

Abstract
Reversible electrochemical intercalation of cations into the interlayer space of 2D materials induces tunable physical and chemical properties in them. In MXenes, a large class of recently developed 2D carbides and nitrides, low intercalation energy, high storage capacitance, and reversible intercalation of various cations have led to their improved performance in sensing and energy storage applications. Herein, a coupled nanopore-actuator system where an ultrathin free-standing MXene film serves as a nanopore support membrane and ionically active actuator is reported. In this system, the contactless MXene membrane in the electric field affects the cation movement in the field through their (de)intercalation between individual MXene flakes. This results in reversible swelling and contraction of the membrane monitored by ionic conductance through the nanopore. This unique nanopore coupled to a mechanical actuation system could provide new insights into designing single-molecule biosensing platforms at the nanoscale.
Keywords: actuation, intercalation, membranes, MXenes, nanopores
1. Introduction
The use of 2D materials such as graphene, molybdenum disulfide (MoS2), tungsten disulfide (WS2), and boron nitride (BN) in nanopore technology for single-molecule sensing of biomolecules has been attractive due to the atomic-level thickness, high strength, and surface tunability of 2D materials.[1] Recently, we demonstrated the potential of an emerging family of 2D materials for nanopore sensing, namely, 2D transition metal carbides and nitrides known as MXenes.[2] MXenes are typically produced from MAX phases, bulk ternary layered carbides and nitrides, through a selective etching process and have a general formula of Mn+1XnTx, where M represents transition metals, X represents carbon/nitrogen, Tx represents functional groups such as oxygen, hydroxyl, and fluorine bound to the transition metal on the basal plane of the MXenes, and n = 1, 2, 3, or 4.[3]
An intriguing property of MXenes (e.g., Ti3C2Tx, Ti2CTx, and V2CTx) is that protons and different cations (Li+, Na+, Mg2+, etc.) can electrochemically (and chemically) intercalate into the interlayer space between individual MXene flakes.[3] This property has enabled the use of MXenes as electrodes in electrochemical energy storage devices such as batteries[4] and supercapacitors;[5,6] tuning MXene films’ electronic,[7] optoelectronic[8] and gas sensing properties;[9] and other exciting applications such as electrochemical actuation.[10,11] In most cases, the improved properties and functionality of MXenes as a result of cation intercalation are due to the accompanying change in their interlayer spacing, as well as charge transfer between the cations and the MXene’s outer layer transition metal.[8,9,12]
To gain more insights and enable tuning and implementing this property of MXenes for different applications, dynamics of cation intercalation between MXene layers have been extensively studied in the past couple of years. Through using various methods such as electrochemical quartz-crystal admittance (EQCA),[13] electrochemical dilatometry,[14] in situ X-ray diffraction (XRD),[5] and electrochemical atomic force microscopy (AFM)[10] it was shown that upon intercalation of cations into thick multilayered MXene electrodes (films), a significant and reversible volume change (expansion or contraction based on the cation charge density and size) occurs.[14] However, less attention has been given to ion intercalation in ultrathin (sub-10 nm thick) MXene membranes, and the resulting actuation property at the nanoscale needs to be demonstrated.
Herein, we study voltage-driven ionic current through individual nanopores (d < 10 nm) in an ultrathin (< 15 layers) free-standing Ti3C2Tx membrane. We find an unusual and unique time-dependent change in the conductance of the pore as a function of voltage, which we attribute to ion-mediated actuation of the pore through reversible voltage-gated swelling and contraction of the membrane. Similar to wireless bipolar electrodes,[15] the electric field across the MXene membrane hosting the nanopore reversibly activates ion permeation through the multilayer, which induces swelling and contraction through ionic (de)intercalation. This is conveniently detected by monitoring the ion current across the pore, which reports on changes to the nanopore thickness as ionic actuation occurs. This nanoscale actuation property could provide new insights into designing sensors at the nanoscale, including nanopores coupled to mechanical actuators.
2. Results and Discussion
We produced dispersions of large-flake monolayer Ti3C2Tx MXene using a previously reported method (see the experimental section for details).[16,17] Few-layer free-standing MXene membranes (films) were assembled and transferred onto silicon chips, each hosting a 30-nm-thick SiNx membrane at its center with a pre-fabricated ≈100 nm aperture, which we have previously developed.[18] Then, following drilling nanopores in the free-standing MXene membrane (the part suspended on the aperture) using a focused electron beam of TEM or STEM, these chips were used in nanopore experiments as we previously reported (see the experimental section for details).[2] Figure 1a shows a schematic of the nanopore experimental setup used in this study. By using Ag/AgCl electrodes immersed in each chamber, a voltage was applied across the MXene nanopore that resulted in an ionic current detected by an electrometer. The close-up look of the nanopore highlights two important nanopore geometry parameters: diameter (dpore) and thickness (Lpore), which affect the ionic current passing through the nanopore. Equation (1)[19] shows the theoretical total ionic conductance (Gtotal) through a nanopore where μi + and μi − are respective electrophoretic mobilities of the cations and anions, n is the number density of anions or cations, and e is the elementary charge. The first term shows the effect of nanopore geometry on the ionic conductance, whereas the second term shows the access resistance term, which is explained as the effect of narrow opening of the pore on the ionic conductance – the effect more noticeable as the pore becomes thinner.
