

Abstract
Extrachromosomal DNA (ecDNA) is prevalent in human cancers and mediates high oncogene expression through gene amplification and altered gene regulation1. Gene induction typically involves cis regulatory elements that contact and activate genes on the same chromosome2,3. Here we show that ecDNA hubs, clusters of ~10–100 ecDNAs within the nucleus, enable intermolecular enhancer-gene interactions to promote oncogene overexpression. ecDNAs encoding multiple distinct oncogenes form hubs in diverse cancer cell types and primary tumors. Each ecDNA is more likely to transcribe the oncogene when spatially clustered with additional ecDNAs. ecDNA hubs are tethered by the BET protein BRD4 in a MYC-amplified colorectal cancer cell line. BET inhibitor JQ1 disperses ecDNA hubs and preferentially inhibits ecDNA-based oncogene transcription. The BRD4-bound PVT1 promoter is ectopically fused to MYC and duplicated in ecDNA, receiving promiscuous enhancer input to drive potent MYC expression. Further, the PVT1 promoter on an exogenous episome suffices to mediate gene activation in trans by ecDNA hubs in a JQ1-sensitive manner. Systematic CRISPRi silencing of ecDNA enhancers reveals intermolecular enhancer-gene activation among multiple oncogene loci amplified on distinct ecDNAs. Thus, protein-tethered ecDNA hubs enable intermolecular transcriptional regulation and may serve as units of oncogene function, cooperative evolution, and potential targets for cancer therapy.
Circular ecDNA encoding oncogenes is a prevalent feature of cancer genomes and potent driver of cancer progression4–8. ecDNAs (including double minutes) are covalently closed, double-stranded, and range from ~100 kilobases to several megabases in size1,9–12. Lacking centromeres, ecDNAs are randomly segregated into daughter cells during cell division, enabling rapid accumulation and selection of ecDNA variants that confer a fitness advantage5,13–15. ecDNAs can re-integrate into chromosomes16–20 and may therefore also act as precursors to some chromosomal amplifications. ecDNAs possess highly accessible chromatin1,21 and co-amplify enhancer elements22,23, suggesting that oncogene amplicons may be shaped by regulatory dependencies to amplify transcription. ecDNAs cluster with one another during cell division or after DNA damage24–26; but the biological consequences of ecDNA clustering and are poorly understood.
ecDNA hubs amplify oncogene expression
We visualized ecDNA localization in interphase nuclei by DNA fluorescence in situ hybridization (FISH)27 using probes targeting ecDNA-amplified oncogenes in multiple cell lines including PC3 (MYC-amplified), COLO320-DM (MYC-amplified), HK359 (EGFR-amplified) and SNU16 (MYC- and FGFR2-amplified)1 (Figure 1a, Extended Data Figure 1a). DNA FISH on metaphase spreads revealed tens to hundreds of individual ecDNAs per cell located outside chromosomes (Figure 1a, Methods). In a subset of cell lines, we employed two-color DNA FISH to interrogate a non-ecDNA neighboring control locus (Extended Data Figure 1a); chromosomal oncogene copies appear as paired dots while ecDNAs have a single color as expected (Figure 1a, Extended Data Figure 1b). In all ecDNA-positive cancer cells we assessed, ecDNA FISH signal was locally concentrated in interphase nuclei despite arising from tens to hundreds of individual ecDNA molecules, suggesting that ecDNAs strongly cluster with one another, a feature we term ecDNA hubs (Figure 1a). ecDNA hubs occupied a much larger space than chromosomal signals and are larger than diffraction limited spots (~0.3 microns), suggesting that they consist of many clustered ecDNA molecules. Quantification using an autocorrelation function g(r) (Methods) showed a significant increase in clustering over short distances (0–40 pixels, 0–1.95 microns, Figure 1b, Extended Data Figure 1c) compared to random distribution. In three primary neuroblastoma tumors with MYCN amplifications, we also observed ecDNA hubs in the vast majority of cancer cells (Figure 1c, Extended Data Figure 1d, e)28. These results suggest that ecDNA clustering occurs across various cancer types with different oncogene amplifications and in primary tumors.
Figure 1. ecDNA imaging correlates ecDNA clustering with transcriptional bursting.
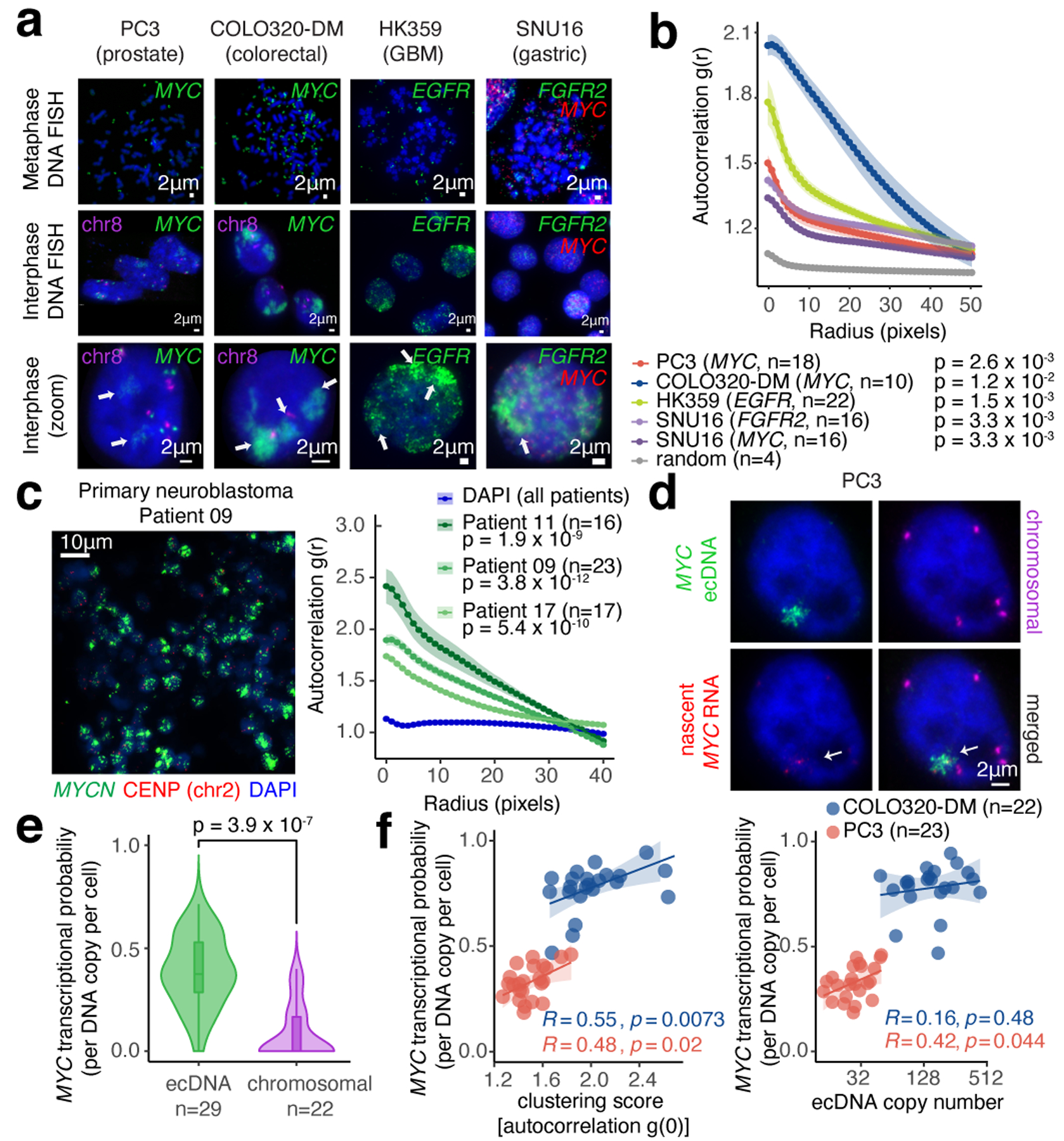
(a) Representative FISH images of interphase ecDNA clustering. A chromosomal control was included for PC3 and COLO320-DM. (b) Interphase ecDNA clustering by autocorrelation g(r) (Methods). Data are mean ± SEM. P-values determined by two-sided Wilcoxon test at r=0 compared to random distribution. (c) Representative FISH image showing ecDNA clustering in a primary neuroblastoma tumor (MYCN ecDNA and chromosomal control, left). ecDNA clustering in three primary tumors using autocorrelation (right). Data are mean ± SEM. P-values determined by two-sided Wilcoxon test at r=0 compared to DAPI. (d) Representative image from combined DNA FISH for ecDNA, chromosomal control, and nascent RNA FISH in PC3 cells. (e) MYC transcription probability measured by joint DNA/RNA FISH (RNA normalized to DNA copy number; box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). P-values determined by two-sided Wilcoxon test. (f) Correlation between MYC transcription probability and ecDNA copy number or clustering (joint DNA/RNA FISH; clustering scores are autocorrelation at r = 0; Pearson’s R, two-sided test).
Next, we visualized actively transcribing MYC alleles by joint DNA and nascent RNA FISH in PC3 and COLO320-DM cells (Figure 1d, Extended Data Figure 1a,f–h) and computed MYC transcription probability from each ecDNA molecule (Methods). The majority of nascent MYC mRNA transcripts came from ecDNA hubs rather than the chromosomal locus even after accounting for copy number (Figure 1d,e). ecDNA clustering is significantly correlated with increased MYC transcription, and ecDNA clustering was a better predictor of MYC transcription probability than copy number (Figure 1f). Further, ecDNAs in hubs are more transcriptionally active compared to singleton ecDNAs (Extended Data Figure 1i). Thus, each ecDNA molecule is more likely to transcribe the oncogene when more ecDNAs are present in hubs.
BRD4 links ecDNA hubs and transcription
MYC is flanked by super enhancers marked by histone H3 lysine 27 acetylation (H3K27ac) and Bromodomain and extraterminal domain (BET) proteins such as BRD429,30. MYC transcription is highly sensitive to BET protein displacement by the inhibitor JQ131,32. To examine MYC ecDNAs in live cells, we inserted a Tet-operator (TetO) array into MYC ecDNAs in COLO320-DM and labeled ecDNAs with TetR-eGFP or TetR-A206K-eGFP to minimize GFP dimerization (Extended Data Figure 2a–d, Methods). Live cell imaging revealed multiple dynamic nuclear foci corresponding to clustered ecDNAs (Extended Data Figure 2e–i, Supplementary Video 1). Epitope tagging of endogenous BRD4 revealed that BRD4 is highly enriched in TetO-labeled ecDNA hubs (Figure 2a, Extended Data Figure 2j–l). Chromatin immunoprecipitation and sequencing (ChIP-seq) of H3K27ac, BRD4, and assay of transposase-accessible chromatin using sequencing (ATAC-seq) showed that H3K27ac peaks, marking active ecDNA enhancers, are indeed also occupied by BRD4 (Figure 2b, Extended Data Figure 3a–c).
Figure 2. BET proteins mediate ecDNA hub formation and transcription.
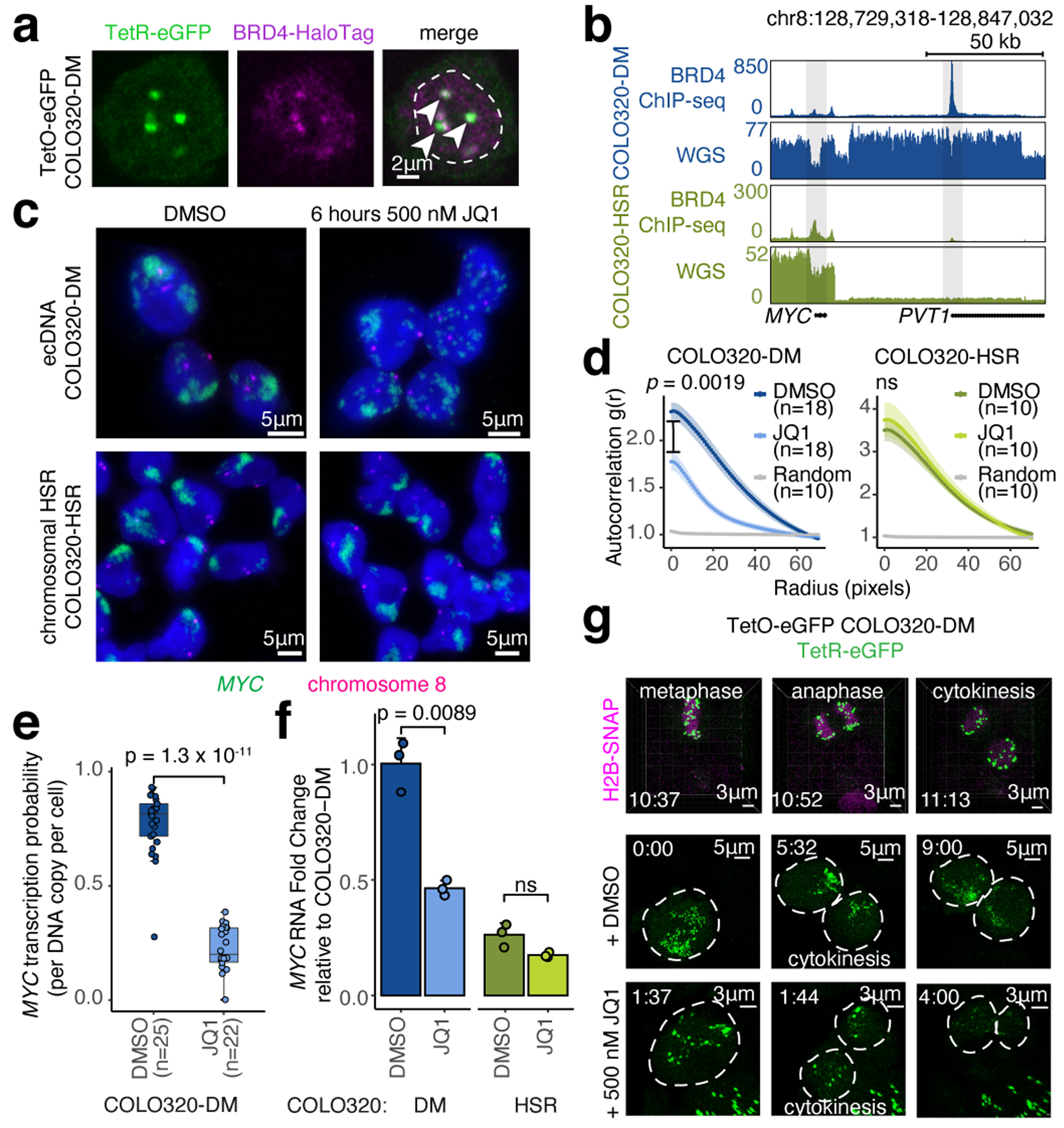
(a) Representative live cell image of ecDNA and BRD4-HaloTag signals in TetO-eGFP COLO320-DM cells (independently repeated twice; dashed line indicates nuclear boundary). (b) BRD4 ChIP-seq and WGS at MYC locus in COLO320-DM and COLO320-HSR cells. (c) Representative DNA FISH images for cells treated with DMSO or 500 nM JQ1 for 6 hours. (d) Clustering measured by autocorrelation g(r) for ecDNAs in COLO320-DM and HSRs in COLO320-HSR treated with DMSO or 500 nM JQ1 for 6 hours. Data are mean ± SEM. P-values determined by two-sided Wilcoxon test at r=0. (e) MYC transcription probability in COLO320-DM treated with DMSO or 500 nM JQ1 for 6 hours (joint DNA/RNA FISH; RNA normalized to ecDNA copy number; box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). P-values determined by two-sided Wilcoxon test. (f) MYC RNA measured by RT-qPCR for COLO320-DM and COLO320-HSR cells treated either with DMSO or 500 nM JQ1 for 6 hours. Data are mean ± SD between 3 biological replicates. P-values determined by two-sided student’s t-test. (g) Representative live cell images of TetR-eGFP-labeled ecDNAs in TetO-eGFP COLO320-DM cells treated with DMSO or 500 nM JQ1 at indicated timepoints through cell division (independently repeated twice for each condition). H2B-SNAP (top) labels histone H2B in mitotic chromosomes.
To determine the role of BET proteins in ecDNA-derived transcription, we focused on isogenic colorectal cancer cell lines COLO320-DM (MYC ecDNA) and COLO320-HSR (chromosomal MYC amplicon or homogeneously staining region; HSR)18, which were derived from the same patient tumor (Extended Data Figure 3a). Treatment with 500 nM JQ1 dispersed ecDNA hubs in COLO320-DM after 6 hours, splitting large ecDNA hubs into multiple small ecDNA signals including singleton ecDNAs and abolishing the most clustered ecDNA hubs [autocorrelation g(r) ≥ 2] (Figure 2c,d, Extended Data Figure 3d–f). JQ1 treatment did not alter the spatial distribution of covalently-linked MYC copies in COLO320-HSR as expected (Figure 2c,d). ecDNA dispersal by JQ1 appears to be highly specific; transcription inhibition by either the RNA polymerase II inhibitor alpha-amanitin or 1,6-hexanediol33 did not affect ecDNA hubs (Extended Data Figure 3g–j).
JQ1 potently inhibited ecDNA-derived oncogene transcription. JQ1 treatment reduced MYC transcription probability per ecDNA copy by four-fold, as shown by joint nascent RNA and DNA FISH (Figure 2e, Extended Data Figure 3g). Because BET proteins are also involved in MYC transcription from chromosomal DNA, we compared the effect of JQ1 on COLO320-DM versus COLO320-HSR. BRD4 ChIP-seq showed that JQ1 treatment equivalently dislodged BRD4 genome-wide in these isogenic cells (Extended Data Figure 3k). Nonetheless, treatment with 500 nM JQ1 preferentially lowered MYC mRNA level in COLO320-DM cells, a dose which had no significant effect on MYC mRNA level in COLO320-HSR cells (Figure 2f). JQ1 dose titration demonstrated a modest preferential killing of COLO320-DM cells over HSR cells (Extended Data Figure 3l–n). A survey of six additional compounds targeting transcription or histone modifications showed that only BET inhibitors selectively inhibited MYC expression in ecDNA+ cells, and MS645, a bivalent BET bromodomain inhibitor34, reduced ecDNA transcription and clustering similar to JQ1 (Extended Data Figure 3o–q). Live cell imaging with TetO-GFP COLO320-DM cells demonstrated that ecDNA hubs condense into smaller particles during mitosis (Figure 2g, Supplementary Video 1–2). After partitioning, ecDNAs re-form large hubs; importantly ecDNA hub assembly following mitosis is blocked by JQ1 (Figure 2g, Supplementary Video 3). Together, these results suggest a unique dependence on bromodomain-H3K27ac interaction of BET proteins for ecDNA hub formation, maintenance, and oncogene transcription in COLO320-DM cells.
PVT1-MYC hijacks ecDNA enhancer input
To link ecDNA structure to regulation of MYC transcription, we reconstructed the COLO320-DM ecDNA using five orthogonal approaches and report the largest ecDNA structure assembled to date. We identified complex structural rearrangements using 1) whole-genome sequencing (WGS)35, 2) nanopore-based single-molecule sequencing, and 3) large DNA contig assembly by optical mapping36 (Extended Data Figure 4a–d). 4) We performed targeted ecDNA digestion using CRISPR-Cas9 followed by pulsed field gel electrophoresis (PFGE) and deep sequencing of megabase-sized DNA fragments to obtain sequence multiplicity information which was highly concordant with optical mapping ecDNA contigs (Extended Data Figure 4e,f). Using these first four methods, we reconstructed a 4.328-megabase ecDNA that contains multiple copies of PVT1-MYC fusion37,38, a canonical MYC sequence, and sequences from multiple chromosomal origins (chromosomes 6, 8, 13, 16) (Extended Data Figure 4e). 5) Finally, we used DNA FISH to confirm colocalization of PLUT, PCAT1, and MYC genes on ecDNAs as predicted by the reconstruction (Extended Data Figure 4g).
The PVT1-MYC fusion makes up >70% of MYC transcripts in COLO320-DM and consists of the promoter and exon 1 of the lncRNA gene PVT1 fused to exons 2 and 3 of MYC (which encode a functional MYC protein isoform39), replacing the promoter and exon 1 of MYC (Figure 3a). Consistently, total MYC RNA transcripts were reduced by CRISPR interference (CRISPRi) of the PVT1 promoter (Extended Data Figure 4h). Multiple PVT1-MYC fusion copies share a common breakpoint, indicative of a common origin (Extended Data Figure 4i). We observed strong BRD4 binding at the PVT1 promoter in COLO320-DM, but not COLO320-HSR (Figure 2b). As the PVT1 promoter can be activated by MYC40, we hypothesize that PVT1-MYC fusion enables positive feedback of MYC expression and circumvents competition between the PVT1 and MYC promoters which is normally observed on the unrearranged chromosome41. Interestingly. PVT1 rearrangement and gene fusion are observed in multiple human cancers and drive gene overexpression42.
Figure 3. Intermolecular activation of an episomal luciferase reporter in ecDNA hubs.
