

Abstract
Herein we disclose an iron-catalyzed method to access skeletal rearrangement reactions akin to the Dowd-Beckwith ring expansion from unactivated C(sp3)-H bonds. Photoinduced ligand-to-metal charge transfer at the iron center generates a chlorine radical, which abstracts electron-rich C(sp3)–H bonds. The resulting unstable alkyl radicals can undergo rearrangement in the presence of suitable functionality. Addition to an electron deficient olefin, recombination with a photoreduced iron complex, and subsequent protodemetallation allows for redox-neutral alkylation of the resulting radical. Simple adjustments to the reaction conditions enable the selective synthesis of the directly alkylated or the rearranged-alkylated products. As a radical clock, these rearrangements also enable the measurement of rate constants of addition into various electron deficient olefins in the Giese reaction.
Keywords: LMCT, primary C(sp3)-H alkylation, iron catalysis, skeletal rearrangement, Dowd-Beckwith, photocatalysis
Graphical Abstract
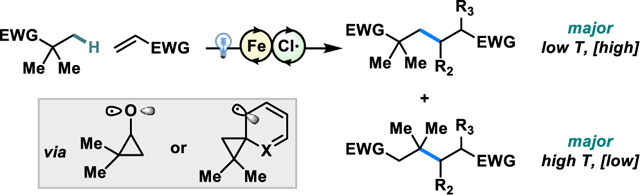
Carbon skeleton rearrangements in which C-C sigma bonds are broken provide powerful bond disconnections in organic synthesis.1,2 Due to the largely inert nature of C-C sigma bonds, formation of high energy intermediates like carbocations or radicals is usually necessary to generate sufficient thermodynamic driving force for their scission. While carbocation-mediated transformations like the Wagner-Meerwein and pinacol rearrangements have been widely studied and utilized in organic synthesis,3–5 there has been less focus on their radical-mediated counterparts.6–8
The Dowd-Beckwith rearrangement is a radical-mediated skeletal rearrangement that has found synthetic applications in ketone ring expansions.9–17 Typically, a primary alkyl radical is generated by halogen abstraction with a stannyl radical. This then adds to a carbonyl, forming a cyclopropyloxy radical which subsequently undergoes β-scission to give a more stable radical that is then quenched with tributyltin hydride (Scheme 1A). Current limitations of the rearrangement include the necessity for preinstallation of a functional handle and the use of stoichiometric tin reagents.
Scheme 1.

Radical rearrangement via FeCl3 ligand-to-metal charge-transfer (LMCT) provides access to divergent C(sp3)–H alkylation products.
The key skeletal rearrangement step in the Dowd-Beckwith ring expansion is related to a family of radical 1,2-rearrangements wherein a primary radical β to a π-system adds to it, forming a transient cyclopropyl intermediate that then undergoes β-scission to form a more stable tertiary radical.18–20 The rate constants of these rearrangements have been measured with various π-systems like alkenes, alkynes, arenes, carbonyls and nitriles, and several have been used as radical clocks.21–25
Hydrogen atom transfer (HAT) has emerged as a powerful mechanism for direct functionalization of C(sp3)-H bonds.26–30 Various reagents and catalysts have been developed to exhibit excellent HAT regioselectivity despite the high energy intermediates required to abstract strong unpolarized C(sp3)-H bonds.31–34 Although much attention has been paid towards the functionalization of C(sp3)-H bonds adjacent to heteroatoms or other radical stabilizing functional groups, less work has focused on the functionalization of compounds bearing electron withdrawing moieties.35 Previous investigations have shown that hydrogen atoms adjacent to electron withdrawing groups are recalcitrant to HAT due to a polarity mismatch,36–41 despite their lower bond dissociation energies compared to unactivated C(sp3)-H bonds.42 We postulated that the directing effect of carbonyls or other electron-withdrawing groups could enable HAT selectively at the beta-position to enable a skeletal rearrangement analogous to the Dowd-Beckwith ring expansion directly from C(sp3)-H bonds (Scheme 1B).
We recently reported the photocatalytic C(sp3)–H alkylation of alkanes using copper (II) chloride. This system mediates intermolecular HAT via the formation of chlorine radical which is readily generated by photoinduced ligand to metal charge transfer (Scheme 1C).43 Herein, we report a redox-neutral, photocatalytic alkylation of unactivated C(sp3)–H bonds distal from electron-withdrawing moieties using an iron (III) chloride catalyst44–49 to enable access to divergent products (Scheme 1D) via a skeletal rearrangement (Scheme 2). Of practical interest is that iron is the most abundant transition metal in the Earth’s crust,50 making it inexpensive and well-suited for use in large-scale syntheses.51 Simple changes to the temperature and concentration of the reaction, as well as the choice of the electron-deficient olefin partner allow for control of the ratio of unrearranged and rearranged products.
Scheme 2.
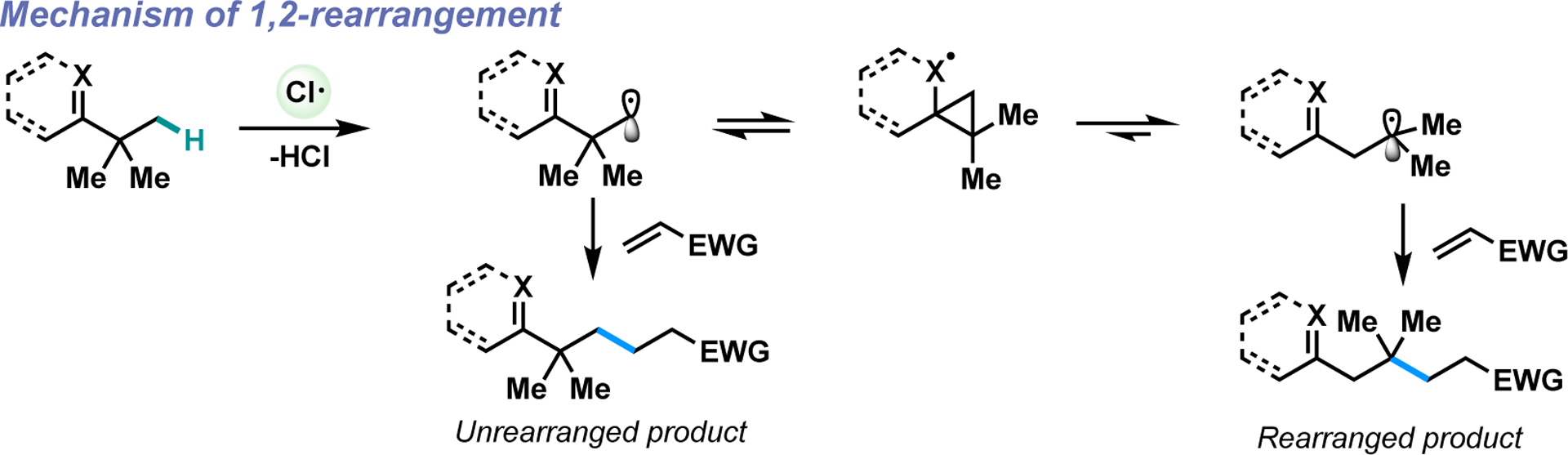
Mechanism of the 1,2-rearrangement.