| (1) |
Since cation intercalation between individual MXene flakes induces a change in their interlayer spacing,[12] MXene nanopores could be ionically activated through reversible swelling and contraction, altering the ionic conductance of the nanopore. Figure 1b shows a dark-field scanning transmission electron microscopy (STEM) image of a transferred MXene film on a SiNx membrane with the arrow indicating the aperture and Figure 1c shows a dark-field STEM image of a fabricated nanopore with a diameter of ≈0.8 nm. All nanopores used in this study are listed in Table S1 (Supporting Information). Figure 1d shows the current-voltage behavior of nanopore A in the ±100 mV voltage window, in three different electrolytes, all buffered with Tris at pH 7.5. The nanopore has an ionic conductance of 70, 58, and 41 nS in 1 m KCl, 1 m NaCl, and 1 m LiCl, respectively. While this graph shows the ohmic behavior of the MXene nanopore at low voltage, at higher voltages, we observed a reversible voltage-gated change in the conductance of the nanopore. Figure 2a shows a current trace of nanopore B at 100, 200, 300, and 400 mV (100 s for each voltage). While for 100 mV the conductance value was stable at ≈47 nS, for higher voltages, we observed a gradual decrease in the conductance, the rate of which increased with increasing voltage. Upon decreasing the voltage back to 100 mV, the conductance value, which had decreased from 47 to 40 nS, started to increase again, and after keeping the voltage at 100 mV for ≈100 s, a partial recovery of the conductance was observed, reaching the value of 44 nS. After about 30 min with no applied voltage across the nanopore, the conductance completely recovered, as shown by the current trace at 100 mV. Interestingly, when the higher voltages were applied for a much shorter time, the reversible conductance change could still be observed. As shown in Equation (1), a change in the nanopore conductance value is possible due to changes in the nanopore geometry (diameter and thickness) or the electrolyte conductivity. Since there is no change in the electrolyte conductivity and it is unlikely that a conductance change is a result of a nanopore diameter change, we hypothesize that the fast voltage-gated change in the conductance is the result of the nanopore thickness change caused by the electrochemical actuation of the MXene nanopore membrane in response to the voltage change.
Figure 1.
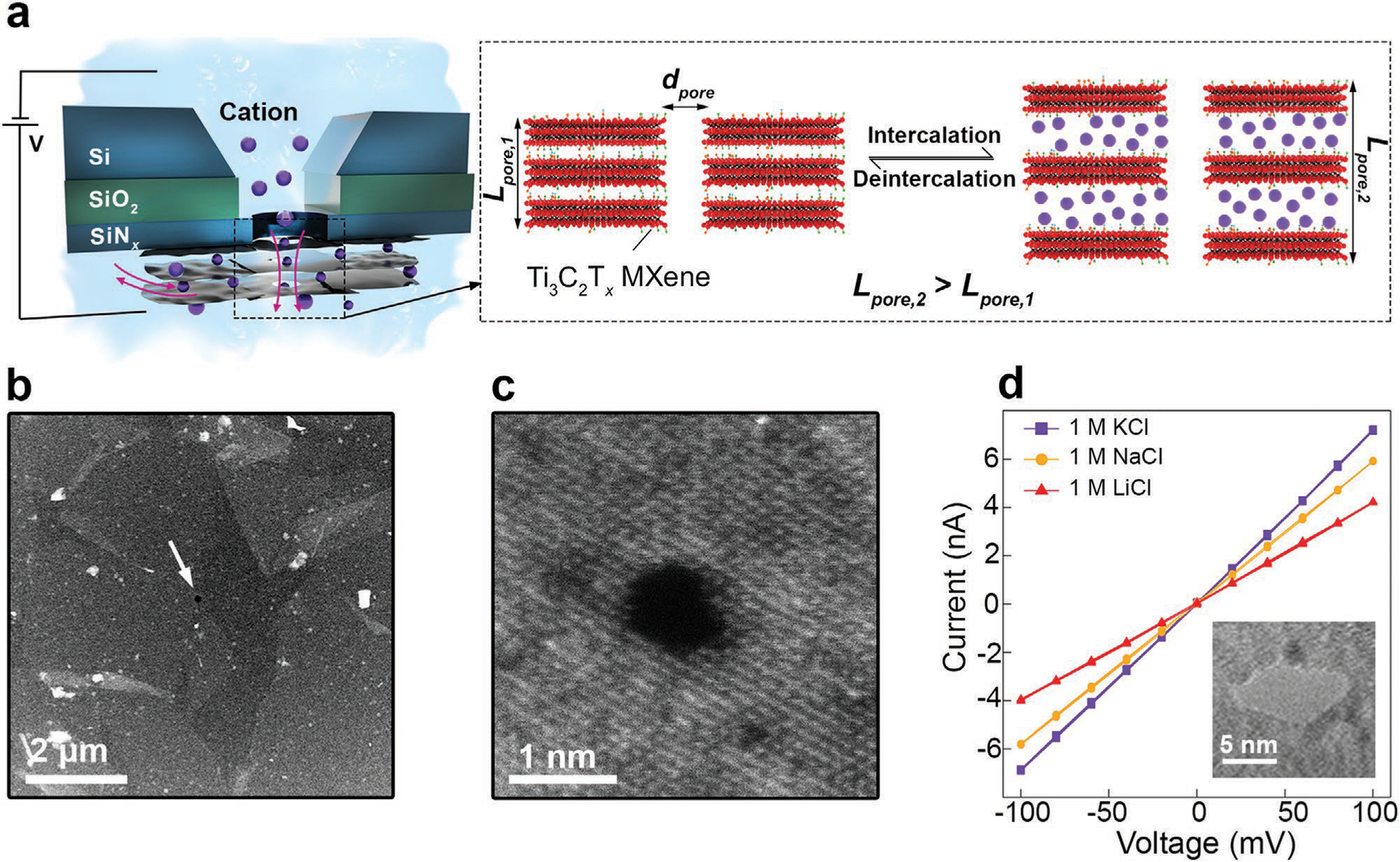
Ionic current detection through MXene nanopores. a) Schematic illustration of MXene nanopore experimental setup (left). A close-up look of the MXene nanopore and the effect of cation intercalation on the nanopore geometry (right). b) HAADF STEM image of a MXene film transferred on a SiNx membrane with a pre-fabricated aperture shown with an arrow. c) HAADF STEM image of a MXene nanopore fabricated with focused electron beam. d) Current–voltage curve of nanopore A in 1 m KCl (purple), 1 m NaCl (yellow), and 1 m LiCl (red). Inset shows the TEM image of the nanopore.
