233 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Markmiller S, Soltanieh S, Server KL, Mak R, Jin W, Fang MY, Luo E-C, Krach F, Yang D, Sen A, Fulzele A, Wozniak JM, Gonzalez DJ, Kankel MW, Gao F-B, Bennett EJ, Lécuyer E, Yeo GW, Context-Dependent and Disease-Specific Diversity in Protein Interactions within Stress Granules. Cell. 172, 590–604.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Youn J-Y, Dunham WH, Hong SJ, Knight JDR, Bashkurov M, Chen GI, Bagci H, Rathod B, MacLeod G, Eng SWM, Angers S, Morris Q, Fabian M, Côté J-F, Gingras A-C, High-Density Proximity Mapping Reveals the Subcellular Organization of mRNA-Associated Granules and Bodies. Mol Cell. 69, 517–-532.e11. (2018). [DOI] [PubMed] [Google Scholar]
- 58.Marmor-Kollet H, Siany A, Kedersha N, Knafo N, Rivkin N, Danino YM, Moens TG, Olender T, Sheban D, Cohen N, Dadosh T, Addadi Y, Ravid R, Eitan C, Cohen BT, Hofmann S, Riggs CL, Advani VM, Higginbottom A, Cooper-Knock J, Hanna JH, Merbl Y, Bosch LVD, Anderson P, Ivanov P, Geiger T, Hornstein E, Spatiotemporal Proteomic Analysis of Stress Granule Disassembly Using APEX Reveals Regulation by SUMOylation and Links to ALS Pathogenesis. Mol Cell. 80, 876–891.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Yang P, Mathieu C, Kolaitis R-M, Zhang P, Messing J, Yurtsever U, Yang Z, Wu J, Li Y, Pan Q, Yu J, Martin EW, Mittag T, Kim HJ, Taylor JP, G3BP1 Is a Tunable Switch that Triggers Phase Separation to Assemble Stress Granules. Cell. 181, 325–345.e28 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Costanzo M, VanderSluis B, Koch EN, Baryshnikova A, Pons C, Tan G, Wang W, Usaj M, Hanchard J, Lee SD, Pelechano V, Styles EB, Billmann M, van Leeuwen J, van Dyk N, Lin Z-Y, Kuzmin E, Nelson J, Piotrowski JS, Srikumar T, Bahr S, Chen Y, Deshpande R, Kurat CF, Li SC, Li Z, Usaj MM, Okada H, Pascoe N, Luis B-JS, Sharifpoor S, Shuteriqi E, Simpkins SW, Snider J, Suresh HG, Tan Y, Zhu H, Malod-Dognin N, Janjic V, Przulj N, Troyanskaya OG, Stagljar I, Xia T, Ohya Y, Gingras A-C, Raught B, Boutros M, Steinmetz LM, Moore CL, Rosebrock AP, Caudy AA, Myers CL, Andrews B, Boone C, A global genetic interaction network maps a wiring diagram of cellular function. Science. 353, aaf1420 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Horlbeck MA, Xu A, Wang M, Bennett NK, Park CY, Bogdanoff D, Adamson B, Chow ED, Kampmann M, Peterson TR, Nakamura K, Fischbach MA, Weissman JS, Gilbert LA, Mapping the Genetic Landscape of Human Cells. Cell. 174, 953–967.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Balcerak A, Trebinska-Stryjewska A, Konopinski R, Wakula M, Grzybowska EA, RNA–protein interactions: disorder, moonlighting and junk contribute to eukaryotic complexity. Open Biol. 9, 190096 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.You K, Huang Q, Yu C, Shen B, Sevilla C, Shi M, Hermjakob H, Chen Y, Li T, PhaSepDB: a database of liquid–liquid phase separation related proteins. Nucleic Acids Res. 48, D354–D359 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Luo Y, Na Z, Slavoff SA, P Bodies: Composition, Properties, and Functions. Biochemistry-us. 57, 2424–2431 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Woodruff JB, Gomes BF, Widlund PO, Mahamid J, Honigmann A, Hyman AA, The Centrosome Is a Selective Condensate that Nucleates Microtubules by Concentrating Tubulin. Cell. 169, 1066–1077.e10 (2017). [DOI] [PubMed] [Google Scholar]
