

Abstract
The HIV-1 Capsid (CA) is considered as a promising target for the development of potent antiviral drugs, due to its multiple roles during the viral life cycle. Herein, we report the design, synthesis, and antiviral activity evaluation of series of novel phenylalanine derivatives as HIV-1 CA protein inhibitors. Among them, 4-methoxy-N-methylaniline substituted phenylalanine (II-13c) and indolin-5-amine substituted phenylalanine (V-25i) displayed exceptional anti-HIV-1 activity with the EC50 value of 5.14 and 2.57 μM respectively, which is slightly weaker than that of lead compound PF-74 (EC50 = 0.42 μM). Besides, surface plasmon resonance (SPR) binding assay demonstrated II-13c and V-25i prefer to combine with CA hexamer rather than monomer, which is similar to PF-74. Subsequently, molecular dynamics simulation (MD) revealed potential interactions between representative compounds with HIV-1 CA hexamer. Overall, this work laid a solid foundation for further structural optimization to discover novel promising HIV-1 CA inhibitors.
Keywords: HIV-1 Capside Protein, PF-74 derivative, Scaffold Hopping, HIV-1 CA inhibitors
1. Introduction
Acquired immune deficiency syndrome (AIDS), mainly caused by the human immunodeficiency virus type 1 (HIV-1), still remains a serious threat to worldwide public health1. Although combination antiretroviral therapy (cART) could suppress the viral load and improve the life quality of the patient living with HIV, the adverse side effects and the emergence of drug resistance etc. restrict its application2,3. Nowadays, HIV-1 infection cannot be completely cured. Consequently, it’s of great significance to continue exploring novel anti-HIV-1 drugs, especially for the unexploited targets with unique mechanism4.
HIV-1 CA is an asymmetric fullerene-shaped cone comprised of about 1500 CA monomer, which is obtained by cutting the gag-pol precursor protein5. CA monomer is composed of an N-terminal domain (NTD, residues 1 to 145) and a C-terminal domain (CTD, residues 150 to 231), which are connected by a flexible linker6,7. HIV-1 CA protein plays multiple roles at the early (uncoating, reverse transcription, nuclear import, integration, etc) and late stages (assembly and maturation) of HIV-1 life cycle8,9. At the early stage, CA protein could interact with host factors, such as cyclophilin A (CypA), nucleoporin 153 (NUP153) and CPSF6 (cleavage and polyadenylation specific factor 6), TNPO3 (transportin-3) and NUP358, promoting the process of uncoating. This is essencial for viral pre-integration complex entrying into the nuclear and escape from host immune surveillance10–12. At the late stage, the recombinant capsid encapsulates the viral gene RNA and gag-pol precursor protein, which are essential for the release of infectious viral particles8. Herein, HIV-1 CA is considered as a promising but underexploited target for the development of small-molecules anti-HIV-1 drugs13–17.
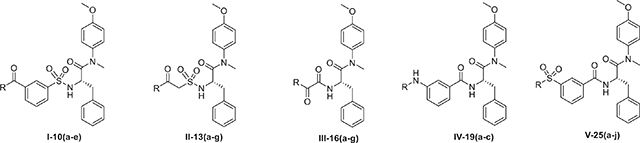
Currently, a series of structurally diverse HIV-1 CA inhibitors has been identified18–22. Among them, PF-74 has attracted a great deal of attention and has been studied extensively23. PF-74 consists of a phenylalanine core targeting the CA NTD, an indole ring targeting the CA CTD and a flexible linker in-between (Figure. 1a)5,24,25. Phenylalanine core could form hydrogen-bond or hydrophobic interactions with Met66, Leu69, Leu56, Asn57, Asn53, Lys70 of the CA NTD, the methylindole could form hydrogen-bond with Gln63, Tyr169 and form a cation-pi interaction with Arg173 and Lys182 in the CTD of the adjacent protomer7,25,26. At the early stage of HIV-1 replication, PF-74 interacts with CA competitively with host factors CPSF6 and NUP153, which disturbs viral uncoating, reverse transcription, nuclear input and integration. At the late stage, PF-74 could interfere with the normal assembly process of CA, which is extremely unfavorable for the stability of CA and the maturation of virus8,23,25. However, relatively low anti-HIV-1 activity (EC50 = 0.45 μM) and poor metabolic stability (t1/2 = 1.3 min) emphasized the significance of continuing to optimize it23,27,28. In our preceding work, lots of structurally novel PF-74 derivatives have been discovered, but they exhibited micromolar antiviral activity (Figure. 1b)29–33. Consequently, the need for continuing identify potential PF-74 derivatives remains. And it is noteworthy that the phenylalanine core is essential for the maintenance of antiviral activity. The linker region and indole substituents of PF-74, which targeting the protein–protein solvent interface, has significant potential to be further modified34. In this work, we continued focusing on the indole substituents and the linker of PF-74, so as to further enrich the structure–activity relationships (SARs) at this section35,36. We retained the privileged phenylalanine fragment, and then we used scaffold hopping strategy to replace the linker with 3-sulfamoylbenzoic acid, 2-sulfamoylacetic acid, 2-amino-2-oxoacetic acid, 3-aminobenzamide or 3-carbamoylbenzenesulfonyl chloride to fully explore the structure–activity relationship of linker. Finally, these key intermediates react with diverse amine fragments to obtain Series I-V respectively (Figure. 1c). As a result, Series I-V, the total of 32 phenylalanine derivatives was obtained.
Fig. 1.

The design of novel phenylalanine derivatives as HIV-1 CA inhibitors (a) Structure and the binding mode of PF-74 in the NTD-CTD interface of CA protein hexamer (PDB code: 5HGL). Red dashed lines indicate H-bond interactions. (b) Several reported phenylalanine derivatives as HIV-1 CA protein inhibitors in our lab. (c) Target compounds designed in this work. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
Herein, we report the design, synthesis and antiviral activity evaluation of 32 novel structural phenylalanine derivatives which divided into five sub-series. Furthermore, we demonstrated that II-13c and V-25i could interact with CA by surface plasmon resonance (SPR) binding assays. In addition, molecular dynamics simulation (MD) of representative compounds II-13c and V-25i was performed to explore the binding mode of CA hexamer. Briefly, this study enriched the SARs of PF-74 derivatives, which would be useful for finding novel and potent HIV-1 CA inhibitors.
2. Results and discussion
2.1. Chemistry
As shown in Scheme 1, (tert-butoxycarbonyl)-L-phenylalanine (5) was selected as the starting material, followed by reacting with 4-methoxy-N-methylaniline to obtain 6, then removal of Boc group of 6 under the trifluoroacetic acid yielded key intermediate 7.
Scheme 1.

The synthetic route of key intermediate 7. Reagents and Conditions: (i) 4-methoxy-N-methylaniline, HATU, DIEA, CH2Cl2, r.t., 8 h; (ii) CF3COOH, CH2Cl2, r.t., 4 h.
Subsequently, as illustrated in Scheme 2, 7 reacted with methyl 3-(chlorosulfonyl)-benzoate or methyl 2-(chlorosulfonyl)-acetate or methyl 2-chloro-2-oxoacetate or 3-nitrobenzoic acid to obtain intermediate 8, 11, 14 or 17, respectively. Then 8, 11 or 14 underwent ester hydrolysis to obtain free acids, which reacted with diverse amine fragments by using HATU and DIEA to produce target compounds (series I, II and III). Then 17 reacted with SnCl2·2H2O to reduce nitro to amino and subsequently, compound 18 was obtained, which could react with sulfonyl chloride substituents to generate the target compounds (series IV).
Scheme 2.

The synthetic route of phenylalanine derivatives (series I-IV). Reagents and Conditions: (i) methyl 3-chlorosulfonylbenzoate or methyl 2-(chlorosulfonyl) acetate or methyl chlorooxoacetate or 3-nitrobenzoyl chloride or amine fragments, TEA, CH2Cl2, 0°C, 4 h; (ii) NaOH, THF/H2O (V:V = 1:1), r.t., 2 h; (iii) amine fragments, HATU, DIEA, CH2Cl2, r.t., 8 h; (iv) SnCl2·2H2O, EtOH, N2, r.t., 8 h.(v) sulfonyl chloride substituents, TEA, CH2Cl2, 0 °C, 4 h.
The synthetic route of series V is shown in Scheme 3. Firstly, 20 reacted with diverse amine fragments in the presence of TEA to obtain 21. Then, their ester bonds were hydrolyzed to get intermediate 22. Next, 22 or commercially available 23, 24 reacted with 7 to gain the target products (series V) by using HATU and DIEA.
Scheme 3.

The synthetic route of phenylalanine derivatives (series v). (i) methyl 3-chlorosulfonylbenzoate or methyl 2-(chlorosulfonyl)acetate or methyl chlorooxoacetate or 3-nitrobenzoyl chloride or amine fragments, TEA, CH2Cl2, 0°C, 4 h; (ii) NaOH, THF/H2O (V:V = 1:1), r.t., 2 h; (iii) 7, HATU, DIEA, CH2Cl2, r.t., 8 h.
2.2. Biological activity
All novel PF-74 derivatives were tested for their antiviral activity and cytotoxicity in MT-4 cell which infected with HIV-1 N L4–3 Nanoluc-sec virus. PF-74 was selected as a positive control. The EC50, evaluated by luciferase gene expression assay37, CC50 and SI which is the ratio of CC50/EC50 are described in Table 1.
Table 1.
Anti-HIV-1 activity and cytotoxicity of the novel phenylalanine derivatives Series I-V.
| ||||
|---|---|---|---|---|
|
| ||||
| Compounds | R | EC50a (μM) | CC50b (μM) | SIc |
|
| ||||
| I-10a |
|
> 9.85 | > 9.85 | NDe |
| I-10b |
|
> 9.30 | > 9.30 | NDe |
| I-10c |
|
> 8.76 | > 8.76 | NDe |
| I-10d |
|
> 8.51 | > 8.51 | NDe |
| I-10e |
|
> 8.32 | > 8.32 | NDe |
| II-13a |
|
8.09 ± 2.76 | > 9.87 | > 1.22 |
| II-13b |
|
> 9.28 | > 9.28 | NDe |
| II-13c |
|
5.14 ± 1.62 | >9.51 | > 1.85 |
| II-13d |
|
> 9.3 | > 9.3 | NDe |
| II-13e |
|
> 9.66 | > 9.66 | NDe |
| II-13f |
|
> 9.87 | > 9.87 | NDe |
| II-13 g |
|
> 9.09 | > 9.05 | NDe |
| III-16a |
|
> 10.23 | > 10.23 | NDe |
| III-16b |
|
> 10.26 | > 10.26 | NDe |
| III-16c |
|
> 10.51 | > 10.51 | NDe |
| III-16d |
|
> 8.81 | > 8.81 | NDe |
| III-16e |
|
> 10.95 | > 10.95 | NDe |
| III-16f |
|
> 10.49 | > 10.23 | NDe |
| III-16 g |
|
> 9.95 | > 9.95 | NDe |
| IV-19a |
|
> 8.91 | > 8.91 | NDe |
| IV-19b |
|
> 7.97 | > 7.97 | NDe |
| IV-19c |
|
> 8.63 | > 8.63 | NDe |
| V-25a |
|
8.68 ± 2.62 | > 10.09 | > 1.12 |
| V-25b |
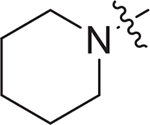
|
4.85 ± 1.53 | > 9.03 | > 1.86 |
| V-25c |
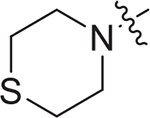
|
5.06 ± 1.43 | > 9.33 | > 1.84 |
| V-25d |
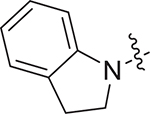
|
> 8.78 | > 8.78 | NDe |
| V-25e |
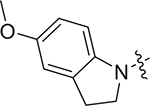
|
> 8.34 | > 8.34 | NDe |
| V-25f |
|
> 8.13 | > 8.13 | NDe |
| V-25 g |
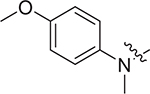
|
5.27 ± 1.65 | > 8.51 | > 1.61 |
| V-25 h |
|
> 8.63 | > 8.63 | NDe |
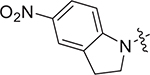
| V-25i |
|
2.57 ± 0.79 | > 8.55 | > 3.33 |
| V-25j |
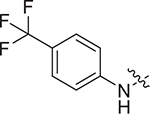
|
> 8.17 | > 8.17 | NDe |
| PF-74 | 0.42 ± 4.11 | > 11.56 | > 27.52 | |
NA: No anti-HIV-1 activity or cytotoxicity at the test concentration.
EC50: the concentration that inhibits HIV-1NL4–3 nanoluc-sec infection of MT-4 cells by 50%.
CC50: the concentration of the compound required to reduce the viability of uninfected cells by 50%, as determined by the MTT method.
SI: selectivity index, the ratio of CC50/EC50
ND: Not determined.
The results exhibited that a lot of compounds in series II and V displayed micromolar antiviral activity, but series I, III and IV didn’t show antiviral activity at the tested concentration (5 μg/mL). Especially for series IV, only the exchange of sulfone and amine positions in sulfonamide of series V, making them lost antiviral activity, which indicted that NTD-CTD dimer interface of CA hexamer is exceedingly sensitive to the linker of PF-74. Among series II and V, the most potent compounds are II-13c (EC50 = 5.14 ± 1.62 μM, CC50 > 9.51 μM) and V-25i (EC50 = 2.57 ± 0.79 μM, CC50 > 8.55 μM), which is slightly weaker than that of PF-74 (EC50 = 0.42 ± 0.11 μM, CC50 > 11.56 μM). Besides, when making a comparison between II and 13c and V-25i, two series of compounds with identical terminal substituents showed similar antiviral activity.
