

Abstract
Arginase deficiency is associated with prominent neuromotor features, including spastic diplegia, clonus, and hyperreflexia; intellectual disability and progressive neurological decline are other signs. In a constitutive murine model, we recently described leukodystrophy as a significant component of the central nervous system features of arginase deficiency. In the present studies, we sought to examine if the administration of a lipid nanoparticle carrying human ARG1 mRNA to constitutive knockout mice could prevent abnormalities in myelination associated with arginase deficiency. Imaging of the cingulum, striatum, and cervical segments of the corticospinal tract revealed a drastic reduction of myelinated axons; signs of degenerating axons were also present with thin myelin layers. Lipid nanoparticle/ARG1 mRNA administration resulted in both light and electron microscopic evidence of a dramatic recovery of myelin density compared with age-matched controls; oligodendrocytes were seen to be extending processes to wrap many axons. Abnormally thin myelin layers, when myelination was present, were resolved with intermittent mRNA administration, indicative of not only a greater density of myelinated axons but also an increase in the thickness of the myelin sheath. In conclusion, lipid nanoparticle/ARG1 mRNA administration in arginase deficiency prevents the associated leukodystrophy and restores normal oligodendrocyte function.
Keywords: MT: Oligonucleotides: Therapies and Applications, arginase deficiency, dysmyelination, lipid nanoparticle, messenger RNA, urea cycle
Graphical abstract
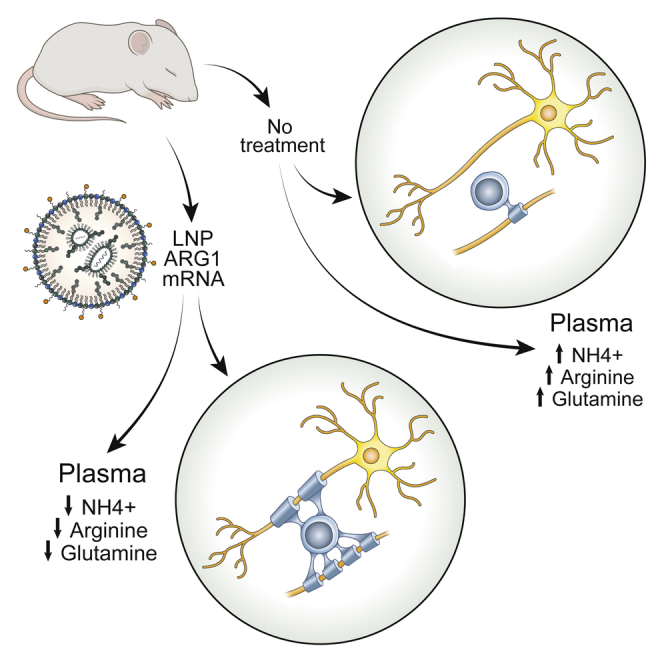
Lipid nanoparticle/ARG1 mRNA administration in a murine model of arginase deficiency prevents the associated leukodystrophy present in this disorder and restores normal oligodendrocyte function.
Introduction
The final step in the urea cycle, the pathway for ammonia detoxification in terrestrial mammals, is mediated by arginase 1 (ARG1), hydrolyzing arginine into ornithine and urea with the latter being excreted in the urine as waste.1, 2, 3 Arginase deficiency, or hyperargininemia, is a rare single-enzyme liver defect in which ARG1 activity is lost from biallelic inactivating mutations. The population of patients with arginase deficiency have multiple heterogeneous missense mutations, nonsense mutations, and deletions, being a pan-ethnic disorder.4, 5, 6, 7, 8, 9, 10, 11 While ARG1 is primarily expressed in the liver, its absence results in an inability to detoxify ammonia and markedly elevated arginine. Disease penetrance is variable, and affected children only uncommonly present neonatally with severe hyperammonemia.7,12 Unlike the other urea cycle disorders where hyperammonemia and related signs often occur within a few days of birth,13,14 the disorder is generally recognized in late infancy or early childhood and is characterized by progressive neurological decline,14,15 with afflicted patients generally being long lived.16
Hyperargininemia is distinct in its presentation from the other urea cycle disorders. The usual course is one of insidious onset, manifesting with microcephaly, spasticity, seizures, failure to thrive, and intellectual disability;17 it is rarely,7,12 if ever, fatal in the neonatal period. The association of the disorder with prominent neuromotor features (including spastic diplegia/tetraplegia, clonus, hyperreflexia, and loss of ambulation) are common in arginase deficiency but uncommon in the other urea cycle disorders.
The cause and mechanism of the unique brain injury that occurs in arginase deficiency have only been partially elucidated.18,19 The neurologic impairment and developmental regression are associated with corticospinal tract (CST)20 and pyramidal tract deterioration. Recently, our group has found that dysmyelination of the CST is a prominent feature of the developing murine brain when arginase 1 is deficient;19 limited clinical case reports21,22 have also suggested that dysmyelination may be a prominent feature in afflicted human patients. While the proximate toxin is not known, the neurologic picture is distinct from those patients who suffer from severe hyperammonemia found in other urea cycle disorders; it is believed that the toxin is arginine or a product derived from arginine such as one or more of the various guanidino compounds found in these patients. Present-day long-term therapy rests on provision of a low-protein diet and administration of sodium benzoate and sodium phenylbutyrate. These therapies, however, are incomplete, being only palliative and not curative.
With the emergence of mRNA therapeutics, we sought to determine if intermittent delivery of mRNA formulated in a lipid nanoparticle (LNP) during a period of critical early myelination could prevent dysmyelination of the CST found as a prominent feature of this disorder.19 Notably, the studies performed herein demonstrated that the CST is not the only long fibers that have myelination affected in the CNS: the striatum, cingulate gyrus, and corpus callosum are also markedly affected in arginase deficiency. Nonetheless, the administration of ARG1 mRNA to mice during the early critical phase of CNS myelination results in near-normal myelination of the corticospinal tract, striatum, cingulum bundle, and corpus callosum, both in myelinated axon density and in myelin thickness. Plasma ammonia, arginine, and guanidino compounds were also improved in their control.
Results
Administration of different nanoparticles results in variable hepatic mRNA expression
Two different lipid nanoparticles comprising different ionizable lipids formulated with ARG1 mRNA were tested in these studies at a dose of 2 mg/kg:15 Lipid A, heptadecan-9-yl 8-((2-hydroxyethyl) (8-(nonyloxy)-8-oxooctyl)amino)octanoate,23 and lipid B, 2-(dinonylamino)-1-(4-(N-(2-(dinonylamino)ethyl)-N-nonylglycyl)piperazin-1-yl)ethan-1-one.24 Each was formulated with firefly luciferase mRNA and administered by intraperitoneal (IP) injection at the right lower quadrant of the abdomen to P12 NIH-Swiss mice (n = 5 per group) before being analyzed by bioluminescent imaging (Figure 1A) and tissue luminometry (Figure 1B) 6 h later. Whole-animal bioluminescent imaging revealed that LNP B demonstrated greater luciferase mRNA-directed protein production in the liver than LNP A (Figure 1A); this is demonstrated by the increased signal intensity (i.e., red area of the pseudocolor scale) in the location of the liver in LNP B mice, whereas expression is primarily in the lower abdomen near the injection site in LNP A-administered mice.
Figure 1.

Intraperitoneal injection of luciferase mRNA demonstrates different tissue exposure patterns depending on lipid nanoparticle formulation
(A and B) Comparative (A) and quantitative (B) bioluminescent imaging of P12 mice intraperitoneally administered LNP A or LNP B formulated with firefly luciferase mRNA demonstrates greater hepatic luciferase protein expression with LNP B; greater abdominal wall injection site expression was detected with LNP A. Low to modest LNP exposure was detected in other organs; such low levels cannot rule out that expression is from contaminated blood cell expression. Data are represented as mean ± SEM (n = 5 per group) (pseudocolor scale: red = highest, purple = lowest). LNP, lipid nanoparticle.
Quantitative examination of selected individual organs and tissues for LNP exposure was performed after mice were euthanized. Following modest perfusion, tissues were homogenized in lysis buffer on ice; supernatants were analyzed by tube luminometry in triplicate and normalized to protein levels. The highest levels of luciferase activity were detected in the liver and at the site of injection in the muscle. Significantly lower levels were detected in kidney, lung, and intestine, and, while luciferase activity was detected in brain tissue homogenates, the very low level could not rule out signal from contamination due to residual blood in the sample. Most organs (brain, heart, intestine, kidney, lung) show similar levels of LNP exposure and are without statistically significant differences between LNPs. LNP A demonstrated the greatest expression locally in the muscle at the site of injection (as also shown in Figure 1A); however, this was not statistically significant compared with LNP B. Conversely, LNP B demonstrated luciferase expression greater than 40 times that of the hepatic expression with LNP A and was statistically significant (p = 0.012).
Administration of ARG1 mRNA formulated with a lipid nanoparticle leads to survival and improved health of arginase 1-deficient mice
Lipid nanoparticle A and lipid nanoparticle B were formulated with ARG1 mRNA as previously described.15 Four groups of mice were subjected to studies with each lipid nanoparticle formulated ARG1 mRNA administered to constitutive Arg1 knockout mice: (1) wild-type Arg1+/+ (n = 26); (2) untreated constitutive Arg1−/− (n = 20); (3) LNP A with ARG1 mRNA daily administration beginning at P8 to constitutive Arg1−/− (n = 12); and (4) LNP B with ARG1 mRNA daily administration beginning at P8 to constitutive Arg1−/− (n = 25). As untreated constitutive arginase 1 mice (Arg1−/−) typically perish around postnatal day (P)12 with few surviving to P14, we examined survival and overall health of mice by weighing daily (as a surrogate for growth and health) from P8 to P28 (Figures 2A–2E). All (100%) untreated Arg1−/− mice (red line) expired by P14 as all (100%) Arg1+/+ mice (black line) survived to P28. The LNP B ARG1 mRNA-treated mice (blue line) fared well, with 24 of 25 surviving (96%) to P14 while 70% survived to P28 (one of the four deceased mice from P14 to P28 died from an injection error); only six (50%) of the LNP A ARG1 mRNA-treated mice (green line) survived to P14, with all remaining mice expiring by P15 (p < 0.0001 by Log rank).
Figure 2.