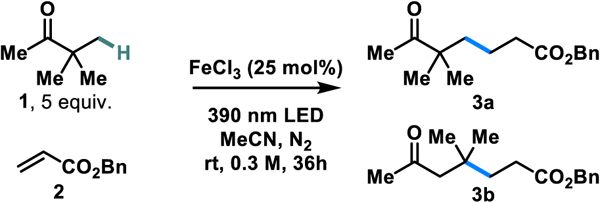
Initial optimization of the reaction was conducted using pinacolone and benzyl acrylate (Table 1). Early investigations found a catalyst loading of 25 mol% FeCl3 and the use of benzyl acrylate in limiting quantities to be effective. At room temperature and a concentration of 0.3 M, we obtain an isomeric ratio (ir) of unrearranged to rearranged product (3a:3b) of 1:1.4. Hypothesizing that an increase in temperature would reduce the rate of intermolecular radical trapping relative to the intramolecular skeletal rearrangement, we performed the reaction at 60 °C, and found that the ir indeed increases to 1:4 in favor of the rearranged product. Decreasing the reaction concentration to 0.1 M also promotes the 1,2-migration, increasing ir to 1:10. Given that reaction yields remain comparable under these conditions, we selected these conditions as optimal for promoting either the unrearranged product (Entry 1 – Conditions A) or the rearranged product (Entry 3 – Conditions B). UV-vis studies of FeCl3 in acetonitrile reveal a peak with λmax = 361 nm, with a tail into the visible region, along with two other maxima in the ultraviolet region (see SI). These peaks closely match the reported absorption spectrum of FeCl4− in acetonitrile, indicating that this is likely to be the photoactive species in our system.52 Addition of lithium chloride to the solution does not change any of the observed λmax values, further supporting this possibility. However, unlike with CuCl2,43 we found that exogenous chloride proved slightly detrimental to the efficiency of the reaction (Entry 4). Control reactions reveal the necessity for light and FeCl3 in order to furnish the desired product. Moreover, irradiating the reaction for 1 hour and continuing the reaction in the dark leads to dramatically reduced yields, disfavoring a radical chain mechanism where FeCl3 acts as an initiator, although we cannot completely rule out short chain processes. 427 nm irradiation is inefficient at promoting the reaction. Finally, we found that the reaction may be performed under air without significant loss in efficiency.
Table 1.
Optimization and Control Studies
| |||
|---|---|---|---|
| Entry | Deviation from Standard Conditions | Yield (%) | ir (3a:3b) |
| 1 | none | 64 | 1:1.4 |
| 2 | 60 °C | 65 | 1:4 |
| 3 | 60 °C, 0.1 M | 67 | 1:10 |
| 4 | added LiCl (62.5 mol%) | 52 | 1:1 |
| 5 | 0% FeCl3 | 0 | - |
| 6 | in the dark | 0 | - |
| 7 | irradiate at 390 nm 1 h, then dark | 2 | n.d. |
| 8 | 427 nm LED | 3 | n.d. |
| 9 | under air | 47 | 1:1 |
Reactions were performed on a 0.3 mmol scale. Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard. n.d.: not determined.
The scope of substrates which undergo this 1,2-migration is summarized in Scheme 3 (see SI for additional substrates). A variety of ketones other than pinacolone were found to be amenable to the transformation. Ketone 4 demonstrates a reduced propensity to undergo the 1,2-migration (12:1 ir), which we propose is due to a smaller Thorpe-Ingold effect compared to pinacolone.19,53–57 As expected, the ir is shifted towards the rearranged product (1.6:1) by performing the reaction under conditions B. Pivaloyl chloride was also observed to undergo 1,2-migration to a small extent, with an ir of 4:1 (5). The product was isolated as the ethyl ester after workup in alkaline ethanol. Aromatic substituents also participate in this 1,2-migration, albeit less efficiently than carbonyl substituents. Tert-butylbenzene gives an ir of 16:1 under conditions A (6), likely because the formation of the cyclopropyl intermediate disrupts aromaticity. The rate constant of this rearrangement is known to be 4 ×102 s−1 (298K),58 which provides a lower limit for the rate of radical addition to ethyl acrylate under our reaction conditions. We found that substituent effects can exert a strong influence on the propensity of the substrate to undergo migration. Electron-withdrawing substituents (-Ac, -CN) in the para-position lead to a complete inversion in ir. Under conditions A, the rearranged products predominate, and under conditions B they are formed exclusively (7–8). As expected, weakly electron-donating substituents such as acetoxy (9) exert only a minor effect on the rate of the 1,2-migration, yielding comparable ir values to the unsubstituted substrate 6. Ketone 10, containing both a phenyl and an acyl substituent adjacent to the site of initial radical formation, acts as a competition experiment between cyclization onto the arene and the carbonyl moieties. We exclusively obtained the rearrangement product resulting from a 1,2-acyl shift (10b). This complete selectivity can be attributed to the fact that the rate of the 1,2-acyl shift is more than two orders of magnitude greater than that of the 1,2-phenyl shift.25 Ring-expansion products similar to the Dowd-Beckwith reaction can be obtained starting with completely unfunctionalized cyclic ketones (11). Complete selectivity for the ring-expanded rearrangement product 11b is observed regardless of the conditions used. Given that electron-deficient arenes (7–8) strongly promote the rearrangement, we wondered whether protonated heteroarenes could function similarly. Indeed, di-tert-butylpyridine 12 reacts in moderate yields, favoring the rearranged product relative to tertbutylbenzene. With benzothiazole 13, the rearranged product is formed exclusively under Conditions B.
Scheme 3.