- 66.Beutel O, Maraspini R, Pombo-García K, Martin-Lemaitre C, Honigmann A, Phase Separation of Zonula Occludens Proteins Drives Formation of Tight Junctions. Cell. 179, 923–936.e11 (2019). [DOI] [PubMed] [Google Scholar]
- 67.Banjade S, Wu Q, Mittal A, Peeples WB, Pappu RV, Rosen MK, Conserved interdomain linker promotes phase separation of the multivalent adaptor protein Nck. Proc National Acad Sci. 112, E6426–E6435 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Riesenberg S, Chintalapati M, Macak D, Kanis P, Maricic T, Pääbo S, Simultaneous precise editing of multiple genes in human cells. Nucleic acids research. 2, 163 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Yang B, Chen X, Wang Y, Feng S, Pessino V, Stuurman N, Cho NH, Cheng KW, Lord SJ, Xu L, Xie D, Mullins RD, Leonetti MD, Huang B, Epi-illumination SPIM for volumetric imaging with high spatial-temporal resolution. Nature methods. 16, 501–504 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cai Y, Hossain MJ, Hériché J-K, Politi AZ, Walther N, Koch B, Wachsmuth M, Nijmeijer B, Kueblbeck M, Martinic-Kavur M, Ladurner R, Alexander S, Peters J-M, Ellenberg J, Experimental and computational framework for a dynamic protein atlas of human cell division. Nature. 561, 411–415 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.von Mering C, Krause R, Snel B, Cornell M, Oliver SG, Fields S, Bork P, Comparative assessment of large-scale data sets of protein-protein interactions. Nature. 417, 399–403 (2002). [DOI] [PubMed] [Google Scholar]
- 72.Schnell U, Dijk F, Sjollema KA, Giepmans BNG, Immunolabeling artifacts and the need for live-cell imaging. Nature methods. 9, 152–158 (2012). [DOI] [PubMed] [Google Scholar]
- 73.Huh W-K, Falvo JV, Gerke LC, Carroll AS, Howson RW, Weissman JS, O’Shea EK, Global analysis of protein localization in budding yeast. Nature. 425, 686–691 (2003). [DOI] [PubMed] [Google Scholar]
- 74.Shaner NC, Lambert GG, Chammas A, Ni Y, Cranfill PJ, Baird MA, Sell BR, Allen JR, Day RN, Israelsson M, Davidson MW, Wang J, A bright monomeric green fluorescent protein derived from Branchiostoma lanceolatum. Nature methods. 10, 407–409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Los GV, Encell LP, McDougall MG, Hartzell DD, Karassina N, Zimprich C, Wood MG, Learish R, Ohana RF, Urh M, Simpson D, Mendez J, Zimmerman K, Otto P, Vidugiris G, Zhu J, Darzins A, Klaubert DH, Bulleit RF, Wood KV, HaloTag: a novel protein labeling technology for cell imaging and protein analysis. ACS chemical biology. 3, 373–382 (2008). [DOI] [PubMed] [Google Scholar]
- 76.Lavis LD, Chemistry Is Dead. Long Live Chemistry! Biochemistry-us. 56, 5165–5170 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Go CD, Knight JDR, Rajasekharan A, Rathod B, Hesketh GG, Abe KT, Youn J-Y, Samavarchi-Tehrani P, Zhang H, Zhu LY, Popiel E, Lambert J-P, Coyaud É, Cheung SWT, Rajendran D, Wong CJ, Antonicka H, Pelletier L, Raught B, Palazzo AF, Shoubridge EA, Gingras A-C, A proximity biotinylation map of a human cell. Biorxiv, 796391 (2019). [Google Scholar]
- 78.Gut G, Herrmann MD, Pelkmans L, Multiplexed protein maps link subcellular organization to cellular states. Science. 361, eaar7042 (2018). [DOI] [PubMed] [Google Scholar]