Intermittent LNP/arginase 1 mRNA administration leads to normal growth and survival of arginase-deficient mice
(A) Kaplan-Meier survival graph comparing arginase 1 wild type (Arg+/+, black line, n = 26) with untreated Arg1−/− (red line, n = 20), Arg1−/− mice treated with ARG1 mRNA formulated with LNP B (blue line, n = 25), and Arg1−/− mice treated with ARG1 mRNA and LNP A (green line, n = 12) (p < 0.0001 by Log rank test). (B) Weights of mouse groups demonstrate similar growth trajectory and weight comparing wild type (Arg1+/+, black line) with ARG1 mRNA LNP B formulation (blue line) (p = 0.999 at P13); however, differences exist compared with ARG1 mRNA LNP A formulation to Arg1−/− (green) (p = 0.009) and untreated Arg1−/− mice (red line) (p = 0.037 at P14). Physical size comparison of representative P14 (C), P18 (D), and P28 (E) LNP B ARG1 formulation with Arg1−/− mouse with an Arg1+/+ wild type. ARG1, arginase 1; Arg1, arginase 1, LNP, lipid nanoparticle. Data in (B) are represented as mean ± SD.
Mouse weight was used as a surrogate for health as previous studies have demonstrated a plateauing of weight at the end of 2 weeks of postnatal life in Arg1−/− mice that will soon expire.25 By day 13, Arg1+/+ mice (n = 26) had substantial and increasing weight gain (7.5 ± 1.4 g [mean ± standard deviation (SD)]) (black line) while untreated Arg1−/− mice (n = 20) did not substantially gain weight after P11 (red line) (Figure 2B); mouse weights plateaued and by P14 were substantially less than wild-type mice (5.2 ± 0.4 g; p = 0.037 versus Arg1+/+). Arg1−/− mice treated with ARG1 mRNA formulated with LNP B (n = 25) (blue line) fared well with P13 weights similar to Arg1+/+ mice (7.5 ± 1.3 g; p = 0.999 versus Arg1+/+) (see also Figure 2C). Similar to survival, ARG1 mRNA formulated with the LNP A (n = 12) (green line) had reduced weight gain by P13 (6.0 ± 0.7 g) compared with wild type and the LNP B group (p = 0.009 versus Arg1+/+). Littermate controls and surviving LNP B/ARG1-injected mice continued to gain weight, albeit with substantially greater weight in the littermates (p < 0.01). Mice were active and appeared healthy up to at least 4 weeks of age (Figures 2D and 2E).
ARG1 mRNA delivery by LNP A and LNP B result in hepatic arginase expression
Hepatic arginase expression (red fluorochrome) and glutamine synthetase (to identify perivenular hepatocytes from periportal hepatocytes; green fluorochrome) after LNP delivery of ARG1 mRNA were examined by immunostaining in comparison with Arg1+/+ wild-type mice (Figure 3A) and untreated Arg1−/− mice (Figure 3B). While substantial pan-hepatic ARG1 expression is detected in the liver by LNP B mRNA delivery (Figure 3C), little ARG1 protein is detected with LNP A delivery (Figure 3D). While hepatic ARG1 mRNA is detected by in situ hybridization by RNAScope 12 h after administration of LNP A ARG1 in hepatocytes (Figure 3 E1), this is markedly reduced or absent by 24 h (Figure 3 E2). LNP B-mediated ARG1 hepatic delivery is detected 12 h after administration and is much more substantial (representative images in Figure 3 F1); however, this is also reduced by 24 h (Figure 3 F2). This in situ hybridization probe is specific in that it does not detect Arg1 in wild-type murine liver (Figure 3G) or in Arg1−/− hepatocytes (Figure 3H).
Figure 3.

Administration of LNP-mRNA results in hepatic arginase 1 expression and resolution of arginase-deficient hepatocyte subcellular abnormalities
Images demonstrate murine arginase 1 expression (Arg1; red), glutamine synthetase (Glu1; green), and DAPI (blue) in (A) wild-type Arg1+/+ hepatocytes; (B) lack of Arg1 expression in murine Arg1−/− hepatocytes; (C) and restored hepatocyte expression of ARG1 when LNP B formulated with ARG1 is administered; this is markedly greater than the LNP A formulation (D). RNA in situ hybridization (E, LNP A; F, LNP B) demonstrates ARG1 mRNA both at 12 h (left images) after administration that is reduced by 24 h (right images). (G) Murine Arg1+/+ liver control demonstrates specificity of human ARG1 probe while (H) demonstrates lack of ARG1 in murine Agr1−/− liver. (I) Low-magnification representative image showing a hepatocyte with a spherical nucleus (N), several glycogen patches are pointed by arrow heads in wild-type Arg1+/+ murine liver. Bar, 5 μm. (J) Low-power representative image showing a hepatocyte with an irregular shape, broken nucleus (N), and many enlarged mitochondria surrounding the nucleus in an untreated Arg1−/− liver. Some mitochondria are seen but no prominent ER and glycogen granules are present. Bar, 2 μm. (K) Low-power representative image showing a hepatocyte having a spherical nucleus (N) that contains a prominent nucleolus; surrounding the nucleus are several patches of glycogen granules (arrowhead) when LNP A formulated mRNA was delivered. Bar, 2 μm. (L) High-power representative image of Figure 3K showing prominent rough ER and several mitochondria close to the nucleus. The ultrastructural features of the hepatocyte demonstrate a complete recovery in the subcellular morphology. ARG1, arginase 1; Arg 1, arginase 1; RNA, ribonucleic acid; LNP, lipid nanoparticle; N, nucleus; M, mitochondria. (A–H) Bar, 50 μm; (I) bar, 5 μm; (J and K) bar, 2 μm; (L) bar, 0.5 μm.
Ultrastructural imaging reveals a complete recovery of hepatocyte morphology in ARG1 mRNA-treated KO mice
To test whether hepatocytes undergo structural or morphological recovery accompanying functional restoration after LNP ARG1 mRNA administration, we carried out electron microscopic (EM) analysis on P14 liver samples from Arg1+/+, untreated Arg1−/−, and LNP A ARG1 mRNA-treated Arg1−/− mice. In Arg1+/+ wild type, as shown in Figure 3I, representative images of hepatocytes show normal fine structural features characterized by having a spherical nucleus with nucleolus and typical granular glycogen patches distributed in the cytoplasm. Prominent rough ER (arrows) and mitochondria (data not shown) are clearly localized. In contrast to wild type, the liver from untreated Arg1−/− animals displayed drastic alterations in hepatocytes at the ultrastructural level; the nuclei often exhibited an abnormal or irregular shape (representative image shown in Figure 3J). The low-magnification EM image clearly shows a broken nucleus with chromatin patches unevenly outlining the nuclear membranes. Mitochondria exhibit an enlarged matrix indicative of the stressful state of the subcellular organelles. No clear rough ER is present in the cytoplasm (data not shown). After LNP A formulated with 2 mg/kg ARG1 mRNA treatment of Arg1−/− mice, the hepatocytes undergo remarkable recovery in their morphology. As demonstrated in Figure 3K the hepatocyte has an easily detected spherical nucleus and a round nucleolus; glycogen granular patches (arrow) are found in the cytoplasm. At high magnification (Figure 3L), the rough ER is prominent and the mitochondria are clearly identified.
ARG1 mRNA delivered by lipid nanoparticle results in functional arginase 1 activity
Western blot was used to detect hepatic ARG1 protein in mice; a functional arginase activity assay was performed to quantitatively assess restored arginase activity after lipid-nanoparticle-mediated ARG1 mRNA delivery. While a dense band demonstrating arginase 1 protein is detected at 37–38 kDa by western blot corresponding to Arg1 in Arg1+/+ mice at P14 (lane 1, Figure 4A), this is absent as expected in Arg1−/− mice (lane 2). Similar to immunostaining, substantial arginase 1 protein is detected in the liver 24 h after ARG1 mRNA formulated with LNP B (lane 4). However, arginase 1 is not strongly detected when LNP A formulation is used (lane 3). Arginase functional activity in hepatocytes was partially restored when livers were tested 24 h after formulated ARG1 mRNA was administered (Figure 4B) (n = 6 per group). While wild-type hepatic Arg1 activity is substantial (12,766 ± 2,652 μg urea/mg protein), there is essentially no activity in untreated Arg1−/− mice (185.3 ± 59.4 μg urea/mg protein, 1.5% of Arg1+/+). Related to ARG1 mRNA levels in hepatocytes mediated by LNP A or LNP B, 4.3% (539.8 ± 245.5 μg urea/mg protein) or 9.4% (1,202.0 ± 555.5 μg urea/mg protein) of wild-type activity can be achieved 24 h after administration.
Figure 4.

Administration of LNP-formulated ARG1 results in functional hepatic arginase activity
Representative images in (A) demonstrate bands of arginase 1 protein in wild-type liver and primarily in Arg1−/− hepatocytes where LNP B-formulated ARG1 mRNA was administered. Functional arginase expression (B) is, while reduced compared with wild type (black bar), reestablished in LNP-administered mice, with the LNP B (blue bar) greater than LNP A formulation (green bar) (n = 6 per group). ARG1, arginase 1; Arg1, arginase 1; LNP, lipid nanoparticle. Data are represented as mean ± SD.
Plasma ammonia and most urea-cycle-related amino acids are improved after ARG1 mRNA is delivered by an LNP B-based formulation
Lack of hepatocyte arginase activity results in urea-cycle-related amino acid derangements in the constitutive murine model2,3 (Figure 5, red data points). This also includes ammonia and the nitrogen-carrying amino acids alanine and glutamine (n = 6–7 per group). While ARG1 mRNA delivery by the LNP A formulation (green data points) results in generally persistent abnormalities compared with age-matched Arg1+/+ wild-type mice (black data points) (alanine, 1,046.0 ± 428.5 μM versus 376.3 ± 59.4 μM, p = 0.091; aspartate, 144.2 ± 112.7 μM versus 22.5 ± 9.1 μM, p = 0.132; arginine, 2,275.0 ± 337.5 μM versus 283.2 ± 61.4 μM, p < 0.0001; citrulline, 218.3 ± 28.3 μM versus 87.8 ± 10.2 μM, p < 0.001; glutamate, 391.6 ± 236.5 μM versus 86.2 ± 30.3 μM, p = 0.050; glutamine, 1,907.0 ± 202.5 μM versus 497.1 ± 136.4 μM, p = 0.008; ornithine, 212.8 ± 79.1 μM versus 186.5 ± 45.1 μM, p = 0.885 [as LNP A formulation versus wild type]), use of the LNP B formulation (blue data points) results in normalization of these amino acids compared with wild-type controls (alanine, 257.6 ± 61.7 μM, p = 0.974; aspartate, 17.8 ± 10.7 μM, p > 0.999; arginine, 511.8 ± 403.8 μM, p = 0.873; citrulline, 88.7 ± 22.8 μM, p > 0.999; glutamate 54.1 ± 27.9 μM, p = 0.992; glutamine 467.4 ± 164.2 μM p > 0.999); the exception is ornithine, which was significantly reduced (58.4 ± 21.2 μM, p = 0.013).