Figure 2.
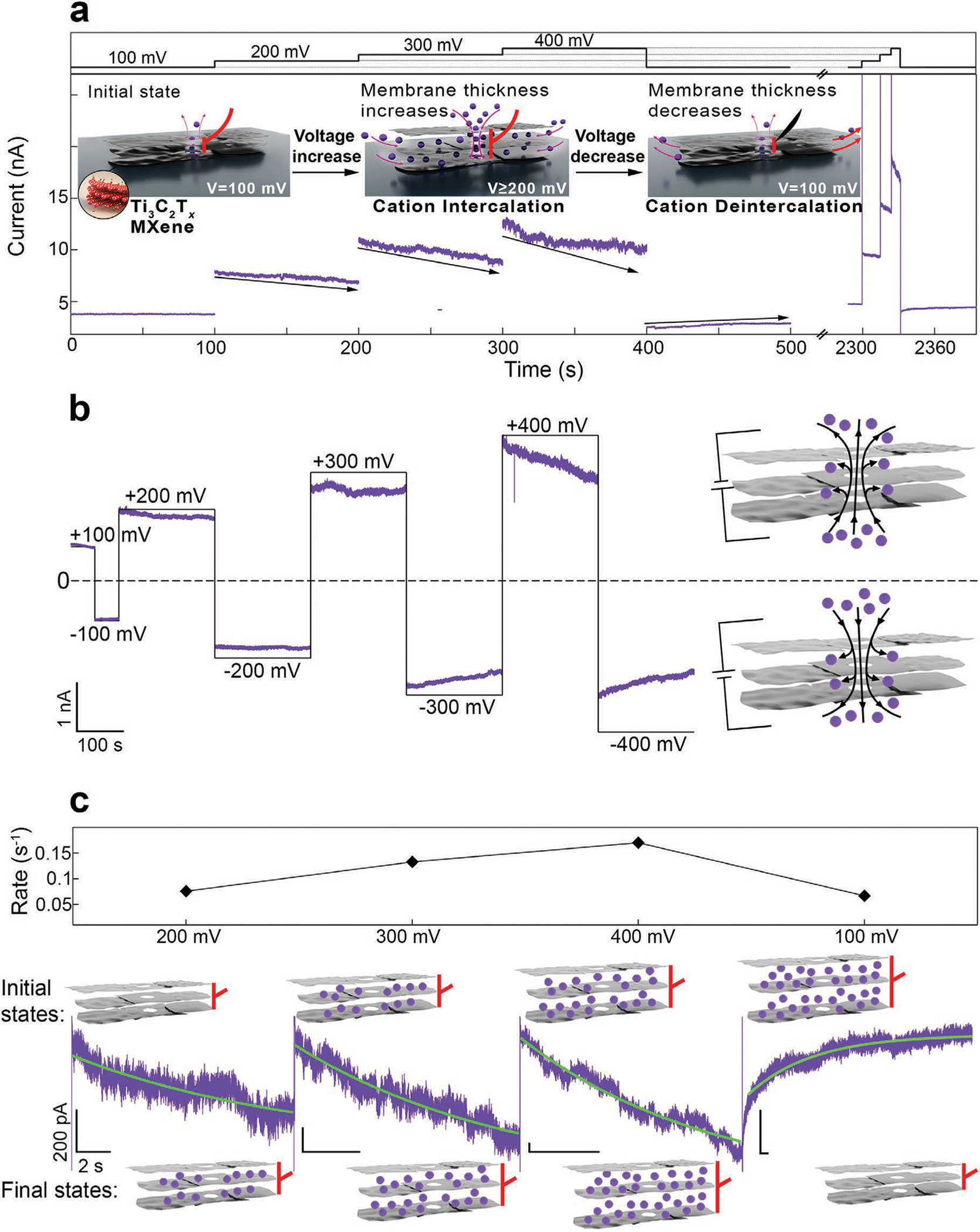
Actuation property of nanopores in few-layer MXene membranes due to K+ (de)intercalation. a) Current trace of nanopore B demonstrates that with increasing voltage, the nanopore conductance value decreases due to cation intercalation. This intercalation process recovers via deintercalation when the voltage is decreased to lower values (e.g., 100 mV). The inset shows schematically how cation (de)intercalation can occur through defects in the MXene multilayer membrane, causing membrane swelling and contraction. b) Current trace of nanopore C at positive and negative voltages showing the actuation properties of MXene pores regardless of the electric field direction (black curve = applied voltage). c) Single-exponential functions were fitted to the current traces shown in (a). As the voltage increases, the rate of the conductance change (τ−1) increases (At 100 mV, the capacitive response of the nanopore to the voltage change is seen as an initial, faster rate that is omitted in our analysis). The time-dependent change in the nanopore thickness is shown schematically. Buffer: 0.4 m KCl, 10 × 10−3 m Tris, pH 7.5. All horizontal and vertical scale bars for the traces are 2 s and 200 pA, respectively.
The proposed ionic actuation mechanism is shown in Figure 2a. In the initial state, V ≤ 100 mV, when a voltage is applied across the MXene nanopore, ion transport occurs only through the nanopore, which results in a steady ionic current, as shown by the current trace at 100 mV. When larger voltages (200, 300, and 400 mV) are applied, cations can permeate through the membrane layers and intercalate into the interlayer space, increasing the interlayer space and thus the total nanopore thickness. Upon decreasing the voltage to 100 mV again, the weaker electric field across the nanopore results in cation deintercalation from the interlayer space, shrinkage of the interlayer space, and partial recovery of the initial membrane thickness. In contrast to previous studies on MXene-cation intercalation, here, no voltage is applied directly to the MXene film, and therefore, the MXene membrane functions as a contactless, floating electrode. We observed this effect for three different electrolytes: LiCl, NaCl, and KCl. Figure S1a,b (Supporting Information) shows current traces of two MXene nanopores tested in 0.4 m LiCl and 0.4 m NaCl and Figure S2 (Supporting Information) shows three current traces of a nanopore tested in all three electrolytes. Figure 2b and Figure S3 (Supporting Information) show that this behavior occurs at both positive and negative bias voltages across the nanopore. Thus, regardless of the direction of the electric field and cation movement, cation intercalation reversibly occurs between the MXene layers in a field-dependent manner.