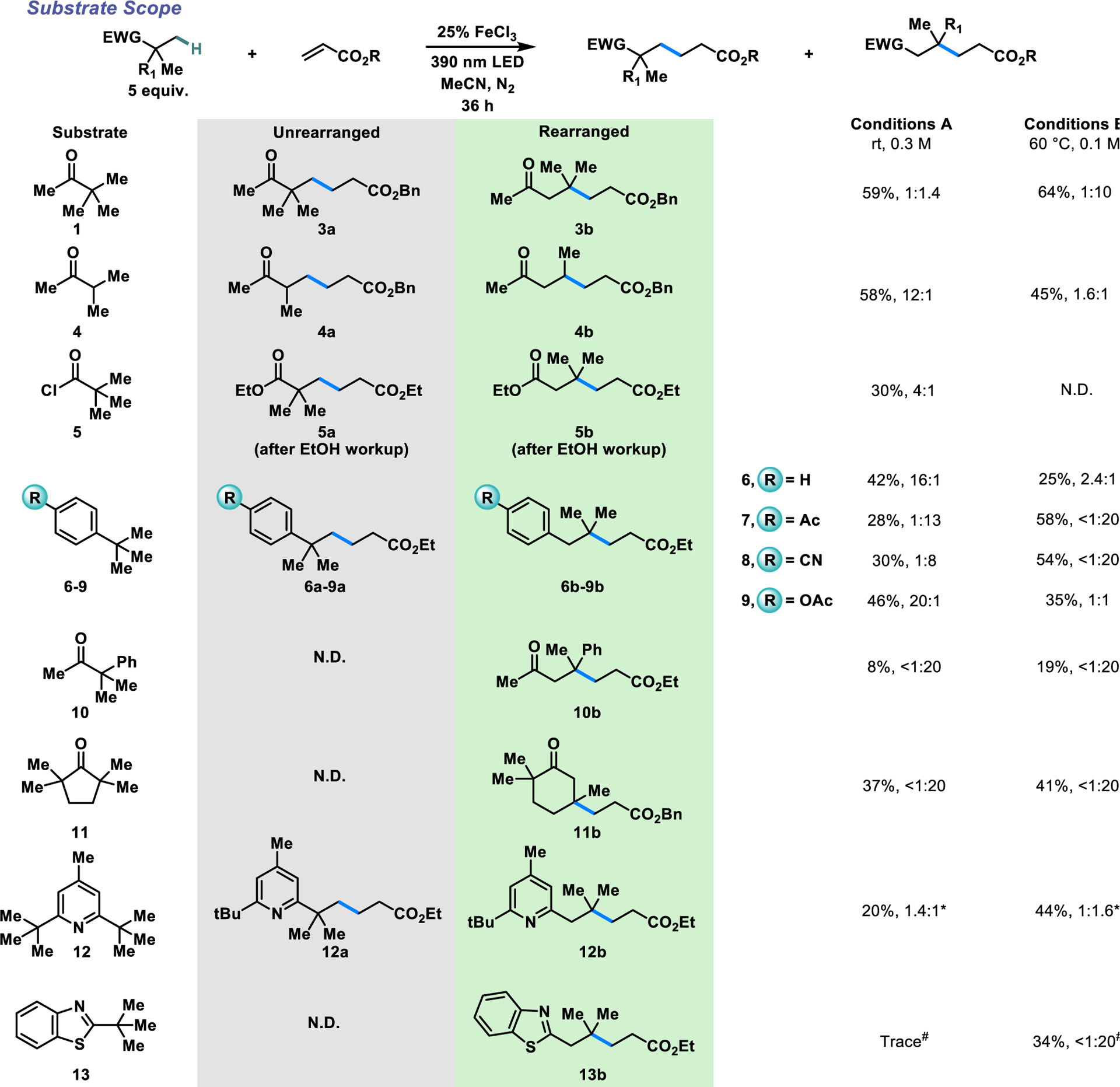
Scope of C-H pronucleophiles for the 1,2-migration. N.D.: not detected. *With 50 mol% FeCl3 and 5 equiv. CF3COOH as an additive. #With 5 equiv. CF3COOH as an additive.
We next examined how the choice of electron-withdrawing olefin would affect the amount of 1,2-migration observed (Scheme 4). Maleic anhydride and N-methylmaleimide react in moderate to good yields. We isolated roughly equimolar mixtures of unrearranged and rearranged product (14–15) under Conditions A, but mostly rearranged product (1:8) under Conditions B. To our surprise, despite being significantly weaker electrophiles than maleic anhydride and N-methylmaleimide in previous studies with closed-shell nucleophiles59,60, benzyl acrylate, acrylonitrile, and ethyl methacrylate react with comparable ir values under the two conditions (3, 16–17). Other electron-withdrawing groups on the olefin are also tolerated, including sulfones, amides and free acids (18–20). These acceptors generally give comparable to slightly higher ratios of rearranged product compared to benzyl acrylate. Reaction with fumaronitrile proceeds in good yields under both reaction conditions (21), with a similarly greater proportion of rearrangement product. Again, this was contrary to our expectations, since fumaronitrile is known to be a stronger electrophile than the acrylates based on previous studies of polar reactions.60 Dimethyl fumarate (22) gives a slightly higher ir in favor of the rearrangement compared to fumaronitrile, while the cis isomer dimethyl maleate (23) heavily favors the rearrangement under both conditions. Small proportions of cyclized rearranged product 22c are observed under conditions B but not under conditions A, suggesting a possible thermally-driven aldol-type reaction. Finally, benzylidenemalononitrile, which displays similar electrophilicity to maleic anhydride in polar reactions,61 also unexpectedly yields high proportions of rearrangement product (24). The alkylated product is isolated exclusively as a cyclized adduct, presumably formed from attack of the bis(cyano)-stabilized anion onto the ketone. The diastereoselectivity of the cyclization was unambiguously confirmed by X-ray crystallography.
Scheme 4.