As for series II, p-methoxy-N-methylaniline (II-13c, EC50 = 5.14 ± 1.62 μM) is better than p-cyanoaniline (II-13a, EC50 = 8.09 ± 2.76 μM). Unfortunately, while the substituents are benzothiazole (II-13b), 5-methoxyindoline (II-13d), 3,5-difluoroaniline (II-13e), indazole (II-13f) and 5-nitroindoline (II-13 g), these compounds lost their activity against HIV-1.
Concerning series V, 5-aminoindoline substituted V-25i (EC50 = 2.57 ± 0.79 μM) has the best antiviral activity. When the substituents are piperidine (V-25b, EC50 = 4.85 ± 1.53 μM), thiomorpholine (V-25c, EC50 = 5.06 ± 1.43 μM) and p-methoxy-N-methylaniline (V-25 g, EC50 = 5.27 ± 1.65 μM), compounds displayed moderate antiviral activity. In addition, the substitution of N, N-dimethylamine (V-25a) caused less antiviral activity with the EC50 value of 8.68 ± 2.62 μM. However, the substitution of indoline (V-25d), 5-methoxyindoline (V-25e), 5-nitroindoline (V-25f), 3,5-difluoroaniline (V-25 h) and p-trifluoromethylaniline (V-25j) lead to loss of antiviral activity at the test concentration.
In conclusion, the preliminary SARs analysis of newly designed compounds showed that the linker of PF-74 derivatives, as well as the substituents part is extremely essential for anti-HIV activities, which would be used for further explore of CTD-NTD interface.
2.3. Binding to HIV-1 CA protein
Surface plasmon resonance (SPR) analysis is employed to ascertain affinity of target compounds with capsid proteins (hexamer, monomer). The representative compounds II-13c, V-25i was chosen for assay. Similarly, PF-74 was selected as a positive control. The result is showed in Table 2 and Figure 2.
Table 2.
SPR results of representative compounds and capsid proteins (hexamer, monomer).
| Compds | Hexamer | Monomer |
|---|---|---|
|
| ||
| PF-74 | 0.12 ± 0.00 μM | 7.15 ± 0.28 μM |
| II-13c | 4.82 ± 0.30 μM | 15.81 ± 0.47 μM |
| V-25i | 4.21 ± 0.57 μM | 11.62 ± 1.63 μM |
Fig. 2.

SPR isotherms of compounds PF-74, II-13c and V-25i.
Firstly, the equilibrium dissociation constant (KD) values of tested compounds showed that they tend to combine with CA hexamer, rather than CA monomer. In addition, the results indicated that the affinity of the three compounds for CA proteins were PF-74 (Hexamer: KD = 0.12 ± 0.00 μM; Monomer: KD = 7.15 ± 0.28 μM) > V-25i (Hexamer: KD = 4.21 ± 0.57 μM; Monomer: KD = 11.62 ± 1.63 μM) > II-13c (Hexamer: KD = 4.82 ± 0.30 μM; Monomer: KD = 15.81 ± 0.47 μM), which is consistent with antiviral activity in vitro (PF-74, EC50 = 0.42 ± 0.11 μM > V-25i, EC50 = 2.57 ± 0.79 μM > II-13d, EC50 = 5.14 ± 1.62 μM). SPR experiments demonstrated these compounds can be defined as HIV-1 CA protein inhibitors.
2.4. Molecular dynamics (MD) simulation
In order to understand the activity results and further explore the binding mode of these compounds with HIV-1 CA hexamer (PDB code: 5HGL, https://www.rcsb.org), II-13c and V-25i were chosen to make molecular dynamics (MD) simulation.
II-13c exhibited docking score, XP Gscore, and binding free energy values of − 3.084 kcal/mol, − 3.084 kcal/mol and − 43.70 kcal/mol, respectively, and those for V-25i were − 3.286, − 3.286 and − 70.63 kcal/mol, respectively. It is shown that the affinity between V and 25i and the action site of HIV-1 CA is higher than that of II-13c, which is consistent with the antiviral activity results at the cell level and the SPR results.
Compound II-13c interacted with the residues Asn53 and Asn57 of the capsid protein through H-bonds, and pi-cation (Lys70) as shown in Fig.3a. Likewise, compound V-25i interacted with the binding site residue Asn57 and Lys70 through H-bond and pi-cation, respectively (Fig.3b).
Fig. 3.

Ligand-interaction to binding site residues of capsid protein (a) II-13c-capsid complex, (b) V-25i-capsid complex.
Next, as a part of the investigation, the binding and conformational stability of both complexes for II-13c and V-25i with CA was analyzed by MD simulation studies. As shown in Fig.4 and Fig.5, the binding conformation of II-13c and V-25i were stable in the binding pocket of capsid (PDB code: 5HGL) during the period of 50 ns MD simulation process. The protein structure stabilized nearly at 20 ns and the average root mean square deviation (RMSD) of Cα was noted as 4.2 Å (Fig.4). The interaction of II-13c and V-25i with binding site residues through H-bond (green), hydrophobic interaction (grey), and water-bridge (blue) are presented in Fig.4 and Fig.5. The residues, Asn57 and Tyr130 interacted to II-13c by water brides (Fig.4b, c). The residues Lys70 and Asn53 interacted to II-13c by pi-cation and H-bond, respectively (Fig.4b, c). Compound II-13c was always bounded with 6–7 contacts during the MD simulations (Fig.4d). The V-25i-capsid complex, protein structure stabilized in the early period of simulation with average Cα-RMSD of 3.24 Å (Fig.5a). And V-25i exhibited interactions to the residues Asn53, Asn57, and Lys70 by water bridge interaction which facilitates the V-25i stability within the binding pocket (Fig.5b, c). The residue Lys70 also interacted to compound V-25i with pi-cation interactions. V-25i remained interacted with 7–8 contacts during the MD simulation process (Fig.5d). The Ramachandran mapping of the residues in the complex with II-13c and V-25i after MD simulation, supported a very acceptable number of residues in favored, additional allowed and generously allowed region (Table 3, entry 1 and 2). There were no residues present in outlier region of the capsid protein in complex with V-25i while 0.5% (Val181) residue observed outlier in II-13c-capsid complex (Fig. 6).
Fig. 4.

Molecular dynamics and simulation of II-13c with capsid protein complex (a) RMSD plot for Cα of capsid protein (PDB code: 5HGL) in complex with II-13c. (b) A Histogram plot showing residues interacting II-13c. (c) The percentage of interactions in molecular dynamic simulations of II-13c complex with capsid. (d) A timeline representation of the interactions and total contacts (H-bonds, hydrophobic interactions, ionic interactions, and water bridges) obtained during the Molecular Dynamic simulations. The panels show the total number of specific contacts the capsid made with the II-13c over the course of the simulation.
Fig. 5.

Molecular dynamics and simulation of V-25i with capsid protein complex (a) RMSD plot for Cα of capsid in complex with V-25i. (b) A histogram plot showing residues interacting V-25i. (c) The percentage of interactions in molecular dynamic simulations of V-25i complex with capsid. (d) A timeline representation of the interactions and total contacts (H-bonds, hydrophobic interactions, ionic interactions, and water bridges) obtained during the molecular dynamic simulations. The panels show the total number of specific contacts the capsid protein made with the V-25i over the course of the simulation.
Table 3.
Occurance of residues in favoured, additional-allowed, generously-allowed, and disallowed region.
| Entry | Complex | Residues in favoured region | Residues in additional allowed region | Residues in generously allowed region | Residues in disallowed region |
|---|---|---|---|---|---|
|
| |||||
| 1 | II-13c-capsid | 88.8% (166) | 10.2% (19) | 0.5% (1) | 0.5% (1) |
| 2 | V-25i-capsid | 86.6% (162) | 12.8% (24) | 0.5% (1) | 0.0% (0) |
Fig. 6.

Ramachandran plot (a) II-13c-capsid complex, (b) V-25i-capsid complex.
In conclusion, in silico study supported the experimental results for compounds II-13c and V-25i active against HIV-1. Compound V-25i showed better docking score, XP-Gscore and binding free energy with capsid protein over II-13c, which further validated by MD simulation for 50 ns that indicated high conformational stability for the former. This could be the reason variations in EC50 of both compounds, however more biochemical assays are needed to be performed to validate.
3. Conclusion
In brief, taking the widely studied HIV-1 CA inhibitor PF-74 as the lead compound, we designed and synthesized 32 novel PF-74 derivatives via scaffold hopping. The antiviral activity of the compounds was significantly affected by the slight change of the linkers and substituents targeting CTD of CA protein. Among them, a lot of compounds of series II and V displayed moderate anti-HIV-1 activity in MT-4 cells. The most potent compounds in these two series are II-13c (EC50 = 5.14 ± 1.62 μM, CC50 > 9.51 μM) and V-25i (EC50 = 2.57 ± 0.79 μM, CC50 > 8.55 μM), though the activity was slightly weaker than PF-74 (EC50 = 0.42 ± 0.11 μM, CC50 > 11.56 μM). Subsequently, the SPR assays indicated that these compounds are more inclined to combine with HIV-1 CA hexamer than monomer. Further molecular dynamics simulation exhibited that the binding conformation between V and 25i and CA hexamer is more stable than II-13c, which is consistent with the results of antiviral activity in vitro and target affinity analysis.
In conclusion, this study enriched the SARs of PF-74 derivatives, and laid a solid foundation for finding novel and potent HIV-1 CA inhibitors.
4. Experimental section
4.1. Chemistry
1H NMR and 13C NMR spectra were recorded on a Bruker Avance-400 NMR spectrometer (Standard G1313A instrument) in DMSO. Chemical shifts were reported in δ values (ppm) and J values were presented in hertz (Hz). High resolution mass spectra (HRMS) were obtained from Thermo Electron LTQ-Orbitrap XL mass spectrometry instrument (Thermo Scientific Inc., USA). Melting points of all the compounds were measured on a micro melting point apparatus and were uncorrected. Flash column chromatography was performed on column packed with Silica Gel 60 (200–300 mesh). Solvents were reagent grade and puried with standard methods when necessary. Sample purity was analyzed on a Shimadzu SPD-20A/20AV high-performance liquid chromatography (HPLC) system with a Inertsil ODS-SP, 5 μm C18 column (150 × 4.6 mm) [HPLC conditions: methanol/water (80/20); flow rate: 1.0 mL/min; UV detection from 210 to 400 nm; temperature: ambient; and injection volume: 20 μL]. Purity of all final compounds was > 95%.
4.1.1. Synthesis of tert-butyl (S)-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)carbamate (6)
A solution of (tert-butoxycarbonyl)-L-phenylalanine (2.65 g, 10.00 mmol) in 15 mL dichloromethane was added PyBop (5.70 g, 15.00 mmol) at 0°C, and the mixture stirred for 0.5 h. Subsequently, DIEA (3.30 mL, 20.00 mmol) and 4-methoxy-N-methylaniline (1.37 g, 10.00 mmol) were added and then the mixture was stirred at room temperature for another 8 h (monitored by TLC), evaporated under reduced pressure and the residue was initially washed by saturated sodium bicarbonate (50 mL) and extracted with ethyl acetate (3 × 10 mL). Then the combined organic layer was washed by 1 N HCl and extracted with ethyl acetate (3 × 10 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford corresponding crude intermediate 6 as yellow oil with a yield of 79%. 1H NMR (400 MHz, DMSO-d6) δ 7.21 (d, J = 8.4 Hz, 3H, Ph-H + NH), 7.15 (d, J = 7.1 Hz, 2H, Ph-H), 7.02 (d, J = 8.4 Hz, 2H, Ph-H), 6.85–6.75 (m, 2H, Ph-H), 4.15 (q, J = 5.4 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 3.12 (s, 3H, NCH3), 2.81–2.54 (m, 2H, CH2), 1.30 (s, 9H, CH3 × 3); 13C NMR (100 MHz, DMSO-d6) δ 172.2, 158.9, 155.7, 138.5, 136.1, 129.2, 128.4, 126.7, 115.2, 78.3, 55.9, 53.5, 37.8, 37.1, 28.6. ESI-MS: m/z 385 [M + H]+, C22H28N2O4 (384.2).