In an effort to understand the rate of cation (de)intercalation between the MXene layers, we fit exponential functions (, where τ−1 is the decay/growth rate) to the current traces. Figure 2c shows a close-up look of the trace shown in Figure 2a with the green graph showing the best single-exponential fit to the trace. Increasing the voltage from 200 to 400 mV resulted in an increase in the rate of the process from 0.076 s−1 at 200 mV, to 0.133 s−1 at 300 mV, and 0.17 s−1 at 400 mV and decreasing the voltage to 100 mV resulted in deintercalation with the rate of 0.067 s−1. The time-dependent cation permeation and thickness change are shown schematically in the inset. At high voltage, while the conductance continues to decrease, more cations permeate through the interlayer space of MXene film and its total thickness continues to increase. At 100 mV, on the other hand, while the conductance continues to increase, cations deintercalate from the interlayer space and the thickness recovers to its initial state. Therefore, we attribute the time-dependent change in nanopore conductance to two phenomena: 1) Cation permeation through the interlayer space results in fewer ions being detected by the electrodes (membrane charging). 2) MXene film thickness change due to cation intercalation results in reduced ionic conductance through the nanopore, as dictated by Equation (1). However, measuring the contribution of each of the two phenomena to the total ionic conductance change is not straightforward. Since we cannot measure the intercalation current directly, we cannot calculate the amount of charge stored in the MXene film. Figure S4 (Supporting Information) shows two other examples of change in the rate of the (de)intercalation process with voltage, in which case the rates are different from pore to pore. We reason that pore-to-pore variation in the (de)intercalation kinetics depends on the pore’s specific features (e.g., MXene flake size, distance of flake edges from the pore, etc.). Moreover, because of the inherent mechanical fluctuation of the ultrathin 2D membranes previously shown as the major factor translating into low-frequency noise in 2D nanopores,[20] fitting the ion current traces to this simplistic model is challenging. Nonetheless, it still provides valuable information about the rate of ion (de) intercalation in MXene membranes, which proceeds with reasonable variation from pore to pore (see Table S2, Supporting Information).