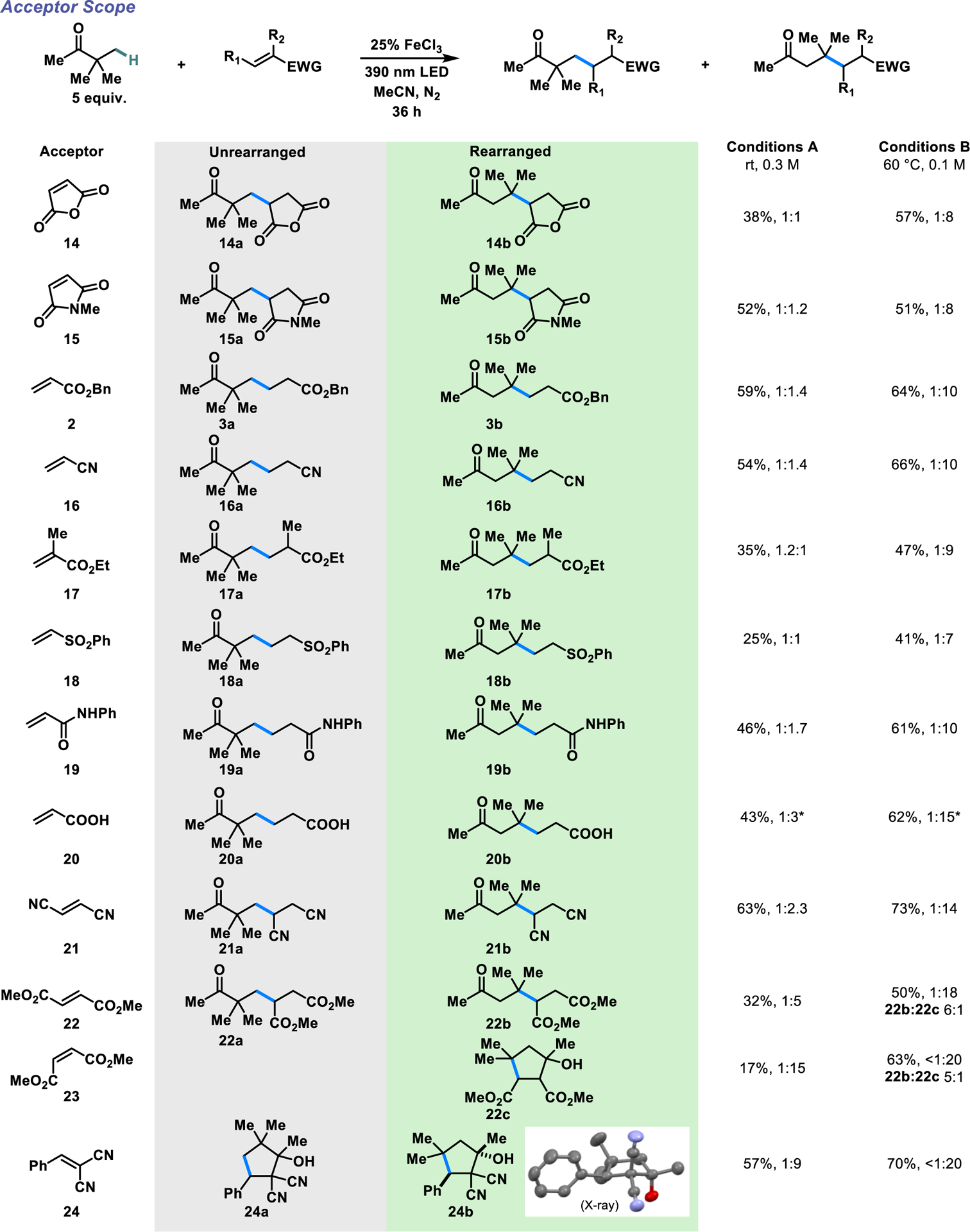
Scope of acceptors for the 1,2-migration. *1 equiv. trifluoroacetic acid used as an additive.
In order to better understand these unexpected observations, we sought to benchmark the rate constants for the addition of radical nucleophiles to these acceptors. Initial experiments demonstrated a linear dependence of the ratio of unrearranged to rearranged product on the concentration of acceptor used (see SI). This linear dependence argues against a Curtin-Hammett scenario62,63 wherein the primary and tertiary radicals are rapidly equilibrating and the product ratio is determined by the energy barriers for the trapping of the two radicals, since the ratio of products would be largely insensitive to acceptor concentration under that regime. Along with the effect of elevated temperature on the proportion of rearranged product, this finding led us to propose a mechanism in which the addition of the initially formed primary radical to the acceptor is in direct competition with the 1,2-migration to form the more stable tertiary radical. While the 1,2-migration is in principle reversible, two factors indicate that the reverse reaction is likely to be heavily outcompeted by trapping of the tertiary radical by the electron-deficient olefin. Tertiary radicals exhibit greater nucleophilicity compared to primary radicals,64 so trapping of the rearranged radical is likely to be significantly more rapid. Moreover, the initial formation of the cyclopropyloxy radical is significantly accelerated by a Thorpe-Ingold effect that is absent in the reverse reaction – the cyclization of 3-butenyl radical to cyclopropylcarbinyl radical is three orders of magnitude slower than the analogous reaction with the 2,2-dimethyl-3-butenyl radical.65 Under this regime, the rate constant for the 1,2-migration can serve as a convenient radical clock to determine the rate constants for addition of the pinacolone primary radical to various acceptors.
Working from the known rate constant for 1,2-migration for di-tert-butyl ketone,25 we determined the corresponding rate constant for pinacolone to be 2.9 × 104 s−1 (see SI). Initial rate experiments were then conducted using the 1,2-migration as a radical clock. The calculated rate constants for the addition of the pinacolone primary radical to the acceptors tested span slightly more than an order of magnitude (Scheme 5A). The values are also generally consistent with the ir values observed in our reactions, with the least reactive acceptors giving the greatest proportions of rearrangement product. Comparison of our rate constants to the known rate constants for methyl and tert-butyl radical addition to similar acceptors64,66–69 (see SI for a detailed table) shows that the pinacolone radical generally adds more slowly than both, reflecting its high steric hindrance and low nucleophilicity (as a primary radical). The fairly low spread of the rate constants is also consistent with literature data sets. Giese-type additions are known to be highly exothermic and therefore have early transition states that are less sensitive to the structure of the electrophilic alkene relative to analogous ionic additions.66 Rate constants for alkyl radical addition to 15, 18, 19, and 24 were not previously known.
Scheme 5.