Figure 5.

Elevated plasma ammonia and altered urea-cycle-related amino acids are improved with LNP-mediated mRNA delivery
The urea-cycle-related plasma amino acids of (A) alanine, (B) aspartate, (C) arginine, (D) citrulline, (E) glutamate, and (F) glutamine are generally improved (LNP A formulation Arg1−/−) or completely corrected (LNP B formulation Arg1−/−; p ≥ 0.796) with mRNA therapy depending on the LNP formulation; ornithine (G), the exception, is reduced with the LNP B formulation. Plasma ammonia (H) is improved with both formulations, albeit better with the LNP B formulation. (N = 6–7 per group). Arg1, arginase 1; mRNA, messenger ribonucleic acid; LNP, lipid nanoparticle. Data are represented as mean ± SD.
While plasma ammonia is markedly elevated in the untreated Arg1−/− mice (1,774.0 ± 329.4 μM [red data points]), there is improvement with both lipid nanoparticles (980.2 ± 261.7 μM, p = 0.001 versus wild type [373.9 ± 110.8 μM] for the LNP A formulation [green data points]), albeit markedly better with the LNP B formulation (493.8 ± 126.9 μM, p = 0.796 versus wild type [blue data points]) (n = 6 per group).
Hepatic urea-cycle-related amino acids are generally improved after ARG1 mRNA delivery by LNP B formulation
A limited amount of hepatic arginase activity results in some improvement of the derangements of urea-cycle-related hepatic tissue amino acids in the constitutive murine model.2,3 Overall, the effect of the ARG1 mRNA formulated to LNP A (green data points) results in no statistically significant improvement of urea-cycle-related hepatic amino acids compared with the untreated constitutive Arg1−/− knockout (red data points) (Figure S1). The LNP B ARG1 mRNA formulation (blue data points) results in improvement with only arginine and ornithine remaining statistically significant in their differences from wild type: alanine, 42.3 ± 25.8 μM versus 31.3 ± 5.8 μM (p = 0.956); arginine, 36.8 ± 39.3 μM versus 0.4 ± 0.14 μM (p = 0.035); aspartate, 18.4 ± 8.6 μM versus 9.3 ± 3.7 μM (p = 0.280); citrulline, 1.0 ± 0.98 μM versus 0.7 ± 0.3 μM (p = 0.976); glutamate, 29.4 ± 18.9 μM versus 22.3 ± 9.4 μM (p = 0.974); glutamine 19.1 ± 8.0 μM versus 24.6 ± 6.7 μM (p = 0.907); and ornithine, 3.9 ± 1.2 μM versus 12.4 ± 3.5 μM (p < 0.0001) (LNP B versus wild type, respectively) (n = 6–7 per group).
LNP formulations of ARG1 result in improvement of plasma guanidino compounds
Five plasma guanidino compounds were examined for alteration with administration of nanoparticles formulated with ARG1 mRNA (Figure 6). Non-statistically significant reductions from untreated Arg1−/− plasma levels were present in four guanidino compounds compared with untreated Arg1−/− mice (n = 6–11 per group). Argininic acid was reduced from 5.1 ± 4.0 μM in untreated Arg1−/− to 1.95 ± 0.99 μM and 2.96 ± 2.14 μM in LNP A (p = 0.117) and LNP B (p = 0.398) formulations, respectively. Plasma creatine was normalized (wild type [190.5 ± 43.9 μM] with LNP A [266.8 ± 158.9 μM, p = 0.591] and LNP B [204.1 ± 53.8 μM, p = 0.996]). Guanidinoacetic acid (GAA) was non-statistically reduced (wild type [10.9 ± 3.0 μM] with LNP A [135.6 ± 22.5 μM, p = 0.687] and LNP B [35.0 ± 37.1 μM, p = 0.997]). While plasma keto-arginine (normally very low at 0.44 ± 0.11 in wild type) is improved over untreated Arg1−/− mice (7.1 ± 5.3 μM) compared with 8.6 ± 7.9 μM in the LNP A group (p = 0.066 versus wild type) and 4.9 ± 4.7 μM in LNP B group (p = 0.471 versus wild type)], homoarginine levels do not show any substantial reduction.
Figure 6.

Abnormalities in plasma guanidino compounds are improved with ARG1 mRNA administration
(A) Argininic acid, (B) creatine, (C) guanidinoacetic acid (GAA), and (E) keto-arginine are all reduced with mRNA ARG1 administration, while (D) homoarginine remains reduced (n = 6–11 per group). ARG1, arginase 1; mRNA, messenger ribonucleic acid. Data are represented as mean ± SD.
Myelin basic protein immunostaining demonstrates diffuse central nervous system dysmyelination in hepatic arginase deficiency and marked recovery with LNP-mediated ARG1 delivery
Murine brains at P14 were sectioned and immunostained with myelin basic protein to assess overall myelination in Arg1+/+ (wild type), Arg1−/− (biallelic mutant), and Arg1−/− mice treated with LNP A and LNP B formulations of ARG1 mRNA (Figures 7, and see S2 for Luxol Fast Blue) (n = 4–5 per group). Figure 7A is a representative section showing myelination in multiple brain regions in the Arg1+/+ mouse, including the cingulum bundle (denoted by red rectangle), striatum, and the associated regions including the internal capsule (consisting of the afferent and efferent axon fibers to the cerebral cortex including the CST axons) (denoted by blue rectangle) and corpus callosum. Conversely, in Figure 7B, there is notably a marked reduction, if not a near-complete absence, of myelination detected in a P14 untreated Arg1−/− mouse. With administration of a lipid nanoparticle carrying ARG1 mRNA, myelination is improved with the LNP A formulation (Figure 7C) and restored with the LNP B formulation (Figure 7D).
Figure 7.

Immunohistochemical staining demonstrates markedly reduced myelination with dramatic improvement in brain myelination when hepatic arginase 1 expression is restored
Representative coronal images demonstrate myelination by MBP (brown) or its paucity at P14: (A) Arg1+/+, (B) Arg1−/−, (C) LNP A-formulated ARG1 mRNA Arg1−/−, and (D) LNP B-formulated ARG1 mRNA Arg1−/−. Representative images of myelination in the striatum (E) Arg1+/+, (F) Arg1−/−, (G) LNP A-formulated ARG1 mRNA Arg1−/−, and (H) LNP B-formulated ARG1 mRNA Arg1−/−. (E) Quantitation of the area fraction of MBP staining in the striatum per group. Representative images of myelination in the cingulum bundle (J) Arg1+/+, (K) Arg1−/−, (L) LNP A-formulated ARG1 mRNA Arg1−/−, and (M) LNP B-formulated ARG1 mRNA Arg1−/−. (N) Quantitation of the area fraction of MBP staining of the cingulum bundle per group (n = 5 per group). (In image A, the circle at lower left is bubble artifact.) ARG1, arginase 1; Arg1, arginase 1; P, postnatal; MBP, myelin basic protein; LNP, lipid nanoparticle; mRNA, messenger ribonucleic acid. Slides counterstained with hematoxylin. Data are presented as mean ± SD. (E–H) Bar, 200 μm; (J–M) bar, 100 μm.
Examination and quantitation of myelination in certain regions allowed for further assessment of the alteration and restoration of myelination (n = 4–6 per group). In Figure 7, the striatum was examined in (E)–(H), with quantitation in (I). Representative sections from Arg1+/+ (E) and untreated Arg1−/− (F) mice demonstrate the marked reduction in myelination at P14 (area fraction myelination wild type 41.63 ± 10.34 versus untreated Arg1−/− 2.095 ± 3.64, p < 0.0001). With ARG1 mRNA lipid nanoparticle administration, we detect limited improvement with the LNP A formulation (G) (5.678 ± 1.797 area fraction of myelination, p < 0.0001 compared with wild type), and a marked improvement is seen with the LNP B formulation (H) (34.41 ± 11.41 area fraction of myelination, p = 0.532 compared with wild type). Similar findings are achieved in the cingulum bundle (imaged in J–M, quantitated in N). While the Arg1+/+ cingulum bundle shows myelination at P14 (J) (area fraction of myelination, 15.76 ± 7.09), myelination is nearly absent in the untreated Arg1−/− brain (K) (1.48 ± 1.58) (p = 0.013). While this lack of myelination is partially improved with ARG1 delivery from the LNP A nanoparticle (L) (area fraction, 10.51 ± 8.15, p = 0.612 compared with wild type), it is markedly improved with the LNP B (M) formulation (N) (area fraction, 28.80 ± 6.84) (p < 0.0001 compared with untreated Arg1−/−).
EM examination of the corticospinal tract reveals recovery of myelinated axon density and myelin sheath layers with intermittent formulated ARG1 mRNA administration
The EM investigation for this study aimed to provide both qualitative and quantitative data for demonstrating the recovery in myelination of CST axons after mRNA treatment in the knockout mice. The EM analysis provided high-resolution EM images and quantitative data for demonstrating that LNP ARG1-treated knockout mice at P14 resulted in remarkable recovery in myelination of CST axons compared with untreated knockout mice at the same age. As shown in Figure 8, control samples from Arg1+/+ P14 mice (A) at low-magnification EM show many axons of variable sizes and myelin layer thickness distributed in the CST region as an oligodendrocyte soma is localized and shown partially to extend processes to form myelin layers with many axons; high magnification of axons is demonstrated (B). In contrast to Arg1+/+ wild type, the untreated Arg1−/− CST region (C) shows a few myelinated axons, some degenerating axons displaying thinner myelin layers, and irregular shapes of axons containing electron dense material accumulated in axoplasm (arrows in C and D), indicating advanced degenerating axons in the region. After ARG1 LNP A-formulated mRNA treatment, a remarkable recovery of myelinated axons was clearly demonstrated (E). Notably, an oligodendrocyte was shown to wrap many axons, some having thick myelin layers as demonstrated under high magnification (F).
Figure 8.