To test the hypothesis that cation intercalation into MXene membrane results in swelling of the membrane, we used the operando AFM as a local dilatometer to measure the thickness change of a 16-layer MXene film during cyclic voltammetry (CV)[21] since direct measurement of the nanopore thickness is not feasible. The 16-layer film was deposited by successive repetition of a monolayer MXene self-assembly method we recently developed.[18] Due to the resolution limit and thermal drift in the AFM measurement, thinner MXene films were not compatible with this method. The 16-layer MXene was selected to ensure the amount of deformation is large enough to overcome the thermal drift. Figure 3a shows a schematic of the operando AFM experimental setup, where the local expansion/contraction of the 16-layer MXene film during cycling in 0.5 m K2SO4 was monitored via an AFM probe. Chloride ions were avoided due to their corrosive nature. The cyclic voltammogram and the 16-layer MXene deformation during cycling at various scan rates are shown in Figure 3b,c, respectively. The same membrane was used to detect the thickness change in 0.5 m Li2SO4 (shown in Figure S5: Supporting Information). The results confirm that for both Li+ and K+ cations, the MXene film expands (deformation > 0) during cation intercalation (negative potential sweep) and reversibly contracts upon potential sweep reversal, also indicated by deintercalation. AFM results are in agreement with our nanopore observations that the intercalation of cations results in reversible swelling of the MXene membrane. One distinct difference between the nanopore experiment and the AFM dilatometry experiment is the contactless charging of the MXene membrane in the nanopore experiment that induces slow, diffusion-limited cation intercalation. Whereas in the AFM experiments, fast cation intercalation into the MXene is induced by using MXenes as a working electrode.
Figure 3.
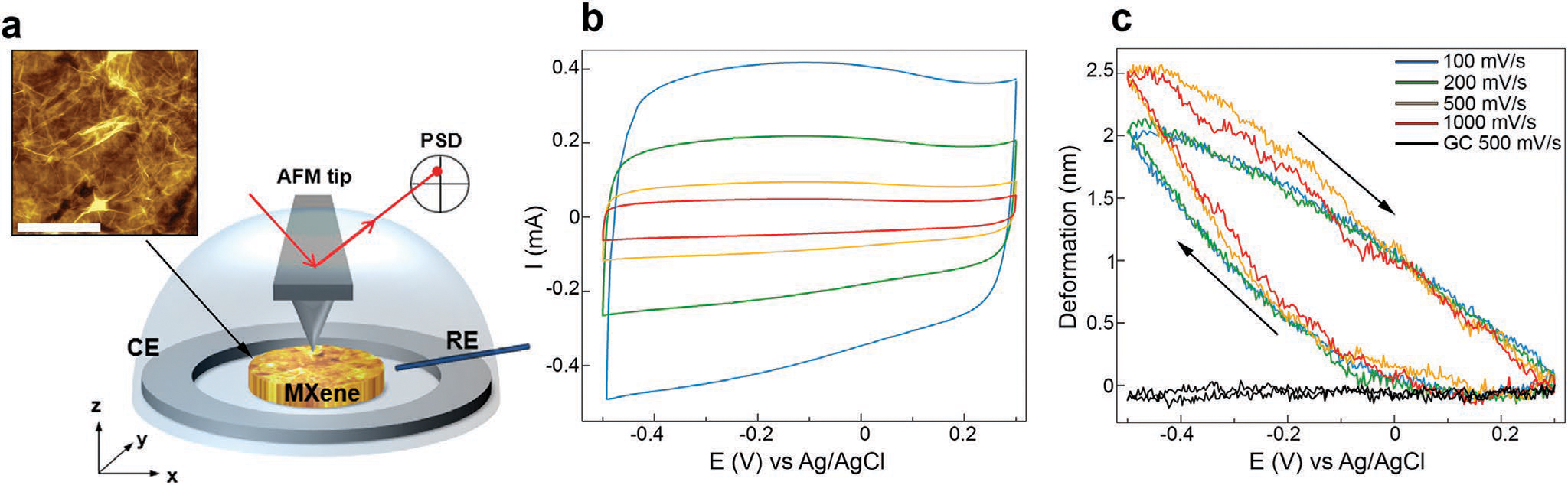
AFM dilatometry experiment of a deformation in a 16-layer MXene membrane. a) Illustration of operando AFM setup. AFM image scale = 4 μm. b) CV and c) deformation of a 16-layer MXene film in 0.5 m K2SO4 at various scan rates.
While our main hypothesis for the observed conductance change in the MXene nanopore is the volumetric swelling/contraction of the MXene film, it is worth noting that double-layer charging/discharging and accumulation of the cations on the MXene membrane surface could be another possible contributing phenomenon. However, regardless of the membrane type, double layer charge/discharge happens within milliseconds in an aqueous electrolyte, therefore its contributions to the overall observed results should be negligible since the timescale for conductance change in our experiments is orders of magnitude longer. For example, Figure S6 (Supporting Information) shows an example of the capacitive response of a MXene nanopore when the voltage was changed from 400 to 100 mV.