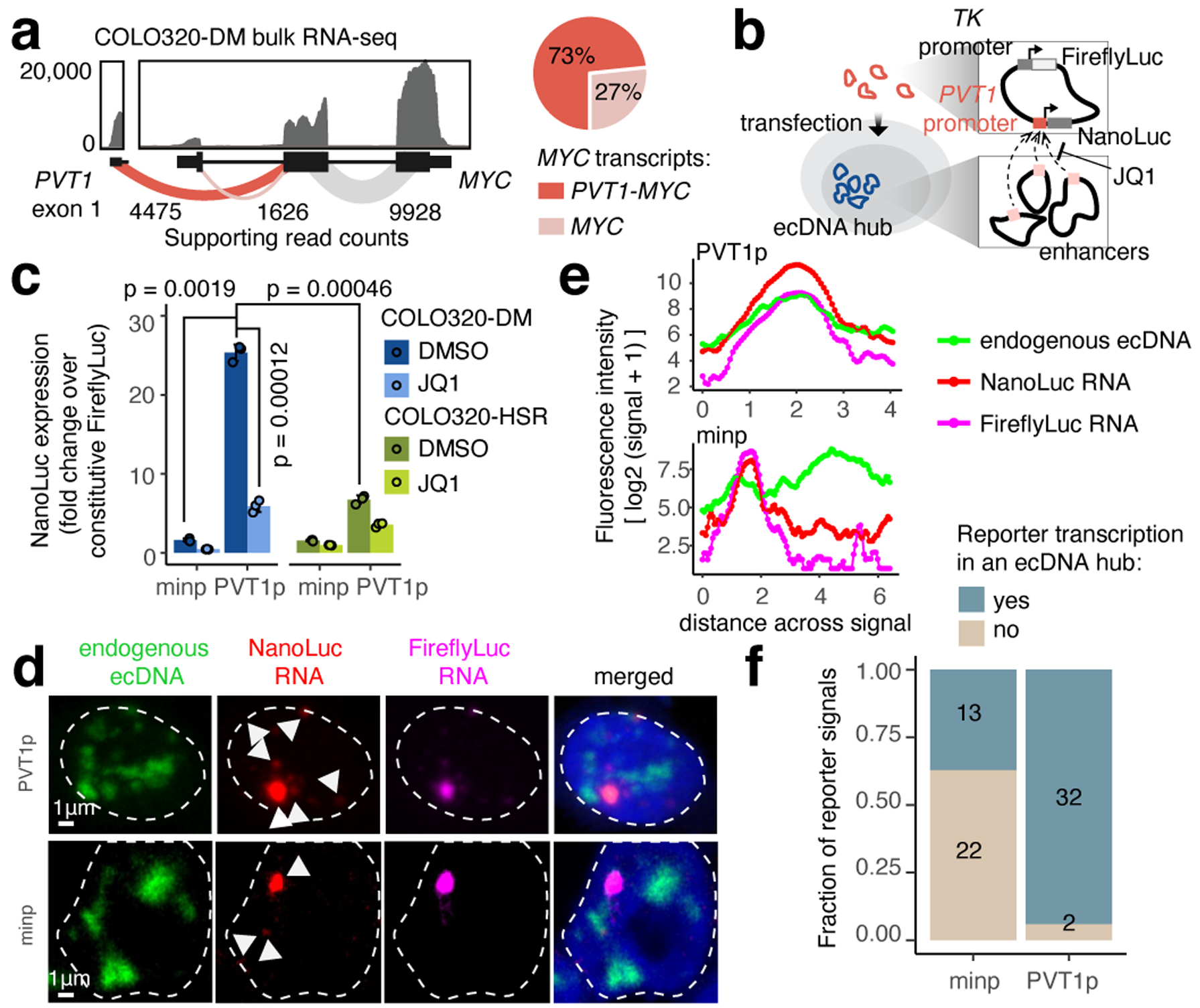
(a) RNA-seq from COLO320-DM with exon-exon junction spanning read counts shown (left). Relative abundance of full-length MYC and fusion PVT1-MYC transcripts using read count supporting either junction (right). (b) PVT1 promoter-driven luciferase reporter system. (c) Luciferase reporter activity driven by either minp or PVT1p with DMSO or JQ1 treatment (500 nM, 6 hours). Data are mean ± SD between 3 biological replicates. P-values determined by two-sided student’s t-test (Bonferroni adjusted). (d) Representative images of PVT1p or minp reporter transcriptional activity and endogenous ecDNA hubs in COLO320-DM visualized by DNA and RNA FISH (independently repeated 3 times). (e) Fluorescence intensities on a line drawn across the center of the largest NanoLuc RNA signal in images in (d). (f) Number of nuclear NanoLuc signals that colocalize with ecDNA hubs.
We next identified ecDNA regulatory elements associated with high oncogene expression. Paired single-cell ATAC-seq and RNA-seq from 72,049 COLO320-DM and COLO320-HSR cells identified 47 ecDNA regulatory elements associated with high MYC expression independent of copy number (Extended Data Figure 5, Methods). Enhancer connectome analysis using H3K27ac HiChIP, a protein-directed 3D genome conformation assay43, revealed multiple enhancers make significant contact with the PVT1/PVT1-MYC promoter (Extended Data Figure 6a,b, Extended Data Figure 5f,g). While the canonical MYC promoter participates in several focal enhancer contacts, HiChIP signal at the PVT1 promoter is elevated across the entire amplified region (Extended Data Figure 6a). CRISPRi targeting of six enhancers individually with high BRD4 occupancy on ecDNA did not significantly reduce bulk MYC mRNA levels (Extended Data Figure 4i) likely due to combinatorial and compensatory enhancer-gene interactions. These results indicate that PVT1 promoter, now driving MYC oncogene expression on ecDNA, receives broad and combinatorial enhancer input within ecDNA hubs.
Gene activation in trans in ecDNA hubs
We next interrogated whether ecDNA molecules cooperate in spatial proximity to achieve gene transcription. We constructed a plasmid containing the 2kb PVT1 promoter driving NanoLuc luciferase (PVT1p-nLuc) and with a constitutive thymidine kinase promoter (TKp) driving Firefly luciferase as an internal control (Figure 3b). In COLO320-DM cells, PVT1p was highly active (~25-fold) compared to TKp or a minimal promoter (minp-nLuc; Figure 3c). Importantly, PVT1p conferred significantly greater (~4-fold) induction in ecDNA+ COLO320-DM cells than in isogenic ecDNA− COLO320-HSR cells (Figure 3c), while minimal promoter and MYC promoter activity was comparable between the isogenic cell lines (Extended Data Figure 6c). Low dose JQ1 treatment that disperses ecDNA hubs strongly reduced PVT1p-mediated transcription in COLO320-DM (~5-fold repression) compared to more modest effect in COLO320-HSR cells (~2 fold) (Figure 3c). Joint DNA FISH and nascent RNA FISH showed that PVT1p conferred increased NanoLuc transcription when colocalized with ecDNA hubs compared to the minimal promoter (Figure 3d–f, Extended Data Figure 6d). Addition of a cis-enhancer to the plasmid increases both PVT1p- or MYCp-driven NanoLuc activity and TKp-driven Firefly luciferase activity (Extended Data Figure 6e,f). Finally, MYCp or incorporation of a cis-enhancer to the plasmid reduced the distinction between reporter activity in COLO320-DM vs. COLO320-HSR cells and sensitivity to JQ1 (Extended Data Figure 6g). Together, these experiments suggest intermolecular enhancer-promoter activation in ecDNA hubs and identify PVT1p as a DNA element capable of activation in ecDNA hubs in trans.
Intermolecular regulation among ecDNAs
We next investigated whether intermolecular enhancer-gene interactions can be precisely mapped and perturbed. We focused on a human gastric cancer cell line, SNU16, which contains two distinct ecDNA types: a MYC amplicon derived from chromosomes 8 and 11 and an FGFR2 amplicon derived from chromosome 10. These ecDNAs intermingle in hubs as demonstrated by two-color interphase FISH (Figure 1a,b, 4a). JQ1 treatment reduced ecDNA-derived transcription of both MYC and FGFR2 (Figure 4b). We generated a subclone, SNU16-dCas9-KRAB, with stable expression of dCas9-KRAB and reduced ecDNA structural heterogeneity as confirmed by metaphase FISH (96.8% distinct MYC and FGFR2 ecDNAs), WGS, and H3K27ac HiChIP analyses (Figure 4c, Extended Data Figure 7a–c). H3K27ac HiChIP demonstrated intermolecular contacts between FGFR2 and MYC ecDNAs with lower contact frequency relative to cis interactions but enriched for focal interactions (Figure 4d, orange). CRISPRi targeting of candidate regulatory elements (20 guides per element; 2,747 guides total; Extended Data Figure 8a–c; Methods)44 identified functional elements linked to expression of MYC or FGFR2 both in cis (oncogene located on the same ecDNA) and in trans (oncogene located on a distinct ecDNA) (Methods, Figure 4e,f, Extended Data Figure 8d). As a positive control, CRISPRi of the MYC and FGFR2 promoters strongly reduced corresponding gene expression. CRISPRi of the FGFR2 promoter had no effect on MYC expression, indicating that downregulation of FGFR2 protein does not affect MYC expression (Figure 4e,f). Importantly, we identified five enhancers on the FGFR2 ecDNA that activate MYC in trans, but no MYC ecDNA enhancers that activate FGFR2 (Figure 4e,f, Extended Data Figure 8e). Perturbations of in-trans interactions resulted in similar significance levels to perturbation of several in-cis interactions on the MYC ecDNA (Figure 4e). We validated that FGFR2 trans-enhancers are not covalently linked to the MYC gene on ~98–100% of ecDNA molecules by dual-color metaphase DNA FISH and in vitro CRISPR-Cas9 digestion (Extended Data Figure 9). CRISPRi of the MYC promoter reduced both MYC and FGFR2 expression, suggesting that the MYC protein may act as a transcriptional activator of FGFR245 (Figure 4e,g, Extended Data Figure 8f). These data suggest that FGFR2 and MYC ecDNAs have been co-selected so that enhancers on both amplicons cooperatively activate MYC expression. The MYC protein then, in turn, activates FGFR2 expression (Figure 4g). Notably, there is little overlap between cis- and trans-regulatory elements, supporting our conclusion that intermolecular enhancer elements directly modify gene expression in trans rather than through downstream effects.
Figure 4. ecDNA hubs mediate intermolecular enhancer-gene interactions.
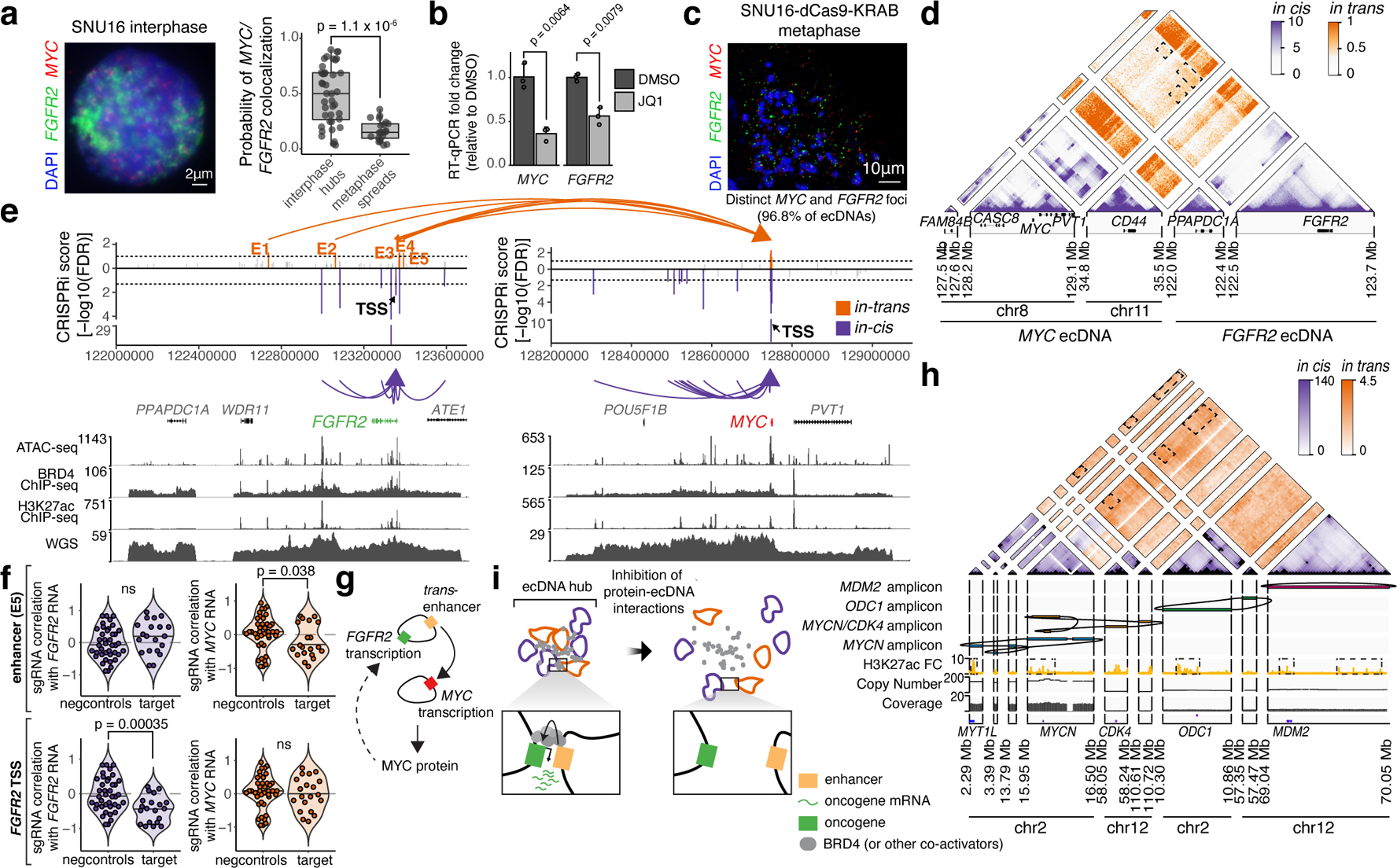
(a) Representative DNA FISH image showing clustering of MYC and FGFR2 ecDNAs in interphase SNU16 (left). MYC and FGFR2 colocalization in SNU16 (right; box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). P-value determined by two-sided Wilcoxon test. (b) Oncogene RNA measured by RT-qPCR in SNU16 treated with DMSO or 500 nM JQ1 for 6 hours. Data are mean ± SD between 3 biological replicates. P-value determined by two-sided student’s t-test. (c) Representative metaphase FISH image in SNU16-dCas9-KRAB. Quantification summarizes 30 cells from one experiment. (d) H3K27ac HiChIP contact matrix (10 kb resolution, KR-normalized read counts) in SNU16-dCas9-KRAB showing cis- and trans- interactions. (e) Top: significance of enhancer CRISPRi effects on oncogene repression (Benjamini-Hochberg adjusted; n=40 negative control sgRNAs, n=20 target sgRNAs; Methods, Extended Data Figure 8). Dashed lines mark FDR < 0.05 for cis-interactions and FDR < 0.1 for trans-interactions; significant enhancers are colored and connected to target genes by loops (E1, FDR = 0.048; E2, FDR = 0.052; E3, FDR = 0.048; E4, FDR = 0.052; E5, FDR = 0.052). All datasets contain two independent experiments except the in-trans dataset for the MYC-targeting sgRNA pool, which contains one independent experiment. Bottom: ATAC-seq, BRD4 ChIP-seq, H3K27ac ChIP-seq, and WGS tracks. (f) Correlations between individual sgRNAs and oncogene expression (Methods). P-values determined by lower-tailed t-test compared to negative controls. Each dot represents an independent sgRNA (n=40 negative control sgRNAs, n=20 target sgRNAs). (g) Cross-regulation between MYC and FGFR2 elements in ecDNA hubs. (h) Top to bottom: Hi-C contact map (KR-normalized read counts in 25kb bins) showing cis- and trans- contacts, reconstructed amplicons, H3K27ac ChIP-seq (mean fold-change over input), copy number and WGS in TR14. (i) ecDNA hub model for intermolecular cooperation.
Finally, to assess intermolecular ecDNA interactions in an independent cancer type, we used nanopore sequencing and WGS to identify four distinct oncogene amplicons in TR14, a neuroblastoma cell line, which also contains ecDNA hubs (Extended Data Figure 10a,b). Hi-C analysis revealed trans interactions, such as those between the MYCN and ODC1 amplicons which are not brought together by structural variants (Figure 4h, Extended Data Figure 10c–e). Trans Hi-C contacts are enriched at sites marked by H3K27ac, which may represent regulatory elements that enable intermolecular cooperation (Figure 4h, Extended Data Figure 10f–h). Together, these results suggest intermolecular enhancer-gene activation in ecDNA hubs occurs for diverse oncogene loci and multiple cancer types.
Discussion
Local ecDNA congregation in ecDNA hubs promotes novel intermolecular enhancer-gene interactions and oncogene overexpression (Figure 4i). Unlike chromosomal transcription hubs which favor local cis regulatory elements and span 100–300 nm46, ecDNA hubs can span >1000 nm and involve trans regulatory elements located on distinct ecDNA molecules. This discovery has profound implications in how ecDNAs undergo selection and how rewiring of oncogene regulation on ecDNA contributes to transcription. First, trans-activation between ecDNAs suggests that oncogene-enhancer co-selection may occur on both individual ecDNAs as well as the repertoire of ecDNAs in a cell. Thus, individual ecDNA molecules may not be required to contain all necessary regulatory elements as a diverse repertoire of regulatory elements are accessible in a hub47. This type of evolutionary dynamics has been documented in viruses, where cooperation of a mixture of specialized variants outperforms a pure wild-type population48,49. Further, mutations on individual molecules may be better tolerated, which may increase ecDNA sequence diversity. Finally, ecDNA hubs promote variable enhancer usage as cluster ecDNA molecules can “sample” various enhancers via novel enhancer-promoter interactions, including ectopic enhancer-promoter interactions between ecDNAs arising from distinct chromosomes as in SNU16.
The recognition that ecDNA hubs promote oncogene transcription may provide new therapeutic opportunities. While chromosomal DNA amplicons such as HSRs are covalently linked, ecDNA hubs are held together by proteins. In COLO320-DM, we show that BET protein inhibition by JQ1 disaggregates ecDNA hubs and reduces ecDNA-derived MYC expression. While MYC and MYCN are regulated by BET proteins31,50, other ecDNA oncogene amplifications may exploit their endogenous enhancer mechanisms in ecDNA hubs and may rely on other gene-specific protein factors. Future studies may identify proteins that mediate ecDNA transcriptional activity in various cancer types and will be highly informative for potential therapeutic efforts.
METHODS
Cell Culture
The TR14 neuroblastoma cell line was a gift from J. J. Molenaar (Princess Máxima Center for Pediatric Oncology, Utrecht, Netherlands). Cell line identity for the master stock was verified by STR genotyping (IDEXX BioResearch, Westbrook, ME). All remaining cell lines used were obtained from ATCC. TR14 cells were cultured in RPMI-1640 medium (Thermo Fisher Scientific, Inc., Waltham, MA) with 1% Penicillin/Streptomycin, and 10% FCS. COLO320-DM, COLO320-HSR and HCC1569 cells were maintained in Roswell Park Memorial Institute 1640 (RPMI; Life Technologies, Cat# 11875-119) supplemented with 10% fetal bovine serum (FBS; Hyclone, Cat# SH30396.03) and 1% penicillin-streptomycin (pen-strep; Thermo Fisher, Cat# 15140-122). PC3 cells were maintained in Dulbecco’s Modified Eagle Medium (DMEM; Thermo Fisher, Cat# 11995073) supplemented with 10% FBS and 1% pen-strep. HK359 cells were maintained in DMEM/Nutrient Mixture F-12 (DMEM/F12 1:1; Gibco, Cat# 11320-082), B-27 Supplement (Gibco, Cat# 17504044), 1% pen-strep, GlutaMAX (Gibco, Cat# 35050061), human epidermal growth factor (EGF, 20 ng/ml; Sigma-Aldrich, E9644), human fibroblast growth factor (FGF, 20 ng/ml; Peprotech) and Heparin (5 ug/ml; Sigma-Aldrich, Cat# H3149-500KU). SNU16 cells were maintained in DMEM/F12 supplemented with 10% FBS and 1% pen-strep. All cells were cultured at 37°C with 5% CO2. All cell lines tested negative for mycoplasma contamination.
Metaphase chromosome spread
Cells in metaphase were prepared by KaryoMAX (Gibco) treatment at 0.1 ug/ml for 3 hr. Single-cell suspension was then collected and washed by PBS, and treated with 75 mM KCl for 15–30 min. Samples were then fixed by 3:1 methanol:glacial acetic acid, v/v and washed for an additional three times with the fixative. Finally, the cell pellet resuspended in the fixative was dropped onto a humidified slide. The distribution of ecDNA counts in metaphase for COLO320-DM, PC3 and HK359 have been described previously1,6. We find that the majority of cells examined in metaphase are ecDNA+, with a small proportion of HSR+ cells:
COLO320-DM: 80% (80/100 cells) ecDNA+, 14% (14/100 cells) HSR+, 6% (6/100 cells) ecDNA+/HSR+
PC3: 80% (43/54 cells) ecDNA+, 11% (6/54 cells) HSR+, 9% (5/54 cells) ecDNA+/HSR+
SNU16-dCas9-KRAB: 100% (29/29 cells) ecDNA+
Metaphase DNA FISH
Slides containing fixed cells in interphase or metaphase were briefly equilibrated by 2X SSC, followed by dehydration in 70%, 85%, and 100% ethanol for 2 min each. FISH probes in hybridization buffer (Empire Genomics) were added onto the slide, and the sample was covered by a coverslip then denatured at 75°C for 1 min on a hotplate, and hybridized at 37°C overnight. The coverslip was then removed, and the sample was washed one time by 0.4X SSC with 0.3% IGEPAL, and two times by 2X SSC with 0.1% IGEPAL, for 2 min each. DNA was stained with DAPI and washed with 2X SSC. Finally, the sample was mounted by mounting media (Molecular Probes) before imaging.