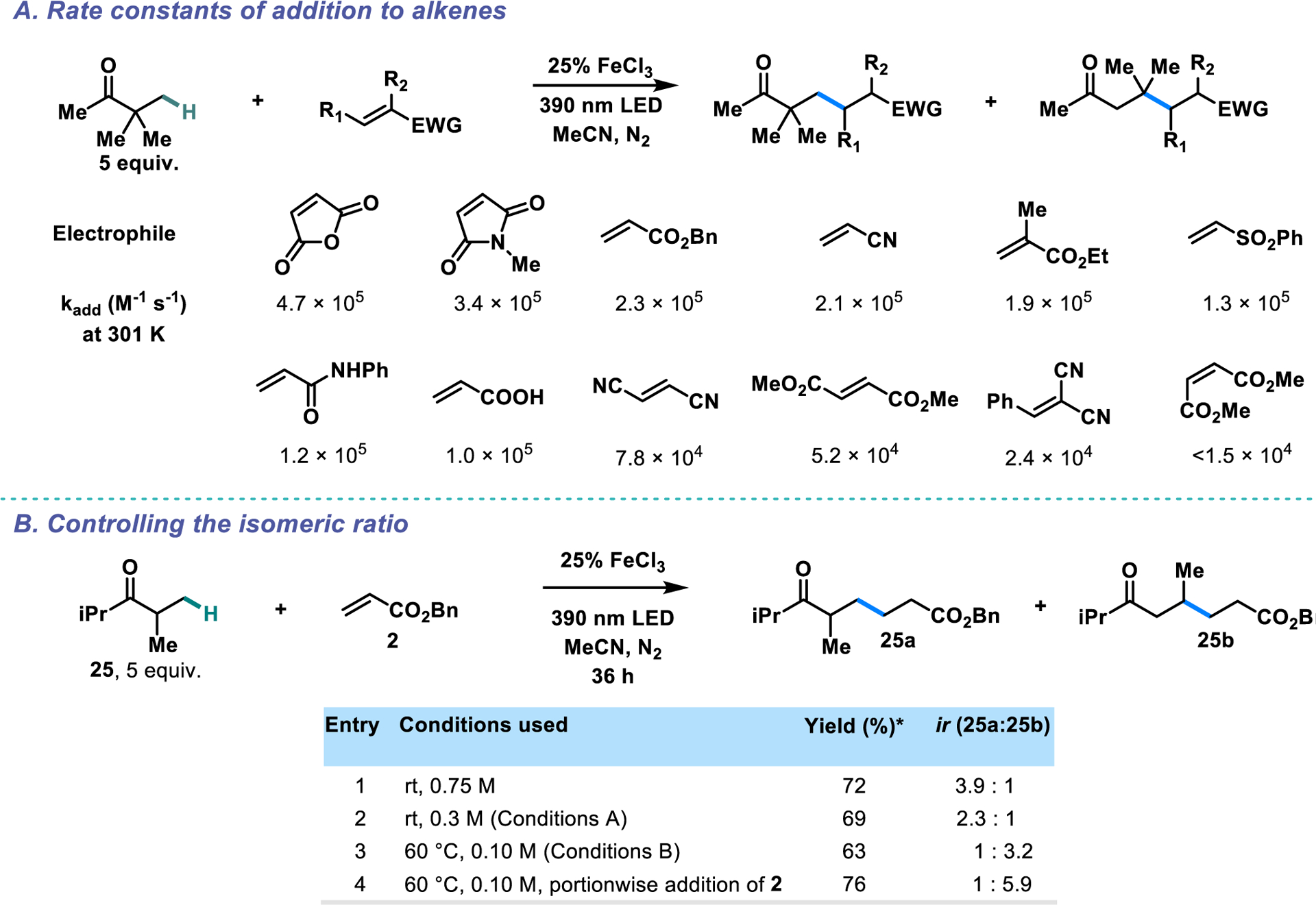
(A) Rate constants of addition to various alkenes. (B) Modifications to the reaction conditions allow for further control of the isomeric ratio (ir). * Yields were determined by 1H NMR using 1,3,5-trimethoxybenzene as the internal standard.
With substrates that only give moderate isomeric ratios, the selectivity can be tuned in either direction with further modifications to the reaction conditions (Scheme 5B). Under Conditions A, diisopropyl ketone 25 gives a moderate ir of 2.3:1. Performing the reaction at a higher concentration increases the ir to almost 4:1 (Entry 3). Similarly, the ir of 1:3.2 under conditions B can be increased to 1:5.9 by addition of benzyl acrylate in five equal portions over 60 h (Entry 4). In line with our proposed mechanism, increasing or decreasing the effective concentration of the acceptor in the reaction mixture yields a correspondingly smaller or greater proportion of rearrangement product.
In conclusion, we report an iron-catalyzed, photocatalytic method for the divergent alkylation of C(sp3)–H bonds mediated by a 1,2-skeletal rearrangement. Unlike typical radical-mediated skeletal rearrangements, no prefunctionalization is required, and control over the ratio of unrearranged to rearranged product can be achieved by simple modifications to the reaction conditions or the choice of acceptor. The 1,2-rearrangement was also utilized as a radical clock to determine the rate constants for the addition of nucleophilic radicals to various electron-deficient olefins.
Supplementary Material
ACKNOWLEDGMENT
We thank NIGMS (GM125206) for support. We thank Dr. Brandon Fowler for HRMS analysis and Dr. Manju Rajeswaran for X-ray crystallography. We are also grateful to Prof. Jack Norton (Columbia University) for valuable discussions about the kinetics experiments.
Footnotes
The authors declare no competing financial interests.
Supporting Information
Experimental details and compound characterization data. The Supporting Information is available free of charge on the ACS Publications website.