Electron microscopy demonstrates markedly reduced myelination with dramatic improvement in brain myelination when hepatic arginase 1 expression is restored
(A and B) Representative images of myelinated axons in CST of a P14 Arg1+/+ mouse. (A) A low-magnification image showing distribution of different sizes of myelinated axons in CST. Note an oligodendrocyte (oligo) localized at the left bottom extending multiple processes to wrap the axons. Bar, 2 μm. (B) A high-magnification image showing a portion of the process from the oligodendrocyte (oligo) surrounding several myelinated axons. Note that one axon at the upper left has thick myelin layers. Bar, 1 μm. (C and D) Lack of myelinated axons and signs of degenerating axons in CST of untreated Arg1−/− mouse. (C) A low-magnification electron micrograph demonstrating a drastic reduction of myelinated axons in CST; note many are small axons. Some degenerating axons with thin myelin layers; electron-dense debris (indicated by two arrows) are identified in the region. Bar, 2 μm. (D) High-power image showing one of the advanced degenerating axons containing electron-dense debris in the axoplasm (indicated by two arrows). Bar, 1 μm. (E and F) Recovery of myelinated axons in CST of the Arg1−/− mouse treated by LNP-formulated ARG1 mRNA. (E) Low-magnification image showing myelinated axons of variable sizes surrounding an oligodendrocyte (oligo) that is extending processes to wrap many axons. Bar, 2 μm. (F) High-power image showing several axons close to the oligodendrocyte (oligo); two axons in the middle have thick myelin layers. Bar, 1 μm. Quantitative data in comparing axon density (G), diameter (H), and G-ratio (I and J) (n = 3 per group). Arg1, arginase 1; CST, corticospinal tract; P, postnatal; oligo, oligodendrocyte; LNP, lipid nanoparticle; mRNA, messenger ribonucleic acid. Data are presented as mean ± SD. (A, C, and E: Bar, 2 μm; B, D, and F: Bar, 1 μm).
The near-complete recovery in myelinated CST axons was quantified as data presented in Figure 8 G–8J. As a first set of data (Figure 8G), the myelinated axon density was compared in Arg1+/+ wild type (n = 3), untreated Arg1−/− (n = 3), and LNP ARG1 mRNA-treated Arg1−/− groups (n = 3). The Arg1+/+ wild-type group (black dataset) shows the highest mean density (0.259 axons/μm2), with the untreated Arg1−/− group (red dataset) demonstrating the lowest (0.085 axons/μm2); the LNP-formulated ARG1 mRNA-treated Arg1−/− group (green dataset) shows a remarkable recovery (0.208/μm2, p = 0.160 versus wild type).
The second set of quantitative data was to compare the axon diameters in three groups as shown in Figure 8H. The histogram compares the mean diameters of myelinated axons. Arg1+/+ group has an average mean diameter of 0.94 μm (black dataset), the untreated Arg1−/− group (red dataset) is 0.75 μm (p < 0.001 compared with Arg1+/+), while the LNP ARG1 mRNA-treated Arg1−/− group showed a complete recovery (0.95 μm, p = 0.975 compared with Arg1+/+). The third set of data was generated from G-ratio analysis that reflects the thickness of the myelin sheath layers. As shown in Figure 8I, the G-ratio values of the three groups all display positive linear correlation with axon diameters (r2 value); the treated Arg1−/− group (green) has the lowest value in r2 (0.129), compared with 0.369 for Arg1+/+ (black) and 0.334 for untreated Arg1−/− (red). Overall, the untreated Arg1−/− group shows a much more restrictive distribution pattern in axon sizes, which is consistent with Figure 8H. Finally, as shown in Figure 8J, the G-ratio value is lowest in Arg1+/+ (0.775) (black dataset) and the highest in untreated Arg1−/− group (0.829) (red dataset); the G-ratio of the treated Arg1−/− group (LNP, green dataset) (0.80) approaches that of the wild type, and while the p value remains significant, the reduction in G-ratio confirms there is a dramatic recovery in axon myelination.
Discussion
Arginase deficiency at present has no completely effective US Food and Drug Administration-approved therapy that remedies the biochemical abnormalities that occur while at the same time averting the development of the neurological dysfunction associated with lack of hepatic arginase. While it is a urea cycle disorder, ammonia, which has been implicated as a neurotoxin in proximal enzyme deficiencies (i.e., hyperammonemic encephalopathy), is likely not a significant contributor to the neurophenotype in arginase-deficient patients; episodes of hyperammonemia are either infrequent or substantially fewer than those that occur with proximal urea cycle enzymopathies. Guanidino compounds have long been suggested to be causative, but to date have remained unproved and their role in the associated progressive neurological decline remains unclear.3,26, 27, 28, 29 As recently described,19 dysmyelination of the corticospinal tract is a prominent feature of the disorder in mice, and likely humans,21,22 and appears to be, at least in part, the cause of spastic diplegia, a characteristic gait abnormality found in these patients. While it remains a metabolic disorder, it has been suggested that arginase deficiency be reclassified as also a leukodystrophy because of the CNS myelination abnormalities present.19
Advances in mRNA synthesis and the development of biodegradable liver-distributing lipid nanoparticles have potentially enabled a new generation of low-risk, albeit transitory, therapeutics to potentially restore liver metabolic function for a whole host of single-enzyme hepatic enzymopathies, including arginase deficiency. We have previously demonstrated in an adult inducible knockout model that longevity could be attained and biochemical control of ammonia, glutamine, and arginine could be achieved with hepatic ARG1 mRNA therapy formulated with LNP A.15 In that study, where hepatic arginase 1 was disrupted with Cre/lox-mediated recombination, mice without treatment perished in 4 weeks; in those studies, we did not examine the control of guanidino compounds, and instead focused on survival, ammonia control, and urea-cycle-related amino acids. In addition, the central nervous system, already developed in these adult mice, was not examined: CNS neurodevelopmental myelination was already complete.
In these present investigations, we studied the constitutive arginase 1 murine model. This model is challenging as therapy must be effective or the mice will perish.3,30 While we focused in part on assessing the biochemical derangement in urea-cycle-related amino acids and ammonia, we also analyzed multiple guanidino compounds that are present in both human patients31 and in the constitutive murine model of arginase deficiency.2,32 With the exception of homoarginine, some control of guanidino compounds is achieved. However, most importantly, we assessed in fine detail and expanded our understanding of the scope of the neurophenotype of dysmyelination in arginase deficiency and examined if it could be prevented with an intermittent mRNA therapeutic approach. Herein we demonstrate that intermittent ARG1 mRNA-formulated lipid nanoparticle administration during a critical period of CNS myelinogenesis results not only in biochemical correction (i.e., ammonia, urea-cycle-related amino acids, and guanidino compound control) but also in the prevention of dysmyelination. The findings of the present study also demonstrate that CNS dysmyelination is not limited to the corticospinal tract in arginase deficiency; instead, it is a global CNS leukodystrophy that, at a minimum, also includes the striatum, internal capsule (including CST), corpus callosum, and cingulum bundle. These abnormalities in myelination are consistent with CNS imaging findings in the limited number of arginase-deficient patients studied19,21,22 and unpublished autopsy data.
To determine if intermittent mRNA administration could avert CNS dysmyelination during early neonatal CNS myelinogenesis, we calculated the G-ratio (smallest G-ratio ∼ thickest myelin sheath layer) in the CST of the cervical spinal cord in P14 mice. In wild-type Arg1+/+ mice, the G-ratio was 0.775, being consistent with our19 and other studies33, 34, 35 performed in similar regions of the rodent CNS; this independent dataset again confirms a theoretical model predicting that the optimal value of the G-ratio from the rat CNS is ∼0.77.19,36 The G-ratio was highest in the untreated Arg1−/− group (0.829; p < 0.001 versus Arg1+/+) with thin myelin layers (i.e., elevated G-ratio) when myelination was present. With the mRNA-treated Arg1−/− group, the G-ratio was 0.80, approaching that of the wild type (p = 0.006, difference between wild type and treated biallelic mutant), a dramatic recovery in axon myelination and thickness. These data indicate that early intermittent mRNA therapy can markedly improve myelination and its quality.
We also closely examined the density of myelinated axons in the two genotypes and in one of the LNP treatment groups. Arg1+/+ mice consistently showed the highest mean density at 0.259 axons/μm2 while untreated Arg1−/− mice showed the lowest at 0.085 axons/μm2 (p < 0.001); the ARG1 mRNA-treated Arg1−/− group demonstrated a remarkable recovery in myelinated axon density to 0.208 axons/μm2 (p = 0.160 versus wild type). Thus, myelinated axons, when present in the untreated Arg1−/− mouse, are reduced in density, number, and thickness of myelin in the cervical CST; following intermittent mRNA administration at a critical time of myelinogenesis, there is not only a reduction in the G-ratio (i.e., an improvement) but also recovery of myelinated axon density and thickness with active axonal wrapping by oligodendrocyte processes.
The neurologic phenotype in arginase deficiency is understood to likely be a bystander to the lack of the functional hepatic enzyme. The relative success of LNPs and hepatocyte expression of exogenously administered mRNA can, in part, be attributed to the favorable physiology of the liver, an organ that is well perfused and with a fenestrated endothelium.37 However, recent advances in both mRNA and lipid carriers greatly added to the efficacy. With systemic administration, free RNA is rapidly degraded by serum RNases with subsequent clearance by the kidneys.38 This physiological barrier has been overcome by formulating RNA into a carrier with adjusted composition, in this case as a lipid nanoparticle. Because of limited arginase mRNA and protein39 half-lives (and in this study working with a murine model with a mutation leading to early mortality unlike the afflicted human patients), this formulated mRNA-based LNP required frequent injections to maintain arginase 1 activity in a therapeutic window to result in survival, urea cycle function, and control of likely hepatic-originating toxins resulting in oligodendrocyte dysfunction. LNPs have been shown to improve the uptake of RNA molecules by cells and facilitate their endosomal release.40 In the case of these investigations, we implemented initially a study of an LNP (i.e., A) with an uptake mechanism that works by an apolipoprotein E (ApoE)-mediated low-density lipoprotein (LDL) receptor-dependent mechanism that has efficient endosomal escape.23 The interaction of LNPs with serum apolipoproteins, including ApoE, enhances uptake into primary hepatocytes.41,42 Multiple receptors have been associated with ApoE-mediated uptake, the LDL receptor being most notable, along with numerous LDL receptor family members.
We had proposed an early intervention to prevent both the biochemical abnormalities and, importantly, dysmyelination that occur in arginase deficiency knowing that postnatal myelination begins to substantially increase in the mouse brain at P8 (data not shown). However, our data indicate a marked disparity in the efficacy of the nanoparticles used in these studies. LNP B provided much greater delivery of ARG1 mRNA to hepatocytes in vivo, resulting in higher arginase hepatic protein (as demonstrated by western blot and functional arginase activity) and greater improvement in control of ammonia, arginase, and guanidino compounds along with a more pronounced effect on quantitative myelination of the cingulum bundle and striatum. It is not clear why LNP A was markedly less efficient than LNP B in hepatocyte mRNA uptake and functional activity for both luciferase and arginase 1. While LNP A is efficient at expressing luciferase protein in muscle/peritoneum near the site of injection, previous investigations have demonstrated that arginase expression in myocytes is ineffective in extending survival and successfully treating arginase deficiency in mice.43 The studies described in Figure 1 did demonstrate exposure of the luciferase mRNA-formulated LNP in extrahepatic tissues; however, outside of the liver for LNP B and the muscle at the site of the injection for LNP A, these were at a low level or modest at best. Very low levels were detected in the brain, with slightly more modest levels detected in the heart, intestine, kidney, and lung. These extrahepatic levels (excluding muscle with LNP A), as much as 3 logs lower than the liver with LNP B, are low enough that we cannot exclude or rule out that they are present from intravascular blood contamination.