Figure S7a–d (Supporting Information) shows current traces of MXene nanopores at V > 200 mV for up to ≈3000 s. These traces show that following the intercalation process that fits the exponential function, the conductance value continues to decrease, however, at a much lower rate. We fitted a double-exponential function to these traces to characterize the processes happening. While we assume the first process is due to the intercalation of cations, we believe that the second process could be any or combination of several processes including rearrangement of the cations between the MXene layers after intercalation and gradual clogging of sub-nm size defects in the MXene membranes over time by ions.
Lastly, while the change in conductance as the result of cation intercalation was observed for nanopores in few-layer MXene membranes, in our experiments, no change in the conductance was observed for a nanopore in a monolayer MXene membrane (as deduced from the low contrast in TEM imaging). Figure S8 (Supporting Information) shows an example of a monolayer MXene membrane and the current trace showing that the conductance does not decrease upon increasing voltage to 200 mV and even 300 mV. Moreover, it is worthwhile to mention that, in addition to Ti3C2Tx MXene, we carried out the same nanopore experiments with Ti2CTx MXene nanopores. This observation was more challenging in Ti2CTx, however, there were still some nanopores that showed this behavior (Figure S9, Supporting Information). Finally, this effect does not occur in other 2D nanopore membranes or ceramic solid-state nanopore membranes, such as silicon nitride (see Figure S10, Supporting Information).
3. Conclusion
When voltage is applied across a single nanopore in a MXene membrane, intercalation between individual flakes of MXenes leads to reversible contraction and swelling of the membrane, which can be detected by change in the ionic conductance through the nanopore. At voltages exceeding 200 mV, cations permeate through the MXene film and intercalate into the interlayer space with the rate increasing with voltage. At voltages below 100 mV, cations deintercalate from the interlayer space. Moreover, by using operando AFM as a local dilatometer, we showed that Li+ and K+ intercalation result in increase in thickness of a 16-layer MXene membrane. By harnessing this intercalation process in combination with the in-plane electrical conductivity of MXenes, novel nanopore platforms with improved performance can be designed and engineered.
4. Experimental Section
MXene Synthesis:
Ti3C2Tx MXene was synthesized using a method similar to previously described works.[17,22] For every 1 g of Ti3AlC2 MAX phase powder (400 mesh, < 38 μm), 20 mL etching solution was prepared with a mixture of 2 mL hydrofluoric acid (HF, Acros Organics, 49%), 12 mL hydrochloric acid (HCl, Fisher Chemical, 37%) and 6 mL DI water. The Ti3AlC2 powder was slowly added to the etching solution in a Polytetrafluoroethylene (PTFE) bottle at room temperature while stirring the mixture with a Teflon-coated magnetic bar at 350 rpm for 24 h at 35°C. The obtained multilayer Ti3C2Tx MXene was repeatedly washed with DI water and centrifuged at 3500 rpm for 5 min until pH reached ≈6. Delamination was achieved by mixing as-obtained multilayer Ti3C2Tx with 1 g of lithium chloride (LiCl, Alfa Aesar, 99%) and 20 mL of DI water. The mixture was stirred with a Teflon-coated magnetic bar at 300 rpm for 24 h at 25°C. Then, the obtained solution was washed with DI water and centrifuged at 3500 rpm for 5 min. The clear supernatant was decanted, and sediment was redispersed in DI water and shaken for 2 min. This process was repeated until a dark supernatant was observed. The sediment was redispersed in DI water and shaken for 2 min, then centrifuged at 3500 rpm for 45 min. After this step, the supernatant containing delaminated single-flake Ti3C2Tx MXene was collected. This process was repeated five times and the supernatants were collected separately. All collected samples were bubbled with argon gas for 1 h to decrease the amount of dissolved oxygen in them.
MXene Nanopore Device Fabrication:
To fabricate free-standing MXene membranes, the previously developed lateral interfacial self-assembly method was used.[2,18] In short, an aqueous MXene dispersion was diluted to the desired concentration and then mixed with methanol in a 1:8 volume ratio (1 part methanol), followed by casting on a chloroform bath in a petri dish. Then, a 5 × 5 mm2 Si/SiO2/SiNx chip hosting 50 nm free-standing SiNx membrane with a pre-fabricated ≈100 nm aperture was submerged in the bath and lifted through the droplet and baked on a hot plate at 80°C for about 5 min to dry. Before transferring the MXene film on the substrate, the chips were cleaned with hot piranha solution and deionized water. Nanopores were fabricated through the free-standing part of the MXene membrane using JEOL 2010FEG at 1.5 Mx magnification, then magnification was lowered to 800 kx for quick imaging to avoid unintentional e-beam induced damage (e.g., pore enlargement or formation of multiple pores).
Experimental Data Acquisition:
MXene nanopore devices were assembled on a PTFE cell with two chambers each containing ≈80 μL of the buffer. Edges of the chips were sealed using silicone elastomer. Afterward, Ag/AgCl electrodes connected to an Axopatch 200B amplifier were immersed in each chamber. The current was measured at 250 kHz sampling rate and low-pass-filtered at 10 kHz. Data processing was done using Igor Pro software.