REFERENCES
- (1).Rojas CM Molecular Rearrangements in Organic Synthesis; John Wiley & Sons, 2015; pp 1–288. [Google Scholar]
- (2).Overman LE Molecular Rearrangements in the Construction of Complex Molecules. Tetrahedron 2009, 65 (33), 6432–6446. 10.1016/j.tet.2009.05.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Hanson JR 3.1 - Wagner–Meerwein Rearrangements. In Comprehensive Organic Synthesis; Trost BM, Fleming I, Eds.; Pergamon: Oxford, 1991; pp 705–719. 10.1016/B978-0-08-052349-1.00077-9. [DOI] [Google Scholar]
- (4).Song Z-L; Fan C-A; Tu Y-Q Semipinacol Rearrangement in Natural Product Synthesis. Chem. Rev 2011, 111 (11), 7523–7556. 10.1021/cr200055g. [DOI] [PubMed] [Google Scholar]
- (5).Gao AX; Thomas SB; Snyder SA Pinacol and Semipinacol Rearrangements in Total Synthesis. In Molecular Rearrangements in Organic Synthesis; John Wiley & Sons, Ltd, 2015; pp 1–34. 10.1002/9781118939901.ch1. [DOI] [Google Scholar]
- (6).Bach T; Hehn JP Photochemical Reactions as Key Steps in Natural Product Synthesis. Angew. Chem. Int. Ed 2011, 50 (5), 1000–1045. 10.1002/anie.201002845. [DOI] [Google Scholar]
- (7).Chen Z-M; Zhang X-M; Tu Y-Q Radical Aryl Migration Reactions and Synthetic Applications. Chem. Soc. Rev 2015, 44 (15), 5220–5245. 10.1039/C4CS00467A. [DOI] [PubMed] [Google Scholar]
- (8).Kärkäs MD; Porco JA; Stephenson CRJ Photochemical Approaches to Complex Chemotypes: Applications in Natural Product Synthesis. Chem. Rev 2016, 116 (17), 9683–9747. 10.1021/acs.chemrev.5b00760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Dowd P; Choi SC A New Tributyltin Hydride-Based Rearrangement of Bromomethyl .Beta.-Keto Esters. A Synthetically Useful Ring Expansion to .Gamma.-Keto Esters. J. Am. Chem. Soc 1987, 109 (11), 3493–3494. 10.1021/ja00245a069. [DOI] [Google Scholar]
- (10).Dowd P; Choi SC Free Radical Ring Expansion by Three and Four Carbons. J. Am. Chem. Soc 1987, 109 (21), 6548–6549. 10.1021/ja00255a071. [DOI] [Google Scholar]
- (11).Beckwith ALJ; O’Shea DM; Westwood SW Rearrangement of Suitably Constituted Aryl, Alkyl, or Vinyl Radicals by Acyl or Cyano Group Migration. J. Am. Chem. Soc 1988, 110 (8), 2565–2575. 10.1021/ja00216a033. [DOI] [Google Scholar]
- (12).Hart DJ; Havas F Complex Examples of the Dowd–Beckwith Rearrangement: A Free Radical Route to 2-Oxabicyclo 〚3.3.0〛octan-3,6-Diones. Comptes Rendus Académie Sci. - Ser. IIC - Chem 2001, 4 (7), 591–598. 10.1016/S1387-1609(01)01274-9. [DOI] [Google Scholar]
- (13).Ardura D; Sordo TL Three-Carbon Dowd−Beckwith Ring Expansion Reaction versus Intramolecular 1,5-Hydrogen Transfer Reaction: A Theoretical Study. J. Org. Chem 2005, 70 (23), 9417–9423. 10.1021/jo051551g. [DOI] [PubMed] [Google Scholar]
- (14).Zerong W Dowd-Beckwith Ring Expansion. In Comprehensive Organic Name Reactions and Reagents; John Wiley & Sons, Inc, 2010; pp 939–941. 10.1002/9780470638859.conrr201. [DOI] [Google Scholar]
- (15).Liu Y; Yeung Y-Y Synthesis of Macrocyclic Ketones through Catalyst-Free Electrophilic Halogen-Mediated Semipinacol Rearrangement: Application to the Total Synthesis of (±)-Muscone. Org. Lett 2017, 19 (6), 1422–1425. 10.1021/acs.orglett.7b00350. [DOI] [PubMed] [Google Scholar]
- (16).Kim YJ; Kim DY Electrochemical Radical Selenylation/1,2-Carbon Migration and Dowd–Beckwith-Type Ring-Expansion Sequences of Alkenylcyclobutanols. Org. Lett 2019, 21 (4), 1021–1025. 10.1021/acs.orglett.8b04041. [DOI] [PubMed] [Google Scholar]
- (17).Duecker FL; Heinze RC; Heretsch P Synthesis of Swinhoeisterol A, Dankasterone A and B, and Periconiastone A by Radical Framework Reconstruction. J. Am. Chem. Soc 2020, 142 (1), 104–108. 10.1021/jacs.9b12899. [DOI] [PubMed] [Google Scholar]
- (18).Urry WH; Kharasch MS Factors Influencing the Course and Mechanism of Grignard Reactions. XV. The Reaction of β,β-Dime-thylphenethyl Chloride with Phenylmagnesium Bromide in the Presence of Cobaltous Chloride. J. Am. Chem. Soc 1944, 66 (9), 1438–1440. 10.1021/ja01237a004. [DOI] [Google Scholar]
- (19).Newcomb M Kinetics of Radical Reactions: Radical Clocks. In Radicals in Organic Synthesis; John Wiley & Sons, Ltd, 2001; pp 316–336. 10.1002/9783527618293.ch16. [DOI] [Google Scholar]
- (20).Newcomb M Radical Kinetics and Clocks. In Encyclopedia of Radicals in Chemistry, Biology and Materials; John Wiley & Sons, Ltd, 2012; pp 1–18. 10.1002/9781119953678.rad007. [DOI] [Google Scholar]
- (21).Maillard B; Ingold KU Kinetic Applications of Electron Paramagnetic Resonance Spectroscopy. XXIV. Neophyl Rearrangements. J. Am. Chem. Soc 1976, 98 (5), 1224–1226. 10.1021/ja00421a028. [DOI] [Google Scholar]
- (22).Effio A; Griller D; Ingold KU; Scaiano JC; Sheng SJ Studies on the Spiro[2.5]Octadienyl Radical and the 2-Phenylethyl Rearrangement. J. Am. Chem. Soc 1980, 102 (19), 6063–6068. 10.1021/ja00539a015. [DOI] [Google Scholar]
- (23).Ingold KU; Warkentin J A Homopropargyl Radical Rearrangement. Kinetics of the Rearrangement of the 2,2,5,5,-Tetramethyl-3-Hexyn-1-Yl Radical. Can. J. Chem 1980, 58 (4), 348–352. 10.1139/v80-056. [DOI] [Google Scholar]