- 79.Hutchins JRA, Toyoda Y, Hegemann B, Poser I, Hériché J-K, Sykora MM, Augsburg M, Hudecz O, Buschhorn BA, Bulkescher J, Conrad C, Comartin D, Schleiffer A, Sarov M, Pozniakovsky A, Slabicki MM, Schloissnig S, Steinmacher I, Leuschner M, Ssykor A, Lawo S, Pelletier L, Stark H, Nasmyth K, Ellenberg J, Durbin R, Buchholz F, Mechtler K, Hyman AA, Peters J-M, Systematic analysis of human protein complexes identifies chromosome segregation proteins. Science (New York, N.Y.). 328, 593–599 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Havugimana PC, Hart GT, Nepusz T, Yang H, Turinsky AL, Li Z, Wang PI, Boutz DR, Fong V, Phanse S, Babu M, Craig SA, Hu P, Wan C, Vlasblom J, Dar V-N, Bezginov A, Clark GW, Wu GC, Wodak SJ, Tillier ERM, Paccanaro A, Marcotte EM, Emili A, A Census of Human Soluble Protein Complexes. Cell. 150, 1068–1081 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Ellenberg J, Swedlow JR, Barlow M, Cook CE, Sarkans U, Patwardhan A, Brazma A, Birney E, A call for public archives for biological image data. Nature Methods. 15, 849–854 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hentze MW, Castello A, Schwarzl T, Preiss T, A brave new world of RNA-binding proteins. Nat Rev Mol Cell Bio. 19, 327–341 (2018). [DOI] [PubMed] [Google Scholar]
- 83.Chujo T, Hirose T, Nuclear Bodies Built on Architectural Long Noncoding RNAs: Unifying Principles of Their Construction and Function. Mol Cells (2017), doi: 10.14348/molcells.2017.0263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Peng Z, Yan J, Fan X, Mizianty MJ, Xue B, Wang K, Hu G, Uversky VN, Kurgan L, Exceptionally abundant exceptions: comprehensive characterization of intrinsic disorder in all domains of life. Cell Mol Life Sci. 72, 137–151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Basile W, Salvatore M, Bassot C, Elofsson A, Why do eukaryotic proteins contain more intrinsically disordered regions? Plos Comput Biol. 15, e1007186 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Shin Y, Brangwynne CP, Liquid phase condensation in cell physiology and disease. Science. 357, eaaf4382 (2017). [DOI] [PubMed] [Google Scholar]
- 87.Qin Y, Winsnes CF, Huttlin EL, Zheng F, Ouyang W, Park J, Pitea A, Kreisberg JF, Gygi SP, Harper JW, Ma J, Lundberg E, Ideker T, bioRxiv, in press, doi: 10.1101/2020.06.21.163709. [DOI] [Google Scholar]
- 88.Hubert L, Arabie P, Comparing partitions. J Classif. 2, 193–218 (1985). [Google Scholar]
- 89.Mészáros B, Erdős G, Dosztányi Z, IUPred2A: context-dependent prediction of protein disorder as a function of redox state and protein binding. Nucleic Acids Res. 46, gky384- (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Emenecker RJ, Griffith D, Holehouse AS, metapredict: a fast, accurate, and easy-to-use predictor of consensus disorder and structure. Biophys J (2021), doi: 10.1016/j.bpj.2021.08.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Young CL, Britton ZT, Robinson AS, Recombinant protein expression and purification: A comprehensive review of affinity tags and microbial applications. Biotechnol J. 7, 620–634 (2012). [DOI] [PubMed] [Google Scholar]
- 92.Dingle G, CrispyCrunch: High-throughput Design and Analysis of CRISPR+HDR Experiments, (available at https://blog.addgene.org/crispycrunch-high-throughput-design-and-analysis-of-crisprhdr-experiments).