As previously reported,23 LNP A is LDL-receptor-mediated, and ARG1 mRNA uptake was effective in our previously published work in adult animals that primarily examined plasma biochemistry.15 While not proved, we speculate that the mechanism of improved ARG1 mRNA expression mediated through LNP B likely occurs through a different mechanism than that of LNP A, which here has reduced efficacy in neonatal and young mice.
There are limitations to the study and the formulated mRNA approach. We have previously demonstrated that, in order to obtain long-term survival in arginase deficiency, ∼10% of wild-type hepatic arginase activity must be present.44 In these studies, while this was accomplished with LNP B, the ability to achieve this minimum level of expression was not reached with LNP A and resulted in a much higher rate of mortality (i.e., related to hyperammonemia). With particular attention directed at LNP B, plasma arginine was nearly normalized; however, hepatic arginine, which is normally nearly undetectable, was elevated, likely due to the half-life of the mRNA and arginase protein, suggesting that only partial restoration was achieved. Lack of complete correction of intracellular arginine levels is likely the cause for some of the guanidino compounds to be incompletely corrected as hepatic arginine is their source. Furthermore, ornithine, a product of the arginase reaction, is reduced in both plasma and hepatocytes, and is further evidence of insufficient hepatic arginase activity. The presence of hypoornithinemia suggests that these treated Arg1−/− mice are in an ornithine auxotrophic state. Finally, these LNPs were not developed specifically for an intraperitoneal delivery approach (note that serial intravenous administration would have been challenging at this age range in mice) and modification potentially could enhance their delivery of mRNA.
In aggregate, we have the following major findings from this study: mRNA-based therapy beginning in the neonatal period, even with restoration of small amounts of hepatic arginase activity, can prevent the dysmyelinating phenotype of the CNS that occurs in arginase deficiency as shown by both light microscopy and EM of the density of myelin and myelinated axons in the cingulum bundle, striatum, and cervical corticospinal tract. Subcellular hepatocyte abnormalities, previously undetected, are also prevented with formulated mRNA administration. In these studies, ARG1 mRNA lipid nanoparticles were administered daily, but it is plausible that less frequent administration may be possible in humans as afflicted patients are long lived, hyperammonemia is an uncommon occurrence, and long-term intravenous access is possible; unlike the typical afflicted human patient with arginase deficiency, hyperammonemia is the cause of mortality in mice. While the proximate toxin(s) that is likely affecting oligodendrocyte function is not known, these studies demonstrate that the likely or putative culprits (i.e., arginine, guanidino compounds) are controlled with intermittent mRNA administration to a level at which oligodendrocyte activity for CNS myelination proceeds normally. Finally, differences in LNPs for hepatic mRNA delivery exist and knowing the mechanism of uptake and endosomal transport will affect the further development of a mRNA regimen as other tissue- and organ-directed therapies progress.
Materials and methods
Mouse procedures
The constitutive arginase knockout mouse30 that has been backcrossed more than 10 times to the NIH-Swiss strain3 was used for these studies. These mice were housed at the University of California, Los Angeles (UCLA) colony under specific pathogen-free conditions; food and water were provided ad libitum and there were no periods of fasting. The procedures conducted were approved by the the UCLA Chancellor’s Animal Research Committee. Mice underwent genotyping by PCR at birth as previously described.3 All attempts were made to include equal numbers of male and female mice with littermates. Mice were fed standard mouse chow (Labdiet/PMI Nutrition International, St Louis, MO, USA [Picolab Rodent diet 20, #5053]). Beginning on postnatal day (P) 8, mice were administered daily, by intraperitoneal route, 2 mg/kg LNP ARG1 mRNA with a 28- or 31-gauge needle. Arg1+/+, untreated Arg1−/− and treated Arg1−/− mice (that survived) were euthanized on either day 14, 2 h after the injection, on day 15, 24 h after the injection for the collection of plasma and liver after the final mRNA/LNP injections, or at the end of the study at day 28.
mRNA synthesis and formulation
Codon-optimized mRNA and synthesis of human ARG1 and firefly luciferase were performed using typical methods as previously described.15,45 Formulation of both LNPs were performed as previously described.24 Briefly, ionizable, fusogenic, structural, and polyethylene glycol (PEG) lipids were dissolved in ethanol and mixed with mRNA at a molar ratio of 3:1 (mRNA:lipid). LNP A and LNP B formulations utilized heptadecan-9-yl 8-((2-hydroxyethyl) (8-(nonyloxy)-8-oxooctyl)amino)octanoate and 2-(dinonylamino)-1-(4-(N-(2-(dinonylamino)ethyl)-N-nonylglycyl)piperazin-1-yl)ethan-1-one ionizable lipids, respectively, with all other lipids as described previously.24
Ammonia analysis
Ammonia determination was performed using Ammonia Assay Kit (Abcam, Cambridge, MA, catalog # ab83360) as per manufacturer’s instructions;46 duplicate plasma samples were processed. Prolonged plasma storage was avoided and sample testing performed with all samples simultaneously to avoid batch effect.
Analysis of metabolites profiling
The concentration of plasma and liver amino acid levels were determined by HPLC utilizing precolumn derivatization with o-phthalaldehyde as in Nissim et al.47 The concentration of guanidino compounds, including creatine (CRE), creatinine (CRN), guanidineacetic acid (GAA), L-homoarginine (L-HA), keto-arginine (KA), and argininic acid (AA) were determined using Agilent 1260 liquid chromatography (LC) combined with triple-quad 6410B mass spectrometry (MS). Briefly, 10 μL of 1 mM internal standard (IS), epsilon amino caproic acid (EACA) was added to 10 μL of plasma. For measuring CRN and KA, samples were reconstituted in 0.1% formate in H2O (solution A) and used for analysis by liquid chromatography (LC)-MS. For measuring CRE, GAA, L-HA, and AA, a separate portion of the plasma samples was derivatized with 3N HCL-butanol and heated for 15 min at 60°C, then dried and reconstituted with 100 μL of solution A for LC-MS analysis. For LC-MS metabolites analysis, we used an Agilent Poroshell 120 EC-C18 column with mobile phase consisting of solution A and solution B (0.1% formate in acetonitrile and 0.005% trifluoroacetic acid). For underivatized compounds, the multiple reaction monitoring (MRM) 132-41; 174-114; and 114-44 were used for measurement of IS, KA, and CRN, respectively. For derivatized compounds, we used MRM 188-69, 232-71, 245-84, 174-101, and 188-44 for measurement of IS, AA, L-HA, GAA, and CRE, respectively.
Functional arginase analysis
Hepatic arginase activity was measured in duplicate from prepared lysates as previously described.3 Briefly, liver tissue was homogenized in 40 μL of 0.1% Triton X-100 and Halt protease inhibitor cocktail (Fisher Scientific, Hampton, NH) per milligram of tissue. Ten microliters of tissue lysate was diluted with water to a final volume of 100 μL; this was added to 100 μL of 25 mM Tris-HCl (pH 7.5) and 20 μL of 10 mM MnCl2. Arginase enzyme was activated by heating the sample to 56°C for 10 min followed by the addition of 100 μL of 0.5 M L-arginine (pH 7.9) and incubation for 1 h at 37°C. Conversion of arginine to urea was stopped with addition of 900 μL of acid mix (1:3:7 mixture of H2SO4 [96%]:H3PO4 [85%]:water). Forty microliters of 9% 1-phenyl-1,2-propanedione-2-oxime (ISPF, Sigma Aldrich, St Louis, MO) dissolved in ethanol, was added, followed by incubating at 95°C for 30 min for color development. Subsequently, samples were diluted 1:6 with water and 200 μL was used to measure absorbance at 540 nm. Urea standards ranging from 0.03 μmol to 0.5 μmol were used creating a calibration curve. To normalize the arginase activity measured above, the total protein content (Bio-Rad, Hercules, CA) was determined.
Western blot
Preparation of the protein samples and western blotting were carried out as described.48 Briefly, liver specimens were homogenized in RIPA buffer containing Halt protease inhibitor cocktail (Thermo Fisher, Waltham, MA) to isolate proteins. Fifty micrograms of the total protein extract, quantified with Bio-Rad Protein Assay Dye (Bio-Rad, Hercules, CA), were separated by SDS-PAGE and probed with ARG1 antibody (Thermo Fisher, catalog # PA5-29645) at 1:1,250, and β-tubulin antibody (Abcam catalog # ab6046; 1:2,500) was utilized as loading control. Targeted proteins were labeled by horseradish peroxidase (HRP)-conjugated goat anti-rabbit immunoglobulin G (IgG) (Santa Cruz Biotechnology, Dallas, TX, catalog # sc-2004; 1:5,000) and detected using SuperSignal West Pico PLUS Chemiluminescent Substrate (Thermo Fisher).
Histology and immunohistochemistry of liver
Explanted livers from euthanized animals were fixed in 10% neutral buffered formalin (v/v) for 48 h and subsequently stored in 70% ethanol. Standard procedures were employed for paraffin embedding of the tissues. Five-micrometer-thick cross sections of these paraffin-embedded tissues were collected on microscope slides and stained using standard immunohistochemistry procedures. Briefly, the sections were deparaffinized in xylene and rehydrated in serial washes of ethanol. Antigen retrieval was performed in 10 mM sodium citrate buffer pH 6.0 in a steamer for 40 min followed by incubation at room temperature (RT) for 30 min. For ARG1 and GLUL, tissue sections were permeabilized with 0.1% Triton X-100 in PBS for 10 min prior to 1-h blocking with 10% normal goat serum. After blocking, sections were incubated with primary antibodies diluted in the blocking buffer overnight at 4°C; hARG1 antibody (Thermo Fisher Scientific PA5-29645) at 1:100, glutamine synthetase (GLUL) (Abcam ab64613) at 1 μg/mL. After incubating with primary antibody, the sections were stained with goat anti-rabbit secondary antibody (Invitrogen A-11012), Goat anti-mouse secondary antibody (Invitrogen A-11001) both at 1:400 or goat anti-rat HRP (Abcam ab97057) at 1:200 for 1 h at RT. For fluorescence staining, the cell nuclei were counterstained with DAPI using VECTASHIELD Antifade Mounting Medium with DAPI (Vector Laboratories) and visualized with LSM 800 confocal microscope (Carl Zeiss, Oberkochen, Germany). For 3, 3'-diaminobenzidine (DAB) staining, sections were treated using DAB Substrate Kit, Peroxidase (HRP) (Vector Laboratories) using manufacturer instructions. Following this, sections were counterstained with hematoxylin (Harris Modified Method Hematoxylin Stain) using standard procedures, mounted with DPX (Fisher Scientific), and visualized using Aperio ScanScope AT and ImageScope software (both Leica Biosystems, Wetzlar, Germany).