Interphase DNA FISH
The Oligopaint FISH probe libraries were constructed as described previously51. Each oligo consists of a 40 nucleotide (nt) homology to the hg19 genome assemble designed from the algorithm developed from the laboratory of Dr. Ting Wu (https://oligopaints.hms.harvard.edu/). Each library subpool consists of a unique sets of primer pairs for orthogonal PCR amplification and a 20 nt T7 promoter sequence for in vitro transcription and a 20 nt region for reverse transcription. Individual Oligopaint probes were generated by PCR amplification, in vitro transcription, and reverse transcription, in which ssDNA oligos conjugated with ATTO488 and ATTO647 fluorophores were introduced during the reverse transcription step. The Oligopaint covered genomic regions (hg19) used in this study are as follows: chr8:116967673–118566852 (hg19_COLO_nonecDNA_1.5Mbp), chr8:127435083–129017969 (hg19_COLO_ecDNA_1.5Mbp), chr8:128729248–128831223 (hg19_PC3_ecDNA1_100kb). A ssDNA oligo pool was ordered and synthesized from Twist Bioscience (San Francisco, CA). 15mm #1.5 round glass coverslips (Electron Microscopy Sciences) were pre-rinsed with anhydrous ethanol for 5min, air dried, and coated with L-poly lysine solution (100ug/mL) for at least 2 hours. Fully dissociated ColoDM320 or PC3 cells were seeded onto the coverslips and recovered for at least 6 hours before experiments. Cells were fixed with 4% (v/v) methanol free paraformaldehyde diluted in 1X PBS at room temperature for 10min. Then cells were washed 2X with 1XPBS and permeabilized in 0.5% Triton-X100 in 1XPBS for 30min. After 2X wash in 1XPBS, cells were treated with 0.1M HCl for 5min, followed by 3X washes with 2XSSC and 30 min incubation in 2X SSC + 0.1% Tween20 (2XSSCT) + 50% (v/v) formamide (EMD Millipore, cat#S4117). For each sample, we prepare 25ul hybridization mixture containing 2XSSCT+ 50% formamide +10% Dextran sulfate (EMD Millipore, cat#S4030) supplemented with 0.5μl 10mg/mL RNaseA (Thermo Fisher Scientific, cat# 12091–021) +0.5μl 10mg/mL salmon sperm DNA (Thermo Fisher Scientific, cat# 15632011) and 20pmol probes with distinct fluorophores. The probe mixture was thoroughly mixed by vortexing, and briefly microcentrifuged. The hybridization mix was transferred directly onto the coverslip which was inverted facing a clean slide. The coverslip was sealed onto the slide by adding a layer of rubber cement around the edges. Each slide was denatured at 78°C for 4 min followed by transferring to a humidified hybridization chamber and incubated at 42°C for 16 hours in a heated incubator. After hybridization, samples were washed 2X for 15 minutes in pre-warmed 2XSSCT at 60 °C and then were further incubated at 2XSSCT for 10min at RT, at 0.2XSSC for 10min at RT, at 1XPBS for 2X5min with DNA counterstaining with DAPI. Then coverslips were mounted on slides with Prolong Diamond Antifade Mountant (Thermo Fisher Scientific Cat#P36961) for imaging acquisition.
DNA FISH of primary neuroblastoma samples was performed on 4 μm sections of FFPE blocks. Slides were deparaffinized, dehydrated and incubated in pre-treatment solution (Dako, Denmark) for 10 minutes at 95–99°C. Samples were treated with pepsin solution for 2 minutes at 37°C. For hybridization, the ZytoLight ® SPEC MYCN/2q11 Dual Color Probe (ZytoVision, Bremerhaven, Germany) was used. Incubation took place overnight at 37°C, followed by counterstaining with 4,6-diamidino-2-phenylindole (DAPI).
Nascent RNA FISH
To quantify the MYC gene expression on the ecDNAs, we ordered the RNA FISH probes conjugated with a Quasar 570 dye (Biosearch Technologies) targeting to the intronic region of human (hg19) MYC gene for detection of nascent RNA transcript. We also ordered the RNA FISH probes conjugated with a Quasar 670 dye targeting to the exonic region of human MYC gene for detection of both mature and nascent RNA transcripts. For simultaneous detection of both ecDNA and MYC transcription, 125nM RNA FISH probes was mixed with the DNA FISH probes (100kb probe instead of the 1.5Mbp probe) together in the hybridization buffer with RNase inhibitor (Thermo Fisher Scientific, cat# AM2694) and incubated at 37°C overnight for ~16 hours. After hybridization, samples were washed 2X for 15 minutes in pre-warmed 2XSSCT at 37 °C and then were further incubated at 2XSSCT for 10min at RT, at 0.2XSSC for 10min at RT, at 1XPBS for 2X5min with DNA counterstaining with DAPI. Then coverslips were mounted on slides with Prolong Diamond Antifade Mountant for imaging acquisition.
Microscopy
DNA FISH images were acquired either with conventional fluorescence microscopy or confocal microscopy. Conventional fluorescence microscopy was performed using an Olympus BX43 microscope, and images were acquired with a QiClick cooled camera. Confocal microscopy was performed using a Leica SP8 microscope with lightning deconvolution (UCSD School of Medicine Microscopy Core). Z-stacks were acquired over an average depth of approximately 8μm, with roughly 0.6μm step size.
DNA/RNA FISH images were acquired on the ZEISS LSM 880 Inverted Confocal microscope attached with an Airyscan 32 GaAsP PMT area detector. Before imaging, the beam position was calibrated centering on the 32 detector array. Images were taken under the Airyscan SR mode with a Plan Apochromat 63X/NA1.40 oil objective in a lens immersion medium having a refractive index 1.515 at 30°C. We used 405nm (Excitation wavelength) and 460nm (Emission wavelength) for the DAPI channel, 488nm (Excitation wavelength) and 525nm (Emission wavelength) for the ATTO488 channel, 561nm (Excitation wavelength) and 579nm (Emission wavelength) for the Quasar570 channel and 633nm (Excitation wavelength) and 654nm (Emission wavelength) for the ATTO647 channel. Z-stacks were acquired with the optimal z sectioning thickness ~200nm, followed by post-processing using the provided algorithm from ZEISS LSM880 platform.
DNA FISH images for primary neuroblastoma samples were collected for 50 non-overlapping tumor cells using a fluorescence microscope (BX63 Automated Fluorescence Microscope, Olympus Corporation, Tokyo, Japan). Computer-based documentation and image analysis was performed with the SoloWeb imaging system (BioView Ltd, Israel) MYCN amplification (MYCN FISH+) was defined as MYCN/2q11.2 ratio > 4.0, as described in the INRG report52. The tumor samples profiled present with multiple MYCN foci visible as in interphase, supporting that amplified MYCN is extrachromosomal in origin, as is the case for approximately 90% of neuroblastoma cases28,53–55.
Metaphase DNA FISH Image Analysis
Colocalization analysis for two-color metaphase FISH data for MYC, PCAT1 and PLUT ecDNAs in COLO320-DM described in Extended Data Figure 4g was performed using Fiji (version 2.1.0/1.53c)56. Images were split into the two FISH colors + DAPI channels, and signal threshold set manually to remove background fluorescence. Overlapping FISH signals were segmented using watershed segmentation. Colocalization was quantified using the ImageJ-Colocalization Threshold program and individual and colocalized FISH signals were counted using particle analysis.
Colocalization analysis for two-color metaphase FISH data for MYC and FGFR2 ecDNAs in SNU16 described in Figure 4c and Extended Data Figure 7a was performed using ecSeg (https://github.com/UCRajkumar/ecSeg, not versioned)57. Briefly, ecSeg takes as input metaphase FISH images containing DAPI and up to two colors of DNA FISH. ecSeg uses the DAPI signal to classify signals as nuclear (arising from interphase nuclei), chromosomal (arising from metaphase chromosome), or extrachromosomal. It then quantifies DNA FISH signal and colocalization segmented by whether the signal is present on chromosomal or extrachromosomal DNA.
Interphase DNA FISH Clustering Analysis
To analyze the clustering of ecDNAs, we applied the autocorrelation function as described previously58 in Matlab (2019). g(r) estimates the probability of detecting another ecDNA signal at increasing distances from the viewpoint of an index ecDNA signal and is equal to 1 for a uniform, random distribution. Specifically, the pair auto-correlation function was calculated by the fast Fourier transform (FFT) method described by the equations below.
is the auto-correlation of a mask matrix that has the value of 1 inside the nucleus used for normalization. The fast Fourier transform and its inverse (FFT and FFT−1) were computed by fft2() and ifft2() functions in Matlab, respectively. Autocorrelation functions were calculated first by converting the Cartesian coordinates to polar coordinates by Matlab cart2pol() function, binning by radius and by averaging within the assigned bins. For comparing auto-correlation with transcription probability, the value of the auto-correlation function at radius of 0 pixels (g(0)) was used to represent the degree of spatial clustering. The g(0) values were also used for calculating statistical significance among groups. For neuroblastoma patient samples, we avoided cells that lack of ecDNA FISH signal (normal cells in the same tissue section may not have ecDNA amplification) for analysis and used the DAPI channel from the same cells as a control.
Colocalization analysis for SNU16 MYC and FGFR2 ecDNAs in Figure 4a was performed using confocal images of both metaphase and interphase nuclei from the same slides. Images were split into the two FISH colors, and background fluorescence was removed manually for each channel. Colocalization for each nucleus was quantified using the ImageJ-Colocalization Threshold program. Analysis was performed across all z-stacks for each nucleus. Manders coefficient (fraction of MYC signal colocalized compared to total MYC signal) was used to quantify colocalization.
ecDNA DNA FISH and nascent RNA FISH Image Analysis
To characterize the ecDNA hub shape and size, we employed the synthetic model—Surfaces object from Imaris (version 9.1, Bitplane) and applied a Gaussian filter (σ = 1 voxel in xy) and background subtraction for optimal segmentation and quantification of ecDNA hubs. ecDNA hubs containing connected voxels were sorted by size and singleton ecDNAs were separated from ecDNA hubs (minimal two ecDNA molecules).
To measure the number of ecDNA or nascent transcripts, we localized the voxels corresponding to the local maximum of identified DNA or RNA FISH signal using the Imaris spots function module. We validated the accuracy of interphase ecDNA counting by comparing to quantification of ecDNA number by metaphase FISH as well as copy number estimated by whole genome sequencing (Extended Data Figure 1f). The copy number distribution from whole genome sequencing is comparable to that from interphase DNA FISH. While copy number estimates from WGS and interphase FISH are slightly higher than those quantified by metaphase FISH imaging, this may reflect the fact that individual ecDNAs can contain multiple copies of MYC.
Whole Genome Sequencing
Whole genome sequencing (WGS) data from COLO320-DM, COLO320-HSR and PC3 cells were generated by a previously published study1 and raw fastq reads obtained from the NCBI Sequence Read Archive, under BioProject accession PRJNA506071. Reads were trimmed of adapter content with Trimmomatic59 (version 0.39), aligned to the hg19 genome using bwa mem (0.7.17-r1188), and PCR duplicates removed using Picard’s MarkDuplicates. WGS data from SNU16 cells was generated by a previously published study60 and aligned reads in bam format from the NCBI Sequence Read Archive, under BioProject accession PRJNA523380. WGS data from HK359 cells was generated by a previously published study6 and aligned reads in bam format obtained from the NCBI Sequence Read Archive, under BioProject accession PRJNA338012. Coverage for WGS was 22X for COLO320-DM, 26X for COLO320-HSR, 1.6X for PC3, 1.2X for HK359, and 7.3X for SNU16.
Generation of ecDNA-TetO array and BRD4-HaloTag knock-in for live cell imaging
sgRNA was designed by E-CRISP (http://www.e-crisp.org/E-CRISP/designcrispr.html) targeting ~0.5kb upstream of MYC transcription start site or N-terminal BRD4 gene. The sgRNA sequences are listed in Supplementary Table 2. The sgRNA was cloned into the modified pX330 (Addgene, Cat# 42230) construct co-expressing wild type SpCas9 and a PGK-Venus cassette. ~500bp homology arms were PCR amplified from COLO320-DM cells and cloned into a pUC19 donor vector together with ~96 copies of TetO array and a blasticidin selection cassette (Addgene #118713) for ecDNA-TetO array or with HaloTag (Addgene #139747) for BRD4. 2 μg of the donor vector and 1 μg of the sgRNA vector were transfected into COLO320-DM cells by lipofectamine 3000. For ecDNA-TetO array, blasticidin (10 μg/ml) selection was applied after 7 days. For BRD4-HaloTag knock-in, 100nM HaloTag ligand JF549 (a kind gift from Luke Lavis’s lab at Janelia Research Campus) was applied to the cells followed by washing and FACS sorting. Individual clones were selected, genotyped by PCR and verified by Sanger sequencing before being tested for imaging. To detect TetO array labeled ecDNA molecules, we used the TetR-eGFP construct as described previously61. To reduce the dimerization potential associated with wild type eGFP, we generated the A206K point mutation according to previous report62. Tet-eGFP labeled hubs have a slightly smaller size compared to monomeric TetR-A206K-eGFP labeled hubs, potentially due to eGFP dimerization effects (Extended Data Figure 2c), but the number of ecDNA hubs per cell is not significantly different with Tet-eGFP vs. TetR-A206K-eGFP (Extended Data Figure 2d).
Live cell imaging microscopy
We transiently expressed TetR-eGFP or TetR-A206K-eGFP61,62 and performed imaging experiments two days after transfection. To image BRD4, we stained the cells with 200nM HaloTag ligand JF646 for 30min followed by 3 times washing in culture medium each for 10 min.
To monitor ecDNA dynamics within the nucleus, the COLO320-DM TetO-eGFP cell line was transfected with the PiggyBac vector expressing H2B-SNAPf and the super PiggyBac transposase (2:1 ratio) as described previously51. Stable transfectants were selected by 500μg/mL G418 and sorted by flow cytometry. Cells were seeded in the 8-well lab-tek chambered coverglass for long-term time lapse imaging throughout the cell cycle. Prior to imaging, COLO320-DM TetO-eGFP cells were stained with 25nM SNAP ligand JF66963 (a kind gift from Luke Lavis’s lab at Janelia Research Campus) at 37°C incubator for 30min followed by 3 washes with regular medium for total 30min. Then cells were transferred to an imaging buffer containing 10% serum in the 1x Opti-Klear live cell imaging buffer pre-warmed at 37°C. Cells were imaged at the Zeiss LSM880 microscope pre-stabilized at 37°C for 2 hours. We illuminated the sample with 1 % 488nm laser and 0.75% 633nm laser with the EC Plan-Neofluar 40x/1.30 Oil lens, beam splitter MBS 488/561/633 and filters BP 495–550 + LP 570. z-stack images were acquired with 0.3μm z step size with 3 minute intervals between each volumetric imaging for up to 12 hours. TetO labeled ecDNA was similarly analyzed as described in previous DNA/RNA FISH section. For BRD4 and PVT1p-nLuc colocalization analysis, a straight line was drawn across the center of the objects in a 2D plane and the fluorescent intensity was profiled along the line path.
JQ1 Treatment
Cells were then treated for 6 hours with 500nM JQ1 in DMSO unless otherwise indicated (Sigma-Aldrich SML1524) or an equivalent volume of DMSO.
ChIP-seq Library Preparation
Three to five million cells per replicate were fixed in 1% formaldehyde for 10–15 minutes at room temperature with rotation and then quenched with 0.125 M glycine for 10 minutes at room temperature with rotation. For COLO320-DM and COLO320-HSR BRD4 ChIP, five million cells per replicate were fixed for 15 minutes, for all conditions three million cells per replicate were fixed for 10 minutes. Fixed cells were pelleted at 800×g for 5 minutes at 4°C and washed twice with cold PBS before storing at −80°C. Pellets were thawed and membrane lysis performed in 5 mL LB1 (50 mM HEPES pH 8.0, 140 mM NaCl, 1 mM EDTA, 10% glycerol, 0.5% NP-40, 0.25% Triton X-100, 1 mM PMSF, Roche protease inhibitors 11836170001) for 10 min at 4°C with rotation. Nuclei were pelleted at 1350×g for 5 min at 4°C and lysed in 5 mL LB2 (10 mM Tris-Cl pH 8.0, 5 M, 200 mM NaCl, 1 mM EDTA, 0.5 mM EGTA, 1 mM PMSF, Roche protease inhibitors) for 10 min at RT with rotation. Chromatin was pelleted at 1350×g for 5 min at 4°C and resuspended in 1 mL of TE Buffer + 0.1% SDS before sonication on a Covaris E220. Samples were clarified by spinning at 16,000×g for 10 min at 4°C. Supernatant was transferred to a new tube and diluted with 1 volume of IP Dilution Buffer (10 mM Tris pH 8.0, 1 mM EDTA, 200 mM NaCl, 1 mM EGTA. 0.2% Na-DOC, 1% Na-Laurylsarcosine, 2% Triton X-100). Following addition of 20 ng spike-in chromatin (Active Motif 61686) and 2 μg spike-in antibody (Active Motif 53083), 50 μL of sheared chromatin was reserved as input and ChIP performed overnight at 4°C with rotation with 7.5 μg of antibody per IP: H3K27Ac (Abcam ab4729), BRD4 (Bethyl Laboratories A301–985A100).
100 μL Protein G Dynabeads per ChIP were washed 3X in 0.5% BSA in PBS and then bound to antibody bound chromatin for 4 hours at 4°C with rotation. Antibody bound chromatin was washed on a magnet 5X with RIPA Wash Buffer (50 mM HEPES pH 8.0, 500 mM LiCl, 1 mM EDTA, 1% NP-40, 0.7% Na-Deoxycholate) and once with 1 mL TE Buffer (10 mM Tris-Cl pH 8.0, 1 mM EDTA) with 500 mM NaCl. Washed beads were resuspended in 200 mL ChIP Elution Buffer (50 mM Tris-Cl pH 8.0, 10 mM EDTA, 1% SDS) and chromatin was eluted following incubation at 65°C for 15 min. Supernatant and input chromatin were removed to fresh tubes and reverse cross-linked at 65°C overnight. Samples were diluted with 200 mL TE Buffer, treated with 0.2 mg/mL RNase A (QIAGEN 19101) for 2 hours at 37°C, then 0.2 mg/mL Proteinase K (New England Biolabs P8107S) for 30 min at 55°C. DNA was purified using the ChIP DNA Clean & Concentrator kit (Zymo Research D5205). ChIP sequencing libraries were prepared using the NEBNext Ultra II DNA Library Prep Kit for Illumina (New England Biolabs E7645S) with dual indexing (New England Biolabs E7600S) following the manufacturer’s instructions. ChIP-seq libraries were sequenced on an Illumina HiSeq 4000 with paired-end 76 bp read lengths.
ChIP-seq Data Processing
Paired-end reads were aligned to the hg19 genome using Bowtie264 (version 2.3.4.1) with the --very-sensitive option following adapter trimming with Trimmomatic59 (version 0.39). Reads with MAPQ values less than 10 were filtered using samtools (version 1.9) and PCR duplicates removed using Picard’s MarkDuplicates (version 2.20.3-SNAPSHOT). MACS265 (version 2.1.1.20160309) was used for peak calling with the following parameters: macs2 callpeak -t chip_bed -c input_bed -n output_file -f BED -g hs -q 0.01 --nomodel --shift 0. A reproducible peak set across biological replicates was defined using the IDR framework (version 2.0.4.2). Reproducible peaks from all samples were then merged to create a union peak set. ChIP-seq signal was converted to bigwig format for visualization using deepTools bamCoverage66 (version 3.3.1) with the following parameters: --bs 5 --smoothLength 105 --normalizeUsing CPM --scaleFactor 10. Enrichment of ChIP signal at peaks was performed using deepTools computeMatrix on ChIP signal in bigwig format containing the ratio of BRD4 ChIP signal over input calculated using deepTools bamCoverage66 (version 3.3.1) with the following parameters: --operation ratio --bs 5 --smoothLength 105.
RT-qPCR
RNA was extracted using RNeasy Plus mini Kit (QIAGEN 74136). Purified RNA was quantified by Nanodrop (Thermo Fisher). For RT-qPCR, 50 ng of RNA, 1X Brilliant II qRT-PCR mastermix with 1 uL RT/RNase block (Agilent 600825), and 200 nM forward and reverse primer were used. Each Ct value was measured using Lightcycler 480 (Roche) and each mean dCt was averaged from duplicate qRT-PCR reaction and performed in biological triplicate. Relative MYC RNA level (RT-qPCR primers MYC_exon3_fw and MYC_exon3_rv) was calculated by ddCt method compared to 18S and GAPDH controls (RT-qPCR primers GAPDH_fw, GAPDH_rv, 18S_fw, 18S_rv). P values were calculated using a Student’s t-test by comparing the relative fold change of biological triplicates. Primer sequences are listed in Supplementary Table 1.
Drug treatments
Approximately 0.6 × 106 COLO320-DM or COLO-320-HSR cells were plated in 6 well plates and cultured under standard conditions for 24 hours. Cells were then treated for 6 hours with one of the following: 500nM JQ1 (Sigma-Aldrich SML1524), 500nM MS645 (Sigma Aldrich SML2549), 1μM THZ-1 (Selleck chemicals S7549), 20μM SGC-SCP30 (Selleck chemicals S7256), 10μM OICR-9429 (Selleck chemicals S7833), 50μM MI-3 (Selleck chemicals S7619), 2μM trichostatin A (Selleck chemicals S1045), or DMSO. Experiments were performed in biological triplicates. RT-qPCR was performed as above in technical triplicates.
Cell Viability Assay
Cells were plated in 96-well plates at 25,000 cells/well in triplicate and incubated either with JQ1 (Sigma-Aldrich SML1524) at the indicated concentrations or an equivalent volume of DMSO for 48 hours. Cell viability was measured using the CellTiterGlo assay kit (Promega G7572) in triplicate with luminescence measured on SpectraMax M5 plate reader with an integration time of 1 second per well. Luminescence was normalized to the DMSO treated controls and p values calculated using a Student’s t-test comparing biological triplicates.
Cell Proliferation Assay
Cells were plated in 96-well plates at 10,000 cells/well and incubated either with JQ1 (Sigma-Aldrich SML1524) at the indicated concentrations or an equivalent volume of DMSO. Every 24 hours, cells were harvested and counted on Countess 3 Automated Cell Counter (Thermo Fisher) with Trypan Blue used to assess cell viability. P values were calculated using a Student’s t-test comparing biological triplicates.
COLO320-DM WGS sequencing and data processing
Genomic DNA was sheared on a Covaris S2 (Covaris Inc.) and libraries were made using the NEBNext Ultra II DNA Library Prep Kit for Illumina (NEB, Inc.). Indexed libraries were pooled, and paired end sequenced (2×75bp) on an Illumina NextSeq 500 sequencer. Read data was processed in BaseSpace (basespace.illumina.com). Reads were aligned to Homo sapiens genome (hg19) using BWA aligner version 0.7.13 (https://github.com/lh3/bwa) with default settings. Coverage for ultra-low WGS for COLO320-DM 0.3X.
COLO320-DM Nanopore sequencing and data processing
Genomic DNA from COLO320-DM cells was extracted using a MagAttract HMW DNA Kit (Qiagen 67563) and prepared for long read sequencing using a Ligation Sequencing Kit (Oxford Nanopore Technologies SQK-LSK109) according to the manufacturer’s instructions. Sequencing was performed on a MinION (Oxford Nanopore Technologies). Coverage for long-read nanopore sequencing for COLO320-DM was 0.5X genome-wide and 50X for the MYC amplicon.
Bases were called from fast5 files using guppy (Oxford Nanopore Technologies, version 2.3.7). Reads were then aligned using NGMLR67 (version 0.2.7) with the following parameters: -x ont --no-lowqualitysplit. Structural variants were called using Sniffles67 (version 1.0.11) using the following parameters: -s 1 --report_BND --report_seq.