4.1.2. Synthesis of (S)-2-amino-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (7)
Triuoroacetic acid (2.36 mL, 31.56 mmol) was added dropwise to intermediate 6 (3.03 g, 7.89 mmol, 1.0 eq) in 30 mL dichloromethane and the resulting mixture solution was stirred at room temperature for 6–7 h. Then, the resulting solution was alkalized to pH 9 with saturated sodium bicarbonate solution, and then extracted with dichloromethane (3 × 20 mL). The combined organic layer was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to yield the crude product 7 as yellow oil with a yield of 94%. ESI-MS: m/z 285 [M + H]+, C17H20N2O2 (284.1)
4.1.3. Synthetic route of methyl (S)-3-(N-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)sulfamoyl)benzoate (8)
Intermediate 7 (0.28 g, 1.00 mmol) was dissolved in 10 mL dichloromethane, and the mixture was added triethylamine (0.21 mL, 1.50 mmol). Subsequently, methyl 3-(chlorosulfonyl)benzoate (0.26 g, 1.10 mmol) was added to the mixture at 0°C for another 4 h (monitored by TLC). Then the mixture was washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the mixture was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford product 8. White solid, yield: 60%. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (d, J = 9.0 Hz, 1H, NH), 8.04 (d, J = 7.7 Hz, 1H, PhH), 7.95 (s, 1H, PhH), 7.67 (d, J = 7.7 Hz, 1H, PhH), 7.53 (t, J = 7.7 Hz, 1H, PhH), 7.12 – 6.96 (m, 7H, PhH), 6.64 (d, J = 4.4 Hz, 2H, PhH), 3.91 (s, 3H, OCH3), 3.81 (s, 3H, OCH3), 3.78 (dd, J = 9.5, 3.6 Hz, 1H, CH), 2.96 (s, 3H, NCH3), 2.81 (dd, J = 13.6, 3.6 Hz, 1H, CH2), 2.55 (s, 1H, CH2). ESI-MS: m/z 483 [M + H]+, C25H26N2O6S (482.15).
4.1.4. Synthetic route of (S)-3-(N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)sulfamoyl)benzoic acid (9)
8 was dissolved in 5 mL tetrahydrofuran. Then aqueous NaOH solution (5 mL) was added to the mixture and stired for another 2 h (monitored by TLC). Subsequently, the resulting mixture solution was alkalized to pH 2 with 1 N HCl to produce white precipitate, filtered, dried to afford 9. White solid, yield: 78%。ESI-MS: 467 m/z [M − H]−, C24H24N2O6S (468.14).
4.1.5. General procedure for the synthesis of target compounds I-10(a–e)
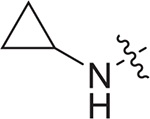
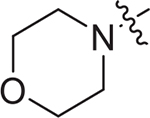
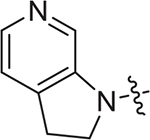
A solution of 9 (1.0 eq) in 15 mL dichloromethane was added PyBop (1.5 eq) at 0 °C, and the mixture stirred for 0.5 h. Subsequently, DIEA (2.0 eq) and amine fragments (2.0 eq) were added to the mixture and then stirred at room temperature for another 8 h (monitored by TLC). The resulting mixture was evaporated under reduced pressure and the residue was initially washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the combined organic layer was washed by 1 N HCl and extracted with dichloromethane (3 × 8 mL), dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford product I-10 (a–e). Yield: 40–70%.
(S)-N-cyclopropyl-3-(N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)sulfamoyl)benzamide (I-10a): white solid, yield: 61%, HPLC purity 98% (tR = 4.02 min), mp: 175–177°C. 1H NMR (400 MHz, DMSO-d6) δ 8.65 (d, J = 3.7 Hz, 1H, NH), 8.38 (d, J = 9.0 Hz, 1H, PhH), 8.03 (s, 1H, NH), 7.95 (d, J = 7.5 Hz, 1H, PhH), 7.54 (d, J = 7.7 Hz, 1H, PhH), 7.47 (t, J = 7.7 Hz, 1H, PhH), 7.10 (d, J = 4.8 Hz, 3H, PhH), 7.03 – 6.90 (m, 4H, PhH), 6.71 (d, J = 4.5 Hz, 2H, PhH), 3.80 (s, 4H, CH + OCH3), 2.96 (s, 1H, CH), 2.93 (s, 3H, NCH3), 2.87 – 2.77 (m, 2H, CH2), 0.74 (d, J = 5.6 Hz, 2H, CH2), 0.64 – 0.58 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 166.5, 159.1, 141.3, 137.2, 135.4, 135.2, 130.9, 129.3, 129.2, 129.0, 128.4, 126.9, 125.9, 115.1, 55.9, 54.8, 38.3, 37.5, 23.6, 6.1. HRMS (ESI) calcd for C27H29N3O5S [M + H]+ 508.1828, found 508.1894.
(S)-N-(4-methoxyphenyl)-N-methyl-2-((3-(morpholine-4-carbonyl) phenyl)sulfonamido)-3-phenylpropanamide (I-10b) : white solid, yield: 57%, HPLC purity 99% (tR = 3.76 min), mp: 141–143°C. 1H NMR (400 MHz, DMSO-d6) δ 8.45 (d, J = 8.9 Hz, 1H, NH), 7.53 (td, J = 17.6, 15.1, 7.4 Hz, 4H, PhH), 7.11 (s, 3H, PhH), 6.95 (d, J = 8.0 Hz, 4H, PhH), 6.80 – 6.69 (m, 2H, PhH), 3.79 (s, 4H, CH + OCH3), 3.68 (d, J = 19.6 Hz, 4H, CH2 × 2), 3.56 (d, J = 8.0 Hz, 2H, CH2), 3.23 (s, 2H, CH2), 2.92 (s, 3H, NCH3), 2.88 – 2.79 (m, 1H, CH2), 2.62 – 2.54 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 168.0, 159.1, 141.6, 137.2, 136.3, 135.4, 130.9, 129.5, 129.3, 129.0, 128.5, 127.7, 126.9, 125.4, 115.1, 66.4, 55.9, 54.8, 38.7, 38.3, 37.5. HRMS (ESI) calcd for C28H31N3O6S [M + H]+ 538.1934, found 538.2010.
(S)-2-((3-(2,3-dihydro-1H-pyrrolo[3,4-c]pyridine-2-carbonyl)phenyl) sulfonamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (I-10c): white solid, yield: 52%, HPLC purity 97% (tR = 4.37 min), mp: 80–82 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.39 (d, J = 8.9 Hz, 1H, pyridinyl-H), 7.66 (dd, J = 13.2, 5.2 Hz, 4H, NH + PhH × 3), 7.57 (d, J = 7.8 Hz, 1H, PhH), 7.46 (t, J = 7.9 Hz, 1H, PhH), 7.14 (s, 3H, PhH), 6.96 – 6.81 (m, 4H, PhH × 2 + pyridinyl-H × 2), 6.82 – 6.70 (m, 4H, PhH), 4.16 (t, J = 8.1 Hz, 2H, CH2), 3.89 – 3.80 (m, 1H, CH), 3.75 (s, 3H, OCH3), 3.16 (t, J = 8.0 Hz, 2H, CH2), 2.93 (s, 3H, NCH3), 2.86 (dd, J = 13.4, 5.4 Hz, 1H, CH2), 2.60 – 2.54 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 167.0, 159.0, 155.6, 145.7, 140.8, 137.3, 137.1, 135.3, 134.3, 132.3, 129.4, 128.9, 128.5, 126.9, 126.7, 126.5, 119.2, 115.0, 55.8, 54.6, 47.7, 38.7, 37.5, 24.7. HRMS (ESI) calcd for C31H30N4O5S [M + H]+ 571.1937, found 571.2009.
(S)-N-(4-methoxyphenyl)-3-(N-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)sulfamoyl)-N-methylbenzamide (I-10d): brown solid, yield: 47%, HPLC purity 98% (tR = 4.84 min), mp: 60–62 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.31 (d, J = 9.0 Hz, 1H, NH), 7.65 (s, 1H, PhH), 7.28 (d, J = 8.0 Hz, 3H, PhH), 7.10 (qd, J = 7.9, 7.4, 4.3 Hz, 5H, PhH), 6.95 (d, J = 8.1 Hz, 2H, PhH), 6.87 (d, J = 8.4 Hz, 2H, PhH), 6.77 (d, J = 8.5 Hz, 2H, PhH), 6.72 (d, J = 6.8 Hz, 2H, PhH), 3.79 (s, 3H, OCH3), 3.77 – 3.72 (m, 1H, CH), 3.64 (s, 3H, OCH3), 3.35 (s, 3H, NCH3), 2.94 (s, 3H, NCH3), 2.82 (dd, J = 13.6, 5.4 Hz, 1H, CH2), 2.49 – 2.40 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 159.0, 158.1, 141.1, 137.3, 137.1, 135.4, 131.7, 129.3, 129.0, 128.7, 128.5, 127.3, 126.9, 115.1, 114.8, 55.9, 55.6, 54.6, 38.4, 37.6. HRMS (ESI) calcd for C32H33N3O6S [M + H]+ 589.2169, found 588.2169.
(S)-N-(benzo[d]thiazol-6-yl)-3-(N-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)sulfamoyl)benzamide (I-10e): white solid, yield: 73%, HPLC purity 98% (tR = 4.81 min)purity: 98%, mp: 190–192 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H, NH), 9.33 (s, 1H, thiazyl-H), 8.71 (s, 1H, NH), 8.47 (d, J = 8.9 Hz, 1H, PhH), 8.16 (s, 1H, PhH), 8.13 – 8.05 (m, 2H, PhH), 7.86 (d, J = 8.8 Hz, 1H, PhH), 7.65 (d, J = 7.7 Hz, 1H, PhH), 7.58 (t, J = 7.7 Hz, 1H, PhH), 7.07 (s, 3H, PhH), 7.01 (d, J = 7.8 Hz, 2H, PhH), 6.95 (d, J = 8.2 Hz, 2H, PhH), 6.76 – 6.67 (m, 2H, PhH), 3.82 (t, J = 10.8 Hz, 1H, CH), 3.74 (s, 3H, OCH3), 2.96 (s, 3H, NCH3), 2.83 (dd, J = 13.5, 3.7 Hz, 1H, CH2), 2.61 – 2.54 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.5, 164.8, 159.1, 155.6, 150.1, 141.5, 137.2, 137.0, 135.5, 135.4, 134.6, 131.5, 129.7, 129.4, 129.2, 129.0, 128.4, 126.8, 126.4, 123.3, 120.4, 115.2, 113.6, 55.8, 54.9, 38.2, 37.6. HRMS (ESI) calcd for C31H28N4O5S2 [M + H]+ 601.1501, found 601.1573.
4.1.6. Synthetic route of methyl (S)-2-(N-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)sulfamoyl)acetate (11)
Intermediate 7 (0.28 g, 1.00 mmol) was dissolved in 15 mL dichloromethane, and the mixture was added triethylamine (0.21 mL, 1.50 mmol). Subsequently, methyl 2-(chlorosulfonyl)acetate (0.12 mL, 1.00 mmol) was added to the mixture at 0°C for 4 h (monitored by TLC). Then the mixture was washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the mixture was dried over anhydrous Na2SO4, filtered, and concentrated under reduced pressure to afford product 11. White solid, yield: 76%. 1H NMR (400 MHz, DMSO-d6) δ 8.06 (d, J = 8.6 Hz, 1H, NH), 7.22 (d, J = 6.2 Hz, 3H, PhH), 7.04 (s, 2H, PhH), 6.99 (d, J = 8.2 Hz, 2H, PhH), 6.86 (d, J = 6.8 Hz, 2H, PhH), 4.08 (td, J = 8.8, 4.9 Hz, 1H, CH), 3.87 – 3.77 (m, 4H, OCH3 + CH2), 3.69 (d, J = 14.7 Hz, 1H, CH2), 3.60 (s, 3H, OCH3), 3.13 (s, 3H, NCH3), 2.87 (dd, J = 13.4, 4.9 Hz, 1H, CH2), 2.66 (dd, J = 13.1, 9.5 Hz, 1H, CH2). ESI-MS: m/z 421 [M + H]+, 443 [M + Na]+, C20H24N2O6S (500.15).
4.1.7. Synthetic route of (S)-2-(N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)sulfamoyl)acetic acid (12)
11 was dissolved in 5 mL tetrahydrofuran. Then aqueous solution containing NaOH (5 mL) was added to the mixture and stired for another 2 h (monitored by TLC). Subsequently, the resulting mixture solution was alkalized to pH 2 with 1 N HCl to produce white precipitate, which was filtered, dried to afford 12. White solid, yield: 89%. ESI-MS: m/z 405 [M − H]−, C19H22N2O6S (406.12).
4.1.8. General procedure for the synthesis of target compounds II-13(a–g)
A solution of 12 (1.0 eq) in 15 mL dichloromethane was added PyBop (1.5 eq) at room temperature, and the mixture stirred for 0.5 h. Subsequently, DIEA (2.0 eq) and amine fragments (2.0 eq) were added to the mixture and then stirred at room temperature for another 8 h (monitored by TLC). The resulting mixture was initially washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the combined organic layer was washed by 1 N HCl and extracted with dichloromethane (3 × 8 mL). Then the combined organic phase was washed with saturated salt water (3 × 10 mL), dried over anhydrous Na2SO4, filtered, and recrystallized with ethyl acetate and petroleum ether to afford product II-13(a–g). Yield: 40–70%.