Luxol fast blue staining
Luxol fast blue staining for myelinated axons was performed by standard methods.
Immunostaining for myelin basic protein
P14 mice were transcardially perfused with 5–10 mL of cold 4% paraformaldehyde (PFA) (Electron Microscopy Services). Whole brains were removed and then post-fixed in 4% PFA for 48 h at 4°C before being transferred to 70% ethanol. Immunostaining for myelin basic protein (MBP) was performed on 5-μm-thick paraffin-embedded coronal tissues sections by standard procedure, as described briefly above. Overnight incubation in rat anti-MBP primary antibody (Abcam, ab7349) at a 1:100 dilution was followed by secondary antibody incubation with a 1:200 dilution of goat anti-rat HRP antibody (Abcam, ab97057) for 1 h at RT and DAB chromogen staining (Vector Lab, SK-4100). A hematoxylin counter stain (Fisher, SH30-500D) was done before mounting and imaging. For quantification, 10× and 20× magnification scans were acquired using Aperio ScanScope AT and Aperio ImageScope software v.12.4.3.5008 was used to frame images with the regions of interest (both Leica Biosystems).
Images of the cingulum bundle were taken at 20× by setting the cingulum bundle in the center of the frame. Images of the striatum were taken at 10× magnification. The region selected was defined by setting the bottom edge of the lateral ventricle to the top of the frame and lateral-most edge of the corpus callosum to the right side of the frame.
ImageJ quantification
MBP-immunostaining semi-quantification was performed using ImageJ with Fiji plugin (NIH) and analyzed by a one-way ANOVA followed by Tukey’s multiple comparisons test with GraphPad Prism v.9.0.1 (GraphPad Software, San Diego, CA). Animals as n = 5 per group were analyzed.
For each area of interest, the images first underwent color deconvolution to separate the DAB staining from the hematoxylin counter stain. This was done through the color deconvolution addition with the Fiji plugin, using the existing H DAB vector option. Using the Color 2 window (R, 0.26814753; G, 0.57031375; B, 0.77642715; this is the channel that corresponds to the DAB staining in an eight-bit scale), threshold values were established and then set across all samples. The threshold value was established by normalizing the value to the wild-type samples. For the DAB channel of each wild-type image, an upper-level threshold value was chosen such that the pixels below this value covered the DAB-stained tissue but not the unstained background (the red highlighted pixels should cover the stained tissue and not the unstained tissue; this is the area that will be counted). The upper-limit values for each wild-type image were averaged together, and that value was used as the upper-limit threshold value across all images for the region of interest. The fraction of pixels below this threshold on the histogram were considered stained tissue, and the pixels above this value were unstained. The percentage area of stained tissue within each image was then measured.
Macro program for running established threshold value on all samples where “name” is the file image name and “X” is the calculated normalized upper-limit threshold value:
run("Color Deconvolution", "vectors = [H DAB]"); selectWindow ("name.tif-(Colour_2)"); setAutoThreshold("Default"); //run("Threshold … "); //setThreshold(0, X); setOption("BlackBackground", false); run("Convert to Mask"); run("Measure").
mRNA in situ hybridization
mRNA in situ hybridization was performed using RNAScope technology on the automated Leica BOND RX autostainer platform using the RNAScope 2.5 LS Reagent Kit-BROWN from Advanced Cell Diagnostics (Newark, CA) to target ARG1 mRNA with control probes to the housekeeping gene Mus musculus peptidylprolyl isomerase B (Ppib); mRNA (catalog # 313918, ACD) as a positive control; the bacterial gene dihydrodipicolinate reductase (DapB) (catalog # 312038, ACD) as a negative control was also used as a quality control check for tissues. This was performed as previously described.15
Electron microscopy
Animals
Three genotype groups of P14 mice were included in the EM study: wild-type group (n = 3), untreated Arg1−/− knockout group (n = 3), and treated Arg1−/− knockout group (n = 3). Mice were anesthetized with pentobarbital (10 mg/mL) and perfused through the heart first with cold 1× PBS solution for 1–2 min until clean, then perfused with 4% paraformaldehyde plus 2.5% glutaraldehyde in 1× PBS solution for at least 5 min. Brains were removed and stored in the fixative solution at 4°C.
Brain sample preparation
Cervical segments of spinal cord containing CST were processed. Cervical segment (C1–C7) spinal cords were identified under a dissecting microscope; sections approximately 0.5–1 mm in thickness were cut coronally with a sharp surgical blade and saved in the cold 1× PBS solution. The dorsal tract of cervical spinal cords was further verified under the dissecting microscope and carefully dissected out for embedding.
EM procedure
Sections containing CST were processed for EM analysis based on the previous studies.18,19 Briefly, sections were fixed in 2% OsO4 PBS for 30 min; after washing in PBS, sections were dehydrated in 70%, 90%, 95%, and 100% ethanol, and then in 100% acetone. Sections were processed in 1:1 pure acetone and Araldite (Ted Pella, Redding, CA) for 1 h and placed in freshly made pure Araldite overnight at 4°C. The next day, sections were transferred to Araldite solution for 3–4 h, and then were mounted on siliconized slides with glass coverslips (Sigmacote, Sigma, St Louis, MO) and polymerized in a 60°C–70°C oven for 48 h. Polymerized sections were examined under a dissecting microscope to confirm dorsal tract of spinal cord containing CST and carefully dissected using a sharp surgical blade. Sections containing these regions were glued to the blank Araldite blocks using superglue and dried in air. Sections were trimmed on a UCT Ultramicrotome (Leica), and a typical cutting surface of the section was about 2 × 2 mm2 including major portion of dorsal tract in spinal cord. To further confirm the selected regions histologically, the semi-thin sections (about 400–600 nm in thickness) were cut on the ultramicrotome using a diamond knife and sections were collected on clean glass slides in drops of water, dried on a hot plate. The slides were stained with toluidine blue; the semithin sections were examined under a light microscope and the regions including CST in spinal cord were confirmed. The blocks were then trimmed and 60- to 70-nm ultrathin sections were cut on the ultramicrotome using a diamond knife. Thin sections were collected on Formvar-coated single-slot copper grids (EMS, Hatfield, PA) and stained with uranyl acetate and lead citrate to enhance contrast.
EM imaging
Stained thin sections were examined in a JEM1200-EX (JEOL) electron microscope at 80 kV. Images were acquired using a wide-angle (top mount) BioScan 600 W 1 K × 1 K digital camera (Gatan, Pleasanton, CA). For axon analysis, EM images were taken at 8,000×, each image covering approximately 274 μm2, which is the unit area for myelinated axon number counting and density calculations. For visualizing the fine structure of axons and myelin sheaths, some images were taken above 20,000×. All the images were saved both as Gatan 3 format and Tiff format, and the files in Tiff format were imported to ImageJ (NIH) for counting and measuring or to Adobe Photoshop CC (Adobe System, San Jose, 2018) for image editing and figure composition.
EM data quantitative analysis
From EM imaging analysis, we generated two sets of data:
-
(1)
Myelinated axon density and comparison in CST of cervical spinal cord in three groups of P14 mice. Myelinated axons are defined as the axon caliber covered by at least one layer of electron-dense myelin sheath regardless of the sagittal- or coronal-sectioned plane. In each genotype group, for each animal, about 15–20 images were taken from the CST region, each image covering approximately 274 μm2. Total myelinated axons were identified and counted in ImageJ (NIH), thus, the density of myelinated axons in each unit area is calculated as the total number of myelinated axons counted/274 μm2 or number of myelinated axons/μm2. For each genotype, three mice were included for quantitative analysis. The myelinated axon density values were scatter plotted using Excel and compared in genotype groups.
-
(2)
Myelin sheath thickness (G-ratio) analysis and a comparison of myelinated axon diameters in CST of three genotype groups. The myelinated axon G-ratio analysis was carried out using EM images taken at 8,000×. For each genotype group, three mice were included. In each animal, about 15 EM images were imported to ImageJ, for each image, approximately 20–50 coronally cut myelinated axons were identified and chosen for measurement based on the method detailed in previous publications49,50. Briefly, for each myelinated axon, its axon diameter (a) and entire myelinated fiber diameter (A) were measured and recorded, and G-ratio = a/A. Thus, at least 50 myelinated axons from each genotype were measured and their G-ratio values were calculated. To analyze the relationship between G-ratio values and myelinated axon diameters, the G-ratio values and axon diameters of myelinated axons from each genotype were plotted in Excel, and a regressive analysis was applied to determine their linear correlations. The axon diameters (a) of all the myelinated axons for G-ratio analysis were calculated for each genotype group and compared.
EM data presentation
All the EM images were saved in Tiff format and imported to Adobe Photoshop CC for adjusting size, contrast, and brightness. The images were saved at a final resolution of 300 dots per inch (dpi). All other histograms or bar graphs were generated in Excel, saved as PDF file format, and imported to Adobe Photoshop. All the figures were composed in Adobe Photoshop CC.
A detailed EM protocol for liver samples was provided in a previous study.15
Assays for luciferase activity
Tissues were removed and placed in flat-bottomed 2.0-mL small capped tubes (USA Scientific, Ocala, FL) with lysis buffer (Promega, Madison, WI) and placed in an ice slush. Tissues were homogenized with a hand-held homogenizer (Omni International, Marietta, GA). Specimens were centrifuged at 4°C at 13,200 rpm for 5 min and then returned to ice. A luminometer (Berthold Detection Systems, Oak Ridge, TN) was used to measure total light emission according to the manufacturer’s instructions. All samples were measured for 10 s after a 5-s delay. Luciferase levels were determined in triplicate by luminometry and recorded as relative light units (RLU).51
Protein assay
Samples were centrifuged at 13,200 rpm for 5 min and dilutions prepared in 96-well plates. Protein determination was performed in duplicate (Bio-Rad, Hercules, CA) and specimens were analyzed on a microplate reader (Bio-Rad, Hercules, CA) at 750 nm. Samples were performed in duplicate and compared with protein standards. Mean and standard error of the mean (SEM) was calculated. Luciferase levels by luminometry were normalized after protein concentrations of the liver samples were determined (RLU/mg protein).51
Bioluminescent imaging
Six hours after lipid-formulated luciferase mRNA injection, mice were placed on a warming table and administered luciferase substrate (D-luciferin, Caliper Life Sciences, Mountain View, CA; 150 mg/kg IP) as previously described.52 Briefly, 5 min later, the isoflurane-anesthetized mice were imaged using the bioluminescence optical imager (IVIS 200; Xenogen, Alameda, CA). Mice were immediately placed in a light-tight chamber and baseline gray-scale body-surface images were taken. Afterward, photons emitted from the firefly luciferase reaction within the animal were acquired to demonstrate the photon diffusion pattern. Maximal luciferase signals were semiquantified using Living Image (Xenogen) and IGOR (Wavemetrics, Lake Oswego, OR) image analysis software. Visual output represents the number of photons emitted per second per square centimeter as a pseudocolor image.