COLO320-DM Optical mapping data collection and processing
Ultra-high molecular weight (UHMW) DNA was extracted from frozen cells preserved in DMSO following the manufacturer’s protocols (Bionano Genomics, USA). Cells were digested with Proteinase K and RNAse A. DNA was precipitated with isopropanol and bound with nanobind magnetic disks. Bound UHMW DNA was resuspended in the elution buffer and quantified with Qubit dsDNA assay kits (ThermoFisher Scientific).
DNA labeling was performed following manufacturer’s protocols (Bionano Genomics, USA). Standard Direct Labeling Enzyme 1 (DLE-1) reactions were carried out using 750 ng of purified UHMW DNA. The fluorescently labeled DNA molecules were imaged sequentially across nanochannels on a Saphyr instrument. A genome coverage of approximately 400X was achieved.
De novo assemblies of the samples were performed with Bionano’s de novo assembly pipeline (Bionano Solve v3.6) using standard haplotype aware arguments. With the Overlap-Layout-Consensus paradigm, pairwise comparison of DNA molecules having 248X coverage against the reference was used to create a layout overlap graph, which was then used to generate the initial consensus genome maps. By realigning molecules to the genome maps (P value cut off of <10−12) and by using only the best matched molecules, a refinement step was done to refine the label positions on the genome maps and to remove chimeric joins. Next, during an extension step, the software aligned molecules to genome maps (P<10−12), and extended the maps based on the molecules aligning past the map ends. Overlapping genome maps were then merged (P<10−16). These extension and merge steps were repeated five times before a final refinement (P<10−12) was applied to “finish” all genome maps.
In-vitro ecDNA digestion and pulsed field gel electrophoresis
Genomic DNA from COLO320-DM cells were embedded in agarose beads as previously described68. Briefly, molten 1% certified low melt agarose (Bio-Rad, 1613112) in PBS and mineral oil (Sigma Aldrich, 69794) was equilibrated to 45°C. 50 million cells were pelleted, washed twice with cold 1X PBS, resuspended in 2 ml PBS, and briefly heated to 45°C. 2 ml agarose solution was added to cells followed by addition of 10 ml mineral oil. The mixture was swirled rapidly to create an emulsion, then poured into cold PBS with continuous stirring to solidify agarose beads. The resulting mixture was centrifuged at 500 × g for 10 minutes; supernatant was removed and beads were resuspended in 10 ml PBS and centrifuged in a clean conical tube. Supernatant was removed, beads were resuspended in buffer SDE (1% SDS, 25mM EDTA at pH 8.0) and placed on shaker for 10 minutes. Beads were pelleted again, resuspended in buffer ES (1% N-laurolsarcosine sodium salt solution, 25 mM EDTA at pH 8.0, 50ug/ml proteinase K) and incubated at 50°C overnight. On the following day, proteinase K was inactivated with 25 mM EDTA with 1 mM PMSF for 1 hour at room temperature with shaking. Beads were then treated with RNase A (1mg/ml) in 25 mM EDTA for 30 minutes at 37°C, and washed with 25 mM EDTA with a 5-minute incubation.
To perform in-vitro Cas9 digestion, 50–100ul agarose beads containing DNA were washed three times with 1X NEBuffer 3.1 (New England BioLabs) with 5-minute incubations. Next, DNA was digested in a reaction with 30nM single-guide RNA (Synthego) and 30nM spCas9 (New England BioLabs, M0386S) after pre-incubation of the reaction mix at room temperature for 10 minutes. Cas9 digestion was performed at 37°C for 4 hours, followed by overnight digestion with 3ul proteinase K (20mg/ml) in a 200ul reaction. Proteinase K was inactivated with 1mM PMSF for 1 hour with shaking. Beads were then washed with 0.5X TAE buffer three times with 10-minute incubations. Beads were loaded into a 1% certified low melt agarose gel (Bio-Rad, 1613112) in 0.5X TAE buffer with ladders (CHEF DNA Size Marker, 0.2–2.2 Mb, S. cerevisiae Ladder: Bio-Rad, 1703605; CHEF DNA Size Marker, 1–3.1 Mb, H. wingei Ladder: Bio-Rad, 1703667) and pulsed field gel electrophoresis (PFGE) was performed using the CHEF Mapper XA System (Bio-Rad) according to the manufacturer’s instructions and using the following settings: 0.5X TAE running buffer, 14°C, two-state mode, run time duration of 16 hours 39 minutes, initial switch time of 20.16 seconds, final switch time of 2 minutes 55.12 seconds, gradient of 6V/cm, included angle of 120°, and linear ramping. Gel was stained with 3X Gelred (Biotium) with 0.1M NaCl on a rocker for 30 minutes covered from light and imaged. Bands were then extracted and DNA was purified from agarose blocks using beta-Agarase I (New England BioLabs, M0392L) following the manufacturer’s instructions.
To sequence the resulting DNA, we first transposed it with Tn5 transposase produced as previously described69, in a 50 ul reaction with TD buffer70, 50ng DNA and 1 ul transposase. The reaction was performed at 37°C for 5 minutes, and transposed DNA was purified using MinElute PCR Purification Kit (Qiagen, 28006). Libraries were generated by 5 rounds of PCR amplification using NEBNext High-Fidelity 2X PCR Master Mix (NEB, M0541L), purified using SPRIselect reagent kit (Beckman Coulter, B23317) at 1.2X volumes and sequenced on the Illumina Miseq platform.
COLO320-DM reconstruction strategy
Due to the large size of the COLO320DM ecDNA (4.3 Mbp), we used a scaffolding strategy based on manual combination of results from multiple data sources. All data which required alignment back to a reference genome used hg19.
The first source of data used was the copy-number aware breakpoint graph detected by AmpliconArchitect (version 1.2)35 (AA) generated from low-coverage WGS data. The AA graph specified copy-numbers of amplicon segments as well as genomic breakpoints between them. AA was run with default settings and seed regions were identified using the PrepareAA pipeline (version 0.931.0, https://github.com/jluebeck/PrepareAA) with CNVKit (version 0.9.6)71. The AA graph file was cleaned with the PrepareAA “graph_cleaner.py” script to remove edges which conform to sequencing artifact profiles - namely, very short everted (inside-out read pair) orientation edges. Such spurious edges appear as numerous short brown ‘spikes’ in the AA amplicon image. Second, we utilized optical map (OM) contigs (Bionano Genomics, USA) which we incorporated with the AA breakpoint graph. We used AmpliconReconstructor (version 1.01)36 (AR) to scaffold together individual breakpoint graph segments against the collection of OM contigs. We ran AR with the --noConnect flag set and otherwise default settings. Third, we utilized the OM alignment tool FaNDOM (version 0.2)72 (default settings) to correct and infer additional OM contig reference alignments and junctions missed by AA and AR. OM contigs identified three additional breakpoint edges, which were subsequently added into the AA graph file. Lastly, we incorporated fragment size and sequencing data from PFGE experiments, identifying from the separated bands the estimated length and identity of genomic segments between CRISPR cut sites.
We explored the various ways the overlapping OM scaffolds could be joined while conforming to the PFGE fragment sizes and identities of the genomic regions suggested from the PFGE data. We selected a candidate structure which was concordant with the PFGE cut data expected fragment sizes, as well as intra-fragment sequence identity and multiplicity of copy count as suggested by AA analysis of the sequenced PFGE bands. The reconstruction used all but five discovered genomic breakpoint edges inside the DM region. The remaining five edges were scaffolded by two different OM contigs and each scaffold individually suggested a separate site of structural heterogeneity within the ecDNA as compared against the reconstruction.
We required that the entirety of the significantly amplified amplicon segments was used in the reconstruction. We estimated that at the baseline, genomic segments appearing once in the reconstruction existed with a copy number between 170–190. In the final structure, all amplicon segments with copy number >40 were used. Additionally, when segments were repeated inside the reconstruction, we ensured that the multiplicities of the amplicon segments suggested the reconstruction matched the multiplicities of the amplicon segments as reported by WGS.
For fine mapping analysis of the PVT1-MYC breakpoint, reads that align to both PVT1 and MYC were extracted from WGS short read sequencing which identified 10 unique reads support the breakpoint. Multiple sequence alignment was performed with ClustalW (version 2.1) for visualization.
RNA-seq Library Preparation
COLO320-DM cells were transfected with Alt-R® S.p. Cas9 Nuclease V3 (IDT, Cat# 1081058) complexed with a non-targeting control sgRNA (Synthego) with a Gal4 sequence following Synthego’s RNP transfection protocol using the Neon Transfection System (ThermoFisher, Cat# MPK5000). 500,000 to 1 million cells were harvested, and RNA was extracted using RNeasy Plus mini Kit (QIAGEN 74136). Genomic DNA was removed from samples using the TURBO DNA-free kit (ThermoFisher, Cat# AM1907), and RNA-seq libraries were prepared using the TruSeq Stranded mRNA Library Prep (Illumina, Cat# 20020595) following the manufacturer’s protocol. RNA-seq libraries were sequenced on an Illumina HiSeq 4000 with paired-end 75 bp read lengths.
RNA-seq Data Processing
Paired-end reads were aligned to the hg19 genome using STAR-Fusion73 (version 1.6.0) and the genome build GRCh37_gencode_v19_CTAT_lib_Mar272019.plug-n-play. Number of reads supporting the PVT1-MYC fusion transcript were obtained from the “star-fusion.fusion_predictions.abridged.tsv” output file and the junction read counts and spanning fragment counts were combined. Reads supporting the canonical MYC exon 1–2 junction were obtained using the Gviz (version 1.30.3) package in R (version 3.6.1)74 in a sashimi plot.
Lentivirus production
Lentiviruses were produced as previously described41. Briefly, 4 million HEK293Ts per 10 cm plate were plated the evening before transfection. Helper plasmids, pMD2.G and psPAX2, were transfected along with the vector plasmid using Lipofectamine 3000 (Thermo Fisher, Cat# L3000) according to the manufacturer’s instructions. Supernatants containing lentivirus were harvested 48 hours later, filtered with a 0.45 um filter and concentrated using Lenti-X concentrator (Clontech, Cat#631232) and stored at 80°C.
Stable CRISPR cell line generation
The pHR-SFFV-dCas9-BFP-KRAB (Addgene, Cat# 46911) plasmid was modified to dCas9-BFP-KRAB-2A-Blast as previously described41. Lentivirus was produced using the modified vector plasmid. Cells were transduced with lentivirus, incubated for 2 days, selected with 1ug/ml blasticidin for 10–14 days, and BFP expression was analyzed by flow cytometry. To generate stable, monoclonal dCas9-KRAB cell lines, single BFP-positive cell clones were sorted into 96-well plates and expanded. Vector expression was validated by flow cytometry.
CRISPR interference in COLO320-DM cells
sgRNAs targeting the MYC and PVT1 promoters were previously published41. sgRNAs targeting enhancers were designed using the Broad Institute sgRNA designer online tool (https://portals.broadinstitute.org/gpp/public/analysis-tools/sgrna-design). An additional guanine was appended to each of the protospacers that do not start with a guanine. sgRNAs were cloned into either mU6(modified)-sgRNA-Puromycin-mCherry or mU6(modified)-sgRNA-Puromycin-EGFP previously generated41 and lentiviruses were produced. To evaluate the effects of CRISPR interference on gene expression, cells were transduced with sgRNA lentiviruses, incubated for 2 days, selected with 0.5ug/ml puromycin for 4 days, and BFP, GFP and/or mCherry expressions were assessed by flow cytometry. Cells were harvested for RT-qPCR assays as described above. All guide sequences are in Supplementary Table 2.
Single-Cell Paired RNA and ATAC-seq Library Preparation
Single-cell paired RNA and ATAC-seq libraries for COLO320-DM and COLO320-HSR were generated on the 10x Chromium Single-Cell Multiome ATAC + Gene Expression platform following the manufacturer’s protocol and sequenced on an Illumina NovaSeq 6000.
Single-cell RNA and ATAC-seq data processing and analysis
A custom reference package for hg19 was created using cellranger-arc mkref (10x Genomics, version 1.0.0). The single-cell paired RNA and ATAC-seq reads were aligned to the hg19 reference genome using cellranger-arc count (10x Genomics, version 1.0.0).
Subsequent analyses on RNA were performed using Seurat (version 3.2.3)75, and those on ATAC-seq were performed using ArchR (version 1.0.1)76. Cells with more than 200 unique RNA features, less than 20% mitochondrial RNA reads, less than 50,000 total RNA reads were retained for further analyses. Doublets were removed using ArchR.
Raw RNA counts were log-normalized using Seurat’s NormalizeData function, scaled using the ScaleData function, and the data were visualized on a UMAP using the first 30 principal components. Dimensionality reduction for the ATAC-seq data were performed using Iterative Latent Semantic Indexing (LSI) with the addIterativeLSI function in ArchR. To impute accessibility gene scores, we used addImputeWeights to add impute weights and plotEmbedding to visualize scores. To compare the accessibility gene scores for MYC with MYC RNA expression, getMatrixFromProject was used to extract the gene score matrix and the normalized RNA data were used.
To identify variable ATAC-seq peaks on COLO320-DM and COLO320-HSR amplicons, we first calculated amplicon copy numbers based on background ATAC-seq signals as previously described, using a sliding window of five megabases moving in one-megabase increments across the reference genome77. We used the copy number z scores calculated for the chr8:124000001–129000000 interval for estimating copy numbers of MYC-bearing ecDNAs in COLO320-DM and MYC-bearing chromosomal HSRs in COLO320-HSR. We then incorporated these estimated copy numbers into the variable peak analysis as follows. COLO320-DM and COLO320-HSR cells were separately assigned into 20 bins based on their RNA expression of MYC. Next, pseudo-bulk replicates for ATAC-seq data were created using the addGroupCoverages function grouped by MYC RNA quantile bins. ATAC-seq peaks were called using addReproduciblePeakSet for each quantile bin, and peak matrices were added using addPeakMatrix. Differential peak testing was performed between the top and the bottom RNA quantile bins using getMarkerFeatures. A false discovery rate cutoff of 1e-15 was imposed. The mean copy number z score for each quantile bin was then calculated and a copy number fold change between the top and bottom bin was computed. Finally, we filtered on significantly differential peaks that are located in chr8:127432631–129010071 and have fold changes above the calculated copy number fold change multiplied by 1.5.
HiChIP Library Preparation
One to four million cells were fixed in 1% formaldehyde in aliquots of one million cells each for 10 minutes at room temperature. HiChIP was performed as previously described43,78 using antibodies against H3K27ac (Abcam ab4729; 2μg antibody for one million cells, 7.5μg antibody for four million cells) with the following optimizations79: SDS treatment at 62°C for 5 min; restriction digest with MboI for 15 min; instead of heat inactivation of MboI restriction enzyme, nuclei were washed twice with 1X restriction enzyme buffer; biotin fill-in reaction incubation at 37°C for 15 minutes; ligation at room temperature for 2 hours. HiChIP libraries were sequenced on an Illumina HiSeq 4000 with paired-end 76 bp read lengths.
HiChIP Data Processing
HiChIP data were processed as described previously43. Briefly, paired end reads were aligned to the hg19 genome using the HiC-Pro pipeline (version 2.11.0)80. Default settings were used to remove duplicate reads, assign reads to MboI restriction fragments, filter for valid interactions, and generate binned interaction matrices. The Juicer (version 1.5) pipeline’s HiCCUPS tool and FitHiChIP (version 8.0) were used to identify loops81,82. Filtered read pairs from the HiC-Pro pipeline were converted into .hic format files and input into HiCCUPS using default settings. Dangling end, self-circularized, and re-ligation read pairs were merged with valid read pairs to create a 1D signal bed file. FitHiChIP was used to identify “peak-to-all” interactions at 10 kb resolution using peaks called from the one-dimensional HiChIP data. A lower distance threshold of 20 kb was used. Bias correction was performed using coverage specific bias. HiChIP contact matrices stored in .hic files were visualized in R (version 4.0.3) using gTrack (version 0.1.0) at 10 kb resolution following Knight-Ruiz normalization. We also compared HiChIP contract matrices following ICE and OneD normalization following copy number correction using the dryhic R package (version 0.0.0.9100)83. Virtual 4C plots were generated from dumped matrices generated with Juicer Tools (1.9.9). The Juicer Tools tools dump command was used to extract the chromosome of interest from the .hic file. The interaction profile of a 10-kb bin containing the anchor was then plotted in R (version 4.0.3) following normalization by the total number of valid read pairs and smoothing with the rollmean function from the zoo package (version 1.8–9).
Reporter plasmid construction and transfection
We constructed a plasmid containing the 2kb PVT1 promoter (chr8:128,804,981–128,806,980, hg19) or the MYC promoter (chr8:128,745,990–128,748,526, hg19) driving NanoLuc luciferase (PVT1p-nLuc) and a constitutive thymidine kinase (TK) promoter driving Firefly luciferase as an internal control (Figure 3b). Briefly, pGL4-tk-luc2 (Promega) was digested with KpnI and PciI. A sequence containing multiple cloning sites (GTACCTGAGCTCGCTAGCCTCGAGAAGATCTGCGTACGGTCGAC), NanoLuc and BGH polyA sequence were inserted in tandem into the vector using Gibson assembly (NEBuilder DNA assembly mix). Next, the PVT1 promoter or the MYC promoter was inserted into the vector via NheI and SalI digestion to generate the final reporter construct. For the negative control, a minimal promoter (TAGAGGGTATATAATGGAAGCTCGACTTCCAGCTT) was used in place of the PVT1 promoter. For constructing plasmids with a cis-enhancer, an enhancer (chr8:128347148–128348310, hg19; positive H3K27ac mark and looping to the PVT1 promoter in HiChIP, overlapping with BRD4 ChIP peak and ATAC-seq peak in COLO320-DM) was inserted directly 5’ to the promoter into the region with multiple cloning sites. To assess luciferase reporter expression, COLO320-DM or COLO320-HSR cells were seeded into a 24-well plate with 75,000 cells per well. Reporter plasmids were transfected into cells the next day with lipofectamine 3000 following the manufacturer’s protocol, using 0.25 μg DNA per well. Two days later, cells were treated with either JQ1 (500nM) or DMSO for 6 hours before collection. Luciferase levels were quantified using Nano-Glo Dual reporter luciferase assay (Promega). The reporter level was calculated as the ratio of NanoLuc reading over firefly reading using Tecan M1000. Mean and standard errors were calculated based on three biological replicates with three technical replicate each.
To analyze the spatial relationship of NanoLuc activity with ecDNA hubs in situ, we designed and ordered the RNA FISH probe sets for NanoLuc luciferase gene (30 probes mix) and Firefly luciferase gene (47 probes mix) conjugated with the Quasar 570 dye and Quasar 670 dye, respectively (Biosearch Technologies). We transfected 0.5 μg PVT1 promoter or minimal promoter reporter plasmid into COLO320-DM cells seeded on the 12mm #1.5 round glass coverslips (Electron Microscopy Sciences). Two days after transfection, DNA/RNA FISH were performed as described in the Nascent RNA FISH section except that a 1.5Mbp probe conjugated with Atto488 was applied together with the NanoLuc Quasar 570 probe and Firefly Quasar 670 probe. We applied the same Gaussian smoothing with Gaussian filter (σ = 1 voxel in xy) and background subtraction in all images for proper segmentation of the active transcription sites of luciferase genes. The size of the active transcription sites was estimated from the diameter of the sphere with identical volume of the segmented objects and the luciferase transcription activity was quantified from the sum of the fluorescence intensity within the segmented transcription sites. The ecDNA hubs were similarly segmented and the binary overlap between the two surfaces were used to determine the spatial relationship between the luciferase gene transcription sites and ecDNA hubs.
SNU16-dCas9-KRAB Whole Genome Sequencing and Data Processing
DNA was extracted from harvested cells using the DNeasy Blood & Tissue Kit (Qiagen) according to the manufacturer’s instructions. Libraries were prepared using a modified Nextera library preparation protocol. 80 ng of input DNA were combined with 1X TD Buffer70, 1 μL transposase69 (40 nM final) in a reaction volume of 50 μL and incubated at 37°C for 5 minutes. Transposed DNA was purified using a MinElute PCR Purification Kit (Qiagen) according to the manufacturer’s instructions. Libraries were generated by 5 rounds of PCR amplification, purified using SPRIselect reagent kit (Beckman Coulter, B23317) at 1.2X volumes and sequenced on an Illumina HiSeq 6000 with paired end 2×150 bp reads. Coverage for SNU16-dCas9-KRAB WGS was 12X.
Reads were trimmed of adapter content with Trimmomatic59 (version 0.39), aligned to the hg19 genome using bwa mem (0.7.17-r1188), and PCR duplicates removed using Picard’s MarkDuplicates (version 2.20.3-SNAPSHOT). Regions of copy number alteration were identified using ReadDepth (version 0.9.8.5) with parameters recommended by AmpliconArchitect (version 1.0), and amplicon reconstruction performed using the default parameters. Structural variant junctions were extracted from the edges_cnseg.txt output files and used for visualization.
ATAC-seq library preparation and data processing
ATAC-seq library preparation was performed as previously described70 and sequenced on the NovaSeq 6000 platform (Illumina, Inc., San Diego, CA) with 2×75bp reads. Adapter-trimmed reads were aligned to the hg19 genome using Bowtie2 (2.1.0). Aligned reads were filtered for quality using samtools (version 1.9), duplicate fragments were removed using Picard (version 2.21.9-SNAPSHOT), and peaks were called using MACS2 (version 2.1.0.20150731) with a q-value cut-off of 0.01 and with a no-shift model. Peaks from replicates were merged, read counts were obtained using bedtools (version 2.17.0) and normalized using DESeq2 (version 1.26.0).
To identify accessible elements in MYC and FGFR2 ecDNAs in SNU16, we filtered on all ATAC-seq peaks within known ecDNA-amplified regions (chr8:128200000–129200000 for the MYC ecDNA, chr10:122000000–123680000 for the FGFR2 ecDNA) whose normalized read counts (using the “counts” function in DESeq2 with normalized = TRUE) exceeded a manually determined threshold (500 for the MYC amplicon, 1000 for the FGFR2 amplicon). Peaks that met all criteria for two technical replicates were included as candidate DNA elements in the CRISPR interference study.