(S)-2-((2-((4-cyanophenyl)amino)-2-oxoethyl)sulfonamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (II-13a): yellow solid, yield: 56%, HPLC purity 97% (tR = 4.30 min), mp: 82–84 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.70 (s, 1H, NH), 8.13 (d, J = 8.4 Hz, 1H, NH), 7.83 (d, J = 8.1 Hz, 2H, PhH), 7.77 (d, J = 8.3 Hz, 2H, PhH), 7.21 (d, J = 5.4 Hz, 3H, PhH), 7.03 (s, 2H, PhH), 6.93 (d, J = 8.3 Hz, 2H, PhH), 6.85 (d, J = 6.5 Hz, 2H, PhH), 4.16 (q, J = 8.0 Hz, 1H, CH), 3.90 (s, 2H, CH2), 3.78 (s, 3H, OCH3), 3.13 (s, 3H, NCH3), 2.89 (dd, J = 13.2, 5.3 Hz, 1H, CH2), 2.73 (d, J = 9.5 Hz, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.4, 161.4, 159.1, 143.1, 137.3, 135.4, 133.7, 129.5, 129.1, 128.6, 127.1, 119.8, 119.4, 115.1, 106.0, 60.6, 55.9, 55.6, 38.7, 38.2. HRMS (ESI) calcd for C26H26N4O5S [M + H]+ 507.1624,found 507.1689. (S)-2-((2-(benzo[d]thiazol-6-ylamino)-2-oxoethyl)sulfonamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (II-13b): white solid, yield: 57%, HPLC purity 97% (tR = 4.18 min), mp: 110–112°C. 1H NMR (400 MHz, DMSO-d6) δ 10.55 (s, 1H, NH), 9.31 (s, 1H, thiazyl-H), 8.51 (s, 1H, NH), 8.06 (d, J = 8.5 Hz, 2H, PhH), 7.61 (d, J = 8.8 Hz, 1H, PhH), 7.20 (d, J = 4.0 Hz, 3H, PhH), 7.02 (s, 2H, PhH), 6.88 (dd, J = 17.9, 6.8 Hz, 4H, PhH), 4.19 (q, J = 7.9 Hz, 1H, CH), 3.91 (s, 2H, CH2), 3.74 (s, 3H, OCH3), 3.13 (s, 3H, NCH3), 2.90 (dd, J = 13.7, 5.7 Hz, 1H, CH2), 2.80 – 2.72 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.4, 160.8, 159.0, 155.5, 149.9, 137.3, 136.7, 135.4, 134.7, 129.6, 129.1, 128.6, 127.0, 123.5, 119.3, 115.1, 112.3, 60.6, 55.8, 55.6, 38.6, 38.1. HRMS (ESI) calcd for C26H26N4O5S2 [M + H]+ 539.1345, found 539.1415.
(S)-N-(4-methoxyphenyl)-2-((2-((4-methoxyphenyl)(methyl)amino)-2-oxoethyl)sulfonamido)-N-methyl-3-phenylpropanamide (II-13c): white solid, yield: 61%, HPLC purity 98% (tR = 6.97 min), mp: 100–102 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.93 (d, J = 4.8 Hz, 1H, NH), 7.67 (d, J = 8.4 Hz, 2H, PhH), 7.15 (p, J = 6.7 Hz, 3H, PhH), 7.11 – 6.94 (m, 4H, PhH), 6.92 (d, J = 9.0 Hz, 2H, PhH), 6.85 (d, J = 6.6 Hz, 2H, PhH), 4.88 (d, J = 14.4 Hz, 1H, CH2), 4.73 (d, J = 14.4 Hz, 1H, CH2), 4.16 (td, J = 8.9, 5.3 Hz, 1H, CH), 3.94 (s, 3H, OCH3), 3.78 (s, 3H, OCH3), 3.05 (s, 6H, NCH3 × 2), 2.83 (dd, J = 13.4, 5.2 Hz, 1H, CH2), 2.65 (dd, J = 13.5, 8.9 Hz, 1H, CH2). 13C NMR (150 MHz, DMSO-d6) δ 171.1, 161.0, 159.0, 155.0, 145.8, 137.5, 135.6, 134.7, 129.6, 129.1, 128.5, 127.0, 126.9, 119.1, 115.1, 57.9, 55.9, 55.4, 46.1, 38.7, 37.9, 23.8. HRMS (ESI) calcd for C27H31N3O6S [M + H]+ 526.1934, found 526.2011.
(S)-2-((2-(5-methoxyindolin-1-yl)-2-oxoethyl)sulfonamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (II-13d): brown solid, yield: 57%, HPLC purity 99% (tR = 5.09 min), mp: 98–100 °C. 1H NMR (400 MHz, DMSO-d6) δ 7.94 (d, J = 8.2 Hz, 2H, PhH + NH), 7.21 (s, 3H, PhH), 7.03 (s, 2H, PhH), 6.93 (d, J = 8.1 Hz, 2H, PhH), 6.86 (s, 3H, PhH), 6.73 (d, J = 8.7 Hz, 1H, PhH), 4.14 (d, J = 6.6 Hz, 1H, CH), 4.02 (t, J = 8.5 Hz, 2H, CH2), 3.91 (d, J = 14.2 Hz, 1H, CH2), 3.79 (d, J = 10.4 Hz, 4H, OCH3 + CH2), 3.72 (s, 3H, OCH3), 3.09 (d, J = 12.4 Hz, 5H, NCH3 + CH2), 2.87 (dd, J = 13.4, 5.4 Hz, 1H, CH2), 2.73 – 2.66 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 159.7, 159.0, 156.6, 137.5, 136.3, 135.5, 134.3, 129.6, 129.2, 128.7, 127.1, 117.4, 115.1, 112.2, 111.2, 59.2, 55.9, 55.8, 55.6, 48.7, 38.7, 37.9, 27.8. HRMS (ESI) calcd for C28H31N3O6S [M + H]+ 538.1934, found 538.2016.
(S)-2-((2-((3,5-difluorophenyl)amino)-2-oxoethyl)sulfonamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (II-13e): white solid, yield: 67%, HPLC purity 98% (tR = 6.03 min), mp: 156–158 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.65 (s, 1H, NH), 8.13 (d, J = 8.3 Hz, 1H, NH), 7.28 (d, J = 8.7 Hz, 2H, PhH), 7.19 (s, 3H, PhH), 7.00 (dd, J = 19.9, 9.0 Hz, 3H, PhH), 6.93 (d, J = 8.3 Hz, 2H, PhH), 6.84 (d, J = 6.3 Hz, 2H, PhH), 4.15 (q, J = 7.9 Hz, 1H, CH), 3.85 (d, J = 4.7 Hz, 2H, CH2), 3.77 (s, 3H, OCH3), 3.12 (s, 3H, NCH3), 2.88 (dd, J = 13.5, 5.3 Hz, 1H, CH2), 2.70 (dd, J = 13.7, 8.8 Hz, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.4, 162.8 (dd, 1JCF = 243.4, 3JCF = 15.2 Hz), 161.3, 159.0, 141.4 (t, 3JCF = 13.7 Hz), 137.4, 135.4, 129.5, 129.1, 128.6, 127.1, 115.1, 102.7 (d, 2JCF = 29.4 Hz), 99.4 (t, 2JCF = 26.2 Hz), 60.6, 55.8, 55.6, 38.5, 38.1. HRMS (ESI) calcd for C25H25F2N3O5S [M + H]+ 518.1483, found 518.1561.
(S)-2-((2-(1H-indazol-1-yl)-2-oxoethyl)sulfonamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropeanamide (II-13f): whit solid, yield: 64%, HPLC purity 99% (tR = 3.76 min), mp: 68–70 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.53 (s, 1H, pyrazoly-H), 8.29 (d, J = 8.3 Hz, 1H, NH), 8.25 (d, J = 8.7 Hz, 1H, PhH), 7.95 (d, J = 7.9 Hz, 1H, PhH), 7.68 (t, J = 7.7 Hz, 1H, PhH), 7.48 (t, J = 7.5 Hz, 1H, PhH), 7.13 (dt, J = 13.1, 6.4 Hz, 5H, PhH), 6.98 (d, J = 8.2 Hz, 2H, PhH), 6.82 (d, J = 7.3 Hz, 2H, PhH), 4.84 (d, J = 14.3 Hz, 1H, CH2), 4.66 (d, J = 14.7 Hz, 1H, CH2), 4.17 (td, J = 9.1, 4.8 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 3.10 (s, 3H, NCH3), 2.86 (dd, J = 13.8, 4.9 Hz, 1H, CH2), 2.70–2.63 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.0, 162.9, 159.1, 141.6, 138.6, 137.4, 135.6, 130.4, 129.5, 128.5, 126.9, 125.7, 122.4, 115.2, 115.1, 57.4, 55.9, 55.7, 38.3, 38.0. HRMS (ESI) calcd for C26H26N4O5S [M + H]+ 507.1624, found 507.1701.
(S)-N-(4-methoxyphenyl)-N-methyl-2-((2-(5-nitroindolin-1-yl)-2-oxoethyl)sulfonamido)-3-phenylpropanamide (II-13 g): brown solid, yield: 57%, HPLC purity 99% (tR = 5.17 min), mp: 92–94 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.14 (s, 3H, NH + PhH × 2), 8.06 (d, J = 8.4 Hz, 1H, PhH), 7.20 (d, J = 6.4 Hz, 3H, PhH), 7.02 (s, 2H, PhH), 6.93 (d, J = 8.2 Hz, 2H, PhH), 6.86 (d, J = 6.8 Hz, 2H, PhH), 4.16 (q, J = 7.4, 6.2 Hz, 3H, CH + CH2), 4.07 (d, J = 14.5 Hz, 1H, CH2), 3.93 (d, J = 14.4 Hz, 1H, CH2), 3.77 (s, 3H, OCH3), 3.25 (t, J = 8.7 Hz, 2H, CH2), 3.07 (s, 3H, NCH3), 2.87 (dd, J = 13.5, 5.4 Hz, 1H, CH2), 2.70 (d, J = 11.5 Hz, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.9, 162.2, 159.0, 148.4, 143.7, 137.4, 135.5, 134.8, 129.6, 129.2, 128.7, 127.1, 124.6, 120.9, 116.1, 115.1, 59.4, 55.9, 55.7, 49.5, 38.6, 37.9, 27.1. HRMS (ESI) calcd for C27H28N4O7S [M + H]+ 553.1679, found 553.1754.
4.1.9. Synthetic route of methyl (S)-2-((1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)amino)-2-oxoacetate (14)
Intermediate 7 (0.28 g, 1.00 mmol) was dissolved in 15 mL dichloromethane, and the mixture was added triethylamine (0.21 mL, 1.50 mmol). Subsequently, methyl 2-chloro-2-oxoacetate (0.21 mL, 1.50 mmol) was added slowly to the mixture at 0°C for another 4 h (monitored by TLC). Then the mixture was washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the combined organic phase was washed with saturated salt water (3 × 10 mL) and was dried over anhydrous Na2SO4, filtered, and recrystallized with ethyl acetate and petroleum ether to afford product 14. White solid, yield: 73%. 1H NMR (400 MHz, DMSO-d6) δ 9.01 (d, J = 7.8 Hz, 1H, NH), 7.16 (t, J = 9.5 Hz, 5H, PhH), 7.00 (d, J = 8.3 Hz, 2H, PhH), 6.85 (d, J = 6.9 Hz, 2H, PhH), 4.48 (q, J = 8.1 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 3.12 (s, 3H, NCH3), 2.88 (dt, J = 13.7, 7.2 Hz, 2H, CH2). ESI-MS: m/z 371 [M + H]+, 393 [M + Na]+, C20H22N2O5 (370.15).
4.1.10. Synthetic route of (S)-2-((1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3- Phenylpropan-2-yl)amino)-2-oxoacetic acid (15)
14 (0.10 g, 0.27 mmol) was dissolved in 5 mL tetrahydrofuran. Then aqueous solution containing NaOH (0.02 g, 0.54 mmol) was added to the mixture and stired for 2 h (monitored by TLC). Subsequently, the resulting mixture solution was alkalized to pH 2 with 1 N HCl to produce white precipitate, filtered, dried to afford 15. White solid, yield: 68%. ESI-MS: m/z 355 [M − H]−, C19H20N2O5 (356.14).
4.1.11. General procedure for the synthesis of target compounds III-16 (a–g)
A solution of 15 (1.0 eq) in 15 mL dichloromethane was added PyBop (1.5 eq) at room temperature, and the mixture stirred for 0.5 h. Subsequently, DIEA (2.0 eq) and amine fragments (2.0 eq) were added to the mixture and then the resulting mixture was stirred at room temperature for another 8 h (monitored by TLC). The mixture was initially washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the combined organic layer was washed by 1 N HCl and extracted with dichloromethane (3 × 8 mL). Then the combined organic phase was washed with saturated salt water (3 × 10 mL), dried over anhydrous Na2SO4, filtered, and recrystallized with ethyl acetate and petroleum ether to afford product III-16(a–g). Yield: 40–80%.
(S)-N1-(benzo[d]thiazol-6-yl)–N2-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)oxalamide (III-16a): white solid, yield: 53%, HPLC purity 99% (tR = 5.81 min), mp: 78–80 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.88 (s, 1H, NH), 9.32 (s, 1H, thiazyl-H), 8.90 (d, J = 7.9 Hz, 1H, NH), 8.63 (s, 1H, PhH), 8.04 (d, J = 8.8 Hz, 1H, PhH), 7.89 (d, J = 8.9 Hz, 1H, PhH), 7.26 (s, 2H, PhH), 7.18 (d, J = 7.2 Hz, 3H, PhH), 7.02 (d, J = 8.3 Hz, 2H, PhH), 6.90 (d, J = 7.0 Hz, 2H, PhH), 4.58 (q, J = 7.8 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 3.15 (s, 3H, NCH3), 3.02 – 2.89 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.6, 159.9, 159.2, 158.5, 156.0, 150.4, 137.8, 135.8, 135.7, 134.4, 129.3, 129.3, 127.0, 123.4, 120.3, 115.2, 113.6, 55.9, 52.6, 37.9, 36.8. HRMS (ESI) calcd for C26H24N4O4S [M + H]+ 489.1518, found 489.1592.