Statistical analysis
Data analysis was performed with Prism v.9.0.1 statistical package (GraphPad Software). Results are expressed as mean ± SD, except in bioluminescent imaging, where SEM was used. p values were considered statistically significant when <0.05 and were determined using one-way ANOVA Dunnet’s multiple comparisons test. Log rank test was utilized for trend to compare survival.
Acknowledgments
The authors thank Oskana Horyn, Ilana Nissim, and Daikhin Evgueni for performing the isotopomer enrichment analysis and metabolite measurements in the Metabolomics Core Facility, The Children’s Hospital of Philadelphia. The authors also thank and acknowledge the Semel Institute for Neuroscience and the Intellectual and Developmental Disabilities Research Center at UCLA for their support. This work was supported by NIH grants R01NS110596, R01NS100979, and R03NS114623, all to G.S.L. The authors acknowledge the use of instruments at the Electron Imaging Center for NanoMachines supported by NIH (1S10RR23057 to Z. Hong Zhou) and CNSI at UCLA. A small grant was also provided by Moderna to support these studies; however, all experimental design and data interpretation were performed independently. The authors also thank Staci Sabnis and Douglas Burdette for helpful discussions.
Author contributions
S.K. designed and performed experiments, and analyzed data. X.-B.L. designed and performed experiments, analyzed data, and wrote the manuscript. B.T. designed and performed experiments, and analyzed data. M.N. performed experiments and analyzed data. J.L. performed experiments, statistically analyzed data, and prepared graphs. A.E. performed experiments. E.N. analyzed data and critically reviewed the manuscript. E.R., K.E.B., and R.W. performed experiments. X.Z. provided critical reagents. P.G.V.M. provided critical reagents and critically reviewed the manuscript. I.N. performed experiments and analyzed data. S.D.C. critically reviewed data and the manuscript. G.S.L. developed concepts, designed experiments, performed experiments, and wrote the manuscript.
Declaration of interests
G.S.L. has served as a consultant to Audentes Therapeutics and on the SAB of Taysha in areas unrelated to this work. S.D.C. is a consultant for Synlogic, Acer, Sana, and Aeglea in areas unrelated to this work. K.E.B., R.W., X.Z., and P.G.V.M. are current or previous employees of Moderna, and receive salary and stock options as compensation for their employment. All other authors declare no competing interests.
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.omtn.2022.04.012.
Supplemental information
References
- 1.Brusilow S.W., Horwich A. In: The Metabolic and Molecular Bases of Inherited Disease. Eighth Edition. Scriver C.R., Beaudet A.L., Sly W.S., Valle D., editors. McGraw-Hill; 2001. The metabolic and molecular bases of inherited disease; pp. 1909–1963. [Google Scholar]
- 2.Lee E.K., Hu C., Bhargava R., Ponnusamy R., Park H., Novicoff S., Rozengurt N., Marescau B., De Deyn P., Stout D., et al. AAV-based gene therapy prevents neuropathology and results in normal cognitive development in the hyperargininemic mouse. Gene Ther. 2013;20:785–796. doi: 10.1038/gt.2012.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Lee E.K., Hu C., Bhargava R., Rozengurt N., Stout D., Grody W.W., Cederbaum S.D., Lipshutz G.S. Long-term survival of the juvenile lethal arginase-deficient mouse with AAV gene therapy. Mol. Ther. 2012;20:1844–1851. doi: 10.1038/mt.2012.129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Carvalho D.R., Brand G.D., Brum J.M., Takata R.I., Speck-Martins C.E., Pratesi R. Analysis of novel ARG1 mutations causing hyperargininemia and correlation with arginase I activity in erythrocytes. Gene. 2012;509:124–130. doi: 10.1016/j.gene.2012.08.003. [DOI] [PubMed] [Google Scholar]
- 5.Edwards R.L., Moseley K., Watanabe Y., Wong L.J., Ottina J., Yano S. Long-term neurodevelopmental effects of early detection and treatment in a 6-year-old patient with argininaemia diagnosed by newborn screening. J. Inherit. Metab. Dis. 2009;32:197–200. doi: 10.1007/s10545-009-1148-2. [DOI] [PubMed] [Google Scholar]
- 6.Grody W.W., Klein D., Dodson A.E., Kern R.M., Wissmann P.B., Goodman B.K., Bassand P., Marescau B., Kang S.S., Leonard J.V., et al. Molecular genetic study of human arginase deficiency. Am. J. Hum. Genet. 1992;50:1281–1290. [PMC free article] [PubMed] [Google Scholar]
- 7.Jain-Ghai S., Nagamani S.C.S., Blaser S., Siriwardena K., Feigenbaum A. Arginase I deficiency: severe infantile presentation with hyperammonemia: more common than reported? Mol. Genet. Metab. 2011;104:107–111. doi: 10.1016/j.ymgme.2011.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lee B.H., Jin H.Y., Kim G.H., Choi J.H., Yoo H.W. Argininemia presenting with progressive spastic diplegia. Pediatr. Neurol. 2011;44:218–220. doi: 10.1016/j.pediatrneurol.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 9.Uchino T., Haraguchi Y., Aparicio J.M., Mizutani N., Higashikawa M., Naitoh H., Mori M., Matsuda I. Three novel mutations in the liver-type arginase gene in three unrelated Japanese patients with argininemia. Am. J. Hum. Genet. 1992;51:1406–1412. [PMC free article] [PubMed] [Google Scholar]
- 10.Uchino T., Snyderman S.E., Lambert M., Qureshi I.A., Shapira S.K., Sansaricq C., Smit L.M., Jakobs C., Joakobs C., Matsuda I., Matsuda L. Molecular basis of phenotypic variation in patients with argininemia. Hum. Genet. 1995;96:255–260. doi: 10.1007/bf00210403. [DOI] [PubMed] [Google Scholar]
- 11.Vockley J.G., Tabor D.E., Kern R.M., Goodman B.K., Wissmann P.B., Kang D.S., Grody W.W., Cederbaum S.D. Identification of mutations (D128G, H141L) in the liver arginase gene of patients with hyperargininemia. Hum. Mutat. 1994;4:150–154. doi: 10.1002/humu.1380040210. [DOI] [PubMed] [Google Scholar]
- 12.Picker J.D., Puga A.C., Levy H.L., Marsden D., Shih V.E., Degirolami U., Ligon K.L., Cederbaum S.D., Kern R.M., Cox G.F. Arginase deficiency with lethal neonatal expression: evidence for the glutamine hypothesis of cerebral edema. J. Pediatr. 2003;142:349–352. doi: 10.1067/mpd.2003.97. [DOI] [PubMed] [Google Scholar]
- 13.Ah Mew N., Simpson K.L., Gropman A.L., Lanpher B.C., Chapman K.A., Summar M.L. In: GeneReviews((R)) Adam M.P., Ardinger H.H., Pagon R.A., Wallace S.E., Bean L.J.H., Gripp K.W., Mirzaa G.M., Amemiya A., editors. 1993. Urea cycle disorders overview. [Google Scholar]
- 14.Carvalho D.R., Brum J.M., Speck-Martins C.E., Ventura F.D., Navarro M.M., Coelho K.E., Portugal D., Pratesi R. Clinical features and neurologic progression of hyperargininemia. Pediatr. Neurol. 2012;46:369–374. doi: 10.1016/j.pediatrneurol.2012.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Truong B., Allegri G., Liu X.B., Burke K.E., Zhu X., Cederbaum S.D., Haberle J., Martini P.G.V., Lipshutz G.S. Lipid nanoparticle-targeted mRNA therapy as a treatment for the inherited metabolic liver disorder arginase deficiency. Proc. Natl. Acad. Sci. U S A. 2019;116:21150–21159. doi: 10.1073/pnas.1906182116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Iyer R., Jenkinson C.P., Vockley J.G., Kern R.M., Grody W.W., Cederbaum S. The human arginases and arginase deficiency. J. Inherit. Metab. Dis. 1998;21:86–100. doi: 10.1023/a:1005313809037. [DOI] [PubMed] [Google Scholar]
- 17.Prasad A.N., Breen J.C., Ampola M.G., Rosman N.P. Argininemia: a treatable genetic cause of progressive spastic diplegia simulating cerebral palsy: case reports and literature review. J. Child Neurol. 1997;12:301–309. doi: 10.1177/088307389701200502. [DOI] [PubMed] [Google Scholar]
- 18.Cantero G., Liu X.B., Mervis R.F., Lazaro M.T., Cederbaum S.D., Golshani P., Lipshutz G.S. Rescue of the functional alterations of motor cortical circuits in arginase deficiency by neonatal gene therapy. J. Neurosci. 2016;36:6680–6690. doi: 10.1523/jneurosci.0897-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu X.B., Haney J.R., Cantero G., Lambert J.R., Otero-Garcia M., Truong B., Gropman A., Cobos I., Cederbaum S.D., Lipshutz G.S. Hepatic arginase deficiency fosters dysmyelination during postnatal CNS development. JCI Insight. 2019;4:e130260. doi: 10.1172/jci.insight.130260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Oldham M.S., VanMeter J.W., Shattuck K.F., Cederbaum S.D., Gropman A.L. Diffusion tensor imaging in arginase deficiency reveals damage to corticospinal tracts. Pediatr. Neurol. 2010;42:49–52. doi: 10.1016/j.pediatrneurol.2009.07.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Segawa Y., Matsufuji M., Itokazu N., Utsunomiya H., Watanabe Y., Yoshino M., Takashima S. A long-term survival case of arginase deficiency with severe multicystic white matter and compound mutations. Brain Dev. 2011;33:45–48. doi: 10.1016/j.braindev.2010.03.001. [DOI] [PubMed] [Google Scholar]
- 22.Gungor S., Akinci A., Firat A.K., Tabel Y., Alkan A. Neuroimaging findings in hyperargininemia. J. Neuroimaging. 2008;18:457–462. doi: 10.1111/j.1552-6569.2007.00217.x. [DOI] [PubMed] [Google Scholar]