CRISPR interference screen
After generation of monoclonal SNU16-dCas9-KRAB cells, MYC and FGFR2 ecDNAs in single clones were assessed using metaphase FISH. A clone with distinct MYC and FGFR2 amplicons on the vast majority of ecDNAs was selected for CRISPR interference experiments.
For the pooled experiments in SNU16-dCas9-KRAB, sgRNAs targeting ATAC-seq peaks were designed using the Broad Institute sgRNA designer online tool. An additional guanine was appended to each of the protospacers. Pooled sgRNA cloning was performed as described previously84. Briefly, sgRNA sequences were designed with flanking Esp3I digestion sites and two nested PCR handles. Oligos were amplified by PCR and then cloned into the lentiGuidePuro vector modified to express a 2A-GFP fusion in frame with puromycin. The vector was pre-digested and then sgRNA cloning was done via one-step digestion/ligation of the insert. 1 uL of this reaction was transformed via electroporation and purified with maxiprep. sgRNA representation was confirmed by sequencing.
SNU16-dCas9-KRAB cells were transduced with the lentiviral guide pool at an effective MOI of 0.2. Cells were incubated for 2 days, selected with puromycin for 4 days, and rested for 3–5 days in culture media without puromycin. 20 million cells were fixed and a two-color RNA flowFISH was performed for ACTB and either MYC or FGFR2 using the PrimeFlow™ RNA Assay Kit (Thermo Fisher) following the manufacturer’s protocol and corresponding probe sets (MYC: VA1–6000107-PF; FGFR2: VA1–14785-PF; ACTB: VA6–10506-PF). ACTB labels a houskeeping control gene to control for noise in RNA flowFISH due to variable staining intensity. Cells were sorted by fluorescence-activated cell sorting (FACS) using the gating strategy shown in Extended Data Figure 8c and as previously described44. The oncogene (MYC/FGFR2) was labeled with Alexa Fluor 647 and ACTB was labeled with Alexa Fluor 750. Based on the assumption that the expression of the housekeeping gene is not correlated with the oncogene, any correlation in fluorescence intensities between the ACTB and the oncogene was attributed to flowFISH staining efficiency and manually regressed using the FACS compensation tool. The degree of compensation was determined so that the top and bottom 25% of cells based on Alexa Fluor 647 signal intensity deviated no more than 15% from the population mean in Alexa Fluor 750 signal intensity. After compensation, we gated on cells with positive ACTB labeling and sorted cells into six bins using Alexa Fluor 647 MFI corresponding to the following percentile ranges: 0–10% (bin 1), 10–20% (bin 2), 35–45% (bin 3), 55–65% (bin 4), 80–90% (bin 5), 90–100% (bin 6). FACS data were analyzed using FlowJo (10.7.0).
Cells were pelleted at 800g for 5 minutes and resuspended in 100ul lysis buffer (50mM Tris-HCl pH 8, 10mM EDTA, 1% SDS). The lysate was incubated at 65°C for 10 minutes for reverse cross-linking and cooled to 37°C. RNase A (10mg/ml) was added at 1:50 by volume and incubated at 37°C for 30 minutes. Proteinase K (20mg/ml) was added at 1:50 by volume and samples were incubated at 45°C overnight. Genomic DNA was extracted using Zymo DNA miniprep kit. Libraries were prepared using 3 rounds of PCR as previously described84. Amplified product sizes were validated on a gel, and the final products were purified using SPRIselect reagent kit (Beckman Coulter, Cat# B23318) at 1.2x sample volumes following the manufacturer’s protocol. Libraries were sequenced on an Illumina Miseq with paired-end 75 bp read lengths. Read 1 was used for downstream analysis.
Relative abundances of sgRNAs were measured using MAGeCK (version 0.5.9.4)85. sgRNA counts were obtained using the “mageck count” command. For samples with PCR replicates, if a PCR replicate has fewer than 1000 total sgRNAs passing filter (raw counts > 20), the replicate was excluded. Next, each sgRNA count was divided by total sgRNA counts for each library and multiplied by one million to give a normalized count (count per million, CPM). For samples with PCR replicates, mean CPM was calculated for each sgRNA. sgRNAs that have CPMs lower than 20 in the unsorted cells were classified as dropouts and removed from the analysis. We then calculated the log2 fold change of each sgRNA in each sorted cell bin over unsorted cells by dividing the respective CPMs followed by log-transformation. sgRNA enrichment was then quantified as previously described84. Briefly, the log2 fold change in the high expression bin was subtracted from that in the low expression bin [log2(low/high)] for each sgRNA. The resulting log2(low/high) values were averaged for each candidate regulatory element and z scores were calculated using the formula z = (x-m)/S.E., where x is the mean log2(low/high) of the candidate element, m is the mean log2(low/high) of negative control sgRNAs, and S.E. is the standard error calculated from the standard deviation of negative control sgRNAs divided by the square root of the number of sgRNAs targeting the candidate element in independent biological replicates. Z scores were used to compute upper-tail p values using the normal distribution function, which were adjusted with p.adjust in R (version 3.6.1) using the Benjamini-Hochberg Procedure to produce false discovery rate (FDR) values. For assessing sgRNA correlations across all six sorted bins for individual elements, we computed Spearman coefficients for all individual sgRNAs across the six fluorescence bins using log2 fold changes over unsorted cells. All sgRNA sequences used in the CRISPRi experiments in SNU16-dCas9-KRAB are listed in Supplementary Table 3.
TR14 Amplicon Reconstruction
We obtained WGS data for TR14 cells as follows. DNA was extracted from harvested cells (NucleoSpin Tissue kit, Macherey-Nagel GmbH & Co. KG, Düren, Germany). Libraries were prepared (NEBNext Ultra II FS DNA Library Prep Kit for Illumina, New England BioLabs, Inc., Ipswich, MA) and sequenced on the NovaSeq 6000 platform (Illumina, Inc., San Diego, CA) with 2×150bp reads. Adapters were trimmed with BBMap 38.58. Reads were then aligned to hg19 using BWA-MEM 0.7.1586 with default parameters and duplicate reads were removed (Picard 2.20.4). Coverage was computed in 20bp bins, normalized as counts per million, using using deepTools 3.3.066. Copy number variation was called using QDNAseq 1.22.087, binning primary alignments with MAPQ≥20 in 10kb bins, default filtering and additional filtering of bins with more than 5% Ns in the reference. Bins were corrected for GC content and normalized. Segmentation was performed using the CBS method with no transformation of the normalized counts and parameter alpha=0.05.
Genomic DNA from TR14 cells was extracted using a MagAttract HMW DNA Kit and fragments >10kb were selected using the Circulomics SRE kit (Circulomics Inc., Baltimore, MD). Libraries were prepared using a Ligation Sequencing Kit and sequenced on a R9.4.1 MinION flowcell (FLO-MIN106). Reads were aligned to hg19 using NGMLR v0.2.7. Structural variants were called using Sniffles v1.0.11 and parameters --min_length 15 --genotype --min_support 3 --report_seq.
To reconstruct the coarse structure of oncogene amplifications in TR14, we compiled all Sniffles structural variants larger than 10kb with a minimum read support of 15 into one genome graph using gGnome 0.188, nodes representing genomic segments connected by reference or structural variant edges. Non-amplified segments (i.e. mean Illumina WGS coverage less than 10-fold the median chromosome 2 coverage) were discarded from the graph. Strong clusters in the genome graph were identified, partitioning the graph into groups of segments that could be reached from one another. We identified the clusters containing the four amplified oncogenes (MYCN, CDK4, MDM2, ODC1) and manually selected circular paths through each cluster that could account for the main copy number steps around the oncogenes. We used gTrack (https://github.com/mskilab/gTrack) for visualization. Hi-C data were used to validate these reconstructions, confirming that all strong off-diagonal signal indicative of structural rearrangements were captured by the reconstruction. Previously studies suggest that the identified amplicons exist as extrachromosomal DNA89,90.
Hi-C
Hi-C libraries were prepared as described previously23. Samples were sequenced with Illumina Hi-Seq according to standard protocols in 100bp paired-end mode at a depth of 433.7 million read pairs. FASTQ files were processed using the Juicer pipeline v1.19.02, CPU version91, which was set up with BWA v0.7.1786 to map short reads to reference genome hg19, from which haplotype sequences were removed and to which the sequence of Epstein-Barr virus (NC_007605.1) was added. Replicates were processed individually. Mapped and filtered reads were merged afterwards. A threshold of MAPQ≥30 was applied for the generation of Hi-C maps with Juicer tools v1.7.591. Knight-Ruiz normalization per hg19 chromosome was used for Hi-C maps82,92, interaction across different chromosome pairs should therefore only carefully be interpreted.
For TR14, we created a custom genome containing additionally the amplicon reconstructions. The sequences of amplicons were composed from hg19 based on the order and orientation of their chromosomal fragments. The original fragment locations on hg19 were masked to allow unambiguous mapping. Note, by this also Hi-C reads from wildtype alleles are mapping to the amplicon sequences leading to a mix of signal, depending on the fraction of amplicons and wildtype allele. After mapping, we kept only amplicons and removed all other chromosomes to create Hi-C maps and apply GW_KR normalization using Juicer Tools v1.19.0291.
TR14 Interaction analysis
TR14 H3K27ac ChIP-seq raw data were downloaded from Gene Expression Omnibus (GSE90683)93. We trimmed adapters with BBMap 38.58 and aligned the reads to hg19 using BWA-MEM 0.7.1586 with default parameters. Coverage tracks were created by extending reads to 200bp, filtering using the ENCODE DAC blacklist and normalizing to counts per million in 10bp bins with deepTools 3.3.066. Enhancers were called using LILY (https://github.com/BoevaLab/LILY, not versioned)93 with default parameters.
The HPCAL1 enhancer region was defined by two LILY-defined boundary enhancers as chr2:10424449–10533951. A virtual 4C track was generated by the mean genome-wide interaction profile (KR-normalized Hi-C signal in 5kb bins) across all overlapping 5kb bins.
For the aggregate analysis of the effect of H3K27 acetylation on interaction, all 5kb bin pairs located on different amplicons were analyzed for their KR-normalized Hi-C signal depending on the mean H3K27ac fold-change over input of each of the two bins. We used 5-fold change threshold to distinguish low- from high-H3K27ac bins.
Extended Data
Extended Data Figure 1. ecDNA FISH strategies and copy number estimation.
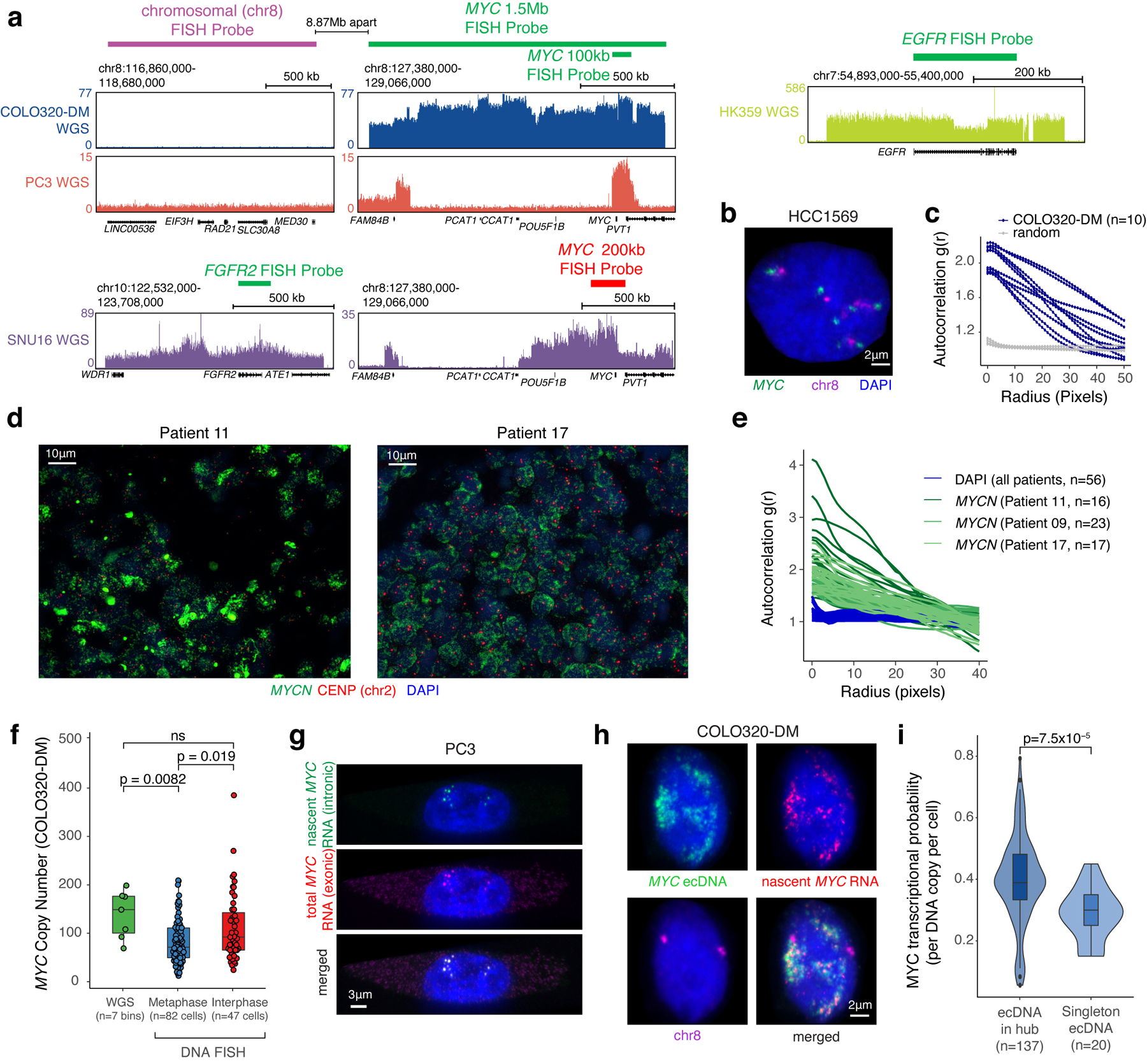
(a) WGS tracks with DNA FISH probe locations. For COLO320-DM and PC3, a 1.5 Mb MYC FISH probe (Figure 1a,b), a 100 kb MYC FISH probe (Figure 1d,e,f), or a 1.5 Mb chromosome 8 FISH probe was used. Commercial probes were used in SNU16 and HK359 cells. (b) Representative DNA FISH image using chromosomal and 1.5 Mb MYC probes in non-ecDNA amplified HCC1569 showing paired signals as expected from the chromosomal loci. (c) ecDNA clustering of individual COLO320-DM cells by autocorrelation g(r). (d) Representative FISH images showing ecDNA clustering in primary neuroblastoma tumors (Patients 11 and 17). (e) ecDNA clustering of individual primary tumor cells from all three patients using autocorrelation g(r). (f) Comparison of MYC copy number in COLO320-DM calculated based on WGS (n=7 genomic bins overlapping with DNA FISH probes), metaphase FISH (n=82 cells) and interphase FISH (n=47 cells). P-values determined by two-sided Wilcoxon test. (g) Representative images of nascent MYC RNA FISH showing overlap of nascent RNA (intronic) and total RNA (exonic) FISH probes in PC3 cells (independently repeated twice). (h) Representative images from combined DNA FISH for MYC ecDNA (100 kb probe) and chromosomal DNA with nascent MYC RNA FISH in COLO320-DM cells (independently repeated four times). (i) MYC transcription probability measured by nascent RNA FISH normalized to DNA copy number by FISH comparing singleton ecDNAs to those found in hubs in COLO320-DM (box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). To control for noise in transcriptional probability for small numbers of ecDNAs, we randomly re-sampled RNA FISH data grouped by hub size and calculated transcription probability. The violin plot represents transcriptional probability per ecDNA hub based on the hub size matched sampling. P-value determined by two-sided Wilcoxon test.
Extended Data Figure 2. Generation of TetR-GFP COLO320-DM cells for ecDNA imaging in live cells.
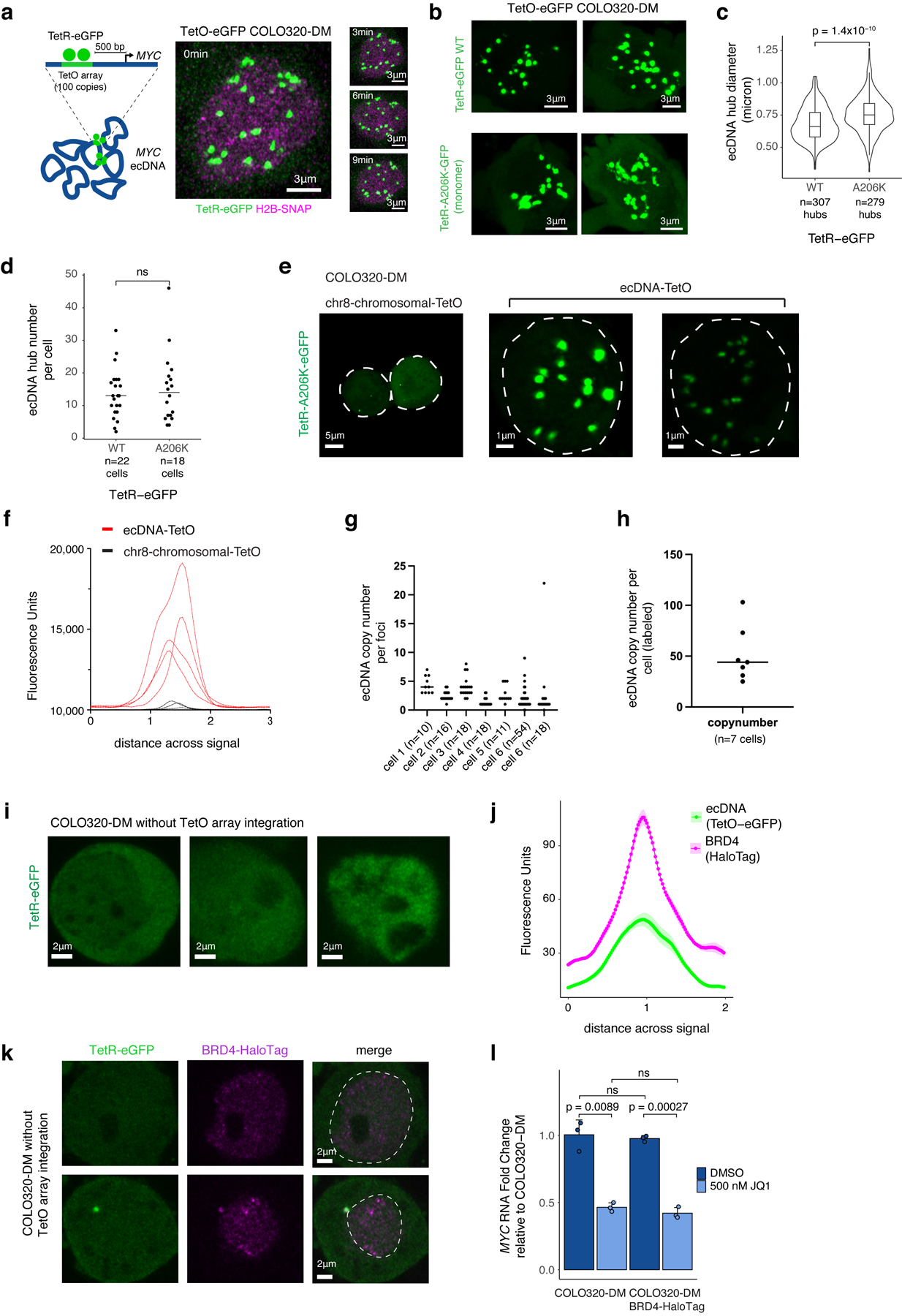
(a) ecDNA imaging based on TetO array knock-in and labeling with TetR-eGFP (left). Representative images of TetR-eGFP signal in TetO-eGFP COLO320-DM cells at indicated timepoints in a time course (right; independently repeated twice). (b) GFP signal in ecDNA-TetO COLO320-DM cells. TetR-eGFP and monomeric TetR-A206K-GFP labeled ecDNA hubs appear to be smaller in living cells than in DNA FISH studies of fixed cells likely because the TetO array is not integrated in all ecDNA molecules and there are potential differences caused by denaturation during DNA FISH and eGFP dimerization. (c) ecDNA hub diameter in microns (box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). P-value determined by two-sided Wilcoxon test. (d) ecDNA hub number per cell. Line represents median. P-value determined by two-sided Wilcoxon test. (e) TetR-eGFP signal in chr8-chromosomal-TetO (chr8:116860000–118680000, left) and ecDNA-TetO (TetO-eGFP COLO320-DM, right) COLO320-DM cells. (f) Fluorescence intensity for chr8-chromosomal-TetO and ecDNA-TetO foci. (g, h) Inferred ecDNA copy number per foci (g; n = number of foci/cell) and per cell (h; n = number of cells) for ecDNA-TetO labeled cells based on summed fluorescence intensity relative to chr8-chromosomal-TetO foci. Line represents median. (i) Representative images of TetR-GFP signal in parental COLO320-DM without TetO array integration which shows minimal TetR-GFP foci. (j) Mean fluorescence intensities for ecDNA (TetO-eGFP) and BRD4 (HaloTag) foci across a line drawn across the center of the largest ecDNA (TetO-eGFP) signal. Data are mean ± SEM for n=5 ecDNA foci. (k) Representative image of TetR-eGFP signal in COLO320-DM cells without TetO array integration overlaid with BRD4-HaloTag signal. Dashed line indicates nucleus boundary. We noted cytoplasmic TetR-eGFP signal in a subset of COLO320-DM cells without TetO array integration but it did not colocalize with BRD4-HaloTag. (l) MYC RNA measured by RT-qPCR for parental COLO320-DM and BRD4-HaloTag COLO320-DM cells treated with DMSO or 500 nM JQ1 for 6 hours which shows similar levels of MYC transcription and sensitivity to JQ1 inhibition following epitope tagging of BRD4. Data are mean ± SD between 3 biological replicates. P-values determined by two-sided student’s t-test.
Extended Data Figure 3. BET inhibition leads to ecDNA hub dispersal.