(S)-2-(2-(5-methoxyindolin-1-yl)-2-oxoacetamido)-N-(4-methoxyphenyl)-N-methyl-3-phenylpropanamide (III-16b): white solid, yield: 50%, HPLC purity 99% (tR = 6.52 min), mp: 82–84 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.00 (d, J = 7.8 Hz, 1H, NH), 7.90 (d, J = 8.8 Hz, 1H, PhH), 7.21 (dt, J = 16.2, 8.4 Hz, 5H, PhH), 7.04 (d, J = 8.1 Hz, 2H, PhH), 6.91 – 6.82 (m, 3H, PhH), 6.76 (d, J = 8.8 Hz, 1H, PhH), 4.63 – 4.50 (m, 1H, CH), 3.89 (q, J = 9.4 Hz, 1H, CH2), 3.82 (s, 3H, OCH3), 3.73 (s, 3H, OCH3), 3.59 (q, J = 10.1, 9.6 Hz, 1H, CH2), 3.15 (s, 3H, NCH3), 3.00 (d, J = 8.2 Hz, 2H, CH2), 2.93 (dd, J = 13.6, 4.2 Hz, 1H, CH2), 2.75 (dd, J = 13.7, 9.9 Hz, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.7, 162.8, 160.7, 159.1, 157.0, 137.8, 135.9, 135.7, 134.7, 129.4, 129.2, 128.6, 126.9, 117.6, 115.3, 112.4, 111.3, 55.9, 55.8, 51.6, 48.6, 37.9, 37.2, 28.3. HRMS (ESI) calcd for C28H29N3O5 [M + H]+ 488.2107, found 488.2186.
(S)-N1-(4-methoxyphenyl)–N2-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)-N1-methyloxalamide (III-16c): yellow solid, yield: 59%, HPLC purity 98% (tR = 4.02 min), mp: 141–143 °C. 1H NMR (400 MHz, DMSO-d6) δ 8.88 (d, J = 8.5 Hz, 1H, NH), 7.20 (p, J = 8.3, 7.8 Hz, 5H, PhH), 7.06 (d, J = 8.4 Hz, 2H, PhH), 6.82 (t, J = 6.9 Hz, 3H, PhH), 6.70 (t, J = 9.1 Hz, 3H, PhH), 4.24 (q, J = 7.6 Hz, 1H, CH), 3.74 (s, 3H, OCH3), 3.68 (s, 3H, OCH3), 3.13 (s, 3H, NCH3), 3.01 (s, 3H, NCH3), 2.77 (dd, J = 13.2, 6.5 Hz, 1H, CH2), 2.53 (s, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.4, 164.9, 163.3, 158.8, 158.4, 137.6, 135.6, 135.1, 129.3, 128.8, 128.6, 127.9, 127.1, 126.9, 114.9, 114.4, 55.7, 55.7, 50.8, 37.8, 37.7, 36.3. HRMS (ESI) calcd for C27H29N3O5 [M + H]+ 476.2107, found 476.2185.
(S)-N1-(3,5-difluorophenyl)–N2-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)oxalamide (III-16d): white solid, yield: 74%, HPLC purity 99% (tR = 8.42 min), mp: 132–134 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.01 (s, 1H, NH), 8.99 (d, J = 7.8 Hz, 1H, NH), 7.58 (d, J = 9.0 Hz, 2H, PhH), 7.21 (dd, J = 24.0, 7.3 Hz, 5H, PhH), 7.02 (d, J = 7.8 Hz, 3H, PhH), 6.89 (d, J = 7.0 Hz, 2H, PhH), 4.55 (q, J = 7.9 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 3.15 (s, 3H, NCH3), 3.06 – 2.88 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.5, 162.7 (dd, 1JCF = 243.0, 3JCF = 15.2 Hz), 159.4, 159.2, 158.9, 140.5 (t, 3JCF = 14.0 Hz), 137.8, 135.8, 129.2, 128.6, 127.0, 115.2, 103.8 (d, 2JCF = 29.7 Hz), 100.2 (t, 2JCF = 26.6 Hz), 55.9, 52.7, 37.9, 36.7. HRMS (ESI) calcd for C25H23F2N3O4 [M + H]+ 468.1798, found 468.1730.
(S)-N1-(4-cyanophenyl)–N2-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)oxalamide (III-16e): white solid, yield: 62%, HPLC purity 98% (tR = 5.55 min), mp: 117–119 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.03 (s, 1H, NH), 9.00 (d, J = 7.9 Hz, 1H, NH), 7.99 (d, J = 8.1 Hz, 2H, PhH), 7.82 (d, J = 8.0 Hz, 2H, PhH), 7.24 (d, J = 7.5 Hz, 2H, PhH), 7.17 (q, J = 6.9, 6.3 Hz, 3H, PhH), 7.01 (d, J = 8.2 Hz, 2H, PhH), 6.88 (d, J = 7.0 Hz, 2H, PhH), 4.55 (q, J = 7.6, 7.1 Hz, 1H, CH), 3.80 (s, 3H, OCH3), 3.14 (s, 3H, NCH3), 2.96 (h, J = 8.8 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.5, 159.5, 159.2, 159.0, 142.2, 137.8, 135.8, 133.6, 129.3, 128.6, 127.0, 120.9, 119.3, 115.2, 106.9, 55.9, 52.7, 39.3, 37.9, 36.7. HRMS (ESI) calcd for C26H24N4O4 [M + H]+ 457.1798, found 457.1872.
(S)-N1-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)–N2-(4-nitrophenyl)oxalamide (III-16f): white solid, yield: 57%, HPLC purity 98% (tR = 6.96 min), mp: 178–180 °C. 1H NMR (400 MHz, DMSO-d6) δ 11.18 (s, 1H, NH), 9.01 (d, J = 7.9 Hz, 1H, NH), 8.25 (d, J = 8.7 Hz, 2H, PhH), 8.06 (d, J = 8.7 Hz, 2H, PhH), 7.24 (d, J = 7.5 Hz, 2H, PhH), 7.17 (t, J = 7.1 Hz, 3H, PhH), 7.02 (d, J = 8.2 Hz, 2H, PhH), 6.89 (d, J = 6.9 Hz, 2H, PhH), 4.56 (q, J = 7.4, 7.0 Hz, 1H, CH), 3.81 (s, 3H, OCH3), 3.15 (s, 3H, NCH3), 3.05 – 2.87 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.5, 159.5, 159.2, 159.1, 144.1, 143.7, 137.8, 135.8, 129.3, 128.6, 127.0, 125.1, 120.8, 115.2, 55.9, 52.7, 37.9, 36.7. EI-HRMS: m/z 477.1765 [M + H]+, C25H24N4O6 (476.1696).
(S)-N-(4-methoxyphenyl)-N-methyl-2-(2-(5-nitroindolin-1-yl)-2-oxoacetamido)-3-phenylpropanamide (III-16 g): orange solid, yield: 56%, HPLC purity 98% (tR = 4.81 min), mp: 70–72 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.20 (d, J = 7.8 Hz, 1H, NH), 8.15 (p, J = 8.9 Hz, 3H, PhH), 7.21 (q, J = 8.3 Hz, 5H, PhH), 7.04 (d, J = 8.3 Hz, 2H, PhH), 6.87 (d, J = 7.0 Hz, 2H, PhH), 4.61 (d, J = 10.4 Hz, 1H, CH), 3.96 (q, J = 9.7 Hz, 1H, CH2), 3.82 (s, 3H, OCH3), 3.64 (q, J = 9.8 Hz, 1H, CH2), 3.16 (s, 5H, NCH3 + CH2), 2.96 (dd, J = 13.9, 4.5 Hz, 1H, CH2), 2.80 – 2.71 (m, 1H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 170.6, 162.6, 162.1, 159.2, 147.8, 144.1, 137.6, 135.8, 135.1, 129.4, 129.2, 128.7, 127.0, 124.7, 121.0, 116.4, 115.3, 55.9, 51.6, 49.1, 38.7, 37.9, 27.5. HRMS (ESI) calcd for C27H26N4O6 [M + H]+ 503.1852, found 503.1924.
4.1.12. Synthetic route of intermediate(S)-N-(1-((4-methoxyphenyl) (methyl)amino)-1-oxo-3-phenylpropan-2-yl)-3-nitrobenzamide (17)
Intermediate 7 (0.10 g, 0.35 mmol) was dissolved in 8 mL dichloromethane, and the mixture was added triethylamine (0.07 mL, 0.53 mmol). Subsequently, 3-nitrobenzoyl chloride (0.08 g, 0.42 mmol) was added slowly to the mixture at 0°C for 4 h (monitored by TLC). Then the mixture was washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the combined organic phase was washed with saturated sodium chloride aqueous solution (3 × 10 mL) and was dried over anhydrous Na2SO4, filtered, and recrystallized with ethyl acetate and petroleum ether to afford product 17. White solid, yield: 61%. 1H NMR (400 MHz, DMSO-d6) δ 9.15 (d, J = 7.8 Hz, 1H, NH), 8.68 (s, 1H, PhH), 8.37 (dd, J = 8.0, 2.3 Hz, 1H, PhH), 8.24 (d, J = 7.8 Hz, 1H, PhH), 7.76 (t, J = 8.0 Hz, 1H, PhH), 7.29 (d, J = 8.1 Hz, 2H, PhH), 7.16 (dt, J = 11.4, 6.8 Hz, 3H, PhH), 7.04 (d, J = 8.8 Hz, 2H, PhH), 6.90 (d, J = 6.9 Hz, 2H, PhH), 4.70 (dt, J = 8.3, 4.3 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.16 (s, 3H, NCH3), 3.01–2.87 (m, 2H, CH2). ESI-MS: m/z 434 [M + H]+, C24H23N3O5 (433.16).
4.1.13. Synthetic route of (S)-3-amino-N-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl) benzamide (18)
Intermediate 17 (0.30 g, 0.60 mmol) was dissolved in 15 mL anhydrous ethanol. Then SnCl2·2H2O (0.56 g, 2.50 mmol) was added to the mixture. The resulting mixture was reacted for 8 h under nitrogen protection at room temperature (monitored by TLC). The mixture was alkalized to pH 9 with 1 N NaOH and then filtered. The filtrate was extracted with ethyl acetate (3 × 10 mL). The combined organic phase was washed with saturated salt water (3 × 10 mL) and dried over anhydrous Na2SO4, filtered, concentrated under reduced pressure to give the corresponding crude product 18. Yellow solid, yield: 88%. 1H NMR (400 MHz, DMSO-d6) δ 8.35 (d, J = 7.6 Hz, 1H, NH), 7.31 (d, J = 7.9 Hz, 2H, PhH), 7.16 (q, J = 10.6, 8.7 Hz, 3H, PhH), 7.06 (t, J = 8.4 Hz, 3H, PhH), 6.96 (d, J = 12.4 Hz, 2H, PhH), 6.89 (d, J = 7.1 Hz, 2H, PhH), 6.68 (d, J = 7.7 Hz, 1H, PhH), 5.19 (s, 2H, NH2), 4.60 (q, J = 7.7 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.15 (s, 3H, NCH3), 2.90 (h, J = 8.8 Hz, 2H, CH2). ESI-MS: m/z 404 [M + H]+, C24H25N3O3 (403.19).
4.1.14. General procedure for the synthesis of target compounds IV-19 (a–c)
Intermediate 18 (1.0 eq) was dissolved in 15 mL dichloromethane, and the mixture was added triethylamine (1.5 eq). Subsequently, fragments containing substituted benzenesulfonyl chlorides (1.1 eq) was added slowly to the mixture at 0°C for 4 h (monitored by TLC). Then the mixture was washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the combined organic phase was washed with saturated sodium chloride aqueous solution (3 × 10 mL) and was dried over anhydrous Na2SO4, filtered, and recrystallized with ethyl acetate and petroleum ether to afford target products IV-19(a–c).
(S)-3-((4-fluorophenyl)sulfonamido)-N-(1-((4-methoxyphenyl) (methyl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (IV-19a): white solid, yield: 60%, HPLC purity 97% (tR = 4.00 min), mp: 140–142 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.45 (s, 1H, NH), 8.66 (d, J = 7.5 Hz, 1H, NH), 7.79 (dd, J = 8.4, 5.1 Hz, 2H, PhH), 7.51 (s, 2H, PhH), 7.38 (t, J = 8.5 Hz, 2H, PhH), 7.31 (d, J = 7.7 Hz, 3H, PhH), 7.23 (d, J = 8.1 Hz, 1H, PhH), 7.15 (d, J = 7.3 Hz, 3H, PhH), 7.05 (d, J = 8.3 Hz, 2H, PhH), 6.87 (d, J = 6.9 Hz, 2H, PhH), 4.58 (q, J = 7.3 Hz, 1H, CH2), 3.83 (s, 3H, OCH3), 3.15 (s, 3H, NCH3), 2.89 (d, J = 7.3 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.7, 166.2, δ 164.7 (d, 1JCF = 251.8 Hz), 159.1, 138.5, 138.0, 136.1 (d, 3JCF = 4.1 Hz), 135.4, 130.1, 130.0, 129.5, 129.3, 129.2, 128.5, 126.8, 123.6, 123.4, 120.3, 117.0 (d, 2JCF = 22.9 Hz), 115.2, 55.9, 53.1, 37.9, 36.7. HRMS (ESI) calcd for C30H28FN3O5S [M + H]+ 562.1734, found 562.1809.