STEM Imaging:
Same Si chips containing MXene free-standing membranes were used for STEM imaging. A probe-corrected FEI Titan Themis 300 STEM with ChemiSTEM technology was used for atomic resolution imaging of MXene sheets.
AFM Dilatometry:
The experimental and data analysis details of AFM dilatometry were reported elsewhere.[21] In short, the AFM dilatometry was conducted via an MFP-3D AFM in a commercial three-electrode in situ electrochemical AFM cell (both from Asylum Research, Oxford Instruments Company, USA). A 16-layer MXene film was transferred onto a glass carbon substrate as the working electrode. A ring-shaped glassy carbon counter electrode and an Ag/AgCl reference electrode were used in this study. 0.5 m K2SO4 (Sigma Aldrich) and 0.5 m Li2SO4 (Sigma Aldrich) were used as electrolytes. Electrochemical measurements were conducted using a Bio-Logic SP-200 potentiostat (Bio-Logic, USA). Cyclic voltammetry measurements were conducted under various scan rates between 100 mV s−1 to 1 V s−1. During the AFM dilatometry measurements, the AFM probe produced a force-distance curve. Once the probe was in contact with the electrode and the setpoint was reached, the AFM probe remained at its position and sent a trigger to the potentiostat to perform CV for ten cycles. The electrode deformation was monitored through a deflection channel. After the CV measurements, the AFM cantilever was withdrawn from the surface. The CVs and deformation curves presented in this work were average responses obtained by averaging over ten cycles. After a test in the K2SO4 electrolyte, the MXene electrode was rinsed several times and immersed in DI water for 10 h before being measured in Li2SO4 electrolytes.
Supplementary Material
Acknowledgements
The authors acknowledge Northeastern’s Kostas Advanced Nano-Characterization Facility (KANCF, Burlington MA, USA) and Wentao Liang for TEM imaging. The synthesis of MXenes (A.VM., Y.G.) and AFM dilatometry work (W.-Y.T., N.B.) were supported by the Fluid Interface Reactions, Structures and Transport (FIRST) Center, an Energy Frontier Research Center (EFRC) funded by the U.S. Department of Energy, Office of Science, Office of Basic Energy Sciences. The authors acknowledge funding by the National Institutes of Health, award # R21HG011689 (M.W.). AFM experiments were performed and supported at the Center for Nanophase Materials Sciences in Oak Ridge National Lab, a DOE Office of Science user facility, through award number CNMS2021-B-00897.
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting Information
Supporting Information is available from the Wiley Online Library or from the author.
Contributor Information
Mehrnaz Mojtabavi, Department of Bioengineering, Northeastern University, Boston, MA 02115, USA.
Wan-Yu Tsai, Chemical Science Division, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA.
Armin VahidMohammadi, A.J. Drexel Nanomaterials Institute and Department of Materials Science & Engineering, Drexel University, Philadelphia, PA 19104, USA.
Teng Zhang, A.J. Drexel Nanomaterials Institute and Department of Materials Science & Engineering, Drexel University, Philadelphia, PA 19104, USA.
Yury Gogotsi, A.J. Drexel Nanomaterials Institute and Department of Materials Science & Engineering, Drexel University, Philadelphia, PA 19104, USA.
Nina Balke, Center for Nanophase Materials Sciences, Oak Ridge National Laboratory, Oak Ridge, TN 37830, USA.
Meni Wanunu, Department of Physics, Northeastern University, Boston, MA 02115, USA; Department of Chemistry and Chemical Biology, Northeastern University, Boston, MA 02115, USA.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.