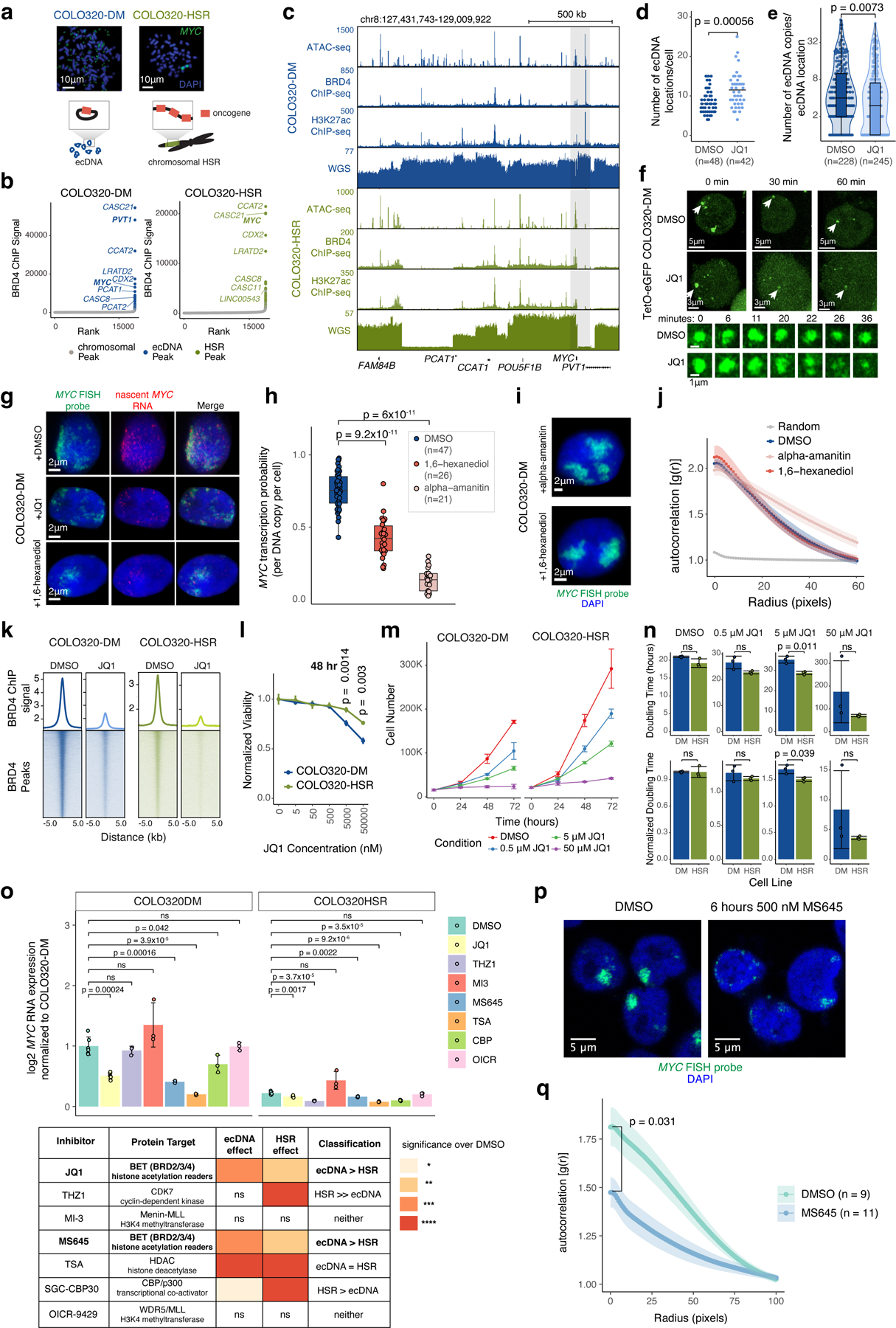
(a) Representative metaphase FISH images and schematic showing ecDNA in COLO320-DM and chromosomal HSRs in COLO320-HSR (independently repeated twice for COLO320-DM and not repeated for COLO320-HSR). (b) Ranked BRD4 ChIP-seq signal. Peaks in ecDNA or HSR amplifications are highlighted and labeled with nearest gene. (c) ATAC-seq, BRD4 ChIP-seq, H3K27ac ChIP-seq and WGS at amplified MYC locus. (d) Number of ecDNA locations (including ecDNA hubs with >1 ecDNA and singleton ecDNAs) from interphase FISH imaging for individual COLO320-DM cells after treatment with DMSO or 500 nM JQ1 for 6 hours. N = number of cells quantified per condition. P-value determined by two-sided Wilcoxon test. (e) ecDNA copies in each ecDNA location from interphase FISH imaging in COLO320-DM after treatment with DMSO or 500 nM JQ1 for 6 hours (box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). N = number of ecDNA locations quantified per condition. P-value determined by two-sided Wilcoxon test. (f) Representative live images of TetR-eGFP-labeled ecDNA after treatment with DMSO or 500 nM JQ1 at indicated timepoints in a time course (top; independently repeated twice) and ecDNA hub zoom-ins (bottom). (g) Representative image from combined DNA/RNA FISH in COLO320-DM cells treated with DMSO, 500 nM JQ1, or 1% 1,6-hexanediol for 6 hours. (h) MYC transcription probability measured by dual DNA/RNA FISH after treatment with DMSO, 1% 1,6-hexanediol, or 100 μg/mL alpha-amanitin for 6 hours (box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range; n = number of cells). P-values determined by two-sided Wilcoxon test. (i) Representative DNA FISH images for MYC ecDNA in interphase COLO320-DM treated with either 1% 1,6-hexanediol or 100 μg/mL alpha-amanitin for 6 hours. (j) ecDNA clustering in interphase cells by autocorrelation g(r) for COLO320-DM treated with DMSO, 1% 1,6-hexanediol, or 100 μg/mL alpha-amanitin for 6 hours. Data are mean ± SEM (n = 10 cells quantified per condition). (k) Averaged BRD4 ChIP-seq signal and heatmap over all BRD4 peaks for cells treated with DMSO or 500 nM JQ1 for 6 hours. (l) Cell viability after treatment with different JQ1 concentrations for 48 hours normalized to DMSO-treated cells. Data are mean ± SD between 3 biological replicates. P-values determined by two-sided student’s t-test. (m) Cell proliferation after treatment with different JQ1 concentrations over 72 hours. Data are mean ± SD between 3 biological replicates. (n) Cell doubling times after treatment with different JQ1 concentrations over 72 hours in hours (top) or after normalization to DMSO-treated cells (bottom). Data are mean ± SD between 3 biological replicates. P-values determined by two-sided student’s t-test. (o) MYC RNA measured by RT-qPCR after treatment with indicated inhibitors for 6 hours (top; each point represents a biological replicate, n=6 for DMSO and JQ1 treatments, n=3 for all other drug treatments). Data are mean ± SD. P-values determined by two-sided student’s t-test. Details of inhibitor panel, protein target, significance of effect on MYC transcription, and comparison of effect on ecDNA and HSR transcription (bottom). (p,q) Representative DNA FISH images (p) and clustering by autocorrelation g(r) (q) for MYC ecDNAs in COLO320-DM treated with DMSO or 500 nM MS645 for 6 hours. Data are mean ± SEM. P-value determined by two-sided Wilcoxon test at radius = 0.
Extended Data Figure 4. Reconstruction of COLO320-DM ecDNA amplicon structure.
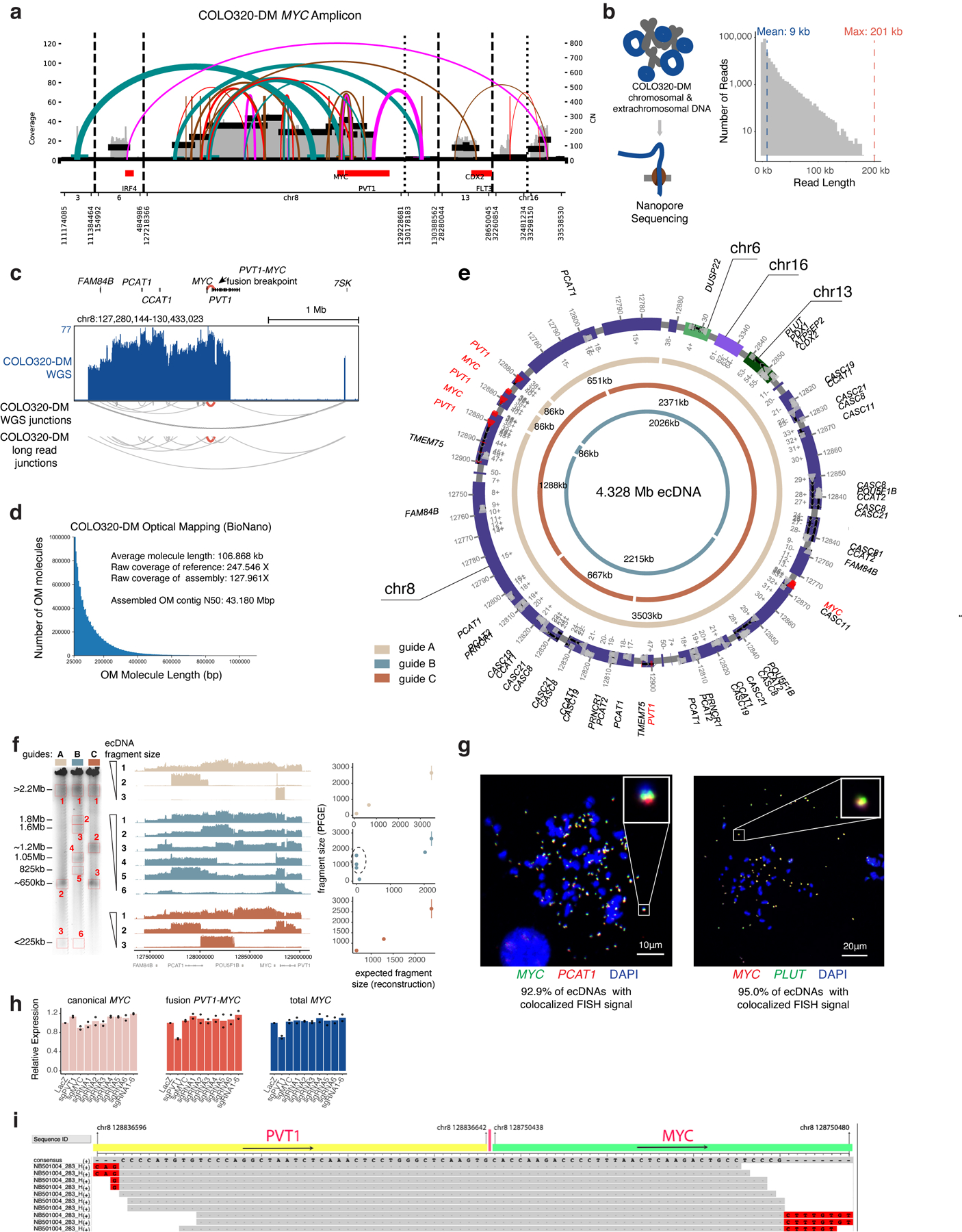
(a) Structural variant (SV) view of AmpliconArchitect (AA) reconstruction of the MYC amplicon in COLO320-DM cells. (b) Nanopore sequencing of COLO320-DM cells (left) and distribution of read lengths. (c) WGS for COLO320-DM with junctions detected by WGS and nanopore sequencing. (d) Molecule lengths used for optical mapping and statistics. (e) Reconstructed COLO320-DM ecDNA after integrating WGS, optical mapping, and in-vitro ecDNA digestion. Chromosomes of origin and corresponding coordinates (hg19) are labeled. Three inner circular tracks (light tan, slate and brown in color; guides A, B and C, respectively) representing expected fragments as a result of Cas9 cleavage using three distinct sgRNAs and their expected sizes. Guide sequences are in Supplementary Table 2 (PFGE_guide_A-C). (f) In-vitro Cas9 digestion of COLO320-DM ecDNA followed by PFGE (left). Fragment sizes were determined based on H. wingei and S. cerevisiae ladders. Uncropped gel image is in Supplementary Figure 1. Middle panel shows short-read sequencing of the MYC ecDNA amplicon for all isolated fragments, ordered by fragment size. Right panel shows concordance of expected fragment sizes by optical mapping reconstruction, and observed fragment sizes by in-vitro Cas9 digestion (discordant fragments circled). Each sgRNA digestion was performed in one independent experiment. (g) Metaphase FISH images showing colocalization of MYC, PCAT1 and PLUT as predicted by optical mapping and in-vitro digestion. N = 20 cells and 1,270 ecDNAs quantified for MYC/PCAT1 DNA FISH and n = 15 cells and 678 ecDNAs for MYC/PLUT DNA FISH from one experiment. (h) RNA expression measured by RT-qPCR for indicated transcripts in COLO320-DM cells stably expressing dCas9-KRAB and indicated sgRNAs (n=2 biological replicates). Canonical MYC was amplified with primers MYC_exon1_fw and MYC_exon2_rv; fusion PVT1-MYC was amplified with PVT1_exon1_fw and MYC_exon2_rv; total MYC was amplified with total_MYC_exon2_fw and total_MYC_exon2_rv. All primer sequences are in Supplementary Table 1 and guide sequences are in Supplementary Table 2. (i) Alignment of junction reads at the PVT1-MYC breakpoint.
Extended Data Figure 5. Single-cell multiomic analysis reveals combinatorial and heterogeneous ecDNA regulatory element activities associated with MYC expression.
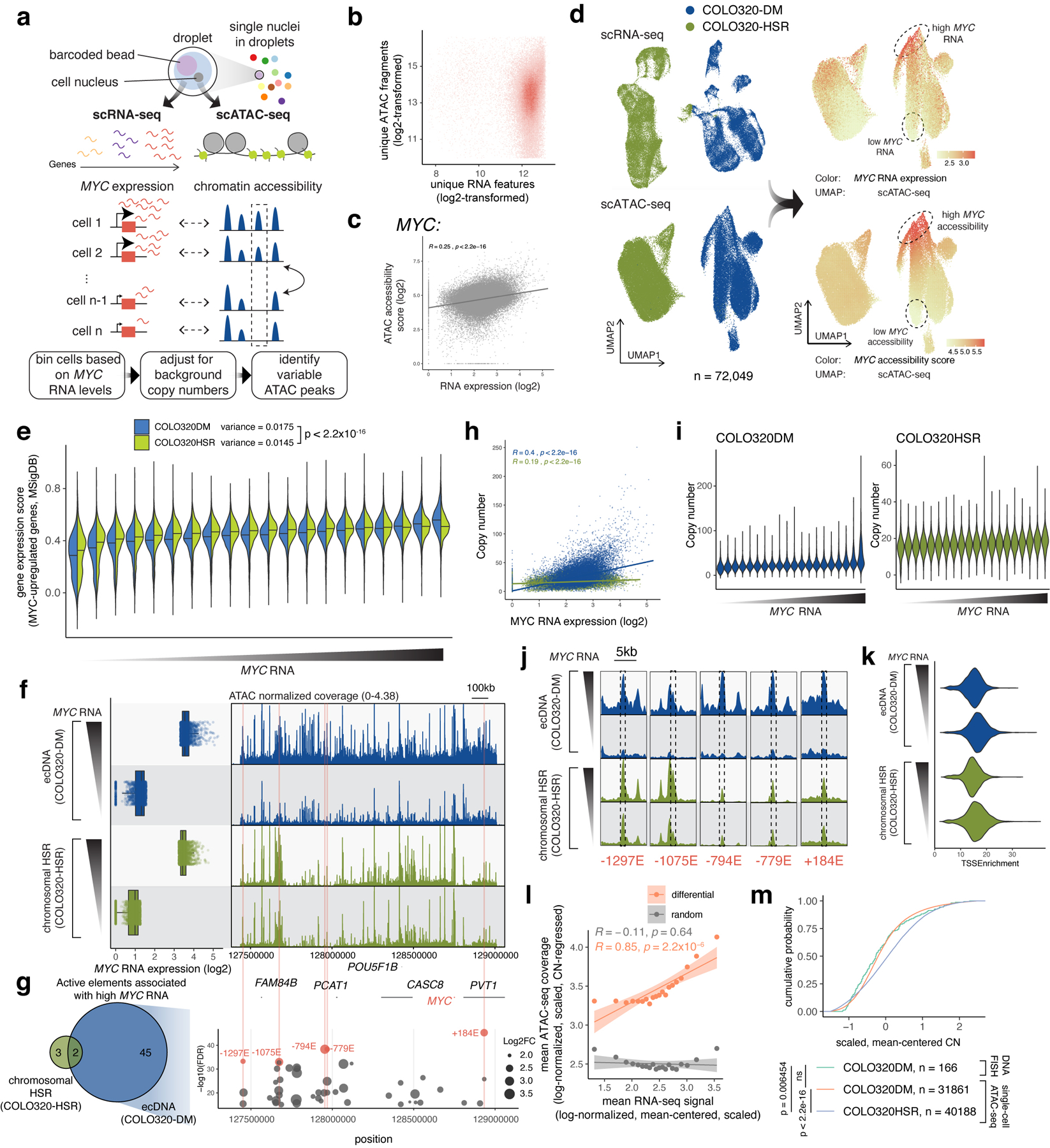
(a) Joint single-cell RNA and ATAC-seq for simultaneously assaying gene expression and chromatin accessibility and identifying regulatory elements associated with MYC expression. (b) Unique ATAC-seq fragments and RNA features for cells passing filter (both log2-transformed). (c) Correlation between MYC accessibility score and normalized RNA expression. (d) UMAP from the RNA or the ATAC-seq data (left). Log-normalized and scaled MYC RNA expression (top right) and MYC accessibility scores (bottom right) were visualized on the ATAC-seq UMAP. (e) Gene expression scores (using Seurat in R) of MYC-upregulated genes (Gene Set M6506, Molecular Signatures Database; MSigDB) across all MYC RNA quantile bins. Horizontal line marks median. Population variances for all individual cells are shown (top). P-value determined by two-sided F-test. (f) MYC expression levels of top and bottom bins (left). Normalized ATAC-seq coverages are shown (right). (g) Number of variable elements identified on COLO320-DM ecDNAs compared to chromosomal HSRs in COLO320-HSR (left). 45 variable elements were uniquely observed on ecDNA. All variable elements on ecDNA are shown on the right (y-axis shows −log10(FDR) and dot size represents log2 fold change. Five most significantly variable elements are highlighted and named based on relative position in kilobases to the MYC TSS (negative, 5’; positive, 3’). (h) Correlation between estimated MYC copy numbers and normalized log2-transformed MYC expression of all individual cells showing a high level of copy number variability. (i) Estimated MYC amplicon copy number of all cell bins. (j) Zoom-ins of the ATAC-seq coverage of each of the five most significantly variable elements identified in (g) (marked by dashed boxes). (k) Similar distributions of TSS enrichment in the high and low cell bins. (l) Mean copy number regressed, log-normalized, scaled ATAC-seq coverage of the differential peaks against mean MYC RNA (log-normalized, mean-centered, scaled) for each cell bin in orange. Same number of random non-differential peaks from the same amplicon interval and shown in grey. Error bands show 95% confidence intervals for the linear models. (m) Cumulative probability of MYC amplicon copy number distributions (mean-centered, scaled) of single-cell ATAC-seq data and DNA FISH data. P-values determined by Kolmogorov-Smirnov test (1000 bootstrap simulations).
Extended Data Figure 6. Endogenous enhancer connectome of COLO320-DM MYC ecDNA amplicon and effect of promoter sequence, cis enhancers, and BET inhibition on episomal reporter activation.
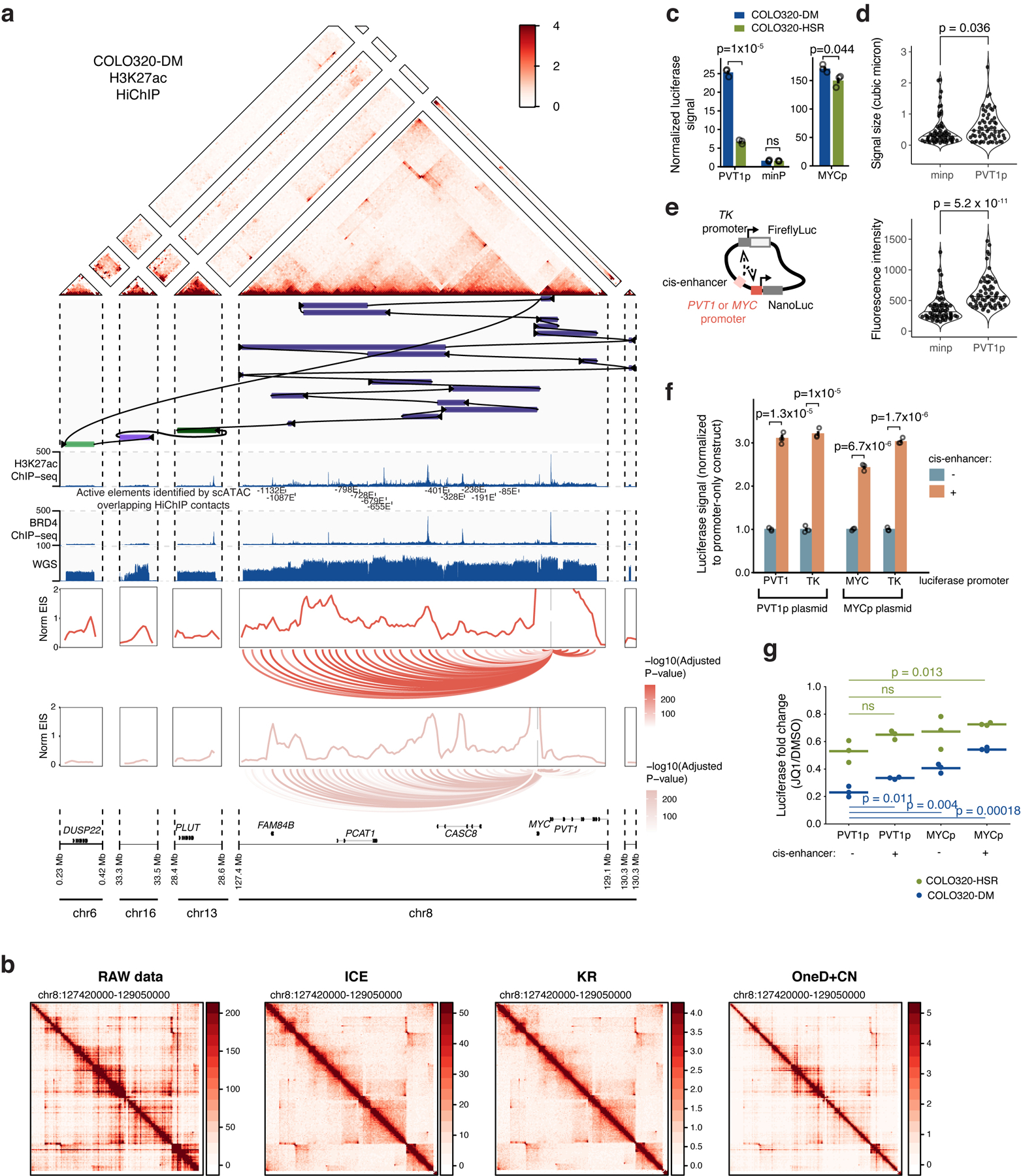
(a) Top to bottom: COLO320-DM H3K27ac HiChIP contact map (KR-normalized read counts, 10 kb resolution), reconstructed COLO320-DM amplicon, H3K27ac ChIP-seq signal, BRD4 ChIP-seq signal, WGS coverage, interaction profile of PVT1 and MYC promoters at 10kb resolution with FitHiChIP loops shown below, colored by adjusted p-value. Active elements identified by scATAC and overlapping H3K27ac HiChIP contacts named by genomic distance to MYC start site: −1132E, −1087E, −679E, −655E, −401E, −328E, −85E. (b) Comparison of HiChIP matrix normalization for COLO320-DM H3K27ac HiChIP at 10kb resolution. HiChIP signal is robust to different normalization methods. (c) Quantification of NanoLuc luciferase signal for plasmids with PVT1p-, minp-, or MYCp-driven NanoLuc reporter expression. Luciferase signal was calculated by normalizing NanoLuc readings to Firefly readings. Bar plot shows mean ± SEM. P values were calculated using a two-sided student’s t-test (n=3 biological replicates). (d) Violin plots showing mean fluorescence intensities and signal sizes of the NanoLuc reporter RNA in PVT1p-reporter and minp-reporter transfected cells. P-values were calculated a two-sided Wilcoxon test. (e) Schematic of PVT1 promoter-driven luciferase reporter plasmid with a cis-enhancer. Details of cis-enhancer are in Methods. (f) Bar plot showing luciferase signal driven by PVT1p, MYCp or the constitutive TKp with or without a cis-enhancer (mean ± SEM). All values are normalized to the corresponding promoter-only construct without a cis-enhancer. P values were calculated using a two-sided student’s t-test (n=3 biological replicates). (g) Dot plots showing fold change in luciferase signal (Firefly-normalized NanoLuc signal) in JQ1-treated over DMSO-treated COLO320-DM and COLO320-HSR cells after transfection with the PVT1p or the MYCp plasmid with or without a cis-enhancer. P values were calculated using a two-sided student’s t-test (n=3 biological replicates).