(S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-3-((4-(trifluoromethoxy)phenyl)sulfonamido)benzamide (IV-19b): white solid, yield: 65%, HPLC purity 98% (tR = 6.95 min), mp: 88–90 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H, NH), 8.69 (d, J = 7.7 Hz, 1H, NH), 7.86 (d, J = 8.0 Hz, 2H, PhH), 7.64 – 7.48 (m, 4H, PhH), 7.32 (d, J = 8.9 Hz, 3H, PhH), 7.23 (d, J = 8.0 Hz, 1H, PhH), 7.15 (q, J = 8.3, 7.2 Hz, 3H, PhH), 7.05 (d, J = 8.2 Hz, 2H, PhH), 6.87 (d, J = 7.1 Hz, 2H, PhH), 4.57 (q, J = 7.3 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.15 (s, 3H, NCH3), 2.88 (d, J = 6.8 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.7, 166.1, 159.1, 151.5, 138.7, 138.5, 137.9, 136.1, 135.5, 129.7, 129.5, 129.3, 129.2, 128.5, 125.2 (d, 1JCF = 307.4 Hz), 123.4, 121.9, 120.4, 115.2, 55.9, 53.1, 40.2, 37.8, 36.7. HRMS (ESI) calcd for C31H28F3N3O6S [M + H]+ 628.1651, found 628.1727.
(S)-3-((3,5-difluorophenyl)sulfonamido)-N-(1-((4-methoxyphenyl) (methyl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (IV-19c): white solid, yield: 51%, HPLC purity 96% (tR = 6.14 min), mp: 172–174 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.57 (s, 1H, NH), 8.64 (d, J = 7.6 Hz, 1H, NH), 7.54 (t, J = 9.7 Hz, 1H, PhH), 7.48 (d, J = 7.6 Hz, 1H, PhH), 7.44 (s, 1H, PhH), 7.34 (d, J = 4.2 Hz, 2H, PhH), 7.29 (d, J = 7.9 Hz, 1H, PhH), 7.27 – 7.21 (m, 2H, PhH), 7.18 (d, J = 8.1 Hz, 1H, PhH), 7.07 (q, J = 8.1, 7.0 Hz, 3H, PhH), 6.97 (d, J = 8.2 Hz, 2H, PhH), 6.79 (d, J = 7.0 Hz, 2H, PhH), 4.50 (q, J = 7.3 Hz, 1H, CH), 3.75 (s, 3H, OCH3), 3.08 (s, 3H, NCH3), 2.81 (d, J = 7.0 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.7, 166.1, 162.5 (dd, 1JCF = 252.1, 3JCF = 12.4 Hz), 159.1, 143.0 (t, 3JCF = 8.3 Hz), 138.5, 137.4, 136.1, 135.6, 129.6, 129.3, 129.2, 128.5, 126.8, 124.1, 123.8, 120.7, 115.2, 110.8 (d, 2JCF = 28.3 Hz), 109.3 (t, 2JCF = 26.2 Hz), 55.9, 53.1, 37.9, 36.7. HRMS (ESI) calcd for C30H27F2N3O5S [M + H]+ 580.1639, found 580.1713.
4.1.15. General procedure for the synthesis of intermediates 21(a–g)
Amine fragments (1.0 eq) were dissolved in 15 mL dichloromethane, and the mixture was added triethylamine (1.5 eq). Subsequently, methyl 3-(chlorosulfonyl) benzoate (20) (1.1 eq) was added to the mixture at 0°C. Then the mixture was stirred for another 4 h (monitored by TLC), washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). Then the combined organic phase was washed with saturated salt water (3 × 10 mL) and was dried over anhydrous Na2SO4, filtered, and then the crude product were recrystallized with ethyl acetate and petroleum ether to afford intermediates 21(a–g).
Methyl 3-(thiomorpholine sulfonyl) benzoate (21a): white solid, yield: 68%. 1H NMR (400 MHz, DMSO-d6) δ 8.28 (d, J = 7.7 Hz, 1H, PhH), 8.20 (s, 1H, PhH), 8.04 (d, J = 7.6 Hz, 1H, PhH), 7.84 (t, J = 7.7 Hz, 1H, PhH), 3.91 (s, 3H, OCH3), 3.29 – 3.14 (m, 4H, CH2 × 2), 2.77 – 2.60 (m, 4H, CH2 × 2). ESI-MS: m/z 302 [M + H]+, 324 [M + Na]+, C12H15NO4S2 (301.04).
Methyl 3-(indole-1-ylsulfonyl) benzoate (21b): white solid, yield: 62%. 1H NMR (400 MHz, DMSO-d6) δ 8.25 (s, 1H, PhH), 8.22 (d, J = 7.8 Hz, 1H, PhH), 8.08 (d, J = 7.8 Hz, 1H, PhH), 7.75 (t, J = 7.8 Hz, 1H, PhH), 7.50 (d, J = 8.1 Hz, 1H, PhH), 7.23 (t, J = 7.8 Hz, 1H, PhH), 7.17 (d, J = 7.3 Hz, 1H, PhH), 7.01 (t, J = 7.4 Hz, 1H, PhH), 3.94 (t, J = 8.3 Hz, 2H, CH2), 3.88 (s, 3H, OCH3), 2.90 (t, J = 8.3 Hz, 2H, CH2). ESI-MS: m/z 318 [M + H]+, 340 [M + Na]+, C16H15NO4S (317.07).
Methyl 3-((5-methoxyindolyl)sulfonyl)benzoate (21c): white solid, yield: 68%. 1H NMR (400 MHz, DMSO-d6) δ 8.21 (d, J = 7.8 Hz, 1H, PhH), 8.18 (s, 1H, PhH), 7.99 (d, J = 7.7 Hz, 1H, PhH), 7.73 (t, J = 7.8 Hz, 1H, PhH), 7.42 (d, J = 8.7 Hz, 1H, PhH), 6.81 (d, J = 8.9 Hz, 1H, PhH), 6.77 (s, 1H, PhH), 3.92 (t, J = 8.2 Hz, 2H, CH2), 3.88 (s, 3H, OCH3), 3.69 (s, 3H, OCH3), 2.75 (t, J = 8.2 Hz, 2H, CH2). ESI-MS: m/z 347 [M + H]+, 369 [M + Na]+, C17H17NO5S (347.08).
Methyl 3-((5-nitroindole-1-yl)sulfonyl)benzoate (21d): brown solid, yield: 76%. 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H, PhH), 8.27 (d, J = 7.7 Hz, 1H, PhH), 8.20 (d, J = 7.9 Hz, 1H, PhH), 8.15 (d, J = 8.8 Hz, 1H, PhH), 8.06 (s, 1H, PhH), 7.80 (t, J = 8.0 Hz, 1H, PhH), 7.64 (d, J = 8.9 Hz, 1H, PhH), 4.08 (t, J = 8.5 Hz, 2H, CH2), 3.89 (s, 3H, OCH3), 3.12 (t, J = 8.5 Hz, 2H, CH2). ESI-MS: m/z 363 [M + H]+, C16H14N2O6S (362.06).
Methyl 3-(N-(4-methoxyphenyl)-N-methylsulfonyl) benzoate (21e): white solid, yield: 62%. 1H NMR (400 MHz, DMSO-d6) δ 8.34 (s, 1H, PhH), 8.27 (d, J = 7.7 Hz, 1H, PhH), 8.20 (d, J = 7.9 Hz, 1H, PhH), 8.15 (d, J = 8.8 Hz, 1H, PhH), 8.06 (s, 1H, PhH), 7.80 (t, J = 8.0 Hz, 1H, PhH), 7.64 (d, J = 8.9 Hz, 1H, PhH), 4.08 (t, J = 8.5 Hz, 2H, CH2), 3.89 (s, 3H), 3.12 (d, J = 17.0 Hz, 2H, CH2). ESI-MS: m/z 336 [M + H]+, C16H17NO5S (335.08).
Methyl 3-(N-(3,5-difluorophenyl) sulfamoyl) benzoate (21f): white solid, yield: 65%. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H, NH), 8.35 (s, 1H, PhH), 8.21 (d, J = 7.5 Hz, 1H, PhH), 8.08 (s, 1H, PhH), 7.77 (t, J = 8.0 Hz, 1H, PhH), 6.93 (t, J = 9.6 Hz, 1H, PhH), 6.78 (d, J = 8.0 Hz, 2H, PhH), 3.90 (s, 3H, OCH3). ESI-MS: m/z 326 [M − H]−, C14H11F2NO4S (327.04).
Methyl 3-(N-(4-(trifluoromethyl)phenyl)sulfamoyl)benzoate (21 g): white solid, yield: 71%. 1H NMR (400 MHz, DMSO-d6) δ 11.02 (s, 1H, NH), 8.36 (s, 1H, PhH), 8.19 (d, J = 8.0 Hz, 1H, PhH), 8.07 (s, 1H, PhH), 7.75 (t, J = 7.7 Hz, 1H, PhH), 7.63 (d, J = 8.2 Hz, 2H, PhH), 7.30 (d, J = 8.3 Hz, 2H, PhH), 3.89 (s, 3H, OCH3). ESI-MS: m/z 358 [M − H]−, C15H12F3NO4S (359.04).
4.1.16. General procedure for the synthesis of intermediates V-25(a–j)
21(a-g) (1.0 eq) was dissolved in 10 mL THF. Then equal volume NaOH solution (2.0 eq) was added to the mixture. The mixture was stirred at room temperature for 2 h (monitored by TLC). Subsequently, the resulting mixture solution was alkalized to pH 2 with 1 N HCl and filtered to obtain the crude product 22(a-g). Then 22(a-g) (1.0 eq) or commercial available 23, 24 and PyBop (1.5 eq) was dissolved in dichloromethane. The mixture was stired at room temperature for 0.5 h. Then DIEA (2.0 eq) and intermediates 7 was added to the mixture for another 8 h (monitored by TLC). The resulting mixture was initially washed by saturated sodium bicarbonate (15 mL) and extracted with dichloromethane (3 × 8 mL). The combined organic layer was washed by 1 N HCl and extracted with dichloromethane (3 × 8 mL). Then the combined organic phase was washed with saturated salt water (3 × 10 mL), dried over anhydrous Na2SO4, filtered, and recrystallized with ethyl acetate and petroleum ether to afford product V-25(a–j).
(S)-3-(N,N-dimethylsulfamoyl)-N-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)benzamide (V-25a): red solid, yield: 68%, HPLC purity 97% (tR = 4.38 min), mp: 80–82 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (d, J = 7.7 Hz, 1H, NH), 8.19 (s, 1H, PhH), 8.15 (d, J = 7.8 Hz, 1H, PhH), 7.89 (d, J = 7.7 Hz, 1H, PhH), 7.74 (t, J = 7.7 Hz, 1H, PhH), 7.33 (d, J = 7.7 Hz, 2H, PhH), 7.15 (q, J = 8.5, 7.2 Hz, 3H, PhH), 7.06 (d, J = 8.4 Hz, 2H, PhH), 6.90 (d, J = 6.8 Hz, 2H, PhH), 4.67 (q, J = 7.4 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.17 (s, 3H, NCH3), 2.95 (d, J = 7.3 Hz, 2H, CH2), 2.63 (s, 6H, CH3 × 2). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 165.1, 159.1, 138.5, 136.0, 135.5, 135.1, 132.3, 130.6, 130.0, 129.3, 129.2, 128.5, 126.8, 126.7, 115.2, 55.9, 53.2, 38.7, 38.0, 36.7. HRMS (ESI) calcd for C26H29N3O5S [M + H]+ 496.1828, found 496.1901.