- 93.Jinek M, Chylinski K, Fonfara I, Hauer M, Doudna JA, Charpentier E, A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science (New York, N.Y.). 337, 816–821 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Bache N, Geyer PE, Bekker-Jensen DB, Hoerning O, Falkenby L, Treit PV, Doll S, Paron I, Müller JB, Meier F, Olsen JV, Vorm O, Mann M, A Novel LC System Embeds Analytes in Pre-formed Gradients for Rapid, Ultra-robust Proteomics*. Mol Cell Proteomics. 17, 2284–2296 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Meier F, Brunner A-D, Koch S, Koch H, Lubeck M, Krause M, Goedecke N, Decker J, Kosinski T, Park MA, Bache N, Hoerning O, Cox J, Räther O, Mann M, Online Parallel Accumulation–Serial Fragmentation (PASEF) with a Novel Trapped Ion Mobility Mass Spectrometer*. Mol Cell Proteomics. 17, i–2545 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Cox J, Mann M, MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nat Biotechnol. 26, 1367–1372 (2008). [DOI] [PubMed] [Google Scholar]
- 97.Prianichnikov N, Koch H, Koch S, Lubeck M, Heilig R, Brehmer S, Fischer R, Cox J, MaxQuant Software for Ion Mobility Enhanced Shotgun Proteomics*. Mol Cell Proteomics. 19, 1058–1069 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Vizcaíno JA, Deutsch EW, Wang R, Csordas A, Reisinger F, Ríos D, Dianes JA, Sun Z, Farrah T, Bandeira N, Binz P-A, Xenarios I, Eisenacher M, Mayer G, Gatto L, Campos A, Chalkley RJ, Kraus H-J, Albar JP, Martinez-Bartolomé S, Apweiler R, Omenn GS, Martens L, Jones AR, Hermjakob H, ProteomeXchange provides globally coordinated proteomics data submission and dissemination. Nat Biotechnol. 32, 223–226 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mertins P, Tang LC, Krug K, Clark DJ, Gritsenko MA, Chen L, Clauser KR, Clauss TR, Shah P, Gillette MA, Petyuk VA, Thomas SN, Mani DR, Mundt F, Moore RJ, Hu Y, Zhao R, Schnaubelt M, Keshishian H, Monroe ME, Zhang Z, Udeshi ND, Mani D, Davies SR, Townsend RR, Chan DW, Smith RD, Zhang H, Liu T, Carr SA, Reproducible workflow for multiplexed deep-scale proteome and phosphoproteome analysis of tumor tissues by liquid chromatography–mass spectrometry. Nat Protoc. 13, 1632–1661 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Drew K, Lee C, Huizar RL, Tu F, Borgeson B, McWhite CD, Ma Y, Wallingford JB, Marcotte EM, Integration of over 9,000 mass spectrometry experiments builds a global map of human protein complexes. Molecular Systems Biology. 13, 932 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Cock PJA, Antao T, Chang JT, Chapman BA, Cox CJ, Dalke A, Friedberg I, Hamelryck T, Kauff F, Wilczynski B, de Hoon MJL, Biopython: freely available Python tools for computational molecular biology and bioinformatics. Bioinformatics. 25, 1422–1423 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Razavi A, van den Oord A, Vinyals O, Generating Diverse High-Fidelity Images with VQ-VAE-2. Arxiv (2019). [Google Scholar]
- 103.Wolf FA, Angerer P, Theis FJ, SCANPY: large-scale single-cell gene expression data analysis. Genome Biol. 19, 15 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Kanehisa M, Sato Y, Kawashima M, Furumichi M, Tanabe M, KEGG as a reference resource for gene and protein annotation. Nucleic Acids Res. 44, D457–D462 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bonald T, Charpentier B, Galland A, Hollocou A, Hierarchical Graph Clustering using Node Pair Sampling. Arxiv (2018). [Google Scholar]
- 106.Mi H, Ebert D, Muruganujan A, Mills C, Albou L-P, Mushayamaha T, Thomas PD, PANTHER version 16: a revised family classification, tree-based classification tool, enhancer regions and extensive API. Nucleic Acids Res. 49, gkaa1106- (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Mass spectrometry raw data and associated MaxQuant output tables are deposited to the ProteomeXchange Consortium via the PRIDEpartner repository (accession PXD024909 for interactome data, and accession PXD029191 for whole-cell abundance data). Bulk RNA-seq raw data and associated kallisto transcript abundance tables are available on GEO (accession GSE186192). Raw microscopy images are hosted by AWS’s Open Datasets Program at https://registry.opendata.aws/czb-opencell/.