Extended Data Figure 7. Generation of monoclonal SNU16-dCas9-KRAB with reduced ecDNA fusions.
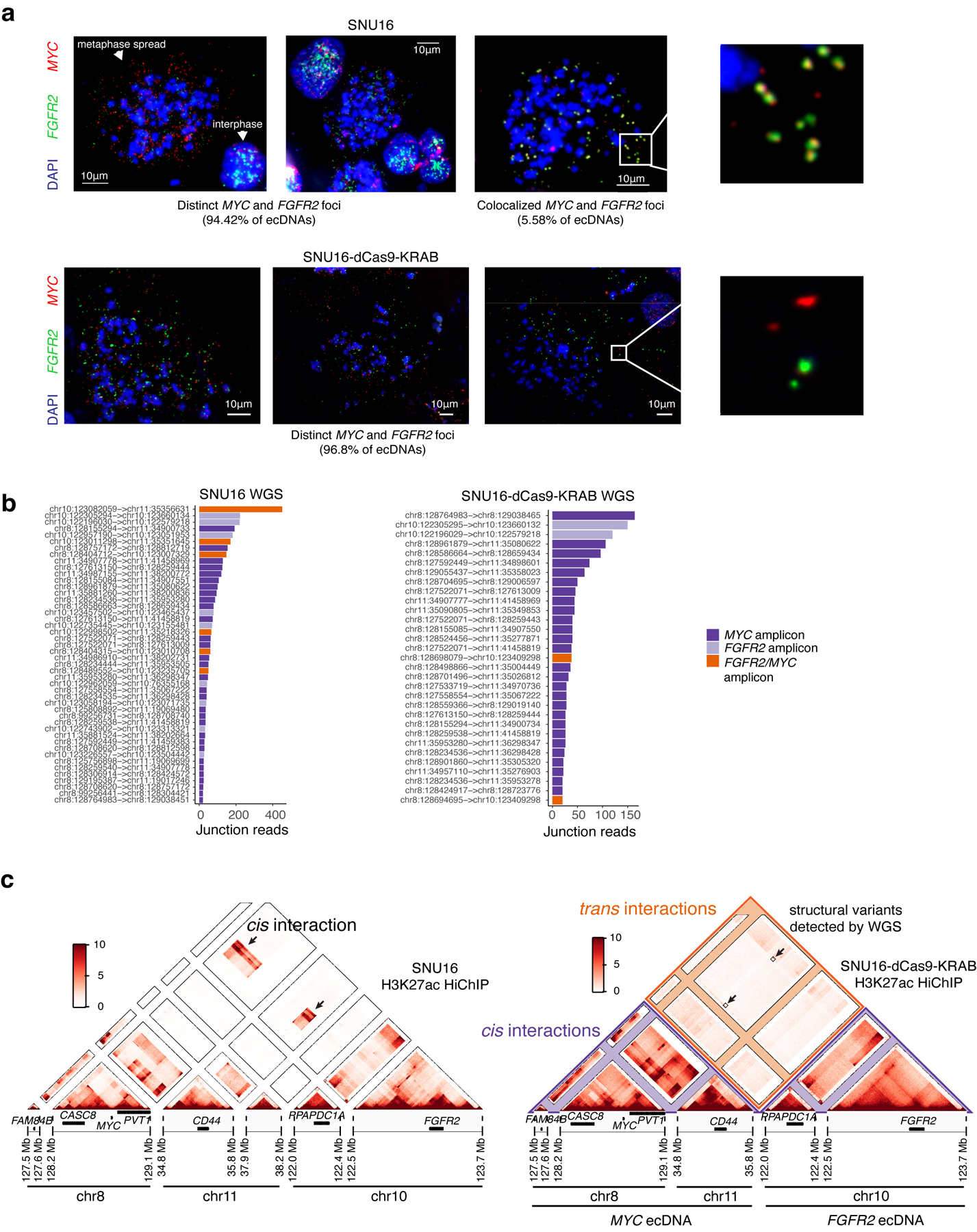
(a) Representative DNA FISH images showing extrachromosomal single-positive MYC and FGFR2 amplifications (top left and top middle) and double-positive MYC and FGFR2 amplifications in metaphase spreads in parental SNU16 cells (top right) with zoom in (top right). N = 42 cells and 8,222 ecDNAs. Representative DNA FISH images showing distinct extrachromosomal MYC and FGFR2 amplifications in metaphase spreads in SNU16-dCas9-KRAB cells (bottom). N = 29 cells and 3,893 ecDNAs. (b) Ranked plot showing number of junction reads supporting each breakpoint in AmpliconArchitect. Breakpoints are colored based on whether they span regions from the same amplicon (MYC/FGFR2) or regions from two distinct amplicons. (c) HiChIP contact matrices at 10kb resolution with KR normalization for parental SNU16 cell line (left) and SNU16-dCas9-KRAB cell line (right). Contact matrix for parental cells contains regions of increased cis contact frequency between chr8 and chr10 as indicated, as compared to SNU16-dCas9-KRAB cells with highly reduced contact cis frequency between chr8 and chr10. Regions of increased focal interaction overlapping low frequency structural rearrangements between chr8 and chr10 described in panel (a) indicated with boxes.
Extended Data Figure 8. Perturbations of ecDNA enhancers via CRISPRi revealed functional intermolecular enhancer-gene interactions.
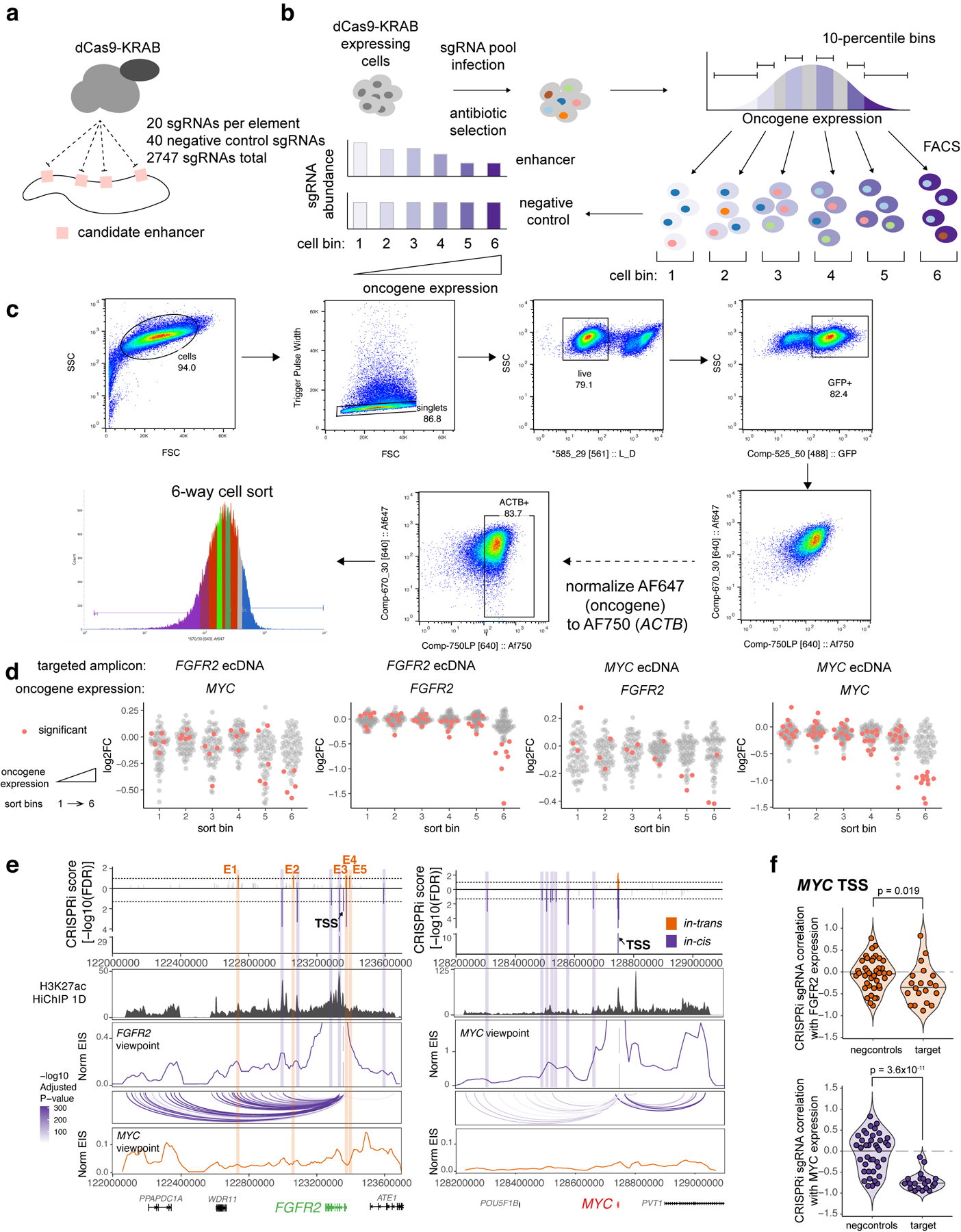
(a) CRISPRi experiments perturbing candidate enhancers in SNU16-dCas9-KRAB cells. Single-guide RNAs (sgRNAs) were designed to target candidate enhancers on FGFR2 and MYC ecDNAs based on chromatin accessibility. (b) Experimental workflow for pooled CRISPRi repression of putative enhancers. Stable SNU16-dCas9-KRAB cells were generated from a single cell clone. Cells were transduced with a lentiviral pool of sgRNAs, selected with antibiotics and oncogene RNA was assessed by flowFISH. Cells were sorted into six bins by fluorescence-activated cell sorting (FACS) based on oncogene expression. sgRNAs were quantified for cells in each bin. (c) FACS gating strategy. (d) Log2 fold changes of sgRNAs for each candidate enhancer element compared to unsorted cells for CRISPRi libraries targeting either MYC or FGFR2 ecDNAs, followed by cell sorting based on expression levels of MYC or FGFR2. Each dot represents the mean log2 fold change of 20 sgRNAs targeting a candidate element. Elements negatively correlated with oncogene expression as compared to the negative control sgRNA distributions in the same pools are marked in red. (e) Barplot showing significance of CRISPRi repression of candidate enhancer elements as in Figure 4e (top). Significant in-trans and in-cis enhancers are colored as indicated. SNU16-dCas9-KRAB H3K27ac HiChIP 1D signal track and interaction profiles of FGFR2 and MYC promoters at 10kb resolution with cis FitHiChIP loops shown below. Interaction profiles in cis shown in purple and in trans shown in orange. (f) Spearman correlations of individual sgRNAs that target MYC TSS across fluorescence bins corresponding to MYC and FGFR2 expression. P values using the lower-tailed t-test comparing target sgRNAs with negative control sgRNAs (negcontrols) are shown. Each dot represents an independent sgRNA.
Extended Data Figure 9. Intermolecular enhancers and MYC are located on distinct molecules for the vast majority of ecDNAs.
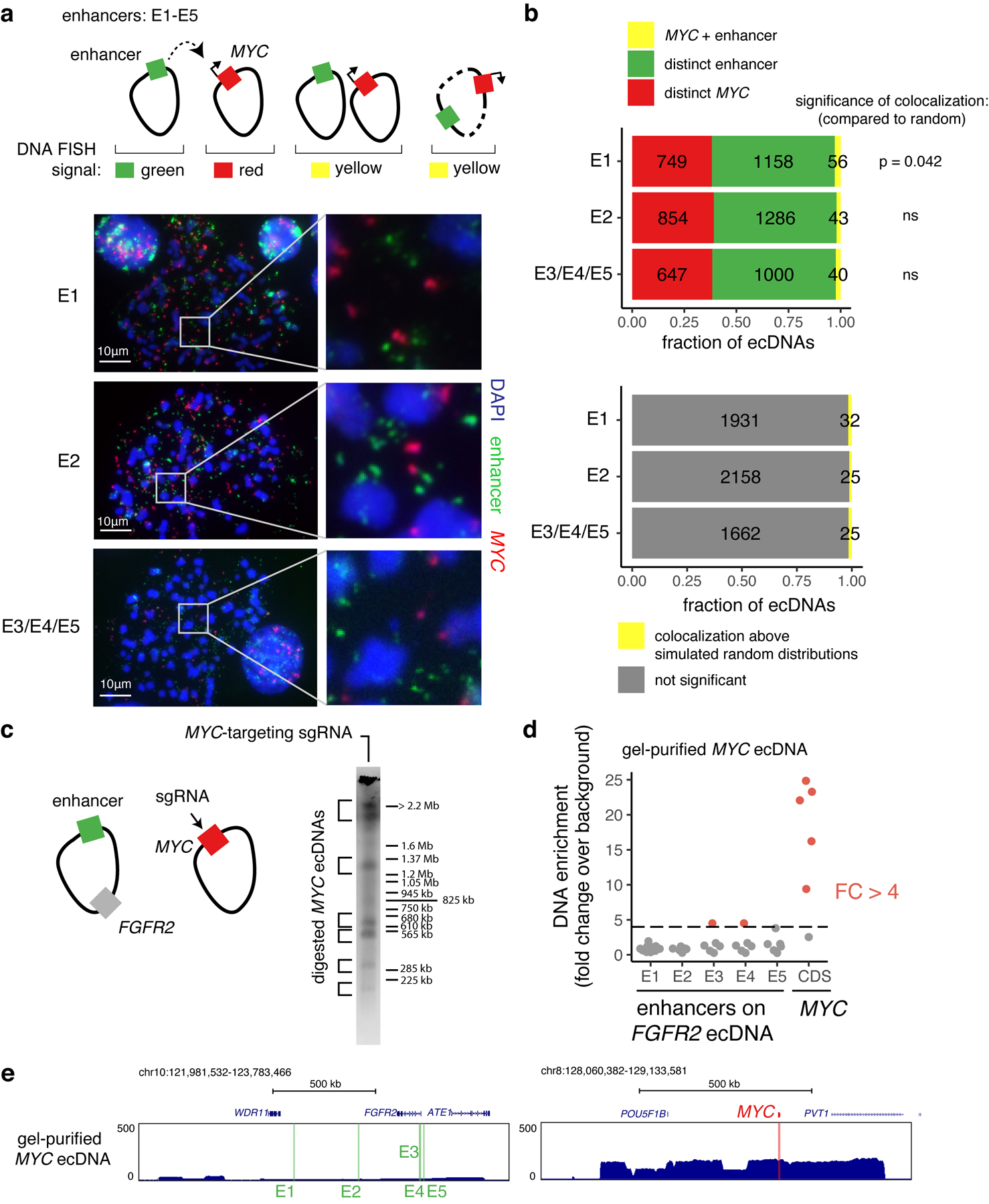
(a) Top: two-color DNA FISH on metaphase spreads for quantifying the frequency of colocalization of the MYC gene and intermolecular enhancers shown in Figure 4e. Above-random colocalization would indicate fusion events. Bottom: representative DNA FISH images. DNA FISH probes target the following hg19 genomic coordinates: E1, chr10:122635712–122782544 (RP11–95I16; n = 11 cells); E2, chr10:122973293–123129601 (RP11-57H2; n = 12 cells); E3/E4/E5, chr10:123300005–123474433 (RP11–1024G22; n = 10 cells). (b) Top: numbers of distinct and colocalized FISH signals. To estimate random colocalization, 100 simulated images were generated with matched numbers of signals and mean simulated frequencies were compared with observed colocalization. P values determined by two-sided t-test (Bonferroni-adjusted). Bottom: number of colocalized signals significantly above random chance. Colocalization above simulated random distributions is the sum of colocalized molecules in excess of random means in all FISH images in which total colocalization was above the random mean plus 95% confidence interval (100 simulated images per FISH image). (c) in-vitro Cas9 digestion of MYC-containing ecDNA in SNU16-dCas9-KRAB followed by PFGE (one independent experiment). Fragment sizes were determined based on H. wingei and S. cerevisiae ladders. Uncropped gel image is in Supplementary Figure 1. MYC CDS guide corresponds to guide B in Supplementary Table 2. (d) Enrichment of enhancer DNA sequences in isolated MYC ecDNAs bands from (c) over background (DNA isolated from a separate PFGE lane in the corresponding size range resulting from undigested genomic DNA) based on normalized reads in 5kb windows. Each dot represents DNA from a distinct gel band. Red indicates fold change above 4. (e) Sequencing track for a gel-purified MYC ecDNA showing enrichment of the MYC amplicon and depletion of the FGFR2 amplicon containing enhancers E1–E5.
Extended Data Figure 10. Reconstruction of four distinct amplicons in TR14 neuroblastoma cell line and intermolecular amplicon interaction patterns associated with H3K27ac marks.
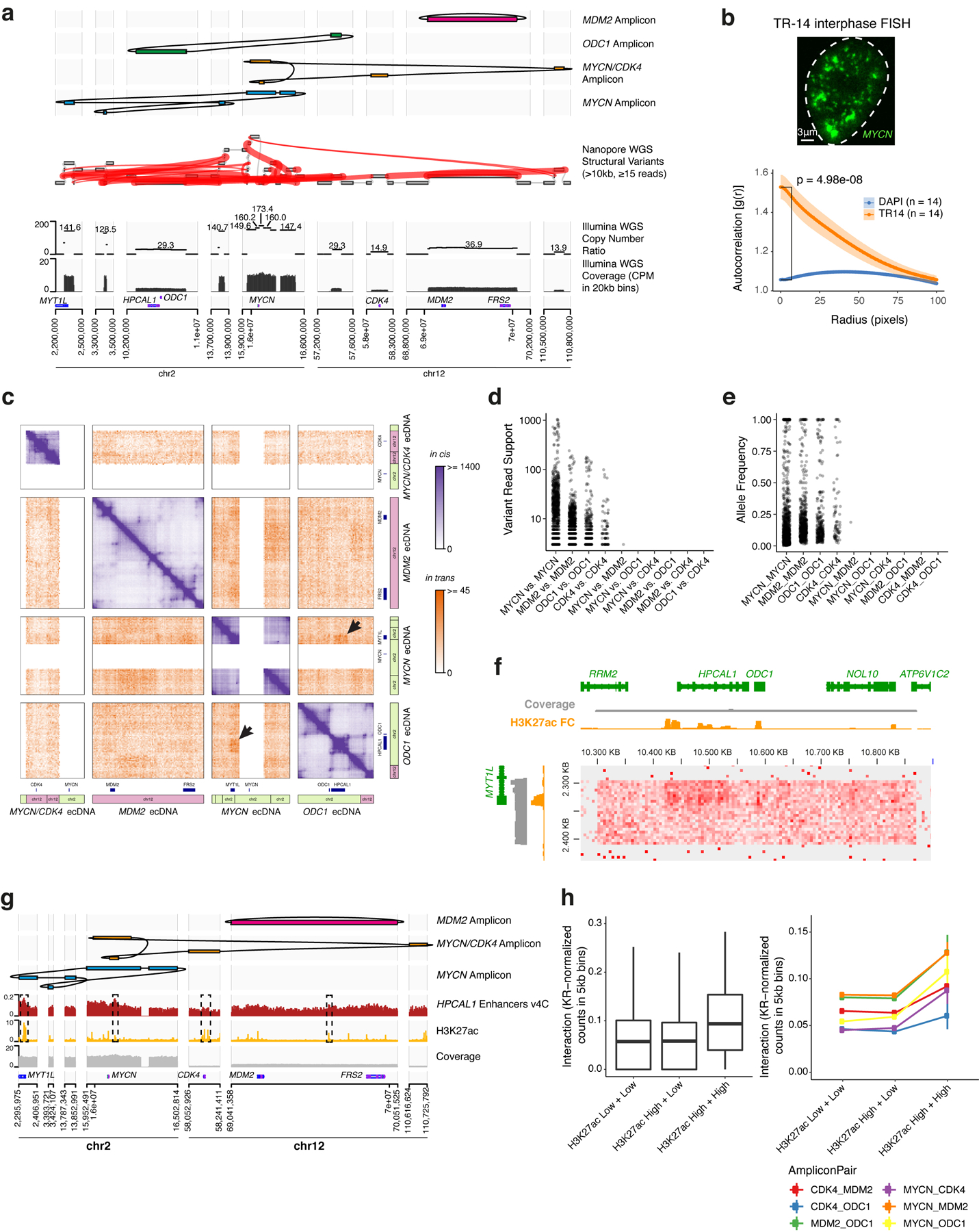
(a) Top to bottom: long read-based reconstruction of four different amplicons; genome graph with long read-based structural variants of >10kb size and >20 supporting reads indicated by red edges; copy number variation and coverage from short-read whole-genome sequencing, positions of the selected genes. (b) A representative DNA FISH image of MYCN ecDNAs in interphase TR14 cells (top) and ecDNA clustering compared to DAPI control in the same cells assessed by autocorrelation g(r) (bottom). Data are mean ± SEM (n = 14 cells). (c) Custom Hi-C map of reconstructed TR14 amplicons. The MYCN/CDK4 amplicon and the MYCN ecDNA share sequences, which prevented an unambiguous short-read mapping in these regions and appear as white areas. Trans interactions appear locally elevated between MYCN ecDNA and ODC1 amplicon (indicated by arrows). Cis and trans contact frequencies are colored as indicated. (d) Read support for structural variants identified by long read sequencing overlapping amplicons. Only one structural variant between distinct amplicons (MYCN and MDM2 amplicons) was identified with 3 supporting reads. (e) Variant allele frequency for structural variants overlapping amplicons. (f) Trans-interaction pattern between enhancers on a MYCN amplicon fragment (vertical) and an ODC1 amplicon fragment (horizontal). Short-read WGS coverage (grey), H3K27ac ChIP-seq track showing mean fold change over input in 1kb bins (yellow) and Hi-C contact map showing (KR-normalized counts in 5kb bins). (g) Top to bottom: three amplicon reconstructions, virtual 4C interaction profile of the enhancer-rich HPCAL1 locus on the ODC1 amplicon with loci on other amplicons (red), and H3K27ac ChIP-seq (fold change over input; yellow). (h) Trans interaction between different amplicons (KR-normalized counts in 5kb bins) depending on H3K27ac signal of the interaction loci (left; box center line, median; box limits, upper and lower quartiles; box whiskers, 1.5x interquartile range). Trans interaction (KR-normalized counts in 5kb bins) separated by amplicon pair (right). H3K27ac High vs. Low denotes at least vs. less than 3-fold mean enrichment over input in 5kb bins. N = 114,636 H3K27ac Low + Low pairs, n = 11,990 H3K27ac High + Low pairs, n = 296 H3K27ac High + High pairs.