(S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-3-(piperidin-1-ylsulfonyl)benzamide (V-25b): white solid, yield: 70%, HPLC purity 96% (tR = 4.01 min), mp: 70–72 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (d, J = 7.6 Hz, 1H, NH), 8.15 (q, J = 8.5, 8.0 Hz, 2H, PhH), 7.86 (d, J = 7.7 Hz, 1H, PhH), 7.72 (t, J = 7.7 Hz, 1H, PhH), 7.32 (d, J = 7.9 Hz, 2H, PhH), 7.14 (d, J = 6.8 Hz, 3H, PhH), 7.05 (d, J = 8.2 Hz, 2H, PhH), 6.89 (d, J = 6.9 Hz, 2H, PhH), 4.66 (q, J = 7.0 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.16 (s, 3H, NCH3), 2.94 (d, J = 7.2 Hz, 2H, CH2), 2.89 (s, 4H, CH2 × 2), 1.54 (s, 4H, CH2 × 2), 1.44 – 1.29 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 165.1, 159.1, 138.5, 136.4, 136.0, 135.1, 132.2, 130.5, 130.0, 129.3, 129.2, 128.5, 126.8, 126.6, 115.2, 55.9, 53.2, 47.0, 38.7, 37.9, 25.1, 23.2. HRMS (ESI) calcd for C29H33N3O5S [M + H]+ 536.2241, found 436.2215. (S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-3-(thio-morpholinosulfonyl)benzamide (V-25c): white solid, yield: 65%, HPLC purity 96% (tR = 5.83 min), mp: 115–117 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (d, J = 7.6 Hz, 1H, NH), 8.17 (s, 1H, PhH), 8.14 (d, J = 7.8 Hz, 1H, PhH), 7.90 (d, J = 7.8 Hz, 1H, PhH), 7.75 (t, J = 7.7 Hz, 1H, PhH), 7.33 (d, J = 8.0 Hz, 2H, PhH), 7.16 (q, J = 8.5, 7.5 Hz, 3H, PhH), 7.06 (d, J = 8.2 Hz, 2H, PhH), 6.90 (d, J = 7.1 Hz, 2H, PhH), 4.67 (q, J = 7.6 Hz, 1H, CH), 3.83 (s, 3H, OCH3), 3.24 (s, 4H, , CH2 × 2), 3.17 (s, 3H, NCH3), 3.03 – 2.90 (m, 2H, CH2), 2.69 (d, J = 6.1 Hz, 4H, CH2 × 2). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 165.0, 159.1, 138.4, 137.1, 136.0, 135.3, 132.5, 130.2, 129.2, 128.5, 126.8, 126.4, 115.2, 55.9, 53.2, 48.2, 37.9, 36.8, 26.8. HRMS (ESI) calcd for C28H31N3O5S2 [M + H]+ 554.1705, found 554.1779. (S)-3-(indolin-1-ylsulfonyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (V-25d): white solid, yield: 58%, HPLC purity 98% (tR = 7.35 min), mp: 82–84 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.10 (d, J = 7.8 Hz, 1H, PhH), 8.31 (s, 1H, NH), 8.08 (d, J = 7.8 Hz, 1H, PhH), 7.91 (d, J = 7.8 Hz, 1H, PhH), 7.63 (t, J = 7.8 Hz, 1H, PhH), 7.49 (d, J = 8.0 Hz, 1H, PhH), 7.31 (d, J = 8.2 Hz, 2H, PhH), 7.16 (dd, J = 17.6, 9.4 Hz, 5H, PhH), 7.04 (d, J = 8.2 Hz, 2H, PhH), 6.97 (t, J = 7.4 Hz, 1H, PhH), 6.87 (d, J = 6.7 Hz, 2H, PhH), 4.64 (q, J = 7.5 Hz, 1H, PhH), 3.97 (dq, J = 16.9, 9.4 Hz, 2H, CH2), 3.81 (s, 3H, OCH3), 3.16 (s, 3H, NCH3), 3.01 – 2.83 (m, 4H, CH2 × 2). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 164.8, 159.1, 141.4, 138.4, 136.9, 136.0, 135.1, 133.0, 132.5, 130.1, 129.3, 129.2, 128.5, 128.0, 126.8, 126.4, 126.0, 124.3, 115.2, 114.5, 55.9, 53.2, 50.5, 37.9, 36.8, 27.6. HRMS (ESI) calcd for C32H31N3O5S [M + H]+ 570.1984, found 570.2055.
(S)-3-((5-methoxyindolin-1-yl)sulfonyl)-N-(1-((4-methoxyphenyl) (methyl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (V-25e): white solid, yield: 72%, HPLC purity 96% (tR = 6.98 min), mp: 90–92 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (d, J = 7.7 Hz, 1H, NH), 8.27 (s, 1H, PhH), 8.07 (d, J = 7.6 Hz, 1H, PhH), 7.81 (d, J = 7.8 Hz, 1H, PhH), 7.62 (t, J = 7.8 Hz, 1H, PhH), 7.41 (d, J = 9.1 Hz, 1H, PhH), 7.30 (d, J = 7.9 Hz, 2H, PhH), 7.15 (d, J = 6.3 Hz, 3H, PhH), 7.04 (d, J = 8.2 Hz, 2H, PhH), 6.89 (d, J = 6.6 Hz, 2H, PhH), 6.75 (s, 2H, PhH), 4.65 (q, J = 7.5 Hz, 1H, CH), 3.97 (dq, J = 16.9, 9.6, 9.1 Hz, 2H, CH2), 3.82 (s, 3H, OCH3), 3.67 (s, 3H, OCH3), 3.17 (s, 3H, NCH3), 2.93 (d, J = 7.4 Hz, 2H, CH2), 2.86 – 2.75 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 164.9, 159.1, 156.9, 138.4, 136.8, 136.0, 135.1, 134.7, 134.6, 132.9, 130.1, 130.0, 129.3, 129.2, 128.5, 126.8, 126.5, 116.1, 115.2, 113.2, 111.5, 55.9, 55.8, 53.2, 50.8, 38.7, 37.9, 28.1. HRMS (ESI) calcd for C33H33N3O6S [M + H]+ 600.2090, found 600.2162.
(S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-3-((5-nitroindolin-1-yl)sulfonyl)benzamide (V-25f): white solid, yield: 71%, HPLC purity 99% (tR = 4.68 min), mp: 98–100 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.12 (d, J = 7.8 Hz, 1H, NH), 8.38 (s, 1H, PhH), 8.13 (t, J = 9.4 Hz, 2H, PhH), 8.06 (s, 2H, PhH), 7.70 (dd, J = 19.3, 8.5 Hz, 2H, PhH), 7.32 (d, J = 8.0 Hz, 2H, PhH), 7.12 (d, J = 6.0 Hz, 3H, PhH), 7.05 (d, J = 8.1 Hz, 2H, PhH), 6.88 (d, J = 6.5 Hz, 2H, PhH), 4.67 (q, J = 7.5 Hz, 1H, CH), 4.11 (dq, J = 18.5, 9.1 Hz, 2H, CH2), 3.82 (s, 3H, OCH3), 3.17 (s, 3H, NCH3), 3.13 (d, J = 8.6 Hz, 2H, CH2), 2.94 (d, J = 6.7 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.5, 164.7, 159.1, 147.3, 143.6, 138.4, 136.6, 136.0, 135.3, 134.2, 133.6, 130.6, 130.1, 129.3, 129.2, 128.5, 126.8, 126.3, 124.9, 121.7, 115.2, 113.2, 55.9, 53.2, 51.3, 38.7, 37.9, 27.0. HRMS (ESI) calcd for C32H30N4O7S [M + H]+ 615.1835, found 615.1907.
(S)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)-3-(N-(4-methoxyphenyl)-N-methylsulfamoyl)benzamide (V-25 g): yellow solid, yield: 58%, HPLC purity 96% (tR = 6.18 min), mp: 81–83 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.06 (d, J = 7.7 Hz, 1H, NH), 8.13 (d, J = 7.7 Hz, 1H, PhH), 8.06 (s, 1H, PhH), 7.66 (t, J = 7.8 Hz, 1H, PhH), 7.57 (d, J = 7.7 Hz, 1H, PhH), 7.30 (d, J = 8.0 Hz, 2H, PhH), 7.16 (d, J = 7.0 Hz, 3H, PhH), 7.05 (d, J = 8.2 Hz, 2H, PhH), 6.98 (d, J = 8.3 Hz, 2H, PhH), 6.89 (t, J = 9.6 Hz, 4H, PhH), 4.65 (q, J = 7.5 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.74 (s, 3H, OCH3), 3.17 (s, 3H, NCH3), 3.14 (s, 3H, NCH3), 2.94 (d, J = 5.2 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 165.1, 159.1, 158.7, 138.4, 137.0, 136.0, 135.0, 133.8, 132.3, 130.5, 129.8, 129.3, 129.2, 128.6, 128.3, 126.8, 115.2, 114.6, 55.9, 55.7, 53.2, 38.7, 37.9, 36.8. HRMS (ESI) calcd for C32H33N3O6S [M + H]+ 588.2090, found 588.2171. (S)-3-(N-(3,5-difluorophenyl)sulfamoyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (V-25 h): white solid, yield: 63%, HPLC purity 97% (tR = 4.37 min), mp: 100–102 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.99 (s, 1H, NH), 9.09 (d, J = 7.7 Hz, 1H, NH), 8.31 (s, 1H, PhH), 8.07 (d, J = 7.8 Hz, 1H, PhH), 7.95 (d, J = 7.8 Hz, 1H, PhH), 7.69 (t, J = 7.7 Hz, 1H, PhH), 7.30 (d, J = 7.8 Hz, 2H, PhH), 7.15 (d, J = 6.9 Hz, 3H, PhH), 7.05 (t, J = 8.4 Hz, 2H, PhH), 6.90 (t, J = 9.3 Hz, 3H, PhH), 6.77 (d, J = 8.1 Hz, 2H, PhH), 4.64 (q, J = 7.7, 7.0 Hz, 1H, CH), 3.82 (d, J = 1.6 Hz, 3H, OCH3), 3.16 (s, 3H, NCH3), 2.93 (t, J = 10.1 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.5, 165.0, 163.0 (dd, 1JCF = 245.3, 3JCF = 15.4 Hz), 159.1, 140.7 (t, 3JCF = 13.4 Hz), 139.6, 138.4, 136.0, 135.3, 132.6, 130.2, 129.7, 129.3, 129.2, 128.5, 126.8, 126.3, 115.2, 102.5 (d, 2JCF = 29.0 Hz), 99.6 (t, 2JCF = 26.0 Hz), 55.9, 53.2, 37.9, 36.8. HRMS (ESI) calcd for C30H27F2N3O5S [M + H]+ 580.1639, found 580.1712. (S)-3-((5-aminoindolin-1-yl)sulfonyl)-N-(1-((4-methoxyphenyl)(methyl)amino)-1-oxo-3-phenylpropan-2-yl)benzamide (V-25i): yellow solid, yield: 72%, HPLC purity 99% (tR = 4.31 min), mp: 95–97 °C. 1H NMR (400 MHz, DMSO-d6) δ 9.07 (d, J = 7.7 Hz, 1H, NH), 8.26 (s, 1H, PhH), 8.04 (d, J = 7.7 Hz, 1H, PhH), 7.70 (d, J = 7.7 Hz, 1H, PhH), 7.58 (t, J = 7.7 Hz, 1H, PhH), 7.31 (d, J = 8.0 Hz, 2H, PhH), 7.22 – 7.11 (m, 4H, PhH), 7.05 (d, J = 8.3 Hz, 2H, PhH), 6.89 (d, J = 6.9 Hz, 2H, PhH), 6.40 (d, J = 8.5 Hz, 1H, PhH), 6.31 (s, 1H, PhH), 4.93 (s, 2H, NH2), 4.65 (q, J = 7.5 Hz, 1H, CH), 3.89 (dd, J = 18.8, 9.6 Hz, 2H, CH2), 3.82 (s, 3H, OCH3), 3.17 (s, 3H, NCH3), 2.93 (d, J = 8.4 Hz, 2H, CH2), 2.57 (q, J = 8.9, 8.0 Hz, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.6, 164.9, 159.1, 146.6, 138.4, 137.1, 136.0, 135.0, 134.2, 132.6, 130.8, 130.1, 129.8, 129.3, 129.2, 128.6, 126.8, 126.5, 116.8, 115.2, 113.1, 111.0, 55.9, 53.1, 50.7, 37.9, 36.8, 28.2. HRMS (ESI) calcd for C32H32N4O5S [M + H]+ 585.2093, found 585.2171. (S)-N-(1-((4-methoxyphenyl)(methyl) amino)-1-oxo-3-phenylpropan-2-yl)-3-(N-(4-(trifluoromethyl)phenyl)sulfamoyl)benzamide (V-25j): white solid, yield: 71%, HPLC purity 97% (tR = 5.55 min), mp: 102–104 °C. 1H NMR (400 MHz, DMSO-d6) δ 10.97 (s, 1H, NH), 9.06 (d, J = 7.7 Hz, 1H, NH), 8.32 (s, 1H, PhH), 8.06 (d, J = 7.6 Hz, 1H, PhH), 7.94 (d, J = 7.9 Hz, 1H, PhH), 7.67 (t, J = 7.7 Hz, 1H, PhH), 7.61 (d, J = 8.2 Hz, 2H, PhH), 7.30 (d, J = 8.2 Hz, 4H, PhH), 7.14 (d, J = 6.8 Hz, 3H, PhH), 7.03 (d, J = 8.1 Hz, 2H, PhH), 6.89 (d, J = 7.0 Hz, 2H, PhH), 4.64 (q, J = 7.4, 5.9 Hz, 1H, CH), 3.82 (s, 3H, OCH3), 3.16 (s, 3H, NCH3), 3.00 – 2.88 (m, 2H, CH2). 13C NMR (100 MHz, DMSO-d6) δ 171.5, 165.0, 159.1, 141.7, 139.9, 138.4, 136.0, 135.2, 132.4, 130.1, 129.6, 129.3, 129.2, 128.5, 127.0 (q, 3JCF = 3.3 Hz), 124.6 (q, 1JCF = 271.4 Hz), 124.3 (q, 2JCF = 32.2 Hz), 119.2, 115.2, 55.9, 53.1, 37.9, 36.8. ESI-MS: m/z 612 [M + H]+, 634 [M + Na]+, C31H28F3N3O5S (611.1702).
4.2. In vitro anti-HIV assay
4.2.1. Assessment of inhibitory activity on HIV-1 replication in MT-4 cells
Inhibitory activity of compounds against HIV-1 infection in MT-4 cells was measured as the reduction in luciferase gene expression after multiple rounds of virus infection of the cells. In brief, 200 TCID50 of virus (HIV-1 NL4–3 Nanoluc-sec) was used to infect MT-4 cells (1 × 105 cells/mL) in the presence of various concentrations of compounds. The mixture incubates at 37°C with 5% CO2 for three days. Then the culture medium was removed from each well and 100 μL of Bright Glo reagent (Promega, Luis Obispo, CA) was added to the cells for measurement of luminescence using a Victor 2 luminometer. The effective concentration (EC50) against HIV-1 strains was defined as the concentration that caused a 50% reduction of luciferase activity (Relative Light Units) compared to virus control wells.