- (24).Chatgilialoglu C; Ingold KU; Tse-Sheepy I; Warkentin J A Homoallyl Radical Rearrangement. Kinetics of the Isomerization of the 2,2-Dimethyl-3-Buten-1-Yl Radical to the 1,1-Dimethyl-3-Buten-1-Yl Radical. Can. J. Chem 1983, 61 (6), 1077–1081. 10.1139/v83-190. [DOI] [Google Scholar]
- (25).Lindsay DA; Lusztyk J; Ingold KU Kinetics of the 1,2-Migration of Carbon-Centered Groups in 2-Substituted 2,2-Dimethylethyl Radicals. J. Am. Chem. Soc 1984, 106 (23), 7087–7093. 10.1021/ja00335a037. [DOI] [Google Scholar]
- (26).Ludwig R Hydrogen-Transfer Reactions.Edited by Hynes JT, Klinman JP, Limbach H-H and Schowen RL. ChemPhysChem 2007, 8 (17), 2539–2539. 10.1002/cphc.200700637. [DOI] [Google Scholar]
- (27).Yi H; Zhang G; Wang H; Huang Z; Wang J; Singh AK; Lei A Recent Advances in Radical C–H Activation/Radical Cross-Coupling. Chem. Rev 2017, 117 (13), 9016–9085. 10.1021/acs.chemrev.6b00620. [DOI] [PubMed] [Google Scholar]
- (28).Hu A; Guo J-J; Pan H; Zuo Z Selective Functionalization of Methane, Ethane, and Higher Alkanes by Cerium Photocatalysis. Science 2018, 361 (6403), 668–672. 10.1126/science.aat9750. [DOI] [PubMed] [Google Scholar]
- (29).Perry IB; Brewer TF; Sarver PJ; Schultz DM; DiRocco DA; MacMillan DWC Direct Arylation of Strong Aliphatic C–H Bonds. Nature 2018, 560 (7716), 70–75. 10.1038/s41586-018-0366-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Chu JCK; Rovis T Complementary Strategies for Directed C(Sp 3)−H Functionalization: A Comparison of Transition-Metal-Catalyzed Activation, Hydrogen Atom Transfer, and Carbene/Nitrene Transfer. Angew. Chem. Int. Ed 2018, 57 (1), 62–101. 10.1002/anie.201703743. [DOI] [Google Scholar]
- (31).Ohkubo K; Fujimoto A; Fukuzumi S Metal-Free Oxygenation of Cyclohexane with Oxygen Catalyzed by 9-Mesityl-10-Methylacridinium and Hydrogen Chloride under Visible Light Irradiation. Chem. Commun 2011, 47 (30), 8515–8517. 10.1039/C1CC12534F. [DOI] [Google Scholar]
- (32).Ohkubo K; Mizushima K; Fukuzumi S Oxygenation and Chlorination of Aromatic Hydrocarbons with Hydrochloric Acid Photosensitized by 9-Mesityl-10-Methylacridinium under Visible Light Irradiation. Res. Chem. Intermed 2013, 39 (1), 205–220. 10.1007/s11164-012-0643-5. [DOI] [Google Scholar]
- (33).Rohe S; Morris AO; McCallum T; Barriault L Hydrogen Atom Transfer Reactions via Photoredox Catalyzed Chlorine Atom Generation. Angew. Chem. Int. Ed 2018, 57 (48), 15664–15669. 10.1002/anie.201810187. [DOI] [Google Scholar]
- (34).Deng H-P; Zhou Q; Wu J Microtubing-Reactor-Assisted Aliphatic C−H Functionalization with HCl as a Hydrogen-Atom-Transfer Catalyst Precursor in Conjunction with an Organic Photoredox Catalyst. Angew. Chem. Int. Ed 2018, 57 (39), 12661–12665. 10.1002/anie.201804844. [DOI] [Google Scholar]
- (35).Newhouse T; Baran PS If C H Bonds Could Talk: Selective C H Bond Oxidation. Angew. Chem. Int. Ed 2011, 50 (15), 3362–3374. 10.1002/anie.201006368. [DOI] [Google Scholar]
- (36).Walling C Free Radicals in Solution; John Wiley & Sons, Inc: New York, NY, USA, 1957. [Google Scholar]
- (37).Zavitsas AA; Pinto JA Meaning of the Polar Effect in Hydrogen Abstractions by Free Radicals. Reactions of the Tert-Butoxy Radical. J. Am. Chem. Soc 1972, 94 (21), 7390–7396. 10.1021/ja00776a021. [DOI] [Google Scholar]
- (38).Roberts BP; Steel AJ An Extended Form of the Evans–Polanyi Equation: A Simple Empirical Relationship for the Prediction of Activation Energies for Hydrogen-Atom Transfer Reactions. J. Chem. Soc. Perkin Trans. 2 1994, No. 10, 2155–2162. 10.1039/P29940002155. [DOI] [Google Scholar]
- (39).Zavitsas A, Factors A Controlling Reactivity in Hydrogen Abstractions by Free Radicals. J. Chem. Soc. Perkin Trans. 2 1996, 0 (3), 391–393. 10.1039/P29960000391. [DOI] [Google Scholar]
- (40).Roberts BP Understanding the Rates of Hydrogen-Atom Abstraction Reactions: Empirical, Semi-Empirical and Ab Initio Approaches. J. Chem. Soc. Perkin Trans. 2 1996, No. 12, 2719–2725. 10.1039/P29960002719. [DOI] [Google Scholar]
- (41).Roberts BP Polarity-Reversal Catalysis of Hydrogen-Atom Abstraction Reactions: Concepts and Applications in Organic Chemistry. Chem. Soc. Rev 1999, 28 (1), 25–35. 10.1039/A804291H. [DOI] [Google Scholar]
- (42).Hudzik JM; Bozzelli JW Thermochemistry and Bond Dissociation Energies of Ketones. J. Phys. Chem. A 2012, 116 (23), 5707–5722. 10.1021/jp302830c. [DOI] [PubMed] [Google Scholar]
- (43).Treacy SM; Rovis T Copper Catalyzed C(Sp3)–H Bond Alkylation via Photoinduced Ligand to Metal Charge Transfer. J. Am. Chem. Soc 2021, 143. [Google Scholar]
- (44).Kochi JK Photolyses of Metal Compounds: Cupric Chloride in Organic Media. J. Am. Chem. Soc 1962, 84 (11), 2121–2127. 10.1021/ja00870a025. [DOI] [Google Scholar]
- (45).Shulpin GB; Kats MM Ferric Chloride Catalyzed Photooxidation of Alkanes by Air in Organic Solvents. React. Kinet. Catal. Lett 1990, 41 (2), 239–243. 10.1007/BF02097875. [DOI] [Google Scholar]
- (46).Shul’pin GB; Kats MM Photochemical Oxidation of Saturated and Alkylaromatic Hydrocarbons by Atmospheric Oxygen in CH3CN or CH2Cl2 Solution, Catalyzed by Iron(III) Halides. Pet. Chem 1991, 31 (5), 647–656. [Google Scholar]
- (47).Shul’pin GB; Nizova GV; Kozlov YN Photochemical Aerobic Oxidation of Alkanes Promoted by Iron Complexes. New J Chem 1996, 20, 1243–1256. [Google Scholar]