- 23.Sabnis S., Kumarasinghe E.S., Salerno T., Mihai C., Ketova T., Senn J.J., Lynn A., Bulychev A., McFadyen I., Chan J., et al. A novel amino lipid series for mRNA delivery: improved endosomal escape and sustained pharmacology and safety in non-human primates. Mol. Ther. 2018;26:1509–1519. doi: 10.1016/j.ymthe.2018.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jiang L., Berraondo P., Jerico D., Guey L.T., Sampedro A., Frassetto A., Benenato K.E., Burke K., Santamaria E., Alegre M., et al. Systemic messenger RNA as an etiological treatment for acute intermittent porphyria. Nat. Med. 2018;24:1899–1909. doi: 10.1038/s41591-018-0199-z. [DOI] [PubMed] [Google Scholar]
- 25.Gau C.L., Rosenblatt R.A., Cerullo V., Lay F.D., Dow A.C., Livesay J., Brunetti-Pierri N., Lee B., Cederbaum S.D., Grody W.W., Lipshutz G.S. Short-term correction of arginase deficiency in a neonatal murine model with a helper-dependent adenoviral vector. Mol. Ther. 2009;17:1155–1163. doi: 10.1038/mt.2009.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Amayreh W., Meyer U., Das A.M. Treatment of arginase deficiency revisited: guanidinoacetate as a therapeutic target and biomarker for therapeutic monitoring. Dev. Med. Child Neurol. 2014;56:1021–1024. doi: 10.1111/dmcn.12488. [DOI] [PubMed] [Google Scholar]
- 27.Delwing-de Lima D., Wollinger L.F., Casagrande A.C.M., Delwing F., da Cruz J.G., Wyse A.T., Delwing-Dal Magro D., Magro D. Guanidino compounds inhibit acetylcholinesterase and butyrylcholinesterase activities: effect neuroprotector of vitamins E plus C. Int. J. Dev. Neurosci. 2010;28:465–473. doi: 10.1016/j.ijdevneu.2010.06.008. [DOI] [PubMed] [Google Scholar]
- 28.Grioni D., Furlan F., Canonico F., Parini R. Epilepsia partialis continua and generalized nonconvulsive status epilepticus during the course of argininemia: a report on two cases. Neuropediatrics. 2013;45:123–128. doi: 10.1055/s-0033-1360479. [DOI] [PubMed] [Google Scholar]
- 29.Silva E.S., Cardoso M.L., Vilarinho L., Medina M., Barbot C., Martins E. Liver transplantation prevents progressive neurological impairment in argininemia. JIMD Rep. 2013;11:25–30. doi: 10.1007/8904_2013_218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Iyer R.K., Yoo P.K., Kern R.M., Rozengurt N., Tsoa R., O'Brien W.E., Yu H., Grody W.W., Cederbaum S.D. Mouse model for human arginase deficiency. Mol. Cell Biol. 2002;22:4491–4498. doi: 10.1128/mcb.22.13.4491-4498.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deignan J.L., De Deyn P.P., Cederbaum S.D., Fuchshuber A., Roth B., Gsell W., Marescau B. Guanidino compound levels in blood, cerebrospinal fluid, and post-mortem brain material of patients with argininemia. Mol. Genet. Metab. 2010;100:S31–S36. doi: 10.1016/j.ymgme.2010.01.012. [DOI] [PubMed] [Google Scholar]
- 32.Deignan J.L., Marescau B., Livesay J.C., Iyer R.K., De Deyn P.P., Cederbaum S.D., Grody W.W. Increased plasma and tissue guanidino compounds in a mouse model of hyperargininemia. Mol. Genet. Metab. 2008;93:172–178. doi: 10.1016/j.ymgme.2007.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Arnett H.A., Mason J., Marino M., Suzuki K., Matsushima G.K., Ting J.P.Y. TNF alpha promotes proliferation of oligodendrocyte progenitors and remyelination. Nat. Neurosci. 2001;4:1116–1122. doi: 10.1038/nn738. [DOI] [PubMed] [Google Scholar]
- 34.Benninger Y., Colognato H., Thurnherr T., Franklin R.J.M., Leone D.P., Atanasoski S., Nave K.A., Ffrench-Constant C., Suter U., Relvas J.B. 1-Integrin signaling mediates premyelinating oligodendrocyte survival but is not required for CNS myelination and remyelination. J. Neurosci. 2006;26:7665–7673. doi: 10.1523/jneurosci.0444-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mason J.L., Langaman C., Morell P., Suzuki K., Matsushima G.K. Episodic demyelination and subsequent remyelination within the murine central nervous system: changes in axonal calibre. Neuropathol. Appl. Neurobiol. 2001;27:50–58. doi: 10.1046/j.0305-1846.2001.00301.x. [DOI] [PubMed] [Google Scholar]
- 36.Chomiak T., Hu B. What is the optimal value of the g-ratio for myelinated fibers in the rat CNS? A theoretical approach. PLoS One. 2009;4:e7754. doi: 10.1371/journal.pone.0007754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Akinc A., Querbes W., De S., Qin J., Frank-Kamenetsky M., Jayaprakash K.N., Jayaraman M., Rajeev K.G., Cantley W.L., Dorkin J.R., et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol. Ther. 2010;18:1357–1364. doi: 10.1038/mt.2010.85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Santel A., Aleku M., Keil O., Endruschat J., Esche V., Fisch G., Dames S., Loffler K., Fechtner M., Arnold W., et al. A novel siRNA-lipoplex technology for RNA interference in the mouse vascular endothelium. Gene Ther. 2006;13:1222–1234. doi: 10.1038/sj.gt.3302777. [DOI] [PubMed] [Google Scholar]
- 39.El-Sayed A.S., Shindia A.A., Diab A.A., Rady A.M. Purification and immobilization of L-arginase from thermotolerant Penicillium chrysogenum KJ185377.1; with unique kinetic properties as thermostable anticancer enzyme. Arch. Pharm. Res. 2014 doi: 10.1007/s12272-014-0498-y. [DOI] [PubMed] [Google Scholar]
- 40.Veiga N., Diesendruck Y., Peer D. Targeted lipid nanoparticles for RNA therapeutics and immunomodulation in leukocytes. Adv. Drug Deliv. Rev. 2020;159:364–376. doi: 10.1016/j.addr.2020.04.002. [DOI] [PubMed] [Google Scholar]
- 41.Bisgaier C.L., Siebenkas M.V., Williams K.J. Effects of apolipoproteins A-IV and A-I on the uptake of phospholipid liposomes by hepatocytes. J. Biol. Chem. 1989;264:862–866. doi: 10.1016/s0021-9258(19)85022-6. [DOI] [PubMed] [Google Scholar]
- 42.Yan X., Kuipers F., Havekes L.M., Havinga R., Dontje B., Poelstra K., Scherphof G.L., Kamps J.A.A.M. The role of apolipoprotein E in the elimination of liposomes from blood by hepatocytes in the mouse. Biochem. Biophys. Res. Commun. 2005;328:57–62. doi: 10.1016/j.bbrc.2004.12.137. [DOI] [PubMed] [Google Scholar]
- 43.Hu C., Kasten J., Park H., Bhargava R., Tai D.S., Grody W.W., Nguyen Q.G., Hauschka S.D., Cederbaum S.D., Lipshutz G.S. Myocyte-mediated arginase expression controls hyperargininemia but not hyperammonemia in arginase-deficient mice. Mol. Ther. 2014;22:1792–1802. doi: 10.1038/mt.2014.99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Kasten J., Hu C., Bhargava R., Park H., Tai D., Byrne J.A., Marescau B., De Deyn P.P., Schlichting L., Grody W.W., et al. Lethal phenotype in conditional late-onset arginase 1 deficiency in the mouse. Mol. Genet. Metab. 2013;110:222–230. doi: 10.1016/j.ymgme.2013.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nelson J., Sorensen E.W., Mintri S., Rabideau A.E., Zheng W., Besin G., Khatwani N., Su S.V., Miracco E.J., Issa W.J., et al. Impact of mRNA chemistry and manufacturing process on innate immune activation. Sci. Adv. 2020;6:eaaz6893. doi: 10.1126/sciadv.aaz6893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Nitzahn M., Allegri G., Khoja S., Truong B., Makris G., Haberle J., Lipshutz G.S. Split AAV-mediated gene therapy restores ureagenesis in a murine model of carbamoyl phosphate synthetase 1 deficiency. Mol. Ther. 2020;28:1717–1730. doi: 10.1016/j.ymthe.2020.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Nissim I., Horyn O., Daikhin Y., Chen P., Li C., Wehrli S.L., Nissim I., Yudkoff M. The molecular and metabolic influence of long term agmatine consumption. J. Biol. Chem. 2014;289:9710–9729. doi: 10.1074/jbc.m113.544726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Khoja S., Nitzahn M., Hermann K., Truong B., Borzone R., Willis B., Rudd M., Palmer D.J., Ng P., Brunetti-Pierri N., Lipshutz G.S. Conditional disruption of hepatic carbamoyl phosphate synthetase 1 in mice results in hyperammonemia without orotic aciduria and can be corrected by liver-directed gene therapy. Mol. Genet. Metab. 2018;124:243–253. doi: 10.1016/j.ymgme.2018.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Cheli V.T., Santiago Gonzalez D.A., Namgyal Lama T., Spreuer V., Handley V., Murphy G.G., Paez P.M. Conditional deletion of the L-type calcium channel Cav1.2 in oligodendrocyte progenitor cells affects postnatal myelination in mice. J. Neurosci. 2016;36:10853–10869. doi: 10.1523/jneurosci.1770-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Liu X.B., Schumann C.M. Optimization of electron microscopy for human brains with long-term fixation and fixed-frozen sections. Acta Neuropathol. Commun. 2014;2:42. doi: 10.1186/2051-5960-2-42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tai D.S., Hu C., Kim E.H., Lipshutz G.S. Augmentation of transgene-encoded protein after neonatal injection of adeno-associated virus improves hepatic copy number without immune responses. Pediatr. Res. 2015;78:239–246. doi: 10.1038/pr.2015.109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hu C., Busuttil R.W., Lipshutz G.S. RH10 provides superior transgene expression in mice when compared with natural AAV serotypes for neonatal gene therapy. J. Gene Med. 2010;12:766–778. doi: 10.1002/jgm.1496. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.