4.2.2. Cytotoxicity assay
Parallel to the antiviral assays, MT-4 cells were cultured in the presence of various concentrations of the compounds and incubate at 37°C with 5% CO2 for three days. Subsequently, 2, 3-bis-(2-methoxy-4-nitro-5-sulfophenyl)–2H- tetrazole-5-formylaniline of phenazine methyl sulfate (XTT) was added to the mixture. Four hours later, the absorbance was measured at 450 nm. The 50% cytotoxic concentration (CC50) was defined as the concentration that caused a 50% reduction in cell viability.
4.3. Binding to CA proteins analysis via surface plasmon resonance (SPR)
Binding of II-13c, V-25i, and PF-74 was examined using a ProteOn XPR36 SPR Protein Interaction Array System (Bio-Rad) at 25 °C. ProteOn GLH sensor chips were preconditioned with two 10 s pulses of 50 mM NaOH, 100 mM HCl, and 0.5 % SDS followed by the system equilibration with the running buffer (20 mM sodium phosphate, 150 mM NaCl, and 0.005% Tween 20, pH 7.4). The surface of a GLH sensorchip was subsequently activated with a 1:100 dilution of a 1:1 mixture of 0.2 M EDC and 0.05 M Sulfo-NHS. Purified HsRAD51 was diluted to 500 μg/ml in 10 mM sodium acetate, pH 5.5, and injected immediately after chip activation across the ligand flow channels at 30 μL/min for 5 min. Unreacted protein was washed out, and the excess of unreacted ester groups on the sensor surface was capped by an injection of 1 M ethanolamine-HCl, pH 8.0, at 5 μL/min for 5 min. A reference surface to correct for nonspecific binding was similarly created by immobilizing an IgG b12 anti-HIV-1 gp120 antibody (IgG b12 anti-HIV-1 gp120; was obtained through the NIH AIDS Reagent Program, Division of AIDS, NIAID, NIH: Anti-HIV-1 gp120 Monoclonal (IgG1 b12) from Drs. Dennis Burton and Carlos Barbas). Serial dilutions of the compounds were prepared in the running buffer supplemented with 3% DMSO and injected at a flow rate of 100 μL/min for a 1 min association phase, followed by 5 min dissociation phase using the “one-shot kinetics” functionality of the ProteOn instrument. Data were analyzed with ProteOn Manager Software version 3.0 (Bio-Rad). The responses from the reference flow cell were subtracted to account for the nonspecific binding and injection artifacts. Experiments were repeated three times. The equilibrium dissociation constants (KD) for the interactions were calculated using a one-site binding model.
4.4. Molecular dynamics simulation
The coordinate structure of capsid protein (PDB code: 5HGL), was collected from RCSB website (https://www.rcsb.org) for ligand–protein complex interaction analysis. The computational work performed using Schrodinger software (Schrodinger release 2020–1 license dated 20 November 2020). As crystal structure possess several structural defects such as missing hydrogen atoms, charge states, side-chain missing, loop missing, inappropriate bond order, and atomic clashes38,39. These issues need to be resolved prior docking study by using protein preparation wizard. The compounds II-13c and V-25i were also prepared by Ligprep tool prior to docking39. Schrödinger suite inbuilt Epik module was also used to predict the ionization states of all compounds at pH 7 ± 2 as well as tautomers generated. This in-silico study was done under OPLS2005 forcefield.
Site specific molecular docking of both compounds against HIV-1 capsid protein performed at XP precision using the Glide module of Schrödinger suite. The Van der Waals radii scaling factor and partial charge cutoff was 0.8 and 0.15 used for docking, respectively. The binding free energy for these three complexes were also calculated by prime MMGBSA.
To validate the docking results, both complexes of compound II-13c and V-25i with capsid protein were selected for extensive 50 ns MD simulation. Both the complexes were studied for the binding stability of both compounds within their respective complex. These complexes were solvated in TIP3P water model and 0.15 M NaCl to mimic a physiological ionic concentration. The stereo-chemical geometry of 5HGL protein residues was measured by Ramachandran map by Procheck40.
Supplementary Material
Acknowledgements
We gratefully acknowledge financial support from the the National Natural Science Foundation of China (NSFC No. 82173677), Shandong Provincial Key research and development project (No. 2019JZZY021011), Science Foundation for Outstanding Young Scholars of Shandong Province (ZR2020JQ31), Foreign cultural and educational experts Project (GXL20200015001), Qilu Young Scholars Program of Shandong University and the Taishan Scholar Program at Shandong Province, NIH grants R01GM125396 (transitioning to R01AI150491) (Cocklin, PI) and T32-MH079785 are gratefully acknowledged.
Footnotes
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Appendix A. Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.bmc.2021.116414.
References
- 1.De Clercq E Antivirals: past, present and future. Biochem Pharmacol. 2013;85(6):727–744. [DOI] [PubMed] [Google Scholar]
- 2.Menéndez-Arias L Molecular basis of human immunodeficiency virus type 1 drug resistance: overview and recent developments. Antiviral Res. 2013;98(1):93–120. [DOI] [PubMed] [Google Scholar]
- 3.Zhang J, Crumpacker C. Eradication of HIV and Cure of AIDS, Now and How? Front Immunol. 2013;4:337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zuo X, Huo Z, Kang D, et al. Current insights into anti-HIV drug discovery and development: a review of recent patent literature (2014–2017). Expert Opin Ther Pat. 2018;28(4):299–316. [DOI] [PubMed] [Google Scholar]
- 5.Zhou J, Price AJ, Halambage UD, James LC, Aiken C, Sundquist WI. HIV-1 resistance to the capsid-targeting inhibitor PF74 results in altered dependence on host factors required for virus nuclear entry. J Virol. 2015;89(17):9068–9079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Sundquist WI, Krausslich H-G. HIV-1 assembly, budding, and maturation. Cold Spring Harb Perspect Med. 2012;2(7):a006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gres AT, Kirby KA, KewalRamani VN, Tanner JJ, Pornillos O, Sarafianos SG. X-ray crystal structures of native HIV-1 capsid protein reveal conformational variability. Science. 2015;349(6243):99–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chen N-Y, Zhou L, Gane PJ, et al. HIV-1 capsid is involved in post-nuclear entry steps. Retrovirology. 2016;13(1). 10.1186/s12977-016-0262-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Thenin-Houssier S, Valente ST. HIV-1 capsid inhibitors as antiretroviral agents. Curr HIV Res. 2016;14(3):270–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lahaye X, Satoh T, Gentili M, et al. The capsids of HIV-1 and HIV-2 determine immune detection of the viral cDNA by the innate sensor cGAS in dendritic cells. Immunity. 2013;39(6):1132–1142. [DOI] [PubMed] [Google Scholar]
- 11.Yamashita M, Engelman AN. Capsid-dependent host factors in HIV-1 infection. Trends Microbiol. 2017;25(9):741–755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rasaiyaah J, Tan CP, Fletcher AJ, et al. HIV-1 evades innate immune recognition through specific cofactor recruitment. Nature. 2013;503(7476):402–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang W, Zhou J, Halambage UD, et al. Inhibition of HIV-1 maturation via small-molecule targeting of the amino-terminal domain in the viral capsid protein. J Virol. 2017;91(9). 10.1128/JVI.02155-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Campbell EM, Hope TJ. HIV-1 capsid: the multifaceted key player in HIV-1 infection. Nat Rev Microbiol. 2015;13(8):471–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Zhang JY, Liu XY, De Clercq E. Capsid (CA) Protein as a novel drug target recent progress in the research of HIV-1 CA inhibitors. Mini Rev Med Chem. 2009;9(4):510–518. [DOI] [PubMed] [Google Scholar]
- 16.Sun L, Zhang XJ, Xu SJ, et al. An insight on medicinal aspects of novel HIV-1 capsid protein. Eur J Med Chem. 2021;217, 113380. [DOI] [PubMed] [Google Scholar]
- 17.Xu SJ, Sun L, Huang BS, et al. Medicinal chemistry strategies of targeting. Future Med Chem. 2020;12(14):1281–1284. [DOI] [PubMed] [Google Scholar]
- 18.Xu JP, Francis AC, Meuser ME, et al. Exploring Modifications of an HIV-1 Capsid Inhibitor: Design, Synthesis, and Mechanism of Action. J Drug Des Res. 2018; 5 (2): 1070. [PMC free article] [PubMed] [Google Scholar]
- 19.Tang C, Loeliger E, Kinde I, et al. Antiviral Inhibition of the HIV-1 Capsid Protein. Journal of Molecular Biology. 2003; 327 (5): 1013–1020. [DOI] [PubMed] [Google Scholar]
- 20.Brian NK, Sampson NK, Isaac K, et al. Structure of the antiviral assembly inhibitor CAP-1 bound to the HIV-1 CA protein. J Mol Biol. 2007;373(2):355–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sticht J, Humbert M, Findlow S, et al. A peptide inhibitor of HIV-1 assembly in vitro. Nat Struct Mol Biol. 2005;12(8):671–677. [DOI] [PubMed] [Google Scholar]
- 22.Lamorte L, Titolo S, Lemke CT, et al. Discovery of novel small-molecule HIV-1 replication inhibitors that stabilize capsid complexes. Antimicrob Agents Chemothe. 2013;57(10):4622–4631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Blair WS, Pickford C, Irving SL, et al. HIV capsid is a tractable target for small molecule therapeutic intervention. PLoS Pathog. 2010;6(12):e1001220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ganser-Pornillos BK, Cheng A, Yeager M. Structure of full-length HIV-1 CA: a model for the mature capsid lattice. Cell. 2007;131(1):70–79. [DOI] [PubMed] [Google Scholar]
- 25.Bhattacharya A, Alam SL, Fricke T, et al. Structural basis of HIV-1 capsid recognition by PF74 and CPSF6. Proc Natl Acad Sci U S A. 2014;111(52):18625–18630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Price AJ, Jacques DA, McEwan WA, et al. Host cofactors and pharmacologic ligands share an essential interface in HIV-1 capsid that is lost upon disassembly. PLoS Pathog. 2014;10(10):e1004459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Jimmy PX, Ashwanth CF, Megan EM, et al. Exploring Modifications of an HIV-1 Capsid Inhibitor Design. J Drug Des Res. 2018;5(2):1070. [PMC free article] [PubMed] [Google Scholar]
- 28.Kortagere S, Madani N, Mankowski MK, et al. Inhibiting early-stage events in HIV-1 replication by small-molecule targeting of the HIV-1 capsid. J Virol. 2012;86(16):8472–8481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Wu G, Zalloum WA, Meuser ME, et al. Discovery of phenylalanine derivatives as potent HIV-1 capsid inhibitors from click chemistry-based compound library. Eur J Med Chem. 2018;158:478–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bester SM, Wei GC, Zhao HY, et al. Structural and mechanistic bases for a potent HIV-1 capsid inhibitor. Science. 2020; 370(6514): 360–364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang XY, Wu GC, Zalloum WA, et al. Discovery of novel 1,4-disubstituted 1,2,3-triazole phenylalanine derivatives as HIV-1 capsid inhibitors. RSC Adv. 2019; 9 (50): 28961–28986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang L, Casey MC, Vernekar SKV, et al. Novel PF74-like small molecules targeting the HIV-1 capsid protein: Balance of potency and metabolic stability. Acta Pharm Sin B. 2021;11(3):810–822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Sun L, Huang T, Dick A, et al. Design, synthesis and structure-activity relationships of 4-phenyl-1H-1,2,3-triazole phenylalanine derivatives as novel HIV-1 capsid inhibitors with promising antiviral activities. Eur J Med Chem. 2020;190:112085. 10.1016/j.ejmech.2020.112085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jiang X, Yu Ji, Zhou Z, et al. Molecular design opportunities presented by solvent-exposed regions of target proteins. Med Res Rev. 2019;39(6):2194–2238. [DOI] [PubMed] [Google Scholar]
- 35.Xu JP, Francis JP, Meuser ME, et al. Exploring Modifications of an HIV-1 Capsid Inhibitor Design, Synthesis, and Mechanism of Action. J Drug Des Res. 2018;5(2):1070. [PMC free article] [PubMed] [Google Scholar]
- 36.Du J, Guo J, Kang D, et al. New techniques and strategies in drug discovery. Chin Chem Lett. 2020;31(7):1695–1708. [Google Scholar]
- 37.Na Liu, Wei L Huang Li, et al. Novel HIV-1 Non-nucleoside reverse transcriptase inhibitor agents: Optimization of diarylanilines with high potency against wild-type and rilpivirine-resistant E138K mutant virus. J Med Chem. 2016;59(8):3689–3704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Madhavi Sastry G, Adzhigirey M, Day T, Annabhimoju R, Sherman W. Protein and ligand preparation: parameters, protocols, and influence on virtual screening enrichments. J Comput Aided Mol Des. 2013;27(3):221–234. [DOI] [PubMed] [Google Scholar]
- 39.Kumar S, Sharma PP, Shankar U, et al. Discovery of New Hydroxyethylamine Analogs against 3CLpro Protein Target of SARS-CoV-2: Molecular Docking, Molecular Dynamics Simulation, and Structure-Activity Relationship Studies. J Comput Aided Mol Des. 2020;60(12):5754–5770. [DOI] [PubMed] [Google Scholar]
- 40.Laskowski RA, MacArthur MW, Moss DS, et al. PROCHECK: a program to check the stereochemical quality of protein structures. J Appl Cryst. 1993;26(2):283–291. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.