References
- [1].a) Garaj S, Hubbard W, Reina A, Kong J, Branton D, Golovchenko JA, Nature 2010, 467, 190; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu S, Lu B, Zhao Q, Li J, Gao T, Chen Y, Zhang Y, Liu Z, Fan Z, Yang F, You L, Yu D, Adv. Mater. 2013, 25, 4549; [DOI] [PubMed] [Google Scholar]; c) Liu K, Feng J, Kis A, Radenovic A, ACS Nano 2014, 8, 2504; [DOI] [PubMed] [Google Scholar]; d) Danda G, Masih Das P, Chou Y-C, Mlack JT, Parkin WM, Naylor CH, Fujisawa K, Zhang T, Fulton LB, Terrones M, Johnson ATC, Drndić M, ACS Nano 2017, 11, 1937; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Gu C, Yu Z, Li X, Zhu X, Cao Z, Ye Z, Jin C, Liu Y, Appl. Phys. Lett. 2019, 115, 223702. [Google Scholar]
- [2].Mojtabavi M, VahidMohammadi A, Liang W, Beidaghi M, Wanunu M, ACS Nano 2019, 13, 3042. [DOI] [PubMed] [Google Scholar]
- [3].VahidMohammadi A, Rosen J, Gogotsi Y, Science 2021, 372, eabf1581. [DOI] [PubMed] [Google Scholar]
- [4].a) Naguib M, Come J, Dyatkin B, Presser V, Taberna P-L, Simon P, Barsoum MW, Gogotsi Y, Electrochem. Commun. 2012, 16, 61; [Google Scholar]; b) Kajiyama S, Szabova L, Sodeyama K, Iinuma H, Morita R, Gotoh K, Tateyama Y, Okubo M, Yamada A, ACS Nano 2016, 10, 3334; [DOI] [PubMed] [Google Scholar]; c) Naguib M, Adams RA, Zhao Y, Zemlyanov D, Varma A, Nanda J, Pol VG, Chem. Commun. 2017, 53, 6883; [DOI] [PubMed] [Google Scholar]; d) VahidMohammadi A, Hadjikhani A, Shahbazmohamadi S, Beidaghi M, ACS Nano 2017, 11, 11135. [DOI] [PubMed] [Google Scholar]
- [5].Lukatskaya MR, Mashtalir O, Ren CE, Dall’Agnese Y, Rozier P, Taberna PL, Naguib M, Simon P, Barsoum MW, Gogotsi Y, Science 2013, 341, 1502. [DOI] [PubMed] [Google Scholar]
- [6].a) Ghidiu M, Lukatskaya MR, Zhao M-Q, Gogotsi Y, Barsoum MW, Nature 2014, 516, 78; [DOI] [PubMed] [Google Scholar]; b) VahidMohammadi A, Mojtabavi M, Caffrey NM, Wanunu M, Beidaghi M, Adv. Mater. 2019, 31, 1806931. [DOI] [PubMed] [Google Scholar]
- [7].Hart JL, Hantanasirisakul K, Lang AC, Anasori B, Pinto D, Pivak Y, van Omme JT, May SJ, Gogotsi Y, Taheri ML, Nat. Commun. 2019, 10, 522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Hantanasirisakul K, Zhao M-Q, Urbankowski P, Halim J, Anasori B, Kota S, Ren CE, Barsoum MW, Gogotsi Y, Adv. Electron. Mater. 2016, 2, 1600050. [Google Scholar]
- [9].Koh H-J, Kim SJ, Maleski K, Cho S-Y, Kim Y-J, Ahn CW, Gogotsi Y, Jung H-T, ACS Sens. 2019, 4, 1365. [DOI] [PubMed] [Google Scholar]
- [10].Come J, Black JM, Lukatskaya MR, Naguib M, Beidaghi M, Rondinone AJ, Kalinin SV, Wesolowski DJ, Gogotsi Y, Balke N, Nano Energy 2015, 17, 27. [Google Scholar]
- [11].Pang D, Alhabeb M, Mu X, Dall’Agnese Y, Gogotsi Y, Gao Y, Nano Lett. 2019, 19, 7443. [DOI] [PubMed] [Google Scholar]
- [12].Lukatskaya MR, Bak S-M, Yu X, Yang X-Q, Barsoum MW, Gogotsi Y, Adv. Energy Mater. 2015, 5, 1500589. [Google Scholar]
- [13].Levi MD, Lukatskaya MR, Sigalov S, Beidaghi M, Shpigel N, Daikhin L, Aurbach D, Barsoum MW, Gogotsi Y, Adv. Energy Mater. 2015, 5, 1400815. [Google Scholar]
- [14].Gao Q, Come J, Naguib M, Jesse S, Gogotsi Y, Balke N, Faraday Discuss. 2017, 199, 393. [DOI] [PubMed] [Google Scholar]
- [15].Koefoed L, Pedersen SU, Daasbjerg K, Curr. Opin. Electrochem. 2017, 2, 13. [Google Scholar]
- [16].Alhabeb M, Maleski K, Anasori B, Lelyukh P, Clark L, Sin S, Gogotsi Y, Chem. Mater. 2017, 29, 7633. [Google Scholar]
- [17].Mathis TS, Maleski K, Goad A, Sarycheva A, Anayee M, Foucher AC, Hantanasirisakul K, Shuck CE, Stach EA, Gogotsi Y, ACS Nano 2021, 15, 6420. [DOI] [PubMed] [Google Scholar]
- [18].Mojtabavi M, VahidMohammadi A, Ganeshan K, Hejazi D, Shahbazmohamadi S, Kar S, van Duin ACT, Wanunu M, ACS Nano 2021, 15, 625. [DOI] [PubMed] [Google Scholar]
- [19].Wanunu M, Dadosh T, Ray V, Jin J, McReynolds L, Drndic M, Nat. Nanotechnol. 2010, 5, 807. [DOI] [PubMed] [Google Scholar]
- [20].Balandin AA, Nat. Nanotechnol. 2013, 8, 549. [DOI] [PubMed] [Google Scholar]
- [21].Tsai W-Y, Wang R, Boyd S, Augustyn V, Balke N, Nano Energy 2021, 81, 105592. [Google Scholar]
- [22].Shuck CE, Sarycheva A, Anayee M, Levitt A, Zhu Y, Uzun S, Balitskiy V, Zahorodna V, Gogotsi O, Gogotsi Y, Adv. Eng. Mater. 2020, 22, 1901241. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.