- (48).Takaki K; Yamamoto J; Matsushita Y; Morii H; Shishido T; Takehira K Oxidation of Alkanes with Dioxygen Induced by Visible Light and Cu(II) and Fe(III) Chlorides. Bull. Chem. Soc. Jpn 2003, 76 (2), 393–398. 10.1246/bcsj.76.393. [DOI] [Google Scholar]
- (49).Takaki K; Yamamoto J; Komeyama K; Kawabata T; Takehira K Photocatalytic Oxidation of Alkanes with Dioxygen by Visible Light and Copper(II) and Iron(III) Chlorides: Preference Oxidation of Alkanes over Alcohols and Ketones. Bull. Chem. Soc. Jpn 2004, 77 (12), 2251–2255. 10.1246/bcsj.77.2251. [DOI] [Google Scholar]
- (50).Yaroshevsky AA Abundances of Chemical Elements in the Earth’s Crust. Geochem. Int 2006, 44 (1), 48–55. 10.1134/S001670290601006X. [DOI] [Google Scholar]
- (51).Chirik P; Morris R Getting Down to Earth: The Renaissance of Catalysis with Abundant Metals. Acc. Chem. Res 2015, 48 (9), 2495–2495. 10.1021/acs.accounts.5b00385. [DOI] [PubMed] [Google Scholar]
- (52).Swanson TB; Laurie VW Electron Magnetic Resonance and Electronic Spectra of Tetrachloroferrate(III) Ion in Nonaqueous Solution1. J. Phys. Chem 1965, 69 (1), 244–250. 10.1021/j100885a036. [DOI] [Google Scholar]
- (53).Jung ME; Piizzi G Gem-Disubstituent Effect: Theoretical Basis and Synthetic Applications. Chem. Rev 2005, 105 (5), 1735–1766. 10.1021/cr940337h. [DOI] [PubMed] [Google Scholar]
- (54).Beesley RM; Ingold CK; Thorpe JF CXIX.—The Formation and Stability of Spiro-Compounds. Part I. Spiro-Compounds from Cyclohexane. J. Chem. Soc. Trans 1915, 107 (0), 1080–1106. 10.1039/CT9150701080. [DOI] [Google Scholar]
- (55).Bruice TC; Pandit UK The Effect of Geminal Substitution Ring Size and Rotamer Distribution on the Intramolecular Nucleophilic Catalysis of the Hydrolysis of Monophenyl Esters of Dibasic Acids and the Solvolysis of the Intermediate Anhydrides. J. Am. Chem. Soc 1960, 82 (22), 5858–5865. 10.1021/ja01507a023. [DOI] [Google Scholar]
- (56).Ringer AL; Magers DH Conventional Strain Energy in Dimethyl-Substituted Cyclobutane and the Gem-Dimethyl Effect. J. Org. Chem 2007, 72 (7), 2533–2537. 10.1021/jo0624647. [DOI] [PubMed] [Google Scholar]
- (57).Bachrach SM The Gem-Dimethyl Effect Revisited. J. Org. Chem 2008, 73 (6), 2466–2468. 10.1021/jo702665r. [DOI] [PubMed] [Google Scholar]
- (58).Weber M; Fischer H Absolute Rate Constants for the β-Scission and Hydrogen Abstraction Reactions of the Tert-Butoxyl Radical and for Several Radical Rearrangements: Evaluating Delayed Radical Formations by Time-Resolved Electron Spin Resonance. J. Am. Chem. Soc 1999, 121 (32), 7381–7388. 10.1021/ja990837y. [DOI] [Google Scholar]
- (59).Mayr’s Database Of Reactivity Parameters - Start page https://www.cup.lmu.de/oc/mayr/reaktionsdatenbank/(accessed Jan 27, 2021).
- (60).Allgäuer DS; Mayr H Electrophilicities of 1,2-Disubstituted Ethylenes. Eur. J. Org. Chem 2014, 2014 (14), 2956–2963. 10.1002/ejoc.201301779. [DOI] [Google Scholar]
- (61).Lemek T; Mayr H Electrophilicity Parameters for Benzylidenemalononitriles. J. Org. Chem 2003, 68 (18), 6880–6886. 10.1021/jo0344182. [DOI] [PubMed] [Google Scholar]
- (62).Seeman JI Effect of Conformational Change on Reactivity in Organic Chemistry. Evaluations, Applications, and Extensions of Curtin-Hammett Winstein-Holness Kinetics. Chem. Rev 1983, 83 (2), 83–134. 10.1021/cr00054a001. [DOI] [Google Scholar]
- (63).Seeman JI The Curtin-Hammett Principle and the Winstein-Holness Equation: New Definition and Recent Extensions to Classical Concepts. J. Chem. Educ 1986, 63 (1), 42–48. 10.1021/ed063p42. [DOI] [Google Scholar]
- (64).Beckwith ALJ; Poole JS Factors Affecting the Rates of Addition of Free Radicals to AlkenesDetermination of Absolute Rate Coefficients Using the Persistent Aminoxyl Method. J. Am. Chem. Soc 2002, 124 (32), 9489–9497. 10.1021/ja025730g. [DOI] [PubMed] [Google Scholar]
- (65).Newcomb M; Glenn AG; Williams WG Rate Constants and Arrhenius Functions for Rearrangements of the 2,2-Dimethyl-3-Butenyl and (2,2-Dimethylcyclopropyl)Methyl Radicals. J. Org. Chem 1989, 54 (11), 2675–2681. 10.1021/jo00272a042. [DOI] [Google Scholar]
- (66).Fischer H; Radom L Factors Controlling the Addition of Carbon-Centered Radicals to Alkenes—An Experimental and Theoretical Perspective. Angew. Chem. Int. Ed 2001, 40 (8), 1340–1371. . [DOI] [Google Scholar]
- (67).Zytowski T; Fischer H Absolute Rate Constants for the Addition of Methyl Radicals to Alkenes in Solution: New Evidence for Polar Interactions. J. Am. Chem. Soc 1996, 118 (2), 437–439. 10.1021/ja953085q. [DOI] [Google Scholar]
- (68).Zytowski T; Fischer H Absolute Rate Constants and Arrhenius Parameters for the Addition of the Methyl Radical to Unsaturated Compounds: The Methyl Affinities Revisited. J. Am. Chem. Soc 1997, 119 (52), 12869–12878. 10.1021/ja973128y. [DOI] [Google Scholar]
- (69).Gilbert BC; Smith JRL; Milne EC; Whitwood AC; Taylor P Kinetic–EPR Studies of the Addition of Aliphatic Radicals to Acrylic Acid and Related Alkenes: The Interplay of Steric and Electronic Factors. J. Chem. Soc. Perkin Trans. 2 1993, No. 11, 2025–2031. 10.1039/P29930002025. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.