Supplementary Material
Acknowledgements
We thank members of the Chang, Liu, Mischel, and Bafna laboratories for discussions, R. Zermeno, M. Weglarz and L. Nichols at the Stanford Shared FACS Facility for assistance with cell sorting experiments, X. Ji, D. Wagh and J. Coller at the Stanford Functional Genomics Facility for assistance with high-throughput sequencing, and A. Pang of Bionano Genomics for assistance with optical mapping. H.Y.C. was supported by NIH R35-CA209919 and RM1-HG007735. K.L.H. was supported by a Stanford Graduate Fellowship. K.E.Y. was supported by the National Science Foundation Graduate Research Fellowship Program (NSF DGE-1656518), a Stanford Graduate Fellowship, and a NCI Predoctoral to Postdoctoral Fellow Transition Award (NIH F99CA253729). Cell sorting for this project was done on instruments in the Stanford Shared FACS Facility. Sequencing was performed by the Stanford Functional Genomics Facility (supported by NIH grants S10OD018220 and 1S10OD021763). Microscopy was performed on instruments in the UCSD Microscopy Core (supported by NINDS NS047101). A.G.H. is supported by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) - 398299703 and the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation programme (grant agreement No. 949172). Z.L. is a Janelia Group Leader and H.Y.C. and R.T. are Investigators of the Howard Hughes Medical Institute.
Footnotes
Code Availability
Custom code used in this study is available at https://github.com/ChangLab/ecDNA-hub-code-2021.
Competing Interests
H.Y.C. is a co-founder of Accent Therapeutics, Boundless Bio, Cartography Biosciences, and an advisor of 10x Genomics, Arsenal Biosciences, and Spring Discovery. P.S.M. is a co-founder of Boundless Bio, Inc. He has equity and chairs the scientific advisory board, for which he is compensated. V.B. is a co-founder and advisor of Boundless Bio. A.T.S. is a founder of Immunai and Cartography Biosciences.
Data Availability
ChIP-seq, HiChIP, Hi-C, RNA-seq, and single cell multiome ATAC + gene expression data generated in this study have been deposited in GEO and are available under accession number GSE159986. Nanopore sequencing data, whole genome sequencing data, sgRNA sequencing data, and targeted ecDNA sequencing data following CRISPR-Cas9 digestion and PFGE generated in this study has been deposited in SRA and are available under accession number PRJNA670737. Optical mapping data generated in this study has been deposited in GenBank with Bioproject code PRJNA731303. The following publicly available data was also used in this study: TR14 H3K27ac ChIP-seq (GEO: GSE90683)93; COLO320-DM, COLO320-HSR and PC3 WGS (SRA: PRJNA506071)1; SNU16 WGS (SRA: PRJNA523380)60; HK359 WGS (SRA: PRJNA338012)6. Microscopy image files are available on figshare at https://doi.org/10.6084/m9.figshare.c.5624713.
REFERENCES
- 1.Wu S et al. Circular ecDNA promotes accessible chromatin and high oncogene expression. Nature 1–5 (2019) doi: 10.1038/s41586-019-1763-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Gorkin DU, Leung D & Ren B The 3D Genome in Transcriptional Regulation and Pluripotency. Cell Stem Cell 14, 762–775 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Zheng H & Xie W The role of 3D genome organization in development and cell differentiation. Nature Reviews Molecular Cell Biology 20, 535–550 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Bailey C, Shoura MJ, Mischel PS & Swanton C Extrachromosomal DNA – relieving heredity constraints, accelerating tumour evolution. Annals of Oncology (2020) doi: 10.1016/j.annonc.2020.03.303. [DOI] [PubMed] [Google Scholar]
- 5.Kim H et al. Extrachromosomal DNA is associated with oncogene amplification and poor outcome across multiple cancers. Nature Genetics 52, 891–897 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Turner KM et al. Extrachromosomal oncogene amplification drives tumour evolution and genetic heterogeneity. Nature 543, 122–125 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Verhaak RGW, Bafna V & Mischel PS Extrachromosomal oncogene amplification in tumour pathogenesis and evolution. Nature Reviews Cancer 19, 283 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cox D, Yuncken C & Spriggs, ArthurI. MINUTE CHROMATIN BODIES IN MALIGNANT TUMOURS OF CHILDHOOD. The Lancet 286, 55–58 (1965). [DOI] [PubMed] [Google Scholar]
- 9.van der Bliek AM, Lincke CR & Borst P Circular DNA of 3T6R50 double minute chromosomes. Nucleic Acids Research 16, 4841–4851 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hamkalo BA, Farnham PJ, Johnston R & Schimke RT Ultrastructural features of minute chromosomes in a methotrexate-resistant mouse 3T3 cell line. Proceedings of the National Academy of Sciences 82, 1126–1130 (1985). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Maurer BJ, Lai E, Hamkalo BA, Hood L & Attardi G Novel submicroscopic extrachromosomal elements containing amplified genes in human cells. Nature 327, 434–437 (1987). [DOI] [PubMed] [Google Scholar]
- 12.VanDevanter DR, Piaskowski VD, Casper JT, Douglass EC & Von Hoff DD Ability of Circular Extrachromosomal DNA Molecules to Carry Amplified MYCN Protooncogenes in Human Neuroblastomas In Vivo. J Natl Cancer Inst 82, 1815–1821 (1990). [DOI] [PubMed] [Google Scholar]
- 13.Nathanson DA et al. Targeted Therapy Resistance Mediated by Dynamic Regulation of Extrachromosomal Mutant EGFR DNA. Science 343, 72–76 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ståhl F, Wettergren Y & Levan G Amplicon structure in multidrug-resistant murine cells: a nonrearranged region of genomic DNA corresponding to large circular DNA. Molecular and Cellular Biology 12, 1179–1187 (1992). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vicario R et al. Patterns of HER2 Gene Amplification and Response to Anti-HER2 Therapies. PLOS ONE 10, e0129876 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Carroll SM et al. Double minute chromosomes can be produced from precursors derived from a chromosomal deletion. Molecular and Cellular Biology 8, 1525–1533 (1988). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kitajima K, Haque M, Nakamura H, Hirano T & Utiyama H Loss of Irreversibility of Granulocytic Differentiation Induced by Dimethyl Sulfoxide in HL-60 Sublines with a Homogeneously Staining Region. Biochemical and Biophysical Research Communications 288, 1182–1187 (2001). [DOI] [PubMed] [Google Scholar]
- 18.Quinn LA, Moore GE, Morgan RT & Woods LK Cell Lines from Human Colon Carcinoma with Unusual Cell Products, Double Minutes, and Homogeneously Staining Regions. Cancer Research 39, 4914–4924 (1979). [PubMed] [Google Scholar]
- 19.Storlazzi CT et al. Gene amplification as double minutes or homogeneously staining regions in solid tumors: Origin and structure. Genome Res. 20, 1198–1206 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wahl GM The Importance of Circular DNA in Mammalian Gene Amplification. Cancer Res 49, 1333–1340 (1989). [PubMed] [Google Scholar]
- 21.Kumar P et al. ATAC-seq identifies thousands of extrachromosomal circular DNA in cancer and cell lines. Science Advances 6, eaba2489 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Morton AR et al. Functional Enhancers Shape Extrachromosomal Oncogene Amplifications. Cell 0, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Helmsauer K et al. Enhancer hijacking determines extrachromosomal circular MYCN amplicon architecture in neuroblastoma. Nature Communications 11, 5823 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Itoh N & Shimizu N DNA replication-dependent intranuclear relocation of double minute chromatin. Journal of Cell Science 111 ( Pt 22), 3275–3285 (1998). [DOI] [PubMed] [Google Scholar]
- 25.Kanda T, Sullivan KF & Wahl GM Histone–GFP fusion protein enables sensitive analysis of chromosome dynamics in living mammalian cells. Current Biology 8, 377–385 (1998). [DOI] [PubMed] [Google Scholar]
- 26.Oobatake Y & Shimizu N Double-strand breakage in the extrachromosomal double minutes triggers their aggregation in the nucleus, micronucleation, and morphological transformation. Genes, Chromosomes and Cancer 59, 133–143 (2020). [DOI] [PubMed] [Google Scholar]
- 27.Beliveau BJ et al. Versatile design and synthesis platform for visualizing genomes with Oligopaint FISH probes. Proceedings of the National Academy of Sciences 109, 21301–21306 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Koche RP et al. Extrachromosomal circular DNA drives oncogenic genome remodeling in neuroblastoma. Nature Genetics 1–6 (2019) doi: 10.1038/s41588-019-0547-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parker SCJ et al. Chromatin stretch enhancer states drive cell-specific gene regulation and harbor human disease risk variants. PNAS 110, 17921–17926 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Whyte WA et al. Master Transcription Factors and Mediator Establish Super-Enhancers at Key Cell Identity Genes. Cell 153, 307–319 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lovén J et al. Selective Inhibition of Tumor Oncogenes by Disruption of Super-Enhancers. Cell 153, 320–334 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Filippakopoulos P et al. Selective inhibition of BET bromodomains. Nature 468, 1067–1073 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sabari BR et al. Coactivator condensation at super-enhancers links phase separation and gene control. Science 361, eaar3958 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ren C et al. Spatially constrained tandem bromodomain inhibition bolsters sustained repression of BRD4 transcriptional activity for TNBC cell growth. PNAS 115, 7949–7954 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Deshpande V et al. Exploring the landscape of focal amplifications in cancer using AmpliconArchitect. Nat Commun 10, 1–14 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Luebeck J et al. AmpliconReconstructor integrates NGS and optical mapping to resolve the complex structures of focal amplifications. Nat Commun 11, 4374 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schwab M, Klempnauer KH, Alitalo K, Varmus H & Bishop M Rearrangement at the 5’ end of amplified c-myc in human COLO 320 cells is associated with abnormal transcription. Mol Cell Biol 6, 2752–2755 (1986). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.L’Abbate A et al. Genomic organization and evolution of double minutes/homogeneously staining regions with MYC amplification in human cancer. Nucleic Acids Res 42, 9131–9145 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hann SR, King MW, Bentley DL, Anderson CW & Eisenman RN A non-AUG translational initiation in c-myc exon 1 generates an N-terminally distinct protein whose synthesis is disrupted in Burkitt’s lymphomas. Cell 52, 185–195 (1988). [DOI] [PubMed] [Google Scholar]
- 40.Carramusa L et al. The PVT-1 oncogene is a Myc protein target that is overexpressed in transformed cells. Journal of Cellular Physiology 213, 511–518 (2007). [DOI] [PubMed] [Google Scholar]
- 41.Cho SW et al. Promoter of lncRNA Gene PVT1 Is a Tumor-Suppressor DNA Boundary Element. Cell 173, 1398–1412.e22 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Tolomeo D, Agostini A, Visci G, Traversa D & Storlazzi CT PVT1: A long non-coding RNA recurrently involved in neoplasia-associated fusion transcripts. Gene 779, 145497 (2021). [DOI] [PubMed] [Google Scholar]
- 43.Mumbach MR et al. HiChIP: efficient and sensitive analysis of protein-directed genome architecture. Nature Methods 13, 919 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fulco CP et al. Activity-by-contact model of enhancer–promoter regulation from thousands of CRISPR perturbations. Nature Genetics 51, 1664–1669 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Park J et al. A reciprocal regulatory circuit between CD44 and FGFR2 via c-myc controls gastric cancer cell growth. Oncotarget 7, 28670–28683 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Furlong EEM & Levine M Developmental enhancers and chromosome topology. Science 361, 1341–1345 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhu Y et al. Oncogenic extrachromosomal DNA functions as mobile enhancers to globally amplify chromosomal transcription. Cancer Cell 39, 694–707.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Xue KS, Hooper KA, Ollodart AR, Dingens AS & Bloom JD Cooperation between distinct viral variants promotes growth of H3N2 influenza in cell culture. eLife 5, e13974 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vignuzzi M, Stone JK, Arnold JJ, Cameron CE & Andino R Quasispecies diversity determines pathogenesis through cooperative interactions in a viral population. Nature 439, 344–348 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Henssen A et al. Targeting MYCN-Driven Transcription By BET-Bromodomain Inhibition. Clin Cancer Res 22, 2470–2481 (2016). [DOI] [PubMed] [Google Scholar]
- 51.Xie L et al. 3D ATAC-PALM: super-resolution imaging of the accessible genome. Nature Methods 1–7 (2020) doi: 10.1038/s41592-020-0775-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ambros PF et al. International consensus for neuroblastoma molecular diagnostics: report from the International Neuroblastoma Risk Group (INRG) Biology Committee. British Journal of Cancer 100, 1471–1482 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Balaban-Malenbaum G & Gilbert F Double minute chromosomes and the homogeneously staining regions in chromosomes of a human neuroblastoma cell line. Science 198, 739–741 (1977). [DOI] [PubMed] [Google Scholar]
- 54.Marrano P, Irwin MS & Thorner PS Heterogeneity of MYCN amplification in neuroblastoma at diagnosis, treatment, relapse, and metastasis. Genes Chromosomes Cancer 56, 28–41 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Villamón E et al. Genetic instability and intratumoral heterogeneity in neuroblastoma with MYCN amplification plus 11q deletion. PLoS One 8, e53740 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Schindelin J et al. Fiji: an open-source platform for biological-image analysis. Nature Methods 9, 676–682 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rajkumar U et al. EcSeg: Semantic Segmentation of Metaphase Images Containing Extrachromosomal DNA. iScience 21, 428–435 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Veatch SL et al. Correlation Functions Quantify Super-Resolution Images and Estimate Apparent Clustering Due to Over-Counting. PLOS ONE 7, e31457 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bolger AM, Lohse M & Usadel B Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Ghandi M et al. Next-generation characterization of the Cancer Cell Line Encyclopedia. Nature 569, 503–508 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Normanno D et al. Probing the target search of DNA-binding proteins in mammalian cells using TetR as model searcher. Nature Communications 6, 7357 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Mirkin EV, Chang FS & Kleckner N Protein-Mediated Chromosome Pairing of Repetitive Arrays. Journal of Molecular Biology 426, 550–557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Grimm JB et al. A general method to optimize and functionalize red-shifted rhodamine dyes. Nature Methods 17, 815–821 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B & Salzberg SL Fast gapped-read alignment with Bowtie 2. Nature Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zhang Y et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramírez F et al. deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sedlazeck FJ et al. Accurate detection of complex structural variations using single-molecule sequencing. Nature Methods 15, 461–468 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Overhauser J Encapsulation of Cells in Agarose Beads. in Pulsed-Field Gel Electrophoresis: Protocols, Methods, and Theories (eds. Burmeister M & Ulanovsky L) 129–134 (Humana Press, 1992). doi: 10.1385/0-89603-229-9:129. [DOI] [PubMed] [Google Scholar]
- 69.Picelli S et al. Tn5 transposase and tagmentation procedures for massively scaled sequencing projects. Genome Res. 24, 2033–2040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Corces MR et al. An improved ATAC-seq protocol reduces background and enables interrogation of frozen tissues. Nature Methods 14, 959–962 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Talevich E, Shain AH, Botton T & Bastian BC CNVkit: Genome-Wide Copy Number Detection and Visualization from Targeted DNA Sequencing. PLoS Comput Biol 12, e1004873 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Raeisi Dehkordi S, Luebeck J & Bafna V FaNDOM: Fast nested distance-based seeding of optical maps. Patterns 2, 100248 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Haas BJ et al. Accuracy assessment of fusion transcript detection via read-mapping and de novo fusion transcript assembly-based methods. Genome Biology 20, 213 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Hahne F & Ivanek R Visualizing Genomic Data Using Gviz and Bioconductor. in Statistical Genomics: Methods and Protocols (eds. Mathé E & Davis S) 335–351 (Springer, 2016). doi: 10.1007/978-1-4939-3578-9_16. [DOI] [PubMed] [Google Scholar]
- 75.Butler A, Hoffman P, Smibert P, Papalexi E & Satija R Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nature Biotechnology 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Granja JM et al. ArchR: An integrative and scalable software package for single-cell chromatin accessibility analysis. bioRxiv 2020.04.28.066498 (2020) doi: 10.1101/2020.04.28.066498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Satpathy AT et al. Massively parallel single-cell chromatin landscapes of human immune cell development and intratumoral T cell exhaustion. Nat Biotechnol 37, 925–936 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Mumbach MR et al. Enhancer connectome in primary human cells identifies target genes of disease-associated DNA elements. Nat. Genet 49, 1602–1612 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Mumbach MR et al. HiChIRP reveals RNA-associated chromosome conformation. Nature Methods 16, 489–492 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Servant N et al. HiC-Pro: an optimized and flexible pipeline for Hi-C data processing. Genome Biol. 16, 259 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Bhattacharyya S, Chandra V, Vijayanand P & Ay F Identification of significant chromatin contacts from HiChIP data by FitHiChIP. Nature Communications 10, 4221 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Rao SSP et al. A 3D Map of the Human Genome at Kilobase Resolution Reveals Principles of Chromatin Looping. Cell 159, 1665–1680 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Vidal E et al. OneD: increasing reproducibility of Hi-C samples with abnormal karyotypes. Nucleic Acids Research 46, e49–e49 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Flynn RA et al. Discovery and functional interrogation of SARS-CoV-2 RNA-host protein interactions. Cell (2021) doi: 10.1016/j.cell.2021.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biology 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Li H & Durbin R Fast and accurate long-read alignment with Burrows–Wheeler transform. Bioinformatics 26, 589–595 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Scheinin I et al. DNA copy number analysis of fresh and formalin-fixed specimens by shallow whole-genome sequencing with identification and exclusion of problematic regions in the genome assembly. Genome Res. 24, 2022–2032 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Hadi K et al. Distinct Classes of Complex Structural Variation Uncovered across Thousands of Cancer Genome Graphs. Cell 183, 197–210.e32 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Blumrich A et al. The FRA2C common fragile site maps to the borders of MYCN amplicons in neuroblastoma and is associated with gross chromosomal rearrangements in different cancers. Hum Mol Genet 20, 1488–1501 (2011). [DOI] [PubMed] [Google Scholar]
- 90.Gogolin S et al. CDK4 inhibition restores G₁-S arrest in MYCN-amplified neuroblastoma cells in the context of doxorubicin-induced DNA damage. Cell Cycle 12, 1091–1104 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Durand NC et al. Juicer Provides a One-Click System for Analyzing Loop-Resolution Hi-C Experiments. cels 3, 95–98 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Knight PA & Ruiz D A fast algorithm for matrix balancing. IMA Journal of Numerical Analysis 33, 1029–1047 (2013). [Google Scholar]
- 93.Boeva V et al. Heterogeneity of neuroblastoma cell identity defined by transcriptional circuitries. Nature Genetics 49, 1408–1413 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
ChIP-seq, HiChIP, Hi-C, RNA-seq, and single cell multiome ATAC + gene expression data generated in this study have been deposited in GEO and are available under accession number GSE159986. Nanopore sequencing data, whole genome sequencing data, sgRNA sequencing data, and targeted ecDNA sequencing data following CRISPR-Cas9 digestion and PFGE generated in this study has been deposited in SRA and are available under accession number PRJNA670737. Optical mapping data generated in this study has been deposited in GenBank with Bioproject code PRJNA731303. The following publicly available data was also used in this study: TR14 H3K27ac ChIP-seq (GEO: GSE90683)93; COLO320-DM, COLO320-HSR and PC3 WGS (SRA: PRJNA506071)1; SNU16 WGS (SRA: PRJNA523380)60; HK359 WGS (SRA: PRJNA338012)6. Microscopy image files are available on figshare at https://doi.org/10.6084/m9.figshare.c.5624713.